
Submitted:
06 October 2023
Posted:
07 October 2023
You are already at the latest version
Abstract
Juvenile myelomonocytic leukemia (JMML) is a deadly pediatric leukemia driven by RAS pathway mutations, of which >35% are gain-of-function in PTPN11. Although DNA hypermethylation portends severe clinical phenotypes, the landscape of histone modifications and chromatin profiles in JMML patient cells have not been explored. Using global mass cytometry, Epigenetic Time of Flight (EpiTOF), we analyzed hematopoietic stem and progenitor cells (HSPCs) from five JMML patients with PTPN11 mutations. These data revealed statistically significant changes in histone methylation, phosphorylation and acetylation marks that were unique to JMML HSPCs when compared with healthy controls. Consistent with these data, assay for transposase-accessible chromatin with sequencing (ATAC-seq) analysis revealed significant alterations of chromatin profiles at loci encoding post translational modification enzymes, strongly suggesting their mis-regulated expression. Collectively, this study reveals histone modification pathways as an additional epigenetic abnormality in JMML patient HSPCs, thereby uncovering a new family of potential druggable targets for the treatment of JMML.
Keywords:
histone modifications
; pediatric leukemia
; chromatin acessibility
; epigenetics
; mass cytometry
; JMML
; novel targets
Key points
- This study reveals alterations in the histone landscape of JMML HSPCs, as putative drivers of leukemogenesis.
- Significantly reduced histone methylation and heterogeneity in histone acetylation, phosphorylation, and ubiquitination in JMML HSPCs was found.
- In correlation, altered accessibility at specific histone modifying enzymes and immunophenotypic markers collectively uncover potential novel therapeutic targets.
1. Introduction
Juvenile myelomonocytic leukemia (JMML) is a deadly pediatric hematologic malignancy characterized by excessive proliferation of monocytes/macrophages, elevated fetal hemoglobin, splenomegaly and hypersensitivity of leukemia cells to granulocyte-macrophage colony-stimulating factor (GM-CSF) (1–5). Hematopoietic stem cell transplantation is the only curative option, but post-transplant relapse occurs in ~50% of patients (6–9). Developing effective therapeutic strategies has been difficult due to the limited understanding of the disease development. Mutations in RAS pathway genes and DNA hypermethylation are major drivers of severe JMML phenotypes (10–13), the latter confirming the key role of epigenetic changes in disease progression and severity (14–16).
Epigenetic changes play an important role in numerous malignant transformations, altering gene expression to promote tumor growth (17–19). In addition to DNA methylation, dysregulation of histone post translational modifications (HPTM) has been extensively linked to reprogramming in numerous cancer subtypes including blood cancers such as acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL) and myelodysplastic syndrome (MDS) (20–22). Histones compress DNA forming chromatin to regulate eukaryotic gene expression. Histone PTMs are added to, or removed from, the N terminal tails of H2B, H3 and H4 histones by a specific set of enzymes; thus, HPTM pathways are key regulators of chromatin structure and transcriptional profiles (23). Within hematopoietic cells, the control of histone modifications is key in regulating cell fate (24–28). However, the landscape of HPTMs in JMML derived hematopoietic stem and progenitor cells (HSPCs), and their chromatin accessibility profile relative to healthy donor (HD) HSPCs, has not been investigated, and to date, there have been no published analyses of the HPTM landscape in JMML patients.
Here we explore the hypothesis that JMML HSPCs carry significant alterations in HPTMs. We have focused this pilot study on JMML patients with gain-of-function mutations in Protein Tyrosine Phosphatase Non-Receptor Type 11 (PTPN11; encodes RAS/MAPK signaling pathway protein, SHP-2), which are found in ~38% of all JMML patients and correlate with aggressive disease and hypermethylated DNA (11,12,29). In addition to JMML, numerous childhood leukemias and solid organ tumors carry gain-of-function PTPN11 mutations (30–33). Further, recent studies have suggested that JMML leukemic initiating cells (LIC) reside within the HSPC compartment (34,35), thus we have examined HPTMs within PTPN11 mutated JMML HSPCs. Finally, as a hallmark of JMML is splenomegaly, we characterize HSPCs isolated from JMML patient spleens.
We provide the first global comparison of HPTMs and chromatin accessibility in PTPN11 mutant JMML HSPCs and healthy controls. Using the highly sensitive mass cytometry epigenetic landscape profiling technology, Epigenetic Time Of Flight (EpiTOF) (36), our data revealed that splenic JMML HSPCs display significant heterogeneity in histone acetylation, phosphorylation and ubiquitination marks, along with significant reduction in several histone methylation marks when compared to control CD34+ cells from healthy donor umbilical cord blood.
In parallel studies, we used the assay for transposase-accessible chromatin with sequencing (ATAC-seq) to examine the genomic impact of the JMML histone landscape. These data revealed altered chromatin profiles at loci encoding post translational modification enzymes in JMML HSPCs, strongly suggesting their mis-regulated expression, and supporting the reduction in HPTMs identified by EpiTOF. This first such analysis of JMML HSPCs reveals histone modification pathways as an additional epigenetic abnormality in JMML patient HSPCs, thereby identifying a new family of potential druggable targets for the treatment of JMML.
2. Material and methods
2.1. Human samples:
Mononuclear cell samples from JMML patients (n=5) were purified from splenectomy preparations and cryopreserved at the biorepository of the EWOG-MDS, located at Freiburg University Medical Center, Germany. Healthy controls were umbilical cord blood (UCB) from Binns program at Stanford School of Medicine. All human samples were collected from donors with informed consent and compliance with relevant ethical regulations. DNA methylation categories were determined as described previously (13).
2.2. Processing cryopreserved samples and CD34 enrichment:
Cryopreserved JMML samples were thawed in pre-warmed RPM1 1640 media (Gibco) supplemented with 20% heat-inactivated FBS (ThermoFisher), 1% each of L-glutamine and Penicillin/streptomycin (Gibco). Two sequential DNase1 treatments during thawing allowed optimal recovery of live mononuclear cells (MNCs). MNCs were filtered through 70µm and CD34+ cells isolated using the CD34 ultrapure microbead kit (Miltenyi Biotec).
2.3. Epigenetic landscape profiling using cytometry by time-of-flight (EpiTOF):
EpiTOF was as previously described (36,39). Briefly, Lin-CD34+ cells from JMML and healthy donors were barcoded using the palladium Cell-ID™ 20-Plex Kit (Fluidigm). All lanthanide labeled immunophenotypic and intracellular antibodies (Figure 1A-Table) were prepared using MAXPAR antibody labeling kit (Fluidigm). Viability staining was 5 minutes at RT with 10uM cisplatin (ENZO Life Sciences), then CyTOF Buffer wash [PBS (ThermoFisher); 1% BSA (Sigma), 2mM EDTA (Fisher), 0.05% sodium azide]. Sample pool was sequentially incubated with lanthanide labeled immunophenotypic antibodies, permeabilized, and labeled with intracellular antibodies (36,39). Cells were washed and stained with 250 nM 191/193Ir DNA intercalator (Fluidigm) in PBS, fixed in 1.6% PFA then washed, filtered (35 μm strainer) and resuspended in ddH2O (ThermoFisher) containing four element calibration beads (Fluidigm) and analyzed on CyTOF2 (Fluidigm) in the Stanford Shared FACS Facility. Raw data were concatenated and normalized using calibration beads following manufacturer protocol.
2.4. EpiTOF data pre-processing and analysis and dimensionality reduction analysis:
EpiTOF raw data (fcs file) were preprocessed on FlowJo software (FlowJo, RRID:SCR_008520). Dead cells were removed, and de-barcoding of the palladium-based mass tags performed to select CD34+ cells and export individual sample fcs files.
2.5. Data normalization:
We applied a two-step process to normalize EpiTOF data. First, we transformed each measured abundance (HPTM, CPM, total histones) in the raw EpiTOF data to reduce the dynamic range and influence of outliers present in the data:
We then applied a linear regression for each HPTM using H3 and H4 as independent variables and using all cells from a single sample for the regression:
Here, and are the transformed abundances of H3 and H4 in cell i and is the transformed abundance of HPTM j in cell i. We used the residual of the regression, , as the normalized abundance of HPTM j in cell i. This was done to regress out the effect of H3 and H4 on the HPTM abundance.
2.6. UMAP projection, clustering of EpiTOF data, and cluster defining HPTMs:
We computed UMAP projections (44) on a subset of 40,000 cells (10,000 randomly sampled cells from each panel and disease group) using and . Phenograph clustering (45) was then performed on the UMAP space using .
We then evaluated the cluster-wise median HPTM abundances and sample-wise cell proportions using these 40,000 cells. We compared the sample-wise cell proportions between the healthy subjects (HCB) and patients with JMML (JSP) and used the FDR-adjusted p-value computed from Wilcoxon rank-sum test to calculate the significance of the difference in proportions.
