
Submitted:
04 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
The MLL1 gene located on chromosome 11q23, is crucial for histone lysine-specific methylation and is frequently implicated in various leukemia types. Despite prior in silico investigations into non-synonymous single nucleotide polymorphisms (nsSNPs) associated with leukemia in the MLL1 gene, there is a dearth of comprehensive studies evaluating their impact on the protein's structure, function, and post-translational modifications. Addressing this, our study screened 2049 nsSNPs from the dbSNP database, identifying 62 high-risk variants. A thorough domain analysis revealed that these nsSNPs are distributed across seven domains of the MLL1 protein, potentially affecting their functions. Our conservation analysis highlighted 31 nsSNPs as deleterious, suggesting their potential effects on protein stability and function. Furthermore, utilizing the MutPred2 tool, we identified mutations affecting metal-binding, protein loop structures, and post-translational modifications, which could alter the MLL1 protein's native functionality. Interaction network analysis underscored the role of MLL1 in hematopoietic cell development, implying that mutations may influence hematological malignancies. Additionally, a subset of non-coding SNPs was found to possess regulatory capabilities, potentially affecting gene expression and chromatin modification. Most significantly, using CScape and CScape-somatic tools, four nsSNPs were predicted to be oncogenic, with two classified as cancer drivers, underscoring their potential role in leukemia development. The oncogenic nsSNP, D2724G, located between the FY-rich N and FY-rich C domain of the MLL1 protein, has raised significant interest due to its potential impact on proteolytic cleavage. Predictive analyses, such as those by Mutpred2, suggest a loss of cleavage at D2724 caused by this mutation, potentially affecting the formation of essential MLL1 fragments, p320 and p180. These fragments play a crucial role in creating a stable multiprotein complex that localizes to a specific subnuclear region. Therefore, mutations like D2724G can potentially alter gene expression patterns, influencing cellular behavior and possibly fostering a cancer-prone environment. In conclusion, our research provides an in-depth assessment of the effects of nsSNPs in the MLL1 gene on leukemia, offering potential avenues for personalized treatment strategies, early detection, prognosis, and a better understanding of hematological malignancy genesis.
Keywords:
MLL1 gene
; leukemia
; computational algorithms
; protein structure
; nsSNPs
1. Introduction
The gene mixed-lineage leukemia 1 (MLL1), also known as KMT2A (Lysine methyltransferase 2A), maps to human chromosome 11 (11q23.3). It spans a sequence of 90,343 base pairs and is composed of 37 exons [1,2].
Numerous species possess a MLL1 counterpart, spanning across fish, birds, amphibians, and mammals. This evolutionary preservation has enabled a thorough exploration of KMT2A’s molecular functions through live experiments on animal models such as Drosophila melanogaster, Danio rerio, and Mus musculus. MLL1 expression is predominantly nuclear and widely distributed in 27 different tissues, with particularly high levels in the ovary, lymph node, endometrium, thyroid, and brain tissues [2]. MLL1 encodes a lysine methyltransferase (KMT) consisting of 3969 amino acids, serving as a transcriptional co-activator with significant roles in hematopoiesis, early developmental gene regulation, and circadian gene expression control. Taspase 1, an endopeptidase, processes MLL1 into two fragments (MLL-C and MLL-N) that form heterodimers and oversee the transcription of specific genes, including HOX genes [3]. The MLL1 protein boasts 18 domains, including the CXXC-type zinc finger, extended PHD domain, and bromodomain. The SET domain, within this protein, possesses methyltransferase activity (mono-, di-, tri-methylation) on lysine 4 of histone 3 (H3K4 me1/2/3), which is a post-transcriptional modification (PTM) responsible for epigenetic transcriptional activation. This efficiency can be enhanced when the protein associates with another component of the MLL1/MLL complex [4].
MLL1 primarily governs the initiation and elongation of transcription through epigenetic modifications within the promoter regions of target genes [5]. Additionally, the MLL1 protein plays a vital role in the regulation of hematopoietic cell proliferation and differentiation, along with the modulation of the Meis homeobox 1 (MEIS1) and homeobox A (HOXA) gene clusters [6]. When these genes experience irregular regulation, it disrupts proper hematopoietic development, frequently leading to the development of leukemia [7].
Rearrangements involving the MLL1 gene and its associated partners are commonly observed in various types of leukemia, including precursor B-cell acute lymphoblastic leukemias (B-ALLs), T-cell acute lymphoblastic leukemias (T-ALLs), acute myeloid leukemias (AMLs), myelodysplastic syndromes (MDSs), mixed lineage (biphenotypic) leukemias (MPALs), and secondary leukemias [1]. Notably, MLL1-rearranged ALL (MLL1-r ALL) is prevalent, affecting over 80% of new ALL diagnoses in infants (under 1 year of age), approximately 5–6% in pediatric patients, and about 15% in adults [1,8].
Several types of tumors have been associated with somatic mutations occurring in the MLL1 gene. The most frequent MLL1 alterations include mutations (3.62%) and fusions (0.13%), as well as losses (0.10%), amplifications (0.07%), and MLL1-EP300 fusions (0.19%). Patient-derived samples commonly exhibit missense mutations (54.36%), synonymous mutations (13.61%), and nonsense mutations (7.34%) as the primary mutations. In contrast, germline MLL1 mutations are predominantly frameshift (41%) and stop mutations (29%), with a minority being missense variants (18%) [2].
Single nucleotide polymorphisms (SNPs) are the most prevalent genetic variations in the human genome, occurring approximately once every 100–300 base pairs. SNPs represent single-nucleotide changes in the DNA sequence, potentially altering the amino acid sequence of proteins and influencing their structure and function [9].
Concerns have been raised, particularly regarding missense non-synonymous SNPs (nsSNPs), as they result in amino acid changes within a protein’s coding sequence, potentially impacting its functionality. Assessing the effects of multiple nsSNPs can be resource-intensive and time-consuming. Nonetheless, by utilizing web-based bioinformatics tools, it becomes both feasible and cost-effective to conduct in silico analyses of numerous SNPs within a gene. Computational genomics has made significant contributions to the investigation of nsSNPs associated with diseases. Various algorithms have been developed to predict the phenotypic consequences of nsSNPs [10].
SNPs have been implicated in numerous diseases, including cancer. Previous studies have established links between SNPs and conditions such as gastric cancer and sporadic prostate cancer [11]. These findings have prompted increased attention to the role of SNPs in cancer diagnosis and risk assessment. SNPs are now recognized as crucial biomarkers for cancer.
A prior study reported an association between polymorphisms in both coding and non-coding regions of the MLL1 gene and leukemia [6]. However, this earlier study employed a limited number of in silico tools compared to the present study. Specifically, it relied on just four computational algorithms: SIFT, PolyPhen, Pupasiute, and UTResource. The previous study identified a deleterious MLL1 gene mutation, Q1198P, linked to acute leukemia. This mutation was believed to disrupt MLL1 protein function, resulting in uncontrolled cell growth and division.
The current investigation aims to identify new deleterious missense nsSNPs within the MLL1 gene, as cataloged in the dbSNP database, and to identify those with particularly detrimental effects on protein structure and function. The screening tools for this study were chosen to encompass a range of technological approaches utilized by various screening tools, facilitating an unbiased, consensus-based classification of deleterious MLL1 variants. Subsequent to SNP screening, additional in silico analyses were conducted, encompassing conservation, interaction, oncogenic potential, phenotypic effects, structural considerations, and post-translational modification (PTM) assessments.
2. Results
2.1. SNP Annotation
We acquired MLL1 nsSNPs from the Ensemble database, encompassing a total of 17,562 SNPs located within the intronic region, 10 SNPs in the 5′UTR region, 1,112 SNPs in the 3′UTR region, and 2,181 SNPs within the coding sequence. Among the SNPs within the coding sequence, 2,097 SNPs were of the missense type (nsSNPs), while 1,024 SNPs were synonymous. For our present study, we focused exclusively on missense nsSNPs as they induce changes in amino acids due to alterations in codons.
2.2. Identification of Harmful nsSNPs
To identify nsSNPs with the potential to detrimentally affect the structure or function of the MLL1 gene, we utilized PredictSNP, which integrates multiple software tools including MAPP, PhD-SNP, PolyPhen1, PolyPhen2, SIFT, and SNAP. Out of the 2,097 nsSNPs, a total of 62 were predicted to be deleterious across all computational algorithms (Table 1).
