Submitted:
07 October 2023
Posted:
08 October 2023
You are already at the latest version
Abstract
Ranging from traditional food packaging, clothing, and furniture to the current small and large electronic devices and automobiles, plastics serve to fulfill diverse demands in our daily lives. However, the global plastic waste generation is dramatically escalating, currently standing at approximately 150 million metric tonnes annually. While some of regenerated plastics recycled by mechanical methods can be used as their parent plastics, cost and energy savings are limited by multiple preliminary processes such as plastic sorting, shredding, washing, and drying. Moreover, the continuous mechanical recycling process degrades the physical properties of the materials. In this context, chemical recycling is emerging as a promising alternative method due to its high efficiency, simple preliminary steps, reducing reliance on fossil resources, and conversion of plastic waste into value-added chemicals. This review provides a state-of-the-art overview of contemporary chemical recycling of polymeric materials via i) depolymerization: “polymers to small valuable molecules” and ii) closed-loop cycles: “polymers to monomers, and/or to polymers”, by encompassing both traditional/advanced depolymerization chemistries and the remaining challenges. These recycling approaches are contextualized within the present industrial technologies, key design principles, and specific recycling case studies related to distinct polymeric materials.
Keywords:
Recycling
; Plastic wastes
; Chemical recycling
; Depolymerization
; Closed-loop recycling
1. Introduction
Plastics have become an essential part of our daily lives, ranging from food packaging, clothing, and housing to the current state-of-the-art technologies found in smart phones, vehicles, and medical equipment. The durability, versatile properties, cost-effectiveness, and light weight of plastics have made them ubiquitous in our daily lives. However, the unintended consequence of this ubiquity is the unprecedented surge in global plastic waste generation[1]. Between 1950 and 2015, cumulative waste generation of primary and secondary (i.e., recycled) plastic waste reached 6300 Mt; with estimates indicating that 2500 Mt of them are currently in use (Figure 1)[2,3]. Within this extensive volume, the bulk of discarded plastics, approximately 4900 Mt (equivalent to 60% of plastics produced during the period), reside in landfills or persist in the natural environment. Among the other portions, 800 Mt (12%) of plastics have been ended in incineration, and only 600 Mt (9%) of them have undergone recycling processes. It is worth to note that only a fraction of this recycled plastic has met more than a single round of recycling[4]. This extensive plastic waste generation has increased worldwide plastic-related environmental concerns and the need for more sustainable plastic recycling approaches[2,5,6,7,8,9,10,11,12].
There are four types of plastic recycling methods based on ASTM and ISO standards (Table 1)[7], offering distinct approaches to dealing with plastic wastes. Mechanical recycling, namely primary or secondary recycling, is the process of recovering plastic waste by mechanical processes such as sorting, washing, drying, grinding, re-granulating, and compounding[7,13,14,15]. Given that this process does not change the chemical structure of the material, multiple re-use/re-cycling of polymeric materials could be possible. In addition, other advantages such as efficiency in terms of time, carbon footprint, and relatively low environmental impact make the mechanical recycling attractive. However, the mechanical process within an extruder applies thermal and physical stresses to polymer melts, leading to oxidative radical formation, chain scission, chain branching, and/or crosslinking, thereby inducing degradation of the materials and/or processing facilities. In this regard, some of recycled materials that exhibit worse physical properties compared to the original materials are used in other fields such as textiles, construction, and low grade packaging (2nd recycling)[16]. The 3rd recycling, i.e., chemical recycling, is defined as “the process of recovering petrochemicals and/or chemical building blocks by changing the chemical structures”. In general, this has been applied to plastic wastes, where mechanical recycling is no longer possible and the resultant products are valuable enough. The last recycling method, quaternary recycling or energy recovery, is applied to plastics that the recycling methods above is not applicable, and is typically used for energy recovery through pyrolysis[17,18,19,20,21,22,23]. The major advantage of this method is a reduction in volume of the plastic wastes, therefore, it retains little value in terms of environmental impact. Furthermore, pyrolysis of some plastics may cause negative environmental impacts, such as greenhouse gas generation and toxic gas emissions.
While industrial chemical recycling has been a minor process compared to mechanical recycling, advanced chemical recycling has emerged as a compelling alternative in response to the inherent limitations of mechanical recycling[6,9,24]. Advanced chemical recycling processes are distinguished by their high efficiency, streamlined preliminary steps, reduced reliance on finite fossil resources, and, in some cases, the achievement of physical properties comparable to those of the original materials. Impartant appealing feature of chemical recycling is to use plastic waste as a carbon source and convert the waste into value-added chemicals or monomers. This approach provides an upcycling strategy for generating value-added chemicals primarily through depolymerization. Furthermore, the recovered monomers obtained by depolymerization can be polymerized again to produce a new polymer that exhibits comparable mechanical properties to the virgin polymer, which is known as closed-loop chemical recycling. This transformative approach holds the potential to redefine the recycling landscape, offering a pathway to not only manage plastic waste more sustainably but also reduce our dependence on virgin fossil resources. However, there remain critical challenges in balancing the polymerizability, depolymerizability, recyclability, and material performance, because there are complicated trade-offs between the factors. Furthermore, achieving mild reaction conditions during chemical recycling is another critical factor to consider, as depolymerization processes often require harsh reaction conditions, including high temperatures, expensive catalysts, and strongly acidic or basic conditions.
This review organizes the latest research efforts on the chemical recycling of the polymeric materials from the polymer life cycle viewpoint: i) polymer to small molecules and ii) polymers to monomers, and monomers to polymers. With this, our objective is to provide a detailed examination of both traditional and state-of-the-art depolymerization chemistries, elucidating their underlying principles, technological implications, and potential advantages. We believe this review provides insight into developing an advanced circular economy for polymeric materials through chemical recycling.
2. Chemical Recycling via Depolymerization: Polymers to Small Valuable Molecules
In a typical chemical recycling process, the major objective is to break the chemical bonds of the polymers (i.e., depolymerization) to produce small organic molecules or oligomers[5,6]. In this process, the reactivity of the depolymerization reaction, as well as the purity and yield of the products, are key parameters to consider. Therefore, polymers that can control reaction selectivity within the polymer backbone have been a primary consideration. The most representative polymer that used for chemical recycling via depolymerization is poly(ethylene terephthalate) (PET) given the regular ester-linkage distributions over the polymer backbone[25,26,27]. Depending on the type of the nucleophiles, PET totally depolymerizes to terephthalic acid derivatives[28,29,30], for example, terephthalic acid (TA), bis(hydroxylethylene) terephthalamide (BHETA), dimethyl terephthalate (DMT), and bis(hydroxylethylene) terephthalate (BHET) (Figure 2a). Among them, glycolysis is the most prevalent chemical recycling approach for PET, first patented in 1965[26,28]. While there are a number of candidates for glycolysis including propylene glycol, 1,4-butanediol, and pentaerythritol, ethylene glycol (EG) has been the most widely used[31,32]. This is because the depolymerization product, BHET, can undergo self-polymerization to PET under the specific heat and low pressure[33,34]. In contrast, the other glycolysis products need additional monomeric part, for example, ethylene glycol[30]. Therefore, extensive researches for the glycolysis of PET to produce BHET have focused on lowering temperature, improving reaction rate, and increasing purity of BHET. Examples include solvent/supercritical fluid assisted-, microwave assisted-, and catalysts assisted- glycolysis[26,34,35,36,37,38,39]. However, many of these examples require a large amount of wasteful/expensive solvents, damand high-energy facilities, use toxic/heavy metal catalysts, require post-purification steps, and/or would likely experience significant technological hurdles with respect to sustainability and scalability. Hydrolysis, aminolysis, and methanolysis of PET are the other well-recognized chemical recycling methods for PET[29,32,40,41,42,43,44]. However, problems associated with use of strong acids or bases, production of many byproducts, and slow reaction rate remain challenges for practical applications.
More sustainable and cost-effective process for plastic wastes including PET have been developed to address problems associated with traditional depolymerization processes[46,47,48,49]. The most representative example is the depolymerization via electrochemical reaction[45,50,51,52,53]. Recently, Behera et al. have demonstrated an electrochemical upcycling process for PET waste (Figure 2b)[45]. Hybrid water electrolysis was adopted to drive the water splitting to generate hydrogen gas at the cathode, while the water oxidation at the anode was replaced by oxidation of PET hydrolysate that contains EG monomer and BHET oligomer. During the electrolysis process, value-added products such as TA and potassium diformate (KDF) are generated along with hydrogen gas under the presence of a cobalt-based one-dimensional (1D) coordination polymer as an electrocatalyst. The polymer catalyst can efficiently perform the oxidation of PET hydrolysate at a low onset potential of 1.27 V (vs. reversible hydrogen electrode (RHE)), which is 160 mV less than the alkaline oxygen evolution reaction under the same conditions. Furthermore, the resulting TA and KDF products can be easily separated with a high yield from the electrolyte solution by simple pH adjustment. Thus, the electrochemical PET upcycling process allows full recyclability of the PET waste with a less energy-intensive strategy. Zhou et al. reported that CoNi0.25P electrocatalyst in KOH electrolyte enhances the selectivity and reactivity upon electrochemical PET depolymerization, which is attributed to easier EG oxidation with low-crystalline CoNi oxyhydroxide, thereby producing highly pure H2, TA, and KDF. Another benefits of the electro-catalyst are found to be high current density (~ 350mA cm-2 at 1.7 V vs. RHE) and high Faradic efficiency and selectivity (> 80%)[52].
Beside PET, depolymerization of polyolefins including polyethylene (PE) and polypropylene (PP) has received considerable attentions given that they constitute the largest portion of the plastic waste, approximately over 40% of all plastic wastes[54,55,56,57,58]. Since they composed of only hydrogen and carbon, common depolymerization chemistries (i.e., addition of nucleophile, strong acid or base) are not applicable. Therefore, developing efficient and highly selective depolymerization chemistries has been the major challenge. Duan et al. have proposed an efficient strategy for low-temperature and noble-metal-free catalytic depolymerization of PE into C3–C6 olefins (Figure 3a)[58]. By mixing PE with rationally designed ZSM-5 zeolite nanosheets at 280 °C in flowing hydrogen as a carrier gas, light hydrocarbons (C1–C7) were produced with a yield of up to 74.6%, where 83.9% of these products were C3–C6 olefins with almost undetectable coke formation. The key technique is to use the ZSM-5 zeolite with a controlled b-axis direction as a catalyst in PE depolymerization, where the ZSM-5 kinetically matches the cascade cracking steps on its external surface and within the micropores by boosting the intermediate diffusion. Compared with the mechanical recycling of thermoplastics, which is mostly considered as downcycling from the resulting material property perspective, the catalytic PE depolymerization modulated by ZSM-5 zeolite provides new opportunities for industrially viable treatment of PE wastes to produce value-added materials.
Gao et al. have developed a cationic d0 metal hydride (M–H+) species supported on sulfated aluminum oxide (SAO) that exhibits increased catalytic activity in hydrogenolysis and alkane metathesis reactions (Figure 3b)[55]. It was found that the cationic tantalum hydrides (Ta(=CHtBu)(CH2tBu)3–H+) sites supported on the SAO are more active in hydrogenolysis and alkane metathesis reactions than the neutral Ta–H sites supported on silica. This reaction chemistry extends to melts of high-density PE (HDPE), where Ta–H+ converts 30% of a low molecular weight HDPE (Mn = 2.5 kg mol–1, Đ = 3.6) to low molecular weight paraffins at moderate temperature, 150 °C, under hydrogenolysis conditions. Under alkane metathesis conditions, Ta–H+ converts the HDPE to a high molecular weight fraction (Mn = 6.2 kg mol–1, Đ = 2.3) and low molecular weight alkane products (C13–C32). This strategy demonstrates that incorporating charge as a design element in supported d0 metal hydrides is viable to catalyze the reactions involving the reorganization of C–C bonds in PE wastes. In their follow-up study, they expanded the Ta-H+ sites supported on SAO catalytic system for the degradation of PP[61]. In this case, low molecular weight branched alkanes with a narrower distribution (C11–C30) were produced in good yield (~70%).
