
Submitted:
29 September 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
Within an amplicon-based metagenomic study of Archaea, Fungi, and Bacteria in Livingston Island, Maritime Antarctica, in many of the samples antagonisms between these three super kingdoms were observed under the form of an inversely proportional dependence of the richnesses of the three types of microorganisms. This was quantified - based on the observed numbers of the total tags and the numbers of the operational taxonomic units (OTUs), as well as based on four alpha diversity parameters – the Shannon, the Simpson, the Chao1, and the ACE indices. We found that the most discriminative results in the antagonism measuring were observed in the comparison of the numbers of the OTUs and the ACE community richness estimator. The antagonism between Archaea and Fungi was strongest, followed by that of Archaea and Bacteria. The Fungi-Bacteria antagonism was slightly detectable. Pearson and Spearman correlation analysis also showed a statistically significant negative correlation between the fungal and archaeal effective tags, while the correlation between archaeal and bacterial diversity was positive. Indications of the order of primary microbial succession in barren ecological niches were also observed, demonstrating that Archaea and Bacteria are the pioneers, followed by Fungi which in time would displace Archaea.
Keywords:
amplicon-based metagenomics
; Maritime Antarctica
; microbial domains antagonism
; Archaea
; Fungi
; Bacteria
1. Introduction
Antarctica, despite being isolated, remote, and difficult to access because of its geographic position and characteristics, can be considered as a major climate generator [1]. Microorganisms are few of ones who can survive in the Antarctic harsh terrestrial environments and form the major component of the biomass within the water ecosystems where they are also at the basis and at the end of every food chain. Antarctic microorganisms possess specific adaptations, allowing them to survive in these challenging environments [2,3]. However, these adaptations render them susceptible to environmental changes, thereby emphasizing the need to investigate them further.
Unfortunately, Antarctic microbial communities are still poorly studied, mostly because of the challenging environments and the remoteness from the needed adequate laboratory infrastructure. However, with the advances of the Next Generation Sequencing (NGS)-based metagenomics techniques, this task became more feasible, relying on in situ direct DNA isolation, followed by the transportation of the extracted DNA samples to the research centers and laboratories. Some good examples of such metagenomic studies are the works of Picazo et al. [4], Kim et al. [5], Coleine et al. [6], and Fernández et al. [7]. Yet, the focus of these studies was put on the characterization of the fungal, archaeal, and bacterial compositions within the soils and the aquatic environments but not on the existing relationships between these three domains.
In January – February 2022 during the 30th Bulgarian polar expedition to Livingston Island (South Shetland Islands, Maritime Antarctica), one of the research projects that members of the team worked on was focused on the taxonomical characterization of some soil and water microbial ecosystems. When we took a first look at the results, we were surprised to find low archaeal richness, as well as an inversely proportional dependence between the richesses of Archaea and Fungi, and to a lesser extent between bacteria and archaea, and fungi and bacteria. Despite an antagonism between Bacteria and Fungi has been reported for agricultural soils [8], no data was available for the Arctic and the Antarctic regions, so we decided to investigate these domains’ antagonisms which could be related to and/or to be more acute in the harsh and in most cases oligotrophic Antarctic environment. To do this we proposed to quantify this antagonism based on some of the metagenomic sequencing data parameters.
2. Materials and Methods
2.1. Sampling sites and sampling methods
All sampling sites are listed in Table 1 and are located around the Bulgarian Antarctic Base “St. Kliment Ohridski” on Livingston Island, Maritime Antarctica. For the solid samples, 150-250 mg of biomass or sediment were collected in as much as possible sterile conditions with a sterile spatula and put into sterile Eppendorf tubes. For the water samples, 2 liters of water were collected in new and unused plastic bottles which were washed 3 times with the water of the sampling site before the collection. All samples were transported immediately to the research station, and DNA was usually isolated within 45-60 minutes, except for the Hannah Point samples whose transportation to Bulgarian Antarctic Base required about 90-120 minutes.
2.2. DNA extraction
To reduce to a minimum possible shift in the microbial compositions of the collected samples, total DNA from them was isolated just after the collection. For the solid samples, the extraction was performed with ZR Soil Microbe DNA MiniPrep Kit (Zymo Research Corp., USA) according to the manufacturer’s Instructions manual. For the water samples, two liters of the water were filtered through a 0.2 µm filter, and then the biomass was washed off with 700 µl of the ZR Soil Microbe DNA MiniPrep Kit’s Bashing Beads buffer. The eluted biomass was transferred within the kit’s bashing beads tubes and further processed as the solid samples. The final elution step was performed in 50 µl of the provided Elution buffer. Two microliters of the eluted DNA were used for the determination of the concentration on a Quantus Fluorimeter (Promega Corp., USA). The eluted DNA samples were kept and transported at -20 °C.
2.3. Metagenomic sequencing
The extracted DNA was shipped on dry ice to the Novogene Company Ltd. (Cambridge, UK) for sequencing on the Illumina HiSeq 2 x 250 paired-end reads platform. The V3-V4 region of the genes encoding the 16S rRNA was chosen for the amplification of eubacterial 16S rRNA with primers 341F (5’-CCTAYGGGRBGCASCAG-3’) and 806R (5’-GGACTACNNGGGTATCTAAT-3’). For fungi, the ITS2 region was amplified using primers ITS3-2024F (5’-GCATCGATGAAGAACGCAGC-3’) and ITS4-2409R (5’-TCCTCCGCTTATTGATATGC-3’), while for archaea the V4-V5 16S rRNA region was amplified using primers Arch519F (5’- CAGCCGCCGCGGTAA-3’) and Arch915R (5’- GTGCTCCCCCGCCAATTCCT-3’). The sequencing was performed to generate 30,000 tags per sample. Novogene Company LTD performed the primary bioinformatic processing.
