Submitted:
08 October 2023
Posted:
09 October 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Several questions regarding the evolution of SARS-CoV-2 remain poorly elucidated. One of these questions is the possible evolutionary impact of SARS-CoV-2 after the infection in domestic animals. In this study, we aimed to evaluate the potential role of cats as generators of relevant SARS-CoV-2 lineages during the pandemic. A total of 105 full-length genome viral sequences obtained from naturally infected cats during the pandemic were evaluated by distinct evolutionary algorithms. Analyses were enhanced, including a set of highly related SARS-CoV-2 sequences recovered from human populations. Our results showed the apparent high susceptibility of cats to the infection SARS-CoV-2 compared with other animal species. Evolutionary analyses indicated that the phylogenomic characteristics displayed by cat populations were influenced by the dominance of specific SARS-CoV-2 genetic groups affecting human populations. However, disparate dN/dS rates at some genes between populations recovered from cats and humans suggested that infection in these two species may suppose a different evolutionary constraint for SARS-CoV-2. Interestingly, the branch selection analysis showed evidence of the potential role of natural selection in the emergence of 5 distinct cat lineages during the pandemic. Although these lineages were apparently irrelevant to public health during the pandemic, our results suggested that additional studies are needed to understand the role of other animal species in the evolution of SARS-CoV-2 during the pandemic.
Keywords:
cats
; SARS-CoV-2
; evolution
; variants
; phylogenomics
1. Introduction
More than three years after the emergence of COVID-19, SARS-CoV-2 has produced officially (9/15/2023) a total of 770,563,467 disease cases and 6,957,216 deaths confirmed around the world (https://covid19.who.int/). During this time, the scientific community has witnessed the exceptional evolutionary dynamics of this pathogen [1,2]. This situation has promoted the diversification of more than 1300 lineages [3]; some of these lineages are associated with variants of concern (VOC) responsible for the multiple contagion waves produced during the pandemic [4]. On 05/05/2023, the World Health Organization announced that COVID-19 was no longer a public health emergency [5]. However, many aspects regarding the biology of SARS-CoV-2 remain poorly understood, like the potential role of distinct animal species in promoting the emergence of lineages with a likely impact on public health.
SARS-CoV-2 infections were initially documented in humans; however, as positive cases increased worldwide, zooanthroponotic transmission events in domestic and wild animals were also reported. Thus, evidence of human-to-animal transmission has been documented in cats, dogs, tigers, lions, puma, snow leopards, gorillas, and mink [6,7,8,9]. Interestingly, in the case of minks, the detection of three mutations (Y453F, F486L, and N501T) located in the receptor binding domain of the spike protein of SARS-CoV-2 may be associated with the adaptation of this virus to this animal species [7]. Furthermore, experimental in-vivo co-infections in white-tailed deer revealed the ability of the variant alpha to outcompete the ancestral lineage A, indicating the potential fitness increase of some variants to infect animal species [10].
Due to the close contact with humans, companion animals are important targets for the infection with SARS-Cov-2. This asseveration is consistent with the most updated report (9/19/2023) from the Animal and Plant Health Inspection Service (USDA), showing the increased number of positive cases detected in cats and dogs (https://www.aphis.usda.gov/aphis/dashboards/tableau/sars-dashboard). However, experimental studies have demonstrated unequal levels of susceptibility to SARS-CoV-2 between both species. Hence, cats show higher susceptibility to infection with SARS-CoV-2 than dogs [11]. While in dogs, SARS-Cov-2 is associated with poor replication and lack of transmissibility to naïve dogs [11,12], cats seem to support higher levels of viral replication, with the ability to promote viral transmission among susceptible cats [11,12,13]. Interestingly, despite the high susceptibility to SARS-CoV-2, multiple experimental trials in cats resulted in subclinical outcomes [12,14,15], highlighting the relevance of cats transmitting this virus in conditions of inapparent signs of disease. This situation may represent a potential risk factor for cat owners.
In contrast, natural infections reported in cats linked to multiple VOC resulted not only in subclinical outcomes but also in clinical infections, including signs of tiredness, lethargy, fever, ocular and nasal discharge, dyspnea, sneezing, cough, vomiting, diarrhea, anorexia, myocarditis, and weight loss [15,16,17,18,19]. The disparate clinical outcomes induced by different lineages of SARS-CoV-2 were demonstrated experimentally in cats, showing that omicron lineage BA.1.1 represents a lower pathogenicity phenotype than B.1 and Delta (B.1.1.529) lineages [15]. The above suggests that diverse lineages of SARS-CoV-2 represent a different phenotype for cats, emphasizing the relevance of evolutionary analysis to understand the dynamics associated with the possible adaptation of SARS-CoV-2 to cat populations and the consequent emergence of new viral lineages.
Interestingly, experimentally, evolutionary analyses in cats showed the rapid evolution of SARS-CoV-2 after infection in this animal species [20,21], suggesting the potential relevance of cats as a generator of new SARS-CoV-2 variants during the pandemic. Furthermore, phylogenetic analyses comparing sequences obtained from natural infections with SARS-CoV-2 in cats and human populations indicated the high susceptibility of cats to the infection with lineages affecting humans and highlighting the unidirectionality of the transmission events between both species (zooanthroponotic transmission) [20].
Based on the above, our study aimed to evaluate the evolutionary dynamics of diverse SARS-CoV-2 lineages related to natural infections in cat populations. For this purpose, we used a combination of multiple evolutionary algorithms to evaluate a set of full-length viral sequences recovered from natural infections in cats during the pandemic. Moreover, including a group of highly related viral sequences obtained from human infections, we developed an interesting evolutionary model to test the possible role of natural selection in the emergence of SARS-CoV-2 due to replication in cat populations. Our results are discussed regarding the relevance of cats as generators of new SARS-CoV-2 lineages and the potential impact of these on public health.
2. Materials and Methods
2.1. Viral Sequences and Metadata Information
A total of 105 complete viral genomes from cats naturally infected with SARS-CoV-2 during the pandemic were retrieved from the GISAID database [22]. Specific information about each sequence is provided in, Supplementary File 1. Additionally, 117 complete viral genomes from human cases showing the highest levels of identity against cat SARS-CoV-2 genomes were obtained from the GenBank database. Metadata information regarding SARS-CoV-2 viral lineages recovered from infections in diverse animal species, including cat (Felis catus), dog (Canis lupus familiaris), white-tailed deer (Odocoileus virginianus), lion (Panthera leo), tiger (Panthera tigris), Western gorilla (Gorilla gorilla), American mink (Neogale vison), house mouse (Mus musculus), Syrian hamster (Mesocricetus auratus), and Pangolin (Manis javanica), was retrieved from the GISAID database [22].
2.2. Hierarchical Cluster Analysis
Hierarchical cluster analysis (HCA) was conducted to assess a comparison between SARS-CoV-2 viral lineages recovered from cats and other animal species. This analysis was performed using the predictive analytic software JMP ® Pro version 16.0.0. Generally, HCA uses a matrix of N x M dimensions, where N corresponds to the multiple viral lineages and M the multiple animal species included in this study. Clusters were defined by the Ward linkage method. Statistical significance between clusters was conducted by analysis of variance (ANOVA) and supported by Tukey's honest significance test (p=0.05).
2.3. Phylogenetic Analysis
The phylogenetic relationship between multiple viral sequences recovered from naturally infected clinical cases from cats and humans was assessed by the maximum likelihood method, using the general time reversible model (GTR) considering gamma distribution and invariable sites. The use of this model was supported by the lowest Bayesian information criterion scores (BIC). Analyses were conducted in the molecular evolutionary genetic analysis software MEGA version 10.2.5. [23]. Additionally, this software was used to perform the pairwise distance analysis included in this study.
2.4. Evolutionary Analysis
To assess the evolutionary dynamics among SARS-CoV-2 viral lineages recovered from cats, we used the evolutionary algorithms tools Fixed Effects Likelihood (FEL) [24,25] and Mixed Effects Model of Evolution (MEME). Both algorithms detect sites under negative or positive selection, acting in a pervasive (MEME and FEL) of episodic (MEME) manner by inferring rates of synonymous and nonsynonymous substitutions in a codon base phylogenetic framework. Moreover, the presence of viral lineages potentially emerging because of natural selection was evaluated by the algorithm Adaptive Branch-Site Random Effects Likelihood (aBSREL) [26]. aBSREL uses the likelihood ratio test statistic for the selection (LRT) of relevant branches in the tree and the Holm-Bonferroni method for the correction p-values. Subsequently, the algorithm Branch-site Unrestricted Statistical Test for Episodic Diversification (BUSTED) [27] was used to identify possible sites under positive selection in the branches detected by aBSREL. BUSTED is a likelihood ratio algorithm to test for evidence of diversifying selection affecting some positions in the alignment along some branches of the tree [28].
3. Results
3.1. Overview of the SARS-CoV-2 Lineages Isolated from Cat Infections Around the World
A description of the cat samples included in this study is provided in Figure 1. The metadata analysis regarding the pangolin lineage classification associated with 179 clinical reports was analyzed by hierarchical cluster analysis to obtain a general perspective about the distinct SARS-CoV-2 lineages isolated from cat infections during the pandemic. Furthermore, metadata reports from nine other animal species were included to enhance the analysis. A total of 179 viral lineages were included in the analysis. Cats were the animal species with the highest number of lineages associated with clinical infections (n=73), followed by dogs and white-tailed deer with 56 and 48, respectively (Figure 2). Hierarchical cluster analysis grouped lineages in two different clusters. While cluster 1 comprised most of the lineages representing single reports in cats, cluster 2 included a specific group of just lineages associated with multiple clinical reports. Interestingly, this situation was consistent for cats and most animal species included in the analysis, suggesting the potential relevance of these lineages to infect animal species during the pandemic (Figure 2). The preponderance of lineages associated with cluster 2 was assessed by analysis of variance (p>0.001) and confirmed by Student’s t-test (p>0.001).
