
Submitted:
05 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
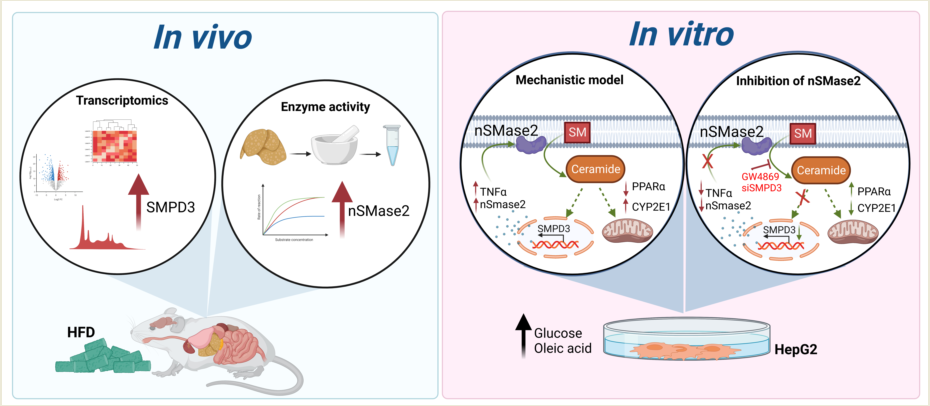
Background: Non-alcoholic fatty liver disease (NAFLD) is manifested by hepatic steatosis, insulin resistance, hepatocyte death, and systemic inflammation. Obesity induces steatosis and chronic inflammation in the liver. However, the precise mechanism underlying hepatic steatosis in the setting of obesity remains unclear. Here we report studies that address this question. Methods: 14 weeks on high-fat diet (HFD) with high sucrose, C57BL/6 mice revealed a phenotype of liver steatosis. Transcriptional profiling analysis of the liver tissues was performed using RNA sequencing (RNA-seq). Results: Our RNA-seq data revealed 692 differentially expressed genes involved in processes of lipid metabolism, oxidative stress, immune responses, and cell proliferation. Notably, the gene encoding neutral sphingomyelinase SMPD3, was predominantly upregulated in the liver tissues of the mice displaying a phenotype of steatosis. Moreover, nSMase2 activity was elevated in these tissues of the liver. Pharmacological and genetic inhibition of nSMase2 prevented intracellular lipid accumulation and TNFα-induced inflammation in in-vitro HepG2-steatosis cellular model. Furthermore, nSMase2 inhibition ameliorates oxidative damage by rescuing PPARα and prevent cell death associated with high glucose/oleic acid-induced fat accumulation in HepG2 cells. Conclusions: Collectively, our findings highlight the prominent role of nSMase2 in hepatic steatosis which could serve as a potential therapeutic target for NAFLD and other hepatic steatosis-linked disorders.
Keywords:
NAFLD
; SMPD3
; Lipotoxicity
; HepG2
; TNF-α
; sphingomyelin pathway
; RNA sequencing
; Oil Red O staining
; fat accumulation
; Obesity
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most commonly occurring chronic liver disease among obese and diabetic individuals [1]. It is characterized by hepatic steatosis caused by lipid accumulation in hepatocytes. The development of hepatic lipotoxicity is considered one of the earliest signs of NAFLD and has been linked to hepatic oxidative stress, inflammatory responses, and insulin resistance [2]. Therefore, therapeutic strategies pursuing the prevention and reduction of lipid content within the hepatocytes are the most sought-after approaches. Although numerous studies have been conducted to understand the pathogenesis of hepatic steatosis, clinical applications remain limited. Alterations in molecular mechanisms that lead to NAFLD, and other disorders related to hepatic steatosis, are partially known. Ceramides are a class of sphingolipids that are abundant in cell membranes. They are generated by three metabolic pathways: de novo synthesis, sphingomyelin hydrolysis, and the salvage pathway [3]. The formation of ceramide can be induced by different stimuli, such as oxidative stress and tumor necrosis factor-α (TNF-α) [4]. The liver is a primary location for ceramide formation and contains significantly more sphingolipids, particularly ceramide and sphingomyelin (SM), than any other organ. As a result, the liver is vulnerable to sphingolipotoxicity [5]. Sphingomyelin synthase (SMS) has two isoforms, SMS1 and SMS2, that metabolize ceramide into SM, while SM-induced ceramide generation occurs via the action of sphingomyelinases (SMases), which can be distinguished by their pH optima [6]. Taking into account the role of the SM pathway in lipid accumulation, inflammation, and proapoptotic signature in the pathogenesis of hepatic steatosis, targeting this pathway may be a valuable approach in treating hepatic steatosis in humans. In the present study, an RNA-seq-based approach was applied to gain a deeper understanding of the influence of high fat diet (HFD) on lipid metabolism and sphingolipid generation in the liver. We present a systematic characterization of liver steatosis at the transcriptional level in mice and in a human-based cell model, along with a sphingomyelin (SM) pathway associated with pathological changes seen in NAFLD.
2. Results
2.1. Transcriptomic Analysis Indicates an Upregulation of Neutral Sphingomyelinase SMPD3/nSMase2 in the Livers with Steatosis
In this study, C57BL/6 male mice were fed HFD for 14 weeks to induce steatosis and NAFLD pathologies. We observed a statistically significant weight gain in HFD-fed mice, as compared to those given chow diet (CD) (Figure 1A,B). In the gross specimen, livers isolated from HFD-fed mice had a significantly enlarged morphology (p = 0.05) and were pale in color, as compared to livers isolated from the CD group (Figure 1C). Further insights into hepatic morphology verified the presence of abnormal hepatic structure, with an increase in both macro- and micro-vesicular steatosis score in livers from HFD-fed mice, as compared to those fed CD (Figure 1D,E). Inflammatory cell infiltration was also observed in liver tissues from HFD-fed mice, as indicated by F4/80 IHC staining (Figure 1F,G), along with obvious significant lipid accumulation that was absent in CD-fed mice (Figure 1H,I).
Next RNA-seq analysis was performed on the liver tissues to explore the mechanisms underlying hepatic steatosis. Principal component analysis (PCA) showed clustering of liver samples according to the progressive morphology of treatment (Figure 2A). To visualize DEGs in the liver of HFD-fed mice, we mapped significantly altered genes in a volcano plot (Figure 2B). In total, we identified 510 upregulated genes and 672 downregulated genes, when comparing mice fed CD. We further performed functional analyses of DEGs using Gene Ontology (GO) and the KEGG database. Next, we conducted GO enrichment analysis to get better insights into the biological role of the up- and down-regulated genes. Livers with steatosis were enriched with upregulated genes involved in processes such as lipid metabolism, oxidative stress responses, immune response, cell proliferation, and apoptosis (Figure 2C). While downregulated genes were majorly associated with triglyceride biosynthetic processes, gluconeogenesis, and glucose response, as well as inflammatory and endoplasmic reticulum responses (Figure 2D). Analysis of the KEGG pathways of upregulated genes revealed potential key roles of genes in cholesterol metabolism, MAPK signaling pathway, steroid biosynthetic processes, positive regulation of NF-kB signaling, Wnt signaling pathway, and bile secretion (Figure 2E). We also observed that pathways of the downregulated genes in livers with steatosis were involved in metabolic pathways, AMPK signaling pathway, fatty acid degradation, PPAR signaling pathway, PI3K-Akt signaling pathway, insulin resistance, and alcoholic liver disease (Figure 2F). The most significantly enriched DEGs were associated with altered functions in cholesterol metabolism, reactive oxygen species, apoptosis, response to ATP, lipid metabolism, and fatty acid metabolism (Figure 2G). Our data show that the livers with steatosis contain abundant mRNAs encoding proteins that are involved in functions associated with lipid and fatty acid metabolism. Interestingly, among the genes that were highly upregulated in livers with steatosis was SMPD3, the gene coding for nSMase2, which catalyzes the hydrolysis of sphingomyelin to form ceramide and phosphocholine [7,8] (Figure 2H). It is well-established that different chronic liver conditions are found to be associated with alterations in SM levels due to subsequent changes in the activity of SMases [9]. To verify our RNA-Seq data, we further assessed the gene expression of SMPD3 and SMPD1 using Taqman analysis. The expression of SMPD3, the gene responsible for nSMase2, was found to be significantly upregulated in livers of mice with hepatic steatosis, with no significant changes in the aSMase expression (Supplementary Figure S1A,B). More importantly, significant elevation in the enzymatic activity of nSMase2 was further confirmed in the livers of HFD-fed mice, as compared to those fed CD (Figure 2I). Collectively, our data highlight dysregulation in sphingolipid metabolism with predominant upregulation of SMPD3/nSMase 2 that has been associated with steatosis.
2.2. Sphingomyelin (SM) Pathway is Involved in Liver Steatosis
To further investigate the mechanistical role of SM in HFD-induced steatosis, we created an in-vitro HepG2 cell model mimicking the characteristics of obesity mediated steatosis. (Supplementary Figure 2). In our HepG2 cell model, we observed significant intracellular lipid accumulation, as determined by Nile red staining and BODIPY flow cytometric analysis (p ≤ 0.05; Figure S2A,B). It is well-documented that the progression of steatosis causes an upregulation of TNF-α and other inflammatory markers [10,11]. To ascertain whether this effect also occurred in our HepG2 model, we investigated the production and secretion of TNF-α. The expression of TNF-α was elevated in response to high glucose plus oleic acid (OA) treatments (p ≤ 0.001), as compared to OA-free and low glucose treatments (Figure S2C–E).
Our data suggest that under high glucose and high fat conditions, both lipid accumulation and inflammation is triggered in HepG2 cells, which is most likely mediated by oxidative stress [12]. To further support our HepG2-based model of liver steatosis, we investigated the expression of PPARα, a nuclear modulator reported to protect against oxidative damage [13,14]. RT-qPCR analysis of PPARα expression showed significant (p ≤ 0.001) inhibition in the high glucose/OA HepG2 cell model (Figure S2F). In concordance to this, a significant reduction in the protein levels of PPARα was also noticed under Western blot analysis (Figure S2G), indicating reduced protection against oxidative damage. Indeed, the gene expression of the cytochrome P450 2E1 (CYP2E1), a contributor to oxidative stress in NAFLD [13], was also found to be elevated in our model (Figure S2H).
