Submitted:
04 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
Depression is a complex neuropsychiatric disorder characterized by pervasive feelings of sadness, hopelessness, and cognitive impairments that can profoundly impact an individual's quality of life. Despite extensive research efforts, the precise mechanisms underlying depression remain elusive. Recent studies have highlighted the intricate interplay between the immune system and the central nervous system in the pathophysiology of depression. In this context, CD4+ T cells, a key component of the adaptive immune system, have emerged as potential players in the neuroinflammatory processes associated with depression. Concurrently, Brain-Derived Neurotrophic Factor (BDNF), a multifunctional neurotrophin, has garnered significant attention for its roles in neural plasticity, mood regulation, and cognitive function. This review paper explores the intriguing intersection between CD4+ T cells and BDNF in the context of depression and delves into the exciting prospects it offers for therapeutic interventions. By examining the intricate crosstalk between the immune system and the neurotrophic factors, we aim to shed light on the potential avenues for novel therapeutic strategies in the treatment of depression.
Keywords:
CD4
; TH17
; TH2
; depression
1. Introduction
Depression stands as a formidable global health challenge, affecting millions of individuals worldwide and imposing a substantial societal burden [1]. While substantial progress has been made in elucidating the intricate neurobiological underpinnings of this debilitating disorder, our understanding of its etiology and pathophysiology remains far from complete [2]. Recent research endeavors have increasingly turned their focus toward the interplay between the immune system and the central nervous system, seeking to uncover novel mechanisms that contribute to the onset and progression of depression. In this emerging landscape, CD4+ T cells, a pivotal component of the adaptive immune response, have come under the spotlight for their potential involvement in mediating neuroinflammatory processes that influence mood and cognition[3,4,5]. Simultaneously, Brain-Derived Neurotrophic Factor (BDNF), a versatile neurotrophin with profound roles in synaptic plasticity, mood regulation, and cognitive function, has been recognized as a key player in depression pathogenesis[6]. This review aims to traverse the intricate terrain where CD4+ T cells intersect with BDNF in the context of depression. By delving into the complex cross-talk between the immune system and neurotrophic factors, we endeavor to provide an encompassing overview of the current state of knowledge and its implications for innovative therapeutic approaches in the management of depression. Through the examination of these converging pathways, we strive to shed light on potential avenues that hold promise for a deeper understanding of depression and the development of more effective treatment modalities.
2. The Role of BDNF in Depression
Brain-Derived Neurotrophic Factor (BDNF) is a vital neurotrophin that plays a pivotal role in the intricate web of neurobiology [7]. It is a protein found predominantly in the brain and is crucial for supporting the growth, differentiation, and maintenance of neurons[8]. BDNF is essential for the processes of synaptic plasticity and neurogenesis, which are fundamental mechanisms for cognitive function, emotional well-being, and overall brain health. The discovery of reduced Brain-Derived Neurotrophic Factor (BDNF) levels in individuals with depression has profound implications for our understanding of the neurobiology of this mood disorder[6]. BDNF is not only a critical factor in brain development but also in the ongoing plasticity and adaptation of the adult brain. In depression, reduced Brain-Derived Neurotrophic Factor (BDNF) levels are closely associated with a several results including hippocampal atrophy, Impaired Neurogenesis, synaptic plasticity disruption.
In depression, several pathways dysregulation contribute toward to a reduced BDNF levels. The CREB (cAMP Response Element-Binding Protein) pathway, for instance, is affected as calcium influx decreases, impairing CREB's ability to bind to cAMP response elements (CREs) in the BDNF gene promoter and leading to decreased BDNF gene transcription [9,10]. Moreover, the self-regulating positive feedback loop, typically facilitated by TrkB receptor activation, loses its efficiency. This decline diminishes the enhancement of BDNF gene transcription through intracellular signaling pathways such as the mitogen-activated protein kinase (MAPK) pathway. Furthermore, the neurotrophin receptor p75NTR, with its dual pro-survival and pro-death effects on neurons, indirectly affects BDNF production, however its signaling becomes perturbed in depression, exacerbating the BDNF insufficiency [11]. Additionally, epigenetic regulation, involving DNA methylation and histone acetylation, undergoes aberrant changes that inhibit BDNF gene expression by disrupting chromatin structure and accessibility. Moreover, microRNAs (miRNAs), such as miR-16, are upregulated in individuals with depression and can potentially inhibit BDNF expression by binding to BDNF mRNA and reducing its stability [12]. Alterations in the levels of miR-132 can affect MeCP2, which, in turn, controls hippocampal BDNF levels in MDD [13]. Also, certain transcription factors, including NF-κB, may have a decreased ability to regulate the expression of the BDNF gene [14]. These complex genetic pathways collectively contribute to the observed reduction in BDNF levels.
One of the most significant consequences of BDNF reduction in depression is the impairment of neurogenesis. This impairment is closely linked to the dysregulation of several signaling pathways that are positively controlled by BDNF. In the context of depression, these pathways may undergo alterations, contributing to the negative impact on neurogenesis and overall brain function. Firstly, BDNF normally activates the MAPK/ERK (Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase) pathway, which is involved in cell growth, differentiation, and survival [15]. Secondly, BDNF can activate STAT proteins, including STAT3, which promote neural growth and differentiation through the JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathway [16]. Thirdly, the Ras pathway, activated by BDNF, influences cell growth, differentiation, and survival[17]. Fourthly, the mTOR (Mammalian Target of Rapamycin) pathway, crucial for protein synthesis, cell growth is normally activated by BDNF [18]. Fifthly, PKA (Protein Kinase A) activation by BDNF influences dendritic growth[19]. Sixthly, BDNF can activate CaMKII (Calcium/Calmodulin-Dependent Protein Kinase II), vital for synaptic plasticity and dendritic spine development[19]. Seventhly, BDNF signaling interacts with the Wnt/β-catenin pathway to influence neural differentiation and synaptic plasticity. Eighthly, the p38 MAPK pathway, involved in cellular stress responses and neural plasticity, can be activated by BDNF. Lastly, BDNF can modulate GSK-3 (Glycogen Synthase Kinase-3) activity, influencing neuronal differentiation, synaptic plasticity, and neuroprotection[20]. In depression, the reduction of BDNF levels can disrupt the fine-tuned control of these pathways, leading to impaired neurogenesis and synaptic plasticity, potentially contributing to the structural and functional alterations observed in the brains of individuals with depression and their associated cognitive and emotional symptoms. Researchers are actively investigating these pathways to develop potential therapeutic interventions that can restore normal BDNF signaling and promote recovery from depression.
