
Submitted:
05 October 2023
Posted:
09 October 2023
You are already at the latest version
Abstract
Understanding the molecular basis of cancer initiation and progression is critical to develop effective treatment strategies. Recently, mutations in genes encoding histone proteins have been identified that drive oncogenesis, converting these essential proteins into “oncohistones”. Understanding how oncohistone mutations, which are commonly missense mutations, subvert the normal function of histones to drive oncogenesis requires defining the functional consequences of such changes. Histones genes are present in multiple copies in the human genome with 15 genes encoding histone H3 isoforms, the histone for which the majority of oncohistone variants have been analyzed thus far. With so many histone genes, engineering human cell lines that assess changes induced by sole expression of the oncohistone variant is technically challenging. In contrast to humans, budding and fission yeast contain only two or three histone H3 genes, respectively. Furthermore, yeast histones share ~90% sequence identity with human H3 protein. The genetic simplicity and evolutionary conservation make yeast an excellent model for characterizing oncohistones. The power of genetic approaches can also be exploited in yeast models to define cellular signaling pathways that could serve as actionable therapeutic targets. In this review, we focus on the value of yeast models to serve as a discovery tool that can inform subsequent translational studies in humans.
Keywords:
Histone
; Oncohistone
; Budding yeast
; Fission yeast
; Epigenetics
; Cancer
Introduction
Cancer is the second leading cause of death in the United States [1]. As a collection of diseases characterized by shared genomic alterations, various cancer types reveal a diversity of mechanisms. Thus, each cancer must be investigated individually to elucidate a detailed mechanistic understanding and enable design of effective therapeutic strategies. Numerous strategies are employed to define mechanisms of cancer initiation and progression with the goal of improving patient outcome. These strategies employ a variety of systems to characterize cancers, from unicellular organisms to computational analyses to animal models. Each of these approaches has strengths and weaknesses, and together, they will enable a full mechanistic and physiological understanding of each cancer type to eventually lead to their eradication. Given the variety of genomic alterations that have been defined as cancer drivers and display a diversity of mechanisms, defining the fundamental cellular processes that underlie those mechanisms is critical. Key discoveries that define our understanding of cellular growth pathways that can contribute to oncogenesis have been made in simple model organisms such as unicellular budding and fission yeast. For example, the basic principles of the cell cycle were initially defined in budding yeast [2]. Many proteins and processes essential to cellular physiology are shared between budding or fission yeast and humans. Therefore, findings in simple eukaryotic model organisms can be readily translated into mammalian systems.
In this review, we highlight the strengths of utilizing yeast models to characterize the molecular basis of cancers caused by mutations that convert critically important histone proteins into oncohistones. We provide a basic introduction to histone function, an overview of what is currently known about oncohistones, how yeast model systems have already contributed to the epigenetics field, and the specific benefits of using these organisms to investigate the molecular disruptions driven by histone mutations in cancers.
Overview of Nucleosomes and Histones
In eukaryotes, DNA is packaged by highly basic histone proteins into the fundamental repeating chromatin unit called a nucleosome [3]. Nucleosomes are nucleoprotein complexes in which ~147 base pairs of DNA is wrapped around a histone octamer consisting of two copies of each core histone protein H2A, H2B, H3, and H4 (Figure 1A) [4,5]. The structure of each histone protein includes a globular domain that interacts with the other histones and associated DNA as well as a flexible N-terminal tail (Figure 1A). Each individual nucleosome is connected by a variable length of linker DNA to form the basic structure of chromatin [6]. The linker histone protein H1 is selectively incorporated into nucleosomes, enabling the repeating nucleosome structures to be further assembled into heterochromatin and compacting the linear DNA into highly condensed chromatin fibers [5,7]. Beyond facilitating the packaging of the genomic material, the structure of chromatin, which largely depends on precise positioning of nucleosomes, dictates the accessibility of cellular machinery to the genome. Hence, chromatin structure determines the ability of this cellular machinery to modulate a wide variety of processes including DNA replication, transcription, cell fate, cell cycle progression, and DNA damage repair via chromatin-mediated regulation [8]. The dynamic and intricate organization of chromatin plays a fundamental role in governing diverse cellular processes, highlighting the essential role of chromatin in the regulation of genomic functions and maintenance within eukaryotic cells.
Histones are among the most evolutionarily conserved proteins in animals, plants, and fungi [9]. These proteins are encoded by multigene families that exhibit modest heterogeneity at the protein level [10]. In humans, the core histone proteins H2A, H2B, H3, and H4 are encoded by 29, 23, 15, and 15 individual genes, respectively, to form 10, 4, 3, and 1 distinct isoforms (Figures 2A and S1) [11]. Though the histone proteins encoded are very similar, these isoforms can perform different functions in regulating biological processes through variable expression and genomic location [12].
One critical regulatory mechanism of chromatin is histone post-translational modification (PTM), which constitutes the “histone code”. The histone code, referring to different spatial or sequential combinations of histone PTMs, choreographs functional activities by modulating the state of the chromatin and maintains homeostasis in response to cellular cues [13]. This code is comprised of molecules covalently bound to histone proteins and include methylation, acetylation, phosphorylation, ubiquitylation, and ADP-ribosylation, among others [8]. The most well studied PTMs occur at the N-terminal tails [8]. Histone PTMs modulate chromatin-dependent functions not only by directly altering the chromatin conformation and DNA-histone interaction, but also by recruiting other cellular machinery. These epigenetic factors play key roles in chromatin remodeling, transcription regulation, replication, and repair, and some are highlighted below [14].
Histones are highly basic proteins with abundant lysine and arginine residues that are subject to various PTMs [15]. Among these PTMs, lysine acetylation, which is a reversible process regulated through the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs), is a well characterized histone modification [14,16]. Several primary acetylation sites on histones have been identified, with the majority of them located within the unstructured N-terminal tail domain. Examples of such sites include H3K14/K18/K23 and H4K5/K8/K12/K16 [17]. Crucial acetylation sites within the globular domain have also been identified, including the extensively studied H3K56ac, which is involved in regulating processes such as DNA-damage response, transcription, and nucleosome packaging [18,19,20]. Acetylation on lysine residues neutralizes the overall positive charge of histones, consequently attenuating interactions between basic histone proteins and negatively charged DNA [21]. Hence, lysine acetylation is generally associated with biological processes dependent on access to DNA, including transcriptional activation and DNA replication, though exceptions exist as well [13,22,23]. In addition to acetylation, methylation is another well characterized PTM. Lysine methylation marks, including mono-, di-, and tri-methylation, and arginine methylation, including mono- and di-methylation, are critical determinants of chromatin state and are involved in processes including regulating DNA damage response and transcription [24,25,26,27,28,29]. In contrast to histone acetylation, methylation does not alter the net charge of histones [14]. Therefore, effects of methylation are thought to be complex, as compared to acetylation, and depend on the location within the chromatin landscape and the degree of methylation [30]. For example, H3K4 methylation is generally associated with transcription activation, yet di-methylation and tri-methylation at this site can trigger transcriptional repression when bound by the co-repressive histone deacetylase complex mSin3a-HDAC1 [31,32]. H3K36 methylation, catalyzed by a set of enzymes in human and exclusively by Set2 in yeasts, is another PTM which modulates diverse cellular processes, including active transcription, DNA damage repair, and splicing, among others [13,33,34,35,36,37,38]. Additionally, many studies have revealed the regulatory roles of a wide range of other histone PTMs, including but not limited to phosphorylation, ubiquitylation, SUMOylation, and ADP-ribosylation [14,39]. The histone code holds tremendous potential and profound implications for biology, highlighting the intricate regulation in place for the epigenome.