2.4. Data processing and operational taxonomic units (OTUs) analyses
The raw files generated from the NGS platform were first subjected to demultiplexing and trimming. Next, the FLASH V1.2.7 software tool [9] was used to merge the paired-end reads. The raw tags quality filtering was performed according to Bokulich et al. [10] and Caporaso et al. [11]. The UCHIME algorithm was used to detect chimera sequences by comparing the tags with the reference database (http://drive5.com/uchime/uchime_download.html) [12]. The effective tags were obtained by removing the chimera sequences, accordingly to Haas et al. [13]. The Uparse v7.0.1001 software [14] was used to assign the effective tags with ≥97% similarity into OTUs. For species annotation at each taxonomic rank, each eubacterial and archaeal representative sequence was compared against the SSUrRNA Database of the SILVA138 Database with the Mothur software [15,16]. For fungi, the comparison was performed again with the Mothur software against the Unite V8.2 database [17]. The alpha diversity analysis was estimated based on the community richness index by the Chao1 estimator (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.chao1.html#skbio.diversity.alpha.chao1) [18] and the ACE estimator (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.ace.html#skbio.diversity.alpha.ace) [19,20]. The community diversity was assessed by the Shanon index (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.shannon.html#skbio.diversity.alpha.shannon) and the Simpson index (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.simpson.html#skbio.diversity.alpha.simpson) [21,22]. Rarefaction curves were constructed in order to assess the reliability of the obtained data [23].
2.5. Antagonism analyses and relative measuring
The mean values of the number of the total tags, the number of the OTUs, the Shannon, the Simpson, The Chao1, and the ACE indices were calculated, then each sample was attributed to “+” or “-“ depending on if its own value was greater or lesser than the mean value for the given type of microorganisms. Then the three types of binome antagonisms in each sample were relatively quantified as the sums of the cases where a discrepancy was observed between the two compared types of microorganisms.
2.6. Pearson and Spearman correlation analysis
Pearson and Spearman correlation analyses were performed between the three groups of microorganisms for each of the alpha diversity metrics, as well as the effective tags. For testing the statistical significance of the results regarding the community as a whole, a significance threshold of p=0.05 was chosen. Pearson correlation assesses the presence of a linear correlation between two variables with normal distribution, while Spearman correlation assesses the presence of a monotonic relationship. These values range from -1 for the presence of a high negative correlation, 0 for the absence of a correlation, to 1 for a presence of a high positive correlation.
3. Results
3.1. Sequencing statistics and alpha diversity indices
3.2. Antagonism study
The numerical values for each index (the number of the total tags, the number of the OTUs, the Shannon, the Simpson, the Chao1, and the ACE indices) in each sample are presented as bar histograms regarding the mean values in Figure 2. To ease the interpretation of the results, each sample was attributed to a “+” (greater) or “-“ (lesser) for each index according to its proper value in comparison to the mean value for this index (Table 3). A summarization of the cases of discrepancies is presented in Table 4 as total numbers as well as percentages.
Pearson linear correlation showed a statistically significant negative correlation between the effective tags of archaea and fungi, but not between any of the other metrics. (Table 5). On the other hand, a very high positive correlation was shown between archaea and bacteria in regard to both the Simpson and Shannon diversity indices.
4. Discussion
The total number of bacterial and archaeal cells on Earth is estimated to be around 1.2×1030 cells, distributed in five major habitats: deep oceanic subsurface, upper oceanic sediment, deep continental subsurface, soil, and oceans [24]. There are no such quantitative estimations for the Fungi, probably because they can exist in both unicellular and multicellular form. However, they also represent a substantial percentage of the Earth’s biomass – 12.7% [25] and show a tremendous species diversity and functional roles in almost all ecosystems [26]. Still, with very few exceptions, the mutual ecological relationships between these three domains of microorganisms as a whole are poorly investigated. Even with the advent of NGS-based metagenomics, most of the studies are focused on research within a single domain in a given ecological niche.
During the Antarctic research season 2021-2022, four water and fourteen terrestrial samples were collected around the Bulgarian Polar Base on Livingston Island in Maritime Antarctica. The major goal of the research was the characterization of some microbiotas in different ecological niches with the means of amplicon-based metagenomics (submerged microbial surface contaminations of rocks and algae, sediments, soils, biomasses from ice melting ponds, lithotelms and glacial lakes (tarns), fresh waters, and marine waters) (Table 1).
The obtained data were considered reliable and informative due to the slopes of the rarefaction curves which were reaching a plateau (Figure 1), meaning that the data were representative [27]. However, before the in-depth analysis of the sequencing data, two observations came into our sight. First, the number of the Archaea (as total tags as well as the number of OTUs) was much lower than those of Fungi and Bacteria. Second, the archaeal alpha diversity indices (Shannon, Simpson Chao1, and ACE) were from the same order as those of the two other studied domains, meaning comparable species richnesses in all three super kingdoms (Table 2) [27]. These two observations suggest that the presence of archaeal cells within the samples was significantly lower in number compared to bacterial and fungal cells, despite all three kingdoms exhibiting similar levels of richness.
