Submitted:
05 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
Apigenin (API) possesses excellent antitumor properties, but its limited water solubility and low bioavailability restrict its therapeutic impact. Thus, a suitable delivery systems is needed to overcome these limitations and improve the therapeutic efficiency. Poly (lactic-co-glycolic acid) (PLGA) is a copolymer extensively utilized in drug delivery. Hyaluronic acid (HA) is a major extracellular matrix and can specifically bind with CD44 on colon cancer cells. Chitosan (CS) can serve as an intermediate binding with HA and PLGA. Herein, we aimed to perpare receptor-selective HA-coated PLGA nanoparticles (NPs) for colon cancer with high expression of CD44. Firstly, API was encapsulated in PLGA to obtain PLGA-API-NPs, which were then sequentially combined with CS and HA to form HA-coated PLGA NPs. HA coated PLGA NPs had a stronger sustained-release capability. Cellular uptake of HA coated PLGA NPs was enhanced in HT-29 cells with high expression of CD44. HA-PLGA-DiR-NPs can target specificity towards the HT-29 ectopic tumor model. Overall, HA coated PLGA NPs was an effectively drug deliver platform for API in treatment of colon cancer with high expression of CD44.

Keywords:
API
; PLGA nanoparticles
; HA
; CD44
; colon cancer
; targeted delivery
1. Introduction
Apigenin (API) is a natural flavone abundant in a range of foods and medicinal herbs, such as oranges, celery, and chamomile [1,2]. Like other flavonoids, API possesses antioxidant, anti-inflammatory and anti-cancer properties, among which, the anti-cancer properties of API have attracted particular attention [3,4]. Studies has revealed that API can inhibit the growth and proliferation of diverse cancer cell lines, ultimatly impeding tumorous growths [4,5]. However, although API holds promising therapeutic potential, its limited water solubility and low bioavailability restrict its ability to effectively reach target tissues, reducing its therapeutic impact [6]. Therefore, there is an urgent need for developing suitable delivery systems to enhance API's solubility and bioavailability, aiming to improve the therapeutic outcomes.
Poly (lactic-co-glycolic acid) (PLGA) is a copolymer extensively studied and utilized in the biomedical field due to its perfect biocompatibility and biodegradability [7]. PLGA nanoparticles are submicron colloids that can encapsulate various therapeutic agents, from small molecules to larger biomolecules like proteins or RNA [8,9]. Compared with other drug delivery systems, PLGA nanoparticles offer several advantages, including perfect biodegradability and biocompatibility, adjustable degradation rates, and the excellent capacity to encapsulate a variety of drugs [10,11]. Thus, PLGA nanoparticles have emerged as a highly popular and widely adopted technology in the field of drug delivery.
Hyaluronic acid (HA) is a major extracellular matrix, characterized as a non-sulfated, linear glycosaminoglycan composed of repeating disaccharide moieties of d-glucuronic acid and N-acetyl-D-glucosamine[12]. Previous studies have shown that HA can specifically bind with the CD44 receptors[13,14], which is notably overexpressed in colon cancer cells and promotes progression and metastasis[15]. The interaction between HA and CD44 offers possibilities for targeted cancer therapy in colon cancer. Zhu C et al. engineered smart nanomicelles by conjugating HA totocopherol succinic acid via disulfide bonds, facilitating precision targeting of paclitaxel delivery to the CD44 receptors. This mechanism markedly inhibited proliferation and metastatic tendencies of colon cancer cells in situ, enhancing therapeutic efficacy while reducing side effects [16,17]. To improve the stability of HA and CD44 binding, chitosan (CS) derived from the crustacean exoskeleton was introduced, which can bind HA and PLGA simultaneously by electrostatic adsorption as an intermediate substance[18].
Herein, we aimed to perpare a receptor-selective drug delivery system (HA-coated PLGA-API-NPs) for colon cancer with high expression of CD44. Dynamic laser scanner (DLS) was used for the characterizion of all PLGA NPs. A drug loading and release assay was employed to evaluate the drug-loading capacity and release properties of HA-coated PLGA-API-NPs. Cell uptake experiments and small animal in vivo imaging techniques were employed to investigate the targeting effect of HA-coated PLGA-API-NPs in vitro and in vivo. The Results revealed that HA-coated PLGA-API-NPs were uniform in size with good dispersibility, stability, and had sustained-release capability than PLGA-API-NPs. Compared with PLGA-API-NPs, HA-coated PLGA-API-NPs showed enhanced targeting capability to HT-29 ectopic tumor model in nude mice, indicating that HA-coated PLGA-API-NPs offered significant promise for the targeted and effective treatment of colon cancer with high expression of CD44.
2. Materials and methods
2.1. Materials
Hyaluronic acid (Molecular weight 50~80 kDa) was from BASF Biotechnology Co,. Ltd. (Shandong, China). PLGA (100 kDa) was from Dai Gang Biological Technology Co., Ltd. (Shandong, China). API was from Yuquan Biological Technology Co., Ltd. (Shanxi, China). Tracking probes including 3,3-dioctadecyloxacarbocyanine perchlorate (DiO), DAPI and DiR were purchased from Beijing Fanbo Science & Technology Co., Ltd (Beijing, China). Methanol (CR) was from Thermo Fisher Technology Co., Ltd (Shanghai, China).
2.2. Preparation and Characterization of PLGA Nanoparticles
According to the prescription screening and previous publication[19], API (6 mg) and PLGA (60 mg) were accurately weighed and dissolved into a mixture of ethanol (3 mL) and dichloromethane (4.5 mL) as the oil phase. The F68 (180 mg), chitosan (4.5 mg) and Tween (0.225 g) were added to 45 mL acetic acid buffer solution as the aqueous phase. All lipid materials were completely dissolved using a magnetic agitator to form clear and transparent solution. The oil and aqueous phases were mixed and stirred for 10 min to form the emulsion. Subsequently, this emulsion was added to 100 mL of ultra-pure water and stirred for 12 h using a high-pressure homogenizer. CS- PLGA-API-NPs were then obtained by centrifuging the mixture for 45 min at 20,000 × g. These obtained nanoparticles were immediately resuspended in 30 mL of acetic acid buffer solution (4 ℃) containing HA (0.25 mg/mL). The mixture was then stirring for 1 h and filtered through a 0.45 μm polycarbonate ester membrane to obtain the HA-coated PLGA-API-NPs. Fluorescence labeling was achieved by substituting API with DiO or DiR to prepare DiO- or DiR-labeled HA-PLGA-NPs. The size distribution and Zeta potential of PLGA-API-NPs, CS-PLGA-API-NPs and HA-coated PLGA-API-NPs were measured by Dynamic laser scanner at room temperature (Malven Zetasizer Nano ZS90, Britain).