Oncohistones are Novel, Cancer-Causing Histone Mutants
The discovery that modest changes to histone sequence can cause a variety of cancers and neurological disorders in humans was surprising [8]. Mutations in histone genes were largely dismissed in earlier epigenetic research because of the genetic redundancy that exists for these genes in humans as histone genes are encoded in clusters resulting in many copies of each histone gene [40]. Focusing on histone H3, which is the protein with the most well-defined variants that contribute to disease, humans have 15 copies of genes encoding H3 variants (Figure 2A). Several other non-canonical H3 encoding genes and isoforms exist (Figure 2A), but their contribution to histone biology is either not well defined or limited by genomic location or cell type specificity. Diploid cells, then, have at least 30 alleles encoding histone H3 protein. A somatic mutation in a single allele was assumed to have low likelihood of producing a phenotype as 29 other wildtype H3 alleles were present, even though not all of these may express protein. Other epigenetic factors, though, have been implicated in cancers [41,42,43,44,45], highlighting the essential roles that dynamic regulation of epigenetic marks play on normal cellular function. Specifically, genomic alterations in enzymes involved in DNA methylation (e.g., DNMT3A) [41], histone marks (e.g., MLL mutations and translocations and histone deacetylases) [42,45], and chromatin remodelers (e.g., SWI/SNF complex members ARID1A and SMARCA4) were identified in various cancers [43]. Underscoring the importance of regular epigenetic programming to promote normal cell function, “nonmutational epigenetic reprogramming” was recently added as a new prospective hallmark of cancer [44]. Clearly, proteins involved in the regulation of chromatin at every level are critical for healthy cellular processes.
A paradigm shift occurred when missense mutations in genes encoding histone proteins were linked to multiple cancers, providing evidence that such mutations could convert critical histone proteins into cancer-driving “oncohistones”. The first oncohistone mutants discovered were H3K27M, K3K36M, and H3G34V/L/R/W [46,47,48]. H3K27M and H3G34R and V were identified in brain tumors such as gliomas and glioblastomas [46,47], while H3G34W and L were discovered in giant cell tumors of bone [48]. H3K36M is present in 95% of chondroblastoma tumors as well as in head and neck squamous cell carcinomas, melanoma, bladder, and colorectal cancers [48,49]. The oncohistone mutants are encountered at higher proportions in pediatric cancer cases as opposed to adults for reasons that are currently unknown. With rare exception, established oncohistone mutations occur in two of the twenty H3 genes, H3-3A and H3-3B, which encode the H3.3 variant [46,47,48] (Figure 2A). Histone variants H3.1 and H3.2 are replication-dependent, meaning they are only incorporated into chromatin during DNA synthesis. Histone H3.3, however, is replication-independent and is deposited throughout the cell cycle, particularly in regions undergoing active transcription and in constitutively heterochromatic regions, such as telomeres [50,51]. The reason for the preference of oncohistones to occur in H3.3 genes is currently not understood, though may be related to the direct impact this histone variant would have on actively transcribed genes.
The major molecular etiology for oncohistone-driven cancers that have been defined thus far appears to be via disruption of key PTMs [52,53]. Both H3K36M and the mutants H3G34R/L/V result in reduced methylation at H3K36 [53,54,55,56]. H3K36M acts as a pseudosubstrate for the H3K36me3 methyl transferase SETD2, outcompeting the wildtype lysine residue by binding the enzyme with high affinity, and sequestering SETD2 to prevent deposition of methyl groups on normal H3 proteins [53,55]. This trapping of SETD2 and the reduction of the enzymatic activity of H3K36me1 and H3K36me2 methyl transferases as well inhibits H3K36 methylation globally [53,55]. The residues H3G33-G34 are also bound by SETD2 and contribute to accurate recognition of the H3K36 target [54,56]. When altered from glycine to arginine, lysine, or valine, these residues impair SETD2 binding to histone H3 and decrease SETD2 enzymatic activity, greatly reducing H3K36 methylation in cis [54,56]. The H3G34 mutants also cause increased H3K27me3 and reduced H3K27ac, though this is secondary to the alterations in H3K36 methylation rather than a direct effect on the H3K27 enzymes [56]. Similar to H3K36M, H3K27M acts as a competitive pseudosubstrate inhibitor for the polycomb repressive complex 2 (PRC2), which contains the H3K27 methyltransferase Ezh2, decreasing Ezh2 activity [52,57,58]. Furthermore, the H3G34 mutants lead to disruptions in gene expression profiles that are similar to those found in cancers [59]. In addition to the oncohistone mutants at H3K27, G34, and K36, there are many other undefined mutations in genes encoding histones that occur at high frequency in cancers [8,60]. The vast majority of these mutants are at residues within the globular domains of the proteins, raising the hypothesis that an additional mode for driving oncogenesis is via disrupted histone-DNA interactions or nucleosome stability [8,60,61].
In each oncohistone mutant, the described disruptions to the histone code lead to alterations in gene expression, changing transcription patterns specifically for H3K36M and H3K27M cells [53,62,63,64]. In the case of H3K36M cells, the reduction in H3K36 methylation causes a redistribution of H3K27me3 from primarily gene to intergenic regions, recruiting the polycomb repressive complex 1 (PRC1) to these new marks and away from typically repressed genes, derepressing them [53,65]. Thus, oncohistones can also cause changes to the epigenetic landscape. Beyond affecting transcription, many other processes may be impacted. H3G34 mutant cells display increased mutation frequencies genome wide, both in engineered in vitro lines and when comparing pediatric glioma whole genome sequencing data sets between H3G34 mutant and non-H3G34 mutant tumors [54], suggesting that DNA repair defects are present in H3G34 tumors. Additionally, methylation at H3K36 is an early signal for recruiting factors for nonhomologous end joining following a double strand break [66,67]. Thus, inhibition of H3K36 methylation in H3K36M cells likely reduces the ability to repair double strand breaks. Various processes are disrupted by oncohistones and may each contribute to cancer development.
In addition to the bona fide oncohistones described above, many other recurrent histone genomic alterations have been associated with cancers, though whether they drive oncogenesis has yet to be fully defined. Various mutations identified in genes encoding the linker histone H1 are primarily associated with lymphomas and are present in other cancer types as well [68,69]. Some of these mutations appear to disrupt the ability of H1 to associate with chromatin [70]. Another histone mutant that clearly improves cellular growth is H2BE76K [61]. This mutation, which lies within the globular domain of histone H2B, destabilizes nucleosome integrity, alters H2B genomic localization, and disrupts gene expression [71]. Further experimentation in a mouse model remains to definitively determine the cancer driving ability of these mutations.
Considering novel oncohistone discovery, analyzing mutation frequencies across all H3 genes may be helpful for identifying patterns that support cancer progression. To explore this idea, we examined mutation frequencies in genes encoding histone H3 across all cancer types using publicly available data from 5406 adult patient tumor samples obtained from COSMIC and cBioPortal databases (Figure 3A). A similar analysis that assessed cancer-associated mutations across all histone genes was performed recently, though with a more limited dataset and a broader scope of genes including all core histones and the linker histone [69]. For the analysis performed here, we focused on the canonical H3 genes and, specifically, the residues that are conserved with S. cerevisiae H3. The top ten residues with the highest mutational burden are H3R2, K27, G34, K36, E50, E73, E97, E105, R116, and R131. As expected, the residues altered in known oncohistone mutants (H3K27, G34, and K36) show the greatest number of changes.