2.3. Identification of nsSNPs within MLL1 Domains
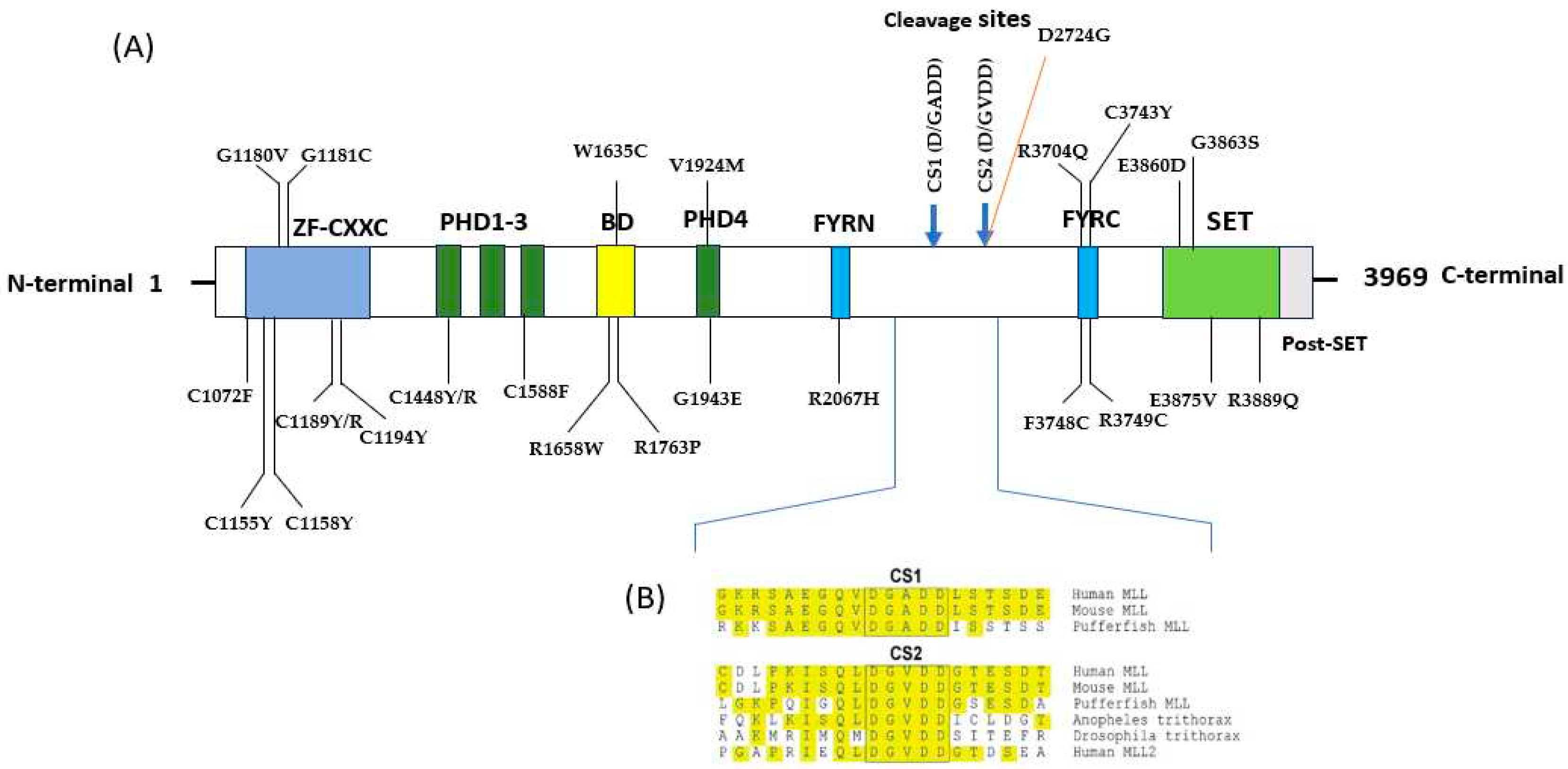
InterPro, a tool for domain identification, conducted a functional analysis of protein families to predict domains and active sites within the MLL1 protein. It identified the following domains: Zinc finger, CXXC (1147-1195); Znf PHD1 finger (1433-1480); Znf PHD2 finger (1481-1531); Znf PHD3 finger (1568-1625); Bromodomain (1633-1767); Znf PHD4 finger (1932-1978); FY rich N (2021-2077); FY rich C (3666-3753); SET domain (3829-3951); and Post SET domain (3953-3969). Notably, 25 out of the 62 nsSNPs were situated within these identified domains (Figure 1, S1 and Table 1).

Table 1.
Deleterious nsSNPs detected through the consensus of six in silico programs.
| Domains | AA change | SNP ID | Polyphen 1 and 2 | PhD-SNP, SIFT, SNAP and MAPP |
Predict SNP |
|---|---|---|---|---|---|
| C1072F | rs1085307947 | Damaging | Deleterious | Deleterious | |
| Zinc finger, CXXC-type | C1155Y | rs1057518074 | Damaging | Deleterious | Deleterious |
| C1158Y | rs1131691503 | Damaging | Deleterious | Deleterious | |
| G1180V | rs1555038115 | Damaging | Deleterious | Deleterious | |
| G1181C | rs1950071303 | Damaging | Deleterious | Deleterious | |
| C1189R | rs886041875 | Damaging | Deleterious | Deleterious | |
| C1189Y | rs1555038125 | Damaging | Deleterious | Deleterious | |
| C1194Y | rs1950106455 | Damaging | Deleterious | Deleterious | |
| PHD1-3 | C1448R | rs863224895 | Damaging | Deleterious | Deleterious |
| C1448Y C1588F |
rs1085307857 rs1555042404 |
Damaging Damaging |
Deleterious Deleterious |
Deleterious Deleterious |
|
| R1630W | rs376776245 | Damaging | Deleterious | Deleterious | |
| Bromodomain | W1635C | rs782594163 | Damaging | Deleterious | Deleterious |
| R1658W | rs373435126 | Damaging | Deleterious | Deleterious | |
| R1763P | rs781944403 | Damaging | Deleterious | Deleterious | |
| W1771R | rs1475344216 | Damaging | Deleterious | Deleterious | |
| R1892C | rs1555044474 | Damaging | Deleterious | Deleterious | |
| Y1895C | rs143373748 | Damaging | Deleterious | Deleterious | |
| G1919R | rs1555044515 | Damaging | Deleterious | Deleterious | |
| Extended PHD 4 | V1924M | rs1555044535 | Damaging | Deleterious | Deleterious |
| G1943E | rs1950444447 | Damaging | Deleterious | Deleterious | |
| L2009W | rs1555044990 | Damaging | Deleterious | Deleterious | |
| R2011W | rs781919638 | Damaging | Deleterious | Deleterious | |
| G2016D | rs1397000127 | Damaging | Deleterious | Deleterious | |
| G2027E | rs1057519403 | Damaging | Deleterious | Deleterious | |
| F/Y-rich N-terminus | R2067H | rs782768278 | Damaging | Deleterious | Deleterious |
| G2277D | rs1555046173 | Damaging | Deleterious | Deleterious | |
| R2519W | rs782129680 | Damaging | Deleterious | Deleterious | |
| R2521H | rs1555046685 | Damaging | Deleterious | Deleterious | |
| D2598V | rs368088982 | Damaging | Deleterious | Deleterious | |
| R2627C | rs1555046878 | Damaging | Deleterious | Deleterious | |
| S2652I | rs782497028 | Damaging | Deleterious | Deleterious | |
| R2659Q | rs1390104203 | Damaging | Deleterious | Deleterious | |
| Y2683H | rs1555046962 | Damaging | Deleterious | Deleterious | |
| L2700R | rs1555047001 | Damaging | Deleterious | Deleterious | |
| D2724G | rs781821970 | Damaging | Deleterious | Deleterious | |
| G2796E | rs1186580008 | Damaging | Deleterious | Deleterious | |
| G2796V | rs1186580008 | Damaging | Deleterious | Deleterious | |
| L2857Q | rs1057520696 | Damaging | Deleterious | Deleterious | |
| G3000R | rs1438502727 | Damaging | Deleterious | Deleterious | |
| S3039C | rs1279929414 | Damaging | Deleterious | Deleterious | |
| P3098L | rs1555047613 | Damaging | Deleterious | Deleterious | |
| G3129R | rs1353151956 | Damaging | Deleterious | Deleterious | |
| G3186D | rs961670105 | Damaging | Deleterious | Deleterious | |
| I3189T | rs782641177 | Damaging | Deleterious | Deleterious | |
| S3211I | rs782372675 | Damaging | Deleterious | Deleterious | |
| L3373H | rs781812638 | Damaging | Deleterious | Deleterious | |
| L3393P | rs1555048205 | Damaging | Deleterious | Deleterious | |
| C3430R | rs373345566 | Damaging | Deleterious | Deleterious | |
| N3459Y | rs782596573 | Damaging | Deleterious | Deleterious | |
| L3617P | rs146191865 | Damaging | Deleterious | Deleterious | |
| FY-rich, C-terminal | R3704Q | rs1555050212 | Damaging | Deleterious | Deleterious |
| C3743Y | rs782658611 | Damaging | Deleterious | Deleterious | |
| F3748C | rs749354451 | Damaging | Deleterious | Deleterious | |
| R3749C | rs782366377 | Damaging | Deleterious | Deleterious | |
| R3789H | rs1555052977 | Damaging | Deleterious | Deleterious | |
| R3810W | rs1555053010 | Damaging | Deleterious | Deleterious | |
| R3822C | rs1591310362 | Damaging | Deleterious | Deleterious | |
| SET domain | E3860D | rs782600332 | Damaging | Deleterious | Deleterious |
| G3863S | rs1591311290 | Damaging | Deleterious | Deleterious | |
| E3875V | rs781980190 | Damaging | Deleterious | Deleterious | |
| R3889Q | rs1555053677 | Damaging | Deleterious | Deleterious |
Figure 2.
