
Submitted:
04 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
In this research, we present a comprehensive methodology to categorize perfumes based on their fragrance profiles and subsequently aid in creating innovative odoriferous molecules using advanced neural networks. Drawing from data on Parfumo and the Good Scents Company webpage (Parfumo, 2008; The Good Scents Company, 2021), the study employs sophisticated web scraping techniques to gather diverse perfume attributes. Following this, a k-means algorithm is applied for perfume clustering, paving the way for recommending similar scents to consumers. The process then bridges customer preferences to molecular design by incorporating their feedback into generating new molecules via graph neural networks (GNNs). Through converting the Simple Molecular Input Line Entry System (SMILES) representation into graph structures, the GNN facilitates the creation of new molecular designs attuned to consumer desires. The proposed approach offers promising avenues for consumers to pinpoint similar perfume choices, incorporating feedback, and for manufacturers to conceptualize new fragrant molecules with a high likelihood of market resonance.
Keywords:
Scientific Machine Learning
; Perfume Engineering
; Graph Neural Networks
; Fragrances
; Consumer Feedback
1. Introduction
The human sense of smell is a potent tool that has contributed to our species' survival for millions of years. It equips us to discern noxious odors, such as cadaverine and skatole, typically linked with spoiled food or potentially harmful substances. Evolutionarily, we've learned to associate these smells with danger, preventing possible health risks from ingestion. Conversely, appealing fragrances like vanillin and linalool can attract, stimulate, and even evoke powerful emotional memories.
Recent discoveries suggest that human olfaction can distinguish over a trillion unique scents (Bushdid et al., 2014). The intricacies of how we identify and interpret these chemicals remain partly a mystery. Nevertheless, our fascination with pleasant scents has spawned a lucrative industry over the centuries (Leffingwell & Associates, 2018). In 2017, the Flavors and Fragrances (F&F) industry was worth over 26 billion US$. Despite a market dip of 8.4% in 2020 due to the COVID-19 pandemic, projections estimate its value at over 36 billion US$ by 2029 (Fortune Business Insight, 2022). This industry caters to various products, ranging from bar soaps to luxury perfumes, the latter being distinguished by unique customer preferences, greater value, and a sophisticated development process.
A perfume is essentially a concoction of chemicals dissolved in a solvent, commonly ethanol. These chemicals create the distinct fragrance notes we perceive. These notes are generally categorized into three tiers: top, middle, and base (Carles, 1961). Top notes, although captivating, are fleeting, giving the first whiff of the perfume and fading within minutes. The heart or middle notes form the perfume's core scent, lingering for several hours. Base notes, enduring from hours to days, can be likened to the aftertaste in food or drink. The longevity and potency of each fragrance note hinge on thermodynamics, transport phenomena, and psychophysics (Rodrigues et al., 2021). For a scent to be registered, molecules must become airborne and reach our nasal receptors. Several studies delve into how psychophysical models can assess human reactions to evolving scents (Almeida et al., 2019; Mata et al., 2005; Teixeira et al., 2009; Wakayama et al., 2019). Within this framework, the budding discipline of Perfume Engineering has taken root. (Rodrigues et al., 2021)
Despite the consistent growth in the perfume industry and the emergence of Perfume Engineering, there remains a noticeable gap in scientific literature offering tools for innovation in this field. The prevailing method for creating new fragrances largely depends on the traditional trial-and-error approach. This is a time-consuming and expensive procedure, possibly rooted in the perception that perfume crafting is an art heavily reliant on manual processes (Santana et al., 2021).
Currently, the F&F industry boasts a repertoire of approximately 10,000 essential oils and scents. The development of a new perfume typically involves a staggering 1,000 tests using the available fragrances. Such an intricate procedure can span up to three years and incur costs reaching US$ 50,000 per kg of the perfume (Rodrigues et al., 2021).
However, the field is not without advancements. Several studies have illuminated the potential of using machine learning in tandem with natural language processing to classify the aroma of unidentified molecules (Debnath & Nakamoto, 2022; Gerkin, 2021; Nozaki & Nakamoto, 2018; Saini & Ramanathan, 2022). While this approach expands the fragrance palette for creators, it doesn't offer insights into optimal fragrance combinations. Santana et al. (2021) touted machine learning as an alternative to phenomenological models for perfume engineering. Their work paves the way for exploring myriad combinations for fragrance creation, yet like the phenomenological models, it leans on a predefined set of compounds. These models primarily determine the concentration of compounds to achieve a specific fragrance spectrum.
Research from other domains reveals that computer-aided molecular design can predict how a molecule relates to a target property (Queiroz et al., 2023c, 2023b, 2023a). This revelation unveils new avenues for the perfume sector. For instance, there's potential to engineer molecules tailor-made to exude desired fragrances. Implementing this methodology would streamline the vast array of molecular choices down to a select few optimal ones. Surprisingly, literature reviews yielded no studies employing this strategy for perfume creation. This gap underscores the significance of our current endeavor: to craft an AI-driven molecular generator tailored for perfume formulation.
Imagine a tool that can craft scent molecules with specific objectives. One pivotal question arises: What if customer acceptance is one of those objectives? This underscores the pressing challenge of bridging the gap between perfume chemistry and consumer perception. Creating such a tool would be a monumental leap forward for the industry. Today, the internet is a rich source of customer feedback on fragrances. Websites like Parfumo, for example, house subjective insights on hundreds of thousands of commercially available perfumes, encompassing user ratings and detailed fragrance notes. Such data sheds light on the relationship between fragrance compositions and public reception, offering unique insights unavailable elsewhere. This information is invaluable, pinpointing the most resonant combinations of fragrance notes. Tapping into this feedback offers a clearer understanding of a scent's market appeal.
Thus, a further aim of this study is to introduce the use of an AI molecule generator that incorporates customer feedback. By doing so, we aim to generate a curated set of molecules ideal for future perfume formulations, crafting a product that seamlessly intertwines chemistry, biological perception, and consumer appeal. In this context this work contributions can be listed as follows:
- A method to quantitatively analyze the relationship between the molecular structure of perfumes and human olfactory perception, advancing understanding in the field of scent psychophysics.
- Systematically extract and utilize consumer feedback from digital platforms, such as Parfumo, to inform the molecular design process, underscoring the importance of integrating market analysis in product design.
- Design and validate an AI-based molecule generator tailored for scent molecule prediction, a first in the domain of fragrance chemistry.
- The proposed AI approach to predict successful scent molecules based on consumer feedback, potentially reduces the lengthy and costly trial-and-error process traditionally associated with perfume creation.
- It is proposed an algorithmic model that incorporates real-world consumer feedback, thereby ensuring the synthesized molecules have a higher likelihood of market success.
2. Methodology
This research is grounded in a methodology that integrates both data analysis and computational methodologies to advance the field of perfume engineering. Our process is comprehensive, aiming to harness the potential of available data and make informed decisions in creating and understanding perfumes. A more in-depth look at the steps involved in our methodology is described as follows:
- Data Collection via Web Scraping: The first step in the proposed methodology involves developing a specialized web scraping program to collect data from the Parfumo forum. This data encompasses consumer feedback, fragrance notes, and ratings on many perfumes. By systematically extracting this information, compiling a comprehensive database serves as a cornerstone for the subsequent step in the methodology.
- In-depth Statistical Analysis: After amassing the database, an analytical phase is proposed to interrogate the data for meaningful patterns and insights. This step sought answers to questions pivotal to the proposed goals: Which fragrance notes appear most frequently across perfumes? Is there a correlation between certain aromatic notes and higher consumer ratings? Are there specific fragrance notes that are commonly paired together? Such queries enabled us to understand consumer preferences and market trends better.
- Database Clustering using the k-means Algorithm: To delve deeper into the interplay of fragrance notes, it is proposed to employ the k-means clustering algorithm. This technique allowed us to group perfumes with similar aromatic profiles, particularly those with recurrent but uniquely combined fragrance notes. The outcome was a categorization of perfumes into distinct clusters, each signifying a particular scent profile.
- Evaluative Analysis of Cluster Ratings: With our clusters defined, the next step focuses on understanding how each cluster fared in consumer ratings. This step provides a granular view of the market reception for different aromatic combinations and provides the necessary information to the AI framework to prioritize which scent profiles resonate most with consumers.
-
Molecule Generation for Desired Perfume Profiles: Central to the proposed methodology is the aspiration to generate a roster of molecules that align with select perfume scents. This process was two-pronged:
- Training Phase: We trained a gated graph neural network (GGNN) on a curated database comprising known molecules and their associated fragrance notes.
- Generation and Refinement: Leveraging the trained GGNN, we embarked on generating a diverse set of molecules. To fine-tune this generation process, we integrated transfer learning techniques, narrowing down to molecules that match our targeted scent profiles.
- VI.
- Assessment of Generated Molecules: Post-generation, an evaluation is proposed to ensure the suitability of the molecules. Hence, we propose cross-referencing the vapor pressure of each synthesized molecule against existing databases. This ensured that the generated molecules matched the aroma profile and conformed to the requisite intensity of the fragrance notes.
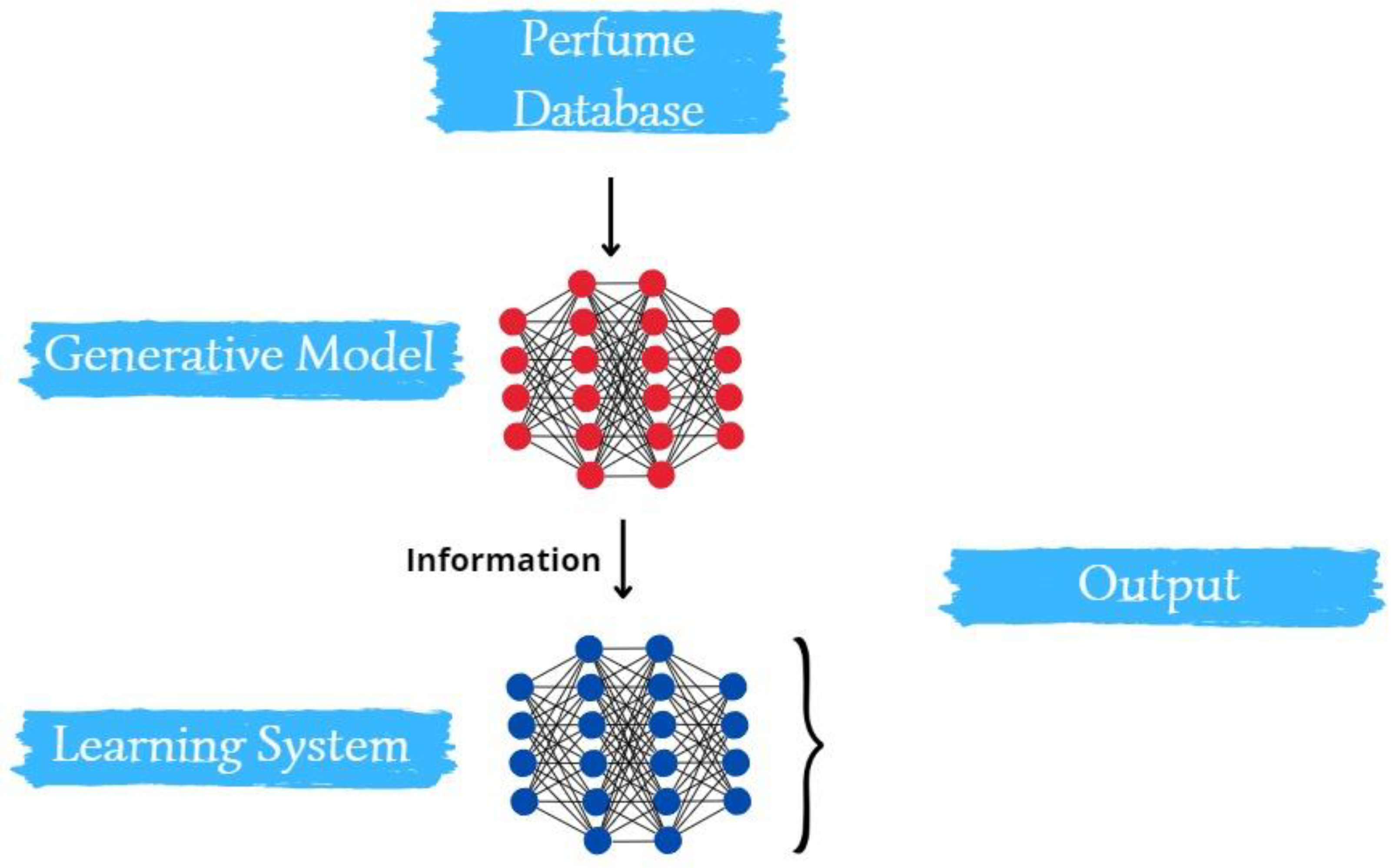
The nuances of this proposed methodology and its implications will be delved into in the subsequent sections of the research. Figure 1 presents a schematical representation of the methodology proposed here and afore described.