The rest of the residues in the top ten are all initially glutamic acid or arginine, which are both charged amino acids. Altering the charge at these residues has the potential to disrupt interactions between histones and other proteins or DNA, possibly to a greater extent than changes at non-charged residues, which may explain the high frequency of these specific amino acid substitutions. The most common type of mutation identified is a premature stop codon, occurring 2905 times across all residues. Other variants only occur with high frequency at a single residue. For example, the primary amino acid change detected at H3K27 is conversion to methionine (994 of 1215 instances), the primary change detected at H3G34 is conversion to tryptophan (403 of 438 instances), and the primary change detected at H3G34 is conversion to arginine (155 of 239 instances). These specific amino acid substitutions could alter cell physiology in a manner that supports oncogenic properties. An additional pattern that emerged from this analysis was that certain amino acids are converted to specific other amino acids with high frequency (>200 instances) but at no particular position within the histone open reading frame (Figure 3B). For example, a common change is glutamic acid to lysine (E>K) (i.e., H3E50K, E59K, E73K, E94K, E97K, E105K, and E133K), switching the charge at this position from positive to negative. In addition, arginine is commonly changed to histidine or cysteine. We also commonly detected a change from alanine to valine, which would maintain the hydrophobic nature of the residue while modestly increasing the size. Each of these recurrent changes that were not position specific can be achieved by a single base change in the original codon, which may contribute to the bias. Functional studies are required to determine whether these amino acid substitutions alter histone function, providing insight into whether the underlying genetic changes are likely to be passenger mutations or oncogenic drivers. Yeast models can provide an invaluable tool to perform such analyses.
Prior Work Investigating Histone Function in Yeast
Histone proteins are highly conserved between the budding yeast Saccharomyces cerevisiae and humans. For histone H3, where the majority of the well characterized oncohistone mutations are located [8], budding yeast histone H3 protein shares 90% identity with human H3.3 protein (Figure 2B). Additionally, budding yeast histones H2A, H2B, and H4 share 73%, 67%, and 92% identity with their human counterparts, respectively, while Schizosaccharomyces pombe fission yeast histones share 78%, 69%, 92% and 91% identity with human H2A, H2B, H3, and H4 (Figures 2B and S2) [72]. This level of conservation together with the conserved functions means that yeast histones can serve as a model for understanding histone function and consequences of changes that occur in oncohistones in higher eukaryotes, including humans.
In Saccharomyces cerevisiae, histone H2A, H2B, H3, and H4 are each encoded by two gene copies clustered at four loci [73]. The variants H2A.Z and centromeric H3 are each encoded by a single gene [74]. Budding yeast histone H3 is encoded by HHT1 and HHT2, and H4 is encoded by HHF1 and HHF2. These genes cluster at two loci (HHT1-HHF1 and HHT2-HHF2) which are slightly different in DNA sequence but produce identical proteins [75,76]. However, HHT2 and HHF2 contribute more than 80% of the mRNA transcripts encoding cellular histone H3 and H4. The level of the HHT1 and the HHF1 transcripts is upregulated when HHT2 and HHF2 genes are deleted [77]. Despite this fact, minor phenotypic changes have been observed in an hht2∆hhf2∆ cells, but no obvious phenotypic defects were observed in hht1∆hhf1∆ cells [77,78]. The other two clustered loci, HTA1-HTB1 and HTA2-HTB2, encodes two slightly different protein copies of histone H2A and H2B [79], yet these two pairs of genes have non-equivalent roles as the deletion of HTA2 and HTB2 does not result in significant growth defects, but the deletion of HTA1 and HTB1 is lethal to cells unless the remaining HTA2-HTB2 locus is duplicated [73,80]. Hence, the simplicity of minimal gene copies encoding histone proteins render budding yeast an effective model organism for the comprehensive investigation of histones.
Epigenetic discoveries in yeast laid the groundwork for initial characterization of histones and their associated factors, enabling these findings to be translated into a mammalian context. Specific examples include modulators of histone acetylation and their role in regulating transcription. The deacetylase Rpd3 was originally identified in budding yeast through a screen designed to identify transcriptional regulators, which then enabled discovery of the related deacetylase in mammalian cells [81,82]. The acetyl transferase Gcn5 was also first discovered and investigated in budding yeast [83,84]. Additionally, the power of yeast genetics is evident in screens that explored critical roles played by specific histone residues. The results from these screens proved to be foundational to our current understanding of histones. Tandem alanine mutants in histone H3 revealed specific sensitivities to DNA damaging agents and that the αN helix is crucial for chaperone function, particularly for nucleosome assembly and disassembly [85]. Another study that examined alanine or PTM mimetic mutants of H3 and H4 provided insight into which histone residues influence chronological lifespan [86]. This analysis identified various residues and epigenetic marks that either extended or reduced lifespan that may correlate with destabilizing histone-DNA or histone-histone interactions [86]. Furthermore, alanine screening of H4 revealed three adjacent C-terminal residues that are required to protect cells from genome instability and ensure proper histone occupancy across the genome [87]. These systematic epigenetic investigations performed in budding yeast helped to provide a foundational understanding of histone protein structure and function and the resulting discoveries propelled mammalian epigenetic research into analysis of disease-relevant mutations, such as oncohistones. Thus, there is great power in employing model organisms to explore mutational landscapes, including oncohistone mutations.
Advantages of Using Yeast Models to Investigate Oncohistones
While much has been uncovered about oncohistones since their discovery a little over a decade ago [46,47,48], many outstanding questions remain to fully understand how the conversion from essential protein to oncogenic driver occurs. There are limitations to investigating these mutants in humans, so taking advantage of model organisms for broad and deep elucidation of the molecular disruptions caused by oncohistone mutants is a logical and productive approach. A major complication to the investigation of how the molecular changes that occur in oncohistone alter histone function in human cells is the large number of histone genes (Figure 4A). Engineering cell lines that faithfully recapitulate the genetic environment found in disease or that solely express mutant histone in the absence of wild type histone is quite technically challenging. While one recent study in mouse embryonic stem cells introduced an H3K27 mutant (H3K27R) into all 28 alleles of the genes encoding histone H3 [88], the engineering required for designing and generating the lines is not conducive to screening a large number of potential oncohistone mutations for functional changes. Therefore, organisms such as budding or fission yeast can circumvent the challenges posed by mammalian cell lines by having much fewer histone genes and allow modeling of oncohistone mutations in a simpler genetic context (Figure 4B) where the histone variant can be readily expressed as the sole histone protein present in the cell. As the majority of oncohistone studies have focused on histone H3, focusing on this core histone protein is of particular interest. Notably, in comparing yeast and human histone H3, the only residues in the N-terminal tail that are not strictly identical are conservative changes (Figure 2B), meaning that the amino acids likely serve similar biochemical functions. This observation is crucial as the N-terminal tail is critical for the dynamic receipt and sending of epigenetic signals via the histone code. Not only is the protein sequence highly similar, but the 3-dimensional structures of human and yeast H3 proteins are also highly conserved [89]. These similarities suggest that histone H3 performs analogous functions in each species and, thus, that molecular disruptions caused by the oncohistone mutants would be similar.
Importantly, many associated epigenetic factors are also highly conserved between humans and budding yeast [90,91,92]. Given the dynamic and layered regulation that occurs at histones, findings uncovered in model organisms are greatly strengthened by the shared structures and functions with humans at various levels. For example, the human acetyl transferases Gcn5 and Tip60 and the methyl transferase SetD2 have conserved counterparts in yeast [90,91,92]. A notable exception is the PRC2 complex, which methylates H3K27 in humans, that is not present in S. cerevisiae or S. pombe [93]. The conservation at multiple layers of epigenetic regulation further supports the use of yeast to model oncohistones.