Hepatocyte apoptosis is a prominent feature of liver damage and NAFLD. Therefore, we evaluated whether the high glucose/high OA model triggers apoptosis and induces cell death. Flow cytometry analyses of annexin V/PI staining confirmed that the generated HepG2 liver steatosis model induced a higher rate of apoptosis in HepG2 cells, when compared to the other applied conditions (Figure S2I). The MFI of annexin V-FITC A was found to be significantly (p ≤ 0.001) upregulated, as compared to that of low glucose or OA-free conditions (Figure S2J). We further assessed cell viability using an MTT assay. We found that the high glucose plus high OA model was associated with a significant reduction in cell survival (p ≤ 0.00001; Figure S2K). Collectively, our data strongly highlight that under the influence of high glucose and OA, steatosis is triggered in HepG2 cells with similar inflammatory and lipotoxic characteristics, as seen in our HFD mice model and the previously reported pathophysiology of NAFLD in humans.
Next, we applied this model to investigate the role of the SM pathway in liver steatosis. We investigated the expression of SM production in our cell model by screening for genes involved in SM metabolism, as presented in Figure 3A. The gene expression of both sphingomyelin synthase 1 (SGMS1) and SGMS2 were found to be downregulated (p ≤ 0.05 and p ≤ 0.01, respectively) in the described liver steatosis cell model, suggesting a reduction in SM synthesis (Figure 3B and Figure 3C, respectively). Interestingly, when we investigated the expression of genes involved in SM hydrolysis, only nSMase2 was induced in the steatosis cell model, with no significant changes found in the transcript levels of aSMase (Figure 3D,E).
To further verify these observations, we measured the enzymatic activity for both nSMase2 and aSMase in our cell model. Notably, steatosis-inducing conditions in HepG2 cells resulted in a significant upregulation in the enzymatic activity of nSMase2 (Figure 3F). On the other hand, aSMase activity was slightly, albeit not significantly, downregulated as observed in Figure 3G. These data suggest that under conditions of steatosis, SM hydrolysis signaling pathway induction is mediated via nSMase2.
2.3. Effect of nSMase2 Inhibition in the HepG2 Liver Steatosis Model
To further evaluate the modulatory effect of nSMase2 in our cell model, we applied loss- or gain-of-function models using the nSMase2 synthetic inhibitor, GW4869, and nSMase2 agonist, Daunorubicin (DNR), respectively. We also tested the influence of aSMase inhibition using the functional (indirect) inhibitor Imip. The activity of nSMase2 was found to be significantly increased (p ≤ 0.0001) under steatosis conditions (high glucose/OA), as compared to normal conditions (low glucose/OA-free). Likewise, nSMase2 activity was significantly elevated in response to DNR treatments; however, Imip had no effect on basal nSMase2 activity, while GW4869 inhibited basal activity (Figure 4A). As anticipated, aSMase enzymatic activity was significantly reduced after treatment with Imip, however, was not affected by GW4869 or DNR (Figure 4B).
To investigate the prospective role of inhibition of nSMase2 activity in liver steatosis, we cultured HepG2 cells under high glucose conditions, and pre-treated the cells with either GW4869, DNR, or Imip prior to OA stimulation to induce steatosis. First, we assessed the level of intracellular lipid accumulation by either flow cytometry analysis (using BIDOPY 493/503; Figure 4C) or confocal microscopy cell imaging (using Nile red staining; Figure 4D). When compared to the level of lipid accumulation under steatosis conditions, pre-treatment with GW4869 or Imip resulted in a significant reduction in total lipid content in HepG2 cells treated with OA. However, nSMase2 inhibition with GW4869 alone was shown to be similar to cells cultivated under normal conditions (Figure 4C,D).
2.4. Inhibition of nSMase2 in HepG2-Steatosis Model Reduces Inflammation and Protects Against Oxidative Stress
The role of SM-derived ceramide inflammatory signaling pathway has been investigated by several groups. Both nSMase2 and aSMase were found to induce the release of various cytokines and chemokines including, among others, TNF-α, indicating that these enzymes are involved in inflammation signaling [15,16]. In our model, the inhibition of nSMase2 using GW4869 significantly downregulated TNF-α expression under steatosis conditions (p ≤ 0.01) (Figure 5A). It is noteworthy that although Imip showed statistically insignificant reduction in the gene expression of TNF-α in steatosis conditions, we found this elevation also non significantly higher to normal conditions. Indeed, analyses of protein expression using Western blot further verified that both GW4869 and Imip inhibitors have a significant effect on TNF-α downregulation (p ≤ 0.001 and p ≤ 0.01, respectively) (Figure 5B); similar findings were observed in TNF-α secretion (Figure 5C). These results are in concordance with those of other groups as they highlight that SM hydrolysis enzymes play a modulatory role in inflammatory signaling.
Furthermore, the inhibition of nSMase2 through its synthetic inhibitor GW4869 showed significant upregulation of PPARα at both gene and protein levels, while further reduction was observed under the inhibition of aSMase and the augmentation of nSMase2 activity through DNR (Figure 5D,E). This observation was further reflected in the decreased expression of CYP2E1 (Figure 5F) and overall increased cell survivability in GW4869-treated cells, as compared to other inhibitors (Figure 5G and Supplementary Figure S3). Together, these results indicate that the inhibition of nSMase2 activity ameliorates oxidative damage, that may subsequently prevent triggered cell death associated with high glucose and OA conditions.
2.5. nSMase2 Deficiency Ameliorates Liver Steatosis Pathologies
To further define the role of nSMase2 in HepG2 liver steatosis cell models, we transfected HepG2 cells with siRNA against nSMase2 (SMPD3), which achieved 30–40% reduction in nSMase2 mRNA levels, as compared to scrambled (control) siRNA (Figure 6A). We cultured the siRNA-transfected HepG2 cells in high glucose media, followed by stimulation with OA. Results showed that the expression of TNF-α, at both the mRNA and protein levels, was significantly reduced in SMPD3 siRNA-transfected HepG2 cells (Figure 6B,C), indicating a role of nSMase2 in the inflammatory response to steatosis. Silencing the nSMase2 gene (SMPD3) was also found to prevent markers of oxidative stress, indicated by the upregulation of PPARα gene and protein expression (Figure 6D,E) and by inhibiting the expression of CYP2E1 (Figure 6F). The influence of silencing the nSMase2 gene, SMPD3, was further examined in its role in modulating lipid accumulation and cell survivability. As expected, our results showed that nSMase2-deficient HepG2 failed to respond to steatosis-inducing conditions (Figure 6G) and improved cell survivability (Figure 6H–J). Overall, the results further support the significant involvement of nSMase2 in modulating liver steatosis in hepatocytes.
3. Discussion
Despite intensive research performed to discover therapeutic solutions for targeting, preventing, or restraining the progression of obesity induced hepatic steatosis, options remain limited. One of the main hurdles in drug discovery studies that target NAFLD development is the absence of a reliable in vitro human model. Although murine models are widely used to identify metabolic pathways that may lead to NAFLD, translating preclinical findings of murine models to human diseases can be slightly disappointing in certain cases. Imbalance in lipid metabolism induced by a modern high fat-consuming lifestyle is often concomitant with an increase in body weight and fat storage, which is in turn associated with the development of metabolic syndrome, such as NAFLD. In our work, mice fed with an HFD for 14 weeks showed a significant increase in body weight, as compared to CD-fed mice. This was shown in the histological results of H&E and lipid staining that indicated the development of fatty liver, with hepatocytes exhibiting higher macrovascular, microvascular, and inflammatory scores, as compared to CD-fed mice. To further investigate these observed changes between HFD- and CD-fed mice livers at a genetic level, we preformed next-generation RNAseq. Through RNAseq analysis we identified transcriptional changes promoted by HFD. It was observed that livers obtained from HFD-fed mice had lower DEGs, as compared to those from CD-fed mice, indicating considerable regulation at the level of mRNA turnover. Furthermore, functional analyses of DEGs using GO biological processes and the KEGG database highlighted a pivotal role of HFD in driving pathologies related to processes of lipid metabolism, oxidative stress responses, immune response, cell proliferation, and apoptosis. More importantly, RNAseq data identified an involvement of SMPD3, the gene coding nSMAse2, as a driver of this process. Indeed, protein lysates obtained from livers of HFD-fed and CD-fed mice further supported the upregulation of SMPD3 expression, along with the enzymatic activity of nSMAse2 in the liver of HFD-fed mice. Sphingolipids are involved in a plethora of biological functions, including, but not limited to, inflammation, immune response, cell life cycle, adhesion, and migration [17,18]. Ceramide is the core product of sphingolipid metabolism and has been shown to play a role in hepatocellular apoptosis and fibrosis [3,19]. In a recent study by Wang et al., the role of epitranscriptomic machinery in postnatal liver development was elucidated. The research highlighted that the RNA methyltransferase enzymes, Mettl3 and Mettl14, see a decline in expression during postnatal liver maturation in male mice. A pivotal finding was the identification of the neutral sphingomyelinase, Smpd3, as a key target of Mettl3. Mettl3 deficiency led to decreased decay of Smpd3 transcripts, resulting in disruptions in sphingolipid metabolism. This disruption was marked by the accumulation of toxic ceramides, causing significant mitochondrial damage and increased endoplasmic reticulum stress, both critical factors in liver damage [9]. Their findings, emphasizing Smpd3's central role in liver health. In our presented work, we observed similar findings regarding SMPD3 expression. However, our observations were made in a high-fat diet (HFD) induced NAFLD mouse model, which emphasis the influence of diet in modulating hepatic nSMase2 activation. Adipose tissue enlargement, caused by excessive energy consumption, may push adipocytes beyond their capacity for lipid storage and can instigate inflammation and oxidative stress, which can lead to variety of metabolic disorders, including diabetes, hypertension, and NAFLD [20,21].