While PP has similar chemical structure with PE, hydrogenolysis activity of PP is unclear. This is because PP has methine carbon in the repeating unit, higher Tm than PE, and different crystallinity from PE[7]. Therefore, finding an optimized condition for hydrogenolysis of PP is a critical issue. Rorrer et al. have demonstrated a catalytic depolymerization of high molecular weight PP (Mw ~ 340,000 Da) to liquid and gaseous hydrocarbons over ruthenium nano-particles supported on carbon (Ru/C) catalyst under mild conditions (200–250 °C, 20–50 bar H2)[54]. Unlike PE hydrogenolysis, which produces linear n-alkanes, PP hydrogenolysis produces a range of iso-alkanes in both the liquid (C5–C32) and gas (C1–C5) with yields above 68% in the absence of solvent. Moreover, the Ru/C catalyst was effective in depolymerizing a mixture of HDPE and PP to produce a mixture of linear and branched liquid alkanes, demonstrating feasibility for the depolymerization of streams of mixed polyolefin waste.
In case of polystyrene (PS), all atoms are connected by strong C–C and C–H bonds, PS is remarkably inert and difficult to degrade under mild conditions[59,62,63,64]. Generally, thermal and catalytic pyrolysis of PS requires high temperature (typically > 300 °C) and expensive catalysts, which limits its practical applications[64,65,66]. To address the issues of the chemical recycling of PS wastes, Huang et al. have proposed light-driven and acid-catalyzed aerobic oxidation for the chemical recycling of PS to valuable chemicals under ambient temperature and 1 bar of O2 (Figure 3d)[59]. Electron paramagnetic resonance (EPR) and density functional theory (DFT) calculations indicate that singlet oxygen (1O2) is involved as the reactive oxygen species in the degradation process, which abstracts a hydrogen atom from a tertiary benzylic C–H bond in PS, leading to hydroperoxidation and subsequent C–C bond cracking via a radical process. Furthermore, an adduct of PS-acid catalyst is formed in situ, acting as a photosensitizer to initiate the formation of singlet oxygen. This simple selective degradation method enables the oxidative cleavage of PS to yield valuable chemicals including benzoic acid, formic acid, and acetophenone under mild conditions. More recently, Rabot et al. converted PS to fungal secondary metabolites (SMs) such as ergothioneine, pleuromutilin, and mutilin by coupling catalytic oxidation with genetic engineering[67]. An oxidative catalytic method degraded PS, rapidly producing distributions of diacids, and these acids are suitable for fungal metabolism. Then, pharmacologically active SMs were produced through the engineering strains of fungi. This result implies a possible pathway for consuming upcycled plastics on an agrochemical scale.
Although polyamides (PAs) can be recycled both chemically and physically, physical recycling generally leads to plastics of lower quality due to impurities and reduced mechanical strength[60,68]. Chemical recycling, on the other hand, enables the valorization of waste into high-value building blocks for the production of virgin PA[69,70]. However, the resonance in the amide group makes PAs much more stable than polyesters, and the strong internal hydrogen bonds in PAs impede their dissolution in common organic solvents. Thus, most PA depolymerization processes often require harsh reaction conditions such as high temperatures (> 250 °C) and strong acidic or basic conditions. Coeck et al. have developed a strategy using ammonolytic hydrogenation of secondary amides for efficient chemical recycling of long-chain PAs (LCPAs) (Figure 3e)[60]. They have demonstrated a heterogeneous catalytic system based on Nb2O5, where the reactions were performed at a relatively low temperature of 200 °C in cyclopentyl methyl ether (CPME) as a green solvent and with limited addition of NH3 and H2. Since ammonolysis is an equilibrium reaction, it was eventually coupled to a hydrogenation process, with the addition of a RuWOx/MgAl2O4 hydrogenation catalyst, in order to achieve complete depolymerization. Industrial LCPA samples were successfully and completely depolymerized to α,ω-diamine monomers, and oligomers, resulting in product distributions of 62% primary amines and 36% secondary amines. Furthermore, the catalytic system was proven to be very robust against a variety of contaminants, e.g., fillers, other plastics, and additives.
Bis-phenol A based polycarbonate (BPA-PC) has been a widely used polymer with environmental concerns given that its traditional synthesis relies on toxic reagents (i.e., BPA, phosgene, or carbon monoxide)[71,72,73]. Therefore, preventing the release of BPA-PC to the environment after disposal is one of the critical concerns. Jehanno et al. proposed organo-catalytic depolymerization approach using the thermally stable and recyclable triazabicyclodecene(TBD)/methanesulfonic acid(MSA) co-catalysts[71]. Relatively low temperature (90 – 160 °C) transesterification produced high-quality six-membered cyclic carbonates with excellent yields due to the thermodynamic stability of the cyclic carbonate structures. This catalytic system is applicable to the complete depolymerization for a large amount of polymers, and can be regenerated and reused for multiple cycles. Upon the depolymerization cycles, they found that steric size of substituents on the six-membered cyclic carbonate determines their ability to ring-open as a consequence of the polymer chain torsion rather than the cyclic monomer conformation.
Biodegradable polyesters such as polylactic acid (PLA) are promising alternatives to conventional plastics[74]. However, complete biodegradation usually takes a few months or even longer and their biodegradation leads to CO2 emissions[74,75,76]. By addressing these issues as well as producing value-added chemicals, Sun et al. proposed a two-step catalytic process to convert polylactic acid waste into methyl methacrylate, a high-value building-block molecule[77]. Polylactic acid is firstly transformed into methyl propionate (MP) with near-quantitative yields (conversion > 99%, selectivity~98%) over the α-MoC catalyst in methanol solution, followed by reacting the obtained MP with formaldehyde to produce methyl methacrylate with a conversion of 81% and selectivity of 90%. Most importantly, the entire transformation process does not require any external high-pressure H2, which adds sustainability benefit to this chemical upcycling process.
3. Chemical Recycling for Closed-loop Cycles: Polymers to Monomers, and/or to Polymers
Chemical recycling that converts polymers to small organics can address plastic waste concerns, however, economical imbalance between polymer and depolymerization products, needs for continuous fossil feedstock, extra sorting process, and ending up with greenhouse gas emissions remain concerns. The development of a circular plastics economy has emerged as a critical goal in addressing the concerns. A key aspect of achieving this goal is the concept of chemical recycling polymers back into their constituent monomers, a process referred to as chemical recycling to monomer (CRM)[78,79]. Theoretically, all the polymerizations are reversible under a certain condition such as temperature, pressure, and concentration. However, CRM process for the most common polymers such as PE, PP, PS, and PVC is practically hard or impossible. The ceiling temperature (Tc) serves as a thermodynamic measure of polymerizability or depolymerizability[80,81,82]. It represents the temperature where polymerization and depolymerization. Most of common polymers have high Tc values that are above the decomposition temperature, therefore, rendering CRM process is hardly achievable (Table 2)[83]. Therefore, designing intrinsically circular polymers is a critical challenge. Balancing the requirements of polymerizability, depolymerizability, recyclability, and material performance often involves complex trade-offs: achieving this balance hinges on meeting thermodynamic, kinetic, and practical capability. Currently, extensive research efforts have focused on developing polymers originating from moderately strained heterocyclic monomer. These monomers exhibit relatively high thermodynamic stability, which in turn moderate their Tc (< 250 °C) (Figure 4a).
Among several families of chemically recyclable polymers that have recently developed, polyesters derived from the ring-opening polymerization (ROP) of lactones are particularly attractive, due to their degradability and ROP reversibility[88,89,90,91]. These features of polyester derivatives provide an efficient strategy to revert the polyesters back to their monomer lactones by carefully manipulating the thermodynamics (such as temperature, concentration, and pressure) of the (de)polymerization, as well as the kinetics and processing conditions. For example, Schneiderman et al. investigated thermodynamic parameters of n-Alkyl-δ-valerolactone monomers[92]. All substituted δ-valactones with arbitrarily long n-alkyl substituents showed thermodynamic- and kinetic- feasibility for polymerization. Among them, β-methyl-δ-valactone (βMVL) exhibited the lowest Tc. In their following study, poly(β-methyl-δ-valactone) (PβMVL) with trihydroxyl end groups has been synthesized, and used as a polyol for the preparation polyurethane (PU) foam (Figure 4b)[84]. In spite of the crosslinked structure of the PU foam, the monomer (i.e., β-methyl-δ-valactone) was easily recovered through a simple distillation set-up given the high tendency of depolymerization. This is beneficial because it does not require the addition of any solvent, alcohol, or water like glycolysis of PET and PU. Other examples for depolymerizable polyester derivatives include poly(2-thiabicyclo[2.2.1]heptan-3-one), polyhydroxyalkanoates (PHA), cyclic strained poly(γ-butyrolactone), PLA, and poly(norcamphor lactone)[82,93,94,95,96,97,98].
For more controllable δ-valerolactone system, Eugene and co-workers have presented a synergistic use of gem-α,α-disubstitution of available at-scale, bio-based δ-valerolactones to yield gem-dialkyl-substituted six-membered lactones[99] . It was found that the α,α-disubstitution of valerolactones monomers (VLR2) exhibits much lower thermodynamic polymerizability-or higher depolymerizability-relative to the parent δVL, leading to greatly reduced Tc upon disubstitution. Note that the Tc values of the PVL and PVLPr2 (Pr: propyl) were found to be 298 °C and 67 °C, respectively. This is primarily due to a more negative entropic change of VLR2, primarily ascribed from a more pronounced loss in conformational (rotational and vibrational) freedom (ΔSc). Thus, the VLR2 allows both the facile polymerization of high-molecular-weight polyesters (PVLR2, Mn up to 333 kg mol-1, Đ = 1.16) under ambient conditions and selective depolymerization to fully recover VLR2 monomers under mild conditions (150 °C with a ZnCl2 catalyst). The recovered monomers can be repolymerized to regenerate the same polymers, thereby closing the monomer-polymer-monomer circular loop. Besides their full chemical recyclability, these polyesters also exhibited better tensile properties, high crystallinity (Tm up to 140 °C), thermal stability (Td up to 330 °C), and oxygen barrier properties compared to the parent PVL, proving that the PVLR2 are good candidates for replacing low density PE (LDPE) in packaging applications. Exploiting the thermodynamic stability of multi-substituted lactones, they synthesized α,α-disubstited PHA in their following work[100]. The straightforward α,α-disubstitution in PHAs significantly enhanced the thermal stability, making PHAs amenable to melt processing. Simultaneously, this structural modification imparts mechanical toughness, intrinsic crystallinity, and the potential for closed-loop chemical recycling to PHAs.
Meanwhile, the design of such circular polymers is challenged by unyielding trade-offs between the monomer’s polymerizability and the polymers’ depolymerizability. For example, the ROP of non-strained five-membered cyclic lactone monomers leads to polylactone that can be completely depolymerized back to pure monomers with low energy input[101,102,103]. However, due to the high stability of monomers, polymerizability and the mechanical properties are very limited. Eugene and co-workers reported the synthesis of the copolymer, i.e., poly(glycolide-co-γ-butyrolactone), through the transesterification at high temperature (160 °C), however, the maximum γ-butyrolactone content in the polymer is only ~20%[104]. To address this trade-off issue, they have introduced a hybrid monomer design strategy in their following work[80,85]. They demonstrated synergistical coupling of a high Tc ε-caprolactone sub-structure for high polymerizability and performance properties with a low Tc γ-butyrolactone sub-structure for high depolymerizability and recyclability within the same monomer structure (Figure 4c). Thus, structural hybridization between two distinct sub-structures leads to an offspring [3.2.1]bicyclic lactone, which exhibits both high polymerizability and depolymerizability, otherwise conflicting properties in a single monomer structure. Furthermore, the resulting polymer became a high-performance material, and thermal transition temperatures are 200 °C higher and tensile modulus 10 times higher than its parent polymers. These results demonstrate that the hybrid monomer design strategy is a powerful approach for designing circular polymers where conflicting properties must be exploited and unified.