2.1. Data Collection via Web Scraping and In-Depth Statistical Analysis
At the outset of our methodology, we aimed to curate a database rich in subjective details about commercially available perfumes. Several data sources were evaluated, but we ultimately gravitated toward the forum Parfumo. This choice was influenced by its consistent updates and the substantial volume of information, with over 140 thousand perfumes listed during the development of our web scraping program.
Figure 2.
Print screen of two perfume pages. To the left, is an example of a page with all the necessary information. To the right, is a page with little information.
Figure 2.
Print screen of two perfume pages. To the left, is an example of a page with all the necessary information. To the right, is a page with little information.
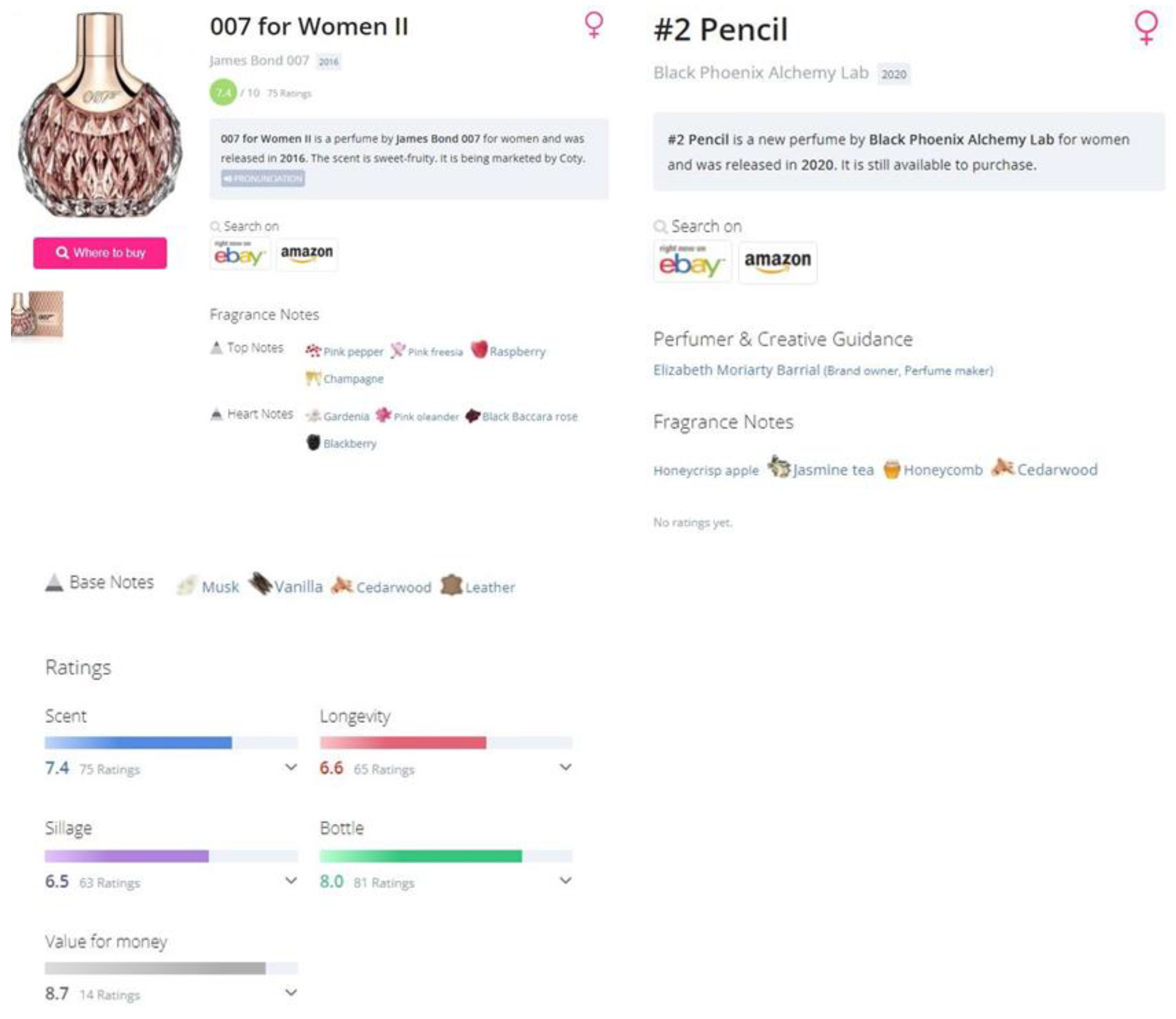
We aim to create a database of commercially available perfumes in the first step of the proposed methodology. Each perfume entry includes baseline details: name, release year, target gender, and brand. Some entries expand to ratings on scent, longevity, sillage, bottle design, and cost-effectiveness. These entries may also have rating counts, user reviews, visuals, community classifications, and fragrance notes, which may be segmented into top, heart, and base notes.
To retrieve data from Parfumo, it is proposed an adaptive web scraper tool. Given the inconsistencies in available data for each perfume, our scraper is designed to be flexible. A preliminary exploration of the forum confirms the consistent presence of name and brand details for every perfume, but other attributes differ. We crafted specific Python functions for each data attribute to cater to this. Exception Handling ensures the scraper manages missing data smoothly. Then, compile the extracted data using the Pandas library into a data frame. These functions employ the Python libraries: Requests, Selenium, and BeautifulSoup. While both Requests and Selenium facilitate data extraction from webpages, they differ in efficiency. The Requests library rapidly reads a page in milliseconds, making it the primary choice. However, for elements embedded in JavaScript, such as numerical ratings, which are not accessible to Requests, the proposed method uses the Selenium library. It emulates a browser environment, albeit at a slightly slower pace of about three seconds per page.
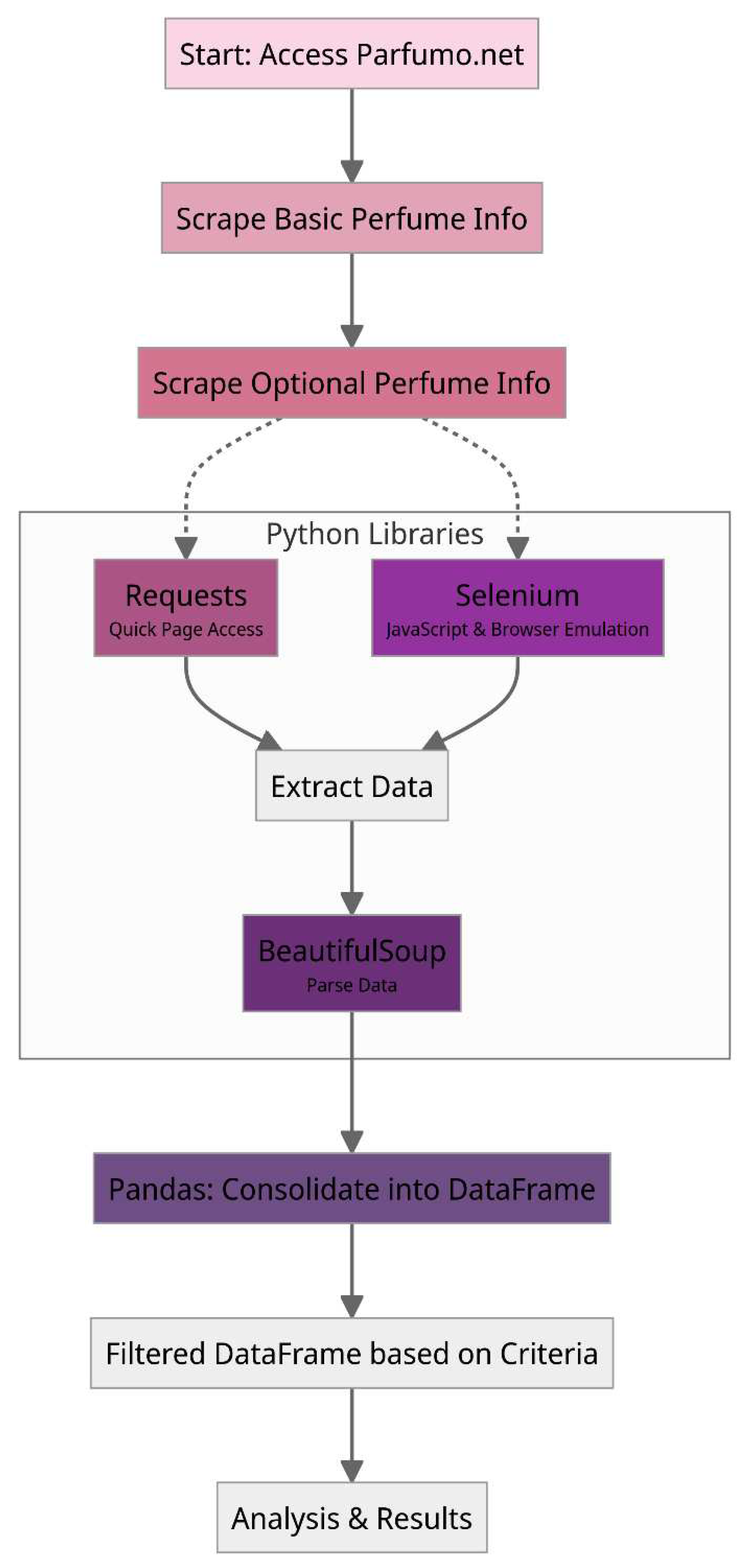
Following successful trials on specific forum pages, the scraper is configured to navigate the entire forum, guided by a link list organized by perfume release year and brand. This methodology phase is illustrated in Figure 3.
Having obtained the database, the information is subject to statistical analysis to better understand the data that is contained, the detailed results of these analyses are presented in the results section of this work. As mentioned, the main objective of this step was to answer pivotal questions that provide a vital comprehension of the data. A few approaches were taken to answer each question:
-
Which fragrance notes appear most frequently across perfumes?To answer this question, it is necessary to count how many perfumes contain a given fragrance note for every fragrance note that is available.
-
Is there a correlation between certain aromatic notes and higher consumer ratings?Here, it is necessary to consider all the perfumes that contain a given fragrance note and calculate the average of the ratings of all perfumes. It is also useful to calculate the sum of ratings for each fragrance note (Sum of the ratings of all the perfumes that contain a given note)
-
Are there specific fragrance notes that are commonly paired together?Firstly, a matrix is created using a technique called one-hot encoding. The matrix contains all the perfumes as rows and all the fragrance notes available as columns. If a perfume contains a given fragrance note, the value of the cell is one, else, it is zero. Next, a co-occurrence of fragrance notes matrix is calculated by multiplying the original matrix with its transpose.