To identify cluster-defining, we compared the single-cell resolution abundance of the HPTM in that cluster to the rest of the clusters. We used effect size, computed using Cohen’s D, and FDR-adjusted p-value computed from Wilcoxon rank-sum test to calculate the significance of the difference in abundance. The computational methods and code used for EpiTOF data analysis are as previously described (46,80).
2.7. Statistical analyses:
Student’s t-test (*p<0.05, **p<0.01, ***p<0.001, not significant p>0.05) using GraphPad Prism software (RRID:SCR_002798).
2.8. Omni ATAC-sequencing on isolated HSPCs:
Chromatin accessibility profiling used Omni ATAC-sequencing (47). CD34+ cells from healthy donor UCBs (n=5) and JMML patient spleen samples (n=4) were washed 3x with 1XPBS (without Ca2+ and Mg2+). Transposition used the Nextera® DNA Sample Preparation Kit (Illumina); transposed DNA was purified using MinElute PCR Purification Kit (Qiagen). Preparation/amplification of ATAC libraries is in Corces et al., 2017. Libraries were quantified by qPCR against KAPA Library Quantification kit (KAPA, Roche) and analyzed using Bioanalyzer High-Sensitivity DNA Analysis kit (Agilent); pmol/µl values between 150bp-1000bp were noted for preparing equimolar sample pool for deep sequencing. Deep sequencing was on NextSeq500 (Illumina) with 50 million reads/sample of sequencing depth.
2.9. Omni ATAC-seq data analysis:
The de-barcoded individual raw data files were extracted from NextSeq500 as fasta.gz files. Data was analyzed on Partek® Flow® software, v10.0 (81). Data processing were as in (82). The mitochondrial and ribosomal DNA were filtered out followed by adapter trimming at <1% per sample. Reads were aligned using Bowtie 2 index against human hg38 assembly on Partek® Flow® software, v10.0 (81). Reads were filtered to remove duplicates and low mapping quality (<20). Peak calling used MACS2 (BAMPE format) and was annotated against annotation assembly and file hg38 ensembl transcripts release 91 to report genes enriched per peak. TSS enrichment was + 5000bp upstream and downstream of promoters, with minimum mapping quality of 30. For comparison of healthy donor to JMML libraries, reads were quantified and normalized. Prior to annotation, unsupervised hierarchical clustering and dimensionality reduction analysis were performed by PCA. Differentially expressed features were identified with Partek® GSA algorithm to account for varying response of each gene to differing data distributions by applying multiple statistical models. Data was filtered with FDR <0.05, minimum fold change of +2-folds; and the high or low peak enrichments per gene locus represented as volcano plots, quantified in bar plots and genome tracks shown per sample group (healthy donor UCBs versus JMML spleens).
3. Results
3.1. A reduction in histone methylation marks and HMT in splenic JMML HSPCs:
DNA hypermethylation is a prognostic feature of JMML, and the DNA demethylating agent, azacytidine, has shown promise (NCT02447666) (37), but there remains an urgent need for targeted, more effective treatment strategies for JMML (7). To understand JMML development within the stem cell compartment and to identify new therapeutic targets, we characterized histone modifications and chromatin accessibility in the CD34+ cells of five PTPN11 mutated JMML spleen derived HSPCs (Figure 1A). We hypothesized that HPTMs are dysregulated in JMML patient derived HSPCs. Therefore, we used EpiTOF (36) to investigate HPTMs in the HSPCs of the five primary PTPN11 mutated JMML spleen samples (38,39). We focused this initial study on the most prevalent and aggressive subtype of JMML (PTPN11 mutated, found in >35% of JMML patients).
Figure 1.
Reduction in histone methylation marks and corresponding HMT in splenic JMML HSPCs. A) Summary of genetic and epigenetic characteristics of EWOG JMML patient samples including presence of PTPN11 mutation, Monosomy 7 status and DNA methylation status (high, intermediate, low). Red = presence; white = absence of characteristics per sample. B) Violin-jitter plots showing total percentage of CD34+ cells isolated from healthy donor (UCB) and JMML (SP) samples. Each patient sample is color coded as defined in the sample legend. C) Dimensionality reduction analysis was performed on normalized datasets using uniform manifold approximation and projection (UMAP) based on histone methylation post translational modification (PTM) marks in Figure 1A-Table. Single-cell level UMAP of UCBs (n=5) or JMML SPs (n=5) are generated with each dot representing a single cell, and each sample is color coded as per the legend. Clustering map is also shown displaying the 13 distinct clusters that were identified with varied distribution of JMML spleens (n=5) and UCBs (n=5) cells within each cluster. D) Each of the 13 clusters had distinct distribution of UCB and JMML cells. Box and whisker plots for clusters with significantly different distribution of JMML and UCB cells are shown. Clusters 5,7 and 11 have significantly higher number of HSPCs from UCB samples. Clusters 3 and 4 have significantly higher number of JMML HSPCs. Each dot represents a single sample, and each patient sample is color coded as per the sample legend. No significant differences were observed in cell-distribution of JMML versus UCBs in clusters 1,2,6,8,9,10,12 and 13. E) Heatmaps were generated for visualization of median abundance of histone methylation marks per UMAP-cluster based on unsupervised hierarchical clustering. Significant abundance or loss of the histone PTM marks that define each cluster distinctly from all the other clusters are marked by an *. The fold change in JMML vs UCB cell proportion per cluster is also highlighted, with JMML-abundant clusters (3 and 4) being marked magenta and UCB-abundant clusters (5,7 and 11) marked green, while no significant distinctions between UCB vs JMML cells proportion being marked gray (1,2,6,8,9,10,12 and 13). Each cluster is defined by distinct histone PTMs signature. Overall differences in histones PTM marks profiles between clusters as well as between individual histone PTMs, are denoted with dendrograms. F) Clustering of bulk ATAC-sequencing [Omni-ATAC protocol (50)] on CD34+ cells from JMML spleens (n=4: JSP6, JSP12, JSP15 and JSP16) and healthy donor controls (UCBs; n=6) shows the two groups are distinct by dimensionality reduction analysis (PCA). Red outlined = JMML spleen samples, blue = heathy UCBs. G) Volcano plot of GSA on normalized reads per UCB and JMML spleen samples; fold change on x-axis, p-value on y-axis. Each dot is a chromosome region; peaks are reads at specific regions per sample. Labeled genes in the volcano plot represent regions at genes of interest that have significantly differential peak enrichments/read counts between controls and JMML spleens. 4378 regions showed higher peak enrichments in UCBs (i.e. Down in JMML), while 2282 regions had increased peak enrichments in JMML (Up in JMML). H) Bar plot of reads at loci encoding the histone modifying enzyme PRDM8, a histone methyltransferase, in control UCB vs. JMML spleen samples and I) corresponding gene tracks representative of peaks/read counts per sample shows absence of peaks at the PRDM8 locus in JMML. Multiple t-tests were performed for statistical analyses (* p<0.05, ** p<0.01, *** p<0.001). HSPC: hematopoietic stem or progenitor cells; UCB: umbilical cord blood; SP: Spleen; H-SNE: Heuristical-stochastic neighbor embedding; PTM: post-translational modifications; UMAP: uniform manifold approximation and projection.
Figure 1.
