
Submitted:
09 October 2023
Posted:
09 October 2023
You are already at the latest version
Abstract
We report an unusual transformation where a transient formation of a nitrene moiety initiates a sequence of steps where leading to a remote oxidative C-H functionalization (R-CH3 to R-CH2OC(O)R’) and concomitant reduction of the nitrene into an amino group. No external oxi-dants or reductants are needed for this formal molecular com-proportionation. Detected and iso-lated intermediates and computational analysis suggest that the process occurs with pyrazole ring opening and recyclization.
Keywords:
furoxan
; 1H-pyrazole
; nitrene
; fragmentation
; cyclization
1. Introduction
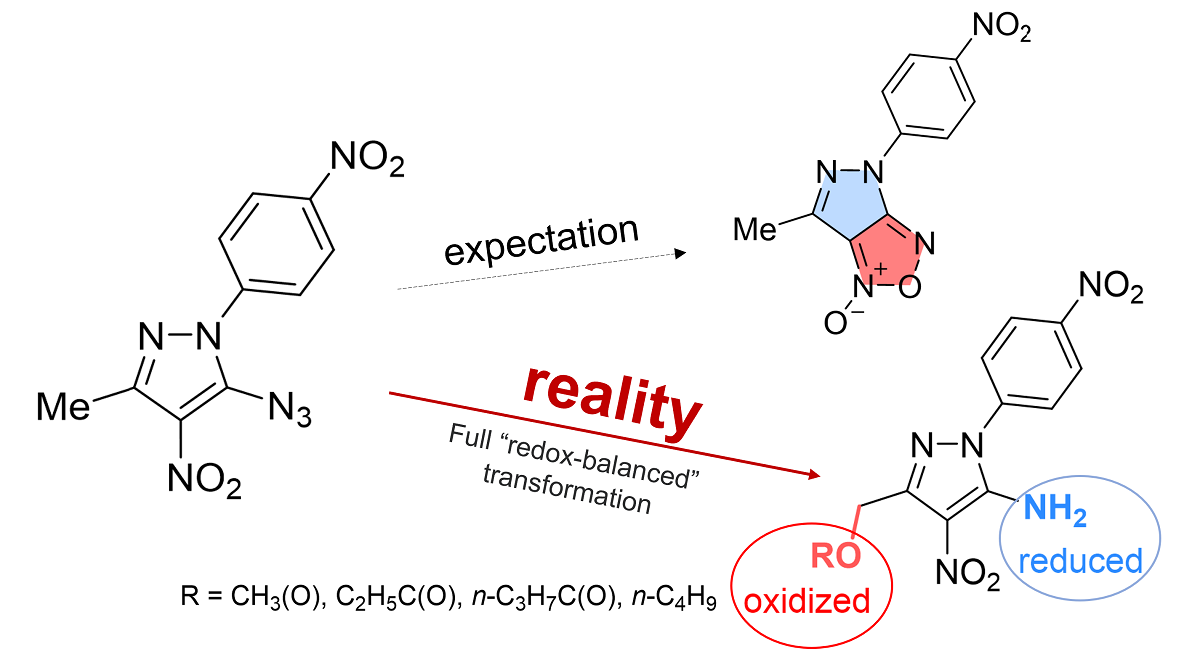
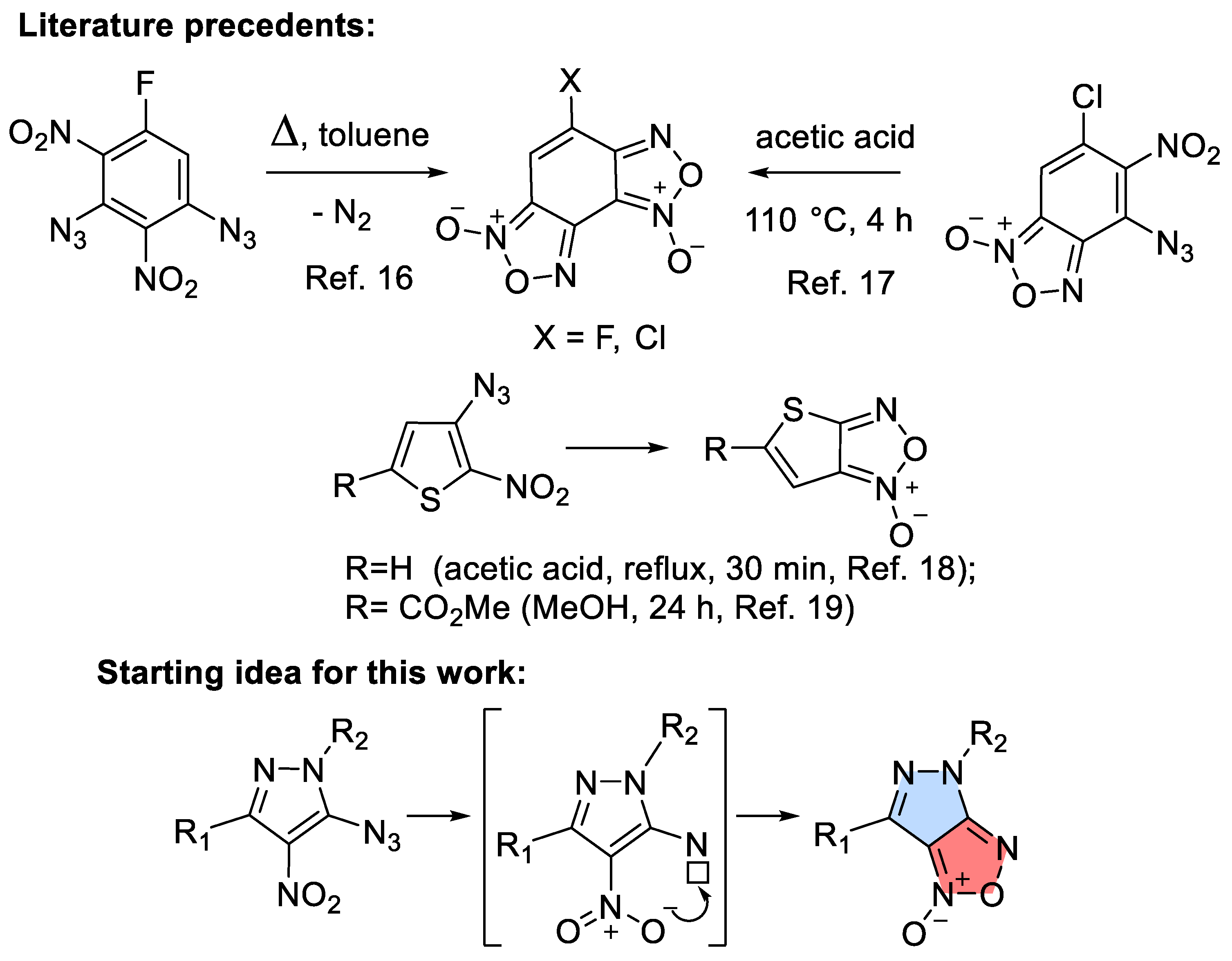
In continuation of our efforts to search for potent anticancer and antimicrobial agents [1,2,3,4], we were interested in synthesizing a previously unknown bis-heterocyclic pyrazolo-furoxan fusion. Pyrazole-based compounds poses antimicrobial, antipyretic, anti-inflammatory and analgesic effects [5,6,7,8]. Furoxan ring-containing molecules exhibit anti-tuberculosis [9], antitumor [10], anti-inflammatory [11], antiaggregant [12], etc. biological activity. For target compounds synthesis we planned to use one of the known methods for obtaining a furoxan cycle – thermolysis of α-nitroazides (Scheme 1) [13,14,15,16,17,18,19] However, when we applied these reaction conditions, the outcome was unexpected. The observed transformation involved a remote methyl group and proceeded as a redox disproportionation where this group was oxidized into a CH2OAc moiety while the azido group was reduced into the amine. Interestingly, the nitro group, which could potentially serve as a relay between the two reacted functionalities, remained unchanged. Considering this surprising outcome, we have explored this new conformation in more detail and wish to report the results of this investigation in this work.