2.3. Drug Loading Amount
The drug loading (DL) amount of HA-coated PLGA-API-NPs was determined by High-performance liquid chromatography (HPLC) after ultrafiltration centrifugation. Briefly, after centrifuging the suspensions at 4 °C and 20,000 × g for 1 h to remove unencapsulated drugs, the filtrate (300 μL) and PLGA-API-NPs solution were added to a 10 mL brown volumetric flask. They were then dispersed in methanol by ultrasonic dispersion for 5 min, calibrated with a mobile phase (Acetonitrile/water: 41/59), followed by ultrasonic dispersion for 15 min, and finally filtered through a 0.45 μm polycarbonate ester membrane. The API concentrations in subsequent filtrate were determined by HPLC. In detail, API standard solution (400 μg/mL in methanol) was prepared by dissolving 10 mg API in 25 mL methanol , and stored at 4 °C for later use. API mixture concentration after demulsification with methanol was measured by HPLC at 210 nm using an Agilent 1100 system with a UV detector and an auto-sampler device. Separation was performed on an analytical column (Welchrom-C18, 5 μm,4.6 × 150 mm) with a guard column at room temperature. The mobile phase was 41 % acetonitrile in water solution (v/v) at a flow rate of 1 mL /min. Calibration curves were constructed and the volume injected into the HPLC system was 20 μL. There were linear relationships from 1 to 40 μg/mL (Y = 76.302X-2.9938,R2 =0.9997). The encapsulation efficiency (EE) and DL were calculated according to the following formula:
Notes: Wt is the total API in PLGA-API-NPs; Wf is the free API in PLGA-API-NPs; Wn is PLGA-API-NPs weight .
2.4. API Release from HA-coated PLGA-API-NPs
1 mL of HA-coated PLGA-API-NPs, PLGA-API-NPs, and free API solutions (matched same API concentrations) were respectively added to three treated dialysis bags, which were clamped and placed in the different sealed beakers with 10 mL of release media (ratio of PBS (pH 7.4) to ethanol by volume: 4/1). The release media was mixed by shaking incubator (37 °C, 100 rpm). At each predetermined time point, 1 mL release medium was removed, filtered by the polycarbonate ester membrane (cutoff: 0.45 μm) and replenished to the container with the same amount of equal temperature medium . API concentration was measured by HPLC, and the cumulative release percentage were calculated according to the following formula (II):
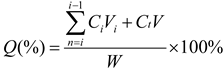
Notes: Ci and Ct are API concentrations before and at time t; Vi is the Sampling volume (mL); V is the total volume of released medium (mL); W is the drug loading of PLGA-NPs (μg).
2.5. In Vitro Cytotoxicity Studies
The anticancer effects of HA-coated PLGA-API-NPs were assessed by MTT assay according to the manufacturer's protocol. HRT-18 and HT-29 cells were treated with free API, PLGA-API-NPs, CS-coated PLGA-API-NPs, HA-coated PLGA-API-NPs or blank HA-coated PLGA-NPs (as a control) for 24 h. The data represented the percentage of surviving cells, calculated as the mean values from 6 replicates.
2.6. In Vitro Cellular Uptake Assay
Cellular uptake of HA-coated PLGA-API-NPs was assessed by fluorescence microscope and flow cytometry using Dio-labeled HA-PLGA-DiO-NPs replaced the drug in vitro. Briefly, HRT-18 and HT-29 cells were seeded in 6-well plates (2 × 106 cells/well) and incubated overnight, then treated with 2 mL of fresh cell medium containing different formulations DiO-labeled PLGA-NPs or HA-PLGA-NPs (2 μg/mL DiO). To quantify CD44-mediated cellular uptake, HRT-18 and HT-29 cells were incubated with DiO-labeled HA-PLGA-NPs (2 μg/mL DiO) for 4 h .The cells were pre-treated with HA (5 mg/mL) for 0.5 h before incubated with DiO-labeled HA-PLGA-NPs. Nnuclei were stained with DAPI and observed by fluorescence microscope. Cells samples were collected, washed and resuspended in PBS by flow cytometry analysis.
2.7. Establishment of Animal Solid Tumor Model
Animal experiments were approved by the Institution Animal Care and Use Committee. Healthy female BALB/C nude mice (4 weeks old) were supplied from Weitong Lihua Laboratory Animal Technology Co., Ltd. HRT-18 or HT-29 cells (1.5 × 107 in 0.1 mL PBS) were injected subcutaneously into mice’s left anterior armpit. The mice were subsequently monitored daily for general health and tumor progression. Once palpable tumors developed, including size and volume, were documented regularly.
2.8. In Vivo Near-Infrared Fluorescence (NIRF) Imaging.
The mice-bearing HRT-18 and HT-29 tumor (reached ∼150 mm3) were divided into four groups randomly (n = 6), and injected through tail vein with DiR-labeled PLGA-NPs, DiR-labeled HA-PLGA-NPs, respectively. At each predetermined time point, anesthetized mice were imaged by in vivo fluorescence imaging. At the end of the study, those mice were dissected, main organs and tumors were collected/excised for fluorescence intensity quantified.
2.9. Statistical Analysis
All dataes were presented as the mean ± standard deviation (SD). Following the variance homogeneity testing, the differences between the groups were analyzed by one-way ANOVA. A P - value < 0.05 was deemed to indicate statistical significance.
3. Results
3.1. Preparation and Characterization of HA-Coated PLGA-API-NPs
As shown in Figure 1A, the average sizes of PLGA-API-NPs, CS-PLGA-API-NPs and HA-coated PLGA-API-NPs were 210.3 nm, 234.3 nm, and 247.2 nm, respectively. The particle size of CS-PLGA-API-NPs were enhanced by about 13 nm after HA coating. PLGA-API-NPs, CS-PLGA-API-NPs, and HA-coated PLGA-API-NPs had zeta potentials of -26.3 mV, +27.9 mV, and -32.7 mV (Figure 1B), indicating CS coating of PLGA nanoparticles followed by HA. The EE and DL of API in the HA-coated PLGA-API-NPs were (90.56 ± 0.57) % and (3.19 ± 0.03) %, respectively. The stability results showed that the nanoparticle suspensions remained stable with no significant changes in particle size and drug loading when 5 days at 4 °C and 25 °C (Table 1).
