Submitted:
02 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
CD8+ cytotoxic T lymphocytes (CTLs) play a crucial role in anti-tumor immunity. In a previous study, we identified a subset of murine effector CTLs expressing the hepatocyte growth factor (HGF) receptor c-Met (c-Met+ CTLs) that are endowed with enhanced cytolytic capacity. HGF directly inhibited the cytolytic function of c-Met+ CTLs, both in 2D in vitro assays and in vivo, leading to reduced T cell responses against metastatic melanoma. To further investigate the role of c-Met+ CTLs in a three-dimensional (3D) setting, we studied their function within B16 melanoma spheroids and examined the impact of cell-cell contact on the modulation of inhibitory checkpoint molecules expression such as KLRG1, PD-1 and CTLA-4. Additionally, we evaluated the cytolytic capacity of human CTLs clones expressing c-Met (c-Met+) and compared it to c-Met− CTLs clones. Our results indicated that, similar to their murine counterparts, c-Met+ human CTLs clones exhibited increased cytolytic activity compared to c-Met− CTLs clones, and this enhanced function was negatively regulated by the presence of HGF. Taken together, our findings highlight the potential of targeting the HGF/c-Met pathway to modulate CTL-mediated anti-tumor immunity. This research holds promises for developing strategies to enhance the effectiveness of CTL-based immunotherapies against cancer.
Keywords:
c-Met
; HGF
; CTLs
; CD8+ cytotoxic T lymphocytes
; spheroids
; tumor
1. Introduction
Malignant tumors exist in a complex microenvironment known as the tumor microenvironment (TME), composed of various cell types, including stromal cells, neutrophils, dendritic cells (DCs), and macrophages. Some of these TME-resident cells release hepatocyte growth factor (HGF) and promote activation of c-Met, the receptor for HGF, within tumor cells (1, 2). c-Met expression has been linked to poor prognosis in different solid tumors (3-5) and overactivation of the HGF/c-Met axis has been shown to promote tumorigenesis and tumor progression in various cancer types, such as gastroesophageal adenocarcinoma, cholangiocarcinoma, colon cancer, kidney cancer, glioblastoma, melanoma, and lung cancer (6-14). Some studies suggest that HGF can act as an immunotolerant factor in autoimmune diseases by affecting DCs (15-18). By contrast, others studies report that HGF can be a potent negative regulator of DC function by i) stimulating T regulatory lymphocytes (Tregs), ii) decreasing interleukin-17-producing lymphocytes (15), and iii) increasing production of both interleukin-10 and transforming growth factor beta (16). In addition, c-Met deletion in neutrophils enhances tumor growth and metastasis, as c-Met is required for chemoattraction and neutrophil-mediated cytotoxicity (19). In blood samples, this phenomenon is represented by a correlation between c-Met deletion and reduced neutrophil infiltration to both primary tumor and distant metastases (19). Finally, HGF acts on myeloid-derived suppressor cells by inducing their expansion and increasing iNOS and ARG1 expression, as well as harboring suppressive function and inducing Tregs (20). Limited studies have reported the expression of c-Met on murine CD4+ and CD8+ thymocytes (21), and T cells (22).
In the context of T lymphocytes, we identified a novel population of highly cytotoxic CTLs expressing c-Met (c-Met+ CTLs) in a murine model of lung metastasis (23). These c-Met+ CTLs exhibited enhanced cytolytic activity with increased production of interferon (IFN-)γ and granzyme (Gr) B compared to their c-Met- counterparts (23). Furthermore, we demonstrated the immunosuppressive role of HGF on c-Met+ CTLs both in vitro and in vivo (23). Adoptive transfer of highly cytotoxic CD8+c-Met+ T cells in tumor-bearing mice induced significantly tumor growth suppression as compared to CD8+c-Met- T cells. Additionally, we identified CD8+c-Met+ tumor-infiltrating lymphocytes (TILs) in human melanoma skin biopsies (23), suggesting that targeting the HGF/c-Met pathway could control CD8+c-Met+ T cell-mediated anti-tumor immunity.
Immune checkpoint inhibitors (ICIs) have shown promising results in cancer treatment, in particular ICIs targeting CTLA-4, PD-1, and PD-L1 have yielded unprecedented and durable responses in a significant percentage of cancer patients in recent years, leading to US Food and Drug Administration (FDA) approval of six ICIs for numerous cancer indications since 2011 (24). However, they are efficient in a subset of patients only and are associated with severe side effects (25). As an alternative approach, cell therapy, such as TILs, engineered T cell receptor (TCR), chimeric antigen receptor (CAR)-T cells, CTLs, and natural killer (NK) cells therapy, have emerged as advanced strategies to overcome limitations faced by ICIs (24, 26). Nevertheless, effective cellular therapies in solid tumors encounter challenges related to target antigen identification, T cell trafficking to the tumor site, and the immunosuppressive TME. Combining cellular therapies with ICIs may improve T cell infiltration and enhance anti-tumor responses (27, 28).
Based on our recent findings (23), we hypothesized that the oncogenic protein c-Met may directly regulate CTL effector functions in the B16 melanoma spheroid model and human CTL clones. In this study, we focused on phenotypic and functional modulation of c-Met+ CTLs generated from Pmel-1 mice in the 3D tumor culture, under stress cell-cell and cell-matrix interaction, which cannot be studied in 2D models. Indeed, cells growing in spheroids display a higher degree of functional and morphological differentiation than monolayer cells. As a result, spheroids can mimic the in vivo spatial architecture, physiological responses, gene expression patterns, mechanisms of drug resistance, and secretion of molecules induced by cell stimuli (29). We thus employed a 3D culture model to mimic partially aspects of the TME and co-cultured B16 melanoma cell-derived spheroids with CTLs c-Met+ to investigate the impact of tumor cells on immune cell effector functions and vice versa (30). We also explored for the first time the role of c-Met expression in human CTLs. We used human CTLs clones to study c-Met expression and its impact on cytolytic function, along with the effect of HGF modulation on their activity. We found that 3D B16 spheroids influenced the expression of negative costimulatory molecules, such as KLRG1, PD-1, and CTLA-4, on c-Met+ CTLs. Finally, we showed, for the first time, that c-Met expression in human CTLs clones conferred higher cytolytic capacity, while HGF treatment dampened this activity by reducing GrB and IFN-γ production.
2. Methods
2.1. Reagents
Human recombinant HGF (hrHGF) was used throughout (#100-39H-25UG; peprotech) as human and murine HGF are cross-reactive. The dose of hrHGF (30 ng/mL) used was chosen based on previous studies analyzing the immunoregulatory effects of hrHGF on CD4+ T cell responses (15). Lower doses were not as effective.
2.2. Spheroids Preparation
Generation of 3D spheroids from B16 melanoma cells can be co-cultured with immune cell population of interest, we cultured directly B16 cells in the wells precoated with agarose, we did not pass by 24-48h of drop hanging step, we obtained similar result concerning cell aggregate, expansion and spheroid formation. Melanoma B16 cells were harvested, counted, and 1000 cells were resuspended in 200 μL of DMEM supplemented with 10% FCS, 40 μg/ml streptomycin, 40 units/ml penicillin, 200 mM l-glutamine, and were plated in each well of 96-well flat-bottom plates (Corning) precoated with 1% agarose (Thermo Fisher) dissolved in double distilled water . Then, the spheroids were incubated for 7 days in a 5% CO2 - humidified incubator at 37°C. At that day, all experiments were performed, and the day was assigned as day 0. Control spheroids were digested with trypsine-0.05 % EDTA (#25300054, Thermo Fisher) and analyzed with flow cytometry to visualize the presence of necrosis in untreated samples (absence of CTLs).
