
Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
Antimicrobial peptides (AMPs) have emerged as a promising solution to tackle bacterial infections and combat antibiotic resistance. However, their clinical application has been hindered by their vulnerability to protease degradation and toxicity towards mammalian cells. To overcome these challenges, our study aims to develop a method to enhance the stability and safety of AMPs, applicable to effective drug-device combination products. KR12 antimicrobial peptide was chosen and in order to further enhance its delivery and efficacy, HIV TAT protein-derived cell-penetrating peptide (CPP) was fused to form CPP-KR12. A new product, CPP-KR12@Si, was developed by forming silica nanoparticles with self-entrapped CPP-KR12 peptide using the biomimetic silica precipitability due to its cationic nature. Peptide delivery from CPP-KR12@Si to bacteria and cells was delivered at a slightly delayed rate with improved stability against trypsin treatment and a reduction in cytotoxicity over CPP-KR12. Finally, the antimicrobial potential of CPP-KR12@Si/bone graft substitute (BGS) combination product was demonstrated, which is coated with CPP-KR12 in the form of nanoparticles on the surface of BGS. Self-entrapped AMP in silica nanoparticles is a safe and effective AMP delivery method that will be useful for developing a drug/device combination product for tissue regeneration.
Keywords:
antimicrobial peptide
; cell penetrating peptide
; silica forming peptide
; biomimetic silica deposition
; drug delivery
; drug device combination
1. Introduction
Implantation of medical devices may be required to treat or repair damaged tissue. When transplanting, perioperative systemic administration of antibiotics is standard practice to prevent infections associated with the implanted device [1]. However, once infection occurs, systemic antibiotic treatment is usually ineffective [2]. Most implant-related infections are antibiotic-resistant and last until the device is removed [2,3]. In addition, infection is the most expensive complication of implantable cardioverter defibrillator therapy [4], and major gastrointestinal resections for organ-specific malignant neoplasia [5]. It is a key contributor to chronic wound healing impairment [6]. Importantly, patients with infections had a greater in-hospital mortality rate than those without infections [7]. One successful approach to prevent device and wound infections is topical administration of antibacterial agents [2]. A drug-device combination strategy to achieve topical antibiotic delivery is to integrate an antibacterial agent with a medical device.
Because biofilms and device-related infections are usually complex, it is advisable to select broad-spectrum antibacterial agents and which have been satisfied by antimicrobial peptides (AMPs) and chitosan in some researches [8,9]. AMPs are a type of peptide that has developed in organisms ranging from prokaryotes to humans to give non-specific immune protection [10]. AMPs are made up of 10 to 60 amino acid residues and have two common and functionally important requirements: an amphipathic structure composed of cationic residues with a net positive charge (+2 to +9) that attracts them to anionic microbial surfaces and hydrophobic residues that preferentially insert into microbial membranes [11]. AMPs have a broad antimicrobial spectrum [12] and are less likely to induce resistance due to their physical membrane disruption mechanism, causing leakage of their contents, and contributes to bacterial killing [13]. In contrast, conventional antibiotics are facing increasing resistance, making the use of AMPs an attractive option for treating bacterial infections [14]. Therefore, antimicrobial peptides (AMPs) are promising therapeutic agents for medical device combination products [2]. Meanwhile, cell-penetrating peptides (CPPs), which are cationic peptides, have been used as vectors to transport oligonucleotides, proteins, peptides, and other materials across bio-membranes into cells due to their ability to cross cell membranes in a non-disruptive manner [15]. Thus, CPPs have received considerable attention for their potential applications in drug delivery to hard to-permeate membrane [16]. The combination of CPPs and AMPs has been investigated as a viable way to treat intracellular infections by providing the dual function of cellular penetration and antimicrobial activity [17,18], and has shown improved antimicrobial activity over AMP alone [17,19]. Despite the advantages of CPP-conjugated AMPs, rapid clearance and cytotoxicity, such as hemolysis, still characteristic of peptide and protein drugs, limit the development of AMPs as systemic antimicrobial agents [20]. Therefore, strategies are needed to incorporate AMPs into medical devices for local release while protecting the peptide from the environment. When developing antimicrobial device combination products, it is important to ensure that the antimicrobial agent remains stable during the manufacturing process and while in storage [21,22]. This may require the use of a biocompatible delivery system that can protect the drug and enable its controlled release without affecting the overall performance of a medical device.
The understanding of how diatoms biologically form their silica skeleton [23] has led to the development of a method to form silica hybrids in a biocompatible manner by mimicking the positively charged peptides involved in silica deposition [24,25,26]. Silica-precipitable peptides have been used for various applications such as enzyme immobilization [27], drug delivery vehicle formation [28], and biosilica coatings on bone graft substitute (BGS) [29] using the biomimetic silicification technique. The potential of AMPs to translocate into cells has led to their use as vectors for drug delivery [30]. Conversely, hydrophobic residues have been incorporated into CPPs to produce antimicrobial peptides with improved permeability [18]. In this study, a hypothesis was proposed regarding the ability of AMP and CPP to precipitate silica due to their cationic nature. To examine this hypothesis, AMP was utilized to create self-encapsulated silica particles, where AMP played a dual function as a silica-precipitable agent and cargo molecule (Scheme 1 a). Furthermore, the feasibility of developing a combination product of AMP-BGS, coated with AMP in the form of nanoparticles on the surface of BGS, was carried out (Scheme 1b).
2. Results and Discussion
2.1. Conjugation of Cell Penetrating Peptide to KR12 Antimicrobial Peptide
2.1.1. CPP Conjugation Enhances the Antibacterial Activity of KR12 over KR12 Alone
KR12 used in this study is a cationic and amphipathic α-helical peptide composed of the 18th-29th residues of LL37 that is a 37-residue, cathelicidin-derived antimicrobial peptide found in humans [31]. KR12 is the shortest LL37-derived peptide identified as the region responsible for LL37’s antibacterial and anti-inflammatory properties [17,32]. In addition, KR12 is known for its ability to promote osteogenic differentiation of bone marrow stem cells [33]. Therefore, it was selected for its potential to perform dual functions - antibacterial and bone regeneration promoting - in the production of an antibacterial bone graft combination. Cell-penetrating peptides (CPPs) are able to cross biological membranes without membrane disruption, so they have been used as a vector for delivery of various cargos [34]. The CPP used in this study is the 49th-57th residues (protein transduction domain) of the TAT protein, a protein involved in the transcription of the human immunodeficiency virus (HIV) [35]. TAT residues 49-57 have been critical in facilitating cellular uptake of linked bioactive macromolecules such as peptides, proteins, oligonucleotides, and pharmacological compounds [36]. First, the amino acid sequences and properties of KR12, CPP, and CPP-KR12, in which CPP is bound to KR12 by a GSS linker, are shown in Table 1. The antimicrobial activity of these peptides was compared by determining the minimum inhibitory concentration (MIC) (Table 2).
KR12 showed antibacterial activity against the Gram-negative bacteria tested, with MIC values in the range of 100-200 μM, indicating low antibacterial potency. It did not show antibacterial activity against the Gram-positive bacteria S. aureus, even at the highest concentration tested, 320 μM. CPP did not show antibacterial activity against any bacterial strain, even at the highest concentration tested, 320 μM (Supplementary Figure S1). On the other hand, the antibacterial activity of CPP-KR12 increased by approximately 8-fold against E. coli and nearly 30-fold against P. aeruginosa. Against S. aureus, it showed MIC (22.80±8.48μM) at a concentration 10 times lower than KR12, indicating improved antibacterial activity against both Gram-negative and positive bacteria. These results were similar to the report by Lee et al., that conjugation of nine arginine residues to AMP significantly enhanced its antimicrobial activity, particularly against Gram-negative bacteria [19]. While CPP itself did not exhibit antimicrobial activity, CPP-fused KR12 inhibited cell growth at lower concentrations, suggesting that the cellular internalization ability of CPP enhanced the delivery of KR12 into bacteria. TAT-KR-12 (YGRKKRRQRRKRIVQRIKDFLR), developed for the treatment of intracellular S. aureus infection in mammals, showed enhanced antibacterial activity against S. aureus [17]. KR12 alone may be limited in its ability to cross thick cell walls and induce cell membrane rupture. However, the incorporation of cell-penetrating peptides leads to an enhancement in the binding affinity to anionic teichoic acid on the cell wall surface due to its increased cationicity [37]. This may result in the accumulation of AMP on the cell wall surface, ultimately facilitating the insertion of peptides into the cell membrane through the somewhat porous reticulated peptidoglycan layer [38].
2.1.2. Comparison of AMP’s Membrane Permeability and Ability to Disrupt Cell Membranes
The SYTOXTM green uptake assay was used to evaluate the permeability of cell membrane induced by the peptides. When AMP induce membrane permeability, SYTOXTM green dye enters the interior of cell and binds to intracellular nucleic acids, resulting in increased fluorescence. The disruption of membrane integrity by peptides leads to an increase in fluorescence intensity, providing an indirect measure of antimicrobial activity as SYTOXTM green cannot pass through an intact membrane. In Figure 1a, it is evident that the fluorescence intensity of CPP and CPP-KR12 increased compared to KR12, and the fluorescence intensity of CPP-KR12 increased 2-fold in Gram-negative bacteria compared to that of KR12. The increased intracellular transport of KR12 was thought to be responsible for the increased antibacterial activity, considering that CPP has no antibacterial activity. The membrane permeability of CPP-KR12 to S. aureus was lower than that of Gram-negative bacteria, suggesting that the lower permeability of CPP-KR12 is related to the lower antibacterial activity in S. aureus. Upon investigating the killing mechanism, we utilized scanning electron microscopy to observe the morphology of E. coli cells after treatment with CPP-KR12. The results, as depicted in Figure 1b, showed that AMP-treated E. coli exhibited fragmented cells or cells with surface holes. We noted an increase in cell membrane disruption at 15 μM CPP-KR12 compared to 30 μM KR12. Additionally, we observed fragmented cells with membrane rupture and aggregated populations in E. coli at 30 μM CPP-KR12. It is suggested that a certain concentration of antimicrobial peptides must adhere to the surface to penetrate the cell membrane and induce cell death.
2.1.3. Comparison of AMP’s DNA Binding Ability
CPP-KR12 had a higher membrane permeability in Gram-negative bacteria than KR12 and appeared to be associated with more potent antibacterial effects in Gram-negative bacteria. However, the membrane permeability of CPP itself does not necessarily correlate with antibacterial efficacy, and there are no significant differences in membrane permeability among the peptides in S. aureus. This suggests that other targets may be involved in the bactericidal effects of KR12 and CPP conjugate. Increasing evidence have shown that antimicrobial peptides (AMPs) may also exert intracellular inhibitory activity as a primary or secondary mechanism for more efficient killing [39]. Certain AMPs have the potential to target intracellular molecules after penetrating the cell membrane. As a result, we investigated the DNA binding properties of the peptides (Figure 2). Plasmid DNA was combined with various peptide concentrations for 30 minutes at room temperature to generate complexes referring to the method of Lee et al. [19]. The delay in plasmid DNA migration on an agarose gel was used to assess peptide binding capacity to DNA.
KR12 only little impacted DNA migration at concentrations up to 32 µM, but CPP-KR12 entirely stopped DNA migration at a concentration of 4 µM. This indicates that CPP-KR12 has a high affinity for binding to DNA. The strength of this binding is heavily influenced by CPP, which inhibits DNA migration at 8 µM. The net charge of KR12, CPP, and CPP-KR12 is +4, +8, and +12, respectively (as shown in Table 1). The capacity for DNA binding appears to grow proportionally to the rise in cationicity. Although CPP, which has a high DNA binding capacity, lacks antibacterial activity, and KR12 has a relatively low DNA binding capacity, it is possible that KR12 has other intracellular targets for bacterial death. The intracellular targets of LL37 and KR12 are not yet fully understood, but several studies have identified their antimicrobial mechanisms. E. coli was used to express LL37 in order to determine its intracellular targets [40]. Under aerobic growth circumstances, LL37's effects were investigated, and it was discovered that intracellular expression of LL37 boosted the formation of reactive oxygen species (ROS) [40]. This led to lethal membrane depolarization , confirming that ROS production is a bactericidal mechanism for LL37 under such conditions [40]. Other study have also found that LL37 mainly increases the production of oxidative molecules that cause cell damage in both Gram-positive and Gram-negative bacteria [41]. Therefore, it is believed that the antibacterial activity of CPP-fused KR12 is enhanced due to increased membrane permeability and affinity for intracellular DNA, in addition to the ROS generation effect of KR12.