When Brain-Derived Neurotrophic Factor (BDNF) levels are reduced, it can have a significant impact on the strengthening of synapses, particularly through mechanisms such as long-term potentiation (LTP). In conditions of reduced BDNF levels, the ability of neurons to induce LTP may be compromised[21,22,23]. This impairment can result in a reduced capacity to enhance synaptic strength in response to learning and neural activity. Moreover, one of the critical ways BDNF contributes to synaptic strengthening is by promoting the insertion of AMPA receptors into the postsynaptic membrane[24]. In situations with lower BDNF levels, this process may be less efficient or less frequent. As a result, there may be fewer AMPA receptors at the synapse, which can weaken synaptic transmission and reduce synaptic strength. Moreover, BDNF's regulation of NMDA receptor subunit composition is disturbed in depression. The balance of subunits, such as NR2A and NR2B, can be altered. This imbalance can affect the properties of NMDA receptors, potentially skewing them toward configurations that are less conducive to synaptic plasticity and learning. Changes in subunit composition can impair the ability of NMDA receptors to participate in the induction and maintenance of LTP, contributing to cognitive deficits observed in depression [25]. These structural changes can hinder the ability of synapses to strengthen and stabilize. Additionally, BDNF is involved in modulating the release and uptake of neurotransmitters, including glutamate[26]. Lower BDNF levels can disrupt the balance of excitatory and inhibitory neurotransmission, which is essential for synaptic strength. Altered neurotransmitter dynamics can result in weaker synaptic transmission [27,28]. Furthermore, BDNF also plays a role in shaping the structure of dendritic spines, which are essential for synaptic connections. Reduced BDNF levels can lead to alterations in spine morphology, potentially resulting in smaller or less stable dendritic spines [29]. Also, BDNF supports structural plasticity by influencing dendritic growth and branching. In conditions of BDNF deficiency, dendritic complexity and spine density may decrease. These structural changes can limit the synaptic contacts that neurons can form and impact the overall strength of synaptic connections[30]. It is interesting to note that In the absence of adequate BDNF signaling, there may be a reduced ability to maintain and strengthen synapses. This could lead to increased synaptic pruning, a process where weaker or less active synapses are eliminated [26]. While synaptic pruning is a natural process, excessive or unregulated pruning can weaken neural circuits. These factors can lead to alterations in mood and emotional regulation.
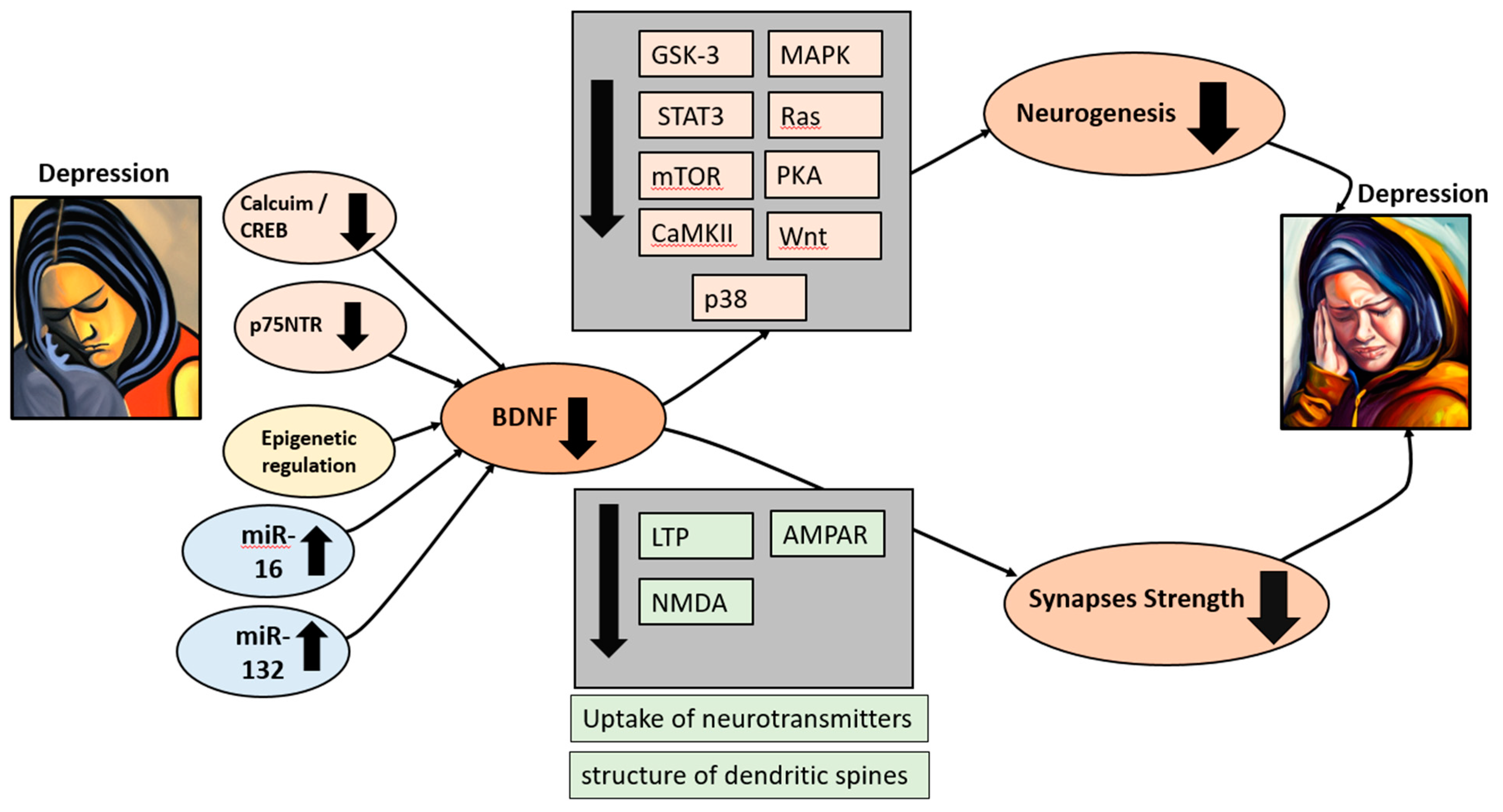
Figure 1.
BDNF plays a crucial role in depression. During depression, BDNF levels are reduced due to a decrease in calcium uptake, leading to subsequent dysregulation of the CREB pathways. Additionally, the decrease in p75NTR, alterations in epigenetic regulation, and the upregulation of microRNAs 16 and 32 also contribute to the reduction in BDNF levels. This reduction in BDNF is followed by a significant decrease in the rate of neurogenesis and synaptic weakening, resulting in the escalation of depression symptoms within a self-perpetuating cycle.
Figure 1.
BDNF plays a crucial role in depression. During depression, BDNF levels are reduced due to a decrease in calcium uptake, leading to subsequent dysregulation of the CREB pathways. Additionally, the decrease in p75NTR, alterations in epigenetic regulation, and the upregulation of microRNAs 16 and 32 also contribute to the reduction in BDNF levels. This reduction in BDNF is followed by a significant decrease in the rate of neurogenesis and synaptic weakening, resulting in the escalation of depression symptoms within a self-perpetuating cycle.