Figure 3.
Flow diagram representing the steps to extract all the information necessary.
2.2. Database Clustering Using the k-Means Algorithm
The next step was to group all similar perfumes into clusters, depending on which fragrance notes they had. The idea behind this clustering is that if a customer likes a perfume, other perfumes in the same cluster will probably be enjoyable as well since they have similar aromas but with different combinations of fragrance notes; it helps customers to buy similar products and manufacturers to produce a similar product to one that was successful in the past. For that effect, three processes were used: one hot encoding to pre-process the data, an autoencoder to compact the data, and finally a k-means clustering algorithm to group the data.
The K-means algorithm is an iterative algorithm that attempts to partition a dataset into K distinct non-overlapping subgroups (clusters), with each data point belonging to only one group. It attempts to keep intra-cluster data points as close as possible while keeping clusters as far as possible. It assigns data points to clusters so that the sum of the squared Euclidean distances between the data points and the cluster's centroid (the arithmetic mean of all the data points in that cluster) is as small as possible. The objective function is given by:
where K is the number of clusters, m is the number of data points, and wik is 1 if the data point belongs to cluster k and 0 if not. First, the objective function is minimized with respect to wik keeping µk fixed, and next, J is minimized with respect to µk keeping wik fixed. The data point is assigned to a cluster based on the closest centroid, then recalculates the centroid (Sinaga & Yang, 2020). The algorithm is applied in a loop to determine the ideal number of clusters. The algorithm is run at each iteration with a sequential number of clusters, and a decision is made based on the silhouette coefficient. This coefficient is a way of measuring the goodness of the clusters; its values range from -1 to 1, with 1 being the ideal value representing a well-defined and contained cluster, as opposed to a scattered cluster represented by a negative silhouette coefficient (Rousseeuw, 1987).
The data relating to fragrance notes in the data frame are all in string format, unusable for the k-means clustering algorithm, which requires numerical inputs. One hot encoding solves this problem by transforming the text strings into binary values: if a perfume contains a given note, its value is one for that note else it is zero. The k-means clustering algorithm loses accuracy at higher dimensions (number of all fragrance notes available in this case) due to the curse of dimensionality problem. The algorithm minimizes the squared Euclidean distance between points. However, at higher dimensions, the Euclidean distance for any two points is nearly the same (Beyer et al., 1999). Therefore, the least common notes were dropped to reduce the dimensions to a manageable number.
To enhance the compactness of the data frame, we suggest two distinct methodologies: Multiple Correspondence Analysis (MCA) and an autoencoder. Both methods yield a data frame with an identical row count as the original yet feature a reduced number of columns with numerical values, in lieu of binary ones, encapsulating the information.
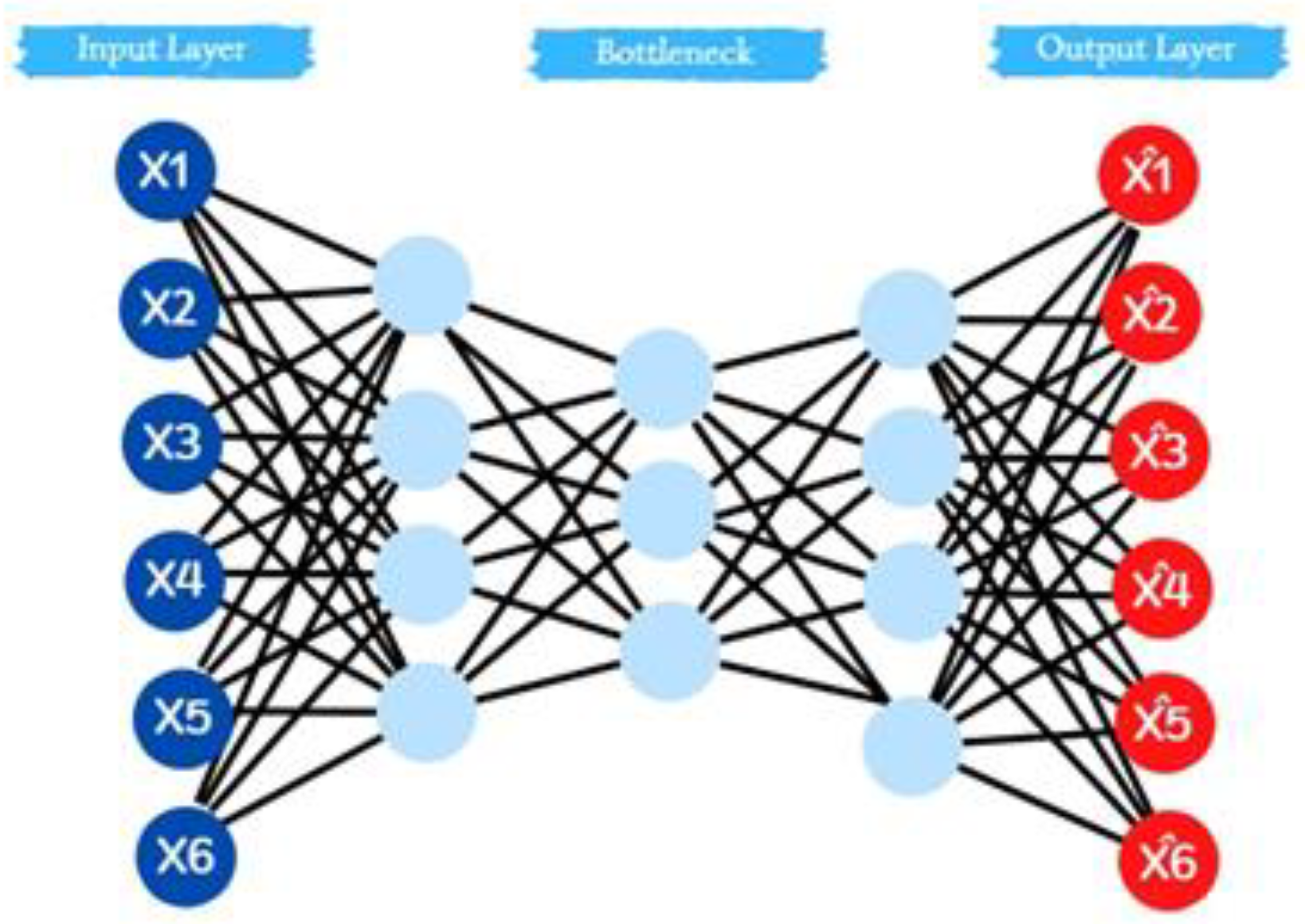
Initially, MCA is deployed due to its straightforward nature. Nevertheless, its data compression capabilities might be limited. To address this potential shortfall, an autoencoder is incorporated into this stage of the methodology. This ensures a more optimal data compression suitable for the k-means algorithm. An autoencoder, in essence, is an unsupervised artificial neural network. It is tailored to learn efficient data compression and subsequently to reconstruct the data to mirror the original as closely as feasible (Wen & Zhang, 2018).
Figure 4.
Graphic representation of the layers of an autoencoder.
The described k-means clustering algorithm is also applied to describe the vapor pressure intervals for the top, heart, and bottom levels. Quantitative information about which molecules can fit at which level is scarce.
2.3. Molecule Generation with GGNN
As highlighted earlier, clustering enables us to identify perfumes similar to a given one. We crafted a straightforward code that accepts a perfume's name and a target gender (male, female, or unisex). This code determines the cluster to which the specified perfume belongs and then pinpoints perfumes within that cluster with the highest weighted scent rating, subsequently revealing their fragrance notes.
The weighted rating is derived by multiplying the scent rating with the number of reviews it has received. To filter out perfumes that might have low quality but high visibility, we set a minimum threshold of 8 for the scent ratings. This ensures that we exclude perfumes that, despite being popular, have garnered negative feedback. The fragrance notes returned from this process are poised to be appealing, given they originate from a perfume that has resonated well with the market.
To ensure a successful perfume formulation, simply knowing the fragrance notes is insufficient. Understanding the specific ingredients that deliver the desired aroma is crucial. In this context, we suggest employing Graph Neural Networks (GNNs) specifically designed for molecular synthesis. By training a Gated Graph Neural Network (GGNN) on a database of established fragrant molecules, the network becomes equipped to generate novel molecules, drawing inspiration from the characteristics of the original compounds.
To train the GGNN model, it is necessary to have a database containing existing molecules that have known fragrance notes, so that the model can learn what aspects of a molecule are likely to result in a specific note. Thus, a database with known molecules and their fragrance notes is used to train the GGNN, sourced from The Good Scents Company, 2021, webpage. Although this new database does not contain identical fragrance notes as those extracted from the Parfumo forum, it is possible to map all the fragrance notes from the new database to those from the forum. This database provides the SMILES (Weininger et al., 1989) representation of molecules and other related information. Given that the GGNN requires graphs as generative inputs, functions from the RDKit library, an open-source cheminformatics software, are employed to transform SMILES into graphs, following the encoding rules set by Weininger et al., 1989. This transformation process interprets atoms as nodes and bonds as edges, embedding chemical information from the atoms and bonds. These embeddings facilitate understanding the relationships within the graph components. Additionally, the graph structure encompasses other features such as the adjacency matrix and edge attributes. The adjacency matrix indicates how nodes interconnect, forming a square matrix based on the number of nodes in the graph, while edge attributes convey the distances between graph edges.
Molecules, now represented as graphs, undergo preprocessing to enable the generative network to effectively reconstruct them during generation. Each node and edge are numerically labeled based on their type. A starting node is determined, and molecules are sequentially deconstructed, adhering to the label order. These processed molecules then serve as inputs for the GGNN training.
The GGNN is a refined version of the graph neural network presented by Li et al. (2015). It incorporates gated recurrent units (GRU) in its propagation phase and utilizes back-propagation through time (BPTT) (Zhou et al., 2019). The integration of GRU and BPTT addresses the vanishing/exploding gradient challenge—a situation where modulating model weights becomes challenging due to the diminishing gradient as one navigates deeper into the recurrent neural network. Specifically, the GRU employs update and reset gates to discern and prioritize pertinent data for predictions. This chosen data is directed to the output and refines its understanding from historical data. Meanwhile, BPTT optimizes performance chronologically, adapting the traditional backpropagation used for systems without memory to those nonlinear systems equipped with memory, as exemplified by the GGNN (Campolucci et al., 1996).