2.1.4. Anti-Inflammatory Effect of AMP on Lipopolysaccharide (LPS)-Induced Inflammation
It has been discovered that some AMPs can successfully neutralize LPS, producing bactericidal and anti-inflammatory actions against Gram-negative bacteria [42,43]. In addition to its antimicrobial properties, AMP's immunomodulatory abilities in infected tissues are important for healing by restoring the host's protective immunity [44]. Since CPP-KR12 displayed strong antibacterial activity against Gram-negative bacteria, it is believed to modulate LPS-induced inflammation. Inflammatory pathways induced by LPS can lead to cytokine production [45]. To investigate the ability of peptides to neutralize LPS, we measured mRNA expression levels of LPS-stimulated tumor necrosis factor-alpha (TNF-α), interleukin 1 beta (IL-1β), and interleukin 6 (IL-6) in RAW264.7 macrophages (Figure 3). The expression of these cytokines increased after LPS treatment, but decreased by about 50% compared to LPS-induced expression by KR12 and CPP-KR12 treatment. The conjugate neutralized LPS more efficiently in IL-6 expression than unconjugated AMP. However, this inhibition was not observed in CPP-treated LPS-stimulated RAW 264.7 cells (Data not shown), suggesting that KR12 is responsible for the immunomodulation.
2.2. Self-Entrapment of AMPs in Silica Particles via AMP-Mediated Silica Deposition
Next, KR12 and CPP-KR12 were investigated to determine their ability to synthesize silica and their potential as antibacterial substances. The R5 peptide (SSKKSGSYSGSKGSRRIL) is generated from silaffin peptides found naturally in the diatom Cylindrotheca fusiformis and has excellent silica precipitation activity [46]. In the presence of negatively charged phosphate ions, the positively charged ε-amino groups of the lysine residues in R5, catalyze forming siloxane bonds resulting in silica deposition from silicic acid (Si(OH)4) in vitro [47]. In our previous study, we demonstrated that cationic BMP2 protein and P4 peptide were efficiently delivered to osteoblast via silica nanoparticles formed by BMP2 or P4 peptide-mediated biomimetic silicification methods [48,49]. The amount of silica formed per 100 μg of each peptide, the loading efficiency of peptide in silica, and self-entrapment efficiency are shown in Table 3. The order of silica forming ability was CPP-KR12 > CPP > KR-12. CPP and CPP-KR12 were entrapped by 95% of the added amount in the as-prepared silica. Co-precipitation of peptide and silica resulted in loading efficiencies of 70% and higher. For CPP and CPP-KR12, silica precipitation occurred as soon as the silica precursor, peptide, and phosphate ion were mixed, and all were seen in particulate form (Figure 4a). KR12, on the other hand, formed more slowly than the other peptides, with a mixture of particle and gel-like forms.
The average particle sizes of 50 randomly selected particles from SEM images were measured and found to be 700, 600, and 500 nm in the order KR12 > CPP > CPP-KR12 (Figure 4b). The zeta potential of each silica particle was measured by dispersion in alcohol (Figure 4c). The peptide-free silica particle showed a zeta potential of 2.15 ± 19.5 mV, while the silica particles formed by CPP and CPP-KR12 showed positive values of 44 ± 21.7 mV and 43.3 ± 8.19 mV, respectively. In contrast, KR12 showed a zeta potential ranging from negative to positive, with a value of 13.5 ± 19.7 mV. The zeta potential values indicated the presence of positively charged properties in the particles attributed to the peptide. It is suggested that the CPP or CPP-KR12 exhibited a cationic nature on the surface of the particle rather than being buried within the silica. The zeta potential of particles is an important characteristic that influences particle stability and cell adhesion [50]. The value of zeta potential, whether positive or negative, is important in particle suspension stabilization [51]. This is because electrostatic repulsion between particles with the same charge produces particle separation. A positive zeta potential value is supposed for particles to adhere to negatively charged surfaces more.
2.3. Characterization of CPP-KR12 in Silica Nanoparticle Form
2.3.1. Minimum Inhibitory Concentration of AMP@Si
The antimicrobial activity of AMP-loaded silica particles was compared by determining MIC (Table 4). Compared to the free form, the antibacterial activity of KR12 in silica resulted in almost similar MIC concentrations against E. coli and decreased against P. aeruginosa. In contrast, it increased against S. aureus. The antibacterial activity of CPP-KR12 loaded silica particles against both Gram-negative and Gram-positive bacteria showed a slight increase in MIC compared to those of the free forms, but there were no significant differences. The antibacterial activity of CPP-KR12@Si was not significantly different from the free form, but KR12@Si showed a difference from the free form. For P. aeruginosa, the charge of the particles was expected to have an effect, and for S. aureus, the particulate form was expected to affect the antibacterial activity.
2.3.2. Comparison of CPP-KR12 Delivery between Free and Immobilized Form
The SYTOXTM green uptake assay was used to compare the release of CPP-KR12 from the immobilized in silica and free forms into E. coli. CPP-KR12@Si showed an approximately 30% decrease in cell membrane permeation rate over 10 minutes compared to CPP-KR12 (Figure 5a). E. coli and Raw264.7 cells were observed after treatment with either silica particles formed by FITC-CPP-KR12 or an equivalent amount of FITC-CPP-KR12 based on peptide concentration. The delivery of FITC-CPP-KR12 into E. coli was observed in both the free and the immobilized form (Figure 5b). No aggregated E. coli fragments were observed in the FITC-CPP-KR12 treated sample, but aggregated particulate forms with a strong FITC fluorescence signal were observed in the FITC-CPP-KR12@Si treated sample. This suggested that the peptide was delivered to the bacteria even in immobilized form. When S. aureus is infected, it is phagocytized by macrophages, in which case the bacteria do not die but survive and persist in the host cells, making it a major cause of post-operative infections, and these cells can act as reservoirs, leading to chronic infections [17]. TAT-KR12 has been reported to enter cells and inhibit the growth of intracellularly infected S. aureus [17]. We observed the delivery of the prepared CPP-KR12@Si to macrophages. Both free and immobilized ones were delivered to macrophages, and the intracellular FITC signal was detected (Figure 5c). Compared to the free form, the particle form delivered more locally to the cells, indicating a more concentrated effect on the cells.
The fluorescence analysis conducted by STEDYCON microscope revealed that the FITC-labeled CPP-KR12 was present at the edges of the silica particles, as shown in Figure 5d. The distribution of the peptide in the silica particle was believed to the cause of the highly cationic nature of CPP-KR12@Si and its comparable antimicrobial activity to the free form. Furthermore, the subcellular localization of CPP-KR12@Si was compared over time, and it was observed that after 2 hours of exposure, the particles were intact and bound to the cell membrane. However, after 24 hours, they were found inside the cell and in a degraded form, as seen in Figure 5e. This suggests that the silica nanoparticles carrying the peptide enter the cell, deliver the peptide, and degrade.
2.3.3. Stability of AMP@Si against Protease Treatment
The absorbance value of the bacteria in the absence of AMP was used as the negative control. The absorbance value of the bacteria grown in the presence of trypsinized AMP was subtracted from that of the negative control, and the resulting difference was expressed as a percentage fraction of the negative control, yielding the residual antibacterial activity of AMP after trypsin treatment (Figure 6a). Free KR12 lost 100% of its antibacterial activity against P. aeruginosa and S. aureus by trypsin treatment. It showed about 5% residual activity against E. coli. CPP-KR12 showed approximately 10% residual antibacterial activity against E. coli and approximately 15% residual activity against P. aeruginosa and S. aureus. Silica particulate AMPs also showed antibacterial activity after trypsin treatment, with KR12@Si showing 40% and CPP-KR12@Si showing 45% residual antibacterial activity against E. coli. In P. aeruginosa, KR12@Si showed 18% and CPP-KR12@Si showed 20% residual antibacterial activity. In S. aureus, KR12@Si lost its antibacterial activity, while CPP-KR12@Si retained more than 60% of its antibacterial activity. When S. aureus was treated with Cpp-KR12@Si, aggregation was observed that was not seen with free AMP. SEM images of CPP-KR12@Si treated staphylococci showed the presence of silica particles between the aggregates of them and the absence of the bacteria (Figure 6b) or membrane disruption around the silica particles (Figure 6b inside). It has been suggested that the silica particles promote aggregation of S. aureus and release antimicrobial peptides at the aggregation site, causing the bacteria to rupture their membranes and die (Figure 6b). The localized aggregation of the silica particles and the release of the antimicrobial peptide were thought to be responsible for the increased killing of S. aureus compared to the free form.
Despite their excellent antimicrobial activity under laboratory conditions and consideration of alternatives to conventional antibiotics, AMPs have been limited in clinical use by their loss of activity through rapid degradation by plasma and bacterial proteases and their short half-life in the body due to rapid hepatic and renal elimination [52]. Several techniques have been employed to improve the stability of AMPs. These strategies involve substituting L-amino acid residues with D- and unnatural amino acids, modifying the N- and/or C-terminus, cyclizing the peptide, using non-peptidic backbones (peptidomimetics), and multimerizing AMP monomers [52,53,54,55,56]. Meanwhile, nanoparticles offer advantages as a delivery system for AMPs, such as a controlled release rate, prevention of protein degradation and inhibition of binding to serum proteins [57,58], which can prolong the circulation of AMPs in the bloodstream and increase their bioavailability [22].
2.4. Cytotoxicity and Hemolytic Activity of AMPs and AMP@Sis
The hemolytic activity (Figure 7a) and cytotoxicity against mammalian cells (Figure 7b) of free AMP and AMP immobilized on silica were compared. KR12 and CPPKR12 showed no significant hemolytic activity against sheep RBC, with less than 1% hemolysis at concentrations up to 77.5 μM. However, KR12@Si and CPP-KR12@Si significantly increased the hemolytic activity to 6.3% and 12.8%, respectively, at the same concentration. Hemolytic activity below 1% was observed at 19.38 μM for KR12@Si and 4.84 μM for CPP-KR12@Si. Higher hemolytic activity was observed for silica particles synthesized without the peptides (data not shown), suggesting that the erythrocyte cell membrane is more sensitive to silica nanoparticles than to AMP itself, resulting in increased hemolysis in the silica particle form compared to the free form.
The study found that KR12 showed no cytotoxicity on day 1 of AMP-treated Raw 264.7 cells at concentrations up to 77.5 μM, as observed in erythrocyte hemolysis. Similarly, KR12@Si showed no cytotoxicity up to 77.5 μM. In contrast, CPP-KR12 showed only 10% cell viability at 77.5 μM and 80% cell viability at 19.38 μM. Interestingly, CPP-KR12@Si showed 80% cell viability at 77.5 µM, indicating that immobilization of AMP on silica can reduce the toxicity of CPP-KR12. While hemolytic activity and cytotoxicity are useful in assessing cytotoxicity to eukaryotic cells, it is important to note that different types of peptide formulations may yield different results depending on the cell type.
While hemolysis refers primarily to the destruction of red blood cells, cytotoxicity refers to the decreased viability of cells. Therefore, even if a substance destroys red blood cells and causes hemolysis, there may be no cytotoxicity if the substance acts only on the outside of the cell and does not penetrate the cell. It is also possible that a substance does not cause membrane disruption on the outside of the cell, but once inside the cell causes cytotoxicity by acting on intracellular targets. Based on the results, CPP-KR12 is more likely to cause cytotoxicity inside the cell than at the cell membrane. According to a study by Yu et al. [59], SiO2 cytotoxicity depends on the cell type and is also affected by surface charge and pore size. After 24 hours of incubation, they found that non-porous or mesoporous SiO2 caused acute toxicity in RAW 264.7 macrophages, leaving only 20–40% of viable cells compared to the control, while amine-modified SiO2 caused little toxicity, leaving 64–85% of viable cells. They postulated that the silanol groups on the surface of the anionic silica particles were toxic because of their strong affinity for the plasma membrane, and that the shielding of the surface silanol groups by amine functionality reduced the cellular accessibility of the silanol groups and consequently the level of cellular binding, resulting in reduced cytotoxicity [60,61]. It is also believed that CPP-KR12@Si shares characteristics with amine-functionalized silica particles.