3. The Role of CD4 T Cells Subsets in Depression.
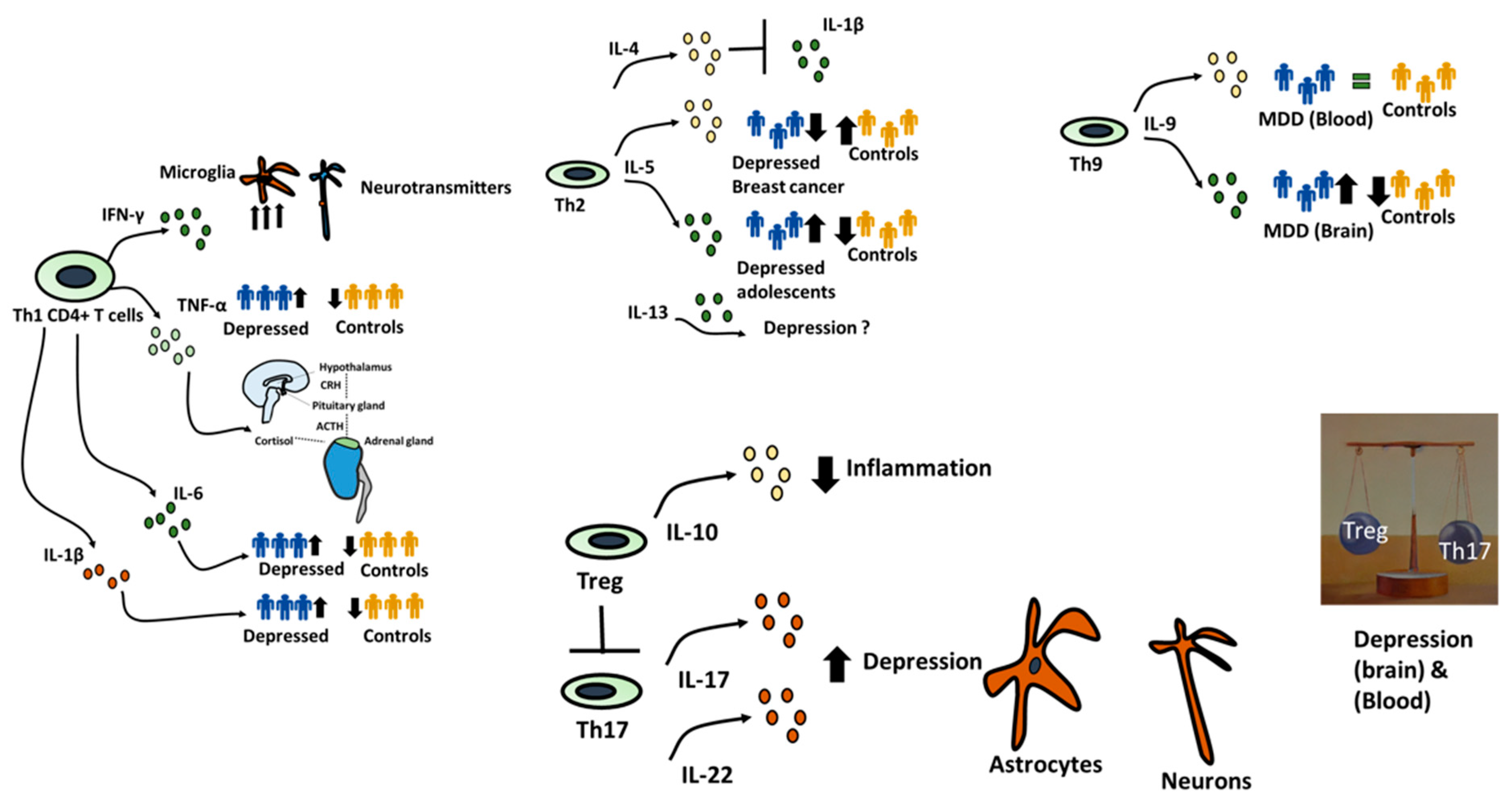
CD4+ T cells, also known as helper T cells, play a critical role in the adaptive immune system, and their remarkable diversity gives rise to various subsets [31]. Among these subsets, Th1 cells, or T Helper 1 cells, are well-known for their involvement in cellular immunity, particularly in the defense against intracellular pathogens such as viruses and intracellular bacteria. Th1 cells produce cytokines like interferon-gamma (IFN-γ), which activate macrophages to enhance their ability to eliminate intracellular invaders, although this response is often associated with inflammation and autoimmune conditions [32]. The impact of Th1 cells on depression may be mediated through the production of proinflammatory cytokines. For example, although IFN-γ, plays a central role in the body's defense against infections, however, its excessive presence is associated with detrimental effects in the context of depression [33]. Individuals with depression often exhibit increased levels of IFN-γ, closely linked to the activation of microglia in the central nervous system (CNS) [34]. This microglial activation can trigger neuroinflammation, which is intricately connected to depressive symptoms. Importantly, IFN-γ has the potential to disrupt the balance of neurotransmitters in the brain, including serotonin and dopamine, which are crucial for mood regulation and whose disturbance is a known contributor to depressive symptoms [35,36]. TNF-α is another pro-inflammatory cytokine primarily produced by Th1 cells, and it possesses potent immune-modulating properties [37]. Individuals with depression often have elevated levels of TNF-α, contributing to neuroinflammation and influencing neural circuits associated with mood regulation. Significantly, TNF-α can also activate the hypothalamic-pituitary-adrenal (HPA) axis, leading to an increase in cortisol levels, which is commonly dysregulated in depression [38,39]. Similarly, IL-1β and IL-6 have been reported to be upregulated in patients with Major Depressive Disorder (MDD), underscoring the role of Th1-associated cytokines in depression [40,41].
Th2 cells, a subset of T Helper cells, are primarily responsible for coordinating humoral immunity and immune responses against extracellular pathogens [42]. They produce a unique set of cytokines that play a crucial role in regulating immune reactions and allergic responses. IL-4, a signature cytokine produced by Th2 cells, is renowned for its role in promoting the differentiation of B cells into antibody-secreting plasma cells[43,44]. Interestingly, IL-4 has been shown to inhibit IL-1β-induced depressive-like behavior and central neurotransmitter alterations IL-5, another cytokine derived from Th2 cells, plays a critical role in the activation and maturation of eosinophils, a type of white blood cell involved in allergic responses and immunity against parasites [45]. Recent findings indicate that patients with Major Depressive Disorder (MDD) and breast cancer patients experiencing depression tend to have lower IL-5 levels in their serum compared to control groups [46] Paradoxically, in adolescents and pregnant women, there appears to be a positive correlation between IL-5 concentration and depression symptoms. These results suggest that other factors, such as pregnancy and gender, could also be influencing cytokine levels [47,48]. Similarly, the role of IL-13, another cytokine produced by Th2 cells and known for its involvement in regulating allergic and immune responses remains a topic of debate with no consensus on its relevance to depression [49,50,51].
There is limited understanding of the role of Th9 cells in depression. T Helper 9 cells, or Th9 cells, are typically associated with allergic responses and immune reactions against extracellular parasites[52]. Th9 cells produce interleukin-9 (IL-9), which enhances the function of mast cells, eosinophils, and other immune components [53]. Interestingly, research has shown that IL-9, a key cytokine produced by Th9 cells, is upregulated in the frontal cortex of patients with Major Depressive Disorder (MDD) [54,55,56]. Notably, a study by Becerril-Villanueva et al. did not find a correlation between IL-9 levels in the serum and the Hamilton Depression Rating Scale score, suggesting that cytokine levels can vary depending on the measurement location, such as the brain or the blood [55].
