
Submitted:
09 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
Methylglyoxal (MGO) is the major compound belonging to reactive carbonyl species (RCS) responsible for the generation of advanced glycation end products (AGEs). Its upregulation followed by deleterious effects at the cellular and systemic level is associated with metabolic disturbances (hyperglycemia/hyperinsulinemia/insulin resistance/hyperlipidemia/inflammatory processes/carbonyl stress/oxidative stress/hypoxia). Therefore, it is implicated in a variety of disorders including metabolic syndrome, diabetes mellitus and cardiovascular diseases. In this review an interplay between pathways leading to MGO generation and scavenging is addressed, in regard to this system’s impairment in pathology. The issues associated with mechanistic MGO involvement in pathological processes, as well as the discussion on its possible causative role in cardiometabolic diseases are enclosed. Finally, the main strategies aimed at MGO and its AGEs downregulation with respect to cardiometabolic disorders treatment are addressed. Potential glycation inhibitors and MGO scavengers are discussed, as well as the mechanisms of their action.
Keywords:
methylglyoxal
; glyoxalase
; advanced glycation end products
; MG-H1
; metabolic syndrome
; insulin resistance
; diabetes mellitus
; cardiovascular disease
; metformin
; methylglyoxal scavengers
1. Methylglyoxal in (patho)physiology
Methylglyoxal (MGO) is the major compound belonging to α-dicarbonyl molecules which are termed “reactive carbonyl species” (RCS) responsible for “carbonyl stress”. They are highly reactive compounds which easily modify biological macromolecules including peptides, proteins, lipoproteins and nucleic acids via the generation of advanced glycation end products (AGEs) [1]. Therefore, together with other irritable molecules like reactive oxygen and nitrogen species (RONS), they disturb the functioning of cellular organelle, thus stimulating their rearrangements leading to autophagy, apoptosis, or proliferation of cells. Such phenomena, when not counteracted by detoxifying mechanisms, stimulate oxidative stress and enhance inflammatory processes contributing to the development of a variety of pathological conditions [1,2,3].
However, since MGO is constantly produced in the organism as a glycolytic by-product, it might be also involved in beneficial processes stimulating and maintaining protective mechanisms to prepare the organism for handling with enhanced/pathological concentrations of RCS and RONS. Such a phenomenon termed hormesis is observed when low vs. high doses of a factor yield opposite effects, e.g. a high concentration of a compound is harmful, whereas at low concentration it works in a beneficial manner [4]. Recent data point to MGO playing such a dual function in organisms [1]. Whereas toxic at high levels [reviewed in ref: [5], when MGO is generated or tested at low concentrations, it seems to stimulate protective mechanisms including the upregulation of heat shock proteins involved in handling with misfolded proteins [6] or the activation of proteasomal system participating in the removal of damaged proteins (yielding the extension of the healthy lifespan of C. elegans) [7].
Normal MGO level in the human blood plasma has been estimated between 0,06-0,25 µM, whereas its cellular concentration reaches 1-5 µM [5]. In metabolic disorders, mostly associated with hyperglycemia (such as metabolic syndrome and diabetes) MGO as well as its main end product (D-lactate) and MGO-derived AGEs (MAGEs) usually undergo upregulation being intertwined in pathological processes [8-24].
1.1. Endogenous sources of MGO
MGO is endogenously produced under physiological conditions, where its main source (around 90%) are trioses derived from glycolysis: dihydroxyacetone phosphate and glyceraldehyde-3-phosphate [5,25-27]. They undergo a non-enzymatic conversion into MGO via an intermediate enediolate phosphate [26]. Around 0.09-0.4 % of glycolytic flux is probably associated with MGO generation [28,29]. This pathway seems to be stimulated under hyperglycemic conditions, due to the fact that the major MGO precursors are glucose (Glc) and fructose (Fru) [30-32]. In their recent report, Zhang et al. [32] have shown that both blood plasma and tissue MGO levels rose in parallel to Glc during oral glucose tolerance test. Additionally, the authors observed the increase in MGO-modified proteins in the circulation, which confirms Glc to be the main source of MGO.
Fructose has drawn attention with respect to its deleterious effects implicated in metabolic syndrome development, including obesity, insulin resistance and hypertension [33-35]. In comparison with glucose, fructose is not so tightly regulated by hormones (e.g. insulin). When it enters glycolytic pathway in the liver (the organ responsible for around 90% of its metabolism), it overcomes regulatory steps limiting glucose degradation (glucokinase and phoshofructokinase), which easily yields trioses accumulation (being converted into diacylglycerol (DAG), triacylglycerol (TAG) and MGO)) [34]. Fructose excess in the liver leads to unfavorable processes, such as uric acid generation, lipogenesis and gluconeogenesis, hence stimulating proinflammatory pathways. On the other hand, an excess of glucose is utilized for glycogen generation - the main carbohydrate energy storage in the liver and muscles. Additionally, fructose is more vulnerable to non-enzymatic oxidation, 8-10 times more active in the formation of AGEs than Glc, and (although present in around 100-time lower level in the blood plasma) under some pathological conditions it may be the main source of MGO [34]. Except for its detrimental effects on the liver, fructose also disturbs the functions of the adipose tissue inducing leptin-resistance, adipogenesis, oxidative stress and inflammation. Such fructose-triggered deleterious pathways are highly probably the consequences of an overload of fructose taken from diet, especially in the form of a high-fructose syrup, being a commonly applied additive in many highly-processed foodstuffs, such as non-alcoholic beverages. Accordingly, the fructose-rich diet has been connected with metabolic disturbances leading to obesity, dyslipidemia, metabolic syndrome, type 2 diabetes (promoting insulin resistance and gluconeogenesis), as well as non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD) [33,34,36]. One of the factors linking fructose overload and many of the above mentioned pathological processes may be an excessive production of MGO. As hypothesized by Gugliucci [37], an excess of dietary Fru (whose increasing intake is paralleled with metabolic syndrome prevalence) would lead to the accumulation of MGO in the liver, which in turn would modify 3 Arg residues in AMP-activated kinases (AMPKs). Since AMPKs are the energy sensors in the cells, they are activated at low energy level (reflected by AMP increase) and stimulate catabolic pathways leading to the energy replenishment. Fru influx into the liver and its entering glycolysis leads to the depletion of ATP (used for its phosphorylation) associated with the increase in AMP. This should stimulate catabolic pathways, and inhibit anabolic ones (via AMPKs activation by AMP). However, experimental data indicate quite opposite regulation accelerating processes of synthesis (lipogenesis, gluconeogenesi) in Fru overload conditions. Hence, Gugliucci has put forward the hypothesis that it might be MGO-modified AMPK that loses its function (since MGO-modification makes it insensitive to AMP regulation), otherwise leading to the acceleration of opposite processes. Finally, instead of degradation/oxidation of macromolecules to gain energy, their synthesis is enhanced yielding hyperglycemia and/or liver steathosis, with further consequences [37].
The minor endogenous sources of MGO include: amino acids, glycerol, ketone bodies, as well as glycated proteins [5,26,38-40]. For example, MGO may be generated from aminoacetone (derived from threonine or glycine catabolism) deamination [41] or degradation of glucose-glycated proteins [38]. Additionally, lipid peroxidation products (aldehydes and ketoaldehydes) give rise to the production of MGO [39]. Therefore, under pathological conditions stimulated by fructose-rich diet and associated with oxidative stress, hyperglycemia, as well as an overproduction of ketone bodies (observed in disturbances connected with metabolic syndrome, diabetes and cardiovascular complications), multiple routes of MGO generation are possible [40].
1.2. Exogenous sources of MGO
MGO and other α-dicarbonyl compounds have been detected in dietary products, especially highly processed and subjected to high temperatures foodstuffs [39,42]. For example, MGO can be found in cookies, alcoholic beverages, soy sauce, coffee and honey [42-46]. However, exogenous MGO sources do not seem to significantly contribute to the total MGO load in the human body due to its putative degradation in the gastrointestinal tract (GI) and detoxification by the glyoxalase system in the epithelial cells lining GI lumen [47]. Nevertheless, deleterious MGO effects can be observed in the GI tract, both via the impact of MGO-glycated foods on the composition of intestinal microbiome, as well as the metabolism of dietary carbohydrates by bacteria, which can lead to MGO formation [39].
1.3. MGO-modification of macromolecules
1.3.1. MGO-derived AGEs (MAGEs)
MGO is the major α-dicarbonyl compound involved in the modification of peptides, proteins, and lipoproteins resulting in AGEs formation (MAGEs). It modifies arginine (Arg), lysine (Lys) and cysteine (Cys) residues in macromolecules, showing the greatest efficiency for Arg alterations [48]. MGO irreversibly reacts with Arg guanidine group, generating several types of derivatives, including 3 cyclic hydroimidazolones: MG-H1, MG-H2, and MG-H3 [5,49,50]. The most prevalent is MG-H1 isoform, which is responsible for more than 90% of MGO alterations [51]. Both MG-H1 and MG-H2 have been detected in the human lens proteins [52], but when antibodies against hydroimidazolones have been tested on human endothelial cells (Ea.hy 926 cells), only MG-H1 and MG-H3 were identified (and their nuclear localization was reported in that study) [53]. Similarly, only MG-H1 and a derivative of MG-H3 (CEA) were detected in the chromatin from human epithelial cells (HEK293), and their presence was observed in the chromatin from several other human cell lines, as well as murine tissues from many organs [54]. Except for hydroimidazolones, other derivatives of MGO-modified Arg include tetrahydropyrimidine (THP) and argpyrimidine (AP) [55,56]. Besides Arg, MGO is able to modify Lys side chain yielding its carboxyethyl derivative (CEL) or forming Lys dimers (MOLD), as well as cross-link Arg with Lys to generate MODIC adducts [5].
A lot of proteins have been reported to undergo MGO-derived Arg modifications, which leads to their impaired functioning. Although in many experimental studies applied concentrations of MGO much exceeded physiological levels of MGO, also low MGO levels seem to alter the functionality of proteins [5]. For example, MGO-modified Arg410 (yielding MG-H1 residues) in albumin [57] probably disturbs a drug-binding function as well as esterase activity of this protein [58]. Additionally, MGO-glycated albumin shows decreased antioxidative potential [59] and seems to stimulate inflammatory processes via the mobilization of such cytokines as TNF-α [60,61] and IL-1β [62]. Other proteins, whose functions can be disturbed upon MGO glycation include collagen [63,64], hemoglobin [65-67], insulin [68], and mitochondrial proteins whose impairment leads to ROS generation [69]. Moreover, MAGEs formation interferes with proteolysis coupled with lysosomal and proteasomal systems [5]. On the one hand, an extensive protein glycation makes proteins resistant to degradation, whereas on the other hand, MGO impact on the ubiquitination process might enhance proteasomal degradation of some proteins (less probable in vivo though – due to greater than physiological MGO levels tested in the experiments) [5]. Therefore, MGO-glycation, especially under hyperglycemic conditions seems rather to impair the functioning of proteolytic systems, leading to the accumulation of misfolded proteins in the cells followed by disturbances in intracellular organelle [5]. Such a phenomenon has been observed in Glo1-knock down mice, where MGO-glycation of a proteasomal subunit decreased proteolytic activity [70]. Furthermore, MAGEs modification of histones seems to affect epigenetic regulation of gene expression. Histones’ side chains of Arg and Lys altered by MGO (yielding MG-H1, MG-H3/CEA, and CEL derivatives) led to the increase or decrease in transcription of multiple genes [54]. Hence, the potential effect of MAGEs on genes expression might lead via multiple pathways to pathology, enhancing deleterious processes especially in metabolic syndrome associated with hyperglycemia, and diabetes, where it might contribute to the development of hyperglycemic legacy effect (metabolic memory) [71].
1.3.2. MGO-derived DNA modifications
In comparison with protein glycations by MGO, much less is known about nucleic acids modifications [5]. The most reactive nucleoside is deoxyguanosine which upon MGO action yields CEdG and MG-dG derivatives [5,27]. CEdG is more abundant and stable, so it seems to play more important role with reference to MGO-associated pathologies [5], mainly metabolic syndrome and diabetes where CEdG increase has been observed in animal models [12,13] and in diabetic patients’ tissues [72], as discussed in the following chapters.
1.4. MGO scavenging system
MGO undergoes detoxification reactions catalyzed by a ubiquitous glyoxalase system composed of glyoxalase 1 (Glo1) and 2 (Glo2), yielding D-lactate [14]. The first enzyme Glo1 requires reduced glutathione (GSH) for the production of an intermediate (lactoylglutathione), whereas Glo2 catalyzes lactoylglutathione conversion into D-lactate, which is coupled with the regeneration of GSH [27]. Abundant in cytosol Glo1 is characterized by a high specificity towards MGO, and catalyzes the rate-limiting reaction in MGO metabolism [27]. Glo2, except for being located in the cytosol, is also present in the mitochondrium [26].
Interestingly, DJ-1 (PARK7 = Parkinson’s disease protein 7) has been also suggested to be involved in MGO detoxification [73]. Whereas mutated DJ-1 gene is implicated in up to 1% of early onset Parkinson’s disease cases [74], its normal product is a multifunctional protein which controls the activity of mitochondria (being engaged in mitophagy) [74]. It is also a sensor of the cellular oxidative stress, upon which it gets activated and in turn switches on protective mechanisms, e.g. controlling the expression of antioxidative enzymes [75]. Additionally, DJ-1 may play a role in MGO degradation due to its glyoxalase activity (less certain because this activity is low in comparison with Glo1), as well as in the repairment of MGO-glycated proteins and nucleic acids, since it also may show deglycase activity [54,76,77] (a more probable function) [5]. However, Pfaff et al. [78] in their DJ-1 knock-down and knock-out fruit flies models have questioned the function of this protein in MGO detoxification.
Overall, this is the glyoxalase system (Glo1 and Glo2) which contributes mainly to MGO scavenging (metabolizing more than 98% of MGO) [5]. Additionally, MGO may also enter other pathways of degradation, yielding pyruvate (when catalyzed by NADPH-dependent aldehyde dehydrogenases - ALDHs) or hydroxyacetone (when catalyzed by aldoketo reductases - AKRs) [5]. The importance of AKRs in MGO scavenging, associated with the protection from AGEs formation and atherosclerotic lesions generation, has been reported by Baba et al. [79]. Therefore, these minor routes of MGO detoxification may play a role in pathological processes partially taking over the functions of glyoxalases whose down-regulation is observed under cellular stress [27]. Such a compensatory mechanism has been shown by Schumacher et al. [80] and Morgenstern et al. [81] in Glo1 knock-out experimental models.
2. MGO and MAGEs in metabolic syndrome and diabetes
2.1. Metabolic syndrome
Metabolic syndrome is a set of disturbances associated with defects in lipid and carbohydrate metabolism. This syndrome is diagnosed in individuals who present any three out of five characteristics; namely enhanced concentration of triacylglycerols, elevated glucose, decreased level of HDL-cholesterol, hypertension, or adiposity connected with an excessive level of visceral/liver fat. A characteristic feature of individuals suffering from metabolic syndrome is insulin resistance and chronic low-grade inflammation [82], which further can develop into such disorders as type-2 diabetes mellitus (T2DM) and cardiovascular conditions [83] including coronary heart disease and stroke [84].
2.2. MGO and MAGEs in metabolic syndrome and diabetes in animal models and cell cultures
Experimental models allowing for the estimation of MGO and MAGEs associations with metabolic disturbances comprise MGO or fructose-fed animals, genetically modified animals which develop obesity, diabetic and atherosclerotic characteristics, as well as glyoxalase 1 deficient or overexpressing animals. Additionally, spontaneously hypertensive rats (SHR) or streptozotocin (STZ)-treated rats have been applied to examine pathological background underlying hypertension or type-1 diabetes (T1DM), respectively. One of such models useful in metabolic syndrome/diabetes studies are genetically modified mice which highly express defective gene coding for leptin receptor (Leprdb/db), hence they develop leptin resistance leading to obesity, hyperinsulinemia and hyperglycemia [85,86]. In search for a diagnostic marker which could be used in the diagnosis of prolonged diabetes, Jaramillo et el. [12] have found that MGO-modified deoxyguanosine - CEdG was significantly elevated in urine and tissues of hyperglycemic Leprdb/db mice in comparison with normoglycemic animals. Similar increase in urinary CEdG has been observed in the diabetic (T1DM) rats [13] (Table 1). Additionally, urinary CEdG was shown to be an independent prognostic factor of hyperglycemia, and was positively correlated with fasting plasma glucose (FPG) in hyperglycemic animals, and with HbA1c in all animals [12]. Furthermore, two protein (M)AGEs - lysine derivatives (CML and CEL) were elevated in the urine of hyperglycemic mice, but they did not correlate with FPG. Nevertheless, a positive correlation was reported between CEdG and both CML and CEL in hyperglycemic mice [12]. Hence, the authors suggested CEdG to be a promising marker in metabolic diseases.
Elevated levels of both MGO and D-lactate (the end product of MGO metabolism by glyoxalases system) have been reported in T1DM rats in the lens and blood [14]. Additionally, MGO concentration was increased in those animals kidneys (Table 1). Furthermore, MGO-treatment has impaired glycemia and lipid profile both in lactating rat mothers (in their blood plasma and breast milk), as well as their adult male offspring who showed features of obesity [87] (Table 1).
To elucidate which metabolic pathways are responsible for the overgeneration of MGO and its deleterious effects in pathology, Liu et al. [88] have examined four different rat models with metabolic syndrome features, complemented with experiments of vascular smooth muscle cells (VSMCs). In both the rats aortas and VSMCs, the authors reported upregulation of enzymes responsible for fructose degradation (aldolase B and fructokinase), as well as fructose-specific transporter (GLUT-5). These effects were stimulated by high Fru level and augmented by insulin, and led to the increase in MGO. Additionally, high Glc level seemed to contribute to MGO generation rather via Fru production (polyol pathway) than glycolysis [88] (Table 1 and Table 2). Thus, the authors underlined the causative[26e importance of Fru associated with MGO generation and further deleterious consequences in obesity, hypertension, and diabetes with cardiovascular impact, especially in light of the Fru-rich diet common in well-developed countries. The same type of VSMCs has been shown to develop oxidative stress upon MGO exposition [89,90] (Table 2). MGO increased the level of RONS through its deleterious effect on the respiratory chain (impairing complex III activity, which was associated with superoxide anion generation and decrease in ATP synthesis), as well as the inhibition of superoxide scavenging enzyme - manganese superoxide dismutase (MnSOD) [90]. As discussed in the following chapters, RONS overgeneration is implicated in pathologic routes leading to cardiometabolic disorders. Similarly, chronic low-grade inflammatory state is associated with metabolic syndrome, diabetes and CVD, and MGO has been shown to mediate macrophages-induced proinflammatory processes, leading to the development/deepening of inflammation [91,92].

Table 1.
Methylglyoxal and its AGEs in cardiometabolic disorders – data from rodent models.
| Experimental model | MGO/MAGEs and associated major findings | Ref./year |
|---|---|---|
| Spontaneously hypertensive rats (SHR) and Wistar Kyoto rats (WKY) | In comparison with normal WKY, in SHR: higher MGO level in blood plasma and kidney (increasing with age), higher CML and CEL staining in the kidney, decreased GSH and GSH/GSSG ratio in the kidney of the oldest 20-week rats. |
[93]/2004 |
| Spontaneously hypertensive rats (SHR) and Wistar Kyoto rats (WKY) | In comparison with normal WKY, in SHR: higher MGO level in blood plasma and aorta (increasing with age), higher MGO level in the liver and kidney (but not in the heart) in 13-week rats, higher CML and CEL staining in the aorta (mostly in endothelial cells, lower in smooth muscle cells), increased oxidative stress (superoxide anion and hydrogen peroxide) in 13-week rats aortas decreased GSH in 13-week rats’ aortas, decreased activities of glutathione peroxidase and reductase in 13-week rats’ aortas, increased activity of SSAO in blood plasma, no difference in blood plasma GSH. |
[94]/2005 |
| aminoguanidine treated SHR, untreated SHR, and WKY rats | In comparison with untreated SHR, in aminoguanidine treated SHR: Attenuation of systolic blood pressure, Correction of MGO level in blood plasma and aorta (raised in untreated SHR) to the level comparable with WKY, Correction of AP and CEL levels in the aorta and mesenteric artery (raised in untreated SHR) to the level comparable with WKY, Attenuation of oxidative stress in aortic tissue (decreased superoxide anion and nitric oxide, increased GSH), Correction of nitric oxide synthases expression in aortic tissue (decrease in iNOS and increase in eNOS) to the level comparable with WKY, Improvement of morphologic changes and endothelium-dependent relaxation in mesenteric artery. |
[95]/2007 |
| Fructose-fed SD rats, Wistar–Kyoto (WKY) rats, SHR rats, and lean, obese, and diabetic Zucker rats |
In aortas of fructose-fed SD rats: increase in MGO and fructose; upregulation of GLUT-5, fructokinase and aldolase B (at mRNA levels). In SHR rats (as compared to WKY control): in the serum: similar Glc, increase in MGO, fructose and insulin; in aortas: increase in MGO and fructose; upregulation of GLUT-5, fructokinase and aldolase B (at mRNA levels). In obese and/or diabetic Zucker rats (as compared to lean Zucker rats control): in the serum: increase in Glc, MGO and fructose increase in insulin in obese rats, but decrease in insulin in diabetic rats. in aortas: increase in MGO and fructose; upregulation of GLUT-5, fructokinase and aldolase B (at mRNA levels); increase in aldose reductase and sorbitol in diabetic rats. |
[88]/2011 |
| aminoguanidine treated SHR, untreated SHR, and WKY rats | In comparison with untreated SHR, in aminoguanidine treated SHR: Attenuation of blood pressure, Correction of AP and CEL levels in the mesenteric artery (raised in untreated SHR) to the level comparable with WKY, Correction of Ang II-induced contraction of the mesenteric artery, Normalization of endothelium-dependent (ACh-induced) relaxation impaired in SHR mesenteric artery, Attenuation of oxidative stress in mesenteric artery, downregulation of NOX1 (but not NOX2) and AT2R expression (upregulated in SHR) in mesenteric artery, no changes in eNOS and SOD-(1-3) Effect of NOX inhibitor on mesenteric artery from SHR: enhanced ACh-induced relaxation, but no effect on Ang II-induced contraction |
[96]/2012 |
| Mesenteric artery isolated from Wistar rats | Upon long-term MGO treatment (42 µM for 3 days): impairment of endothelium-dependent (ACh-induced) relaxation, no effect on endothelium-independent (SNP-induced) relaxation, decrease in ACh-induced: NO production and VASP phosphorylation, increase in apoptosis of endothelial cells associated with superoxide radical elevation, Upon long-term MGO treatment (42 µM for 3 days) and a NOX inhibitor: reversal of MGO-impaired endothelium-dependent (ACh-induced) relaxation, reversal of MGO-caused eNOS downregulation. |
[97]/2013 |
| Endothelium-denuded thoracic aorta and superior mesenteric artery isolated from Wistar rats | Upon MGO treatment (420 µM for 30 min): Inhibition of noradrenaline-induced contraction of aorta and mesenteric artery, in aorta prevented by a large conductance Ca2+-activated K+ (BKCa)-channel inhibitor. |
[98]/2009 |
| Wistar rats infused with MGO, or treated with MGO and retinoic acid (RA). | Upon MGO treatment: increase in CML in blood plasma; In heart tissue: decrease in catalase, SOD and GSH; increase in cardiac fibrosis; up-regulated expression of RAGE (3.5 fold), TGF-β (4.4 fold), SMAD2 (3.7 fold), SMAD3 (6.0 fold), IL-6 (4.3 fold) and TNF-α (5.5 fold). Attenuation of the above effects by RA co-treatment. |
[99]/2017 |
| Lactating Wistar rats treated by gavage with MGO and they adult male offspring | Upon MGO treatment in mother rats: In blood plasma: increase in Glc and fructosamine; decrease in insulin and the functionality of pancreatic β-cells; increase in total cholesterol, triglycerides, cholesterol (LDL and VLDL); decrease in HDL cholesterol. In breast milk: increase in Glc, TAG, cholesterol, fructosamine, decrease in insulin. In the offspring: increase in body weight and adipose tissue; increase in Glc, insulin and fructosamine; decrease in the functionality of pancreatic β-cells; increase in total plasma cholesterol and LDL and VLDL cholesterol, TAG; decrease in HDL cholesterol. |
[87]/2018 |
| Sprague Dawley (SD) rats: untreated, fructose treated, N-acetyl cysteine (NAC)-treated, fructose+ NAC treated. | In comparison with untreated SD, in Fru-treated SD rats: Increase in blood pressure, increase in blood serum TAG (attenuated by NAC co-treatment), increase in blood serum insulin (attenuated by NAC co-treatment),, decrease in insulin-induced Glc uptake by visceral adipose tissue (improved by NAC co-treatment), increase in MGO in the adipose tissue and serum (attenuated by NAC co-treatment), increase in PI3K protein in the adipose tissue (counteracted by NAC co-treatment), decreased PI3K recruitment to phosphorylated IRS-1 (restored by NAC co-treatment), no changes in IR, IRS-1 expression and phosphorylation in the adipose tissue, no changes in total cholesterol, HDL-cholesterol, HbA1c, Glc in the blood serum |
[100]/2007 |
| Sprague Dawley (SD) rats: untreated, fructose treated, metformin treated, fructose+metformin treated. | In comparison with untreated SD, in fructose treated SD rats: increase in systolic blood pressure (attenuated by metformin co-treatment), increase in MGO in the serum and aorta (reduced by metformin co-treatment), increased CEL staining in the aorta (normalized by metformin co-treatment), decreased eNOS staining in endothelial cells of the aorta (increased by metformin co-treatment), increased hydrogen peroxide in the aorta, and decreased GSH in the serum (corrected by metformin co-treatment), no difference in aorta GSH levels. damaged mesenteric artery (increased wall thickness, decreased lumen) (corrected by metformin co-treatment), increased CEL and CML staining in the mesenteric artery (corrected by metformin co-treatment). |
[31]/2008 |
| Leprdb/db murine model of metabolic syndrome | Higher CEdG (238.4±112.8 pmol/24 h) in urine of hyperglycemic mice (FPG, ≥200 mg/dL) than normoglycemic mice (16.1±11.8 pmol/24 h). Enhanced CEL in urine of hyperglycemic mice |
[12]/2017 |
| STZ-treated rats (T1DM) | Around 4-fold increase in CEdG in urine of diabetic rats. | [13]/2008 |
| STZ-treated rats (T1DM) | Increased MGO and D-lactate in the lens and blood. Increased MGO in the kidney of diabetic rats. |
[14]/1993 |
| Glo1 KO mice | In comparison with Glo1+/+ mice; MG-H1 elevation in murine liver; but not in the brain. |
[101]/2017 |
| STZ-treated Glo1 KO mice | Neither MGO nor MG-H1 elevation in hyperglycemic mice. | [80]/2018 |
| STZ-treated Glo1 overexpressing rats | Reduction of hyperglycemia-elevated MGO/AGEs. | [102]/2011 |
| STZ-treated normal and Glo1 overexpressing rats | Effects of diabetes: increase in 3DG and CML, but not MGO and CEL in the heart. Effects of Glo1 overexpression: decrease in diabetes-enhanced cardiac mRNA profile associated with oxidative stress and fibrosis, partial attenuation of diabetes-upregulated cardiac RAGE |
[103]/2013 |
| STZ-treated normal and Glo1 overexpressing Wistar rats | Endothelium-dependent NO-mediated relaxation in mesenteric arteries isolated from the rats: impaired in diabetic normal rats, improved in diabetic Glo1 overexpressing rats. MGO-exposed mesenteric arteries: increase in MG-H1 (in adventitia and endothelium) in arteries from diabetic normal rats, but not from diabetic Glo1 overexpressing rats, impaired vasoreactivity corrected by Glo1 overexpression, increase in nitrotyrosine (a peroxynitrite marker). |
[104]/2010 |
| STZ-treated normal and Glo1 overexpressing Wistar rats | Effects of Glo1 overexpression: prevention of diabetes-stimulated MG-H1 and CML increase in mesenteric arteries isolated from the rats; correction of diabetes-impaired endothelium-dependent relaxation of mesenteric arteries; prevention of diabetes-increased VCAM-1 and ICAM-1 in mesenteric arteries isolated from the rats; attenuation of diabetes-cause markers of early damage in the kidney; no impact on diabetes-increased ICAM-1 in blood plasma no impact on eNOS expression, as well as mesenteric arteries morphology and collagen between groups. |
[105]/2014 |
| STZ-treated Glo1 overexpressing ApoE KO mice |
Effects of Glo1 overexpression: prevention of diabetes-stimulated MG-H1 modification of proteins in aortas and kidneys, no impact on diabetes-stimulated aortal collagen glycation (FL, CML, 3DG-H, MG-H1), no impact on diabetes-induced serum fasting glucose level, no impact on diabetes-induced serum cholesterol and TAG levels, no impact on diabetes-induced atherosclerotic lesions in aortas. |
[106]/2014 |
| Glo1 underexpressing ApoE KO mice | Effects of Glo1 underexpression (around 75% activity decrease): increase in MG-H1 protein modifications in aortas and kidneys, no impact on aortal collagen glycation (e.g. FL, CML, 3DG-H, MG-H1), no impact on atherosclerotic lesions in aortas, no impact on serum fasting glucose, cholesterol and TAG levels |
[106]/2014 |
| STZ-treated ApoE KO and RAGE/ApoE DKO mice; MGO-fed ApoE KO and RAGE/ApoE DKO mice; Glo1 inhibited (BBGC-treated) apoE KO mice |
Increase in MGO level in diabetic mice comparably to MGO-fed and BBGC-treated mice. Increase in atherosclerotic plaques in aortas from MGO-fed apoE KO and RAGE/apoE DKO mice, and BBGC-treated apoE KO mice (similarly to diabetic mice). Upregulation of adhesive and proinflammatory molecules (ICAM-1, tetherin, MCP-1, mac-1,2) in aortas of MGO- and BBGC-treated ApoE KO. Upregulation of ICAM-1, tetherin, and mac-1 in aortas of MGO-fed RAGE/ApoE DKO mice. |
[107]/2014 |
| STZ-treated ApoE KO mice (DM); STZ-treated ApoE KO mice fed with high-lipid diet (DM + HLD); STZ-treated ApoE KO mice fed with high-lipid diet and NAC (DM + HLD + NAC) |
All of the below effects were attenuated by NAC (in DM + HLD + NAC mice). In the serum from the mice: increase in MDA in DM mice, and more enhanced increase in DM + HLD mice; decrease in NO in DM mice, and more enhanced decrease in DM + HLD mice; In aortas extracted from the mice: increase in atherosclerotic plaque lesion in the aortic root from DM mice and more enhanced increase in DM + HLD; increase in MGO and protein carbonyls in DM and DM + HLD; decrease in SOD-1 and GPX-1 protein expression in DM and more enhanced decrease in DM + HLD; decrease in phosphorylated forms of Akt and eNOS in DM and more enhanced decrease in DM + HLD; decrease in GSH in DM and DM + HLD. |
[108]/2021 |
| MGO-treated C57/BL6 male mice | Increase in systemic insulin resistance. Reduction in insulin-induced activation of Akt and eNOS in murine aortas, reflected by reduction of insulin-stimulated increase in serum NO. Induction of ERK ½ phosphorylation in murine aortas and endothelin-1 release (comparable to insulin effect). |
[109]/2014 |
| Glo1 overexpressing ApoE KO mice | No effect of Glo1 overexpression on the size and severity of atherosclerotic plaques in the murine aortas. No effect of Glo1 overexpression on inflammatory markers (e.g. MCP-1, IL-6) in murine aortas, neither systemic inflammation (blood plasma lymphocytes T and B, cytokines - MCP-1, IL-1β,6,10, IFN-γ). No improvement of oxidative stress markers worsened in apoE KO mice by Glo1 overexpression. No differences in MGO and AGE markers (CML, CEL, MG-H1) in blood plasma and aortas between the murine groups. |
[110]/2014 |
| STZ-treated ApoE KO and Glo1 overexpressing ApoE KO-mice | No effect of Glo1 overexpression on: diabetes-induced increase in AGE markers (CML, GO), diabetes-enhanced atherosclerotic plaque lesions in murine aortas, inflammatory phenotype (MCP-1, monocytes), diabetes-induced plasma fasting glucose level, diabetes-induced plasma fasting cholesterol level. |
[[110]/2014 |
| MGO-fed normal Wistar rats and (non-obese) T2DM Goto-Kakizaki (GK) rats | In both Wistar and diabetic rats upon MGO treatment: decline in NO-dependent vascular relaxation, increase in superoxide and nitrotyrosine, upregulation of aortal MCP-1, AGEs and RAGE, increase in plasma MGO and urinary 8-OHdG levels. |
[111]/2012 |
| MGO-fed normal Wistar rats and (non-obese) T2DM Goto-Kakizaki rats | In MGO-fed normal Wistar rats (in comparison with normal Wistar rats): increase in plasma free fatty acids, decrease in serum adiponectin, increase in plasma and tissue MGO and urinary 8-OHdG levels. In the adipose tissue from MGO-fed Wistar rats: increase in AGEs, glycoconjugates and fibrosis, higher expression of TGF-β (but not its cleaved form), increase in proapoptotic factors (decreased Bcl2/Bax ratio and upregulation of caspase 3), decrease in VEGF level, but unchanged angiopoietin 2, increase in MCP-1 and F4/80 |
[112]/2012 |
| MGO-fed Wistar rats | In MGO-fed Wistar rats (in comparison with control Wistar rats): increase in plasma free fatty acids; no impact on glycemia (fasting and 2 h after glucose administration), glycated haemoglobin, insulinemia and serum total cholesterol, triglycerides and adiponectin levels. In the adipose tissue from MGO-fed Wistar rats: increase in CEL and fibrosis. In the adipose tissue of MGO-fed Wistar rats after blood supply reduction: increase of ERK1/2 phosphorylation (p-ERK1 plus p-ERK2); increase in perilipin A degradation (due to MGO-induced glycation); decrease in IkBa; decrease in PPARγ expression; decrease in Akt activation. |
[113]/2013 |
| Wistar rats: control (C), MGO-fed (MGO), high-fat diet-fed (HFD), high-fat diet group with MG supplementation (HFDMGO), and T2DM (non-obese) Goto-Kakizaki (GK) rats |
In the circulation of HFDMGO rats (as compared with control): increase in FFAs, insulin, Glc intolerance development. In the adipose tissue of HFDMGO and GK rats (as compared with control): increase in CEL; no change in GLO1 levels; increase in hypoxia; no change in HIF-1α, but decrease in HIF-2α expression; decrease in IR phosphorylation; no changes in phosphorylated Akt, PGC1α and the differentiation factors PPAR-γ and C/EBPα. In the adipose tissue of HFDMG and MGO rats (as compared with control): increase in (proinflammatory) M1 macrophages and CD31 (endothelial cell marker) In the adipose tissue of HFDMG, MGO and GK rats (as compared with control): decrease in blood flow; decrease in VEGF/Ang-2 ratio. In the adipose tissue of HFDMGO rats (as compared with control): decrease in perilipin A. Upon MGO exposition (50-1000 µM) and/or Glo1 inhibition in adipose tissue explants: inhibition of capillarization. In the skeletal muscles of HFDMGO rats (as compared with control): decrease in IR protein (but not phosphorylated IR), active Akt (phosphorylated Akt) and GLUT-4. |
[114]/2017 |
| MGO-treated (intragastrically) hereditary hypertriglyceridaemic rats (HHTg) | Upon MGO treatment: increase in non-fasting Glc and insulin in blood serum; increase in proinflammatory MCP-1 and TNFα in the serum; decrease in the conversion of Glc into lipids upon insulin-stimulation in white adipose tissue (WAT); increase in adrenaline-stimulated lipolysis in WAT; shift in components of phospholipids: increase in saturated fatty acids (e.g. palmitic and myristic) and decrease in polyunsaturated fatty acids (especially ω-3; e.g. α-linelenic and docosahexaenoic acids) in WAT; no effect on Glo1 expression in WAT; decrease in Nrf2 expression in WAT; increase in MCP-1 and TNFα expression in WAT; no effect on HIF-1 expression in WAT |
[115]/2020 |
| MGO-fed normal Wistar rats | Upon MGO treatment: no change in serum glucose, increase in serum cholesterol, creatinine and fructosamine, proinflammatory and profibrotic response (increased IL-1β, TNF-α, CTGF, TGF-β; disturbances in wound healing), upregulation of AGEs and RAGE in skin vasculature, progressive thickening of skin blood vessel wall followed by its detachment from matrix, luminal occlusion and endothelial cells death ending up with vessel destruction, no vasodilation upon nitroglycerine treatment. |
[116]/2005 |
| MGO-fed normotensive Sprague-Dawley rats | Upon MGO treatment: Decrease in insulin sensitivity (improved by NAC and TM2002 (AGEs inhibitor). Increase in CEL and nitrotyrosine in the kidney from the rats. |
[117]/2009 |
| Sprague-Dawley (SD) rats | Thoracic aortic rings isolated from SD rats upon MGO treatment: inhibition of ACh-induced endothelium-dependent relaxation (prevented by aminoguanidine (AG) and N-acetyl cysteine (NAC), but not restored by NOX inhibitor), no effect on endothelium-independent relaxation |
[118]/2010 |
| MGO-treated Sprague-Dawley (SD) rats | Upon MGO treatment (and attenuated by alagebrium) in SD rats: impairment in Glc tolerance, increase in plasma insulin, decrease in plasma glutathione. In the visceral adipose tissue isolated from the studied rats: decrease in insulin-stimulated glucose uptake, reduced plasma membrane GLUT-4 and IRS-1 tyrosine phosphorylation, no change in insulin receptor and IRS-1 protein expression. |
[119]/2010 |
| Fru-fed, and continuously MGO-treated Sprague-Dawley (SD) rats | In Fru-fed SD rats (as compared to SD control): increase in blood pressure and vascular remodeling; increase in MGO in plasma and aorta tissue (attenuated by metformin); increase in Akt1 phosphorylation at Ser-473 in aorta (attenuated by metformin). In MGO-treated SD rats (as compared to SD control): increase in Akt1 phosphorylation at Ser-473 in aorta (attenuated by alagebrium). |
[120]/2011 |
| Continuously MGO-treated Sprague-Dawley (SD) rats | Upon constant MGO treatment (and attenuated by alagebrium) in SD rats: increase in fasting plasma glucose, total cholesterol, TAG and free fatty acids decrease in fasting plasma insulin and HDL, and plasma and tissue glutathione, enhanced formation of CML and increased apoptosis of pancreatic β-cells. In the adipose tissue isolated from the studied rats: decrease in insulin-stimulated glucose uptake, reduced plasma membrane GLUT-4, IRS-1 phosphorylation and PI3K activity, no change in insulin receptor and IRS-1 protein expression, In the pancreatic islets (β-cells) isolated from the studied rats: reduced GLUT-2 (= decreased Glc uptake) and glucokinase, lowered insulin secretion – down-regulation of factors promoting insulin expression (PDX-1 and MafA), and up-regulation of the factor inhibiting insulin expression (C/EBPβ), upregulation of NF-kB and RAGE. |
[121]/2011 |
| Continuously MGO-treated Sprague-Dawley (SD) rats | Upon constant MGO treatment (and attenuated by alagebrium) in SD rats: increase in blood pressure; increase in plasma norepinephrine, epinephrine, dopamine, angiotensin, renin, and aldosterone. In aortas from the rats: elevated adrenergic α1D receptor, angiotensin AT1 receptor, and angiotensin protein and mRNA. In the kidney from the rats: Increase in angiotensin AT1 receptor, renin, and angiotensin protein and mRNA. In aortas and kidney from the rats: increase in phosphorylated Erk 1/2 (p-Erk 1/2) and NFATc expression. |
[122]/2014 |
| C57BL/6J mice, diabetic Akita and Leprdb/db mice |
Reduced aortic endothelial outgrowth in both diabetic mice clones, normalized by inhibitors of lysosomal enzymes/autophagy. Decreased VEGFR-2 in diabetic mice aortas. |
[123]/2012 |
| STZ-treated Glo1 overexpressing C57BL/6 mice |
Preventive effect of Glo1 overexpression on: diabetes-upregulated circulating inflammatory markers in mice, diabetes-reduced endothelial cell number in murine hearts, diabetes-caused deterioration in cardiomyocytes function associated with their enhanced death (via the stabilization of neuregulin, NOS, Bcl-2), diabetes-induced RAGEs and TNF-α in the murine hearts, diabetes-caused cardiac functions loss. |
[124]/2016 |
| STZ-treated Glo1 overexpressing C57BL/6 mice |
Effects of Glo1 overexpression: increased survival of BMCs (extracted from Glo1 diabetic mice) cultured in hyperglycemic (20 mM Glc) and proapoptotic conditions, associated with upregulation of anti-apoptotic Bcl-2 and Bcl-XL, and decrease in oxidative stress markers (protein carbonyls), maintenance of migratory potential of diabetic BMCs, recovery of neovascularization and blood flow in diabetic mice. |
[125]/2014 |
| Glo1 knockdown mice (C57BL/6J mice treated with Glo1 inhibitor - BBGC) | Observations in aortas extracted from the mice: increase in MG-H1, decrease in aortas sprouting (impaired angiogenesis). |
[126]/2019 |
| High-fat diet fed C57BL/6 mice | Increase in body weight, glycaemia, glucose intolerance and insulin resistance. Increased expression of NF-κB-p65 and HoxA5 in aortas. |
[127]/2019 |
| MGO and/or metformin (MET) treated C57BL/6 mice | Upon MGO treatment in murine blood/serum (partially restored with MET pretreatment): decrease in the levels of SOD, CAT, and GPX; increase in MDA; increase in proinflammatory cytokines (IL-1β and IL-6) and the anti-inflammatory cytokine IL-10. Upon MGO treatment in murine aortas (partially restored with MET co-treatment): increase in aortas thickness, apoptosis; decrease in Nrf2 expression and Akt phosphorylation. |
[128]/2022 |
| Mouse aortic tissue isolated from non-diabetic Glo1KD mice after insulin injection | Decrease in miR-190a expression and insulin sensitivity |
[129]/2017 |
| Mouse aortic tissue isolated from non-diabetic Glo1KD mice | Decrease in miR-214 expression |
[130]/2018 |
Table 2.
