
Submitted:
04 October 2023
Posted:
04 October 2023
You are already at the latest version
Abstract
Iron, a crucial element in our environment, plays a vital role in numerous natural processes. Understanding the presence and concentration of iron in the environment is very important, as it impacts various aspects of our planet’s health. On-site detection and speciation of iron are significant for several reasons. In this context, the present work aims to evaluate the applicability of voltammetry for on-site determination of iron and its possible speciation using a portable voltammetric analyzer. Voltammetry offers the advantage of convenience and cost-effectiveness. For iron (III) determination a modification of a glassy carbon electrode (GCE) with an antimony-bismuth film (SbBiFE) using acetate buffer (pH=4) as supporting electrolyte was used. The technique adopted was Square Wave Adsoptive Cathodic Stripping Voltammetry (SW-AdCSV) and we used 1-(2-piridylazo)-2-naphthol (PAN) as iron (III) ligand. Linearity, repeatability, detection limit and accuracy were determined using synthetic solutions, then a Standard Reference Material (SRM) 1643f - Trace Elements in Water was used for validation measurements in real matrix. The procedure was finally applied to real samples (tap, lake and sea water) and the results obtained were compared by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES). The results obtained demonstrated the excellent applicability of the proposed on-site voltammetric procedure for the determination of iron and its speciation in water.
Keywords:
iron
; Speciation
; Voltammetry
; Water
; Antimony-Bismuth Film
1. Introduction
Iron has both positive and negative impacts on the environment, depending on its form, concentration, and distribution [1,2]. Iron is an essential micronutrient for living organisms, including humans, plants, and animals. As for humans, it plays a crucial role in various biological processes, such as oxygen transport, energy production, and enzyme function. Furthermore, it is a component of soil minerals and plays a vital role in maintaining soil fertility [3,4]. It affects soil structure, pH balance, and nutrient availability [1]. Adequate iron levels promote healthy plant growth and development. On the other hand, high concentrations of iron in water can lead to contamination. Iron-rich groundwater or surface water can discoloration, present a reddish or brownish tint. This iron-contaminated water may be unsuitable for certain uses, such as drinking, irrigation, or industrial processes.
Iron is present in waters in concentrations that vary greatly among sea, lake or drinking water. Soluble iron is found in seawater in very low concentrations (nanomolar or picomolar) due to some phenomena, such as particle scavenging, low solubility and effective removal caused by biological absorption. These processes are less present in other waters, such as lake water or drinking water, in which iron concentrations are higher [5,6,7,8].
Iron also has a relevant influence on human health: whereas it is an essential nutrient required for various physiological processes, its dysregulation can contribute to the pathogenesis of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease [9,10,11,12,13]. In recent years many studies have been carried out in order to understand its role and facilitate its determination, also as a marker of such diseases [10].
Electrochemical methods are widely used for iron determination and speciation due to their sensitivity, selectivity, and ability to provide real-time measurements [4,14]. Over the years, different voltammetric techniques and different iron complexants, able to enhance the selectivity and sensitivity of the response, have been tested. Differential Pulse Voltammetry (DPV), Cyclic Voltammetry (CV) and Square Wave Voltammetry (SWV) are the most commonly used techniques [15,16,17,18,19]. A wide range of complexing agents were used, such as 1-nitroso-2-naphthol (NN) [20], 4,4′-[3-(2-pyridyl)1,2,4-triazine-5,6-diyl]bis(benzene sulfonic acid) disodium salt hydrate (ferrozine, FZ) [21], dihydroxynaphthalene (DHN) [22,23], 2-(5-bromo-2-pyridylazo)-5-diethylaminophenol (5-Br-PADAP) [24] and 2-(2-thiazolylazo)-p-cresol (TAC) [25]. Another fundamental aspect is the use of suitably modified working electrodes (WE): in the literature there are some important examples, such as chemically-modified carbon-paste electrodes (CMCPEs) [26,27], nafion coated-electrodes (NCE) [28], thick-film graphite-containing electrode modified with calomel (TFGME) [29], bismuth-coated glassy carbon electrode (BiFE) [30,31], gold and bismuth bimetallic nanoparticles decorated with L-cysteine-functionalized graphene oxide nanocomposites (Au-BiNPs/SH-GO) [32] and gold 2-mercaptosuccinic acid self-assembled monolayers (AuMSA SAM) [33]. The use of electrode-specific modifiers and complexants greatly helps increase the selectivity and sensitivity of these voltammetric methods.
An advantage of voltammetry is the availability of portable instruments, which enables on-site measurements, useful in various fields, such as clinical practice, quality control and environmental monitoring.
The aim of this study was to develop a new portable voltammetric method for the on-site determination of iron (III) in different types of water. The work is focused on the use of a new antimony-bismuth film modified glassy carbon electrode (SbBiFE-GCE), coupled with 1-(2-piridylazo)-2-naphthol (PAN) as ligand for iron (III). All the analysis were carried out in triplicate, with the use of a portable potentiostat and tested outside laboratories.
2. Material and method
2.1. Instruments
Square Wave Adsoptive Cathodic Stripping Voltammetry (SW-AdCSV) was carried out with a Palmsens 4 portable potentiostat (PalmSens, Houten, the Netherlands). A classic electrochemical cell was equipped with a glassy carbon WE (GCE, 2-mm diameter, Metrohm, Herisau, Switzerland), Ag/AgCl/KCl 3 mol L−1 reference electrode (RE) and auxiliary platinum electrode. A mechanical stirrer (IKA-Topolino, Staufen, Germany) was connected to the PalmSens4, powered by a portable battery for on-site analysis. A Cyberscan 2100 pH meter (Eutech Instruments srl, Thermo Fisher Scientific, Waltham, Massachusetts, USA) was used to adjust solution pH, the pH meter was calibrated daily with pH 4 and pH 7 buffers. An Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES), in particular Perkin Elmer Optima 7000 (Perkin Elmer, Norwalk, Connecticut, USA), was used for the measurement of total iron in the test analysis.