The yeast model systems offer flexibility in how to analyze the functional consequences of oncohistone mutations. In a human cell line, the 15 copies of histone H3 makes generating cells that express the oncohistone protein as the sole histone protein is technically quite challenging (Figure 4A), although this task has been accomplished for H3K27 [88]. However, in the budding yeast system, where each histone is encoded by only two genes, one can readily engineer cells that express an oncohistone as the sole histone protein present. In fact, offering flexibility, one copy of a histone gene can be altered using genome editing and then the other histone gene can be retained intact or deleted (Figure 4B). Studies can then be performed using cells where a single allele is edited and the other allele is wild type, for example hht2-K36M HHT1, to more closely model what happens in cancer cells where the oncohistone allele is dominant to the numerous wild type alleles of any histone gene (Figure 4B). Alternatively, if the locus encoding the second histone gene is deleted, this creates a model where the oncohistone variant is the sole histone protein expressed in cells (for example hht2-K36M hht1Δ) (Figure 4B). This latter experimental design allows direct analysis of how a specific oncohistone mutant alters histone function. These two scenarios provide the flexibility to assess 1) dominant phenotypes in the presence of the other wildtype histone gene, which aligns with what happens in oncohistone-driven cancers where only a single allele of many is altered and 2) recessive phenotypes in the absence of any wildtype histone gene. The ease of generating these models allows rapid screening of a large number of potential oncohistone mutations to assess whether they are likely to alter histone function.
Another advantage of the yeast model system is the ability to screen for numerous phenotypes by plating oncohistone models on plates under a variety of conditions, including different temperatures, nutrient sources, and drugs that disrupt different pathways [94] (Figure 4C). Such growth assays have been exploited extensively to systematically analyze the functional importance of different histone residues for specific PTMs and cell fitness [95,96,97]. As an example of this modeling, we performed a serial dilution assay of the oncohistone H3K36M and the mutant H3K36R, which has been identified in T-cell acute lymphoblastic leukemia, though its oncogenicity is uncertain [98], expressed both in the presence of wildtype H3 (hht2-K36M/R HHT1 cells) and as the sole copy of H3 (hht2-K36M/R hht1Δ) (Figure 4C). To illustrate this experimental approach, results of such an experiment are presented in Figure 4C. For this experiment, yeast cells were grown in culture, serially diluted and plated on control plates, plates that contain caffeine, which induces cellular stress (ref), or plates that contain phleomycin, which induces double strand breaks (ref). H3K36M cells show sensitivity to cellular stress and double strand breaks at similar levels in the presence or absence of wildtype protein. On the other hand, H3K36R cells show an increase in sensitivity to cellular stress and double strand breaks when H3K36R is expressed as the sole copy of histone H3 (Figure 4C). Analyses such as these can provide insight into the genetic and biochemical properties of oncohistone mutants. Yeast oncohistone models could be valuable to extend such an analysis to explore specific amino acid changes identified in cancer patients such as those displayed in Figure 3 to identify those mutations that alter histone function. The power of yeast genetics means that when phenotypes are linked to specific amino acid changes, those phenotypes can be exploited as the basis for a genetic screen to identify specific pathways that could intersect with the particular oncohistone mutant [99]. These pathways have the potential to represent therapeutically actionable targets in oncohistone-driven cancers.
Despite the fact that yeast do not develop cancer, taking advantage of these approaches has revealed altered growth patterns associated with yeast oncohistone models. In budding yeast, expression of either H3K36M or H3K36R as the sole copy of H3 results in reduced growth in the presence of caffeine, suggesting sensitivity of these oncohistone model cells to cellular stress [99]. The caffeine sensitivity is also observed when methylation at H3K36 is prevented by deleting the SET2 gene, encoding the H3K36 methyltransferase [27]. As described above, one of the greatest strengths of using yeast as a model system is the ability to perform unbiased screens. To identify factors and pathways upon which H3K36 mutants are specifically sensitive, our group took advantage of the caffeine-sensitive growth of H3K36 mutant cells in the absence of wild type protein to perform a high copy suppressor screen [99]. The goal of such a screen is to identify suppressors that may identify therapeutically actionable pathways in the corresponding cancers. We identified various suppressors of growth defects on caffeine [99], some of which have known roles in regulating histone function, such as the lysine acetyltransferase Esa1 [100] and a putative transcriptional regulator that interacts with Rpd3 and Set3 histone deacetylase complexes, Tos4 [101]. Further experimentation to elucidate the mechanism of suppression has the potential to provide insight into mechanisms that underlie altered histone function in cancer.
Many other investigations of oncohistone mutants in yeast have provided insight into the physiological changes that could contribute to oncogenesis. H3G34R fission yeast mutants result in reduced levels of methylation and acetylation at H3K36 [52]. H3G34V mutants in fission yeast reveal sensitivity to induced DNA double strand breaks, while H3G34R mutants are sensitive to DNA replication stress and defective in homologous recombination [63,102]. These results are congruent with what is found in humans, that cells expressing mutant H3G34 display heightened frequency of genomic mutations [54]. H3K27 methylation is absent in S. cerevisiae and S. pombe, possibly because their mechanisms for inhibiting gene transcription is distinct from the heterochromatin present in mammalian systems [93]. Acetylation at this site does occur in yeast, however [103]. Due to this inconsistency with human epigenetic regulation, as well as the absence of the PRC2 complex as described above, yeast have not been employed as a model for the H3K27M oncohistone.
The strengths of employing yeast models to characterize novel oncohistones are exemplified through investigations of a putative H2B oncohistone. The variant H2BE76K, which occurs in bladder and head and neck cancers, is found in the globular domain of histone H2B [61]. In budding yeast, engineering one H2B gene to harbor the H2BE76K mutation while maintaining one wildtype copy causes temperature sensitivity and reduced nucleosome stability [61]. Additionally, an array of variants in histone residues located in the acidic patch or histone-DNA interface that are common in cancers displayed increased chromatin remodeling processes and lethality growth defects in fission yeast, suggesting that cancer-related mutants at these residues could also be investigated in the model organism [104]. Yeast model organisms have proven to be useful not only for initial fundamental histone investigations, but also for ongoing characterization of clinically relevant oncohistone mutants. The genetic simplicity of these models, ability to readily screen for phenotypes, and the power of genetic screens will continue to provide insight into disease etiology that can be readily extended to humans.
Conclusions
Much of the field of epigenetics was initially discovered in model organisms such as yeast. While key fundamental discoveries have emerged from studies in simple model organisms, yeast can model human disease-causing mutations, including oncohistones, in ways that facilitate discovery. In recent years, budding yeast oncohistone models were used in a screen to identify potentially therapeutically-actionable suppressors of growth alterations [99], and the impact of oncohistone mutants on DNA repair was characterized in fission yeast [63]. Analyzing candidate oncohistones, here we present recurrent changes that occur at amino acids conserved between human H3.3 and budding yeast, which may be worthy of further investigation to assess their contribution to disrupted growth patterns. We also provide a comprehensive comparison of gene number, protein variants, and protein sequences between human and yeast histones that should be a valuable resource for the scientific community. In summary, yeast serves as an excellent model for the analysis of oncohistone mutants, providing a simple system to assay function either in the presence or the absence of wild type histone protein, which is technically demanding in a mammalian system. The findings revealed in yeast will drive the field forward, enabling molecular insights to initiate translational studies and, hopefully, lead to improved treatment paradigms for patients with oncohistone-driven cancers.
Supplementary Materials
Figure S1: (A) Schematic of gene encoding Histone H2A in Yeast and Humans; (B) Schematic of gene encoding Histone H2B in Yeast and Humans; (C) Schematic of gene encoding Histone H4 in Yeast and Humans. Figure S2: (A) H2A protein sequence alignments between Yeast and Humans; (B) H2B protein sequence alignments between Yeast and Humans; (C) H4 protein sequence alignments between Yeast and Humans.