In a similar manner, Shi et al. further developed the monomer structural hybridization strategy toward a “one monomer-two polymers-one monomer” loop via orthogonal (de)polymerization of a lactone/olefin hybrid monomer (Figure 4d)[86]. Two well-known low Tc polymers, γ-butyrolactone (γ-BL) toward ROP to polyester and cyclohexene toward ring-opening metathesis polymerization (ROMP) to poly(cyclic olefin), are notoriously non-polymerizable. The authors have demonstrated a strategy to render not only the polymerizability of both the γ-BL and cyclohexene sites, orthogonally, but also complete and orthogonal depolymerization, through creating a bicyclic lactone/olefin (BiL=) hybrid. The produced polyester, P-(BiL=)ROP, by ROP exhibited a high Tm of 141–239 °C and Tg of 103–115 °C, while the poly(cyclic olefin), P-(BiL=)ROMP, by ROMP has a Tg of 113 °C. Despite their excellent thermomechanical properties, both P-(BiL=)ROP and P-(BiL=)ROMP showed full chemical recyclability to regenerate the same monomer under mild conditions (25-40 °C) in the presence of a catalyst. These findings demonstrated that orthogonal polymerization of a bifunctional monomer offers an attractive strategy to produce two different classes of polymers from a single monomer, but a more significant, modern challenge is to additionally enable both types of the orthogonally produced polymers with orthogonal depolymerizability to recover the same monomer.
Besides polylactone derivatives, polyacetals are promising candidates for chemical recycling to monomer, but lack useful tensile strengths due to the low molecular weights produced using current uncontrolled cationic ROP (CROP) methods[70,105,106]. Abel et al. have presented reversible-deactivation CROP (RD-CROP) of cyclic acetals using a commercial halomethyl ether initiator and an indium bromide catalyst (Figure 4e)[87]. The RD-CROP method affords molecular weight control and living chain-end retention for the polymerization of cyclic acetal monomers, enabling the synthesis of high molecular weight polymers. By adopting this RD-CROP, they have synthesized poly(1,3-dioxolane) (PDXL), which exhibited tensile strength comparable to some commodity polyolefins. Furthermore, depolymerization of PDXL using strong acid catalysts returns monomer in near-quantitative yield and even proceeds from a commodity plastic waste mixture. This result demonstrated that the RD-CROP method affords a tough thermoplastic that can undergo selective depolymerization to monomers. The highest molecular weight of PDXL is ~220 kDa high in this study, however, its properties could be further improved by reaching to the ultrahigh molecular weight (UHMW). In the following study, Hester et al. reported the synthesis of UHMW PDXL by employing 2,6-di-tert-butylpyridine (DTBP) as a proton trap agent and [Et3O+][BF4–] as an initiator[107]. In general, protic impurities cause a several unwanted reactions such as initiation of new polymer chain, protonation of an existing chain, reaction with polymerization counter anion. This results in macrocyclization, transacetalization, and degradation of counter anion. A small amount of DTBP scavenges protic impurities upon the polymerization, making the polymerization controllable and predictable. Ultra-low loading of initiator is another key factor to achieve UHMW. Triethyl oxonium salts, also known as Meerwein salts[108], which are composed of an oxonium cation and a noncoordinating counterion such as BF4−, PF6−, and SbF6− found to be effective and robust. With them, the molecular weight of PDXL was over 1000 kDa, and the resultant polymer exhibited competitive mechanical properties to the commodity polymers such as UHMWPE and HDPE.
As presented above, there are a number of examples that can construct CRM process, however, most examples are limited within polymers synthesized from cyclic monomer containing heteroatoms. Other common polymers such as polyolefins, polyesters from low stability monomers, thermosets are not suitable for CRM process due to the thermodynamic reasons[109,110]. Therefore, there is an additional research requirement to develop chemical recycling system by chain scission/reconstruction process. There have been extensive research efforts for developing the chemical recycling system by adding key materials or chemistries into the reprocessing periods.
The primary reason that depolymerization and/or degradation of conventional polyolefin materials normally happens at high temperatures of 350~800 °C is primarily due to the inherently large bond strength of C–C bonds with standard lengths of 1.53~1.54 Å[111,112]. To address this challenge, Luo et al. demonstrated a topochemical approach for creating reversible elongated C–C bonds with a bond length of 1.57~1.63 Å between repeating units in the solid state with decreased bond dissociation energies (Figure 5a)[113]. In solid-state topochemical polymerization, the condensed-phase interactions of the crystal lattice favorably position monomer molecules to react. In this way, adding extra steric hindrance to fine-tune the C–C bond length is possible without inhibiting polymerization. The resulting polymers exhibited rapid depolymerization via breakage of the elongated bond within a desirable temperature range of 140~260 °C while otherwise remaining remarkably stable under harsh conditions. Furthermore, the topochemical polymerization produces high-quality stereo-regular and ultra-high-molecular-weight crystalline polymers. Thus, this strategy potentially enables polymer systems that perform similarly to traditional hydrocarbon-based polymers while also easily depolymerizing to monomers through thermal cleavage of the relatively weak C–C bonds. In addition, the resulting polymers can be further processed to useful forms using techniques applied to other conventional polymers such as molding, extrusion, and 3D printing.
Despite the successful examples of developing chemically recyclable synthetic polymers, studies regarding the upcycling of biopolymers into new degradable polymers are rare. Poly(3-hydroxybutyrate) (P3HB), as one type of PHAs, is a complete biopolymer that is generally produced via microbial fermentation[118,119]. Although P3HB takes several months to fully degrade into CO2 and H2O, chemical upcycling of P3HB is required given that the annual output of PHAs has been increasing in recent years[120]. However, chemical upcycling of P3HB is not easy because simple thermal pyrolysis usually produces undesired products, such as low molecular weight oligomers with ketene, propylene, acetaldehyde, and CO2. In this context, Li et al. have proposed a chemical upcycling of poly(3-hydroxybutyrate) (P3HB) into bicyclic ether-ester monomers toward value-added, degradable, and recyclable poly(ether ester)[114]. The upcycling of P3HB involved four steps: efficient conversion of P3HB into ethyl 3-hydroxybutyrate (HBEt), selective ring-opening with cyclohexene oxide (CHO), cyclization to form the new bicyclic monomer 4-methyloctahydro-2H-benzo[b][1,4]dioxepin-2-one (4-MOHB), and its ROP. Thermal analysis revealed that the resulting polymers exhibited an amorphous state with moderate Tg and great thermal stability. Most importantly, the polymers can be efficiently recycled back to monomers through solution depolymerization catalyzed by TsOH or pyrolysis using Sn(Oct)2 or ZnCl2 as a catalyst. This work provides a feasible approach for the chemical upcycling of P3HB bioplastics to produce new recyclable polymers with a closed-loop life cycle.
Arroyave et al. have reported a chemical recycling route for the conversion of post-consumer HDPE into telechelic macromonomers suitable for circular reprocessing (Figure 5c)[115]. Unsaturated C-C bond was introduced into HDPE waste by catalytic dehydrogenation using an Ir-POCOP catalyst. The dehydrogenated HDPE waste undergoes cross-metathesis to form telechelic macromonomers with molecular weights dependent on the degree of unsaturation. While the direct re-polymerization of the macromonomers yields a brittle material primarily due to the low molecular weight, aminolysis of telechelic macromonomers with a small amount of diethanolamine increased the overall mechanical properties. After hydrogenation and partial aminolysis, these macromonomers can be polymerized to yield a material with comparable mechanical and thermal properties to HDPE. This strategy demonstrated that the incorporation of main-chain ester functionalities in PE enables the chemical recycling of PE waste and envision that this methodology will offer a pathway to functionalize and recycle PE waste.
Reversible bond reorganization of various covalent bonds has enabled the regeneration of polymers, which are inherently depolymerizable to form pristine monomers under certain conditions[121,122,123,124,125]. However, the breakage of covalent bonds within polymeric networks often requires an energy-intensive process and undergoes side reactions, resulting in mixed products rather than clean reusable monomers. Thus, it remains a major challenge to design dynamic bonds, capable of effective bonding and reversible cleaving, for preparing chemically recyclable cross-linked polymers. To resolve this issue, Qin et al. have developed chemically recyclable cross-linked polyamic acid materials through the incorporation of dynamic maleic acid tertiary amide bonds (Figure 5d)[116]. The dynamic maleic acid tertiary amide bond is based upon a reversible amidation reaction between maleic anhydrides and secondary amines, enabling reversible chemical control over depolymerization of the resulting polymer networks. This dynamic bond allows for the construction of polymer networks with tailorable and robust mechanical properties, covering strong elastomers with a tensile strength of 22.3 MPa and rigid plastics with a yield strength of 38.3 MPa. Furthermore, these robust polymeric materials can be completely depolymerized in an acidic aqueous solution at ambient temperature, leading to efficient monomer recovery with >94% separation yields. The recovered monomers can be used to remanufacture cross-linked polymeric materials without losing their original mechanical performance. This reversible amidation chemistry unveils a general approach to designing polymer networks with tunable mechanical performance and closed-loop recyclability.
PU is the sixth most produced polymer worldwide, with polyurethane foams (PUFs) accounting for about 67% of global polyurethane consumption[124,126,127]. However, existing methods of chemical recycling of PUF waste suffer from incomplete degradation of urethane groups and lack of selectivity for degradation of urethane bonds alone, resulting in differently functionalized polyols containing various side products. Grdadolnik et al. have addressed this issue by introducing microwave-assisted aminolysis with amine reagents that contain primary and tertiary amino groups in the structure[117]. These reagents enable complete degradation of the urethane groups in the structure of the flexible PUFs with a much lower amount of degradation reagent than is typically required for solvolysis reactions. The purified, recovered polyols are close equivalents to the corresponding virgin polyols in terms of their structural and molar mass characteristics. Furthermore, the PUFs made from the recovered polyols have comparable mechanical properties to those made from virgin polyols.
4. Conclusion & Perspectives
This review has covered an up-to-date overview of chemical recycling strategies for addressing plastic wastes related concerns related to sustainability and practical feasibility. These include i) conventional hydrolytic degradation, ii) electrochemical depolymerization, iii) catalytic depolymerization of commodity plastics to produce valuable small organics, iv) thermodynamics modulation with stable heterocyclic compounds for molecular circularity, and v) oligomerization and re-polymerization via addition of junctional compounds. These methods not only enhance depolymerization efficiency but also simplify initial sorting procedures, generate more valuable products, and diminish our dependence on finite fossil resources. As we have reviewed, a key aspect found in some of the most important works in the field is the collaborative effort of a diverse group of scientists on the fundamental questions. While this review deals primarily with depolymerization technologies of polymeric materials, an essential overarching perspective is the effect of macromolecular science on plastic related environmental challenges.
As we have reviewed, there have been several critical challenges, opportunities, and emerging efforts are on the horizon. It still remains difficult to create perfect plastic recycling system totally addressing plastic wastes challenges within a practical manner. A primary example of this difficulty is the preparation of a readily monomer-recoverable polymer while concurrently exhibiting good polymerizability to achieve high molecular weight and mechanical robustness. Prioritizing factors or parameters is also challenging due to the complexity of kinetics, thermodynamics, economics, and materials properties. We believe that expanding polymer design principles into a more innovative methodology, exploring uncharted directions in the context of chemical recycling, will yield significant advancement in resolving the plastic wastes issues. The challenges are substantial, but with concerted efforts, innovative solutions, and a shared commitment to a more sustainable future, we can realize the vision of a world where plastics are not a burden but a valuable resource in continuous circulation.
Funding
This research was supported by the Institutional Research Program of KRICT (KK2311-10) and Inha University Research Grant (69932-01).