The high energy stored in the azide functionality makes it a powerful tool for many organic transformations [20,21]. In addition, the N3-group can be considered as a convenient amine surrogate, i.e., a form of amine protection, as this group can be easily installed into a molecule and converted into corresponding NH2 later in the synthetic sequence. Several reductive strategies for azide-to-amine transformations include the use of strong reducing agents such as LiAlH4 [22], NaBH4 [23], Zn(BH4)2 [24]. The search for milder conditions led to the development of sulfide chemistry [25,26] or copper(I) catalysis [27]. However, Staudinger reaction with the use of phosphine or phosphite reagents remains the most frequent choice for this reduction [28,29,30,31]. Nevertheless, each of these approaches has its disadvantages, including poor group tolerance, additional synthesis of ligands, or phosphorus(V) waste.
On the other hand, C–H activation in aliphatic functional groups remains challenging and usually requires oxidative conditions. Furthermore, direct esterification of C–H bonds is often limited by the nature of substrates [32,33]. Several oxidative acetoxylation protocols with the in situ generation of hypervalent iodine species have been described [34,35]. The combination of copper- [36,37] or palladium-catalysts [38,39,40,41,42] with external oxidants or electrochemical oxidation [43] also provided a useful tool for AcO-group insertion into alkane moiety. However, these approaches are either incompatible with sensitive functional groups (e.g., unprotected amines) or demand the presence of directing groups. Using acetic acid itself for C–H acetoxylation is unusual and conceptually appealing.
2. Results and Discussion
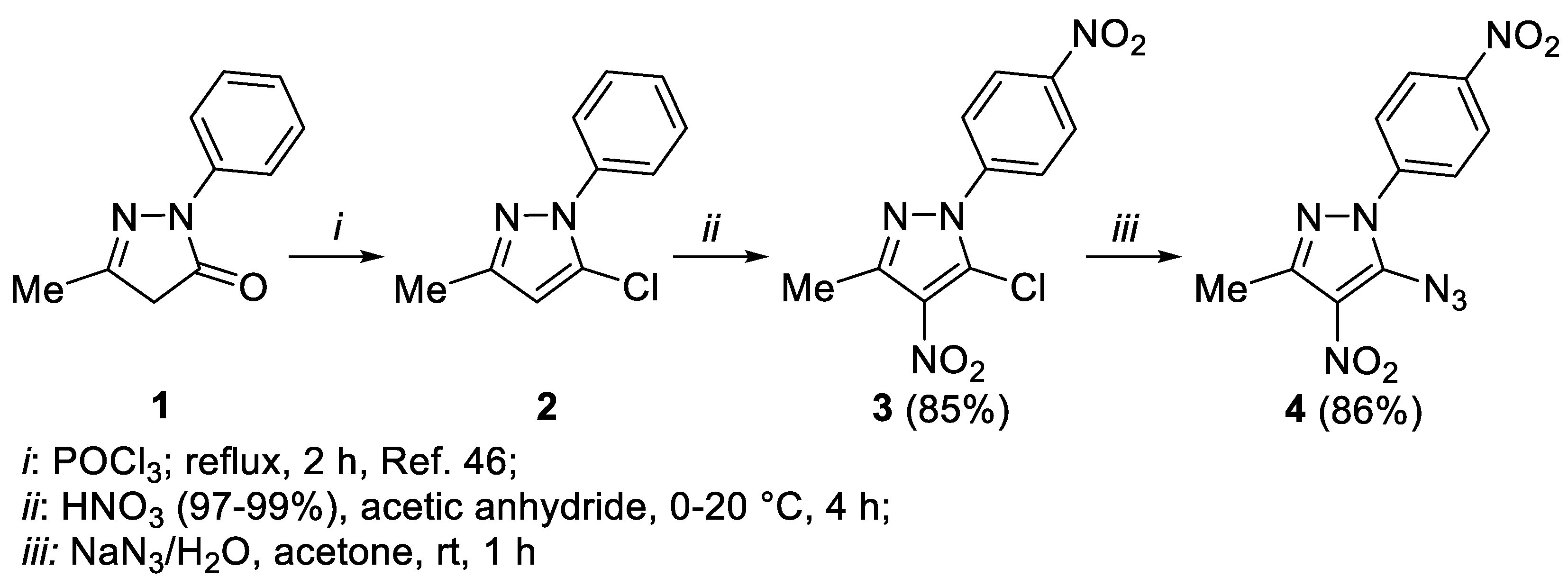
Synthesis of reactants and analysis of products. We started by preparing 5-chloro-3-methyl-1-phenyl-1H-pyrazole 2 from edaravone 1, a medication used to treat stroke and amyotrophic lateral sclerosis [44,45] (Scheme 2) according to the literature method [46]. It was reported that nitration of compound 2 with concentrated nitric acid in acetic anhydride led to nitration at the 4th position of the pyrazole ring with the formation of 5-chloro-3-methyl-4-nitro-1-phenyl-1H-pyrazole [46]. When we used fuming nitric acid (97-99%) it resulted in incorporation of the additional nitro group at the para-position of the phenyl group, allowing us to obtain compound 3.
Reaction of 5-chloro-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole 3 with sodium azide resulted in the facile replacement of chlorine by the azido group. We have shown earlier that the presence of adjacent azide and nitro groups usually leads to cyclization to the furoxan ring upon refluxing in high-boiling solvents [16,17]. In some cases, this reaction proceeds spontaneously at the stage of azide preparation [47].
However, to our surprise, thermolysis of o-azidonitro derivative 4 in acetic acid led to 5-amino-4-nitro-1-(4-nitrophenyl)-1H-pyrazol-3-yl)methyl acetate 5a, instead of the expected cyclization product furoxan. The new product is formed as a result of the reduction of the azido group to amino group and the oxidative conversion of the methyl group to an CH2OAc moiety (Scheme 3). Aminopyrazole derivatives are widely used in organic synthesis as convenient starting reagents for obtaining new annelated heterocycles, which may be of interest as potentially physiologically active compounds [48]. For example, 4-amino-pyrazole-3-carboxylic esters are used as intermediates for the synthesis of Sildenafil (Viagra) and Allopurinol [49].
The reaction of azide 4 with propionic, butyric acids and butanol leads to similar products 5b-d. In all cases, compound 6 was isolated as a by-product in trace amounts.
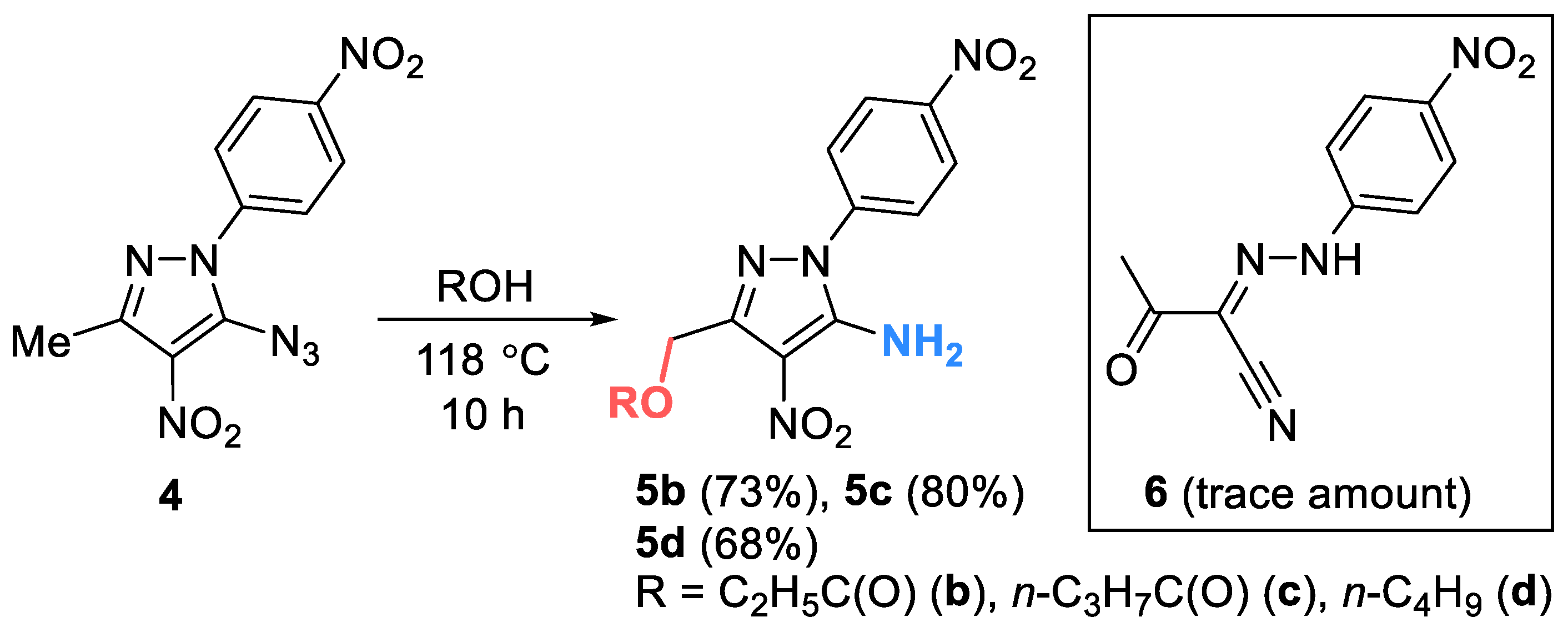
Scheme 4.