One of the most probable explanations of this disparity is the assumption that some antagonism between Archaea and the other two domains should exist, in turn probably caused by the harsh, and in some cases oligotrophic, Antarctic conditions. So, we decided to relatively measure this antagonistic relationship most simply - by comparing in binomes: in how many samples when one of the domains is more than the observed average, the other is less than the observed average. These results, graphically presented in Figure 2, were difficult to quantify, so, a simpler comparison matrix composed of only “+” and “-“ was constructed (Table 3) which in turn was used in our quantitative analysis of the antagonism relationships (Table 4).
The number of the total tags, which represents the number of effective tags after merging and filtering the sequencing reads, is the most primary parameter because it directly reflects the distribution of the number of the sequenced DNA molecules within the sample. This antagonism was observed in 10 of 18 cases (56%) in all three binomial comparison groups: Fungi vs Archaea; Fungi vs Bacteria and Archaea vs Bacteria.
The next parameter which was analyzed was the number of OTUs in each sample. Analyzing the OTUs numbers has the advantage to reflect the taxonomic groups [28]. In this case the discrepancy between Archaea and Fungi was more distinct – it was observed in 2/3 of all samples and even more in the water samples - 3/4 of the cases. Concerning the binome Fungi-Bacteria, the discrepancy was less distinct than in the case of the total number of the tags (in only 44% of the samples) while between Archaea and Bacteria, discrepancies were observed within the same percentage as within the total number of the tags.
Concerning the four observed alpha diversity indices, no such clear tendencies could be observed. Still, in our opinion, they should also be taken into account because they reflect the microbial communities’ structures. The observed discrepancies in all the three binomes concerning the Shanon and the Simposon indices [21,22,29] varied between 17% and 50%. Both are used as estimators of the species richness and evenness, so the low percentages could be explained by the high OTUs values for the three domains within the analyzed samples. On the other hand, more interesting results were obtained for the Chao1 and the ACE richness estimators which are abundance-based [18,19,20]. Regarding the Chao1 index, a discrepancy in more than 50% of the cases was observed only for the binome Archaea-Bacteria. This can be explained by the fact that this index is based on the singletons and the doubletons, thus estimating the “missed” species, Bacteria being largely the most species-abundant superkingdom. On the other hand, discrepancies in 56% and 61% of the cases were observed respectively for the binomes Fungi-Archaea and Archaea-Bacteria. This tendency corresponds more to those observed for the numbers of the total tags and OTUs because the ACE richness estimator takes into account both the abundant and the rare species, and thus should be more informative when domains’ antagonisms are investigated.
The presence of a moderate, negative correlation between the effective tags of fungi and archaea, coupled with the lack of such a distinct correlation in the rest of the metrics hints at a possible explanation for these results. Firstly, it is important to note that the effective tags represent the sequencing reads before they are grouped into OTUs based on a 97% similarity threshold. If the majority of the effective tags that are associated with the presence of a correlation between two groups are also assigned to a small number of OTUs, it is most likely that any potential interactions are limited within a subset of the species in the sample, and thus will not be as apparent in the alpha diversity metrics. On the other hand, if no correlation is observed between the number of effective tags, yet such a correlation is present within the alpha diversity metrics, this could point to a large number of species contributing to the interaction. We see a similar trend in some of the interactions between archaea and bacteria, where the lack of correlation in effective tags is followed by a very high correlation in diversity.
Additionally, it is crucial to outline that the given samples represent vastly different ecological niches, encompassing sediment, salt water, fresh water, biomass, and surface contamination samples. In fact, roughly half of the samples are from a type of surface contamination, and thus the correlation results will inevitably be skewed in favor of the specific interactions within those types of communities. The annotated taxonomy, which is not included in this paper, also points to the existence of microbial profiles that are specific to each sampling location. Thus, the interactions that take place on the scale of the sampling locations will inevitably overlap with the presence of any macro-scale interactions that could be attributed to the Antarctic climate as a whole. However, due to the lack of samples for a large number of the sampling sites, it is difficult to exclude them from the dataset, without compromising how representative the sample is of the Antarctic community on Livingston Island as a whole.
An interesting case is sample S22 which was obtained from biomass on a submerged rock in the new nameless tarn located some several hundred meters North-Est of the Bulgarian Polar Base “St. Kliment Ohridski”. This is the only sample where the numbers of the total tags and the OTUs in Archaea exceed those of Fungi, and approach those of Bacteria. More interestingly, the number of the archaeal total tags is some 16% greater than the fungal one. However, the archaeal OTUs are more than 6 times more numerous than the fungal ones. Not surprisingly, the greatest Chao1 and ACE community richness estimators among Archaea are observed in this sample in comparison to all other samples we analyzed. At this stage of research, it would be difficult and to a somewhat extent speculative to explain this observation, still, it is worth noting that this tarn was formed only very recently – during the last pre-Covid19 Bulgarian Antarctic expedition in 2018-2019 permanent glacier existed in its location, and the tarn discovered only during the 2020-2021 expedition when the scientists returned to the Bulgarian Polar Base (the 2019-2020 expedition being only logistical for base maintenance). So, we cannot say if the glacier retracted to form the tarn one or two years before the sample was taken off, but surely this tarn represents a very new ecosystem. This fact makes plausible the speculation of primary succession of the super kingdoms in newly colonized areas. It has been observed that microorganisms are the first colonizers after the receding glaciers in the mountains [30] so, it is logical that the same process occurs in Antarctica. Unfortunately, the primary microbial succession in barren lands and other ecological niches is poorly investigated [31]. According to our hypothesis, Archaea and Bacteria are the first, while for Fungi to colonize the new environment more time is needed. If this hypothesis is true, it would explain why in the “old” ecological niches in our study in general Fungi dominate over Archaea.