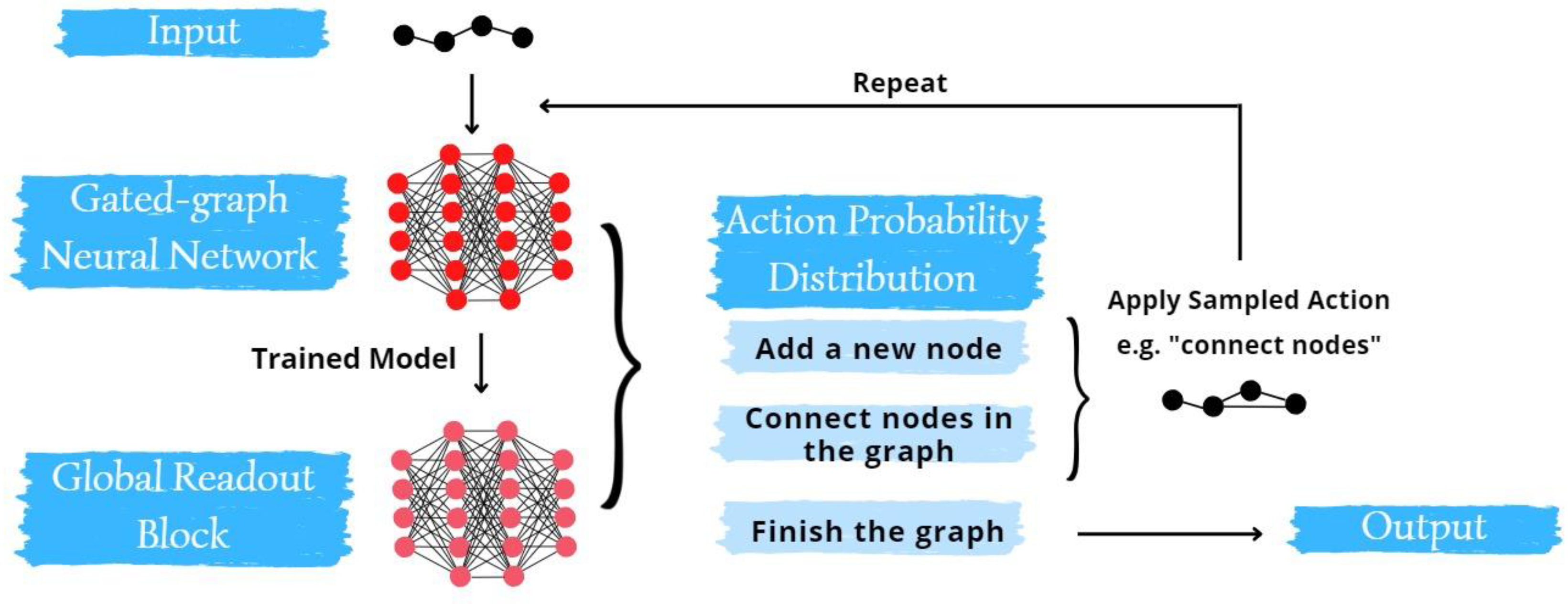
The GGNN yields two primary outputs: the graph embedding (g) and the final transformed node feature matrix (HL). These outputs subsequently become inputs for the global readout block (GRB). The GRB, structured as a tiered multi-layered perceptron (MLP) architecture, is a distinct feedforward artificial neural network. It calculates the graph's action probability distribution (APD), a vector comprising probabilities for every conceivable action to evolve a graph. APD samples guide the model in graph creation. The potential actions are threefold: introducing a new node to the graph, linking the recent node to a pre-existing one, or completing the graph. It's crucial to note that certain actions might be unsuitable for specific graphs, necessitating the model's ability to assign zero probabilities to such invalid actions. The cumulative probabilities of all actions must equal 1, establishing the target vectors that the model strives to learn during training. The following equations describe the operations undertaken by the GGNN.
where is the node feature vector for the initial node 𝑣 at the GGNN layer and is equal to its node feature vector in the graph, is a GRU gate in the specific MLP layer t, and relative to the node 𝑣, are normalization constants, is the set of neighbor nodes for 𝑣; u is a specific node in the graph, is a trainable weight tensor in r regarding the edge label le, b is a learnable parameter, z is also a GRU gate, 𝜌 is a non-linear function, ⨀ is an element-wise multiplication. The functional form of these equations is translated by the following:
where and are the incoming messages and hidden states of node vi, eij is the edge feature vector between vi and vj, l is a GNN layer index and L is the final GNN layer index. g, the final graph embedding is given by:
The processes undertaken by the global readout block are translated by the following equations. The SOFTMAX function is the activation function of the block, it converts a vector of numbers into a vector of probabilities.
The training phase is executed in small batches and the activation function of the model is the scaled exponential linear unit (SELU), translated by the following two equations; the function is applied after every linear layer in the MLP. The loss in this phase is given by the Kullback-Leibler divergence (Kullback & Leibler, 1951) between the target APD and predicted APD. Additionally, the model uses the Adam optimizer in several stages. Developed by (Kingma & Ba, 2014), it is a straightforward first-order gradient-based optimization algorithm.
During the training phase, graph samples are taken at consistent intervals for evaluation. The metric of choice for this evaluation is the uniformity-completeness Jensen-Shannon divergence (UC-JSD), as introduced by Arús-Pous et al. (2019). UC-JSD serves as a measure of similarity for the distributions of the negative log-likelihood (NLL) per action sampled. Ideally, values should approach zero.
The culminating phase is dedicated to graph generation. Here, the APD, which was formed in the GRB, is sampled to construct the graphs. A graph will persist in its growth either until the 'terminate' action is chosen from the APD or when an invalid action transpires. Invalid actions include adding a node to a non-existent node in a graph (unless the graph is vacant), linking an already connected pair, or appending a node to a graph that's already reached its node capacity, as determined during preprocessing. It's worth noting that hydrogens are excluded during both the training and generation stages. They are later incorporated using RDKit functions, depending on the valency of each atom.
Figure 5.
Schematic representation of the methodology of the GGNN platform.
2.4. Molecule Generation for Desired Perfume Profiles and Assessment of Vapor Pressure
The molecules generated previously were all fragrant since the GGNN model was trained with a database containing only fragrant molecules. However, the output of the model has no information on the fragrance notes of each molecule. Since the objective of this work is to present ingredients that can be used to formulate a perfume, it is necessary to generate only molecules that correspond to the desired fragrance notes. For that effect, a technique called transfer learning was used.
The established definition of transfer learning was presented by Xue et al. in 2019. It is a machine learning technique applied to solve problems with a lack of data through transferring knowledge from another related problem or data set. In this case, the knowledge obtained by the GGNN platform during training with all the molecules in the database was transferred to another model being trained only with molecules that are known to have the desired fragrance note. The model could not be trained only with the known molecules a priori since there are not enough molecules per fragrance note in the database to generate accurate results. Instead, the already trained model was trained again with a new set of molecules. This process was made in a loop to generate molecules for all fragrance notes available in the database.
Figure 6.
Schematic representation of the transfer learning process.
Finally, the thermo library, an open-source software available for use in Python, was used to consult the vapor pressure of all generated molecules. The library consults thermodynamic databases to check if the vapor pressure of the generated molecules is known, if not it returns an error. To define the type of note that each generated molecule is, the k-means clustering algorithm was also used to define vapor pressure intervals for the top, heart, and bottom levels. Quantitative information about which molecules can fit at which level is scarce. A sample of 1,000 fragrant molecules was scraped from the Good Scents company webpage, with information about the molecules’ duration (how long the smell lasts) and vapor pressure in mmHg at 25ºC. The algorithm was executed to calculate three clusters, and the centroid of each cluster was used to define the interval. Each centroid has a unique value of vapor pressure. At 95% confidence, the values are presented as intervals. At 25ºC, molecules with a vapor pressure between 0 and 0.0183 mmHg fall in the bottom category; molecules between 0.0183 and 0.0833 mmHg are classified as heart, and the molecules with a vapor pressure above 0.0833 mmHg can be used as top notes. Thus, all necessary information for the proposed methodology is achieved.
3. Results and discussion
3.1. Database and Statistics
We execute the data scraping step of the proposed methodology on Google Colaboratory. Google Colaboratory is a free jupyter notebook environment for prototyping code on powerful hardware options such as graphical process units (GPUs) and tensor process units (TPUs) (Bisong, 2019). The execution took three days to complete. The resulting DataFrame contains 72,754 unique perfume entries across 18 columns. Given our study's focus on fragrance notes, target gender, and user ratings, we filter the dataset to include only relevant entries. This leaves us with 27,443 perfumes. Detailed analyses of this refined data will be presented in the subsequent sections of this manuscript.
With this data frame, the next step was to evaluate it statistically. The following tables represent simple statistical analysis made on the database that was created after the Parfumo forum was scraped. Table 1 represents the number of perfumes that contain a given fragrance note, separated into the three target genders available in the forum. Since it is difficult to show the data for all the fragrance notes used, only the fourteen most common notes are shown here. This table shows which fragrance notes are preferred by perfume manufacturers and are most used. Table 2 shows the sum of the ratings of all perfumes that contain the given note. For example, if there are two different perfumes with the fragrance note ‘rose’, one of them has 15 ratings and the other has 10 ratings, then the sum of the ratings would be 25. This table builds on the information obtained in Table 1; it again shows which fragrance notes are the most popular, but also if the perfumes that contain that fragrance note are popular or not. For instance, the ‘iris’ note has a total of 26,082 ratings shared between 553 perfumes. This gives an average of 47.16 ratings per perfume. The ‘musk’ note, on the other hand, has 60,448 ratings shared between 1,794 perfumes, resulting on an average of 33.69 ratings per perfume. While the musk rating is more common to be found in fragrances, the products containing the ‘iris’ note tend to be more popular amongst consumers. Additionally, Table 3 shows the average rating of the perfumes that contain a given fragrance note. The ratings of the perfumes containing the note ‘iris’ tend are higher in average than the perfumes containing the note ‘musk’, again indicating that the note is more well-accepted by consumers.
These tables shine light on the topic of which fragrance notes should be considered on the manufacturing process of a new product. Even if a manufacturer chooses to maintain the traditional method of selecting fragrances for a new perfume through trial and error, it is useful to know which fragrance notes appear to be more widely accepted by the market, reducing the potential candidate ingredients to a more manageable number.
To better understand the data included in the database, a co-occurrence matrix was created to visualize what fragrance notes tend to occur with other fragrance notes. This could be useful in the formulation of perfumes since it shows which notes work well together; two fragrance notes could have a synergetic combination that wouldn’t be initially thought of while designing a new smell. While this data was not directly used in this work to generate fragrance molecules, it could be useful in future works. The co-occurrence matrix is simply obtained by multiplying the matrix with perfumes and their fragrance notes (obtained using the one-hot encoding technique) with its transpose; Figure 7 represents this as a heatmap. From it, it is possible to observe which fragrance notes tend to appear more often together. For example, the notes musk and bergamot appear frequently in the same perfumes while musk and white musk appear infrequently. Additionally, a distribution of the scent ratings given to all perfumes and to only the perfumes containing the Saffron fragrance note are represented in Figure 8. Ratings in both cases follow a normal distribution with a negative skew, but it is visible that the ratings for the perfumes with the Saffron note have a higher mode.
3.2. Clusters
Next, the perfumes were clustered into groups using the k-means algorithm as described in the methods section of this document. There are a total of 4,788 different fragrance notes in the filtered data frame. After developing a code to perform one hot encoding and executing it, the result was a binary matrix with 4788 columns and 27,443 rows, which was used for this step. It was found that the 100 most common notes were present in 97.6% of perfumes, meaning that thousands of notes were highly specific and appeared only on very few perfumes, many of those being proprietary aromatic notes.
Still, 100 dimensions proved too high for the k-means algorithm; the clusters obtained by running the algorithm were scattered, meaning they contained very different perfumes in the same cluster. It was necessary to compact the data frame even further following the proposed methodology. As previously mentioned, the compression capability of this method was not sufficient. Reducing the columns from 100 to 11 meant that 70% of the variance in the data was lost, an unacceptable deficit. Thus, an autoencoder was activated to compress the data frame into 15 columns, an adequate number for the k-means algorithm.
The devised neural network encompasses five layers. Starting with the initial layer, the data is compacted from 100 columns down to 80. Subsequently, it is further condensed to 60 columns in the second layer. The centerpiece of this network, the third layer known as the "bottleneck", shrinks the input to its smallest form of 15 columns. Post this compression, the subsequent layers work to reverse this process. The data expands from 15 to 60 in the fourth layer, progresses to 80 in the next, and eventually restores back to the initial 100 columns.
Upon reconstruction, the re-expanded data frame is juxtaposed with its original version to evaluate the efficacy of the autoencoder. Training was undertaken for 1,000 epochs using the perfumes data frame, leveraging backpropagation to optimize layer weights and curtail losses. By the concluding epoch, the training loss settled at a minute 4 × 10−5, while the area under the precision-recall curve reached 0.8602, approaching the maximum value of 1.
Post validation of the autoencoder's proficient performance, the compressed data from the final epoch—without undergoing decompression—was utilized. Specifically, the data from the "bottleneck" layer became the foundation for the k-means algorithm. This clustering process was iteratively performed, revealing that the optimal cluster count stands at 23, attributable to its peak silhouette score of 0.1356.