Additionally, no correlation was found between the prevalence of these lineages in human populations and the number of reports in cats and other species (Figure 2). For example, in the case of cats, except B.1.1.7 (Alpha variant) representing 7.66% of the total number of SARS-CoV-2 sequences reported in the GISAID database and the second most prevalent in cats (7.79%), B.1, the most prevalent lineage reported in cats associated with 10.38% of the reports, represented just the 0.88% of the sequences in GISAID related to human infections. In this context, the highest prevalent lineages in humans (BA.2, BA.1.1, AY.4, BA.1, and AY.43) were associated with main cluster 1 (Figure 2).
Clinical reports showing cat deaths were related to single lineages, including B.1.526, P.1, B.1.369, B.1.36.35, B.1.234, and B.1.2 (the only one included in cluster 2) (Figure 2).
3.2. Evolutionary Characteristics of Viral Lineages Affecting Cat Populations
The viral lineages affecting cat populations were classified into different genetic groups based on the GISAID classification to assess the evolutionary dynamics of these viral. The prevalence of diverse genetic clades in cats was compared with those predicted for human populations (Figure 3A). Generally, between 2020 and 2022, we found a positive correlation between the genetic groups affecting humans and cat populations, indicating that viral genetic groups affecting cats during the pandemic were highly influenced by the circulation dynamics of these groups in humans. The latest can be exemplified by the dominance of the GK and GRA genetic groups in human populations and the high correlation (R2=0.99) in the prevalence of these genetic groups in cat infections during 2022 (Figure 3A). GH and GK genetic groups were associated with most clinical reports evaluated in this study (Figure 3B).
Subsequently, to get more insights into the phenotypic characteristics and the evolutionary dynamics of viral lineages affecting cats during the pandemic, we conducted a phylogenomic analysis using a representative set of 105 sequences (Figure 4). This number was defined based on sequence quality. Thus, we favored sequences that preserved the integrity of multiple viral genes. The pairwise analysis showed an identity between 99.62% and 100% (~99.86%) and 99.20% and 100% (~99.72%) at the nucleotide and amino acid levels, respectively, indicating the high conservation among SARS-CoV-2 viral lineages affecting cat populations. Furthermore, 100% of identity was found between some viral sequences from genetic groups GH (9 B.1.497 GH 21-11 B.1.497 GH 21 and 55 B.1 GH 20-56 B.1 GH 20), GR (72 B.1.1.254 GR 20-73 B.1.254 GR 20 and 87 B.1.1.298 GR 20-89 B.1.1.298 GR 20), and GK (100 AY.3 GK 21-101 AY.3 GK 21). Considering our results regarding the differences in the prevalence of different viral lineages during the infection in cats (Figure 2), our phylogenetic inference showed the existence of multiple events of divergence within some of the most prevalent lineages (B.1.497, B.1, B.1,2, AY.69, AY.103, AY.3, B.1.1.7) (Figure 4), indicating that cats were infected with potentially distinct subtypes of each lineage. In addition, dN/dS rates equal to 0.6584 obtained by FEL analysis indicated that purifying selection was the main force driving the evolution of SARS-CoV-2 among cat populations. However, dN/dS calculations within different lineages evidenced that purifying selection is dissimilarly acting among them (Figure 4). Thus, it was possible to observe the contrasting dN/dS rates between B.1.497 (dN/dS= 0.905) and AY.3 (dN/dS= 0.346), suggesting that lineages infecting cats were subjected to different evolutionary pressures during the pandemic.
An additional phenotypic characteristic of viral lineages affecting cats during the pandemic was the presence of premature stop codons at the ORF8 gene at codon positions 27 and 64 of 9 viruses (Figure 4), suggesting that ORF8 is not an essential protein for the infection in cats.
To identify potential phenotypic differences among multiple lineages, we conducted an evolutionary analysis to identify codon sites under positive selection using the algorithm MEME. Ten codon sites were identified along the genome, involving six different genes, the S gene accounting for the highest number of codons under positive selection (n=4) (Figure 5A). On the other hand, the most frequent substitutions in the population were mutations at NSP2-85 (n=28), NSP4-492 (n=22), and S-452 (n=32) gene codons. Furthermore, most of the codons identified under positive selection were wildly spread among diverse genetic groups, even when some were present in the population in lower frequencies (NSP16-216, S-95, S-477, S-501, and N-13). When we evaluated the dataset by the algorithm FEL, we identified codons NSP4-492, NSP16-216, S-95, S-452, S-477, and N-13 under positive selection (p-value 0.1), suggesting that these codons were evolving under pervasive diversifying selection; hence, highlighting the relevance of these codons among different genetic groups. Conversely, codons identified under positive selection just by MEME analysis (NSP2-85, NSP2-280, S-501, and N-3) might be associated with an evolutionary pattern of episodic selection.
Once we identified potentially relevant codon positions during the infection of SARS-CoV-2 in cat populations, we analyzed the evolutionary dynamic of those positions during infections of this pathogen in human populations. For this purpose, we attempted to recreate a dataset of viral sequences associated with human infections highly related to viral sequences recovered from cats. We conducted a BLAST analysis, using each of the 105 cat sequences used in this study as a query. As a result, a total of 117 representative viral sequences recovered from human infections were obtained for this analysis. Phylogenetic analysis using this new dataset evidenced the close genetic relationship between both viral populations (Figure 5). No evidence of specific SARS-CoV-2 host adaptation was observed. However, based on the branch topology and the ancestral relationship between cat and human lineages displayed in the tree, it is possible to predict the existence of multiple divergence events that occurred during the infection of SARS-CoV-2 in cats, suggesting that human-cat transmission events promoted the evolution of SARS-COV-2.
Furthermore, MEME analysis showed that all positions identified in cat populations (Figure 5A) appeared under positive selection in human populations (Figure 5B). Overall, we found that mutations associated with these codons have been found in SARS-CoV-2 lineages, affecting humans in numerous countries globally (Figure 5B). When we assessed the Pearson correlation between the allele frequency found in cats at different codons under positive selection and the frequency of these alleles in viral sequences recovered from humans at GISAID database (Figure 5B), we found a significant positive correlation (p-value=0.0159; r=0.5955) in the frequency between both populations, indicating that the viral circulation dynamics of SARS-CoV-2 in human population influenced the phenotypic characteristics of lineages affecting cats. Suitable examples of the latter can be observed in codon NSP2-280, where the mutation AAC-TAC (present in a single viral sequence in cats) represents just 0.53% of the mutations reported in the GISAID database in the human population and in the polymorphic codon sites NSP16-216 and S-452 where codons with low frequent substitutions appeared in frequencies lower than 1% in human populations (Figure 5B).
In contrast, when we compared dN/dS rates between multiple gene segments from viral populations recovered from humans and cats, we observed a disparity between both populations in the dN/dS rates from genes ORF3a, E, ORF7b, and ORF10 (Figure 5C), suggesting that replication of SARS-CoV-2 in these hosts may represent a dissimilar selective environment.
3.3. Evaluating the Hypothesis about Independent Evolution of SARS-CoV-2 in Cat Populations
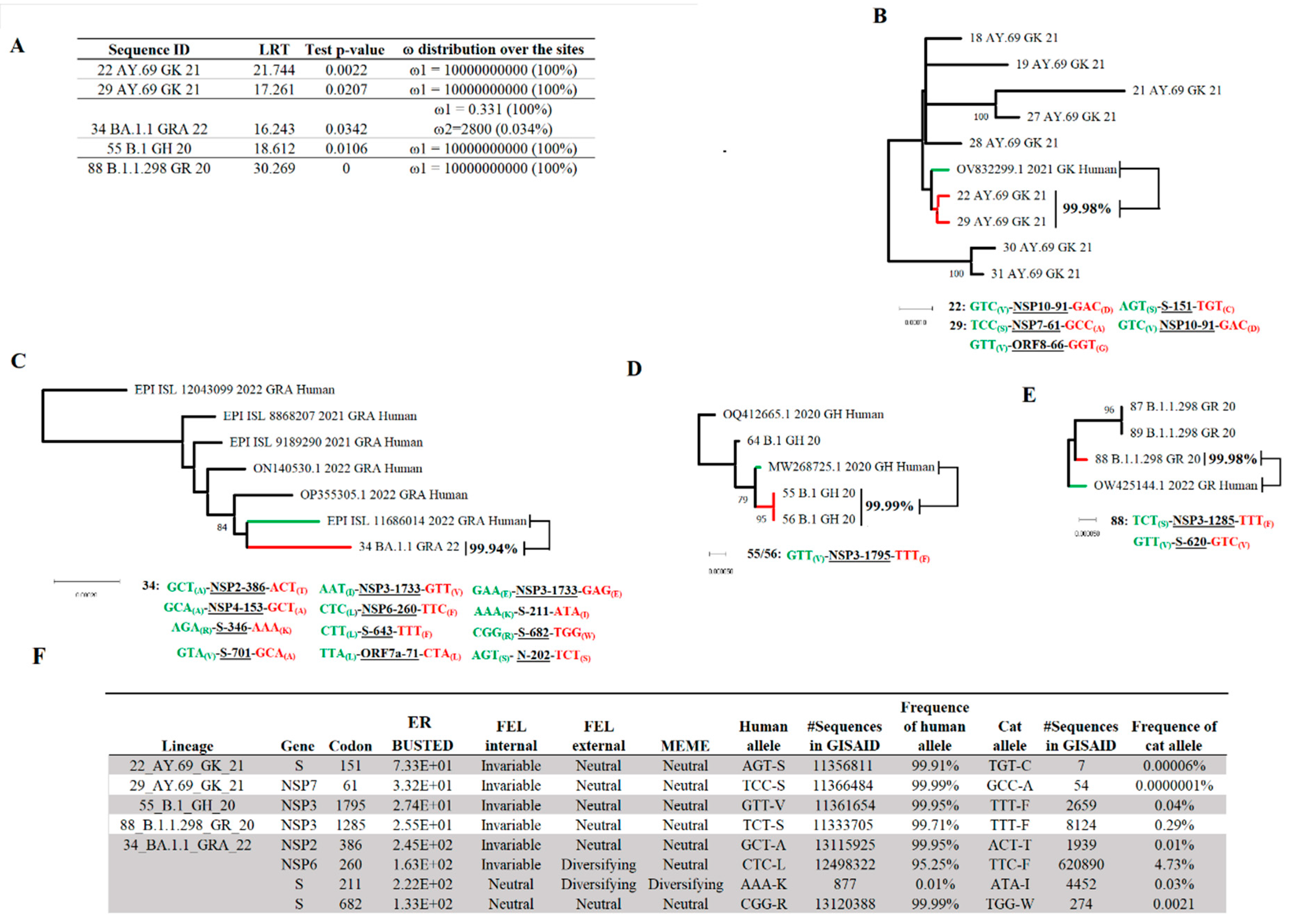
We tested the hypothesis about the potential independent evolution of SARS-CoV-2 in cat populations. To this end, we used data comprising 105 viral sequences recovered from naturally infected cats and a set of 117 highly related viral sequences recovered from human infections. The algorithm aBSREL evaluated this dataset to attempt to discover potential cat lineages evolving because of natural selection. Interestingly, five different cat lineages were found under positive selection (Figure 4A). Three of these lineages were associated with the pangolin lineages B.1 (55 B.1 GH 20), AY.69 (22 AY.69 GK 21 and 29 AY.69 GK 21), some of the most prevalent lineages in cats. The other two cat lineages were associated with the pangolin lineages BA.1.1 (34 BA.1.1 GRA 22) and B.1.1.298 (88 B.1.1.298 GR 20). Phylogenetic analysis showed the close genetic relationship of these lineages with viral lineages associated with human populations, with a nucleotide identity calculated between 99.94% and 99.99% (Figure 4B–E). Overall, with exception of cat lineage 34 BA.1.1 GRA 22, where a total of 12 SNPs were identified among seven different genes (Figure 6C) between this lineage and the closet human lineage, for the rest of the lineages between 1 and 3 SNPs were identified in lineages: 22 AY.69 GK 21, 29 AY.69 GK 21, 55 B.1 GH 20, and 88 B.1.1.298 GR 20, impacting a minimal number of genes (Figure 6B,D,E).