Reduction in histone methylation marks and corresponding HMT in splenic JMML HSPCs. A) Summary of genetic and epigenetic characteristics of EWOG JMML patient samples including presence of PTPN11 mutation, Monosomy 7 status and DNA methylation status (high, intermediate, low). Red = presence; white = absence of characteristics per sample. B) Violin-jitter plots showing total percentage of CD34+ cells isolated from healthy donor (UCB) and JMML (SP) samples. Each patient sample is color coded as defined in the sample legend. C) Dimensionality reduction analysis was performed on normalized datasets using uniform manifold approximation and projection (UMAP) based on histone methylation post translational modification (PTM) marks in Figure 1A-Table. Single-cell level UMAP of UCBs (n=5) or JMML SPs (n=5) are generated with each dot representing a single cell, and each sample is color coded as per the legend. Clustering map is also shown displaying the 13 distinct clusters that were identified with varied distribution of JMML spleens (n=5) and UCBs (n=5) cells within each cluster. D) Each of the 13 clusters had distinct distribution of UCB and JMML cells. Box and whisker plots for clusters with significantly different distribution of JMML and UCB cells are shown. Clusters 5,7 and 11 have significantly higher number of HSPCs from UCB samples. Clusters 3 and 4 have significantly higher number of JMML HSPCs. Each dot represents a single sample, and each patient sample is color coded as per the sample legend. No significant differences were observed in cell-distribution of JMML versus UCBs in clusters 1,2,6,8,9,10,12 and 13. E) Heatmaps were generated for visualization of median abundance of histone methylation marks per UMAP-cluster based on unsupervised hierarchical clustering. Significant abundance or loss of the histone PTM marks that define each cluster distinctly from all the other clusters are marked by an *. The fold change in JMML vs UCB cell proportion per cluster is also highlighted, with JMML-abundant clusters (3 and 4) being marked magenta and UCB-abundant clusters (5,7 and 11) marked green, while no significant distinctions between UCB vs JMML cells proportion being marked gray (1,2,6,8,9,10,12 and 13). Each cluster is defined by distinct histone PTMs signature. Overall differences in histones PTM marks profiles between clusters as well as between individual histone PTMs, are denoted with dendrograms. F) Clustering of bulk ATAC-sequencing [Omni-ATAC protocol (50)] on CD34+ cells from JMML spleens (n=4: JSP6, JSP12, JSP15 and JSP16) and healthy donor controls (UCBs; n=6) shows the two groups are distinct by dimensionality reduction analysis (PCA). Red outlined = JMML spleen samples, blue = heathy UCBs. G) Volcano plot of GSA on normalized reads per UCB and JMML spleen samples; fold change on x-axis, p-value on y-axis. Each dot is a chromosome region; peaks are reads at specific regions per sample. Labeled genes in the volcano plot represent regions at genes of interest that have significantly differential peak enrichments/read counts between controls and JMML spleens. 4378 regions showed higher peak enrichments in UCBs (i.e. Down in JMML), while 2282 regions had increased peak enrichments in JMML (Up in JMML). H) Bar plot of reads at loci encoding the histone modifying enzyme PRDM8, a histone methyltransferase, in control UCB vs. JMML spleen samples and I) corresponding gene tracks representative of peaks/read counts per sample shows absence of peaks at the PRDM8 locus in JMML. Multiple t-tests were performed for statistical analyses (* p<0.05, ** p<0.01, *** p<0.001). HSPC: hematopoietic stem or progenitor cells; UCB: umbilical cord blood; SP: Spleen; H-SNE: Heuristical-stochastic neighbor embedding; PTM: post-translational modifications; UMAP: uniform manifold approximation and projection.
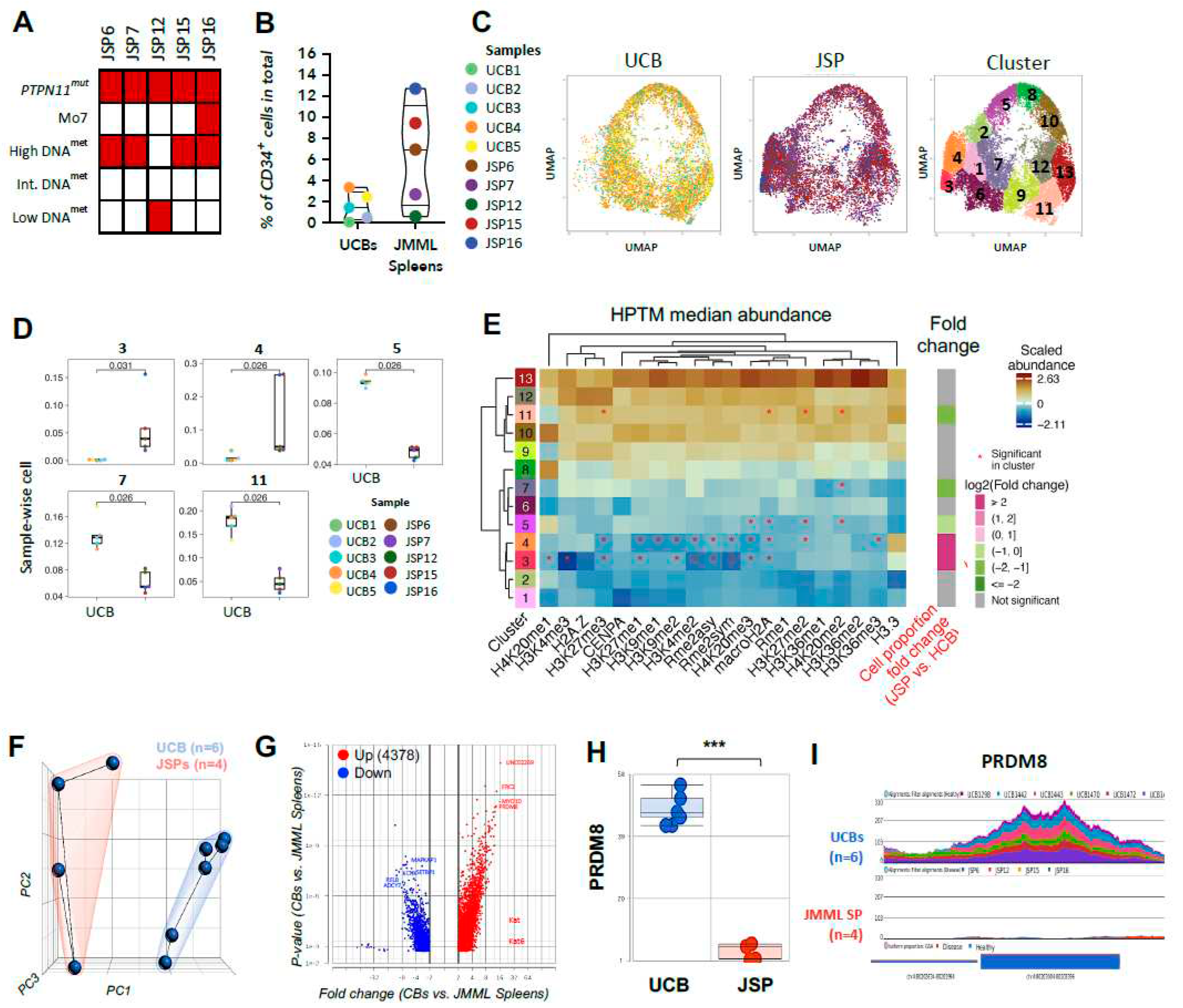
JMML patients present with splenomegaly for which they often undergo splenectomy; thus, the spleen provides a rich source of leukemia cells, as evident from the high number of CD34+ cells we recovered from most JMML patients (2-12% in JMML spleens compared to 0.3-3.5% in healthy donor umbilical cord blood (HD UCBs) (Figure 1B). The leukemogenic capacity of JMML splenic CD34+ cells has been previously confirmed in humanized mouse models through serial transplantation (40,41). Further, as healthy pediatric spleen tissue is unavailable, and comparable adult spleens contain <0.1% CD34+ cells (42,43), we used HD UCBs as controls for primary JMML spleens, consistent with previous studies (35,40). We investigated histone methylation (Figure 1), acetylation, ubiquitination, and phosphorylation PTMs (Figure 2) in the CD34+ compartment from HSPCs of five JMML patient (pt) samples alongside five HD UCB controls.
To compare the distribution of the histone methylation marks in isolated CD34+ cells from JMML vs HD UCBs, we first performed dimensionality reduction analyses using uniform manifold approximation and projection (UMAPs; (44)), and then clustered the cells using PhenoGraph (45) (Figure 1C). These analyses were performed as previously described (46). The histone methylation profiles grouped the CD34+ cells from JMML and HD UCB controls into 13 distinct clusters (Figure 1C; right panel).
Clusters 3 and 4 had significantly higher proportions of JMML derived HSPCs than HD UCBs, whereas clusters 5, 7 and 11 had significantly lower proportions of JMML HSPCs (Figure 1D). The remaining clusters (1, 2, 6, 8, 9, 10, 12, and 13) showed no significant differences in cell proportions. Overall, comparative analysis of the median abundance of each individual methylation mark in clusters 3, 4, 5, 7 and 11 revealed that several were significantly different between JMML HSPCs and HD UCBs and when compared to all other clusters combined (Figure 1E). This analysis was not performed for clusters 1, 2, 4, 6 and 8 as there was no significant difference in JMML vs HD UCB cell proportions. In Figure 1E, the heatmap shows the median abundance of individual HPTM marks at the single-cell level in each cluster. Additionally, the fold-change between JMML vs HD UCB HSPC proportions per cluster is shown to the right.