2.2. Reagents
All solutions were prepared with high-purity water (HPW-Milli-Q, Millipore, 18.2 MΩ cm, Milford, MA, USA). Antimony and bismuth standard solutions were obtained by dilution from concentrated (1000 mg L−1) Merck TraceCERT stock solutions (Merck, Darmstadt, Germany). Ammonium iron (II) sulfate hexahydrate and iron (III) nitrate nonahydrate (Sigma Aldrich-Merck, Darmstadt, Germany) were used for the preparation respectively of iron (II) and iron (III) standards. Standard solutions were prepared for both analytes at 1000 mg L-1 concentration and subsequently diluted to 1 mg L-1. Acetate buffers (0.1 mol L-1, pH 3.0–6.0) were prepared using potassium acetate and acetic acid (Sigma Aldrich-Merck, Darmstadt, Germany). A 0.001 mol L−1 solution of PAN (Sigma Aldrich-Merck, Darmstadt, Germany) was prepared in ethanol. Standard Reference Material (SRM) 1643f Trace Elements in Water (NIST, National Institute of Standards and Technology, Gaithersburg, Maryland, USA) with a total iron certified concentration of 93.44 ± 0.78 µg L-1 was used for validation measurements.
2.3. Preparation of –SbBiFE-GCE
Before use the GCE was sequentially polished with three different alumina powders, having different granulometry, namely 0.05, 0.3 and 1 µm (CH Instruments, Austin, Texas, USA) on a microcloth pad (BASi, West Lafayette, Indiana, USA) and then rinsed with HPW and ethanol. This mechanical cleaning of the GCE was carried out daily.
After that, GCE was modified by the formation of a double film of bismuth and antimony in two successive steps. All parameters for film deposition and the amount of bismuth and antimony have been optimised and are reported below.
The first step is the deposition of the antimony film; in particular, a cell containing 0.1 mol L-1 acetate buffer at pH 4.5 and 10 mL of solution with antimony concentration of 50 mg L-1 was prepared. A -1.0 V deposition potential was applied for 300 seconds.
The second step, analogously to the first one, allows the subsequent deposition of bismuth onto the WE surface in the same supporting electrolyte, but with a bismuth concentration equal to 100 mg L-1 in the 10 mL cell. The parameters for the deposition were the same as those adopted for antimony.
2.4. Parameters for iron determination
For iron determination in synthetic solution a cell containing 10 mL of 0.1 mol L-1 acetate buffer (pH 4) with an optimized and known concentration of PAN (5 µmol L-1) was used.
For the analysis of the certified sample, the cell consisted of 0.5 mL of sample, 9.5 mL of acetate buffer 0.1 mol L-1 (pH 4) and an amount 50 µL of PAN (final concentration 5 µmol L-1 in cell). The concentration was quantified with the standard addition method, by adding two successive aliquots of 5 μg L-1 of iron (III). The test was carried out in triplicate.
The lake water was collected in Avigliana Lake (TO, Italy), while the sea water was sampled in Savona (Italy). The water samples were sampled using 1L plastic bottles, storing them in the fridge and carrying out the analysis of the sample within 24h of collection.
For these samples analyses were carried out using a voltammetric cell containing 0.25 mL of sample, 9.75 mL of 0.1 mol L-1 acetate buffer (pH 4) and 5 µmol L-1 PAN. Then two standard additions of iron (III) (10 μg L-1 each) were made, only two standard additions were made to the samples due to the limited durability of the film.
Table 1 shows the SWAdSV parameters optimised for the determination of iron (III) and which will be used in all subsequent tests.
3. Results and discussion
The aim of this paper was to develop a method that would allow to selectively analyze iron (III) present in different matrices directly in the field.
The decision to modify the electrode with a film of Sb-Bi was made starting from the study of Segura et al [30], where only Bi was used, integrating with other recent studies of using Sb for the modification of WE.
The optimization of the conditions for film formation was carried out by testing different concentrations of Bi and Sb and different deposition times. As for the potential of deposition and the support electrolyte, it was decided to use those reported in some papers regarding only the bismuth film [30,31]. The tests were carried out by evaluating the accuracy for the determination of a concentration of 5 µg L-1 of iron (III) in a synthetic solution (10 mL of 0.1 mol L-1 acetate buffer (pH 4) and 5 µmol L-1 PAN).
In particular, the conditions reported in Table 2 were tested.
In the tests 2-3, using 300 and 200 mg L-1 of Bi and Sb respectively, with a deposition time of 120 and 30 s, the film does not stick to the WE surface, and iron has not been determined (n.d. in Table 2); with 300 seconds (test 1) the results are better, but the film is thicker and the sensitivity (data not showed) is worse than the analogue with lower concentrations of Bi and Sb (test 4), as well as the obtained recovery.
Tests 7-8-9 have similar problems: with short deposition times iron was not determinable. The best results were obtained with test number 4, with a percentage recovery of 95.3 %.
All the preliminary tests were made first on synthetic solutions (Table 2) and then on a SRM 1643f using the SWAdSW parameters reported in Table 1.
To obtain good results in term of sensitivity different concentrations of PAN were tested, namely 1, 5 and 10 µmol L-1. The concentration that permits the best response was 5 µmol L-1 in cell, confirming the findings by Segura et al [30]; tests made at higher and lower concentrations (10 µmol L-1 and 1 µmol L-1) gave rise to a worsening of the performance of the method. In particular, with a higher concentration we lost sensitivity (87.5 % recovery) and with a lower concentration iron was not displayed.
Tests were also made using a standard of iron (II), showing an absence of signal and consequently certifying the selectivity of the method and of the complexant used for iron (III) in the experimental conditions adopted.
In the second step of the work samples were analysed for total iron, as described in sections 3.2 and 3.3, after addition of nitric acid in order to oxidize any iron (II) present.
The results obtained for total iron were also confirmed by another analytical technique, namely ICP - OES.
3.1. Synthetic solutions
The first tests were carried out on synthetic solutions. The electrochemical cell was composed of 10 mL of 0.1 mol L-1 acetate buffer (pH 4) and 5 µmol L-1 PAN to which subsequent aliquots of iron (III) were added.