Figure 9.
2D representation of the clustered perfumes.
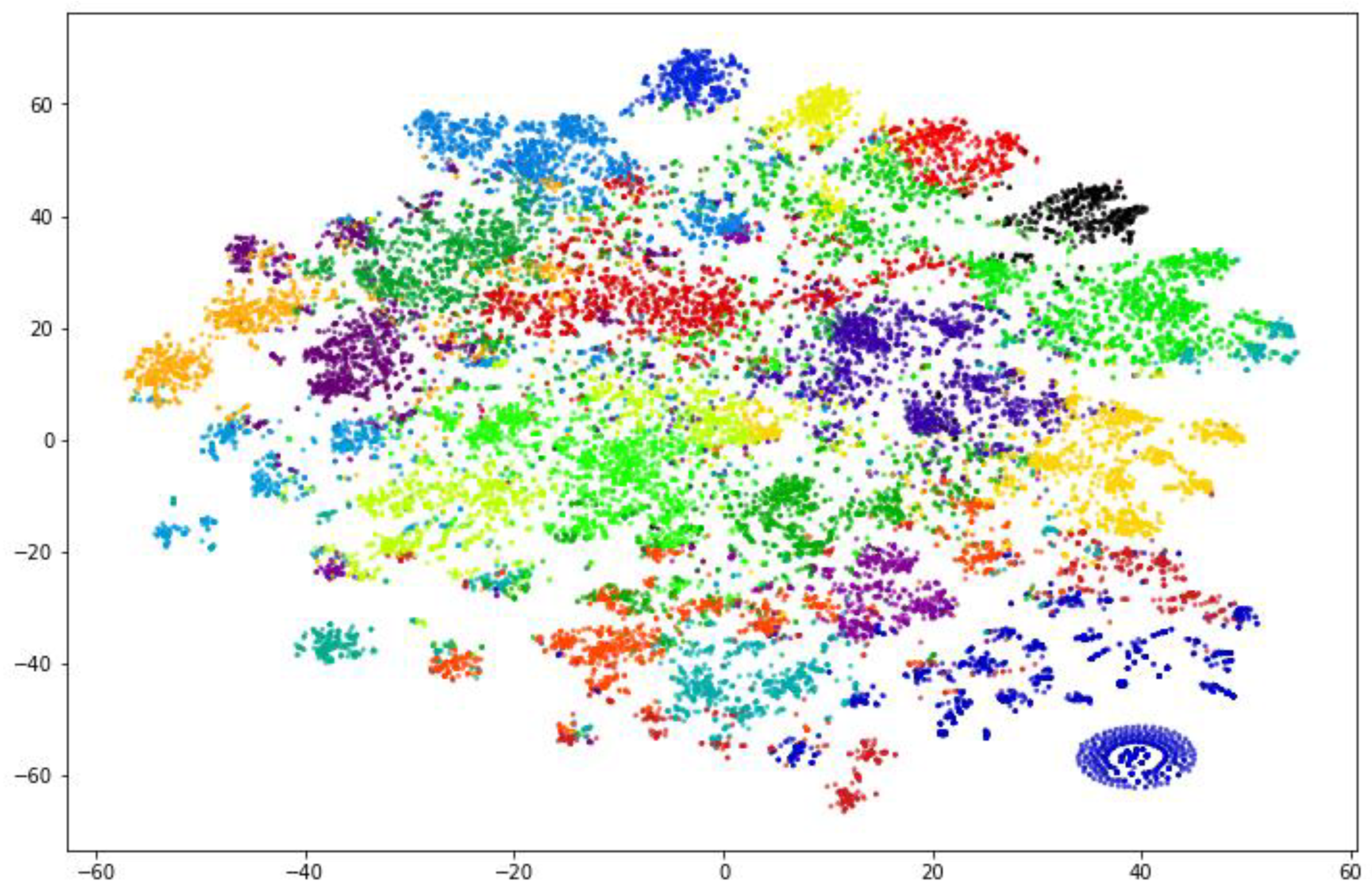
Each point in the graph is a perfume, and its position on the graph depends solely on which fragrance notes the perfume contains. All points with the same color belong to one cluster of similarly grouped perfumes. Note that the values in the axes have no physical meaning, what matters is the proximity of adjacent points. Most clusters seem to be well defined and concise, but a few areas contain multi-colored points, meaning that a few perfumes that belong to one cluster could also fit into another cluster. A very concise group can be observed in the bottom right corner of the graph, this group contains all the perfumes that had no fragrance notes included in the analysis (only the 100 most common notes were used), meaning that to the k-means algorithm they were identical and that is why the group is so concise.
To assess if the clusters are significantly different to the set of all perfumes, a series of p-tests were made. For each cluster and for each fragrance note inside that cluster, the set of all perfumes following those restrictions were compared to the set of all perfumes containing the same fragrance note. The p-values for each of those p-tests are shown in the figure below.
The white spots in the graph represent p-values above 0.05. It is visible that most of the p-values obtained were inferior to 0.05, meaning that the clusters are significantly different to the set of all perfumes. This test does not compare the clusters with each other, but from Figure 10 it is visible that the clusters are generally well-separated, and thus likely to be different from each other, with a few exceptions as mentioned earlier.
Having obtained clusters of similar perfumes, it was now possible to find similar perfumes. As a case study, the perfume Armani Privé – Rose Milano was chosen as a starting perfume. This perfume is popular and comes from a well-known brand, being classified as unisex and having a scent rating of 7.8. It was verified that the perfume belongs to cluster number 14, and contains the following fragrance notes: pear, bergamot, lemon, jasmine, rose, white musk, patchouli and amber. The perfume with the highest rating from the same cluster and the same target gender was found to be Tuscan Leather, by Tom Ford in 2007 with 1147 scent ratings at an average value of 8.5. The fragrance notes of this perfume were then extracted to be used as target fragrances for the molecules generated by the GGNN model. The top fragrance notes are raspberry and saffron; the heart fragrance notes are jasmine and frankincense; the bottom notes are leather, amber, and wood. As mentioned in the methods section, several molecules were generated for each of the target fragrance notes; the vapor pressure of all molecules was estimated using the Thermo library in Python and then the molecules that did not correspond to the appropriate level were excluded.
3.3. Generated Molecules
The GGNN model underwent training over a span of 1000 epochs. This process was executed within a Linux environment (Ubuntu 64-bit) using the Oracle VM VirtualBox software hosted on a Windows 10 system. The utilized machine boasted an AMD Ryzen 9 5900X 12-Core Processor clocked at 3.79 GHz, 32.0 GB of RAM, a 64-bit operating system, and an NVIDIA GeForce RTX 3060 GPU. The entire training process spanned a week. Epoch 660 stood out and was selected for generation, given its lowest UC-JSD value.
The efficacy of the model's architecture is significantly influenced by its hyperparameters. Their precise values can greatly impact the model's accuracy and thus demand optimization tailored to specific cases. For this study, the hyperparameters were fine-tuned using a trial-and-error approach, with their finalized values detailed in the subsequent table.

Table 4.
Hyperparameters used on the platform.
| Parameters | Value | Parameters | Value | Parameters | Value |
|---|---|---|---|---|---|
| A | 0.5 | GGNN width | 100 | MLP activation function | SOFTMAX |
| Batch size | 20 | Initial learning rate | 1x10−4 | MLP depth | 4 |
| Block size | 1000 | Learning rate decay factor | 0.99 | MLP droupout probability | 0 |
| Epochs | 500 | Learning rate decay interval | 10 | MLP hidden dimensions | 500 |
| Generation epoch | 1040 | Loss function | Kullback-Leibler divergence | Number of samples | 200 |
| GGNN activation function | SELU | Maximum relative learning rate | 1 | Optimizer | Adam |
| GGNN depth | 4 | Message passing layers | 3 | Sigma | 20 |
| GGNN dropout probability | 0 | Input size of GRU | 100 | Weight decay | 0 |
| GGNN hidden dimension | 250 | Minimum relative learning rate | 1x10−4 | Weight initialization | Uniform |
To define the type of note that each generated molecule is, the k-means clustering algorithm also defined vapor pressure intervals for the top, heart, and bottom levels. Quantitative information about which molecules can fit at which level is scarce. A sample of 1,000 fragrant molecules was scraped from the Good Scents company webpage, with information about the molecules’ duration (how long the smell lasts) and vapor pressure in mmHg at 25ºC. The algorithm was executed to calculate three clusters, and the centroid of each cluster was used to define the interval. Each centroid has a unique value of vapor pressure. At 95% confidence, the values are presented as intervals. At 25ºC, molecules with a vapor pressure between 0 and 0.0183 mmHg fall in the bottom category; molecules between 0.0183 and 0.0833 mmHg are classified as heart, and the molecules with a vapor pressure above 0.0833 mmHg can be used as top notes.
Presented below are the molecules generated that fit the vapor pressure requirement. Using the pubchem website, it was found that all molecules exist and are already in use by the fragrance industry (and other industries like the flavor industry in some cases). The GGNN model generated 30 molecules for each fragrance note. However, since only the already-existing molecules can have their vapor pressure discovered by the thermo library used, all the non-existing molecules do not fit the vapor pressure requirement for each fragrance note and are dropped. Additionally, by consulting the good scents company website, it was found that most molecules are used by the perfume industry for fragrance notes that are similar to the target fragrance note (for example, ‘raspberry’ is similar to ‘fruity’).
For the ‘raspberry’ note, 6 molecules fit the requirements and can be seen in Figure 11. Out of these, 4 are mainly used in industry to generate a ‘fruity’ smell. Two other molecules are normally used for a ‘nutty’ or ‘jasmine’ scent. For the ‘saffron’ note, 7 molecules were found (Figure 12); their uses in the industry are more varied than the previous molecules. Each molecule is used for different smells, these are: ‘berry’, ‘vegetable’, ‘spicy’, ‘fruity’, ‘floral’, ‘meaty’ and ‘green’.
Regarding the ‘frankincense’ target note, only one molecule is suggested (Figure 13), and it is listed in the good scents company website as having a ‘spicy and anisic’ scent. For ‘jasmine’, 2 molecules are suggested (Figure 14), and they are usually used by the industry to generate a ‘floral’, ‘sweet’ and ‘spicy’ smell.
4 molecules are suggested for the ‘amber’ target note (Figure 15). Out of these, 2 are described as ‘fatty’, ‘sweet’ and ‘fruity’. One is described as ‘earthy’ and the last one is used to generate the ‘ambergris’ smell. For the ‘leather’ target note, 6 molecules are given by the model (Figure 16). One is used for the ‘leather’ smell, 2 are described as ‘waxy’, one is described as ‘sweet apricot kernel’ and one is used in ‘floral’ perfumes. The last molecule for this target fragrance note is called ‘skatole’. This molecule is infamously known to have a strong ‘fecal’ scent at high concentrations, but at low concentrations it is used to give an ‘over-mature flower’ or ‘forest-floor’ smell. Finally, for the ‘wood’ target note, 2 molecules are suggested (Figure 17). Both are used in industry to generate a ‘wood’ smell.
While a few molecules generated by the model might not seem the most appropriate for the target fragrance notes, most of the molecules generated are either used by the industry to generate the desired target smell or are used to give a similar scent. It reinforces the capacity of the proposed methodology to suggest relevant molecules targeted at a desired olfactory experience.