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gibb, B.C. Plastics are forever. Nat. Chem. 2019, 11, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.L.; Wang, S.; Jambeck, J.R. The Chinese import ban and its impact on global plastic waste trade. Sci. Adv. 2018, 4, eaat0131. [Google Scholar] [CrossRef]
- Antonopoulos, I.; Faraca, G.; Tonini, D. Recycling of post-consumer plastic packaging waste in the EU: Recovery rates, material flows, and barriers. Waste Manage. 2021, 126, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manage. 2017, 69, 24–58. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; García, J.M. Chemical recycling of waste plastics for new materials production. Nat. Rev. Chem. 2017, 1, 0046. [Google Scholar] [CrossRef]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef]
- Thiounn, T.; Smith, R.C. Advances and approaches for chemical recycling of plastic waste. J. Polym. Sci. 2020, 58, 1347–1364. [Google Scholar] [CrossRef]
- Vollmer, I.; Jenks, M.J.F.; Roelands, M.C.P.; White, R.J.; van Harmelen, T.; de Wild, P.; van der Laan, G.P.; Meirer, F.; Keurentjes, J.T.F.; Weckhuysen, B.M. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem. Int. Ed. 2020, 59, 15402–15423. [Google Scholar] [CrossRef]
- Haque, F.M.; Ishibashi, J.S.A.; Lidston, C.A.L.; Shao, H.; Bates, F.S.; Chang, A.B.; Coates, G.W.; Cramer, C.J.; Dauenhauer, P.J.; Dichtel, W.R.; Ellison, C.J.; Gormong, E.A.; Hamachi, L.S.; Hoye, T.R.; Jin, M.; Kalow, J.A.; Kim, H.J.; Kumar, G.; LaSalle, C.J.; Liffland, S.; Lipinski, B.M.; Pang, Y.; Parveen, R.; Peng, X.; Popowski, Y.; Prebihalo, E.A.; Reddi, Y.; Reineke, T.M.; Sheppard, D.T.; Swartz, J.L.; Tolman, W.B.; Vlaisavljevich, B.; Wissinger, J.; Xu, S.; Hillmyer, M.A. Defining the Macromolecules of Tomorrow through Synergistic Sustainable Polymer Research. Chem. Rev. 2022, 122, 6322–6373. [Google Scholar] [CrossRef]
- Korley, L.T.J.; Epps, T.H.; Helms, B.A.; Ryan, A.J. Toward polymer upcycling—adding value and tackling circularity. Science 2021, 373, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.M.; Robertson, M.L. The future of plastics recycling. Science 2017, 358, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Damayanti, D.; Saputri, D.R.; Marpaung, D.S.S.; Yusupandi, F.; Sanjaya, A.; Simbolon, Y.M.; Asmarani, W.; Ulfa, M.; Wu, H.-S. Current Prospects for Plastic Waste Treatment. Polymers 2022, 14, 3133. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, G.; Uchida, N.; Tuyen, L.H.; Tanaka, K.; Matsukami, H.; Kunisue, T.; Takahashi, S.; Viet, P.H.; Kuramochi, H.; Osako, M. Mechanical recycling of plastic waste as a point source of microplastic pollution. Environ. Pollut. 2022, 303, 119114. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.; Bourdon, S.; Brossard, J.-M.; Cauret, L.; Fontaine, L.; Montembault, V. Mechanical recycling: Compatibilization of mixed thermoplastic wastes. Polym. Degrad. Stab. 2018, 147, 245–266. [Google Scholar] [CrossRef]
- Damayanti, D.; Wulandari, L.A.; Bagaskoro, A.; Rianjanu, A.; Wu, H.-S. Possibility Routes for Textile Recycling Technology. Polymers 2021, 13, 3834. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.B.; Arora, J.S.; Chew, J.W.; Dauenhauer, P.J.; Mushrif, S.H. Effect of Temperature and Transport on the Yield and Composition of Pyrolysis-Derived Bio-Oil from Glucose. Energy Fuels 2018, 32, 6008–6021. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Giridharan, K.; Stalin, B.; Kumaran, S.; Kavimani, V.; Nagaprasad, N.; Tesfaye Jule, L.; Krishnaraj, R. Energy recovery of waste plastics into diesel fuel with ethanol and ethoxy ethyl acetate additives on circular economy strategy. Sci.Rep. 2022, 12, 5330. [Google Scholar] [CrossRef]
- Zolghadr, A.; Sidhu, N.; Mastalski, I.; Facas, G.; Maduskar, S.; Uppili, S.; Go, T.; Neurock, M.; Dauenhauer, P.J. On the Method of Pulse-Heated Analysis of Solid Reactions (PHASR) for Polyolefin Pyrolysis. ChemSusChem 2021, 14, 4214–4227. [Google Scholar] [CrossRef]
- Antelava, A.; Jablonska, N.; Constantinou, A.; Manos, G.; Salaudeen, S.A.; Dutta, A.; Al-Salem, S.M. Energy Potential of Plastic Waste Valorization: A Short Comparative Assessment of Pyrolysis versus Gasification. Energy Fuels 2021, 35, 3558–3571. [Google Scholar] [CrossRef]
- Wulandari, Y.R.; Chen, S.S.; Hermosa, G.C.; Hossain, M.S.A.; Yamauchi, Y.; Ahamad, T.; Alshehri, S.M.; Wu, K.C.W.; Wu, H.-S. Effect of N2 flow rate on kinetic investigation of lignin pyrolysis. Environ. Res. 2020, 190, 109976. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.R.; Avasarala, S.; Murali, D.; Rajagopalan, N.; Sharma, B.K. Materials and Energy Recovery from E-Waste Plastics. ACS Sustainable. Chem. Eng. 2018, 6, 4594–4602. [Google Scholar] [CrossRef]
- Briassoulis, D.; Hiskakis, M.; Babou, E.; Antiohos, S.K.; Papadi, C. Experimental investigation of the quality characteristics of agricultural plastic wastes regarding their recycling and energy recovery potential. Waste Manage. 2012, 32, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Tullo, A.H. Plastic has a problem; is chemical recycling the solution. Chem. Eng. News 2019, 97, 39. [Google Scholar]
- Benyathiar, P.; Kumar, P.; Carpenter, G.; Brace, J.; Mishra, D.K. Polyethylene Terephthalate (PET) Bottle-to-Bottle Recycling for the Beverage Industry: A Review. Polymers 2022, 14, 2366. [Google Scholar] [CrossRef] [PubMed]
- Sheel, A.; Pant, D. Recycling of Polyethylene Terephthalate Bottles. 4 – Chemical Depolymerization of PET Bottles via Glycolysis, William Andrew Publishing, United States, 2019; pp. 61–84.
- Damayanti; Wu, H. -S. Strategic Possibility Routes of Recycled PET. Polymers 2021, 13, 1475. [Google Scholar] [CrossRef]
- Ghosal, K.; Nayak, C. Recent advances in chemical recycling of polyethylene terephthalate waste into value added products for sustainable coating solutions – hope vs. hype. Mat. Adv. 2022, 3, 1974–1992. [Google Scholar] [CrossRef]
- Štrukil, V. Highly Efficient Solid-State Hydrolysis of Waste Polyethylene Terephthalate by Mechanochemical Milling and Vapor-Assisted Aging. ChemSusChem 2021, 14, 330–338. [Google Scholar] [CrossRef]
- Hofmann, M.; Sundermeier, J.; Alberti, C.; Enthaler, S. Zinc(II) acetate Catalyzed Depolymerization of Poly(ethylene terephthalate). ChemistrySelect 2020, 5, 10010–10014. [Google Scholar] [CrossRef]
- Mohammadi, S.; Enayati, M. Dual catalytic activity of antimony (III) oxide: The polymerization catalyst for synthesis of polyethylene terephthalate also catalyze depolymerization. Polym. Degrad. Stab. 2022, 206, 110180. [Google Scholar] [CrossRef]
- Chandra, A.; Siddiqua, S. Sustainable utilization of chemically depolymerized polyethylene terephthalate (PET) waste to enhance sand-bentonite clay liners. Waste Manage. 2023, 166, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.H.; Bhatty, J.I.; Gamlen, G.A.; Dollimore, D. Aspects of the chemistry of poly(ethylene terephthalate): 5. Polymerization of bis(hydroxyethyl)terephthalate by various metallic catalysts. Polymer 1984, 25, 1333–1336. [Google Scholar] [CrossRef]
- Imran, M.; Kim, B.-K.; Han, M.; Cho, B.G.; Kim, D.H. Sub- and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis(2-hydroxyethyl) terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693. [Google Scholar] [CrossRef]
- Le, N.H.; Ngoc Van, T.T.; Shong, B.; Cho, J. Low-Temperature Glycolysis of Polyethylene Terephthalate. ACS Sustainable. Chem. Eng. 2022, 10, 17261–17273. [Google Scholar] [CrossRef]
- Luo, Y.; Selvam, E.; Vlachos, D.G.; Ierapetritou, M. Economic and Environmental Benefits of Modular Microwave-Assisted Polyethylene Terephthalate Depolymerization. ACS Sustainable. Chem. Eng. 2023, 11, 4209–4218. [Google Scholar] [CrossRef]
- Selvam, E.; Luo, Y.; Ierapetritou, M.; Lobo, R.F.; Vlachos, D.G. Microwave-assisted depolymerization of PET over heterogeneous catalysts. Cat. Today 2023, 418, 114124. [Google Scholar] [CrossRef]
- López-Fonseca, R.; Duque-Ingunza, I.; de Rivas, B.; Flores-Giraldo, L.; Gutiérrez-Ortiz, J.I. Kinetics of catalytic glycolysis of PET wastes with sodium carbonate. Chem. Eng. J. 2011, 168, 312–320. [Google Scholar] [CrossRef]
- Xin, J.; Zhang, Q.; Huang, J.; Huang, R.; Jaffery, Q.Z.; Yan, D.; Zhou, Q.; Xu, J.; Lu, X. Progress in the catalytic glycolysis of polyethylene terephthalate. J. Environ. Manage. 2021, 296, 113267. [Google Scholar] [CrossRef]
- Motonobu, G.; Hiroshi, K.; Akio, K.; Tsutomu, H.; Shoji, N. Depolymerization of polyethylene terephthalate in supercritical methanol. J. Phys.: Condens. Matter 2002, 14, 11427. [Google Scholar]
- Attallah, O.A.; Janssens, A.; Azeem, M.; Fournet, M.B. Fast, High Monomer Yield from Post-consumer Polyethylene Terephthalate via Combined Microwave and Deep Eutectic Solvent Hydrolytic Depolymerization. ACS Sustainable. Chem. Eng. 2021, 9, 17174–17185. [Google Scholar] [CrossRef]
- Shukla, S.R.; Harad, A.M. Aminolysis of polyethylene terephthalate waste. Polym. Degrad. Stab. 2006, 91, 1850–1854. [Google Scholar] [CrossRef]