3.2. Cumulative Release Rate In Vitro
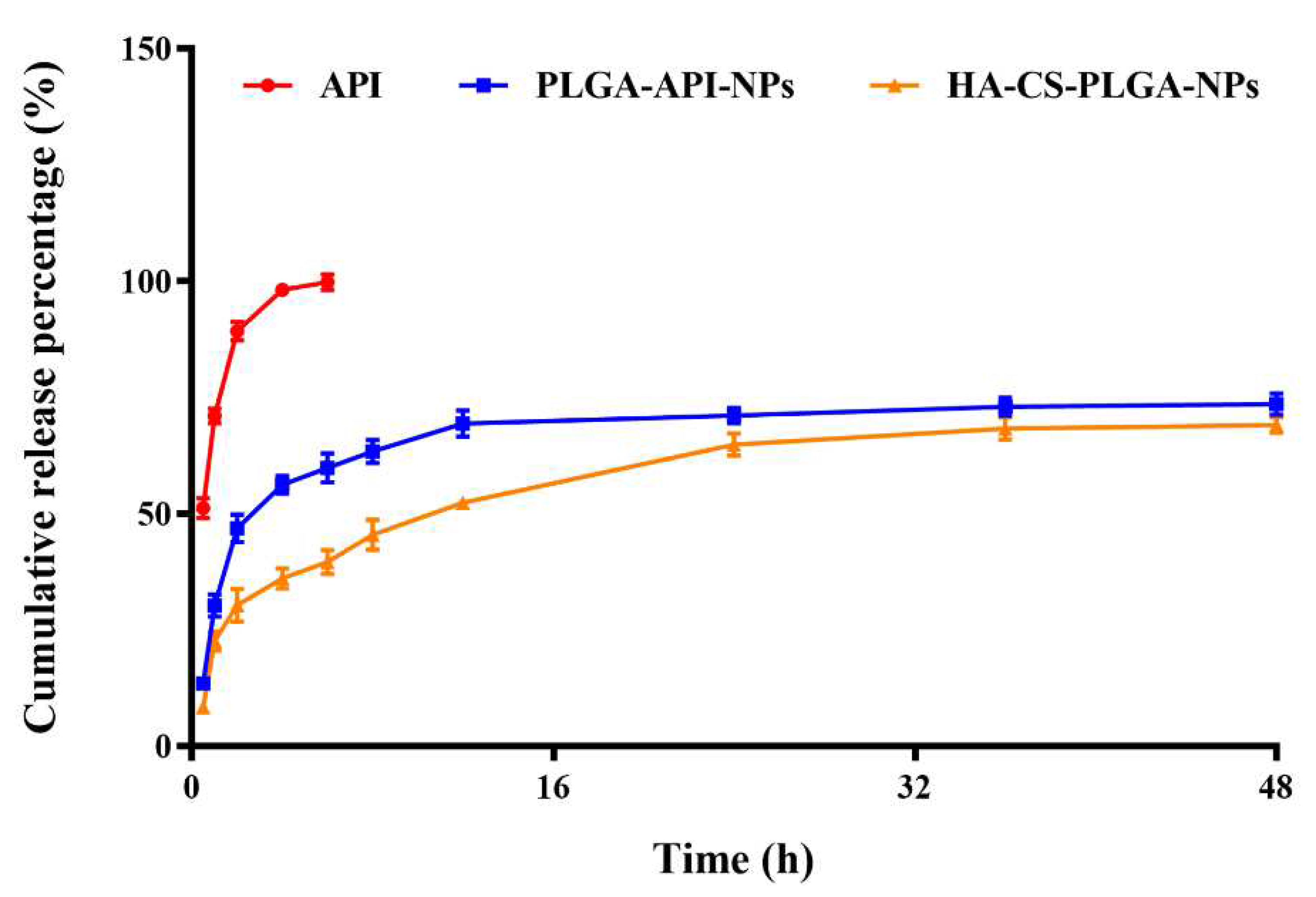
The in vitro cumulative release rates of CS-PLGA-API-NPs and HA-coated PLGA-API-NPs were calculated according to the formula (II) mentioned in the method. The uncoated PLGA-API-NPs released over 70% within 24 hours, whereas HA-coated PLGA-API-NPs required 48 h to release 68% of API. The release curve results indicated that uncoated PLGA-API-NPs exhibited rapid release, while HA-coated PLGA-API-NPs displayed sustained and controlled release over an extended period. This suggested that the HA coating acted as a barrier, effectively modulating and controlling the release of API. Such controlled release may offer therapeutic advantages when prolonged drug release is desired.
Figure 2.
The release of API from API, PLGA-API-NPs, and HA-coated PLGA-API-NPs in release media (pH 7.4 phosphate buffer contain 20 % ethanol). The data are presented as means ± SD (n = 3).
Figure 2.
The release of API from API, PLGA-API-NPs, and HA-coated PLGA-API-NPs in release media (pH 7.4 phosphate buffer contain 20 % ethanol). The data are presented as means ± SD (n = 3).
3.3. In Vitro Cellular Uptake Assay.
The expression of the CD44 receptor on HRT-18 and HT-29 cells were examined using immunofluorescence staining (Figure 3A). HT-29 cells displayed a significantly higher CD44 receptor expression compared to HRT-18 cells (p < 0.01). Thus, in subsequent experiments, HT-29 cells and HRT-18 cells were used as high and low CD44 expression models, respectively. To assess CD44 receptors mediated cellular internalization, those cells were incubated with HA-coated PLGA-API-NPs for 4 h. As shown in Figure 3B and C, HT-29 cells uptake of DiO was significantly higher than in HRT-18 cells. The preincubation of HT-29 cells with free HA significantly decreased DiO cellular uptake at 4 h incubation, indicating CD44-mediated cellular internalization of HA-coated PLGA-API-NPs into HT-29 cells. In contrast, the pretreatment of HRT-18 cells with free HA caused no a notable change in DiO uptake, as CD44 receptors-mediated internalization were not the major pathway.
3.4. In Vitro Cytotoxicity of HA-Coated PLGA-API-NPs.
The capability of a drug to induce cytotoxicity in cancer cells frequently serves as a benchmark for its anticancer potential, with greater cytotoxicity conferring stronger anticancer activity. The cytotoxicity of HA-coated PLGA-API-NPs, CS-PLGA-API-NPs, PLGA-API-NPs and free API were assessed by MTT assay after 24 h incubation, revealing dose-dependent responses (Figure 4). Compared with PLGA-API-NPs, HA-coated PLGA-API-NPs showed greater cytotoxic effect on HT-29 cells. Notably, HA-coated PLGA-API-NPs induced significantly higher killing of HT-29 cells compared with HRT-18 cells, which should be attributed to the efficient CD44 receptors-mediated API internalization into HT-29 cells.