2.3. Cells and cultures
Splenocytes were derived from Pmel-1 T cell receptor (TCR) transgenic mice (The Jackson Laboratory), whose TCR recognizes an H-2Db-restricted epitope corresponding to amino acids 25-33 of murine and human gp100 (gp10025-33), a self-tumor antigen. Approximately 90% of splenic CD3+ T cells in Pmel-1 TCR mice are TCRVβ13+ CD8+ T cells and demonstrate specificity for gp100 (31).
2.4. CTL stimulation
Single-cell suspensions of pooled spleens from Pmel-1 mice were prepared by gently homogenizing the tissues and passing them through a 70 μm Nylon cell strainer (Falcon, BD Biosciences). Red blood cells were depleted with ACK lysing buffer (BioWhittaker). Cells were resuspended in RPMI and activated with 10 μg/mL Gp10025-33 peptide (KVPRNQDWL) for 1 h at 37°C. After being washed and counted, IL-2 (50 U/mL; PeproTech) was added to cultures at days 2 and 4. Cells cultured for 5 days were tested for functional and phenotypic assays.
2.5. Human CTLs clones and T2 cells
Human CTLs clones and T2 cells were maintained in RPMI 1640 supplemented with 200 mM l-glutamine, 50 μM β-mercaptoethanol, 100 mM sodium pyruvate, MEM vitamins, 40 μg/ml streptomycin, 40 units/ml penicillin (standard medium M′), and 8% human FCS (Laboratoires Jacques Boy). Human CTL clones targeting different antigens were kindly proposed by the laboratory of Pierre Yves Dietrich (Geneva University Hospitals). Briefly, human clones where isolated using peptide-MHC tetramers and cultured in limiting dilution condition as previously described (32). T cell clones were periodically restimulated as previously described (32). We used the following clones: clones GE 450 1E8 and GE 522 5E3 specific for the HLA-A2-restricted Melan-A (MLA)-derived peptide (26-35 A27L) from patients with melanoma and clone GE 549 1F10 specific for the HLA-A2-restricted Brevican (BCA2)-derived peptide (478-486) from patients with glioblastoma (32). T2 cells are HLA-A*0201 human lymphoid cells that are defective in antigen processing but effectively present exogenously supplied peptides (33).
2.6. Flow cytometry
Single-cell suspensions from Pmel-1 spleens generated in vitro were incubated in blocking solution (PBS with 1% of fetal calf serum) for 20 min on ice prior to staining to block non-specific Fc-mediated interactions. Then they were stained for 30 min at 4 °C with appropriate fluorochrome-conjugated antibodies (Abs, 1:100) (Table S1) or isotype-matched irrelevant Abs to determine background fluorescence. For intracellular cytokines and molecular staining of IFN-γ, TNF-α and GrB, T cells were stimulated with phorbol myristate acetate (PMA, 50 ng/ml) and ionomycin (500 ng/ml) in the presence of brefeldin A (10 μg/ml; Sigma-Aldrich) and then fixed and permeabilized using BD Cytofix/Cytoperm Plus Kit (BD Biosciences). Samples were processed on a FACS LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo analysis software (version 10.3). Live, apoptotic, and dead populations were defined based on 7-AAD Viability Staining Solution from eBioscience, or with 0.2ug/ml Dapi (#D1306) and Annexin V (#17-8007-72) according to the manufacturer’s instructions.
Cytotoxicity was determined as level of necrotic (Dapi+AnnexinV-) cells relative to negative control condition (necrotic B16 cultured alone in absence of CTLs). Percent specific lysis was calculated according to the formula: (1-(control /test))*100. To determine the viability of cells prior to fixation and permeabilization, we used the LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific).
For cell-separation, at the end of culture, the cells were harvested, stained at 4°C for 30 min with CD3, CD8, and c-Met, and purified as CD8+ c-Met− and CD8+ c-Met+ by cell sorting using a FACS Aria SORP II cell sorter (BD Biosciences). Live cells were gated based on forward scatter (FSC) and side scatter (SSC) and by DAPI exclusion. Ag-activated c-Met− and c-Met+CD8+ T cell populations showed similar physical properties of size and internal complexity. Cell viability (routinely > 95%) at the end of the FACS sorting procedure was determined using the trypan blue dye exclusion method.
Human Fc-block (BD Pharmingen) was used to block non-specific binding of Fc-receptors prior to the extracellular staining of human CTL clones. They were then stained for 30 min at 4 °C with appropriate fluorochrome-conjugated Abs (1:100) (Table S1) or isotype-matched irrelevant Abs to determine background fluorescence. For intracellular cytokines and molecular staining of IFN-γ and GrB, T cells were stimulated and processed as explained before.
2.7. RT-PCR
Human CTL clones were treated with hHGF (30ng/ml) for 72h. Total RNA was isolated using the RNeasy micro kit (Qiagen) and quantitative real–time duplex PCR analysis (ABI 7500; Applied Biosystems) was conducted after reverse transcription with the iScript cDNA Synthesis Kit (Bio–Rad). Primers and probes were obtained from Applied Biosystems. The levels of c-Met, IFNγ, GrB, perforin, TNFα, FasL and PD-1 mRNA expressions were normalized with the expression of a simultaneously analyzed housekeeping gene (β–actin). All measurements were conducted in triplicate.
2.8. In vitro cytotoxicity assay
Cytotoxicity of antigen-specific human CTLs clones were analyzed with a DELFIA® cell cytotoxicity kit (PerkinElmer) according to manufacturer’s instructions. Briefly, CTL clones were incubated or not with HGF for 72h (30ng/ml) before use in the assay. T2 target cells (105 cells) were incubated or not with the cognate CEF3, CEF5, MLA or BCA2 peptides for 1 h at 37°C, and then washed and labelled with 50 μM of fluorescence-enhancing ligand bis(acetoxymethyl)2,2′:6′,2′′-terpyridine-t,6′′-dicarboxylate (BATDA) for 30 min at 37°C. After washing, 5 × 103 labelled cells/well were mixed with antigen-specific CTLs at the indicated ratio in 96-well plates. Plates were incubated for 4 h at 37°C. A total of 20 μL of supernatant were harvested from each well and added to wells containing 200 μL of 50 μM Europium solution (Aldrich Chemical) in 0.3 M acetic acid (pH 4). Plates were shaken for 15 min at room temperature and the fluorescence of the Europium-TDA (Eu) chelates formed was quantitated in a time-resolved fluorometer (DELFIA 1234). All assays were performed in triplicates. Spontaneous release was determined as Eu detected in the supernatant of targets incubated in the absence of effector cells. Maximum release was determined as Eu detected in the supernatants of target cells incubated with lysis buffer (provided in the kit, solution containing 0.03% digitoninand 19% dimethylsulphaxide (DMSO)) instead of effectors. Percent specific lysis was calculated according to the formula: (experimental release - spontaneous release)/(maximum release - spontaneous release) × 100.