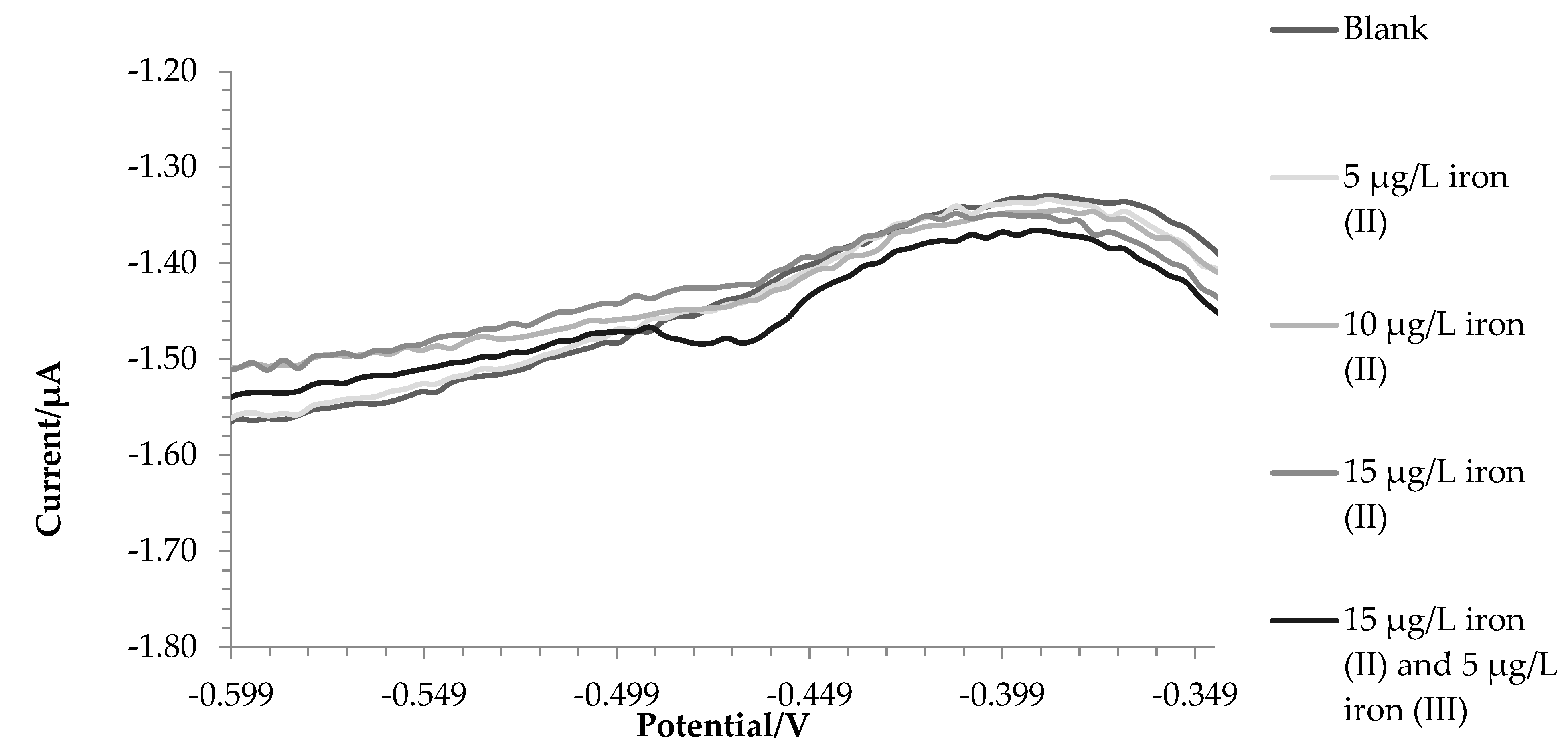
Figure 1 reports voltammograms for the blank and 4 successive additions of iron (III). The analytical signal was the current intensity registered for the potential of -0.475 V.
The results obtained are shown in Table 3.
Figure 2.
Voltammograms of the blank solution, successive additions of iron (II) and an addition of iron (III).
Figure 2.
Voltammograms of the blank solution, successive additions of iron (II) and an addition of iron (III).
The true iron concentration of the home-made standards used for the preparation of synthetic solutions have been checked by ICP-OES by the analysis of the total iron present in solution. The concentrations were computed by means of an external calibration obtained by dilution from concentrated (1000 mg L−1) Sigma-Aldrich standard solution. Results obtained, shown in Table 4 confirmed the reliability of the iron (III) and iron (II) standards.
3.2. Certified reference material
After that, the accuracy of the method was tested on a Standard Reference Material (SRM) 1643f - Trace Elements in Water, with total iron certified concentration of 93.44 ± 0.78 µg L-1.
The iron (III) concentration in SRM was estimated by standard addiction method as reported before. The accuracy of the technique was found to be excellent, since we found 96.40 ± 2.45 µg L-1 of iron with a recovery of 103.16%. The SRM 1643f contain HNO3 as stabilizer, therefore it is not surprising to find all the iron as iron (III).
Figure 3 shows the voltammogram for this test.
3.3. Water samples
After optimization and verification of effectiveness of the method, it was tested on real samples of water (tap water, sea water and lake water).
For lake water, the same sample was analysed both in the laboratory and in the field to demonstrate the applicability of the method outside the laboratory.
For lake and tap water the method responded excellently, in detail the tests were carried out in triplicate and the comparison with ICP-OES was also made. Before the analysis, nitric acid (final concentration0.01 mol L-1) was added to the water samples. In this way, all the iron in solution is present as iron (III) and a direct comparison can be performed between voltametric and ICP-OES analysis.