Methylglyoxal and its AGEs in cardiometabolic disorders – data from cell line experiments.
| Experimental model | MGO/MAGEs and associated major findings | Ref./year |
|---|---|---|
| Single aortic VSMCs from spontaneously hypertensive rats (SHR) and Wistar Kyoto rats (WKY) | In comparison with normal WKY, in VSMCs from SHR: higher MGO and AGEs levels, increased AGEs formation upon MGO exposition, increased oxidative stress (enhanced further by MGO exposition), decreased GSH/GSSG ratio, greater activation of NF-κB and expression of ICAM-1 (both enhanced further by MGO exposition). Increase in GSSG content in both WKY and SHR VSMCs upon MGO exposition. |
[131]/2002 |
| Rat L6 myoblasts | Upon MGO treatment (the myoblasts exposed to 0.5-2.5, 2.5 or 5 mM MGO for 10-30 min. – with 3% of MGO entering the cells) and insulin stimulation: reduced Glc uptake not mediated by ROS generation (improved by aminoguanidine), no changes in insulin receptor autophosphorylation, reduced IRS-1 tyrosine phosphorylation, no changes in serine/threonine IRS-1 phosphorylation, abolished IRS-1–associated PI3K activity (reversed by aminoguanidine), decreased PKB Ser/Thr phosphorylation (attenuated by aminoguanidine). Upon MGO treatment (the myoblasts exposed to 2.5 mM MGO for 30 min): increased p-ERK (partially mediated by ROS generation; prevented by aminoguanidine). Upon MGO treatment (the myoblasts exposed to 5 mM MGO up to 3h): ROS generation (reversed by aminoguanidine). |
[132]/2006 |
| L6 GLUT4 myc -tagged myoblasts | Upon MGO treatment (the myoblasts exposed to 100-400 µM for 24 h) (and previous insulin stimulation): increase in GLUT-4 on the plasma membrane Upon MGO treatment (the myoblasts exposed to 400 µM for 24 h) (without previous insulin stimulation): increase in MG-H1 (prevented by NAC but not by tiron); increase in GLUT-4 on the plasma membrane (prevented by NAC but not by tiron); increase in MG-H1 on GLUT-4; increase in ROS (prevented by NAC and tiron); decrease in Akt1 protein expression and increase in apoptosis; no effect on Akt2 and total Akt phosphorylation. |
[133]/2014 |
| 3T3-L1 cells (cell line from mouse adipose tissue); L8 cells (rat skeletal muscle cell line), H4-II-E cells (rat hepatocyte cell line), cloned INS-1E cells (derived from rat insulinoma) | Upon MGO-modified insulin treatment (3T3-L1 and L8 cells) in comparison with unmodified insulin stimulation: reduced Glc uptake. Upon insulin and MGO co-treatment (3T3-L1 cells exposed to 3-300 µM MGO) or MGO pretreatment (3T3-L1 cells exposed to 1-30 µM MGO for 24/48h) and insulin stimulation: no effect on Glc uptake. Upon MGO treatment (3T3-L1 cells exposed to 3 or 30 µM MGO for 24h): no effect on insulin receptor expression (at mRNA level). Upon MGO-modified insulin treatment (H4-II-E and INS-1E cells) in comparison with unmodified insulin: abolishing of C-peptide release by INS-1E cells and decrease in modified insulin clearance by H4-II-E cells. |
[68]/2006 |
| insulin-secreting INS-1E rat beta cells | Upon MGO treatment (the cells exposed to 0.25-1.0 mM MGO for up to 60 min – with 12.5% of MGO entering the cells): no impact on ROS generation. Upon MGO treatment (the cells exposed to different MGO levels within 0.25-1.0 mM MGO for 30 min, and insulin stimulation): no effect on IR Tyr phosphorylation; decrease in insulin-dependent IRS phosphorylation (prevented by aminoguanidine (AG)); decrease in insulin-dependent complex formation between IRS and PI3K p85 subunit (prevented by AG); decrease in insulin-dependent PKB phosphorylation at Thr 308 (prevented by AG); decrease in insulin-dependent (and PKB-catalyzed) GSK-3 phosphorylation at Ser (prevented by AG). Upon MGO treatment (the cells exposed to 0.5 mM MGO for 30 min, and 0.05 or 0.1 mM for 24 h): formation of CEL and argpyrimidine (AP) on IRS (prevented by AG); decrease in insulin-induced Pdx1, Ins1 and Gck mRNA expression (restored by AG). Upon Glc and MGO treatment (the cells exposed to 0.5 mM MGO for 30 min. and 0.05 or 0.1 mM for 24 h): decrease in Glc-stimulated insulin secretion (prevented by AG); decrease in Glc-stimulated PKB phosphorylation at Thr 308 (prevented by AG); decrease in Glc-induced Pdx1, Ins1 and Gck mRNA expression |
[134]/2011 |
| mouse insulinoma cells (MIN6) and rat insulinoma cells (INS-1) | Upon MGO treatment (0.05 mM or 0.1 mM for 3 h) in both cell lines: decrease in Glc-stimulated insulin secretion (prevented by NAC); increase in ROS (prevented by NAC). Upon MGO treatment (0.05 or 0.1 mM for 3 h) in MIN6 cells: increase in apoptosis (prevented by NAC); decrease in mitochondrial membrane potential (prevented by NAC); decrease in ATP synthesis (prevented by NAC); up-regulation of uncoupling protein 2 (UCP-2) (mRNA and protein) (prevented by NAC); increased expression of p-JNK, JNK, p-P38, and P-38. |
[135]/2016 |
| 3T3-L1 cells (cell line from mouse adipose tissue) | Upon MGO treatment (20 µM): reduced Glc uptake (improved by NAC), no changes in IR, IRS-1 and PI3K expression, reduced IRS-1 tyrosine phosphorylation and PI3K kinase activity (reversed by NAC). |
[100]/2007 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | Upon MGO treatment (3-300 µM for 45min.-18h): increase in hydrogen peroxide, nitric oxide, superoxide anion and peroxynitrite |
[89]/2005 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | Upon MGO treatment (30 µM for 18h) (and attenuated by alagebrium): increase in CEL, nitric oxide, nitrotyrosine in the cells; In the cells’ mitochondria: increase in ROS and superoxide anion; decrease in MnSOD activity; decrease in complex III activity of the respiratory chain, and decrease in ATP production. |
[90]/2009 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | VSMC upon MGO treatment (10, 30, 50 µM for 24 and/or 72 h): increase in DNA synthesis and cells proliferation (abolished by Akt inhibitor and in Akt1 knock-down VSMC); increase in Akt1 phosphorylation at Ser-473, and GSK-3α/β phosphorylation; decrease in total p21; increase in phosphorylated p21 and p27; increase in CDK2 activity. VSMC upon MGO treatment (100 µM for 24h): increase in cells apoptosis. |
[120]/2011 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | Upon fructose or Glc treatment (25 mM for 6, 12 or 24h): increase in MGO; upregulation of Glo1 and Glo2 (at the protein level); Upon fructose treatment (25 mM for 6, 12 or 24h): upregulation of GLUT-5 and fructokinase (at mRNA levels), and aldolase B (at mRNA and protein levels) – further enhanced by fructose + insulin co-treatment Inhibition of fructose-induced MGO increase by aldolase B knock-down. Upon Glc treatment (25 mM for 12h): downregulation of aldolase A and upregulation of aldolase B. Inhibition of Glc-induced MGO increase by aldolase B knock-down and inhibitors of polyol pathway. |
[88]/2011 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | Upon MGO (30 µM for 24 h) or Glc treatment (25 mM for 24 h): increase in adrenergic α1D receptor, angiotensin AT1 receptor, and angiotensin protein and mRNA (attenuated by alagebrium); Upon MGO (30 µM for 24 h): increase in phosphorylated Erk 1/2 (p-Erk 1/2) and NFATc expression (attenuated by alagebrium); increase in the protein and mRNA for NF-κB, angiotensin, AT1 receptor, and adrenergic α1D receptor (attenuated by alagebrium); Upon RAGE siRNA: attenuation of the increase in RAGE and NF-kB p65 protein expression (induced by MGO). Upon angiotensinogen siRNA: attenuation of the increase in NF-kB p65, angiotensin, AT1 receptor, and adrenergic α1D receptor protein expression (induced by MGO). |
[122]/2014 |
| A-10 cells: rat thoracic aortic SMC line (VSMC) | Upon Fru treatment (15 and/or 30 mM): increase in MGO, peroxynitrite, nitric oxide and superoxide anion. Upon MGO treatment (10 µM, 6h): increase in nitric oxide and superoxide anion, increase in iNOS staining |
[136]/2006 |
| VSMCs isolated from the thoracic aorta of male Wistar rats | Upon MGO treatment (10 µM, 3 or 9 h): upregulation of ER stress markers (induction of PERK phosphorylation, increase in IRE1α and ATF6 expression); no effect on apoptosis |
[137]/2022 |
| Human immortalised endothelial cells (ECRF-24) | Effects of Glo1 silencing in ECRF-24: decrease in the cells viability; upregulation of pro-inflammatory factors (MCP-1, IL-6, TNF); upregulation of vascular activating factors (VCAM-1 and ICAM-1). |
[105]/2014 |
| Mouse aortic endothelial cells (MAECs) | Upon 0.5 Mm MGO (up to 16 h) or Glo1 inhibitor (BBGC) treatment, followed by insulin stimulation): suppression of insulin-stimulated IRS-1 tyrosine phosphorylation and Akt activation; suppression of insulin-stimulated eNOS activation (reduction of Ser-1177 phosphorylation and threonine-497 dephosphorylation) and NO production. Upon 0.5 Mm MGO treatment (up to 16 h): Induction of ERK ½ phosphorylation and endothelin-1 release (comparable to insulin effect). Upon Glo1 inhibitor (BBGC) treatment: Induction of ERK½ phosphorylation(comparable to insulin effect). Reversal of the above MGO effects by ERK½ inhibitors. |
[109]/2014 |
| Human umbilical vein endothelial cells (HUVECs) treated with 0.56 mM (1-24 h) MGO or 0.56 mM MGO and NAC | The below effect was attenuated by Telmisartan. Upon MGO treatment: increase in the cells apoptosis |
[138]/2008 |
| Human umbilical vein endothelial cells (HUVECs) treated with 1 mM MGO or 1 mM MGO and NAC | All of the below effects were attenuated by NAC. Upon MGO treatments: increase in ROS, decrease in SOD-1 and GPX-1 protein expression; decrease in phosphorylated forms of Akt and eNOS. |
[108]/2021 |
| Human umbilical vein endothelial cells (HUVECs) | Upon MGO treatment (200 μM for 24 h) (reversed by metformin pretreatment): increase in apoptosis (increase in cleaved caspase-3 and increase in the Bax/Bcl-2 ratio); decrease in Akt phosphorylation, and Nrf2 and HO-1 expression; Upon MGO treatment (200 μM for 1 h) (reversed by metformin pretreatment): increase in ROS and MDA, decrease in SOD, CAT, and GPX-1 activities; decrease in mitochondrial membrane potential and structural damage to the mitochondrial membranes. |
[128]/2022 |
| Primary aortic endothelial cells isolated from C57BL/6 mice, cultured with Glo1 inhibitor (BBGC) | Increased expression of adhesion molecules (VCAM, ICAM-1, tetherin, MCP-1) followed by increased adhesion of monocytes to endothelium. |
[107]/2014 |
| Primary aortic endothelial cells isolated from RAGE KO mice, cultured with Glo1 inhibitor (BBGC) | Increased expression of adhesion molecules (VCAM, ICAM-1, tetherin) followed by increased adhesion of monocytes to endothelium |
[107]/2014 |
| Rat aortic endothelial cells (RAECs) from SD rats, Human umbilical vein endothelial cells (HUVECs) |
Upon 25 mM Glc exposition for 24 h: increase in MGO level in both RAECs and HUVECs. Upon MGO exposition (30 µM for 3 or 24 h): increase in NOX activity, ROS generation in both cell lines, decrease in NO production by eNOS and decrease in GSH in both cell lines, decrease in Ser-1177 phosphorylation in eNOS in HUVECs (prevented by AG) |
[118]/2010 |
| MGO-exposed bone marrow-derived EPCs (from C57BL/6 mice) | MGO effect: decrease in EPCs viability, downregulation of VEGFR-2 (mRNA and protein decrease), impairment in blood vessel tube formation in EPCs, restoration of MGO-induced EPCs dysfunctions by RAGE antagonist. |
[139]/2018 |
| MGO-exposed bovine aortic endothelial cells (BAEC) and human umbilical cord vein endothelial cells (HUVEC) | MGO effect: downregulation of VEGFR-2 (protein decrease) in both cell lines in a dose- and time-dependent manner (enhanced in BAEC by Glo1 downregulation, and abolished by Glo1 up-regulation), around 80% reversal of VEGFR-2 reduction in RAGE-knock-down cells, decrease in tube formation and BAEC cell migration. |
[123]/2012 |
| Human U937 monocytes |
Hypoxia increased MGO level. TNF decreased Glo1 activity and increased MGO, MG-H1 and CML levels. TNF increased IL-8, MCP-1 and MMP-9. Hydrogen peroxide increased CML. High Glc (30 mM) had no effect on Glo1, MGO, AGEs. Exposition of the cells on MGO/CML had no effect on IL-8, MCP-1 and MMP-9. MGO exposition increased MG-H1 and CML. MGO exposition induced apoptosis. Glo1 attenuation (with siRNA) had no effect on MGO production. |
[140]/2014 |
| Human vascular endothelial cells (HVECs) obtained from patients with coronary heart disease | Upon MGO, GO or combined MGO + GO exposition (150 – 300 µM for 72 h): induction of senescence by increasing ROS production and p21 expression; increase in CML and MG-H1; arrest of the cells in the G2 cell cycle phase. The above MGO/GO induced effects were prevented by aminoguanidine. Reduction of Glo1 expression. |
[141]/2017 |
| Human aortic endothelial cells (HAECs) | Upon MGO (1-100 μmol/L MGO for 20 min) treatment: increase in superoxide production from mitochondria; partial stimulation of NOS. |
[142]/2010 |
| Human aortic endothelial cells (HAECs) | Upon MGO (1 or 5 mM for 2-8 h) treatment: decrease in cell viability; decrease in thioredoxin protein and mRNA levels (abolished by metformin); oxidative damage of peroxiredoxin; increase in ROS and mitochondrial-dependent apoptosis. |
[143]/2012 |
| Human aortic endothelial cells (HAECs) | Upon Glc treatment (20 mM for 2-6 days): increase in MGO, D-lactate and MG-H1 in the cells and cell culture; down-regulation of Glo1 (activity and protein, but not mRNA); up-regulation of (among others) UPR pathways (e.g. heat shock proteins); down-regulation of a mediator of EC migration (annexin-A1), an anti-coagulatory factor (annexin-A5); an anti-inflammatory factor (chromobox protein homolog-5; (CBX5); increase in hexokinase-2 (protein but not mRNA) and glycogen. |
[144]/2019 |
| Human aortic endothelial cells (HAECs) from healthy (H-HAECs) and T2DM (D-HAECs) donors |
D-HAECs – impaired network formation, proliferation, increased apoptosis (in comparison with H-HAECs). MGO (10µM)-treated H-HAECs – impaired network formation, proliferation, increased apoptosis – reversed by KATP channel inhibition. D-HAECs – upregulation of three MAPK pathways (p-JNK, p-p38, and p-ERK) (in comparison with H-HAECs). MGO (10µM)-treated H-HAECs – upregulation of three MAPK pathways (p-JNK, p-p38, and p-ERK) – reversal of JNK activation by KATP channel inhibition. |
[126]/2019 |
| Mouse aortic endothelial cells (MAECs) isolated from Glo1KD and WT mice; MGO or Glo1 inhibitor treated mouse coronary artery endothelial cells (MCECs) |
Glo1KD MAECs (in comparison with WT MAECs): decreased cell growth, proliferation, migration and Matrigel invasion; 2-fold upregulation of NF-κB-p65 and antiangiogenic HoxA5 (prevented by AG) associated with downregulation of VEGFR-2 (no change in VEGF expression); improvement of migration and invasion by HoxA5 silencing. Upon 0.5 Mm MGO (for 16 h) or Glo1 inhibitor (BBGC) treatment of MCECs: decreased migration; upregulation of NF-κB-p65 and HoxA5. |
[127]/2019 |
| MGO-treated mouse aortic endothelial cells (MAECs); MGO-treated human endothelial cells (HUVECs) | Upon 0.5 Mm MGO (for 16 h) treatment of MAECs and HUVECs: downregulation of miR-190a. Upon miR-190a inhibition: decrease in insulin-induced Tyr phosphorylation of IRS1, and AKT phosphorylation at Ser473 and Thr308 (no effect on IR); impairment of insulin-dependent eNOS activation/NO release; increase in ERK1/2 phosphorylation. Upon miR-190a over-expression: prevention of MGO impairment of insulin stimulated IRS1/Akt/eNOS/NO release pathway. Upon MGO exposure and miR-190 inhibition: Upregulation of GTPase Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS). |
[129]/2017 |
| MGO-treated mouse aortic endothelial cells (MAECs) | Upon 0.5 Mm MGO (for 16 h): downregulation of miR-214. Upon MGO exposure and miR-214 inhibition: 4-fold upregulation of PH domain leucine-rich repeat protein phosphatase 2 (PHLPP2). Upon miR-214 overexpression: downregulation of PHLPP2. Upon miR-214 overexpression in MGO treated and insulin-stimulated MAECs: Reversal of MGO-impaired Ser473 phosphorylation on Akt |
[130]/2018 |
| Human endothelial cells (HUVECs and microvascular endothelial cells from human foreskin) | No binding neither activation of endothelial cells by MGO-modified albumin and CML-modified albumin (no increase in adhesion molecules – VCAM-I, ICAM-I and E-selectin) | [56]/2006 |
| Human endothelial cells (HUVECs) | Hyperglycemic conditions (30 mM Glc) caused: increase in MGO and argpyrimidine (AP) in the cells (no impact on 2-deoxyglucosone, glyoxal, CML and CEL), Hsp-27 was the major AP-modified protein. decrease in cells proliferation. |
[145]/2006 |
| Human endothelial cells (HUVECs) | Upon 0.5 Mm MGO (for 24 h) treatment (reversed by phosphocreatine and NAC): increase in apoptosis (caspases upregulation and Bcl-2/Bax decrease); increase in ROS and calcium; upregulation of NOX4; decrease in mitochondrial membrane potential; decrease in Akt and eNOS phosphorylation; decrease in cGMP and NO; induction of NF-κB. |
[146]/2017 |
| Human endothelial cells (HUVECs) | Upon TNF-α (12.5ng/ml) and MGO (800 µM for 24 h) treatment: down-regulation of genes mainly associated with cell cycle (topoisomerase (DNA) II alpha, marker of proliferation Ki-67, cyclin A2, etc.); up-regulation of heme oxygenase-1, insulin like growth factor binding protein 3, plasminogen activator inhibitor 2, and others; decrease of VCAM-1. Some MGO-induced effects prevented by L-carnosine (20 mM). |
[147]/2019 |
| Human endothelial cells (HUVECs) | Upon MGO (800 µM for 5 h) treatment: increase in DNA damage and p53 phosphorylation (at Ser15); decrease in mTORC1 targets phosphorylation (4EBP1 and p70S6K); increase in autophagy; increase in protein carbonylation; no effect on GSH/ GSSG. |
[148]/2021 |
| Human endothelial cells (HUVECs) |
Upon MGO (500 µM) and MGO-modified Hb: increase in HUVECs apoptosis; decrease in HUVECs proliferation and migration; Upon MGO-modified Hb: increase in ROS, decrease in mitochondrial membrane potential; decrease in phosphorylated JNK and p-38; Upon MGO (500 µM): increase in phosphorylated JNK. |
[66]/2021 |
| eNOS overexpressing human endothelial cells (HUVECs) | No inhibition of eNOS activity by MG-H1 and AP | [149]/2008 |
| Human endothelial cells (HUVECs and EA.hy926) | MGO exposition induced apoptosis via ROS generation and c-FLIPL downregulation (c-FLIPL downregulation was probably mediated by inactivation of NF-κB pathway through p65 downregulation). | [150]/2017 |
| Human endothelial cells (EA.hy926) | Upon MGO treatment (50 – 200 μM for 2-8 h): increase in superoxide radical (probably produced by eNOS through MGO-induced eNOS uncoupling); Upon MGO treatment (100 μM for 8 h): decrease in eNOS phosphorylation (at Ser-1177) |
[151]/2013 |
| Glo1 KO human cell line (Glo1-/- HEK293 cells) | No elevation of MGO. |
[54]/2018 |
| Glo1 knockdown human cell line (GLO1-siRNA-transfected HAECs) incubated at high Glc concentration (25 mM) | MGO increase. No increase in MG-H1 of cellular proteins. Increase in MG-H1 free adduct in the culture medium. No impact on eNOS Downregulation of some structural proteins and enzymes metabolizing collagen. Increase in endothelin-1, collagen 1 and 5 expression. Induction of apoptosis. Increase in markers of inflammation and endothelial dysfunction (IL-6, RAGE, MCP-1, sVCAM-1, sICAM-1). |
[152]/2016 |
| Glo1 overexpressing human cell line (GLO1-transfected human dermal microvascular ECs (HMEC-1 cells) incubated at high Glc concentration (20 mM) | 4-fold increased GLO1 activity improved hyperglycemia diminished tube formation. | [153]/2008 |
| Glo1 overexpressing human cell line (GLO1-transfected human cardiac ECs (HCEC cells) exposed to MGO (5 µM), high glucose (30 mM), TNF-α (10 ng/mL) | 9-fold increased GLO1 expression protected from cell death induced by MGO, high glucose, TNF-α. | [124]/2016 |
2.2.1. Enzymatic mechanisms compensating for deficient glyoxalases
In search for the effects of Glo1 deficiency on the phenotype associated with MGO level and MAGEs generation, Glo1 knockout (Glo1 KO) models have been designed [27,101]. Experiments on such models have shown the upregulation of both MGO and MG-H1, or either of them in yeasts (S. cerevisiae) [6], worms (C. elegans) [69,154], flies (D. melanogaster) [155], and fish (D. rerio) [156]. However, when Glo1 KO mice, as well as murine and human Glo1 KO cell lines have been applied, no alterations in MGO/MG-H1 were reported [54,80,81], or the increase in MG-H1 was observed in the murine liver but not in the brain [101]. In Glo1 KO mice, alternative MGO scavenging pathways seem to compensate for the deficient glyoxalase system, which is not observed in less-organized animals [27]. Such a phenomenon has been reported by Schumacher et al. [80] and Morgenstern et al. [81]. The first team observed that the total deletion of Glo1 did not seem to impair the animals condition, both with and without diabetes induction [80]. In turn, they reported the upregulation of aldoketo reductases (AKR1b3) and aldehyde dehydrogenases (ALDH1a3), as well as the shift in percentage of MGO metabolites from D-lactate towards hydroxyacetone (mainly in the kidney) and pyruvate (in the liver). Therefore, the authors suggest the compensatory function of AKRs (showing much greater upregulation when compared to ALDHs) in MGO detoxification, especially that neither MGO nor MG-H1 were increased in their experimental model. Similarly, neither MGO nor its MAGEs (MG-H1 and CEL) elevations have been reported by Morgenstern et al. [81] in murine Glo1 KO Schwann cells, which instead showed an increased expression of several subtypes of AKRs and ALDHs. Therefore, in pathological conditions associated with excessive MGO generation, including cardiometabolic disturbances, the function of glyoxalases system might be partially compensated by respective reductases and dehydrogenases in the human organism.
2.2.2. MGO/MAGEs in insulin resistance development
Insulin resistance, a condition observed in metabolic syndrome as well as in T2DM, and implicated in cardiometabolic disorders, is characterized by the impairment of insulin-triggered signaling pathways which leads to disturbances in insulin-controlled metabolism of carbohydrates and lipids, as well as endothelial dysfunction. The main organs affected by insulin resistance include the liver, adipose tissue, skeletal muscles, endothelium, as well as pancreas.
As discussed by Nigro et al. [157] and Shamsaldeen et al. [158], MGO accumulation in pathology is implicated in insulin resistance development both through the modification of this hormone molecule itself as well as the components of its signaling pathways.
In skeletal muscles insulin resistance is mainly featured by decreased Glc uptake caused by an inefficient mobilization of Glc transporters (GLUT-4) which are normally increased upon insulin induction. MGO has been shown to accumulate in metabolically impaired skeletal muscles as a result of lowered efficiency of its main scavenging system (glyoxalases) [159]. En excess of MGO disturbs insulin signaling and promotes oxidative and inflammatory processes, which is associated with mitochondrial damage (including mitochondrial DNA), MAGE formation (MG-H1) and structural changes of muscle proteins [159 and references therein].
In search for the effect of MGO on insulin resistance in skeletal muscles, MGO-exposed and insulin-stimulated rat myoblasts have been examined [132,133]. Whereas a short-term exposition to high concentration of MGO decreased Glc uptake by the cells (probably by the modification of IRS-1, which lowered its insulin-induced tyrosine phosphorylation, followed by the impairment in PI3K mobilization and PKB phosphorylation) [132], longer exposition to low MGO levels caused increased Glc uptake [133] (Table 2). In the latter study, MGO was shown to interfere with GLUT-4 transporters translocation, diminishing their endocytosis and hence increasing their number on the myocytes surface. Although MGO-induced ROS generation was observed in these cells, MGO effect on GLUT-4 seemed to be independent of oxidative stress. Additionally, MGO-induced apoptosis of myocytes as well as GLUT-4 modification (with MG-H1 formation) was reported in this study [133]. These observations indicate at MGO interference with Glc uptake by skeletal muscle cells [132,133] (Table 2). However, neither an impact on insulin receptor autophosphorylation, serine/threonine phosphorylation of IRS-1, nor Akt phosphorylation were found upon MGO treatment [132,133].
