
Submitted:
04 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
Osteoclasts (OCs) are the multinucleated, bone-resorbing giant cells originally differentiated from a monocyte/macrophage lineage. OC differentiation and maturation, also called osteoclastogenesis, are strictly regulated by a variety of signaling pathways primarily induced by the interactions of two prerequisite cytokines, macrophage colony-stimulating factor (M-CSF) and the receptor activator of nuclear factor-κB ligand (RANKL) to their respective surface receptors, c-fms and RANK, in OC precursors (pre-OCs). Rab11 has emerged as the spatiotemporal regulators of intracellular vesicular transport in the endosomal recycling system; however, how it regulates osteoclastogenesis is incompletely understood. OC-triggered bone resorption is best characterized to be immensely dependent upon lysosomal function, the intracellular acidic organelles containing more than 50 acid hydrolases secreted into bone matrix microenvironment (BME). On the contrary, it is little known about the lysosomal function on modulating the turnover of c-fms and RANK surface receptors via Rab GTPase-mediated vesicular transport, thereby dictating osteoclastogenesis. In this review, I briefly describe the mechanism underlying lysosome-induced osteoclastogenesis via the Rab11-mediated modulation of the surface receptors in OCs.
Keywords:
c-fms
; RANK
; Rab11
; lysosomes and osteoclastogenesis
1. Introduction
Osteoclasts (OCs) that are the multinucleated cells differentiated and formed by the fusion of hematopoietic cells of the monocyte/macrophage lineage are responsible for bone tissue destruction, which is essential for bone remodeling and morphogenesis during development [1,2]. OC differentiation and maturation, also referred to as osteoclastogenesis, is primarily induced by binding of receptor activator of nuclear factor Kappa-B ligand (RANKL) to the cell surface receptor, RANK [3,4]. In principle, this interaction mechanistically stimulates six signaling cascades critical for promoting osteoclastogenesis including (i) nuclear factor of activated T cells cytoplasmic-1 (NFATc-1); (ii) nuclear factor kappa B (NF-кB); (iii) phosphatidylinositol 3-kinase (PI3K/Akt); (iv) Jun N-terminal kinase (JNK); (v) extracellular signal-regulated kinase (ERK); and (vi) p38 mitogen-activated protein kinase (MAPK) [4,5,6,7,8], thereby leading to secretion of the lysosomal enzymes such as, for instance, the tartrate-resistant acid phosphatase (TRAP), Cathepsin K (CTSK), and the matrix metalloproteinase (MMP9), through the ruffle borders into the mineralized bone extracellular matrix (ECM) for bone destruction [9,10]. In addition, binding of the monocyte/macrophage colony stimulating factor (M-CSF), to its specific receptors, c-fms receptors, is also essential for survival and differentiation of pre-mature to mature OCs [11,12].
In order to acidify bone ECM and resorb bone as well, mature OCs firstly attach to bone surface, resulting in establishing a sealing zone (actin ring) required for creating the ruffled borders [13], and secondly secreting lysosomes through the ruffled borders [13]. Interestingly, the proton (H+) pumps and chloride (Cl-) channels are highly expressed in the ruffled borders so as to acidify the resorption lacuna [10,13]; moreover, the lysosomal proteins are abundantly founded at the ruffled borders, suggesting that lysosomes are the major sources for secreting the bone-resorbing components into bone ECM [10]. Specifically, lysosomes break up hydroxyapatite (the mineral component of the bone ECM), the decarboxylation, and osteocalcin (a bone-derived hormone), i.e. Besides, lysosomes synthesize and secrete specific proteases into the resorption lacuna to destruct the organic ECM constituents, mainly comprising the type I collagen (90%) [10]. Generally, lysosomal enzymes required for OC-mediated bone degradation could be functionally branched into 2 classes consisting of (i) the enzymes such as the vacuolar-type H+ATPases (V-ATPases) and chloride (Cl-) channels responsible for acidifying the resorption lacuna and (ii) the proteases such as Cathepsin K (CTSK), the matrix metalloproteinases (MMPs, noticeably MMP9 and MMP13) and tartrate acid-resistant phosphatase (TRAP or ACP5) necessitating the degradation of the bone ECM [9].