2.9. Western blotting
Human BCA2-specific CTLs were homogenized using a polytron in lysis buffer (50 mM Tris–HCl [pH 7.5], 250 mM NaCl, 1% Triton X-100, 1 mM EDTA, and 1 mM DTT) containing complete protease inhibitors (Roche). Equal amounts (20 μg) of total protein from each sample were transferred to 8% sodium dodecyl sulfate (SDS)-polyacrylamide gel and blotted onto an Immobilon-P polyvinylidene difluoride (PVDF) membrane (Millipore). Antibodies for Phospho-c-Met (Tyr1234/5, #3077, 1:500), c-Met (#3127, 1:400), β-actin (#8457, 1:1000) were all obtained from Cell Signaling Technology (Danvers, MA). The membranes were cut and the two parts were incubated overnight at 4 °C with either the c-Met (~ 145-170 kDa) or the β-actin (~ 42 kDa) primary antibody. The same procedure was followed to detect the phosphor c-Met (~ 140 kDa). Next, the membranes were washed with TBS/T and incubated with anti-mouse IgG (Cell Signaling #7076) or anti-rabbit IgG (Cell Signaling #7074) horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Bands were developed by adding horseradish peroxidase substrate (Thermo Fisher) and recorded with X-ray film or ChemiDocTM digital imaging system from Bio-Rad.
2.10. Statictical analysis
Statistical comparisons were performed using paired and unpaired Student’s t-tests. P < 0.05 were considered statistically significant. All statistical analyses were performed with GraphPad Prism software, version 9.2.
3. Results
3.1. Using 3D B16 spheroids to study the phenotype and function of tumor-specific c-Met+ CTLs
Here, we provide a detailed workflow used for the generation of 3D spheroids from B16 melanoma cells that can be co-cultured with cytotoxic cells from Pmel-1 mice. Using agarose pre-coated wells and seeding B16 cells on the top, spheroids were generated over a period of 7 days, as shown in (Figure 1A). After in vitro activation, CTLs were sorted into c-Met+ and c-Met- cells (Figure S1) and co-cultured with B16 spheroids for 4 hours to determine their cytotoxic capacity and phenotype modification in contact with the spheroids. We named adherent CTLs (c-Met+ or c-Met-), CTLs that were in contact with the spheroids even after washing with PBS, and non-adherent CTLs, CTLs staying in suspension (Figure 1B).
3.2. Phenotypic and function of c-Met+ CTLs in contact with 3D B16 spheroids
Our previous data indicated that cytotoxic T cells expressing c-Met (c-Met+CTLs) are highly cytotoxic compared to their c-Met− counterpart (23). We anticipated similar cytolytic performance on 3D spheroid culture. In particular, 3D spheroids were shown to be markedly more susceptible to cell death than cells grown in a monolayer (2D) conformation (34). We tested the lysis function by flow cytometry and by quantifying necrotic B16 DAPI+ as shown in the gating strategy (Figure 1B, right panel). We found that the rate of B16 spheroids lysis was significantly increased when co-cultured with c-Met+ CTLs compared with their c-Met- counterpart (Figure 2A). We then assessed the proportion of CTLs producing IFN-γ, GrB and TNF-α in contact with spheroids, and showed that c-Met+ T cells expressed higher level of these three molecules compared to their c-Met− counterpart (Figure 2B and Figure S2A for isotype controls). Interestingly, it was also observed in the non-adherent CTLs compartment. Notably, in contact with B16 spheroids, c-Met+ CTLs displayed increased CD107a expression, a marker commonly used to measure CTLs activity (Figure 2C). Analysis of FasL (CD178) expression did not show any difference between both populations in contact with B16 spheroids (Figure S3).
3.3. Modulation of immune checkpoint expression by c-Met+ CTLs in contact with 3D B16 spheroids
We next assessed whether cell contact between CTLs and B16 3D spheroids modulated immune checkpoint molecules such as KLRG1, PD-1, CTLA-4, Tim-3 and LAG-3. We observed a significant increase of KLRG1 expression on c-Met+ CTLs compared to their c-Met- counterpart (Figure 3A and Figure S2B for isotype controls). This observation is in line with another publication showing that, during acute infections or vaccinations, naïve CD8+ T cells become activated and differentiate into effector T cells containing KLRG1Hi terminal effector cells (35). In contrast, we detected decreased expression of inhibitory receptors such as PD-1 and CTLA-4 on c-Met+ CTLs compared to their c-Met- counterpart upon contact with B16 spheroids (Figure 3B,C and Figure S2B for isotype controls). Additionally, other checkpoint molecules such as Tim-3 and LAG-3 were not modulated in neither population upon contact with B16 spheroids (Figure S4).
3.4. Modulation of c-Met expression, phenotype and function by HGF on human T cell clones
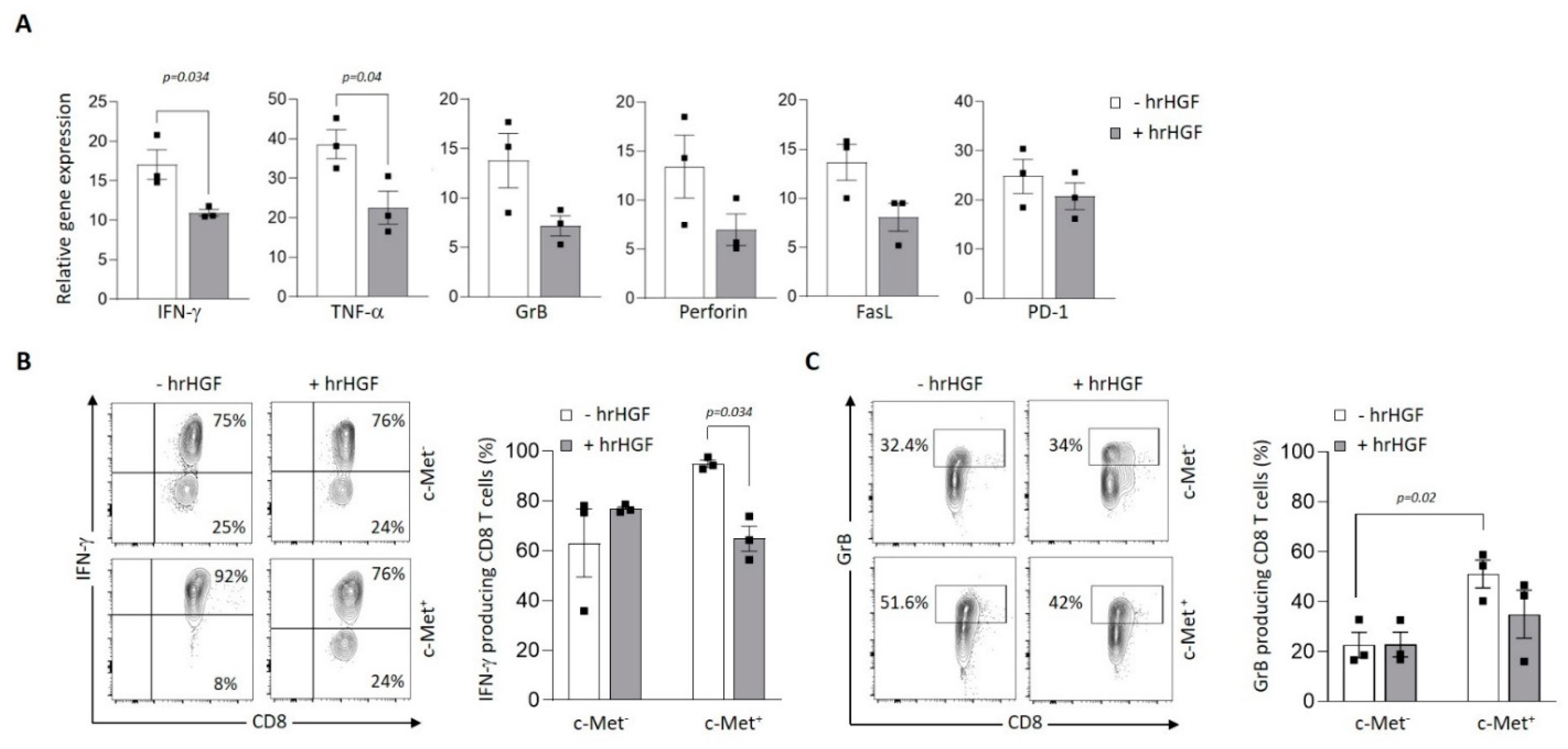
Then, we assessed expression of c-Met on tumor antigen-specific human CD8+ T cell clones generated from cancer patients. We observed variable expression of c-Met in the different clones both at the mRNA and protein levels (Figure 4A,B). Interestingly, the clones that displayed significant c-Met expression (GE 522 5E3 and GE 549 1F10) had an efficient killing capacity of T2 target cells loaded with the specific antigen (MLA or BCA2), which was reduced after addition of human recombinant (hr)HGF (Figure 4C). To further study the biological function of hrHGF on c-Met expressing CD8 clones, we selected the GE 549 1F10 clone which showed the highest reduction of killing activity in presence of hrHGF (Figure 4C), and we confirmed the presence of phospho-c-Met (P-c-Met) one hour after hrHGF treatment (Figure 4D). Additionally, using RT-PCR on the clone of interest, we assessed its phenotypic profile in presence of hrHGF. We noticed a significant decrease of IFN-γ and TNF-α expression levels and although less striking, a similar tendency was observed for GrB, perforin, FasL and PD-1 levels (Figure 5A). At the protein level, production of IFN-γ and GrB were reduced after incubation with hrHGF (Figure 5B,C). Altogether, our data suggest the presence of cytotoxic c-Met+ CD8 clones derived from cancer patients and these cells have a higher cytotoxic capacity compared to c-Met- CD8 clones, which is decreased upon in vitro treatment with hrHGF.