(A) Conserved domain structure of human MLL11 with cleavage sites (CS1 and CS2). Domain and homologous superfamily of MLL1 gene produced by InterPro. The MLL1 protein features a CXXC domain, which consists of four cysteine residues and two zinc ions. In the N-terminal region, it possesses four plant homeotic domains (PHD), alongside a FY-rich N-terminal (FYRN) domain. Additionally, there is a FY-rich C-terminal (FYRC) domain and a catalytically active SET domain situated at the C-terminal. (B) Conservation of CS1 (D/GADD) and CS2 (D/GVDD) among MLL family members.
Figure 2.
(A) Conserved domain structure of human MLL11 with cleavage sites (CS1 and CS2). Domain and homologous superfamily of MLL1 gene produced by InterPro. The MLL1 protein features a CXXC domain, which consists of four cysteine residues and two zinc ions. In the N-terminal region, it possesses four plant homeotic domains (PHD), alongside a FY-rich N-terminal (FYRN) domain. Additionally, there is a FY-rich C-terminal (FYRC) domain and a catalytically active SET domain situated at the C-terminal. (B) Conservation of CS1 (D/GADD) and CS2 (D/GVDD) among MLL family members.
2.4. Evolutionary Conservation Analysis
ConSurf analysis identified that among the 62 deleterious nsSNPs found in the native MLL1 gene, 50 were associated with highly conserved residues. The assessment of evolutionary conservation and solvent accessibility helped pinpoint structural and functional residues affected by nsSNPs in the MLL1 gene (Figure S2 and Table 2).
Table 2.
Amino acid conservation scores, confidence intervals, and conservation colors produced by ConSurf.
Table 2.
Amino acid conservation scores, confidence intervals, and conservation colors produced by ConSurf.
| POS | SEQ | SCORE (normalized) |
COLOR | CONFIDENCE INTERVAL |
CONFIDENCE INTERVAL COLOR |
B/E | FUNCTION |
|---|---|---|---|---|---|---|---|
| 1072 | C | -0.851 | 9 | -0.937, -0.817 | 9,9 | b | s |
| 1155 | C | -0.851 | 9 | -0.937, -0.817 | 9,9 | e | f |
| 1158 | C | -0.851 | 9 | -0.937, -0.817 | 9,9 | b | s |
| 1180 | G | -0.861 | 9 | -0.937, -0.842 | 9,9 | e | f |
| 1181 | G | -0.861 | 9 | -0.937, -0.842 | 9,9 | e | f |
| 1189 | C | -0.851 | 9 | -0.937, -0.817 | 9,9 | b | s |
| 1194 | C | -0.541 | 8 | -0.760, -0.398 | 9,7 | b | / |
| 1448 | C | -0.856 | 9 | -0.937, -0.817 | 9,9 | b | s |
| 1588 | C | -0.712 | 9 | -0.864,-0.611 | 9,8 | b | s |
| 1635 | W | -0.799 | 9 | -0.937,-0.760 | 9,9 | b | s |
| 1658 | R | -0.893 | 9 | -0.943,-0.883 | 9,9 | e | f |
| 1763 | R | -0.635 | 8 | -0.790,-0.565 | 9,8 | e | f |
| 1771 | W | -0.565 | 8 | -0.817,-0.398 | 9,7 | b | / |
| 1892 | R | -0.893 | 9 | -0.943,-0.883 | 9,9 | e | f |
| 1895 | Y | -0.861 | 9 | -0.937,-0.842 | 9,9 | b | s |
| 1919 | G | -0.740 | 9 | -0.883,-0.653 | 9,8 | e | f |
| 1924 | V | -0.901 | 9 | -0.943,-0.883 | 9,9 | b | s |
| 1943 | G | -0.866 | 9 | -0.937,-0.842 | 9,9 | e | f |
| 2009 | L | -0.492 | 7 | -0.692,-0.332 | 8,7 | b | / |
| 2011 | R | -0.809 | 9 | -0.900,-0.760 | 9,9 | e | f |
| 2016 | G | -0.866 | 9 | -0.937,-0.842 | 9,9 | e | f |
| 2027 | G | -0.866 | 9 | -0.937,-0.842 | 9,9 | b | s |
| 2067 | R | -0.893 | 9 | -0.943,-0.883 | 9,9 | e | f |
| 2519 | R | -0.393 | 7 | -0.611,-0.258 | 8,6 | e | / |
| 2521 | R | -0.497 | 7 | -0.692,-0.398 | 8,7 | e | / |
| 2627 | R | -0.893 | 9 | -0.943,-0.883 | 9,9 | e | f |
| 2652 | S | -0.646 | 8 | -0.790,-0.565 | 9,8 | e | f |
| 2659 | R | -0.810 | 9 | -0.900,-0.760 | 9,9 | e | f |
| 2683 | Y | -0.728 | 9 | -0.864,-0.653 | 9,8 | b | s |
| 2700 | L | -0.497 | 7 | -0.692,-0.398 | 8,7 | b | / |
| 2724 | D | -0.893 | 9 | -0.943,-0.883 | 9,9 | e | f |
| 2857 | L | -0.864 | 9 | -0.937,-0.842 | 9,9 | b | s |
| 3039 | S | -0.842 | 9 | -0.915,-0.817 | 9,9 | e | f |
| 3098 | P | -0.752 | 9 | -0.883,-0.692 | 9,8 | e | f |
| 3129 | G | -0.313 | 7 | -0.565,-0.175 | 8,6 | b | / |
| 3189 | I | -0.827 | 9 | -0.915,-0.790 | 9,9 | b | s |
| 3373 | L | -0.610 | 8 | -0.790,-0.514 | 9,8 | b | / |
| 3393 | L | -0.729 | 9 | -0.883,-0.653 | 9,8 | e | f |
| 3430 | C | -0.269 | 6 | -0.565,-0.083 | 8,5 | b | / |
| 3704 | R | -0.886 | 9 | -0.943,-0.864 | 9,9 | e | f |
| 3743 | C | -0.846 | 9 | -0.937,-0.817 | 9,9 | b | s |
| 3748 | F | -0.856 | 9 | -0.937,-0.817 | 9,9 | b | s |
| 3749 | R | -0.698 | 8 | -0.842,-0.611 | 9,8 | e | f |
| 3789 | R | -0.887 | 9 | -0.943,-0.864 | 9,9 | e | f |
| 3810 | R | -0.887 | 9 | -0.943,-0.864 | 9,9 | e | f |
| 3822 | R | -0.794 | 9 | -0.900,-0.728 | 9,9 | b | s |
| 3860 | E | -0.882 | 9 | -0.943,-0.864 | 9,9 | e | f |
| 3863 | G | -0.857 | 9 | -0.937,-0.817 | 9,9 | b | s |
| 3875 | E | -0.882 | 9 | -0.943,-0.864 | 9,9 | e | f |
| 3889 | R | -0.793 | 9 | -0.900,-0.728 | 9,9 | b | s |
Abbreviations: B, buried; E, Exposed; S, structural; F, functional; POS, position.
2.5. Assessment of Protein Structural Stability
For the assessment of changes in MLL1 stability, considering relative solvent accessibility (RI) and alterations in free energy (DDG), we employed the I-Mutant 2.0 tool, which introduced point mutations into the MLL1 protein. The results indicated that 32 out of the 50 deleterious nsSNPs led to a decrease in stability (Table 3). Additionally, we utilized the MUpro web server to further evaluate the 32 missense substitutions that were predicted to be harmful in the previous steps (Table 3).
Table 3.
Effect of nsSNPs on protein stability predicted by I-Mutant 2.0 and MuPro.