- Fukushima, K.; Lecuyer, J.M.; Wei, D.S.; Horn, H.W.; Jones, G.O.; Al-Megren, H.A.; Alabdulrahman, A.M.; Alsewailem, F.D.; McNeil, M.A.; Rice, J.E.; Hedrick, J.L. Advanced chemical recycling of poly(ethylene terephthalate) through organocatalytic aminolysis. Polym. Chem. 2013, 4, 1610–1616. [Google Scholar] [CrossRef]
- Chan, K.; Zinchenko, A. Conversion of waste bottles’ PET to a hydrogel adsorbent via PET aminolysis. J. Environ. Chem. Eng. 2021, 9, 106129. [Google Scholar] [CrossRef]
- Behera, S.; Dinda, S.; Saha, R.; Mondal, B. Quantitative Electrocatalytic Upcycling of Polyethylene Terephthalate Plastic and Its Oligomer with a Cobalt-Based One-Dimensional Coordination Polymer Having Open Metal Sites along with Coproduction of Hydrogen. ACS Cat. 2023, 13, 469–474. [Google Scholar] [CrossRef]
- Delle Chiaie, K.R.; McMahon, F.R.; Williams, E.J.; Price, M.J.; Dove, A.P. Dual-catalytic depolymerization of polyethylene terephthalate (PET). Polym. Chem. 2020, 11, 1450–1453. [Google Scholar] [CrossRef]
- Yang, B.; Li, W.; Zhang, M.; Wang, L.; Ding, X. Recycling of High-Value-Added Aramid Nanofibers from Waste Aramid Resources via a Feasible and Cost-Effective Approach. ACS Nano 2021, 15, 7195–7207. [Google Scholar] [CrossRef] [PubMed]
- Navarre, N.; Mogollón, J.M.; Tukker, A.; Barbarossa, V. Recycled plastic packaging from the Dutch food sector pollutes Asian oceans. Resour. Conserv. Recycl. 2022, 185, 106508. [Google Scholar] [CrossRef]
- Hong, M.; Chen, E.Y.-X. Chemically recyclable polymers: a circular economy approach to sustainability. Green Chem. 2017, 19, 3692–3706. [Google Scholar] [CrossRef]
- Cho, J.; Kim, B.; Kwon, T.; Lee, K.; Choi, S.-I. Electrocatalytic upcycling of plastic waste. Green Chem. 2023. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Li, X.; Deng, K.; Yu, H.; Xu, Y.; Wang, H.; Wang, Z.; Wang, L. Electrocatalytic upcycling of polyethylene terephthalate plastic to formic acid coupled with energy-saving hydrogen production over hierarchical Pd-doped NiTe nanoarrays. Appl. Catal., B 2024, 340, 123236. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, Y.; Li, Z.; Xu, M.; Wang, Y.; Ge, R.; Kong, X.; Zheng, L.; Duan, H. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 2021, 12, 4679. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.S.; Ramasamy, K.K. Electrochemical Oxidation of Lignin and Waste Plastic. ACS Omega 2020, 5, 27735–27740. [Google Scholar] [CrossRef] [PubMed]
- Rorrer, J.E.; Troyano-Valls, C.; Beckham, G.T.; Román-Leshkov, Y. Hydrogenolysis of Polypropylene and Mixed Polyolefin Plastic Waste over Ru/C to Produce Liquid Alkanes. ACS Sustainable. Chem. Eng. 2021, 9, 11661–11666. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, L.; Conley, M.P. Cationic Tantalum Hydrides Catalyze Hydrogenolysis and Alkane Metathesis Reactions of Paraffins and Polyethylene. J. Am. Chem. Soc. 2023, 145, 4964–4968. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Gorbea, G.D.; Danielson, E.; Cui, S.; Ellison, C.J.; Bates, F.-S. Threading-the-Needle: Compatibilization of HDPE/i-PP blends with butadiene-derived polyolefin block copolymers. PNAS 2023, 120, e2301352120. [Google Scholar] [CrossRef] [PubMed]
- Eagan, J.M.; Xu, J.; Di Girolamo, R.; Thurber, C.M.; Macosko, C.W.; LaPointe, A.M.; Bates, F.-S.; Coates, G.W. Combining polyethylene and polypropylene: Enhanced performance with PE/i-PP multiblock polymers. Science 2017, 355, 814–816. [Google Scholar] [CrossRef]
- Duan, J.; Chen, W.; Wang, C.; Wang, L.; Liu, Z.; Yi, X.; Fang, W.; Wang, H.; Wei, H.; Xu, S.; Yang, Y.; Yang, Q.; Bao, Z.; Zhang, Z.; Ren, Q.; Zhou, H.; Qin, X.; Zheng, A.; Xiao, F.-S. Coking-Resistant Polyethylene Upcycling Modulated by Zeolite Micropore Diffusion. J. Am. Chem. Soc. 2022, 144, 14269–14277. [Google Scholar] [CrossRef]
- Huang, Z.; Shanmugam, M.; Liu, Z.; Brookfield, A.; Bennett, E.L.; Guan, R.; Vega Herrera, D.E.; Lopez-Sanchez, J.A.; Slater, A.G.; McInnes, E.J.L.; Qi, X.; Xiao, J. Chemical Recycling of Polystyrene to Valuable Chemicals via Selective Acid-Catalyzed Aerobic Oxidation under Visible Light. J. Am. Chem. Soc. 2022, 144, 6532–6542. [Google Scholar] [CrossRef]
- Coeck, R.; De Bruyne, A.; Borremans, T.; Stuyck, W.; De Vos, D.E. Ammonolytic Hydrogenation of Secondary Amides: An Efficient Method for the Recycling of Long-Chain Polyamides. ACS Sustainable. Chem..Eng. 2022, 10, 3048–3056. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, L.; Conley, M.P. Polypropylene Degradation Catalyzed by Tantalum Hydrides Supported on Sulfated Alumina. ACS Catal. 2023, 13, 10765–10769. [Google Scholar] [CrossRef]
- Li, R.; Zhang, Z.; Liang, X.; Shen, J.; Wang, J.; Sun, W.; Wang, D.; Jiang, J.; Li, Y. Polystyrene Waste Thermochemical Hydrogenation to Ethylbenzene by a N-Bridged Co, Ni Dual-Atom Catalyst. J. Am. Chem. Soc. 2023, 145, 16218–16227. [Google Scholar] [CrossRef] [PubMed]
- Kiel, G.R.; Lundberg, D.J.; Prince, E.; Husted, K.E.L.; Johnson, A.M.; Lensch, V.; Li, S.; Shieh, P.; Johnson, J.A. Cleavable Comonomers for Chemically Recyclable Polystyrene: A General Approach to Vinyl Polymer Circularity. J. Am. Chem. Soc. 2022, 144, 12979–12988. [Google Scholar] [CrossRef] [PubMed]
- Ukei, H.; Hirose, T.; Horikawa, S.; Takai, Y.; Taka, M.; Azuma, N.; Ueno, A. Catalytic degradation of polystyrene into styrene and a design of recyclable polystyrene with dispersed catalysts. Cat. Today 2000, 62, 67–75. [Google Scholar] [CrossRef]
- Maafa, I.M. Pyrolysis of Polystyrene Waste: A Review. Polymers 2021, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Aguado, R.; Olazar, M.; x; Gaisán, B. ; Prieto, R.; Bilbao, J. Kinetics of polystyrene pyrolysis in a conical spouted bed reactor. Chem. Eng. J. 2003, 92, 91–99. [Google Scholar] [CrossRef]
- Rabot, C.; Chen, Y.; Lin, S.-Y.; Miller, B.; Chiang, Y.-M.; Oakley, C.E.; Oakley, B.R.; Wang, C.C.C.; Williams, T.J. Polystyrene Upcycling into Fungal Natural Products and a Biocontrol Agent. J. Am. Chem. Soc. 2023, 145, 5222–5230. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Figueira, R.; Hofmann, M.; Koschke, S.; Enthaler, S. Chemical Recycling of End-of-Life Polyamide 6 via Ring Closing Depolymerization. ChemistrySelect 2019, 4, 12638–12642. [Google Scholar] [CrossRef]
- Kamimura, A.; Yamamoto, S. An Efficient Method To Depolymerize Polyamide Plastics: A New Use of Ionic Liquids. Org. Lett. 2007, 9, 2533–2535. [Google Scholar] [CrossRef]
- Češarek, U.; Pahovnik, D.; Žagar, E. Chemical Recycling of Aliphatic Polyamides by Microwave-Assisted Hydrolysis for Efficient Monomer Recovery. ACS Sustainable Chem. Eng. 2020, 8, 16274–16282. [Google Scholar] [CrossRef]
- Jehanno, C.; Demarteau, J.; Mantione, D.; Arno, M.C.; Ruipérez, F.; Hedrick, J.L.; Dove, A.P.; Sardon, H. Synthesis of Functionalized Cyclic Carbonates through Commodity Polymer Upcycling. ACS Macro Lett. 2020, 9, 443–447. [Google Scholar] [CrossRef]
- Kim, J.G. Chemical recycling of poly(bisphenol A carbonate). Polym. Chem. 2020, 11, 4830–4849. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.-B. Chemical recycling to monomers: Industrial Bisphenol-A-Polycarbonates to novel aliphatic polycarbonate materials. J. Polym Sci. 2022, 60, 3256–3268. [Google Scholar] [CrossRef]
- Haider, T.P.; Völker, C.; Kramm, J.; Landfester, K.; Wurm, F.R. Plastics of the Future? The Impact of Biodegradable Polymers on the Environment and on Society. Angew. Chem. Int. Ed. 2019, 58, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hillmyer, M.A.; Ellison, C.J. Enhanced Polyester Degradation through Transesterification with Salicylates. J. Am. Chem. Soc. 2021, 143, 15784–15790. [Google Scholar] [CrossRef]
- Kim, H.J.; Reddi, Y.; Cramer, C.J.; Hillmyer, M.A.; Ellison, C.J. Readily Degradable Aromatic Polyesters from Salicylic Acid. ACS Macro Lett. 2020, 9, 96–102. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, J.; Wang, M.; Yu, S.; Xu, Y.; Tian, S.; Gao, Z.; Xiao, D.; Liu, G.; Zhou, W.; Wang, M.; Ma, D. Valorization of waste biodegradable polyester for methyl methacrylate production. Nat. Sustainability 2023, 6, 712–719. [Google Scholar] [CrossRef]
- Coates, G.W.; Getzler, Y.D.Y.L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Vora, N.; Christensen, P.R.; Demarteau, J.; Baral, N.R.; Keasling, J.D.; Helms, B.A.; Scown, C.D. Leveling the cost and carbon footprint of circular polymers that are chemically recycled to monomer. Sci. Adv. 2021, 7, eabf0187. [Google Scholar] [CrossRef]
- Shi, C.; Reilly, L.T.; Phani Kumar, V.S.; Coile, M.W.; Nicholson, S.R.; Broadbelt, L.J.; Beckham, G.T.; Chen, E.Y.-X. Design principles for intrinsically circular polymers with tunable properties. Chem 2021, 7, 2896–2912. [Google Scholar] [CrossRef]
- Diesendruck, C.E.; Peterson, G.I.; Kulik, H.J.; Kaitz, J.A.; Mar, B.D.; May, P.A.; White, S.R.; Martínez, T.J.; Boydston, A.J.; Moore, J.S. Mechanically triggered heterolytic unzipping of a low-ceiling-temperature polymer. Nat. Chem. 2014, 6, 623–628. [Google Scholar] [CrossRef]
- Cederholm, L.; Wohlert, J.; Olsén, P.; Hakkarainen, M.; Odelius, K. “Like Recycles Like”: Selective Ring-Closing Depolymerization of Poly (L-Lactic Acid) to L-Lactide. Angew. Chem. 2022, 134, e202204531. [Google Scholar] [CrossRef]
- Stevens, M.P. , Polymer chemistry, Oxford university press New York, 1990.