Finally, to identify potential codon sites linked to the emergence of cat lineages, we evaluated previously relevant branches identified by aBSREL using the algorithm BUSTED. Evidence ratios (site level likelihood ratios) for ω>1 were found in eight different codon sites among incident branches of identified cat lineages (Figure 6F). Four were identified in the lineage 34 BA.1.1 GRA 22 from these sites, while single sites were identified in the remaining lineages. Subsequently, we assessed the evolutionary significance of these sites, using specialized algorithms to identify natural selection at codon sites (FEL and MEME). Evidence of diversified selection in just two sites associated with isolation 34 BA.1.1 GRA 22 (NSP6 codon 260 and S codon 211) (Figure 6F) was found. In both cases, FEL identified evidence of diversifying selection in the external nodes, suggesting that these codon sites did not give an adaptive advantage to SARS-CoV-2 at the cat population level.
Afterward, to assess the impact during the pandemic of the alleles present in codon sites potentially linked to the emergence of cat lineages, we evaluated the frequency of these alleles at specific codon sites in the GISAID database. Our results indicated that in all cases, the occurrence of the alleles associated with multiple cat lineages was present at a low frequency in the GISAID database (Figure 6F). The latter suggests the lack of relevance of these alleles in promoting the adaptation of dominant SARS-CoV-2 lineages during the pandemic. Conversely, at these codon sites, alleles present in viral sequences isolated from humans, including the ones associated with the possible ancestral lineages predicted in our model, matched with the most frequent allele for this codon site described in the GISAID database. The only exception was the allele present in the predicted ancestral sequence for cat lineage 34 BA.1.1 GRA 22, where at gene S codon 211, the human isolate has the allele AAA that showed an overall frequency at this codon position of 0.01% (Figure 6F).
4. Discussion
At this point, we consider that deciphering the evolutionary dynamics of SARS-CoV-2 appears as an imperative task to understand the future impact of SARS-CoV-2 in the world. As a main host of this pathogen, during the pandemic, multiple research subjects involving COVID-19 have focused mainly on understanding this disease from the human standpoint. However, as mentioned before, multiple animal species were affected by SARS-CoV-2 during the pandemic, which shows this viral agent's complex biology. Currently, limited information is known about the potential role of animal species in the epidemiological triad of this disease. In this sense, a recent study conducted on white-tailed deer in the US showed the relevance of this animal species as a potential reservoir of SARS-CoV-2, promoting not only the evolution of this virus but also probably spillovers to human populations [29], stressing the preponderance of some animal species in the evolution of SARS-CoV-2, and the potential consequences for public health.
Herein, we focused on SARS-CoV-2 infections in domestic cats. We considered cats a potentially relevant animal species for the evolution of SARS-CoV-2 based on their high susceptibility to this virus and its ability to transmit the infection to susceptible cats [11,12,13]. In this context, to obtain more insights about the conceivable consequences of the evolution of SARS-CoV-2 produced by natural infections in cat populations, we aimed to describe the evolutionary dynamics of viral lineages affecting cat populations during the pandemic. The results of our study provide a different but complementary perspective to previous studies [20,21], supporting the potential role of cats as generators of new variants of SARS-CoV-2.
Our study has diverse limitations to be considered in interpreting the results presented here. The most important limitation is the reduced number of available full-length sequences obtained from naturally infected cats during the pandemic, representing equally multiple geographical regions worldwide. This fact may have hindered the detection of additional codon sites under positive selection and lineages evolving from cat populations due to natural selection. Moreover, it is crucial to consider that the results from this study were obtained entirely by in-silico approaches, warning that experimental evidence is necessary to validate the relevance of our predictions. Therefore, sequencing mistakes affecting the quality of the viral sequences used in this study are another factor to consider in interpreting the results from this study.
To assess the susceptibility of cats to the infection with SARS-CoV-2 in nature, first, we compared clinical reports available in the GISAID database between cats and multiple other animal species. Our results indicated that compared with other animal species, the infections in cats were associated with a higher number of viral lineages (Pangolin lineage classification), indicating the increased susceptibility of this animal species in nature. However, although the results of our analysis might have been biased by the limited available metadata information from cats and the other animal species, our results were consistent with the differences in susceptibility experimentally evidenced between cats and dogs [11,12], supporting the view about cats as a highly susceptible animal species, with a potential role for the transmission of SARS-CoV-2 in nature [30,31].
Furthermore, we identified specific lineages of SARS-CoV-2 associated with the highest prevalence of cases in different animal species. Remarkably, the detection of these lineages was coherent among cats and other multiple animal species, supporting the accuracy of our results, thus highlighting the potential ability of these lineages to infect other species different than humans efficiently. In this context, our last asseveration may be supported by our result showing that except for lineage B.1.1.7 (alpha), most of the highly prevalent lineages found infecting multiple animal species did not correspond with the high prevalence of these lineages in human populations.
Contrariwise, our analysis identified some lineages that may have more affinity for specific animal species. Although this result could have been highly influenced by the reduced metadata information about different species, it may be consistent with the experimental evidence showing the disparate levels of pathogenicity associated with diverse SARS-CoV-2 lineages in cats [15].
Consequently, we focused on understanding if the circulation dynamics of SARS-CoV-2 in cats were influenced by the dynamic in human populations. We conduct this analysis using the GISAID genetic clade classification, which congregates multiple pangolin SARS-CoV-2 lineages in phylogenetic clusters based on the statistical distribution of the genome distances among lineages [32]. The positive correlation found in this study between the circulation dynamics of different SARS-CoV-2 genetic groups in cats and human populations during the pandemic suggested that natural infections in cats were influenced by the circulation dynamics of SARS-CoV-2 in human populations. The latter evidences the unlikely role of cats as reservoirs of SARS-CoV-2 in nature. On the other hand, despite there is a discrepancy between these and our previous results that showed the lack of correlation between the most prevalent pangolin lineages in cats and the dominant ones in human populations, it makes it possible to suggest that regardless of infections in cats were influenced for the dominant genetic groups in human populations, specific pangolin lineages within these groups might have had more affinity to infect cat populations. This condition may explain the discrepancy between both analyses.
The evaluation of phylogenomics of viral lineages that affected cat populations throughout the pandemic denoted the existence of multiple events of divergence within different lineages during the infection in cat populations. The evolutionary significance of these events was more evident when we reconstructed the phylogenetic analysis along with a set of highly related viral genomes recovered from human populations. Thus, two different conclusions can be made from this analysis: i) consistently with a previous study evaluating the phylogenetic relationship between SARS-CoV-2 lineages associated with the delta variant recovered from cats and human populations [20], our analysis did not show any topological host pattern association among different lineages recovered from both species, indicating the lack of host-specific adaptation patterns; ii) despite the lack of host-specific adaptation patterns, the branch topology observed (denoting multiple events of divergence) among diverse viruses within the same lineage recovered from both hosts evidenced the evolution of SARS-CoV-2 in cat populations.