4. Discussion
Evidence supporting HGF/c-Met signaling pathway as relevant targets for personalized cancer treatment have been widely described, mostly when expressed on tumor cells (39). Currently, there are three main methods used to inhibit the kinase activity of c-Met: i) by preventing extracellular binding of HGF through neutralizing antibodies or biological antagonists; ii) by blocking phosphorylation of tyrosine residues in the kinase domain using small-molecule inhibitors; or iii) by blocking c-Met kinase-dependent signaling through relevant signal transducers or downstream signaling components (36). Several small-molecule inhibitors and monoclonal antibodies recognizing c-Met have been evaluated in preclinical studies (37-39), however, albeit the major advancement they provide, only patients with tumors overexpressing c-Met are eligible for HGF/c-Met targeting therapy, since these molecules inhibit typical c-Met-associated biological processes, such as oncogenesis, cancer metastasis, and drug resistance (40). Previous findings from our group demonstrated that in a murine model of CTL-mediated killing, HGF treatment decreased the effector capacity of tumor specific CD8+ T cells (33). Based on these results, we demonstrated that expression of c-Met on tumor infiltrating CD8+ T cells favored their anti-tumoral function and ultimately limited tumor growth (23). Nonetheless, it remains unclear which mechanisms underly the generation of c-Met+ CTLs and how does HGF produced by the tumor microenvironment (TME) regulate their functions?
The current study sough to bring forth some insights, using the B16 melanoma spheroid in vitro model, a relevant and adaptable system to investigate the crosstalk between c-Met+ CTLs and tumor cells. We observed the existence of tumorigenic CD8+ T cells subtype expressing c-Met with higher activation levels when in direct contact with B16 spheroid compared to c-Met−CD8+ T cells, which is in line with our previous observations related to murine c-Met+ CTLs (23, 41). Concurrently, when in contact with B16 spheroids, c-Met+ CTLs had decreased levels of inhibitory receptors (i.e., PD-1, CTLA-4) compared to their c-Met negative CTLs homologs. In addition, we demonstrated that human CD8+ T cell clones derived in vitro from cancer patients PBMCs, express the c-Met receptor albeit at different levels upon TCR activation. More importantly, c-Met+ CD8 clones produced higher level of effector molecules (i.e., IFN-γ and GrB) compared to their c-Met- counterpart which was decreased upon treatment with HGF, suggesting a direct effect of HGF on dual GrB/IFN-γ-based CTL-mediated cytotoxicity. Altogether, these data emphasize the potential therapeutic implications of targeting the HGF/c-Met signaling axis, particularly in tumors that overexpress c-Met. While several approaches have been explored to inhibit c-Met activity such as capmatinib a highly potent and selective Met inhibitor that was recently approved by the FDA for treatment of metastatic non-small-cell lung cancer (34), the results of this study shed light on the importance of considering the potential impact of these therapies on CTLs expressing c-Met in the TME.
5. Conclusions
In conclusion, our study provides compelling evidence for the high cytotoxicity of c-Met+ CTLs in murine and human in vitro models and highlights the potential influence of HGF on the function of these CTLs. These findings underscore the importance of further investigating the role of c-Met+ TILs in anti-tumor immunity and the implications of HGF/c-Met targeting therapies in the context of CTL-mediated anti-tumoral responses. Shedding light on these mechanisms will certainly pave the way for development of more effective and targeted immunotherapies for cancer treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: Flow cytometry gating strategy for the identification of Pmel-1 CD8+Vβ13+ T cells; Table S1: Antibodies for flow cytometry; Figure S2: Isotypes controls presentation; Figure S3: FasL expression by CTLs in contact with B16 Spheroids; Figure S4: LAG-3 and Tim-3 expression by CTLs in contact with B16 Spheroids.
Author Contributions
Writing original draft and supervision of the manuscript (MB, NLT and PHL); designing and drafting of the manuscript (MB, NLT and PHL); acquisition of data (MB, NLT, IS, VD) and editing (IS, NLT, GB, HF, VD, DM and PHL). All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the Swiss National Science Foundation (Grant 310030_176078 and 310030_214931) and the Private Foundation of the HUG (PHL).
Institutional Review Board Statement
Patients from which the human CTL clones were obtained were enrolled in a research protocol accepted by the local ethics committee (CCER protocol 03-126) and patients signed an informed consent form.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
We thank Catherine Juillard for the RT-PCR on human clones. We thank the personnel of the flow cytometry core facility of the University of Geneva for their technical assistance. We would like to thank Professor Pierre Yves Dietrich for stimulating discussions.