The amount of iron found was 171.7 ± 3.8 µg L-1 using voltammetry in the first one and 187.5 ±- 5.7 µg L-1 with ICP-OES. For tap water, 207.8 ± 6.6 µg L-1 and 200.9 ± 1.5 µg L-1 were found by voltammetry and ICP-OES respectively. A T test has been done to assess whether the results obtained with the two techniques are significantly different. For tap water, there are not significant differences between the two data (p value = 0.14). In the case of lake water the values are statistically different (p value = 0.03), this is probably due to the fact that being a more complex matrix not all iron (II) has been oxidized to iron (III), in fact the value at the ICP-OES, which evaluates the total iron, turns out to be higher.
However, it was not possible to determine the concentration of iron in sea water, probably due to the fact that the concentration of iron is presumably lower than the LOQ of the proposed technique. As an example, seawater from eastern Mediterranean sea contains around 83 ng L-1 of iron [34]. In any case standard additions are displayed and the instrumental LOQ is 2.07 μg L-1.
The technique was found to be very accurate for water analysis even in the field; the lake water was also analyzed directly on-field and the result was 169.8 ± 4.1 µg L-1 compared to the 171.7 ± 3.8 µg L-1 of the laboratory. However, the greatest limit of the procedure is the scarce duration of the antimony and bismuth film. In fact, it was seen that it is necessary to regenerate it every three samples in order not to lose sensitivity.
4. Conclusion
The proposed method has been shown to be excellent for the determination of iron (III) in water, using a method also applicable in the field. This can allow a potential increase in water controls in case of iron contamination even directly on site by optimizing the method is the need to regenerate the Bi/Sb film after the analysis of three samples. Anyway, the time required for a complete voltammetric analysis, including film deposition, is 30 minutes: this value is acceptable, and a throughput of 25 samples for 8 hours of operation is expected.
The method is suitable also for speciation studies, considering that iron (II) is not detected using the proposed experimental conditions The quantification of iron (II) can be performed indirectly by difference of the results obtained by the analysis of two aliquots of the same sample with and without the spike of an oxidant reagent that convert all the iron in the iron (III) form (e.g., HNO3).
Future development of this study will be testing the applicability of the procedure also to the analysis of biological samples (such as plasma or serum). Even more challenging is the detection of iron in the CerebroSpinal Fluid (CSF), ranging from about 30 in controls and 70 μg L-1 in AD patients [35]. The determination of iron in CSF samples is carried out by means of Graphite Furnace Atomic Absorption Spectrometer (GF-AAS), which is a very precise but time requiring method. The possibility of on-site analysis will enable researchers to have rapid and immediate monitoring of iron concentration, testing it suitability as a possible marker for early detection of neurodegenerative diseases.
Author Contributions
Authors contribution: Conceptualization: Paolo Inaudi (P.I.), Agnese Giacomino (A.G.); Formal analysis and investigation: Paolo Inaudi (P.I.), Monica Argenziano (M.A.), Writing - original draft preparation: Paolo Inaudi (P.I.), Laura Favilli (L.F.); Writing – review and editing: Stefano Bertinetti (S.B.), Mery Malandrino (M.M.), Caterina Guiot (C.G.), Ornella Abollino (o.A.); Supervision: Agnese Giacomino (A.G.), Ornella Abollino (O.A.).
Funding
This work is financially supported by CRT Foundation - Ordinary Demand Disbursements 2021.
Data Availability Statement
Data, associated metadata, and calculation tools are available from the corresponding author (paolo.inaudi@unito.it).
Conflicts of Interest
the authors declare no conflict of interest.
References
- Nagajyoti, P.C.; Lee, K.D.; Sreekanth, T.V.M. Heavy metals, occurrence and toxicity for plants: a review. Environ. Chem. Lett. 2010, 8, 199–216. [Google Scholar] [CrossRef]
- Statham, P.J.; Jacobson, Y.; Berg, C.v.D. The measurement of organically complexed FeII in natural waters using competitive ligand reverse titration. Anal. Chim. Acta 2012, 743, 111–116. [Google Scholar] [CrossRef]
- Hopwood, M.J.; Birchill, A.J.; Gledhill, M.; Achterberg, E.P.; Klar, J.K.; Milne, A. A Comparison between Four Analytical Methods for the Measurement of Fe(II) at Nanomolar Concentrations in Coastal Seawater. Front. Mar. Sci. 2017, 4, 192. [Google Scholar] [CrossRef]
- Lu, M.; Rees, N.V.; Kabakaev, A.S.; Compton, R.G. Determination of Iron: Electrochemical Methods. Electroanalysis 2012, 24, 1693–1702. [Google Scholar] [CrossRef]
- Liu, X.; Millero, F.J. The solubility of iron in seawater. Mar. Chem. 2002, 77, 43–54. [Google Scholar] [CrossRef]
- Bowie, A.R.; Maldonado, M.T.; Frew, R.D.; Croot, P.L.; Achterberg, E.P.; Mantoura, R.C.; Worsfold, P.J.; Law, C.S.; Boyd, P.W. The fate of added iron during a mesoscale fertilisation experiment in the Southern Ocean. Deep. Sea Res. Part II: Top. Stud. Oceanogr. 2001, 48, 2703–2743. [Google Scholar] [CrossRef]
- Wells, M.L. The level of iron enrichment required to initiate diatom blooms in HNLC waters. Mar. Chem. 2003, 82, 101–114. [Google Scholar] [CrossRef]
- Boyd, P.W.; Ellwood, M.J. The biogeochemical cycle of iron in the ocean. Nat. Geosci. 2010, 3, 675–682. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef]