Visceral adiposity associated with metabolic syndrome and further complications leads to initiation of chronic inflammation (connected with a shift towards proinflammatory macrophages yielding secretion of IL-6 and TNF-α), disturbances in adipokines profile (augmented leptin secretion paralleled by decreased adiponectin), coupled with insulin resistance development [82]. An impact of MGO on these processes has been studied in rodent adipocytes, where its inhibitory effects on Glc uptake, IRS-1 tyrosine phosphorylation and PI3K kinase activity were observed [100,119,121] (Table 1 and Table 2). Additionally, MGO-fed rats developed some pathological features characteristic for insulin resistance and diabetes, such as lowered insulin sensitivity, enhanced free fatty acids levels and decreased adiponectin in the circulation, as well as pro-apoptotic, pro-fibrotic and pro-inflammatory characteristics in the adipose tissue [112,114,115,117] (Table 1). However, not all of the data coming from experiments on MGO-fed rats reported the impairment of glycemia or insulinemia [113] (Table 1). Other MGO-induced disturbances in adipose tissue included impairment of blood vessels formation associated with increased hypoxia [114] (Table 1). Additionally, MGO seemed to stimulate (adrenaline-induced) lipolysis [115] which might be mediated by MGO-caused degradation of perilipin A [113,114] (Table 1). Since perilipins are proteins stabilizing lipid droplets and protecting them from lipases [160], their decrease would lead to enhanced hydrolysis of triacylglycerols, yielding efflux of free fatty acids to the circulation.
Similarly, MGO-injected mice have developed systemic insulin resistance resulting from the impairment of insulin-triggered signaling pathway, as observed in the murine aortas and endothelial cells [109] (Table 1 and Table 2). MGO treatment caused the suppression of insulin-induced pathway leading through the activation of IRS-1, Akt and eNOS, probably partially via the induction of ERK ½ which inhibited IRS-1. In this way MGO seems to disturb the balance between the processes yielding vasorelaxation (NO production), and the routes ending up with vasoconstriction (endothelin-1 production) in favor of the latter [109].
MicroRNA oligonucleotides (miRNAs) are responsible for the post-transcriptional regulation of components of multiple signaling pathways, including insulin-triggered routes, hence are implicated in different disorders encompassing cardiometabolic diseases [161,162]. In search for the elucidation of miRNAs role in MGO-induced insulin resistance in endothelial cells, Mirra et al. [129] have performed diabetes-associated miRNAs profiling in murine endothelial cells exposed to MGO. They found four miRNAs downregulation, from which two: miR-190a and miR-214 affected MGO-induced insulin resistance in endothelium leading to its dysfunction [129,130]. MGO seems to impair insulin-triggered pathway (IRS1/Akt/eNOS/NO release) through downregulation of miR-190a, which in turn is associated with KRAS GTPase upregulation. Inhibitory effect of MGO on miR-190a may be connected with its modification/activation of histone deacetylase (HDAC), thus epigenetically restraining miR-190a synthesis[129] (Table 1 and Table 2). Similarly, MGO-caused miR-214 downregulation is associated with the disturbance in insulin signaling, as shown by its effect on Akt activity [130] (Table 1 and Table 2). Namely, miR-214 seems to be a negative regulator of PH domain leucine-rich repeat protein phosphatase 2 (PHLPP2) – the enzyme which inactivates Akt via its dephosphorylation. miR-214 inhibition (comparably with MGO exposition) led to 4-fold increase in PHLPP2, which attenuated insulin-induced Akt pathway in murine endothelial cells [130] (Table 2). Therefore, it might be suggested that MGO effect on endothelial cells is mediated by down-regulation of two miRNAs (miR-190a and 214), followed by the inhibition of insulin-triggered Akt pathway, shifting the balance from vasodilation towards vasoconstriction due to decreased NO generation.
Except for disturbing downstream components of insulin signaling pathway, MGO seems to modify insulin molecule itself, as has been reported by Jia et al. [68]. In light of these authors’ findings it is rather MGO-modified insulin which impairs Glc uptake both by adipocytes and skeletal muscle cells, and not free MGO. Additionally, MGO-modified insulin lost the ability for attenuation of insulin release by pancreatic β-cells (impaired feedback inhibition) and was inefficiently cleared by hepatocytes [68] (Table 2).
The impact of MGO on insulin- or Glc-stimulated signaling and its consequences in rat pancreatic β-cells has been investigated by Fiory et al. [134]. The authors observed that MGO inhibited insulin secretion by Glc-induced pancreatic β-cells, which was associated with the attenuation of PKB activation. Additionally, MGO-caused inhibition of several components of insulin-triggered signaling pathway was found (IRS, PI3K, PKB, GSK-3), as well as the reversed by MGO insulin- and Glc-induced up-regulation of three genes coding for pancreatic duodenal homeobox-1, insulin and glucokinase. Impairment of insulin signaling pathway was probably associated with MGO modification of IRS, since CEL and AP adducts were detected on this protein upon MGO exposition [134] (Table 2). Similar inhibitory effect of MGO on insulin secretion by pancreatic β-cells upon Glc induction has been observed by Bo et al. [135]. However, different signaling routes were analyzed in the latter study; namely these leading through ROS generation and MAPK pathway upregulation. MGO induced oxidative stress and apoptosis, and these effects were associated with up-regulation of uncoupling protein 2 (UCP-2), decrease in mitochondrial membrane potential and ATP synthesis, as well as increased expression and activation of JNK and P-38 kinases, finally resulting in insulin secretion inhibition [135] (Table 2).
Therefore, MGO seems to be involved in insulin resistance development through the modification of the hormone molecule in the circulation, as well as alteration of insulin-triggered signaling components intracellularly (in endothelial cells, adipocytes, myocytes and pancreatic β-cells), which impairs pancreas functionality, as well as insulin-regulated vascular homeostasis and metabolism of lipids and carbohydrates in the adipose tissue and skeletal muscles. MGO-affected components probably include IRS-1, PI3K and PKB/Akt, but not insulin receptor [68,100,119,121,132,134]. Moreover, MAPK pathway, oxidative stress and UCP-2 up-regulation coupled with mitochondrial dysfunction and apoptosis triggering – all caused by MGO - probably lead to pancreatic β-cells impairment [135].
Due to the inhibitory effects on Glc and insulin removal from the circulation, MGO/MAGE seem to be important factors contributing to hyperglycemia and hyperinsulinemia. On the other hand, MGO might also contribute to the reduction of insulin in the circulation, the phenomenon observed in later stages of T2DM. Such an effect has been reported by Dhar et al. [121] who found out that MGO treatment of rats diminished Glc uptake by pancreatic cells, enhanced their apoptosis and inhibited insulin secretion (Table 1).
2.3. MGO, its metabolic products and MAGEs in patients with metabolic syndrome and diabetes
Studies on MGO impact on pathomechanisms of disease development mostly focus on its participation in the development and perpetuation of metabolic syndrome and diabetes with their macro- as well as micro-complications.
In vitro studies on red blood cell suspension indicated that the level of MGO, S-D-lactoylglutathione and their end-metabolite - D-lactate in culture had been elevated under hyperglycemic conditions [28]. At the same time the activities of Glo1 and Glo2 did not exhibit any elevation. This led to the conclusion that periodic hyperglycemia may lead to the development of complications associated with diabetes. Indeed, subsequent studies revealed that the systemic concentrations of MGO, S-D-lactoylglutathione and D-lactate are elevated in diabetic patients, as compared to healthy subjects, pointing to the increased flux of metabolites through glyoxalase system [10,16,18,19]. It has been demonstrated that plasma MGO was able to discriminate between patients with both T1DM [Ref. 21 in [9]] and T2DM and healthy subjects [9]. However, MGO is very reactive and therefore the end-product of its metabolism - D-lactate is often measured, as a surrogate marker reflecting MGO concentration [16]. Nevertheless, it has been pointed out that it would be worth elucidation to which degree D-lactate is the product of MGO conversion or the result of gut bacteria metabolism, since they are also able to produce this compound [16]. The concentration of MGO metabolites has been increased several fold both in insulin-dependent as well as non-insulin-dependent diabetes, however there were differences between those two types of diabetes in respect of glyoxalase system enzymes – while Glo1 activity has been upregulated in both types of diabetes, Glo2 exhibited elevation in non-insulin-dependent diabetes only [10]. This results in the accumulation of S-D-lactoylglutathione in the circulation of insulin-dependent diabetes and negative correlation between D-lactate and GSH. Moreover, a positive correlation had been noted between the level of D-lactate and HbA1c [10].
Scheijen at al. [16] studying blood and urine samples of T2DM patients also observed a positive correlation between D-lactate and HbA1c. The same correlation has been found in the study conducted by Beisswenger et al. [17], although only in patients who were not treated with metformin. Administration of metformin, commonly used in diabetes treatment, caused that this relationship was no longer observed. Additionally T2DM patients treated with metformin had lower systemic levels of MGO and higher levels of D-lactate [17]. The authors proposed two possible explanations – either metformin binds α-dicarbonyl group of MGO or intensifies MGO detoxification through glyoxylase pathway and thus increases the concentration of D-lactate [17].
Hyperglycemia leads to the increased formation of triosephosphate metabolites and MGO, but what is important, in the case of diabetic patients, these toxic metabolites are accumulating even at normal glucose levels [11]. It may be in part due to the fact that metabolism in diabetic subjects is faster. As a result high concentrations of fructose-1,6-bisphosphate (FBP) are being produced, the compound, which after splitting leads to the formation of glyceraldehyde-3-phosphate (GAP), which should be further processed by GAP dehydrogenase (GAPDH). Unfortunately, GAPDH could be downregulated by ROS, which can possibly lead to the elevation of GAP, which after conversion to DHAP can be a source of MGO [11]. This hypothesis, however, requires further studies.
Since the diabetic patients are exposed to the higher levels of MGO precursors, also coming from sources other than glucose, therefore, they are more susceptible to the development of diabetic complications even if their glycemic status is under control [11,163].
Because D-lactate is a relatively stable end-product of MGO metabolism, Scheijen at al. [16] proposed that it could be considered as a possible indicator of diabetic complications.
Later Schumacher et al. [80] have reported the disturbances of an alternative pathway of MGO scavenging in diabetic patients. Whereas the components of the glyoxalase pathway; Glo1 activity and D-lactate concentration did not discriminate between diabetic patients with and without complications, AKRs activity and hydroxyacetone level showed such a potential. T2DM patients without complications had the highest concentration of hydroxyacetone and the greatest activity of MGO-dependent AKR in erythrocytes. As the authors suggest, in the case of advanced diabetes it might be the minor pathway of MGO detoxification (leading via AKRs) which compensates for the faulty glyoxalase system. These findings are in agreement with the authors observations obtained in Glo1 -/- murine model mentioned earlier [80].
McLellan et al. [10] observed that duration of diabetes positively correlated with diabetic complications such as retinopathy, nephropathy and neuropathy occurrence. They found elevated concentrations of MGO in the blood of diabetic patients and observed that the development of complications is the result of chronic exposure to high doses of this compound. They indicated that patients with such complications had higher age, HbA1c concentrations and Glo1 activity than patients without the complications. They concluded that patient’s age and duration of the disease are risk factors for the development of diabetic complications. The authors found HbA1c to be a risk factor for diabetic complications development and D-lactate as a risk factor for retinopathy. However, in the latter case low D-lactate levels is posing higher risk, possibly due to the slow conversion of increased levels of MGO to D-lactate and hence longer and higher exposure.
Exposition to high glucose levels leads to the production of MGO and to intensification of MAGEs production that changes the properties of proteins. Tubular cells are responsible for restoration of proteins and peptides and this function is impaired in diabetes since in hyperglycemia tubular cells have lower ability to handle RCS-modified proteins [164]. RCS--mediated nucleoside modifications have also been demonstrated in the kidneys of diabetic patients [72]. Elevated accumulation of CEdG has been noted in the kidneys of these patients and it has been suggested that it may lead to the loss of genetic integrity in the kidneys of diabetic patients [72].
Chou et al. [15] observed that in the early stages of kidney damage the level of D-lactate is elevated, while in the advanced stages it is declining. It was reflecting the fluctuations in MGO production. The authors suggested that in the early stages of renal dysfunction self-reparatory mechanisms require huge energy input and hence the glycolytic flux is intensified. Later, when reparative processes are no longer possible, fibrosis of tissue is progressing and less energy is needed, hence lower MGO and subsequent D-lactate production [15].
Diabetic patients with neuropathy can suffer from pain and hyperalgesia. Glo1 activity in peripheral nerves is low which leads to accumulation of MGO [165,166]. Bierhaus et al. [9] observed increased concentrations of plasma MGO in patients with pain and that MGO can discriminate between diabetic patients with pain and without pain. They demonstrated that MGO is causing depolarization of sensory neurons and is leading to the changes in voltage-gated sodium channel Nav1.8. In a very interesting experiment they subjected wild-type mice dorsal root ganglion neurons to plasma of diabetic patients with and without pain and measured COX-2 as a surrogate marker of neural function. They observed that plasma from pain-suffering patients induced higher COX-2 transcription than plasma from patients without pain. What is interesting spiking of plasma from diabetic patients without pain with MGO caused COX-2 expression elevation. The mechanism by which MGO can influence channel function is so far unknown and it is opening new research avenues connected to pain perception and therapeutic goals.
3. MGO and MAGEs in cardiovascular disorders
3.1. Pathological routes linking metabolic syndrome and diabetes with cardiovascular complications
Pathological features characteristic for metabolic syndrome and diabetes (insulin resistance, hyperglycemia, hypertension, dyslipidemia and obesity) increase the risk of cardiovascular disease (CVD) [167-169]. Micro- and macrovascular complications are typical for the patients suffering from diabetes, and macrovascular pathologies mostly conditioned by atherosclerosis development yield cardiovascular diseases including myocardial infarction [5,30, 167,170,171]. Angiopathy, underlying these events, is associated with the dysfunction of vascular endothelium caused by oxidative stress, inflammatory processes, and ER stress, which impairs its vasodilatory functions, increases permeability, and enhances proatherogenic and prothrombotic features [169,171,172]. Both insulin resistance and hyperglycemia have been shown to decrease the generation of nitric oxide in endothelial cells, and stimulate the production of plasminogen activator inhibitor-1 (PAI-1) [Rev. in [30], what would lead to impaired blood flow and hypertension as well as disturbances in thrombolytic functions. Additionally, insulin resistance and hyperglycemia are implicated in dyslipidemia associated with increase in lipoproteins/triacylglycerols as well as free fatty acids in the circulation [30]. For example diabetic mice have been reported to show decreased clearance of apo-B-48 lipoprotein remnants, which was associated with the dysfunction of extracellular matrix (ECM) components (perlecan HSPG) impairing the lipoproteins removal from the circulation [173].
The mechanisms which link increased glycolytic flux with endothelial and hence cardiovascular impairment, consider the stimulation of side pathways of glycolysis leading to the overproduction of sorbitol (polyol pathway), generation of glucosamine-6-phosphate (hexosamine pathway), and overproduction of trioses due to the inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Trioses accumulation leads to the activation of protein kinase C (PKC) (by overproduced DAG) as well as MAGEs generation (due to the excessive generation of MGO) [5,30,169]. Sorbitol production catalyzed by aldose reductase is associated with the depletion of NADPH (exhausted in this reaction) and hence the decrease in GSH. Consequently, the polyol pathway would exacerbate the oxidative stress [30]. Hyperglycemia-induced hexosamine pathway leads to the generation of UDPGlcNAc molecules, whose accumulation enhances binding of GlcNAc moieties to many proteins, what modifies their functions. For example GlcNAc-glycosylated transcription factor Sp1 seems to induce the expression of PAI-1 gene, whereas GlcNAc binding with eNOS impairs its activity. Such mechanisms result in PAI-1 increase and NO decrease [30]. Similar effects on PAI-1 and eNOS are observed upon PKC activation. Additionally, DAG-PKC-mediated signaling would upregulate endothelin-1 (ET-1), VEGF, TGF-β, NF-κB, and NADPH oxidases (NOXs). Such effects are associated with the induction of oxidative stress (via NOXs), inflammatory processes (NF-κB signaling), blood flow disturbances (eNOS decrease and ET-1 increase), angiogenesis and vascular permeability (VEGF), as well the occlusion of blood vessels resulting from an impairment of fibrinolysis (PAI-1 impact) and an excessive production of ECM components (type IV collagen and fibronectin possibly upregulated by TGF-β) [30,174]. Trioses-derived dicarbonyl molecules (mainly MGO) show much greater efficiency in AGEs formation in comparison to glucose [30]. Therefore, they modify intra- and extracellular proteins, the latter being components both of ECM and plasma proteins. These actions impair the functionality of blood vessels; e.g. via decreasing their elasticity through the disturbances in collagens structure, or stimulating pro-oxidative, pro-inflammatory and pro-coagulatory processes (induced by blood plasma AGEs binding with their receptors on macrophages or endothelial cells) [30]. The upregulation of AGEs receptors (RAGEs) and their ligands being observed under hyperglycemic conditions, further adds to the ROS and inflammation enhancement, contributing to vascular endothelium destruction [175].
As proposed by Brownlee and Giacco [30,170,175], all five pathological pathways mentioned above (polyol, hexosamine, DAG-PKC, MGO-MAGEs and RAGEs) are initiated by hyperglycemia-induced ROS generation by the mitochondrial respiratory chain. Enhanced reactive oxygen species would damage nuclear DNA, which in turn would activate PARP. Subsequently, active PARP would modify GAPDH via ADP-ribosylation (using NAD+). This would lead to the inhibition of GAPDH and the obstruction of glycolytic pathway at trioses level. Consequently, the accumulation of the above mentioned side products and their detrimental effects are observed. Since most of these pathological pathways further stimulate ROS generation, auto-augmentation of such routes would deepen the metabolic disturbances in the vicious circle mode. This mechanism seems to be similar in the case of both micro- and macrovascular complications. However, the causative relationship between hyperglycemia and cardiovascular disorders is not so obvious as in the case of microvascular complications [71]. Actually, CVD seem to be more conditioned by insulin resistance which (due to the lack of insulin-mediated inhibition) stimulates the release of free fatty acids (FFAs) from the adipose tissue. These FFAs are taken up by endothelial cells and undergo (uncontrolled by insulin) excessive β-oxidation yielding the substrates for the respiratory chain. Consequently, as in the case of hyperglycemia-conditioned accelerated aerobic glycolysis, an excessive ROS production is observed, which triggers most of the discussed earlier pathological pathways [175,176].
3.2. MGO/MAGEs contribution to blood vessels wall impairment, hypertension, dyslipidemia and atherosclerosis
3.2.1. Blood vessels focusing on endothelium – impairment of angiogenesis
In light of the above mentioned mechanism, MGO and its glycation end products comprise an important causative pathway contributing to vascular pathologies conditioned by hyperglycemia, hyperlipidemia and insulin resistance [171]. AGEs are involved in the induction of oxidative stress in the endothelial progenitor cells (EPCs), as well as down-regulation of antioxidative and anti-inflammatory enzymes (catalase, superoxide dismutase and paraoxonase 2) and eNOS, but up-regulation of NOXs in human endothelial cells, which impairs the functioning and healing of endothelium [118,171,177,178]. Increased glycolytic/FFAs flux in mammalian endothelial cells has been shown to elevate ROS generation, which in turn raises MGO and MAGEs levels [30,118,170,175,176,179]. On the other hand, Glo1 upregulation attenuates these effects [102,179]. For example, high-Glc exposed Glo1-knockdown human aortic endothelial cells (HAECs) have shown the increase in MGO, followed by the upregulation of inflammatory processes, endothelial dysfunction and disturbances in ECM components [152]. Additionally, MGO-treatment of HAECs have induced ROS generation [142], as well as the cells apoptosis associated with oxidative stress connected (at least partially) with the impairment of antioxidative thioredoxin/peroxiredoxin system [143] (Table 2). The effects leading to oxidative stress development might be associated/augmented by NOS activation, as observed by Miyazawa et al. [142]. However, in other studies increased Glc/MGO demonstrated no effect on eNOS in HAECs [152], in accordance with the lack of eNOS inhibition by MG-H1 and AP observed in other experiments on human endothelium [149] (Table 2). Unlike in HAECs, in human umbilical vein endothelial cells (HUVECs) MGO exposition led to the inhibition of eNOS activity and NO production, probably via the attenuation of eNOS Ser-1177 phosphorylation and Akt phosphorylation [108,118]. In turn, ROS accumulation was observed in MGO-treated HUVEC cells, probably being the consequence of NOX upregulation and/or SOD-1/CAT/GPX downregulation [108,118,128] (Table 2). Similarly in human endothelial EA.hy926 cells, MGO treatment caused the decrease in eNOS Ser-1177 phosphorylation associated with the uncoupling of this enzyme and superoxide radical generation [151] (Table 2). Signaling pathways engaged in MGO-mediated vascular impairment associated with ROS generation and mitochondrial-dependent apoptosis of endothelial cells, have been proposed by Wang et al. [128]. In their experiments on MGO-treated HUVECs and mice, the authors reported the involvement of PI3K/Akt/Nrf2/HO-1 routes, which upon MGO inhibition led to down-regulation of antioxidative enzymes and up-regulation of oxidative stress coupled with mitochondrial dysfunction, as well as pro-apoptotic and pro-inflammatory events. These pathological routes were attenuated by metformin both in the endothelial cells and in mice [128]] (Table 1 and Table 2). Similarly, in other experiments on HUVECs, MGO treatment has induced mitochondrial-dependent apoptosis, impairment of Akt/eNOS/NO pathway and upregulation of prooxidative (NOX4/ROS) and proinflammatory (NF-κB) routes, all of which were reversed by phosphocreatine and NAC [146] (Table 2).
Many MGO-affected routes may be mediated by its impact on p53 protein which is induced in response to cellular stress [147,148]. Upon accumulation of DNA damages, p53 inhibits cell cycle diverting the cell towards apoptosis. However, p53 also controls a variety of “non-classical” pathways, such as metabolic homeostasis, ferroptosis, autophagy, or senescence [180]. MGO has been shown to alter genomic profile associated with cell cycle regulation, especially p53 pathway [147] (Table 2) in HUVECs. Additionally, MGO exhibited the stimulatory effect on p53 signaling in the same type of HUVEC cell line, where MGO caused DNA damage and induced p53 phosphorylation associated with the inhibition of mTORC1 and stimulation of autophagy [148] (Table 2). Moreover, a prolonged MGO exposition can divert endothelial cells into senescence phenotype, as has been reported in human vascular endothelial cells (HVECs) from the patients suffering from coronary heart disease. HVECs exposition to combined MGO and GO action led to the cells senescence through the increase in ROS and upregulation of p21 [141]. (Table 2).
In search for other signaling pathways presumably engaged by MGO/MAGE, human aortic endothelial cells (HAECs) derived from healthy and T2DM individuals have been examined. In comparison with untreated healthy cells, both diabetic and MGO-exposed healthy cells were dysfunctional and showed upregulation of three MAPK pathways. Additionally, it seems that MGO-induction of KATP channel contributed partially to MGO-caused deleterious effects on endothelial cells (via JNK pathway) [126] (Table 2).
These and other experiments conducted on endothelial or endothelial progenitor cells (EPCs) exposed to MGO suggest its causative effect on endothelium dysfunction [66,108,118,123,124,139,146,150] (Table 2). Mentioned above pro-oxidative and pro-inflammatory pathways induced by MGO can also affect angiogenesis process. For example, MGO-treated murine EPCs [139], as well as human and bovine endothelial cells [123] have demonstrated decreased VEGFR-2 level, probably mediated by MAGEs induction of RAGE. This was associated with the impaired capability of blood vessel tube formation [123,139] (Table 1 and Table 2). Hence, MGO seems to impair angiogenesis process which partially might be corrected by Glo1 overexpression, as has been shown in diabetic rats [181]. In endothelial cells the mechanism responsible for MGO/RAGE-induced VEGFR-2 degradation (leading to decrease in tube formation) was peroxynitrite (ONOO-)-mediated autophagy process (this finding was supported on diabetic mice aortas experiments) [123] (Table 1 and Table 2). Therefore, the authors suggested the mechanism leading to a decrease in angiogenesis, which starts from hyperglycemia associated with MGO increase. Further, MGO induces RAGE which leads to peroxynitrite generation and VEGFR-2 degradation through autophagic pathway with the final result in lowered angiogenesis. Another route associated with MGO-impaired angiogenesis has been proposed by Nigro et al. [127]. In their in vitro and in vivo experiments, the authors observed the involvement of NF-κB-p65 and HoxA5 in MGO-stimulated effects leading to the downregulation of VEGFR-2 and decreased capability of new vessels formation in both MAECs from Glo1KO mice and MGO-stimulated MCECs [127]. Both NF-κB-p65 and HoxA5 were also upregulated in high-fat diet fed mice which developed diabetic characteristics. Judging from these observations, it might be suggested that MGO induces NF-κB-p65 which further binds with promoter region of HoxA5 activating this transcription factor. In turn, HoxA5 stimulates signaling pathways preventing angiogenesis [127]] (Table 1 and Table 2). In line with these findings, Glo1 overexpression in bone marrow-derived circulating angiogenic cells extracted from diabetic mice, has demonstrated protective actions on blood vessels, restoring the cells viability and potential towards neovascularization (impaired under hyperglycemic/hypoxic conditions) [125] (Table 1). Reversely, Glo1 silencing in human endothelial cells has caused upregulation of pro-inflammatory and pro-adhesive factors [105] (Table 2).
MGO/MAGE-weakened vasodilatory and/or angiogenic capacity both in endothelial and EPC cells would enhance the risk of CVD in diabetic patients. Especially, EPCs circulating in the blood plasma, play important functions in repairing blood vessels, hence their impairment may diminish healing forces of the organism with respect to the cardiovascular system.
3.2.2. Cardiovascular system in animal models
MGO treatment of both normal (Wistar and Sprague-Dawley rats) and diabetic rats (GK rats) has impaired or worsened the condition of the animals cutaneous vasculature or aortas; namely their vasodilatory functions, or increased cardiac fibrosis, which was accompanied by the deterioration of oxidative status, inflammation, and glycation [99,111,116,118] (Table 1). Similarly, vasorelaxation has been impaired by diabetes and/or Glc/MGO treatment in mesenteric arteries (from STZ-treated or normal Wistar rats) [97,104,105]. This effect was accompanied by intracellular elevation of MG-H1, CML, VCAM-1, ICAM-1 and nitrosative stress, and was corrected by antioxidants and/or Glo1 overexpression [104,105] (Table 1). MGO increase in murine models (MGO-fed or Glo1 inhibitor-treated apoE KO mice) to the level characteristic for diabetes, has enhanced vascular adhesive properties as well as atherogenesis. Those effects were comparable to the diabetic ones, although the mice had normal glucose level [107] (Table 1). Some of the effects leading to increased adhesiveness and inflammation were RAGE independent (observed in RAGE-deficient mice), however others required the presence of RAGE (e.g. MCP-1 upregulation) [107] (Table 1 and Table 2). In C57BL/6 mice MGO treatment impaired oxidative status (through the decrease in antioxidative enzymes and increase in lipid peroxidation marker) and increased cytokines level in the circulation. This was associated with disturbances of aorta structure reflected by its increased thickness and apoptosis level, and down-regulation of Akt/Nrf2 route in aorta (as simultaneously observed in HUVECs) [128]. Metformin pretreatment attenuated most of these deleterious MGO effects both in the mice and human endothelial cells (HUVECs) [128] (Table 1 and Table 2). In STZ-treated rats, diabetes has lowered Glo1 activity, upregulated some AGE markers (3-deoxyglucosone (3-DG) and CML), but had no impact on MGO and CEL levels in the rat hearts [103]. Additionally, diabetes altered the expression of genes associated with oxidative stress, DNA damage, heart fibrosis and inflammation, which was partially attenuated by Glo1 overexpression [103] (Table 1). A protective effect of Glo1 overexpression against diabetes-induced cardiovascular impairment has been demonstrated in diabetic mice, where it improved vascular inflammation and heart muscle condition [124] (Table 1). However, Glo1 overexpression has not protected from diabetes-associated atherosclerosis as well as endothelial dysfunction [106,110], neither Glo1 underexpression enhanced atherosclerosis in a murine model [106] (Table 1). Although Glo1 expression manipulation was expectedly correlated with the overall aortal MG-H1 level, it did not show any impact on the degree of aortal atherosclerotic lesions neither aortal collagen glycation (including MG-H1) [106,110], (Table 1). Nevertheless, since these studies were conducted on apoE deficient mice models, it might be suggested that characteristic for them dyslipidemia may have interfered with the obtained results. For example, as the authors presumed, intracellularly working Glo1 metabolizes mainly glycolysis-derived MGO, whereas under conditions of extracellular lipid overload accompanied by inflammatory/oxidative stress, it may be lipid peroxidation which is the additional source of MGO/MAGE not handled by Glo1, and thus enhancing pathological routes [110]. A causative input of MGO/(M)AGE in development of atherosclerosis in a diabetes murine model accompanied by human endothelial cells experiments, has been observed by Fang et al. [108]. The authors reported elevated levels of MGO and protein carbonyls in aortas of diabetic ApoE deficient mice in which atherosclerotic lesions were increased. These effects were associated with increased ROS, the downregulation of antioxidative enzymes as well lowering of Akt and eNOS phosphorylation, and decrease in aortal GSH and serum nitric oxide. Since all of these pathologies were attenuated by NAC, it might be supposed that NAC enhancing GSH synthesis would contribute to more efficient scavenging of MGO by glyoxalases system [108] (Table 1 and Table 2).
3.2.3. Cardiovascular disorders in patients
As discussed earlier, MGO, its metabolic as well as glycation end products are elevated in diabetic individuals. Enhanced (M)AGEs levels have been observed in blood plasma/serum both in T1DM [21] and T2DM patients [22,23], and in T2DM individuals MG-H1 concentration was increased especially in retinopathy cases [24]. AGEs have been associated with higher risk of incident cardiovascular events in both T2DM (CEL and CML) [23] and T1DM (CEL, CML and pentosidine) [182]. However, no independent association was observed between AGEs (CEL, CML and pentosidine) and prior cardiovascular events in diabetic (T2DM) [23,183] and non-diabetic individuals [183], what might be connected with AGEs-scavenging effects of medicines applied in CVD treatment (as discussed in chapter 4).
When CML concentrations were estimated in association with CVD mortality risk in older individuals, such a connection was observed in non-diabetic subpopulation in whom greater CML levels in blood serum raised the risk of dying, especially of CVD [184,185]. In addition to CML, in older non-diabetic women, increased soluble RAGE forms (sRAGE and esRAGE) were connected with higher CVD-caused mortality rates [185]. In line with these findings, in an 18-year follow-up study comparing the association of MG-H1 with CVD mortality in diabetic and non-diabetic cohorts, only non-diabetic women showed positive correlation; those who died of CVD had higher MG-H1 serum levels [186,187]. Other studies have shown no association or even inverse dependance between AGE/CML and CVD events/mortality [188,189]. In T2DM individuals with nephropathy, serum CML levels were not correlated with cardiovascular events [188], whereas in hemodialysis patients low CML levels were connected with higher all-cause mortality rates and showed tendency towards greater CVD mortality [189].
Although the above observations seem to be inconsistent, showing either the association of AGEs with CVD or the lack of such a dependance, it is probably connected with differences in studied populations and applied methodology (Ab-based ELISA vs. chromatography/mass spectrometry, as well as statistical approach, as discussed by Hanssen et al. [23]). Therefore, a stronger evidence speaks in favor of greater CVD risk in individuals with higher (M)AGEs not only in older people, but also in diabetics. Similar uncertainty exists regarding blood plasma MGO level association with CVD occurrence. Hanssen et al. have reported positive association between higher fasting MGO level in plasma and increased risk of incident CVD both in T1DM [190] and T2DM [191] patients. Also, in T2DM individuals blood plasma MGO has been estimated as a predictor of intima media thickening, vascular stiffening, and blood pressure elevation [192]. However, in their other study, Hanssen et al. [193] have reported no correlation between higher fasting and post-OGTT plasma MGO levels and prior CVD in cohorts with normal, prediabetic and diabetic (T2DM) conditions. Therefore, supposedly unlike macrovascular, rather microvascular complications (CKD and retinopathy) seem to be connected with elevated MGO [193].
3.2.4. Atherosclerosis
(M)AGEs have been detected in atherosclerotic plaques and atherosclerotic-like lesions [21,55,140,194]. CML accumulation has been observed in macrophages and at calcification sites in degenerated aortic valves [194], as well as CML colocalization with THP has been found in macrophages within atherosclerotic plaques extracted from coronary arteries derived from control and diabetic individuals [21]. Additionally, THP which is an MGO-derived AGE, was elevated in T1DM patients’ serum and positively associated with soluble vascular cell adhesion molecule 1 (sVCAM-1) (unlike other AGEs: CML, CEL and pentosidine) and secreted phospholipase A 2 (sPLA2) [21]. Since elevated sVCAM-1 and sPLA2 have been associated with atherosclerotic processes running in the organism [195,196], THP might be involved in CVD development.
When (M)AGEs have been estimated in human carotid endarterectomy specimens, higher levels of CML and MG-H1 were observed in rupture-prone plaques (in comparison with stable ones). CML and MG-H1 were localized mainly in macrophages around the necrotic core (but also in endothelial cells), and they were correlated with inflammatory (IL-8, MCP-1) and pro-apoptotic (cleaved caspase 3) markers, as well as matrix metalloproteinases (MMP-9) [140]. These findings were accompanied by lowered Glo1 mRNA/protein in ruptured plaques (present in all cells of the plaque, except for the necrotic core), but no change in RAGE expression was observed. Also, no associations were found between the studied AGEs and plasma glucose levels, neither between plaques coming from diabetic vs. non-diabetic patients. In search for the cause and effect relationship between MGO/MAGE and inflammation, the authors performed experiments on human monocytes. They observed that although TNF/hypoxia decreased Glo1 and upregulated MGO/MAGEs, MGO/MAGEs did not mediate TNF-induced secretion of IL-8, MCP-1 and MMP-9. Nevertheless, Glo1 knockdown worsened the viability of MGO-exposed cells, what suggests Glo1 protective function in this system [140] (Table 2). The lack of MGO engagement in proinflammatory pathways stimulation has been also observed in TNF-α induced HUVECs, where MGO led to VCAM-1 decrease [147] (Table 2).