Additionally, to obtain more insights into the evolutionary dynamics of SARS-CoV-2 in cat populations, our dataset was evaluated by multiple evolutionary tests previously used in SARS-CoV-2 studies [33]. We aimed to understand if multiple divergence events observed in our phylogenetic analysis might be linked to natural selection or genetic drift. The overall dN/dS rate <1 calculated in the viral lineages from the cat population indicated that negative selection was the main force shaping the evolution of SARS-CoV-2 in cats. This result suggests that in nature, human-cat transmission events were characterized by a strong evolutionary constraint in cats, favoring the preservation of the viral phenotypes circulating in humans. Our result was consistent with an experimental study in cats, showing the relevance of purifying selection during the infection of SARS-CoV-2 in this animal species [34]. However, when we calculated dN/dS ratios within different lineages, we observed disparate levels of purifying selection among them, suggesting that different lineages were subjected to different evolutionary constraints. This situation may be consistent with the phenotypic differences depicted in diverse SARS-CoV-2 lineages during experimental infections in cats [15].
In evaluating the presence of positive selection at different codon sites of the viral genomes recovered from cat populations, we described multiple sites under positive selection. However, when we analyzed the dataset from human populations, we found that all these sites were already selected because of the evolution of SARS-CoV-2 in human populations, supporting our last statement about the essential role of purifying selection during the evolution of SARS-CoV-2 in cat populations. In this context, some of the codon sites predicted under positive selection in both populations, including NSP4- 492 (increased infectivity and evasion of the immune response [35], S-95 (increased infectivity and transmissibility) [36], S-452 (decreased sensitivity to monoclonal antibodies) [37,38,39], S-477 (decreased sensitivity to monoclonal antibodies) [40,41], S-501(decreased sensitivity to monoclonal antibodies, enhance infection and transmission) [42], and N-13 (Affect CD8+ response) [43], have shown their biological relevance for the immune host evasion produced by SARS-CoV-2, suggesting that these positions may provide adaptive advantages for this virus during the infection in both hosts. However, despite the failure to infer putative specific codon sites evolving under positive selection exclusively in the viral population recovered from cats, our comparison among genes between viral populations recovered from both hosts, suggested disparate evolutionary rates between viruses recovered from different hosts, indicating the possible different evolutionary contrast imposed by humans and cats during the infection. In this sense, further experiments are required to explain the increased levels of positive selection found in E and ORF10 genes in cat populations. Based on previous publications, it may be possible to speculate about probable repercussions in controlling the immune host response [44,45,46].
Finally, according to the algorithm used in aBSREL, our results showed evidence of the potential action of natural selection in the emergence of SARS-CoV-2 lineages due to replication in cat populations. However, considering the minimal number of cat lineages found evolving by the action of natural selection and the topology showing the divergence events that occurred between human and cat lineages, it is possible to suggest that genetic drift is another significant force shaping the evolution of SARS-Cov-2 in cats. This result was consistent with a previous experimental study in cats [34]. Also, we found that specific mutations linked to the identified cat lineages under positive selection had minimal impact in human populations, suggesting the unconnected role of cats as generators of relevant SARS-CoV-2 variants during the pandemic.
In summary, our study shows a perspective on the evolution of SARS-CoV-2 in cat populations during the pandemic. Our findings were consistent with previous publications using experimental approaches in cats, indicating the rapid evolution of SARS-CoV-2 in this domestic animal, with purifying selection and genetic drift as the main evolutionary forces acting on this virus during the infection in cats. Although the main conclusion of our study is the possible lack of relevance of cats as generators of relevant variants for public health, considering the high susceptibility of cats to the infection of SARS-CoV-2, it is important to warn about the possible role of cats in the transmission of this pathogen. Considering the phenotypic differences observed experimentally among SARS-CoV-2 lineages in cats [15], it is essential to state that our results may be regarded as inconclusive based on the limited dataset used in this study. In this sense, more studies are needed to understand the potential role of cats in the generation of relevant SARS-CoV-2 lineages for the public health, especially in the context of the Omicron sub-lineages infections currently dominating the infections in human populations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: SARS-CoV-2 viral sequences from cats. Figure S2: Phylogenomic dynamics between cat and human SARS-CoV-2 lineages. Figure S3: The phylodynamics of multiple codon positions identified by BUSTED.
Author Contributions
Conceptualization, N-GR F-BA, and L-VS.; methodology, N-GR, and L-VS.; validation, N-GR and L-VS, formal analysis, N-GR, and L-VS; investigation, N-GR and L-VS; resources, N-GR F-BA and L-VS; data curation, N-GR, and L-VS .; writing—original draft preparation, N-GR and L-VS .; writing—review and editing, N-GR F-BA, and L-VS.; visualization, N-GR F-BA and L-VS; supervision, L-VS; project administration, F-BA, funding acquisition, F-BA. All authors have read and agreed to the published version of the manuscript.”
Funding
Please add: “This research was funded by The National Autonomous University of Mexico (UNAM), grant number: FMVZ/SEC/OSR/435/2022.
Data Availability Statement
During this study were not generated new SARS-CoV-2 sequences. All sequences used for this study are publicly available in the GISAID and GenBank databases.
Acknowledgments
Authors thankfully acknowledge Kathleen Apicelli for her valuable assistance in improving the quality of the figures presented in this manuscript. In addition, we gratefully acknowledge all data contributors responsible for obtaining the specimens and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. Supplementary files one and two included all accession numbers from which specific information about contributors and laboratories responsible for generating these sequences can be found in the GISAID database.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front Microbial. 2020, 11, 550674. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200. [Google Scholar] [CrossRef]
- O'Toole, A.; Pybus, O.G.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genomics 2022, 23, 121. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L. The complex evolutionary dynamics of SARS-CoV-2, a big challenge to control the pandemic of COVID-19. J. Med. Virol. 2022, 94, 5082–5085. [Google Scholar] [CrossRef]
- Lenharo, M. WHO declares end to COVID-19's emergency phase. Nature 2023. [Google Scholar] [CrossRef]
- Mahdy, M.A.A.; Younis, W.; Ewaida, Z. An Overview of SARS-CoV-2 and Animal Infection. Front. Vet. Sci. 2020, 7, 596391. [Google Scholar] [CrossRef]
- Prince, T.; Smith, S.L.; Radford, A.D.; Solomon, T.; Hughes, G.L.; Patterson, E.I. SARS-CoV-2 Infections in Animals: Reservoirs for Reverse Zoonosis and Models for Study. Viruses 2021, 13, 494. [Google Scholar] [CrossRef]
- Navarro-Lopez, R.; Solis-Hernandez, M.; Rocha-Martinez, M.K.; Eberl, S.; Gomez-Romero, N.; Velazquez-Salinas, L.; Estrada-Franco, J.G. Near-Full-Length Genome Sequences Representing an Event of Zooanthroponotic Transmission of SARS-CoV-2 Lineage B.1.189 in Mexico during 2020. Microbiol Resour Announc 2022, 11, e0049722. [Google Scholar] [CrossRef]
- Koeppel, K.N.; Mendes, A.; Strydom, A.; Rotherham, L.; Mulumba, M.; Venter, M. SARS-CoV-2 Reverse Zoonoses to Pumas and Lions, South Africa. Viruses 2022, 14, 120. [Google Scholar] [CrossRef]