Conflicts of Interest
The authors declare no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Matsumoto, K.; Date, K.; Ohmichi, H.; Nakamura, T. Hepatocyte growth factor in lung morphogenesis and tumor invasion: role as a mediator in epithelium-mesenchyme and tumor-stroma interactions. Cancer Chemother Pharmacol 1996, 38, S42–S47. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Matsumoto, K.; Kiritoshi, A.; Tano, Y.; Nakamura, T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions. Cancer Res 1997, 57, 3305–3313. [Google Scholar] [PubMed]
- Lee, W.Y.; Chen, H.H.; Chow, N.H.; Su, W.C.; Lin, P.W.; Guo, H.R. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin Cancer Res 2005, 11, 2222–2228. [Google Scholar] [CrossRef]
- Sawada, K.; Radjabi, A.R.; Shinomiya, N.; Kistner, E.; Kenny, H.; Becker, A.R.; Turkyilmaz, M.A.; Salgia, R.; Yamada, S.D.; Vande Woude, G.F.; et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res 2007, 67, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Masuya, D.; Huang, C.; Liu, D.; Nakashima, T.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer 2004, 90, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Ang, A.; Liao, W.L.; Shen, J.; O'Day, E.; Loberg, R.D.; Cecchi, F.; Hembrough, T.; Ruzzo, A.; Graziano, F. MET tyrosine kinase receptor expression and amplification as prognostic biomarkers of survival in gastroesophageal adenocarcinoma. Cancer 2017, 123, 1061–1070. [Google Scholar] [CrossRef]
- Heo, M.H.; Kim, H.K.; Lee, H.; Kim, K.M.; Lee, J.; Park, S.H.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Park, Y.S.; et al. The Clinical Impact of c-MET Over-Expression in Advanced Biliary Tract Cancer (BTC). J Cancer 2017, 8, 1395–1399. [Google Scholar] [CrossRef]
- Gayyed, M.F.; Abd El-Maqsoud, N.M.; El-Hameed El-Heeny, A.A.; Mohammed, M.F. c-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J Gastrointest Oncol 2015, 6, 618–627. [Google Scholar] [CrossRef]
- Peltola, K.J.; Penttila, P.; Rautiola, J.; Joensuu, H.; Hanninen, E.; Ristimaki, A.; Bono, P. Correlation of c-Met Expression and Outcome in Patients With Renal Cell Carcinoma Treated With Sunitinib. Clin Genitourin Cancer 2017, 15, 487–494. [Google Scholar] [CrossRef]
- Reis, H.; Metzenmacher, M.; Goetz, M.; Savvidou, N.; Darwiche, K.; Aigner, C.; Herold, T.; Eberhardt, W.E.; Skiba, C.; Hense, J.; et al. MET Expression in Advanced Non-Small-Cell Lung Cancer: Effect on Clinical Outcomes of Chemotherapy, Targeted Therapy, and Immunotherapy. Clin Lung Cancer 2018, 19, e441–e463. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, K.Y.; Giubellino, A. The Role of MET in Melanoma and Melanocytic Lesions. Am J Pathol 2019, 189, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Hack, S.P.; Bruey, J.M.; Koeppen, H. HGF/MET-directed therapeutics in gastroesophageal cancer: a review of clinical and biomarker development. Oncotarget 2014, 5, 2866–2880. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Santiago-Raber, M.L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A 2010, 107, 6424–6429. [Google Scholar] [CrossRef]
- Okunishi, K.; Dohi, M.; Nakagome, K.; Tanaka, R.; Mizuno, S.; Matsumoto, K.; Miyazaki, J.; Nakamura, T.; Yamamoto, K. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol 2005, 175, 4745–4753. [Google Scholar] [CrossRef]
- Baek, J.H.; Birchmeier, C.; Zenke, M.; Hieronymus, T. The HGF receptor/Met tyrosine kinase is a key regulator of dendritic cell migration in skin immunity. J Immunol 2012, 189, 1699–1707. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Molnarfi, N.; Dunand-Sauthier, I.; Merkler, D.; Schneiter, G.; Bruscoli, S.; Riccardi, C.; Tabata, Y.; Funakoshi, H.; Nakamura, T.; et al. Hepatocyte growth factor limits autoimmune neuroinflammation via glucocorticoid-induced leucine zipper expression in dendritic cells. J Immunol 2014, 193, 2743–2752. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef]
- Yen, B.L.; Yen, M.L.; Hsu, P.J.; Liu, K.J.; Wang, C.J.; Bai, C.H.; Sytwu, H.K. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Reports 2013, 1, 139–151. [Google Scholar] [CrossRef]
- Jin, J.; Goldschneider, I.; Lai, L. In vivo administration of the recombinant IL-7/hepatocyte growth factor beta hybrid cytokine efficiently restores thymopoiesis and naive T cell generation in lethally irradiated mice after syngeneic bone marrow transplantation. J Immunol 2011, 186, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Komarowska, I.; Coe, D.; Wang, G.; Haas, R.; Mauro, C.; Kishore, M.; Cooper, D.; Nadkarni, S.; Fu, H.; Steinbruchel, D.A.; et al. Hepatocyte Growth Factor Receptor c-Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity 2015, 42, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Benkhoucha, M.; Molnarfi, N.; Kaya, G.; Belnoue, E.; Bjarnadottir, K.; Dietrich, P.Y.; Walker, P.R.; Martinvalet, D.; Derouazi, M.; Lalive, P.H. Identification of a novel population of highly cytotoxic c-Met-expressing CD8(+) T lymphocytes. EMBO Rep 2017, 18, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol Adv 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Filipiak-Duliban, A.; Brodaczewska, K.; Kajdasz, A.; Kieda, C. Spheroid Culture Differentially Affects Cancer Cell Sensitivity to Drugs in Melanoma and RCC Models. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Theoret, M.R.; Finkelstein, S.E.; Surman, D.R.; de Jong, L.A.; Vyth-Dreese, F.A.; Dellemijn, T.A.; Antony, P.A.; Spiess, P.J.; Palmer, D.C.; et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 2003, 198, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Salter, R.D.; Howell, D.N.; Cresswell, P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics 1985, 21, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Moreno, K.; Chavez-Garcia, D.; Hirata, G.; Vazquez-Duhalt, R. Monolayer (2D) or spheroids (3D) cell cultures for nanotoxicological studies? Comparison of cytotoxicity and cell internalization of nanoparticles. Toxicol In Vitro 2022, 85, 105461. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis Transl Med 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Agwa, E.S.; Ma, P.C. Targeting the MET receptor tyrosine kinase in non-small cell lung cancer: emerging role of tivantinib. Cancer Manag Res 2014, 6, 397–404. [Google Scholar] [CrossRef]
- Robinson, K.W.; Sandler, A.B. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist 2013, 18, 115–122. [Google Scholar] [CrossRef]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 2006, 66, 1721–1729. [Google Scholar] [CrossRef]
- Toschi, L.; Janne, P.A. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin Cancer Res 2008, 14, 5941–5946. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Molnarfi, N.; Schneiter, G.; Walker, P.R.; Lalive, P.H. The neurotrophic hepatocyte growth factor attenuates CD8+ cytotoxic T-lymphocyte activity. J Neuroinflammation 2013, 10, 154. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of protocol for B16 spheroid formation and cytotoxic in vitro test. (A) Representative images on the right show the aggregates formed by B16 melanoma cells (1,000 cells seeded) over 7 days. The images were acquired with a Zeiss Observer Z1 microscope. Scale bar, 200 µm. (B) Diagram illustrating the different steps of 3D B16 culture process model with c-Met+ and c-Met- CTLs (left panel), and the gating strategy for B16 cells and CTLs (right panel).