Although Th17 and Treg cells share a significant portion of their transcriptome, they differ considerably in their functions. Th17 cells, or T Helper 17 cells, play a critical role in defending against extracellular bacteria and fungi by producing cytokines like interleukin-17 (IL-17) and interleukin-22 (IL-22), which facilitate neutrophil recruitment and tissue repair [57,58]. However, Th17 responses can become dysregulated, contributing to autoimmune disorders and chronic inflammation. Notably, Th17 cells have the capacity to target NMDAR and counter the effects of BDNF [59]. Our research, along with others, has revealed that Th17 cells accumulate in the brains of depressed mice across various depression models [4,60,61]. Once in the brain, they interact with the brain environment, leading to increased astrogliosis, demyelination, and the formation of lesion-like structures [4]. Additionally, they exhibit a pro-inflammatory profile by producing IFNγ and TNFα and are regulated by RORγt expression. IL-17 has been shown to disrupt tight junctions of the blood-brain barrier (BBB) both in vitro and in vivo in the context of conditions like multiple sclerosis (MS), promoting inflammation within the central nervous system [62].
Regulatory T cells, or Treg cells, play a crucial role in maintaining immune tolerance and preventing autoimmune responses [63]. They achieve this by suppressing excessive immune activation and inflammation and by producing anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) Dysfunction in Treg cells can lead to the development of autoimmune diseases and allergies [64,65,66]. An imbalance in the Th17 to Treg cell ratio has been observed in both animal models and patients with depression characterized by an elevated frequency of Th17 cells and a reduced frequency of Treg cells [67,68,69,70]. This same imbalance in Th17 and Treg cell numbers has been identified in mouse models, where minimal Treg cell infiltration into the brain was observed. Importantly, on a therapeutic level, injecting Treg cells in co-transfer mouse models of depression led to a significant improvement in depression symptoms [4]. Furthermore, it has been reported that antidepressant treatment results in an increase in the number of Treg cells [71].
T Follicular Helper cells, or Tfh cells, play a central role in shaping antibody responses and facilitating germinal center formation in lymphoid tissues [72]. These cells provide crucial signals to B cells, encouraging class switching and affinity maturation, thereby enhancing the generation of high-affinity antibodies during infections and vaccinations[73]. Lastly, Their involvement is often associated with conditions like asthma and allergic inflammation. It's important to emphasize that our understanding of the precise role of Tfh cells in depression is still evolving, and more research is needed to elucidate the specific mechanisms involved. Additionally, depression is a complex condition influenced by multiple factors, including genetic, environmental, and neurobiological elements. Tfh cells represent one facet of the intricate interplay between the immune system and mood regulation. Further research may provide more insights into the functions of Tfh cells and their potential relevance in the development and treatment of depression.
Figure 2.
CD4+ T cells role in depression. Diverse CD4+ T cell subtypes exhibit significant variations in their impact on depression. Th1 and Th17 subtypes appear to promote inflammation and potentially contribute to depressive symptoms, while Treg subtypes exhibit anti-inflammatory properties and may aid in reducing depression symptoms. There is a clear call for further research in this area, specifically to explore the roles of Th9 and Tfh cells and to establish a consensus regarding the effects of cytokines associated with each cell subtype. Additionally, it is crucial to consider various factors such as the measurement location (CNS vs. blood), age (young vs. old), gender, and the presence of other underlying pathological conditions in future investigations.
Figure 2.
CD4+ T cells role in depression. Diverse CD4+ T cell subtypes exhibit significant variations in their impact on depression. Th1 and Th17 subtypes appear to promote inflammation and potentially contribute to depressive symptoms, while Treg subtypes exhibit anti-inflammatory properties and may aid in reducing depression symptoms. There is a clear call for further research in this area, specifically to explore the roles of Th9 and Tfh cells and to establish a consensus regarding the effects of cytokines associated with each cell subtype. Additionally, it is crucial to consider various factors such as the measurement location (CNS vs. blood), age (young vs. old), gender, and the presence of other underlying pathological conditions in future investigations.
4. The Interaction between CD4+ and BDNF
The interaction between various T cell subsets, such as Th1, Th2, Th17, Tfh, Th9, and brain-derived neurotrophic factor (BDNF) in depression is a complex and emerging area of research. These interactions involve both direct and indirect mechanisms. Direct mechanisms can occur through interactions between peripheral CD4+ T cells and BDNF, with these interactions taking place in two potential locations: (i) within the brain, between cells that have successfully infiltrated the blood-brain barrier and brain BDNF, and (ii) within the vessels of the blood-brain barrier. A previous study suggested a positive relationship between CD4+ T cell frequency and BDNF levels in the brain [74,75]. However, this study did not account for the high variation within CD4+ T cells, which include highly proinflammatory cells like Th17 and Th1, as well as anti-inflammatory cells like Tregs. Our research and that of others have shown that CD4+ T cells exhibit differences in their ability to infiltrate the BBB during pathological conditions. Th17 cells use a paracellular diapedesis mechanism and seem to have more favorable conditions for infiltrating the BBB compared to Tregs. Additionally, our unpublished data indicates that Th1 cells can enter the brain in depressive-like models through a transcellular route. Direct interactions may also occur in the bloodstream. It has been reported that serum BDNF levels negatively correlate with the Th1/Th2 ratio and Th17/Treg ratio in patients with cardiovascular diseases [76]. Furthermore, it has been suggested that BDNF can increase T cell proliferation and influence the differentiation of T cells into various subsets [77]. However, the exact impact of BDNF on the differentiation of CD4+ T cells remains an open question.
Indirect interactions could encompass interactions between BDNF and the gut-brain axis, primarily influenced by the interaction of CD4+ T cells with the microbiota and the release of cytokines that might impact BDNF levels [78]. In the case of anti-inflammatory cytokines like IL-4, they have been associated with stimulating astrocytic BDNF production, shifting microglial immune responses toward a neuroprotective M2 phenotype, and increasing hippocampal neurogenesis [79,80,48]. Conversely, pro-inflammatory cytokines appear to have the potential to decrease BDNF levels, but this effect can be reversed by administering the TNF-α inhibitor infliximab [81]. Indirect interactions also involve various cytokines associated with CD4+ T cells and BDNF. Elevated IL-1 expression in neurodegenerative disorders may render neurons susceptible to degeneration by disrupting BDNF-induced neuroprotection, primarily through inhibiting the PI3-K/Akt and MAPK/ERK pathways and suppressing CREB activation. This interference with BDNF signaling appears to involve ceramide-associated mechanisms and can be partially corrected by ceramide production inhibitors [82]. Paradoxically, IL-17 appears to enhance BDNF production by astrocytes. Similarly, in the case of IFNγ, there have been reports of IFNγ inducing BDNF production [83,84]. It is possible that these effects could be related to the nature of the cells induced, as astrocytes are considered the guardians of the blood-brain barrier (BBB). Therefore, IL-17 induction might logically prompt them to respond by producing more BDNF. Furthermore, this influence could be time-dependent, with initial IL-17 induction leading to BDNF induction, but studies on how chronic IL-17 induction affects astrocyte BDNF production are lacking. Additionally, in our depression model, we observed that Th17 cells appear to be associated with higher levels of astrocyte activation, which seems to be chronic. We hypothesize that a vicious cycle occurs involving IL6, IL1, and IL17 production by Th17 cells and the overactivation of astrocytes [85]. This area of research remains largely uninvestigated, with specific questions about the roles of various cytokines, such as IL9 and IL22, in relation to BDNF levels still unanswered.