One of the reasons we consider our data informative and reflecting the actual situation is that we chose a non-discriminative method for DNA isolation based on the physical destruction of the microbial cells. Another one is that we used the same DNA samples for the three types of metagenomic analyses in combination with equal sequencing efforts – the generation of at least 30,000 tags per sample, which resulted in 3-4 times more tags except for Archaea (meaning that the archaeal DNA is indeed less represented within the sample). This means that comparisons between the three investigated domains are possible, especially in light of the plateaued refraction curves, prerequisites for correct enough quantitative analyses in amplicon-based metagenomics where primer annealing biases could be a concern.
Our study, as a pioneering one, has some drawbacks. First, we acknowledge that physicochemical parameters in the different ecological niches studied could impact the observed antagonisms. Still, we believe that the tendencies we observed are visible enough. Unfortunately, studying the exact mechanisms of the antagonistic interactions was far beyond the scope of the research project. At present, we can only suspect that both types of antagonistic interactions are involved – active, due to the synthesis of different types of antimicrobials, and passive, resulting from the concurrence for nutrients and energy between the different types of microorganisms. The lack of replicas should be considered as another drawback. Nevertheless, replicating these experiments is not possible because of the location of the area where the study was performed – Livingston Island in Maritime Antarctica. Additionally, the polar bases are seasonal, meaning that they are accessible only during the Antarctic summer. Still, because of the seasonal landscape erosion caused by the retreat of the glaciers every summer, the small glacial ponds where most of the biomass samples were taken off form in different locations every year. Finally, the third major drawback of the research is the lack of a well-established methodology for quantification of the antagonisms between the three domains. Unfortunately, this drawback could not be adequately addressed because of the very scarce publications of similar studies. Here, we only propose a very basic methodology that worked well for a field study of a location with limited seasonal accessibility. For these reasons, this manuscript should be regarded as a preliminary communication that could be the basis for future research projects.
5. Conclusions
We could only speculate about the causes of the observed antagonism between the three major microbial phyla in Maritime Antarctica. However, our data suggest that some antagonisms exist, especially between Fungi and Archaea and between Archaea and Bacteria, based on the values of the numbers of the total tags and the OTUs, as well as on the values of the Chao1 and the ACE community richness indices. Furthermore, we observed some indications about the order of colonization of new barren ecological niches, according to which Archaea and Bacteria are the first, followed by Fungi. Based on our findings the best indicators of antagonism are the numbers of the OTUs and the ACE estimator, giving the most discriminative results. Unfortunately, because of the general lack of similar studies, not only for Antarctica but also worldwide, we cannot either compare our findings or explain the causes of the antagonisms.
Our results of the studies of pioneer microbial communities are only preliminary thus further studies including inter-domains functional relationships undoubtedly are needed. Yet, we demonstrated the power of NGS-based metagenomics in research on inter-domain relationships, as well as in the investigation of the primary colonization of new ecological recesses.
Author Contributions
Conceptualization, S.G.D. and N.N.; methodology, S.G.D. and V.D.; software, S.G.D.; validation, S.G.D., R.I. and M.I.; formal analysis, S.G.D., V.D., D.G.-M. and S.P; investigation, S.G.D., V.D., R.I., M.K. and S.P.; resources, S.G.D. and N.N.; data curation, S.G.D.; writing—original draft preparation, S.G.D., N.N. and D.G.-M.; writing—review and editing, S.G.D., N.N. and M.K.; visualization, S.G.D.; supervision, S.G.D.; project administration, S.G.D., L.K. and M.I.; funding acquisition, S.G.D.
Funding
This research was funded by Grant № 70-25-72 from 03.08.2021 г. of the National Center for Polar Studies - Sofia University “St. Kliment Ohridski”.
Data Availability Statement
The raw sequencing reads of this study have been deposited in the Nacional Center for Biotechnology Information of the National Library of Medicine as sequence reads archives under the following accession numbers: PRJNA979344 (for Archaea), PRJNA979782 (for Fungi) and PRJNA979776 (for Bacteria).