Figure 1.
Schematic diagram of protocol for B16 spheroid formation and cytotoxic in vitro test. (A) Representative images on the right show the aggregates formed by B16 melanoma cells (1,000 cells seeded) over 7 days. The images were acquired with a Zeiss Observer Z1 microscope. Scale bar, 200 µm. (B) Diagram illustrating the different steps of 3D B16 culture process model with c-Met+ and c-Met- CTLs (left panel), and the gating strategy for B16 cells and CTLs (right panel).
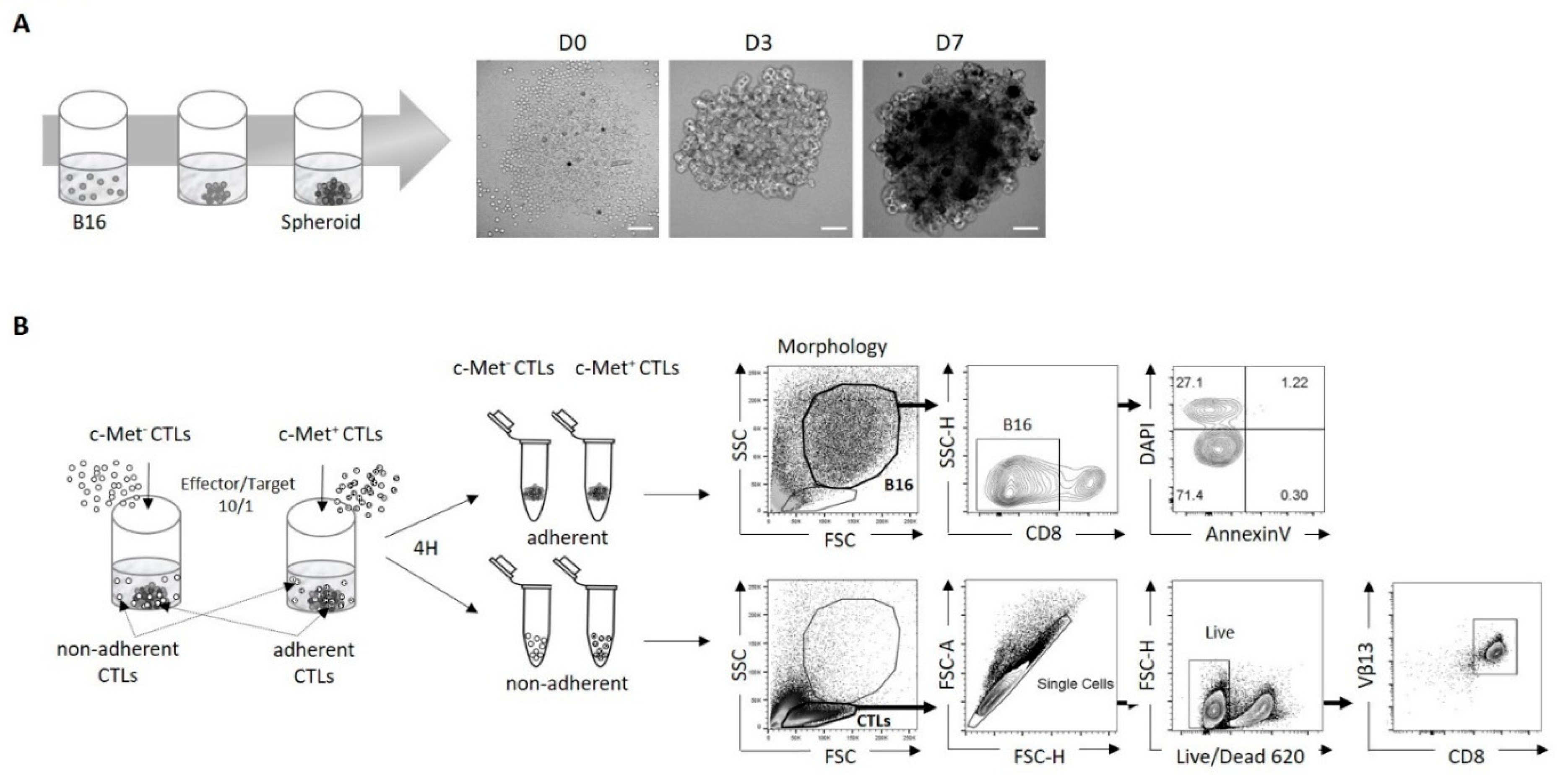
Figure 2.
Phenotypic and functional characterization of murine melanoma c-Met+ and c-Met- CTLs in 3D B16 spheroid culture. (A) In vitro cytolytic activity of day 5-activated c-Met+ and c-Met− Pmel-1 CD3+CD8+ T cells. Effector/target ratios of 10/1 were tested for the killing assay, B16 alone were used as control. Annexin V and DAPI were used to determine the percentage of necrotic B16 cells as shown with contour plot (left panel) and data are represented as paired samples between c-Met- vs c-Met+ CTLs for each mouse (right panel). Cytotoxicity was determined as level of necrotic (Dapi+AnnexinV-) cells relative to negative control condition (necrotic B16 in absence of CTLs). Values represent mean ± SEM of six mice (n=6) per group. P-values were calculated using unpaired, 2-tailed Student’s t-test. (B) Representative density plots (left panel) and paired flow cytometry quantifications (right panel) of IFN-γ , GrB and TNFα in CD8+c-Met+ vs CD8+c-Met- T cells from CTLs adherent to spheroids or CTLs in suspension (non-adherent) (n=6), data are representative of three different experiments. P-values were calculated using a paired t-test, and unpaired t-test for intergroup comparison. (C) Representative histogram plots and average geometric mean (right panel) of CD107a 4h after co-culture of spheroids B16 and CTLs c-Met+ vs c-Met- from six experiments of (n=6). P-values were calculated using a paired t-test and unpaired t-test for intergroup comparison.
Figure 2.
Phenotypic and functional characterization of murine melanoma c-Met+ and c-Met- CTLs in 3D B16 spheroid culture. (A) In vitro cytolytic activity of day 5-activated c-Met+ and c-Met− Pmel-1 CD3+CD8+ T cells. Effector/target ratios of 10/1 were tested for the killing assay, B16 alone were used as control. Annexin V and DAPI were used to determine the percentage of necrotic B16 cells as shown with contour plot (left panel) and data are represented as paired samples between c-Met- vs c-Met+ CTLs for each mouse (right panel). Cytotoxicity was determined as level of necrotic (Dapi+AnnexinV-) cells relative to negative control condition (necrotic B16 in absence of CTLs). Values represent mean ± SEM of six mice (n=6) per group. P-values were calculated using unpaired, 2-tailed Student’s t-test. (B) Representative density plots (left panel) and paired flow cytometry quantifications (right panel) of IFN-γ , GrB and TNFα in CD8+c-Met+ vs CD8+c-Met- T cells from CTLs adherent to spheroids or CTLs in suspension (non-adherent) (n=6), data are representative of three different experiments. P-values were calculated using a paired t-test, and unpaired t-test for intergroup comparison. (C) Representative histogram plots and average geometric mean (right panel) of CD107a 4h after co-culture of spheroids B16 and CTLs c-Met+ vs c-Met- from six experiments of (n=6). P-values were calculated using a paired t-test and unpaired t-test for intergroup comparison.
