Submitted:
03 October 2023
Posted:
04 October 2023
You are already at the latest version
Abstract
A single geological carbon dioxide (CO2) storage within salinity aquifer as a paramount stratagem in the context of forthcoming CO2 sequestration initiatives. Within the expansive tapestry of terrestrial salinity aquifers in China, replete with nuances in mineral composition and structural diversity, a paradigm distinct from the marine sedimentary basins of North America, a labyrinthine stratigraphic environment of elevated complexity unfolds. Consequently, conspicuous lacunae and insufficiencies manifest within the existing knowledge reservoir pertaining to CO2-water-rock (mineral) reaction mechanisms. Notably, a conspicuous dearth of insights prevails concerning the dissolution kinetics inherent to individual minerals embroiled in these reactions. The present investigation undertakes a scrupulous exploration into the dissolution and erosive processes manifest within four emblematic terrestrial minerals in conjunction with CO2. Furthermore, the study meticulously scrutinizes the evolution of ionic composition and pH within the formation water throughout the trajectory of the geochemical reactions. To accomplish this, a simulation apparatus is configured to emulate conditions germane to temperature, pressure, and mineralization characteristics characteristic of a prototypical salinity aquifer reminiscent of the Daqing oilfield in China. The findings that emerge from this scrutiny reveal that the erosive action of CO2 fluid begets varying degrees of dissolution within the four minerals under investigation. Moreover, our meticulous inquiry lays bare that, in the terminal phases of the reaction process, feldspar precipitates secondary minerals, characterized primarily by dawsonite, in response to the aquatic milieu typifying the profound salinity stratum of terrestrial sedimentation. This milieu is notably enriched with Na+ ions and NaHCO3. This phenomenon suggests that feldspar, under these specific conditions, assumes the mantle of a mineral amenable to carbon sequestration, thus enriching the reservoir for CO2 mineralization within the salinity stratum of terrestrial sedimentation.
Keywords:
salinity aquifer
; CO2-water-rock (mineral) reaction
; CO2 storage
; dawsonite
1. Introduction
The World Meteorological Organization, in conjunction with other esteemed agencies, has formally declared the month of July in the year 2023 as an epoch-making moment, signifying an unprecedented zenith in global mean temperatures since the inception of meteorological data archives. Indeed, it portends a climatic extremity, possibly rendering it the warmest month witnessed in the annals of Earth's climatic history over the expanse of a staggering 120,000 years. Seasoned experts within the field assert that within the context of our burgeoning global warming milieu, the manifestation of these record-breaking temperature elevations should be regarded as a foreseeable consequence. These stark developments further underscore the inescapable veracity of a shifting climate regime, typified by the onset of increasingly severe weather phenomena. It is, therefore, imperative that a concerted and exigent effort is mounted for the abatement of greenhouse gas emissions.
Since the historic moment of ratification, encapsulated within the Paris Agreement, during the 21st United Nations Climate Change Conference in 2015, nations across the globe have vigilantly and fervently embarked upon the crafting of robust strategies to usher in the era of CO2 emission peak and ultimate neutrality [1,2,3]. Amidst the myriad of avenues available to expedite the trajectory toward carbon neutrality, the sphere of negative emission technologies looms large, standing as a pivotal and indispensable component, vital for the realization of carbon neutrality, particularly within the ambit of profound decarbonization [4,5,6,7,8,9].
In this landscape, Carbon Dioxide Capture, Utilization, and Storage (CCUS) emerges as a preeminent archetype, endowed with profound potential. This technology bears the capacity to amass an appreciable share, approximately 15%, of the overarching imperative of total emission reduction. CCUS, in essence, serves as a cornerstone, playing a decisive role in underpinning the pursuit of carbon neutrality targets. Within the gamut of geological CO2 storage typologies, the deep salinity aquifer repository beckons as the archetype replete with unrivaled potential. It has been postulated by eminent scholars that the deep salinity aquifers resident within the principal sedimentary basins of China possess an awe-inspiring CO2 storage potential, estimated at a prodigious 119.2 billion metric tons [10,11,12]. This estimation has been made through the meticulous and highly precise CSLF calculation methodology.
The methodology underpinning salinity aquifer sequestration encompasses a panoply of stratagems, including tectonic sequestration, residual gas sequestration, dissolution sequestration, and mineralization sequestration. Mineralization storage, a salient facet of this paradigm, entails the dissolution of CO2 within formation water, thus inducing a consequential reduction in its pH. This catalyzes an intricate sequence of water-rock geochemical reactions, precipitating the dissolution of select minerals and concomitant secondary mineral precipitation, chiefly comprising carbonate minerals. Within this intricate matrix, an important facet unfolds wherein a fraction of metal cations, such as Ca2+ and Mg2+, enter into reactions with carbonates, culminating in precipitation and thereby effectuating the enduring sequestration of CO2. Significantly, mineralization storage epitomizes the quintessential storage modality, representing the culminating and definitive transformational outcome of the initial three storage mechanisms. However, it should be noted that the temporal cycle inherent to mineralization storage is protracted, spanning timeframes ranging from centuries to millennia [13,14,15].
Moreover, it is imperative to acknowledge the propensity for CO2 injected into subterranean strata to engage in reactions with subsurface rock minerals, thereby engendering the potential for leakage and other untoward incidents, particularly within reservoirs exhibiting suboptimal capping integrity. This predicament inherently impinges upon the assured safety of long-term storage ventures [16,17]. Thus, the imperative emerges for a comprehensive and penetrating exploration of the intricate geochemical reaction mechanisms governing the interplay between CO2 and formation minerals. Such a scientific endeavor assumes pivotal import, as it underpins the accurate prognostication of sequestration volumes and constitutes the bedrock for the evaluation of long-term storage safety.
The pursuit of Carbon Capture and Storage/Carbon Capture Utilization and Storage (CCS/CCUS) has yielded significant research contributions across North America and various international domains, particularly with respect to comprehending the mechanisms underlying CO2 mineralization and storage within diverse geological reservoirs such as oil reservoirs, oil-water transition zones, and deep salinity aquifers. Ueda et al. [18] conducted meticulous investigations into the intricacies of CO2-water-rock (mineral) interactions within rock formations via controlled laboratory experiments. Their work, which delved into the effects of mineral composition, pressure, and temperature, corroborated the feasibility and substantial potential inherent in CO2 mineralization and subsequent storage. Ketzer et al. [19], in their endeavors within the Rio Bonito Formation of Brazil, probed the CO2-water-rock (mineral) interplay during CO2 sequestration. Their findings unveiled the dissolution of calcite cement within sandstone, accompanied by the observation of calcite and iron calcite precipitation. Farquhar et al. [20] completed an exhaustive 16-day indoor simulation experiment, conducted to fathom the geochemical dynamics of sandstone reservoirs located in Queen's Island, Australia, subjected these formations to CO2 dissolution. This investigation discerned the dissolution of various sandstone minerals, encompassing carbonate minerals, chlorite, and a minor fraction of feldspars. Li et al. [21], employing modeling techniques involving PHREEQC calculations, embarked upon an inquiry into the interaction dynamics between the liquid phase and rock strata. Their analyses illuminated a nuanced relationship wherein the dissolution phenomena underwent alterations commensurate with burial depth. Balashov et al. [22] leveraging the MK76 code, engineered a comprehensive simulation framework aimed at elucidating the dissolution and precipitation phenomena afflicting sandstone minerals over protracted temporal horizons. Their research unveiled calcite as the preeminent carbon-fixing mineral and underscored the influence exerted by initial mineral dissolution upon the precipitation of secondary minerals. These pioneering investigations collectively contribute to a deeper comprehension of the intricate realm of CO2 mineralization and storage within geological reservoirs, further accentuating the salience of these processes within the purview of carbon management strategies.
However, in general, research outcomes have predominantly centered on the characteristics of marine sedimentary basins. Yet, for the widely distributed terrestrial sedimentary salinity aquifer basins in China, substantial disparities arise due to differences in mineral composition, structural maturity, and related factors in comparison to marine sedimentary basins. Additionally, the stratigraphic environment in these terrestrial basins is notably more complex. Consequently, there exist significant gaps and deficiencies in our understanding of CO2-water-rock (minerals) reaction mechanisms [23,24,25,26] within these contexts, necessitating further in-depth investigations.