To understand how HFD induced hepatic steatosis and influenced the generation of ceramide via the SM pathway at a mechanistic level, we developed a cell-based hepatic steatosis model to perform a more in-depth analysis of conditions such as NAFLD. The model was based on applying constant stress to the HepG2 cells by switching their culture media from normal (5.6 mmol) to high (25.2 mmol) glucose, followed by treatment with fatty acids using oleic acid. The level of intracellular lipid accumulation within HepG2 cells increased when the cellular metabolism was switched from low glucose/low fat to high abundance of both. This phenomenon has previously been demonstrated in thyroid hormone receptor knockout mice, where changes in whole-body energy metabolism affected hepatic liver balance [22]. Diverse glucose fluctuation is a characteristic of badly managed diabetes and a known factor that triggers diabetic complications [23,24,25]. Previously, we showed that constant fluctuation in glucose conditions from low to high can trigger pro-inflammatory production in macrophages [25]. Indeed, implementing these conditions on HepG2 cells was also found to enhance the inflammatory response and induce the expression and secretion of the inflammatory marker, TNF-α. Further, the high glucose/high fatty acid model induced pathologies of hepatic steatosis and mediated oxidative stress and cellular apoptosis. Together, these phenotypic alterations are not only observed in patients with type 2 diabetes mellitus, but are also seen in the hepatic manifestation of those with metabolic syndromes, such as NAFLD [26,27,28,29], indicating a significant resemblance presented in our cell model in mimicking these phenotypes.
In our HepG2-steatosis model, we found that the gene expressions of both SMSG1 and SMSG2 were significantly downregulated, indicating a reduction in SM synthesis. This effect was previously revealed by Li et al, as the group showed that fat-fed female mice lacking Sms1/Sms2 (double knockout) had significantly higher liver triglyceride and cholesterol levels [30]. We also found a significant upregulation in nSMase2 expression and activity under steatosis conditions, similar to the observed nSMase2 activity in the liver of HFD-fed mice. During the onset of diabetes, the circulation of several pro-inflammatory cytokines, such as TNF-α, are elevated. Our results, along with others, have shown that the upregulation in circulatory TNF-α can initiate ceramide synthesis by binding to TNFR1 and activating nSMases2, which hydrolyze SM to ceramide [15,31]. Moreover, in our cell model, loss- or gain-of-function of nSMase2 revealed that the inhibition of this enzyme significantly prevented the accumulation of intracellular lipid accumulation in hepatocytes, while its agonist presented a trend of increase intracellular lipid accumulation. Interestingly, although we found no remarkable differences during altered aSMase expression, the indirect inhibition of this enzyme also showed a significant reduction in intracellular lipid accumulation. It also showed a significant reduction in hepatic inflammation induced by lipotoxicity, similar to the effect observed under the inhibition of nSMase2. This observation is in concordance with other studies that established the influence of inflammation on the degradation action of SMase for converting SM into ceramide [32,33,34,35]. Oxidative stress is a major factor that leads to inflammation, especially during aging [36]. The activation of PPARα has a protective role for both fatty acid-induced oxidative stress and apoptosis [37,38,39]. In a study by Montagner A. et al, it was shown that hepatocyte PPARα deletion in PPARαhep−/− mouse model represented an impaired fatty acid homeostasis in the liver and hepatic lipid buildup [40]. Indeed, in our steatosis model, the lipotoxic effect of OA stimulation was associated with a downregulation of PPARα and upregulation of CYP2E1 proteins. Remarkably, the inhibition and deficiency of nSMase2 increased PPARα activity, reduced CYP2E1 expression, and strengthened cell survival. This interesting observation was limited to nSMase2, and not aSMase. However, it is important to note that, while Imip is well-known as an aSMase inhibitor, this impact is indirectly via disrupting the intracellular lysosomal membrane and inhibiting aSMase binding to the membrane, a process that results in aSMase degradation and ultimately deactivation. For that reason, it is impossible to interrupt aSMase function within the scope of this study, without gene silencing or knock-down of the aSMase gene expression. Altogether, we identified a critical function of nSMase2 in mediating hepatic inflammation, oxidative stress, and apoptosis, all of which are established pathogenic features of liver steatosis.
4. Materials and Methods
Animal Studies
We performed the experiments according to the protocols approved by the Institutional Animal Care and Use Committee of Dasman Diabetes Institute (RAAM- 2016-007). We ensured that all efforts were made to minimize animal discomfort, and the animals were treated humanely. All mice were housed under a 12-h day/night cycle at constant temperature (approximately 22°C) and suitable humidity (45–70%).
For the induction of liver steatosis, we fed 8-weeks-old C57BL/6 mice (Jackson Laboratories, Maine, United States) with either standard chow diet (CD; n=5) (8664; Harlan Teklad; 6.4% w/w fat) or HFD (n=5) (58% Kcal from fat; #D12331I, Research Diets, Inc.) for 14 weeks to induce liver steatosis. We measured the body weight (BW) of the animals at the beginning of the experiment and then weekly throughout the study. Mice were sacrificed upon completion of the 14-week feeding period and isolated their livers, which were then dissected into several sections for RNA extraction, SMase activity assays, and histology analysis.
Histopathological Studies of Mice Liver
To perform hematoxylin and eosin (H&E) and macrophage infiltration marker F4/80 staining, we fixed and washed dissected tissues, subsequently dehydrated them in ethanol, and embedded them in paraffin wax. For staining, we deparaffinized the paraffin-embedded sections (4 μm thick) of liver tissue in xylene and rehydrated them through descending grades of ethanol (100, 95, and 75%) to water. At this point, the slides were considered ready for H&E or F4/80 immunohistochemistry (IHC) staining.
F4/80 IHC Staining and Quantification
We performed antigen retrieval by placing slides in target retrieval solution (pH 6.0; Dako, Glostrup, Denmark) in the pressure cooker, boiling for 8 min and cooling for 15 min. After washing in phosphate-buffered saline (PBS), we blocked endogenous peroxidase activity with 3% H2O2 for 30 min and non-specific antibody binding with 5% nonfat milk for 1 h, followed by 1% bovine serum albumin solution for 1 h. We incubated the slides were overnight at room temperature with a primary antibody (1:800 dilution of rabbit polyclonal anti-F4/80 antibody; Abcam® ab100790). After washing with PBS (0.5 % Tween), we incubated the slides for 1 h with a secondary antibody (goat anti-rabbit conjugated with horseradish peroxidase (HRP) polymer chain; EnVision™ Kit from Dako, Glostrup, Denmark), and developed color using the 3,3ʹ-diaminobenzidine (DAB) chromogen substrate. For analysis, we captured digital photomicrographs of four different regions to assess the regional heterogeneity in tissue samples in 20x using PannoramicScan (3DHistech, Hungry). All samples were analyzed using the imageJ software (NIH, USA). For the quantification of F4/80 % area score, we captured 10 random fields of the sample in 20x magnification. We then imported the Jpg images into the Fiji image J application. The images were then color deconvoluted into the H-Dab form. We set the max threshold value and applied it to all Figure ure ures. In the Analysis box, we selected the area percentage along with the mean gray value, which displayed the size of the IHC image and staining percentage. Once the reading was done for all fields, we exported the analysis file, averaged the readings, and applied appropriate statistical measures [41].
H&E Staining and Quantification
We placed thin, 4–5 μM, sections on frosted slides and stained them with hematoxylin for 5 min, followed by a 1-min wash under tap water. Then, we added the counterstain eosin for 30 sec. We then re-dehydrated the slides and mounted them in a xylene-based mounting medium. For obtaining the macro- and micro-vesicular steatosis score, we converted images from the RGB format to a 16-bit grayscale image, and then set the image threshold to the internode parameter and highlight all vacuoles to be counted; we applied this to all slides. We analyzed the binary image of the particles according to their circularity and compared them side-by-side with their original H&E image for accuracy. In the Analysis box, we selected the area percentage along with mean gray value, which displayed the size of the H&E image and staining percentage of the selected cells. Once the reading was done for all fields, we exported the analysis file, averaged the readings, and applied appropriate statistical measures [42].
Nile Red Staining in Mouse Liver
For intracellular neutral lipid detection, we used the fluorescent Nile red (9-diethylamino5H-benzo[a]phenoxazine-5-one) dye. We embedded liver tissues in optimal cutting temperature (OCT) and dissected them into 5 mm slices using a cryostat and placed them on frosted slides. Before staining, we fixed the hepatic sections with 4% (v/v) paraformaldehyde (PFA) for 10 min, followed by washing for 5 min. We stained the tissues with Nile Red for 15 min, followed by three washes in PBS. To visualize the nucleus, we counterstained the liver tissues and mounted them with coverslips using mounting-containing DAPI (Vectashield, Vectorlab, H1500).
RNA Sequencing and Data Analysis
We performed RNA isolation from the treated and untreated cell lines using the Rneasy kit (Qiagen, Hilden, Germany), following standard protocols. We used 40 ng of purified RNA for whole transcriptome sequencing. Next, we prepared transcriptome libraries using the Truseq stranded mRNA kit (Illumina Inc. USA), following manufacturers protocols. We validated the prepared libraries and quantified them using a bioanalyzer (Agilent, California, United States) and qubit fluorometer (Thermofisher Scientific, Massachusetts, United States), respectively. We then performed paired-end sequencing on the Novaseq 6000 system (Illumina Inc. USA). We converted the bcl files to fastq using the bcl2fastq program version 2.20. We carried out quality control of the fastq files using FastQC (v0.11.9) [43]. Next, we used Trimmomatic (v0.39) [44] for adaptor and quality trimming and for the removal of very short reads, and HISAT2 (v2.1.0) [45] for data alignment. To enumerate the number of reads associated with the genes, we used htseq-count tool in HTSeq (v 0.9.1) [46]. Lastly, we performed differential gene expression (DEGs) analysis using Bioconductor package edgeR [47], adopting the default setting.
Identification of Enriched Biological Process and Hepatic Pathways Comparing CD- and HFD-Fed Mice
Pathway enrichment and biological process on liver transcriptome comparing CD and HFD. To identify pathways altered in mice on HFD, as compared to those on CD, we used the DEGs with a P value <0.05 obtained from edgeR paired analysis comparing HFD- and CD-fed mice. We used Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/home.jsp) to perform gene ontology enrichment of biological processes. We also performed enriched pathways using the KEGG pathway database [48], and carried out the visualization of networks using cytoscape [49].