3.5. In Vivo NIRF Imaging
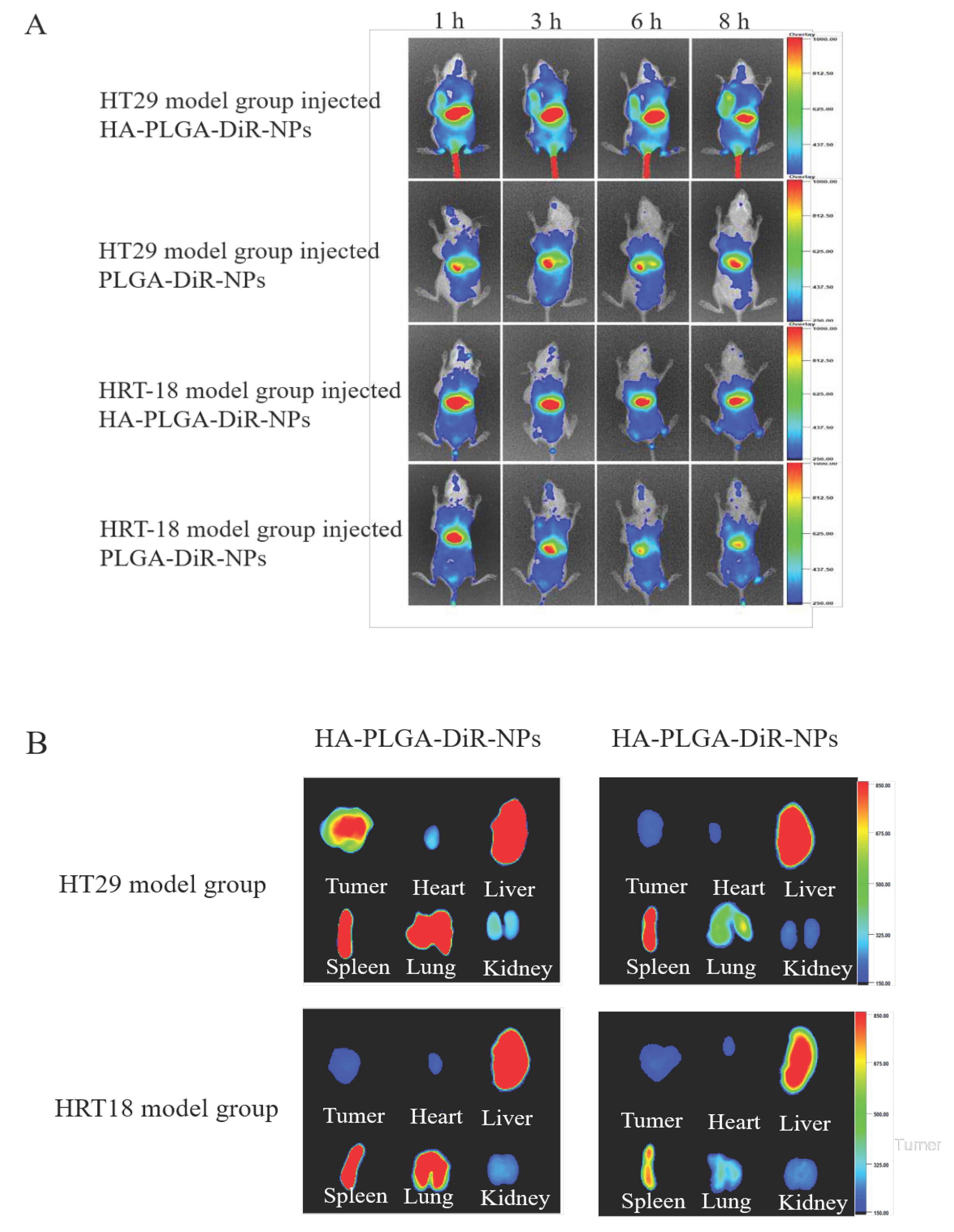
In order to predict the antitumor efficacy of nanoparticles, the biodistribution studies of HA-coated PLGA-DiR-NPs were conducted to investigate their in vivo targeted behavior. PLGA-DiR-NPs and HA-coated PLGA-DiR-NPs were injected intravenously into the two tumor-bearing mice, respectively, and then monitored by near-infrared optical imaging. Figure 6A showed the fluorescent images of mice at 1, 3, 6, and 8 h postinjection. Compared with PLGA-DiR-NPs mice group, stronger fluorescence signals was observed at the HT-29 tumor sites of HA-coated PLGA-DiR-NPs group throughout the experiment, while both formulations showed less distribution in HRT-18 model mice. These results demonstrated greater accumulation of HA-coated PLGA-DiR-NPs at the HT-29 tumor site, due to HA-mediated active targeting effect, since HRT-18 cells expressed less CD44 receptors and DIR. The fluorescence intensity at tumor sites of both groups peaked at 8 h postinjection then decreased gradually, indicating this was the optimal laser irradiation timepoint. However, tumor fluorescence intensity was significantly stronger for HA-coated PLGA-DiR-NPs group than PLGA-DiR-NPs group at 8 h postinjection, indicating satisfactory targeting ability of HA-coated PLGA-DiR-NPs. The excised organs and tumors were measured for fluorescence intensity at 8 h postinjection (Figure 6B). Tumor of HA-coated PLGA-DiR-NPs mice group showed significantly stronger fluorescence than that of PLGA-DiR-NPs group. Notably, kidneys and hearts of both groups displayed only weak fluorescence, suggesting minimal side effects. These results demonstrated that HA-coated PLGA-DiR-NPs could efficiently accumulate into the tumor site via folate-mediated active targeting effect, maximizing antitumor effects and minimizing side effects.
Figure 5.
CD44 mediated tumor-targeting delivery of PLGA-NPs coated HA. (A) In vivo fluorescent small animal imaging in vivo of two model rats injected with. (B) Visceral fluorescence images of two model rats after injection of different drugs. The data are presented as means ± SD (n = 3).
Figure 5.
CD44 mediated tumor-targeting delivery of PLGA-NPs coated HA. (A) In vivo fluorescent small animal imaging in vivo of two model rats injected with. (B) Visceral fluorescence images of two model rats after injection of different drugs. The data are presented as means ± SD (n = 3).
4. Discussion
Despite promising therapeutic properties, the poor water solubility and swift metabolic degradation of API hamper its delivery and efficacy[20]. To overcome these limitations, numerous drug delivery systems have been investigated to enhance the therapeutic efficiency of API. Notably, PLGA NPs are an effective carrier due to biocompatibility and biodegradability, small size and particulate structure, and lack of toxic metabolites[9]. However, Unmodified PLGA NPs are primarily taken up by organs with significant blood flow, limiting absorption by colon cancer cells[21]. To enhance the uptake by colon cancer cells, particularly those expressing high levels of CD44, a receptor-selective drug delivery system (HA-coated PLGA-API-NPs) were developed in this study, which specifically transports the hydrophobic antitumor drug API to colon cancer cells overexpressing CD44.
The are designed to enhance or promote its entry into the target tissues by ligands-receptor binding , while safeguarding the active drug payload against non-specific delivery[22,23]. Recent investigation on targeted NPs also have demonstrated that targeting ligands primarily enhance selective NPs uptake into tumor cells, minimizing accumulation in normal tissues[16]. The targeting ligands provides specific interactions between NPs and the surface of target cells, playing a pivotal role in nanoparticulate delivery. These targeting ligands facilitate the binding of NPs to cell surface receptors and their penetration into cells via receptor-mediated endocytosis[24,25]. In our studies, we functionalized PLGA NPs with CD44 receptor ligand HA to target the overexpressed CD44 on colon tumor cell membranes. As a linear polysaccharide of D-glucuronic acid and N-acetylglucosamine, HA plays a vital role in cell adhesion, growth, and migration[26]. HA internalization occurs via CD44 and other matrix receptors[27]. Thus, HA conjugated with the NPs system can serve as a valuable ligand for targeting overexpressed CD44 receptors. In this context, CS acted as a bridging agent, adhering to both HA and PLGA under electrostatic interactions, thereby enhancing the targeting ligand binding efficiency[28].