In the realm of experimental investigations into CO2-water-rock interactions, emphasis is commonly placed on indoor experiments conducted under elevated temperature and pressure conditions. These experiments are often facilitated using high-temperature, high-pressure reactors or core replacement methods. The primary distinction between these approaches lies in the presence or absence of fluid transport within the experimental setup. While these experiments have significantly contributed to our understanding of CO2-water-rock interactions, they do have inherent limitations. One notable limitation is the substitution of pure water for authentic formation water in the experimental design, leading to deviations in reaction outcomes compared to those encountered in real geological formations. Additionally, the intricate mineral composition of the selected geological cores poses challenges in precisely studying the dissolution mechanisms of individual minerals involved in the reactions. Notably, significant disparities in mineral composition can result in more pronounced variations in reaction outcomes [27,28,29].
To comprehensively analyze the complex mechanisms involved in CO2-water-rock (mineral) reactions within terrestrial deep salinity aquifers, this study employs the Daqing oilfield in China as a representative example. The research involves core sampling to investigate the mineral composition of salinity aquifer rocks, including sandstone minerals and clay minerals, within three distinct blocks. Four specific minerals—potassium feldspar, plagioclase feldspar, calcite, and kaolinite—have been selected for detailed examination due to their significance within the geological context. These minerals are subjected to simulated conditions of temperature, pressure, and mineralization typical of the salinity aquifer in the Daqing oilfield. Under these controlled conditions, the study explores the dissolution and erosion processes affecting these minerals, as well as the resulting changes in ionic composition and pH of the formation water throughout the geochemical reaction process.
2. Experiments
2.1. Experimental Sample
Table 1 and Table 2 present the mineral composition data for sandstone minerals and clay minerals in three distinct blocks of the Daqing Oilfield, respectively. A cursory examination of the mineral content data reveals that quartz predominates in abundance. However, it is noteworthy that quartz exhibits remarkable chemical stability, even when subjected to the temperature and pressure conditions encountered within salinity aquifers. Consequently, in the short-term indoor experiments, discerning significant dissolution phenomena in quartz proves challenging. Therefore, the experimental focus narrows to three primary minerals: potassium feldspar, plagioclase feldspar, and calcite. Among these, potassium feldspar and plagioclase feldspar assume prominence due to their substantial content within the feldspar mineral category. These minerals are particularly susceptible to dissolution and erosion when subjected to CO2 injection conditions. On the other hand, calcite, a quintessential carbonate mineral, while relatively less abundant in terrestrial sedimentary strata (comprising less than 10% of the mineral content), exhibits a comparatively higher dissolution rate than feldspar minerals. This property renders calcite influential in the context of CO2 mineralization and storage. Kaolinite, a clay mineral, emerges as an additional subject of interest, as it is an outcome of feldspar dissolution. The intensity of the dissolution reaction involving kaolinite holds significance, as it directly impacts the stratum's stability over protracted durations under CO2 injection conditions.
It is worth noting that acquiring high-purity feldspar minerals and clay minerals under natural conditions proves challenging. Consequently, the mineral groups utilized in the experiments unavoidably encompass trace impurities. However, the precision of the experiments is diligently maintained through meticulous analysis, including monitoring changes in water ion composition during the reaction process. The experimental fluid employed in the study was meticulously prepared in the laboratory to align with the composition of formation water in a designated block within Aonan. This fluid exhibited a mineralization level of 11636.3 mg/L. For reference, Table 3 presents a comprehensive breakdown of the ionic composition of the formation water and pertinent details concerning the prepared reagents utilized in the experiments.
2.2. Experimental Setups
The experimental device consists of a stainless steel high-temperature and high-pressure reactor, an ISCO constant-speed piston pump, a thermostat box, a return valve, etc. The experimental system device is shown in Figure 1. In the experiment, CO2 was injected into the reactor through the ISCO piston pump to simulate the stratum pressure, and the thermostat box controlled the temperature of the whole reaction system to be the simulation of the stratum temperature all the time. The experimental parameters simulate the temperature and pressure conditions of formation water and ionic composition in a block of Aonan. The partial pressure of CO2 in the reactor is 16MPa; the temperature of the thermostat is 60℃.
2.3. Experimental Process
Prior to the commencement of the reaction, it is necessary to crush and mill the minerals. This process enables the minerals to undergo a more pronounced dissolution reaction within a shorter timeframe. Pre and post-reaction, samples of the minerals were obtained for comparative analysis through compositional analysis, surface morphology examination, and specific surface area testing. The mineral composition analysis employed X-ray diffraction (XRD), while the mineral surface morphology analysis utilized scanning electron microscopy (SEM). The specific surface area of the minerals was determined using a specific surface area adsorption instrument. The mass of the four groups of reacted minerals equaled 20g.
To prevent clogging the pipeline and affecting fluid sampling, a layer of stainless steel mesh was placed at the bottom of the reactor. Mineral powder was added, followed by injecting 400mL of prepared formation water into the kettle. CO2 was then injected into the kettle using an ISCO piston pump until the pressure reached 16 MPa. Once the preset pressure was reached, the valve was closed, and the thermostat temperature was set to 60℃ to initiate the reaction. Over the course of 15 days, liquid samples were taken at five different time points: 1,3, 6, 10, and 15 days after the start of the reaction. The sampling interval gradually increased, in accordance with the decreasing rate of mineral dissolution. After the five sampling events, the pH and ionic concentration were measured along with the initial preparation of formation water. Following the reaction, the mineral powder was removed, rinsed with deionized water, and then dried in an oven at 60℃ for 24 hours. Subsequently, mineral composition, surface morphology analysis, and specific surface area measurement were conducted.
Eqs (1) to (4) represent the primary reaction equations governing the dissolution of potassium feldspar, sodium feldspar, calcite, and kaolinite, respectively.
In the actual stratigraphic environment, various factors such as the complex compo sition of reservoir minerals, differences in temperature, pressure, and other environmental parameters contribute to the generation of different products from similar minerals. For instance, sodium feldspar can generate different secondary minerals like kaolinite and sodalite (NaAlCO3(OH)2) through dissolution, depending on the ionic composition of formation water, as well as the temperature and pressure conditions. Consequently, the reaction formula for these minerals is not unique. To understand the mechanism of mineral erosion, it is necessary to consider the reaction environment and the time span involved. Furthermore, in the mineralization sequestration process, which occurs over an extended period, the mechanism of action varies at different stages.
3. Results and Discussion
3.1. pH Change of the Reaction System
Under the experimental conditions, CO2 is in a supercritical state, making it challenging to determine pH in situ due to high temperature, pressure, and a closed system. To overcome this, we chose to analyze liquid samples taken after CO2 dissolution equilibrium to determine pH. Additionally, we utilized the PR-HV equation of state [30] to estimate the CO2 solubility under the reaction conditions. This approach helps minimize experimental error by allowing for comparative analysis.
The Peng-Robinson equation of state is a useful tool for calculating CO2 solubility under varying temperatures, pressures, and mineralization of formation water. The equation is as follows:
In the given equation, represents the pressure of the reaction system in MPa. denotes the ideal gas constant, while signifies the temperature of the reaction system in Kelvin. represents the molar volume in cm3/mol. The variables and correspond to the gravitational and repulsive constants of the mixing system, respectively. The variable represents the number of moles of the total components in the mixing system. denotes the mole fraction of component i in the liquid phase. The parameters and represent the equation of state for the i-component of the pure substance. Additionally, is a temperature-dependent function. represents the excess Gibbs free energy as the system's pressure approaches infinity. and denote the critical temperature and pressure of component i, respectively. is defined as the ratio of to . Lastly, is a function of the eccentricity factor , which is given by: 0.37646+1.5426-0.26992;。
Many researchers have discovered that the Peng-Robinson calculation model is not entirely suitable for strong polar material water conditions in numerous experiments and practical applications. As a result, a new PR-HV prediction model was subsequently proposed based on it, in conjunction with the Huron-Vidal fugacity coefficient model.
In the given equation, represents the mixture fugacity coefficient, denotes the mixture deviation factor, signifies the mixture molar volume in cubic meters per mole, and represents the activity coefficient of the component.
Utilizing the PR-HV model and the provided conditions, the fugacity coefficient of the gas phase can be determined within the mixed system. Subsequently, the fugacity coefficient of the liquid phase and the H2CO3 content can be iterated to calculate the solubility of CO2. It is important to note that this paper's CO2 solubility calculation model assumes that all mineralization in the formation water is due to NaCl. The parameters used in the model are presented in Table 4.
Based on the given conditions, the solubility of CO2 in water was calculated to be 1.26 mol/kg when CO2 dissolution reached equilibrium in the experimental simulation of stratigraphic conditions. Since carbonic acid is a weak acid, its ionization determines the concentration of H+ in the solution. This concentration can be obtained using the ionization equilibrium constant and the solubility.