The network of intracellular vesicle trafficking amongst subcellular compartments is evolutionarily conserved in eukaryotic cells. Rab GTPases, the largest subfamily of small GTPases (approx. 70 members) in human genome [14], specifically localize to integral membrane of intracellular organelles, and are best characterized to be crucial for delivering cargos to accurate destination via the vesicular delivery system, including budding, motility, docking and fusion of vesicles, cooperatively coupled to receptor signaling pathways [15,16,17]. Dynamic modification of Rab GTPases between inactive (GDP-bound) and active (GTP-bound) forms are catalyzed by specific enzymes. More particularly, guanine exchange factors (GEFs) catalyze conversion of GDP-bound to GTP-bound form to switch on various cellular signals whereas GTPase activating proteins (GAPs) inactivate Rab GTPases via catalyzing GTP hydrolysis of GTP-bound form [15]. Rab11 family, consisting of three members Rab11a, Rab11b and Rab11c/Rab25, accumulated to perinuclear recycling endosomes and post-Golgi vesicles, is cardinal for regulating recycling of endocytosed cargos via the endosomal membrane recycling system. Rab11 deficiency resulted in accumulation of recycling carriers comprising endocytosed transferrin and transferrin receptor beneath the cell surface [18]. Besides, Rab11 connected with recycling carriers moved along microtubules to the cell periphery, and directly regulated vesicle exocytosis at the cell surface in concert with the exocyst [19]. Rab11a is expressed ubiquitously whereas Rab11b is exclusively abundance in the brain, heart and testes. Structurally, Rab11b shares 89% amino acid sequence homology with Rab11a. Their structural differences are based on their C-terminal regions [20]. The active (GTP-bound) state of Rab11 is able to recruit its effector proteins such as Rab11-family of interacting proteins (Rab11FIPs) [21], motor proteins, such as myosin Va/b [22], the octameric exocyst tethering complex subunit Sec15 (also known as EXOC6 in mammals) [23], and heat shock proteins 90 (HSP90) [24,25]. Previous reports have elaborately highlighted the critical roles of Rab GTPases in regulating OC-mediated bone resorption [26,27] and autophagy [26] though still questionably; therefore, it will not be detailed here. In this review, I endeavor to discuss a novel role of Rab11 on modulating the turnover of cell surface receptors through a lysosome-dependent mechanism, therefore contributing to maintain bone remodeling and homeostasis.
2. Cell surface receptors (c-fms and RANK) and osteoclastogenesis
2.1. Colony-stimulating factor 1 receptor (c-fms)
c-fms, also known as CSF1R, is a receptor tyrosine kinase interacting either with macrophage factor (M-CSF) or with IL-34 ligand to activate downstream signaling cascades prerequisite for the survival, function, proliferation and differentiation of myeloid lineage cells, thereby inducing osteoclastogenesis [28]. Consistently, the expression level of c-fms is limited in pre-OCs as compared to this in mature OCs. M-CSF- or IL-34-induced c-fms-mediated signaling activation results in the elevated expression level of receptor activator of NF-кB ligand (RANKL), which is crucial for inducing OC differentiation, maturation and activity. Besides, the M-CSF/c-fms signaling activation also induces the expression of receptor activator of NF-кB (RANK), a specific receptor for RANKL [29]. Genetically, Csf1r gene comprises 21 introns and 22 exons, and located on human chromosome 5 (5q32) [30] and in a syntonic region on mouse chromosome 18 (18D) [31]. To date, a plenty of transcription factors required for the transcriptional activation of Csf1r have been identified such as Ets (the E26 transformation-specific family of transcription factors), PU.1, ATF, C/EBP, RUX, AP-1, IRF, STAT, KLF, REL, and FUS/TLS [32]. c-fms receptor, of which conformation structure is highly conserved in human and mouse, principally consists of two distinct regions: (1) the extracellular domain and (2) the intracellular cytoplasmic domain [33,34]. Of which, the extracellular domain contains immunoglobulin (Ig)-like domains directly binding to their specific ligands, a linker region, and a single-pass trans-membrane helix [11]. In the absence of ligands, c-fms receptor is abundantly present in an inactive auto-inhibitory state. The binding of M-CSF or IL-34 results in c-fms receptor dimerization and subsequent auto-phosphorylation of the dimer in trans on selected tyrosine residues. Specifically, it was reported that six tyrosine residues, comprising Y559, Y697, Y706, Y721, Y807 and Y974, positioned in the c-fms cytoplasmic domain, and two tyrosine residues, positioned in an oncogenic form of c-fms receptor, were phosphorylated, and therefore being as the binding platforms for SH2 and/or PTB domain-containing proteins, which activate the downstream signaling events [35,36].