We designed HA coated PLGA NPs to target the hydrophobic anti-tumor drug API to CD44 overexpressing colon cancer cells. HA coated PLGA NPs were successfully prepared with a mean particle size of 254 ± 6.23 nm, an DL of 3.19 ± 0.03%, and an EE of 90.56 ± 0.57%, indicating good reproducibility. The stability and in vitro release of the nanoparticles were thoroughly evaluated in this study. It exhibited satisfactory stability over 5 days with no significant changes in particle size and drug loading. However, enhanced stability is needed for clinical applications. Lyophilization could further improve nanoparticles stability in future studies. The PLGA-API-NPs exhibited a rapid initial release rate due to surface-adsorbed API quickly detaching in medium. The PLGA with a lactic acid to glycolic acid ratio of 75:25 also resulted in relatively quickly degradation. Thus, most of the encapsulated API was released within 12 h. In contract, HA coated PLGA NPs exhibited slower initial release followed by sustained release after 12 h, attributed to the outer HA and CS coating delaying drug release[29]. Neither nanoparticle achieved a cumulative release rate of 80% within 48 hours, suggesting a fraction of API remains tightly entrapped in PLGA matrix for gradual release over time[30]. Future studies are needed to characterize the prolonged release profile and determine in vitro release kinetics.
MTT assay was utilized to assess cytotoxicity of HA coated PLGA NPs, and flow cytometry was used to quantitatively analyze cellular uptake. HA coated PLGA NPs exhibited strong targeting to CD44-overexpressing HT-29 cells with minimal cytotoxicity towards HRT-18 cells but higher for HT-29 cells. This suggested that HA modification significantly enhanced the nanoparticle's targeting selectivity for high CD44 expression. Flow cytometry results also showed the highest fluorescence intensity in HT-29 cells after HA-PLGA-DiO-NPs treatment, further corroborating specific CD44 receptors binding. Fluorescence imaging clearly demonstrated greater uptake of HA-PLGA-DiO-NPs in CD44-high HT-29 cells, confirming effective HA targeting colon cancer cells. However, the ligands modification can alter the nanoparticles-receptors interactions, influencing the in vitro and in vivo behaviors of the nanoparticulate system[31]. Consequently, further studies should investigate PLGA nanoparticles with varying HA molecular weights and density to fully elucidate effects on cytotoxicity and cellular uptake, providing a solid theoretical basis for targeted therapy development.
Small animal in vivo fluorescence imaging technique was employed to monitor nanoparticles distribution in tumor-bearing mice. The HA coated PLGA NPs exhibited significant targeting in high CD44 expressing mice models, while uncoated PLGA nanoparticles lacked specificity. PLGA-DiR-NPs fluorescence intensity peaked at 4 hours then declined, possibly attributed to the rapid initial DiR release followed by slower release and metabolic processes. However, the fluorescence intensity of HA-PLGA-DiR-NPs continuously increased, reaching its maximum at 8-hour. The delayed DiR release can be attributed to HA modification resulting in a more controlled release, consisting with the in vitro results. Furthermore, a significant accumulation of nanoparticles was observed in the liver potentially stems from hepatic uptake of larger nanoparticles[32]. Subsequent investigations in this project should focus on devising methods to decrease PLGA nanoparticles size without compromising drug-loading capacity significantly, which may minimize nanoparticle uptake and improve the accumulation of the drug at the liver tumor sites. Overall, data confirmed that HA coated PLGA NPs might be an excellent platform for targeted delivery API.
5. Conclusions
In this study, we developed HA-coated PLGA-API-NPs to improve API stability and enable targeted delivery specifically to colon cancer tumors with high CD44 expression. HA coated PLGA NPs were uniform in size, with good dispersibility and stability by particle size and zeta potential. The results of drug release assay confirmed that HA coated PLGA NPs had a stronger sustained-release capability than PLGA NPs after 12 h. Cellular uptake of HA coated PLGA NPs was enhanced in HT-29 cells as compared to HRT-18 cells due to their specificity towards the CD44 receptors on the membrane of HT-29 colon cancer cells. In vivo, compared with PLGA-DiR-NPs, HA-PLGA-DiR-NPs enhanced targeting specificity towards the HT-29 ectopic tumor model in nude mice. These results confirmed that HA coated PLGA NPs was an effectively drug deliver system for API, which elevated the therapeutic concentration of API at the tumor site, exhibited excellent specificity, and minimized concerns about off-target effects. This approach holds extensive potential for selectively targeting and effectively treating chemotherapy-resistant tumor cells.
Ethical Compliance
Research experiments conducted in this article with animals were approved by the Ethical Committee and responsible authorities of our research organization(s) following all guidelines, regulations, legal, and ethical standards as required for animals.
Acknowledgments
This work was supported by the “Harbin University of Commerce Youth Top Talent Program” (Grant No: 2020CX41).