The H+ concentration was calculated to be 10-3.8 mol/L, resulting in a pH of 3.8. However, during the experiment, the measured pH of the solution after dissolution equilibrium was found to be 4.1, slightly higher than the calculated value. The author attributes this discrepancy to two main factors. Firstly, the experiment used NaHCO3 water, which contains that inhibits the ionization of H2CO3 to some extent. Additionally, the rapid decompression of the solution during the test caused some CO2 to escape, leading to a slightly higher pH.
By comparing and analyzing the two methods, it is evident that the water type of the stratum water has an impact on the dissolution of CO2 during the process of CO2 salinity aquifer sequestration. Additionally, the presence of NaHCO3 in the water has a negative effect on the dissolution and potential mineralization of CO2.
The pH changes of the four reaction systems are depicted in Figure 2. The curves, from highest to lowest, correspond to the calcite group, plagioclase group, potassium feldspar group, and kaolinite group, respectively. The pH of the calcite group remains stable between 5.0 and 5.2 during the first 1 to 15 days after the reaction, suggesting a relatively quick attainment of dissolution-precipitation equilibrium. On the other hand, both the plagioclase feldspar group and the potassium feldspar group exhibit an increasing pH trend throughout the reaction process, with values lower than those of the calcite group. This implies that feldspar minerals have a slower reaction rate compared to carbonate minerals, requiring more time to reach equilibrium. Furthermore, the pH of the plagioclase feldspar group surpasses that of the kaolinite group, which in turn exceeds the pH of the potassium feldspar group. The higher pH of the plagioclase group indicates a faster reaction rate and a more intense dissolution process. Conversely, the kaolinite group displays the lowest pH, indicating its ability to remain stable in weakly acidic environments. This finding holds significant implications for studying the stability of strata under long-term CO2 injection conditions, as kaolinite is the primary product of dissolved and eroded feldspar minerals. Additionally, it aids in determining the chemical properties of kaolinite under weakly acidic conditions.
When comparing the pH changes during the reaction process of various mineral groups, it becomes apparent that there is a significant difference in the sensitivity of different types of minerals to CO2 fluids. The sensitivity rankings, from highest to lowest, are carbonate minerals, feldspar minerals, and clay minerals. In China, terrestrial sedimentary basins are widely distributed, with the formation minerals primarily consisting of feldspar and clay minerals. The content of carbonate minerals is relatively low in these basins. As a result, the interaction between CO2, water, and rock (minerals) is a lengthy and intricate process, leading to slow changes in the pH of the formation water.
3.2. Changes in Cation Mass Concentractions
The relationship between the mass concentration of major cations and time during the reaction process of each mineral group is illustrated in Figure 3. Specifically, Figure 3(a) depicts the concentration change of major cations during the dissolution process of potassium feldspar group minerals. The concentration of K+ gradually increases during the reaction, albeit at a slow rate, and does not exceed 5 ppm. This indicates that potassium feldspar is undergoing dissolution, but the rate of dissolution is slow. The concentration of Na+ initially increases and then slightly decreases as the reaction progresses. This can be attributed to the presence of plagioclase feldspar in the mineral group, where the increase in Na+ concentration may result from the dissolution of plagioclase feldspar. The slight decrease in concentration towards the end of the reaction suggests that the minerals are approaching equilibrium, leading to a decrease in the rate of dissolution. The concentration of Ca2+ and Mg2+ exhibits a small decrease overall, indicating their involvement in carbonate minerals such as CaCO3, MgCO3, CaMg(CO3)2, etc. Additionally, XRD analysis conducted before and after the reaction of the minerals (refer to section 2.4 for more details) confirms their participation in the transformation process from illite to ilmenite.
Figure 3(b) illustrates the changes in the concentration of main cations during the dissolution process of the plagioclase feldspar group. The Na+ concentration in this group is significantly higher compared to the other groups, primarily due to the dissolution of sodium feldspar. The overall Na+ concentration exhibits an initial rapid increase followed by a gradual decline. This trend can be attributed to several factors. Firstly, the sodium feldspar gradually reaches a saturated state, leading to a decrease in the reaction rate. Additionally, there may be natrolithiation of calcium feldspar or the precipitation of Na-containing minerals. Conversely, the dissolution of sodium feldspar may also result in the precipitation of Na-containing minerals. The Ca2+ concentration is noticeably higher than that of the potassium feldspar group, primarily due to the dissolution of calcium feldspar. In the first fifteen days after the reaction, the Ca2+ concentration shows no significant trend and exhibits small fluctuations. This suggests that calcium feldspar has a higher dissolution rate than sodium feldspar and reaches the saturation state more quickly. The concentration of Mg2+ shows no obvious trend and remains largely unaffected by any reaction.
Figure 3(c) illustrates the variations in the concentration of major cations during the dissolution of minerals belonging to the kaolinite group. It indicates that there are minimal changes in the concentration of major cations, suggesting a weak reaction and chemical stability of kaolinite in a weakly acidic stratigraphic environment. Specifically, the concentration of Na+ slightly decreased in the later stages of the reaction, although to a lesser extent compared to the first two groups of feldspar minerals. Additionally, the concentrations of Ca2+ and Mg2+ initially decreased, then increased, and finally stabilized in the later stages of the reaction. Kaolinite, being the primary product of feldspar minerals undergoing dissolution, is formed in an acidic environment with low cation concentration. The main factor contributing to its chemical stability in acidic conditions is the presence of hydrogen bonding as an intermolecular force. Furthermore, the edges of its crystals serve as the primary areas of charge distribution, and the cation exchange effect is weak.
Figure 3(d) illustrates the variations in the concentration of major cations during the dissolution process of minerals in the calcite group. The concentration of Ca2+ is notably higher compared to other groups, averaging around 400 ppm. There is a slight change in concentration within 1 to 15 days after the reaction, followed by a gradual decrease in the later stages. This indicates that calcite achieves dissolution equilibrium relatively quickly compared to feldspar minerals and clay minerals. The equilibrium time for calcite is an order of magnitude shorter than that of the other two minerals. The slight decrease in concentration in the later stages can be attributed to the re-precipitation effect of calcite. On the other hand, the concentration of Na+ and Mg2+ shows minimal variation and has a negligible impact on the dissolution of calcite.
When comparing the changes in mass concentration of major cations during the dissolution of various mineral groups, it is evident that the time to reach equilibrium varies significantly among different classes of minerals. Carbonate minerals exhibit the highest reaction rate, while feldspar minerals and clay minerals have lower rates. Based on data analysis, it is apparent that the dissolution of these two mineral types takes several years to reach equilibrium, indicating a slow dissolution process. By comparing Figure 3(a) and Figure 3(b), it can be observed that plagioclase feldspar reacts at a higher rate than potassium feldspar, resulting in a shorter time required to reach saturation.
3.3. Changes in Cation Mass Concentractions
The scanning electron microscopy images in Figure 4 reveal the variations in surface dissolution among different mineral groups before and after the reaction. Specifically, Figure 4 (a) and Figure 4(b) compare the surface images of the potassium feldspar group before and after the reaction. Prior to the reaction, the mineral surface of the potassium feldspar group appeared relatively smooth and flat, with only a small portion showing depression. However, after the reaction, the mineral surface became rougher, exhibiting more pronounced dissolution steps and unevenness. This indicates that the minerals underwent partial dissolution. Additionally, the reaction led to an increase in the specific surface area of the minerals, further confirming their partial dissolution.
Figure 4(c) and Figure 4(d) depict a comparison of scanning electron microscope images illustrating the surface of plagioclase group minerals before and after a reaction. It is worth noting that, prior to the experiments discussed in this paper, the plagioclase feldspar had large particles, necessitating some grinding. As a result, the scanning electron microscope image of the plagioclase group minerals before the reaction exhibited poor homogeneity and numerous small crystalline grains on the surface. However, the scanning electron microscope image after the reaction reveals a more pronounced dissolution of the plagioclase group. Almost all of the small crystal grains present on the surface before the reaction have dissolved, leaving behind a fish scale-like surface with attached secondary mineral precipitates. This observation, combined with the analysis of the change in Na+ concentration during the reaction and the XRD analysis (for more details, refer to section 2.4), indicates the formation of secondary sodium alumina (NaAlCO3(OH)2) precipitates on the surface.