In light of the above data, MGO/MAGE seem to be involved in the processes accelerating atherogenesis. However, the pathological events mediated by α-dicarbonyl compounds and their glycation end products occurring in the blood vessels and atherosclerotic plaques are not necessarily reflected by their levels in the blood plasma or serum, as has been recently reported by Berge et al. [197]. In search for prognostic markers which could be applied in the evaluation of coronary artery disease (CAD) risk conditioned by coronary atherosclerosis in middle-aged or older male athletes, the authors found no associations between plasma concentrations of α-dicarbonyl compounds (MGO, GO, 3-DG) or (M)AGEs (CML, CEL, MG-H1), and the number and type of coronary artery plaques, or the level of coronary arteries calcification [197]. However, as reported in an earlier study, serum CML level was elevated in CAD patients (especially in diabetics), but it was pentosidine which was correlated with CAD [198]. Causative involvement of MGO/MAGE in CAD has been suggested in integrative genomics study, where GLO1 gene was found to be associated with CAD pathology [199].
3.2.5. Endoplasmic reticulum stress (ER stress) followed by unfolded protein response (UPR) in blood vessels
MGO intracellular accumulation upon hyperglycemia and associated cardiometabolic disturbances leads to modification of multiple proteins, which impairs proper proteins folding and trafficking. As a consequence, endoplasmic reticulum stress (ER stress) may develop, which further can induce unfolded protein response (UPR) to restore protein homeostasis. UPR can trigger three pathways mediated by protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1α (IRE1α), and transcription factor 6 (ATF6). However, when pathological changes exceed the capacity of UPR, harmful pathways get accelerated leading to oxidative stress, inflammation and cells death via apoptosis. As recently reviewed by Ren et al. [200], ER stress with UPR and other routes initiation, leading to such deleterious effects, is associated with cardiovascular diseases. MGO involvement in these processes has been shown in endothelial (HAECs) and vascular smooth muscle cells (VSMCs). Glc-treatment (or Glo1 silencing) of HAECs led to the accumulation of MGO/MAGE in the cells, as well as up-regulation of UPR pathways associated with heat shock proteins increase and stimulation of pro-inflammatory and pro-thrombotic routes. Protective effects of trans-resveratrol and hesperetin combination (tRES-HESP) was shown in this model; tRES-HESP increased the expression of Glo1 and decreased the expression of hexokinase-2, in this way correcting Glc metabolism and diminishing MGO level/effects [144] (Table 2). Similarly, experiments on MGO-treated rat aortal VSMCs pointed to MGO involvement in the induction of ER stress, since MGO caused the upregulation of three UPR pathways (PERK, IRE1α, and ATF6). However, no apoptosis was triggered in MGO-treated VSMCs [137] (Table 2).
3.2.6. Hypertensive and pro-coagulatory properties of MGO/MAGE
Hypertension, being one of metabolic syndrome components, is the leading risk factor of CVD [201]. MGO and MAGEs involvement in hypertension has been addressed in experiments conducted on spontaneously hypertensive rats (SHR) which develop genetically-conditioned hypertension, not associated with insulin resistance neither hyperglycemia [202,203]. Elevated levels of MGO in blood plasma, aorta, liver and kidney (but not heart) and (M)AGEs in aorta, mesenteric artery and kidney have been demonstrated in these animals [93-96]. These effects were accompanied by oxidative stress indicative by the increase in superoxide radical and hydrogen peroxide and decrease in GSH or GSH/GSSG ratio [93-95] (Table 1). Similar findings have been reported in Sprague Dawley (SD) rats which, due to being fructose fed or MGO treated, developed hypertension associated with MGO up-regulation and initiation of pro-hypertensive routes [31,100,122] (Table 1).
Additionally, MGO/MAGE-mediated dysfunction of blood vessels reflected by morphological changes in mesenteric arteries was shown. This was associated with eNOS down-regulation, eutrophic inward vascular remodeling and impairment of endothelium-dependent relaxation in hypertensive rats [31,95,96] (Table 1). These disturbances seem to be mediated by MGO/(M)AGE-triggered oxidative and/or (NF-κB-mediated) inflammatory pathways, as has been demonstrated in SHR-derived VSMCs [131] and rat’s VSMCs exposed to Fru or MGO [122,136] (Table 2). The pathological events connected with hypertension, MGO/(M)AGE upregulation, oxidative stress and blood vessels impairment have been corrected by MGO/(M)AGEs scavengers: aminoguanidine [95,96], metformin [31] or alagebrium [122] (Table 1 and Table 2). Moreover, proapoptotic MGO effect on HUVECs has been inhibited by telmisartan (a selective angiotensin II type 1 receptor (AT1R) blocker) [138]. These observations point to MGO/(M)AGE being involved in the development of genetically-conditioned hypertension (independent of hyperglycemia/diabetes), as well as hypertension caused by dietary fructose/glucose overload. The mechanism underlying MGO-induced hypertension probably involves activation of RAAS system through MGO/RAGE/NF-κB route [122] (Table 1 and Table 2). Additionally, MGO-induced activation of aortal smooth muscle cells proliferation might contribute to vascular impairment and hypertension, as suggested by Chang et al. [120]. In their experiments on Fru or MGO-treated rats, as well as aorta-derived vascular smooth muscle cells (VSMC), they have observed MGO-stimulated proliferation of VSMC, which effect was mediated by MGO activation of Akt1. Namely, MGO was shown to form adduct with Cys-77 residue at Akt1, probably changing the protein conformation. This in turn led to Akt1 activation via Ser-473 phosphorylation yielding the cells proliferation [120] (Table 1 and Table 2).
On the other hand, a short-term MGO-exposition of aorta and mesenteric artery (devoid of endothelial layer) has exerted an inhibitory effect on noradrenalin-induced contraction of VSMCs [98]. This seemed to have been mediated by MGO opening of one type of calcium activated potassium channels [98] (Table 1).
Disbalance between blood coagulation cascade and thrombolytic system, which promotes thrombosis, increases the risk of cardiovascular incidents associated with blood vessels occlusion such as myocardial infarction. MGO has been shown to be able to form adducts with antithrombin III (ATIII). MGO modification of ATIII at Arg 393 led to ATIII inhibition reflected by its inefficient blocking of thrombin and factor Xa [204]. Hence MGO might contribute to the impairment of the processes responsible for the inhibition of thrombus formation, and in this way enhance the risk of CVD, especially in patients with metabolic syndrome/diabetes characterized by elevated plasma MGO level.
3.2.7. Dyslipidemia
Qualitative and quantitative disturbances in lipids and lipoproteins in the circulation (observed in metabolic syndrome) are strongly associated with the induction and development of CVD. Especially pathologically altered LDL particles such as oxidized LDL (oxLDL) and small dense LDL (sdLDL) are involved in the process of atherogenesis [205]. MGO seems to play an important role in LDLs modifications making them more prone to accumulate in blood vessel wall, which is associated with the induction of proinflammatory events, formation of foam cells and hence atherosclerotic plaques development. MGO involvement in LDL particles alteration has been demonstrated by Rabbani et al. [206,207]. The authors observed enhanced LDL apoB100 glycation at Arg and Lys residues in T2DM patients [207]. Increased apoB100 AGEs included MGO-derived MG-H1, CEL and MOLD, as well as other α-dicarbonyl-derived AGEs (G-H1, 3DG-H and pentosidine). Minimally MGO-modified LDL particles tended to change their features; their size dropped (resembling sdLDL) and they showed greater binding to proteoglycans associated with atherosclerotic plaques (biglycan, aggrecan and perlecan) [206]. Additionally, the authors reported that Arg18 modified by MGO leads to the conformational change in apoB100 which enhances LDL binding with proteoglycans. Thus minimally MGO-modified LDL particles increased adhesiveness to the aorta wall through binding with heparan sulfate containing proteoglycans [206]. Therefore, it probably is MG-H1 altered Arg18 on apoB100 which changes LDL particles’ properties enhancing their binding with blood vessels’ proteoglycans containing heparan sulfate. This in turn would extend the time of LDL particles attachment to endothelium and their exposition to RONS/α-dicarbonyl stressors, further enabling the modifications of LDL particles towards more proatherogenic characteristics [206,207]. However, minimal MGO modification did not alter LDLs affinity for their receptors on hepatocyte-like cells and fibroblasts, as well as did not make them recognizable by scavenger receptors on macrophages. Additionally, their clearance from the murine organism was not changed [206]. Beside apoprotein modification, MGO may also take part in lipids oxidation, as has been demonstrated by Lankin et al. [208], who reported MGO contribution to LDLs lipoperoxidation (mediated by ROS generation) under hyperglycemic conditions. Metformin treatment of diabetic patients has been shown to inhibit MGO-mediated LDLs modifications [207, 208].
Similarly, MGO and other dicarbonyls seem to modify HDL particles exacerbating their cholesterol scavenging, as well as antioxidative and anti-atherogenic functions, especially in diabetics [209,210]. MGO-derived MG-H1 in HDL particle mainly seems to modify Arg residues in apolipoprotein A1 (apoA1) which alters its conformation. The observed consequences include the conversion of HDL particles into smaller and more dense ones which tend to be easier removed from the circulation, hence their concentration falls [121,209]. Additionally, HDL-mediated reverse cholesterol transport as well as this lipoprotein protective actions may be impaired, such as cholesterol esterification (through weakened LCAT binding), cholesteryl esters exchanging for TAGs (via CETP inhibition), and antioxidative/anti-poisonous properties (weakened PON1 binding) [209].
Therefore, MAGEs seem to participate in the conversion of both LDL and HDL particles into proatherogenic ones, mainly contributing to the impairment of their apoproteins functionality.
Generally, as recently discussed by Schalkwijk et al. [211], whereas the involvement of AGEs in the pathomechanism of cardiovascular complications in diabetes is well known, less scientific data are available which explain the exact function of MGO. MGO, being the major precursor of AGEs, contributes to endothelial dysfunction through the induction of oxidative stress, inflammation, ER stress, as well as apoptosis. Together with AGEs it impairs angiogenesis and promotes atherosclerosis-associated inflammation. However, its role in AGEs-stimulated cholesterol accumulation in macrophages, vascular smooth muscle cells (VSMCs) phenotype switch into macrophage-like, as well as VSMCs calcification, is not elucidated [211].
4. Potential glycation inhibitors and MGO scavengers – therapeutic strategies
Several therapeutic approaches are considered to reduce or prevent MGO-induced toxicity (Figure 1 and Figure 2). The first is an activation of the glyoxalase system (Glo1/2). However, this approach is limited to conditions where the amount of enzyme or glutathione is insufficient. Unfortunately, we currently do not know of many inducers of glyoxalase. The reduced activity of Glo1 is restored, for example, by candesartan [212,213] and pyridoxamine [214]. The scientific literature also more extensively describes this effect for a combination of two plant polyphenols trans-resveratrol, and hesperetin (tRES-HESP, a combination of stilbenoid and flavonoid, respectively) [215,216]. However, no effect on Glo1 was confirmed for the other flavonoid isoquercitrin (= quercetin-3-O-glucoside) [217]. Nevertheless, glycation inhibitors are usually characterized by more than one mechanism of action determining the overall anti-AGE potential. Further strategies assume that highly reactive MGO can be neutralized by mechanisms independent of the glyoxalase system, such as uptake (chemical binding) using small molecules, contributing to its removal from the extracellular and intracellular environment. The uptake of MGO in statu nascendi is thought to prevent its direct toxicity but also to reduce glucotoxicity induced by ROS and RNS (together known as RONS), the formation of AGEs, and the interaction of AGEs with the trans-membrane receptor RAGE. The binding of AGEs to RAGE on the surface of cells, including immune, endothelial and vascular smooth muscle cells or platelets, induces an intracellular response to carbonyl stress, oxidative stress, and nitrosative stress, characterized by activation of transcription factors such as NFκB. Vascular wall stress induced by RCS and RONS is characteristic of atherosclerosis, and the resulting activation of NFκB and MAPK systems may be a possible mechanism for AGE-induced angiopathy [218-220]. Excessive production of RONS generates a flux of reactive carbonyls, whose accumulation is positively correlated with the development of autophagy [220]. In this context, a strategy oriented toward the molecular cascade of the AGE-RAGE axis, or more accurately the MAGE-RAGE axis, opens up a new pharmacological approach [221,222].
In general, molecular mechanisms for inhibiting excessive non-enzymatic glycation include any biological or chemical reaction that can reduce or prevent the generation of glycated macromolecules (peptides, proteins, lipoproteins, and nucleic acids) in vivo to reduce the formation of AGEs and interrupt the sequence of adverse events resulting from their deposition and leading to cell, tissue and organ dysfunction. These reactions can occur both intracellularly and extracellularly, possibly simultaneously at multiple sites. Seven molecular pathways (Figure 2) and related mechanisms have been proposed by which low-molecular-weight compounds known as glycation inhibitors reduce the levels of RCS and AGEs (mainly MGO and MAGEs) in the body:
(1) Restoring activity or normal levels of Glo1 RNA/protein.
(2) Trapping/scavenging of reactive dicarbonyls (anti-RCS, anti-MGO, etc.), for example, methylglyoxal, glyoxal, malonyl dialdehyde, or others from both carbohydrate, lipid, and some amino acid (threonine) metabolism, resulting in lowering of carbonyl stress.
(3) Trapping/scavenging reactive oxygen and nitrogen species (RONS) yielding direct or indirect antioxidant effect (e.g., by quenching radicals, earlier termination of radical reactions), as well as up-regulating the antioxidant protection system (superoxide dismutase, catalase, glutathione peroxidase, glutathione etc.) and down-regulating pro-oxidative enzymes (e.g. NOX and iNOS), leading to a reduction in oxidative stress and nitrosative stress.
(4) Chelation of transition metal cations catalyzing the oxidation of monosaccharides, fatty acids, cholesterol, amino acids, nucleotides and secondary reactions of glycated macromolecules.
(5) Protection of functional groups of macromolecules vulnerable to non-enzymatic glycation, e.g., by reversible non-covalent binding to functional groups of intra- and extracellular components subject to the glycation reaction or oxidative transformation (for example, serum albumin, lens crystalline, collagen or Amadori products). Various interactions, such as hydrogen bonds formation, electrostatic forces, and hydrophobic and polar interactions, are usually responsible for such interactions.
(6) Inhibition of AGE/RAGE pathway - down-regulation of membrane RAGE (RAGE antagonists) and induction of secretory RAGE represents attractive target for treating pathogenic glycation-related diseases. The interaction of AGEs with trans-membrane RAGE results in the activation of pro-inflammatory genes (mediated by NF-ĸB) and ROS generation, hence it is involved in the pathophysiology of many age-associated diseases, including cardiovascular disease. Some isoforms of this receptor lack a transmembrane domain and are therefore secreted from cells as sRAGE (soluble RAGE produced by alternative splicing) or esRAGE (endogenous secretory RAGE proteolytically exfoliated by metalloproteinases). The most circulating RAGE comes from sRAGE, and esRAGE accounts for only a minor fraction (about 20%). Circulating sRAGE and esRAGE compete with RAGE for ligand binding, and they act as a decoy to eliminate existing AGEs. Thus, increasing the level of circulating RAGE can reduce the activation of the AGE/RAGE pathway [223].
(7) Breaking AGEs cross-links; the mechanism attributed to the action of alagebrium (ALT-711).
Mechanisms (1)-(5) essentially prevent and limit the accumulation of AGEs and their precursors (MAGEs and MGO) in biological systems, pathway (6) inhibits RAGE-mediated signaling. On the other hand, mechanism (7) offers the removal of already formed AGE cross-links, which would be suitable for reducing existing cellular and tissue burdens associated with protein cross-linking, such as blood vessel wall stiffness contributing to hypertension. Results from clinical trials indicate that alagebrium may be the first representative of agents with this specific action in diseases associated with AGE accumulation [224].
MGO (methylglyoxal) is generated as a result of spontaneous fragmentation of two intermediates of glycolysis and fructolysis: G3P (glyceraldehyde 3-phosphate) and DHAP (dihydroxyacetone phosphate). Additionally, its minor quantities can be derived from the metabolism of acetone, aminoacetone and threonine, as well as from highly processed foods characterized by high content of glucose and fructose. An excessive consumption of simple sugars has been associated with the development of insulin resistance, hyperglycemia (enhanced by gluconeogenesis) and dyslipidemia (enhanced by de novo lipogenesis resulting in increased level of free fatty acids, as well as hypercholesterolemia and hypertriglyceridemia). These metabolic disturbances lead to such disorders as NAFLD (non-alcoholic fatty liver disease), obesity, metabolic syndrome and T2DM (diabetes type 2). Hyperglycemia observed in diabetics enhances the metabolism of glucose accelerating the generation of MGO and its advanced glycation end products – (M)AGEs. Moreover, stimulated aerobic glycolysis increases reactive oxygen species and reactive nitrogen species (RONS) production (induces oxidative stress and nitrosative stress) which leads to the inhibition of GAPDH (glyceraldehyde 3-phosphate dehydrogenase). Finally, as glycolysis and fructolysis are inhibited at the level of trioses, more G3P and DHAP are produced, resulting in more MGO (carbonyl stress). Thus, considering MGO-caused damage to blood vessels, T2DM patients show increased risk for cardiovascular complications (angiopathy and cardiomyopathy), which might result from inefficient control of postprandial glycemia. Increased in diabetes MGO and (M)AGEs show deleterious impact on endothelium functioning, which is associated with the promotion of oxidative stress, low-grade inflammation, atherogenesis, and CVD development. MGO and MAGEs accumulation is observed when the key system responsible for MGO detoxyfication (Glo1/Glo2) is overloaded. Then MGO reacts with arginine or lysine residues of peptides, proteins and lipoproteins yielding stable adducts (e.g., MG-Hs, CEA, AP, THP, CEL and MOLD). Additionally, it causes the formation of macromolecule cross-links with the involvement of arginine and lysine (MODIC). Moreover, MGO participates in the DNA and RNA modification, reacting with deoxyguanosine and inducing nucleic acids cross-linking. Hence, MGO can lead to epigenetic changes through the alteration of genetic material (nucleic acids and/or histones), and induce metabolic memory comprising prolonged up-regulation of pro-oxidative (ROS increase) and pro-inflammatory (AGE/RAGE and NF-κB-mediated) pathways. To date, several groups of small molecules have been evaluated for possible MGO-trapping effects, inhibiting glycation or (M)AGE/RAGE signaling through various mechanisms. The glycation inhibitors exhibit the ability to activate the Glo1/Glo2 system, capture MGO and RONS, chelate transition metals that catalyze oxidation, and they can inhibit the MAGE-RAGE axis by inducing circulating RAGE (sRAGE and esRAGE), and also by other mechanisms (see Figure 2).
4.1. Overview of the potential glycation inhibitors and MGO scavengers
Glycemic control and inhibition of non-enzymatic glycation are at the heart of strategies to prevent MGO-induced vascular complications in diabetes. Methylglyoxal at the micromolar level causes rapid injury to endothelial cells and contributes directly to their inflammation and dysfunction. The repeated increase in MGO associated with spikes in hyperglycemia also impairs angiogenesis in vivo. For this reason, MGO appears to be a critical precursor of AGEs (MAGEs) and AGE-associated disturbances [5,17,126,190,225]. Its neutralization (removal) is probably the principal way in which a number of antiglycation agents inhibit the formation of AGEs (MAGEs). Aminoguanidine (syn. pimagedine) is the first known MGO-trapping glycation inhibitor that effectively prevented the formation of AGEs and reduced cross-linking of arterial wall proteins in hyperglycemia. In experimental models of diabetes complications, it was effective in inhibiting disease progression, including preventing vascular dysfunction, reducing renal basement membrane thickening and albuminuria, and normalizing endothelial cell proliferation in retinopathy. The clinical effect of aminoguanidine was studied in two randomized placebo-controlled trials on more than 600 patients with T2DM and T1DM accompanied by nephropathy or retinopathy. However, these studies were discontinued prematurely due to several side effects [226,227]. Therefore, there is an intense search for new glycation inhibitor candidates, including among widely used medications.
To date, several groups of small molecules have been evaluated for possible MGO-trapping effects, inhibiting glycation or AGE/RAGE (MAGE/RAGE) signaling through various mechanisms, including oral antihyperglycemic agents (biguanides, sulfonylureas, thiazolidinediones) used to treat T2DM and insulin resistance; angiotensin II receptor antagonists (angiotensin II receptor blockers) and angiotensin-converting enzyme inhibitors; calcium antagonists (calcium channel blockers) and arterial smooth muscle agents (hydrazinophthalazine derivatives) used to treat hypertension; hypocholesterolemic agents (statins) and phlebotropic agents (vasodilators, anti-varicose, and capillary stabilizing agents) used to treat peripheral vascular disease (including in patients with diabetes), and anti-inflammatory, analgesic and antipyretic agents used to treat inflammatory joint disease, cold, flu and headache (including non-steroidal anti-inflammatory drugs - NSAIDs); as well as some vitamins (including B1, B6, and others).
A list of potential and known glycation inhibitors and MGO scavengers, along with suggested mechanisms of antiglycation action, is presented in Table 3. However, only preliminary data derived from in vitro or animal model studies are available to date for most of these inhibitors. Studies involving healthy volunteers or patients have been conducted for metformin [228], atorvastatin [229], cerivastatin [230], benfotiamine [231], pyridoxamine [232], hesperidin [233], and isoquercitrin [217]. Among the compounds discussed in this section, only biguanides, hydrazinophthalazines and bioflavonoids show the ability to trap MGO with the formation of adducts having a different chemical structure from the precursor (MGO attaches to the scavenger via covalent bonds) [228,234-236]. In turn, Voziyan et al. [237] and Colzani et al. [235], confirmed the ability of pyridoxamine to uptake RCS in a test with GO and MDA, respectively. The inhibitor's ability to capture RCS results in the disposal of MGO and other carbonyls, just as circulating RAGE captures and eliminates AGEs. An MGO-metformin metabolite (an imidazolinone derivative) is excreted in the urine of metformin-treated patients [228]; nevertheless, the ultimate fate of the adducts of other scavengers remains unknown.
Since the agents in the discussion are commonly used in primary therapeutic ranges due to their known mechanisms of action, the possibility of their broader use in pharmacotherapy due to inhibition of the MAGE-RAGE axis is considered. This approach may reduce or slow down the effect of MAGE-RAGE on the development or progression of CVD.
4.1.1. Oral antihyperglycemic agents
Blood glucose-lowering agents are known to reduce vascular dysfunction in preclinical models through a combination of mechanisms that apparently act independently of the glucose-lowering benefits. In the group of antihyperglycemic medicines, the effect of lowering AGEs and cross-linking macromolecules by directly reducing MGO levels has been proven only for biguanides (metformin, buformin) [238,239,240,241]. Metformin treatment of patients with T2DM and atherosclerosis, besides its hypoglycemic effect, is associated with increased Glo1 activity. However, there was no effect on Glo1 protein levels. Besides, Glo1 activity correlates with HbA1c levels [242]. Human studies have also shown that metformin can trap MGO (Table 3). The reaction produces an imidazolinone-like metabolite via nucleophilic addition involving a biguanide group ([228]. The latter mechanism presumably contributes to a significant reduction in MGO levels and appears to offer therapeutic value in inhibiting MAGE-RAGE (and also AMPK activation).
Sulfonylureas (glibenclamide = glyburide, gliclazide, glipizide, glimepiride) restore cellular antioxidant levels and reduce oxidative stress, resulting in the inhibition of AGE production; however, the antiglycation mechanism for them is not fully known. In the in vitro models (HSA or BSA-glucose, HSA or BSA-MGO), glibenclamide, gliclazide and glipizide directly inhibited the formation of AGEs (i.e. N6-(carboxymethyl)-L-lysine and argpyrimidine), and the strength of this effect was comparable to aminoguanidine [241,243,244]. In the presence of glibenclamide [243], modification of human plasma albumin by glucose and MGO decreased by 70% (early and advanced glycation products), and the possible mechanism was attributed to interaction with proteins (mechanism (5), protection of glycation sites). Nevertheless, the antiglycation effect of sulfonylurea derivatives in vivo is probably related to their principal direction of action. The hypoglycemic effect of sulfonylurea derivatives results from binding to the sulfonylurea receptor (SUR) and closing ATP-sensitive potassium channels (KATP channel - KATP, potassium channels activated by a decrease in intracellular ATP and an increase in ADP) in pancreatic β-cells. Depolarization of the cell membrane occurs by impairing potassium excretion and increasing intracellular K+, followed by the opening of voltage-gated Ca2+ channels and increasing Ca2+ influx, which induces insulin secretion [277]. SUR subunits act as sensors of cellular metabolism. KATP has been identified in the membranes of numerous cell types, including endothelium, cardiomyocytes, and subcellular membranes (mitochondrial, nuclear, sarcolemmal). After an energy crisis in a cell, mitochondrial activity tends to deteriorate. In such a situation, mitochondrial KATP channels open and close, leading to unbalanced trans-membrane ion transport and overproduction of ROS. In a human aortic endothelial cell (HAEC) line, MGO caused sustained abnormal activation of KATP channels and increased their conductance. In addition, MGO exposure potentiated three mitogen-activated protein kinase (MAPK) pathways in HAEC, and glibenclamide reversed the activation of JNK (stress-activated protein kinase) by blocking KATP (KATP antagonist). Therefore, it seems that through this mechanism, KATP blockers may prevent MGO-induced endothelial (and other) cell dysfunction [126]. Depletion of ATP stores and an elevation in AMP levels are also associated with the activation of AMPK (adenosine 5′-monophosphate-activated protein kinase), which plays a pivotal role in the cell's energy metabolism. Once activated, AMPK inhibits ATP consumption pathways and turns on catabolic ATP production pathways, and through down-regulatory signaling pathways and target molecules, modulates carbohydrate and lipid metabolism (glucose uptake, gluconeogenesis, fatty acid oxidation, cholesterol synthesis, lipid synthesis, etc.). In addition, AMPK is involved in the alleviation of oxidative stress, the regulation of autophagy, and the countering of apoptosis. In a study by Lee, Kim, and Choi [245], gliclazide increased the level of phosphorylated AMPK in vascular smooth muscle cells (VSMCs) and inhibited platelet-derived growth factor (PDGF)-induced VSMC proliferation by increasing intracellular Ca2+ concentration, which is beneficial for CVD risk reduction. Gliclazide also increased the level of Ca2+/calmodulin-dependent protein kinase β (CaMKKβ), an upstream kinase of AMPK. These results suggest that the effect of KATP channels on AMPK activity was achieved through the regulation of intracellular Ca2+ levels. Oral administration of gliclazide induced activation of CaMKKβ and AMPK in vivo, demonstrating that gliclazide suppressed VSMC proliferation through the CaMKKβ-AMPK signaling pathway. Elucidating the physiological functions of KATP and AMPK in the cardiovascular system remains an active topic of study [278]. In a randomized controlled trial (PioRAGE) involving patients with T2DM, glimepiride increased plasma esRAGE and decreased RAGE expression in peripheral mononuclear cells, but to a lesser extent than pioglitazone [246].
Thiazolidinediones (pioglitazone, rosiglitazone) inhibited the AGE/RAGE pathway and NF-κB as well as alleviated cellular oxidative stress in an in vitro model on isolated rat platelets, human umbilical vein endothelial cells, and human embryonic kidney cells. These effects were associated with a down-regulation of RAGE and RAGE mRNA expression and restoration of cellular antioxidants [249,279,280]. The low sRAGE level is associated with the incidence of vascular complications in patients with T2DM [281]. Preliminary evidence suggests that some thiazolidinediones in a hyperglycemic environment also can modulate soluble RAGE levels. A Gul et al. [250] study showed that pioglitazone (but not rosiglitazone) in T2DM patients significantly increased circulating sRAGE levels, which may contribute to its antiatherosclerotic effect. Similar results were obtained by Koyama et al. [246]. On the other hand, Liu and co-authors [248] confirmed that pioglitazone and rosiglitazone inhibit platelet aggregation via AMPK activation. Pioglitazone, moreover, preferentially binds to proteins and mitigates structural changes, maintaining their integrity [249].
The potential to prevent hyperglycemia-induced RCS and ROS (or RONS) accumulation can presumably be attributed to other oral blood glucose-lowering agents, including sodium-glucose cotransporter 2 (SGLT2) inhibitors [282].
4.1.2. Angiotensin II receptor antagonists, and angiotensin-converting enzyme inhibitors
Angiotensin II receptor antagonists (candesartan, irbesartan, losartan, olmesartan, telmisartan, valsartan) and angiotensin-converting enzyme inhibitors (captopril, enalaprilat, perindoprilat, temocaprilat) reduce the formation of AGEs by several mechanisms, including activation of the glyoxalase system, antioxidant activity and chelation of transition metal cations, which leads to a decrease in the levels of RONS, MGO, and MAGEs [212,213,251,252,253]. The anti-AGE effect of olmesartan and temocaprilat (the active metabolite of temocapril) is not mediated by RCS trapping [252]. Studies by Miller et al. [212,213] revealed that in vascular cells of the retina, angiotensin II is a negative regulator of Glo1 activity and expression. In bovine retinal endothelial cells, bovine retinal pericytes, and an animal model, candesartan attenuated vascular damage in diabetic retinopathy by restoring Glo1 activity and mRNA for Glo1 to at least control levels. In contrast, it reduced mRNA for TNF-α and iNOS, cellular •NO (nitric oxide) levels below controls, and mRNA for both ICAM-1 (intercellular adhesion molecule 1, which is an endothelium- and leukocyte-associated transmembrane protein and is involved in stabilizing cell-cell interactions and facilitating leukocyte transendothelial migration) and VEGF (vascular endothelial growth factor, which is involved in vasculogenesis and angiogenesis) to non-diabetic control levels. The anti-AGE efficacy of candesartan has been confirmed in Ren 2 rats. As a result, the authors observed a reduction in retinal argpyrimidine levels and total AGEs in plasma compared to the control group [213].
4.1.3. Calcium channel blockers
Calcium channel blockers (amlodipine, lacidipine, nifedipine, diltiazem, semotiadil) inhibit glycation and delay AGE formation through mechanisms related to preventing macromolecule oxidation. They generally act as antioxidants and protect, for example, lipoproteins from further modification, but the effect on Amadori product generation is weak. Sobal, Menzel, and Sinzinger [254] compared the antioxidant efficacy of calcium antagonists in preventing copper-catalyzed oxidation of non-glycated and glycated LDL. The authors showed significant antioxidant activity of calcium channel blockers during long-term LDL glycation. Oxidation of native LDL was inhibited most efficiently by lacidipine and semotiadil, but only lacidipine significantly inhibited the oxidation of glycated LDL.
4.1.4. Arterial smooth muscle agents
Hydralazine is an antihypertensive agent (from hydrazinophthalazine derivatives) used in the treatment of essential hypertension or severe hypertension associated with conditions requiring immediate action, e.g., heart failure. The mechanism of hydralazine anty-AGE activity was documented by Nangaku et al. [253] and Colzani et al. [235]. Unlike olmesartan, but similar to aminoguanidine, hydralazine effectively captures RCS (i.e., MGO and GO) in vitro. It also impairs oxidative metabolism. Hydralazine reduces the level of carbon-centered radicals in a dose-dependent manner and the concentration of hydroxyl radicals. Finally, hydralazine interrupts the Fenton reaction because it chelates copper and inhibits the autoxidation of ascorbic acid. Inhibiting LDL glycation by trapping reactive carbonyls that induce LDL modification prevents lipid loading and foam cell formation in macrophage cells. RCS scavengers, such as biguanides and hydrazinophthalazines, can inhibit LDL glycation and prevent diabetes-induced atherosclerosis at concentrations equivalent to or above the glycating agent [255].
4.1.5. Lipid modifying agents (statins)
Statins (atorvastatin, cerivastatin, fluvastatin, pitavastatin, pravastatin, rosuvastatin, simvastatin) are used to lower cholesterol levels by inhibiting hydroxymethylglutaryl-CoA (HMG-CoA) reductase, which is involved in hepatic cholesterol synthesis. The beneficial effects of statin therapy on reducing the pathogenesis of the cardiovascular system, arteriosclerosis, and diabetic complications are commonly known. Studies in animal models have shown that RAGE is the best-known target for AGEs in the cardiovascular system, and the AGE/RAGE pathway contributes to the progression of atherosclerosis [223]. Statins can lower serum AGE levels in a manner independent of but also related to, the hypocholesterolemic effect. The anti-AGE effect of statins may be linked to increased sRAGE levels, decreased RAGE expression (6), and a slight increase in PPAR-γ receptor expression, leading to reduced ROS production and neutrophil adhesion in vitro. Indeed, data obtained in a group of T2DM patients treated with simvastatin showed a significant reduction in ROS production and neutrophil adhesion[218]. Statins appear to activate PPAR-γ by stimulating cyclooxygenase-2 and modulating Wnt signaling pathway (Wnt proteins are secreted glycoproteins that regulate diverse developmental processes) by inhibiting Dickkopf-related protein 1 (DKK-1), which acts antagonistically to Wnt. PPAR-γ is involved in the modulation of gene transcription and has protective effects on the endothelium by inhibiting endothelin-1 release and mitigating/preventing the inflammatory response [259]. Blocking the AGE/RAGE pathway was also confirmed for pravastatin and rosuvastatin in a model of diabetic nephropathy [260]. Quade-Lyssy et al. [223] connected the anti-AGE activity of statins at low concentrations to their cholesterol-lowering effects. Lovastatin in mouse alveolar epithelial cells endogenously expressing RAGE and human embryonic kidney cells overexpressing RAGE, induced sRAGE secretion but did not affect esRAGE secretion. Secretion of sRAGE was also evident after the restoration of the isoprenylation pathway, confirming the correlation between sterol biosynthesis and activation of RAGE excretion. In contrast, the lovastatin-stimulated RAGE secretion was completely abrogated by the metalloproteinase inhibitor [223]. Similarly, in another model, atorvastatin produced an anti-AGE effect in diabetic nephropathy by increasing the sRAGE level [257]. Furthermore, statins can reduce the level of AGEs by increasing the expression of NAD(P)H dehydrogenase (quinone) 1 (NQO-1) and heme oxygenase 1 (HO-1) genes in the ERK5-dependent Nrf2 (nuclear factor erythroid 2-related factor 2 dependent on extracellular signal-regulated kinase 5) signaling pathway [258,283]. The Nrf2 transcription factor regulates the expression of antioxidant proteins [162], and ERK5/NRF2 signaling plays a significant role in vascular protection against oxidative stress and the maintenance of endothelial integrity [284].