4. Conclusion
The traditional way of creating new scents by trial and error is still very labor intensive and time consuming. Although there have been innovations in the area in the last years, there is still a lot of room for new solutions that improve the efficiency of the creation process. The method presented in this work is intended to be used as a tool in the manufacturing of new perfumes. First, a database was created with information about commercially available perfume hosted on the Parfumo website. Next, similar perfumes were grouped into clusters using the k-means algorithm. After selecting an existing perfume in the market, a similar and better evaluated perfume was found and its fragrance notes were taken as targets for a new scent. Several fragrant molecules were then generated for each note. Additionally, the k-means algorithm was used to define a range of intervals of vapor pressures for each of the three fragrance levels (top, heart and bottom); the ranges obtained were 0-0.01833 mmHg for bottom notes, 0.01833-0.0833 mmHg for heart notes and 0.0833+ for top notes. The molecules were then filtered, based on the estimated vapor pressure of each molecule, to ensure that the molecule would not only have the appropriate smell, but also the correct duration corresponding to the level where the fragrance note belongs. At least one molecule was found for all fragrance notes that fit all requirements.
The method presented here is not all-encompassing, the molecules generated by the GGNN are likely to have the desired scent, but experiments are still necessary to guarantee that the smell is pleasant and to determine the relative composition of each molecule as well as which solvent should be used. Nevertheless, this new tool can be very useful for perfume makers since it can save time and money by narrowing the amount of ingredients that a perfume maker must test to achieve the desired smell, making the overall creation process more efficient.
References
- Almeida, R. N., Costa, P., Pereira, J., Cassel, E., & Rodrigues, A. E. (2019). Evaporation and Permeation of Fragrance Applied to the Skin. Industrial & Engineering Chemistry Research, 58(22), 9644–9650. [CrossRef]
- Arús-Pous, J., Johansson, S. V., Prykhodko, O., Bjerrum, E. J., Tyrchan, C., Reymond, J.-L., Chen, H., & Engkvist, O. (2019). Randomized SMILES strings improve the quality of molecular generative models. Journal of Cheminformatics, 11(1), 71. [CrossRef]
- Bell, C. and C. (2023). Thermo: Chemical properties component of Chemical Engineering Design Library (ChEDL). https://github.com/CalebBell/thermo.
- Beyer, K., Goldstein, J., Ramakrishnan, R., & Shaft, U. (1999). When Is “Nearest Neighbor” Meaningful?.
- Bisong, E. (2019). Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform (pp. 59–64). Apress. [CrossRef]
- Bushdid, C., Magnasco, M. O., Vosshall, L. B., & Keller, A. (2014). Humans can discriminate more than 1 trillion olfactory stimuli. Science, 343(6177), 1370–1372. [CrossRef]
- Campolucci, P., Uncini, A., & Piazza, F. (1996). Causal back propagation through time for locally recurrent neural networks. 1996 IEEE International Symposium on Circuits and Systems. Circuits and Systems Connecting the World. ISCAS 96, 531–534. [CrossRef]
- Carles, J. (1961). A Method of Creation & Perfumery.
- Debnath, T., & Nakamoto, T. (2022). Predicting individual perceptual scent impression from imbalanced dataset using mass spectrum of odorant molecules. Scientific Reports, 12(1). [CrossRef]
- Fortune Business Insight. (2022, May). Flavors and Fragrances Market Size, Share & COVID-19 Impact Analysis.
- Gerkin, R. C. (2021). Parsing Sage and Rosemary in Time: The Machine Learning Race to Crack Olfactory Perception. In Chemical Senses (Vol. 46). Oxford University Press. [CrossRef]
- Kingma, D. P., & Ba, J. (2014). Adam: A Method for Stochastic Optimization.
- Kullback, S., & Leibler, R. A. (1951). On Information and Sufficiency. The Annals of Mathematical Statistics, 22(1), 79–86. http://www.jstor.org/stable/2236703.
- Leffingwell & Associates. (2018). Flavor & Fragrance Industry - Top 10. Flavor & Fragrance Industry - Top 10.
- Li, Y., Tarlow, D., Brockschmidt, M., & Zemel, R. (2015). Gated Graph Sequence Neural Networks.
- Mata, V. G., Gomes, P. B., & Rodrigues, A. E. (2005). Engineering perfumes. AIChE Journal, 51(10), 2834–2852. [CrossRef]
- Nozaki, Y., & Nakamoto, T. (2018). Predictive modeling for odor character of a chemical using machine learning combined with natural language processing. PLoS ONE, 2008, 13(6). https://www.parfumo.com/. [CrossRef]
- Parfumo.
- Queiroz, L. P., Rebello, C. M., Costa, E. A., Santana, V. V., Rodrigues, B. C. L., Rodrigues, A. E., Ribeiro, A. M., & Nogueira, I. B. R. (2023a). A Reinforcement Learning Framework to Discover Natural Flavor Molecules. Foods, 12(6), 1147. [CrossRef]
- Queiroz, L. P., Rebello, C. M., Costa, E. A., Santana, V. V., Rodrigues, B. C. L., Rodrigues, A. E., Ribeiro, A. M., & Nogueira, I. B. R. (2023b). Generating Flavor Molecules Using Scientific Machine Learning. ACS Omega, 8(12), 10875–10887. [CrossRef]
- Queiroz, L. P., Rebello, C. M., Costa, E. A., Santana, V. V., Rodrigues, B. C. L., Rodrigues, A. E., Ribeiro, A. M., & Nogueira, I. B. R. (2023c). Transfer Learning Approach to Develop Natural Molecules with Specific Flavor Requirements. Industrial & Engineering Chemistry Research, 62(23), 9062–9076. [CrossRef]
- RDKit: Open-source cheminformatics. (n.d.). https://www.rdkit.org.
- Richardson, L. (2007). Beautiful soup documentation. April.
- Rodrigues, A. E., Nogueira, I., & Faria, R. P. V. (2021). Perfume and Flavor Engineering: A Chemical Engineering Perspective. Molecules, 26(11), 3095. [CrossRef]
- Rousseeuw, P. J. (1987). Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. Journal of Computational and Applied Mathematics, 20, 53–65. [CrossRef]
- Saini, K., & Ramanathan, V. (2022). A Review of Machine Learning Approaches to Predicting Molecular Odor in the Context of Multi-Label Classication. [CrossRef]
- Santana, V. V., Martins, M. A. F., Loureiro, J. M., Ribeiro, A. M., Rodrigues, A. E., & Nogueira, I. B. R. (2021). Optimal fragrances formulation using a deep learning neural network architecture: A novel systematic approach. Computers & Chemical Engineering, 150, 107344. [CrossRef]
- Sinaga, K. P., & Yang, M.-S. (2020). Unsupervised K-Means Clustering Algorithm. IEEE Access, 8, 80716–80727. [CrossRef]
- Teixeira, M. A., Rodríguez, O., Mata, V. G., & Rodrigues, A. E. (2009). The diffusion of perfume mixtures and the odor performance. Chemical Engineering Science, 64(11), 2570–2589. [CrossRef]
- The Good Scents Company. (2021). http://www.thegoodscentscompany.com/.
- The Pandas Development Team (2020). pandas-dev/pandas: Pandas. Zenodo. [CrossRef]
- Wakayama, H., Sakasai, M., Yoshikawa, K., & Inoue, M. (2019). Method for Predicting Odor Intensity of Perfumery Raw Materials Using Dose–Response Curve Database. Industrial & Engineering Chemistry Research, 58(32), 15036–15044. [CrossRef]
- Weininger, D., Weininger, A., & Weininger, J. L. (1989). SMILES. 2. Algorithm for generation of unique SMILES notation. Journal of Chemical Information and Computer Sciences, 29(2), 97–101. [CrossRef]
- Wen, T., & Zhang, Z. (2018). Deep Convolution Neural Network and Autoencoders-Based Unsupervised Feature Learning of EEG Signals. IEEE Access, 6, 25399–25410. [CrossRef]
- Xue, D., Gong, Y., Yang, Z., Chuai, G., Qu, S., Shen, A., Yu, J., & Liu, Q. (2019). Advances and challenges in deep generative models for de novo molecule generation. WIREs Computational Molecular Science, 9(3). [CrossRef]
- Zhou, Z., Kearnes, S., Li, L., Zare, R. N., & Riley, P. (2019). Optimization of Molecules via Deep Reinforcement Learning. Scientific Reports, 9(1), 10752. [CrossRef]
Figure 1.
Schematic representation of the methodology proposed in this work.
Figure 7.
Heatmap showing co-occurrence of fragrance notes.
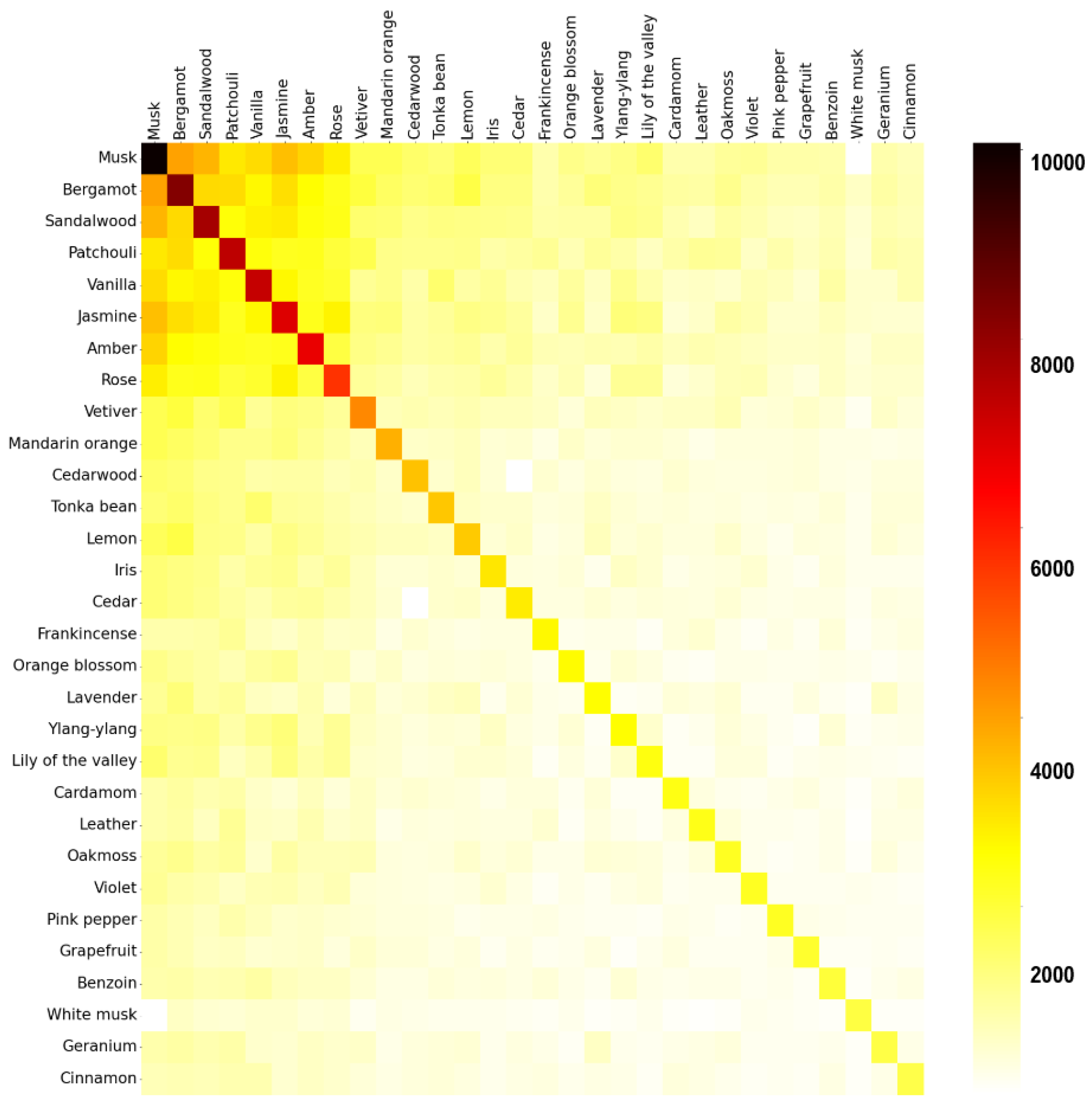
Figure 8.