Reaction of nitroazide 4 with selected carboxylic acids and butanol.
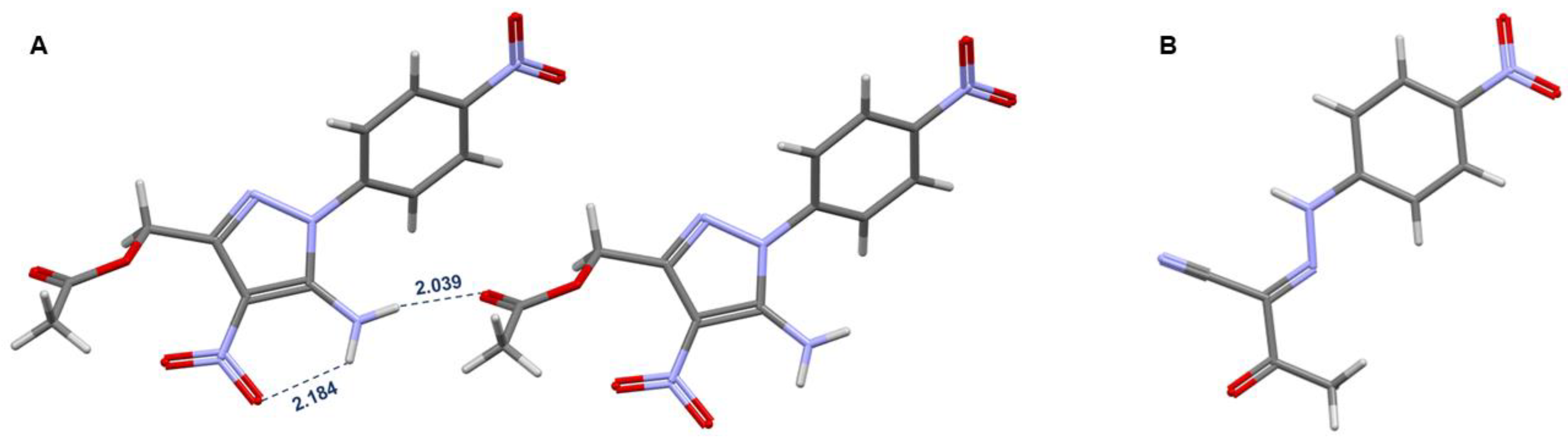
The structures of compounds 3, 5a, and 6 were confirmed by X-Ray Analysis (Figure S13,S14 and Figure 1). Bihetaryl scaffold of 5a is twisted due to steric repulsion in ortho-positions (Figure 1, A). Interplanar and torsion angles between p-nitro-phenylene and pyrazole rings are 37.91º and 36.37º, respectively. Acetoxy-group lies in “gauche”-conformation to diaza-cycle (torsion angle C10C9C12O13 59.42º) where the acceptor C-O bond aligns with the donor heterocycle π-system. Interestingly, NH2 substituent participates in both intra- and intermolecular hydrogen bonding with oxygens of NO2 and C=O groups, respectively.
Hydrazide 6 has a completely planar structure with two cross-conjugated fragments at the NH-moiety (Figure 1, B). A lone pair of the NH nitrogen can participate in conjugation with both substituents, and as a result, the central N-atom is sp2-hybridized. The sum of valence angles at the NH nitrogen equals 360.0º.
Possible mechanism. Considering that this process involves a metal-free C–H activation at relatively mild conditions, its mechanism is intrinsically interesting at it may pave the way to similar C–H activations of a broader range of substrates. The key step is likely to be the formation of the nitro-substituted nitrene. Aryl nitrenes are capable of complex transformations including a variety of ring expansions, fragmentations, and bond insertions [50,51,52,53]. Singlet aryl nitrenes can also be trapped by reactions with internal nucleophiles [54,55,56]. Although Ph-nitrene is known to be a triplet (~15 kcal/mol lower than the open-shell singlet state), the singlet/triplet gap in pyrazol nitrene has not been studied.
Once the pyrazole nitrene 7 is formed, it can potentially react in several ways. Direct intramolecular attack of an oxygen from the nitro-group leads to neutral bicyclic species 8, which can be converted into 9 by protonation. Alternatively, pyrrole nitrogen can increase electron density at the nitrene via resonance, facilitating protonation with the formation of a nitrenium ion (not shown) which may also cyclize with the formation of fused bicyclic cation 9. When 9 is formed, the mechanistic path can diverge. Here, we have considered two possibilities. First, intramolecular activation of methyl substituent by electron-deficient oxygen in 9 may enable hydride transfer, allowing for the formation of a stabilized carbocation 10. The nucleophilic attack of ROH at the cationic carbon, followed by proton transfer and the furoxan ring opening, would form the final product 5. An interesting feature of this path would be that nitrene would provide transient activation of the nitro group, rendering it an even stronger hydride acceptor. However, computational analysis at the M062x/6-31+G (d,p) level (with AcOH as solvent) suggested that the barrier for such hydride shift is prohibitively large (67 kcal/mol) and the carbocationic intermediate 10 is nearly 25 kcal/mol higher in energy than 9.
In an alternative intramolecular activation path, prototropic tautomerization of cation 9 in acidic media would lead to the dearomatizing methyl-methylene tautomeric transformation 9→13. ROH attack on alkene fragment can synchronously open the furoxan ring and unmask the nitro and imine groups of 14. Subsequent proton transfers reestablish aromaticity with the formation of pyrazole 15. Deprotonation of 15 would lead to the observed product 5. We consider this path unfavorable due to the >15 kcal/mol penalty for the loss of aromaticity in 13. Furthermore, the formation of the key precursor, i.e., the bicyclic cation 9 by protonation of neutral 8 by acetic acid is also predicted to be >50 kcal/mol uphill.
Based on these considerations and the formation of an acyclic side product 6, we explored the possibility of ring opening as the first step in the reaction sequence. Here, we were guided by the literature precedent as 5-azidopyrazoles are known to undergo ring opening processes with the formation of cyano-group after nitrogen loss upon heating [57,58]. Furthermore, our attempts to optimize the singlet state of nitrene 7 led to its barrierless Grob fragmentation with the formation of acyclic nitrile 16.
Protonation of intermediate 16 can lead to the formation of hydrazonium cation 17 which may undergo cyclization to N-amino-substituted aziridine cation 18. Due to hybridization effects [59] and inverse α-effect [60,61,62], such cations are expected to be quite reactive and should undergo quick capture by an external nucleophile (18→19). Collapse of amino acetal with the concomitant aziridine ring opening in 19 relieves the transient strain. Hydrazine lone pair then assists in elimination of the NO2 group to reestablish conjugation and, after deprotonation, forms the isolated byproduct 6.
Furthermore, the same intermediate 16 can undergo isomerization into alkene 20 which after nucleophilic addition of ROH can undergo a 5-exo-trig ring closure that recreates the heterocyclic moiety. Protonation of the cyclic nitronate 21 is coupled with aromatization and formation of the final product 5.