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The Therapeutic Potential of Apigenin. Int J Mol Sci 2019, 20. [CrossRef]
- Majma Sanaye, P.; Mojaveri, M.R.; Ahmadian, R.; Sabet Jahromi, M.; Bahramsoltani, R. Apigenin and its dermatological applications: A comprehensive review. Phytochemistry 2022, 203, 113390. [CrossRef]
- Imran, M.; Aslam Gondal, T.; Atif, M.; Shahbaz, M.; Batool Qaisarani, T.; Hanif Mughal, M.; Salehi, B.; Martorell, M.; Sharifi-Rad, J. Apigenin as an anticancer agent. Phytother Res 2020, 34, 1812-1828. [CrossRef]
- Singh, D.; Gupta, M.; Sarwat, M.; Siddique, H.R. Apigenin in cancer prevention and therapy: A systematic review and meta-analysis of animal models. Crit Rev Oncol Hematol 2022, 176, 103751. [CrossRef]
- Yoon, J.H.; Kim, M.Y.; Cho, J.Y. Apigenin: A Therapeutic Agent for Treatment of Skin Inflammatory Diseases and Cancer. Int J Mol Sci 2023, 24. [CrossRef]
- Tang, D.; Chen, K.; Huang, L.; Li, J. Pharmacokinetic properties and drug interactions of apigenin, a natural flavone. Expert Opin Drug Metab Toxicol 2017, 13, 323-330. [CrossRef]
- Su, Y.; Zhang, B.; Sun, R.; Liu, W.; Zhu, Q.; Zhang, X.; Wang, R.; Chen, C. PLGA-based biodegradable microspheres in drug delivery: recent advances in research and application. Drug Deliv 2021, 28, 1397-1418. [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater Sci Eng C Mater Biol Appl 2018, 92, 1041-1060. [CrossRef]
- Rocha, C.V.; Gonçalves, V.; da Silva, M.C.; Bañobre-López, M.; Gallo, J. PLGA-Based Composites for Various Biomedical Applications. Int J Mol Sci 2022, 23. [CrossRef]
- Chereddy, K.K.; Payen, V.L.; Préat, V. PLGA: From a classic drug carrier to a novel therapeutic activity contributor. J Control Release 2018, 289, 10-13. [CrossRef]
- Shariati, A.; Chegini, Z.; Ghaznavi-Rad, E.; Zare, E.N.; Hosseini, S.M. PLGA-Based Nanoplatforms in Drug Delivery for Inhibition and Destruction of Microbial Biofilm. Front Cell Infect Microbiol 2022, 12, 926363. [CrossRef]
- Hoarau, A.; Polette, M.; Coraux, C. Lung Hyaluronasome: Involvement of Low Molecular Weight Ha (Lmw-Ha) in Innate Immunity. Biomolecules 2022, 12. [CrossRef]
- Krolikoski, M.; Monslow, J.; Puré, E. The CD44-HA axis and inflammation in atherosclerosis: A temporal perspective. Matrix Biol 2019, 78-79, 201-218. [CrossRef]
- Guo, Q.; Yang, C.; Gao, F. The state of CD44 activation in cancer progression and therapeutic targeting. Febs j 2022, 289, 7970-7986. [CrossRef]
- Siddiqui, L.; Hasan, N.; Mishra, P.K.; Gupta, N.; Singh, A.T.; Madaan, A.; Jaggi, M.; Saad, S.; Ekielski, A.; Iqbal, Z.; et al. CD44 mediated colon cancer targeting mutlifaceted lignin nanoparticles: Synthesis, in vitro characterization and in vivo efficacy studies. Int J Pharm 2023, 123270. [CrossRef]
- Babu, A.; Amreddy, N.; Muralidharan, R.; Pathuri, G.; Gali, H.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. Chemodrug delivery using integrin-targeted PLGA-Chitosan nanoparticle for lung cancer therapy. Sci Rep 2017, 7, 14674. [CrossRef]
- Zhu, C.; Zhang, H.; Li, W.; Luo, L.; Guo, X.; Wang, Z.; Kong, F.; Li, Q.; Yang, J.; Du, Y.; et al. Suppress orthotopic colon cancer and its metastasis through exact targeting and highly selective drug release by a smart nanomicelle. Biomaterials 2018, 161, 144-153. [CrossRef]
- Karabiyik Acar, O.; Kayitmazer, A.B.; Torun Kose, G. Hyaluronic Acid/Chitosan Coacervate-Based Scaffolds. Biomacromolecules 2018, 19, 1198-1211. [CrossRef]
- Kumar, S.; Lather, V.; Pandita, D. A facile green approach to prepare core-shell hybrid PLGA nanoparticles for resveratrol delivery. Int J Biol Macromol 2016, 84, 380-384. [CrossRef]
- Wang, M.; Firrman, J.; Liu, L.; Yam, K. A Review on Flavonoid Apigenin: Dietary Intake, ADME, Antimicrobial Effects, and Interactions with Human Gut Microbiota. Biomed Res Int 2019, 2019, 7010467. [CrossRef]
- Hashemi, M.; Shamshiri, A.; Saeedi, M.; Tayebi, L.; Yazdian-Robati, R. Aptamer-conjugated PLGA nanoparticles for delivery and imaging of cancer therapeutic drugs. Arch Biochem Biophys 2020, 691, 108485. [CrossRef]
- Graf, N.; Bielenberg, D.R.; Kolishetti, N.; Muus, C.; Banyard, J.; Farokhzad, O.C.; Lippard, S.J. α(V)β(3) integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano 2012, 6, 4530-4539.
- Nethi, S.K.; Bhatnagar, S.; Prabha, S. Synthetic Receptor-Based Targeting Strategies to Improve Tumor Drug Delivery. AAPS PharmSciTech 2021, 22, 93. [CrossRef]
- Farokhzad, O.C.; Karp, J.M.; Langer, R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv 2006, 3, 311-324. [CrossRef]
- Kumar Shukla, M.; Parihar, A.; Karthikeyan, C.; Kumar, D.; Khan, R. Multifunctional GQDs for receptor targeting, drug delivery, and bioimaging in pancreatic cancer. Nanoscale 2023, 15, 14698-14716. [CrossRef]
- Almalik, A.; Day, P.J.; Tirelli, N. HA-coated chitosan nanoparticles for CD44-mediated nucleic acid delivery. Macromol Biosci 2013, 13, 1671-1680.
- Maytin, E.V. Hyaluronan: More than just a wrinkle filler. Glycobiology 2016, 26, 553-559. [CrossRef]
- Guelfi, G.; Stefanetti, V.; Zampini, D.; Oommen, O.P.; Brecchia, G.; Dall'Aglio, C.; Arcelli, R.; Cochetti, G.; Boni, A.; Mearini, E. Gold nanoparticles approach to detect chondroitin sulphate and hyaluronic acid urothelial coating. Sci Rep 2017, 7, 10355. [CrossRef]
- Bayer, I.S. Hyaluronic Acid and Controlled Release: A Review. Molecules 2020, 25. [CrossRef]
- Singh, S.; Singha, P. Effect of Modifications in Poly (Lactide-co-Glycolide) (PLGA) on Drug Release and Degradation Characteristics: A Mini Review. Curr Drug Deliv 2021, 18, 1378-1390. [CrossRef]
- Ramezani, M.; Ebrahimian, M.; Hashemi, M. Current Strategies in the Modification of PLGA-based Gene Delivery System. Curr Med Chem 2017, 24, 728-739. [CrossRef]
- Park, J.K.; Utsumi, T.; Seo, Y.E.; Deng, Y.; Satoh, A.; Saltzman, W.M.; Iwakiri, Y. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine 2016, 12, 1365-1374. [CrossRef]
Figure 1.
The size distribution (A) and Zeta potential (B) of PLGA-API-NPs, CS-coated PLGA-API-NPs and HA-coated PLGA-API-NPs. The data are presented as means ± SD, n = 3.
Figure 1.
The size distribution (A) and Zeta potential (B) of PLGA-API-NPs, CS-coated PLGA-API-NPs and HA-coated PLGA-API-NPs. The data are presented as means ± SD, n = 3.