c-fms receptor-induced signaling pathways are crucial for differentiating pre-OCs into mature OCs. Csf1r-silencing mice or op/op mice exhibit a significant reduction of OCs, disorganized matrix, reduced mineralization, weakened long bones and abnormal OBs [37,38]. During in vitro osteoclastogenesis, c-fms receptor-induced signaling pathways lead to a remarkable increase in the expression of RANK receptors in pre-OCs. In addition to its vital role in proliferating and differentiating pre-OCs into mature OCs, c-fms-mediated signaling also regulates OC motility and cytoskeletal reorganization via Tyr559 phosphorylation-induced c-Src-dependent manner [39]. Furthermore, crosstalk between c-fms and other specific molecules such as integrin β3 and DAP12 contributes to regulate cytoskeletal reorganization and adhesion of mature OCs. Integrin β3 is induced by RANKL-mediated stimulation and binds to extracellular matrix proteins such as vitronectin, osteoponin, and bone sialoprotein [40,41]. Indeed, it was revealed that integrin β3-deficient mice exhibited remarkably dysfunctional OCs and simultaneously developed an osteoclerotic phenotype [41], and enhancement of M-CSF/c-fms-mediated signaling rescued the defectively functional OCs in integrin β3-deficient mice [41]. Besides, it was shown that DAP12 that is an adaptor molecule containing an immunoreceptor tyrosine-based activation motif (ITAM) was indispensable for osteoclastogenesis. Activation of DAP12-mediated c-fms receptor triggered ITAM phosphorylation mediated by Src family kinases, thereby contributing to regulate cytoskeletal reorganization in OCs [42].
2.2. RANK receptors
As above-mentioned, the binding of RANKL to its receptor RANK, which is encoded by Tnfrsf11a, leads to generate downstream RANKL-RANK signaling cascades, which trigger pre-OCs to differentiate into mature OCs [4]. RANKL is expressed in two distinct forms including (1) a membrane-bound form expressed in cell surface of osteoblasts (OBs) and a soluble form. The membrane-bound RANKL is cleaved into the soluble form by metalloproteinases such as matrix metalloproteinase (MMP)-14 [43,44]. Although both forms function as agonistic ligands for RANK receptor, the membrane-bound RANKL is believed to be more efficient. Mice with the genetic deletions of Tnfsf11 or Tnfsf11a exhibit severe osteopetrosis accompanied by defective tooth eruption owing to the mortality of their OCs [43,45]. On the other hand, RANKL-RANK signaling is also suppressed by Tnfsf11b- encoded osteoprotegerin, which is a soluble decoy receptor for RANKL preventing RANKL from binding to RANK receptor [4]. RANK is a member of the tumor necrosis factor receptor (TNFR) superfamily that lacks intrinsic enzymatic