- Cool, K.; Gaudreault, N.N.; Morozov, I.; Trujillo, J.D.; Meekins, D.A.; McDowell, C.; Carossino, M.; Bold, D.; Mitzel, D.; Kwon, T.; et al. Infection and transmission of ancestral SARS-CoV-2 and its alpha variant in pregnant white-tailed deer. Emerg. Microbes Infect. 2022, 11, 95–112. [Google Scholar] [CrossRef]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef]
- Bosco-Lauth, A.M.; Hartwig, A.E.; Porter, S.M.; Gordy, P.W.; Nehring, M.; Byas, A.D.; VandeWoude, S.; Ragan, I.K.; Maison, R.M.; Bowen, R.A. Experimental infection of domestic dogs and cats with SARS-CoV-2: Pathogenesis, transmission, and response to reexposure in cats. Proc. Natl. Acad. Sci. USA 2020, 117, 26382–26388. [Google Scholar] [CrossRef]
- Neira, V.; Brito, B.; Agüero, B.; Berrios, F.; Valdés, V.; Gutierrez, A.; Ariyama, N.; Espinoza, P.; Retamal, P.; Holmes, E.C.; et al. A household case evidences shorter shedding of SARS-CoV-2 in naturally infected cats compared to their human owners. Emerg. Microbes Infect. 2021, 10, 376–383. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Trujillo, J.D.; Carossino, M.; Meekins, D.A.; Morozov, I.; Madden, D.W.; Indran, S.V.; Bold, D.; Balaraman, V.; Kwon, T.; et al. SARS-CoV-2 infection, disease and transmission in domestic cats. Emerg. Microbes Infect. 2020, 9, 2322–2332. [Google Scholar] [CrossRef]
- Martins, M.; do Nascimento, G.M.; Nooruzzaman, M.; Yuan, F.; Chen, C.; Caserta, L.C.; Miller, A.D.; Whittaker, G.R.; Fang, Y.; Diel, D.G. The Omicron Variant BA.1.1 Presents a Lower Pathogenicity than B.1 D614G and Delta Variants in a Feline Model of SARS-CoV-2 Infection. J. Virol. 2022, 96, e0096122. [Google Scholar] [CrossRef]
- Zoccola, R.; Beltramo, C.; Magris, G.; Peletto, S.; Acutis, P.; Bozzetta, E.; Radovic, S.; Zappulla, F.; Porzio, A.M.; Gennero, M.S.; et al. First detection of an Italian human-to-cat outbreak of SARS-CoV-2 Alpha variant - lineage B.1.1.7. One Health 2021, 100295. [Google Scholar] [CrossRef]
- Schiaffino, F.; Ferradas, C.; Jara, L.M.; Salvatierra, G.; Dávila-Barclay, A.; Sanchez-Carrion, C.; Ulloa, A.; Mascaro, L.; Pajuelo, M.J.; Guevara Sarmiento, L.; et al. First Detection and Genome Sequencing of SARS-CoV-2 Lambda (C.37) Variant in Symptomatic Domestic Cats in Lima, Peru. Front. Vet. Sci. 2021, 8, 737350. [Google Scholar] [CrossRef]
- Kuhlmeier, E.; Chan, T.; Agüí, C.V.; Willi, B.; Wolfensberger, A.; Beisel, C.; Topolsky, I.; Beerenwinkel, N.; Stadler, T. ; Swiss Sars-CoV-Sequencing Consortium; Jones, S.; et al. Detection and Molecular Characterization of the SARS-CoV-2 Delta Variant and the Specific Immune Response in Companion Animals in Switzerland. Viruses. [CrossRef]
- Ferasin, L.; Fritz, M.; Ferasin, H.; Becquart, P.; Corbet, S.; Ar Gouilh, M.; Legros, V.; Leroy. E.M. Infection with SARS-CoV-2 variant B.1.1.7 detected in a group of dogs and cats with suspected myocarditis. Vet. Rec. 2021, 189, e944. [Google Scholar] [CrossRef]
- Bashor, L.; Gagne, R.B.; Bosco-Lauth, A.; Stenglein, M.; VandeWoude, S. Rapid evolution of SARS-CoV-2 in domestic cats. Virus Evol. 2022, 8, veac092. [Google Scholar] [CrossRef]
- Bashor, L.; Gagne, R.B.; Bosco-Lauth, A.M.; Bowen, R.A.; Stenglein, M.; VandeWoude, S. SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection. Proc. Natl. Acad. Sci. USA 2021, 118, e2105253118. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W. GISAID's Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Hale, V.L.; Dennis, P.M.; McBride, D.S.; Nolting, J.M.; Madden, C.; Huey, D.; Ehrlich, M.; Grieser, J.; Winston, J.; Lombardi, D.; et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature 2022, 602, 481–486. [Google Scholar] [CrossRef]
- Gonzales, J.L.; de Jong, M.C.M.; Gerhards, N.M.; Van der Poel, W.H.M. The SARS-CoV-2 Reproduction Number R0 in Cats. Viruses 2021, 13, 2480. [Google Scholar] [CrossRef]
- Gerhards, N.M.; Gonzales, J.L.; Vreman, S.; Ravesloot, L.; van den Brand, J.M.A.; Doekes, H.P.; Egberink, H.F.; Stegeman, A.; Oreshkova, N.; van der Poel, W.H.M.; et al. Efficient Direct and Limited Environmental Transmission of SARS-CoV-2 Lineage B.1.22 in Domestic Cats. Microbiol. Spectr. 2023, 11, e0255322. [Google Scholar] [CrossRef]
- Han, A.X.; Parker, E.; Scholer, F.; Maurer-Stroh, S.; Russell, C.A. Phylogenetic Clustering by Linear Integer Programming (PhyCLIP). Mol. Biol. Evol. 2019, 36, 1580–1595. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L. Deciphering the evolutionary mechanisms of SARS-CoV-2: Absence of ORF8 protein and its potential advantage in the emergence of viral lineages. J. Med. Virol. 2023, 95, e29002. [Google Scholar] [CrossRef]
- Braun, K.M.; Moreno, G.K.; Halfmann, P.J.; Hodcroft, E.B.; Baker, D.A.; Boehm, E.C.; Weiler, A.M.; Haj, A.K.; Hatta, M.; Chiba, S.; et al. Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Pathog. 2021, 17, e1009373. [Google Scholar] [CrossRef]
- Lin, X.; Sha, Z.; Trimpert, J.; Kunec, D.; Jiang, C.; Xiong, Y.; Xu, B.; Zhu, Z.; Xue, W.; Wu, H. The NSP4 T492I mutation increases SARS-CoV-2 infectivity by altering non-structural protein cleavage. Cell Host Microbe 2023, 31, 1170–1184. [Google Scholar] [CrossRef]
- Abduljaleel, Z.; Melebari, S.; Athar, M.; Dehlawi, S.; Udhaya Kumar, S.; Aziz, S.A.; Dannoun, A.I.; Malik, S.M.; Thasleem, J.; George Priya Doss, C. SARS-CoV-2 vaccine breakthrough infections (VBI) by Omicron variant (B.1.1.529) and consequences in structural and functional impact. Cell Signal 2023, 109, 110798. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe. [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Crawford, K.H.D.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; Veesler, D.; Bloom, J.D. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Q.; Song, S.; Shen, S.; Zhou, B.; Wang, H.; Cheng, L.; Ge, X.; Ju, B.; Zhang, Z. Increased resistance of SARS-CoV-2 Lambda variant to antibody neutralization. J. Clin. Virol. 2022, 150–151, 105162. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, L.; Zhang, Y.; Wang, H.; Ding, R.; Nie, J.; Li, Q.; Liu, S.; Yu, Y.; Yang, X.; et al. The Antigenicity of Epidemic SARS-CoV-2 Variants in the United Kingdom. Front. Immunol. 2021, 12, 687869. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhang, L.; Liang, Z.T.; Nie, J.H.; Wu, J.J.; Li, Q.Q.; Ding, R.X.; Zhang, Y.; Chen, G.Q.; Wang, Y.C.; et al. Infectivity and antigenicity of pseudoviruses with high-frequency mutations of SARS-CoV-2 identified in Portugal. Arch. Virol. 2022, 167, 459–470. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- de Silva, T.I.; Liu, G.; Lindsey, B.B.; Dong, D.; Moore, S.C.; Hsu, N.S.; Shah, D.; Wellington, D.; Mentzer, A.J.; Angyal, A.; et al. COVID-19 Genomics UK (COG-UK) Consortium; Maini, M.K.; Ogg. G.; Knight, J.C.; ISARIC4C Investigators; Peng, Y.; Rowland-Jones, S.L.; Dong, T. The impact of viral mutations on recognition by SARS-CoV-2 specific T cells. iScience 2021, 24, 103353. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by Sars-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Zhou, S.; Lv, P.; Li, M.; Chen, Z.; Xin, H.; Reilly, S.; Zhang, X. SARS-CoV-2 E protein: Pathogenesis and potential therapeutic development. Biomed. Pharmacother. 2023, 159, 114242. [Google Scholar] [CrossRef]
Figure 1.
General overview of the metadata information associated with the SARS-CoV-2 sequences from cats used in this study. A total of 179 reports related to cat sequences submitted to the GISAID database were considered. Reports included sequences submitted in 2020 (65 sequences), 2021 (103 sequences), and 2022 (11 sequences). A) Viral sequences recovered from cat infections are described in 29 countries, covering multiple geographic regions. B) The gender of the cats was reported in half of the reports (n=90), of which 67.77% came from male cats. C) The age of the cats ranged from 3 months to up to 18 years. Opened bars represent ages where cat deaths were reported (one report for each age). In this case, age was recorded in just 84 reports.
Figure 1.
General overview of the metadata information associated with the SARS-CoV-2 sequences from cats used in this study. A total of 179 reports related to cat sequences submitted to the GISAID database were considered. Reports included sequences submitted in 2020 (65 sequences), 2021 (103 sequences), and 2022 (11 sequences). A) Viral sequences recovered from cat infections are described in 29 countries, covering multiple geographic regions. B) The gender of the cats was reported in half of the reports (n=90), of which 67.77% came from male cats. C) The age of the cats ranged from 3 months to up to 18 years. Opened bars represent ages where cat deaths were reported (one report for each age). In this case, age was recorded in just 84 reports.
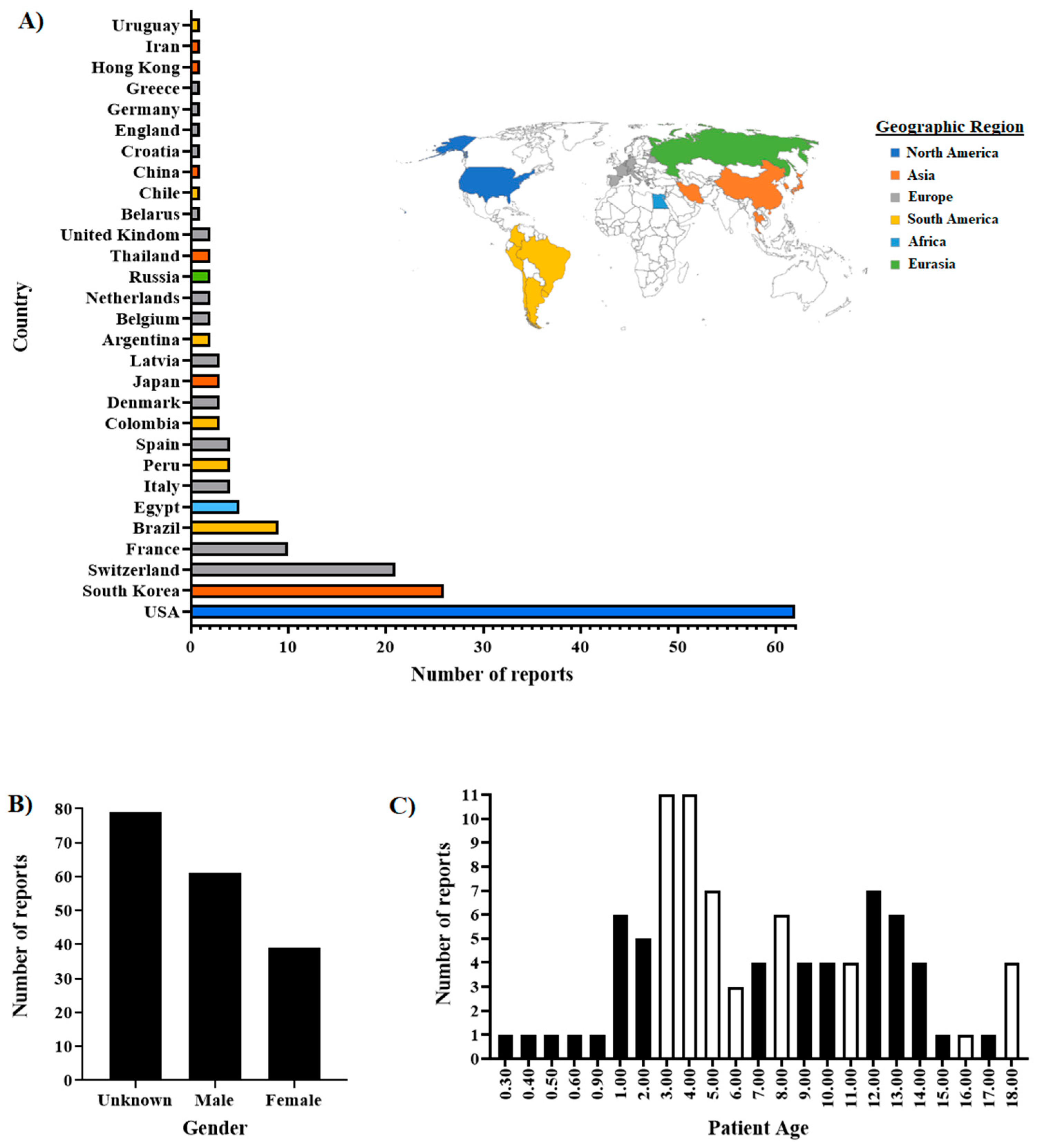
Figure 2.