Figure 4(e) and Figure 4(f) depict a comparison of scanning electron microscope images of the surface of kaolinite group minerals before and after the reaction. Overall, the surface morphology of kaolinite remains relatively unchanged, indicating a weak dissolution reaction. However, there is a slight degree of dissolution observed at the crystal's edges, resembling petals. The majority of the crystal's middle section remains unaltered. The weak etching effect on kaolinite is primarily attributed to its molecular structure. In the presence of a high concentration of H+ ions, Al-OH readily combines with H+ ions, resulting in a positive charge. This charge is evenly distributed in the aluminum-oxygen octahedron, enhancing the stability of the silica-oxygen tetrahedron compared to the aluminum-oxygen octahedron. The chemical stability of kaolinite in an acidic environment suggests that various clay minerals may convert to kaolinite under the influence of CO2 dissolution.
Figure 4(g) and Figure 4(h) illustrate a comparison of scanning electron microscope images of the surface of calcite group minerals before and after the reaction. The dissolution effect on calcite is more pronounced, as evidenced by the transformation of intact large grains into fine microcrystals or complete dissolution after the reaction. The surface of calcite becomes rough, exhibiting dissolution steps. Analysis of the change in Ca2+ concentration during the reaction process reveals that most of the Ca2+ in the dissolving grains exists in a free state within the solution. A small portion of Ca2+ participates in reprecipitation and reconverts into CaCO3, which attaches to the mineral surface.
Comparing the scanning electron microscope images of the surface corrosion changes before and after the reaction for each mineral group, it is evident that the corrosion intensity varies significantly among different mineral types. Feldspar and calcite minerals exhibit more noticeable corrosion, while kaolinite shows minimal corrosion. Additionally, the surfaces of all mineral groups become rougher after the reaction. The determination of the specific surface area of the minerals before and after the reaction corroborates these observations, indicating a gradual increase in specific surface area with the mineral reaction. This increase in surface area provides favorable conditions for further mineral corrosion by CO2.
3.4. Changes in Cation Mass Concentractions
The XRD diffraction analysis was conducted to examine the changes in mineral composition before and after the reaction in each group. The results revealed that the feldspar minerals experienced significant alterations in mineral composition, while the changes in kaolinite and calcite were relatively minor. Specifically, Figure 5(a) illustrates the XRD diffraction characteristics of potassium feldspar group minerals before and after the reaction. The spectrum changes, combined with quantitative analysis, indicated a decrease in potassium feldspar content and an increase in quartz content. This suggests that the dissolution of potassium feldspar resulted in the formation of secondary quartz. Additionally, there was a slight increase in clay minerals. The quantitative analysis of clay minerals revealed a small amount of illite in the potassium feldspar group before the reaction, which decreased by 43% after the reaction and transformed into an illite-illite mixed layer with a mixed layer ratio of 45%.
Figure 5(b) displayed the XRD diffraction characteristics of plagioclase group minerals before and after the reaction. A new peak emerged in the diffraction pattern after the reaction, identified as sodium alumina (NaAlCO3(OH)2) through comparison with the standard card of mineral diffraction. This discovery demonstrated that plagioclase feldspar can generate secondary carbonate minerals when exposed to CO2 fluid, thus providing a new direction for CO2 mineralization and storage. Regarding the final form of carbon sequestration in the brackish water layer, most scholars believe that CO32- combines with divalent metal cations to produce precipitates such as CaCO3 and MgCO3. However, the concentration of divalent metal cations in the brackish water layer is not very high, which limits the efficiency of this process. Sodalite, a secondary carbonate mineral, contains Na+ and Al3+ as its metal cations. The dissolution of feldspar minerals, particularly sodium feldspar, and the high Na+ concentration in the stratigraphic fluids of the brackish water layer can provide ample Na+ and Al3+. Therefore, sodalite holds great promise as a carbon sequestration mineral. The reaction equation for the production of sodalumite through the CO2 dissolution of sodium feldspar is as follows:
The generation of sodalite requires high temperature and high CO2 partial pressure, a weak acidic fluid environment rich in Na+ and Al3+, and the injection of supercritical CO2 into the feldspar-rich brackish water layer. These conditions facilitate the mineralization and sequestration of CO2.
Figure 5(c) depicted the XRD diffraction characteristics of kaolinite group minerals before and after the reaction. No significant changes were observed after the reaction. Quantitative analysis revealed a 0.5% dissolution of kaolinite and a corresponding 0.5% increase in quartz content, indicating that kaolinite dissolves to generate secondary quartz. The stability of kaolinite in an acidic environment was further confirmed by comparing the XRD diffraction patterns of the kaolinite group before and after the reaction. the reaction equation is as follows:
Figure 5(d) displayed the XRD diffraction features of calcite group minerals before and after the reaction. A comparison of the maps revealed almost no change in the mineral composition of the calcite group. Although calcite exhibited the highest reaction rate and intensity in the early stage of the reaction, its mineral composition remained largely unchanged. This can be attributed to the high purity of minerals in the calcite group, with few other cations available to combine with CO32- and form precipitates.
The analysis of XRD profiles before and after the reaction indicated that feldspar minerals underwent more complex composition changes upon dissolution, particularly in the plagioclase group. The abundance of Na+ and Al3+ in plagioclase feldspar makes it highly suitable for CO2 mineralization and sequestration.
3.5. Mineral Specific Surface Area Variations
The mineral dissolution rate is significantly influenced by the size of the mineral specific surface area. The equation for the mineral dissolution rate, based on the classical transition state theory, can be expressed as follows:
In this equation, represents the mineral dissolution rate, represents the mineral specific surface area, is the mineral dissolution rate constant at room temperature, is the activation energy of the mineral dissolution reaction, is the ideal gas constant, is the reaction temperature, is the activity of a specific component in the solution, represents the number of reaction stages, is the mineral saturation, and and are experimental fitting parameters.
When other conditions remain constant, the specific surface area of minerals has a linear relationship with the mineral dissolution rate. However, the reaction rate varies during the mineral dissolution process due to changes in the specific surface area. Various literature reports show significant variations in the specific surface area of the same mineral, attributed to differences in mineral structure maturity and determination methods. This paper employed the liquid nitrogen adsorption method using a specific surface area adsorption meter (BET) to measure the changes in specific surface area before and after the mineral dissolution reaction. The results of these tests are presented in Table 5.
The test results indicate that the specific surface area of each mineral group increases after the reaction. This increase is primarily due to the minerals' rougher surface resulting from dissolution, which is beneficial for long-term CO2 mineralization and sequestration. However, the formation of secondary mineral precipitates after primary mineral dissolution may diminish this advantage if they adhere to the surface. The specific surface area of similar minerals varies significantly due to the low maturity and poor homogeneity of mineral structure in terrestrial sedimentary strata. In future studies, it is crucial to thoroughly investigate and analyze the homogeneity of the strata and the maturity of minerals in the target area. Additionally, testing the specific surface area of minerals using various methods should be conducted to minimize errors. This will provide valuable references for predicting the amount of CO2 mineralization storage and the efficiency of storage.
4. Conclusions
In this research paper, we have undertaken an investigation into the dissolution behavior of four commonly found terrestrial sedimentary minerals in the presence of CO2. By simulating the temperature, pressure, and mineralization conditions of the brackish water layer, we have utilized a block from the Daqing oilfield in China as a representative example. It is worth noting that this study is the first of its kind to examine the mineral compositions as independent entities. Our findings can be summarized as follows:
- (1)
- The nature of formation water in the CO2 brackish water layer has a direct influence on the degree of CO2 ionization. In the extensively distributed deep brackish water layer of terrestrial sedimentation in our country, the predominant water type is primarily NaHCO3, which acts as an inhibitor to some extent, affecting the ionization of CO2 and consequently influencing the pH of the formation water.
- (2)
- The erosion induced by CO2 fluid exhibits variations among different mineral types. Carbonate minerals undergo the most significant erosion, whereas feldspar minerals and clay minerals erode at a slower rate. Furthermore, plagioclase feldspar demonstrates a higher erosion rate compared to potassium feldspar.
- (3)
- The dissolution of feldspar minerals in the Na+-rich brackish water layer promotes the formation of secondary minerals such as dawsonite. These minerals possess the potential to be utilized for carbon sequestration purposes.
- (4)
- With the progression of dissolution and erosion, the specific surface area of minerals gradually increases. This phenomenon decelerates the reaction rate and facilitates the long-term mineralization and sequestration of CO2.