| SNP ID | AA substitution | SVM2 | RI | DDG kcal/mol | SVM3 | RI | MUpro | DDG |
|---|---|---|---|---|---|---|---|---|
| rs1085307947 | C1072F | Decrease | 5 | -0.49 | Large decrease | 1 | Decrease | -0.49142 |
| rs886041875 | C1189R | Decrease | 5 | -1.05 | Large decrease | 2 | Decrease | -1.01449 |
| rs863224895 | C1448R | Decrease | 9 | -0.72 | Large decrease | 2 | Decrease | -1.05828 |
| rs373435126 | R1658W | Decrease | 1 | -0.09 | Large decrease | 1 | Decrease | -1.51963 |
| rs143373748 | Y1895C | Decrease | 6 | -0.85 | Large decrease | 0 | Decrease | -1.03143 |
| rs1555044990 | L2009W | Decrease | 4 | -1.32 | Large decrease | 1 | Decrease | -1.35469 |
| rs1397000127 | G2016D | Decrease | 8 | -0.99 | Large decrease | 3 | Decrease | -0.45411 |
| rs1057519403 | G2027E | Decrease | 5 | -1.61 | Large decrease | 4 | Decrease | -0.50805 |
| rs782768278 | R2067H | Decrease | 8 | -1.39 | Large decrease | 7 | Decrease | -1.13602 |
| rs1555046685 | R2521H | Decrease | 8 | -0.83 | Large decrease | 4 | Decrease | -1.04097 |
| rs1555046878 | R2627C | Decrease | 3 | -1.11 | Large decrease | 4 | Decrease | -0.73453 |
| rs782497028 | S2652I | Decrease | 3 | 0.13 | Large decrease | 1 | Decrease | -0.10003 |
| rs1390104203 | R2659Q | Decrease | 4 | -0.70 | Large decrease | 2 | Decrease | -0.59524 |
| rs1555047001 | L2700R | Decrease | 7 | -0.69 | Large decrease | 1 | Decrease | -1.82806 |
| rs781821970 | D2724G | Decrease | 6 | -0.89 | Large decrease | 6 | Decrease | -1.41038 |
| rs1186580008 | G2796E | Decrease | 6 | -0.75 | Large decrease | 1 | Decrease | -0.42903 |
| rs118658000 | G2796V | Decrease | 6 | -0.55 | Large decrease | 3 | Decrease | -0.47996 |
| rs1057520696 | L2857Q | Decrease | 9 | -2.31 | Large decrease | 5 | Decrease | -1.82752 |
| rs1279929414 | S3039C | Decrease | 4 | -2.02 | Large decrease | 6 | Decrease | -0.43857 |
| rs1555047613 | P3098L | Decrease | 7 | -1.11 | Large decrease | 3 | Decrease | -0.38588 |
| rs1353151956 | G3129R | Decrease | 9 | -1.35 | Large decrease | 1 | Decrease | -0.28198 |
| rs782641177 | I3189T | Decrease | 8 | -1.31 | Large decrease | 5 | Decrease | -2.11623 |
| rs781812638 | L3373H | Decrease | 7 | -1.34 | Large decrease | 5 | Decrease | -1.74765 |
| rs1555048205 | L3393P | Decrease | 3 | -1.12 | Large decrease | 2 | Decrease | -1.58189 |
| rs782658611 | C3743Y | Decrease | 1 | -0.23 | Large decrease | 3 | Decrease | -1.13086 |
| rs749354451 | F3748C | Decrease | 6 | -1.7 | Large decrease | 4 | Decrease | -1.82151 |
| rs782366377 | R3749C | Decrease | 3 | -0.85 | Large decrease | 2 | Decrease | -1.00862 |
| rs1555052977 | R3789H | Decrease | 9 | -1.44 | Large decrease | 6 | Decrease | -0.70688 |
| rs1591310362 | R3822C | Decrease | 0 | -0.85 | Large decrease | 3 | Decrease | -0.95916 |
| rs782600332 | E3860D | Decrease | 3 | -0.39 | Large decrease | 1 | Decrease | -1.01581 |
| rs1591311290 | G3863S | Decrease | 9 | -1.27 | Large decrease | 5 | Decrease | -1.34469 |
| rs1555053677 | R3889Q | Decrease | 9 | -1.25 | Large decrease | 5 | Decrease | -1.25798 |
Abbreviations: AA, amino acids; SVM 2 and 3, Support vector machines; RI, Reality Index; DDG, change in Gibbs free energy.
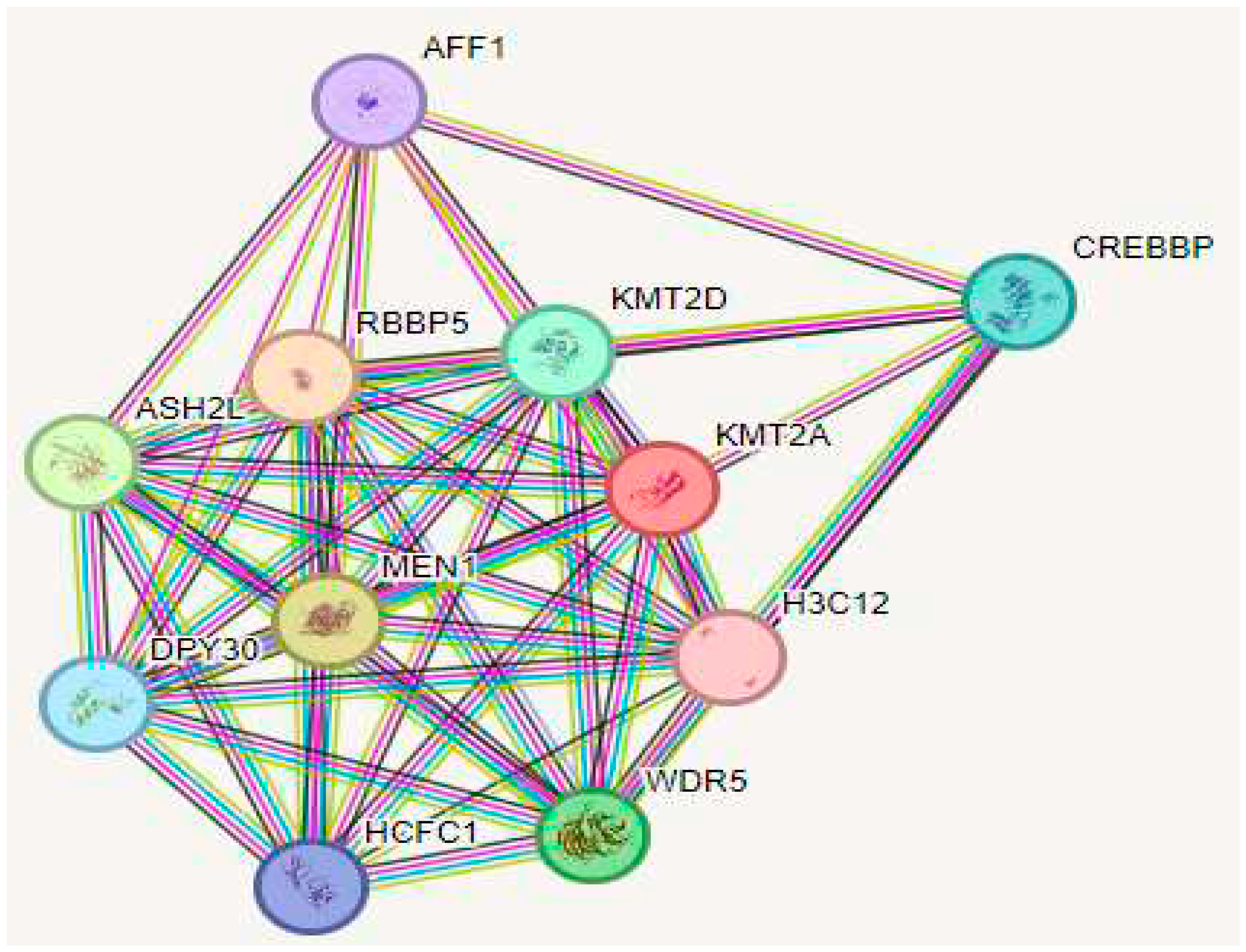
2.6. Analysis of Protein-Protein Interactions and Functional Associations
We utilized the STRING tool to predict the proteins closely interacting with MLL1. The findings revealed that MLL1 exhibits close interactions with several proteins, including Retinoblastoma-binding protein 5 (RBBP5), Menin (MEN1), Set1/Ash2 histone methyltransferase complex subunit ASH2 (ASH2), WD repeat-containing protein 5 (WDR5), CREB-binding protein (CREBBP), Host cell factor 1 (HCFC1), Protein dpy-30 homolog (DPY30), PC4 and SFRS1-interacting protein (PSIPI1), Chromodomain-helicase-DNA-binding protein 8 (CHD8), and Transcriptional activator Myb (MYB) (Figure 4)
Figure 4.
The STRING protein-protein interaction network was queried using MLL1. The colored lines connecting the proteins represent different types of interaction evidence.
Figure 4.
The STRING protein-protein interaction network was queried using MLL1. The colored lines connecting the proteins represent different types of interaction evidence.
2.7. Assessment of the Functional Implications of Non-coding SNPs
Based on the analysis conducted using the Regulome DB database, it was observed that four out of the five SNPs were assigned a ranking of 2b. This ranking suggests that a variety of data types, such as TF binding, motif information, DNase Footprint, and DNase peak, were accessible for the specific chromosomal location. Furthermore, a probability score approaching 1 strongly indicates that these particular SNPs are highly likely to be regulatory variants. Table 4 shows the results obtain from regulome DB database analysis.
Table 4.
RegulomeDB results of the functional consequences of non-coding SNPs analysis.
| Chromosome location | dbSNP IDs | Rank | Score |
|---|---|---|---|
| chr11:118522268..118522269 | rs188913109 | 2b | 0.64591 |
| chr11:118523887..118523888 | rs539251803 | 2b | 0.6751 |
| chr11:118524166..118524167 | rs141036837 | 2b | 0.50723 |
| chr11:118524283..118524284 | rs190548021 | 2b | 1.0 |
| chr11:118524539..118524540 | rs147772025 | 3a | 0.37421 |
2.8. Prediction of Pathogenicity of nsSNPs
The outcome from MutPred2 analysis indicated that 32 nsSNPs were identified as deleterious, including variants such as C1072F, C1189R, C1448R, R1658W, Y1895C, L2009W, G2016D, G2027E, R2067H, R2521H, R2627C, S2652I, R2659Q, L2700R, D2724G, G2796E, G2796V, L2857Q, S3039C, P3098L, G3129R, I3189T, L3373H, L3393P, C3743Y, F3748C, R3749C, R3789H, R3822C, E3860D, G3863S, and R3889Q. The potential molecular mechanisms disrupted by these variants are summarized in Table 5, presenting the results obtained from the MutPred2 server. MutPred2 score is the average of scores from all the neutral networks of MutPred2. A score threshold of 0.05 would suggest pathogenicity.
Table 5.
Result of MutPred2 analysis of the 32 nsSNPs, including their MutPred2 score and their impact on the different molecular mechanism.
Table 5.