There are several groundbreaking therapeutic interventions exploring the potential of BDNF interaction in neurodegenerative diseases, including depression. For instance, in a notable study, CD4+ T cells were genetically modified to produce BDNF and then injected intracerebroventricularly into the 5XFAD mouse model of Alzheimer's disease, resulting in a reduction in AD symptoms Additionally, patients with Treg cells expressing BDNF showed improved outcomes in stroke cases [86]. There is also speculation that controlling food intake could impact BDNF levels through the gut-brain axis, primarily mediated by the function of CD4+ T cells [87]. Furthermore, compounds like ellagic acid have been shown to increase BDNF levels in the plasma by 21% and are known to promote the differentiation of CD4+ T cells into an anti-inflammatory subtype [88,89]. Exercise has also been associated with an increased BDNF/IL6 ratio in neurodegenerative diseases, highlighting its potential role in enhancing the symptoms of depression [90]. These diverse approaches underscore the promising avenues for harnessing BDNF in the treatment of various neurodegenerative conditions.
5. Open Questions and Conclusions.
While we have gained some knowledge regarding the interaction between CD4+ T cells and BDNF production, many aspects remain elusive. Questions persist concerning how Th17 cells, which do not specifically target myelin sheaths, engage with neurons and how this dynamic unfolds over time and space within the brain. Additionally, the impact of Th17 cells on AMPA receptors, closely linked to BDNF production, remains an enigma. Speculation suggests that prolonged activation by Th17 cells may diminish BDNF levels. Conversely, therapeutic strategies involving Treg cells, which generate anti-inflammatory cytokines, hold promise for augmenting BDNF production and potentially benefiting depression therapy. Moreover, does BDNF influence the differentiation of CD4+ T cells into distinct subtypes? It is conceivable that it may favor anti-inflammatory types such as Tregs, though empirical evidence is still required. Overall, the interaction between BDNF and subpopulations of CD4+ T cells represents a crucial area of investigation that could potentially enhance the prognosis for depression patients.
References
- Herrman, H.; Patel, V.; Kieling, C.; Berk, M.; Buchweitz, C.; Cuijpers, P.; Furukawa, T.A.; Kessler, R.C.; Kohrt, B.A.; Maj, M.; et al. Time for United Action on Depression: A Lancet–World Psychiatric Association Commission. Lancet 2022, 399, 957–1022. [CrossRef]
- Nemeroff, C.B. The State of Our Understanding of the Pathophysiology and Optimal Treatment of Depression: Glass Half Full or Half Empty? Am. J. Psychiatry 2020, 177, 671–685. [CrossRef]
- Kubick, N.; Lazarczyk, M.; Strzałkowska, N.; Charuta, A.; Horbańczuk, J.O.; Sacharczuk, M.; Mickael, M.E. Factors Regulating the Differences in Frequency of Infiltration of Th17 and Treg of the Blood–Brain Barrier. Immunogenetics 2023. [CrossRef]
- Mickael, M.E.; Bhaumik, S.; Chakraborti, A.; Umfress, A.A.; van Groen, T.; Macaluso, M.; Totenhagen, J.; Sorace, A.G.; Bibb, J.A.; Standaert, D.G.; et al. RORγt-Expressing Pathogenic CD4+ T Cells Cause Brain Inflammation during Chronic Colitis. J. Immunol. 2022, 208, 2054–2066. [CrossRef]
- Mickael, M. Th17 Contributes to Brain Inflammation in Depression. Eur. J. Immunol. 2022, 52, 261–262.
- Cavaleri, D.; Moretti, F.; Bartoccetti, A.; Mauro, S.; Crocamo, C.; Carrà, G.; Bartoli, F. The Role of BDNF in Major Depressive Disorder, Related Clinical Features, and Antidepressant Treatment: Insight from Meta-Analyses. Neurosci. Biobehav. Rev. 2023.
- Arosio, B.; Guerini, F.R.; Voshaar, R.C.O.; Aprahamian, I. Blood Brain-Derived Neurotrophic Factor (BDNF) and Major Depression: Do We Have a Translational Perspective? Front. Behav. Neurosci. 2021.
- Zuccato, C.; Cattaneo, E. Brain-Derived Neurotrophic Factor in Neurodegenerative Diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [CrossRef]
- Yan, T.; Xu, M.; Wan, S.; Wang, M.; Wu, B.; Xiao, F.; Bi, K.; Jia, Y. Schisandra Chinensis Produces the Antidepressant-like Effects in Repeated Corticosterone-Induced Mice via the BDNF/TrkB/CREB Signaling Pathway. Psychiatry Res. 2016. [CrossRef]
- Shen, X.; Gu, X.; Liu, Y.Y.; Yang, L.; Zheng, M.; Jiang, L. Association between Dietary Calcium and Depression among American Adults: National Health and Nutrition Examination Survey. Front. Nutr. 2023. [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New Insights into BDNF Function in Depression and Anxiety. Nat. Neurosci. 2007.
- Bai, M.; Zhu, X.; Zhang, Y.; Zhang, S.; Zhang, L.; Xue, L.; Yi, J.; Yao, S.; Zhang, X. Abnormal Hippocampal BDNF and MiR-16 Expression Is Associated with Depression-Like Behaviors Induced by Stress during Early Life. PLoS One 2012. [CrossRef]
- Su, M.; Hong, J.; Zhao, Y.; Liu, S.; Xue, X. MeCP2 Controls Hippocampal Brain-Derived Neurotrophic Factor Expression via Homeostatic Interactions with MicroRNA-132 in Rats with Depression. Mol. Med. Rep. 2015. [CrossRef]
- Caviedes, A.; Lafourcade, C.; Soto, C.; Wyneken, U. BDNF/NF-ΚB Signaling in the Neurobiology of Depression. Curr. Pharm. Des. 2017. [CrossRef]
- Gudasheva, T.A.; Povarnina, P.Y.; Tarasiuk, A. V.; Seredenin, S.B. Low-Molecular Mimetics of Nerve Growth Factor and Brain-Derived Neurotrophic Factor: Design and Pharmacological Properties. Med. Res. Rev. 2021.