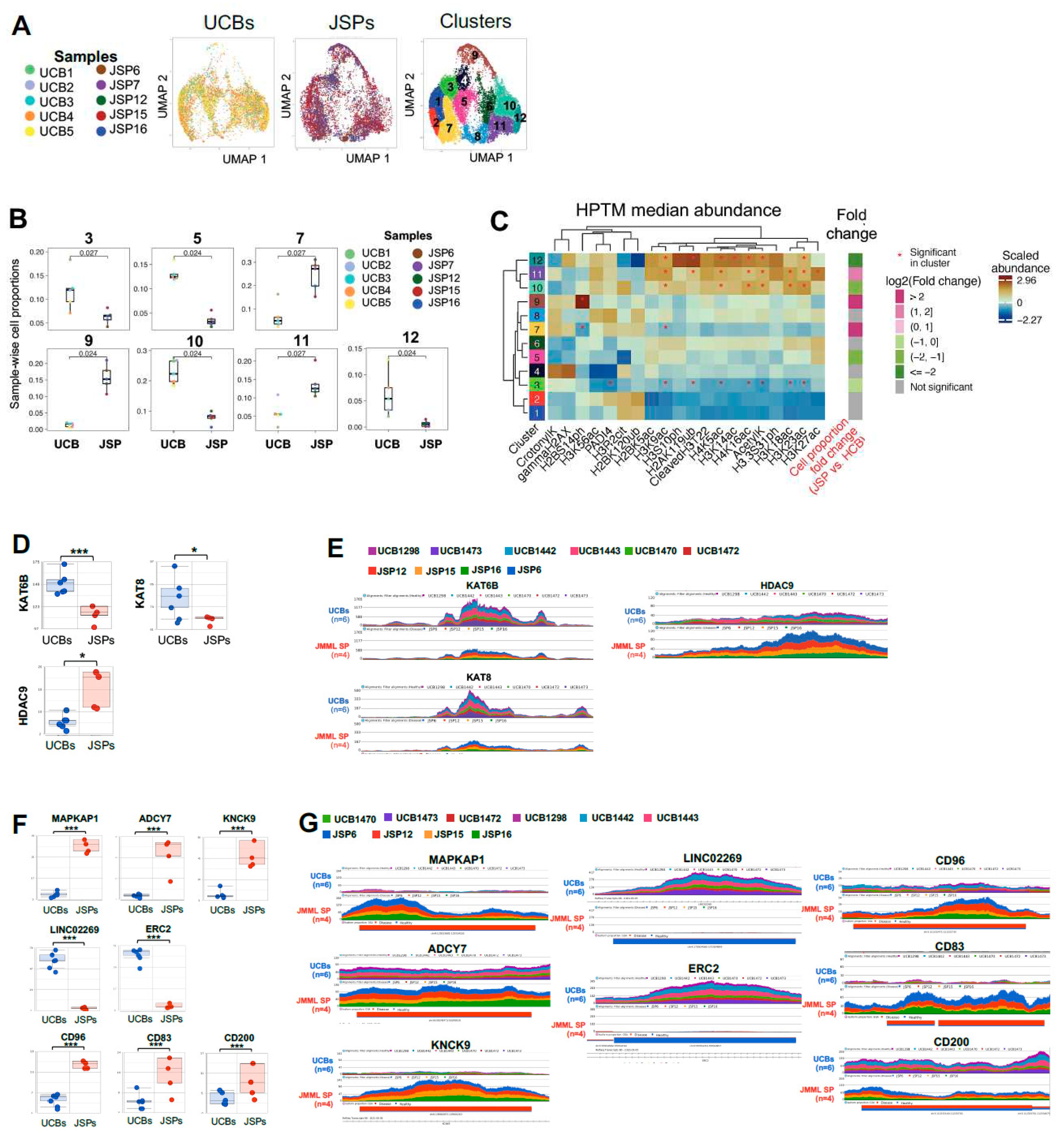
Figure 2.
Loss of histone acetylation and phosphorylation markers and corresponding HATs in primary JMML splenic-HSPCs. EpiTOF performed on HSPCs (CD34+ cells) from healthy donor controls (UCB n=5) and JMML patient samples (SP n=5) at the single-cell level. A) Dimensionality reduction analysis was performed on normalized datasets using uniform manifold approximation and projection (UMAP) based on histone acetylation, phosphorylation, and ubiquitination post translational modification (PTM) marks in Figure 1A-Table. Single-cell level UMAP of UCBs (n=5) or JMML SPs (n=5) are generated with each dot representing a single cell, and each sample is color coded as per the legend. Clustering map is also shown displaying the 12 distinct clusters that were identified with varied distribution of JMML spleens (n=5) and UCBs (n=5) cells within each cluster. B) Each of the 12 UMAP clusters had distinct distribution of UCB and JMML cells. Box and whisker plots for the clusters with significantly different distribution of JMML vs UCB cells are shown. Clusters 3,5,10 and 12 have significantly higher number of HSPCs from UCB samples. While clusters 7,9 and 11 have significantly higher number of JMML HSPCs. Each dot represents a single sample, and each sample is color coded as per the legend. No significant differences in cell-distribution between JMML versus UCBs were observed in clusters 1,2,4,6 and 8. C) Heatmaps were generated for visualization of median histone PTMs abundance per UMAP-cluster based on unsupervised hierarchical clustering. Significant abundance or loss of the histone PTM marks that define each cluster distinctly from all the other clusters are marked by an *. The fold change in JMML vs UCB cells proportion in each cluster is also highlighted, with JMML-abundant clusters (7,9 and 11) being marked in magenta and UCB-abundant clusters (3,5,10 and 12) marked in green, while no significant distinctions between UCB vs JMML cells proportion are marked gray (1,2,4,6 and 8). Each cluster is defined by a distinct trend of histone PTMs signatures. The overall differences in histones PTM marks profiles between clusters as well as between individual histone PTMs, are denoted with dendrograms. D) Bulk ATAC-seq was performed on CD34+ cells from JMML spleens (n=4: JSP6, JSP12, JSP15 and JSP16) and healthy donor controls (UCBs; n=6). Bar plots of reads at loci encoding histone modifying enzymes Kat6B, Kat8, HDAC5 and HDAC9 in control UCB vs. JMML spleen samples and E) corresponding gene tracks representative of peaks/read counts per sample. F) The most significantly distinct loci between UCBs and JMML SPs summarized with LS mean read counts per sample. Bar plots High read counts in JMML include MAPK-AP1, ADCY7, KNCK9, CD96, CD83 and CD200. Low read counts in JMML at loci encoding LINC02269 and ERC2. Bar plots of reads at these loci along with their respective G) Gene tracks in JMML spleens compared to UCB controls are shown. Multiple t-tests were performed for statistical analyses (* p<0.05, ** p<0.01, *** p<0.001). Each sample was color coded consistently in the entire Figure 1, as shown in the legends of Figure 1B and 1C. HSPC: hematopoietic stem or progenitor cells; HD: healthy donor; UCB: umbilical cord blood; SP: Spleen; UMAP: uniform manifold approximation and projection.
Figure 2.
Loss of histone acetylation and phosphorylation markers and corresponding HATs in primary JMML splenic-HSPCs. EpiTOF performed on HSPCs (CD34+ cells) from healthy donor controls (UCB n=5) and JMML patient samples (SP n=5) at the single-cell level. A) Dimensionality reduction analysis was performed on normalized datasets using uniform manifold approximation and projection (UMAP) based on histone acetylation, phosphorylation, and ubiquitination post translational modification (PTM) marks in Figure 1A-Table. Single-cell level UMAP of UCBs (n=5) or JMML SPs (n=5) are generated with each dot representing a single cell, and each sample is color coded as per the legend. Clustering map is also shown displaying the 12 distinct clusters that were identified with varied distribution of JMML spleens (n=5) and UCBs (n=5) cells within each cluster. B) Each of the 12 UMAP clusters had distinct distribution of UCB and JMML cells. Box and whisker plots for the clusters with significantly different distribution of JMML vs UCB cells are shown. Clusters 3,5,10 and 12 have significantly higher number of HSPCs from UCB samples. While clusters 7,9 and 11 have significantly higher number of JMML HSPCs. Each dot represents a single sample, and each sample is color coded as per the legend. No significant differences in cell-distribution between JMML versus UCBs were observed in clusters 1,2,4,6 and 8. C) Heatmaps were generated for visualization of median histone PTMs abundance per UMAP-cluster based on unsupervised hierarchical clustering. Significant abundance or loss of the histone PTM marks that define each cluster distinctly from all the other clusters are marked by an *. The fold change in JMML vs UCB cells proportion in each cluster is also highlighted, with JMML-abundant clusters (7,9 and 11) being marked in magenta and UCB-abundant clusters (3,5,10 and 12) marked in green, while no significant distinctions between UCB vs JMML cells proportion are marked gray (1,2,4,6 and 8). Each cluster is defined by a distinct trend of histone PTMs signatures. The overall differences in histones PTM marks profiles between clusters as well as between individual histone PTMs, are denoted with dendrograms. D) Bulk ATAC-seq was performed on CD34+ cells from JMML spleens (n=4: JSP6, JSP12, JSP15 and JSP16) and healthy donor controls (UCBs; n=6). Bar plots of reads at loci encoding histone modifying enzymes Kat6B, Kat8, HDAC5 and HDAC9 in control UCB vs. JMML spleen samples and E) corresponding gene tracks representative of peaks/read counts per sample. F) The most significantly distinct loci between UCBs and JMML SPs summarized with LS mean read counts per sample. Bar plots High read counts in JMML include MAPK-AP1, ADCY7, KNCK9, CD96, CD83 and CD200. Low read counts in JMML at loci encoding LINC02269 and ERC2. Bar plots of reads at these loci along with their respective G) Gene tracks in JMML spleens compared to UCB controls are shown. Multiple t-tests were performed for statistical analyses (* p<0.05, ** p<0.01, *** p<0.001). Each sample was color coded consistently in the entire Figure 1, as shown in the legends of Figure 1B and 1C. HSPC: hematopoietic stem or progenitor cells; HD: healthy donor; UCB: umbilical cord blood; SP: Spleen; UMAP: uniform manifold approximation and projection.