Preparation of Mice Liver Tissue Lysates and Determination of aSMase and nSMase2 Activity
We suspended the animal liver homogenates in 0.1% NP-40 detergent in PBS, followed by sonication for 30 s on ice at 20 watt. We allowed the samples to rest on ice for 30 min, followed by centrifugation for 10 min at 16,000 g. We then used cell lysates for acidic SMase (aSMase) and neutral SMase (nSMase2) activity assays. We assayed the enzyme activity using 50 µg total protein / 10 µL Tris-MgCl2, with a pH of 5.0 for aSMase and a pH of 7.4 for nSMase2 using the Amplex Red Sphingomyelinase assay kit (Invitrogen, Monza, Italy), in accordance with the manufacturer’s instructions. We measured the reaction at Ex/Em= 535/587 in kinetic mode for 30 min at 37oC. The maximum signal was reached in 13 min.
Cell Culture and Two Stress Factor Administration
We then cultured human hepatoma HepG2 cells in the DMEM medium, supplemented with 1 mM sodium pyruvate, 10 mM HEPES, 100 g/ml Normocin, 50U/ml penicillin, and 50 g/ml streptomycin (P/S; (Gibco, Invitrogen, Grand Island, NY, USA). We performed the experiments when cells reached around 60% confluence after being cultured in 5 mmol/L D-glucose supplemented DMEM-based medium for at least 24 hrs. All cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2. Once the cells reached approximately 60% confluence, we switched them to a high glucose medium (25.2 mmol D-Glucose) for 24 hours and treated them with 0.1 mM of oleate acid (OA) solution for another 24 hours. We treated the control “vehicle” cells with an OA-free medium containing albumin.
Cell Stimulation
We plated HepG2 in 12-well plates (Costar, Corning Incorporated, Corning, NY, USA) at 5 x 105 cells/well, unless indicated otherwise. After switching the cells to high glucose media, we directly treated the cells for 24 hours with either the nSMase2 synthetic inhibitor GW4869 (10 μM; Chem Cruz, SC-218578), the nSMase2 inducer daunorubicin hydrochloride (DN; 1μM; Chem Cruz, SC-200921), or the aSMase synthetic inhibitor imipramine (Imip) hydrochloride (10 μM; Sigma-Aldrich, 113-52-0). Then, we stimulated the cells with 150 μM of OA, to trigger steatosis in the presence of inhibitors.
Cell Proliferation Assay
We determined cell proliferation by performing an MTT assay. Briefly, we plated 5 103 cells into a 96-well plate and incubated at 37 °C in 5% CO2 for 12 hours. Cells were cultured under different conditions to evaluate the influence of high glucose high OA conditions. for stimulation studies, cells were treated with different inhibitors prior to treatment with 0.1 mM of OA once cultured are treated, 1 mg/ml of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium, Promega Bio Sciences LLC, San Luis Obispo, CA, USA) was added to each well. The developed Formazan crystals were subsequently dissolved in 10% SDS/0.04 eq/L HCl solution for 1 h at 37 °C. Absorbance was measured at 490 nm using a microplate reader (DTX880; Beckman Coulter, Brea, CA, USA). Cell survival was expressed as the percentage of control cells as follows: CS (%) = (mean Atreated well/mean Acontrol well) × 100.
Flow Cytometric Analysis of Apoptosis Using Annexin V versus Propidium Iodide (PI)
We used an annexin V apoptosis detection kit for flow cytometry (Sigma-Aldrich, MO, USA), and performed the annexin V assay in conjunction with propidium iodide (PI) staining. We cultured HepG2 cells, as mentioned above. We harvested the cells by trypsinization and centrifugation at 1000 rpm for 5 min and then re-suspended them in 1x binding buffer prior to staining with 5 μl of annexin V and 10 μl of propidium iodide solution for 15 min at room temperature. We then performed the fluorescence-activated cell sorting (FACS) analysis using a FACSCanto II (BD Bioscience, San Jose, CA, USA), according to the manufacturer’s instructions. We recorded approximately 20,000 counts in each analysis. We used unstained cells to set the quadrant of the negative versus positive gates. We used BD FACSDivaTM Software 8 (BD Biosciences) to analyze FACS data.
Flow Cytometry-BODIPY Lipid Staining and Quantification
We used the BODIPY 495/503 stain (Cat # D3922, Life Technologies) to quantify lipid content in the cell. We washed the cells thrice with FACS buffer and resuspended them in 2% PFA. We then centrifuged the cells and resuspended them in FACS buffer for FACS analysis (FACSCanto II). We used BD FACSDiva Software 8 (BD Biosciences) to analyze FACS data. We then used unstained cells to set the quadrant of the negative versus positive gates. The stain index (SI) was calculated as the difference between the mean fluorescence intensity (MFI) of the positive and negative populations, divided by two times the standard deviation of the negative populations (unstained cells), represented as light grey in histograms, as previously described.
Confocal Microscopy
To visualize lipid accumulation by confocal microscopy, we seeded and treated HepG2 cells on a coverslip for 4 h. At the end of the experiment, we treated the cells with 4% PFA for 10 min and stained them with Nile red for 15 min. We then washed the cells thrice with PBS, for 5 min each, and counterstained and mounted them over slides using a mounting-containing DAPI (Vectashield, Vectorlab, H1500).
We captured the confocal images on the inverted Zeiss LSM710 spectral confocal microscope (Carl Zeiss, Gottingen, Germany) and EC Plan-Neofluar 40×/1.30 oil DIC M27 objective lens. After exciting samples with 543 nm HeNe laser and 405 nm line of an argon ion laser, we conFigure ure ured optimized emission detection bandwidths using Zeiss Zen 2010 control software. We analyzed all samples using the same parameters and the resulting color markup of analysis was confirmed for each sample. The Correlated Total Cell Fluorescence (CTCF) was calculated using the following equation:
CTCF = IntDen − (Area of selected cells X background mean grey value (BMGV))
Real-Time Quantitative Reverse Transcriptase Polymerase Chain Reaction (PCR)
We extracted total RNA using RNeasy Mini Kit (Qiagen, Valencia, CA, USA), as per the manufacturer’s instructions. We synthesized cDNA from 1 μg of the total RNA using high-capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA, USA). Real-time quantitative PCR (RT-qPCR) was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) using the TaqMan Gene Expression Master Mix (Applied Biosystems). Each reaction contained 50 ng of cDNA amplified with inventoried TaqMan Gene Expression Assay products (TNF-α: Assay Hs01113624_g1; peroxisome proliferators activated receptors-alpha (PPARα): Assay Hs00947536_m1; cytochrome P450 2E1 (CYP2E1): Assay ID: HS00559367_M1; GAPDH: Assay ID: Hs03929097_g1, SMPD1: Assay ID: Hs03679346_g1/ Mm00488318_m1, SGMS1: Assay ID: HS00983630_m1, SGMS2: Assay ID: Hs00380453_m1, SMPD3: Assay ID: Hs00920354_m/ Mm00491359_m1). We normalized the threshold cycle (Ct) values to the housekeeping gene GAPDH. Relative mRNA expression was presented as fold expression over the average of control gene expression [15]. The expression level in the control treatment was 1. Values are presented as mean ± SEM. Results with a p value of < 0.05 were considered statistically significant.
Western Blotting
We harvested HepG2 cultures and incubated them for 30 min with lysis buffer (62.5 mM Tris-HCl, pH 7.5, 1% Triton X-100 and, 10% glycerol). Next, we centrifuged lysates at 14,000 g for 10 min and collected the supernatant. We measured protein concentrations using the Quickstart Bradford Dye Reagent, 1x Protein Assay kit (Bio-Rad Labora-tories, Inc, CA). We mixed the protein samples (25 µg) with sample loading buffer, heated them for 5 min at 95 °C and resolved on 12% polyacrylamide gels using SDS-PAGE. Next, we transferred cellular proteins to Immuno-Blot PVDF membranes (Bio-Rad Laboratories) by electroblotting. We then blocked the membranes with 5% nonfat milk in PBS for 1 h, followed by overnight incubation at 4 °C with primary antibodies against TNF-α (Cell signalling technology; cat # 6945), PPARα (Abcam; cat# ab3484), or β-actin (Cell signalling technology; cat # 3700T) at a 1:1000 dilution. We purchased all primary antibodies from Cell Signaling (Cell Signaling Technology, Inc, USA). We then washed the blots four times with Tris-buffered saline (TBS) and incubated them for 2 h with HRP-conjugated secondary antibody (Promega, Madison, WI, USA). Next, we developed immune re-active bands using an Amersham ECL plus Western Blotting Detection System (GE Health Care, Buckinghamshire, UK) and visualized them using the Molecular Imager ChemiDoc MP Imaging Systems (Bio-Rad Laboratories, state, country).
Sandwich Enzyme-Linked Immunosorbent Assay (ELISA)
We quantified the secreted interleukin (IL)-1β and TNFα protein concentrations in the media using sandwich ELISA, in accordance with the manufacturer’s instructions (R&D systems, Minneapolis, USA).
5. Conclusions
In this study, we presented a cell-based liver steatosis model mimicking the pathologies associated with liver steatosis. Both excess glucose and fatty acid (OA) recapitulated the hepatic NAFLD phenotype, which was found in the HFD-induced steatosis mice models, providing an easy human-based in-vitro model that can be further utilized in translational medicine and drug assessments. Furthermore, HFD-induced steatosis was found to be associated with elevated nSMase2. Targeting the gene expression of nSMase2 or its enzymatic activity using siRNA or GW4869, respectively, restored hepatic steatosis damage. Therefore, our data supports nSMAse2 as a potential novel target for treating NAFLD.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Increase of nSMase gene expression livers of mice fed with diet-induced steatosis. Figure S2: Inducing hepatic steatosis with high glucose and high oleate acid culture in HepG2. Figure S3: Effect of nSMase inhibition in HepG 2 liver steatosis model.