- Lin, G.; Zhang, H.; Sun, F.; Lu, Z.; Reed-Maldonado, A.; Lee, Y.C.; Wang, G.; Banie, L.; Lue, T.F. Brain-Derived Neurotrophic Factor Promotes Nerve Regeneration by Activating the JAK/STAT Pathway in Schwann Cells. Transl. Androl. Urol. 2016. [CrossRef]
- Cunha A Simple Role for BDNF in Learning and Memory? Front. Mol. Neurosci. 2010. [CrossRef]
- An, X.; Yao, X.; Li, B.; Yang, W.; Cui, R.; Zhao, G.; Jin, Y. Role of BDNF-MTORC1 Signaling Pathway in Female Depression. Neural Plast. 2021, 2021, 1–8. [CrossRef]
- Spencer, T.K.; Mellado, W.; Filbin, M.T. BDNF Activates CaMKIV and PKA in Parallel to Block MAG-Mediated Inhibition of Neurite Outgrowth. Mol. Cell. Neurosci. 2008. [CrossRef]
- Yang, J. wei; Ru, J.; Ma, W.; Gao, Y.; Liang, Z.; Liu, J.; Guo, J. hui; Li, L. yan BDNF Promotes the Growth of Human Neurons through Crosstalk with the Wnt/β-Catenin Signaling Pathway via GSK-3β. Neuropeptides 2015. [CrossRef]
- Diógenes, M.J.; Costenla, A.R.; Lopes, L. V.; Jerónimo-Santos, A.; Sousa, V.C.; Fontinha, B.M.; Ribeiro, J.A.; Sebastio, A.M. Enhancement of LTP in Aged Rats Is Dependent on Endogenous BDNF. Neuropsychopharmacology 2011. [CrossRef]
- Bramham, C.R.; Messaoudi, E. BDNF Function in Adult Synaptic Plasticity: The Synaptic Consolidation Hypothesis. Prog. Neurobiol. 2005.
- Escobar, M.L.; Figueroa-Guzmán, Y.; Gómez-Palacio-Schjetnan, A. In Vivo Insular Cortex LTP Induced by Brain-Derived Neurotrophic Factor. Brain Res. 2003. [CrossRef]
- Caldeira, M. V.; Melo, C. V.; Pereira, D.B.; Carvalho, R.; Correia, S.S.; Backos, D.S.; Carvalho, A.L.; Esteban, J.A.; Duarte, C.B. Brain-Derived Neurotrophic Factor Regulates the Expression and Synaptic Delivery Ofα-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid Receptor Subunits in Hippocampal Neurons. J. Biol. Chem. 2007, 282, 12619–12628. [CrossRef]
- Afonso, P.; De Luca, P.; Carvalho, R.S.; Cortes, L.; Pinheiro, P.; Oliveiros, B.; Almeida, R.D.; Mele, M.; Duarte, C.B. BDNF Increases Synaptic NMDA Receptor Abundance by Enhancing the Local Translation of Pyk2 in Cultured Hippocampal Neurons. Sci. Signal. 2019, 12. [CrossRef]
- Hu, B.; Nikolakopoulou, A.M.; Cohen-Cory, S. BDNF Stabilizes Synapses and Maintains the Structural Complexity of Optic Axons in Vivo. Development 2005, 132, 4285–4298. [CrossRef]
- Ninan, I.; Bath, K.G.; Dagar, K.; Perez-Castro, R.; Plummer, M.R.; Lee, F.S.; Chao, M. V. The BDNF Val66Met Polymorphism Impairs NMDA Receptor-Dependent Synaptic Plasticity in the Hippocampus. J. Neurosci. 2010, 30, 8866–8870. [CrossRef]
- Carvalho, A.L.; Caldeira, M. V.; Santos, S.D.; Duarte, C.B. Role of the Brain-Derived Neurotrophic Factor at Glutamatergic Synapses. In Proceedings of the British Journal of Pharmacology; 2008.
- Kellner, Y.; Gödecke, N.; Dierkes, T.; Thieme, N.; Zagrebelsky, M.; Korte, M. The BDNF Effects on Dendritic Spines of Mature Hippocampal Neurons Depend on Neuronal Activity. Front. Synaptic Neurosci. 2014. [CrossRef]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-Derived Neurotrophic Factor and the Development of Structural Neuronal Connectivity. Dev. Neurobiol. 2010.
- Abbas, A.; Lichtman, A.; Pillai, S. Cellular and Mollecular Immunology 9th Edition. Elsevier 2018.
- Cao, H.; Diao, J.; Liu, H.; Liu, S.; Liu, J.; Yuan, J.; Lin, J. The Pathogenicity and Synergistic Action of Th1 and Th17 Cells in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2023, 29, 818–829. [CrossRef]
- Zhang, J.; He, H.; Qiao, Y.; Zhou, T.; He, H.; Yi, S.; Zhang, L.; Mo, L.; Li, Y.; Jiang, W.; et al. Priming of Microglia with <scp>IFN</Scp> -γ Impairs Adult Hippocampal Neurogenesis and Leads to Depression-like Behaviors and Cognitive Defects. Glia 2020, 68, 2674–2692. [CrossRef]
- Myint, A.M.; Leonard, B.E.; Steinbusch, H.W.M.; Kim, Y.K. Th1, Th2, and Th3 Cytokine Alterations in Major Depression. J. Affect. Disord. 2005. [CrossRef]
- Foley, K.F.; Pantano, C.; Ciolino, A.; Mawe, G.M. IFN-γ and TNF-α Decrease Serotonin Transporter Function and Expression in Caco2 Cells. Am. J. Physiol. - Gastrointest. Liver Physiol. 2007. [CrossRef]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of Interferon-γ in Microglial-Mediated Loss of Dopaminergic Neurons. J. Neurosci. 2007. [CrossRef]
- Chen, J.; Xiang, X.; Nie, L.; Guo, X.; Zhang, F.; Wen, C.; Xia, Y.; Mao, L. The Emerging Role of Th1 Cells in Atherosclerosis and Its Implications for Therapy. Front. Immunol. 2023.
- Dunn, A.J. Cytokine Activation of the HPA Axis. In Proceedings of the Annals of the New York Academy of Sciences; 2000.
- Dziurkowska, E.; Wesolowski, M. Cortisol as a Biomarker of Mental Disorder Severity. J. Clin. Med. 2021.