- Schneiderman, D.K.; Vanderlaan, M.E.; Mannion, A.M.; Panthani, T.R.; Batiste, D.C.; Wang, J.Z.; Bates, F.S.; Macosko, C.W.; Hillmyer, M.A. Chemically Recyclable Biobased Polyurethanes. ACS Macro Lett. 2016, 5, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Li, Z.-C.; Caporaso, L.; Cavallo, L.; Falivene, L.; Chen, E.Y.-X. Hybrid monomer design for unifying conflicting polymerizability, recyclability, and performance properties. Chem 2021, 7, 670–685. [Google Scholar] [CrossRef]
- Shi, C.; Clarke, R.W.; McGraw, M.L.; Chen, E.Y.-X. Closing the “One Monomer–Two Polymers–One Monomer” Loop via Orthogonal (De)polymerization of a Lactone/Olefin Hybrid. J. Am. Chem. Soc. 2022, 144, 2264–2275. [Google Scholar] [CrossRef]
- Abel, B.A.; Snyder, R.L.; Coates, G.W. Chemically recyclable thermoplastics from reversible-deactivation polymerization of cyclic acetals. Science 2021, 373, 783–789. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.; Yan, Q.; Liu, F.; Shen, Y.; Li, Z. Rapid and Controlled Polymerization of Bio-sourced δ-Caprolactone toward Fully Recyclable Polyesters and Thermoplastic Elastomers. Angew. Chem. Int. Ed. 2022, 61, e202201407. [Google Scholar] [CrossRef]
- Shen, Y.; Xiong, W.; Li, Y.; Zhao, Z.; Lu, H.; Li, Z. Chemoselective ring-opening polymerization of bio-renewable α-methylene-γ-butyrolactone via an organophosphazene/urea binary synergistic catalytic system toward a sustainable polyester. CCS Chem. 2020, 3, 620–630. [Google Scholar] [CrossRef]
- Li, X.-L.; Clarke, R.W.; An, H.-Y.; Gowda, R.R.; Jiang, J.-Y.; Xu, T.-Q.; Chen, E.Y.-X. Dual Recycling of Depolymerization Catalyst and Biodegradable Polyester that Markedly Outperforms Polyolefins. Angew. Chem. Int. Ed. 2023, 62, e202303791. [Google Scholar] [CrossRef]
- Gallin, C.F.; Lee, W.-W.; Byers, J.A. A Simple, Selective, and General Catalyst for Ring Closing Depolymerization of Polyesters and Polycarbonates for Chemical Recycling. Angew. Chem. 2023, 135, e202303762. [Google Scholar] [CrossRef]
- Schneiderman, D.K.; Hillmyer, M.A. Aliphatic Polyester Block Polymer Design. Macromolecules 2016, 49, 2419–2428. [Google Scholar] [CrossRef]
- Shi, C.; McGraw, M.L.; Li, Z.-C.; Cavallo, L.; Falivene, L.; Chen, E.Y.-X. High-performance pan-tactic polythioesters with intrinsic crystallinity and chemical recyclability. Sci. Adv. 2020, 6, eabc0495. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Westlie, A.H.; Watson, E.M.; Chen, E.Y.-X. Stereosequenced crystalline polyhydroxyalkanoates from diastereomeric monomer mixtures. Science 2019, 366, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Westlie, A.H.; Caporaso, L.; Cavallo, L.; Falivene, L.; Chen, E.Y.-X. Biodegradable polyhydroxyalkanoates by stereoselective copolymerization of racemic diolides: stereocontrol and polyolefin-like properties. Angew. Chem. 2020, 132, 7955–7964. [Google Scholar] [CrossRef]
- Zhu, J.-B.; Watson, E.M.; Tang, J.; Chen, E.Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science 2018, 360, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Cederholm, L.; Olsén, P.; Hakkarainen, M.; Odelius, K. Chemical recycling to monomer: thermodynamic and kinetic control of the ring-closing depolymerization of aliphatic polyesters and polycarbonates. Polym. Chem. 2023, 14, 3270–3276. [Google Scholar] [CrossRef]
- Bruckmoser, J.; Remke, S.; Rieger, B. Ring-Opening Polymerization of a Bicyclic Lactone: Polyesters Derived from Norcamphor with Complete Chemical Recyclability. ACS Macro Lett. 2022, 11, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Clarke, R.W.; Jiang, J.-Y.; Xu, T.-Q.; Chen, E.Y.-X. A circular polyester platform based on simple gem-disubstituted valerolactones. Nat. Chem. 2023, 15, 278–285. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Z.; Shi, C.; Scoti, M.; Barange, D.K.; Gowda, R.R.; Chen, E.Y.-X. Chemically circular, mechanically tough, and melt-processable polyhydroxyalkanoates. Science 2023, 380, 64–69. [Google Scholar] [CrossRef]
- Hong, M.; Chen, E.Y.-X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of γ-butyrolactone. Nat. Chem. 2016, 8, 42–49. [Google Scholar] [CrossRef]
- Falconnet, A.; Nicolas, M.; Vollgraff, T.; Konradi, R.; Bruchmann, B.; Rodewald, D.; Hashmi, A.S.K.; Schaub, T. Facile preparation of biodegradable poly(γ-butyrolactone) via base-assisted ring-opening polymerization. Green Chem. 2023, 25, 3624–3632. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, X.; Wu, J.; Hu, X.; Zhu, N.; Guo, K. Access to high-molecular-weight poly(γ-butyrolactone) by using simple commercial catalysts. Polym. Chem. 2022, 13, 439–445. [Google Scholar] [CrossRef]
- Liu, X.; Hong, M.; Falivene, L.; Cavallo, L.; Chen, E.Y.-X. Closed-Loop Polymer Upcycling by Installing Property-Enhancing Comonomer Sequences and Recyclability. Macromolecules 2019, 52, 4570–4578. [Google Scholar] [CrossRef]
- Shen, T.; Chen, K.; Chen, Y.; Ling, J. Ring-Opening Polymerization of Cyclic Acetals: Strategy for both Recyclable and Degradable Materials. Macromol. Rapid Commun. 2023, 44, 2300099. [Google Scholar] [CrossRef] [PubMed]
- Kaya, K.; Debsharma, T.; Schlaad, H.; Yagci, Y. Cellulose-based polyacetals by direct and sensitized photocationic ring-opening polymerization of levoglucosenyl methyl ether. Polym. Chem. 2020, 11, 6884–6889. [Google Scholar] [CrossRef]
- Hester, H.G.; Abel, B.A.; Coates, G.W. Ultra-High-Molecular-Weight Poly(Dioxolane): Enhancing the Mechanical Performance of a Chemically Recyclable Polymer. J. Am. Chem. Soc. 2023, 145, 8800–8804. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications. Meerwein’s salt 5th Ed. Springer International Publishing, Switzerland, 2014; pp. 384–385.
- Westlie, A.H.; Chen, E.Y.-X.; Holland, C.M.; Stahl, S.S.; Doyle, M.; Trenor, S.R.; Knauer, K.M. Polyolefin Innovations toward Circularity and Sustainable Alternatives. Macromol. Rapid Commun. 2022, 43, 2200492. [Google Scholar] [CrossRef] [PubMed]
- Morici, E.; Dintcheva, N.T. Recycling of Thermoset Materials and Thermoset-Based Composites: Challenge and Opportunity. Polymers 2022, 14, 4153. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, Y.; Jia, X.; Huang, Z. Selective Catalytic Transfer Dehydrogenation of Alkanes and Heterocycles by an Iridium Pincer Complex. Angew. Chem. Int. Ed. 2014, 53, 1390–1394. [Google Scholar] [CrossRef]
- Bresciani, G.; Zacchini, S.; Pampaloni, G.; Marchetti, F. Carbon–Carbon Bond Coupling of Vinyl Molecules with an Allenyl Ligand at a Diruthenium Complex. Organometallics 2022, 41, 1006–1014. [Google Scholar] [CrossRef]
- Luo, X.; Wei, Z.; Seo, B.; Hu, Q.; Wang, X.; Romo, J.A.; Jain, M.; Cakmak, M.; Boudouris, B.W.; Zhao, K.; Mei, J.; Savoie, B.M.; Dou, L. Circularly Recyclable Polymers Featuring Topochemically Weakened Carbon–Carbon Bonds. J. Am. Chem. Soc. 2022, 144, 16588–16597. [Google Scholar] [CrossRef]
- Li, Z.; Shen, Y.; Li, Z. Chemical Upcycling of Poly(3-hydroxybutyrate) into Bicyclic Ether–Ester Monomers toward Value-Added, Degradable, and Recyclable Poly(ether ester). ACS Sustainable Chem. Eng. 2022, 10, 8228–8238. [Google Scholar] [CrossRef]
- Arroyave, A.; Cui, S.; Lopez, J.C.; Kocen, A.L.; LaPointe, A.M.; Delferro, M.; Coates, G.W. Catalytic Chemical Recycling of Post-Consumer Polyethylene. J. Am. Chem. Soc. 2022, 144, 23280–23285. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Liu, S.; Huang, Z.; Zeng, L.; Xu, J.-F.; Zhang, X. Closed-loop chemical recycling of cross-linked polymeric materials based on reversible amidation chemistry. Nat. Commun. 2022, 13, 7595. [Google Scholar] [CrossRef]
- He, H.; Su, H.; Yu, H.; Du, K.; Yang, F.; Zhu, Y.; Ma, M.; Shi, Y.; Zhang, X.; Chen, S.; Wang, X. Chemical Recycling of Waste Polyurethane Foams: Efficient Acidolysis under the Catalysis of Zinc Acetate. ACS Sustainable Chem. Eng. 2023, 11, 5515–5523. [Google Scholar] [CrossRef]
- Chen, G.-Q. A microbial polyhydroxyalkanoates (PHA) based bio- and materials industry. Chem. Soc. Rev. 2009, 38, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Wang, Y.; Tong, Y.; Chen, G.-Q. Grand Challenges for Industrializing Polyhydroxyalkanoates (PHAs). Trends Biotechnol. 2021, 39, 953–963. [Google Scholar] [CrossRef]
- Zhao, D.; Li, Z.; Shen, Y.; Li, Z. Crystalline Stereoregular Poly(ether-ester) via MeAl[Salen]-Catalyzed Well-Controlled Ring-Opening Polymerization of Enantiopure Cyclic Ether-Ester Monomer. Macromolecules 2023, 56, 6019–6026. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef]
- Heo, Y.; Sodano, H.A. Self-Healing Polyurethanes with Shape Recovery. Adv. Funct. Mater. 2014, 24, 5261–5268. [Google Scholar] [CrossRef]
- Jin, K.; Banerji, A.; Kitto, D.; Bates, F.S.; Ellison, C.J. Mechanically Robust and Recyclable Cross-Linked Fibers from Melt Blown Anthracene-Functionalized Commodity Polymers. ACS Appl. Mater. Interfaces 2019, 11, 12863–12870. [Google Scholar] [CrossRef]
- Sheppard, D.T.; Jin, K.; Hamachi, L.S.; Dean, W.; Fortman, D.J.; Ellison, C.J.; Dichtel, W.R. Reprocessing Postconsumer Polyurethane Foam Using Carbamate Exchange Catalysis and Twin-Screw Extrusion. ACS Cent. Sci. 2020, 6, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Alfarhan, S.; Brown, J.; Liu, B.; Long, T.; Jin, K. Chemically recyclable crosslinked thiol-ene photopolymers via thiol-disulfide exchange reactions. J. Polym. Sci. 2022, 60, 3379–3390. [Google Scholar] [CrossRef]
- Sternberg, J.; Pilla, S. Chemical recycling of a lignin-based non-isocyanate polyurethane foam. Nat. Sustainability 2023, 6, 316–324. [Google Scholar] [CrossRef]
- Fortman, D.J.; Sheppard, D.T.; Dichtel, W.R. Reprocessing Cross-Linked Polyurethanes by Catalyzing Carbamate Exchange. Macromolecules 2019, 52, 6330–6335. [Google Scholar] [CrossRef]
Figure 1.
Global production, use, recycling, and end-up of plastics (1950 to 2015; in million metric tons).[2] Reproduced with permission. Copyright 2017, American Association for the Advancement of Science (AAAS).
Figure 1.
Global production, use, recycling, and end-up of plastics (1950 to 2015; in million metric tons).[2] Reproduced with permission. Copyright 2017, American Association for the Advancement of Science (AAAS).