activity required for directly activating its downstream signaling molecules. Consequently, following the RANKL-RANK interaction, RANK transduces intracellular signals by recruiting adaptor molecules such as TNFR-associated factors (TRAFs), which then trimerize cytosolic tail of RANK receptor, and activate MAPKs, NF-кB, activator protein-1 (AP-1) and finally nuclear factor of activated T-cells cytosolic 1 (NFATc1), resulting in the commitment of monocyte/macrophage precursor cells to the OC lineage and the activation of mature OCs [7]. In the late stage of OC differentiation, the osteoclastogenic genes required for bone resorption are transcribed by NFATc1 [7]. More particularly, activation of TRAF6 leads to upregulate the NF-кB transcriptional activity by enhancing IкB kinase (IKK) complex that is mediated by either atypical protein kinase C (PKC) or TGFβ-activated kinase 1 (TAK1)-dependent phosphorylation [46]. The interaction of scaffolding protein p62 with TRAF6 and aPKCs brings out the establishment of a multimeric protein complex that regulates IKKβ [47]. In addition, TRAF6 also forms complexes with TAK1 and the adaptor proteins TAK1-binding protein 1 (TAB1) and TAB2, which enables TAK1 to phosphorylate IKK complex and subsequently activate NF-κB downstream signaling cascades [48]. It is best characterized that NF-κB signaling is indispensable for osteoclastogenesis. That is, NF-κB p50/p52 double-knockout mice exhibit considerable osteopetrosis owing to the deterioration in OC formation [49]. AP-1 transcription factor comprising Fos (c-Fos, FosB, Fra-1, and Fra-2), Jun (c-Jun, JunB, and JunD), and ATF (ATFa, ATF, ATF2, ATF4, and B-ATF) family members is also activated via the induction of c-Fos by the adaptor proteins [50]. c-Fos knockout mice and transgenic mice that overexpress dominant negative c-Jun exhibit severe osteopetrosis, indicating that the transcription factors, NF-κB and AP-1, are crucial for activating the downstream targets of RANKL signaling pathway at the early stage of OC differentiation [7]. Furthermore, the recruitment of adaptor proteins activates MAPKs such as c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase, and Akt/PKB [51].
3. Rab11, lysosomes and osteoclastogenesis
3.1. Lysosomes
As mentioned above, mature OCs are responsible for resorbing the bone extracellular matrix (ECM). Bone destruction highly depends upon lysosomes, membrane-enclosed organelles containing an array of approx. 50 hydrolases and are capable of breaking down all types of biological polymers including nucleic acids, carbohydrates and lipids [52]. Several published articles including ours thoroughly described the functional roles of lysosomes on in mediating OC-induced bone resorption [10,52,53]; therefore, it should not be mentioned in this review.