Author Contributions
Conceptualization, Fatema Al-Rashed, Yusuf Hannun and Rasheed Ahmad; Data curation, Ashraf Al Madhoun, Fatemah Bahman, Halemah AlSaeed, Texy Jacob, Reeby Thomas, Areej Al-Roub, Sardar Sindhu, Fawaz Alzaid, Thangavel Alphonse Thanaraj and Fahd Al-Mulla; Formal analysis, Reeby Thomas, Fahd Al-Mulla, Yusuf Hannun and Rasheed Ahmad; Funding acquisition, Fatema Al-Rashed; Investigation, Fatema Al-Rashed, Ashraf Al Madhoun and Rasheed Ahmad; Methodology, Fatema Al-Rashed, Hossein Arefanian, Ashraf Al Madhoun, Fatemah Bahman, Halemah AlSaeed, Texy Jacob, Reeby Thomas, Areej Al-Roub, Sardar Sindhu, Fawaz Alzaid, Md. Zubbair Malik and Rasheeba Nizam; Project administration, Fahd Al-Mulla; Resources, Rasheed Ahmad; Software, Md. Zubbair Malik, Rasheeba Nizam and Thangavel Alphonse Thanaraj; Supervision, Fahd Al-Mulla and Yusuf Hannun; Validation, Hossein Arefanian, Ashraf Al Madhoun, Fatemah Bahman, Halemah AlSaeed, Texy Jacob, Reeby Thomas, Areej Al-Roub, Sardar Sindhu, Md. Zubbair Malik, Rasheeba Nizam, Thangavel Alphonse Thanaraj, Fahd Al-Mulla, Yusuf Hannun and Rasheed Ahmad; Visualization, Rasheed Ahmad; Writing – original draft, Fatema Al-Rashed, Hossein Arefanian and Thangavel Alphonse Thanaraj; Writing – review & editing, Fatema Al-Rashed, Ashraf Al Madhoun, Fatemah Bahman, Halemah AlSaeed, Sardar Sindhu, Fawaz Alzaid, Md. Zubbair Malik, Fahd Al-Mulla, Yusuf Hannun and Rasheed Ahmad.
Funding
This work was supported and funded by the Kuwait Foundation for the Advancement of Sciences (KFAS) grant No. (RA -040-2021) (to F.A.R) and National Institutes of Health (NIH, USA) Grant P01 CA97132 (to Y.A.H.).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. All animal experimental protocols were approved by the Dasman Diabetes Institute Institutional Animal Care and Use Committee (RAAM- 2016-007). All institutional and national guidelines for the care and use of laboratory animals were followed.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
Acknowledgments
We would like to thank Ms. Lubaina Koti for editing the manuscript for language, structure, and accuracy.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Stefan, N.; Cusi, K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. The Lancet Diabetes & Endocrinology 2022. [Google Scholar]
- Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Current diabetes reports 2010, 10, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog Lipid Res 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Ahmad, Z.; Snider, A.J.; Thomas, R.; Kochumon, S.; Melhem, M.; Sindhu, S.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A. , et al. Ceramide kinase regulates tnf-α-induced immune responses in human monocytic cells. Sci Rep 2021, 11, 8259. [Google Scholar] [CrossRef]
- Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in non-alcoholic fatty liver disease and hepatocellular carcinoma: Ceramide turnover. Int J Mol Sci 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Goñi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett 2002, 531, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Khavandgar, Z.; Murshed, M. Sphingolipid metabolism and its role in the skeletal tissues. Cell Mol Life Sci 2015, 72, 959–969. [Google Scholar] [CrossRef]
- Chatterjee, S. Neutral sphingomyelinase: Past, present and future. Chem Phys Lipids 1999, 102, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, S.; Sun, J.; Han, P.; Xu, B.; Li, X.; Zhong, Y.; Xu, Z.; Zhang, P.; Mi, P.; et al. M. Nat Metab 2023, 5, 842–860.
- Wandrer, F.; Liebig, S.; Marhenke, S.; Vogel, A.; John, K.; Manns, M.P.; Teufel, A.; Itzel, T.; Longerich, T.; Maier, O.; et al. Tnf-receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in nafld mice. Cell Death Dis 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Potoupni, V.; Georgiadou, M.; Chatzigriva, E.; Polychronidou, G.; Markou, E.; Zapantis Gakis, C.; Filimidou, I.; Karagianni, M.; Anastasilakis, D.; Evripidou, K.; et al. Circulating tumor necrosis factor-α levels in non-alcoholic fatty liver disease: A systematic review and a meta-analysis. J Gastroenterol Hepatol 2021, 36, 3002–3014. [Google Scholar] [CrossRef]
- Biswas, P.; Datta, C.; Rathi, P.; Bhattacharjee, A. Fatty acids and their lipid mediators in the induction of cellular apoptosis in cancer cells. Prostaglandins Other Lipid Mediat 2022, 160, 106637. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. Cyp2e1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome proliferator-activated receptors (ppars) and oxidative stress in physiological conditions and in cancer. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Al-Rashed, F.; Ahmad, Z.; Thomas, R.; Melhem, M.; Snider, A.J.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. Neutral sphingomyelinase 2 regulates inflammatory responses in monocytes/macrophages induced by tnf-α. Sci Rep 2020, 10, 16802. [Google Scholar] [CrossRef]
- Li, C.; Guo, S.; Pang, W.; Zhao, Z. Crosstalk between acid sphingomyelinase and inflammasome signaling and their emerging roles in tissue injury and fibrosis. Front Cell Dev Biol 2019, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.G.; Way, G.W.; Zhou, H. Bile acids and sphingolipids in non-alcoholic fatty liver disease. Chin Med J (Engl) 2022, 135, 1163–1171. [Google Scholar] [CrossRef]
- Muralidharan, S.; Torta, F.; Lin, M.K.; Olona, A.; Bagnati, M.; Moreno-Moral, A.; Ko, J.H.; Ji, S.; Burla, B.; Wenk, M.R.; et al. Immunolipidomics reveals a globoside network during the resolution of pro-inflammatory response in human macrophages. Front Immunol 2022, 13, 926220. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.D.; Wang, J.W. Ceramide de novo synthesis in non-alcoholic fatty liver disease: Pathogenic mechanisms and therapeutic perspectives. Biochem Pharmacol 2022, 202, 115157. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Bickel, P.E.; Scherer, P.E. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 2016, 15, 639–660. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Lee, H.Y.; Jurczak, M.J.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.A.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid hormone receptor-α gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 2012, 153, 583–591. [Google Scholar] [CrossRef]
- Sachwani, G.R.; Jaehne, A.K.; Jayaprakash, N.; Kuzich, M.; Onkoba, V.; Blyden, D.; Rivers, E.P. The association between blood glucose levels and matrix-metalloproteinase-9 in early severe sepsis and septic shock. J Inflamm (Lond) 2016, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Shinke, T.; Otake, H.; Sugiyama, D.; Takaya, T.; Takahashi, H.; Terashita, D.; Uzu, K.; Tahara, N.; Kashiwagi, D.; et al. Effects of daily glucose fluctuations on the healing response to everolimus-eluting stent implantation as assessed using continuous glucose monitoring and optical coherence tomography. Cardiovascular diabetology 2016, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Sindhu, S.; Arefanian, H.; Al Madhoun, A.; Kochumon, S.; Thomas, R.; Al-Kandari, S.; Alghaith, A.; Jacob, T.; Al-Mulla, F.; et al. Repetitive intermittent hyperglycemia drives the m1 polarization and inflammatory responses in thp-1 macrophages through the mechanism involving the tlr4-irf5 pathway. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Tsuchiya, A.; Natsui, K.; Koseki, Y.; Takeda, N.; Tomiyoshi, K.; Yamazaki, F.; Yoshida, Y.; Shimbo, T.; Tamai, K.; et al. Synthesized hmgb1 peptide prevents the progression of inflammation, steatosis, fibrosis, and tumor occurrence in a nonalcoholic steatohepatitis mouse model. Hepatol Res 2022. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C. Pathogenesis and therapeutic strategies related to non-alcoholic fatty liver disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Meng, L.C.; Zheng, J.Y.; Qiu, Y.H.; Zheng, L.; Liu, Y.Q.; Miao, X.L.; Lu, X.Y. Salvianolic acid b ameliorates non-alcoholic fatty liver disease by inhibiting hepatic lipid accumulation and nlrp3 inflammasome in ob/ob mice. Int Immunopharmacol 2022, 111, 109099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, T.; Wang, T.; Liu, X.; Hamada, K.; Sun, D.; Sun, Y.; Yang, Y.; Wang, J.; Takahashi, S.; et al. Crosstalk between cyp2e1 and ppar. Acta Pharm Sin B 2022, 12, 2224–2238. [Google Scholar] [CrossRef]
- Li, Z.; Chiang, Y.P.; He, M.; Worgall, T.S.; Zhou, H.; Jiang, X.C. Liver sphingomyelin synthase 1 deficiency causes steatosis, steatohepatitis, fibrosis, and tumorigenesis: An effect of glucosylceramide accumulation. iScience 2021, 24, 103449. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective tnf-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J Clin Invest 2003, 111, 197–208. [Google Scholar] [CrossRef]
- Kim, M.H.; Ahn, H.K.; Lee, E.J.; Kim, S.J.; Kim, Y.R.; Park, J.W.; Park, W.J. Hepatic inflammatory cytokine production can be regulated by modulating sphingomyelinase and ceramide synthase 6. Int J Mol Med 2017, 39, 453–462. [Google Scholar] [CrossRef]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol Res 2012, 42, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M.; Karakashian, A.; Rutkute, K. Role of neutral sphingomyelinases in aging and inflammation. Subcell Biochem 2008, 49, 469–486. [Google Scholar] [PubMed]
- Jin, J.; Zhang, X.; Lu, Z.; Perry, D.M.; Li, Y.; Russo, S.B.; Cowart, L.A.; Hannun, Y.A.; Huang, Y. Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am J Physiol Endocrinol Metab 2013, 305, E853–E867. [Google Scholar] [CrossRef] [PubMed]
- Tibbles, L.A.; Woodgett, J.R. The stress-activated protein kinase pathways. Cell Mol Life Sci 1999, 55, 1230–1254. [Google Scholar] [CrossRef]
- Moran, E.; Ding, L.; Wang, Z.; Cheng, R.; Chen, Q.; Moore, R.; Takahashi, Y.; Ma, J.X. Protective and antioxidant effects of pparα in the ischemic retina. Invest Ophthalmol Vis Sci 2014, 55, 4568–4576. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Q.; Li, H.; Lan, X.; Kan, M.; Lin, J.; Wang, J.; Zhang, Z.; Ming, S.; Li, Z.; et al. The anti-aging effect of velvet antler polypeptide is dependent on modulation of the gut microbiota and regulation of the pparα/apoe4 pathway. J Integr Neurosci 2021, 20, 573–583. [Google Scholar]
- Kim, M.J.; Kim, D.H.; Bang, E.; Noh, S.G.; Chun, P.; Yokozawa, T.; Moon, H.R.; Chung, H.Y. Pparα agonist, mhy3200, alleviates renal inflammation during aging via regulating ros/akt/foxo1 signaling. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver pparα is crucial for whole-body fatty acid homeostasis and is protective against nafld. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative determination of protein expression using immunohistochemistry staining and analysis: An integrated protocol. Bio Protoc 2019, 9. [Google Scholar] [CrossRef]
- Yen, K.; Le, T.T.; Bansal, A.; Narasimhan, S.D.; Cheng, J.X.; Tissenbaum, H.A. A comparative study of fat storage quantitation in nematode caenorhabditis elegans using label and label-free methods. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. Fqc dashboard: Integrates fastqc results into a web-based, interactive, and extensible fastq quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq--a python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to cytoscape plugins. Nat Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef]
Figure 1.