CPP-KR12 was found to have a higher degree of internalization compared to KR12, which could potentially lead to intracellular toxicity. The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) can regulate the expression of antioxidant proteins such as superoxide dismutase (SOD) and glutathione peroxidase (GPx), which can scavenge ROS and protect cells from damage [62]. In Raw264.7 cells without LPS, CPP-KR12 treatment increased the mRNA expression levels of NRF-2, SOD1, and GPx1, indicating increased ROS generation. KR12 is known to have antimicrobial properties through ROS generation (Supplementary Figure S2) [40,41]. The treatment of LPS or KR12 resulted in a decrease of near 50% in the expression levels of antioxidant proteins in Raw264.7 cells, as compared to the control. However, the concentration of CPP-KR12 used in the experiment (6 µM) was not cytotoxic. This suggests that the increase in mRNA levels of antioxidant proteins may be a protective mechanism activated by CPP-KR12 (Supplementary Figure S2). It is worth noting that NRF-2 is considered as a double-edged sword [63]. Therefore, CPP-KR12 can induce cytotoxicity in mammalian cells at high concentrations, but at low concentrations, it may have immunomodulatory and cytoprotective effects. At high concentrations, CPP-KR12's increased permeability and DNA binding capacity, combined with intracellular ROS production, may lead to potential cytotoxicity. In contrast, the cytotoxicity of CPP-KR12 was found to be reduced when it was entrapped in silica, as its intracellular delivery was delayed. After a 5-day incubation period, both the free and immobilized forms showed similar cell viability at 77.5 μM, with a 40% survival rate (Supplementary Figure S3). The increased cell viability observed in the free form on day 5 seemed to be due to the growth of surviving cells. In summary, when delivered in a silica entrapped form, it becomes more resistant to proteases and less cytotoxic while retaining its antimicrobial activity.
2.5. AMP-Device Combination Products
To determine the surface composition of the AMP@Si coated bone graft substitute (BGS), X-ray photoelectron spectroscopy (XPS) was employed. Survey spectra are often recorded in the broad binding energy range of 0 to >1000 eV for elemental analysis to collect signatures of all species present on the surface of samples [64]. XPS survey scan spectra exhibited the silica peak at 154 and 103 eV and the nitrogen peak at 398.08 eV both in KR12@Si/BGS and CPP-KR12@Si/BGS (Figure 8a). Comparing the atomic ratios among the BGSs (Figure 8a), the surface coating decreased the atomic ratios of Ca and P, while increasing the atomic ratios of O, N, and Si. This indicated that silica (SiO2) and peptide are present both in KR12@Si or CPP-KR12@Si coated BGS. SEM images revealed that the control had a smooth surface, while the KR12@Si coated surface had a thin film of silica, and the CPP-KR12@Si coated surface had a particulate silica coating (Figure 8b). XPS analysis showed little difference in composition, but differences in surface morphology were observed due to differences in the silica deposition patterns of KR12 and CPP-KR12. To evaluate the efficacy of the antimicrobial BGS combination, E. coli was inoculated with the formulated BGS in the medium and incubated overnight. Antibacterial activity of AMP@Si/BGS was determined through LIVE/DEAD BacLight™ bacteria viability kits. Independent of the integrity of the cell membrane, SYTO 9 enters all cells and binds to DNA and RNA, generating green fluorescence [65]. In contrast, propidium iodide (PI) only enters cells with damaged cell membranes and binds to nucleic acids, generating red fluorescence [66]. Since PI has a higher affinity for nucleic acids than SYTO9, PI will bind to nucleic acids instead of SYTO9, when DNA is exposed to the two dyes at the same time [67]. Based on the combination of these two DNA binding and membrane permeability-dependent stains, the red signal is regarded as non-viable cells and the green signal as viable cells [65]. While E. coli exposed to control or KR12@Si/BGS showed similar viability with a high ratio of live bacteria, many dead cells stained red were observed in CPP-KR12@Si/BGS (Figure 8c), indicating that CPP-KR12@Si/BGS exhibited effective antibacterial activity compared to KR12@Si/BGS. It has been reported that the release rate of biomolecules immobilized on gel-like silica was slower than that on particulate silica [68]. Therefore, the difference in the silica formulation as well as the lower cell permeability compared to CPP-KR12 was thought to be the reason why KR12@Si/BGS did not exhibit antibacterial activity in the current formulation. The loading efficiency of CPP-KR12 per BGS was determined to be 0.1%. This indicates that 50 µg of AMP was loaded into 50 mg of BGS per 1 mL of medium. Converting this to a molar concentration yields an AMP concentration of approximately 16 µM. This concentration is close to the MIC for E. coli (14.54 µM), but it is not toxic to eukaryotic cells. The antimicrobial bone graft device was prepared by coating CPP-KR12 in the form of silica particles on the surface of bone graft material. This showed antimicrobial effect at non-toxic doses to host cells. In the future, we will evaluate whether this composite not only delivers antimicrobial agents locally, but also has a positive effect on promoting bone regeneration by parallel immobilization of bone regeneration-promoting factors on silica.
3. Materials and Methods
3.1. Materials
The peptides named KR12 and Cpp-KR12 were synthesized by GenScript (Piscataway, NJ, U.S.) and CPP peptide and fluorescein isothiocyanate (FITC)-labeled CPP-KR12 was synthesized by Peptron (Peptron, Dajeon, Korea). FITC was conjugated to the N-terminus of the peptide through a 6-aminohexanoic acid (Ahx) linker. After purification by HPLC and subsequent LC/mass analysis, the synthetic peptides used in this work were received from each stated provider. The peptide had a purity of 95%. Table 1 shows the obtained mass data as well as the peptide sequences. The tetramethyl orthosilicate (TMOS), L-ascorbic acid, ammonium heptamolybdate, and Lipopolysaccharides (LPS) from Escherichia coli O111:B4 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Bone graft substitute based on Beta tricalcium phosphate was provided by CG-Bio (CG Bio Inc., Korea), which is granule type bone graft substitute which has a 0.6–1 mm sized with average 300 μm sized interconnected pores and porosity of 70–80% [29]. Pierce Quantitative Fluorometric Peptide Assay Kit, CellTracker Red CMTPX (Invitrogen), SYTOXTM Green Nucleic Acid Stain, and Live/DeadTM BacLightTM Bacterial Viability Kit were purchased from Thermo Fisher Scientific Co. (Thermo Fisher Scientific Korea, Seoul). CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) was purchased from Promega (Promega Korea Ltd.). All the other reagents were of analytical grade.
3.2. Bacterial Strains
Two gram-negative bacterial strains, namely Escherichia coli (wild type W2110) and Pseudomonas aeruginosa (wild type PK01) and gram-positive strain, Staphylococcus aeruginosa (ATCC 29213) were obtained from the department of Biotechnology and Bioinformatics in Korea University.
3.3. Minimum Inhibitory Concentration (MIC) of Antimicrobial Peptides (AMPs)
The broth microdilution assay was used to test the antimicrobial activity of AMPs, as previously reported [69]. Briefly, bacteria were raised to mid-logarithmic growth phase in Mueller Hinton Broth (MHB) media at 37°C. Then, in 96-well plates, 50 µL of peptides solution (0-320 µM) diluted using a two-fold serial dilution method were mixed with an equal volume of bacterial solution (3 106 CFU/ml) and incubated for 18 h at 37°C under 200 rpm. The negative control was the culture medium itself. The lowest antimicrobial peptide concentration that exhibited the same turbidity (< 0.1) as the negative control after measurement with a microplate reader (Infinite M200 PRO NanoQuant; TECAN, Männedorf, Switzerland) at 600 nm was considered the MIC. The assay was performed in triplicate.
3.4. Silica Deposition and Quantification
According to Luckarift et al. [24], AMP-catalyzed silica deposition was started by adding 10 µL of 0.5 M mM sodium phosphate (pH 7.6) buffer to 90 µL ddH2O containing 100 µg of AMP and 10 µmoles of hydrolyzed TMOS. The reaction was performed at room temperature for 5 minutes with stirring. The silica precipitate was washed by centrifugation three times with ddH2O (14000g, 5 min, 4oC). Silica precipitation was determined using a modified molybdenum blue assay as previously described [49]. Briefly, the washed silica precipitate was dried, dissolved in 1 M NaOH solution, and diluted 5- to 10-fold. The total reaction solution was 50 μL. Equal volumes of 1% ammonium heptamolybdate, 1% oxalic acid and 100 mM ascorbic acid were added sequentially to this mixture. Optical density was measured at 810nm.
3.5. Measurement of Entrapping and Loading Efficiency of AMP in Silica Particles
A total of 100 μg of peptide was used for the precipitation of silica particles loaded with the corresponding peptides, as described above. The amount of co-precipitated peptide was estimated by subtracting the amount of AMP that remained in the supernatant after the precipitates were removed from the original feeding amount. The quantification of peptide was determined using the Pierce Quantitative Fluorometric Peptide Assay kit (Thermo Fisher Scientific, USA).The amount of silica was determined using the above-mentioned modified molybdenum blue method. The entrapment efficiency (%) and loading efficiency (%) of AMP were estimated using the following equations:
3.6. SYTOXTM Green Uptake Assay
SYTOXTM Green Nucleic Acid Stain was used to fluorometrically determine the relative sensitivity of suspensions of E. coli, P. aeruginosa and S. aureus to different AMPs [70]. The relative sensitivity of free CPP-KR12 and CPP-KR12@Si to E. coli suspension was also determined fluorometrically by SYTOX Green staining. E. coli (108 CFU/mL) was incubated with 5 μM SYTOX Green in 1 x PBS at room temperature for 20 min, and a 100-fold diluted E. coli suspension was recorded in the absence and presence of 30 µM CPP-KR12 or CPP-KR12@Si for 10 min. The fluorescence emission spectrum was excited at 490 nm.
3.7. Stability of AMP@Si against Protease Attack
Free AMP or AMP@Si was dissolved in 1×PBS at a concentration of 300 μM and left at 37oC for 3 min in the presence of 0.025% trypsin, followed by the addition of FBS to stop the activity of trypsin. Bacteria were incubated overnight in the presence of each trypsinized peptide formula and broth absorbance measured at 600 nm. As a negative control, the absorbance of the strain grown in the absence of the peptide was set. To determine the remaining antimicrobial activity of AMP after treatment of trypsin, the absorbance of the negative control was subtracted from the absorbance of the sample treated with AMP. The resulting difference was then expressed as a percentage of the absorbance of the negative control.
3.8. Cytotoxicity and Hemolytic Activity Assay
Free peptides and silica particles loaded with them (AMP@Si) were compared for their cytotoxicity against mouse macrophages (Raw264.7 cells) and their hemolytic activity against red blood cells (RBCs). Free AMP or AMP@Si was added to each well of a 96-well plate in two-fold serial dilutions to 50 μL with DMEM medium containing 10% FBS and 1% antibiotics. Raw 264.7 macrophages at a concentration of 4 × 104 cells per 50 μL were added to the wells containing the peptide to give a final volume of 100 μL. Cell proliferation was confirmed by MTS assay (Promega, USA) after 24 h or 5 days of incubation. The MTS tetrazolium compound is reduced by viable cells to generate a colored formazan product. The intensity of the color at 490 nm is proportional to the number of viable cells in the sample. To calculate cell viability, the absorbance of cells treated with the peptide was divided by the absorbance of cells that were not treated with the peptide. The resulting quotient was then expressed as a percentage. Fresh RBCs from sheep blood (KisanBio, Seoul, Korea) were recovered by centrifugation at 150 g for 5 min and washed three times with PBS. Peptides and silica particles diluted in two-fold serial dilutions with PBS were added to each well of a 96-well plate, giving a total volume of 100 μL. A 2% RBCs suspension (2% w/v RBC in PBS) was added to each concentration of peptide in 100 μL to give a final volume of 200 μL and the RBCs was incubated at 37°C for 1 hour. The supernatant (100 μL) was collected after centrifugation at 1000 g for 5 minutes and the absorbance (A) was measured at 570 nm [69]. The value of "zero hemolysis" was determined by the absorbance of PBS alone, and 100% hemolysis was determined by the absorbance of the supernatant obtained from 0.1% (v/v) Triton X-100 treated RBCs. The percentage of hemolysis was calculated as hemolysis (%) = (AAMP-APBS) / (ATriton-APBS) ×100.
3.9. Effect of AMPs on mRNA Expression of Pro-Inflammatory Cytokines
Quantitative PCR (qPCR) was performed as previously described [71] to evaluate the mRNA expression of pro-inflammatory cytokines, including TNF-alpha (forward: ATGGCCTCCCTCTCATCAGT; reverse: TGGTTTGCTACGACGTGGG), IL-1beta (forward: TGCCACCTTTTGACAGTGATG; reverse: AAGGTCCACGGGAAAGACAC), and IL-6 (forward: GTCCTTCAGAGAGATACAGAAACT; reverse: AGCTTATCTGTTAGGAGAGCATTG) in Raw264.7 cells in the absence or presence of 100 ng/mL LPS. The amount of target genes was expressed as relative quantification (RQ) over the expression level of LPS alone-treated cells. The quantitative gene expression data were normalized to the expression levels of Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward: CCTGGCCAAGGTCATCCATG; reverse: GCAGGAGACAACCTGGTCCT) [72].