3.2. The axis of c-fms and RANK receptors-Rab GTPases-Lysosomes
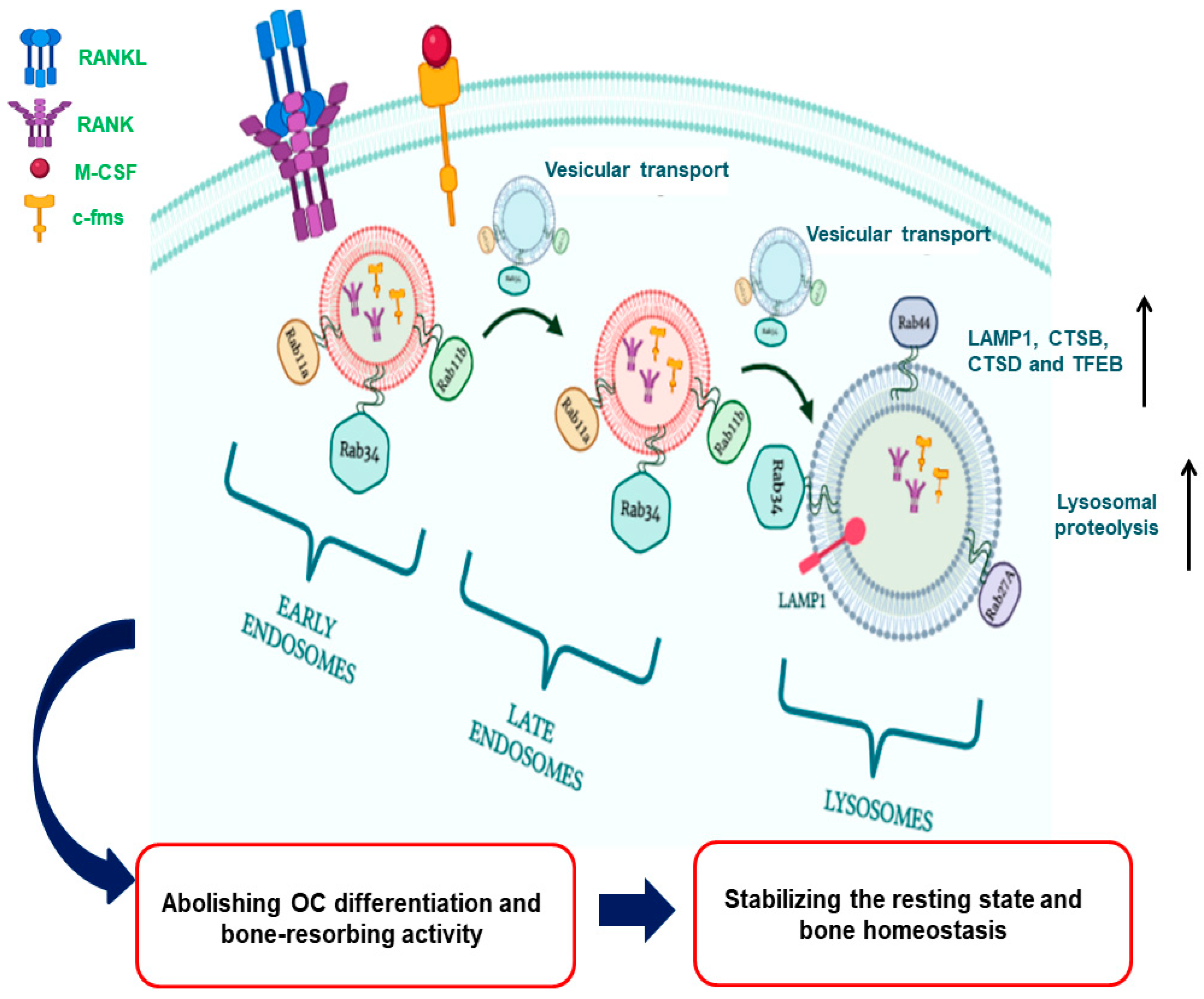
As mentioned above, osteoclastogenesis is essentially regulated by RANK/c-fms-mediated signaling pathways, following the binding of specific ligands to these receptors. Besides, it is well-known that the ligand-induced stimulation of c-fms/RANK receptors promotes lysosomal function that is crucial for bone resorption; however, little evidence unravels how lysosomes provide a feedback to diminish the turnover of these receptors on cell surface to debilitate osteoclastogenesis and bone-resorbing activity thereof, thereby stabilizing the resting state and maintaining bone homeostasis. In our previous studies, it was obvious that Rab11a and Rab11b seemed not to be involved in the regulation of the expression of c-fms and RANK receptors via the endosomal membrane recycling system, but via the endosomal-lysosomal system in OCs [54,55]. To date, two endosomal recycling pathways described are fast and slow recycling routes. Of which, transferrin (TfR) receptor is a representative candidate recycled to cell surface via the slow recycling pathways; however, we demonstrated that Rab11a-mediated transport route of c-fms and RANK surface receptors is distinct from that of TfR receptor [55]. Instead of it, it was interestingly noted that Rab11a and Rab11b upregulated during OC differentiation caused size-based enlargement of early and late endosomes and enhanced lysosomal activity, prompting us that Rab11a and Rab11b are involved in cargo internalization into early and late endosomes, and mediate the vesicular transport of internalized cargos to lysosomes via the axis of early endosomes-late endosomes-lysosomes in OCs. Later, inhibiting lysosomal activity by a specific inhibitor, chloroquine (CLQ), we revealed that lysosomes play a crucial role in degrading these receptors in OCs, and Rab11a and Rab11b accelerate the delivery of internalized cargo to lysosomes for their proteolysis [54,55]. Additionally, later we revealed Rab34 also participated in regulating the expression of c-fms and RANK receptors; however, it is not clear whether Rab11 and Rab34 regulate vesicular transport of these receptors independently or independently [56] to lysosomes via this axis (Figure 1). From these observations, we propose a regulatory mechanism that lysosomes play a bi-directional role in (1) regulating bone resorption via secreting specific proteases such as TRAP, Cathepsin K and MMP9 into BME [24,25], and (2) reversing osteoclastogenesis via abolishing the surface expression of c-fms and RANK receptors in OCs, thereby balancing bone remodeling.