Some metabolites of statins have extra anti-AGE effects. The hydroxyl metabolites of atorvastatin acquire unusual antioxidant properties [256]. They prevent lipoprotein oxidation, and their effect on HDL and LDL is related to the protection of paraoxonase activity (paraoxonase is an enzyme associated with high-density lipoproteins, capable of hydrolyzing lipid peroxides). An accessible multicenter, double-blind, randomized clinical trial of cerivastatin in patients with T2DM confirmed significantly lowered endogenous AGEs (by 21 % after 12 weeks, CML was determined using ELISA assay) correlated with reduced LDL cholesterol and apolipoprotein B levels in all LDL subfractions, without effect on HbA1c [230]. After 12 weeks of treatment with cerivastatin the concentration of oxidized LDL was lowered by 23%. The mechanisms outlined above may underlie the anti-inflammatory and anticoagulant effects of statins related to AGEs. They also reinforce the thesis that their use in diabetic angiopathy is warranted to improve endothelial homeostasis [261]. However, further studies are needed to clarify whether the discount of serum AGEs by statins and their metabolites can reduce the risk of future cardiovascular events. The effect of statins on AGEs was summarized by Niedzielski et al. [285].
4.1.6. Peripheral vasodilators and vasoprotectives
For phlebotropic and angioprotective agents (pentoxifylline, calcium dobesylate and bioflavonoids), mechanisms (1)-(5) have been reported [233,236,239]. Pentoxifylline is a competitive, non-selective inhibitor of phosphodiesterases that increases intracellular cAMP concentrations and possesses anti-inflammatory and antioxidative properties. It exhibits potential for slowing the progression of atherosclerosis, stabilizing plaque, reducing risk and improving the outcome of vascular events, providing benefits in intermittent claudication and angina, enhancing cerebral blood flow in patients with cerebrovascular disease, improving prognosis in congestive heart failure, and aiding diabetes control. The effect of pentoxifylline on vascular health is summarized by McCarty et al. [286]. In the BSA-glucose model, pentoxifylline, like metformin and pioglitazone, showed a moderate inhibitory effect on the early glycation stage and the ability to inhibit AGE-crosslinking [239].
Calcium dobesylate is a well-known vasoactive agent (in use for more than 40 years) that exhibits a multidirectional mode of action confirmed in laboratory studies, animal models, and clinical trials [263,287]. Several studies have shown positive effects of dobesylate on endothelial dysfunction, microinflammation, vasoconstriction and increased vascular permeability. It likely reduces endothelial damage by acting through multiple pathogenic pathways involved in the progression of angiopathy. Nevertheless, the mechanism responsible for this effect has not been definitively elucidated. At the cellular level, dobesylate reduces oxidative stress and inflammation. It can also protect the endoplasmic reticulum and mitochondria and reduce excessive calcium loading, as reflected in a decrease in endothelial cell damage [288]. Deng and co-workers [262] also confirmed that at both high glucose and lipid levels, dobesylate decreases calcium impairment in the endoplasmic reticulum in cultured rat cardiomyocytes. Its anti-glycation and anti-AGEs potential (higher than metformin) was confirmed in vitro in a model with bovine serum albumin and MGO. Although dobesylate does not capture RCS, it protects the albumin protein from modification under carbonyl stress conditions through direct antioxidant activity [236]. On the other hand, the reduction of angiogenesis and capillary permeability, crucial in microvascular disease, is associated with inhibition of vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). Dobesylate recognizes both growth factors, changes their three-dimensional structure at the site of recognition by the receptors (VEGFR and FGFR), and is therefore capable of dissociating the receptor-growth factor signaling complex [289]. Finally, the systematic reviews and meta-analyses have confirmed that dobesylate therapy is significantly associated with reduced symptoms of diabetic retinopathy and nephropathy, both at the overall and local levels [287,289,290].
Plant flavonoids (a.k.a. bioflavonoids, included in a broad class of polyphenols with antioxidant properties) like rutin, isoquercitrin, quercetin, hesperidin, hesperetin, diosmin, diosmetin, and their semi-synthetic derivatives, e.g., troxerutin in experimental glycation models have shown the ability to scavenge ROS and RCS - mainly MGO [234, 236]. Generally, compounds in this chemical group capture one or two molecules of MGO and reduce the accumulation of MAGEs. However, some 7-O-substituted derivatives, such as diosmin and troxerutin, did not scavenge MGO in vitro but protected the model protein from modifications and AGE formation through antioxidant action. Interestingly, quercetin adducts with MGO retain antioxidant activity and scavenge radicals in a dose-dependent manner [264]. The results of Bhuiyan et al [234] further suggest that the ability of flavonoids to chelate transition metal cations is significant for their overall anti-AGE activity. Flavonoids are also known to interact with macromolecules; e.g., quercetin forms complexes with albumin protein involving hydrogen bonds and hydrophobic interactions [291]. The formation of non-persistent complexes in the area of glycation sites can protect the structure of the macromolecule from modifications. Besides biguanides and hydrazinophthalazines, bioflavonoids are practically the only glycation inhibitors with the ability to trap MGO (and other RCS) and the formation of stable adducts [234]. To date, results from two placebo-controlled clinical trials have been published that confirmed the ability of flavonoids to lower plasma MGO levels (changes in other RCS were not statistically significant). In a study by Van den Eynde et al. [217] involving subjects with (pre)hypertension, isoquercitrin (quercetin-3-O-glucoside 160 mg/day) decreased MGO levels significantly by about 11%; however, there was no significant change in Glo1 expression. A similar result (about a 10% reduction in MGO) was obtained for hesperidin (450 mg/day) in a study by Bednarska et al. [233]. Although the biochemical effect of MGO-trapping flavonoids usually does not exceed a dozen percent, long-term observational studies suggest that even such a slight reduction in blood methylglyoxal concentrations can be clinically significant [190,233]. Nevertheless, further work is needed to determine the exact pharmacological effects and optimize dosage.
The positive effects of flavonoids on cardiometabolic health are further confirmed by epidemiological studies. Wang et al. [292], in a systematic review and meta-analysis of 14 prospective cohort studies, found that the intake of flavonoids from different subgroups (flavonols, flavones, flavanones, flavan-3-ols, and others) is inversely associated with cardiovascular disease risk. Liu et al. [265], Kim and Je [293], Grosso et al. [294], and Micek et al. [295] conducted meta-analyses of prospective cohort studies evaluating the effect of flavonoids on the risk of mortality from any cause and cardiovascular disease in the general population. In a study by Liu et al. [265], flavonoids significantly reduced the risk of all-cause mortality by 18% in all adult subjects. A dose-response analysis showed that those ingesting 200 mg/day of total flavonoids had the lowest risk of all-cause mortality. The same authors found a marginally significant association between flavonoid intake and the risk of death from CVD and coronary heart disease (but the studies evaluating the effects of CVD and CHD deaths were in limited numbers). Previous meta-analyses have indicated that a high dietary intake of flavonols (a subgroup of flavonoids, including quercetin derivatives) can reduce the risk of coronary heart disease mortality by 20% [296] and total flavonoids by 15% [297]. According to Kim and Je [293], those with the highest intake of flavonoids had a 14% lower risk of cardiovascular and all-cause death compared to those with the least, while Grosso et al. [294] showed a 26% lower risk from any cause, and increasing the amount of flavonoids by 100 mg/day led to a linear risk reduction of 6% and 4% for overall mortality and CVD, respectively. Micek and coworkers [295] included 39 prospective cohort studies involving 1,501,645 people and a total of 33,637 cases of cardiovascular disease. In the last meta-analysis, the increase in total flavonoid intake was also linearly associated with lower cardiovascular disease risk. Among the flavonoid subgroups, however, a higher intake of flavonols and flavones was inversely correlated with coronary heart disease risk.
4.1.7. Anti-inflammatory, analgesic and antipyretic agents
Nonsteroidal anti-inflammatory agents (acetylsalicylic acid, diclofenac, ibuprofen) and paracetamol in older preclinical and human studies prevented modification of lens proteins by carbonylation and non-enzymatic glycation [266-268]. Protection by diclofenac is based on the non-covalent interaction of the medication with serum albumin. There is evidence that diclofenac blocks specifically at least one of the principal glycation sites of human serum albumin [266]. However, these results need to be verified by modern methods. On the other hand, Indurthi, Leclerc, and Vetter [298] suggest that glycation in hyperglycemic patients can significantly alter the pharmacokinetics of diclofenac with possible negative implications for patients. Instead, Rasheed et al. [270] demonstrated the anti-AGEs, antioxidant, and transition metal cation chelating potential of oxicams (meloxicam, piroxicam), nimesulide, and mefenamic acid in in vitro tests with glucose and MGO as glycation agents.
4.1.8. Selected B vitamins
Small clinical trials have shown that B vitamins (thiamine, benfotiamine, pyridoxamine) can help delay the progression of end-stage renal failure due to diabetic kidney disease by inhibiting vascular inflammation and endothelial cell damage. However, a 2015 Cochrane systematic review of clinical trials does not support this evidence. Cochrane experts also do not recommend B vitamins or combinations of B vitamins to delay the progression of end-stage renal disease. One study found thiamine to be beneficial for reducing albuminuria; however, there was no improvement in renal function or blood pressure after B vitamin preparations [299].
Benfotiamine (S-benzoyl thiamine O-monophosphate, a lipid-soluble vitamin B1) is a known NADPH oxidase inhibitor that prevents tissue damage in many experimental models. It has been confirmed that benfotiamine not only directly inhibits NADPH oxidase activity but also prevents/inactivates the protein kinase C (PKC) pathway, thereby blocking NF-κB activation in diabetic patients. In addition, the inhibitory effect of benfotiamine on NADPH oxidase may occur indirectly through the activation of transketolase, which finally inhibits NADPH oxidase production and activates antioxidant defense mechanisms [272]. In mice with streptozotocin-induced diabetes, benfotiamine significantly reduced the elevation of MGO, AGEs (MAGEs), RAGE, and collagen cross-linking without affecting hypertriglyceridemia and hypercholesterolemia [273]. A clinical trial in T2DM patients without complications confirmed that it significantly reduces CML-AGEs (AGEs with N6-(carboxymethyl)-L-lysine) and maintains normal sRAGE levels (sRAGE was decreased in the placebo group) [231].
The activity of pyridoxamine (vitamin B6) against RCS and AGEs has been confirmed in experimental models such as BSA-glucose, ribonuclease A-glucose, human hemoglobin-glucose and in the GO uptake assay [237,271,274,300]. In a study on obese mice, pyridoxamine improved glucose tolerance, insulin metabolism and vascular dysfunction. Furthermore, it reduced fasting insulin levels and improved insulin sensitivity in obese mice and mice with type 2 diabetes [275,276,301]. Its effects on glycation, metabolic and vascular risk parameters in humans were evaluated in a randomized, placebo-controlled, double-blind study involving subjects with abdominal obesity [232]. Daily doses of 25 and 200 mg of pyridoxamine were metabolically active. The higher dose reduced levels of MGO (9% reduction), AGEs (mainly MAGEs, like MG-H1), and soluble intercellular adhesion molecule-1 (sICAM-1). Both doses decreased the levels of endothelial dysfunction marker and soluble vascular cell adhesion molecule-1 (sVCAM-1) but had no effect on insulin sensitivity or vascular function. The molecular mechanism of the above properties is likely related to MGO uptake (without impact on GO and 3-DG) and inhibition of MAGE formation. The reduction of adhesion markers seems particularly promising, as they are involved in the pathogenesis of endothelial damage and atherosclerosis. A summary of scientific data for B vitamins is presented in [302,303].
4. Conclusions and remarks for future research
Cardiovascular diseases (CVD) are presently the main cause of death in the world, and type-2 diabetes is one of the most frequently recognized ch,ronic illnesses. Therefore, they severely deteriorate population health and overload the heath care system. Typical cardiovascular complications in T2DM patients comprise coronary heart disease, ischemic stroke, peripheral artery disease, and heart failure. Therefore, there is an urgent need to elucidate mechanisms for lowering the risk of cardiometabolic disorders, which would enable to design efficient ways in prophylacsy and therapy.
Methylglyoxal (MGO) is generated in the human organism mainly as a result of spontaneous fragmentation of two unstable intermediates of glycolysis: glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP). Additionally, minor quantities of MGO can be derived from the metabolism of acetone, aminoacetone and threonine, as well as from highly processed foods characterized by high content of glucose and fructose. An excessive consumption of fructose-glucose syrup has been associated with the development of insulin resistance, hyperglycemia (enhanced by gluconeogenesis) and dyslipidemia (enhanced by de novo lipogenesis resulting in increased level of free fatty acids, as well as hypercholesterolemia and hypertriglyceridemia). These metabolic disturbances lead to such disorders as non-alcoholic fatty liver disease, obesity, metabolic syndrome and type-2 diabetes. Hyperglycemia observed in diabetics enhances the metabolism of glucose accelerating the generation of MGO and its advanced glycation end products – MAGEs. Moreover, stimulated aerobic glycolysis increases reactive oxygen species (ROS) production which leads to the inhibition of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Finally, since glycolysis is inhibited at trioses level, more G3P and DHAP, and consequently MGO is produced. Thus, considering MGO-caused damage to blood vessels, T2DM patients show increased risk for cardiovascular complications (angiopathy and cardiomyopathy), which might result from inefficient control of postprandial glycemia. Only early and intensive glycemia control shows long-term beneficial effects with respect to cardiovascular complications. Increased in diabetes MGO and (M)AGEs show deleterious impact on endothelium functioning, which is associated with the promotion of oxidative stress, low-grade inflammation, atherogenesis, and CVD development. Therefore, the elucidation of mechanisms underlying these pathological processes would allow for the establishment of better prophylactic and therapeutic approaches to deal with these diseases.
MGO and MAGEs accumulation is observed when the main system responsible for MGO detoxyfication (Glo1/Glo2) is overloaded. Then MGO reacts with arginine or lysine yielding stable hydroimidazolone adducts (MG-H1, MG-H2, MG-H3), CEA, AP, THP, CEL and MOLD. Additionally, it causes the formation of cross-links with the involvement of arginine and lysine (MODIC). Moreover, MGO participates in the modification of DNA and RNA, reacting with deoxyguanosine and yielding e.g. CEdG, as well as inducing nucleic acids cross-linking. Hence, MGO can lead to epigenetic changes through the alteration of genetic material (nucleic acids and/or histones), and induce metabolic memory comprising prolonged up-regulation of pro-oxidative (ROS increase) and pro-inflammatory (NF-κB-mediated) pathways. Also, persistent MGO accumulation in the organism would impair actions of important regulators of metabolic processes, especially proteins containing arginine residues in their functional units. Such a modification of guanidine groups in Arg side chains of AMP-activated kinase (AMPK) (causing its dysfunction) would explain the shift in balance between catabolic and anabolic processes in favor of these latter (observed in cardiometabolic disorders). However, further experiments are required to test the hypothesis that this is MGO which modifies AMPK.
Experiments aimed at the identification of factors preventing glycation and AGEs accumulation in tissues, as well as reversing already generated modifications and cross-links of macromolecules, have been conducted since the beginning of nineties of the previous century. However, despite a significant development of experimental techniques, anly a few compounds with the ability of MGO trapping, have been reported within these 30 years. They include some of the biguanides, hydrazinophthalazines, bioflavonoids and pyridoxamine. A bit larger group comprises glycation inhibitors (anti-AGEs factors), such as some biguanides, sulfonylureas, thiazolidinediones, angiotensin II receptor antagonists, angiotensin-converting enzyme inhibitors, calcium channel antagonists, hydrazinophthalazines, statins, vasodilators, vasoprotectives, anti-inflammatory and analgesic agents, as well as B vitamins. These medicines are commonly applied in pharmacotherapy of hyperglycemia, insulin resistance, atherosclerosis, hypertension, peripheral artery disorder, as well as inflammatory conditions. They are applied as the first choice medicines (blood glucose lowering agents, or agents for the treatment of cardiovascular conditions). or complementary therapeutics in the cardiometabolic conditions (e.g. bioflavonoides, calcium dobesylate, pirydoxamine, benfotiamine). However, studies on factors capable of degradation of cross-links in modified macromolecules are in their infancy. Therefore, considering their potential in reversing MGO-caused damage of biological components, such experiments should be intensified to yield new therapeutics which would help in MAGE scavenging.
The main mechanisms involved in antiglycation and MGO trapping actions presented in this review are meant to chart a path in search for new molecules characterized with high therapeutic potential. Furthermore, considering available scientific data, a well-designed clinical trials should be performed, which would confirme the anti-MGO/MAGEs effects of reported glycation inhibitors. Finally, it will be interesting to see whether MGO-capture agents such as metformin, hydralazine, pyridoxamine, quercetin, and hesperidin can compete with Glo1 in the same compartments and how the kinetics of the trapping reaction affects the binding efficiency of methylglyoxal.
Author Contributions
Conceptualization, I.B and I.F.; resources, I.B., I.F. M.M.; writing—original draft preparation, I.B., I.F. M.M.; writing—review and editing; I.B., I.F. M.M.; visualization, I.F.; supervision, I.B., I.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACh | acetylcholine |
| AG | aminoguanidine |
| AGEs | advanced glycation end products |
| AKRs | Aldoketo reductases |
| Akt | PKB = protein kinase B (serine/threonine kinase) |
| ALDHs | Aldehyde dehydrogenases |
| AMPK | AMP-activated kinase |
| Ang II (Ang-2) | angiotensin II |
| AP | argpyrimidine |
| apoA1 | apolipoprotein A1 |
| apoB100 | apolipoprotein B100 |
| ApoE KO | apolipoprotein E knockout |
| ATF6 | activating transcription factor 6 |
| AT2R | angiotensin II receptor type 2 |
| BBGC | bromobenzyl-glutathione cyclopentyl diester (glyoxalase-1 inhibitor) |
| BMCs | bone marrow cells |
| CAD | coronary artery disease |
| CAT | catalase |
| CEA | N7 -carboxyethyl arginine |
| C/EBP | transcription factor C/EBP |
| CEdG | N2 -carboxyethyl-20 –deoxyguanosine |
| CEL | Nε -(1-carboxyethyl)lysineB |
| C. elegans | Caenorhabditis elegans |
| CETP | cholesteryl ester transfer protein |
| CKD | chronic kidney disease |
| CML | Nε-(1-carboxymethyl)lysine |
| CTGF | connective tissue growth factor |
| CHD | coronary heart diseases |
| CVD | cardiovascular diseases |
| DAG | diacylglycerol |
| DJ-1 (PARK7) | Parkinson’s disease protein 7 |
| DKO | double knock-out |
| 3-DG | 3-deoxyglucosone |
| 3DG-H | 3-DG-derived hydroimidazolones |
| EA.hy926 | hybrid human umbilical vein endothelial cell line |
| ECM | extracellular matrix |
| eNOS | endothelial nitric oxide synthase |
| EPCs | endothelial progenitor cells |
| ER | endoplasmic reticulum |
| esRAGE | endogenous secretory RAGE proteolytically exfoliated by metalloproteinases |
| FFAs | free fatty acids |
| FL | Nε-fructosyl-lysine |
| FPG | fasting plasma glucose |
| Fru | fructose |
| Glc | glucose |
| GlcNAc | N-acetylglucosamine |
| Glo1 | Glyoxalase 1 |
| Glo1 KO | Glo1 knockout |
| Glo2 | Glyoxalase 2 |
| GLUT | glucose transporter |
| GO | glyoxal |
| GPX | glutathione peroxidase |
| GSH | reduced glutathione |
| GSK-3 | Glycogen synthase kinase-3 |
| GSSG | oxidized glutathione |
| HAECs | human aortic endothelial cells |
| HbA1c | hemoglobin A1c |
| HEK293 | human embryonic kidney cells |
| HIF | hypoxia-inducible factor |
| HoxA5 | homeobox A5 transcription factor |
| HO-1 | heme oxygenase 1 |
| HSPG | heparan sulfate proteoglycan |
| HUVECs | human umbilical cord vein endothelial cells |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFN-γ | interferon gamma |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8 |
| IL-1β | interleukin-1 β |
| IR | insulin receptor |
| IRE1 | inositol-requiring enzyme-1 |
| IRS-1 | insulin receptor substrate 1 |
| KATP channel | ATP-sensitive potassium channel |
| KRAS | GTPase Kirsten Rat Sarcoma Viral Oncogene Homolog |
| LCAT | lecithin-cholesterol acyltransferase |
| Mac-1 | macrophage-1 antigen |
| Mac-2 | macrophage-2 antigen |
| MAECs | mouse aortic endothelial cells |
| MafA | musculoaponeurotic fibrosarcoma oncogene family A |
| MAGEs | MGO-derived AGEs |
| MCP-1 | monocyte chemoattractant peptide-1 |
| MDA | malondialdehyde |
| MG-dG | 3-(20–deoxyribosyl)-6,7-dihydro-6,7-dihydroxy-6/7-methylimidazo-[2,3-b]purin-9(8)one |
| MG-H1-3 | MGO-derived hydroimidazolones 1-3 |
| MG-H1 | [Nδ -(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine] |
| MG-H2 | 2-amino-5-(2-amino-5-hydro-5-methyl-4- imidazolon-1-yl)-pentanoic acid |
| MG-H3 | 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)-pentanoic acid |
| MGO | methylglyoxal |
| MMP-9 | matrix metalloproteinase 9 |
| MnSOD | manganese superoxide dismutase |
| MODIC | 2-ammonio-6-((2-[(4- ammonio-5-oxido-5-oxopentyl)amino]-4-methyl-4,5- dihydro-1H-imidazol-5-ylidene)amino)hexanoate |
| MOLD | 1,3-di(Nε-lysino)-4-methyl-imidazolium |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NAC | N-acetyl cysteine |
| NFATc | Nuclear factor of activated T-cells, cytoplasmic |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2 related factor 2 |
| OGTT | oral glucose tolerance test |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PAI-1 | plasminogen activator inhibitor 1 |
| PARP | poly(ADP-ribose) polymerase |
| Pdx1 | gene coding for pancreatic duodenal homeobox-1 |
| PDX-1 | homeodomain (HD)-containing transcription factor (syn: IPF-1 (insulin promoter factor 1) |
| PERK | double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase |
| PGC1α | transcriptional coactivator PGC1-α |
| PI3K | phosphatidylinositol (PI) 3-kinase |
| PKB/Akt | protein kinase B (serine/threonine kinase) |
| PKC | protein kinase C |
| p-JNK | phosphorylated c-Jun NH2 - terminal kinase |
| p-p38 | phosphorylated p38 kinase |
| p-ERK | phosphorylated extracellular signal-regulated kinase |
| PHLPP2 | PH domain leucine-rich repeat protein phosphatase 2 |
| PON1 | paraoxonase 1 |
| PPAR | peroxisome proliferation-activated receptor |
| RAGE | AGEs receptor |
| RAAS | renin-angiotensin-aldosterone system |
| RCS | Reactive carbonyl species |
| RONS | Reactive oxygen and nitrogen species |
| sdLDL | small dense low density lipoproteins |
| SD rats | Sprague Dawley rats |
| SHR | Spontaneously hypertensive rats |
| SNP | sodium nitroprusside |
| SSAO | semicarbazide-sensitive amine oxidase |
| STZ | streptozotocin |
| sICAM-1 | soluble intercellular adhesion molecule 1 |
| SOD-(1-3) | superoxide dismutase (1-3) |
| sPLA2 | secreted phospholipase A 2 |
| sRAGE | soluble RAGE produced by alternative splicing |
| sVCAM-1 | soluble vascular cell adhesion molecule 1 |
| TAG | triacylglycerol |
| TAK1 | transforming growth factor-β-activated kinase 1 |
| T1DM | type 1 diabetes |
| T2DM | type 2 diabetes |
| TGF-β | transforming growth factor β |
| THP | tetrahydropyrimidine |
| TNF-α | tumor necrosis factor α |
| UCP-2 | uncoupling protein 2 |
| UDPGlcNAc | uridine diphosphate N-acetylglucosamine |
| UPR | unfolded protein response |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR-2 | vascular endothelial growth factor receptor 2 |
| VSMCs | vascular smooth muscle cells |
| 8-OHdG | 8-hydroxy-2-deoxyguanosine |
| WKY | Wistar Kyoto rats |
References
- Nigro C, Leone A, Fiory F, Prevenzano I, Nicolò A, Mirra P, Beguinot F, Miele C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells. 2019, 8, 749. [CrossRef]
- Stratmann, B. Dicarbonyl Stress in Diabetic Vascular Disease. Int J Mol Sci. 2022 May 31;23(11):6186. [CrossRef]
- Kosmachevskaya, O.V.; Novikova, N.N.; Topunov, A.F. Carbonyl Stress in Red Blood Cells and Hemoglobin. Antioxidants (Basel). 2021, 10, 253. [CrossRef] [PubMed]
- Hoffmann, G.R. A perspective on the scientific, philosophical, and policy dimensions of hormesis. Dose Response. 2009, 7, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Zemva, J.; Fink, C.A.; Fleming, T.H.; Schmidt, L.; Loft, A.; Herzig, S.; Knieß, R.A.; Mayer, M.; Bukau, B.; Nawroth, P.P. Hormesis enables cells to handle accumulating toxic metabolites during increased energy flux. Redox Biol. 2017, 13, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M.; Priebe, S.; Grigolon, G.; Rozanov, L.; Groth, M.; Laube, B.; Guthke, R.; Platzer, M.; Zarse, K.; Ristow, M. Impairing L-Threonine Catabolism Promotes Healthspan through Methylglyoxal-Mediated Proteohormesis. Cell Metab. 2018, 27, 914–925. [Google Scholar] [CrossRef]
- Masania J, Malczewska-Malec M, Razny U, Goralska J, Zdzienicka A, Kiec-Wilk B, Gruca A, Stancel-Mozwillo J, Dembinska-Kiec A, Rabbani N, Thornalley PJ. Dicarbonyl stress in clinical obesity. Glycoconj J. 2016, 33, 581–9. [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauerj, S.K.; Eberhardt, M.; Schnölzer, M.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933, Erratum in: Nat Med. 2012 Sep;18(9):1445. Lasischka, Felix [corrected to Lasitschka, Felix]. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. (Lond). 1994, 87, 21–29. [Google Scholar] [CrossRef]
- Fleming, T.; Cuny, J.; Nawroth, G.; Djuric, Z.; Humpert, P.M.; Zeier, M.; Bierhaus, A.; Nawroth, P.P. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates?. Diabetologia. 2012, 55, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, R.; Shuck, S.C.; Chan, Y.S.; Liu, X.; Bates, S.E.; Lim, P.P.; Tamae, D.; Lacoste, S.; O'Connor, T.R.; Termini, J. DNA Advanced Glycation End Products (DNA-AGEs) Are Elevated in Urine and Tissue in an Animal Model of Type 2 Diabetes. Chem. Res. Toxicol. 2017, 30, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Synold, T.; Xi, B.; Wuenschell, G.E.; Tamae, D.; Figarola, J.L.; Rahbar, S.; Termini, J. Advanced glycation end products of DNA: quantification of N2-(1-Carboxyethyl)-2'-deoxyguanosine in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.A.; Mirrlees, D.; Thornalley, P.J. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor Statil. Biochem. Pharmacol. 1993, 46, 805–811. [Google Scholar] [CrossRef]
- Chou, C.K.; Lee, Y.T.; Chen, S.M.; Hsieh, C.W.; Huang, T.C.; Li, Y.C.; Lee, J.A. Elevated urinary D-lactate levels in patients with diabetes and microalbuminuria. J. Pharm. Biomed. Anal. 2015, 116, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.; Hanssen, N.M.; van de Waarenburg, M.P.; Jonkers, D.M.; Stehouwer, C.D.; Schalkwijk, C.G. L(+) and D(-) lactate are increased in plasma and urine samples of type 2 diabetes as measured by a simultaneous quantification of L(+) and D(-) lactate by reversed-phase liquid chromatography tandem mass spectrometry. Exp. Diabetes Res. 2012, 2012, 234812. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999, 48, 198–202. [Google Scholar] [CrossRef]
- McLellan, A.C.; Phillips, S.A.; Thornalley, P.J. The assay of methylglyoxal in biological systems by derivatization with 1,2-diamino-4,5-dimethoxybenzene. Anal. Biochem. 1992, 206, 17–23. [Google Scholar] [CrossRef]
- McLellan, A.C.; Phillips, S.A.; Thornalley, P.J. Fluorimetric assay of D-lactate. Anal. Biochem. 1992, 206, 12–16. [Google Scholar] [CrossRef]
- Christopher, M.M.; Broussard, J.D.; Fallin, C.W.; Drost, N.J.; Peterson, M.E. Increased serum D-lactate associated with diabetic ketoacidosis. Metabolism. 1995, 44, 287–290. [Google Scholar] [CrossRef]
- van Eupen MG, Schram MT, Colhoun HM, Hanssen NM, Niessen HW, Tarnow L, Parving HH, Rossing P, Stehouwer CD, Schalkwijk CG. The methylglyoxal-derived AGE tetrahydropyrimidine is increased in plasma of individuals with type 1 diabetes mellitus and in atherosclerotic lesions and is associated with sVCAM-1.. Diabetologia. 2013, 56, 1845–55. [CrossRef]
- Kilhovd BK, Giardino I, Torjesen PA, Birkeland KI, Berg TJ, Thornalley PJ, Brownlee M, Hanssen KF. Increased serum levels of the specific AGE-compound methylglyoxal-derived hydroimidazolone in patients with type 2 diabetes. Metabolism. 2003, 52, 163–7. [CrossRef]
- Hanssen, N.M.; Beulens, J.W.; van Dieren, S.; Scheijen, J.L.; van der A, D.L.; Spijkerman, A.M.; van der Schouw, Y.T.; Stehouwer, C.D.; Schalkwijk, C.G. Plasma advanced glycation end products are associated with incident cardiovascular events in individuals with type 2 diabetes: a case-cohort study with a median follow-up of 10 years (EPIC-NL). Diabetes. 2015, 64, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Fosmark DS, Torjesen PA, Kilhovd BK, Berg TJ, Sandvik L, Hanssen KF, Agardh CD, Agardh E. Increased serum levels of the specific advanced glycation end product methylglyoxal-derived hydroimidazolone are associated with retinopathy in patients with type 2 diabetes mellitus. Metabolism. 2006, 55, 232–6. [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: the first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.; Campos Campos, M.; Nawroth, P.; Fleming, T. The Glyoxalase System-New Insights into an Ancient Metabolism. Antioxidants (Basel). 2020, 9, 939. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar] [CrossRef]
- Martins, A.M.; Cordeiro, C.A.; Ponces Freire, A.M. In situ analysis of methylglyoxal metabolism in Saccharomyces cerevisiae. FEBS Lett. 2001, 499, 41–44. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Wang X, Jia X, Chang T, Desai K, Wu L.. Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J Hypertens. 2008, 26, 765–72. [CrossRef]
- Zhang X, Scheijen JLJM, Stehouwer CDA, Wouters K, Schalkwijk CG. Increased methylglyoxal formation in plasma and tissues during a glucose tolerance test is derived from exogenous glucose. Clin Sci (Lond). 2023, 137, 697–706. [CrossRef]
- Mortera, R.R.; Bains, Y.; Gugliucci, A. Fructose at the crossroads of the metabolic syndrome and obesity epidemics. Front. Biosci. (Landmark Ed). 2019, 24, 186–211. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef]
- Vasdev S, Ford CA, Longerich L, Gadag V, Wadhawan S. Role of aldehydes in fructose induced hypertension. Mol Cell Biochem. 1998, 181, 1–9. [CrossRef]
- Naomi, N.D.; Ngo, J.; Brouwer-Brolsma, E.M.; Buso, M.E.C.; Soedamah-Muthu, S.S.; Pérez-Rodrigo, C.; Harrold, J.A.; Halford, J.C.G.; Raben, A.; Geleijnse, J.M.; et al. Sugar-sweetened beverages, low/no-calorie beverages, fruit juice and non-alcoholic fatty liver disease defined by fatty liver index: the SWEET project. Nutr. Diabetes. 2023, 13, 6. [Google Scholar] [CrossRef]
- Gugliucci, A. Fructose surges damage hepatic adenosyl-monophosphate-dependent kinase and lead to increased lipogenesis and hepatic insulin resistance. Med Hypotheses. 2016, 93, 87–92. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012, 42, 1133–1142. [Google Scholar] [CrossRef]
- Hernandez-Castillo, C.; Shuck, S.C. Diet and Obesity-Induced Methylglyoxal Production and Links to Metabolic Disease. Chem. Res. Toxicol. 2021, 34, 2424–2440. [Google Scholar] [CrossRef]
- Kalapos, M.P. Where does plasma methylglyoxal originate from?. Diabetes Res. Clin. Pract. 2013, 99, 260–271. [Google Scholar] [CrossRef]
- Lyles, G.A.; Chalmers, J. The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amine oxidase in human umbilical artery. Biochem. Pharmacol. 1992, 43, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Guo, H.; Ou, J.; Liu, P.; Huang, C.; Wang, M.; Simal-Gandara, J.; Battino, Jafari, S.M.; Zou, L.; et al.; et al. Benefits, deleterious effects and mitigation of methylglyoxal in foods: A critical review. Trends Food Sci. Technol. 2021, 107, 201–212. [Google Scholar] [CrossRef]
- Degen, J.; Hellwig, M.; Henle, T. 1,2-dicarbonyl compounds in commonly consumed foods. J. Agric. Food. Chem. 2012, 60, 7071–7079. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-H.; Shin, H.-S. In-solution derivatization and detection of glyoxal and methylglyoxal in alcoholic beverages and fermented foods by headspace solid-phase microextraction and gas chromatography−mass spectrometry. J. Food Compos. Anal. 2020, 92, 103584. [Google Scholar] [CrossRef]