Distribution of scent ratings for all perfumes and for perfumes containing the saffron fragrance note.
Figure 8.
Distribution of scent ratings for all perfumes and for perfumes containing the saffron fragrance note.
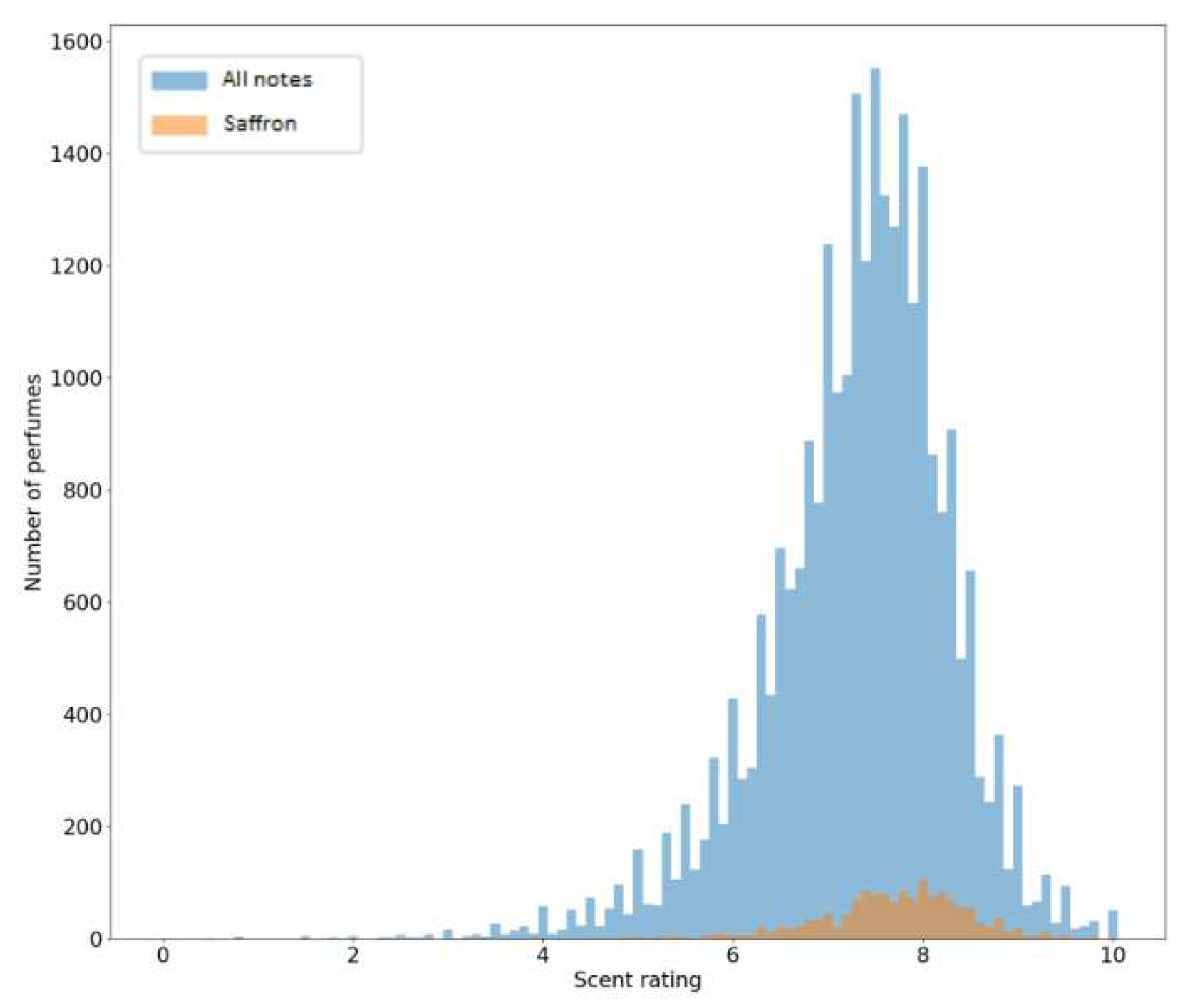
Figure 10.
p-tests for each note inside each cluster.
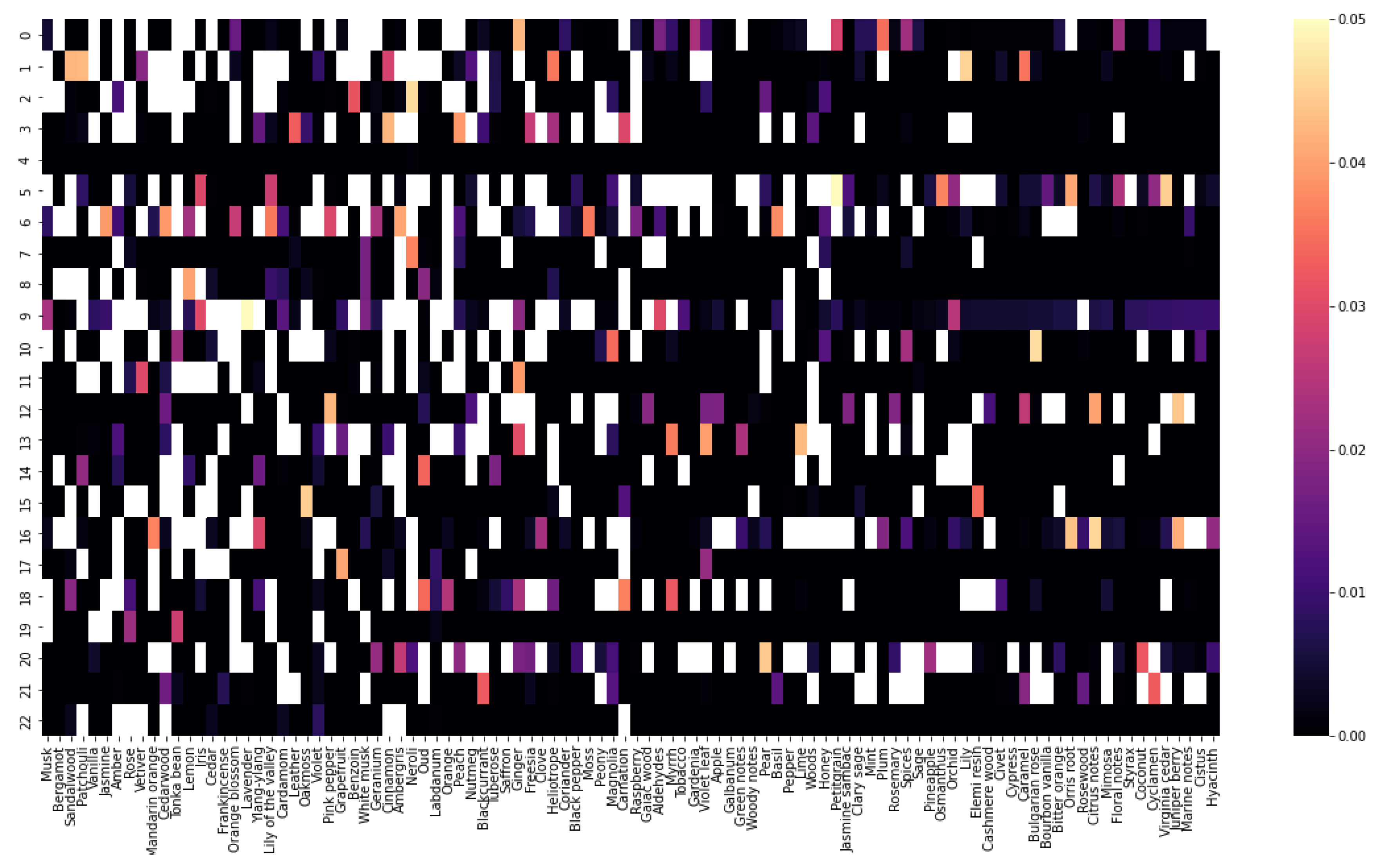
Figure 11.
Molecules for the fragrance note raspberry (top note).
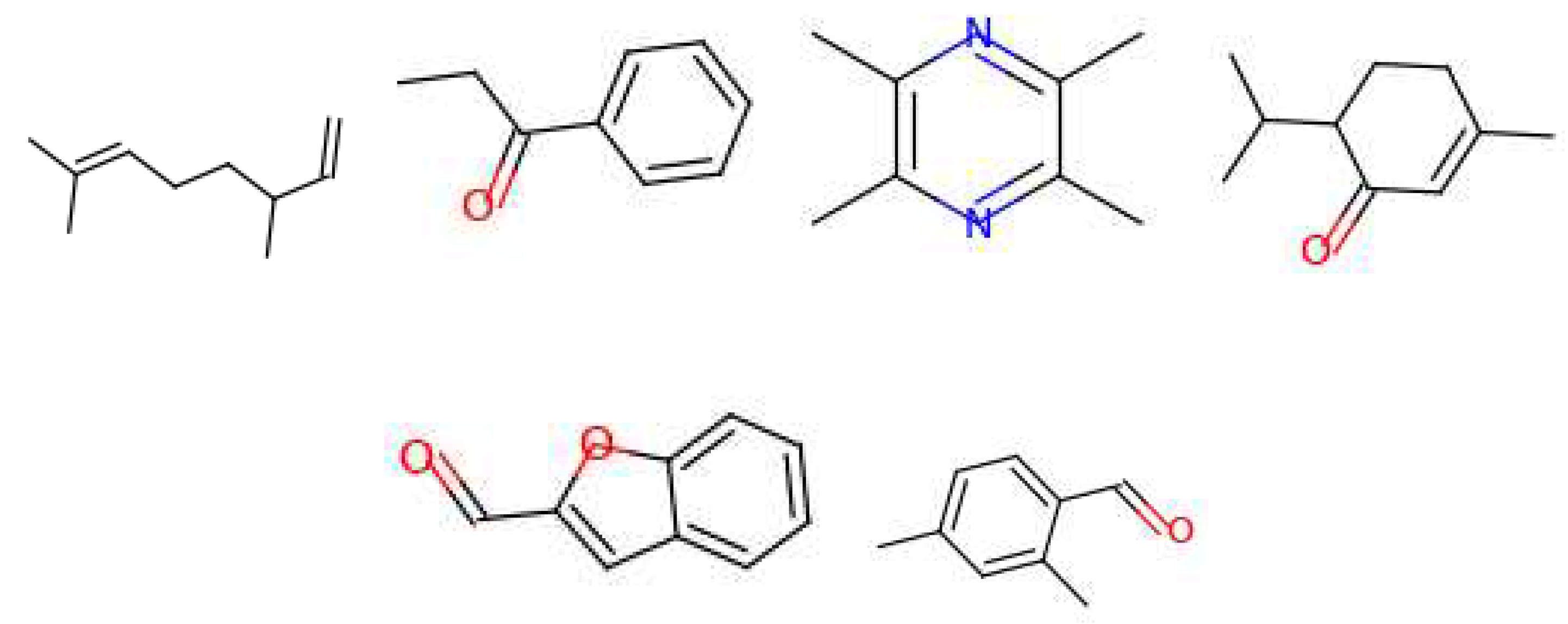
Figure 12.
Molecules for the fragrance note saffron (top note).

Figure 13.
Molecules for the fragrance note frankincense (heart note).