- Min, X.; Wang, G.; Cui, Y.; Meng, P.; Hu, X.; Liu, S.; Wang, Y. Association between Inflammatory Cytokines and Symptoms of Major Depressive Disorder in Adults. Front. Immunol. 2023, 14. [CrossRef]
- Brown, S.J.; Christofides, K.; Weissleder, C.; Huang, X.-F.; Shannon Weickert, C.; Lim, C.K.; Newell, K.A. Sex- and Suicide-Specific Alterations in the Kynurenine Pathway in the Anterior Cingulate Cortex in Major Depression. Neuropsychopharmacology 2023. [CrossRef]
- Zhang, M.; Jin, H.; Li, Y.; Jiao, C.; Huang, P.; Bai, Y.; Gong, Z.; Zhang, H.; Liu, S.; Wang, H. Genetically Engineered Bacterial-like Particles Induced Specific Cellular and Humoral Immunity as Effective Tick-borne Encephalitis Virus Vaccine. Aggregate 2023, 4. [CrossRef]
- Rengarajan, J.; Mowen, K.A.; McBride, K.D.; Smith, E.D.; Singh, H.; Glimcher, L.H. Interferon Regulatory Factor 4 (IRF4) Interacts with NFATc2 to Modulate Interleukin 4 Gene Expression. J. Exp. Med. 2002. [CrossRef]
- Chakma, C.R.; Good-Jacobson, K.L. Requirements of IL-4 during the Generation of B Cell Memory. J. Immunol. 2023, 210, 1853–1860. [CrossRef]
- Allgire, E.; Ahlbrand, R.; Nawreen, N.; Ajmani, A.; Hoover, C.; McAlees, J.; Lewkowich, I.; Sah, R. Altered Fear Behavior in Aeroallergen House Dust Mite Exposed C57Bl/6 Mice: A Model of Th2-Skewed Airway Inflammation. Neuroscience 2023, 528, 75–88. [CrossRef]
- Xu, Y.; Liang, J.; Sun, Y.; Zhang, Y.; Shan, F.; Ge, J.; Xia, Q. Serum Cytokines-Based Biomarkers in the Diagnosis and Monitoring of Therapeutic Response in Patients with Major Depressive Disorder. Int. Immunopharmacol. 2023. [CrossRef]
- Karlsson, L.; Nousiainen, N.; Scheinin, N.M.; Maksimow, M.; Salmi, M.; Lehto, S.M.; Tolvanen, M.; Lukkarinen, H.; Karlsson, H. Cytokine Profile and Maternal Depression and Anxiety Symptoms in Mid-Pregnancy—the FinnBrain Birth Cohort Study. Arch. Womens. Ment. Health 2017, 20, 39–48. [CrossRef]
- Pérez-Sánchez, G.; Becerril-Villanueva, E.; Arreola, R.; Martínez-Levy, G.; Hernández-Gutiérrez, M.E.; Velasco-Velásquez, M.A.; Alvarez-Herrera, S.; Cruz-Fuentes, C.; Palacios, L.; De La Peña, F.; et al. Inflammatory Profiles in Depressed Adolescents Treated with Fluoxetine: An 8-Week Follow-up Open Study. Mediators Inflamm. 2018. [CrossRef]
- Liu, C.; Liu, L.; Huang, Y.; Shi, R.; Wu, Y.; Hakimah Binti Ismail, I. Contribution of IL-33/ILC2-Mediated Th2 Cytokines during the Progression of Minimal Change Disease. Int. Immunopharmacol. 2023. [CrossRef]
- Ho, H.Y.; Chin-Hung Chen, V.; Tzang, B.S.; Hsieh, C.C.; Wang, W.K.; Weng, Y.P.; Hsu, Y.T.; Hsaio, H.P.; Weng, J.C.; Chen, Y.L. Circulating Cytokines as Predictors of Depression in Patients with Breast Cancer. J. Psychiatr. Res. 2021. [CrossRef]
- Zhu, J. T Helper 2 (Th2) Cell Differentiation, Type 2 Innate Lymphoid Cell (ILC2) Development and Regulation of Interleukin-4 (IL-4) and IL-13 Production. Cytokine 2015.
- Licona-Limón, P.; Arias-Rojas, A.; Olguín-Martínez, E. IL-9 and Th9 in Parasite Immunity. Semin. Immunopathol. 2017.
- Kaplan, M.H. Th9 Cells: Differentiation and Disease. Immunol. Rev. 2013. [CrossRef]
- Shelton, R.C.; Claiborne, J.; Sidoryk-Wegrzynowicz, M.; Reddy, R.; Aschner, M.; Lewis, D.A.; Mirnics, K. Altered Expression of Genes Involved in Inflammation and Apoptosis in Frontal Cortex in Major Depression. Mol. Psychiatry 2011. [CrossRef]
- Becerril-Villanueva, E.; Pérez-Sánchez, G.; Alvarez-Herrera, S.; Girón-Pérez, M.I.; Arreola, R.; Cruz-Fuentes, C.; Palacios, L.; De La Penã, F.R.; Pavón, L. Alterations in the Levels of Growth Factors in Adolescents with Major Depressive Disorder: A Longitudinal Study during the Treatment with Fluoxetine. Mediators Inflamm. 2019. [CrossRef]
- Harsanyi, S.; Kupcova, I.; Danisovic, L.; Klein, M. Selected Biomarkers of Depression: What Are the Effects of Cytokines and Inflammation? Int. J. Mol. Sci. 2023.
- Mickael, M.-E.; Basu, R.; Bhaumik, S. Retinoid-Related Orphan Receptor RORγt in CD4+T Cell Mediated Intestinal Homeostasis and Inflammation. Am. J. Pathol. 2020, 02.
- Mickael, M.E.; Kubick, N.; Łazarczyk, M.; Sacharczuk, M.; Marchewka, J.; Urbański, P.; Horbańczuk, J.O. Transcriptome Analysis of the Th17/Treg Axis Reveals Multiple Pathways That Ensure Distinct Differentiation Patterns. Anim. Sci. Pap. Reports 2023, 41, 79–93.
- Zeng, C.; Li, L.; Chen, L.; Li, P.; Chen, M.; Wu, X.; Chen, C. Th17 Cells Regulate the Progress of Anti-NMDAR Encephalitis. Am. J. Transl. Res. 2022, 14, 6268–6276.
- Beurel, E.; Lowell, J.A. Th17 Cells in Depression. Brain. Behav. Immun. 2018, 69, 28–34. [CrossRef]
- Cui, M.; Dai, W.; Kong, J.; Chen, H. Th17 Cells in Depression: Are They Crucial for the Antidepressant Effect of Ketamine? Front. Pharmacol. 2021, 12. [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 Lymphocytes Promote Blood-Brain Barrier Disruption and Central Nervous System Inflammation. Nat. Med. 2007, 13, 1173–1175. [CrossRef]
- Łazarczyk, M.; Mickael, M.E.; Skiba, D.; Kurzejamska, E.; Ławiński, M.; Horbańczuk, J.O.; Radziszewski, J.; Fraczek, K.; Wolinska, R.; Paszkiewicz, J.; et al. The Journey of Cancer Cells to the Brain: Challenges and Opportunities. Int. J. Mol. Sci. 2023, 24, 3854. [CrossRef]
- Edwar-Mickael, M.; Kubick, N. CD4 + Tregs May Be Essential for Solving Astrocyte Glial Scar Deadlock. Neural Regen. Res. 2021, 16, 2563. [CrossRef]
- Kubick, N.; Klimovich, P.; Flournoy, P.H.; Bieńkowska, I.; Łazarczyk, M.; Sacharczuk, M.; Bhaumik, S.; Mickael, M.-E.; Basu, R. Interleukins and Interleukin Receptors Evolutionary History and Origin in Relation to CD4+ T Cell Evolution. Genes (Basel). 2021, 12, 813. [CrossRef]
- Bhaumik, S.; Łazarczyk, M.; Kubick, N.; Klimovich, P.; Gurba, A.; Paszkiewicz, J.; Teodorowicz, P.; Kocki, T.; Horbańczuk, J.O.; Manda, G.; et al. Investigation of the Molecular Evolution of Treg Suppression Mechanisms Indicates a Convergent Origin. Curr. Issues Mol. Biol. 2023, 45, 628–648. [CrossRef]
- Huang, C.; Zhang, F.; Li, P.; Song, C. Low-Dose IL-2 Attenuated Depression-like Behaviors and Pathological Changes through Restoring the Balances between IL-6 and TGF-β and between Th17 and Treg in a Chronic Stress-Induced Mouse Model of Depression. Int. J. Mol. Sci. 2022. [CrossRef]
- Ghosh, R.; Kumar, P.K.; Mitra, P.; Purohit, P.; Nebhinani, N.; Sharma, P. Circulating T Helper 17 and IFN-γ Positive Th17 Cells in Major Depressive Disorder. Behav. Brain Res. 2020. [CrossRef]
- Harkin, A.; Corrigan, M.; O’Rourke, A.M.; Moran, B.; Fletcher, J. Inflammation in the Pathogenesis of Depression; a Disorder of Neuroimmune Origin? Neuronal Signal. 2023. [CrossRef]
- Ambrée, O.; Ruland, C.; Zwanzger, P.; Klotz, L.; Baune, B.T.; Arolt, V.; Scheu, S.; Alferink, J. Social Defeat Modulates T Helper Cell Percentages in Stress Susceptible and Resilient Mice. Int. J. Mol. Sci. 2019. [CrossRef]
- Grosse, L.; Carvalho, L.A.; Birkenhager, T.K.; Hoogendijk, W.J.; Kushner, S.A.; Drexhage, H.A.; Bergink, V. Circulating Cytotoxic T Cells and Natural Killer Cells as Potential Predictors for Antidepressant Response in Melancholic Depression. Restoration of T Regulatory Cell Populations after Antidepressant Therapy. Psychopharmacology (Berl). 2016. [CrossRef]
- Crotty, S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity 2014.