- Papetti, A.; Mascherpa, D.; Gazzani, G. Free α-dicarbonyl compounds in coffee, barley coffee and soy sauce and effects of in vitro digestion. Food Chem. 2014, 164, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Lorenzo, G.; Morales, F.J. Analysis, distribution, and dietary exposure of glyoxal and methylglyoxal in cookies and their relationship with other heat-induced contaminants. J. Agric. Food Chem. 2010, 58, 2966–2972. [Google Scholar] [CrossRef]
- Degen, J.; Vogel, M.; Richter, D.; Hellwig, M.; Henle, T. Metabolic transit of dietary methylglyoxal. J. Agric. Food. Chem. 2013, 61, 10253–10260. [Google Scholar] [CrossRef]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [CrossRef]
- Ahmed, N.; Argirov, O.K.; Minhas, H.S.; Cordeiro, C.A.; Thornalley, P.J. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nepsilon-carboxymethyl-lysine- and Nepsilon-(1-carboxyethyl)lysine-modified albumin. Biochem. J. 2002, 364, 1–14. [Google Scholar] [CrossRef]
- Klöpfer, A.; Spanneberg, R.; Glomb, M.A. Formation of arginine modifications in a model system of Nα-tert-butoxycarbonyl (Boc)-arginine with methylglyoxal. J. Agric. Food Chem. 2011, 59, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J. Biol. Chem. 2005, 280, 5724–5732. [Google Scholar] [CrossRef]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Invest. Ophthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef]
- Wang, T.; Streeter, M.D.; Spiegel, D.A. Generation and characterization of antibodies against arginine-derived advanced glycation endproducts. Bioorg. Med. Chem. Lett. 2015, 25, 4881–4886. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; Marnett, L.J. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. U S A. 2018, 115, 9228–9233. [Google Scholar] [CrossRef]
- Oya T, Hattori N, Mizuno Y, Miyata S, Maeda S, Osawa T, Uchida K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J Biol Chem. 1999, 274, 18492–502. [CrossRef]
- Lieuw-a-Fa ML, Schalkwijk CG, Engelse M, van Hinsbergh VW. Interaction of Nepsilon(carboxymethyl)lysine- and methylglyoxal-modified albumin with endothelial cells and macrophages. Splice variants of RAGE may limit the responsiveness of human endothelial cells to AGEs. Thromb Haemost. 2006, 95, 320–8. [CrossRef]
- Ahmed, N.; Thornalley, P.J. Peptide mapping of human serum albumin modified minimally by methylglyoxal in vitro and in vivo. Ann. N. Y. Acad. Sci. 2005, 1043, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tanase, S.; Nakajou, K.; Maruyama, T.; Kragh-Hansen, U.; Otagiri, M. Role of arg-410 and tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 2000, 349, 813–819. [Google Scholar] [CrossRef]
- Faure, P.; Troncy, L.; Lecomte, M.; Wiernsperger, N.; Lagarde, M.; Ruggiero, D.; Halimi, S. Albumin antioxidant capacity is modified by methylglyoxal. Diabetes Metab. 2005, 31, 169–177. [Google Scholar] [CrossRef]
- Abordo, E.A.; Thornalley, P.J. Synthesis and secretion of tumour necrosis factor-alpha by human monocytic THP-1 cells and chemotaxis induced by human serum albumin derivatives modified with methylglyoxal and glucose-derived advanced glycation endproducts. Immunol. Lett. 1997, 58, 139–147. [Google Scholar] [CrossRef]
- Fan, X.; Subramaniam, R.; Weiss, M.F.; Monnier, V.M. Methylglyoxal-bovine serum albumin stimulates tumor necrosis factor alpha secretion in RAW 264.7 cells through activation of mitogen-activating protein kinase, nuclear factor kappaB and intracellular reactive oxygen species formation. Arch. Biochem. Biophys. 2003, 409, 274–286. [Google Scholar] [CrossRef]
- Westwood, M.E.; Thornalley, P.J. Induction of synthesis and secretion of interleukin 1 beta in the human monocytic THP-1 cells by human serum albumins modified with methylglyoxal and advanced glycation endproducts. Immunol. Lett. 1996, 50, 17–21. [Google Scholar] [CrossRef]
- Dobler, D.; Ahmed, N.; Song, L.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes. 2006, 55, 1961–1969. [Google Scholar] [CrossRef]
- Monnier, V.M.; Sell., D.R.; Strauch, C.; Sun, W.; Lachin, J.M.; Cleary, P.A.; Genuth, S.; DCCT Research Group. The association between skin collagen glucosepane and past progression of microvascular and neuropathic complications in type 1 diabetes. J. Diabetes Complications. 2013, 27, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Bose, T.; Bhattacherjee, A.; Banerjee, S.; Chakraborti, A.S. Methylglyoxal-induced modifications of hemoglobin: structural and functional characteristics. Arch. Biochem. Biophys. 2013, 529, 99–104. [Google Scholar] [CrossRef]
- Lee JH, Samsuzzaman M, Park MG, Park SJ, Kim SY. Methylglyoxal-derived hemoglobin advanced glycation end products induce apoptosis and oxidative stress in human umbilical vein endothelial cells. Int J Biol Macromol. 2021, 187, 409–421. [CrossRef] [PubMed]
- Chen HJ, Chen YC, Hsiao CF, Chen PF. Mass Spectrometric Analysis of Glyoxal and Methylglyoxal-Induced Modifications in Human Hemoglobin from Poorly Controlled Type 2 Diabetes Mellitus Patients. Chem Res Toxicol. 2015, 28, 2377–89. [CrossRef]
- Jia X, Olson DJ, Ross AR, Wu L. Structural and functional changes in human insulin induced by methylglyoxal. FASEB J. 2006, 20, 1555–7. [CrossRef]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008, 7, 260–269. [Google Scholar] [CrossRef]
- Queisser, M.A.; Yao, D.; Geisler, S.; Hammes, H.P.; Lochnit, G.; Schleicher, E.D.; Brownlee, M.; Preissner, K.T. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010, 59, 670–678. [Google Scholar] [CrossRef]
- Folz, R.; Laiteerapong, N. The legacy effect in diabetes: are there long-term benefits?. Diabetologia. 2021, 64, 2131–2137. [Google Scholar] [CrossRef]
- Li, H.; Nakamura, S.; Miyazaki, S.; Morita, T.; Suzuki, M.; Pischetsrieder, M.; Niwa, T. N2-carboxyethyl-2'-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int. 2006, 69, 388–392. [Google Scholar] [CrossRef]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson's disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson's disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P. et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science. 2017, 357, 208–211. [CrossRef]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef]
- Baba, S.P.; Barski, O.A.; Ahmed, Y.; O'Toole, T.E.; Conklin, D.J.; Bhatnagar, A.; Srivastava, S. Reductive metabolism of AGE precursors: a metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes. 2009, 58, 2486–2497. [Google Scholar] [CrossRef]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef]
- Fahed G, Aoun L, Bou Zerdan M, Allam S, Bou Zerdan M, Bouferraa Y, Assi HI. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int J Mol Sci. 2022, 23, 786. [CrossRef]
- Lemieux, I.; Després, J.P. Metabolic Syndrome: Past, Present and Future. Nutrients. 2020, 12, 3501. [Google Scholar] [CrossRef]
- Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration (BMI Mediated Effects), Lu, Y.; Hajifathalian, K.; Ezzati, M.; Woodward, M.; Rimm, E.B.; Danaei, G. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1·8 million participants. Lancet. 2014, 383, 970–983. [CrossRef]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996, 379, 632–635. [Google Scholar] [CrossRef]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Francisco FA, Barella LF, Silveira SDS, Saavedra LPJ, Prates KV, Alves VS, Franco CCDS, Miranda RA, Ribeiro TA, Tófolo LP, Malta A, Vieira E, Palma-Rigo K, Pavanello A, Martins IP, Moreira VM, de Oliveira JC, Mathias PCF, Gomes RM. Methylglyoxal treatment in lactating mothers leads to type 2 diabetes phenotype in male rat offspring at adulthood. Eur J Nutr. 2018, 57, 477–486. [CrossRef]
- Liu J, Wang R, Desai K, Wu L. Upregulation of aldolase B and overproduction of methylglyoxal in vascular tissues from rats with metabolic syndrome. Cardiovasc Res. 2011, 92, 494–503. [CrossRef] [PubMed]
- Chang T, Wang R, Wu L. Methylglyoxal-induced nitric oxide and peroxynitrite production in vascular smooth muscle cells. Free Radic Biol Med. 2005, 38, 286–93. [CrossRef]
- Wang H, Liu J, Wu L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem Pharmacol. 2009, 77, 1709–16. [CrossRef] [PubMed]
- Tsokanos FF, Muley C, Khani S, Hass D, Fleming T, Wolff G, Bartelt A, Nawroth P, Herzig S. Methylglyoxal Drives a Distinct, Nonclassical Macrophage Activation Status. Thromb Haemost. 2021, 121, 1464–1475. [CrossRef]
- Prantner D, Nallar S, Richard K, Spiegel D, Collins KD, Vogel SN.Classically activated mouse macrophages produce methylglyoxal that induces a TLR4- and RAGE-independent proinflammatory response. J Leukoc Biol. 2021, 109, 605–619. [CrossRef]
- Wang X, Desai K, Clausen JT, Wu L. Increased methylglyoxal and advanced glycation end products in kidney from spontaneously hypertensive rats. Kidney Int. 2004, 66, 2315–21. [CrossRef]
- Wang X, Desai K, Chang T, Wu L. Vascular methylglyoxal metabolism and the development of hypertension. J Hypertens. 2005, 23, 1565–73. [CrossRef]
- Wang X, Chang T, Jiang B, Desai K, Wu L. Attenuation of hypertension development by aminoguanidine in spontaneously hypertensive rats: role of methylglyoxal. Am J Hypertens. 2007, 20, 629–36. [CrossRef]
- Mukohda M, Okada M, Hara Y, Yamawaki H. Methylglyoxal accumulation in arterial walls causes vascular contractile dysfunction in spontaneously hypertensive rats. J Pharmacol Sci. 2012, 120, 26–35. [CrossRef]
- Mukohda M, Morita T, Okada M, Hara Y, Yamawaki H. Long-term methylglyoxal treatment causes endothelial dysfunction of rat isolated mesenteric artery. J Vet Med Sci. 2013, 75, 151–7. [CrossRef]
- Mukohda M, Yamawaki H, Nomura H, Okada M, Hara Y. Methylglyoxal inhibits smooth muscle contraction in isolated blood vessels. J Pharmacol Sci. 2009, 109, 305–10. [CrossRef]
- Subramanian U, Nagarajan D. All-Trans Retinoic Acid supplementation prevents cardiac fibrosis and cytokines induced by Methylglyoxal. Glycoconj J. 2017, 34, 255–265. [CrossRef]
- Jia X, Wu L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol Cell Biochem. 2007, 306, 133–9. [CrossRef]
- Jang, S.; Kwon, D.M.; Kwon, K.; Park, C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490, 460–465. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef]
- Brouwers, O.; de Vos-Houben, J.M.; Niessen, P.M.; Miyata, T.; van Nieuwenhoven, F.; Janssen, B.J.; Hageman, G.; Stehouwer, C.D.; Schalkwijk, C.G. Mild oxidative damage in the diabetic rat heart is attenuated by glyoxalase-1 overexpression. Int. J. Mol. Sci. 2013, 14, 15724–15739. [Google Scholar] [CrossRef]
- Brouwers O, Niessen PM, Haenen G, Miyata T, Brownlee M, Stehouwer CD, De Mey JG, Schalkwijk CG. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia. 2010, 53, 989–1000. [CrossRef]
- Brouwers O, Niessen PM, Miyata T, Østergaard JA, Flyvbjerg A, Peutz-Kootstra CJ, Sieber J, Mundel PH, Brownlee M, Janssen BJ, De Mey JG, Stehouwer CD, Schalkwijk CG. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia. 2014, 57, 224–35. [CrossRef]
- Geoffrion, M.; Du, X.; Irshad, Z.; Vanderhyden, B.C.; Courville, K.; Sui, G.; D'Agati, V.D.; Ott-Braschi, S.; Rabbani, N.; Thornalley, P.J.; et al. Differential effects of glyoxalase 1 overexpression on diabetic atherosclerosis and renal dysfunction in streptozotocin-treated, apolipoprotein E-deficient mice. Physiol. Rep. 2014, 2, e12043. [Google Scholar] [CrossRef]
- Tikellis, C.; Pickering, R.J.; Tsorotes, D.; Huet, O.; Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes. 2014, 63, 3915–3925. [Google Scholar] [CrossRef]
- Fang X, Liu L, Zhou S, Zhu M, Wang B. N-acetylcysteine inhibits atherosclerosis by correcting glutathione-dependent methylglyoxal elimination and dicarbonyl/oxidative stress in the aorta of diabetic mice. Mol Med Rep. 2021, 23, 201. [CrossRef]
- Nigro C, Raciti GA, Leone A, Fleming TH, Longo M, Prevenzano I, Fiory F, Mirra P, D'Esposito V, Ulianich L, Nawroth PP, Formisano P, Beguinot F, Miele C. Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia. 2014, 57, 1485–94. [CrossRef]
- Hanssen, N.M.; Brouwers, O.; Gijbels, M.J.; Wouters, K.; Wijnands, E.; Cleutjens, J.P.; De Mey, J.G.; Miyata, T.; Biessen, E.A.; Stehouwer, C.D.; et al. Glyoxalase 1 overexpression does not affect atherosclerotic lesion size and severity in ApoE-/- mice with or without diabetes. Cardiovasc. Res. 2014, 104, 160–170. [Google Scholar] [CrossRef]
- Sena, C.M.; Matafome, P.; Crisóstomo, J.; Rodrigues, L.; Fernandes, R.; Pereira, P.; Seiça, R.M. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol. Res. 2012, 65, 497–506. [Google Scholar] [CrossRef]
- Matafome P, Santos-Silva D, Crisóstomo J, Rodrigues T, Rodrigues L, Sena CM, Pereira P, Seiça R. Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch Physiol Biochem. 2012, 118, 58–68. [CrossRef]
- Rodrigues T, Matafome P, Seiça R. Methylglyoxal further impairs adipose tissue metabolism after partial decrease of blood supply. Arch Physiol Biochem. 2013, 119, 209–18. [CrossRef]
- Rodrigues T, Matafome P, Sereno J, Almeida J, Castelhano J, Gamas L, Neves C, Gonçalves S, Carvalho C, Arslanagic A, Wilcken E, Fonseca R, Simões I, Conde SV, Castelo-Branco M, Seiça R. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci Rep. 2017, 7, 1698. [CrossRef]
- Hüttl M, Markova I, Miklankova D, Makovicky P, Pelikanova T, Šeda O, Šedová L, Malinska H. Adverse Effects of Methylglyoxal on Transcriptome and Metabolic Changes in Visceral Adipose Tissue in a Prediabetic Rat Model. Antioxidants (Basel). 2020, 9, 803. [CrossRef]
- Berlanga, J.; Cibrian, D.; Guillén, I.; Freyre, F.; Alba, J.S.; Lopez-Saura, P.; Merino, N.; Aldama, A.; Quintela, A.M.; Triana, M.E.; et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. (Lond). 2005, 109, 83–95. [Google Scholar] [CrossRef]
- Guo Q, Mori T, Jiang Y, Hu C, Osaki Y, Yoneki Y, Sun Y, Hosoya T, Kawamata A, Ogawa S, Nakayama M, Miyata T, Ito S. Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague-Dawley rats. J Hypertens. 2009, 27, 1664–71. [CrossRef]
- Dhar A, Dhar I, Desai KM, Wu L.. Methylglyoxal scavengers attenuate endothelial dysfunction induced by methylglyoxal and high concentrations of glucose. Br J Pharmacol. 2010, 161, 1843–56. [CrossRef]
- Dhar A, Desai KM, Wu L.. Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague-Dawley rats. Br J Pharmacol. 2010, 159, 166–75. [CrossRef]
- Chang T, Wang R, Olson DJ, Mousseau DD, Ross AR, Wu L.. Modification of Akt1 by methylglyoxal promotes the proliferation of vascular smooth muscle cells. FASEB J. 2011, 25, 1746–57. [CrossRef]
- Dhar A, Dhar I, Jiang B, Desai KM, Wu L.. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes. 2011, 60, 899–908. [CrossRef]
- Dhar I, Dhar A, Wu L, Desai KM. Methylglyoxal, a reactive glucose metabolite, increases renin angiotensin aldosterone and blood pressure in male Sprague-Dawley rats. Am J Hypertens. 2014, 27, 308–16. [CrossRef]
- Liu, H.; Yu, S.; Zhang, H.; Xu, J. Angiogenesis impairment in diabetes: role of methylglyoxal-induced receptor for advanced glycation endproducts, autophagy and vascular endothelial growth factor receptor 2. PLoS One. 2012, 7, e46720. [Google Scholar] [CrossRef]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes. 2016, 65, 1699–1713. [Google Scholar] [CrossRef]
- Vulesevic, B.; McNeill, B.; Geoffrion, M.; Kuraitis, D.; McBane, J.E.; Lochhead, M.; Vanderhyden, B.C.; Korbutt, G.S.; Milne, R.W.; Suuronen, E.J. Glyoxalase-1 overexpression in bone marrow cells reverses defective neovascularization in STZ-induced diabetic mice. Cardiovasc. Res. 2014, 101, 306–316. [Google Scholar] [CrossRef]
- Wang, Y.; Hall, L.M.; Kujawa, M.; Li, H.; Zhang, X.; O'Meara, M.; Ichinose, T.; Wang, J.M. Methylglyoxal triggers human aortic endothelial cell dysfunction via modulation of the KATP/MAPK pathway. Am. J. Physiol. Cell. Physiol. 2019, 317, C68–C81. [Google Scholar] [CrossRef]
- Nigro C, Leone A, Longo M, Prevenzano I, Fleming TH, Nicolò A, Parrillo L, Spinelli R, Formisano P, Nawroth PP, Beguinot F, Miele C. Methylglyoxal accumulation de-regulates HoxA5 expression, thereby impairing angiogenesis in glyoxalase 1 knock-down mouse aortic endothelial cells. Biochim Biophys Acta Mol Basis Dis. 2019, 1865, 73–85. [CrossRef]
- Wang G, Wang Y, Yang Q, Xu C, Zheng Y, Wang L, Wu J, Zeng M, Luo M. Metformin prevents methylglyoxal-induced apoptosis by suppressing oxidative stress in vitro and in vivo. Cell Death Dis. 2022, 13, 29. [CrossRef]
- Mirra P, Nigro C, Prevenzano I, Procopio T, Leone A, Raciti GA, Andreozzi F, Longo M, Fiory F, Beguinot F, Miele C. The role of miR-190a in methylglyoxal-induced insulin resistance in endothelial cells. Biochim Biophys Acta Mol Basis Dis. 2017, 1863, 440–449. [CrossRef]
- Nigro C, Mirra P, Prevenzano I, Leone A, Fiory F, Longo M, Cabaro S, Oriente F, Beguinot F, Miele C. miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. Int J Mol Sci. 2018, 19, 522. [CrossRef]
- Wu L, Juurlink BH. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension. 2002, 39, 809–14. [CrossRef]
- Riboulet-Chavey A, Pierron A, Durand I, Murdaca J, Giudicelli J, Van Obberghen E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes. 2006, 55, 1289–99. [CrossRef]
- Engelbrecht B, Mattern Y, Scheibler S, Tschoepe D, Gawlowski T, Stratmann B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm Metab Res. 2014, 46, 77–84. [CrossRef]
- Fiory F, Lombardi A, Miele C, Giudicelli J, Beguinot F, Van Obberghen E. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia. 2011, 54, 2941–52, Erratum in: Diabetologia. 2012, 55, 272. [CrossRef]
- Bo J, Xie S, Guo Y, Zhang C, Guan Y, Li C, Lu J, Meng QH. Methylglyoxal Impairs Insulin Secretion of Pancreatic β-Cells through Increased Production of ROS and Mitochondrial Dysfunction Mediated by Upregulation of UCP2 and MAPKs. J Diabetes Res. 2016;2016:2029854. [CrossRef]
- Wang H, Meng QH, Chang T, Wu L.. Fructose-induced peroxynitrite production is mediated by methylglyoxal in vascular smooth muscle cells. Life Sci. 2006, 79, 2448–54. [CrossRef]
- Kırça M, Yeşilkaya A. Methylglyoxal stimulates endoplasmic reticulum stress in vascular smooth muscle cells. J Recept Signal Transduct Res. 2022, 42, 279–284. [CrossRef]
- Baden T, Yamawaki H, Saito K, Mukohda M, Okada M, Hara Y. Telmisartan inhibits methylglyoxal-mediated cell death in human vascular endothelium. Biochem Biophys Res Commun. 2008, 373, 253–7. [CrossRef]
- Kim, J.H.; Kim, K.A.; Shin, Y.J.; Kim, H.; Majid, A.; Bae, O.N. Methylglyoxal induced advanced glycation end products (AGE)/receptor for AGE (RAGE)-mediated angiogenic impairment in bone marrow-derived endothelial progenitor cells. J. Toxicol. Environ. Health A. 2018, 81, 266–277. [Google Scholar] [CrossRef]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef]
- Navarrete Santos A, Jacobs K, Simm A, Glaubitz N, Horstkorte R, Hofmann B. Dicarbonyls induce senescence of human vascular endothelial cells. Mech Ageing Dev. 2017 Sep;166:24-32. [CrossRef]
- Miyazawa N, Abe M, Souma T, Tanemoto M, Abe T, Nakayama M, Ito S. Methylglyoxal augments intracellular oxidative stress in human aortic endothelial cells. Free Radic Res. 2010, 44, 101–7. [CrossRef]
- Oba T, Tatsunami R, Sato K, Takahashi K, Hao Z, Tampo Y. Methylglyoxal has deleterious effects on thioredoxin in human aortic endothelial cells. Environ Toxicol Pharmacol. 2012, 34, 117–126. [CrossRef]
- Irshad Z, Xue M, Ashour A, Larkin JR, Thornalley PJ, Rabbani N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci Rep. 2019, 9, 7889. [CrossRef]
- Schalkwijk CG, van Bezu J, van der Schors RC, Uchida K, Stehouwer CD, van Hinsbergh VW. Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. FEBS Lett. 2006, 580, 1565–70, Erratum in: FEBS Lett. 2006, 580, 2400. [CrossRef]
- Chu P, Han G, Ahsan A, Sun Z, Liu S, Zhang Z, Sun B, Song Y, Lin Y, Peng J, Tang Z. Phosphocreatine protects endothelial cells from Methylglyoxal induced oxidative stress and apoptosis via the regulation of PI3K/Akt/eNOS and NF-κB pathway. Vascul Pharmacol. 2017 Apr;91:26-35. [CrossRef]
- Braun JD, Pastene DO, Breedijk A, Rodriguez A, Hofmann BB, Sticht C, von Ochsenstein E, Allgayer H, van den Born J, Bakker S, Hauske SJ, Krämer BK, Yard BA, Albrecht T. Methylglyoxal down-regulates the expression of cell cycle associated genes and activates the p53 pathway in human umbilical vein endothelial cells. Sci Rep. 2019, 9, 1152. [CrossRef]
- Zhang X, Rodriguez-Niño A, Pastene DO, Pallavi P, van den Born J, Bakker SJL, Krämer BK, Yard BA. Methylglyoxal induces p53 activation and inhibits mTORC1 in human umbilical vein endothelial cells. Sci Rep. 2021, 11, 8004. [CrossRef]
- Brouwers O, Teerlink T, van Bezu J, Barto R, Stehouwer CD, Schalkwijk CG. Methylglyoxal and methylglyoxal-arginine adducts do not directly inhibit endothelial nitric oxide synthase. Ann N Y Acad Sci. 2008 Apr;1126:231-4. [CrossRef]
- Jang, J.H.; Kim, E.A.; Park, H.J.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Woo, C.H.; Doh, K.O.; Kim, K.H.; Lee, T.J. Methylglyoxal-induced apoptosis is dependent on the suppression of c-FLIPL expression via down-regulation of p65 in endothelial cells. J. Cell. Mol. Med. 2017, 21, 2720–2731. [Google Scholar] [CrossRef]
- Su Y, Qadri SM, Wu L, Liu L. Methylglyoxal modulates endothelial nitric oxide synthase-associated functions in EA.hy926 endothelial cells. Cardiovasc Diabetol. 2013 Sep 19;12:134. [CrossRef]
- Stratmann, B.; Engelbrecht, B.; Espelage, B.C.; Klusmeier, N.; Tiemann, J. Gawlowski, T.; Mattern, Y.; Eisenacher, M.; Meyer, H.E.; Rabbani, N. et al. Glyoxalase 1-knockdown in human aortic endothelial cells - effect on the proteome and endothelial function estimates. Sci. Rep. 2016, 6, 37737. [Google Scholar] [CrossRef]
- Ahmed, U.; Dobler, D.; Larkin, S.; Rabbani, N.; Thornalley, P.J. Reversal of hyperglycemia-induced angiogenesis deficit of human endothelial cells by overexpression of glyoxalase 1 in vitro. Ann. N. Y. Acad. Sci. 2008, 1126, 262–264. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bose, N.; Gong, J.; Hall, D.; Rifkind, A.; Bhaumik, D.; Peiris, T.H.; Chamoli, M.; Le, C.H.; Liu, J.; et al. A Caenorhabditis elegans Model Elucidates a Conserved Role for TRPA1-Nrf Signaling in Reactive α-Dicarbonyl Detoxification. Curr. Biol. 2016, 26, 3014–3025. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934.e8. [Google Scholar] [CrossRef]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight. 2019, 4, e126154. [Google Scholar] [CrossRef]
- Nigro C, Leone A, Raciti GA, Longo M, Mirra P, Formisano P, Beguinot F, Miele C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int J Mol Sci. 2017, 18, 188. [CrossRef]
- Shamsaldeen YA, Mackenzie LS, Lione LA, Benham CD. Methylglyoxal, A Metabolite Increased in Diabetes is Associated with Insulin Resistance, Vascular Dysfunction and Neuropathies. Curr Drug Metab. 2016, 17, 359–67. [CrossRef]
- Mey JT, Haus JM. Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front Cardiovasc Med. 2018 Sep 10;5:117. [CrossRef]
- Sztalryd C, Brasaemle DL. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim Biophys Acta Mol Cell Biol Lipids. 2017 Oct;1862(10 Pt B):1221-1232. [CrossRef]
- Mirra P, Nigro C, Prevenzano I, Leone A, Raciti GA, Formisano P, Beguinot F, Miele C. The Destiny of Glucose from a MicroRNA Perspective. Front Endocrinol (Lausanne). 2018 Feb 26;9:46. [CrossRef]
- Minjares M, Wu W, Wang JM. Oxidative Stress and MicroRNAs in Endothelial Cells under Metabolic Disorders. Cells. 2023, 12, 1341. [CrossRef]
- Matafome P, Rodrigues T, Sena C, Seiça R. Methylglyoxal in Metabolic Disorders: Facts, Myths, and Promises. Med Res Rev. 2017, 37, 368–403. [CrossRef]
- Ozdemir AM, Hopfer U, Rosca MV, Fan XJ, Monnier VM, Weiss MF. Effects of advanced glycation end product modification on proximal tubule epithelial cell processing of albumin. Am J Nephrol. 2008, 28, 14–24. [CrossRef]
- Bierhaus A, Nawroth PP. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia. 2009, 52, 2251–63. [CrossRef]
- Jack MM, Ryals JM, Wright DE. Characterisation of glyoxalase I in a streptozocin-induced mouse model of diabetes with painful and insensate neuropathy. Diabetologia. 2011, 54, 2174–82. [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Eringa, E.C.; Serne, E.H.; Meijer, R.I.; Schalkwijk, C.G.; Houben, A.J.; Stehouwer, C.D.; Smulders, Y.M.; van Hinsbergh, V.W. Endothelial dysfunction in (pre)diabetes: characteristics, causative mechanisms and pathogenic role in type 2 diabetes. Rev. Endocr. Metab. Disord. 2013, 14, 39–48. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin. Sci. (Lond). 2005, 109, 143–159. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Micali, L.R.; Wouters, K. Advanced glycation endproducts in diabetes-related macrovascular complications: focus on methylglyoxal. Trends Endocrinol. Metab. 2023, 34, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature. 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Ebara, T.; Conde, K.; Kako, Y.; Liu, Y.; Xu, Y.; Ramakrishnan, R.; Goldberg, I.J.; Shachter, N.S. Delayed catabolism of apoB-48 lipoproteins due to decreased heparan sulfate proteoglycan production in diabetic mice. J. Clin. Invest. 2000, 105, 1807–1818. [Google Scholar] [CrossRef]
- Koya, D.; Jirousek, M.R.; Lin, Y.W; Ishii, H.; Kuboki, K.; King, G.L. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J. Clin. Invest. 1997, 100, 115–126. [Google Scholar] [CrossRef]
- Giacco, F. ; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Invest. 2006, 116, 1071–1080. [Google Scholar] [CrossRef]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Ragavachetty Nagaraj, N.; Subramaniam Rajesh, B. Effect of advanced glycation end product on paraoxonase 2 expression: Its impact on endoplasmic reticulum stress and inflammation in HUVECs. Life Sci. 2020, 246, 117397. [Google Scholar] [CrossRef]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 1998, 101, 1142–1147. [Google Scholar] [CrossRef]
- Wang H, Guo M, Wei H, Chen Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct Target Ther. 2023, 8, 92. [CrossRef]
- Brouwers O, Yu L, Niessen P, Slenter J, Jaspers K, Wagenaar A, Post M, Miyata T, Backes W, Stehouwer C, Huijberts M, Schalkwijk C. Glyoxalase-1 overexpression partially prevents diabetes-induced impaired arteriogenesis in a rat hindlimb ligation model. Glycoconj J. 2016, 33, 627–30. [CrossRef]
- Nin JW, Jorsal A, Ferreira I, Schalkwijk CG, Prins MH, Parving HH, Tarnow L, Rossing P, Stehouwer CD. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12-year follow-up study. Diabetes Care. 2011, 34, 442–7. [CrossRef]
- Hanssen NM, Engelen L, Ferreira I, Scheijen JL, Huijberts MS, van Greevenbroek MM, van der Kallen CJ, Dekker JM, Nijpels G, Stehouwer CD, Schalkwijk CG. Plasma levels of advanced glycation endproducts Nε-(carboxymethyl)lysine, Nε-(carboxyethyl)lysine, and pentosidine are not independently associated with cardiovascular disease in individuals with or without type 2 diabetes: the Hoorn and CODAM studies. J Clin Endocrinol Metab. 2013, 98, E1369–E1373. [CrossRef]
- Semba RD, Bandinelli S, Sun K, Guralnik JM, Ferrucci L.. Plasma carboxymethyl-lysine, an advanced glycation end product, and all-cause and cardiovascular disease mortality in older community-dwelling adults. J Am Geriatr Soc. 2009, 57, 1874–80. [CrossRef]
- Semba RD, Ferrucci L, Sun K, Beck J, Dalal M, Varadhan R, Walston J, Guralnik JM, Fried LP. Advanced glycation end products and their circulating receptors predict cardiovascular disease mortality in older community-dwelling women. Aging Clin Exp Res. 2009, 21, 182–90. [CrossRef]
- Kilhovd BK, Juutilainen A, Lehto S, Rönnemaa T, Torjesen PA, Hanssen KF, Laakso M. Increased serum levels of methylglyoxal-derived hydroimidazolone-AGE are associated with increased cardiovascular disease mortality in nondiabetic women. Atherosclerosis. 2009, 205, 590–4. [CrossRef]
- Kilhovd BK, Juutilainen A, Lehto S, Rönnemaa T, Torjesen PA, Birkeland KI, Berg TJ, Hanssen KF, Laakso M. High serum levels of advanced glycation end products predict increased coronary heart disease mortality in nondiabetic women but not in nondiabetic men: a population-based 18-year follow-up study. Arterioscler Thromb Vasc Biol. 2005, 25, 815–20. [CrossRef]
- Busch M, Franke S, Wolf G, Brandstädt A, Ott U, Gerth J, Hunsicker LG, Stein G; Collaborative Study Group. The advanced glycation end product N(epsilon)-carboxymethyllysine is not a predictor of cardiovascular events and renal outcomes in patients with type 2 diabetic kidney disease and hypertension. Am J Kidney Dis. 2006, 48, 571–9. [CrossRef]
- Schwedler SB, Metzger T, Schinzel R, Wanner C. Advanced glycation end products and mortality in hemodialysis patients. Kidney Int. 2002, 62, 301–10. [CrossRef]
- Hanssen NMJ, Scheijen JLJM, Jorsal A, Parving HH, Tarnow L, Rossing P, Stehouwer CDA, Schalkwijk CG. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease in Individuals With Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes. 2017, 66, 2278–2283. [CrossRef]
- Hanssen NMJ, Westerink J, Scheijen JLJM, van der Graaf Y, Stehouwer CDA, Schalkwijk CG; SMART Study Group. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease and Mortality in Individuals With Type 2 Diabetes. Diabetes Care. 2018, 41, 1689–1695. [CrossRef]
- Ogawa S, Nakayama K, Nakayama M, Mori T, Matsushima M, Okamura M, Senda M, Nako K, Miyata T, Ito S. Methylglyoxal is a predictor in type 2 diabetic patients of intima-media thickening and elevation of blood pressure. Hypertension. 2010, 56, 471–6. [CrossRef]
- Hanssen NMJ, Scheijen JLJM, Houben AJHM, van de Waarenburg M, Berendschot TTJM, Webers CAB, Reesink KD, van Greevenbroek MMJ, van der Kallen C, Schaper NC, Schram MT, Henry RMA, Stehouwer CDA, Schalkwijk CG. Fasting and post-oral-glucose-load levels of methylglyoxal are associated with microvascular, but not macrovascular, disease in individuals with and without (pre)diabetes: The Maastricht Study. Diabetes Metab. 2021, 47, 101148. [CrossRef]
- Baidoshvili A, Niessen HW, Stooker W, Huybregts RA, Hack CE, Rauwerda JA, Meijer CJ, Eijsman L, van Hinsbergh VW, Schalkwijk CG. N(omega)-(carboxymethyl)lysine depositions in human aortic heart valves: similarities with atherosclerotic blood vessels. Atherosclerosis. 2004, 174, 287–92. [CrossRef]
- Varona JF, Ortiz-Regalón R, Sánchez-Vera I, López-Melgar B, García-Durango C, Castellano Vázquez JM, Solís J, Fernández-Friera L, Vidal-Vanaclocha F. Soluble ICAM 1 and VCAM 1 Blood Levels Alert on Subclinical Atherosclerosis in Non Smokers with Asymptomatic Metabolic Syndrome. Arch Med Res. 2019, 50, 20–28. [CrossRef]
- Boekholdt SM, Keller TT, Wareham NJ, Luben R, Bingham SA, Day NE, Sandhu MS, Jukema JW, Kastelein JJ, Hack CE, Khaw KT. Serum levels of type II secretory phospholipase A2 and the risk of future coronary artery disease in apparently healthy men and women: the EPIC-Norfolk Prospective Population Study. Arterioscler Thromb Vasc Biol. 2005, 25, 839–46. [CrossRef]
- Berge K, Aengevaeren VL, Mosterd A, Velthuis BK, Lyngbakken MN, Omland T, Schalkwijk CG, Eijsvogels TMH. Plasma Advanced Glycation End Products and Dicarbonyl Compounds Are Not Associated with Coronary Atherosclerosis in Athletes. Med Sci Sports Exerc. 2023, 55, 1143–1150. [CrossRef]
- Kerkeni M, Weiss IS, Jaisson S, Dandana A, Addad F, Gillery P, Hammami M. Increased serum concentrations of pentosidine are related to presence and severity of coronary artery disease. Thromb Res. 2014, 134, 633–8. [CrossRef]
- Mäkinen VP, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segrè AV, Ghosh S, Vivar J, Nikpay M, Stewart AF, Nelson CP, Willenborg C, Erdmann J, Blakenberg S, O'Donnell CJ, März W, Laaksonen R, Epstein SE, Kathiresan S, Shah SH, Hazen SL, Reilly MP; Coronary ARtery DIsease Genome-Wide Replication And Meta-Analysis (CARDIoGRAM) Consortium, Lusis AJ, Samani NJ, Schunkert H, Quertermous T, McPherson R, Yang X, Assimes TL.. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014, 10, e1004502. [CrossRef]
- Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. 2021, 18, 499–521. [CrossRef]
- Amponsah-Offeh M, Diaba-Nuhoho P, Speier S, Morawietz H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants (Basel). 2023, 12, 281. [CrossRef]
- Natalucci S, Ruggeri P, Cogo CE, Picchio V, Burattini R. Insulin sensitivity and glucose effectiveness estimated by the minimal model technique in spontaneously hypertensive and normal rats. Exp Physiol. 2000, 85, 775–81. [CrossRef]
- Natalucci S, Ruggeri P, Cogo CE, Picchio V, Brunori A, Burattini R. Age-related analysis of glucose metabolism in spontaneously hypertensive and normotensive rats. Exp Physiol. 2003, 88, 399–404. [CrossRef]
- Jacobson R, Mignemi N, Rose K, O'Rear L, Sarilla S, Hamm HE, Barnett JV, Verhamme IM, Schoenecker J. The hyperglycemic byproduct methylglyoxal impairs anticoagulant activity through covalent adduction of antithrombin III. Thromb Res. 2014, 134, 1350–7. [CrossRef]
- Qiao YN, Zou YL, Guo SD. Low-density lipoprotein particles in atherosclerosis. Front Physiol. 2022 Aug 30;13:931931. [CrossRef]
- Rabbani N, Godfrey L, Xue M, Shaheen F, Geoffrion M, Milne R, Thornalley PJ. Glycation of LDL by methylglyoxal increases arterial atherogenicity: a possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes. 2011, 60, 1973–80. [CrossRef]
- Rabbani N, Chittari MV, Bodmer CW, Zehnder D, Ceriello A, Thornalley PJ. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes. 2010, 59, 1038–45. [CrossRef]
- Lankin, V.; Konovalova, G.; Tikhaze, A.; Shumaev, K.; Kumskova, E.; Viigimaa, M. The initiation of free radical peroxidation of low-density lipoproteins by glucose and its metabolite methylglyoxal: a common molecular mechanism of vascular wall injure in atherosclerosis and diabetes. Mol. Cell. Biochem. 2014, 395, 241–252. [Google Scholar] [CrossRef]
- Godfrey L, Yamada-Fowler N, Smith J, Thornalley PJ, Rabbani N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr Diabetes. 2014, 4, e134. [CrossRef]
- Kurosaki Y, Tsukushi T, Munekata S, Akahoshi T, Moriya T, Ogawa Z. Semiquantitative analysis of apolipoprotein A-I modified by advanced glycation end products in diabetes mellitus. J Clin Lab Anal. 2013, 27, 231–6. [CrossRef]
- Schalkwijk, C.G.; Micali, L.R.; Wouters, K. Advanced glycation endproducts in diabetes-related macrovascular complications: focus on methylglyoxal. Trends Endocrinol. Metab. 2023, 34, 49–60. [Google Scholar] [CrossRef]
- Miller, A. G. , Tan, G., Nagaraj, R. H., Cooper, M. E., & Wilkinson-Berka, J. L. (2009). Candesartan Attenuates Vascular Injury in Diabetic Retinopathy by Restoring Glyoxalase I Function. Investigative Ophthalmology & Visual Science, 50(13), 4806-4806.