Author Contributions
Conceptualization, K.W. and W.L.; methodology, K.W. and Z.J.; validation, K.W.and S.N.; formal analysis, K.W. and N.J.; investigation, J.C. and M.Z; resources, W.L. and Z.J.; data curation, N.J. and W.J.; writing—original draft preparation,K.W.; writing—review and editing, W.L. and Z.J.; visualization, K.W. and J.C.; supervision, W.L. and Z.J.; project administration, W.L.; funding acquisition, W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Major Science and Technology project of the CNPC in China (grant No. 2021ZZ05).
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time, as the data also forms part of an ongoing study.
Acknowledgments
The authors are grateful for the financial support of the CNPC in China.
Conflicts of Interest
The authors declare that the publication of this paper has no conflict of interest.
References
- Dou, L.-R.; Sun, L.-D.; Lv, W.-F.; Wang, M.-Y.; Gao, F.; Gao, M.; Jiang, H. The Development Trend of Global Carbon Dioxide Capture, Utilization and Storage Industry and the Challenges and Countermeasures Faced by China. Pet. Explor. Dev. 2023, 50, 1083–1096. [Google Scholar] [CrossRef]
- Schleussner, C.-F.; Rogelj, J.; Schaeffer, M.; Lissner, T.; Licker, R.; Fischer, E.M.; Knutti, R.; Levermann, A.; Frieler, K.; Hare, W. Science and Policy Characteristics of the Paris Agreement Temperature Goal. Nat. Clim. Change 2016, 6, 827–835. [Google Scholar] [CrossRef]
- Guo, J.-X.; Huang, C.; Wang, J.-L.; Meng, X.-Y. Integrated Operation for the Planning of CO2 Capture Path in CCS–EOR Project. J. Pet. Sci. Eng. 2020, 186, 106720. [Google Scholar] [CrossRef]
- Zou, C.-N.; Wu, S.-T.; Yang, Z.; Pan, S.; Wang, G. Progress, Challenges and Significance of Building Carbon Industrial System in the Context of Carbon Neutrality Strategy. Pet. Explor. Dev. 2023, 50, 190–205. [Google Scholar] [CrossRef]
- Yuan, S.-Y.; Ma, D.-S.; Li, J.-S.; Zhou, T.-Y.; Ji, Z.-M.; Han, H.-S. Progress and Prospect of Industrialization of Carbon Dioxide Capture, Oil Displacement and Storage. Pet. Explor. Dev. 2022, 49, 828–834. [Google Scholar] [CrossRef]
- Liu, H.-J.; Were, P.; Li, Q.; Gou, Y.; Hou, Z. Worldwide Status of CCUS Technologies and Their Development and Challenges in China. Geofluids 2017, 2017. [Google Scholar] [CrossRef]
- Jiang, K.; Ashworth, P.; Zhang, S.; Liang, X.; Sun, Y.; Angus, D. China’s Carbon Capture, Utilization and Storage (CCUS) Policy: A Critical Review. Renew. Sustain. Energy Rev. 2020, 119, 109601. [Google Scholar] [CrossRef]
- Jiang, K.; Ashworth, P. The Development of Carbon Capture Utilization and Storage (CCUS) Research in China: A Bibliometric Perspective. Renew. Sustain. Energy Rev. 2021, 138, 110521. [Google Scholar] [CrossRef]
- Hasan, M.F.; First, E.L.; Boukouvala, F.; Floudas, C.A. A Multi-Scale Framework for CO2 Capture, Utilization, and Sequestration: CCUS and CCU. Comput. Chem. Eng. 2015, 81, 2–21. [Google Scholar] [CrossRef]
- Lewis, D.; Bentham, M.; Cleary, T.; Vernon, R.; O’Neill, N.; Kirk, K.; Chadwick, A.; Hilditch, D.; Michael, K.; Allinson, G. Assessment of the Potential for Geological Storage of Carbon Dioxide in Ireland and Northern Ireland. Energy Procedia 2009, 1, 2655–2662. [Google Scholar] [CrossRef]
- Sang, S.-X.; Liu, S.-Q.; Zhu, Q.L.; Han, S.; Zhang, S. Research Progress of CO2 Geological Sequestration Potential and Synergy of Energy Resources. J. China Coal Soc. 2023, 48, 2700–2761. [Google Scholar]
- Zhang, Q.; Lin, J.; She, Y.-N.; Ma, C.; Hao, J. Research Progress on Evaluation of Carbon Dioxide Geological Sequestration Potential in China. China Pet. Chem. Ind. Stand. Qual. 2022, 42, 144–146. [Google Scholar] [CrossRef]
- Liu, B.; Pan, Y. Review on Analysis of CO2 Burial Mechanism and Influencing Factors in Deep Salt Water. Suppl. Environ. Eng. 2017, 5. [Google Scholar]
- Cao, L.; Bian, L.-H. Research Progress of CO2 Geological Storage Technology and Storage Potential Evaluation Method. Undergr. Water 2018, 35, 211–213. [Google Scholar]
- Liu, T.; Diao, Y.-J.; Jin, X.-L. Research Status of Evaluation Methods of Carbon Dioxide Geological Storage Potential at Home andAbroad. Coal Sci. Technol. 2019, 33, 116–121. [Google Scholar]
- Bacon, D.H.; Qafoku, N.P.; Dai, Z.; Keating, E.H.; Brown, C.F. Modeling the Impact of Carbon Dioxide Leakage into an Unconfined, Oxidizing Carbonate Aquifer. Int. J. Greenh. Gas Control 2016, 44, 290–299. [Google Scholar] [CrossRef]
- Zheng, L.; Spycher, N.; Varadharajan, C.; Tinnacher, R.M.; Pugh, J.D.; Bianchi, M.; Birkholzer, J.; Nico, P.S.; Trautz, R.C. On the Mobilization of Metals by CO2 Leakage into Shallow Aquifers: Exploring Release Mechanisms by Modeling Field and Laboratory Experiments. Greenh. Gases Sci. Technol. 2015, 5, 403–418. [Google Scholar] [CrossRef]
- Ueda, A.; Kato, K.; Ohsumi, T.; Yajima, T.; Ito, H.; Kaieda, H.; Metcalfe, R.; Takase, H. Experimental Studies of CO2-Rock Interaction at Elevated Temperatures under Hydrothermal Conditions. Geochem. J. 2005, 39, 417–425. [Google Scholar] [CrossRef]
- Ketzer, J.M.; Iglesias, R.; Einloft, S.; Dullius, J.; Ligabue, R.; De Lima, V. Water–Rock–CO2 Interactions in Saline Aquifers Aimed for Carbon Dioxide Storage: Experimental and Numerical Modeling Studies of the Rio Bonito Formation (Permian), Southern Brazil. Appl. Geochem. 2009, 24, 760–767. [Google Scholar] [CrossRef]
- Farquhar, S.M.; Pearce, J.K.; Dawson, G.K.W.; Golab, A.; Sommacal, S.; Kirste, D.; Biddle, D.; Golding, S.D. A Fresh Approach to Investigating CO2 Storage: Experimental CO2–Water–Rock Interactions in a Low-Salinity Reservoir System. Chem. Geol. 2015, 399, 98–122. [Google Scholar] [CrossRef]
- Li, J.; Ahmed, R.; Zhang, Q.; Guo, Y.; Li, X. A Geochemical Model of Fluids and Mineral Interactions for Deep Hydrocarbon Reservoirs. Geofluids 2017, 2017. [Google Scholar] [CrossRef]
- Balashov, V.N.; Guthrie, G.D.; Hakala, J.A.; Lopano, C.L.; Rimstidt, J.D.; Brantley, S.L. Predictive Modeling of CO2 Sequestration in Deep Saline Sandstone Reservoirs: Impacts of Geochemical Kinetics. Appl. Geochem. 2013, 30, 41–56. [Google Scholar] [CrossRef]
- Diao, Y.; Zhu, G.; Cao, H.; Zhang, C.; Li, X.; Jin, X. Mesoscale Assessment of CO2 Storage Potential and Geological Suitability for Target Area Selection in the Sichuan Basin. Geofluids 2017, 2017. [Google Scholar] [CrossRef]
- Leung, D.Y.; Caramanna, G.; Maroto-Valer, M.M. An Overview of Current Status of Carbon Dioxide Capture and Storage Technologies. Renew. Sustain. Energy Rev. 2014, 39, 426–443. [Google Scholar] [CrossRef]
- Guo, J.-Q.; Wen, D.-G.; Zhang, S.; Xu, T.; Li, X. Evaluation and Demonstration Project of Carbon Dioxide Geological Storage Potential in China. Geol. Surv. China 2015, 2, 36–46. [Google Scholar]
- Zhang, S.-Q. Geological Characteristics of Continental Sedimentary Basins in China. Hydrogeol. Environ. Geol. Surv. Cent. China Geol. Surv. 2010. [Google Scholar]
- Tutolo, B.M.; Luhmann, A.J.; Kong, X.-Z.; Saar, M.O.; Seyfried Jr, W.E. CO2 Sequestration in Feldspar-Rich Sandstone: Coupled Evolution of Fluid Chemistry, Mineral Reaction Rates, and Hydrogeochemical Properties. Geochim. Cosmochim. Acta 2015, 160, 132–154. [Google Scholar] [CrossRef]
- Wigand, M.; Carey, J.W.; Schütt, H.; Spangenberg, E.; Erzinger, J. Geochemical Effects of CO2 Sequestration in Sandstones under Simulated in Situ Conditions of Deep Saline Aquifers. Appl. Geochem. 2008, 23, 2735–2745. [Google Scholar] [CrossRef]
- Huq, F.; Haderlein, S.B.; Cirpka, O.A.; Nowak, M.; Blum, P.; Grathwohl, P. Flow-through Experiments on Water–Rock Interactions in a Sandstone Caused by CO2 Injection at Pressures and Temperatures Mimicking Reservoir Conditions. Appl. Geochem. 2015, 58, 136–146. [Google Scholar] [CrossRef]
- Huron, M.-J.; Vidal, J. New Mixing Rules in Simple Equations of State for Representing Vapour-Liquid Equilibria of Strongly Non-Ideal Mixtures. Fluid Phase Equilibria 1979, 3, 255–271. [Google Scholar] [CrossRef]
Figure 1.