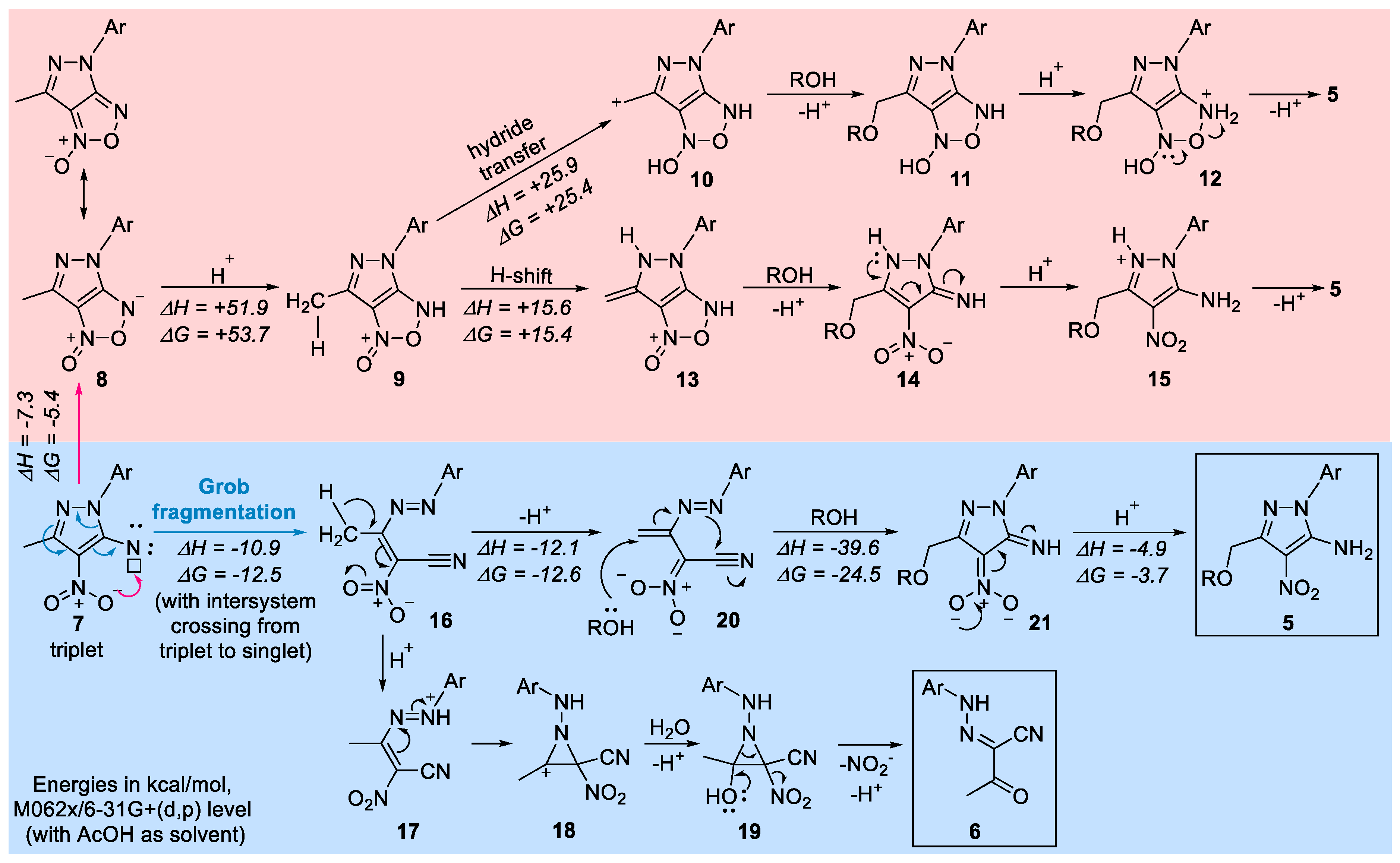
Scheme 5.
Suggested mechanism for the formation of unusual products in the attempted synthesis of pyrazole/furoxan hybrid. Pathways discarded from the computational evidence are shown at the red background. The suggested plausible path is shown in blue.
Scheme 5.
Suggested mechanism for the formation of unusual products in the attempted synthesis of pyrazole/furoxan hybrid. Pathways discarded from the computational evidence are shown at the red background. The suggested plausible path is shown in blue.
In conclusion, we report an interesting “redox-balanced” transformation where two functional groups separated in space undergo simultaneous redox transformations in the opposite directions: the Me group is oxidized while the nitrene moiety is reduced. The absence of usual furoxan products in this case can be attributed to the combination of two factors: the lower aromaticity of pyrazole relative to benzene [63,64] and accumulation of strain upon fusion of the two five-membered rings [65]. The interplay of electronic effects due to the presence of multiple nitrogen atoms in pyrazole activates the fast Grob fragmentation into functionally rich acyclic nitro nitrile 16 which can recyclize after prototropic isomerization and Michael-like addition of AcOH.
3. Materials and Methods
Chemistry
IR spectra were recorded as emulsion in vaseline oil (sample concentration 0.25%) on a Tensor 37 Vertex 70 RAM II spectrometer (Bruker Optik GmbH, Germany) in the range 400–4000 сm−1; given are the most intense absorption bands. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE 400 spectrometer (Bruker BioSpin, Rheinstetten, Germany) operating at 400 MHz (for 1H NMR) and 101 MHz (for 13C NMR) and on Brucker spectrometers AVANCEIII-500 (Bruker BioSpin, Rheinstetten, Germany) operating at 500.1 MHz for 1Hat 303 K and 126 MHz (for 13C NMR). Chemical shifts were measured in δ (ppm) with reference to the solvent (δ = 7.27 ppm and 77.00 ppm for CDCl3, δ = 2.06 ppm and 28.94 ppm for (CD3)2CO for 1H and 13C NMR, respectively). Electrospray ionization (ESI) mass spectra were obtained on an Amazon X mass spectrometer from Bruker Daltonics (Bremen, Germany) with an ion trap. The measurements were carried out in the mode of recording negative ions in the m/z range from 100 to 2000. Elemental analysis was performed on a CHNS-O Elemental Analyser EuroEA3028-HT-OM (EuroVector S.p.A., Milan, Italy) with an accuracy ±0.4% for C, H, Cl and N. The melting point was determined in glass capillaries on a Stuart SMP 10 instrument (Keison Products, Chelmsford, UK). The progress of reactions and the purity of product were monitored by TLC on Sorbfil UV-254 plates (Sorbpolimer, Krasnodar, Russia); the chromatograms were developed under UV light.
X-ray crystallography data. The data set for the single crystal 3 and 6 were collected on a Bruker Quest diffractometer using graphite monochromated MoKα (0.71073 Å) radiation and ω-scan rotation. Data collection: images were indexed, integrated, and scaled using the APEX2 [66] data reduction package and corrected for absorption using SADABS [67]. The structure 3 were solved by the direct methods and refined using SHELX [68]. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were calculated on idealized positions and refined as riding atoms. The X-ray analysis was performed on the equipment of Spectral-Analytical Center of FRC Kazan Scientific Center of RAS.
Crystallographic data for compound 3: C10H7ClN4O4, M 282.65, monoclinic, P21/c, a 3.7604(1), b 32.2193(11), c 9.0673(3) Å,β93.985(1)°, V 1095.92(6) Å3, Z 4, Dcalcd 1.713g·cm–3, μ(Mo-Kα) 0.367 mm–1, F(000) 576, (θ 1.3- 27.9°, completeness 99.9%), T 100(2) К, orange prism, (0.11 x 0.17 x 0.56) mm3,transmission 0.6946 - 0.7456, 39950 measured reflections, 2597 independent (Rint 0.044), 173 parameters, R1 = 0.0367 (for 2379 observed I> 2σ(I)) , wR2 = 0.1435 (all data), GOOF 1.05, largest diff. peak and hole 0.50 and -0.40 e.A-3. CCDC number 2299127.
Crystallographic data for compound 6: C10H8N4O3, M 232.20, monoclinic, P21/n, a 7.0578(14), b 13.434(3), c 11.160(2) Å,β 96.736(6)°, V 1050.8(4) Å3, Z 4, Dcalcd 1.468g·cm–3, μ(Mo-Kα) 0.113 mm–1, F(000) 480, (θ 2.4 - 27.9°, completeness 99.8%), T 162(2) К, orange needle, (0.04 x 0.05 x 0.15) mm3, transmission 0 0.6291 - 0.7456, 39950 measured reflections, 23472 independent (Rint 0.273), 155 parameters, R1 = 0.0852 (for 1013 observed I> 2σ(I)) , wR2 = 0.2504 (all data), GOOF 0.941, largest diff. peak and hole 0.31 and -0.28 e.A-3. CCDC number 2299128.