4. Discussion
In this review, we briefly described a novel effect of lysosomes on degrading the cell surface receptors (c-fms and RANK receptors), and Rab11 play a housekeeping role in facilitating lysosome-induced degradation of these receptors in OCs, thereby stabilizing osteoclastogenesis and bone remodeling.
Funding
Research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest
References
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. Journal of Cell Science 2000, 113, 377-381. [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337-342. [CrossRef]
- Tobeiha, M.; Moghadasian, M.H.; Amin, N.; Jafarnejad, S. RANKL/RANK/OPG Pathway: A Mechanism Involved in Exercise-Induced Bone Remodeling. Biomed Res Int 2020, 2020, 6910312. [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 2008, 473, 139-146. [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 2006, 12, 17-25. [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab 2021, 39, 19-26. [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol Cells 2017, 40, 706-713. [CrossRef]
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Suzuki, H. Regulatory mechanisms of RANKL presentation to osteoclast precursors. Curr Osteoporos Rep 2014, 12, 115-120. [CrossRef]
- Vääräniemi, J.; Halleen, J.M.; Kaarlonen, K.; Ylipahkala, H.; Alatalo, S.L.; Andersson, G.; Kaija, H.; Vihko, P.; Väänänen, H.K. Intracellular machinery for matrix degradation in bone-resorbing osteoclasts. J Bone Miner Res 2004, 19, 1432-1440. [CrossRef]
- Lacombe, J.; Karsenty, G.; Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 2013, 12, 2744-2752. [CrossRef]
- Mun, S.H.; Park, P.S.U.; Park-Min, K.-H. The M-CSF receptor in osteoclasts and beyond. Experimental & Molecular Medicine 2020, 52, 1239-1254. [CrossRef]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. cmj 2016, 52, 12-17. [CrossRef]
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. International Journal of Molecular Sciences 2018, 19, 984.
- Bhuin, T.; Roy, J.K. Rab proteins: the key regulators of intracellular vesicle transport. Exp Cell Res 2014, 328, 1-19. [CrossRef]
- Zhen, Y.; Stenmark, H. Cellular functions of Rab GTPases at a glance. J Cell Sci 2015, 128, 3171-3176. [CrossRef]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr Opin Cell Biol 2013, 25, 414-419. [CrossRef]
- Lürick, A.; Gao, J.; Kuhlee, A.; Yavavli, E.; Langemeyer, L.; Perz, A.; Raunser, S.; Ungermann, C. Multivalent Rab interactions determine tether-mediated membrane fusion. Mol Biol Cell 2017, 28, 322-332. [CrossRef]
- Takahashi, S.; Kubo, K.; Waguri, S.; Yabashi, A.; Shin, H.W.; Katoh, Y.; Nakayama, K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J Cell Sci 2012, 125, 4049-4057. [CrossRef]
- Takahashi, S.; Kubo, K.; Waguri, S.; Yabashi, A.; Shin, H.-W.; Katoh, Y.; Nakayama, K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. Journal of Cell Science 2012, 125, 4049-4057. [CrossRef]
- Scapin, S.M.N.; Carneiro, F.R.G.; Alves, A.C.; Medrano, F.J.; Guimarães, B.G.; Zanchin, N.I.T. The crystal structure of the small GTPase Rab11b reveals critical differences relative to the Rab11a isoform. Journal of Structural Biology 2006, 154, 260-268. https://doi.org/.