High fat diet (HFD) induces chronic liver injury similar to NAFLD mice model. C57BL/6 male mice were given either HFD or chow diet (CD) for 14 consecutive weeks followed by liver isolation and processing. (A) Timeline weight accumulation for both groups. (B) Liver weight at 14 weeks. (C) Representative photomicrographs of liver (D) Representative H&E staining shows tissue morphology. (E) Calculated macrovascular and microvascular score. (F) Representative immunohistology staining for specific antibodies against F4/80 macrophage infiltration marker. (G) Calculated percentage of area of expression of F4/80 (H) Representative nile red staining to visualize intracellular neutral-lipid accumulation (yellow). (I) Calculated CTCF for nile red staining. A minimum of n=5 mice in each group. For histology analysis 10 random fields of the sample are taken in 20x magnification. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20x magnifications: Scale bar = 50 um.
Figure 1.
High fat diet (HFD) induces chronic liver injury similar to NAFLD mice model. C57BL/6 male mice were given either HFD or chow diet (CD) for 14 consecutive weeks followed by liver isolation and processing. (A) Timeline weight accumulation for both groups. (B) Liver weight at 14 weeks. (C) Representative photomicrographs of liver (D) Representative H&E staining shows tissue morphology. (E) Calculated macrovascular and microvascular score. (F) Representative immunohistology staining for specific antibodies against F4/80 macrophage infiltration marker. (G) Calculated percentage of area of expression of F4/80 (H) Representative nile red staining to visualize intracellular neutral-lipid accumulation (yellow). (I) Calculated CTCF for nile red staining. A minimum of n=5 mice in each group. For histology analysis 10 random fields of the sample are taken in 20x magnification. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20x magnifications: Scale bar = 50 um.
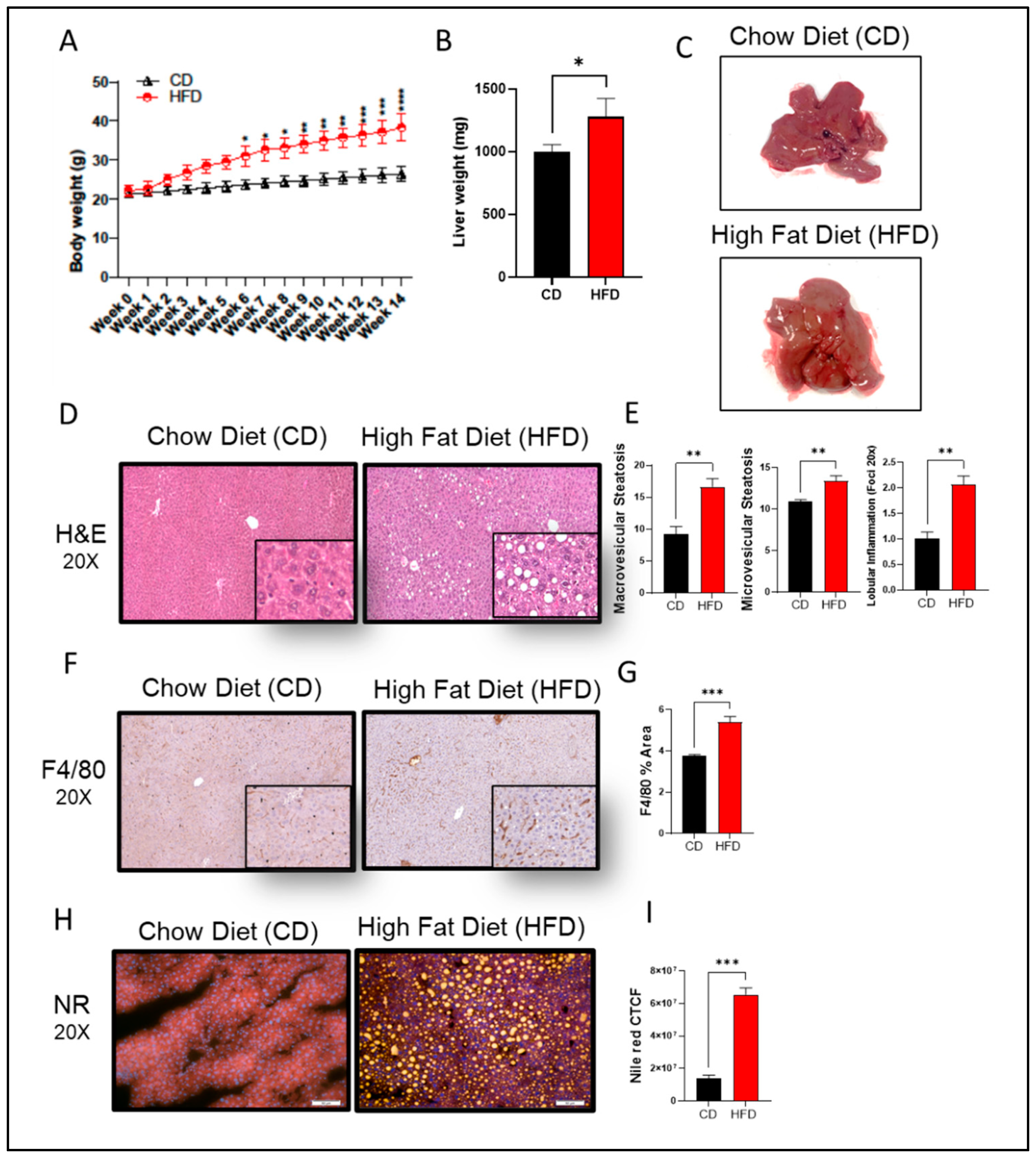
Figure 2.
Changes in transcriptomic profile of the steatosis in liver of high-fat chow diet fed mice. (A) PCA analysis shows the expression correlation based on count per millions (CPM) value in each sample of CD and HFD conditions. (B) Volcano plot distribution highlighting differentially expressed genes (DEGs) between CD and HFD fed mice samples. The orange and cyan colored points denote the up (510) and downregulated (672) DEGs respectively. Brown colored points represent non significands DEGs of transcriptome of HFD and CD. (C, D) Gene Ontology (GO) enrichment biological process significantly alters HFD compared to CD fed mice in steatosis in liver. Bar plot of significant biological process of UP and down-regulated DEGs of transcriptome respectively. X-axes represent Fold Enrichment with significance (P < 0.05) indicated by order and color trend. (E, F) Significant enriched KEGG pathway of the up and down-regulated DEGs respectively. Dot size indicates count. Count represents the number of genes associated with each pathway. Dot color denotes the p values of pathways and x-axis represent fold enrichment. (G) Gene networks of Lipid metabolic process, cholesterol metabolism, fatty acid metabolic process, apoptosis, reactive oxidative stress (ROS), apoptosis and response to ATP. Orange and cyan color nodes are represented up-regulated and down-DEGs in liver of HFD fed mice respectively. (H) Chord plot to show the relationship between genes and top significant GO term that include cholesterol, lipid and fatty acid metabolism. The genes involved to that enrichment (Left) arranged in order of their expression level (-log fold change). (I) nSMase2 activity level measured in steatosis in liver of high-fat diet fed mice. Significance marked with * P ≤ 0.05 refers to comparisons to the CD and HFD mice.
Figure 2.
Changes in transcriptomic profile of the steatosis in liver of high-fat chow diet fed mice. (A) PCA analysis shows the expression correlation based on count per millions (CPM) value in each sample of CD and HFD conditions. (B) Volcano plot distribution highlighting differentially expressed genes (DEGs) between CD and HFD fed mice samples. The orange and cyan colored points denote the up (510) and downregulated (672) DEGs respectively. Brown colored points represent non significands DEGs of transcriptome of HFD and CD. (C, D) Gene Ontology (GO) enrichment biological process significantly alters HFD compared to CD fed mice in steatosis in liver. Bar plot of significant biological process of UP and down-regulated DEGs of transcriptome respectively. X-axes represent Fold Enrichment with significance (P < 0.05) indicated by order and color trend. (E, F) Significant enriched KEGG pathway of the up and down-regulated DEGs respectively. Dot size indicates count. Count represents the number of genes associated with each pathway. Dot color denotes the p values of pathways and x-axis represent fold enrichment. (G) Gene networks of Lipid metabolic process, cholesterol metabolism, fatty acid metabolic process, apoptosis, reactive oxidative stress (ROS), apoptosis and response to ATP. Orange and cyan color nodes are represented up-regulated and down-DEGs in liver of HFD fed mice respectively. (H) Chord plot to show the relationship between genes and top significant GO term that include cholesterol, lipid and fatty acid metabolism. The genes involved to that enrichment (Left) arranged in order of their expression level (-log fold change). (I) nSMase2 activity level measured in steatosis in liver of high-fat diet fed mice. Significance marked with * P ≤ 0.05 refers to comparisons to the CD and HFD mice.