The data set for the single crystal 5a were collected on a Rigaku Synergy S instrument (Rigaku Oxford diffraction, Tokyo, Japan) with a HyPix detector and a PhotonJet microfocus X-ray tube using Cu Kα (1.54184 Å) radiation at a low temperature. Images were indexed and integrated using the CrysAlisPro data reduction package. Data were corrected for systematic errors and absorption using the ABSPACK module: numerical absorption correction based on Gaussian integration over a multifaceted crystal model and empirical absorption correction based on spherical harmonics according to the point group symmetry using equivalent reflections. The GRAL module was used for the analysis of systematic absences and space group determination. The structure was solved by direct methods using SHELXT [69] and refined by the full-matrix least-squares on F2 using SHELXL [70]. Non-hydrogen atoms were refined anisotropically. The hydrogen atoms were inserted at the calculated positions and refined as riding atoms. The figures were generated using the Mercury v4.1 [71] program. Crystals were obtained by the slow evaporation method.
Crystallographic data for compound 5a: C12H11N5O6 (M =321.26 g/mol): triclinic, space group P-1 (no. 2), a = 7.7774(2) Å, b = 9.7999(2) Å, c = 9.96110(10) Å, α = 102.833(2)°, β = 111.543(2)°, γ = 92.620(2)°, V = 681.72(3) Å3, Z = 2, T = 110.0(5) K, μ(Cu Kα) = 1.107 mm-1, Dcalc = 1.565 g/cm3, 7375 reflections measured (9.348° ≤ 2Θ ≤ 153.212°), 2745 unique (Rint = 0.0229, Rsigma = 0.0227) which were used in all calculations. The final R1 was 0.0347 (I > 2σ(I)) and wR2 was 0.0938 (all data). CCDC number 2294752.
CCDC 2299127, 2299128, 2294752 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or deposit@ccdc.cam.uk).
5-Chloro-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole (3). Acetic anhydride (6 mL) was added to 5-chloro-3-methyl-1-phenyl-1H-pyrazole 2 (0.44 g, 2.3 mmol), reaction mixture was cooled to 0 °C and then fuming nitric acid (97–99%, 4 mL) was added dropwise. Reaction mixture was stirred at room temperature for 4 h, then poured over crushed ice. The obtained precipitate was filtered off, washed with cold water (100 mL) and dried under vacuum (0.06 mm Hg) at 40 °C to constant weight. Crude product was recrystallized from acetone to give target compound. Yellow powder, yield 0.55 g (85%), m.p.: 148–150 °С. IR (ν, cm–1): 1346 (NO2 symm), 1530 (NO2 аsymm). 1H NMR (500 MHz, CDCl3): δ = 8.41 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.6 Hz, 2H), 2.64 (s, 3H). 13C NMR (126 MHz, CDCl3): δ = 148.7, 147.9, 141.7, 131.2, 128.2, 126.0, 125.0, 14.7. Anal. calcd (%) for C10H7ClN4O4: C, 42.50; H, 2.50; Cl, 12.54; N, 19.82. Found: C, 42.54; H, 2.48; Cl, 12.53; N, 19.85.
5-Azido-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole (4). To a solution of 5-chloro-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole 3 (0.50 g, 1.8 mmol) in acetone (5 mL) at room temperature was added a solution of sodium azide (0.15 g, 2.3 mmol) in 1 mL of water. The reaction mixture was stirred for 1 h (the reaction was monitored by thin layer chromatography, eluent: toluene – ethylacetate (2:1, v/v)). After completion of the reaction, the solvent was removed under reduced pressure, washed with cold water and dried in vacuum (0.06 mm Hg) at 40 ˚С to constant weight. Light brown powder, yield 0.45 g (86%), m.p.: 104–106 °С. IR (ν, cm–1): 1349 (NO2 symm), 1557 (NO2 аsymm), 2151 (N3). 1H NMR (500 MHz, Acetone-d6): δ = 8.40–8.43 (m, 2H), 8.07–8.11 (m, 2H), 2.55 (s, 3H). 13C NMR (101 MHz, Acetone-d6): δ = 147.9, 147.6, 142.5, 138.5, 126.2, 125.2, 125.1, 14.4. Anal. calcd (%) for C10H7N7O4: C, 41.53; H, 2.44; N, 33.90. Found: C, 41.58; H, 2.47; N, 33.87.
Synthesis of compounds 5a-d (general method). 5-Azido-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole 4 (0.1 g, 0.34 mmol) was heated in 3 mL of acid/butanol at 118 ˚С for 5 h (for acetic acid) or 10 h (for propionic, butyric acids and butanol). Then solvent was removed under reduced pressure. In case of propionic, butyric acids and butanol сrude product was purified by column chromatography on silica gel (eluent: toluene – ethylacetate (10:1, v/v)) to give the target compound (the side-product 6 was isolated in trace amount).
5-Amino-4-nitro-1-(4-nitrophenyl)-1H-pyrazol-3-yl)methyl acetate (5a). Gray pearlescent solid (0.08 g) was obtained in 78% yield. M.p.: 198–199 °С. IR (ν, cm–1): 1346 (NO2 symm), 1599 (NO2 аsymm), 1637 (CO), 1721 (С=O), 3408 (NH2). 1H NMR (400 MHz, Acetone-d6): δ = 8.46 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.35 (br.s, 2H), 5.33 (s, 2H), 2.08 (s, 3H). 13C NMR (101 MHz, Acetone-d6): δ = 169.6, 147.1, 144.7, 142.3, 125.1, 124.9, 116.9, 58.7, 19.6. Anal. calcd (%) for C12H11N5O6: C, 44.87; H, 3.45; N, 21.80. Found: C, 44.83; H, 3.52; N, 21.85. ESI, m/z for C12H11N5O6: 319.99 [M-H]-.
(5-Amino-4-nitro-1-(4-nitrophenyl)-1H-pyrazol-3-yl)methyl propionate (5b). Orange oil, yield 0.085 g (73%). IR (ν, cm–1): 1347 (NO2 symm), 1598 (NO2 аsymm), 1635 (CO), 1738 (С=O), 3430 (NH2). 1H NMR (400 MHz, Acetone-d6): δ = 8.38–8.42 (m, 2H), 7.91–7.97 (m, 2H), 7.36 (br.s, 2H), 5.31 (s, 2H), 2.38 (q, J = 7.6 Hz, 2H), 1.11 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, Acetone-d6): δ = 173.9, 147.9, 145.7, 143.1, 125.9(4), 125.9(1), 125.6, 117.7, 59.5, 27.7, 9.4. Anal. calcd (%) for C13H13N5O6: C, 46.57; H, 3.91; N, 20.89. Found: C, 46.72; H, 4.02; N, 20.92. ESI, m/z for C13H13N5O6: 334.04 [M-H]-.
(5-Amino-4-nitro-1-(4-nitrophenyl)-1H-pyrazol-3-yl)methyl butyrate (5c). Orange oil, yield 0.096 g (80%). IR (ν, cm–1): 1347 (NO2 symm), 1598 (NO2 аsymm), 1634 (CO), 1734 (С=O), 3434 (NH2). 1H NMR (600 MHz, Acetone-d6): δ = 8.42–8.48 (m, 2H), 7.95–8.01 (m, 2H), 7.36 (br.s, 2H), 5.33 (s, 2H), 2.35 (t, J = 7.4 Hz, 2H), 1.65 (q, J = 7.4 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, Acetone-d6): δ = 173.1, 148.0, 147.9, 145.7, 143.2, 125.9(5), 125.8(9), 125.7, 59.4, 36.3, 19.1, 13.8. Anal. calcd (%) for C14H15N5O6: C, 48.14; H, 4.33; N, 20.05. Found: C, 48.20; H, 4.37; N, 20.01. ESI, m/z for C14H15N5O6: 348.05 [M-H]-.