We focused our attention on methylation marks that were significantly different in clusters enriched for JMML HSPCs (clusters 3 and 4) or clusters significantly enriched for HD UCB HSPCs (clusters 5, 7 and 11), reasoning that these would reveal disease-specific features.
Several methylation marks were uniquely lower in clusters 3 and 4, which were enriched for JMML HSPCs (Figure 1D, 1E-Fold change). Specifically, we observed significantly lower abundance of H3K27me3, H3K27me1, H3K9me2, H3K4me2, Rme2asy, and Rme2sym, when compared to all other clusters (Figure 1E, S1-2). The UMAP cluster 3 also had significantly lower H4K20me1 and H3K4me3, whereas cluster 4 had significantly lower H3K9me1 and H3K36me3 (Figure 1E, S1-2). These data suggest these specific methylation marks are uniquely and significantly reduced in JMML HSPCs.
In contrast, clusters 5 and 7, which were significantly enriched for HD UCB HSPCs showed a specific and significant reduction in median abundance of the histone methylation mark H4K20me2 (Figure 1E, S1-2). Curiously however, cluster 11, which was also enriched for HD UCB HSPCs, showed a significantly higher median abundance of H4K20me2 (Figure 1E, S1-2). We note that overall, cluster 11 has higher median abundance of several methylation marks. For example, while the JMML enriched cluster 3 and 4 were significantly lower for H3K27me3, the HD UCB HSPC enriched cluster 11 was significantly higher for this methylation mark (see column 4; Figure 1E).
To identify whether the histone hypomethylation patterns that we observed in the overall JSPs vs HD UCBs might be driven by specific HSPC subsets, we performed dimensionality reduction analysis on isolated HSPC subsets (see Supplementary Table 2) from each JMML HSPC and HD UCB sample. Phenotypically, we observed considerable differences in the distribution of distinct HSPC subsets in JMML vs HD UCBs (Figure S3) with significantly higher hematopoietic stem cells (HSCs), CD34+CD38-CD45R+CD90+ (Leukemic-MPP; Figure S3B) and CD34+CD38+CD45RA+CD90+ (Figure S3C) subsets. The latter two subsets were found almost exclusively in JMML samples compared to HD UCBs (Figure S3B-C). Though not statistically significant, the JMML samples also had a higher percentage of granulocyte-monocyte progenitors (GMPs) and lower Lymphoid-myeloid primed progenitor (LMPPs) when compared to HD UCBs (Figure S3B-C). However, we found heterogenous clustering of the HSPC subsets from both JMML and HD UCBs in the UMAP space generated based on the histone methylation panel (Figure S4) making it difficult to identify any unique HPTM signatures that was specific to a JMML or HD HSPC subset.
Our data shows that specific methylation marks are reduced in JMML HSPCs suggesting alterations in the chromatin structure. Thus, we next analyzed chromatin accessibility profiles using bulk ATAC-seq on CD34+ cells from JMML (n=4; pts #6, #12, #15 and #16), and HD UCB (n=6) samples (47). Dimensionality reduction using principal component analysis revealed distinct clustering of healthy versus JMML samples indicative of differential chromatin accessibility in these two groups (Figure 1F). The heterogeneity within disease samples (red outline-Figure 1F) likely reflects the diversity in patient characteristics including disease stage at diagnosis, age, gender, and outcome (Supplementary Table 1). Differential accessibility analysis identified 4378 loci with significantly decreased, and 2282 loci with significantly increased chromatin accessibility in JMML vs UCBs (Figure 1G). These data confirm that there are significant global changes to chromatin structure in JMML v’s HD UCB CD34+ cells.
We specifically examined loci for genes encoding histone methyltransferases (HMT) and demethylases (DNMT). Figure 1H and 1I show comparison of the chromatin accessibility profiles of JMML vs UCB HSPCs at the PRDM8 locus. These data revealed a complete loss of peaks at the PRDM8 locus that was unique to the JMML HSPCs (Figure 1H-I). PRDM8 catalyzes H3K9me (48) and our observation of the absence of peaks at the PRDM8 locus in all JMML samples is consistent with the significant reduction of H3K9me1 and H3K9me2 methylation we observed in JMML HSPC enriched clusters 3 and 4 (columns 7 and 8; Figure 1E). We conclude that the reduction of H3K9me marks observed in JMML enriched clusters 3 and 4 is likely due to a loss of PRDM8 expression.
3.2. Heterogenous histone acetylation, ubiquitination, and phosphorylation profiles in primary JMML splenic-HSPCs:
Next, we compared the histone acetylation, phosphorylation and ubiquitination marks in JMML HSPCs (n=5) to control HD UCB HSPCs (n=5) by again performing dimensionality reduction with UMAPs followed by clustering with PhenoGraph (Figure 2) as previously described (46). Histone acetylation, phosphorylation and ubiquitination data grouped the CD34+ cells from JMML HSPCs and HD UCB controls into 12 distinct clusters (Figure 2A). We observed that JMML HSPCs were significantly enriched in clusters 7, 9 and 11. In contrast, clusters 3, 5, 10 and 12 had were enriched for HD UCB HSPCs with significantly lower proportions of JMML HSPCs (Figure 2B).
As for our analysis of the methylation marks in Figure 1, we analyzed which HPTMs were uniquely affected in JMML enriched, or HD UCB enriched clusters, and whether their median abundance was significantly up or down regulated relative to all other clusters (Figure 2C). We found that while JMML enriched cluster 7 and 9 had significantly less H2BS14ph, JMML enriched cluster 9 was significantly increased at this phosphorylation site indicating strong heterogeneity across patients at this site. Cluster 11 showed significantly more HPTM at H3K27ac. Although trends were apparent, we observed no other statistically significant changes in HPTM marks that were uniquely affected in JMML enriched HSPCs.
We then evaluated HPTM signatures unique to HD UCB HSPC clusters 3, 5, 10 and 12. Figure 2B, Figure 2C-Fold change) and found only two changes were unique to the HD UCB cells. In cluster 3, we observed significantly reduced levels of PAD14 (Figure 2C, S5-6). While cluster 12 had significantly higher proportion of H3K14ac (Figure 2C, S56).
To determine whether the heterogenous histone acetylation, phosphorylation and ubiquitination patterns that we observed in JMML HSPCs vs HD UCBs might be driven by specific HSPC subsets, we performed dimensionality reduction analysis on isolated HSPC subsets (Table 2) from each JSP and UCB sample. As for the histone methylation panel (Figure S4), we found heterogenous clustering of all the HSPC subsets from JSPs and UCBs in the UMAP space generated based on the histone acetylation, phosphorylation and ubiquitination panel (Figure S7). We identified 14 distinct clusters in the UMAP space (Figure S7A), however, each cluster consisted of some proportion of every HSPC subset from UCBs as well as JSPs (Figure S7B-C). Thus, we could not assign the unique HPTM signatures to any specific disease or healthy HSPC subsets.
We next used ATAC-seq to examine gene loci encoding HPTM enzymes that catalyze histone acetylation, phosphorylation and ubiquitination marks, as we did for the methylating enzymes. These analyses revealed that HD UCB HSPCs had significant peak enrichments, at histone acetyltransferases (HATs) Kat6b (catalyzes acetylation of H3K23 (49,50)), and Kat8 (catalyzes H4K16ac (51)), when compared to JMML samples (Figure 2D-E). These significantly higher open chromatin regions at these HATs loci in UCB HSPCs were consistent with the significantly higher abundance of H3K23ac and H4K16ac in all 3 UCB HSPC enriched clusters (Figure 2C). Interestingly, the JMML enriched cluster 11 also demonstrated an upregulation of these acetylation marks. Overall, however, these data suggest reduced Kat6b and Kat8 HAT activities in JMML HPSCs leading to a reduction in HPTM acetylation. Consistent with this conclusion, we also observed significant peak enrichments at histone deacetylases HDAC9 and HDAC5 in the JMML samples (Figure 2D-E). Collectively, we conclude that there is reduced histone acetylation in the majority of JMML HSPCs when compared to HD UCBs (Figure 2).