3.10. Gel Retardation Assay
Gel retardation assays were used to determine the ability of peptides to bind and interact with DNA, following the method of Lee et al., [19]. In brief, increasing amounts of peptides were mixed with 200 ng plasmid DNA (pQE80L) in reaction buffer, which contained 10 mM Tris-HCl, 5% glycerol, 50 µg/ml BSA, 1 mM dithiothreitol, and 1 mM ethylenediaminetetraacetic acid (EDTA). As negative controls, wells containing only plasmid were used. The peptide-plasmid mixture was incubated for 30 minutes in a 37°C water bath before being separated by agarose gel electrophoresis (1% agarose) in 0.5% TAE buffer. Migration of DNA was detected by RedSafe™ Nucleic Acid Staining Solution (iNtRON Biotechnology, Inc. Seongnam, Korea).
3.11. STED Microscopy and Confocal Images of Cells
Raw264.7 cells on a coverslip were incubated in DMEM with free FITC-CPP-KR12 or FITC-CPP-KR12@Si in a humidified atmosphere containing 5% CO2 at 37°C for 2 h or overnight. The cells were rinsed three times with 1× PBS and fixed with 4% paraformaldehyde at 37°C for 30 minutes. After three washes with 1× PBS, cells were immersed in PBS with CellTracker Red CMTPX (Thermo Fisher Scientific) (5000:1 dilution) for 30 minutes for cytoplasm staining. The stained cells were washed three times with PBS and stained with 1 ug/mL of DAPI (4',6-diamidino-2-phenylindole) to detect nuclei. The logarithmic growth of E. coli was washed three times with 1× PBS and exposed to each peptide formula for a period of 20 min to 1 h. The cells were then fixed with 2.5% glutaraldehyde and observed under a fluorescence microscope after three PBS washes. Confocal images were taken along the XY plane using a STEDYCON microscope (Abberior Instruments GmbH, Germany) with a 100× oil immersion objective. Raw264.7 cells show rhodamine-labeled cytoplasm (red), FITC-labeled CPP-KR12 or CPP-KR12@Si (green), and DAPI-labeled nucleic acids (blue). E. coli shows green fluorescence due to FITC-labeled AMP. The laser power was set to 10% and the pixel size was 30 nm.
3.12. Live/Dead Cell Assay
A fluorescence microscope (CytationTM 7, BioTek) was used to observe the results of the BacLight live/dead bacterial viability kit according to the manufacturer's method (ThermoFisher Scientific Korea, Seoul, Korea). E. coli grown overnight was added to the culture medium containing the prepared AMP@Si/β-TCP BGS (50 mg/mL) at a concentration of 100-fold dilution. An equal amount of E. coli inoculated into media containing β-TCP without AMP was used as a control to compare antibacterial activity. After incubation at 37 °C for 12 h, the grown E. coli was recovered by centrifugation and washed with PBS to remove medium components, then SYTO 9 (3.34 mM) and propidium iodide (PI) solutions (20 mM) were mixed in an equal 1:1 volume ratio and 3 µL of the mixture was added per 1 mL of the washed bacterial suspension and stained under dark conditions for 15 min. 5 µL of the stained E. coli suspension was carefully applied to a microscope slide. A coverslip was then placed over the sample. The sample was observed under a fluorescence microscope.
3.13. Zeta Potential Measurement
For the zeta potential studies, AMP@Si were suspended in ethanol. The suspensions were injected into zeta cells and allowed to equilibrate at 25 °C for 15 min, and measurements were acquired on a Zetasizer Nano ZS (Marvern Instrument Ltd. ZEN3600). Zeta potential was calculated from the mean of 10 measurements, using the Smoluchowski equation as described previously (55). Experiments were performed in triplicate.
3.14. Sample Preparation of Bacterial Cells for Scanning Electron Microscopy (SEM)
Mid-logarithmic E. coli or S. aureus cells ( 1108 CFU/mL) were cultured in 3 ml of culture medium at 37°C for 6 h in the presence or absence of AMP. The bacterial suspension was centrifuged, washed three times with PBS, and fixed overnight at 4°C with 1.5 ml PBS containing 2.5% glutaraldehyde [69]. After fixation, the bacterial cells were washed three times with sterile ultrapure water. Resulting cells were subjected to a graded ethanol series (30, 50, 70, 80, 90, 95, and 100 %) at 25°C for 10 minutes each. A smear of 10 µL of the bacterial suspension in 100% ethanol was applied to a glass slide and allowed to air dry.
3.15. Preparation of Bone Graft Substitute (BGS) Coated with AMP@Si (AMP@Si /β-TCP)
The 500 mg of granular β-tri-calcium phosphate (β-TCP) having a size of 0.6 to 1 mm was added to 1 mL of 1×PBS containing 1 mg of AMP and 20 μmol of hydrolyzed TMOS, gently agitated with a rotary mixer overnight at 4°C, followed by 10 washes with ddH2O to remove unreacted TMOS and peptides. The final BGS was dried in a biosafety cabinet. All procedures were performed under aseptic conditions.
3.16. Scanning Electron Microscopy (SEM)
The dried sample was coated with platinum using an ion coater (COXEM Co., Ltd. Daejeon, Korea) for SEM observation. The structure of bacteria, AMP@Si and BGS were analyzed using scanning electron microscopy (FE-SEM, JSM-6700F, JEOL, Japan).
3.17. High-Performance X-ray Photoelectron Spectrometer
The surface chemical compositions of as prepared β-TCP BGS were analyzed by High-performance X-ray Photoelectron Spectrometer (HP-XPS, Thermo Scientific ESCALAB 250) with an Al-Kα X-ray source at the Busan Center of the Korea Basic Science Institute.
3.18. Statistical Analysis
Statistical analyses were performed using Student’s t-test to determine the differences between the two groups. Statistical significance was set at p < 0.05.
4. Conclusions
The CPP-KR12 peptide is a potent antimicrobial with broad-spectrum activity. Compared to KR12, CPP-KR12 has enhanced cell permeability and DNA binding, resulting in greater antimicrobial activity. In addition, it can reduce inflammation through LPS neutralization. When CPP-KR12 was mineralized with silica with a 95% entrapment efficiency through its excellent silica precipitation ability, it showed more resistant to protease attack and reduced cytotoxicity toward Raw264.7 macrophage while retaining antimicrobial activity compared to those of free form CPP-KR12. This silica mineralized CPP-KR12 (CPP-KR12@Si) binds to the surface of S. aureus and induces aggregation, leading to cell death more effectively than the free form, which showed low antibacterial activity against the corresponding strain. Despite its potent antimicrobial properties, CPP-KR12 is not cytotoxic at minimally inhibitory concentrations for bacteria. Finally, bone grafts treated with CPP-KR12 as silica nanoparticles were shown to be effective against E. coli infections. Overall, CPP-KR12@Si is a highly versatile and effective tool in the fight against microbial infections.
5. Patents
This section is not mandatory but may be added if there are patents resulting from the work reported in this manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Antibacterial activity of indicated peptide depending on concentration; Figure S2: Anti-oxidant protein expression in response of AMPs in the presence or absence of lipopolysaccharide (LPS) in RAW264.7 cells; Supplementary Figure S3: Comparison of cell viability after 5 days of AMP treatment.
Author Contributions
S. P. Pack contributed to the conception, design, data acquisition, analysis, and interpretation, and drafted and critically revised the manuscript; M. -R. Ki contributed to conception, design, data acquisition, analysis, and interpretation, performed all statistical analyses, and drafted and critically revised the manuscript; S.H.Kim and T.I. Park contributed to data acquisition, analysis, and drafted the manuscript; all authors gave final approval and agreed to be accountable for all aspects of the work..
Funding
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (NRF-2021R1A5A8032895 and NRF-2021R1A2C2011564). This work was also supported by the National Research Foundation of Korea (NRF), funded by the Korean Ministry of Education (NRF-2021R1I1A3046565).
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
The data presented in this study are available for publication else-where.
Acknowledgments
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Uçkay, I.; Hoffmeyer, P.; Lew, D.; Pittet, D. Prevention of surgical site infections in orthopaedic surgery and bone trauma: state-of-the-art update. Journal of Hospital Infection 2013, 84, 5-12. [CrossRef]
- Bayramov, D.F.; Neff, J.A. Beyond conventional antibiotics — New directions for combination products to combat biofilm. Advanced Drug Delivery Reviews 2017, 112, 48-60. [CrossRef]
- Schierholz, J.M.; Beuth, J. Implant infections: a haven for opportunistic bacteria. Journal of Hospital Infection 2001, 49, 87-93. [CrossRef]
- van Barreveld, M.; Verstraelen, T.E.; Buskens, E.; van Dessel, P.F.H.M.; Boersma, L.V.A.; Delnoy, P.P.H.M.; Tuinenburg, A.E.; Theuns, D.A.M.J.; van der Voort, P.H.; Kimman, G.P.; Zwinderman, A.H.; Wilde, A.A.M.; Dijkgraaf, M.G.W.; van Barreveld, M.; Verstraelen, T.E.; Buskens, E.; van Dessel, P.F.H.M.; Boersma, L.V.A.; Delnoy, P.P.H.M.; Tuinenburg, A.E.; Theuns, D.A.M.J.; van der Voort, P.H.; Kimman, G.P.; Zwinderman, A.H.; Wilde, A.A.M.; Dijkgraaf, M.G.W. Hospital utilisation and the costs associated with complications of ICD implantation in a contemporary primary prevention cohort. Netherlands Heart Journal 2022, 31, 244-253. [CrossRef]
- Zogg, C.K.; Ottesen, T.D.; Kebaish, K.J.; Galivanche, A.; Murthy, S.; Changoor, N.R.; Zogg, D.L.; Pawlik, T.M.; Haider, A.H. The Cost of Complications Following Major Resection of Malignant Neoplasia. J Gastrointest Surg 2018, 22, 1976-1986. [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int J Mol Sci 2016, 17. [CrossRef]
- Komori, A.; Iriyama, H.; Kainoh, T.; Aoki, M.; Naito, T.; Abe, T. The impact of infection complications after trauma differs according to trauma severity. Sci Rep 2021, 11, 13803. [CrossRef]
- Dostert, M.; Trimble, M.J.; Hancock, R.E.W. Antibiofilm peptides: overcoming biofilm-related treatment failure. RSC Advances 2021, 11, 2718-2728. [CrossRef]
- Carpa, R.; Farkas, A.; Dobrota, C.; Butiuc-Keul, A. Double-Network Chitosan-Based Hydrogels with Improved Mechanical, Conductive, Antimicrobial, and Antibiofouling Properties. Gels 2023, 9. [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. Eur J Biochem 2001, 268, 5589-5600. [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front Microbiol 2020, 11, 582779. [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Frontiers in Microbiology 2020, 11. [CrossRef]
- Zhang, Q.-Y.; Yan, Z.-B.; Meng, Y.-M.; Hong, X.-Y.; Shao, G.; Ma, J.-J.; Cheng, X.-R.; Liu, J.; Kang, J.; Fu, C.-Y. Antimicrobial peptides: mechanism of action, activity and clinical potential. Military Medical Research 2021, 8. [CrossRef]
- Xuan, J.; Feng, W.; Wang, J.; Wang, R.; Zhang, B.; Bo, L.; Chen, Z.-S.; Yang, H.; Sun, L. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resistance Updates 2023, 68. [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972-1994. [CrossRef]
- Ghorai, S.M.; Deep, A.; Magoo, D.; Gupta, C.; Gupta, N. Cell-Penetrating and Targeted Peptides Delivery Systems as Potential Pharmaceutical Carriers for Enhanced Delivery across the Blood–Brain Barrier (BBB). Pharmaceutics 2023, 15. [CrossRef]
- Huo, S.; Chen, C.; Lyu, Z.; Zhang, S.; Wang, Y.; Nie, B.e.; Yue, B. Overcoming Planktonic and Intracellular Staphylococcus aureus-Associated Infection with a Cell-Penetrating Peptide-Conjugated Antimicrobial Peptide. ACS Infectious Diseases 2020, 6, 3147-3162. [CrossRef]
- Tang, Q.; Tan, P.; Dai, Z.; Wang, T.; Xu, S.; Ding, Y.; Jin, J.; Zhang, X.; Zhang, Y.; Zhou, C.; Yue, Z.; Fu, H.; Yan, J.; Ma, X. Hydrophobic modification improves the delivery of cell-penetrating peptides to eliminate intracellular pathogens in animals. Acta Biomaterialia 2023, 157, 210-224. [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694-15701. [CrossRef]
- Ngambenjawong, C.; Chan, L.W.; Fleming, H.E.; Bhatia, S.N. Conditional Antimicrobial Peptide Therapeutics. ACS Nano 2022, 16, 15779-15791. [CrossRef]
- Hupcey, M.A.Z.; Ekins, S. Improving the drug selection and development process for combination devices. Drug Discovery Today 2007, 12, 844-852. [CrossRef]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Advances in Colloid and Interface Science 2017, 242, 17-34. [CrossRef]
- Poulsen, N.; Sumper, M.; Kröger, N. Biosilica formation in diatoms: Characterization of native silaffin-2 and its role in silica morphogenesis. Proceedings of the National Academy of Sciences 2003, 100, 12075-12080. [CrossRef]
- Luckarift, H.R.; Spain, J.C.; Naik, R.R.; Stone, M.O. Enzyme immobilization in a biomimetic silica support. Nature Biotechnology 2004, 22, 211-213. [CrossRef]
- Pamirsky, I.; Golokhvast, K. Silaffins of Diatoms: From Applied Biotechnology to Biomedicine. Marine Drugs 2013, 11, 3155-3167. [CrossRef]
- Abdelhamid, M.A.A.; Pack, S.P. Biomimetic and bioinspired silicifications: Recent advances for biomaterial design and applications. Acta Biomaterialia 2021, 120, 38-56. [CrossRef]
- Ryu, Y.H.; Yeo, K.B.; Ki, M.-R.; Kim, Y.J.; Pack, S.P. Improved stability and reusability of endoglucanase from Clostridium thermocellum by a biosilica-based auto-encapsulation method. Biochemical Engineering Journal 2016, 105, 144-149. [CrossRef]
- Ki, M.-R.; Kim, J.K.; Kim, S.H.; Nguyen, T.K.M.; Kim, K.H.; Pack, S.P. Compartment-restricted and rate-controlled dual drug delivery system using a biosilica-enveloped ferritin cage. Journal of Industrial and Engineering Chemistry 2020, 81, 367-374. [CrossRef]
- Lee, D.K.; Ki, M.-R.; Kim, E.H.; Park, C.-J.; Ryu, J.J.; Jang, H.S.; Pack, S.P.; Jo, Y.K.; Jun, S.H. Biosilicated collagen/β-tricalcium phosphate composites as a BMP-2-delivering bone-graft substitute for accelerated craniofacial bone regeneration. Biomaterials Research 2021, 25. [CrossRef]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. European Biophysics Journal 2011, 40, 387-397. [CrossRef]
- Dürr, U.H.N.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 2006, 1758, 1408-1425. [CrossRef]
- Jacob, B.; Park, I.-S.; Bang, J.-K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. Journal of Peptide Science 2013, 19, 700-707. [CrossRef]
- Li, H.; Zhang, S.; Nie, B.e.; Du, Z.; Long, T.; Yue, B. The antimicrobial peptide KR-12 promotes the osteogenic differentiation of human bone marrow stem cells by stimulating BMP/SMAD signaling. RSC Advances 2018, 8, 15547-15557. [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomedicine & Pharmacotherapy 2018, 108, 1090-1096. [CrossRef]
- Ryu, J.; Han, K.; Park, J.; Choi, S.Y. Enhanced uptake of a heterologous protein with an HIV-1 Tat protein transduction domains (PTD) at both termini. Mol Cells 2003, 16, 385-391.