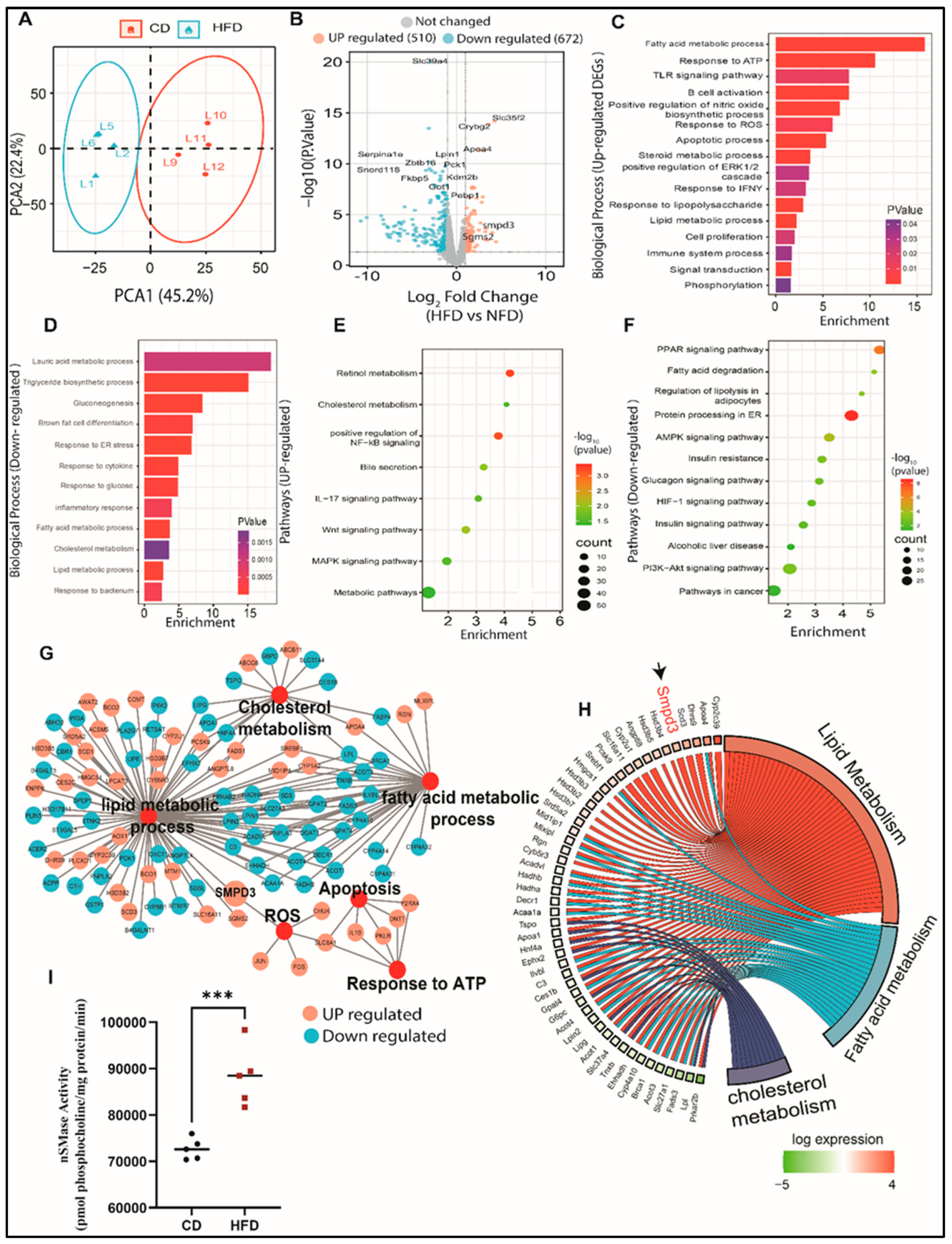
Figure 3.
Increase of nSMase Hydrolysis in HepG2 liver steatosis model. HepG2 cells were cultured in either low 5.6mmol D-glucose or high 25.2mmol D-glucose in the presence or absence of OA stimulation. (A) Schematic representation of main sphingomyelin (SM) metabolic pathway. (B) qRT-PCR analysis of SGMS1 gene expression. (C) qRT-PCR analysis of SGMS2 gene expression. (D) qRT-PCR analysis of SMPD3 gene expression. (E) qRT-PCR analysis of SMPD1 gene expression. (F) nSMase activity level measured in HepG2 steatosis model. (G) aSMase activity level measured in HepG2 steatosis model. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20 magnifications: Scale bar = 50 um.
Figure 3.
Increase of nSMase Hydrolysis in HepG2 liver steatosis model. HepG2 cells were cultured in either low 5.6mmol D-glucose or high 25.2mmol D-glucose in the presence or absence of OA stimulation. (A) Schematic representation of main sphingomyelin (SM) metabolic pathway. (B) qRT-PCR analysis of SGMS1 gene expression. (C) qRT-PCR analysis of SGMS2 gene expression. (D) qRT-PCR analysis of SMPD3 gene expression. (E) qRT-PCR analysis of SMPD1 gene expression. (F) nSMase activity level measured in HepG2 steatosis model. (G) aSMase activity level measured in HepG2 steatosis model. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20 magnifications: Scale bar = 50 um.
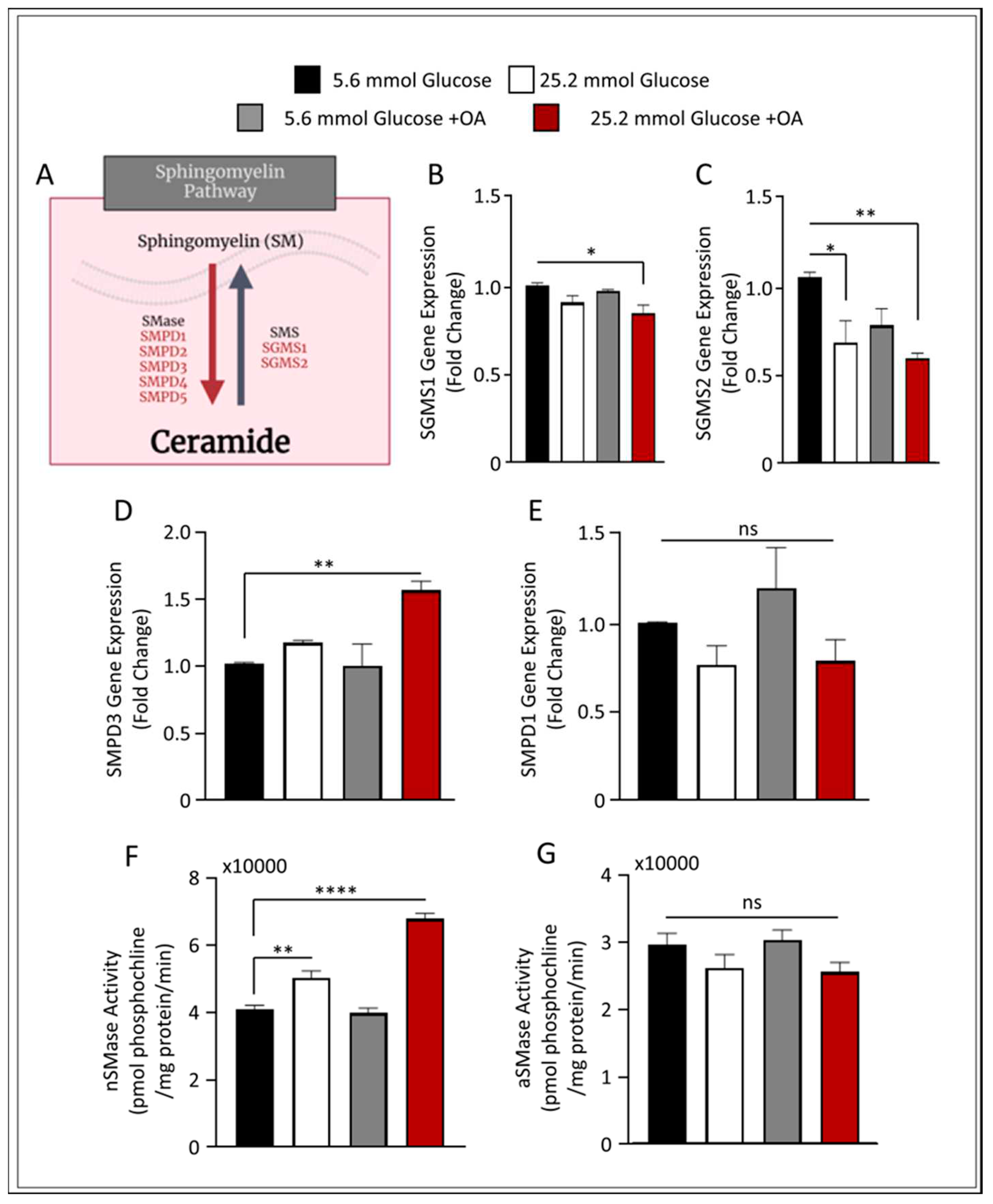
Figure 4.
Effect of nSMase2 inhibition in HepG2 liver steatosis model. HepG2 cells were pretreated with GW4869 (10μM), Imipramine (10μM) or Daunorubicin (1μM) for 1 hour followed by 150μM stimulation with Oleic acid. (A) nSMase2 activity level measured in HepG2 steatosis model. (B) aSMase activity level measured in HepG2 steatosis model. (C) Bar graph of median fluorescence intensity (MFI) of BODIPY 493/503 calculated from three independent determinations, with a similar result presented in the histogram. (D) Nile red fluorescence staining. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20 magnifications: Scale bar = 50 um.
Figure 4.
Effect of nSMase2 inhibition in HepG2 liver steatosis model. HepG2 cells were pretreated with GW4869 (10μM), Imipramine (10μM) or Daunorubicin (1μM) for 1 hour followed by 150μM stimulation with Oleic acid. (A) nSMase2 activity level measured in HepG2 steatosis model. (B) aSMase activity level measured in HepG2 steatosis model. (C) Bar graph of median fluorescence intensity (MFI) of BODIPY 493/503 calculated from three independent determinations, with a similar result presented in the histogram. (D) Nile red fluorescence staining. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant. Images are shown at 20 magnifications: Scale bar = 50 um.