We also identified other loci for which the ATAC-seq data showed enriched peaks specifically in JMML HSPCs. Genes at these loci included mitogen-activated protein kinase associated protein 1 (MAPK-AP1) a component of the mTOR2 pathway (52), potassium channel gene KCNK9 and adenylate cyclase 7 (ADCY7) (Figure 2F-G). Both KCNK9 and ADCY7 are often found overexpressed in human cancers (53–55). In contrast, peaks at the neurotransmitter release associated gene ERC2; and long intergenic non-coding RNA 2269 (LINC02269) were significantly reduced in the JMML HSPCs (Figure 2F-G). JMML HSPC specific significant peak enrichments were also observed at the immunophenotypic markers: CD96, CD83 and CD200, although there was heterogeneity between patient samples at CD83 and CD200 (Figure 2F-G). These data are in agreement with Louka et al’s finding of CD96 expression on putative JMML LICs (35).
4. Discussion
The biggest hurdles in treating JMML patients is the lack of druggable targets (7). As reversible features, epigenetic changes have great potential for therapeutic intervention. However, while DNA hypermethylation is a prognostic feature of aggressive disease, and the DNA demethylating agent, 5-azacitidine, has shown promise (NCT02447666) (37), there is an urgent need for targeted, more effective treatment strategies to cure JMML. To understand JMML development within the stem cell compartment and to identify new therapeutic targets, we have characterized the histone modifications and chromatin accessibility in the CD34+ compartment of five PTPN11 mutated JMML spleen derived HSPCs. To the best of our knowledge, these data represent the first such analyses.
We found the spleen-derived PTPN11 mutated JMML HSPCs exhibited significantly reduced histone methylation marks that are unique to JMML HSPC at H3K27me1-3, H3K9me1-2, H3K4me2, Rme2asy, Rme2sym, H4K20me1, and H3K36me3. Indeed, the JMML-enriched UMAP clusters 3 and 4 had overall significantly reduced histone methylation marks when compared to all the other clusters suggesting a consistent histone hypomethylation signature in JMML samples. We also found significant - although heterogeneous, changes at H2BS14ph, whereby one JMML enriched cluster contained cells that were significantly increased for phosphorylation marks at this site, while another JMML enriched cluster was significantly reduced. Additional JMML HSPC specific changes were observed at H3K27ac, which was significantly increased in a cluster enriched for JMML cells. We similarly identified HD UCB HSPC specific HPTMs and observed: i) a significant reduction in median abundance of the histone methylation mark H4K20me2 in 2 out of the 3 clusters (the third HD UCB enriched cluster showed an increase at this site); ii) significantly reduced levels of PAD14 and significantly higher proportion of H3K14ac. Taken together these data demonstrate significant differences in HPTMs between the 2 sample groups.
Loss of HPTM marks such as those observed in our analysis of JMML HSPCs are signature events in numerous malignancies including: H3K23ac in solid tumor models such as breast cancer, KRAS mutated colorectal cancer and non-small cell lung cancer (49,56); H3S10ph in aggressive solid tumors including colon cancer (57,58); and, H3K36me2, H3K36me2 and H3K27me2 in leukemic transformation in AML (59) and poor AML patient prognosis (60,61). Consistent with this loss in JMML, our ATACseq analysis showed decreased peak enrichments at histone acetyltransferases Kat6b and Kat8 and methyltransferase PRDM8 but increased peak enrichments at histone deacetylases HDAC9 and HDAC5. Kat6b and Kat8 are specific for H4K16ac and H3K23ac, respectively. The concurrent loss of Kat8 and H4K16ac has been shown in aberrant gene expression in AML (62), suggestive of a similar mechanism in JMML. Similarly, Kat6b plays a tumor suppressor role in non-small cell lung cancer wherein its loss leads to cancer growth (63). PRDM8 is also a strong tumor suppressor that is downregulated in hepatocellular carcinoma (64,65); thus, loss of peaks at this locus supports an analogous role in JMML onset. Indeed, a recent study showed that knockout of PRDM8 in iPSCs led to impaired hematopoietic differentiation and myeloid-biased malignant transformation (66).
Increases in HDAC activity, as our ATACseq data suggests for HDAC5 and HDAC9 in JMML spleen HSPCs, have been identified in a variety of tumor types. Several class I HDAC inhibitors are approved for cancer treatment (i.e., vorinostat, romidepsin and belinostat). However, these drugs do not inhibit class II HDACs, such as HDAC5 and HDAC9 (67). Instead, the pan-HDAC inhibitor, panobinostat, which has shown promise treating patients with multiple myeloma (68), has also demonstrated targeted killing of primary JMML HSPCs, interestingly showing greater efficacy than 5-azacytidine in vitro (69). We also note that TMP195, a first-in-class highly selective class II HDAC inhibitor, has shown potent activity in breast cancer mouse model studies (70), making it an attractive candidate to test in JMML patients who display increased HDAC5 and HDAC9 activity.
We also observed reduced peaks at numerous other loci. Indeed, overall, we observed largely closed, heterochromatic regions in JMML HSPCs suggesting significant repression of gene expression. However, specific loci such as MAPKAP1 displayed peak enrichments. MAPKAP1/Sin1 is linked to RAS and mTOR2 pathways and is known to drive Akt phosphorylation (71,72). RAS pathway mutations, including elevation of pAKT drive JMML, and preclinical evidence suggests a therapeutic role for mTOR inhibition in JMML (35,73–75). Thus, our data suggests a possible link between RAS and mTOR2 pathways in JMML via MAPKAP1, making this pathway a putative therapeutic target. We also found highly enriched peaks at the potassium channel gene KCNK9 in all JMML samples. Others have shown that overexpression of this gene promotes malignancy in several cancers (76). In addition, several immunophenotypic gene regions were differentially regulated in JMML. CD83 is a marker of AML LICs and therapeutic targeting using CD83-CAR T cells has shown preliminary success in humanized mouse models of AML (77), suggesting it could be a potential therapeutic target in JMML. Interestingly, another known marker of AML LICs, CD200 had significantly higher read counts in JMML HSPCs when compared to UCBs. CD200 is upregulated in the most resistant pediatric AMLs and confers immunosuppressive properties when co-cultured with engineered T regulatory 1 cells (78,79). CD200 protein was also upregulated in HSPCs of all our JMML samples (data not shown).
Collectively, our data identifies aberrant histone PTMs in splenic JMML HSPCs and implicates the activity of PTM modifying enzymes as responsible for the observed changes. We are currently exploring these changes including confirming transcriptional profiles using RNAseq and isolating specific HSPC cell subsets to identify specific cell lineages in which these changes occur.
5. Conclusion
In conclusion, we have identified aberrant histone PTMs in splenic PTPN11 mutated JMML HSPCs and revealed differential accessibilities at the loci of the respective HPTM modifying enzymes. Additionally, we have shown by ATAC-seq a significant heterochromatin (closed) structure at key genomic regions of known tumor suppressors, HATs, and HMTs, which explains an extensive gene repression. The newly identified signatures of aberrant histone PTMs - such as the targeted histone hypoacetylation and hypomethylation, may be used in the future as prognostic or predictive epigenetic markers in patients with JMML. Most importantly, we have identified surface markers such as CD96, CD83, and CD200 as targets for cell therapy. The data obtained in this pilot study will be extended and validated in other JMML genotypes (NF1, KRAS, NRAS and CBL). Further, we are validating the observed HPTMs changes - including the transcriptional profiles, by single cell RNAseq to identify whether these changes occur in a specific cell lineage or not.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
R.S. contributed to conceptualization, methodology, investigation, funding acquisition, data curation, analysis, and validation, wrote the first draft of the manuscript and prepared figs. 1, 2 and supplementary tables and figures. M.D. contributed to EpiTOF methodology and data curation. A.G. contributed to bioinformatic analysis, prepared figs. 1C-E, 2A-C and supplementary figures S1, 2, 4-7. A.G. also contributed to methodology and editing the manuscript. L.K. contributed towards supervision of bioinformatic analysis in figs 1C-E, 2A-C. C.N. and C.F. provided patient samples for the study from their biobank (EWOG-MDS cohort, Germany). C.F. also helped in editing the methodology section of the manuscript. K.M.S. and N.L. provided feedback on the manuscript. R.Pa. contributed to data curation, project administration, funding acquisition and review of the manuscript. R.Pe. extensively edited and reviewed the manuscript and provided scientific feedback data interpretation. A.M.C. provided feedback on ATAC-seq methodology and analysis sections of the manuscript. Y.L.L. provided feedback on the study and reviewed the manuscript. A.K. contributed to conceptualization, EpiTOF methodology development, funding acquisition, data curation, interpretation, investigation feedback, and manuscript editing and review. P.J.U. provided supervision, resources, and funding for the study. P.K. provided guidance on data analysis and interpretation for the figures, along with feedback on manuscript. A.B. is the principal investigator and corresponding author of the paper. A.B. has contributed through study design, funding acquisition, supervision, editing and review of the manuscript. All authors reviewed the manuscript.