- Jasiecki, J.; Targońska, M.; Wasąg, B. The Role of Butyrylcholinesterase and Iron in the Regulation of Cholinergic Network and Cognitive Dysfunction in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2033. [Google Scholar] [CrossRef]
- Kim, N.; Lee, H.J. Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7697. [Google Scholar] [CrossRef] [PubMed]
- Laglera, L.M.; Monticelli, D. Iron detection and speciation in natural waters by electrochemical techniques: A critical review. Curr. Opin. Electrochem. 2017, 3, 123–129. [Google Scholar] [CrossRef]
- Cuculić, V.; Pižeta, I.; Branica, M. Voltammetry of Dissolved Iron(III)–Nitrilotriacetate–Hydroxide System in Water Solution. Electroanalysis 2005, 17, 2129–2136. [Google Scholar] [CrossRef]
- Abualhaija, M.M.; Berg, C.M.v.D. Chemical speciation of iron in seawater using catalytic cathodic stripping voltammetry with ligand competition against salicylaldoxime. Mar. Chem. 2014, 164, 60–74. [Google Scholar] [CrossRef]
- Demir, E.; Göktug, Ö.; İnam, R.; Doyduk, D. Development and characterization of iron (III) phthalocyanine modified carbon nanotube paste electrodes and application for determination of fluometuron herbicide as an electrochemical sensor. J. Electroanal. Chem. 2021, 895, 115389. [Google Scholar] [CrossRef]
- Vukosav, P.; Mlakar, M. Speciation of biochemically important iron complexes with amino acids: L-aspartic acid and L-aspartic acid - glycine mixture. Electrochimica Acta 2014, 139, 29–35. [Google Scholar] [CrossRef]
- van Huis, A. Edible insects: future prospects for food and feed security; in FAO forestry paper, no. 171; Food and Agriculture Organization of the United Nations: Rome, 2013. [Google Scholar]
- Gledhill, M.; Berg, C.M.v.D. Measurement of the redox speciation of iron in seawater by catalytic cathodic stripping voltammetry. Mar. Chem. 1995, 50, 51–61. [Google Scholar] [CrossRef]
- Iwai, H.; Fukushima, M.; Yamamoto, M. Determination of Labile Fe(II) Species Complexed with Seawater Extractable Organic Matter Under Seawater Conditions Based on the Kinetics of Ligand-exchange Reactions with Ferrozine. Anal. Sci. 2013, 29, 723–728. [Google Scholar] [CrossRef]
- Laglera, L.M.; Caprara, S.; Monticelli, D. Towards a zero-blank, preconcentration-free voltammetric method for iron analysis at picomolar concentrations in unbuffered seawater. Talanta 2016, 150, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Sanvito, F.; Pacileo, L.; Monticelli, D. Fostering and Understanding Iron Detection at the Ultratrace Level by Adsorptive Stripping Voltammetry with Catalytic Enhancement. Electroanalysis 2019, 31, 212–216. [Google Scholar] [CrossRef]
- Ghoneim, M.M.; Hassanein, A.M.; Hammam, E.; Beltagi, A.M. Simultaneous determination of Cd, Pb, Cu, Sb, Bi, Se, Zn, Mn, Ni, Co and Fe in water samples by differential pulse stripping voltammetry at a hanging mercury drop electrode. Anal. Bioanal. Chem. 2000, 367, 378–383. [Google Scholar] [CrossRef]
- Croot, P. L.; Johansson, M. Determination of Iron Speciation by Cathodic Stripping Voltammetry in Seawater Using the Competing Ligand 2-(2-Thiazolylazo)-p-cresol (TAC). Electroanalysis 2000, 12, 565–576. [Google Scholar] [CrossRef]
- Gao, Z.; Li, P.; Zhao, Z. Determination of iron(II) with chemically-modified carbon-paste electrodes. Talanta 1991, 38, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Gholivand, M.B.; Geravandi, B.; Parvin, M.H. Anodic Stripping Voltammetric Determination of Iron(II) at a Carbon Paste Electrode Modified with Dithiodianiline (DTDA) and Gold Nanoparticles (GNP). Electroanalysis 2011, 23, 1345–1351. [Google Scholar] [CrossRef]
- Ugo, P.; Moretto, L.; De Boni, A.; Scopece, P.; Mazzocchin, G. Iron(II) and iron(III) determination by potentiometry and ion-exchange voltammetry at ionomer-coated electrodes. Anal. Chim. Acta 2002, 474, 147–160. [Google Scholar] [CrossRef]
- Stozhko, N.Y.; Inzhevatova, O.V.; Kolyadina, L.I. Determination of Iron in Natural and Drinking Waters by Stripping Voltammetry. J. Anal. Chem. 2005, 60, 668–672. [Google Scholar] [CrossRef]
- Segura, R.; Toral, M.I.; Arancibia, V. Determination of iron in water samples by adsorptive stripping voltammetry with a bismuth film electrode in the presence of 1-(2-piridylazo)-2-naphthol. Talanta 2008, 75, 973–977. [Google Scholar] [CrossRef]
- Bobrowski, A.; Nowak, K.; Zarębski, J. Application of a bismuth film electrode to the voltammetric determination of trace iron using a Fe(III)–TEA–BrO3− catalytic system. Anal. Bioanal. Chem. 2005, 382, 1691–1697. [Google Scholar] [CrossRef]
- Zhou, N.; Li, J.; Wang, S.; Zhuang, X.; Ni, S.; Luan, F.; Wu, X.; Yu, S. An Electrochemical Sensor Based on Gold and Bismuth Bimetallic Nanoparticles Decorated L-Cysteine Functionalized Graphene Oxide Nanocomposites for Sensitive Detection of Iron Ions in Water Samples. Nanomaterials 2021, 11, 2386. [Google Scholar] [CrossRef] [PubMed]
- Shervedani, R.K.; Hatefi-Mehrjardi, A.; Asadi-Farsani, A. Sensitive determination of iron(III) by gold electrode modified with 2-mercaptosuccinic acid self-assembled monolayer. Anal. Chim. Acta 2007, 601, 164–171. [Google Scholar] [CrossRef]
- Statham, P.J.; Hart, V. Dissolved iron in the Cretan Sea (eastern Mediterranean). Limnol. Oceanogr. 2005, 50, 1142–1148. [Google Scholar] [CrossRef]
- Ficiarà, E.; Boschi, S.; Ansari, S.; D'Agata, F.; Abollino, O.; Caroppo, P.; Di Fede, G.; Indaco, A.; Rainero, I.; Guiot, C. Machine Learning Profiling of Alzheimer's Disease Patients Based on Current Cerebrospinal Fluid Markers and Iron Content in Biofluids. Front. Aging Neurosci. 2021, 13, 607858. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Voltammograms of the blank solution and successive additions of iron (III).