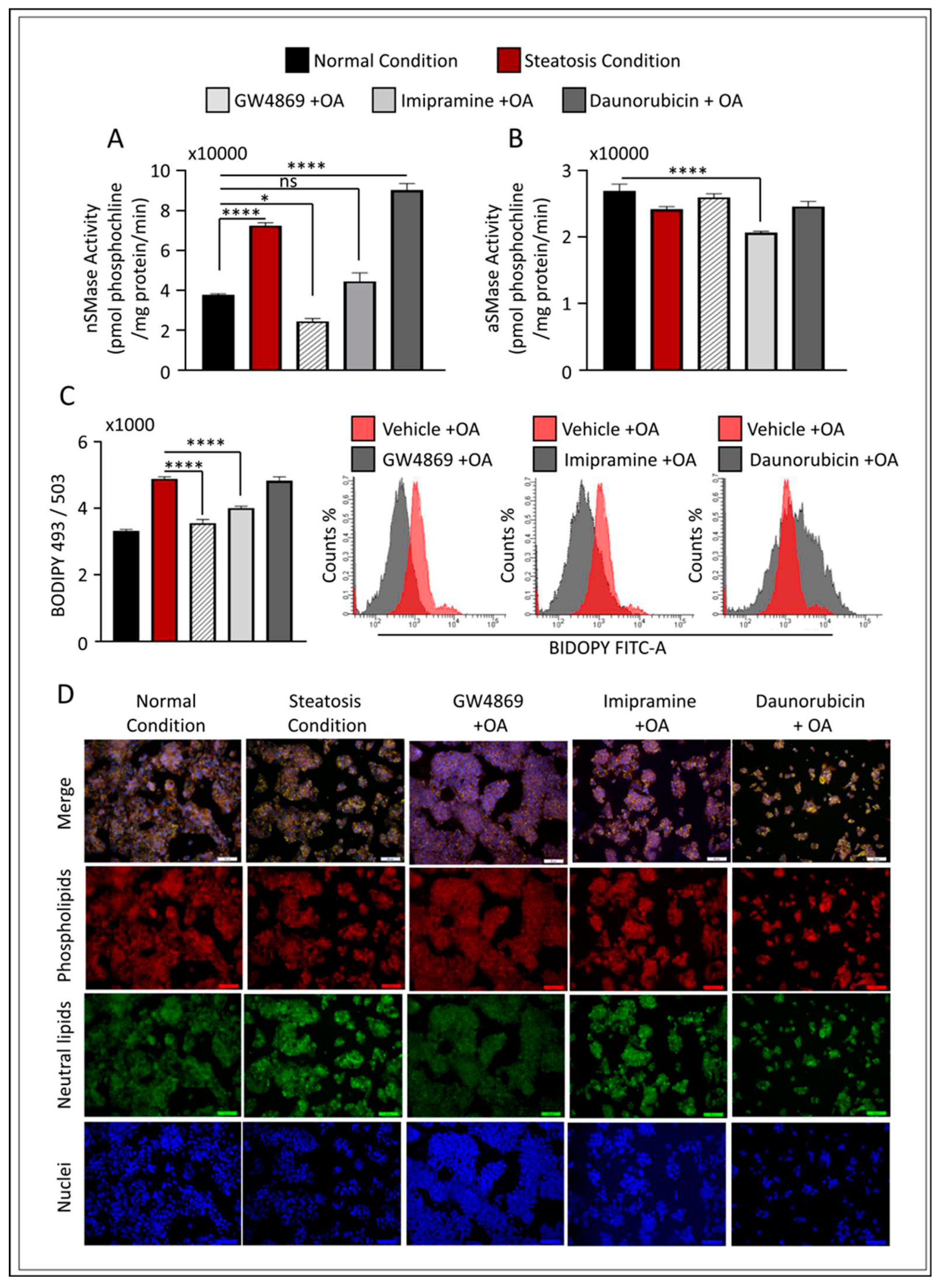
Figure 5.
Effect of nSMase2 inhibition in HepG2 liver steatosis-induced inflammation and oxidative stress. (A) qRT-PCR analysis of TNF-α expression. (B) TNF-α protein analysis with representative immunoblot. (C) TNF-α protein secretion in the media measured by enzyme-linked immunosorbent assay. (D) qRT-PCR analysis of PPARα expression. (E) PPARα protein analysis with representative immunoblot (F) qRT-PCR gene expression analysis of Cyp2E1 expression. (G) Bar graph for percentage of viable cells through MTT assay. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant.
Figure 5.
Effect of nSMase2 inhibition in HepG2 liver steatosis-induced inflammation and oxidative stress. (A) qRT-PCR analysis of TNF-α expression. (B) TNF-α protein analysis with representative immunoblot. (C) TNF-α protein secretion in the media measured by enzyme-linked immunosorbent assay. (D) qRT-PCR analysis of PPARα expression. (E) PPARα protein analysis with representative immunoblot (F) qRT-PCR gene expression analysis of Cyp2E1 expression. (G) Bar graph for percentage of viable cells through MTT assay. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant.
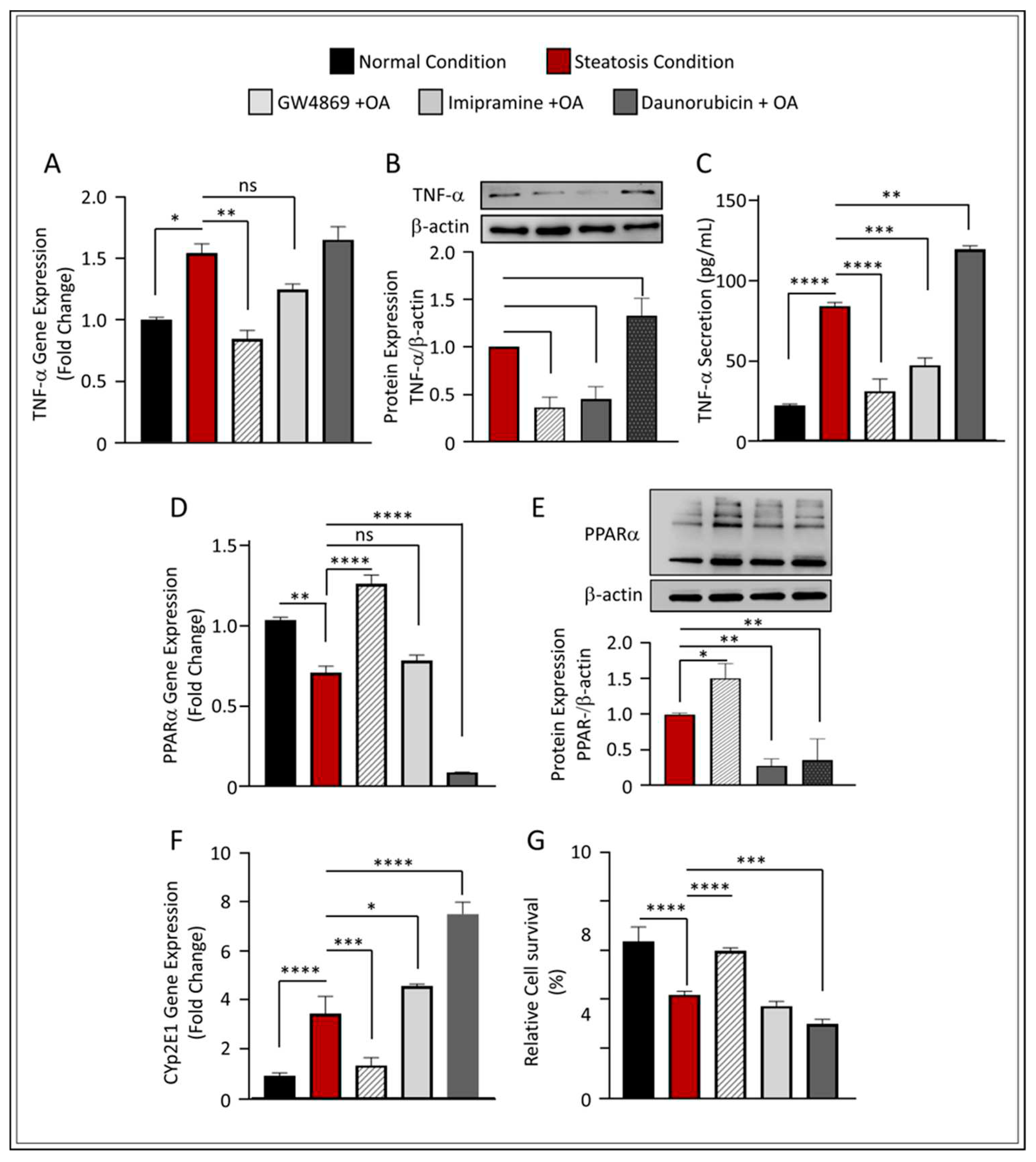
Figure 6.
Effect of nSMase2 deficiency in HepG2 liver steatosis model. (A) qRT-PCR analysis of SMPD3 gene expression. (B) qRT-PCR analysis of TNF-α expression. (C) TNF-α protein analysis with representative immunoblot. (D) qRT-PCR analysis of PPARα expression. (E) PPARα protein analysis with representative immunoblot. (F) qRT-PCR gene expression analysis of Cyp2E1 expression. (G) Bar graph of median fluorescence intensity (MFI) of BODIPY 493/503 calculated from three independent determinations, with the similar result presented in the histogram. (H) Representative flow cytometry plots using Annexin V-FITC/PI staining for apoptosis. (I) Bar graph of median fluorescence intensity (MFI) of Annexin V-FITC calculated from three independent determinations, with the similar result presented in the histogram. (J) Bar graph for percentage of viable cells through MTT assay. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant.
Figure 6.
Effect of nSMase2 deficiency in HepG2 liver steatosis model. (A) qRT-PCR analysis of SMPD3 gene expression. (B) qRT-PCR analysis of TNF-α expression. (C) TNF-α protein analysis with representative immunoblot. (D) qRT-PCR analysis of PPARα expression. (E) PPARα protein analysis with representative immunoblot. (F) qRT-PCR gene expression analysis of Cyp2E1 expression. (G) Bar graph of median fluorescence intensity (MFI) of BODIPY 493/503 calculated from three independent determinations, with the similar result presented in the histogram. (H) Representative flow cytometry plots using Annexin V-FITC/PI staining for apoptosis. (I) Bar graph of median fluorescence intensity (MFI) of Annexin V-FITC calculated from three independent determinations, with the similar result presented in the histogram. (J) Bar graph for percentage of viable cells through MTT assay. Results were obtained from a minimum of three independent experiments. All data are expressed as mean ± SEM. * p ≤ 0.5, ** p ≤ 0.1 *** p ≤ 0.001, **** p ≤ 0.0001 and ns: non-significant.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