Result of MutPred2 analysis of the 32 nsSNPs, including their MutPred2 score and their impact on the different molecular mechanism.
| AA variation | MutPred2 score | Molecular mechanism with P value less than 0.05 |
|---|---|---|
| C1072F | 0.926 | Altered Disordered interface Altered DNA binding Gain of Strand Altered Metal binding |
| C1189R | 0.825 | Altered Disordered interface Gain of Acetylation at K1185 |
| C1448R | 0.956 | Altered Metal binding Gain of Strand Altered Transmembrane protein Gain of Pyrrolidone carboxylic acid at Q1449 Loss of Sulfation at Y1447 |
| R1658W | 0.685 | Loss of Intrinsic disorder Loss of Helix Gain of Loop |
| Y1895C | 0.729 | Altered Ordered interface Loss of Proteolytic cleavage at R1892 Altered Transmembrane protein |
| L2009W | 0.662 | Altered Transmembrane protein |
| G2016D | 0.578 | Altered DNA binding Altered Transmembrane protein |
| G2027E | 0.911 | Gain of Helix |
| R2067H | 0.809 | Altered Ordered interface Altered Transmembrane protein Loss of Disulfide linkage at C2068 |
| R2521H | 0.150 | - |
| R2627C | 0.782 | Altered Disordered interface Loss of Intrinsic disorder Altered DNA binding Gain of Proteolytic cleavage at R2622 |
| S2652I | 0.321 | - |
| R2659Q | 0.290 | - |
| L2700R | 0.848 | Gain of Intrinsic disorder Gain of B-factor |
| D2724G | 0.720 | Altered Disordered interface Altered Metal binding Loss of Proteolytic cleavage at D2724 Gain of O-linked glycosylation at T2727 |
| G2796E | 0.260 | - |
| G2796V | 0.274 | - |
| L2857Q | 0.832 | Gain of Intrinsic disorder Altered Disordered interface Loss of Helix |
| S3039C | 0.249 | - |
| P3098L | 0.572 | Loss of Strand Altered Transmembrane protein Gain of Pyrrolidone carboxylic acid at Q3103 |
| G3129R | 0.546 | Loss of Strand Gain of ADP-ribosylation at G3129 Loss of B-factor |
| I3189T | 0.540 | Gain of O-linked glycosylation at S3185 |
| L3373H | 0.707 | Gain of Intrinsic disorde |
| L3393P | 0.740 | Gain of Intrinsic disorder Altered Disordered interface |
| C3743Y | 0.814 | Altered Metal binding Gain of Loop Altered Transmembrane protein |
| F3748C | 0.898 | Altered Metal binding Loss of SUMOylation at K3752 Altered Transmembrane protein |
| R3749C | 0.770 | Loss of Intrinsic disorder Loss of SUMOylation at K3752 Altered Transmembrane protein |
| R3789H | 0.512 |
Loss of Phosphorylation at Y3794 Loss of ADP-ribosylation at R3789 Gain of Sulfation at Y3794 |
| R3822C | 0.627 | Altered Disordered interface Loss of Intrinsic disorder |
|
E3860D |
0.903 |
Gain of Allosteric site at I3859 Altered Metal binding Altered Ordered interface Gain of Relative solvent accessibility Altered DNA binding Loss of Catalytic site at E3860 |
| G3863S | 0.907 | Altered Ordered interface Gain of Allosteric site at E3860 Altered Disordered interface Loss of Strand Gain of Relative solvent accessibility Altered Metal binding Altered DNA binding Gain of Catalytic site at E3860 |
|
R3889Q |
0.876 |
Altered Metal binding Altered Transmembrane protein Altered Ordered interface Loss of Allosteric site at M3887 Gain of Relative solvent accessibility Altered DNA binding Gain of Catalytic site at R3889 |
2.9. Predicting the Association of nsSNPs with Cancer Susceptibility
We employed CScape and CScape-somatic to assess the oncogenic potential of the screened nsSNPs. The CScape results assigned oncogenic scores of 0.650128, 0.552600, 0.766650, and 0.627944 to D2724G, L3393P, E3860D, and G3863S, respectively. Conversely, F3748C and R3789H received benign scores of 0.455280 and 0.192980, respectively, indicating a lack of association with cancer susceptibility.
CScape-somatic differentiated between mutations contributing to cancer as driver or passenger variants. Driver variants are involved in tumor initiation, while passenger variants accumulate after tumor initiation and typically exhibit low or no tumorigenic potential. According to CScape-somatic, D2724G, L3393P, F3748C, and R3789H mutants were classified as driver cancer variants with scores of 0.552102, 0.736249, 0.612477, and 0.881861, respectively. In contrast, E3860D and G3863S mutants were categorized as passenger cancer variants with scores of 0.249088 and 0.307779, respectively. These classifications were based on p-value scores ranging from 0 to 1, where values above 0.5 indicated driver oncogenic potential, while values below 0.5 indicated passenger benign variants.
Table 6.
Oncogenic nature mutation predicted using Cscape and Cscape-somatic software.
| CScape | CScape- somatic | ||||||
|---|---|---|---|---|---|---|---|
| Variant ID | SNP | Input | Coding score |
Message | Input | Coding score |
Message |
| rs781821970 | D2724G | 11,118504063,A,G | 0.650128 | Oncogenic | 11,118504063,A,G | 0.552102 | Driver |
| rs1555048205 | L3393P | 11,118506070,T,C | 0.5526 | Oncogenic (*HC) |
11,118506070,T,C | 0.736249 | Driver |
| rs749354451 | F3748C | 11,118519714,T,G | 0.45528 | Benign | 11,118519714,T,G | 0.612477 | Driver |
| rs1555052977 | R3789H | 11,118520001,G,A | 0.19298 | Benign | 11,118520001,G,A | 0.881861 | Driver |
| rs782600332 | E3860D | 11,118521354,G,T | 0.76665 | Oncogenic | 11,118521354,G,T | 0.249088 | Passenger |
| rs1591311290 | G3863S | 11,118521361,G,A | 0.627944 | Oncogenic | 11,118521361,G,A | 0.307779 | Passenger |
*HC = High Confidence.
3. Discussion
The MLL1 gene, alternatively known as KMT2A, codes for the histone lysine-specific N-methyltransferase 2A protein, which is located on chromosome 11q23, this gene often undergoes rearrangements in several types of leukemia, including acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and mixed lineage leukemia (MLL) [29]. Previous in silico study identified several nsSNPs within the MLL1 gene that are associated with leukemia, with Q1198P being one such variant that may disrupt the normal function of the MLL1 protein, leading to uncontrolled cell growth and division. However, this prior study utilized only four computational algorithms—SIFT, PolyPhen, Pupasiute, and UTResource. Furthermore, it did not delve into the comprehensive effects of nsSNPs on protein structure, stability, functional consequences, or post-translational modifications (PTMs). The present study addresses this research gap by identifying the most deleterious nsSNP variants within the MLL1 gene and assessing their impact on protein structure, stability, and function. Additionally, this study provides a comprehensive characterization of both coding and non-coding nsSNPs associated with MLL1.
Various tools were employed to estimate the likelihood of detrimental effects caused by nsSNPs within the MLL1 gene. Initially, the nsSNPs sourced from the dbSNP database underwent a screening process based on their predicted functional significance. The PredictSNP tool was utilized to pinpoint potentially harmful nsSNPs capable of inducing substantial alterations in the structure or function of the MLL1 gene. This comprehensive analysis identified a total of 62 nsSNPs as high-risk candidates out of the initial pool of 2049 nsSNPs.
The InterPro tool was utilized to determine the positions of the nsSNPs within the various domains of the MLL1 protein. Analysis indicated that, out of the 62 nsSNPs identified, 25 are situated within these domains. Surprisingly, those nsSNPs are spread across seven unique protein domains. Such a distribution suggests that the presence of nsSNPs in these specific domains might disrupt their normal functions and activities. It’s essential to emphasize that each domain is associated with distinct functions. Therefore, the presence of nsSNPs in these areas could potentially alter their structural integrity and subsequent functional behaviors, leading to unforeseen biological consequences
The CXXC domains, also known as zinc finger (ZF)-CXXC domains, have the ability to bind and identify non-methylated CpG DNA of target genes through the coordination of two zinc ions with four cysteine residues (Cys4) [30]. The presence of these nsSNPs within this domain holds the potential to compromise its functionality. Furthermore, the maintenance of the structural integrity of the CXXC domain protein is contingent upon the availability of zinc ions; mutations or nsSNPs affecting any cysteine residues involved in zinc coordination can lead to the protein’s unfolding [31].
In addition to the ZF-CXXC domain, the MLL1 protein encompasses four PHD fingers, namely PHD1 to PHD4. Each finger exhibits a Cys4-His-Cys3 motif, which is orchestrated by two zinc ions and plays a pivotal role in binding to methylated histone. This domain is instrumental in inhibiting the development of leukemia. The incorporation of the PHD2-PHD3 finger in the chimeric MLL1-AF9 has been demonstrated to inhibit the transformation of mouse bone marrow, foster the differentiation of hematopoietic cells, and diminish Hoxa9 expression [32,33]. Consequently, nsSNPs hold the potential to negatively impact the functionality of this domain and thereby facilitate the progression of leukemia.