- Zou, L.; Peng, Q.; Wang, P.; Zhou, B. Progress in Research and Application of HIV-1 TAT-Derived Cell-Penetrating Peptide. The Journal of Membrane Biology 2016, 250, 115-122. [CrossRef]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochimica et Biophysica Acta (BBA) - Biomembranes 2016, 1858, 936-946. [CrossRef]
- Melo, M.N.; Ferre, R.; Castanho, M.A.R.B. Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nature Reviews Microbiology 2009, 7, 245-250. [CrossRef]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrobial Agents and Chemotherapy 2017, 61. [CrossRef]
- Liu, W.; Dong, S.L.; Xu, F.; Wang, X.Q.; Withers, T.R.; Yu, H.D.; Wang, X. Effect of Intracellular Expression of Antimicrobial Peptide LL-37 on Growth of Escherichia coli Strain TOP10 under Aerobic and Anaerobic Conditions. Antimicrobial Agents and Chemotherapy 2013, 57, 4707-4716. [CrossRef]
- Rowe-Magnus, D.A.; Kao, A.Y.; Prieto, A.C.; Pu, M.; Kao, C.; Davies, J.E. Cathelicidin Peptides Restrict Bacterial Growth via Membrane Perturbation and Induction of Reactive Oxygen Species. mBio 2019, 10. [CrossRef]
- Pulido, D.; Nogués, M.V.; Boix, E.; Torrent, M. Lipopolysaccharide Neutralization by Antimicrobial Peptides: A Gambit in the Innate Host Defense Strategy. Journal of Innate Immunity 2012, 4, 327-336. [CrossRef]
- Sun, Y.; Shang, D. Inhibitory Effects of Antimicrobial Peptides on Lipopolysaccharide-Induced Inflammation. Mediators of Inflammation 2015, 2015, 1-8. [CrossRef]
- Dong, J.; Wang, W.; Zhou, W.; Zhang, S.; Li, M.; Li, N.; Pan, G.; Zhang, X.; Bai, J.; Zhu, C. Immunomodulatory biomaterials for implant-associated infections: from conventional to advanced therapeutic strategies. Biomaterials Research 2022, 26. [CrossRef]
- Noori, M.S.; Courreges, M.C.; Bergmeier, S.C.; McCall, K.D.; Goetz, D.J. Modulation of LPS-induced inflammatory cytokine production by a novel glycogen synthase kinase-3 inhibitor. European Journal of Pharmacology 2020, 883. [CrossRef]
- Lechner, C.C.; Becker, C.F.W. A sequence-function analysis of the silica precipitating silaffin R5 peptide. Journal of Peptide Science 2014, 20, 152-158. [CrossRef]
- Lechner, C.; Becker, C. Silaffins in Silica Biomineralization and Biomimetic Silica Precipitation. Marine Drugs 2015, 13, 5297-5333. [CrossRef]
- Ki, M.R.; Kim, S.H.; Nguyen, T.K.M.; Son, R.G.; Jun, S.H.; Pack, S.P. BMP2-Mediated Silica Deposition: An Effective Strategy for Bone Mineralization. ACS Biomater Sci Eng 2022. [CrossRef]
- Ki, M.-R.; Nguyen, T.K.M.; Park, T.-I.; Park, H.-M.; Pack, S.P. Biomimetic Silica Particles with Self-Loading BMP-2 Knuckle Epitope Peptide and Its Delivery for Bone Regeneration. Pharmaceutics 2023, 15. [CrossRef]
- Nanomaterials for Drug Delivery and Therapy; 2019; p.^pp.
- Németh, Z.; Csóka, I.; Semnani Jazani, R.; Sipos, B.; Haspel, H.; Kozma, G.; Kónya, Z.; Dobó, D.G. Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives. Pharmaceutics 2022, 14. [CrossRef]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Frontiers in Microbiology 2020, 11. [CrossRef]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Advanced Drug Delivery Reviews 2014, 78, 28-45. [CrossRef]
- Qiu, S.; Zhu, R.; Zhao, Y.; An, X.; Jia, F.; Peng, J.; Ma, Z.; Zhu, Y.; Wang, J.; Su, J.; Wang, Q.; Wang, H.; Li, Y.; Wang, K.; Yan, W.; Wang, R. Antimicrobial activity and stability of protonectin withD-amino acid substitutions. Journal of Peptide Science 2017, 23, 392-402. [CrossRef]
- Oliva, R.; Chino, M.; Pane, K.; Pistorio, V.; De Santis, A.; Pizzo, E.; D’Errico, G.; Pavone, V.; Lombardi, A.; Del Vecchio, P.; Notomista, E.; Nastri, F.; Petraccone, L. Exploring the role of unnatural amino acids in antimicrobial peptides. Scientific Reports 2018, 8. [CrossRef]
- Khara, J.S.; Priestman, M.; Uhía, I.; Hamilton, M.S.; Krishnan, N.; Wang, Y.; Yang, Y.Y.; Langford, P.R.; Newton, S.M.; Robertson, B.D.; Ee, P.L.R. Unnatural amino acid analogues of membrane-active helical peptides with anti-mycobacterial activity and improved stability. Journal of Antimicrobial Chemotherapy 2016, 71, 2181-2191. [CrossRef]
- Häffner, S.M.; Parra-Ortiz, E.; Browning, K.L.; Jørgensen, E.; Skoda, M.W.A.; Montis, C.; Li, X.; Berti, D.; Zhao, D.; Malmsten, M. Membrane Interactions of Virus-like Mesoporous Silica Nanoparticles. ACS Nano 2021, 15, 6787-6800. [CrossRef]
- Singh, S.; Papareddy, P.; Mörgelin, M.; Schmidtchen, A.; Malmsten, M. Effects of PEGylation on Membrane and Lipopolysaccharide Interactions of Host Defense Peptides. Biomacromolecules 2014, 15, 1337-1345. [CrossRef]
- Yu, T.; Malugin, A.; Ghandehari, H. Impact of Silica Nanoparticle Design on Cellular Toxicity and Hemolytic Activity. ACS Nano 2011, 5, 5717-5728. [CrossRef]
- Slowing, I.I.; Wu, C.W.; Vivero-Escoto, J.L.; Lin, V.S.Y. Mesoporous Silica Nanoparticles for Reducing Hemolytic Activity Towards Mammalian Red Blood Cells. Small 2009, 5, 57-62. [CrossRef]
- Zhao, Y.; Sun, X.; Zhang, G.; Trewyn, B.G.; Slowing, I.I.; Lin, V.S.Y. Interaction of Mesoporous Silica Nanoparticles with Human Red Blood Cell Membranes: Size and Surface Effects. ACS Nano 2011, 5, 1366-1375. [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2018, 1865, 721-733. [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med 2019, 8, 2252-2267. [CrossRef]
- Greczynski, G.; Hultman, L. X-ray photoelectron spectroscopy: Towards reliable binding energy referencing. Progress in Materials Science 2020, 107. [CrossRef]
- Stiefel, P.; Schmidt-Emrich, S.; Maniura-Weber, K.; Ren, Q. Critical aspects of using bacterial cell viability assays with the fluorophores SYTO9 and propidium iodide. BMC Microbiology 2015, 15. [CrossRef]
- Rosenberg, M.; Azevedo, N.F.; Ivask, A. Propidium iodide staining underestimates viability of adherent bacterial cells. Scientific Reports 2019, 9. [CrossRef]
- Stocks, S.M. Mechanism and use of the commercially available viability stain,BacLight. Cytometry 2004, 61A, 189-195. [CrossRef]
- Abdelhamid, M.A.A.; Yeo, K.B.; Ki, M.-R.; Pack, S.P. Self-encapsulation and controlled release of recombinant proteins using novel silica-forming peptides as fusion linkers. International Journal of Biological Macromolecules 2019, 125, 1175-1183. [CrossRef]
- Lu, Y.; Tian, H.; Chen, R.; Liu, Q.; Jia, K.; Hu, D.-L.; Chen, H.; Ye, C.; Peng, L.; Fang, R. Synergistic Antimicrobial Effect of Antimicrobial Peptides CATH-1, CATH-3, and PMAP-36 With Erythromycin Against Bacterial Pathogens. Frontiers in Microbiology 2022, 13. [CrossRef]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl Environ Microbiol 1997, 63, 2421-2431. [CrossRef]
- Ki, M.-R.; Kim, S.H.; Nguyen, T.K.M.; Son, R.G.; Jun, S.H.; Pack, S.P. BMP2-Mediated Silica Deposition: An Effective Strategy for Bone Mineralization. ACS Biomaterials Science & Engineering 2022, 9, 1823-1833. [CrossRef]
- Lee, S.; Lee, T.A.; Song, S.J.; Park, T.; Park, B. Hyperproduction of IL-6 caused by aberrant TDP-43 overexpression in high-fat diet-induced obese mice. FEBS Letters 2015, 589, 1825-1831. [CrossRef]
Scheme 1.
Self-entrapment of AMPs in silica matrix through CPP-KR12-mediated silica deposition (a) and antimicrobial peptide-device combination product (b).
Scheme 1.
Self-entrapment of AMPs in silica matrix through CPP-KR12-mediated silica deposition (a) and antimicrobial peptide-device combination product (b).
Figure 1.
(a) SYTOXTM green uptake assay analyzing membrane permeability in the indicated bacterial cells over KR12, CPP, or CPP-KR12. Relative fluorescence unit (RFU) value represents fluorescence ratio relative to that measured in absence of peptide in each strain after 10 min exposure of indicated peptide (30 μM) . Values are presented as mean ± SE (N = 3). *p <0.05 vs KR12 in each strain. (b) SEM image of E. coli treated with indicated AMP. The image is a 10,000× magnification, and the scale bar represents 1 µm.