Acknowledgments
The authors would like to thank the logistics team of the Bulgarian Polar Base in Livingston Island for their valuable help and support which made this research possible. Furthermore, we owe special thanks to Prof. Hristo Pimpirev, chairman of the Bulgarian Antarctic Institute, and Mr. Dragomior Mateev, Science and Logistic coordinator of the Bulgarian polar expeditions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barker, P.F.; Filippelli, G.M.; Florindo, F.; Martin, E.E.; Scher, H.D. Onset and role of the Antarctic Circumpolar Current. Deep Sea Research Part II: Topical Studies in Oceanography 2007, 54, 2388–2398. [Google Scholar] [CrossRef]
- Bej, A.K.; Mojib, N. Cold adaptation in Antarctic biodegradative microorganisms. Polar microbiology: the ecology, biodiversity and bioremediation potential of microorganisms in extremely cold environments 2009, 157–177. [Google Scholar]
- Robinson, C.H. Cold adaptation in Arctic and Antarctic fungi. New Phytologist 2001, 151, 341–353. [Google Scholar] [CrossRef]
- Picazo, A.; Rochera, C.; Villaescusa, J.A.; Miralles-Lorenzo, J.; Velázquez, D.; Quesada, A.; Camacho, A. Bacterioplankton community composition along environmental gradients in lakes from Byers peninsula (Maritime Antarctica) as determined by next-generation sequencing. Frontiers in microbiology 2019, 10, 908. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.-H.; Lim, J.-H.; Jeong, J.-H.; Heo, J.-M.; Kim, I.-N. Distribution and Control of Bacterial Community Composition in Marian Cove Surface Waters, King George Island, Antarctica during the Summer of 2018. Microorganisms 2020, 8, 1115. [Google Scholar] [CrossRef]
- Coleine, C.; Biagioli, F.; de Vera, J.P.; Onofri, S.; Selbmann, L. Endolithic microbial composition in Helliwell Hills, a newly investigated Mars-like area in Antarctica. Environmental Microbiology 2021, 23, 4002–4016. [Google Scholar] [CrossRef]
- Fernández, G.C.; Lecomte, K.; Vignoni, P.; Rueda, E.S.; Coria, S.H.; Lirio, J.M.; Mlewski, E.C. Prokaryotic diversity and biogeochemical characteristics of benthic microbial ecosystems from James Ross Archipelago (West Antarctica). Polar Biology 2022, 45, 405–418. [Google Scholar] [CrossRef]
- Li, X.; Garbeva, P.; Liu, X.; klein Gunnewiek, P.J.A.; Clocchiatti, A.; Hundscheid, M.P.J.; Wang, X.; de Boer, W. Volatile-mediated antagonism of soil bacterial communities against fungi. Environmental Microbiology 2020, 22, 1025–1035. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. , et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome research 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Applied and Environmental Microbiology 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M. , et al. Towards a unified paradigm for sequence-based identification of fungi. Molecular Ecology 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Chao, A. Non-parametric estimation of the classes in a population. Scandinavian Journal of Statistics 1984, 11, 265–270. [Google Scholar]
- Chao, A.; Lee, S.M. Estimating the Number of Classes Via Sample Coverage. J Am Stat Assoc 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A.; Ma, M.C.; Yang, M.C.K. Stopping Rules and Estimation for Recapture Debugging with Unequal Failure Rates. Biometrika 1993, 80, 193–201. [Google Scholar] [CrossRef]
- Lemos, L.N.; Fulthorpe, R.R.; Triplett, E.W.; Roesch, L.F.W. Rethinking microbial diversity analysis in the high throughput sequencing era. Journal of microbiological methods 2011, 86, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Magurran, A. Measursuring biological diversity. In Blackwelll Publ; Company United Kingdom: New Jersey, US, 2004. [Google Scholar]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nature Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nature Reviews Microbiology 2019, 17, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.M.; Phillips, R.; Milo, R. The biomass distribution on Earth. Proceedings of the National Academy of Sciences 2018, 115, 6506–6511. [Google Scholar] [CrossRef]
- Peay, K.G.; Kennedy, P.G.; Talbot, J.M. Dimensions of biodiversity in the Earth mycobiome. Nature Reviews Microbiology 2016, 14, 434–447. [Google Scholar] [CrossRef]
- Kim, B.R.; Shin, J.; Guevarra, R.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. Journal of microbiology and biotechnology 2017, 27, 2089–2093. [Google Scholar] [CrossRef]
- He, Y.; Caporaso, J.G.; Jiang, X.-T.; Sheng, H.-F.; Huse, S.M.; Rideout, J.R.; Edgar, R.C.; Kopylova, E.; Walters, W.A.; Knight, R. , et al. Stability of operational taxonomic units: an important but neglected property for analyzing microbial diversity. Microbiome 2015, 3, 20. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688–688. [Google Scholar] [CrossRef]
- Ciccazzo, S.; Esposito, A.; Borruso, L.; Brusetti, L. Microbial communities and primary succession in high altitude mountain environments. Annals of Microbiology 2016, 66, 43–60. [Google Scholar] [CrossRef]
- Ni, G.; Lappan, R.; Hernández, M.; Santini, T.; Tomkins, A.G.; Greening, C. Functional basis of primary succession: Traits of the pioneer microbes. Environ Microbiol 2023, 25, 171–176. [Google Scholar] [CrossRef]
Figure 1.
Rarefaction curves of Fungi (A), Archaea (B), and Bacteria (C) in the samples.