- Baetz, N.W.; Goldenring, J.R. Rab11-family interacting proteins define spatially and temporally distinct regions within the dynamic Rab11a-dependent recycling system. Molecular Biology of the Cell 2013, 24, 643-658. [CrossRef]
- Lapierre, L.A.; Kumar, R.; Hales, C.M.; Navarre, J.; Bhartur, S.G.; Burnette, J.O.; Provance, D.W., Jr.; Mercer, J.A.; Bähler, M.; Goldenring, J.R. Myosin vb is associated with plasma membrane recycling systems. Mol Biol Cell 2001, 12, 1843-1857. [CrossRef]
- Zhang, X.M.; Ellis, S.; Sriratana, A.; Mitchell, C.A.; Rowe, T. Sec15 is an effector for the Rab11 in mammalian cells. J Biol Chem 2004, 279, 43027-43034. [CrossRef]
- Tran, M.T.; Okusha, Y.; Htike, K.; Sogawa, C.; Eguchi, T.; Kadowaki, T.; Sakai, E.; Tsukuba, T.; Okamoto, K. HSP90 drives the Rab11a-mediated vesicular transport of the cell surface receptors in osteoclasts. Cell Biochem Funct 2022, 40, 838-855. [CrossRef]
- Tran, M.T.; Okusha, Y.; Feng, Y.; Sogawa, C.; Eguchi, T.; Kadowaki, T.; Sakai, E.; Tsukuba, T.; Okamoto, K. A novel role of HSP90 in regulating osteoclastogenesis by abrogating Rab11b-driven transport. Biochim Biophys Acta Mol Cell Res 2021, 1868, 119096. [CrossRef]
- Roy, M.; Roux, S. Rab GTPases in Osteoclastic Bone Resorption and Autophagy. International Journal of Molecular Sciences 2020, 21, 7655.
- Roy, M.; Roux, S. Rab GTPases in Osteoclastic Endomembrane Systems. Biomed Res Int 2018, 2018, 4541538. [CrossRef]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. Journal of Leukocyte Biology 2016, 100, 481-489. https://doi.org/.
- Arai, F.; Miyamoto, T.; Ohneda, O.; Inada, T.; Sudo, T.; Brasel, K.; Miyata, T.; Anderson, D.M.; Suda, T. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 1999, 190, 1741-1754. [CrossRef]
- Le Beau, M.M.; Pettenati, M.J.; Lemons, R.S.; Diaz, M.O.; Westbrook, C.A.; Larson, R.A.; Sherr, C.J.; Rowley, J.D. Assignment of the GM-CSF, CSF-1, and FMS genes to human chromosome 5 provides evidence for linkage of a family of genes regulating hematopoiesis and for their involvement in the deletion (5q) in myeloid disorders. Cold Spring Harb Symp Quant Biol 1986, 51 Pt 2, 899-909. [CrossRef]
- Hoggan, M.D.; Halden, N.F.; Buckler, C.E.; Kozak, C.A. Genetic mapping of the mouse c-fms proto-oncogene to chromosome 18. J Virol 1988, 62, 1055-1056. [CrossRef]
- Rojo, R.; Pridans, C.; Langlais, D.; Hume, D.A. Transcriptional mechanisms that control expression of the macrophage colony-stimulating factor receptor locus. Clin Sci (Lond) 2017, 131, 2161-2182. [CrossRef]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol 2014, 6. [CrossRef]
- Liu, H.; Leo, C.; Chen, X.; Wong, B.R.; Williams, L.T.; Lin, H.; He, X. The mechanism of shared but distinct CSF-1R signaling by the non-homologous cytokines IL-34 and CSF-1. Biochim Biophys Acta 2012, 1824, 938-945. [CrossRef]
- Yu, W.; Chen, J.; Xiong, Y.; Pixley, F.J.; Yeung, Y.G.; Stanley, E.R. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J Biol Chem 2012, 287, 13694-13704. [CrossRef]