3-(Butoxymethyl)-4-nitro-1-(4-nitrophenyl)-1H-pyrazol-5-amine (5d). Orange oil, yield 0.075 g (68%). IR (ν, cm–1): 1346 (NO2 symm), 1598 (NO2 аsymm), 1634 (CO), 1702 (С=O), 3430 (NH2). 1H NMR (400 MHz, Acetone-d6): δ = 8.41–8.45 (m, 2H), 7.94–7.99 (m, 2H), 7.29 (br.s, 2H), 4.68 (s, 2H), 3.59 (t, J = 6.5 Hz, 2H), 1.62–1.53 (m, 2H), 1.48–1.32 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, Acetone-d6): δ = 147.9, 147.7(4), 147.6(8), 143.4, 125.9, 125.6, 117.9, 71.3, 65.9, 32.5, 19.9, 14.1. Anal. calcd (%) for C14H17N5O5: C, 50.15; H, 5.11; N, 20.89. Found: C, 50.23; H, 5.17; N, 20.82. ESI,) m/z for C14H17N5O5: 334.08 [M-H]-.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, p. 2-3 – experimental section; Figures S1–S12 (p. 4-9) – copies of NMR spectra of all synthesized compounds; S13-S14 (p. 10) – the X-ray diffraction data of compound 3; p. 11-18 – computational data; p. 19 – references for computational data.
Author Contributions
E.C., A.G. — investigation (chemistry), writing—original draft preparation, supervision (chemistry), N.Ak., V.M. — investigation (chemistry), R.Z., N.Ap. — funding acquisition, A.B. — project administration, D.I., A.D. — investigation (X-Ray), D.T — writing—original draft preparation, B.K.C., K.C. — investigation (quantum-chemical computations), I.A. — writing—original draft preparation, project administration. All authors have read and agreed to the published version of the manuscript.
Funding
Research was funded by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan (Grant No. AP19677249). The synthesis was carried out at Arbuzov Institute of Organic and Physical Chemistry and was supported by the Ministry of Science and Higher Education of the Russian Federation at FRC Kazan Scientific Center (grant No. 075-15-2022-1128). X-Ray Analysis was partially funded by the government assignment for the FRC Kazan Scientific Center of RAS.
Acknowledgments
The authors are grateful to the Assigned Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for technical assistance in research. B. K. Chabuka acknowledges the support by the ACM SIGHPC computational and data science fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of all described compounds are available from the author E. Chugunova.
References
- Sondhi, S.M.; Kumar, S.; Kumar, N.; Roy, P. Synthesis anti-inflammatory and anticancer activity evaluation of some pyrazole and oxadiazole derivatives. Med. Chem. Res. 2012, 21, 3043–3052. [Google Scholar] [CrossRef]
- Wei, Z.-Y.; Liu, J.-C.; Zhang, W.; Li, Y.-R.; Li, C.; Zheng, C.-J.; Piao, H.-R. Synthesis and Antimicrobial Evaluation of (Z)-5-((3-phenyl-1H-pyrazol-4-yl)methylene)-2-thioxothiazolidin-4-one Derivatives. Med. Chem. 2016, 12, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Chandrakantha, B.; Isloor, A.M.; Shetty, P.; Isloor, S.; Malladi, S.; Fun, H.K. Synthesis, characterization and antimicrobial activity of novel ethyl 1-(N-substituted)-5-phenyl-1H-pyrazole-4-carboxylate derivatives. Med. Chem. Res. 2012, 21, 2702–2708. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. ; Shamsuzzaman Review: biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Bennani, F.E.; Doudach, L.; El Rhayam, Y.; Karrouchi, K.; Cherrah, Y.; Tarib, A.; Ansar, M.; Faouzi, M.E.A. Identification of the new progress on Pyrazole Derivatives Molecules as Antimicrobial and Antifungal Agents. West Afr. J. Med. 2022, 39, 1217–1244. [Google Scholar] [PubMed]
- Pasin, J.S.M.; Ferreira, A.P.O.; Saraiva, A.L.L.; Ratzlaff, V.; Andrighetto, R.; Machado, P.; Marchesan, S.; Zanette, R.A.; Bonacorso, H.G.; Zanatta, N.; et al. Antipyretic and antioxidant activities of 5-trifluoromethyl-4,5-dihydro-1H-pyrazoles in rats. Brazilian J. Med. Biol. Res. 2010, 43. [Google Scholar] [CrossRef]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; Dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Saad, H.A.; Osman, N.A.; Moustafa, A.H. Synthesis and Analgesic Activity of Some New Pyrazoles and Triazoles Bearing a 6,8-Dibromo-2-methylquinazoline Moiety. Molecules 2011, 16, 10187–10201. [Google Scholar] [CrossRef]
- dos Santos Fernandes, G.F.; de Souza, P.C.; Marino, L.B.; Chegaev, K.; Guglielmo, S.; Lazzarato, L.; Fruttero, R.; Chung, M.C.; Pavan, F.R.; dos Santos, J.L. Synthesis and biological activity of furoxan derivatives against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2016, 123, 523–531. [Google Scholar] [CrossRef]
- Zhang, Z.; Bai, Z.-W.; Ling, Y.; He, L.-Q.; Huang, P.; Gu, H.-X.; Hu, R.-F. Design, synthesis and biological evaluation of novel furoxan-based coumarin derivatives as antitumor agents. Med. Chem. Res. 2018, 27, 1198–1205. [Google Scholar] [CrossRef]
- Abdelall, E.K.A. Synthesis and biological evaluations of novel isoxazoles and furoxan derivative as anti-inflammatory agents. Bioorg. Chem. 2020, 94, 103441. [Google Scholar] [CrossRef] [PubMed]
- Ustyuzhanina, N.E.; Fershtat, L.L.; Gening, M.L.; Nifantiev, N.E.; Makhova, N.N. New insight into the antiaggregant activity of furoxans. Mendeleev Commun. 2016, 26, 513–515. [Google Scholar] [CrossRef]
- Ritter, H.; Licht, H.H. Synthesis and reactions of dinitrated amino and diaminopyridines. J. Heterocycl. Chem. 1995, 32, 585–590. [Google Scholar] [CrossRef]
- Fogel’zang, A.E.; Egorshev, V.Y.; Sinditskii, V.P.; Dutov, M.D. Organic azide structure and combustion trends. Combust. Explos. Shock Waves 1990, 26, 558–564. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Yao, Q. Synthesis and quantitative structure–activity relationship (QSAR) analysis of some novel oxadiazolo[3,4-d]pyrimidine nucleosides derivatives as antiviral agents. Bioorg. Med. Chem. Lett. 2015, 25, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Jovené, C.; Jacquet, M.; Chugunova, E.A.; Kharlamov, S.V.; Goumont, R. Synthesis and 1-oxide/3-oxide interconversion of 4-substituted benzodifuroxans: A thorough NMR and theoretical study of the structure of 4-fluoro- and 4-chloro-benzodifuroxan. Tetrahedron 2016, 72. [Google Scholar] [CrossRef]
- Chugunova, E.A.; Voloshina, A.D.; Mukhamatdinova, R.E.; Serkov, I.V.; Proshin, A.N.; Gibadullina, E.M.; Burilov, A.R.; Kulik, N.V.; Zobov, V.V.; Krivolapov, D.B.; et al. The Study of the Biological Activity of Amino-Substituted Benzofuroxans. Lett. Drug Des. Discov. 2014, 11, 502–512. [Google Scholar] [CrossRef]
- Boulton, A.J.; Middleton, D. Furazans and furazan oxides. V. Tropono[4,5-c]-, thieno[2,3-c]-, and biphenyleno[2,3-c]furazan oxides. J. Org. Chem. 1974, 39, 2956–2962. [Google Scholar] [CrossRef]
- Noto, R.; Rainieri, R.; Arnone, C. Effect of the nature of the starting aromatic ring on the cyclization of o-nitroaryl azides: kinetic and thermodynamic studies of the conversion of two azido(methoxycarbonyl)nitrothiophenes into methoxycarbonylthienofurazan oxides. J. Chem. Soc.{,} Perkin Trans. 2 1989, 127–130. [CrossRef]
- Rostovtsev, V. V; Green, L.G.; Fokin, V. V; Sharpless, K.B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Pearson, T.J.; Shimazumi, R.; Driscoll, J.L.; Dherange, B.D.; Park, D.-I.; Levin, M.D. Aromatic nitrogen scanning by ipso-selective nitrene internalization. Science (80-. ). 2023, 381, 1474–1479. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.H. Reduction of Organic Azides to Primary Amines with Lithium Aluminum Hydride. J. Am. Chem. Soc. 1951, 73, 5865–5866. [Google Scholar] [CrossRef]
- Rolla, F. Sodium borohydride reactions under phase-transfer conditions: reduction of azides to amines. J. Org. Chem. 1982, 47, 4327–4329. [Google Scholar] [CrossRef]
- Ranu, B.C.; Sarkar, A.; Chakraborty, R. Reduction of Azides with Zinc Borohydride. J. Org. Chem. 1994, 59, 4114–4116. [Google Scholar] [CrossRef]
- Becher Krystian; Krake, Niels; Brøndum, Klaus; Christensen, Nils Just; Vinader, Maria Victoria, J.P. Syntheses of o-Aminohetarenecarbaldehydes via Azides. Synthesis (Stuttg). 1989, 1989, 530–533. [Google Scholar] [CrossRef]
- Kale, A.; Medishetti, N.; Kanugala, S.; C, G.K.; Atmakur, K. Na2S-promoted reduction of azides in water: synthesis of pyrazolopyridines in one pot and evaluation of antimicrobial activity. Org. Biomol. Chem. 2019, 17, 3186–3194. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, B.; Besora, M.; Monasterio, Z.; Ventura-Espinosa, D.; White, A.J.P.; Maseras, F.; Díez-González, S. Copper-mediated reduction of azides under seemingly oxidising conditions: catalytic and computational studies. Catal. Sci. Technol. 2018, 8, 5763–5773. [Google Scholar] [CrossRef]
- Staudinger, H.; Meyer, J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 2, 635–646.