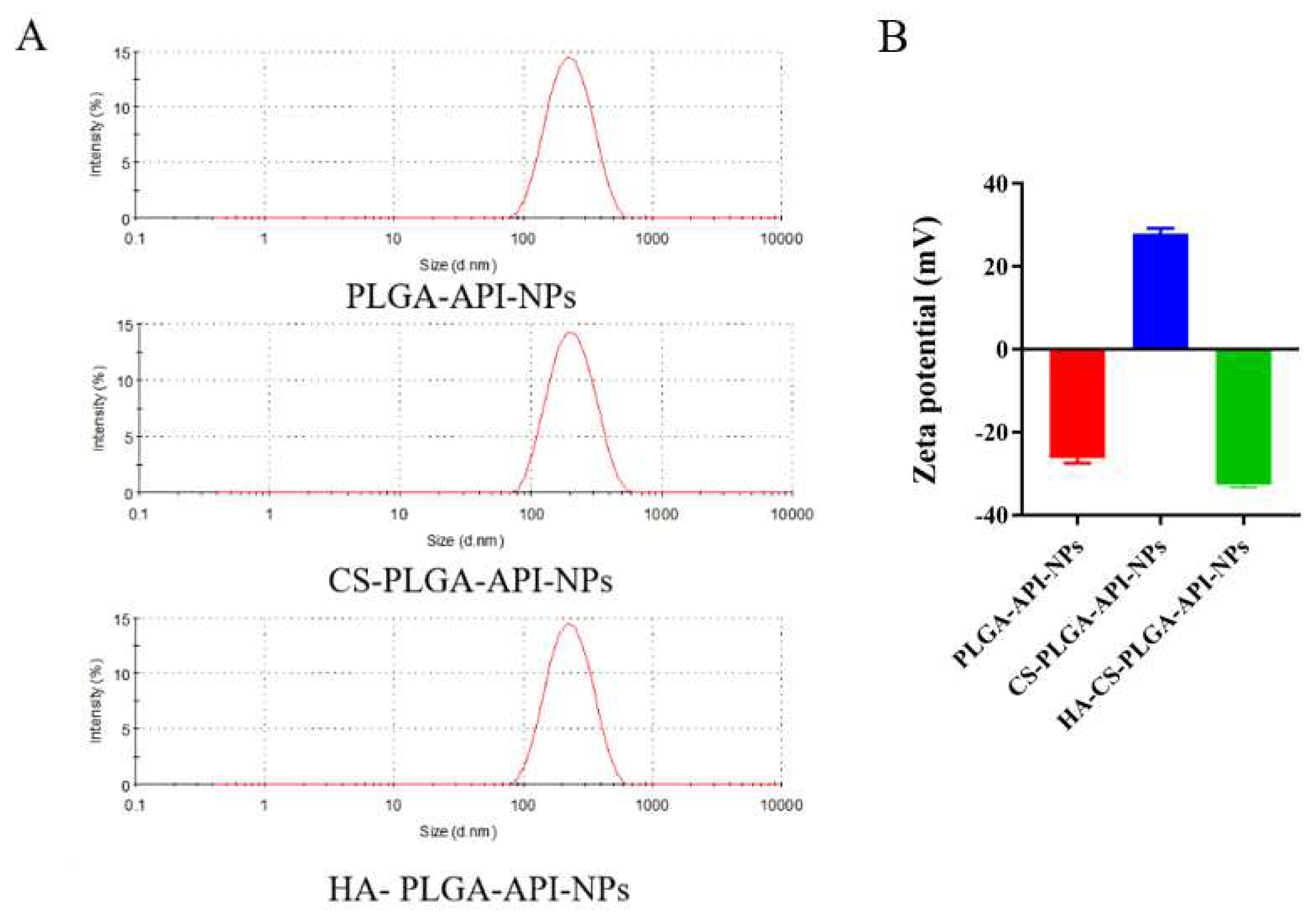
Figure 3.
Cell uptake. (A) Uptake of HA-PLGA-DiO-NPs by HT-29 and HRT-18 under different conditions. (B) Effects of different modified nanoparticles on the uptake of HT-29 cells. (C) Drug uptake in HT-29 and HRT-18 cells following the administration of HA-PLGA-DiO-NPs. The data are presented as means ± SD (n = 3), *P < 0.05, **P < 0.01.
Figure 3.
Cell uptake. (A) Uptake of HA-PLGA-DiO-NPs by HT-29 and HRT-18 under different conditions. (B) Effects of different modified nanoparticles on the uptake of HT-29 cells. (C) Drug uptake in HT-29 and HRT-18 cells following the administration of HA-PLGA-DiO-NPs. The data are presented as means ± SD (n = 3), *P < 0.05, **P < 0.01.
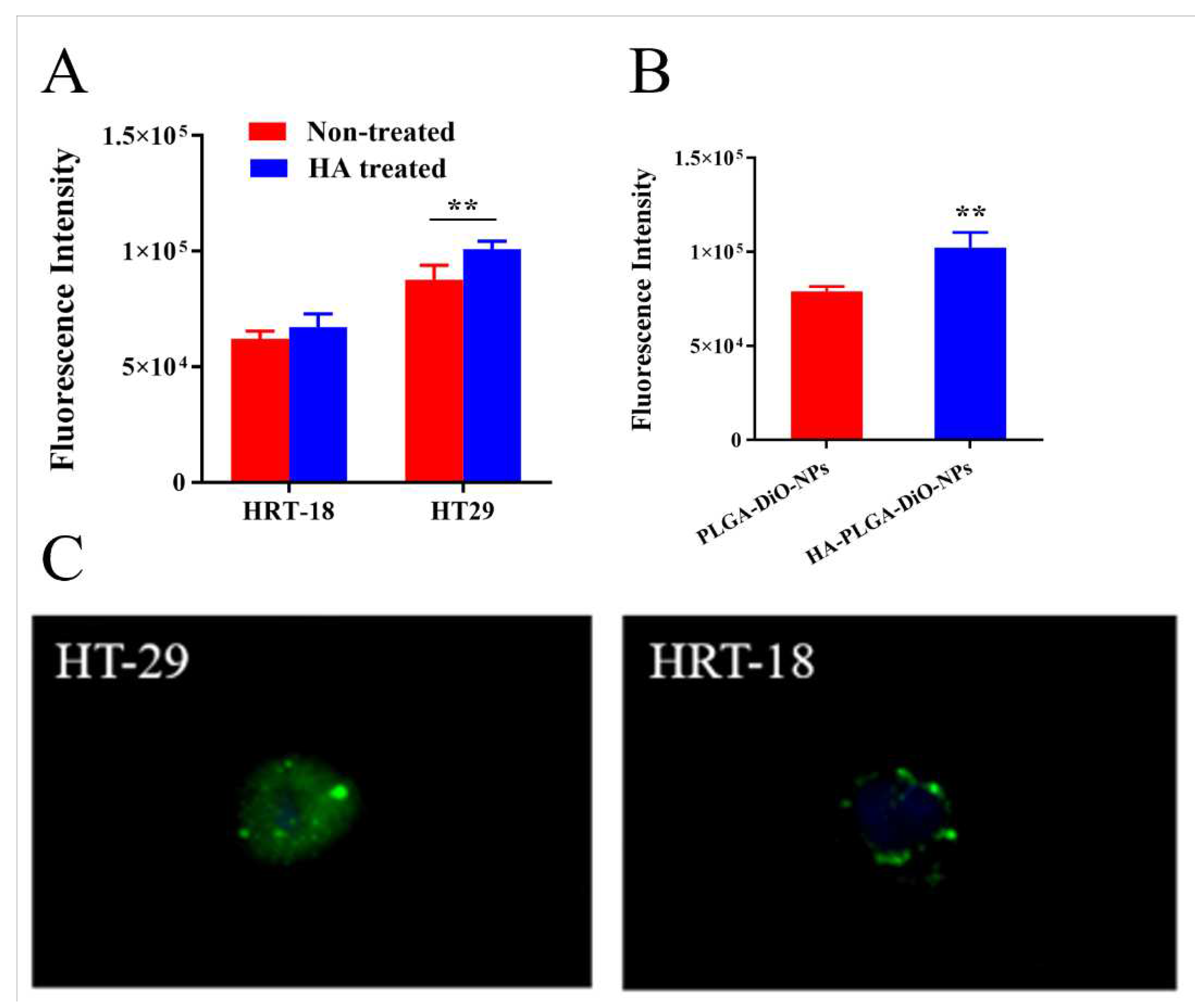
Figure 4.