Figure 1.
(a) SYTOXTM green uptake assay analyzing membrane permeability in the indicated bacterial cells over KR12, CPP, or CPP-KR12. Relative fluorescence unit (RFU) value represents fluorescence ratio relative to that measured in absence of peptide in each strain after 10 min exposure of indicated peptide (30 μM) . Values are presented as mean ± SE (N = 3). *p <0.05 vs KR12 in each strain. (b) SEM image of E. coli treated with indicated AMP. The image is a 10,000× magnification, and the scale bar represents 1 µm.
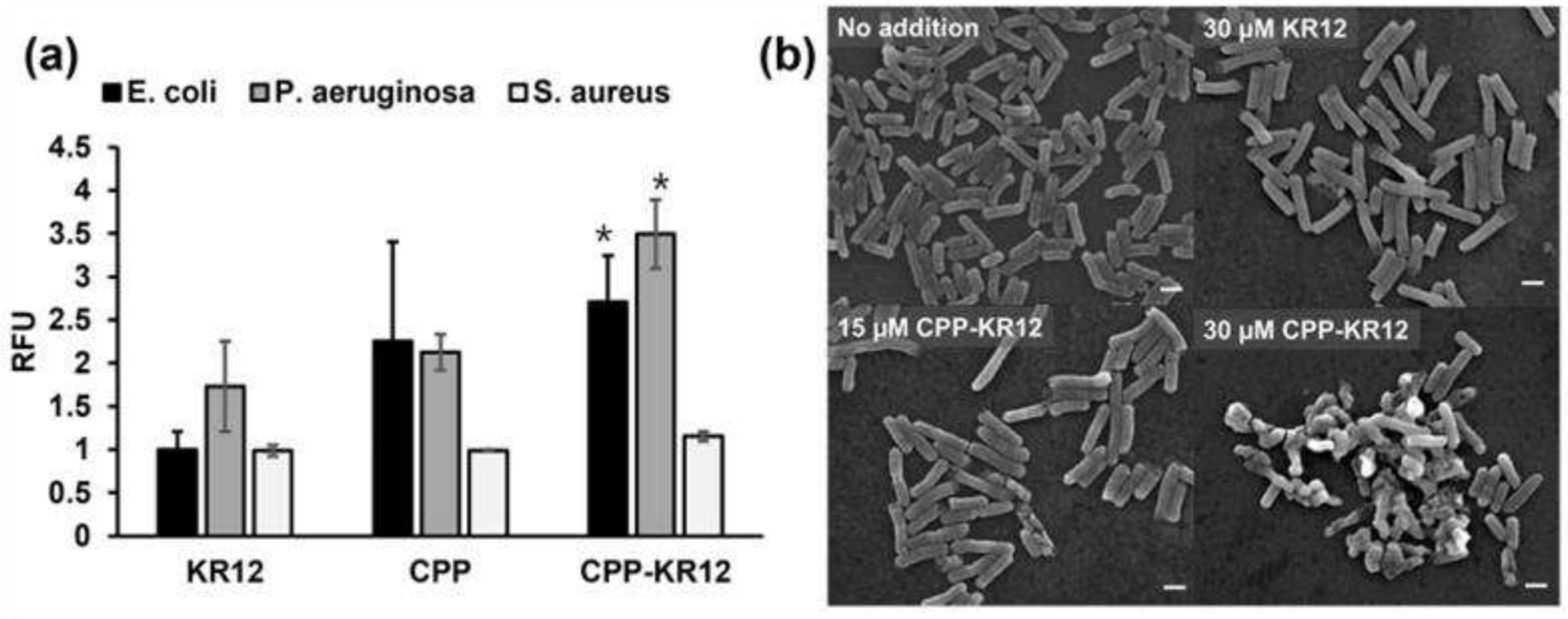
Figure 2.
Gel retardation assay of Peptide-DNA complexes was performed using peptides of varying concentrations. The concentration of peptide used for complexation with DNA is expressed in micromolar units.
Figure 2.
Gel retardation assay of Peptide-DNA complexes was performed using peptides of varying concentrations. The concentration of peptide used for complexation with DNA is expressed in micromolar units.
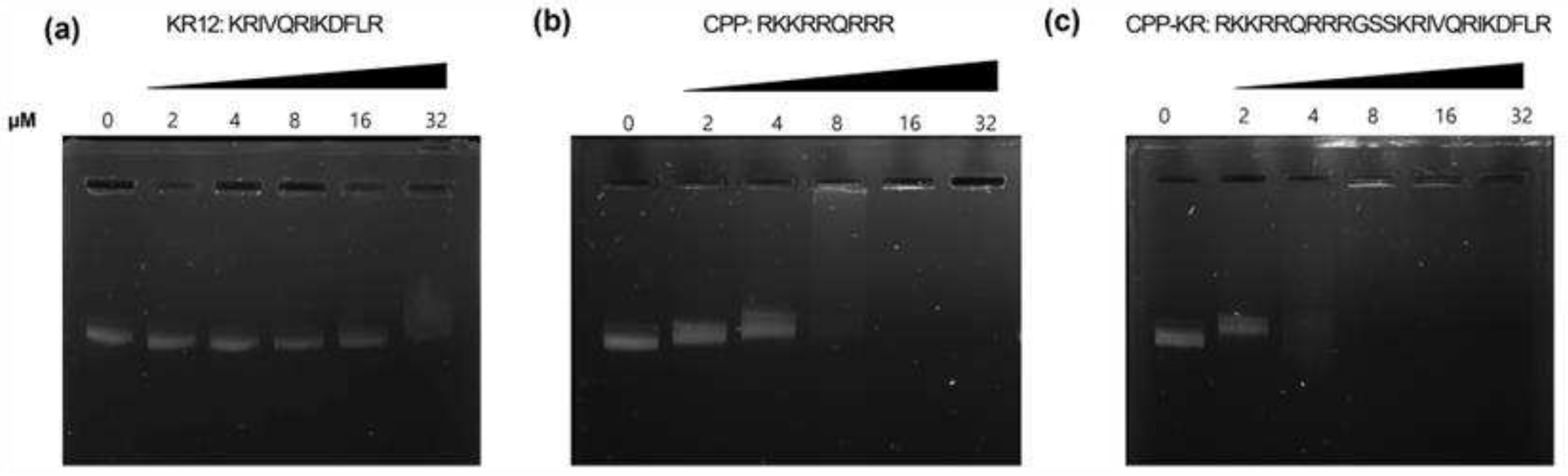
Figure 3.
Anti-inflammatory response of AMPs in LPS-stimulated RAW264.7 cells. The mRNA expression levels of inflammatory cytokines, namely TNF-α, IL-1β, and IL-6 were measured by qRT-PCR. Glyceraldehyde-3-Phosphate-Dehydrogenase (GAPDH) gene was used for normalization in gene expression. Gene levels in each treated cell were calculated relative to those in cells exposed to LPS without AMP. IL-6 was not detected in the control without both LPS and AMP, so it was indicated as n.d (not detected). Values are presented as mean ± SE (N = 3). ***p < 0.001, ****p <0.0001.
Figure 3.
Anti-inflammatory response of AMPs in LPS-stimulated RAW264.7 cells. The mRNA expression levels of inflammatory cytokines, namely TNF-α, IL-1β, and IL-6 were measured by qRT-PCR. Glyceraldehyde-3-Phosphate-Dehydrogenase (GAPDH) gene was used for normalization in gene expression. Gene levels in each treated cell were calculated relative to those in cells exposed to LPS without AMP. IL-6 was not detected in the control without both LPS and AMP, so it was indicated as n.d (not detected). Values are presented as mean ± SE (N = 3). ***p < 0.001, ****p <0.0001.
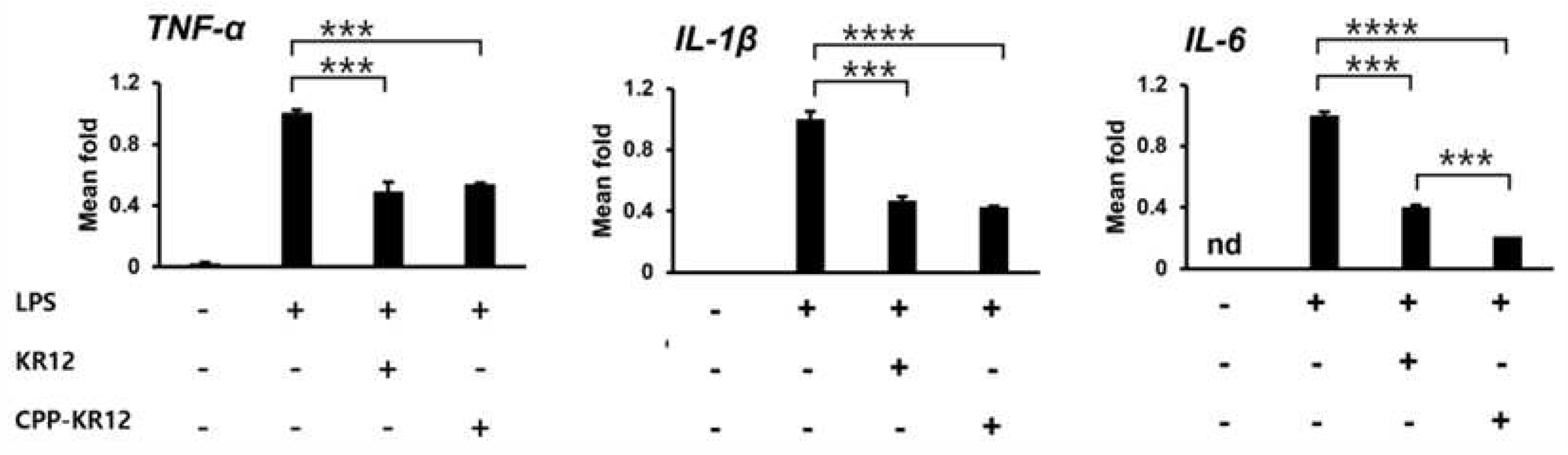
Figure 4.
Shape and properties of silica particles formed by each AMP. SEM image at 10,000x magnification (a) and size distribution (b) of silica particles formed by each AMP. The dispersion of particle size was obtained by measuring the diameter of 50 particles from SEM images. Middle line on the figure represents the average sizes for the corresponding particles and the upper and lower line represent standard deviation. ****p <0.0001. (c) Zeta potential of each silica particle dispersed in alcohol. The zeta potential value is the mean and standard deviation of the 3 measurements. ***p <0.001, ****p <0.0001 vs. Si, ## p<0.01, ###p <0.001 vs. KR12@Si.
Figure 4.
Shape and properties of silica particles formed by each AMP. SEM image at 10,000x magnification (a) and size distribution (b) of silica particles formed by each AMP. The dispersion of particle size was obtained by measuring the diameter of 50 particles from SEM images. Middle line on the figure represents the average sizes for the corresponding particles and the upper and lower line represent standard deviation. ****p <0.0001. (c) Zeta potential of each silica particle dispersed in alcohol. The zeta potential value is the mean and standard deviation of the 3 measurements. ***p <0.001, ****p <0.0001 vs. Si, ## p<0.01, ###p <0.001 vs. KR12@Si.