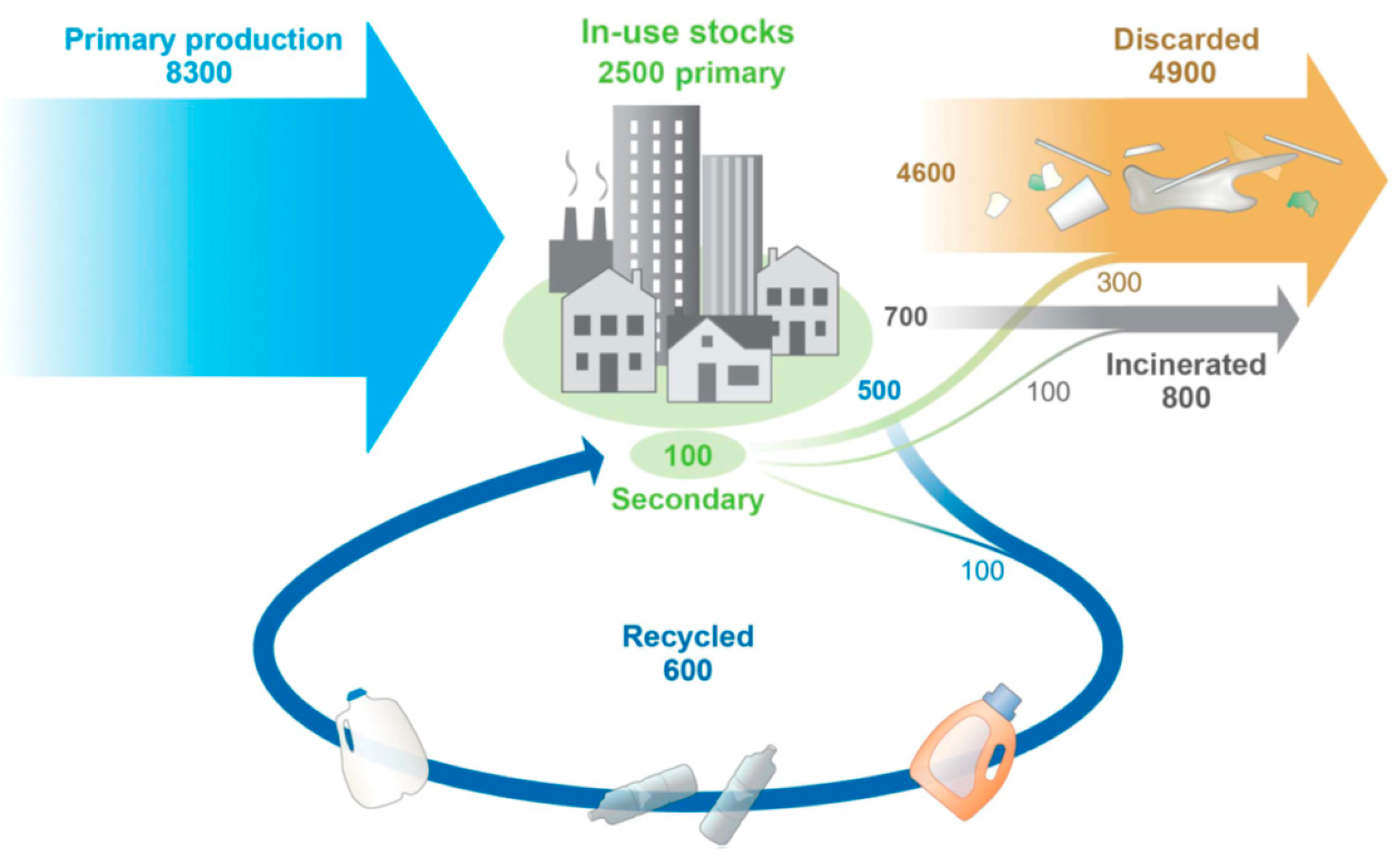
Figure 2.
Chemical recycling methods of PET. (a) Traditional depolymerization pathways of PET and depolymerized products. (b) depolymerization of PET by electro-catalytic reaction[45]. Reproduced with permission. Copyright 2023, American Chemical Society (ACS).
Figure 2.
Chemical recycling methods of PET. (a) Traditional depolymerization pathways of PET and depolymerized products. (b) depolymerization of PET by electro-catalytic reaction[45]. Reproduced with permission. Copyright 2023, American Chemical Society (ACS).
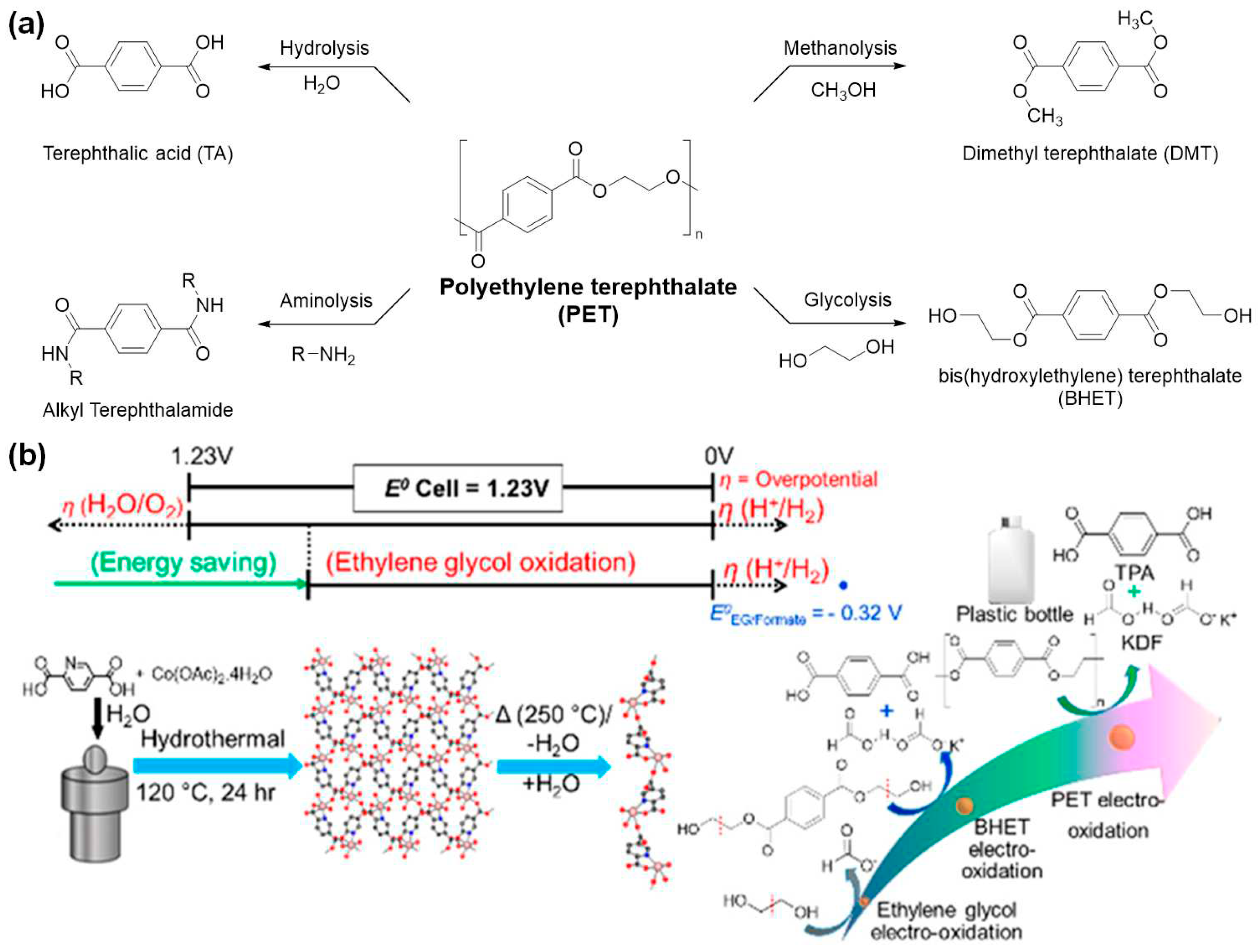
Figure 3.
Chemical recycling via depolymerization of commodity plastics to produce valuable small organic molecules. (a) Schematic illustration of the carbon flow upon the s-ZSM-5 catalyzed depolymerization of PE and possible utilization of the products[58]. Copyright 2022, American Chemical Society (ACS). (b) Hydrogenolysis of a generic alkane Ta–H+ supported on sulfated aluminum oxide to convert high density PE (HDPE) to low molecular weight hydrocarbons[55]. Copyright 2023, American Chemical Society (ACS). (c) Hydrogenolysis of high molecular weight PP (Mw ~ 340 kDa) over ruthenium nanoparticles (5 wt %) supported on carbon.[54] Copyright 2021, American Chemical Society (ACS). (d) Depolymerization of PS in photo-catalyzed flow process yielding highly pure formic acid and benzoic acid[59]. Copyright 2022, American Chemical Society (ACS). (e) Chemical recycling of polyamide via catalyzed ammonolytic hydrogenation[60]. Copyright 2022, American Chemical Society (ACS). All the figures are reproduced with permission.
Figure 3.
Chemical recycling via depolymerization of commodity plastics to produce valuable small organic molecules. (a) Schematic illustration of the carbon flow upon the s-ZSM-5 catalyzed depolymerization of PE and possible utilization of the products[58]. Copyright 2022, American Chemical Society (ACS). (b) Hydrogenolysis of a generic alkane Ta–H+ supported on sulfated aluminum oxide to convert high density PE (HDPE) to low molecular weight hydrocarbons[55]. Copyright 2023, American Chemical Society (ACS). (c) Hydrogenolysis of high molecular weight PP (Mw ~ 340 kDa) over ruthenium nanoparticles (5 wt %) supported on carbon.[54] Copyright 2021, American Chemical Society (ACS). (d) Depolymerization of PS in photo-catalyzed flow process yielding highly pure formic acid and benzoic acid[59]. Copyright 2022, American Chemical Society (ACS). (e) Chemical recycling of polyamide via catalyzed ammonolytic hydrogenation[60]. Copyright 2022, American Chemical Society (ACS). All the figures are reproduced with permission.
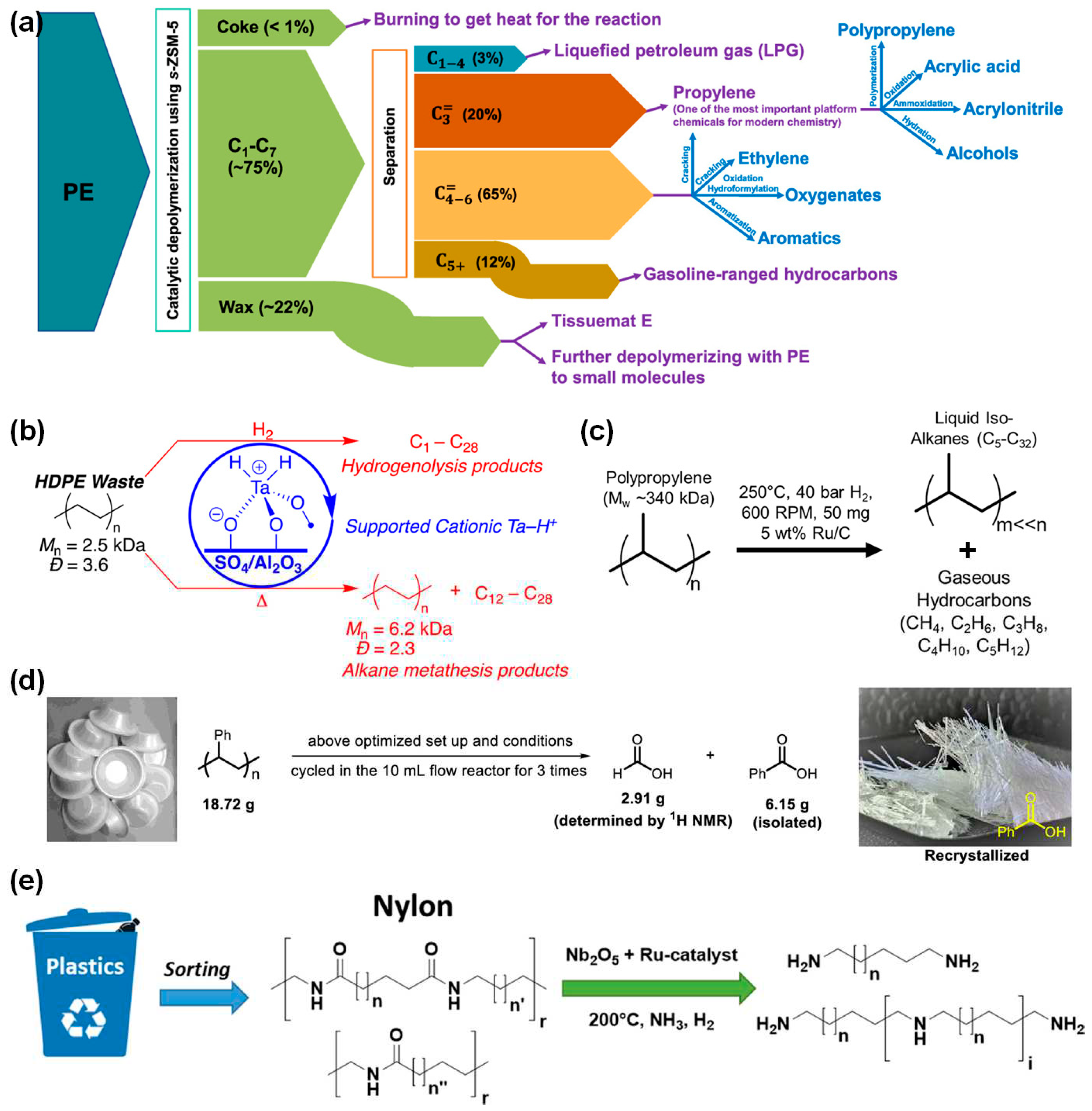
Figure 4.