- Garg, A.K.; Mitra, T.; Schips, M.; Bandyopadhyay, A.; Meyer-Hermann, M. Amount of Antigen, T Follicular Helper Cells and Affinity of Founder Cells Shape the Diversity of Germinal Center B Cells: A Computational Study. Front. Immunol. 2023. [CrossRef]
- Wolf, S.A.; Steiner, B.; Akpinarli, A.; Kammertoens, T.; Nassenstein, C.; Braun, A.; Blankenstein, T.; Kempermann, G. CD4-Positive T Lymphocytes Provide a Neuroimmunological Link in the Control of Adult Hippocampal Neurogenesis. J. Immunol. 2009. [CrossRef]
- Chen, Z.; Huang, Y.; Wang, B.; Peng, H.; Wang, X.; Wu, H.; Chen, W.; Wang, M. T Cells: An Emerging Cast of Roles in Bipolar Disorder. Transl. Psychiatry 2023.
- Huo, Y.; Feng, Q.; Fan, J.; Huang, J.; Zhu, Y.; Wu, Y.; Hou, A.; Zhu, L. Serum Brain-Derived Neurotrophic Factor in Coronary Heart Disease: Correlation with the T Helper (Th)1/Th2 Ratio, Th17/Regulatory T (Treg) Ratio, and Major Adverse Cardiovascular Events. J. Clin. Lab. Anal. 2023. [CrossRef]
- Tian, B.; Yang, C.; Wang, J.; Hou, X.; Zhao, S.; Li, Y.; Yang, P. Peripheral Blood Brain-Derived Neurotrophic Factor Level and Tyrosine Kinase B Expression on T Lymphocytes in Systemic Lupus Erythematosus: Implications for Systemic Involvement. Cytokine 2019. [CrossRef]
- Li, J.H.; Liu, J.L.; Li, X.W.; Liu, Y.; Yang, J.Z.; Chen, L.J.; Zhang, K.K.; Xie, X.L.; Wang, Q. Gut Microbiota from Sigma-1 Receptor Knockout Mice Induces Depression-like Behaviors and Modulates the CAMP/CREB/BDNF Signaling Pathway. Front. Microbiol. 2023. [CrossRef]
- Mucci, F.; Marazziti, D.; Vecchia, A. Della; Baroni, S.; Morana, P.; Carpita, B.; Mangiapane, P.; Morana, F.; Morana, B.; Dell’osso, L. State-of-the-Art: Inflammatory and Metabolic Markers in Mood Disorders. Life 2020.
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the Brain: A Cytokine To Remember. J. Immunol. 2012. [CrossRef]
- Zhang, C.; Liu, B.; Pawluski, J.; Steinbusch, H.W.M.; Kirthana Kunikullaya, U.; Song, C. The Effect of Chronic Stress on Behaviors, Inflammation and Lymphocyte Subtypes in Male and Female Rats. Behav. Brain Res. 2023, 439, 114220. [CrossRef]
- Tong, L.; Balazs, R.; Soiampornkul, R.; Thangnipon, W.; Cotman, C.W. Interleukin-1β Impairs Brain Derived Neurotrophic Factor-Induced Signal Transduction. Neurobiol. Aging 2008. [CrossRef]
- Litteljohn, D.; Nelson, E.; Hayley, S. IFN-Î3 Differentially Modulates Memory-Related Processes under Basal and Chronic Stressor Conditions. Front. Cell. Neurosci. 2014, 8. [CrossRef]
- Bergström, A.; Jayatissa, M.N.; Mørk, A.; Wiborg, O. Stress Sensitivity and Resilience in the Chronic Mild Stress Rat Model of Depression; an in Situ Hybridization Study. Brain Res. 2008. [CrossRef]
- Kubick, N.; Flournoy, P.C.H.; Enciu, A.-M.; Manda, G.; Mickael, M.-E. Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease. Pharmaceutics 2020, 12, 880. [CrossRef]
- Chan, A.; Yan, J.; Csurhes, P.; Greer, J.; McCombe, P. Circulating Brain Derived Neurotrophic Factor (BDNF) and Frequency of BDNF Positive T Cells in Peripheral Blood in Human Ischemic Stroke: Effect on Outcome. J. Neuroimmunol. 2015. [CrossRef]
- Bhatt, S.; Kanoujia, J.; Mohana Lakshmi, S.; Patil, C.; Gupta, G.; Chellappan, D.K.; Dua, K. Role of Brain-Gut-Microbiota Axis in Depression: Emerging Therapeutic Avenues. CNS Neurol. Disord. - Drug Targets 2022. [CrossRef]
- Liu, Y.; Yu, S.; Wang, F.; Yu, H.; Li, X.; Dong, W.; Lin, R.; Liu, Q. Chronic Administration of Ellagic Acid Improved the Cognition in Middle-Aged Overweight Men. Appl. Physiol. Nutr. Metab. 2018. [CrossRef]
- Hajiluian, G.; Karegar, S.J.; Shidfar, F.; Aryaeian, N.; Salehi, M.; Lotfi, T.; Farhangnia, P.; Heshmati, J.; Delbandi, A.-A. The Effects of Ellagic Acid Supplementation on Neurotrophic, Inflammation, and Oxidative Stress Factors, and Indoleamine 2, 3-Dioxygenase Gene Expression in Multiple Sclerosis Patients with Mild to Moderate Depressive Symptoms: A Randomized, Triple-Blind,. Phytomedicine 2023, 155094. [CrossRef]
- Devasahayam, A.J.; Kelly, L.P.; Williams, J.B.; Moore, C.S.; Ploughman, M. Fitness Shifts the Balance of BDNF and IL-6 from Inflammation to Repair among People with Progressive Multiple Sclerosis. Biomolecules 2021, 11, 504. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