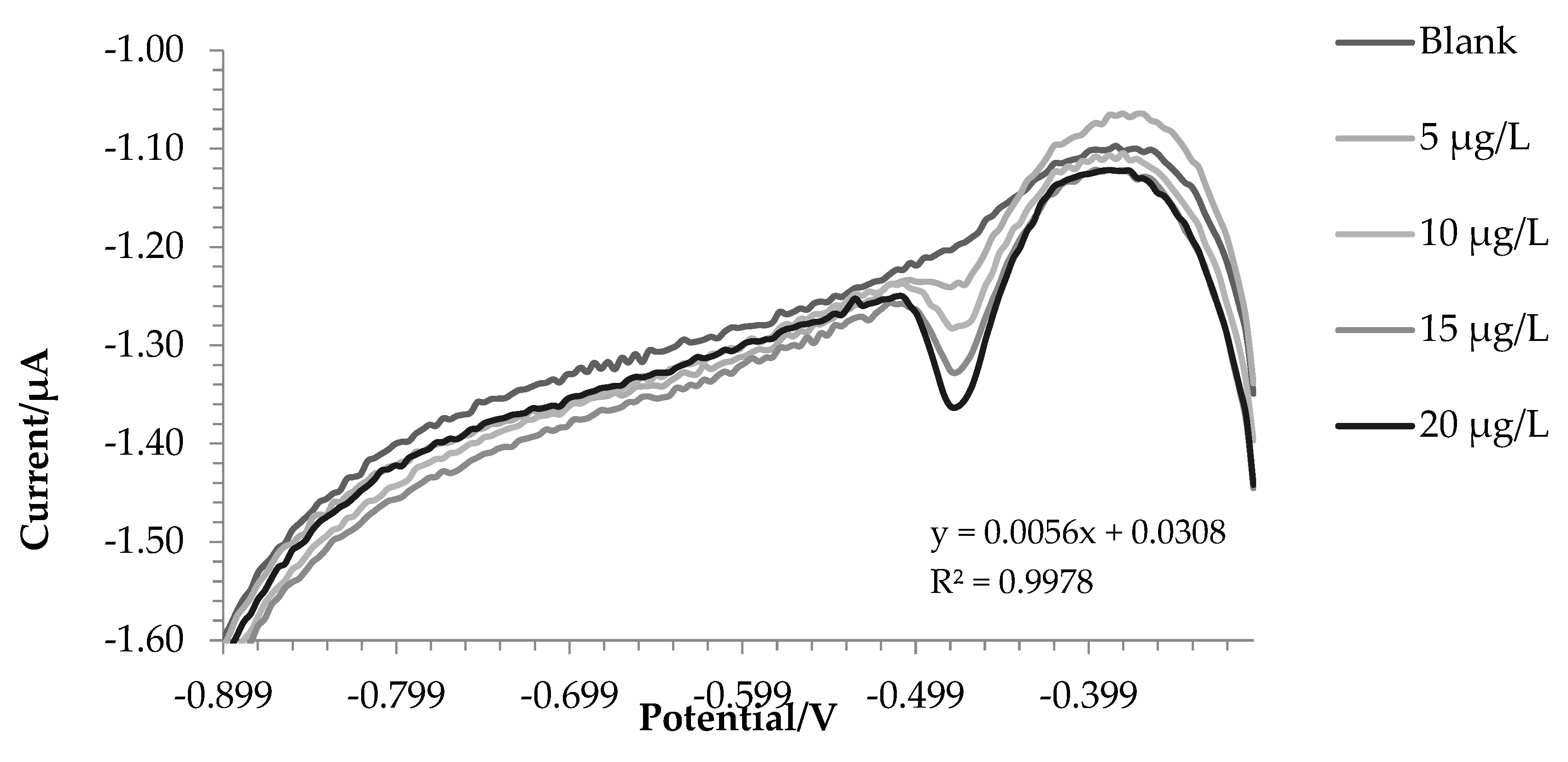
Figure 3.
Voltammograms of SRM and two standard additions of Fe(III).