Experimental system setup diagram.
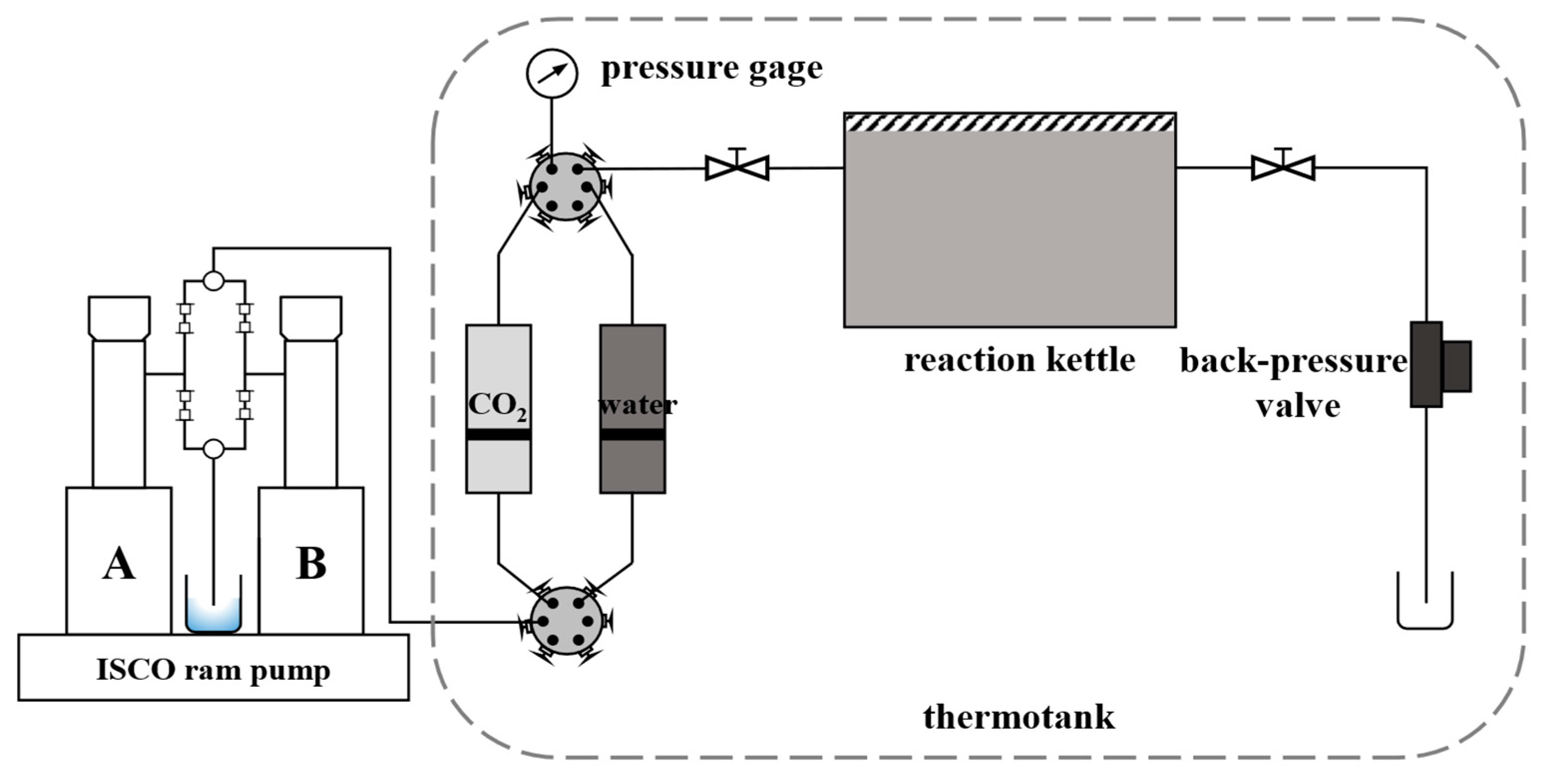
Figure 2.
Variation of aqueous solution pH with time for different mineral groups.
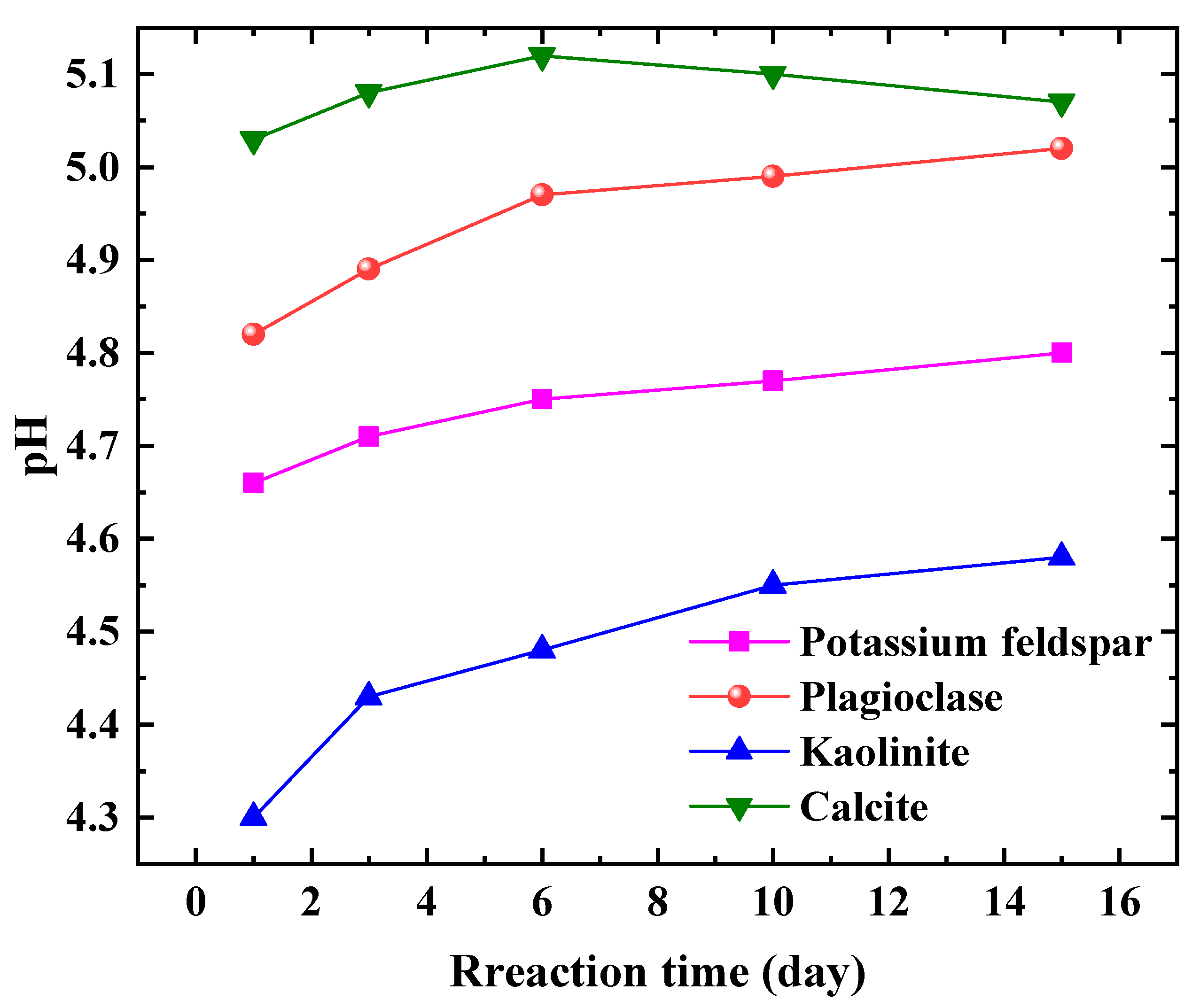
Figure 3.