SARS-CoV-2 viral lineages affecting cats (Felis catus) and other animal species. A total of 179 SARS-CoV-2 were analyzed by hierarchical cluster analysis to assess the relationship between lineages that affected Felis catus and other animal species during the pandemic. These species include dog (Canis lupus familiaris), white-tailed deer (Odocoileus virginianus), lion (Panthera leo), tiger (Panthera tigris), Western gorilla (Gorilla gorilla), American mink (Neogale vison), house mouse (Mus musculus), Syrian hamster (Mesocricetus auratus), and Pangolin (Manis javanica). Two main clusters were identified in the analysis. Cluster 2 (green) comprises the 12 more prevalent lineages among different species. The Y left axe shows the SARS-CoV-2 pangolin lineages included in the analysis. The number in the parenthesis next to each lineage indicates the proportion of this specific lineage among the total number of sequences reported in the GISAID database. Black asterisks reflect lineages where mortality was reported in cats. Numbers below animal species in the X axe show the number of lineages reported during the infection in different species. At the same time, green asterisks indicate species where the prevalence of positive cases was statistically significantly higher in the lineages associated with cluster 2.
Figure 2.
SARS-CoV-2 viral lineages affecting cats (Felis catus) and other animal species. A total of 179 SARS-CoV-2 were analyzed by hierarchical cluster analysis to assess the relationship between lineages that affected Felis catus and other animal species during the pandemic. These species include dog (Canis lupus familiaris), white-tailed deer (Odocoileus virginianus), lion (Panthera leo), tiger (Panthera tigris), Western gorilla (Gorilla gorilla), American mink (Neogale vison), house mouse (Mus musculus), Syrian hamster (Mesocricetus auratus), and Pangolin (Manis javanica). Two main clusters were identified in the analysis. Cluster 2 (green) comprises the 12 more prevalent lineages among different species. The Y left axe shows the SARS-CoV-2 pangolin lineages included in the analysis. The number in the parenthesis next to each lineage indicates the proportion of this specific lineage among the total number of sequences reported in the GISAID database. Black asterisks reflect lineages where mortality was reported in cats. Numbers below animal species in the X axe show the number of lineages reported during the infection in different species. At the same time, green asterisks indicate species where the prevalence of positive cases was statistically significantly higher in the lineages associated with cluster 2.

Figure 3.
Circulation dynamics of SARS-COV-2 lineages during the pandemic. A) The graphic shows the circulation dynamics of different genetic groups of SARS-CoV-2 (based on GISAID classification) described during the pandemic and their relationship with those circulating in cat populations. Correlation analysis is presented between the circulation of specific genetic groups affecting humans and cat populations during the same pandemic year. Asterisks indicate significant correlations. Analyses were conducted in the software GraphPad Prism 9.5.0. B) The graphic shows the overall number of clinical reports in cat populations associated with specific genetic groups of SARS-COV-2 (GISAID classification).
Figure 3.
Circulation dynamics of SARS-COV-2 lineages during the pandemic. A) The graphic shows the circulation dynamics of different genetic groups of SARS-CoV-2 (based on GISAID classification) described during the pandemic and their relationship with those circulating in cat populations. Correlation analysis is presented between the circulation of specific genetic groups affecting humans and cat populations during the same pandemic year. Asterisks indicate significant correlations. Analyses were conducted in the software GraphPad Prism 9.5.0. B) The graphic shows the overall number of clinical reports in cat populations associated with specific genetic groups of SARS-COV-2 (GISAID classification).
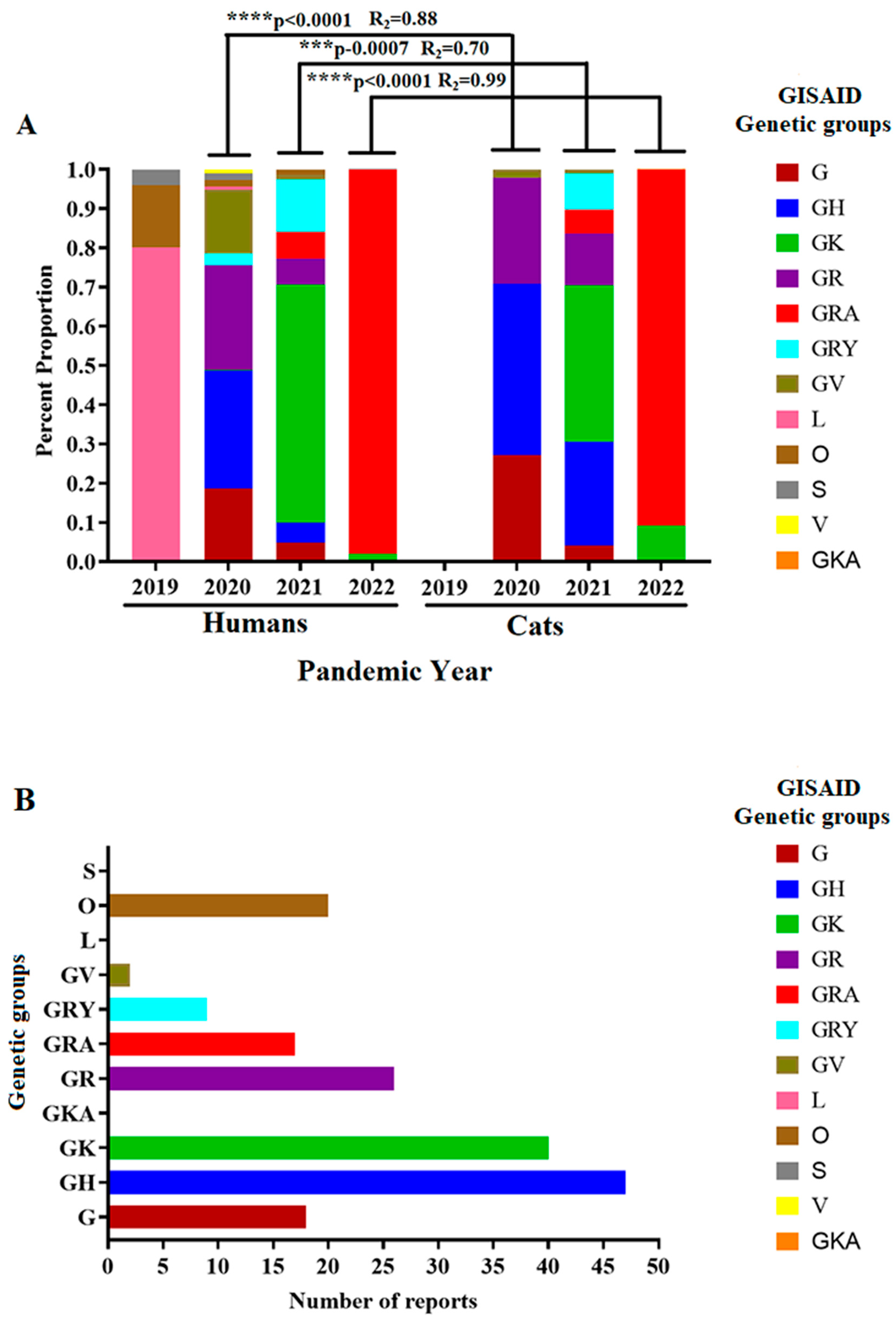
Figure 4.
Phylogenomics of SARS-CoV-2 lineages affecting cats. A phylogenetic analysis was conducted using 105 full-length viral sequences retrieved from the GISAID database (specific details about the sequences can be found in Supplementary File 1). The analysis was performed using the maximum likelihood method and the general reversible model. In the tree, labels B.1.497, B.1, B.1.2, B.1.177, AY.69, AY.20, AY.103, AY.44, AY.3, B.1.1.7 identify clusters associated with the most prevalent SARS-CoV-2 pangolin lineages related with cat infections. The right pangolin lineages are grouped in the context of GISAID genetic group classification. The dN/dS ratios are indicated for the most prevalent cat lineages. Calculations were conducted using the FEL algorithm. Asterisks from different colors reflect specific codon sites at different genes evolving under positive selection in SARS-CoV-2 lineages affecting cats. Information about the codon sites associated with specific asterisk colors is presented in Figure 6. Black bars label lineages carrying premature stop codons at gen ORF8 codon positions 27 and 64.