Funding
This work was supported by a Stanford Maternal and Child Health Research Institute (MCHRI) pilot grant as well as the Children's Leukemia Research Association (CLRA) grant (#137046). The Sequencing funding, NIH S10OD021763 was used by the Human Immune Monitoring Center for the sequencing of ATAC-seq libraries.
Ethical Approval
All cryopreserved primary JMML samples were provided to us deidentified from the
biorepository of the EWOG-MDS, located at Freiburg University Medical Center, Germany. These de-identified
JMML cryopreserved patient samples were obtained from EWOG following guidelines under approved IRB
protocol 46055 at Stanford University. Healthy controls were also de-identified umbilical cord blood (UCB)
samples obtained from the Binns program at Stanford School of Medicine. All human samples were collected
from donors with informed consent and compliance with relevant ethical regulations.
Availability of data and materials
The raw data files that support the findings of this study will be made openly
available on code ocean.
Acknowledgments
We thank the Human Immune Monitoring Center for sequencing of ATAC-seq libraries (Sequencing funding, NIH S10OD021763), the Stanford Binns Program for healthy donor umbilical cord samples and Dr. Robertson Parkman for valuable advice. This work was supported by a Stanford Maternal and Child Health Research Institute (MCHRI) pilot grant and the Children's Leukemia Research Association (CLRA).
Disclosure of Conflicts of Interest
The authors declare no potential conflict of interests of any financial or personal nature.
References
- Mayerhofer C, Niemeyer CM, Flotho C. Current Treatment of Juvenile Myelomonocytic Leukemia. J Clin Med. J Clin Med; 2021;10. [CrossRef]
- de Vries ACH, Zwaan CM, van den Heuvel-Eibrink MM. Molecular basis of juvenile myelomonocytic leukemia. Haematologica. Ferrata Storti Foundation; 2010;95:179–82. [CrossRef]
- Niemeyer CM. JMML genomics and decisions. Hematology (United States). American Society of Hematology; 2018;2018:307–12.
- Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: who’s the driver at the wheel? Blood. 2019;133:1060–70.
- Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS, Jacks T, et al. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. American Society of Hematology; 1991;77:925–9.
- Locatelli F, Nöllke P, Zecca M, Korthof E, Lanino E, Peters C, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105:410–9. [CrossRef]
- Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125:1083–90. [CrossRef]
- Chan RJ, Cooper T, Kratz CP, Weiss B, Loh ML. Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium. Leuk Res. 2009;33:355–62. [CrossRef]
- Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. Nature Publishing Group; 2008. page 1335–42.
- Poetsch AR, Lipka DB, Witte T, Claus R, Nöllke P, Zucknick M, et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics. 2014;9:1252–60.
- Olk-Batz C, Poetsch AR, Nöllke P, Claus R, Zucknick M, Sandrock I, et al. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood. Blood; 2011;117:4871–80. [CrossRef]
- Stieglitz E, Mazor T, Olshen AB, Geng H, Gelston LC, Akutagawa J, et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun. Nature Publishing Group; 2017;8:1–8. [CrossRef]
- Schonung M, Meyer J, Nollke P, Olshen AB, Hartmann M, Murakami N, et al. International Consensus Definition of DNA Methylation Subgroups in Juvenile Myelomonocytic Leukemia. Clin Cancer Res. Clin Cancer Res; 2021;27:158–68. [CrossRef]
- Liu YL, Lensing SY, Yan Y, Cooper TM, Loh ML, Emanuel PD. Deficiency of CREB and over expression of miR-183 in juvenile myelomonocytic leukemia. Leukemia. Leukemia; 2013. page 1585–8. [CrossRef]
- Leoncini PP, Bertaina A, Papaioannou D, Flotho C, Masetti R, Bresolin S, et al. MicroRNA fingerprints in juvenile myelomonocytic leukemia (JMML) identified miR-150-5p as a tumor suppressor and potential target for treatment. Oncotarget. 2016;7:55395–408. [CrossRef]
- Helsmoortel HH, Bresolin S, Lammens T, Cavé H, Noellke P, Caye A, et al. LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood. American Society of Hematology; 2016;127:1163–72. [CrossRef]
- Fluhr S, Krombholz CF, Meier A, Epting T, Mücke O, Plass C, et al. Epigenetic dysregulation of the erythropoietic transcription factor KLF1 and the β-like globin locus in juvenile myelomonocytic leukemia. Epigenetics. Taylor and Francis Inc.; 2017;12:715–23. [CrossRef]
- Sun XJ, Man N, Tan Y, Nimer SD, Wang L. The role of histone acetyltransferases in normal and malignant hematopoiesis. Front Oncol. Frontiers Media S.A.; 2015. page 1. [CrossRef]
- Li X-Q, Guo Y-Y, De W. DNA methylation and microRNAs in cancer. World J Gastroenterol. Baishideng Publishing Group Inc; 2012;18:882–8.
- Dhall A, Zee BM, Yan F, Blanco MA. Intersection of epigenetic and metabolic regulation of histone modifications in acute myeloid leukemia. Front Oncol. Frontiers Media S.A.; 2019;9:432. [CrossRef]
- Fathi E, Farahzadi R, Montazersaheb S, Bagheri Y. Epigenetic Modifications in Acute Lymphoblastic Leukemia: From Cellular Mechanisms to Therapeutics. Curr Gene Ther. Curr Gene Ther; 2021;21:60–71. [CrossRef]
- Issa JP. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am. Hematol Oncol Clin North Am; 2010;24:317–30. [CrossRef]
- Cavalieri V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes (Basel). Genes (Basel); 2021;12. [CrossRef]
- Taylor P, Ballabio E, Milne T a. Epigenetic control of gene expression in and MLL fusion proteins Epigenetic control of gene expression in leukemogenesis : Cooperation between wild type MLL and MLL fusion proteins. 2014;3556:37–41.
- Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26:6697–714. [CrossRef]
- Hu G, Zhao K. Correlating histone modifi cation patterns with gene expression data during hematopoiesis. Methods in Molecular Biology. Humana Press Inc.; 2014;1150:175–87.
- Rodrigues CP, Shvedunova M, Akhtar A. Epigenetic Regulators as the Gatekeepers of Hematopoiesis. Trends in Genetics. Elsevier Ltd; 2021. page 125–42. [CrossRef]
- Jiang P, Wang H, Zheng J, Han Y, Huang H, Qian P. Epigenetic regulation of hematopoietic stem cell homeostasis. Blood Science. Ovid Technologies (Wolters Kluwer Health); 2019;1:19–28. [CrossRef]
- Lipka DB, Witte T, Toth R, Yang J, Wiesenfarth M, Nöllke P, et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat Commun. Nature Publishing Group; 2017;8:2126. [CrossRef]
- Liu X, Zheng H, Li X, Wang S, Meyerson HJ, Yang W, et al. Gain-of-function mutations of Ptpn11 (Shp2) cause aberrant mitosis and increase susceptibility to DNA damage-induced malignancies. Proc Natl Acad Sci U S A. National Academy of Sciences; 2016;113:984–9. [CrossRef]
- Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. Nat Genet; 2003;34:148–50.
- Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating Mutations of the Noonan Syndrome-Associated SHP2/PTPN11 Gene in Human Solid Tumors and Adult Acute Myelogenous Leukemia. Cancer Res. American Association for Cancer Research; 2004;64:8816–20.
- Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lleuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. American Society of Hematology; 2004;103:2325–31.
- Caye A, Rouault-Pierre K, Strullu M, Lainey E, Abarrategi A, Fenneteau O, et al. Despite mutation acquisition in hematopoietic stem cells, JMML-propagating cells are not always restricted to this compartment. Leukemia. Springer Nature; 2019;1–11.