- van Kalkeren, H.A.; Bruins, J.J.; Rutjes, F.P.J.T.; van Delft, F.L. Organophosphorus-Catalysed Staudinger Reduction. Adv. Synth. Catal. 2012, 354, 1417–1421. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Lenting, P.E.; Mecinović, J. Sustainable organophosphorus-catalysed Staudinger reduction. Green Chem. 2018, 20, 4418–4422. [Google Scholar] [CrossRef]
- Lenstra, D.C.; Wolf, J.J.; Mecinović, J. Catalytic Staudinger Reduction at Room Temperature. J. Org. Chem. 2019, 84, 6536–6545. [Google Scholar] [CrossRef] [PubMed]
- Vil’, V.A.; Barsegyan, Y.A.; Kuhn, L.; Terent’ev, A.O.; Alabugin, I. V Creating, Preserving, and Directing Carboxylate Radicals in Ni-Catalyzed C(sp3)–H Acyloxylation of Ethers, Ketones, and Alkanes with Diacyl Peroxides. Organometallics 2023. [Google Scholar] [CrossRef]
- Kuhn, L.; Vil’, V.A.; Barsegyan, Y.A.; Terent’ev, A.O.; Alabugin, I. V Carboxylate as a Non-innocent L-Ligand: Computational and Experimental Search for Metal-Bound Carboxylate Radicals. Org. Lett. 2022, 24, 3817–3822. [Google Scholar] [CrossRef]
- Sheng Xiaolong; Tang, Mingfang; Gao, Botao; Huang, Guosheng, J.L. An Efficient Method for the α-Acetoxylation of Ketones. Synthesis (Stuttg). 2007, 2007, 1165–1168. [Google Scholar] [CrossRef]
- Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. Iodobenzene-Catalyzed α-Acetoxylation of Ketones. In Situ Generation of Hypervalent (Diacyloxyiodo)benzenes Using m-Chloroperbenzoic Acid. J. Am. Chem. Soc. 2005, 127, 12244–12245. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, F.; Yao, L.; Shi, T.; Tang, B.; Kuninobu, Y.; Wang, Z. C–N and C–O Bond Formation in Copper-Catalyzed/Mediated sp3 C–H Activation: Mechanistic Studies from Experimental and Computational Aspects. J. Org. Chem. 2020, 85, 9713–9726. [Google Scholar] [CrossRef]
- Wang, Z.; Kuninobu, Y.; Kanai, M. Copper-Mediated Direct C(sp3)–H and C(sp2)–H Acetoxylation. Org. Lett. 2014, 16, 4790–4793. [Google Scholar] [CrossRef]
- Dick, A.R.; Hull, K.L.; Sanford, M.S. A Highly Selective Catalytic Method for the Oxidative Functionalization of C−H Bonds. J. Am. Chem. Soc. 2004, 126, 2300–2301. [Google Scholar] [CrossRef]
- Desai, L. V; Hull, K.L.; Sanford, M.S. Palladium-Catalyzed Oxygenation of Unactivated sp3 C−H Bonds. J. Am. Chem. Soc. 2004, 126, 9542–9543. [Google Scholar] [CrossRef]
- Wang, D.-H.; Hao, X.-S.; Wu, D.-F.; Yu, J.-Q. Palladium-Catalyzed Oxidation of Boc-Protected N-Methylamines with IOAc as the Oxidant: A Boc-Directed sp3 C−H Bond Activation. Org. Lett. 2006, 8, 3387–3390. [Google Scholar] [CrossRef]
- Desai, L. V; Malik, H.A.; Sanford, M.S. Oxone as an Inexpensive, Safe, and Environmentally Benign Oxidant for C−H Bond Oxygenation. Org. Lett. 2006, 8, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khaskin, E.; Anderson, N.P.; Zavalij, P.Y.; Vedernikov, A.N. Catalytic aerobic oxidation of substituted 8-methylquinolines in PdII-2,6-pyridinedicarboxylic acid systems. Chem. Commun. 2008, 3625–3627. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Lee, M.; Dunn, A.L.; Sanford, M.S. Palladium-Catalyzed C–H Bond Acetoxylation via Electrochemical Oxidation. Org. Lett. 2018, 20, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, M.; Ricardo Pires, J.; Viseu, I.; Magalhães, P.; Gregório, H.; Afreixo, V.; Gregório, T. Edaravone for acute ischemic stroke - Systematic review with meta-analysis. Clin. Neurol. Neurosurg. 2022, 219, 107299. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Kuhad, A.; Kuhad, A. Edaravone: a new hope for deadly amyotrophic lateral sclerosis. Drugs Today (Barc). 2018, 54, 349–360. [Google Scholar] [CrossRef]
- Purohit, M.K.; Chakka, S.K.; Scovell, I.; Neschadim, A.; Bello, A.M.; Salum, N.; Katsman, Y.; Bareau, M.C.; Branch, D.R.; Kotra, L.P. Structure–activity relationships of pyrazole derivatives as potential therapeutics for immune thrombocytopenias. Bioorg. Med. Chem. 2014, 22, 2739–2752. [Google Scholar] [CrossRef]
- Chugunova, E.A.; Timasheva, R.E.; Gibadullina, E.M.; Burilov, A.R.; Goumont, R. First Synthesis of Benzotrifuroxan at Low Temperature: Unexpected Behavior of 5,7-Dichloro-4,6-dinitrobenzo-furoxan with Sodium Azide. Propellants, Explos. Pyrotech. 2012, 37, 390–392. [Google Scholar] [CrossRef]
- dos Santos, M.S.; Oliveira, M.L. V; Bernardino, A.M.R.; de Léo, R.M.; Amaral, V.F.; de Carvalho, F.T.; Leon, L.L.; Canto-Cavalheiro, M.M. Synthesis and antileishmanial evaluation of 1-aryl-4-(4,5-dihydro-1H-imidazol-2-yl)-1H-pyrazole derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 7451–7454. [Google Scholar] [CrossRef]
- Hassaneen, H.M.E.; Hassaneen, H.M.; Elnagdi, M.H. Enamines in Heterocyclic Synthesis: A Route to 4-Substituted Pyrazoles and Condensed Pyrazoles. Zeitschrift für Naturforsch. B 2004, 59, 1132–1136. [Google Scholar] [CrossRef]
- Kvaskoff, D.; Lüerssen, H.; Bednarek, P.; Wentrup, C. Phenylnitrene, phenylcarbene, and pyridylcarbenes. Rearrangements to cyanocyclopentadiene and fulvenallene. J. Am. Chem. Soc. 2014, 136, 15203–15214. [Google Scholar] [CrossRef]
- Sankaranarayanan, J.; Rajam, S.; Hadad, C.M.; Gudmundsdottir, A.D. The ability of triplet nitrenes to abstract hydrogen atoms. J. Phys. Org. Chem. 2010, 23, 370–375. [Google Scholar] [CrossRef]
- Voskresenska, V.; Wilson, R.M.; Panov, M.; Tarnovsky, A.N.; Krause, J.A.; Vyas, S.; Winter, A.H.; Hadad, C.M. Photoaffinity Labeling via Nitrenium Ion Chemistry: Protonation of the Nitrene Derived from 4-Amino-3-nitrophenyl Azide to Afford Reactive Nitrenium Ion Pairs. J. Am. Chem. Soc. 2009, 131, 11535–11547. [Google Scholar] [CrossRef] [PubMed]
- Falvey, D.E.; Gudmundsdottir, A.D. Nitrenes and Nitrenium Ions; Wiley Series of Reactive Intermediates in Chemistry and Biology; Wiley, 2013; ISBN 9780470390597.