Author Contributions
Xinran Zhang: Conceptualization, Writing – Original Draft; Dorelle V. Fawwal: Data Curation; Jennifer M. Spangle: Writing – Reviewing and Editing; Anita H. Corbett: Conceptualization, Writing – Original Draft, Writing – Reviewing and Editing, Supervision; Celina Y. Jones: Conceptualization, Writing – Original Draft, Writing – Reviewing and Editing, Visualization, Supervision.
Funding
R21 (R21CA256456) to AHC and JMS, NSF (NSF CAREER AS32477) to JMS, MIRA (R35GM150587) to JMS, T32 (NIH 5T32GM135060-04) to AHC and DVF, IRACDA (NIH 5K12GM000680-24) to AHC and CYJ.
Acknowledgments
We would like to thank Dr. Milo Fasken for his work generating yeast strains used in our prior and current oncohistone investigations and his insight in conceptualizing the direction of oncohistone projects. We would also like to thank Dr. Kirti Sad for helpful discussions bridging the yeast and human oncohistone data.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Mortality in the United States, 2021. NCHS Data Brief 2022, 1–8. [Google Scholar]
- Hartwell, L.H. Yeast and cancer. Biosci Rep 2004, 24, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M. Histone structure and function. Curr Opin Cell Biol 1991, 3, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Grunstein, M. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol 2006, 7, 657–666. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Stützer, A.; Liokatis, S.; Kiesel, A.; Schwarzer, D.; Sprangers, R.; Söding, J.; Selenko, P.; Fischle, W. Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol Cell 2016, 61, 247–259. [Google Scholar] [CrossRef]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of 'oncohistone' mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Albers, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P.; Wilson, J.; Hunt, T. Molecular Biology of the Cell, 5th edn.(Garland Science, New York, 2008).
- Flaus, A.; Downs, J.A.; Owen-Hughes, T. Histone isoforms and the oncohistone code. Curr Opin Genet Dev 2021, 67, 61–66. [Google Scholar] [CrossRef]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A standardized nomenclature for mammalian histone genes. Epigenetics Chromatin 2022, 15, 34. [Google Scholar] [CrossRef]
- Singh, R.; Bassett, E.; Chakravarti, A.; Parthun, M.R. Replication-dependent histone isoforms: a new source of complexity in chromatin structure and function. Nucleic Acids Res 2018, 46, 8665–8678. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta 2009, 1789, 45–57. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Thorne, A.W.; Kmiciek, D.; Mitchelson, K.; Sautiere, P.; Crane-Robinson, C. Patterns of histone acetylation. Eur J Biochem 1990, 193, 701–713. [Google Scholar] [CrossRef]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. Embo j 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem 2006, 281, 37270–37274. [Google Scholar] [CrossRef]
- Bernier, M.; Luo, Y.; Nwokelo, K.C.; Goodwin, M.; Dreher, S.J.; Zhang, P.; Parthun, M.R.; Fondufe-Mittendorf, Y.; Ottesen, J.J.; Poirier, M.G. Linker histone H1 and H3K56 acetylation are antagonistic regulators of nucleosome dynamics. Nat Commun 2015, 6, 10152. [Google Scholar] [CrossRef]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 "tail" to DNA. J Biol Chem 1993, 268, 305–314. [Google Scholar] [CrossRef]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 2006, 23, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M.; O'Neill, L.P. Histone acetylation in chromatin and chromosomes. Semin Cell Biol 1995, 6, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; Yue, W.W.; Oppermann, U.; Klose, R.J. Dynamic protein methylation in chromatin biology. Cell Mol Life Sci 2009, 66, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat Res Rev Mutat Res 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gogol, M.; Carey, M.; Pattenden, S.G.; Seidel, C.; Workman, J.L. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev 2007, 21, 1422–1430. [Google Scholar] [CrossRef]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 2006, 75, 243–269. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A 2002, 99, 8695–8700. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Lam, U.T.F.; Tan, B.K.Y.; Poh, J.J.X.; Chen, E.S. Structural and functional specificity of H3K36 methylation. Epigenetics Chromatin 2022, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.K.; Nguyen, T.T.T.; Li, A.Y.; Yeo, Y.P.; Chen, E.S. Histone H3 lysine 36 methyltransferase mobilizes NER factors to regulate tolerance against alkylation damage in fission yeast. Nucleic Acids Res 2018, 46, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.C.; Deegan, R.S.; Subramanian, L.; Gal, C.; Sarkar, S.; Blaikley, E.J.; Walker, C.; Hulme, L.; Bernhard, E.; Codlin, S.; et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat Commun 2014, 5, 4091. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Fong, N.; Erickson, B.; Bentley, D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A 2011, 108, 13564–13569. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, M.R.; Jha, D.K.; Ucles, S.A.; Flood, D.M.; Strahl, B.D.; Stevens, S.W.; Kress, T.L. Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol 2016, 13, 412–426. [Google Scholar] [CrossRef]
- Yuan, H.; Li, N.; Fu, D.; Ren, J.; Hui, J.; Peng, J.; Liu, Y.; Qiu, T.; Jiang, M.; Pan, Q.; et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest 2017, 127, 3375–3391. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Cheung, W.L.; Hsu, J.Y.; Diaz, R.L.; Smith, M.M.; Allis, C.D. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 2005, 120, 25–36. [Google Scholar] [CrossRef]
- Sierra, F.; Lichtler, A.; Marashi, F.; Rickles, R.; Van Dyke, T.; Clark, S.; Wells, J.; Stein, G.; Stein, J. Organization of human histone genes. Proc Natl Acad Sci U S A 1982, 79, 1795–1799. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R. Sophisticated Conversations between Chromatin and Chromatin Remodelers, and Dissonances in Cancer. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet 2017, 49, 180–185. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res 2011, 21, 421–434. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc Natl Acad Sci U S A 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shan, C.M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep 2017, 7, 43906. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J Mol Biol 2018, 430, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 2016, 7, 11316. [Google Scholar] [CrossRef]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc Natl Acad Sci U S A 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Mitchener, M.M.; Muir, T.W. Oncohistones: Exposing the nuances and vulnerabilities of epigenetic regulation. Mol Cell 2022, 82, 2925–2938. [Google Scholar] [CrossRef]
- Bennett, R.L.; Bele, A.; Small, E.C.; Will, C.M.; Nabet, B.; Oyer, J.A.; Huang, X.; Ghosh, R.P.; Grzybowski, A.T.; Yu, T.; et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov 2019, 9, 1438–1451. [Google Scholar] [CrossRef]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol 2019, 137, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J Biol Chem 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci U S A 2011, 108, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Hu, X.; Jing, Q.; Zeng, X.; Chan, K.M.; Han, J. Mechanism of cancer: Oncohistones in action. J Genet Genomics 2018, 45, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Bonner, E.R.; Dawood, A.; Gordish-Dressman, H.; Eze, A.; Bhattacharya, S.; Yadavilli, S.; Mueller, S.; Waszak, S.M.; Nazarian, J. Pan-cancer atlas of somatic core and linker histone mutations. NPJ Genom Med 2023, 8, 23. [Google Scholar] [CrossRef]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014, 46, 176–181. [Google Scholar] [CrossRef]