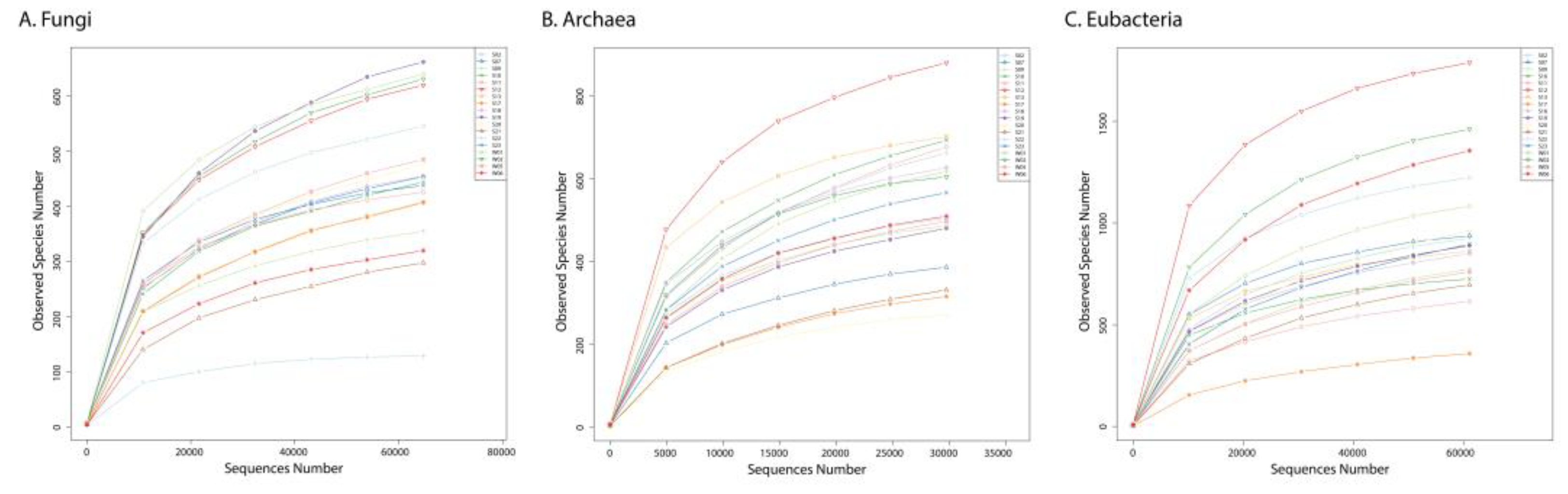
Figure 2.
Discrepancies between the three domains in the observed numbers of the total tags (A), and OTUs (B), as well as in the calculated Shannon (C), Simpson (D), Chao1 (E), and ACE (F) alpha diversity community richness estimators.
Figure 2.
Discrepancies between the three domains in the observed numbers of the total tags (A), and OTUs (B), as well as in the calculated Shannon (C), Simpson (D), Chao1 (E), and ACE (F) alpha diversity community richness estimators.
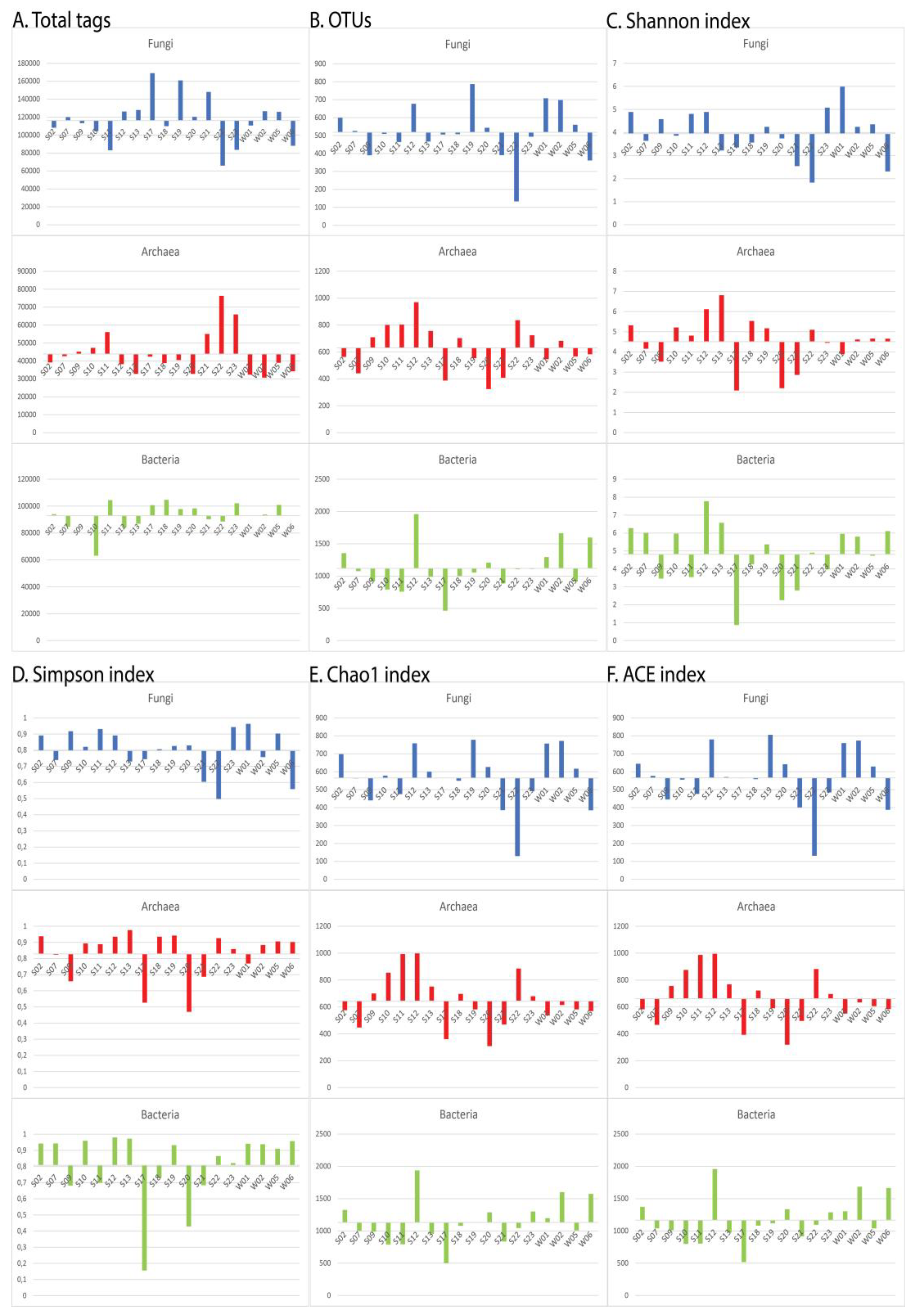

Table 1.