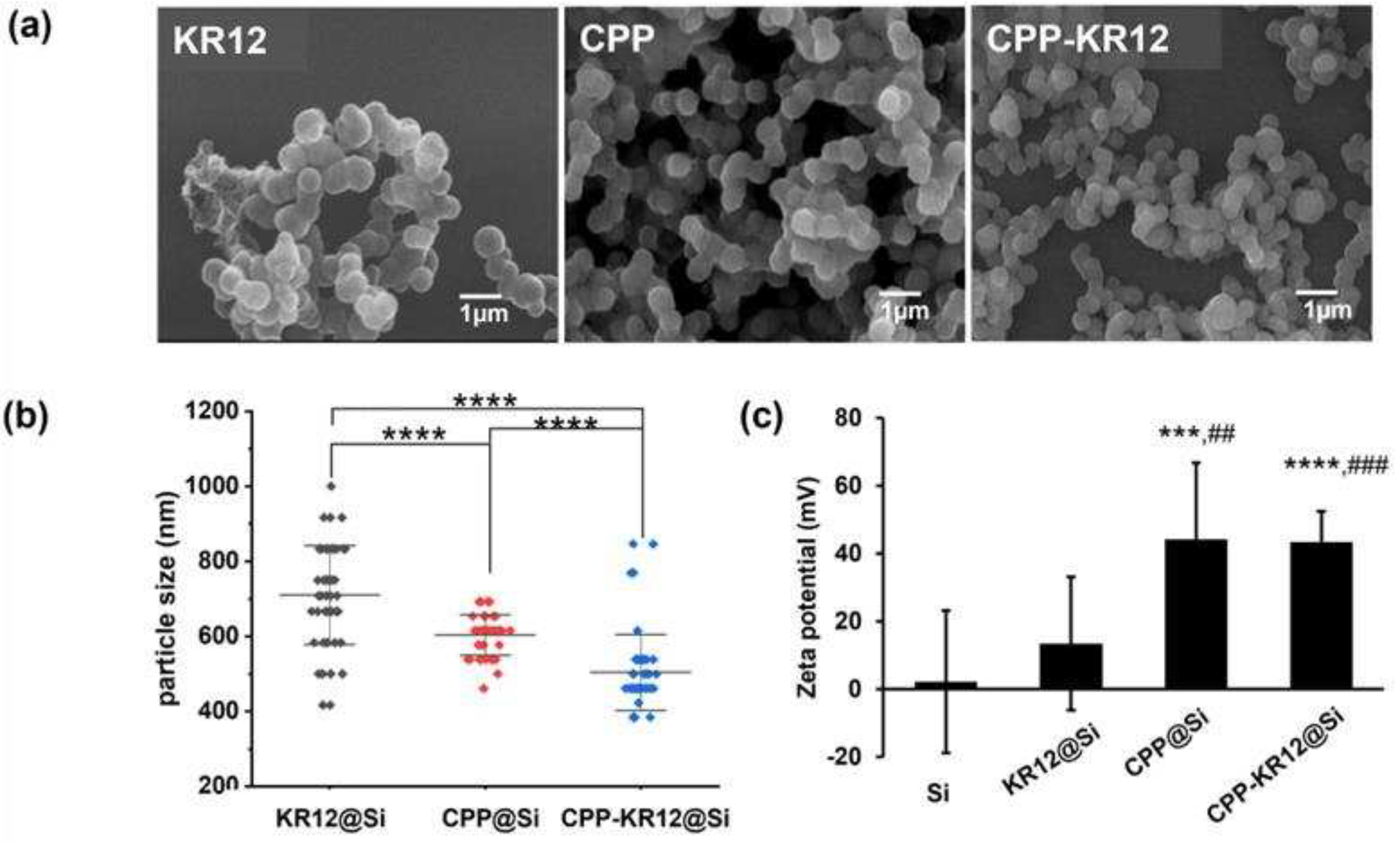
Figure 5.
Fluorometric determination of the relative sensitivity of suspensions of E. coli to free CPP-KR12 and CPP-KR12@Si with SYTOXTM green stain. E. coli (108 CFU/mL) was incubated with 5μM of SYTOXTM green at room temp for 20 min in 1 x PBS. The fluorescence emission spectrum of each 490 nm-excited 100-fold diluted E. coli suspension in the absence and presence of 30 μM of CPP-KR12 or CPP-KR12@Si was acquired for 10 min (a). Delivery of FITC-CPP-KR12 to E. coli (b) and to Raw 264.7 cells (c). Silica particles were labeled by FITC-conjugated corresponding peptides. Cytosol and nuclei of Raw264.7 were stained by CellTracker Red and DAPI, respectively. Raw264.7 cells exposed to FITC-CPP-KR12@Si for 2h (d) and 24h (e). In both (d) and (e), the left panel displays the merged fluorescence of DAPI, CellTracker, and FITC, which helps identify nuclei, cytosol, and particles. On the other hand, the right panel only shows FITC fluorescence, emphasizing the distribution of particles.
Figure 5.
Fluorometric determination of the relative sensitivity of suspensions of E. coli to free CPP-KR12 and CPP-KR12@Si with SYTOXTM green stain. E. coli (108 CFU/mL) was incubated with 5μM of SYTOXTM green at room temp for 20 min in 1 x PBS. The fluorescence emission spectrum of each 490 nm-excited 100-fold diluted E. coli suspension in the absence and presence of 30 μM of CPP-KR12 or CPP-KR12@Si was acquired for 10 min (a). Delivery of FITC-CPP-KR12 to E. coli (b) and to Raw 264.7 cells (c). Silica particles were labeled by FITC-conjugated corresponding peptides. Cytosol and nuclei of Raw264.7 were stained by CellTracker Red and DAPI, respectively. Raw264.7 cells exposed to FITC-CPP-KR12@Si for 2h (d) and 24h (e). In both (d) and (e), the left panel displays the merged fluorescence of DAPI, CellTracker, and FITC, which helps identify nuclei, cytosol, and particles. On the other hand, the right panel only shows FITC fluorescence, emphasizing the distribution of particles.
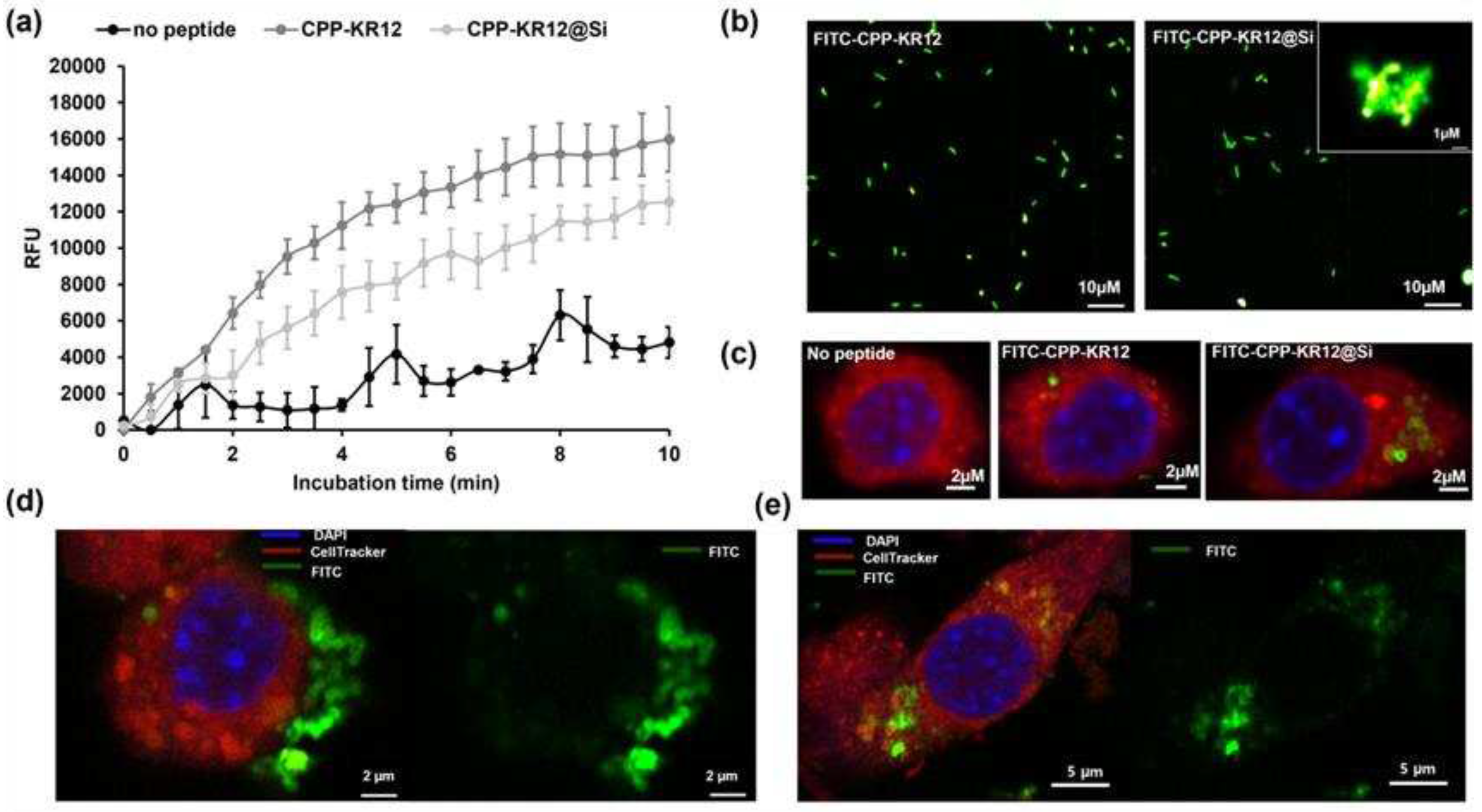
Figure 6.
Comparison of residual antibacterial activity of AMP and AMP entrapped in silica after trypsin treatment and SEM image of S. aureus treated with CPP-KR12@Si. (a) The residual antibacterial activity was calculated by taking the difference in optical density at 600 nm between the negative control and the sample containing the trypsinized AMP. It was then divided by the optical density of the negative control and multiplied by 100. Each bacterial solution grown in a medium without AMP is used as a corresponding negative control. Values are presented as mean ± SE (N = 3). * p <0.05, **p <0.01, or ***p <0.001 vs corresponding free form of AMP in each strain. (b) SEM image of S. aureus treated with CPP-KR12@Si. The image is a 10,000× magnification, the inside image is a 20,000× magnification, and the scale bar represents 1 µm.
Figure 6.
Comparison of residual antibacterial activity of AMP and AMP entrapped in silica after trypsin treatment and SEM image of S. aureus treated with CPP-KR12@Si. (a) The residual antibacterial activity was calculated by taking the difference in optical density at 600 nm between the negative control and the sample containing the trypsinized AMP. It was then divided by the optical density of the negative control and multiplied by 100. Each bacterial solution grown in a medium without AMP is used as a corresponding negative control. Values are presented as mean ± SE (N = 3). * p <0.05, **p <0.01, or ***p <0.001 vs corresponding free form of AMP in each strain. (b) SEM image of S. aureus treated with CPP-KR12@Si. The image is a 10,000× magnification, the inside image is a 20,000× magnification, and the scale bar represents 1 µm.
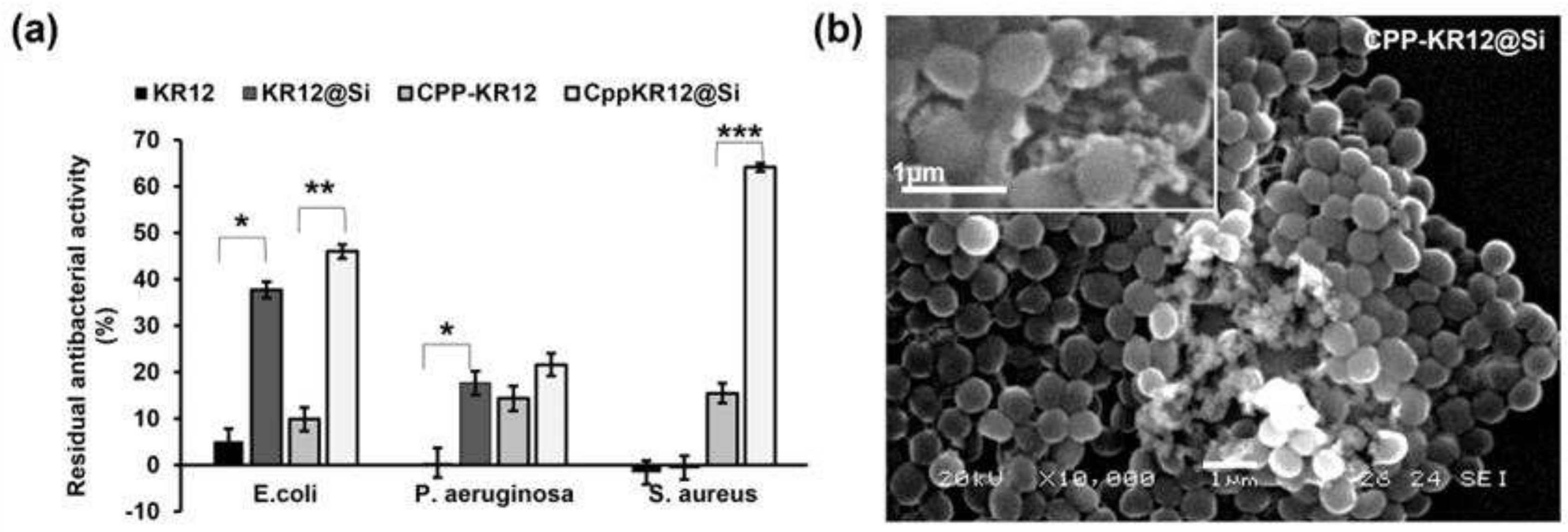
Figure 7.