- Kang, T.Z.E.; Zhu, L.; Yang, D.; Ding, D.; Zhu, X.; Wan, Y.C.E.; Liu, J.; Ramakrishnan, S.; Chan, L.L.; Chan, S.Y.; et al. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J Biol Chem 2021, 296, 100374. [Google Scholar] [CrossRef]
- McBurney, K.L.; Leung, A.; Choi, J.K.; Martin, B.J.; Irwin, N.A.; Bartke, T.; Nelson, C.J.; Howe, L.J. Divergent Residues Within Histone H3 Dictate a Unique Chromatin Structure in Saccharomyces cerevisiae. Genetics 2016, 202, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Libuda, D.E.; Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature 2006, 443, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.R.; Ganguli, D.; Nagarajavel, V.; Clark, D.J. Regulation of histone gene expression in budding yeast. Genetics 2012, 191, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M.; Murray, K. Yeast H3 and H4 histone messenger RNAs are transcribed from two non-allelic gene sets. J Mol Biol 1983, 169, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, Y.; Wang, A.; Qin, Y.; Luo, M.; Wu, Q.; Boeke, J.D.; Dai, J. Construction of Comprehensive Dosage-Matching Core Histone Mutant Libraries for Saccharomyces cerevisiae. Genetics 2017, 207, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.L.; Smith, M.M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol 1988, 8, 945–954. [Google Scholar] [CrossRef]
- Liang, D.; Burkhart, S.L.; Singh, R.K.; Kabbaj, M.H.; Gunjan, A. Histone dosage regulates DNA damage sensitivity in a checkpoint-independent manner by the homologous recombination pathway. Nucleic Acids Res 2012, 40, 9604–9620. [Google Scholar] [CrossRef]
- Hereford, L.; Fahrner, K.; Woolford, J., Jr.; Rosbash, M.; Kaback, D.B. Isolation of yeast histone genes H2A and H2B. Cell 1979, 18, 1261–1271. [Google Scholar] [CrossRef]
- Norris, D.; Osley, M.A. The two gene pairs encoding H2A and H2B play different roles in the Saccharomyces cerevisiae life cycle. Mol Cell Biol 1987, 7, 3473–3481. [Google Scholar] [CrossRef]
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol Cell Biol 1991, 11, 6317–6327. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, T.; Thireos, G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. Embo j 1992, 11, 4145–4152. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Brownell, J.E.; Sobel, R.E.; Ranalli, T.A.; Cook, R.G.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 1996, 383, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Kim, H.J.; Yoo, J.K.; Park, H.J.; Cho, E.J. Analysis of Saccharomyces cerevisiae histone H3 mutants reveals the role of the alphaN helix in nucleosome function. Biochem Biophys Res Commun 2008, 374, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Ngubo, M.; Reid, J.L.; Patterton, H.G. Distinct structural groups of histone H3 and H4 residues have divergent effects on chronological lifespan in Saccharomyces cerevisiae. PLoS One 2022, 17, e0268760. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Srinivasan, M.; Nakanishi, S.; Leatherwood, J.; Shilatifard, A.; Sternglanz, R. A conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol Cell Biol 2011, 31, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Sankar, A.; Mohammad, F.; Sundaramurthy, A.K.; Wang, H.; Lerdrup, M.; Tatar, T.; Helin, K. Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat Genet 2022, 54, 754–760. [Google Scholar] [CrossRef]
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. Embo j 2001, 20, 5207–5218. [Google Scholar] [CrossRef]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol 2004, 24, 1884–1896. [Google Scholar] [CrossRef]
- Spedale, G.; Timmers, H.T.; Pijnappel, W.W. ATAC-king the complexity of SAGA during evolution. Genes Dev 2012, 26, 527–541. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Xue, H.; Cao, M.; Bai, G.; Mu, Z.; Yao, Y.; Sun, S.; Fang, D.; Huang, J. Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discov 2021, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; Sengupta, R.; Schotta, G.; Jenuwein, T. Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome. Cold Spring Harb Symp Quant Biol 2004, 69, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Hyland, E.M.; Yuan, D.S.; Huang, H.; Bader, J.S.; Boeke, J.D. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 2008, 134, 1066–1078. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: a database for histone mutations and their phenotypes. Genome Res 2009, 19, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Sanderson, B.W.; Delventhal, K.M.; Bradford, W.D.; Staehling-Hampton, K.; Shilatifard, A. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol 2008, 15, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Collord, G.; Martincorena, I.; Young, M.D.; Foroni, L.; Bolli, N.; Stratton, M.R.; Vassiliou, G.S.; Campbell, P.J.; Behjati, S. Recurrent histone mutations in T-cell acute lymphoblastic leukaemia. Br J Haematol 2019, 184, 676–679. [Google Scholar] [CrossRef]
- Lemon, L.D.; Kannan, S.; Mo, K.W.; Adams, M.; Choi, H.G.; Gulka, A.O.D.; Withers, E.S.; Nurelegne, H.T.; Gomez, V.; Ambrocio, R.E.; et al. A Saccharomyces cerevisiae model and screen to define the functional consequences of oncogenic histone missense mutations. G3 (Bethesda) 2022, 12. [Google Scholar] [CrossRef]
- Smith, E.R.; Eisen, A.; Gu, W.; Sattah, M.; Pannuti, A.; Zhou, J.; Cook, R.G.; Lucchesi, J.C.; Allis, C.D. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc Natl Acad Sci U S A 1998, 95, 3561–3565. [Google Scholar] [CrossRef]
- Cooke, S.L.; Soares, B.L.; Müller, C.A.; Nieduszynski, C.A.; Bastos de Oliveira, F.M.; de Bruin, R.A.M. Tos4 mediates gene expression homeostasis through interaction with HDAC complexes independently of H3K56 acetylation. J Biol Chem 2021, 296, 100533. [Google Scholar] [CrossRef]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- D'Arcy, S.; Luger, K. Understanding histone acetyltransferase Rtt109 structure and function: how many chaperones does it take? Curr Opin Struct Biol 2011, 21, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat Chem Biol 2021, 17, 403–411. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Oncohistone mutants confer oncogenic growth. (A) Nucleosomes are comprised of two copies of each of the four core histone proteins: H2A, H2B, H3, and H4. When wild type histones are present, cells show normal growth. (B) When an oncohistone missense mutation occurs, such as H3K36M (red), various functional consequences may result. Even with many wildtype histone H3 proteins present, a single mutation in one allele can confer oncogenic growth through a variety of mechanisms, including altered post translational modifications, modification of the genomic landscape, and/or disruption of gene expression.
Figure 1.
Oncohistone mutants confer oncogenic growth. (A) Nucleosomes are comprised of two copies of each of the four core histone proteins: H2A, H2B, H3, and H4. When wild type histones are present, cells show normal growth. (B) When an oncohistone missense mutation occurs, such as H3K36M (red), various functional consequences may result. Even with many wildtype histone H3 proteins present, a single mutation in one allele can confer oncogenic growth through a variety of mechanisms, including altered post translational modifications, modification of the genomic landscape, and/or disruption of gene expression.