Sampling sites.
| Samples | Description | Coordinates |
|---|---|---|
| Solid samples (S) | ||
| S02 | Microbial surface contamination from a rock submerged in the lagoon. | -62.641324, -60.368854 |
| S09, S10, S11, S21, S23 | Microbial surface contamination from rocks submerged in meltwater ponds close to the research base. | -62.641450, -60.356733 |
| S07, S19 | Microbial surface contamination from a rock submerged in the littoral zone of Sea Lion Tarn. | -62.647727, -60.353677 |
| S17 | Microbial surface contamination from a rock inside the area of the research base. | -62.641241, -60.361171 |
| S20 | Microbial surface contamination from algae within the littoral zone of Sea Lion Tarn. | -62.647727, -60.353677 |
| S13 | Biomass from a lithotelm in Hannah Point | -62.653262, -60.607902 |
| S22 | Biomass from a rock, submerged in an unnamed lake near the research base. | -62.640819, -60.350725 |
| S12 | Soil from underneath a patch of vegetation near the nameless lake. | -62.640819, -60.350725 |
| S18 | Sediment from the littoral zone of Sea Lion Tarn. | -62.647727, -60.353677 |
| Water samples (W) | ||
| W01 | Water from the littoral zone of the lagoon. | -62.641324, -60.368854 |
| W02 | Water from the littoral zone of Sea Lion Tarn | -62.647727, -60.353677 |
| W05 | Water from the pelagic zone of Johnson Dock | -62.659572, -60.370434 |
| W06 | Water from the littoral zone of South Bay, near the base. | -62.638681, -60.367835 |
Table 2.
Sequencing statistics and alpha diversity indices values.
| Sample | Total tags | OTUs | Shannon | Simpson | Chao1 | ACE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | |
| S02 | 108303 | 39141 | 94003 | 599 | 563 | 1355 | 4,888 | 5,320 | 6,275 | 0,891 | 0,938 | 0,942 | 697,250 | 575,562 | 1325,202 | 644,266 | 582,952 | 1371,986 |
| S07 | 119964 | 42644 | 84660 | 526 | 441 | 1077 | 3,639 | 4,157 | 6,010 | 0,739 | 0,823 | 0,943 | 562,544 | 447,338 | 1003,306 | 576,516 | 466,759 | 1041,073 |
| S09 | 113287 | 45224 | 93056 | 390 | 709 | 920 | 4,579 | 3,521 | 3,455 | 0,917 | 0,659 | 0,682 | 440,429 | 700,038 | 989,007 | 445,363 | 756,005 | 1010,778 |
| S10 | 104399 | 47252 | 63121 | 509 | 801 | 792 | 3,857 | 5,213 | 5,967 | 0,821 | 0,894 | 0,959 | 578,200 | 854,182 | 789,629 | 555,830 | 875,150 | 798,890 |
| S11 | 83007 | 56071 | 104444 | 464 | 803 | 758 | 4,803 | 4,807 | 3,541 | 0,931 | 0,889 | 0,699 | 474,684 | 993,260 | 794,219 | 476,334 | 986,739 | 803,751 |
| S12 | 126350 | 38224 | 84187 | 677 | 970 | 1959 | 4,892 | 6,117 | 7,776 | 0,891 | 0,935 | 0,980 | 758,261 | 997,560 | 1936,242 | 779,822 | 994,997 | 1957,808 |
| S13 | 127940 | 32805 | 86878 | 467 | 756 | 989 | 3,218 | 6,811 | 6,573 | 0,730 | 0,976 | 0,972 | 599,820 | 751,688 | 958,425 | 570,035 | 767,540 | 975,776 |
| S17 | 169009 | 42381 | 100712 | 506 | 387 | 464 | 3,356 | 2,091 | 0,872 | 0,744 | 0,527 | 0,156 | 566,125 | 360,000 | 501,726 | 563,556 | 392,774 | 518,494 |
| S18 | 109955 | 38800 | 104705 | 508 | 703 | 999 | 3,566 | 5,533 | 4,262 | 0,806 | 0,935 | 0,729 | 549,721 | 696,966 | 1079,174 | 558,794 | 722,050 | 1083,829 |
| S19 | 160873 | 40505 | 97775 | 787 | 554 | 1055 | 4,247 | 5,170 | 5,366 | 0,826 | 0,942 | 0,932 | 778,059 | 581,346 | 1136,078 | 805,678 | 590,759 | 1119,067 |
| S20 | 120389 | 32799 | 98326 | 543 | 324 | 1209 | 3,742 | 2,203 | 2,252 | 0,829 | 0,470 | 0,428 | 626,217 | 308,983 | 1288,044 | 641,587 | 319,270 | 1336,556 |
| S21 | 148086 | 55083 | 90215 | 391 | 408 | 887 | 2,545 | 2,862 | 2,799 | 0,604 | 0,687 | 0,684 | 385,549 | 469,600 | 836,520 | 401,038 | 496,508 | 915,860 |
| S22 | 65967 | 76305 | 88563 | 133 | 836 | 1105 | 1,827 | 5,095 | 4,903 | 0,498 | 0,926 | 0,865 | 129,441 | 885,269 | 1044,691 | 131,693 | 882,408 | 1094,848 |
| S23 | 83584 | 65897 | 102063 | 494 | 724 | 1111 | 5,074 | 4,448 | 3,978 | 0,943 | 0,859 | 0,821 | 491,774 | 679,129 | 1301,684 | 484,776 | 696,083 | 1288,961 |
| W01 | 110722 | 32349 | 92749 | 708 | 546 | 1294 | 5,987 | 3,897 | 5,955 | 0,963 | 0,769 | 0,941 | 756,450 | 536,670 | 1196,763 | 759,495 | 550,773 | 1304,621 |
| W02 | 126520 | 30704 | 93799 | 698 | 682 | 1662 | 4,246 | 4,616 | 5,799 | 0,757 | 0,884 | 0,938 | 771,759 | 613,806 | 1600,936 | 773,188 | 634,555 | 1685,559 |
| W05 | 125937 | 38998 | 100912 | 560 | 567 | 915 | 4,355 | 4,660 | 4,740 | 0,903 | 0,906 | 0,910 | 616,724 | 581,394 | 1008,050 | 629,018 | 605,718 | 1040,591 |
| W06 | 88052 | 34246 | 92969 | 361 | 583 | 1597 | 2,312 | 4,655 | 6,110 | 0,559 | 0,902 | 0,957 | 384,800 | 568,860 | 1574,371 | 387,693 | 584,415 | 1665,36 |
| Average value | 116241 | 43857 | 92952 | 518 | 631 | 1119 | 3,952 | 4,510 | 4,81 | 0,797 | 0,829 | 0,808 | 564,878 | 644,536 | 1131,337 | 565,816 | 661,414 | 1167,434 |
Table 3.