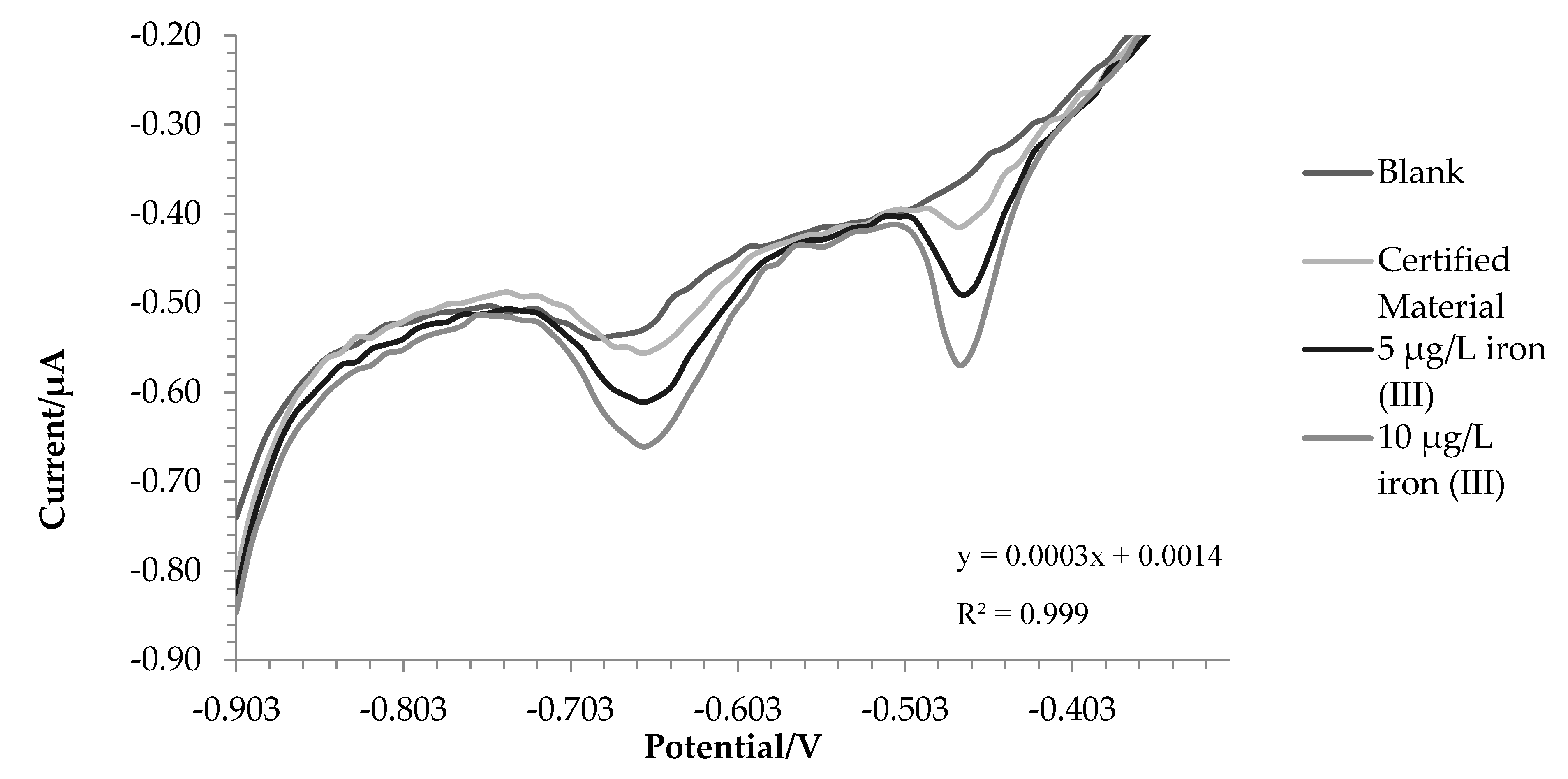
Figure 4.
Voltammograms recorded for lake water analysis.
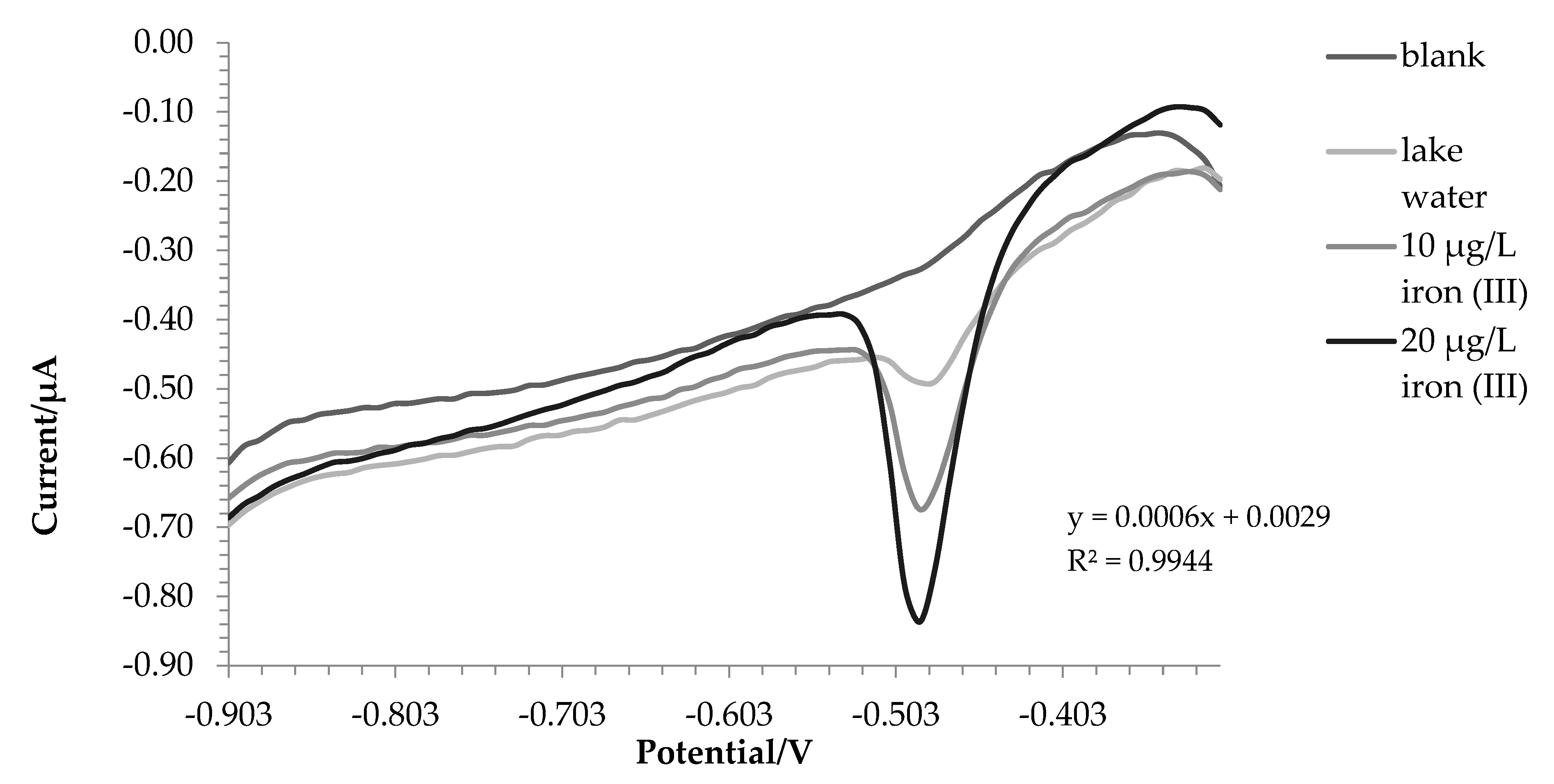
Figure 5.