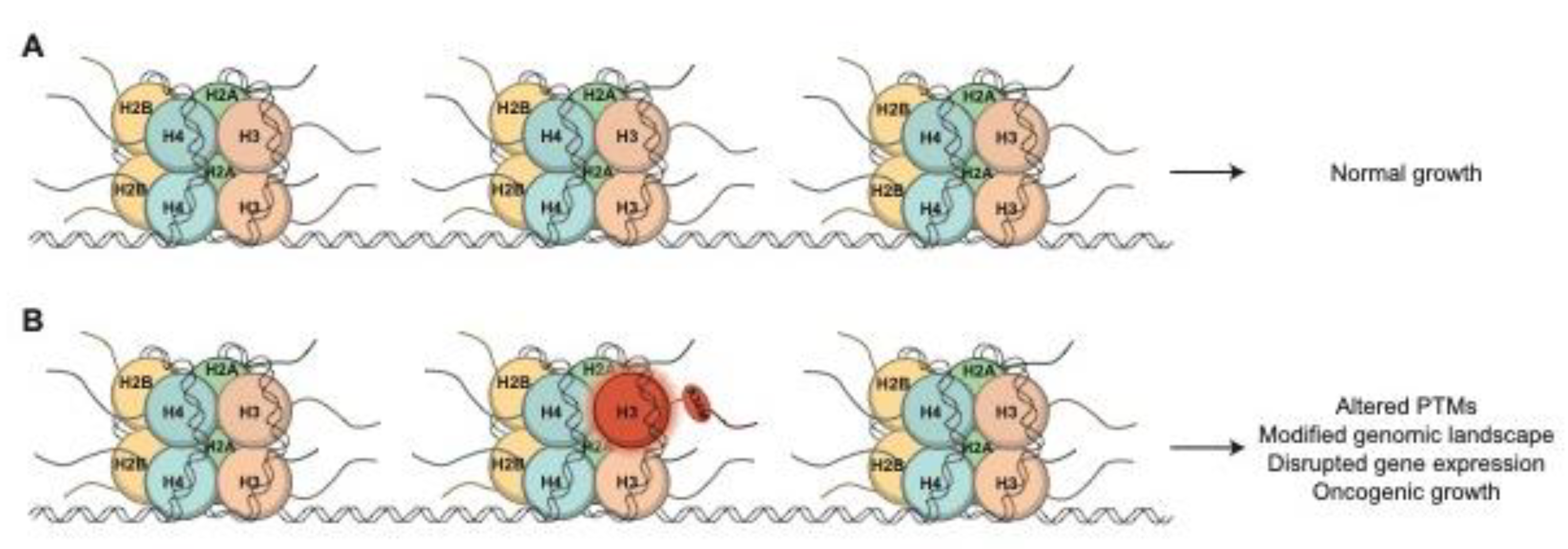
Figure 2.
A comparison of histone H3 from S. cerevisiae, S. pombe, and H. sapiens. (A) The number of gene copies and protein variants differs greatly between yeast species (S. cerevisiae and S. pombe), which are termed HHT genes and humans (H. sapiens), which are termed H3XX, with the exception of the specialized centromeric histone CENPA, for histone H3. The genes that encode each variant are color coordinated with their respective Histone Variants. *Non-canonical H3 genes and variants. (B) Protein sequence alignments for histone H3 in S. cerevisiae and the three canonical H3 variants in H. sapiens. Orange residues represent conservative changes, where the biochemical properties of the amino acid are maintained, and blue residues represent non conservative changes, where the biochemical properties are altered. (C) Protein sequence alignments for histone H3 in S. pombe and the three canonical H3 variants in H. sapiens. Color coding is same as for (B).
Figure 2.
A comparison of histone H3 from S. cerevisiae, S. pombe, and H. sapiens. (A) The number of gene copies and protein variants differs greatly between yeast species (S. cerevisiae and S. pombe), which are termed HHT genes and humans (H. sapiens), which are termed H3XX, with the exception of the specialized centromeric histone CENPA, for histone H3. The genes that encode each variant are color coordinated with their respective Histone Variants. *Non-canonical H3 genes and variants. (B) Protein sequence alignments for histone H3 in S. cerevisiae and the three canonical H3 variants in H. sapiens. Orange residues represent conservative changes, where the biochemical properties of the amino acid are maintained, and blue residues represent non conservative changes, where the biochemical properties are altered. (C) Protein sequence alignments for histone H3 in S. pombe and the three canonical H3 variants in H. sapiens. Color coding is same as for (B).
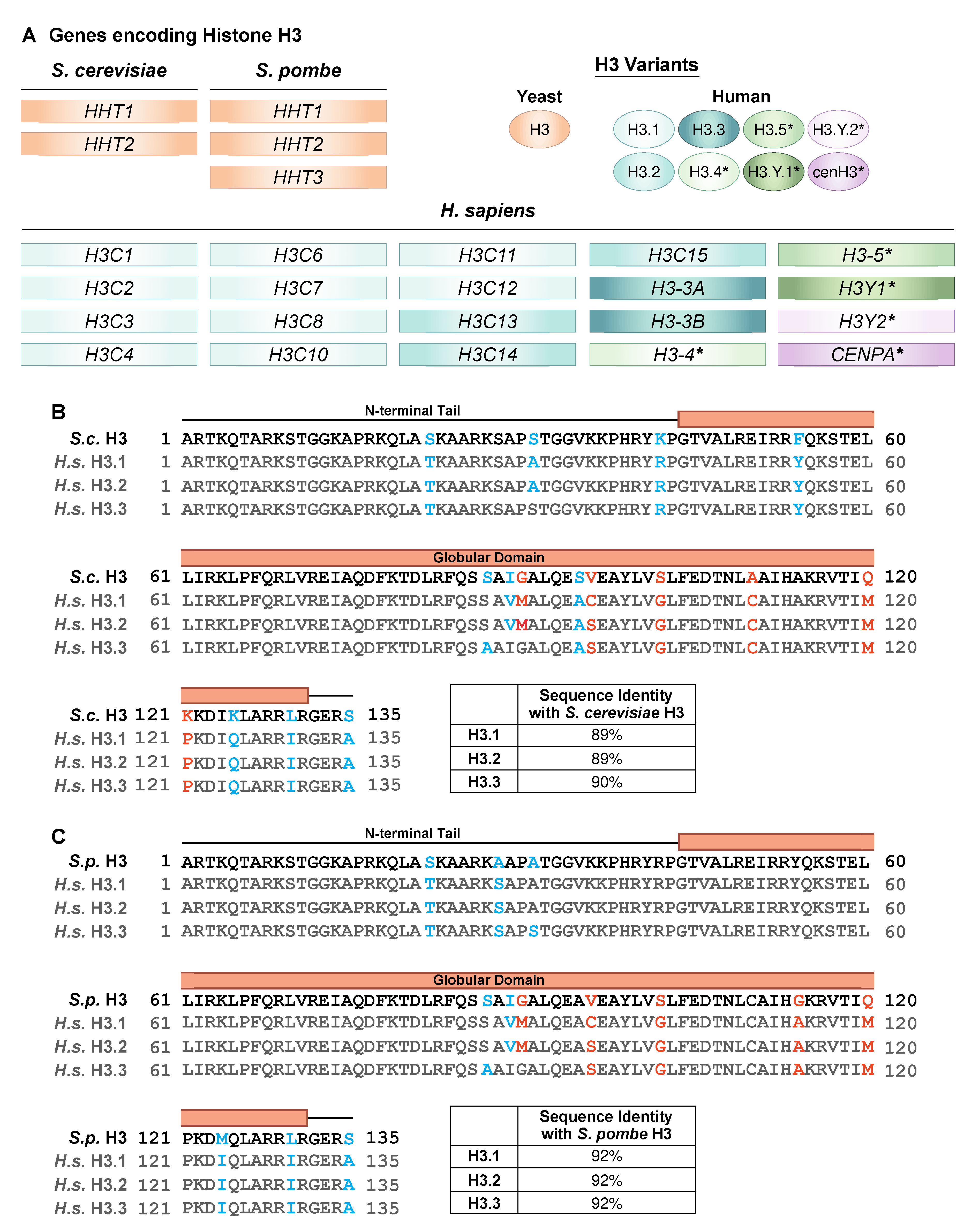
Figure 3.
Mutations that alter histone H3 sequence recur in human cancers. (A) A cross cancer mutation analysis was performed utilizing 5,406 adult patient samples obtained from the COSMIC and cBioPortal databases. Samples from the two databases were cross-referenced and duplicate entries were removed. The data were then visualized using PRISM GraphPad. This summary includes missense mutations found in H3C1, H3C2, H3C3, H3C4, H3C5, H3C6, H3C7, H3C8, H3C10, H3C11, H3C12, H3C13, H3C14, H3C15, H3-3A, H3-3B, and H3-4. The number of patients that have a mutation (missense or premature termination codon as indicated by *) located in the codon corresponding to each specific amino acid residue along the protein is indicated on the Y-axis (#Mutations across Cancers) with the residues that the amino acid is altered to indicated by the colors or the asterisk (*). Only residues that are conserved in S. cerevisiae H3 are shown along the X-axis as these represent potential oncohistones that could be modeled in budding yeast. (B) The number of events for which a given amino acid is converted into all other amino acids was assessed, regardless of position in the protein sequence. Premature termination codons were excluded to focus on missense mutants. Same dataset as in (A).