Situated between PHD3 and PHD4 is a bromodomain (BRD), which is vital for regulating gene transcription through interaction with protein acetyl-lysine [34]. It is not uncommon for proteins with BRDs or chromatin loci, such as enhancers that recruit BRD proteins, to be deregulated in cancer, leading to aberrant oncogene expression. The recent advent of potent BRD inhibitors, including those targeting the Bromo, has proven to be markedly efficacious in murine cancer models, thus offering a strong basis for further drug development [35]. In this scenario, it is plausible to suggest that the identified nsSNP such as W1635C, R1658W and R1763P in this domain could contribute to the emergence of blood cancer and resistance to therapeutics.
Following the BRD, the protein structure showcases an additional PHD, complemented by an FY-rich N-terminal domain (FYRN) and an FY-rich C-terminal domain (FYRC). These specific domains are integral for the non-covalent dimerization of the protein’s N- and C-terminal fragments after they undergo proteolytic cleavage. One can postulate that the presence of nsSNPs within these domains might interfere with this dimerization process, potentially compromising the protein’s function or stability.
The final identified domain is the SET domain, which plays a crucial role in the protein’s histone mono-methylation, di-methylation, or tri-methylation activities [36]. Alterations in the conformation of the SET domain, brought about by somatic cancer mutations in MLL1, lead to an elevation in methyltransferase activity and a reduction in dependency on complex partners. There is a likelihood that mutant enzymes experience a loss of control over their activities, culminating in modifications to cellular MLL1 activity [37]. The presence of mutations in the SET domain has been documented in human cancers, indicating their potential widespread influence on chromatin modifications regulated by genes [38]. Consequently, nsSNPs located within this domain could be of significant importance in the onset of cancer.
Through the application of ConSurf, 50 out of 62 nsSNPs were identified as deleterious, having high conservation values; among these, 24 are functional (exposed) and 17 are structural (buried). Conservation profiles of proteins are instrumental in assessing the impact of significant mutations. Upon examining ConSurf’s outcomes, 31 out of the 59 nsSNPs were discerned as deleterious, namely C1072F, C1189R, C1448R, R1658W, Y1895C, L2009W, G2016D, G2027E, R2067H, R2521H, R2627C, S2652I, S2652I, R2659Q, L2700R D2724G, L2857Q, S3039C, P3098L, G3129R, I3189T,L3373H, L3393P,C3743Y, F3748C, R3749C, R3789H, R3822C, E3860D, G3863S, R3889Q. Owing to their high conservancy, nsSNPs are potentially susceptible and possess the capability to influence biological mechanisms, notably protein-protein interactions, and to diminish protein stability. It is commonly observed that buried residues are situated at the protein’s core, and alterations in these residues predominantly impact the function of the protein [39].
Intramolecular interactions of various kinds play a crucial role in determining protein stability and folding. These include interactions that are hydrophobic, electrostatic, and involve hydrogen bonding. The stability state of a protein is a significant factor that dictates its functionality. Tools such as I-Mutant and MuPro have indicated that 32 out of the 50 high-risk nsSNPs could potentially reduce protein stability. The implications of diminished protein stability are becoming clearer, often leading to elevated levels of protein degradation, misfolding, and aggregation [40]. Additionally, pathogenic non-synonymous mutations may cause incorrect protein folding or a reduction in stability [41]. There is growing speculation that around 25% of nsSNPs present in human populations might impact protein function through modifications in protein stability [42].
This study took advantage of the MutPred2 tool to refine predictions, elucidating the molecular mechanisms that potentially underline the pathogenicity of amino acid variations or nsSNPs. Remarkably, eight SNPs (C1072F, C1448R, D2724G, C3743Y, F3748C, E3860D, G3863S, and R3889Q) were identified to modify metal-binding, which is noteworthy given the protein’s lack of metal-binding properties at these specific positions. Substitutions R1658W and C3743Y were observed to influence the transmembrane function, inducing a gain in loop structure. These structural loops have the potential to modify the inherent functionality and transmembrane properties of proteins [43,44]. Additionally, five mutations were found to result in altered PTM sites, including the loss of SUMOylation at K3752, the gain of Sulfation at Y3794, the gain of O-linked glycosylation at T2727, the loss of Sulfation at Y1447, and Acetylation at K1185. In total, the mutations led to alterations in three PTM sites. From these findings, it can be speculated that the deleterious nsSNPs hold the potential to impact significantly the native structure and functionality of the MLL1 protein.
Based on STRING, the functional network of MLL1 interactions with ten different proteins revealed metabolic pathways involved in developing hematopoietic cells. The presence of a mutation or nsSNPs may disrupt MLL1 expression. Therefore, abnormal MLL1 may play a significant role in developing hematological malignancies.
The Regulome DB is instrumental in predicting DNA properties and identifying regulatory elements within the human genome. Among the identified non-coding SNPs, rs188913109, rs539251803, rs141036837, rs190548021, and rs147772025 are projected to exert regulatory control over the MLL1 protein. This is attributed to their predicted association with transcription binding sites, matched or unmatched motifs, and the presence of a DNase footprint with a DNase peak. There is a plausible speculation that these nsSNPs could play a significant role in disease pathogenesis by inducing alterations in gene expression, transcription factor binding, chromatin modification, and DNA methylation. Such alterations have the potential to disrupt the normal functioning and biochemical regulation of the human genome.
In this research, the tools CScape and CScape-somatic were employed to evaluate the potential oncogenic nature of the screened SNPs. All four of the discovered nsSNPs, including L3393P, were predicted to possess oncogenic properties, with L3393P being identified with high confidence. CScape-somatic played a pivotal role in this study, differentiating between oncogenic nsSNPs that have the potential to act as cancer drivers and those likely to be passenger variants. Notably, the oncogenic variants D2724G and L3393P were categorized as cancer drivers, whereas E3860D and G3863S were classified as passenger variants. Cancer driver mutations typically emerge relatively early in tumor development, while passenger variants tend to accumulate as the tumor progresses, typically exhibiting lower levels of oncogenic activity [45]. The presence of the oncogenic nsSNP, D2724G, situated between the FY-rich N and FY-rich C domain, is intriguing. Given that Mutpred2 indicates a loss of proteolytic cleavage at D2724 due to this SNP, one might speculate that the cleavage process of the MLL1 protein could be disrupted by this mutation. This cleavage is pivotal for the formation of the N-terminal p320 (N320) and C-terminal p180 (C180) fragments [46]. Should this cleavage be hindered, the subsequent formation and stability of the complex these fragments constitute might be compromised. This particular complex is essential as it is sequestered to a distinct subnuclear region.
Further, given the importance of the threonine aspartase 1 enzyme in post-translationally cleaving MLL at specific cleavage points to yield the two crucial subunits (p320 and p180) [46], any obstruction in this process can potentially disrupt the formation of the robust multiprotein structure. This structure, as we know, plays a pivotal role in regulating the transcriptional activity of a gamut of genes, notably the Hox genes. Thus, any deviation from this intricate process, such as the one possibly caused by the D2724G mutation, could have wide-ranging implications, possibly altering gene expression patterns and, consequently, cellular behavior. Given its oncogenic nature, this mutation might foster an environment conducive to cancer development or progression. This raises speculative yet significant implications that these novel variants might play a pivotal role in the genesis of hematological malignancies, potentially influencing the progression and development of these disorders.
The clinical implications of understanding mutations in MLL1 protein including, the understanding of mutations and their positions on the MLL1 protein has profound implications for targeted therapeutic approaches. Recognizing these mutations, particularly in areas like the bromodomain (BRD) that are critical for gene transcription regulation, provides a tailored treatment path. Specifically, patients with BRD mutations might benefit significantly from BRD inhibitors, given their efficacy in murine cancer models.
Moreover, nsSNPs can offer insights into disease prognosis. Those with high conservation values and proven deleterious effects can serve as reliable biomarkers. Identifying these mutations can equip clinicians with a clearer understanding of disease severity and its probable trajectory. Building on this knowledge, the era of personalized medicine beckons, where treatments are no longer one-size-fits-all but are customized based on an individual’s unique genetic makeup, ensuring more effective outcomes.
Furthermore, the distinction between oncogenic nsSNPs, especially between cancer drivers and passenger variants, is paramount for understanding cancer development and progression. Some mutations, like the nsSNP D2724G, have the potential to alter numerous gene transcription activities, cultivating an environment conducive to cancer. Early identification of such critical mutations is pivotal for prompt diagnosis and intervention.
Delving deeper, non-coding SNPs that might regulate MLL1 protein paint a more complex genetic picture, emphasizing the intricate control mechanisms that govern MLL1’s function. Acknowledging how these SNPs modulate gene expression and other genomic functions can provide a comprehensive understanding of the genetic mechanisms at play, especially in disease scenarios.
Lastly, insights into specific mutations, especially in domains like ZF-CXXC or SET, are invaluable for drug development. By targeting these mutations, novel drugs can be formulated. Additionally, grasping these mutations can elucidate potential drug resistance pathways, steering research towards combination therapies or advanced drugs to counteract resistance effectively.
This study presents several potential limitations. It predominantly depends on computational techniques and in silico analyses to forecast the effects of nsSNPs on protein structure and function, which may not consistently mirror the actual biological context and may exhibit limitations in precision. Confirmation of the computational findings through experimental validation is essential for future investigations. Conducting experimental studies using cell lines, animal models, or clinical samples can yield more dependable and rigorous results. Although the study offers important insights into the potential consequences of nsSNPs in the MLL1 gene on leukemia, addressing the aforementioned limitations could considerably reinforce the study, enhancing its relevance and applicability in a clinical context.