- Miller AG, Tan G, Binger KJ, Pickering RJ, Thomas MC, Nagaraj RH, Cooper ME, Wilkinson-Berka JL.. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes. 2010, 59, 3208–15. [CrossRef]
- Oh S, Ahn H, Park H, Lee JI, Park KY, Hwang D, Lee S, Son KH, Byun K. The attenuating effects of pyridoxamine on adipocyte hypertrophy and inflammation differ by adipocyte location. J Nutr Biochem. 2019 Oct;72:108173. [CrossRef]
- Rabbani N, Xue M, Weickert MO, Thornalley PJ. Reversal of Insulin Resistance in Overweight and Obese Subjects by trans-Resveratrol and Hesperetin Combination-Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients. 2021, 13, 2374. [CrossRef]
- Salazar J, Navarro C, Ortega Á, Nava M, Morillo D, Torres W, Hernández M, Cabrera M, Angarita L, Ortiz R, Chacín M, D'Marco L, Bermúdez V. Advanced Glycation End Products: New Clinical and Molecular Perspectives. Int J Environ Res Public Health. 2021, 18, 7236. [CrossRef]
- Van den Eynde MDG, Geleijnse JM, Scheijen JLJM, Hanssen NMJ, Dower JI, Afman LA, Stehouwer CDA, Hollman PCH, Schalkwijk CG. Quercetin, but Not Epicatechin, Decreases Plasma Concentrations of Methylglyoxal in Adults in a Randomized, Double-Blind, Placebo-Controlled, Crossover Trial with Pure Flavonoids. J Nutr. 2018, 148, 1911–1916. [CrossRef]
- Yoon SJ, Yoon YW, Lee BK, Kwon HM, Hwang KC, Kim M, Chang W, Hong BK, Lee YH, Park SJ, Min PK, Rim SJ. Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp Mol Med. 2009, 41, 802–11. [CrossRef]
- Recabarren-Leiva D, Burgos CF, Hernández B, Garcïa-García FJ, Castro RI, Guzman L, Fuentes E, Palomo I, Alarcón M. Effects of the age/rage axis in the platelet activation. Int J Biol Macromol. 2021 Jan 1;166:1149-1161. [CrossRef]
- Wang F, Yuan Q, Chen F, Pang J, Pan C, Xu F, Chen Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front Cell Dev Biol. 2021 Sep 20;9:742483. [CrossRef]
- Yamagishi S, Nakamura K, Matsui T, Ueda S, Fukami K, Okuda S. Agents that block advanced glycation end product (AGE)-RAGE (receptor for AGEs)-oxidative stress system: a novel therapeutic strategy for diabetic vascular complications. Expert Opin Investig Drugs. 2008, 17, 983–96. [CrossRef]
- Fishman SL, Sonmez H, Basman C, Singh V, Poretsky L.. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Mol Med. 2018, 24, 59. [CrossRef]
- Quade-Lyssy P, Kanarek AM, Baiersdörfer M, Postina R, Kojro E. Statins stimulate the production of a soluble form of the receptor for advanced glycation end products. J Lipid Res. 2013, 54, 3052–61. [CrossRef]
- Toprak C, Yigitaslan S. Alagebrium and Complications of Diabetes Mellitus. Eurasian J Med. 2019, 51, 285–292. [CrossRef]
- Hanssen NMJ, Tikellis C, Pickering RJ, Dragoljevic D, Lee MKS, Block T, Scheijen JL, Wouters K, Miyata T, Cooper ME, Murphy AJ, Thomas MC, Schalkwijk CG. Pyridoxamine prevents increased atherosclerosis by intermittent methylglyoxal spikes in the aortic arches of ApoE-/- mice. Biomed Pharmacother. 2023 Feb;158:114211. [CrossRef]
- Thornalley, PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, Spinowitz BS, Whittier FC, Wuerth JP; ACTION I Investigator Group. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004, 24, 32–40. [CrossRef]
- Kinsky OR, Hargraves TL, Anumol T, Jacobsen NE, Dai J, Snyder SA, Monks TJ, Lau SS. Metformin Scavenges Methylglyoxal To Form a Novel Imidazolinone Metabolite in Humans. Chem Res Toxicol. 2016, 29, 227–34. [CrossRef]
- Jinnouchi Y, Yamagishi S, Takeuchi M, Ishida S, Jinnouchi Y, Jinnouchi J, Imaizumi T. Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin Exp Med. 2006, 6, 191–3. [CrossRef]
- Scharnagl H, Stojakovic T, Winkler K, Rosinger S, März W, Boehm BO. The HMG-CoA reductase inhibitor cerivastatin lowers advanced glycation end products in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2007, 115, 372–5. [CrossRef]
- Contreras, C. L. Guzman-Rosiles, I., Del Castillo, D., Gomez-Ojeda, A., & Garay-Sevilla, M. E. (2017). Advanced glycation end products (AGEs) and sRAGE levels after benfotiamine treatment in diabetes mellitus type 2. The FASEB Journal, 2017, 31, 646–32. [Google Scholar] [CrossRef]
- Van den Eynde MDG, Houben AJHM, Scheijen JLJM, Linkens AMA, Niessen PM, Simons N, Hanssen NMJ, Kusters YHAM, Eussen SJMP, Miyata T, Stehouwer CDA, Schalkwijk CG. Pyridoxamine reduces methylglyoxal and markers of glycation and endothelial dysfunction, but does not improve insulin sensitivity or vascular function in abdominally obese individuals: A randomized double-blind placebo-controlled trial. Diabetes Obes Metab. 2023, 25, 1280–1291. [CrossRef]
- Bednarska K, Fecka I, Scheijen JLJM, Ahles S, Vangrieken P, Schalkwijk CG. A Citrus and Pomegranate Complex Reduces Methylglyoxal in Healthy Elderly Subjects: Secondary Analysis of a Double-Blind Randomized Cross-Over Clinical Trial. Int J Mol Sci. 2023, 24, 13168. [CrossRef]
- Bhuiyan MN, Mitsuhashi S, Sigetomi K, Ubukata M. Quercetin inhibits advanced glycation end product formation via chelating metal ions, trapping methylglyoxal, and trapping reactive oxygen species. Biosci Biotechnol Biochem. 2017, 81, 882–890. [CrossRef]
- Colzani M, De Maddis D, Casali G, Carini M, Vistoli G, Aldini G. Reactivity, Selectivity, and Reaction Mechanisms of Aminoguanidine, Hydralazine, Pyridoxamine, and Carnosine as Sequestering Agents of Reactive Carbonyl Species: A Comparative Study. ChemMedChem. 2016, 11, 1778–89. [CrossRef]
- Bednarska K, Fecka I. Potential of Vasoprotectives to Inhibit Non-Enzymatic Protein Glycation, and Reactive Carbonyl and Oxygen Species Uptake. Int J Mol Sci. 2021, 22, 10026. [CrossRef]
- Voziyan PA, Metz TO, Baynes JW, Hudson BG. A post-Amadori inhibitor pyridoxamine also inhibits chemical modification of proteins by scavenging carbonyl intermediates of carbohydrate and lipid degradation. J Biol Chem. 2002, 277, 3397–403. [CrossRef]
- Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol. 1999, 58, 1765–73. [CrossRef]
- Rahbar S, Natarajan R, Yerneni K, Scott S, Gonzales N, Nadler JL. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin Chim Acta. 2000 Nov;301(1-2):65-77. [CrossRef]
- Kiho T, Kato M, Usui S, Hirano K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta. 2005 Aug;358(1-2):139-45. [CrossRef]
- Adeshara K, Tupe R. Antiglycation and cell protective actions of metformin and glipizide in erythrocytes and monocytes. Mol Biol Rep. 2016, 43, 195–205. [CrossRef]
- Peters AS, Wortmann M, Fleming TH, Nawroth PP, Bruckner T, Böckler D, Hakimi M. Effect of metformin treatment in patients with type 2 diabetes with respect to glyoxalase 1 activity in atherosclerotic lesions. Vasa. 2019, 48, 186–192. [CrossRef]
- Qais FA, Sarwar T, Ahmad I, Khan RA, Shahzad SA, Husain FM. Glyburide inhibits non-enzymatic glycation of HSA: An approach for the management of AGEs associated diabetic complications. Int J Biol Macromol. 2021 Feb 1;169:143-152. [CrossRef]
- Li W, Ota K, Nakamura J, Naruse K, Nakashima E, Oiso Y, Hamada Y. Antiglycation effect of gliclazide on in vitro AGE formation from glucose and methylglyoxal. Exp Biol Med (Maywood). 2008, 233, 176–9. [CrossRef]
- Lee KY, Kim JR, Choi HC. Gliclazide, a KATP channel blocker, inhibits vascular smooth muscle cell proliferation through the CaMKKβ-AMPK pathway. Vascul Pharmacol. 2018 Mar;102:21-28. [CrossRef]
- Koyama H, Tanaka S, Monden M, Shoji T, Morioka T, Fukumoto S, Mori K, Emoto M, Shoji T, Fukui M, Fujii H, Nishizawa Y, Inaba M. Comparison of effects of pioglitazone and glimepiride on plasma soluble RAGE and RAGE expression in peripheral mononuclear cells in type 2 diabetes: randomized controlled trial (PioRAGE). Atherosclerosis. 2014, 234, 329–34. [CrossRef]
- Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, Hombach V, de Caterina R, Basta G, Wautier MP, Wautiers JL.. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004, 53, 2662–8. [CrossRef]
- Liu Y, Park JM, Chang KH, Huh HJ, Lee K, Lee MY. AMP-Activated Protein Kinase Mediates the Antiplatelet Effects of the Thiazolidinediones Rosiglitazone and Pioglitazone. Mol Pharmacol. 2016, 89, 313–21. [CrossRef]
- Adeshara KA, Agrawal SB, Gaikwad SM, Tupe RS. Pioglitazone inhibits advanced glycation induced protein modifications and down-regulates expression of RAGE and NF-κB in renal cells. Int J Biol Macromol. 2018 Nov;119:1154-1163. [CrossRef]
- Oz Gul O, Tuncel E, Yilmaz Y, Ulukaya E, Gul CB, Kiyici S, Oral AY, Guclu M, Ersoy C, Imamoglu S. Comparative effects of pioglitazone and rosiglitazone on plasma levels of soluble receptor for advanced glycation end products in type 2 diabetes mellitus patients. Metabolism. 2010, 59, 64–9. [CrossRef]
- Jakuš V, Hrnciarová M, Cársky J, Krahulec B, Rietbrock N. Inhibition of nonenzymatic protein glycation and lipid peroxidation by drugs with antioxidant activity. Life Sci. 1999;65(18-19):1991-3. [CrossRef]
- Miyata T, van Ypersele de Strihou C, Ueda Y, Ichimori K, Inagi R, Onogi H, Ishikawa N, Nangaku M, Kurokawa K. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: biochemical mechanisms. J Am Soc Nephrol. 2002, 13, 2478–87. [CrossRef]
- Nangaku M, Miyata T, Sada T, Mizuno M, Inagi R, Ueda Y, Ishikawa N, Yuzawa H, Koike H, van Ypersele de Strihou C, Kurokawa K. Anti-hypertensive agents inhibit in vivo the formation of advanced glycation end products and improve renal damage in a type 2 diabetic nephropathy rat model. J Am Soc Nephrol. 2003, 14, 1212–22. [CrossRef]
- Sobal G, Menzel EJ, Sinzinger H. Calcium antagonists as inhibitors of in vitro low density lipoprotein oxidation and glycation. Biochem Pharmacol. 2001, 61, 373–9. [CrossRef]
- Brown BE, Mahroof FM, Cook NL, van Reyk DM, Davies MJ. Hydrazine compounds inhibit glycation of low-density lipoproteins and prevent the in vitro formation of model foam cells from glycolaldehyde-modified low-density lipoproteins. Diabetologia. 2006, 49, 775–83. [CrossRef]
- Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis. 1998, 138, 271–80. [CrossRef]
- Lu L, Peng WH, Wang W, Wang LJ, Chen QJ, Shen WF. Effects of atorvastatin on progression of diabetic nephropathy and local RAGE and soluble RAGE expressions in rats. J Zhejiang Univ Sci B. 2011, 12, 652–9. [CrossRef]
- Hwang AR, Nam JO, Kang YJ. Fluvastatin inhibits advanced glycation end products-induced proliferation, migration, and extracellular matrix accumulation in vascular smooth muscle cells by targeting connective tissue growth factor. Korean J Physiol Pharmacol. 2018, 22, 193–201. [CrossRef]
- Fukuda K, Matsumura T, Senokuchi T, Ishii N, Kinoshita H, Yamada S, Murakami S, Nakao S, Motoshima H, Kondo T, Kukidome D, Kawasaki S, Kawada T, Nishikawa T, Araki E. Statins meditate anti-atherosclerotic action in smooth muscle cells by peroxisome proliferator-activated receptor-γ activation. Biochem Biophys Res Commun. 2015, 457, 23–30. [CrossRef]
- Ishibashi Y, Yamagishi S, Matsui T, Ohta K, Tanoue R, Takeuchi M, Ueda S, Nakamura K, Okuda S. Pravastatin inhibits advanced glycation end products (AGEs)-induced proximal tubular cell apoptosis and injury by reducing receptor for AGEs (RAGE) level. Metabolism. 2012, 61, 1067–72. [CrossRef]
- Spadaccio C, De Marco F, Di Domenico F, Coccia R, Lusini M, Barbato R, Covino E, Chello M. Simvastatin attenuates the endothelial pro-thrombotic shift in saphenous vein grafts induced by Advanced glycation endproducts. Thromb Res. 2014, 133, 418–25. [CrossRef]
- Deng J, Cai X, Hao M, Liu X, Chen Z, Li H, Liu J, Liao Y, Fu H, Chen H, Qin G, Yan D. Calcium Dobesilate (CaD) Attenuates High Glucose and High Lipid-Induced Impairment of Sarcoplasmic Reticulum Calcium Handling in Cardiomyocytes. Front Cardiovasc Med. 2021 Feb 2;8:637021. [CrossRef]
- Ribeiro ML, Seres AI, Carneiro AM, Stur M, Zourdani A, Caillon P, Cunha-Vaz JG; DX-Retinopathy Study Group. Effect of calcium dobesilate on progression of early diabetic retinopathy: a randomised double-blind study. Graefes Arch Clin Exp Ophthalmol. 2006, 244, 1591–600. [CrossRef]
- Liu G, Xia Q, Lu Y, Zheng T, Sang S, Lv L.. Influence of Quercetin and Its Methylglyoxal Adducts on the Formation of α-Dicarbonyl Compounds in a Lysine/Glucose Model System. J Agric Food Chem. 2017, 65, 2233–2239. [CrossRef]
- Liu XM, Liu YJ, Huang Y, Yu HJ, Yuan S, Tang BW, Wang PG, He QQ. Dietary total flavonoids intake and risk of mortality from all causes and cardiovascular disease in the general population: A systematic review and meta-analysis of cohort studies. Mol Nutr Food Res. 2017 Jun;61(6). [CrossRef]
- van Boekel MA, van den Bergh PJ, Hoenders HJ. Glycation of human serum albumin: inhibition by Diclofenac. Biochim Biophys Acta. 1992, 1120, 201–4. [CrossRef]
- Rendell, M., Nierenberg, J., Brannan, C., Valentine, J. L., Stephen, P. M., Dodds, S., Mercer P.; Smith P.K.; Walder, J. (1986). Inhibition of glycation of albumin and hemoglobin by acetylation in vitro and in vivo. The Journal of Laboratory and Clinical medicine. 1986, 108(4), 277–285. [CrossRef]
- Harding JJ, Egerton M, Harding RS. Protection against cataract by aspirin, paracetamol and ibuprofen. Acta Ophthalmol (Copenh). 1989, 67, 518–24. [CrossRef]
- Roberts KA, Harding JJ. Ibuprofen, a putative anti-cataract drug, protects the lens against cyanate and galactose. Exp Eye Res. 1990, 50, 157–64. [CrossRef]
- Rasheed S, Sánchez SS, Yousuf S, Honoré SM, Choudhary MI. Drug repurposing: In-vitro anti-glycation properties of 18 common drugs. PLoS One. 2018, 13, e0190509. [CrossRef]
- Booth AA, Khalifah RG, Hudson BG. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem Biophys Res Commun. 1996, 220, 113–9. [CrossRef]
- Ahmed LA, Hassan OF, Galal O, Mansour DF, El-Khatib A. Beneficial effects of benfotiamine, a NADPH oxidase inhibitor, in isoproterenol-induced myocardial infarction in rats. PLoS One. 2020, 15, e0232413. [CrossRef]
- Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, Li J, Ren J. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med. 2009 Aug;13(8B):1751-1764. Retraction in: J Cell Mol Med. 2023 May 7. Retraction in: J Cell Mol Med. 2023 May 7. [CrossRef]
- Metz TO, Alderson NL, Chachich ME, Thorpe SR, Baynes JW. Pyridoxamine traps intermediates in lipid peroxidation reactions in vivo: evidence on the role of lipids in chemical modification of protein and development of diabetic complications. J Biol Chem. 2003, 278, 42012–9. [CrossRef]
- Hagiwara S, Gohda T, Tanimoto M, Ito T, Murakoshi M, Ohara I, Yamazaki T, Matsumoto M, Horikoshi S, Funabiki K, Tomino Y. Effects of pyridoxamine (K-163) on glucose intolerance and obesity in high-fat diet C57BL/6J mice. Metabolism. 2009, 58, 934–45. [CrossRef]
- Unoki-Kubota H, Yamagishi S, Takeuchi M, Bujo H, Saito Y. Pyridoxamine, an inhibitor of advanced glycation end product (AGE) formation ameliorates insulin resistance in obese, type 2 diabetic mice. Protein Pept Lett. 2010, 17, 1177–81. [CrossRef]
- Takahashi H, Shibasaki T, Park JH, Hidaka S, Takahashi T, Ono A, Song DK, Seino S. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes. 2015, 64, 1262–72. [CrossRef]
- Lu Q, Li X, Liu J, Sun X, Rousselle T, Ren D, Tong N, Li J. AMPK is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci Rep. 2019, 39, BSR20181995. [CrossRef]
- Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, Hombach V, de Caterina R, Basta G, Wautier MP, Wautiers JL.. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes. 2004, 53, 2662–8. [CrossRef]
- Liu Y, Park JM, Chang KH, Huh HJ, Lee K, Lee MY. AMP-Activated Protein Kinase Mediates the Antiplatelet Effects of the Thiazolidinediones Rosiglitazone and Pioglitazone. Mol Pharmacol. 2016, 89, 313–21. [CrossRef]
- Reyaz A, Alam S, Chandra K, Kohli S, Agarwal S. Methylglyoxal and soluble RAGE in type 2 diabetes mellitus: Association with oxidative stress. J Diabetes Metab Disord. 2020, 19, 515–521. [CrossRef]
- Semo D, Obergassel J, Dorenkamp M, Hemling P, Strutz J, Hiden U, Müller N, Müller UA, Zulfikar SA, Godfrey R, Waltenberger J. The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms. J Clin Med. 2023, 12, 1356. [CrossRef]
- Hwang AR, Han JH, Lim JH, Kang YJ, Woo CH. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PLoS One. 2017, 12, e0178278. [CrossRef]
- Tusa I, Menconi A, Tubita A, Rovida E. Pathophysiological Impact of the MEK5/ERK5 Pathway in Oxidative Stress. Cells. 2023, 12, 1154. [CrossRef]
- Niedzielski M, Broncel M, Gorzelak-Pabiś P, Woźniak E. New possible pharmacological targets for statins and ezetimibe. Biomed Pharmacother. 2020 Sep;129:110388. [CrossRef]
- McCarty MF, O'Keefe JH, DiNicolantonio JJ. Pentoxifylline for vascular health: a brief review of the literature. Open Heart. 2016, 3, e000365. [CrossRef]
- Zhang X, Liu W, Wu S, Jin J, Li W, Wang N. Calcium dobesilate for diabetic retinopathy: a systematic review and meta-analysis. Sci China Life Sci. 2015, 58, 101–7. [CrossRef]
- Deng, J. , Feng, J., Sun, L., Suo, X., Yao, L., LI, Z., Wang Y., Zhou J., Wang, L. (2009). Aristolochic acid-induced endothelial cell injury and the mechanism of calcium dobesilate antagonism. Journal of Chinese Physician, 913-916.
- Haller H, Ji L, Stahl K, Bertram A, Menne J. Molecular Mechanisms and Treatment Strategies in Diabetic Nephropathy: New Avenues for Calcium Dobesilate-Free Radical Scavenger and Growth Factor Inhibition. Biomed Res Int. 2017;2017:1909258. [CrossRef]
- Zhou Y, Qi C, Li S, Shao X, Mou S, Ni Z. Diabetic Nephropathy Can Be Treated with Calcium Dobesilate by Alleviating the Chronic Inflammatory State and Improving Endothelial Cell Function. Cell Physiol Biochem. 2018, 51, 1119–1133. [CrossRef]
- Holovko, O. O. , Dmytrenko, O. P., Lesiuk, A. I., Kulish, M. P., Pavlenko, O. L., Naumenko, A. P., Pinchuk-Rugal, T. M.; Kaniuk M. I.; Veklich, T. O. (2023). Mechanisms of the interaction of bovine serum albumin with quercetin. -15. [CrossRef]
- Wang X, Ouyang YY, Liu J, Zhao G. Flavonoid intake and risk of CVD: a systematic review and meta-analysis of prospective cohort studies. Br J Nutr. 2014, 111, 1–11. [CrossRef]
- Kim Y, Je Y. Flavonoid intake and mortality from cardiovascular disease and all causes: A meta-analysis of prospective cohort studies. Clin Nutr ESPEN. 2017 Aug;20:68-77. [CrossRef]
- Grosso G, Micek A, Godos J, Pajak A, Sciacca S, Galvano F, Giovannucci EL.. Dietary Flavonoid and Lignan Intake and Mortality in Prospective Cohort Studies: Systematic Review and Dose-Response Meta-Analysis. Am J Epidemiol. 2017, 185, 1304–1316. [CrossRef]
- Micek A, Godos J, Del Rio D, Galvano F, Grosso G. Dietary Flavonoids and Cardiovascular Disease: A Comprehensive Dose-Response Meta-Analysis. Mol Nutr Food Res. 2021, 65, e2001019. [CrossRef]
- Huxley RR, Neil HA. The relation between dietary flavonol intake and coronary heart disease mortality: a meta-analysis of prospective cohort studies. Eur J Clin Nutr. 2003, 57, 904–8. [CrossRef]
- Jiang W, Wei H, He B. Dietary flavonoids intake and the risk of coronary heart disease: a dose-response meta-analysis of 15 prospective studies. Thromb Res. 2015, 135, 459–63. [CrossRef]
- Indurthi VS, Leclerc E, Vetter SW. Calorimetric investigation of diclofenac drug binding to a panel of moderately glycated serum albumins. Eur J Pharm Sci. 2014 Aug 1;59:58-68. [CrossRef]
- Raval AD, Thakker D, Rangoonwala AN, Gor D, Walia R. Vitamin B and its derivatives for diabetic kidney disease. Cochrane Database Syst Rev. 2015 Jan 12;1:CD009403. [CrossRef]
- Ramis R, Ortega-Castro J, Caballero C, Casasnovas R, Cerrillo A, Vilanova B, Adrover M, Frau J. How Does Pyridoxamine Inhibit the Formation of Advanced Glycation End Products? The Role of Its Primary Antioxidant Activity. Antioxidants (Basel). 2019, 8, 344. [CrossRef]
- Maessen DE, Brouwers O, Gaens KH, Wouters K, Cleutjens JP, Janssen BJ, Miyata T, Stehouwer CD, Schalkwijk CG. Delayed Intervention With Pyridoxamine Improves Metabolic Function and Prevents Adipose Tissue Inflammation and Insulin Resistance in High-Fat Diet-Induced Obese Mice. Diabetes. 2016, 65, 956–66. [CrossRef]
- Borg DJ, Forbes JM. Targeting advanced glycation with pharmaceutical agents: where are we now? Glycoconj J. 2016, 33, 653–70. [CrossRef]
- Sambon M, Wins P, Bettendorff L.. Neuroprotective Effects of Thiamine and Precursors with Higher Bioavailability: Focus on Benfotiamine and Dibenzoylthiamine. Int J Mol Sci. 2021, 22, 5418. [CrossRef]
Figure 1.
The main actors in the process of non-enzymatic glycation, cellular defense mechanisms against excessive glycation and accumulation of MGO and MAGEs, as well as known glycation inhibitors and MGO scavengers.
Figure 1.
The main actors in the process of non-enzymatic glycation, cellular defense mechanisms against excessive glycation and accumulation of MGO and MAGEs, as well as known glycation inhibitors and MGO scavengers.
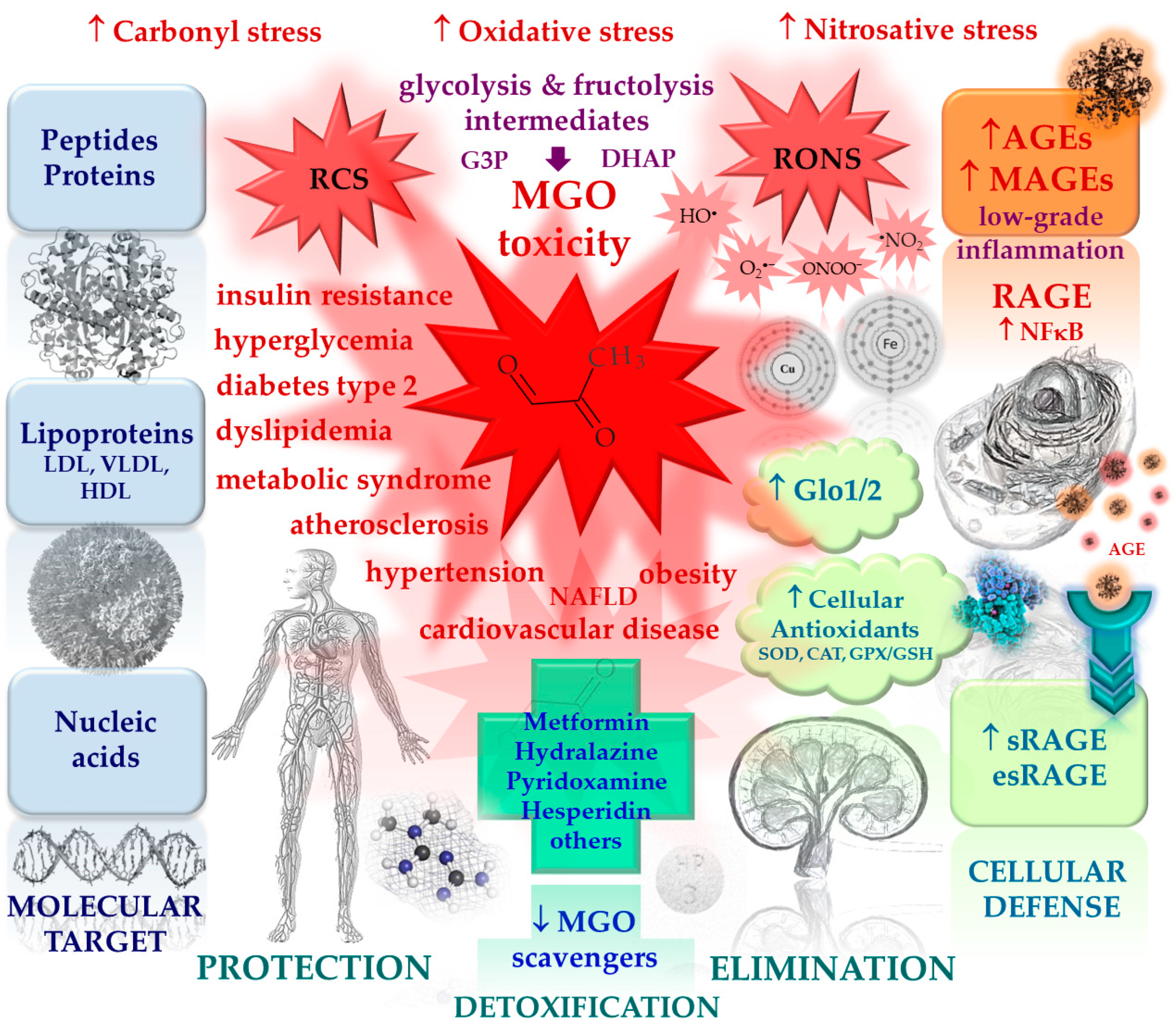
Figure 2.
Assumed mechanisms of glycation inhibitors, and related pathways.