Figure 3.
Mutations that alter histone H3 sequence recur in human cancers. (A) A cross cancer mutation analysis was performed utilizing 5,406 adult patient samples obtained from the COSMIC and cBioPortal databases. Samples from the two databases were cross-referenced and duplicate entries were removed. The data were then visualized using PRISM GraphPad. This summary includes missense mutations found in H3C1, H3C2, H3C3, H3C4, H3C5, H3C6, H3C7, H3C8, H3C10, H3C11, H3C12, H3C13, H3C14, H3C15, H3-3A, H3-3B, and H3-4. The number of patients that have a mutation (missense or premature termination codon as indicated by *) located in the codon corresponding to each specific amino acid residue along the protein is indicated on the Y-axis (#Mutations across Cancers) with the residues that the amino acid is altered to indicated by the colors or the asterisk (*). Only residues that are conserved in S. cerevisiae H3 are shown along the X-axis as these represent potential oncohistones that could be modeled in budding yeast. (B) The number of events for which a given amino acid is converted into all other amino acids was assessed, regardless of position in the protein sequence. Premature termination codons were excluded to focus on missense mutants. Same dataset as in (A).
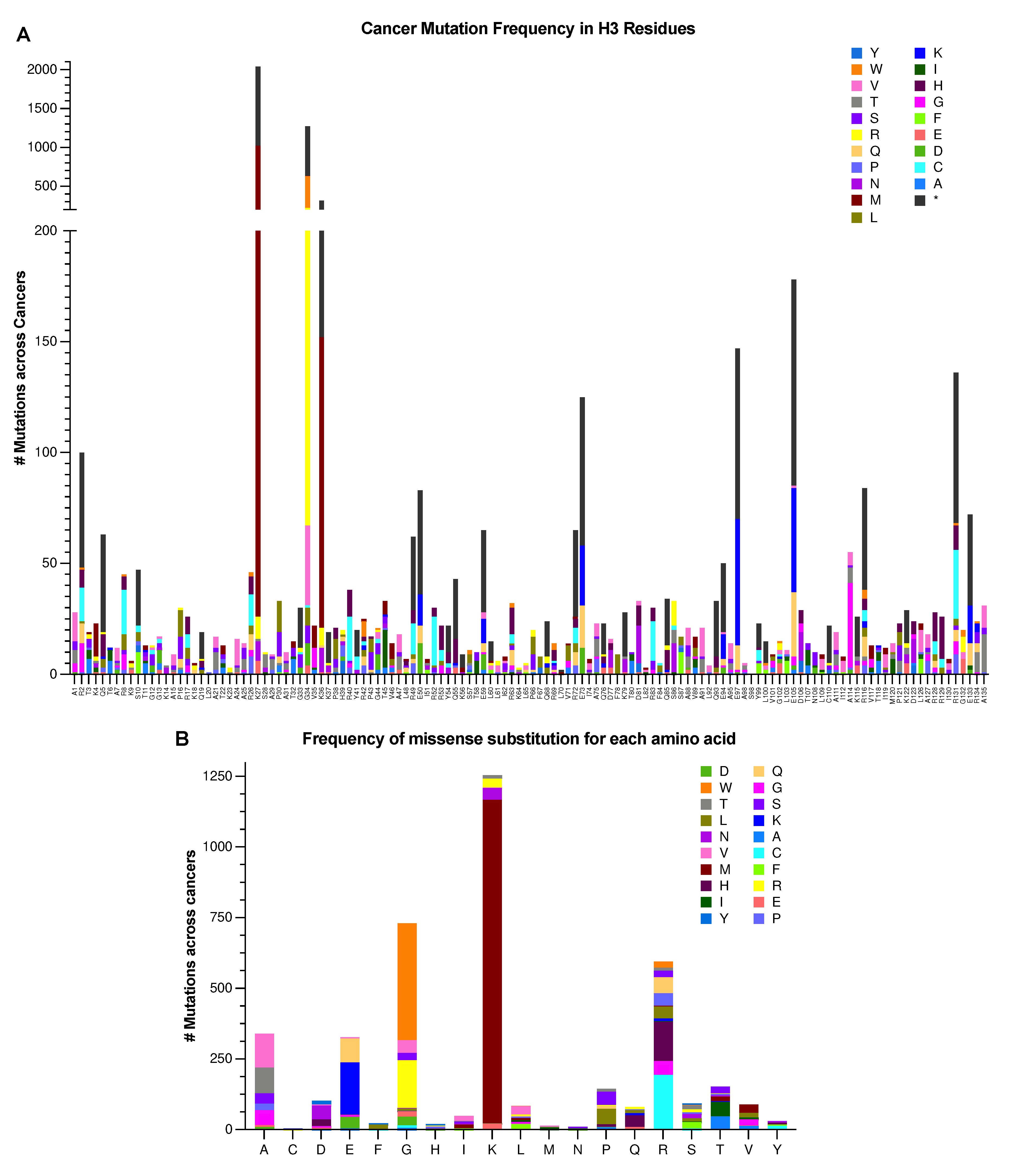
Figure 4.
The strength of modeling oncohistone mutants in budding yeast. (A) The human genome encodes 15 copies of histone H3, making creating cells that express an oncohistone as the sole histone protein a very technically challenging task. (B) Budding yeast can be easily engineered to maintain wildtype H3 genes (top), express one mutant and one wildtype H3 (middle), or express only mutant H3 (bottom). The resulting ratio of wildtype to mutant H3 proteins in a pair of nucleosomes for the given genotypes is depicted to the right. (C) In this serial dilution growth assay, budding yeast cultures were diluted to OD=5 and serially diluted ten-fold before plating onto control or drug plates. The analysis compares H3K36 mutant cells that either have one mutant and one wildtype histone protein (hht2-K36M/R HHT1) and cells that contain mutant histone as the sole copy of histone H3 (hht2-K36M/R hht1Δ) to control wildtype (WT) cells. Cells on control plates were grown for two days, cells on phleomycin were grown for three days, and cells on caffeine were grown for five days. The cellular damage caused by the drugs is indicated below the plates, and the ratio of wildtype to mutant H3 in the nucleosomes within each model is depicted to the right.
Figure 4.
The strength of modeling oncohistone mutants in budding yeast. (A) The human genome encodes 15 copies of histone H3, making creating cells that express an oncohistone as the sole histone protein a very technically challenging task. (B) Budding yeast can be easily engineered to maintain wildtype H3 genes (top), express one mutant and one wildtype H3 (middle), or express only mutant H3 (bottom). The resulting ratio of wildtype to mutant H3 proteins in a pair of nucleosomes for the given genotypes is depicted to the right. (C) In this serial dilution growth assay, budding yeast cultures were diluted to OD=5 and serially diluted ten-fold before plating onto control or drug plates. The analysis compares H3K36 mutant cells that either have one mutant and one wildtype histone protein (hht2-K36M/R HHT1) and cells that contain mutant histone as the sole copy of histone H3 (hht2-K36M/R hht1Δ) to control wildtype (WT) cells. Cells on control plates were grown for two days, cells on phleomycin were grown for three days, and cells on caffeine were grown for five days. The cellular damage caused by the drugs is indicated below the plates, and the ratio of wildtype to mutant H3 in the nucleosomes within each model is depicted to the right.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