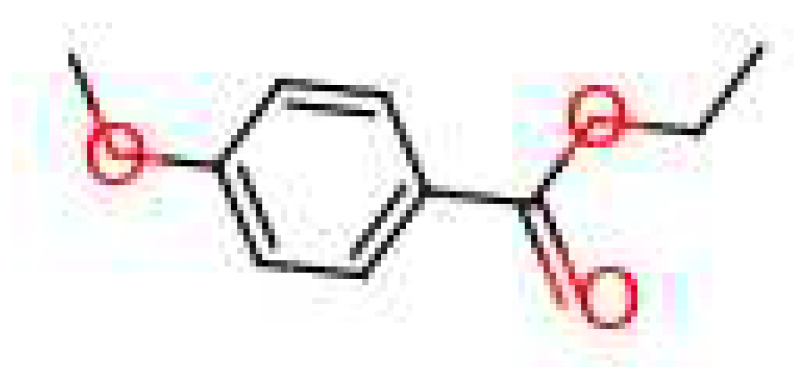
Figure 14.
Molecules for the fragrance note jasmine (heart note).
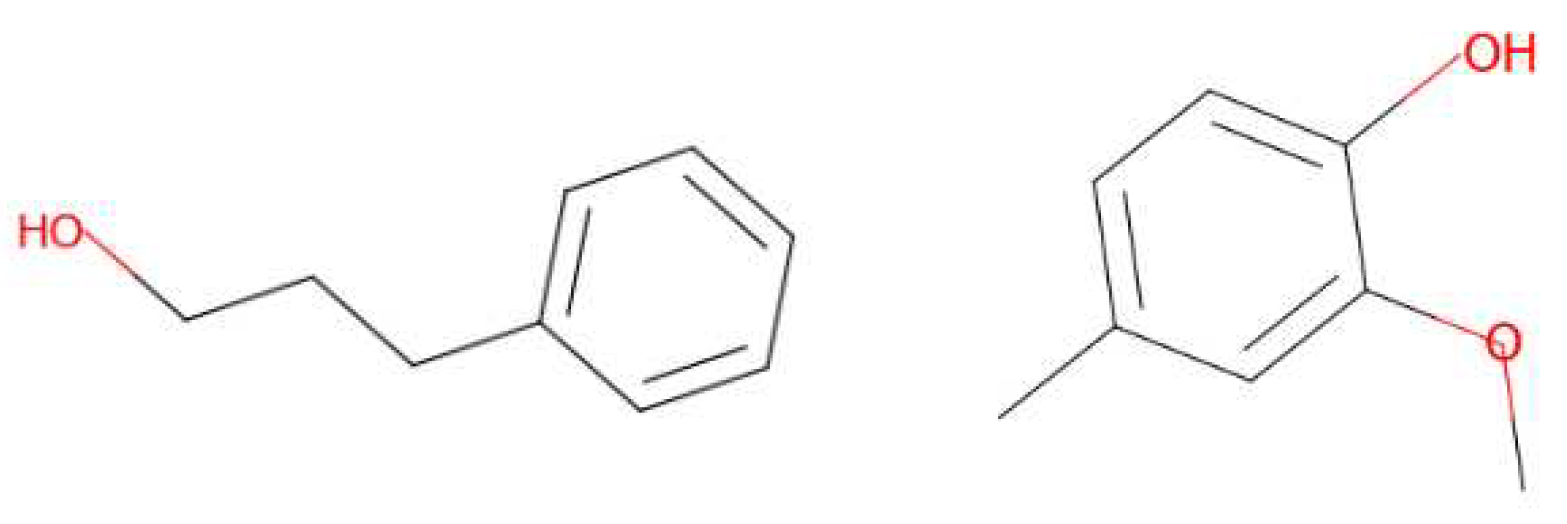
Figure 15.
Molecules for the fragrance note amber (bottom note).
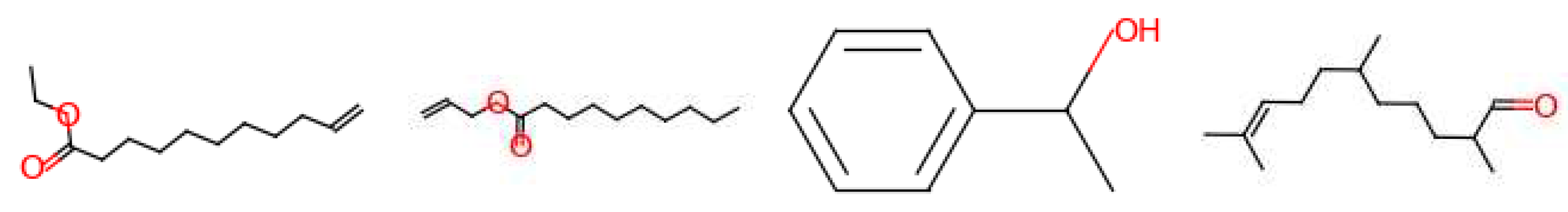
Figure 16.
Molecules for the fragrance note leather (bottom note).
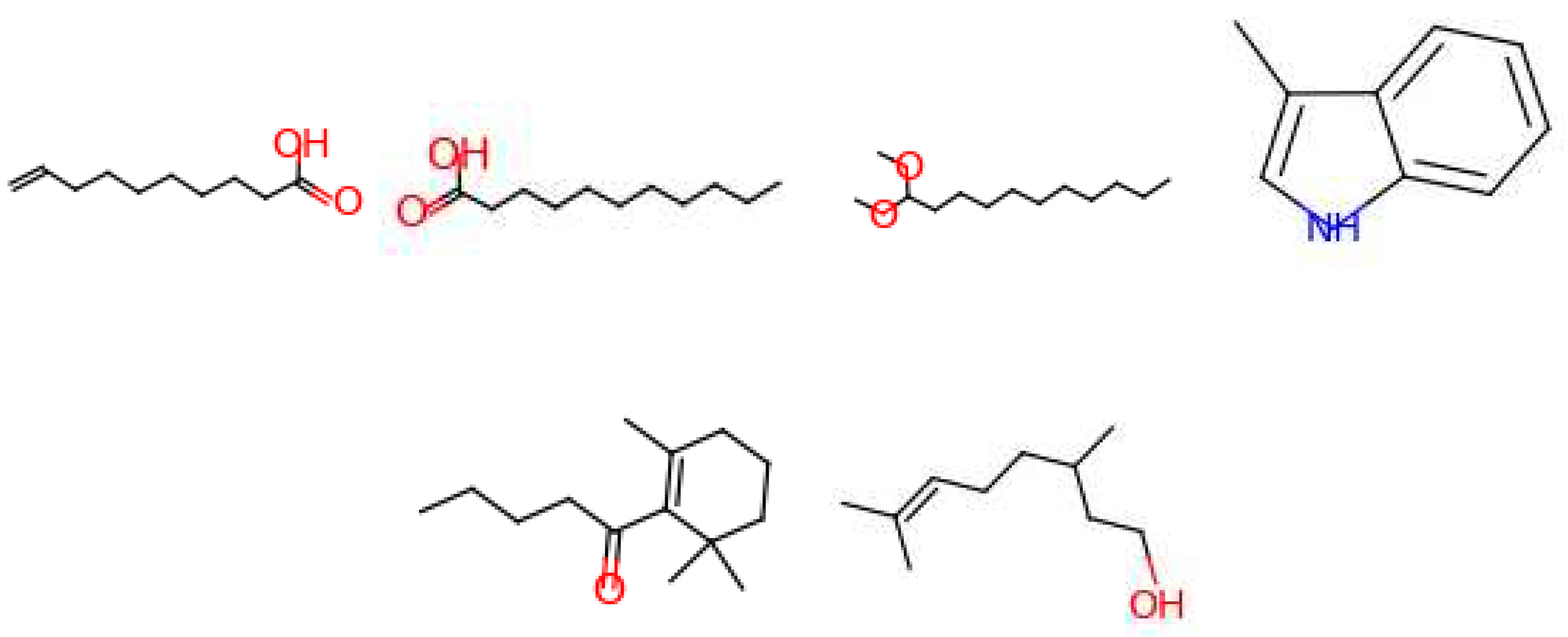
Figure 17.
Molecules for the fragrance note wood (bottom note).
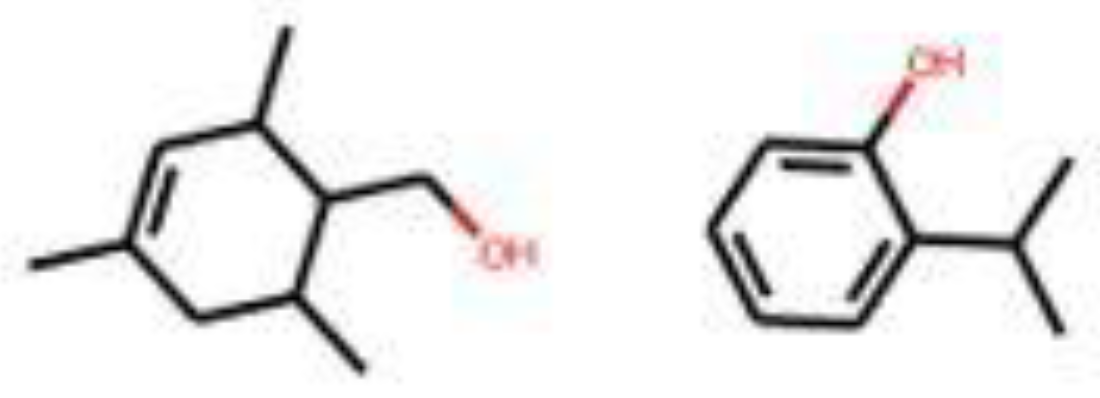
Table 1.
Frequency of fragrance notes.
| Leather | Iris | Cedarwood | Tonka bean | Frankincense | Vetiver | Rose | Jasmine | Bergamot | Patchouli | Amber | Sandalwood | Vanilla | Musk | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unisex | 354 | 297 | 375 | 342 | 535 | 497 | 479 | 480 | 656 | 769 | 715 | 741 | 823 | 947 |
| Male | 137 | 54 | 100 | 92 | 80 | 202 | 48 | 77 | 256 | 213 | 230 | 213 | 133 | 242 |
| Female | 48 | 202 | 92 | 151 | 83 | 121 | 451 | 558 | 266 | 292 | 366 | 393 | 582 | 605 |
Table 2.
Number of ratings per fragrance note.
| Leather | Iris | Cedarwood | Tonka bean | Frankincense | Vetiver | Rose | Jasmine | Bergamot | Patchouli | Amber | Sandalwood | Vanilla | Musk | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unisex | 10978 | 13436 | 16039 | 14513 | 19407 | 18097 | 15865 | 14268 | 21439 | 23906 | 28377 | 24897 | 30168 | 34312 |
| Male | 4935 | 5860 | 5517 | 3969 | 4360 | 10468 | 1577 | 1823 | 13677 | 10051 | 10051 | 5219 | 7445 | 7816 |
| Female | 2485 | 6786 | 3514 | 7105 | 3338 | 5913 | 13263 | 15201 | 11273 | 13691 | 13691 | 10636 | 17570 | 18320 |
Table 3.
Average rating of each fragrance note.
| Leather | Iris | Cedarwood | Tonka bean | Frankincense | Vetiver | Rose | Jasmine | Bergamot | Patchouli | Amber | Sandalwood | Vanilla | Musk | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unisex | 7,75 | 7,65 | 7,84 | 7,83 | 7,80 | 7,82 | 7,62 | 7,61 | 7,72 | 7,81 | 7,78 | 7,78 | 7,81 | 7,69 |
| Male | 7,52 | 8,15 | 7,65 | 7,65 | 7,82 | 7,69 | 7,70 | 7,66 | 7,55 | 7,89 | 7,53 | 7,53 | 7,79 | 7,64 |
| Female | 7,95 | 7,68 | 7,77 | 7,77 | 7,55 | 7,80 | 7,60 | 7,47 | 7,57 | 7,53 | 7,53 | 7,53 | 7,56 | 7,51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