Voltammograms recorded for tap water analysis.
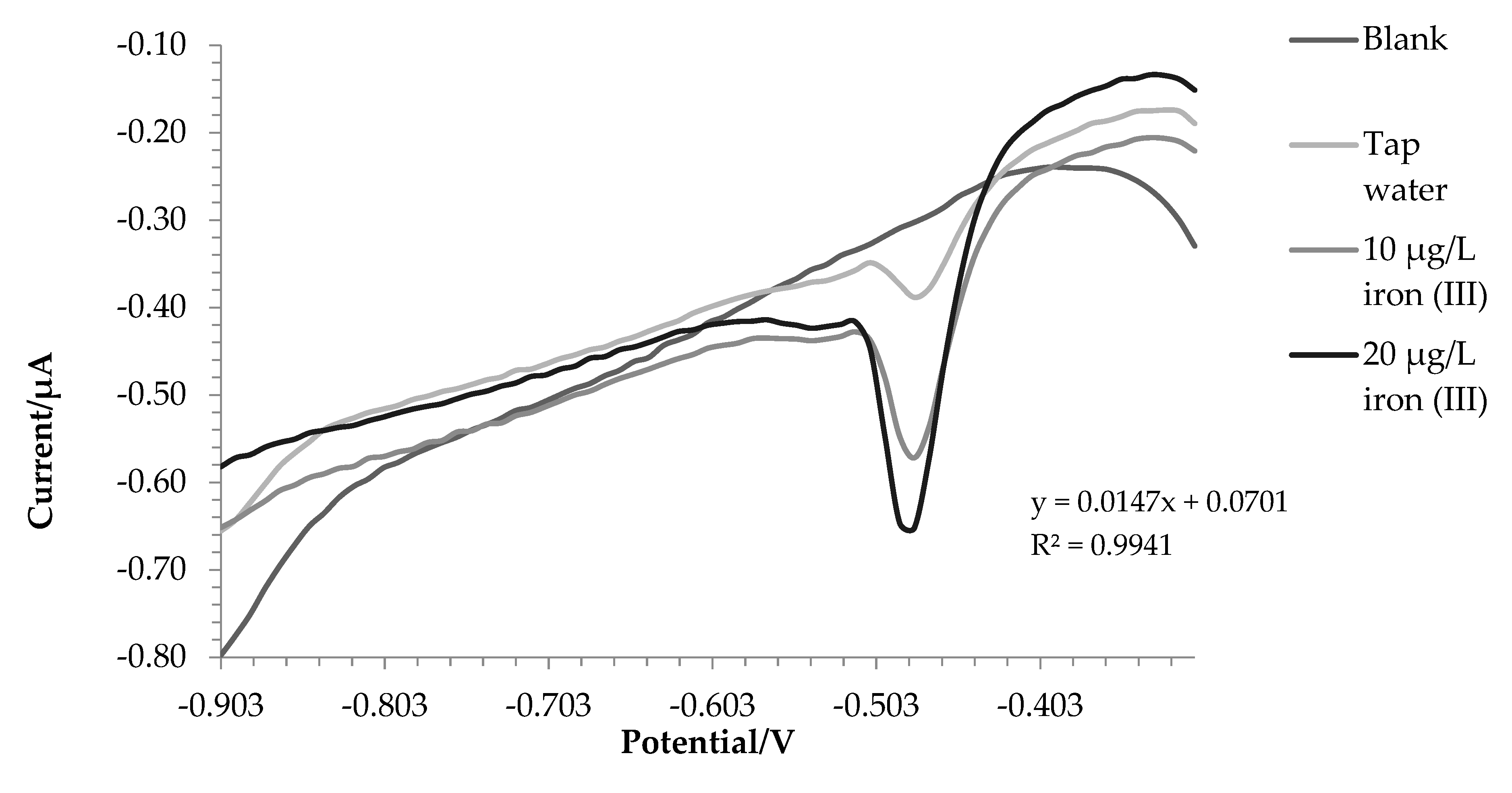

Table 1.
SWV optimized parameters.
| Analyte deposition settings | |
| Edep | -0.4 V |
| tdep | 180 s |
| SWV Settings | |
| teq | 10 s |
| Ebegin | -0.3 V |
| Eend | -0.9 V |
| Estep | 0.004 V |
| Amplitude | 0.025 V |
| Frequency | 10.0 Hz |
Table 2.
Tests for best SbBiFE performance.
| Test | Bi concentration (mg L-1) | Sb concentration (mg L-1) | Deposition time (s) | Accuracy (% of recovery) |
| 1 | 300 | 200 | 300 | 76.7 |
| 2 | 300 | 200 | 120 | n.d. |
| 3 | 300 | 200 | 30 | n.d. |
| 4 | 100 | 50 | 300 | 95.3 |
| 5 | 100 | 50 | 120 | 89.8 |
| 6 | 100 | 50 | 30 | 88.7 |
| 7 | 20 | 10 | 300 | 80.5 |
| 8 | 20 | 10 | 120 | n.d. |
| 9 | 20 | 10 | 30 | n.d. |
Table 3.
Figures of merit of the developed method.
| WE | Analyte | Repeatability (RSD %, n=3) | Linearity | LOD* (μg L-1) | LOQ * (μg L-1) | Accuracy (% recovery) |
|---|---|---|---|---|---|---|
| SbBiFE | Iron (III) | 3.05 | y = 0.056x + 0.0308 R² = 0.9978 | 0.54 | 1.78 | 95.4 |
* Limit of Detection (LOD) and Limit of Quantification (LOQ) were estimated as 3 and 10 times the standard deviation of the blank (n=3), respectively.
Table 4.
Analysis of prepared iron standards vs ICP-OES.
| Analyte | Theoretical concentration prepared (μg L-1) | Results with ICP-OES (μg L-1) | Recovery (%) |
| Iron (II) | 1000 | 1058 | 105.8 |
| Iron (III) | 1000 | 1034 | 103.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