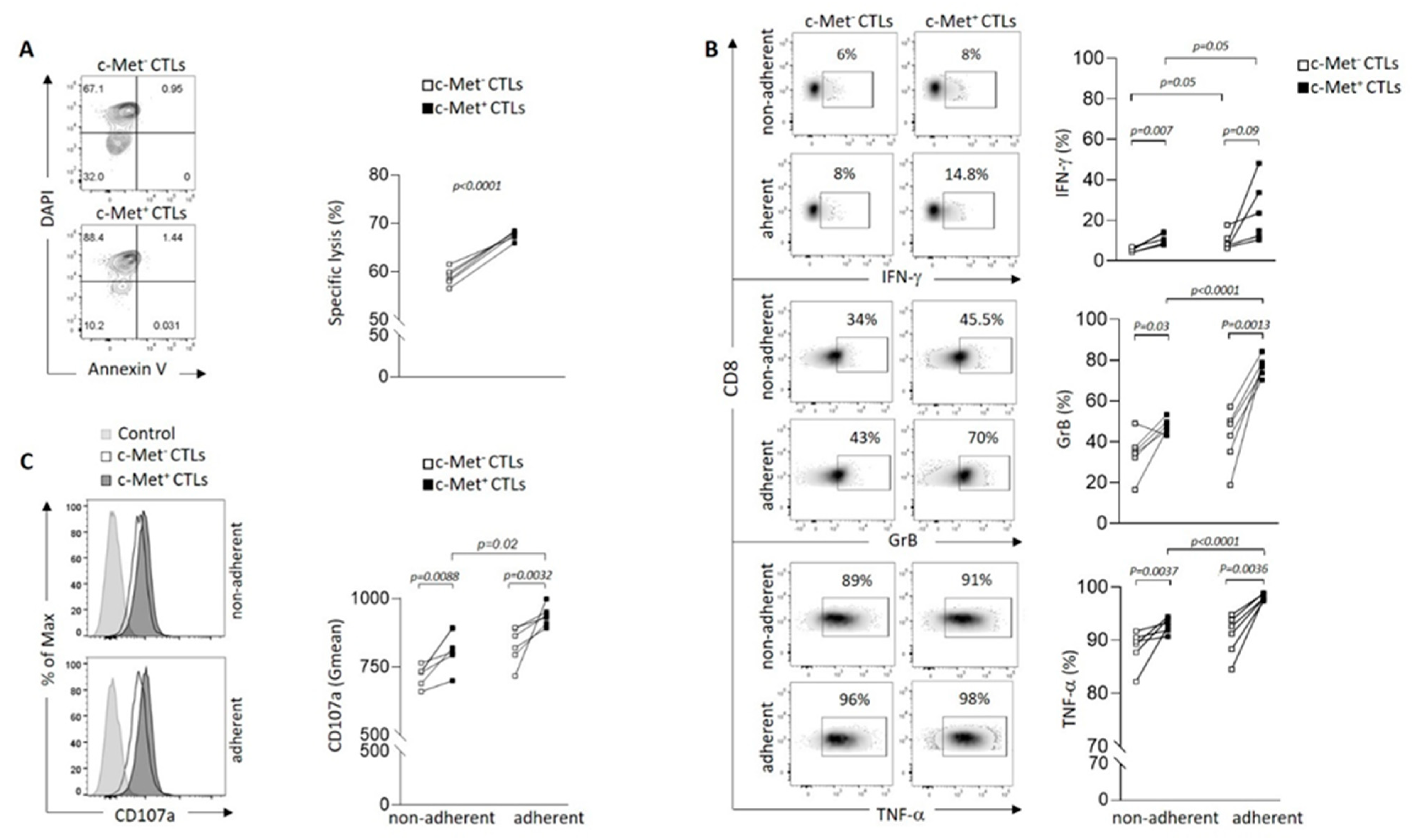
Figure 3.
Immune checkpoint molecules modulation of c-Met+ vs c-Met- CTLs in contact with 3D B16 spheroid. (A, B, C) Representative contour plots (left panel) and paired flow cytometry quantifications (right panels) of KLRG1, PD1 and CTLA-4 expression on c-Met+ vs c-Met- CTLs, 4h after co-culture with B16 spheroids (n=6 mice). P-values were calculated using a paired t-test.
Figure 3.
Immune checkpoint molecules modulation of c-Met+ vs c-Met- CTLs in contact with 3D B16 spheroid. (A, B, C) Representative contour plots (left panel) and paired flow cytometry quantifications (right panels) of KLRG1, PD1 and CTLA-4 expression on c-Met+ vs c-Met- CTLs, 4h after co-culture with B16 spheroids (n=6 mice). P-values were calculated using a paired t-test.
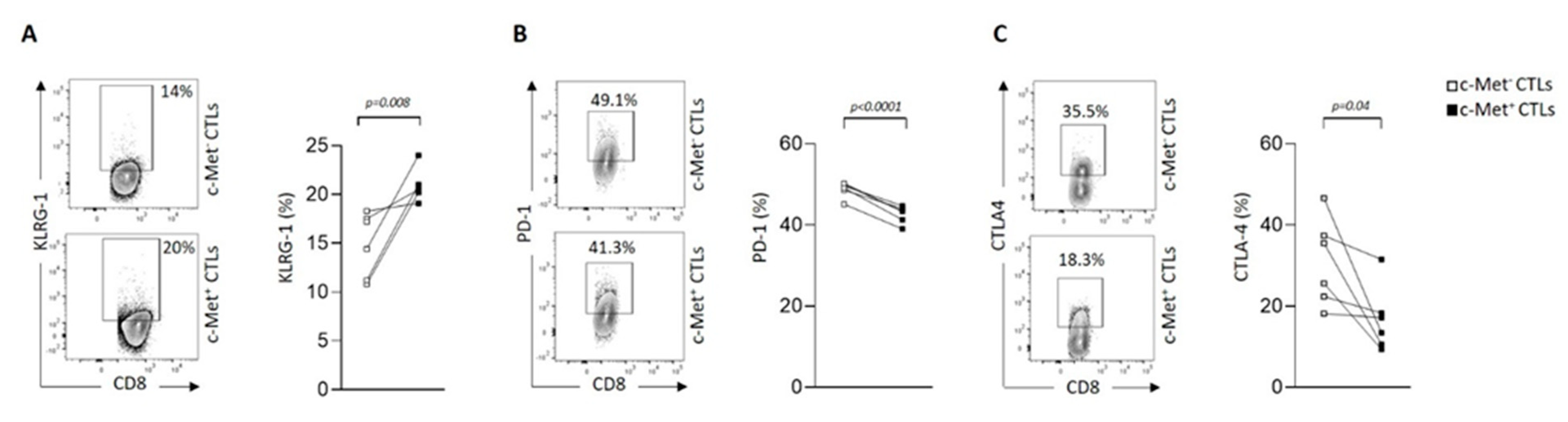
Figure 4.
c-Met expression on human CTL clones and modulation of killing capacity by HGF in vitro. (A) c-Met mRNA expression by qRT-PCR from CTL clones isolated from cancer patients compared to Hacat and T2 cell lines, used as positive and negative controls respectively. (B) Flow cytometry histograms showing c-Met protein expression (light gray) on CTL clones and control cell lines compared to the isotype control (dark gray). Percentage of c-Met+ CTLs are indicated. (C) In vitro killing assays were performed using DELFIA® cell cytotoxicity kit (PerkinElmer). T2 target cells were cultured with indicated ratio of CTL clones in presence (squares) or not (triangles) of the cognate peptide. Clones were incubated or not with HGF for 72h (black symbols, 30 ng/ml) before use in the assay. MLA: HLA-A2-restricted Melan-A-derived peptide (26-35 A27L), BCA2: HLA-A2-restricted Brevican-derived peptide (478-486). (D) Protein expression levels of c-Met and phospho c-Met from BCA2 clone 1h after treatment or not with hrHGF, as measured by Western blot..