4. Materials and Methods
4.1. Retrieval of SNP Data
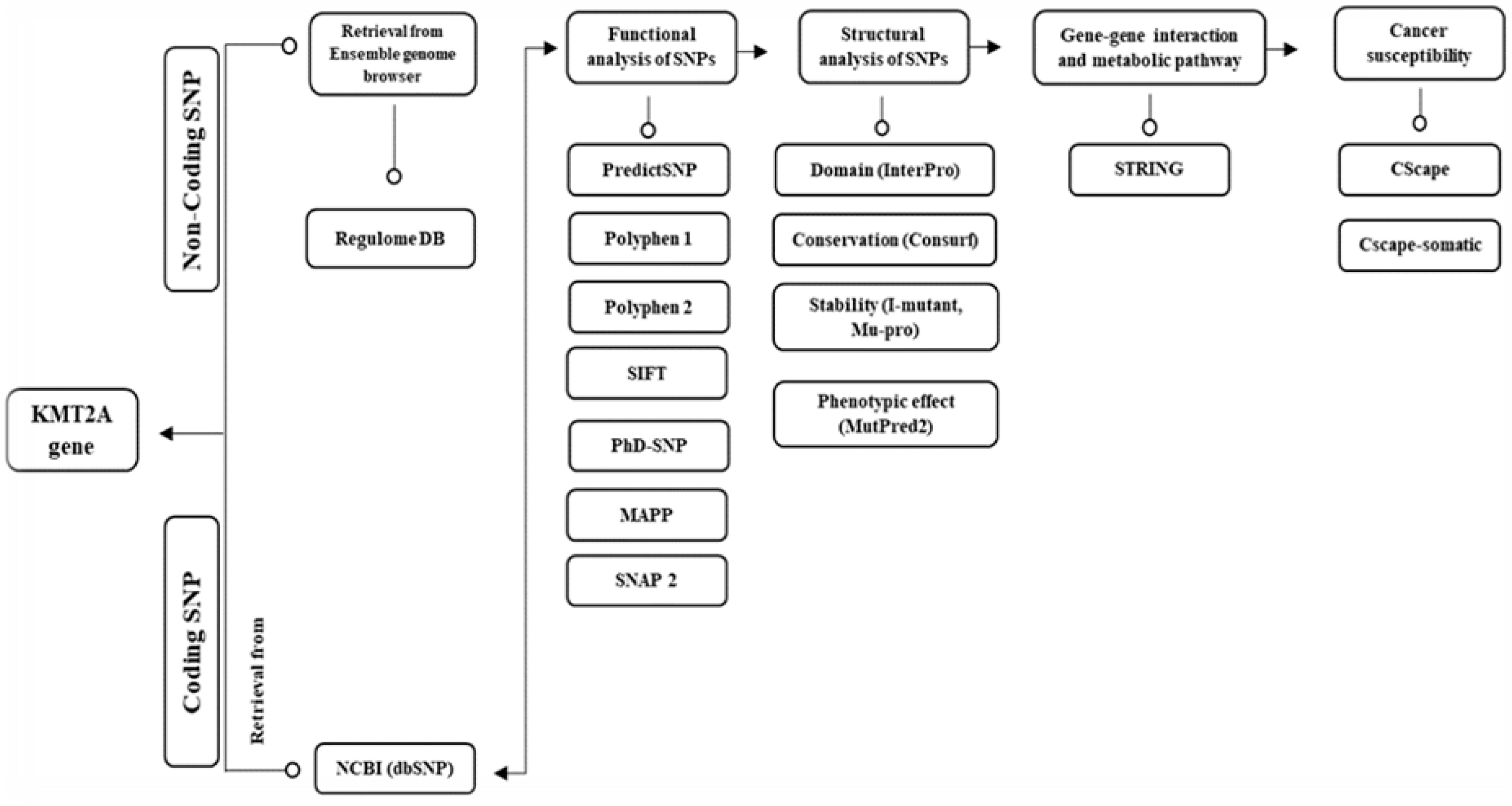
We obtained the genetic data for the human MLL1 gene from the ENSEMBL genome browser, specifically using accession number ENST00000691053.1 [12]. To capture missense nsSNPs within both the coding and non-coding regions, we selected the transcript encoding the entire human MLL1 protein, consisting of 3,969 amino acids. Additionally, we acquired the sequence for the MLL1 protein from the NCBI database with accession number NP_001184033.1 (accessed on December 26, 2022) [13]. An overview of the research protocol implemented in this study is depicted in Figure 1.
Figure 1.
illustrates the comprehensive methodological framework utilized in this study.
4.2. Prediction of Functional Effects of nsSNPs
To assess the functional impact of nsSNPs, we utilized PredictSNP1.0 (http:// loschmidt.chemi.muni.cz/predictsnp1/), accessed on December 26, 2022. PredictSNP1.0 is a consensus classifier resource that integrates predictions from six high-performing tools: SIFT, PolyPhen-1, PolyPhen-2, MAPP, PhD-SNP, and SNAP [14,15,16,17,18,19].
4.3. Identification of nsSNPs on MLL1 Protein Domains
The InterPro software located nsSNPs within conserved domains of the MLL1 protein. InterPro (https://www.ebi.ac.uk/interpro/), accessed on December 30, 2022, identifies protein motifs and domains, aiding in functional characterization through a database of protein families, domains, and functional sites [20].
4.4. Analysis of Protein Evolutionary Conservation
A ConSurf web server (http://consurf.tau.ac.il/), accessed on December 30, 2022 [21], assessed amino acid sequence conservation. This web-based algorithm estimated the degree of amino acid conservation based on multiple sequence alignment, assigning grades ranging from 1 to 9. Grade 9 indicated the most highly conserved residue, while descending numbers represented decreasing conservation levels. ConSurf also considered nucleotide and amino acid conservation and analyzed phylogenetic relationships between homologous sequences. The conserved nsSNPs in MLL1 were further examined.
4.5. Analysis of nsSNP Effects on Protein Stability
To evaluate the impact of deleterious nsSNPs on MLL1 protein structure and stability, we utilized I-Mutant 2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html, accessed on December 30, 2022). I-Mutant 2.0 predicted changes in protein stability by measuring the change in free energy Delta Delta G (DDG) upon mutation. A DDG value of 0 indicated reduced protein stability, while a DDG value greater than 0 signified increased stability [22].
MuPro, an additional software employed in this study, was utilized to assess the impact of nsSNPs on protein stability. This tool belongs to a category of machine learning programs that rely on support vector machines to predict the consequences of single-site amino acid substitutions on protein stability [23]. Specifically, MUpro (accessible at http://mupro.proteomics.ics.uci.edu/, accessed on December 30, 2022) utilizes the primary protein sequence of MLL1 and employs an algorithm to forecast changes in energy, assigning a confidence score that ranges from -1 to 1. A score below 0 suggests that a single mutation is likely to reduce protein stability, while a score exceeding 0 indicates that a mutation is likely to enhance protein stability.
4.6. Protein-Protein Interaction and Functional Analysis
The STRING online tool (https://string-db.org, accessed on December 30, 2022), examined the interaction of MLL1 with other proteins by predicting the top ten interacting proteins based on gene fusion, co-expression, function, and experimental data. The interactions were quantified using combined scores ranging from 0 to 1, with higher values indicating stronger interactions [24].
4.7. Analysis of the Functional Consequences of Non-Coding SNPs
RegulomeDB provided an annotation service for regulatory SNPs, amalgamating information derived from experimental datasets, computational forecasts, and manual annotations sourced from ENCODE. This utility assigned scores to genetic variants, enabling the differentiation of functional SNPs from a substantial pool of variants. To evaluate the impact of non-coding SNPs, the rsIDs of individual variants were submitted to the RegulomeDB database. These variants were categorized into ranks as follows: 1a-1f, indicating a high likelihood of affecting transcription factor binding and being associated with gene target expression; 2a-2c, signifying a reasonable probability of affecting binding; and 3a-3b, suggesting a lower likelihood of affecting binding. Additionally, variants classified as 4, 5, or 6 indicated minimal evidence of binding impact (Table S3) [25].
4.8. Prediction of Molecular Pathogenicity of nsSNPs
We employed MutPred2 (http://mutpred.mutdb.org/, accessed on December 3, 2022) to predict the structural and functional consequences of amino acid substitutions. The effects may encompass destabilizing the protein and disrupting its structure, interfering with macromolecular binding, and excising post-translational modification (PTM) sites, among others. These effects can result in significant changes in the protein’s phenotypic characteristics. Within this server, the input consisted of the FASTA sequence of the MLL1 protein and the specific amino acid variations of interest. The P-value threshold was maintained at its default value of 0.05.
MutPred2 provided general scores (g) and property scores (p) to assess the likelihood of an amino acid substitution being deleterious or disease-associated. Missense mutations with MutPred2 (g) scores above 0.5 were considered harmful, while scores exceeding 0.75 indicated high-confidence harmful predictions [26].