- Louka E, Povinelli B, Rodriguez-Meira A, Buck G, Wen WX, Wang G, et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. Journal of Experimental Medicine. Rockefeller University Press; 2021;218. [CrossRef]
- Cheung P, Vallania F, Dvorak M, Chang SE, Schaffert S, Donato M, et al. Single-cell epigenetics – Chromatin modification atlas unveiled by mass cytometry. Clinical Immunology. Academic Press Inc.; 2018;196:40–8. [CrossRef]
- Niemeyer CM, Flotho C, Lipka DB, Stary J, Rössig C, Baruchel A, et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv. American Society of Hematology; 2021;5:2901–8. [CrossRef]
- Cheung P, Vallania F, Dvorak M, Chang SE, Schaffert S, Donato M, et al. Single-cell epigenetics – Chromatin modification atlas unveiled by mass cytometry. Clinical Immunology. Academic Press Inc.; 2018;196:40–8. [CrossRef]
- Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell. Cell Press; 2018;173:1385-1397.e14. [CrossRef]
- Krombholz CF, Aumann K, Kollek M, Bertele D, Fluhr S, Kunze M, et al. Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2-/-γc-/- mice. Haematologica. Haematologica; 2016;101:597–606.
- Yoshimi A, Balasis ME, Vedder A, Feldman K, Ma Y, Zhang H, et al. Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood. 2017;130:397–407. [CrossRef]
- Barosi G, Vittorio R, Margherita M, Luca VG, Alessandro P, Vittorio N, et al. Spleen neoangiogenesis in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. Br J Haematol; 2004;124:618–25. [CrossRef]
- Wang X, Prakash S, Lu M, Tripodi J, Ye F, Najfeld V, et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. Journal of Clinical Investigation. American Society for Clinical Investigation; 2012;122:3888–99. [CrossRef]
- McInnes L, Healy J, Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. 2018;
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. Cell; 2015;162:184–97. [CrossRef]
- Ganesan A, Dermadi D, Kalesinskas L, Donato M, Sowers R, Utz PJ, et al. Inferring direction of associations between histone modifications using a neural processes-based framework. iScience. Elsevier; 2023;26:105756.
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. NIH Public Access; 2017;14:959–62. [CrossRef]
- Eom GH, Kim K, Kim SM, Kee HJ, Kim JY, Jin HM, et al. Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun. Biochem Biophys Res Commun; 2009;388:131–6. [CrossRef]
- Simó-Riudalbas L, Pérez-Salvia M, Setien F, Villanueva A, Moutinho C, Martínez-Cardús A, et al. KAT6B is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Res. American Association for Cancer Research Inc.; 2015;75:3936–44.
- Khokhar ES, Borikar S, Eudy E, Stearns T, Young K, Trowbridge JJ. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp Hematol. Elsevier Inc.; 2020;82:43-52.e4. [CrossRef]
- J F, MA L-D, N H, W L, RB S, Q M, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. Nature; 2013;500:468–71.
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. Genes Dev; 2006;20:2820–32. [CrossRef]
- Li C, Xie J, Lu Z, Chen C, Yin Y, Zhan R, et al. ADCY7 supports development of acute myeloid leukemia. Biochem Biophys Res Commun. Academic Press Inc.; 2015;465:47–52. [CrossRef]
- Zeng Y, Li N, Zheng Z, Chen R, Liu W, Zhu J, et al. A Pan-Cancer Analysis of the Prognostic Value and Expression of Adenylate Cyclase 7 (ADCY7) in Human Tumors. Int J Gen Med. Dove Press; 2021;14:5415. [CrossRef]
- Mu D, Chen L, Zhang X, See LH, Koch CM, Yen C, et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell. Cancer Cell; 2003;3:297–302. [CrossRef]
- LeRoy G, DiMaggio PA, Chan EY, Zee BM, Blanco MA, Bryant B, et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics & Chromatin 2013 6:1. BioMed Central; 2013;6:1–14. [CrossRef]
- Y Z, Q L, H C. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis. Carcinogenesis; 2013;34:1756–63.
- Khan SA, Reddy D, Gupta S. Global histone post-translational modifications and cancer: Biomarkers for diagnosis, prognosis and treatment? http://www.wjgnet.com/. Baishideng Publishing Group Inc.; 2015;6:333–45. [CrossRef]
- J H, AT N, Y Z. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. Blood; 2011;117:3869–80.
- Ferrari KJ, Scelfo A, Jammula SG, Cuomo A, Barozzi I, Stützer A, et al. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol Cell. Cell Press; 2014;53:49–62. [CrossRef]
- AD van D, FW H, YH Q, J C, E J, ESJM de B, et al. Loss of H3K27 methylation identifies poor outcomes in adult-onset acute leukemia. Clin Epigenetics. Clin Epigenetics; 2021;13.
- Zhao W-B, Wang M, Gao S, Shaikh AS, Chen J, Li X-Z. The histone acetyltranseferase KAT8 regulates cell differentiation by suppression of MN1 in AML. Br J Haematol. John Wiley & Sons, Ltd; 2018;182:276–9. [CrossRef]
- Simó-Riudalbas L, Pérez-Salvia M, Setien F, Villanueva A, Moutinho C, Martínez-Cardús A, et al. KAT6B is a tumor suppressor histone H3 lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Research. American Association for Cancer Research Inc.; 2015;75:3936–44.
- Chen Z, Gao W, Pu L, Zhang L, Han G, Zuo X, et al. PRDM8 exhibits antitumor activities toward hepatocellular carcinoma by targeting NAP1L1. Hepatology. John Wiley and Sons Inc.; 2018;68:994–1009. [CrossRef]
- Casamassimi A, Rienzo M, Di Zazzo E, Sorrentino A, Fiore D, Proto MC, et al. Multifaceted Role of PRDM Proteins in Human Cancer. Int J Mol Sci. Multidisciplinary Digital Publishing Institute (MDPI); 2020;21.
- Cypris O, Eipel M, Franzen J, Rösseler C, Tharmapalan V, Kuo C-C, et al. PRDM8 reveals aberrant DNA methylation in aging syndromes and is relevant for hematopoietic and neuronal differentiation. Clinical Epigenetics 2020 12:1. BioMed Central; 2020;12:1–14. [CrossRef]
- Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. Arch Pharm Res; 2015;38:933–49. [CrossRef]
- Eleutherakis-Papaiakovou E, Kanellias N, Kastritis E, Gavriatopoulou M, Terpos E, Dimopoulos MA. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J Oncol. Hindawi Limited; 2020;2020. [CrossRef]
- Kurata T, Matsuda K, Hirabayashi K, Shigemura T, Sakashita K, Nakahata T, et al. Panobinostat inhibits the proliferation of CD34 + CD38 − cells under stimulation of hematopoietic growth factors on AGM-S3 cells in juvenile myelomonocytic leukemia. Pediatric Blood and Cancer. John Wiley and Sons Inc.; 2018;65. [CrossRef]
- Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017 543:7645. Nature Publishing Group; 2017;543:428–32. [CrossRef]
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes and Development. Genes Dev; 2006;20:2820–32. [CrossRef]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell. Elsevier B.V.; 2006;127:125–37.
- Caye A, Strullu M, Guidez F, Cassinat B, Gazal S, Fenneteau O, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. Nature Publishing Group; 2015;47:1334–40. [CrossRef]
- De Vries ACH, Zwaan CM, Van Den Heuvel-Eibrink MM. Molecular basis of juvenile myelomonocytic leukemia. Haematologica. Haematologica; 2010. page 179–82.
- Liu YL, Castleberry RP, Emanuel PD. PTEN deficiency is a common defect in juvenile myelomonocytic leukemia. Leuk Res. Pergamon; 2009;33:671–7.
- D M, L C, X Z, LH S, CM K, C Y, et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer cell. Cancer Cell; 2003;3:297–302.
- Shrestha B, Walton K, Reff J, Sagatys EM, Tu N, Boucher J, et al. Human CD83-targeted chimeric antigen receptor T cells prevent and treat graft-versus-host disease. J Clin Invest. American Society for Clinical Investigation; 2020;130:4652–62. [CrossRef]
- Cieniewicz B, Uyeda MJ, Chen PP, Sayitoglu EC, Liu JMH, Andolfi G, et al. Engineered type 1 regulatory T cells designed for clinical use kill primary pediatric acute myeloid leukemia cells. Haematologica. 2020;Online ahe. [CrossRef]
- JM H, SM D, V V, J M, JA K, A M, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. Blood Adv; 2020;4:5402–13.
- Bai L, Dermadi D, Kalesinskas L, Dvorak M, Chang SE, Ganesan A, et al. Mass-cytometry-based quantitation of global histone post-translational modifications at single-cell resolution across peripheral immune cells in IBD. Journal of Crohn’s and Colitis. 2022;
- Partek Inc. (2020). Partek® Flow® (Version 10.0) [Computer software]. [Internet]. Available from: https://www.partek.com/partek-flow/.
- Smith JP, Sheffield NC. Analytical Approaches for ATAC-seq Data Analysis. Current Protocols in Human Genetics. Blackwell Publishing Inc.; 2020;106:e101. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