Figure 4.
Phylogenomics of SARS-CoV-2 lineages affecting cats. A phylogenetic analysis was conducted using 105 full-length viral sequences retrieved from the GISAID database (specific details about the sequences can be found in Supplementary File 1). The analysis was performed using the maximum likelihood method and the general reversible model. In the tree, labels B.1.497, B.1, B.1.2, B.1.177, AY.69, AY.20, AY.103, AY.44, AY.3, B.1.1.7 identify clusters associated with the most prevalent SARS-CoV-2 pangolin lineages related with cat infections. The right pangolin lineages are grouped in the context of GISAID genetic group classification. The dN/dS ratios are indicated for the most prevalent cat lineages. Calculations were conducted using the FEL algorithm. Asterisks from different colors reflect specific codon sites at different genes evolving under positive selection in SARS-CoV-2 lineages affecting cats. Information about the codon sites associated with specific asterisk colors is presented in Figure 6. Black bars label lineages carrying premature stop codons at gen ORF8 codon positions 27 and 64.
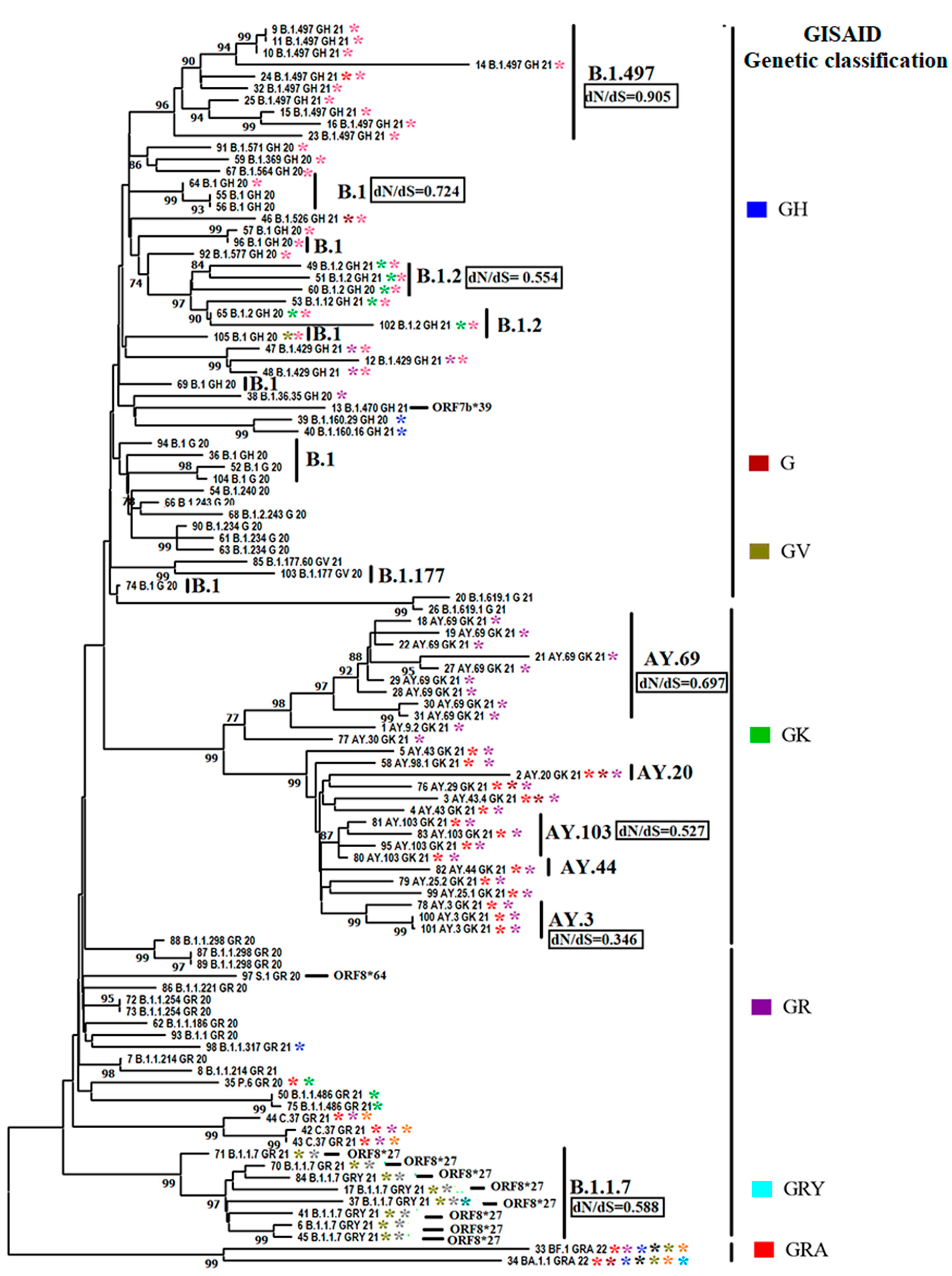
Figure 5.
Phylogenomic dynamics between cat and human SARS-CoV-2 lineages. The phylogenetic analysis was conducted using 222 SARS-CoV-2 lineages (Cats n=105, Humans n=117). The SARS-CoV-2 lineages associated with cats and humans were identified in red and green, respectively. Because of size restrictions, the version of this phylogenetic tree showing the ID’s of cats and human recovered viral lineages, is shown in supplementary figure 2. This figure was created using BioRender.com, under the academic license number: WW25XVH794.
Figure 5.
Phylogenomic dynamics between cat and human SARS-CoV-2 lineages. The phylogenetic analysis was conducted using 222 SARS-CoV-2 lineages (Cats n=105, Humans n=117). The SARS-CoV-2 lineages associated with cats and humans were identified in red and green, respectively. Because of size restrictions, the version of this phylogenetic tree showing the ID’s of cats and human recovered viral lineages, is shown in supplementary figure 2. This figure was created using BioRender.com, under the academic license number: WW25XVH794.
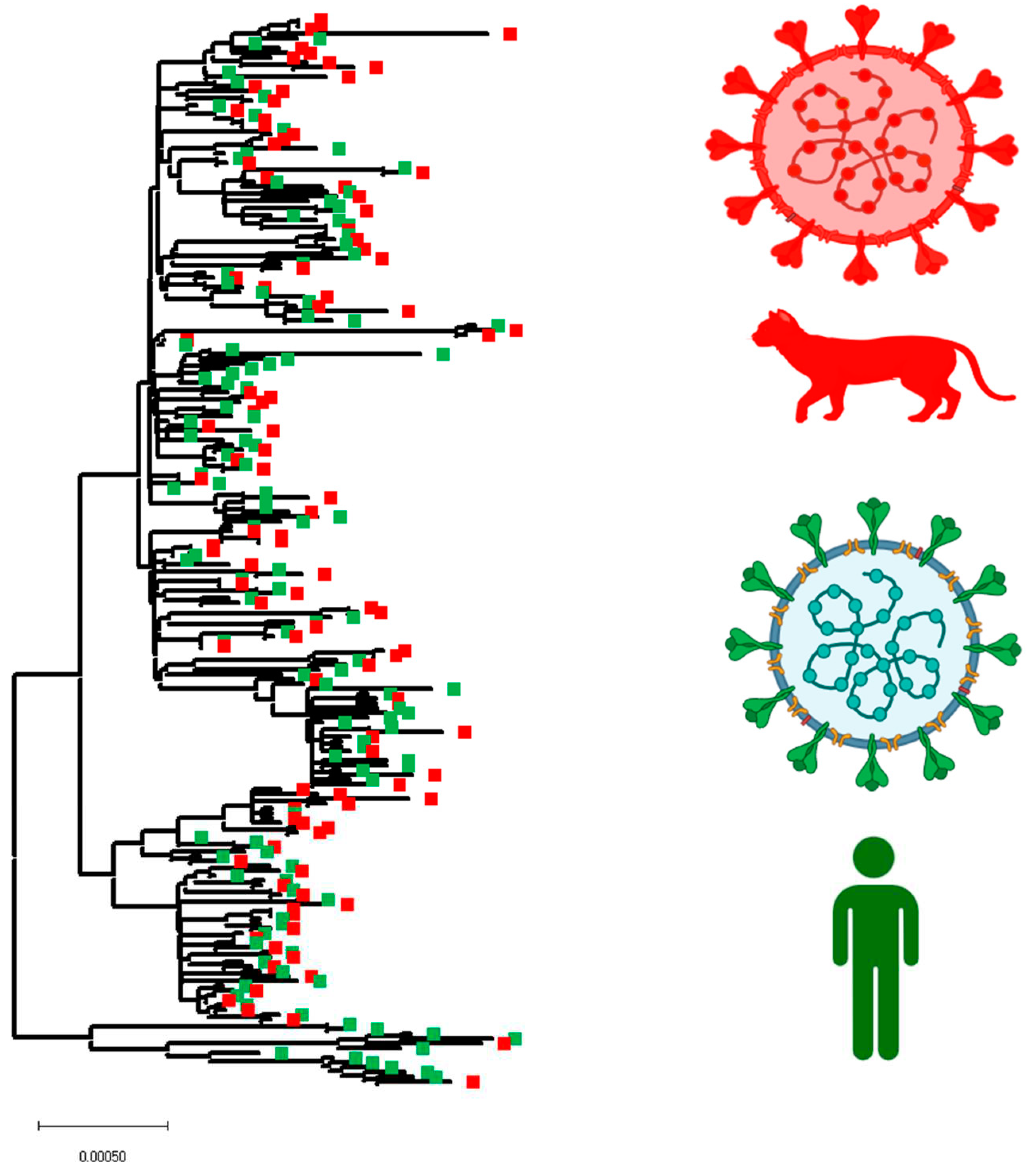
Figure 6.