Comparisons matrix based on the results represented in Figure 2.
Table 3.
Comparisons matrix based on the results represented in Figure 2.
| Sample | Total tags | OTUs | Shannon | Simpson | Chao1 | ACE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | Fungi | Archaea | Bacteria | |
| S02 | - | - | + | + | - | - | + | + | + | + | + | + | + | + | + | + | + | + |
| S07 | + | + | - | + | - | + | - | + | + | + | + | + | - | - | - | + | - | - |
| S09 | - | + | + | - | + | - | + | - | - | + | - | - | - | + | - | - | + | - |
| S10 | - | + | - | + | + | - | - | + | + | + | + | + | + | + | - | + | + | - |
| S11 | - | + | + | - | + | - | + | + | - | + | + | - | - | + | - | - | + | - |
| S12 | + | + | - | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| S13 | + | - | - | - | + | - | - | + | + | - | + | + | + | + | - | + | + | - |
| S17 | + | + | + | + | - | - | - | - | - | + | - | - | + | - | - | - | - | - |
| S18 | - | - | + | + | + | - | - | + | - | + | + | - | + | + | + | + | + | - |
| S19 | + | - | + | + | - | + | + | + | + | + | + | + | + | - | + | + | - | + |
| S20 | + | - | + | + | - | + | - | - | - | + | - | - | + | - | + | + | - | + |
| S21 | + | + | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| S22 | - | + | - | - | + | + | - | + | + | - | + | + | - | + | - | - | + | - |
| S23 | - | + | + | + | + | - | + | + | - | + | + | + | + | + | + | - | + | + |
| W01 | + | - | - | + | - | + | + | - | + | + | - | + | + | - | + | + | - | + |
| W02 | + | - | + | + | + | + | + | + | + | - | + | + | + | - | + | + | - | + |
| W05 | + | + | + | + | - | - | + | + | + | + | + | + | - | - | - | + | - | - |
| W06 | - | - | + | - | + | + | - | + | + | - | + | + | - | - | + | - | - | + |
Table 4.
Discrepancies between the three microorganisms’ domains.
| Total tags | OTUs | Shannon | Simpson | Chao1 | ACE | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of cases of discrepancies | |||||||||||
| Fungi vs Archaea | 10 | 12 | 8 | 8 | 8 | 10 | |||||
| Fungi vs Bacteria | 10 | 8 | 8 | 9 | 4 | 7 | |||||
| Archaea vs Bacteria | 10 | 10 | 4 | 3 | 10 | 11 | |||||
| Percentages of cases of discrepancies | |||||||||||
| Fungi vs Archaea | 56% | 67% | 44% | 44% | 44% | 56% | |||||
| Fungi vs Bacteria | 56% | 44% | 44% | 50% | 22% | 39% | |||||
| Archaea vs Bacteria | 56% | 56% | 22% | 17% | 56% | 61% | |||||
Table 5.
Correlation coefficients between the different groups of microorganisms and the different metrics.
Table 5.
Correlation coefficients between the different groups of microorganisms and the different metrics.
| Community Correlation | Effective Tags | OTUs | Shannon | Simpson | Chao1 | ACE |
|---|---|---|---|---|---|---|
| Fungi – Archaea | -0.481 | -0.137 | 0.071 | -0.029 | -0.179 | -0.202 |
| -0.389 | -0.290 | 0.055 | -0.151 | -0.115 | -0.201 | |
| Archaea – Bacteria | -0.003 | 0.290 | 0.802 | 0.833 | 0.170 | 0.108 |
| 0.034 | 0.057 | 0.688 | 0.618 | -0.022 | -0.057 | |
| Fungi – Bacteria | 0.060 | 0.310 | 0.187 | 0.020 | 0.344 | 0.341 |
| -0.084 | 0.375 | 0.110 | -0.136 | 0.395 | 0.455 | |
| The upper shows the Pearson correlation, while the number below presents the Spearman correlation. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