Figure 4.
c-Met expression on human CTL clones and modulation of killing capacity by HGF in vitro. (A) c-Met mRNA expression by qRT-PCR from CTL clones isolated from cancer patients compared to Hacat and T2 cell lines, used as positive and negative controls respectively. (B) Flow cytometry histograms showing c-Met protein expression (light gray) on CTL clones and control cell lines compared to the isotype control (dark gray). Percentage of c-Met+ CTLs are indicated. (C) In vitro killing assays were performed using DELFIA® cell cytotoxicity kit (PerkinElmer). T2 target cells were cultured with indicated ratio of CTL clones in presence (squares) or not (triangles) of the cognate peptide. Clones were incubated or not with HGF for 72h (black symbols, 30 ng/ml) before use in the assay. MLA: HLA-A2-restricted Melan-A-derived peptide (26-35 A27L), BCA2: HLA-A2-restricted Brevican-derived peptide (478-486). (D) Protein expression levels of c-Met and phospho c-Met from BCA2 clone 1h after treatment or not with hrHGF, as measured by Western blot..
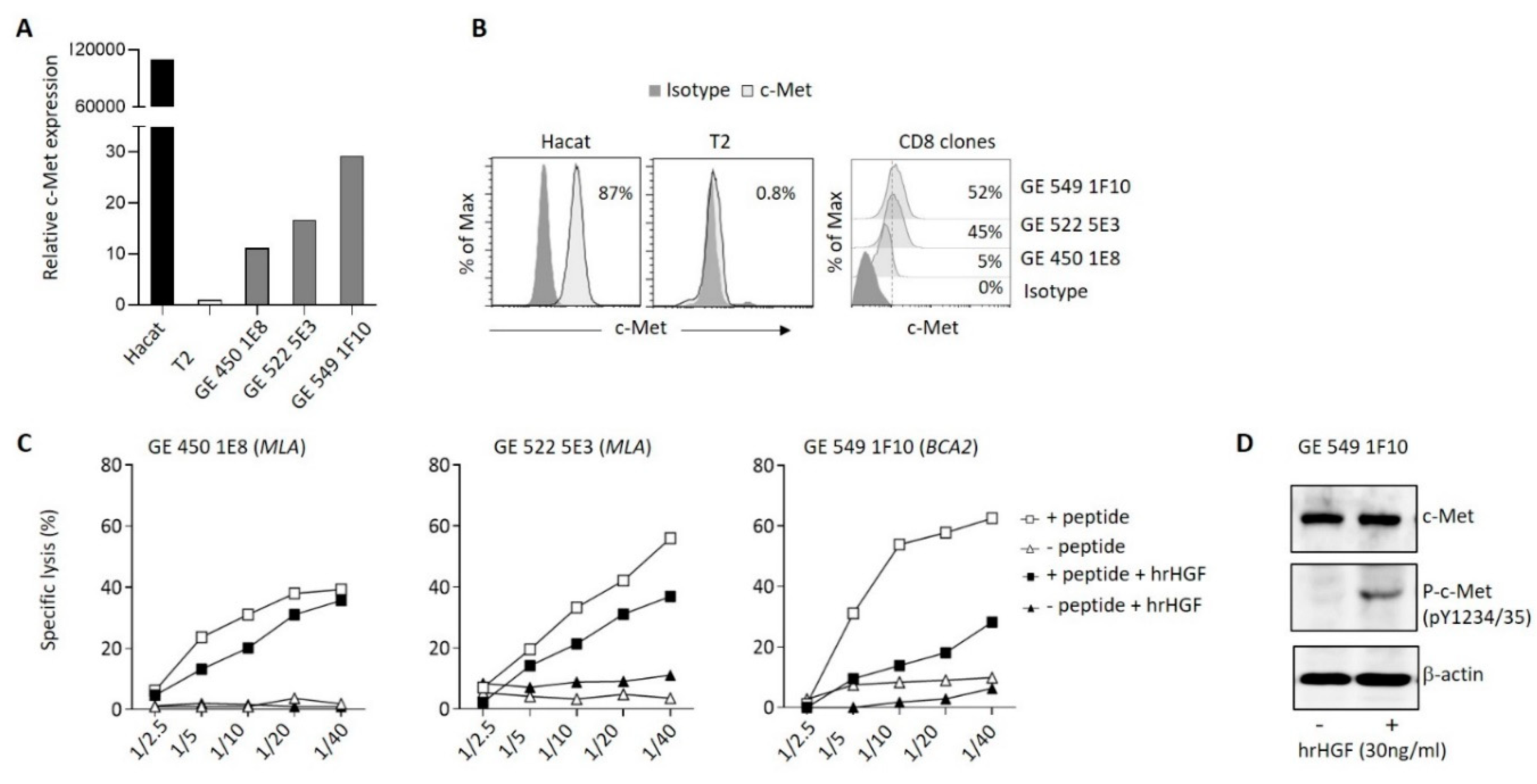
Figure 5.
HGF modulation of GBM-specific clone function. (A) Relative expression of IFNγ, TNFα, GrB, FasL and PD-1 mRNA in the BCA2-specific clone GE 549 1F10 72h after treatment or not with hHGF, as measured by qRT–PCR. Data are representative of three independent experiments of (n=3) carried out in triplicate. Error bars show mean ± SEM, P-values were calculated using unpaired 2-tailed Student’s t-test. (B, C) Representative contour plots of IFNγ and GrB (left panels) and mean frequencies (bar graphs) of the BCA2-specific clone GE 549 1F10 expressing c-Met or not after treatment or not with hrHGF for 72h (right panel). P-values were calculated using paired t-test.
Figure 5.
HGF modulation of GBM-specific clone function. (A) Relative expression of IFNγ, TNFα, GrB, FasL and PD-1 mRNA in the BCA2-specific clone GE 549 1F10 72h after treatment or not with hHGF, as measured by qRT–PCR. Data are representative of three independent experiments of (n=3) carried out in triplicate. Error bars show mean ± SEM, P-values were calculated using unpaired 2-tailed Student’s t-test. (B, C) Representative contour plots of IFNγ and GrB (left panels) and mean frequencies (bar graphs) of the BCA2-specific clone GE 549 1F10 expressing c-Met or not after treatment or not with hrHGF for 72h (right panel). P-values were calculated using paired t-test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