4.9. Association of nsSNPs with Cancer Susceptibility
CScape and CScape-somatic were employed to forecast the oncogenic potential of the scrutinized SNPs [27,28]. CScape-somatic boasts an impressive accuracy rate of 92% and specializes in predicting the oncogenic character of somatic point mutations within the coding regions of cancer genomes. The input format adhered to the following structure: chromosome, position, reference, and mutant, aligning with the GRCh38 assembly. The output yielded p-values, which could range from 0 to 1. A p-value exceeding 0.5 denoted a detrimental effect, while a value below 0.5 indicated benignity. Notably, CScape-somatic had the capacity to discern whether these cancer-related mutations acted as drivers or passengers. Cancer drivers typically manifest in the initial stages of tumor development, whereas passenger variants accumulate during later stages of tumor growth and typically exhibit low or negligible oncogenic potential.
5. Conclusions
This study presents a comprehensive analysis of the effects of various nsSNPs within the MLL1 gene, a critical player in several leukemia types. Employing a myriad of analytical tools, the research identified several high-risk nsSNPs and assessed their impact on protein structure, stability, and function, thereby addressing gaps in previous in silico studies. The distribution of these nsSNPs across essential domains of the MLL1 protein indicates their potential to interfere with the protein’s multifaceted functions, thereby contributing to leukemia progression.
In particular, the study illuminated the susceptibility of critical domains, such as the CxxC, PHD fingers, BRD, and SET domain, to disruptions by nsSNPs, which could compromise their respective functionalities and induce aberrations in cellular activities and genomic regulation. Furthermore, the findings suggest that these deleterious nsSNPs, especially those affecting metal-binding and transmembrane functions, could significantly alter the native structure and functionality of the MLL1 protein.
In summary, this research provides valuable insights into the multifarious implications of nsSNPs within the MLL1 gene, paving the way for a deeper understanding of their role in leukemia and offering a foundation for future studies and therapeutic developments. The identification of potential cancer driver mutations in the MLL1 protein underscore the necessity for further investigations into their mechanistic contributions to disease and their potential as targets for novel therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, H.A. and H.Y.; methodology, H.A.; software, H.A.,O.S., M.K., R.A. and G.A validation, O.S., M.K., R.A. and G.A.; formal analysis, H.A.,O.S., M.K., R.A. and G.A.; investigation, H.A.,O.S., M.K., R.A. and G.A.; resources, H.A.; data curation, H.A.; writing—original draft preparation, H.A.; writing—review and editing, H.A.; visualization, H.Y.; supervision, H.A and H.Y .; project administration, H.A.; funding acquisition, H.A. and H.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Winters AC, Bernt KM. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Frontiers in pediatrics. 2017;5:4. [CrossRef]
- Castiglioni S, Di Fede E, Bernardelli C, Lettieri A, Parodi C, Grazioli P, et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes. 2022 Mar;13(3). [CrossRef]
- Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992 Nov;71(4):691–700. [CrossRef]
- Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome biology. 2005;6(8):227. [CrossRef]
- Bochyńska A, Lüscher-Firzlaff J, Lüscher B. Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin. Cells. 2018 Mar;7(3). [CrossRef]
- Górecki M, Kozioł I, Kopystecka A, Budzyńska J, Zawitkowska J, Lejman M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines. 2023 Mar;11(3). [CrossRef]
- Collins CT, Hess JL. Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Current opinion in hematology. 2016 Jul;23(4):354–61.
- Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annual review of pathology. 2012;7:283–301. [CrossRef]
- Robert F, Pelletier J. Exploring the Impact of Single-Nucleotide Polymorphisms on Translation. Frontiers in genetics. 2018;9:507. [CrossRef]
- Kumar A, Rajendran V, Sethumadhavan R, Shukla P, Tiwari S, Purohit R. Computational SNP analysis: current approaches and future prospects. Cell biochemistry and biophysics. 2014 Mar;68(2):233–9. [CrossRef]
- Allemailem KS, Almatroudi A, Alrumaihi F, Makki Almansour N, Aldakheel FM, Rather RA, et al. Single nucleotide polymorphisms (SNPs) in prostate cancer: its implications in diagnostics and therapeutics. American journal of translational research. 2021;13(4):3868–89.
- Hunt SE, McLaren W, Gil L, Thormann A, Schuilenburg H, Sheppard D, et al. Ensembl variation resources. Database [Internet]. 2018 Jan 1;2018:bay119. Available from: https://doi.org/10.1093/database/bay119. [CrossRef]
- Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Research [Internet]. 2001;29(1):308–11. Available from: https://doi.org/10.1093/nar/29.1.308. [CrossRef]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, et al. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLOS Computational Biology [Internet]. 2014;10(1):1–11. Available from: https://doi.org/10.1371/journal.pcbi.1003440. [CrossRef]
- Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012 Jul;40(Web Server issue):W452-7. [CrossRef]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic acids research. 2007;35(11):3823–35. [CrossRef]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics. 2013 Jan;Chapter 7:Unit7.20. [CrossRef]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic acids research. 2002 Sep;30(17):3894–900.
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics (Oxford, England). 2006 Nov;22(22):2729–34. [CrossRef]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic acids research. 2001 Jan;29(1):37–40.
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic acids research. 2016 Jul;44(W1):W344-50. [CrossRef]
- Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic acids research. 2005 Jul;33(Web Server issue):W306-10. [CrossRef]
- Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins: Structure, Function, and Bioinformatics [Internet]. 2006;62(4):1125–32. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/prot.20810. [CrossRef]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research. 2019 Jan;47(D1):D607–13.
- Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome research. 2012 Sep;22(9):1790–7. [CrossRef]
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel K, Lin G, Nam H-J, et al. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. 2017.
- Rogers MF, Gaunt TR, Campbell C. CScape-somatic: distinguishing driver and passenger point mutations in the cancer genome. Bioinformatics (Oxford, England). 2020 Jun;36(12):3637–44.
- 28. Rogers MF, Shihab HA, Gaunt TR, Campbell C. CScape: a tool for predicting oncogenic single-point mutations in the cancer genome. Scientific Reports [Internet]. 2017;7(1):11597. Available from: https://doi.org/10.1038/s41598-017-11746-4. [CrossRef]
- George Priya Doss C, Rajasekaran R, Sethumadhavan R. Computational identification and structural analysis of deleterious functional SNPs in MLL gene causing acute leukemia. Interdisciplinary sciences, computational life sciences. 2010 Sep;2(3):247–55. [CrossRef]
- Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochemical Society transactions. 2013 Jun;41(3):727–40. [CrossRef]
- Allen MD, Grummitt CG, Hilcenko C, Min SY, Tonkin LM, Johnson CM, et al. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. The EMBO journal. 2006 Oct;25(19):4503–12.
- Muntean AG, Giannola D, Udager AM, Hess JL. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood. 2008 Dec;112(12):4690–3. [CrossRef]
- Ali M, Hom RA, Blakeslee W, Ikenouye L, Kutateladze TG. Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochimica et biophysica acta. 2014 Feb;1843(2):366–71. [CrossRef]
- Josling GA, Selvarajah SA, Petter M, Duffy MF. The role of bromodomain proteins in regulating gene expression. Genes. 2012 May;3(2):320–43. [CrossRef]
- Filippakopoulos P, Knapp S. Chapter 10 - Bromodomains as Anticancer Targets. In: Egger G, Arimondo P, editors. Drug Discovery in Cancer Epigenetics [Internet]. Boston: Academic Press; 2016. p. 239–71. Available from: https://www.sciencedirect.com/science/article/pii/B9780128022085000102.
- Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Experimental & Molecular Medicine [Internet]. 2017;49(4):e324–e324. Available from: https://doi.org/10.1038/emm.2017.11. [CrossRef]
- Weirich S, Kudithipudi S, Jeltsch A. Somatic cancer mutations in the MLL1 histone methyltransferase modulate its enzymatic activity and dependence on the WDR5/RBBP5/ASH2L complex. Molecular oncology. 2017 Apr;11(4):373–87.
- Zhang Z-L, Yu P-F, Ling Z-Q. The role of KMT2 gene in human tumors. Histology and histopathology. 2022 Apr;37(4):323–34. [CrossRef]
- Malleshappa Gowder S, Chatterjee J, Chaudhuri T, Paul K. Prediction and analysis of surface hydrophobic residues in tertiary structure of proteins. TheScientificWorldJournal. 2014;2014:971258.
- Singh SM, Kongari N, Cabello-Villegas J, Mallela KMG. Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-beta aggregates. Proceedings of the National Academy of Sciences of the United States of America. 2010 Aug;107(34):15069–74.
- Bross P, Corydon TJ, Andresen BS, Jørgensen MM, Bolund L, Gregersen N. Protein misfolding and degradation in genetic diseases. Human mutation. 1999;14(3):186–98.
- Yue P, Moult J. Identification and analysis of deleterious human SNPs. Journal of molecular biology. 2006 Mar;356(5):1263–74. [CrossRef]
- Nagi AD, Regan L. An inverse correlation between loop length and stability in a four-helix-bundle protein. Folding & design. 1997;2(1):67–75. [CrossRef]
- Tastan O, Klein-Seetharaman J, Meirovitch H. The effect of loops on the structural organization of alpha-helical membrane proteins. Biophysical journal. 2009 Mar;96(6):2299–312. [CrossRef]
- Kumar S, Warrell J, Li S, McGillivray PD, Meyerson W, Salichos L, et al. Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences. Cell. 2020 Mar;180(5):915-927.e16.
- Stauber, R. H., Hahlbrock, A., Knauer, S. K. & Wünsch, D. Cleaving for growth: threonine aspartase 1--a protease relevant for development and disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 30, 1012–1022 (2016).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