The hemolytic activity and cytotoxicity of AMPs. (a) Hemolysis (%) is the difference in optical density at 570 nm of sheep red blood cells (RBCs) between sample containing the indicated AMP and the negative control as a percentage of the difference in optical density between the positive and negative controls. RBCs present in PBS alone serve as a negative control, while RBCs treated with 1% Triton X-100 in PBS serve as a positive control. Values are presented as mean ± SE (N = 3). **p <0.01, ***p <0.001 or ****p <0.0001 vs. corresponding free form of AMP in each concentration. (b) The cytotoxicity of AMP was measured 24 h after the addition of each indicated concentration of AMP to Raw 264.7 cells grown overnight. Cell survival was expressed as a percentage of the negative control grown without AMP. Values are presented as mean ± SE (N = 3). ***p <0.001, or ****p <0.0001 vs corresponding free form of AMP in each concentration.
Figure 7.
The hemolytic activity and cytotoxicity of AMPs. (a) Hemolysis (%) is the difference in optical density at 570 nm of sheep red blood cells (RBCs) between sample containing the indicated AMP and the negative control as a percentage of the difference in optical density between the positive and negative controls. RBCs present in PBS alone serve as a negative control, while RBCs treated with 1% Triton X-100 in PBS serve as a positive control. Values are presented as mean ± SE (N = 3). **p <0.01, ***p <0.001 or ****p <0.0001 vs. corresponding free form of AMP in each concentration. (b) The cytotoxicity of AMP was measured 24 h after the addition of each indicated concentration of AMP to Raw 264.7 cells grown overnight. Cell survival was expressed as a percentage of the negative control grown without AMP. Values are presented as mean ± SE (N = 3). ***p <0.001, or ****p <0.0001 vs corresponding free form of AMP in each concentration.
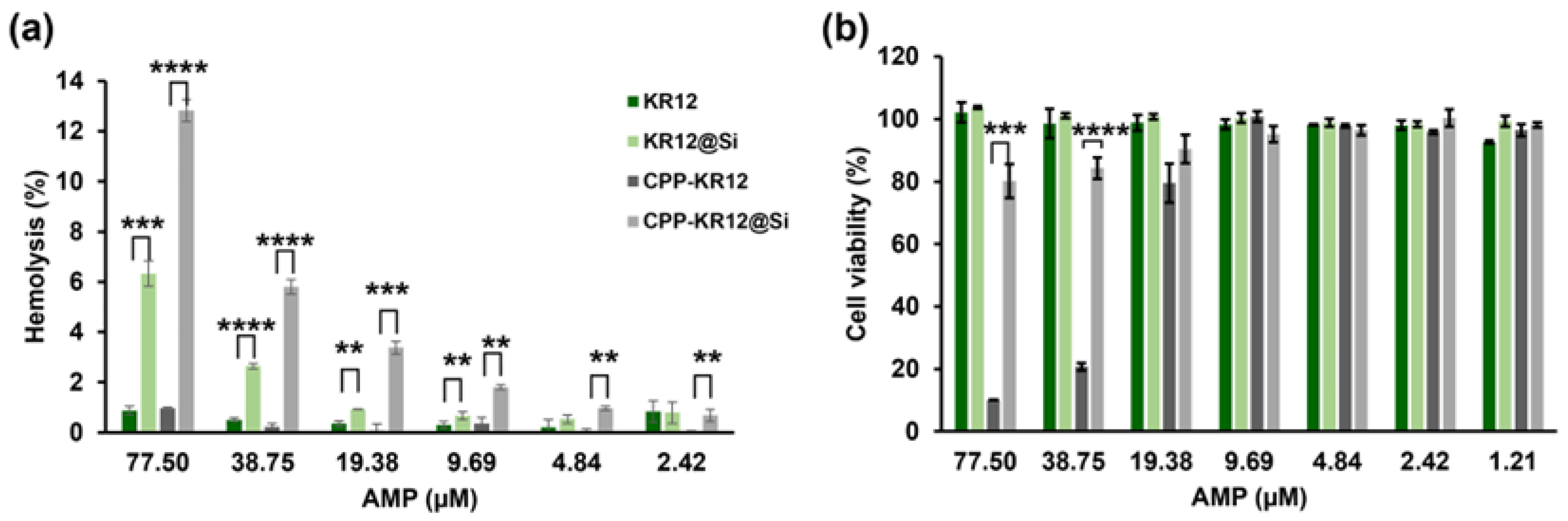
Figure 8.
AMP-device combination and its antimicrobial activity. (a) X-ray photoelectron spectroscopy of as-prepared carriers. Atomic percentages of main elements observed in carriers. The atomic percentages of the constituent were calculated as the ratio of each atom to the sum of the main compounds except carbon. (b) SEM images of AMP-device combination. The image is a 10,000× magnification. Scale bar: 1 μm. and (c) Fluorescence microscope images of E. coli stained with BacLight live/dead kit, which displays green for the live bacteria and red for dead bacteria in medium solutions with the indicated device. Scale bar: 20 μm.
Figure 8.
AMP-device combination and its antimicrobial activity. (a) X-ray photoelectron spectroscopy of as-prepared carriers. Atomic percentages of main elements observed in carriers. The atomic percentages of the constituent were calculated as the ratio of each atom to the sum of the main compounds except carbon. (b) SEM images of AMP-device combination. The image is a 10,000× magnification. Scale bar: 1 μm. and (c) Fluorescence microscope images of E. coli stained with BacLight live/dead kit, which displays green for the live bacteria and red for dead bacteria in medium solutions with the indicated device. Scale bar: 20 μm.
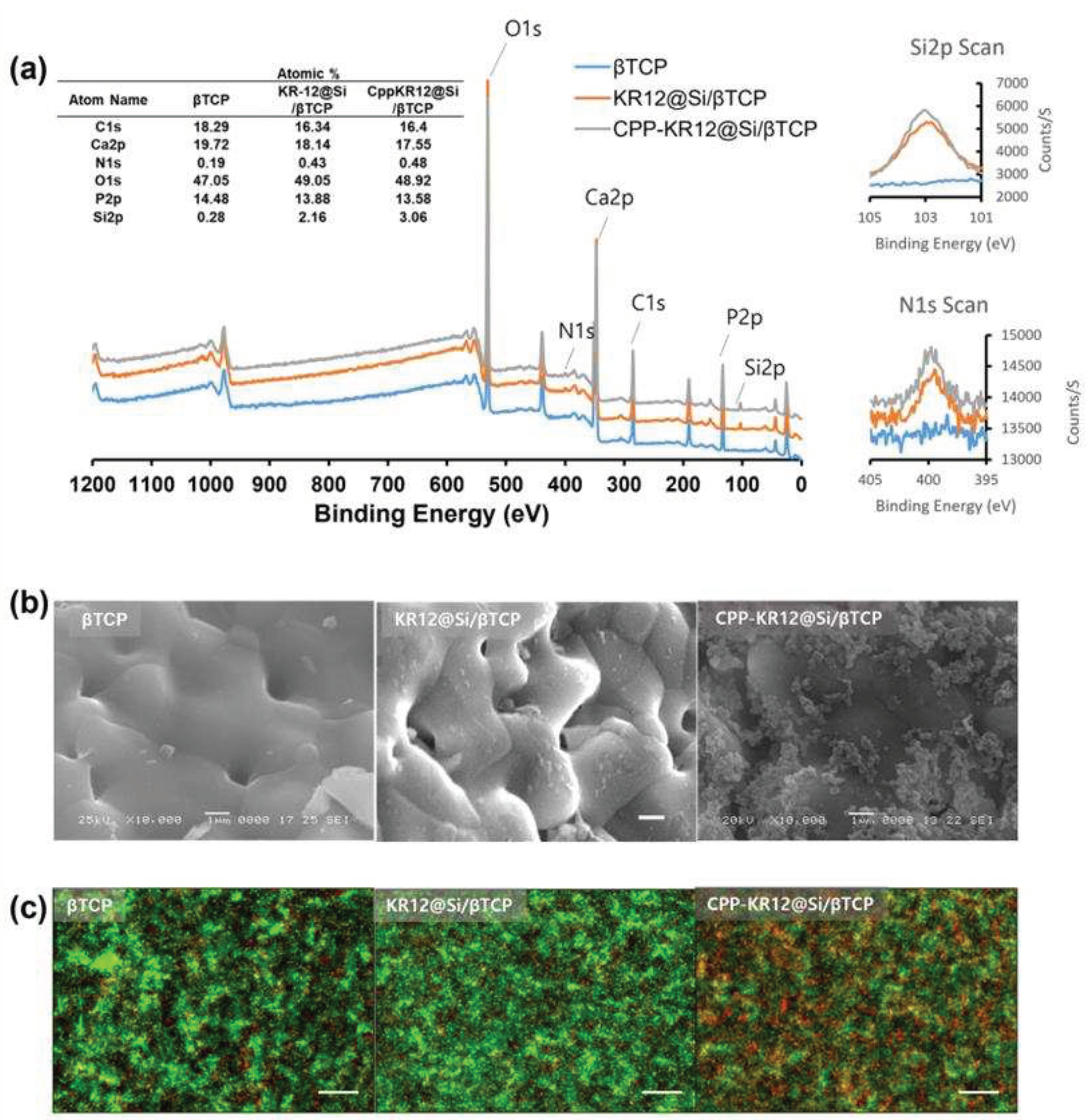

Table 1.
Peptide sequences and their characteristics.
| Name | Sequence (N-C) | AA # | Calculated Mass (Da)1 |
Observed Mass (Da)2 |
pI1 | Net Charge1 |
|---|---|---|---|---|---|---|
| KR12 | KRIVQRIKDFLR | 12 | 1612.00 | 1612.80 | 12.79 | +4 |
| CPP | RKKRRQRRR | 9 | 1379.89 | 1380.00 | 14.00 | +8 |
| CPP-KR12 | RKKRRQRRRGSSKRIVQRIKDFLR | 24 | 3163.94 | 3165.00 | 13.39 | +12 |
| FITC-CPP-KR12 | FITC-Ahx- RKKRRQRRRGSSKRIVQRIKDFLR | 24 | 3624.05 | 3625.92 | 13.39 | +12 |
1The pI value and net charge were calculated by Pepdraw (https://pepdraw.com/). The N-terminus of each peptide was acetylated and the C-terminus was amidated. 2The observed mass was obtained by Mass Spectrum analysis by company. The N-terminus of each peptide was acetylated and the C-terminus was amidated. KR12 and CPP consist of residues 18 to 29 of the human cathelicidin LL-37 [32] and residues 49 to 57 (protein transduction domain) of the HIV Tat proteins [35], respectively. Ahx: 6-aminohexanoic acid; FITC: Fluorescein isothiocyanate.
Table 2.
Minimum Inhibitory Concentration (MIC) of peptides.
| Peptide | E. coli | P. aeruginosa | S. aureus |
|---|---|---|---|
| KR12 | 103.36±25.82 | 181.98±68.20 | > 320a |
| CPP-KR12 | 12.09±4.97 | 6.12±3.72 | 22.80±8.48 |
Minimum Inhibitory Concentration (MIC) was calculated in triplicate and values are expressed as mean ± standard error. The unit is micromoles/L (µM). CPP did not exhibit antibacterial activity against three kinds of bacteria at the highest concentration of 320 µM. aKR12 did not have antibacterial activity against S. aureus at a concentration of 320 µM.
Table 3.
Comparison of LE% and EE%.
| Initial AMP (μg) | Entrapped AMP (μg) | Silica deposition (μg) |
LEa (%) | EEb (%) | |
| KR12@Si | 100 | 56.70±4.71 | 25.14±2.08 | 69.16 | 56.70 |
| CPP@Si | 100 | 95.45±2.06 | 30.29±2.44 | 76.31 | 95.45 |
| CPP-KR12@Si | 100 | 94.45±1.93 | 33.70±2.86 | 73.83 | 94.45 |
a LE is the peptide loading efficiency, which is defined as the percentage of the amount of peptide in the nanoparticle to the total amount of peptide combined with the silica nanoparticle. b EE is the peptide entrapment efficiency, defined as the percentage of the amount of peptide in the nanoparticle to the total amount of peptide initially applied to the formulation.
Table 4.
MIC of AMP@Si.
| Peptide | E. coli | P. aeruginosa | S. aureus |
| KR12@Si | 105.27±28.62 | N.da | 58.16±19.39 |
| CPP-KR12@Si | 14.54±4.89 | 8.40±2.81 | 28.75±16.87 |
MIC was calculated in triplicate and values are expressed as mean ± standard error. The unit is micromolesL-1 (µM). KR12@Si did not exhibit antibacterial activity against P. aeruginosa at the highest concentration of 180 µM. aN.d means ‘not determined’.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