Comparison between the evolutionary dynamics of SARS-CoV-2 lineages that affected cat and human populations during the pandemic. A) A Total of 105 sequences were evaluated by MEME analysis to infer potentially relevant codon sites evolving under positive selection in SARS-COV-2 lineages that affected cat populations during the pandemic. Sites under positive selection were identified with asterisks of different colors (See tree in Figure 4 to identify lineages displaying specific sites under positive selection). In the column identified as “codon population composition at this site“ numbers in parenthesis represent the number of viral sequences in the population carrying this specific codon. Letters in the parenthesis indicate amino acids encoded by specific codons. B) MEME analysis conducted on 117 viral sequences recovered from humans during the pandemic. Information about the countries where mutations at specific codon sites under positive selection were found, and the overall percentage of human sequences carrying these mutations was obtained from the GISAID database. For MEME analysis α= synonymous substitution rate at site, β+=Nonsynonymous substitution rate at site for the positive/neutral evolution component, LRT=likelihood ratio test statistic for diversifying selection. C) Comparison of dN/dS ratios at different gene segments of SARS-CoV-2 between cat and human populations. Calculations were conducted using MEME.
Figure 6.
Comparison between the evolutionary dynamics of SARS-CoV-2 lineages that affected cat and human populations during the pandemic. A) A Total of 105 sequences were evaluated by MEME analysis to infer potentially relevant codon sites evolving under positive selection in SARS-COV-2 lineages that affected cat populations during the pandemic. Sites under positive selection were identified with asterisks of different colors (See tree in Figure 4 to identify lineages displaying specific sites under positive selection). In the column identified as “codon population composition at this site“ numbers in parenthesis represent the number of viral sequences in the population carrying this specific codon. Letters in the parenthesis indicate amino acids encoded by specific codons. B) MEME analysis conducted on 117 viral sequences recovered from humans during the pandemic. Information about the countries where mutations at specific codon sites under positive selection were found, and the overall percentage of human sequences carrying these mutations was obtained from the GISAID database. For MEME analysis α= synonymous substitution rate at site, β+=Nonsynonymous substitution rate at site for the positive/neutral evolution component, LRT=likelihood ratio test statistic for diversifying selection. C) Comparison of dN/dS ratios at different gene segments of SARS-CoV-2 between cat and human populations. Calculations were conducted using MEME.
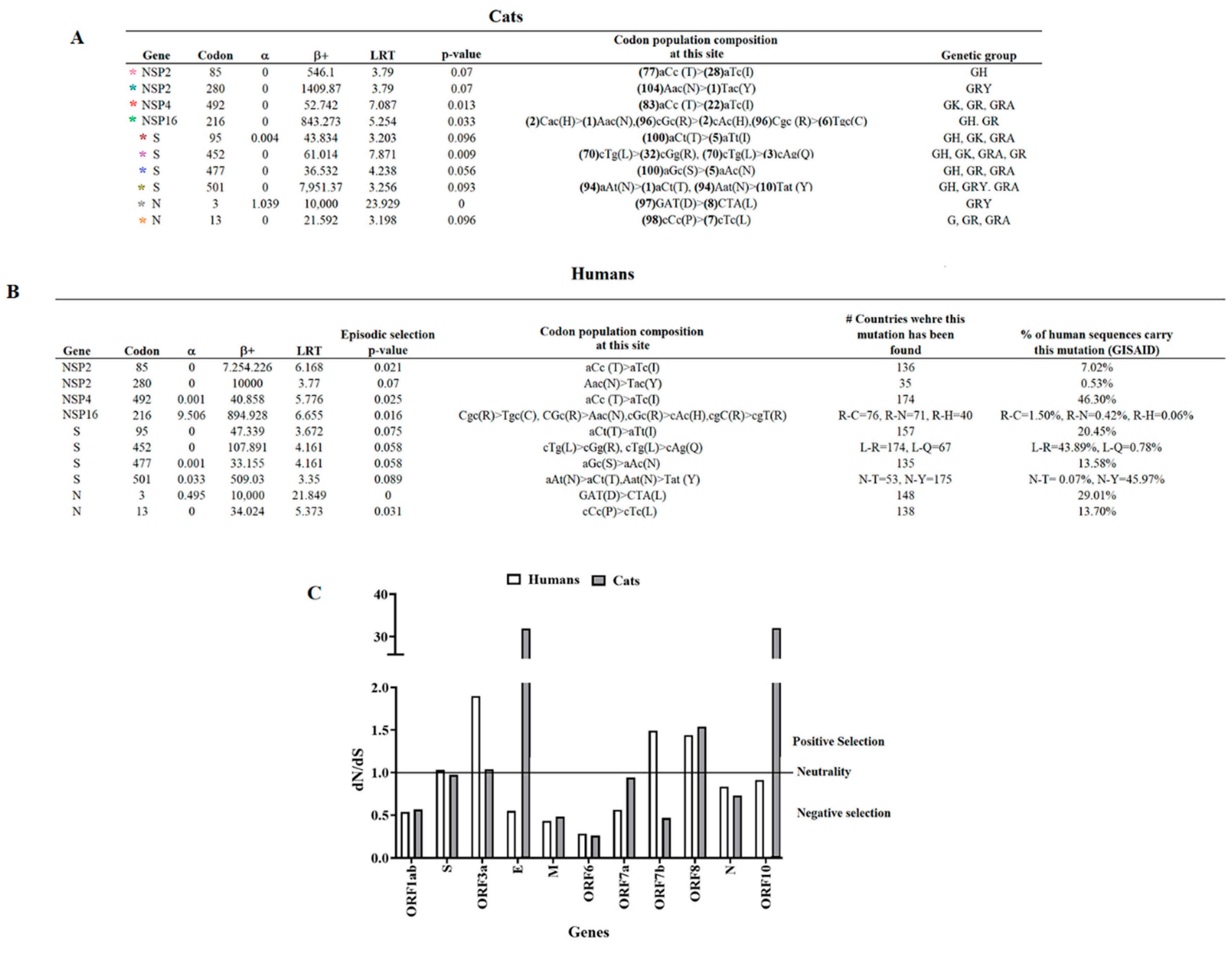
Figure 7.
Identification of potential SARS-CoV-2 lineages evolving under positive selection from cat populations. A) Results of the aBSREL analyses identifying potential cat lineages evolving because of natural selection. B) Phylogenetic relationship between identified cat SARS-CoV-2 lineages (red branches) 22 AY.69 GK 21, 29 AY.69 GK 21, and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence OV832299.1 2021 GK human). C) Phylogenetic relationship between identified cat SARS-CoV-2 lineages (red branch) 34 BA.1.1 GRA 22, and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence EPI ISL 11686014 2022 GRA human). D) Phylogenetic relationship between identified cat SARS-CoV-2 lineage (red branch) 55 B.1 GH 20 (similar to 56 B.1 GH 20) and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence MW268725.1 2020 GH human). E) Phylogenetic relationship between identified cat SARS-CoV-2 lineage (red branches) 88 B.1.1.298 GR 20 and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence OW425144.1 2022 GR Human). The percentage in bold between cat and human sequences reflects their nucleotide identity. Below, multiple trees display the polymorphic codon sites at different genes impacting the identity between human (information in green) and cat (information in red) sequences. All these short trees were obtained from the phylogenetic analysis in supplementary figure 2. F) Busted analysis was conducted to evidence ratios (ER Busted) for specific codon sites associated with the branches linked to the emergence of cat lineages showing ω>1. The relevance of these sites was assessed by FEL and MEME (cutoff 0.1). The overall number of sequences in the GISAID database showing the alleles in cat lineages and human ancestral sequences at specific codon positions are shown. Frequencies represented by different alleles in humans and cats at specific codon positions were obtained from the GISAID database. The phylodynamics of multiple codon positions identified by BUSTED analysis are presented in supplementary file 3.
Figure 7.
Identification of potential SARS-CoV-2 lineages evolving under positive selection from cat populations. A) Results of the aBSREL analyses identifying potential cat lineages evolving because of natural selection. B) Phylogenetic relationship between identified cat SARS-CoV-2 lineages (red branches) 22 AY.69 GK 21, 29 AY.69 GK 21, and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence OV832299.1 2021 GK human). C) Phylogenetic relationship between identified cat SARS-CoV-2 lineages (red branch) 34 BA.1.1 GRA 22, and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence EPI ISL 11686014 2022 GRA human). D) Phylogenetic relationship between identified cat SARS-CoV-2 lineage (red branch) 55 B.1 GH 20 (similar to 56 B.1 GH 20) and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence MW268725.1 2020 GH human). E) Phylogenetic relationship between identified cat SARS-CoV-2 lineage (red branches) 88 B.1.1.298 GR 20 and its closest related ancestral SARS-CoV-2 human lineage (green branch, sequence OW425144.1 2022 GR Human). The percentage in bold between cat and human sequences reflects their nucleotide identity. Below, multiple trees display the polymorphic codon sites at different genes impacting the identity between human (information in green) and cat (information in red) sequences. All these short trees were obtained from the phylogenetic analysis in supplementary figure 2. F) Busted analysis was conducted to evidence ratios (ER Busted) for specific codon sites associated with the branches linked to the emergence of cat lineages showing ω>1. The relevance of these sites was assessed by FEL and MEME (cutoff 0.1). The overall number of sequences in the GISAID database showing the alleles in cat lineages and human ancestral sequences at specific codon positions are shown. Frequencies represented by different alleles in humans and cats at specific codon positions were obtained from the GISAID database. The phylodynamics of multiple codon positions identified by BUSTED analysis are presented in supplementary file 3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