Cell and cytotoxicity (n=6). (A) Cytotoxicity assay against HT-29 and HRT-18 cells treated with blank nanoparticles. (B) Cytotoxicity assay against HT-29, HRT-18 cells treated with HA-coated PLGA-API-NPs. (C) Cytotoxicity of free API and different modified nanoparticles on HT-29. The data are presented as means ± SD, n = 3, *P < 0.05, **P < 0.01.
Figure 4.
Cell and cytotoxicity (n=6). (A) Cytotoxicity assay against HT-29 and HRT-18 cells treated with blank nanoparticles. (B) Cytotoxicity assay against HT-29, HRT-18 cells treated with HA-coated PLGA-API-NPs. (C) Cytotoxicity of free API and different modified nanoparticles on HT-29. The data are presented as means ± SD, n = 3, *P < 0.05, **P < 0.01.
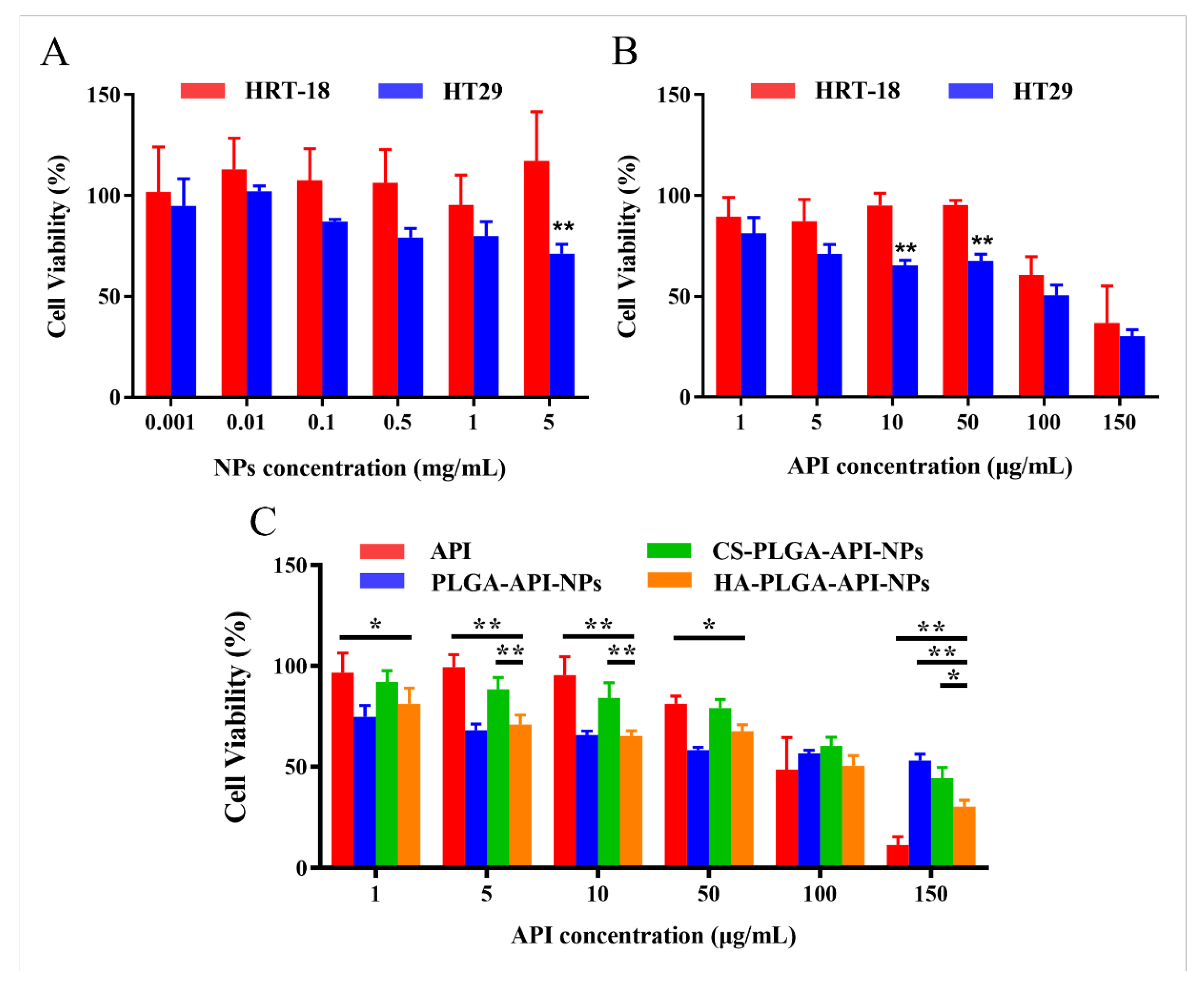

Table 1.
Stability of HA-coated PLGA-API-NPs at 4 °C and 25 °C (n=3).
| Temperature (°C) | Time (d) | Particle Size (nm) | Drug Loading (%) |
|---|---|---|---|
| 0 | 245±4.55 | 3.27±0.04 | |
| 4 | 5 | 255±3.25 | 3.06±0.05 |
| 10 | 249±5.67 | 2.89±0.05 | |
| 0 | 245±3.22 | 3.27±0.06 | |
| 25 | 5 | 258±2.87 | 2.99±0.06 |
| 10 | 267±4.55 | 2.66±0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