Changes of major cations in aqueous solutions of different mineral groups with time.
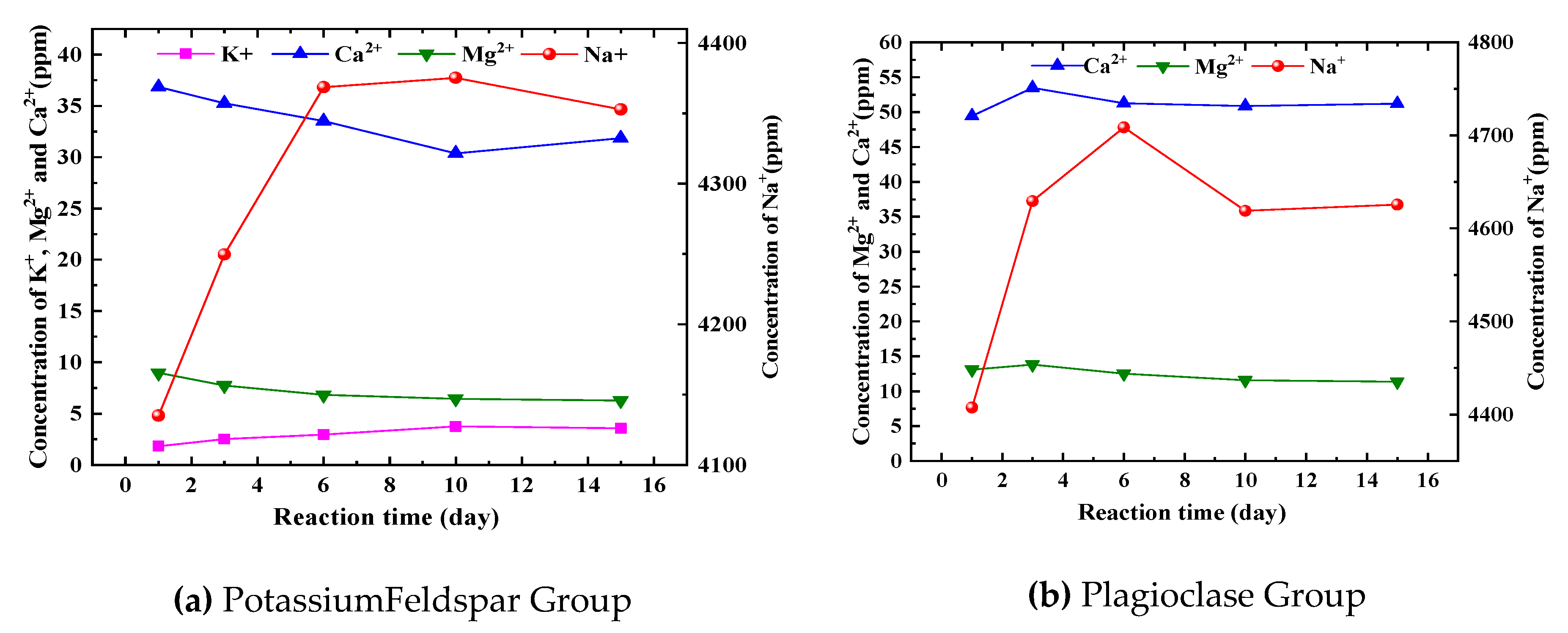
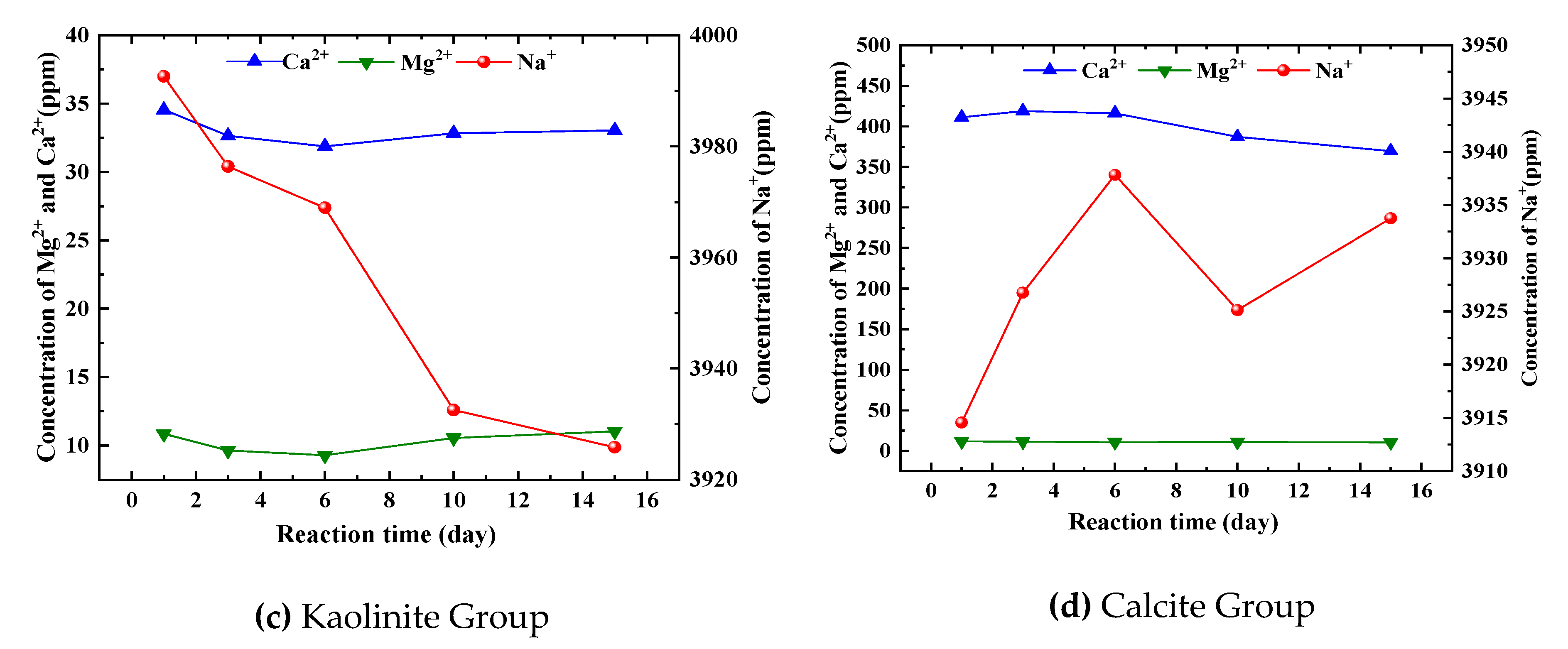
Figure 4.
Scanning electron microscope images of the surface of each group of minerals before and after reaction.
Figure 4.
Scanning electron microscope images of the surface of each group of minerals before and after reaction.


Figure 5.
XRD characteristics observed prior to and subsequent to the retion of each mineral group.