- Carra, C.; Bally, T.; Albini, A. Role of conformation and electronic structure in the chemistry of ground and excited state o-pyrazolylphenylnitrenes. J. Am. Chem. Soc. 2005, 127, 5552–5562. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bettinetti, G.; Minoli, G. Photodecomposition of Some Para-Substituted 2-Pyrazolylphenyl Azides. Substituents Affect the Phenylnitrene S−T Gap More Than the Barrier to Ring Expansion. J. Am. Chem. Soc. 1999, 121, 3104–3113. [Google Scholar] [CrossRef]
- Tomioka, H.; Ichikawa, N.; Komatsu, K. Photochemistry of 2-(methoxycarbonyl)phenyl azide studied by matrix-isolation spectroscopy. A new slippery energy surface for phenylnitrene. J. Am. Chem. Soc. 1993, 115, 8621–8626. [Google Scholar] [CrossRef]
- Becher, J.; Brøndum, K.; Krake, N.; Pluta, K.; Simonsen, O.; Molina, P.; Begtrup, M. An unexpected ring opening–ring closure reaction of 5-azido-4-formylpyrazole. J. Chem. Soc.{,} Chem. Commun. 1988, 541–542. [CrossRef]
- Dehaen, W.; Becher, J. Synthesis of 5-amino-4-cyanopyrazoles via ring opening-ring closure of 5-azido-4-iminomethylpyrazoles isolation of the intermediate. Tetrahedron Lett. 1991, 32, 3565–3568. [Google Scholar] [CrossRef]
- Alabugin, I. V.; Bresch, S.; Dos Passos Gomes, G. Orbital hybridization: A key electronic factor in control of structure and reactivity. J. Phys. Org. Chem. 2015, 28, 147–162. [Google Scholar]
- Juaristi, E.; dos Passos Gomes, G.; Terent’ev, A.O.; Notario, R.; Alabugin, I. V Stereoelectronic Interactions as a Probe for the Existence of the Intramolecular α-Effect. J. Am. Chem. Soc. 2017, 139, 10799–10813. [Google Scholar] [CrossRef]
- Nigst, T.A.; Antipova, A.; Mayr, H. Nucleophilic Reactivities of Hydrazines and Amines: The Futile Search for the α-Effect in Hydrazine Reactivities. J. Org. Chem. 2012, 77, 8142–8155. [Google Scholar] [CrossRef]
- Yaremenko, I.A.; Belyakova, Y.Y.; Radulov, P.S.; Novikov, R.A.; Medvedev, M.G.; Krivoshchapov, N. V; Korlyukov, A.A.; Alabugin, I. V; Terent′ev, A.O. Inverse α-Effect as the Ariadne’s Thread on the Way to Tricyclic Aminoperoxides: Avoiding Thermodynamic Traps in the Labyrinth of Possibilities. J. Am. Chem. Soc. 2022, 144, 7264–7282. [Google Scholar] [CrossRef] [PubMed]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2019, 25. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Liebman, J.F. The annular tautomerism of imidazoles and pyrazoles: The possible existence of nonaromatic forms. Struct. Chem. 2006, 17, 439–444. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Gold, B.; Mikhailovskaya, T.F.; Alabugin, I. V Strain control in nucleophilic cyclizations: reversal of exo-selectivity in cyclizations of hydrazides of acetylenyl carboxylic acids by annealing to a pyrazole scaffold. J. Phys. Org. Chem. 2012, 25, 998–1005. [Google Scholar] [CrossRef]
- APEX2 (Version 2.1), SAINTPlus. Data Reduction and Correction Program (Version 7.31A), Bruker Advansed X-ray Solutions, BrukerAXS Inc.,Madison, Wisconsin, USA, 2006.
- G.M. Sheldrick. SADABS, Program for empirical X-ray absorption correction, Bruker-Nonius, 1990 - 2004.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
Scheme 1.
The original goal of this work: fusion of two dinitrogen heterocycles, pyrazole and furoxan.
Scheme 1.
The original goal of this work: fusion of two dinitrogen heterocycles, pyrazole and furoxan.
Scheme 2.
Reaction of edaravone 1 with POCl3, following nitration and azidation with formation of 5-azido-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole 4.
Scheme 2.
Reaction of edaravone 1 with POCl3, following nitration and azidation with formation of 5-azido-3-methyl-4-nitro-1-(4-nitrophenyl)-1H-pyrazole 4.
Scheme 3.
Unexpected redox disproportionation of α-nitroazide 4.
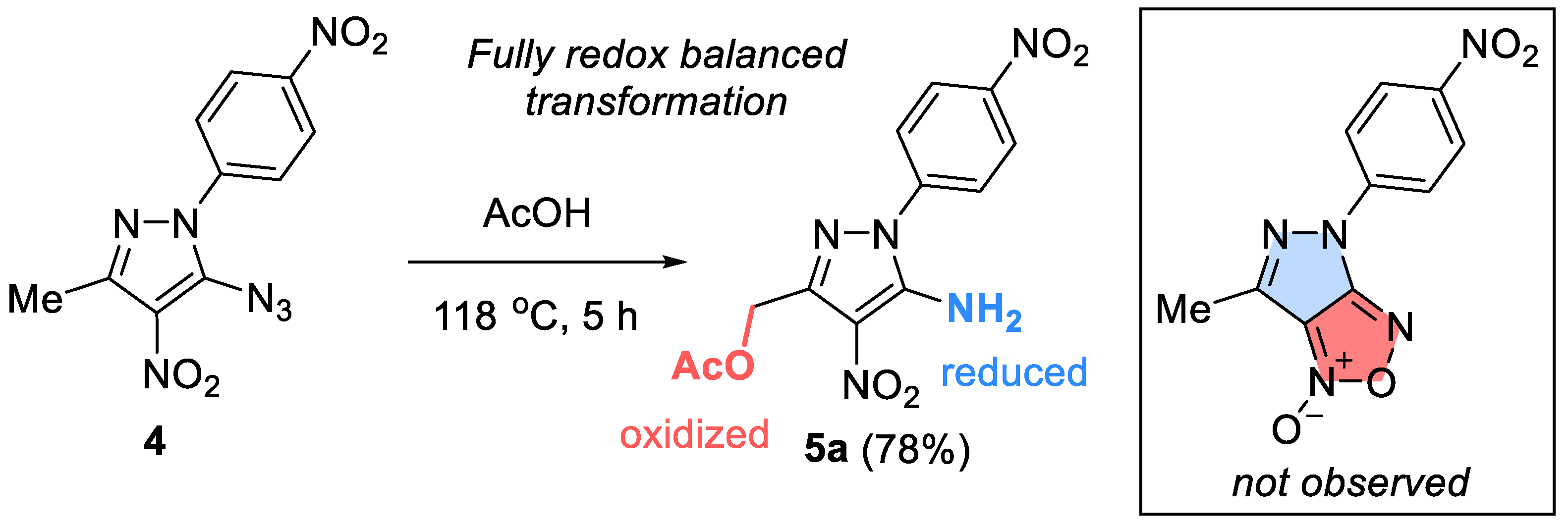
Figure 1.
(A) – fragment of crystal packing of compound 5a, (B) – molecular structure of compound 6.
Figure 1.
(A) – fragment of crystal packing of compound 5a, (B) – molecular structure of compound 6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