Chemical recycling systems for closed-loop circular polymers. (a) Thermodynamic preferences for depolymerization to monomers based on the free energy differences between monomers and polymers. (b) Synthesis of PβMVL polyol, PU foam, and chemical recycling to βMVL from the crosslinked PU foam[84]. Copyright 2016, American Chemical Society (ACS). (c) Hybrid 5-/7- membered lactone system for balancing polymerizability and recyclability[85]. Copyright 2021, Elsevier Inc. (d) “One Monomer−Two Polymers−One Monomer” strategy by hybrid lactone/olefin monomers[86]. Copyright 2022, American Chemical Society (ACS). (e) Closed-loop chemical recycling of cyclic acetals and monomer recovery from a mixed commodity plastics feedstock[87]. Copyright 2021, American Association for the Advancement of Science (AAAS). All the figures are reproduced with permission.
Figure 4.
Chemical recycling systems for closed-loop circular polymers. (a) Thermodynamic preferences for depolymerization to monomers based on the free energy differences between monomers and polymers. (b) Synthesis of PβMVL polyol, PU foam, and chemical recycling to βMVL from the crosslinked PU foam[84]. Copyright 2016, American Chemical Society (ACS). (c) Hybrid 5-/7- membered lactone system for balancing polymerizability and recyclability[85]. Copyright 2021, Elsevier Inc. (d) “One Monomer−Two Polymers−One Monomer” strategy by hybrid lactone/olefin monomers[86]. Copyright 2022, American Chemical Society (ACS). (e) Closed-loop chemical recycling of cyclic acetals and monomer recovery from a mixed commodity plastics feedstock[87]. Copyright 2021, American Association for the Advancement of Science (AAAS). All the figures are reproduced with permission.
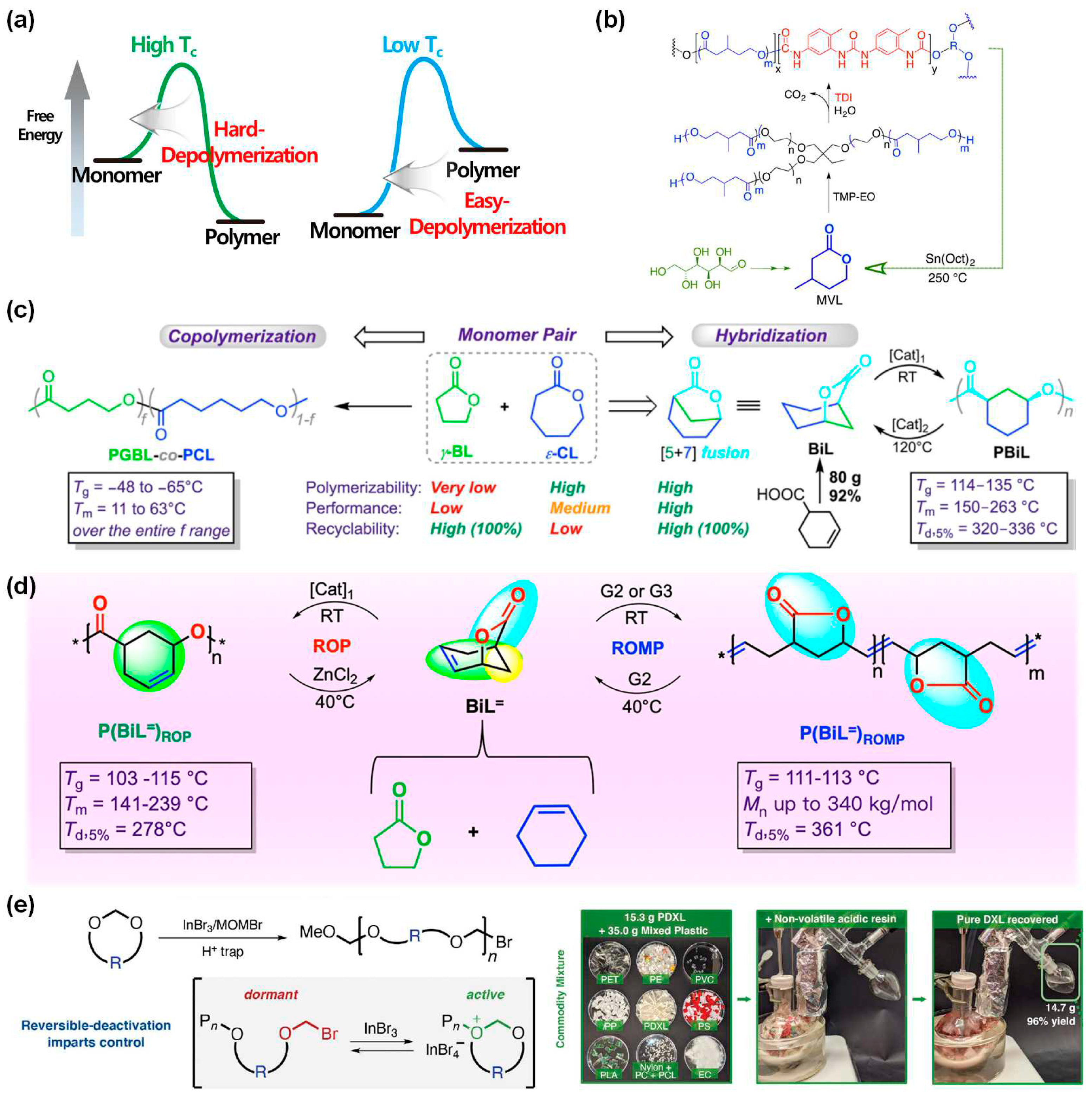
Figure 5.
Chemical recycling via chain scission/reconstruction by adding key chemicals and/or chemistries. (a) Circularly recyclable polymers by topochemically weakened C−C bonds[113]. Copyright 2022, American Chemical Society (ACS). (b) Chemical upcycling of P3HB toward the bicyclic monomer 4-MOHB, which can be polymerized to afford P(4-MOHB) with a closed-loop cycle[114]. Copyright 2022, American Chemical Society (ACS). (c) Chemical recycling of post-consumer waste HDPE into a chemically recyclable material[115]. Copyright 2022, American Chemical Society (ACS). (d) Preparation of polymer networks from bifunctional maleic anhydrides and multifunctional secondary amines, their depolymerization in acid aqueous solution, and circularity[116]. Copyright 2022, Springer Nature. (e) Chemical recycling of PUFs by catalytic acidolysis process[117]. Copyright 2023, American Chemical Society (ACS). All the figures are reproduced with permission.
Figure 5.
Chemical recycling via chain scission/reconstruction by adding key chemicals and/or chemistries. (a) Circularly recyclable polymers by topochemically weakened C−C bonds[113]. Copyright 2022, American Chemical Society (ACS). (b) Chemical upcycling of P3HB toward the bicyclic monomer 4-MOHB, which can be polymerized to afford P(4-MOHB) with a closed-loop cycle[114]. Copyright 2022, American Chemical Society (ACS). (c) Chemical recycling of post-consumer waste HDPE into a chemically recyclable material[115]. Copyright 2022, American Chemical Society (ACS). (d) Preparation of polymer networks from bifunctional maleic anhydrides and multifunctional secondary amines, their depolymerization in acid aqueous solution, and circularity[116]. Copyright 2022, Springer Nature. (e) Chemical recycling of PUFs by catalytic acidolysis process[117]. Copyright 2023, American Chemical Society (ACS). All the figures are reproduced with permission.
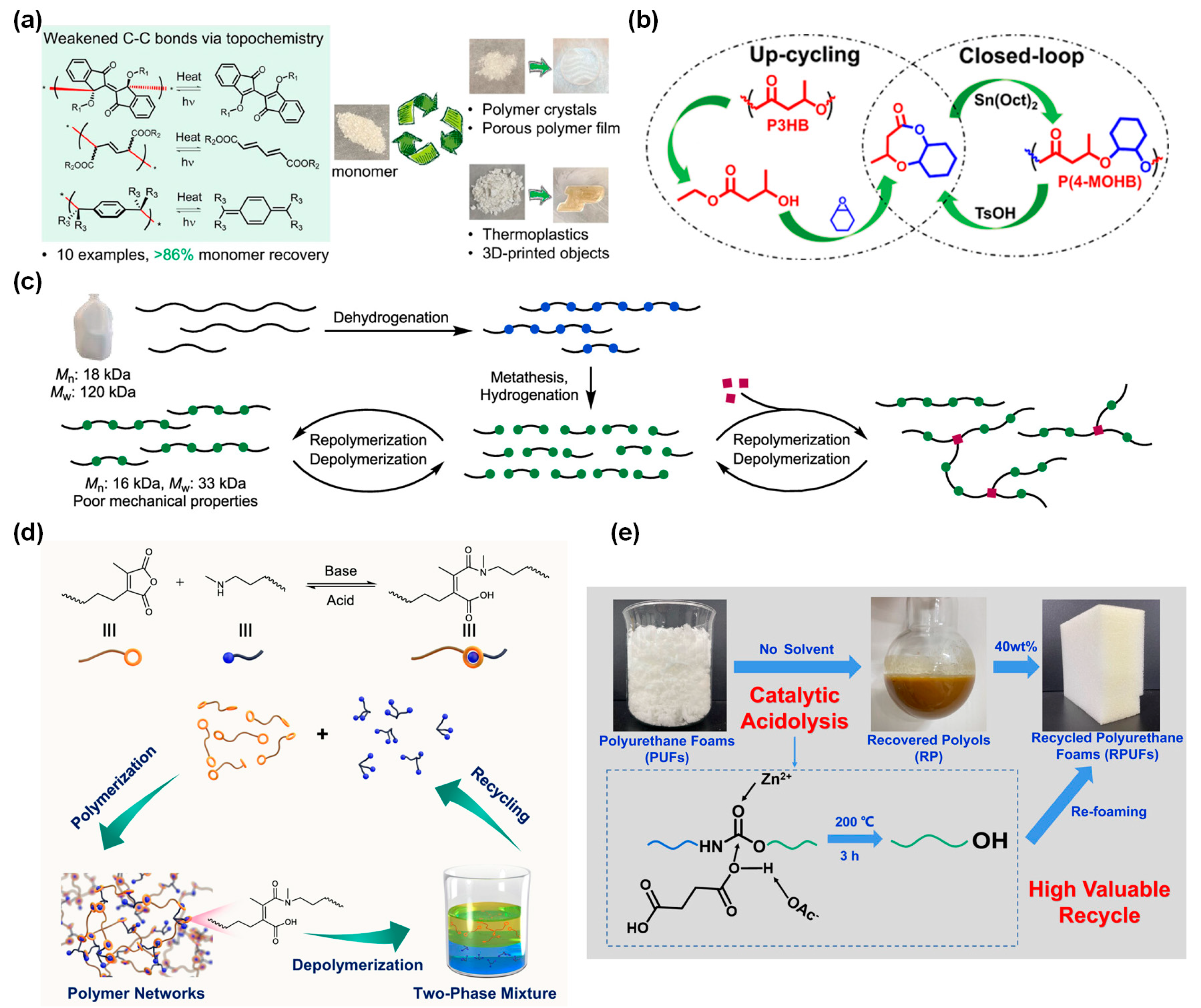

Table 1.
Common definitions of plastic recycling methods and their examples.
| ASTM D7209 | ISO 15270:2008 | Example |
| Primary (1st) recycling | Mechanical recycling | PET bottle to PET bottle |
| Secondary (2nd) recycling | Mechanical recycling | PET bottle to PET fiber |
| Tertiary (3rd) recycling | Chemical recycling | Glycolysis of PET |
| Quaternary (4th) recycling | Energy recovery | Pyrolysis of PET |
Table 2.
Ceiling temperature of common polymers.
| Monomer | Polymer | Tc (°C) |
| Ethylene | Polyethylene | 610 |
| Isoprene | Polyisoprene | 466 |
| 1,3-butadiene | Polybutadiene | 585 |
| Methyl methacrylate | Polymethyl methacrylate | 198 |
| Styrene | Polystyrene | 395 |
| Tetrafluoroethylene | Polytetrafluoroethylene (Teflon™) |
1100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