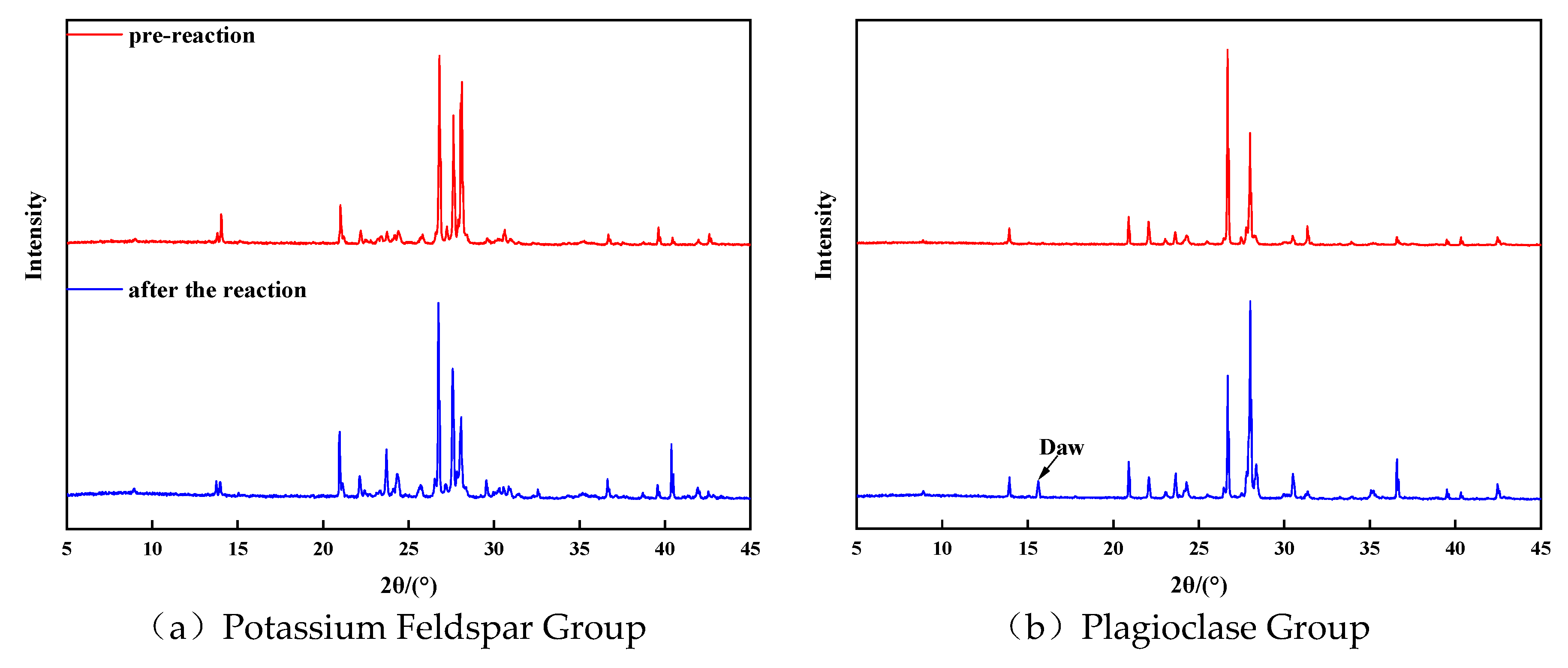
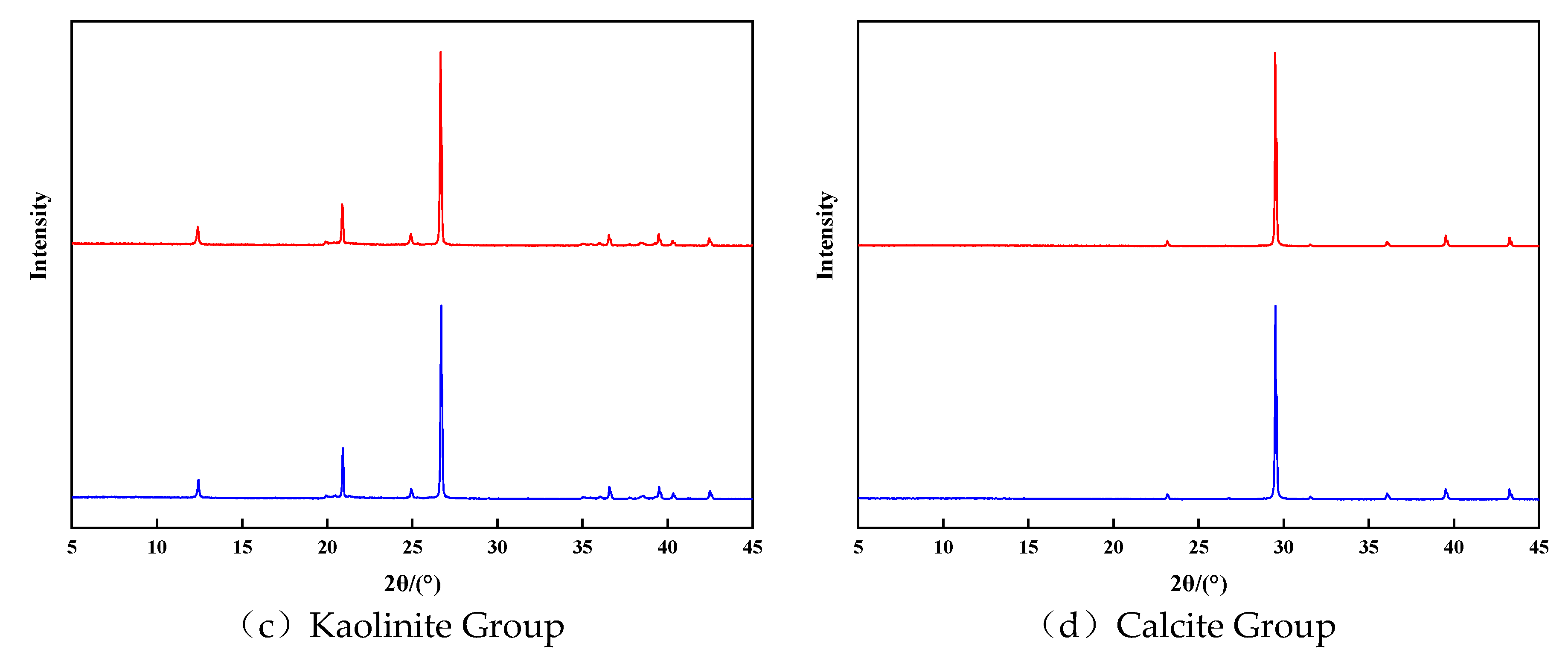

Table 1.
Typical sandstone mineral composition of Daqing oilfield.
| Block | Core Count | Quartz(%) | Potassium Feldspar(%) | Plagioclase(%) | Calcite(%) |
|---|---|---|---|---|---|
| AoNan | 18 | 25.28 | 22.00 | 7.33 | 0.67 |
| ChaoYanggou | 105 | 24.86 | 20.52 | 7.89 | 5.66 |
| ChangGeng | 276 | 25.74 | 21.80 | 6.45 | 3.67 |
Table 2.
Typical clay mineral composition of Daqing oilfield.
| Block | Core Quantity | Illite(%) | Chlorite(%) | Smectite(%) | Kaolinite(%) |
|---|---|---|---|---|---|
| AoNan | 198 | 62.43 | 14.26 | 1.66 | 1.02 |
| ChaoYanggou | 246 | 45.89 | 17.35 | 2.75 | 4.20 |
| ChangGeng | 441 | 31.55 | 15.81 | 33.48 | 7.96 |
Table 3.
Information on formation water composition and formulated medicines in Aonan 6 block of Daqing Oilfie.
Table 3.
Information on formation water composition and formulated medicines in Aonan 6 block of Daqing Oilfie.
| Compound | Mass Concentration | Specification | Manufacturer |
|---|---|---|---|
| MgCl2-6H2O | 0.1032 | Analytically Pure | Sinopharm Group Chemical Reagent Co.,LTD |
| CaCl2 NaHCO3 Na2SO4 NaCl |
0.1113 4.1449 0.3018 6.9751 |
Table 4.
Critical parameters for each component of the PR-HV model.
| Component | Pc/MPa | Tc/K | ω | M/(g∙mol-1) | Normal Boiling Point |
|---|---|---|---|---|---|
| CO2 | 7.38 | 31.05 | 0.225 | 44.0 | -78.5 |
| H2O | 22.09 | 374.15 | 1.000 | 18.0 | 100.0 |
| NaCl | 3.55 | 426.85 | 0.344 | 58.5 | 1413.0 |
Table 5.
Alterations in Specific Surface Area Preceding and Subsequent to Mineral Reaction.
| Mineral Group | Pre-reaction BET(cm2/g-1) | After-reaction BET(cm2/g-1) | Rate of Change(%) |
|---|---|---|---|
| Potassium feldspar Plagioclase Kaolinite Calcite |
185.57 | 251.69 | 35.63 |
| 74.19 | 103.46 | 39.45 | |
| 633.94 350.14 |
862.83 445.79 |
36.11 27.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