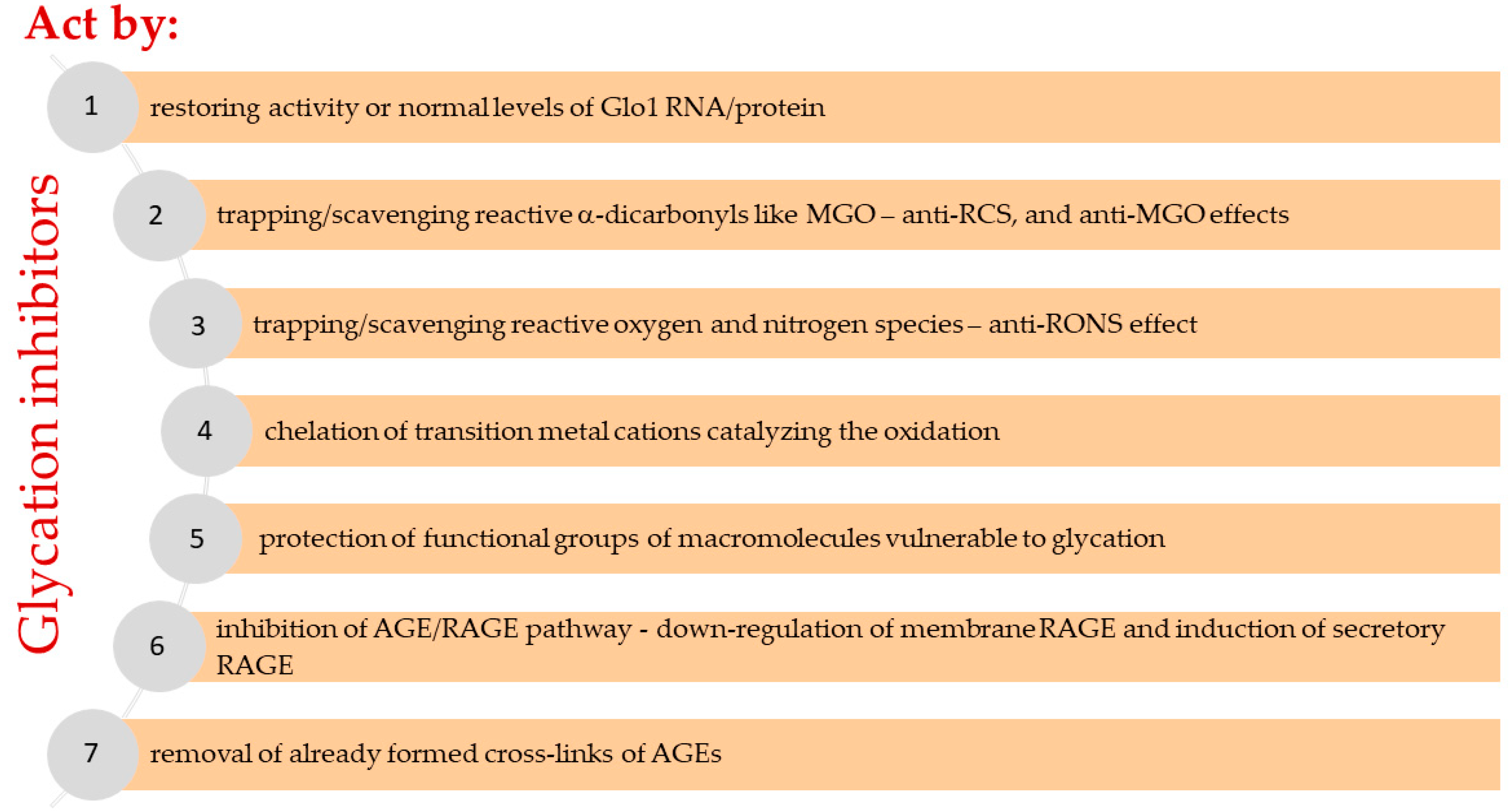
Table 3.
Potential and known glycation inhibitors and MGO scavengers with their possible mechanism of action.
Table 3.
Potential and known glycation inhibitors and MGO scavengers with their possible mechanism of action.
| Medication | AGE-RAGE/MAGE-RAGE axis grip points, and biochemical and physiological effects | Research models and methods |
|---|---|---|
| 1. Antihyperglycemic agents used in the management of type 2 diabetes (blood glucose lowering agents) | ||
| 1.1. Biguanides | ||
|
Metformin |
In vitro: MGO scavenger, inhibits carbonyl stress; reduces ↓ cross-linking, ↓ AGE, and ↓ HbA1c formation; restores the level of antioxidants in THP-1 cells and erythrocytes; diminishes mitochondrial complex I activity; activates ↑ AMPK; Human studies: reduces ↓ MGO in plasma in a dose-dependent manner; enhances ↑ Glo1 activity in peripheral blood cells and atherosclerotic lesions; scavenges MGO to form imidazolinone metabolite excreted in the urine |
BSA-MGO; LC-MS, and spectroscopic analysis of MGO adducts [238]; BSA-glucose, models of the early stage of glycation - Amadori products, and late glycation products - AGEs, ELISA [239]; BSA-MGO, RNase-MGO; ELISA, and western blot with the anti-AGE antibody [240]; BSA-MGO; glycation markers (fructosamine, carbonyls, free thiols, β-amyloid aggregation), and protein structural markers (absorption spectroscopy, gel electrophoresis) were examined; monocytes (THP-1 cells) and erythrocytes were treated with medication; and anti-oxidant indices (CAT, SOD, GSH, NO), cell viability, lipid peroxidation, and others were determined [241]; Case-control study: two groups of subjects with T2DM who were either treated with metformin (500 to 2500 mg/day for ≥ 3 months) or not treated with metformin and nondiabetic control subjects; HPLC, GC-MS [17]; patients with T2DM and carotid artery disease were included into the study prospectively; Glo1-activity and protein expression was measured by a spectrophotometric assay and western blot [242]; Proteomic and Metabolomic Biomarker Investigation of T2DM (project no. 07-0812-01), subjects recruited into the study included nondiabetic, pre-type-2 diabetic, and diabetic patients; NMR, LC-MS [228] |
| Buformin | In vitro: reduces ↓ AGE formation (more potent inhibitor than metformin) and ↓ cross-linking | BSA-MGO, RNase-MGO; ELISA, and western blot with the anti-AGE antibody; LC-MS analysis of MGO adducts [240] |
| 1.2. Sulfonylureas | ||
|
Glibenclamide (= glyburide) |
In vitro and Animal studies: reduces ↓ AGE formation; KATP channel antagonist; activates the JNK/p38 MAPK pathway; this effect arises partly through activation of KATP | HSA-glucose, HSA-MGO; AGEs, fructosamine, carbonyl groups, free lysine, and free thiol groups were determined; interaction studies, molecular docking [243]; male C57BL/6J mice; HAECs from healthy and T2DM donors; PCR and western blot analyses; Glo1 activity; immunofluorescence staining of MGO-AGEs in mouse aorta [126] |
| Gliclazide | In vitro and Animal studies: reduces ↓ AGE formation; KATP channel antagonist; inhibits vascular smooth muscle cell proliferation through the CaMKKβ–AMPK pathway; effects of KATP on AMPK activity is mediated by the regulation of intracellular Ca2+ levels | BSA-MGO, BSA-glucose; AGEs were assessed by fluorescence, ELISA, and western blot [244]; male C57BL/6 mice; LKB1-deficient A549 cells; VSMCs were isolated from the Sprague–Dawley rat thoracic aortas; cell proliferation assays using MTT; intracellular Ca2+ concentrations in VSMCs were assessed by a Ca2+ indicator dye; RT-PCR and western blot [245] |
| Glipizide | In vitro: reduces ↓ AGE formation; restores the level of antioxidants in THP-1 cells and erythrocytes | BSA-MGO; glycation markers (fructosamine, carbonyls, free thiols, β-amyloid aggregation) and protein structural markers (absorption spectroscopy, gel electrophoresis) were studied; THP-1 cells (monocytes) and erythrocytes were treated with medication; and antioxidant indicators (CAT, SOD, GSH, NO) were determined; cell viability, lipid peroxidation, and erythrocyte hemolysis were determined [241] |
| Glimepiride | Human studies: increases plasma ↑ esRAGE and reduces ↓ RAGE expression in peripheral mononuclear cells less than pioglitazone | RCT: a single-center randomized trial of pioglitazone versus glimepiride (clinical trial no. UMIN000002055); participants naive to glucose-lowering therapy at screening or taking 2 mg/day or less glimepiride or the equivalent dosage of another sulfonylurea; levels in the plasma of sRAGE and esRAGE, and RAGE expression in peripheral mononuclear cells were determined [246] |
| 1.3. Thiazolidinediones | ||
| Pioglitazone |
In vitro: reduces ↓ AGE and ↓ HbA1c formation; reduces ↓ RAGE and ↓ RAGE mRNA expression in human endothelial cells, thus limiting the EC susceptibility toward proinflammatory AGE effects; suppresses RAGE and NF-κB levels, hence alleviating cellular oxidative stress and inflammation; preferentially binds to protein and relieving protein structural changes; pioglitazone restores cellular antioxidants and reduces levels of IL-6 and TNF-α by declining expression of membrane RAGE and NF-κB; pioglitazone and rosiglitazone inhibit platelet aggregation by activating ↑ AMPK; Human studies: there is a significant increase in circulating ↑ sRAGE or sRAGE/esRAGE in the pioglitazone group; this effect is not observed in the rosiglitazone and medical nutrition therapy groups; pioglitazone suppresses RAGE expression and increases circulating sRAGE/esRAGE, and those activities are not necessarily dependent on plasma glucose or insulin resistance levels |
BSA-glucose; Amadori products, and late glycation products – AGEs; ELISA [239]; HUVECs, AGE-BSA, flow cytometry, western and northern blot, RT-PCR [247]; rat platelets; AMPK was examined by western blot with a specific antibody; PPAR-γ DNA binding with a PPAR-γ transcription factor assay kit; cGMP using a cGMP assay kit [248]; BSA-MGO; human embryonic kidney – 293 (HEK-293) cells; ROS was measured by fluorophotometric method; structural modification of albumins was analyzed by electrophoresis (native-PAGE) and HPLC; proteins were visualized with a chemiluminescence kit, and densitometry analysis of scanned western blot images; cytokines by ELISA; fructosamine, protein carbonyls, β-amyloid, free thiols in glycated albumin and AOPP were determined [249]; RCT: T2DM subjects were randomly assigned to receive pioglitazone (30 mg/day), rosiglitazone (4 mg/day), or placebo (medical nutrition therapy) for 12 weeks; changes in plasma glucose, HbA1c, insulin resistance (homeostasis model assessment), total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides, and sRAGE were evaluated at baseline and after 12 weeks [250]; a single-center randomized trial of pioglitazone versus glimepiride (clinical trial no. UMIN000002055); participants naive to glucose-lowering therapy at screening or taking 2 mg/day or less glimepiride or the equivalent dosage of another sulfonylurea; levels in the plasma of sRAGE and esRAGE, and RAGE expression in peripheral mononuclear cells were determined [246] |
| Rosiglitazone | ||
| 2.Agents for the treatment of cardiovascular conditions | ||
| 2.1. Angiotensin II receptor antagonists (blockers, ARBs) and angiotensin-converting enzyme inhibitors | ||
|
Candesartan Irbesartan Losartan Olmesartan Telmisartan Valsartan (ARBs) |
In vitroand Animal studies: reduces ↓ AGE (argpyrimidine, pentosidine and CML) formation; chelates transition metal cations, acts as an antioxidant, and inhibits the formation of ↓ ROS and ↓ RCS; the effect on AGE formation is common to all tested ARBs; a similar but milder effect is observed with ACE inhibitors (IC50 of pentosidine formation in BSA-arabinose model: valsartan > candesartan > olmesartan > temocaprilat > enalaprilat > irbesartan = losartan = telmisartan > captopril > perindoprilat); candesartan attenuates vascular injury in diabetic retinopathy by restoring Glox1 function and reducing ↓ ∙NO; reinstates both ↑ Glo1 activity and ↑ Glo1 mRNA level; reduces ↓ mRNA levels of ICAM-1, VEGF, TNF-α and iNOS; decreases ↓ total AGEs, MAGEs, and argpyrimidine in retina and plasma; olmesartan, in rats with type 2 like diabetes, reduces in a dose-dependent manner the development of diabetic nephropathy as evidenced by a decrease in proteinuria and pathologic evidence of diabetic glomerulosclerosis | HSA-glucose; Amadori products, and fluorescent AGEs were determined [251]; BSA-arabinose; pentosidine measurement by LC; CML by GC-MS; inhibition on leukocyte-derived superoxide production; RCS trapping; LC analysis of GO adducts; metal chelating activity test [252]; male subline of spontaneously hypertensive/NIH-corpulent rat - SHR/NDmc-cp (fat/fat); biochemical measurements in blood and urine; morphologic analysis; immunohistochemistry; pentosidine and CML were determined by LC and GC/MS, respectively [253]; bovine retinal endothelial cells (BREC), and bovine retinal pericytes (BRP); diabetic Sprague-Dawley rats; Ren-2 rats with an enhanced renin-angiotensin system; Glo1 activity was assessed by measuring the formation of S-lactoylglutathione; •NO with using 2,3-diaminonaphthalene; mRNA for Glo1, intercellular adhesion molecule (ICAM-1), vascular endothelial growth factor (VEGF), TNF-α, iNOS, and RAS component were quantitated by RT-PCR; argpyrimidine, MAGEs, and total AGEs (all AGEs including MAGEs) by ELISA [212,213] |
|
Captopril Enalaprilat (active metabolite of enalapril) Perindoprilat (active metabolite of perindopril) Temocaprilat (active metabolite of temocapril) (ACE inhibitors) | ||
| 2.2. Calcium channel antagonists (blockers) | ||
|
Amlodipine Isradipine Lacidipine Nifedipine (with vascular effects) |
In vitro: acts as an antioxidant (lacidipine > semotiadil > amlodipine > nifedipine > diltiazem), inhibits ↓ glycation and ↓ glycoxidation; inhibits the copper-mediated oxidation of non-glycated and glycated LDL | LDL-glucose; AGEs by ELISA; early glycation products were determined with the fructosamine test, and lipid peroxidation by TBARS quantitation [254] |
|
Diltiazem (with direct cardiac effects) | ||
| Semotiadil (experimental) | ||
| 2.3.Arterial smooth muscle agents, hydrazinophthalazine derivatives | ||
| Hydralazine |
In vitro: MGO scavenger, inhibits carbonyl stress; inhibits the formation of AGEs (pentosidine and CML); chelates transition metal cations, acts as an antioxidant, and inhibits the formation of ↓ ROS; inhibits the glycation of LDL and prevents the formation of model foam cells from RCS-modified low-density lipoproteins; Animal studies: the effect of hydralazine (5 mg) is similar to that of olmesartan (1 mg) but reached statistical significance only for kidney pentosidine content |
Ubiquitin-RCS; reaction products of MGO were generated in vitro and characterized by ESI-MS [235]; LDL-MGO; LDL modification was characterized by changes in mobility (agarose gel electrophoresis), cross-linking (SDS-PAGE) and loss of amino acid residues (LC); accumulation of cholesterol and cholesteryl esters in murine macrophages was assessed by LC [255]; male subline of spontaneously hypertensive/NIH-corpulent rat - SHR/NDmc-cp (fat/fat); biochemical measurements in blood and urine; morphologic analysis; immunohistochemistry; pentosidine and CML by LC and GC/MS, respectively [253] |
| 2.4. Statins (lipid modifying agents, HMG-CoA reductase inhibitors) | ||
| Atorvastatin |
In vitro: atorvastatin o- and p-OH metabolites are potent antioxidants and protect LDL, VLDL, and HDL from oxidation; the inhibitory effects of these metabolites on HDL oxidation are associated with the protection of paraoxonase activity; Animal studies: a serum and renal ↑ sRAGE level was up-regulated and associated with a reduction of AGEs, though renal esRAGE mRNA expression was not significantly increased; Human studies: decreases serum levels of ↓ AGEs in hypercholesterolemic T2DM patients without CVD but do not lower fasting glucose or HbA1c levels; AGEs changes do not correlate with lipid parameters; atorvastatin tends to decrease the serum level of 8-OHdG, but not significantly |
VLDL, LDL, and HDL were isolated from fasted normolipidemic volunteers; lipoprotein oxidation was measured by TBARS assay and photometrically by the lipid peroxidation test; LDL oxidation by macrophages; lipoprotein electrophoresis; DPPH assay; paraoxonase activity measurements [256]; Sprague-Dawley rats after streptozotocin-induced diabetes with or without atorvastatin treatment (10 mg/kg for 24 weeks); sRAGE and glycated albumin levels were measured by ELISA and bromocresol purple methods; renal AGEs, RAGE, sRAGE, and esRAGE were determined with PCR and western blot [257]; RTC: patients were treated with 10 mg atorvastatin by 4 weeks, control subjects were treated with diet therapy by 4 weeks; blood pressure, serum AGEs, serum 8-hydroxydeoxy-guanosine (8-OHdG), oxidative stress markers and others were measured before and after the treatment; AGEs by ELISA; others with commercially laboratory tests [229] |
| Lovastatin | In vitro: significantly increases the level of ↑ sRAGE by enhancement of full-length RAGE shedding but did not influence the secretion of esRAGE | Cell lines HEK/RAGE (stably expressing full-length RAGE) and HEK/esRAGE (stably expressing esRAGE being a recombinant version of the splice variant esRAGE); mouse alveolar epithelial (MLE-12) cells; immunoblot analysis; RT-PCR, and others [223] |
| Cerivastatin | Human studies: significantly lowered the concentration of ↓ CML-derived AGEs (compared to the placebo group); the effect on CML-AGEs is correlated with the reduction of LDL cholesterol and LDL apolipoprotein B; HbA1c is not changed | RCT: a multicenter, double-blind, randomized, parallel-group comparison of cerivastatin 0.4 mg daily for 12 weeks vs. placebo; the primary objective of the study was the effect of cerivastatin on dense LDL subfractions; CML-AGEs was assessed in patients with elevated fasting glucose, impaired glucose tolerance, or diabetes [230] |
| Fluvastatin |
In vitro: inhibits mitogen-activated protein kinase kinase ↓ MEK (MAPK/ERK kinase also known as MAP2K, MAPKK), which downregulated the transcription of EGR-1 (early growth response protein 1) and leads to decreased levels of CTGF (connective tissue growth factor), and consequently reduces proliferation, migration, and ECM (extracellular matrix) accumulation in AGE-induced VSMCs; activates ↑ PPAR-γ in HASMCs, but not in HUVECs; induces COX-2 expression in HASMCs, but not in HUVECs; suppresses migration and proliferation of HASMCs, and inhibited lipopolysaccharide-induced expression of MCP-1 (monocyte chemoattractant protein-1) and TNF-α in HASMCs; Animal studies: statins suppress atherosclerotic lesion formation in Apoe−/− mice; transcriptional activity of ↑ PPAR-γ was increased; and the expression of ↓ MCP-1 and ↓ TNF-α was decreased in the aorta of statin-treated Apoe−/− mice |
Primary cell culture of rat VSMCs; mRNA for CTGF as amplified cDNA was analyzed by qPCR; protein levels by western blot; cell proliferation by MTT assay; cell cycle by flow cytometry [258]; human aortic smooth muscle cell (HASMC) and human umbilical vein endothelial cell (HUVECs) cultures; ELISA assay kits of human MCP-1 and TNF-α; thymidine incorporation assay; cell migration assay; in an animal model (Apoe−/− mice) plasma total cholesterol, LDL cholesterol, HDL cholesterol, and triglyceride concentrations were measured [259] |
| Pitavastatin | ||
| Pravastatin | In vitro: inhibits ↓ AGE-induced up-regulation of RAGE mRNA level; inhibits ROS generation, and apoptosis in human renal proximal tubular cells | AGE-BSA in cultures of human renal proximal tubule epithelial cells ex vivo; gene expression was evaluated by qRT-PCR; ROS with dihydroethidium staining; apoptosis by ELISA; and others [260] |
| Rosuvastatin | ||
| Simvastatin |
In vitro: significantly reduces ↓ AGE-induced oxidative stress (ROS overproduction) in ECs and decreases neutrophil adhesion to endothelium; decreases ↓ RAGE mRNA expression, and non-statistically increases ↑ PPAR-γ mRNA expression (PPAR -γ has a protective effect on ECs by inhibiting endothelin-1 release and attenuating/preventing the endothelial inflammatory response); Animal studies: 12-week administration attenuates AGE-induced proliferation of aortic smooth muscle cells in Sprague-Dawley rats and reduces ↓ NF-κβ and ↓ MAPK activation in those cells |
AGE-BSA in cultures of endothelial cells (EC) ex vivo; EC were isolated from healthy subjects and diabetic patients (Hb1Ac< 6%, AGE > 7 μg/mL); fluorescently labeled neutrophils were counted manually under a microscope; ROS accumulation was estimated by spectrofluorimetric analysis; mRNA for RAGE and PPAR-γ as amplified cDNA was analyzed by RT-PCR [261]; male Sprague-Dawley rats; streptozocin-induced hyperglycemia; rat aortic smooth muscle cells (RASMCs); cultured RASMCs were co-incubated with AGE-BSA; measurement of intracellular ROS generation [218] |
| 2.5. Peripheral vasodilators (purine derivatives) | ||
| Pentoxifylline | In vitro: reduces ↓ AGE and ↓ HbA1C formation | BSA-glucose; models of the early stage of glycation - Amadori products, and late glycation products – AGEs; ELISA [239] |
| 2.6. Vasoprotectives (e.g. for the treatment of peripheral vascular disease) | ||
| 2.6.1. Antivaricose agents | ||
| Calcium dobesylate |
In vitro: reduces ↓ AGE formation; ROS scavenger; inhibits the formation of ↓ ROS, acts as an antioxidant; protects glycation reaction substrates from ROS and MGO-induced modifications; reduces the impairment of calcium handling in the sarcoplasmic reticulum and ↓ ROS formation in rat cardiomyocytes caused by high glucose and high lipid levels; Human studies: significantly reduces the permeability of the blood-retinal barrier (BRB) as measured by the posterior vitreous penetration ratio (PVPR); the effect is manifest regardless of the degree of metabolic control and the use of anti-hypertensive and lipid-lowering agents; has a significant beneficial effect in controlling the hemorrhages and the global evolution of diabetic retinopathy |
BSA-MGO; dicarbonyl trapping; LC-MS analysis of MGO and GO adducts [236]; neonatal rat ventricular myocytes (NRVMs); measurement of Action Potential (AP)-Elicited Ca2+ transient and SR Ca2+ content; confocal microscopy [262]; RCT: a double-blind placebo-controlled study, subjects with T2DM and early diabetic retinopathy, treatment was 2 g daily for 24 months, the primary parameter - posterior vitreous penetration ratio (PVPR), was measured every 6-months by fluorophotometry; secondary parameters were fundus photography, fluorescein angiography, and safety assessments [263] |
| 2.6.2. Capillary stabilizing agents (bioflavonoids) | ||
|
Diosmin (diosmetin – aglycone) |
In vitro: MGO scavenger (but not diosmin and troxerutin), ROS scavenger, inhibits the formation of ↓ ROS and ↓ RCS, acts as an antioxidant, chelates transition metal cations; reduces ↓ AGE formation; Human studies: quercetin-3-O-glucoside and hesperidin in (pre)hypertensive and healthy subjects decreased plasma ↓ MGO concentration (by ~10-11%); there was no significant change in Glo1 expression; the hesperetin and trans-resveratrol combination (tRES-HESP) induces expression of Glo1 countering the accumulation of MGO in overweight and obese subjects; produces a reversal of insulin resistance, improving dysglycemia, and low-grade inflammation; MGO metabolism-related variables correlated with BMI, dysglycemia, vascular inflammation, blood pressure, and dyslipidemia; in the meta-analysis: dose-response analysis showed that those consuming 200 mg/day of total flavonoids had the lowest risk of all-cause mortality |
BSA-glucose, BSA-ribose, BSA-MGO, lysine-glucose; LC-MS, ESI-MS and NMR analyses of MGO adducts, effect of metal cations on AGE formation [234,236,264]; RCT (clinica trial No. NCT01691404): a randomized, double-blind, placebo-controlled crossover study of quercetin-3-O-glucoside 160 mg/d for 4 weeks separated by 4-week washout periods; RNA was isolated from the PBMCs; MGO was evaluated by LC-MS; the gene expression of Glo1 by qRT-PCR [217]; RCT (clinical trial no. NCT03781999 ): a randomized, double-blind, placebo-controlled crossover study of hesperidin 450 mg/d (and punicalagin 60 mg/d) for 4 weeks, with a 4 weeks washout; MGO was evaluated by LC-MS [233]; RCT (clinicaltrial no. NCT02095873): a randomized, double-blind, placebo-controlled crossover study of 120 mg hesperetin + 90 mg trans-resveratrol/d, for 8 weeks, with 6 weeks washout; the primary endpoint was insulin sensitivity measured by oral glucose insulin sensitivity index (OGIS) in oral glucose tolerance tests (OGTTs) at the start and end of each treatment period; secondary endpoints were brachial artery flow-mediated dilatation response, also including brachial artery dilatation response to a sub-therapeutic dose of glyceryl trinitrate (FMD-GTN); Glo1 activity and mRNA of PBMCs were assayed by spectrophotometric and Nanostring methods, respectively; plasma MGO, plasma protein and urine MGO-derived AGE (MG-H1), were measured by LC-MS; plasma D-lactate was assayed by endpoint enzymatic assay by microplate fluorimetry; urinary metabolites by LC-MS [215]; meta-analysis of cohort studies; the random-effect model was used to calculate the summary risk estimates and dose-response analysis was performed; ten studies were included in the meta-analysis [265] |
|
Hesperidin (hesperetin – aglycone) | ||
|
Rutin Troxerutin Isoquercitrin (= quercetin-3-O-glucoside) (quercetin – aglycone) | ||
| 3. Anti-inflammatory, analgesic and antipyretic agents (including non-steroidal anti-inflammatory drugs NSAIDs) | ||
|
Acetylsalicylic acid (= aspirin) |
In vitro: inhibits ↓ albumin and hemoglobin glycation (but not salicylic acid), blocks at least one of the main glycation sites of HSA; Animal studies: decreases glycohemoglobin and glycoalbumin levels in diabetic rats; Human studies: low doses protect against cataracts |
HSA-glucose 6-phosphate [266]; albumin and hemoglobin glycation, diabetic rats in a long-term experiment, the affinity aminophenyl boronic acid procedure for determination of glycosylated protein was used [267]; a case-control study involving 423 cataract patients and 608 control subjects on the protective effect against cataracts associated with the consumption of painkillers (aspirin, paracetamol, and ibuprofen family) [268] |
| Diclofenac | In vitro: reduces ↓ albumin and hemoglobin glycation, blocks at least one of the main glycation sites of HSA | HSA-glucose 6-phosphate [266] |
| Ibuprofen |
In vitro: prevents modification of lens proteins by carbonylation and non-enzymatic glycation; reduces cyanate and galactose binding but not glucose 6-phosphate; protects against opacities; it appears to have a different mechanism of action from that of aspirin; Human studies: low doses protect against cataracts |
Animal lenses were incubated with 14C-labelled galactose, 14C-labelled glucose 6-phosphate and 14C-labelled potassium cyanate [269]; A case-control study involving 423 cataract patients and 608 control subjects on the protective effect against cataracts associated with the consumption of analgesics (aspirin, paracetamol, and ibuprofen family) [268] |
| Nimesulide | In vitro: reduces ↓ AGE formation; acts as an antioxidant, chelates transition metal cations | BSA-MGO, BSA-glucose, DPPH test; Fe+2 chelation test [270] |
| Mefenamic acid | ||
| Meloxicam | ||
| Piroxicam | ||
| Paracetamol | Human studies: low doses protect against cataracts | A case-control study involving 423 cataract patients and 608 control subjects on the protective effect against cataracts associated with the consumption of analgesics (aspirin, paracetamol, ibuprofen) [268] |
| 4. Selected B vitamins | ||
| Thiamine pyrophosphate (B1) | In vitro: reduces ↓ AGE formation (thiamine and thiamine monophosphate are not inhibitors); is essential for sustaining cellular defenses against oxidative stress | BSA-glucose, ribonuclease A-glucose, human hemoglobin-glucose; AGEs by ELISA [271] |
|
Benfotiamine (a lipid soluble thiamine derivative) |
Animal studies: reduces ↓ AGE formation; activates antioxidant defense mechanisms, an ↓ NADPH oxidase inhibitor (this enzyme plays an essential role in ROS production and myocardial cytotoxicity); improves markers of oxidative stress, inflammation, and apoptosis; inhibits ↓ NF-κB by activating transketolase in diabetic animals, prevents experimental diabetic retinopathy; significantly attenuates or ablates diabetes-induced elevation in cardiac levels of ↓ MGO, ↓ AGEs (MAGEs), ↓ RAGE, and ↓ cross-linked collagen without affecting hypertriglyceridemia and hypercholesterolemia; Human studies: significantly reduces ↓ CML-derived AGE levels in the subject group and sRAGE in the placebo group |
Wistar rats; a myocardial injury was induced by isoproterenol hydrochloride; the NADPH oxidase activity in cardiac tissue was measured by lucigenin chemiluminescence method; oxidative stress markers were assessed calorimetrically; inflammatory markers (PKC, NF-κB and metalloproteinase-9), and apoptotic markers (p53 and caspase-8) by ELISA; histopathologic assessment of myocardial damage [272]; STZ-induced diabetic mice; cardiomyocytes with MAGEs; MGO was measured by o-phenylenediamine-based assay; AGEs by immunohistochemical staining and ELISA; western blot was applied to measure of RAGE, GSK-3β, phospho-GSK-3β; qPCR quantification of RAGE, immunofluorescence detection of MAGEs and RAGE in isolated cardiomyocytes; myocardial collagen cross-linking; measurement of mitochondrial membrane potential [273]; RCT: a randomized, controlled, double-blind, clinical trial (clinical trial no. NCT02772926); T2DM subjects with no complications; benfotiamine treatment (900 mg/day) or placebo for 12 weeks; basal and final anthropometric data, blood pressure, glucose, HbA1c, and lipid profile were measured; CML-AGEs and sRAGE were measured by ELISA kits [231] |
| Pyridoxamine, pyridoxal, pyridoxal phosphate, pyridoxine (B6) |
In vitro: GO and MDA scavenger, reduces ↓ AGE formation; reduces ↓ ALE formation (but pyridoxine is slightly effective at the highest concentrations); Animal studies: increases ↑ Glo1 expression in visceral and perivascular adipose tissue; inhibits the ↓ AGE/RAGE pathway; pyridoxamine reduces atherosclerosis and inflammation induced by MGO; in obese mice improves glucose tolerance and insulin metabolism; prevents adipose tissue inflammation and vascular dysfunction; decreases fasting insulin levels and improved insulin sensitivity in obese and type 2 diabetic mice, most likely by trapping MGO and inhibiting AGE formation; Human studies: reduces ↓ MGO (9%), ↓AGEs (MG-H1), ↓ sVCAM-1, and SOCOM-1, but does not affect insulin sensitivity and vascular function in abdominally obese individuals; the reduction in adhesion markers is promising because these are important in the pathogenesis of endothelial damage and atherosclerosis |
BSA-glucose, ribonuclease A-glucose, human hemoglobin-glucose; ubiquitin-RCS; AGEs were assessed by ELISA; adducts with MGO, GO and MDA were analyzed by ESI-MS, 1H and 13C NMR [235,237,271,274]; Sprague-Dawley rats; murine macrophage cultures (Raw 264.7); Mus musculus pre-adipocyte cells (3T3-L1-MBX); interactions between AGEs and RAGE were investigated by ELISA; qPCR, western blot, histological assessment, and others [214]; Male C57BL/6J mice; serum insulin, hydrogen peroxide, MDA, AGEs, and urinary 8-hydroxy-2′-deoxyguanosine were measured; antioxidant enzymes and adipocytokine messenger RNA expressions in the adipose tissues, and Akt/protein kinase B activity and glucose transporter 4 translocation in skeletal muscle were also measured [275]; KK-Ay mice, a model animal of obese, type 2 diabetes; fasting blood glucose, serum levels of insulin and AGEs were elevated [276]; Wild-type C57Bl6 mice; AGEs and AGE precursors were assessed by LC-MS; metabolic, urinary and atherosclerotic parameters were analyzed; and flow cytometry was performed to identify circulating immune cells [225]; RCT (clinical trial no. NCT02954588): a randomized, double-blind, placebo-controlled crossover study pyridoxamine of 25 or 200 mg per day; insulin sensitivity, β-cell function, insulin-mediated microvascular recruitment, skin microvascular function, flow-mediated dilation, plasma inflammation and endothelial function markers were assessed; pyridoxamine metabolites, RCS and AGEs were measured using LC-MS [232] |
AGEs, advanced glycation end products; AMPK, AMP-activated protein kinase; AOPP, advanced oxidation protein products; BSA, bovine serum albumin; cDNA, complementary DNA; cGMP, cyclic GMP; CML, N6-(carboxymethyl)-L-lysine; CML-AGEs, AGEs with N6-(carboxymethyl)-L-lysine; ELISA, enzyme-linked immunosorbent assay; Glo1, glyoxalase 1; GO, glyoxal; HAECs, human aortic endothelial cells; HbA1c, glycated hemoglobin; HPLC, high-performance liquid chromatography; HUVECs, human umbilical vein endothelial cells; HSA, human serum albumin; IL-6, interleukin-6; KATP, ATP-sensitive potassium channel; LC-MS, liquid chromatography–mass spectrometry; MAPK, mitogen-activated protein kinase; MDA, malonyl dialdehyde; MG-H1, methylglyoxal-derived Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine; MGO, methylglyoxal; MS, mass spectrometry; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa-B; NMR, nuclear magnetic resonance spectroscopy; iNOS, inducible nitric oxide synthase; PCR, polymerase chain reaction; PPAR-γ, peroxisome proliferator-activated receptors-gamma; qPCR, quantitative real-time polymerase chain reaction; RAGE, receptor for advanced glycation end products; sRAGE, soluble RAGE; esRAGE, endogenous secretory RAGE; RCS, reactive carbonyl species; RCT, randomized clinical trial; ROS, reactive oxygen species; RT-PCR, reverse transcription polymerase chain reaction; TNF-α, tumor necrosis factor-α; T2DM, type 2 diabetes mellitus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