- Mancini, A.; Niedenthal, R.; Joos, H.; Koch, A.; Trouliaris, S.; Niemann, H.; Tamura, T. Identification of a second Grb2 binding site in the v-Fms tyrosine kinase. Oncogene 1997, 15, 1565-1572. [CrossRef]
- Dai, X.M.; Zong, X.H.; Akhter, M.P.; Stanley, E.R. Osteoclast deficiency results in disorganized matrix, reduced mineralization, and abnormal osteoblast behavior in developing bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2004, 19, 1441-1451. [CrossRef]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442-444. [CrossRef]
- Insogna, K.L.; Sahni, M.; Grey, A.B.; Tanaka, S.; Horne, W.C.; Neff, L.; Mitnick, M.; Levy, J.B.; Baron, R. Colony-stimulating factor-1 induces cytoskeletal reorganization and c-src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J Clin Invest 1997, 100, 2476-2485. [CrossRef]
- Nakamura, I.; Duong, L.T.; Rodan, S.B.; Rodan, G.A. Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab 2007, 25, 337-344. [CrossRef]
- McHugh, K.P.; Hodivala-Dilke, K.; Zheng, M.H.; Namba, N.; Lam, J.; Novack, D.; Feng, X.; Ross, F.P.; Hynes, R.O.; Teitelbaum, S.L. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest 2000, 105, 433-440. [CrossRef]
- Faccio, R.; Zou, W.; Colaianni, G.; Teitelbaum, S.L.; Ross, F.P. High dose M-CSF partially rescues the Dap12-/- osteoclast phenotype. J Cell Biochem 2003, 90, 871-883. [CrossRef]
- Tsuda, E.; Goto, M.; Mochizuki, S.; Yano, K.; Kobayashi, F.; Morinaga, T.; Higashio, K. Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun 1997, 234, 137-142. [CrossRef]
- Nakashima, T.; Kobayashi, Y.; Yamasaki, S.; Kawakami, A.; Eguchi, K.; Sasaki, H.; Sakai, H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun 2000, 275, 768-775. [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175-179. [CrossRef]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep 2014, 15, 46-61. [CrossRef]
- McManus, S.; Roux, S. The adaptor protein p62/SQSTM1 in osteoclast signaling pathways. J Mol Signal 2012, 7, 1. [CrossRef]
- Kishida, S.; Sanjo, H.; Akira, S.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1-binding protein 2 facilitates ubiquitination of TRAF6 and assembly of TRAF6 with IKK in the IL-1 signaling pathway. Genes Cells 2005, 10, 447-454. [CrossRef]
- Jimi, E.; Katagiri, T. Critical Roles of NF-κB Signaling Molecules in Bone Metabolism Revealed by Genetic Mutations in Osteopetrosis. Int J Mol Sci 2022, 23. [CrossRef]
- Garces de Los Fayos Alonso, I.; Liang, H.C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers (Basel) 2018, 10. [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 2011, 75, 50-83. [CrossRef]
- Tsukuba, T.; Sakai, E.; Nishishita, K.; Kadowaki, T.; Okamoto, K. New functions of lysosomes in bone cells. Journal of Oral Biosciences 2017, 59, 92-95. https://doi.org/.
- Toyomura, T.; Murata, Y.; Yamamoto, A.; Oka, T.; Sun-Wada, G.-H.; Wada, Y.; Futai, M. From Lysosomes to the Plasma Membrane: LOCALIZATION OF VACUOLAR TYPE H+-ATPase WITH THE a3 ISOFORM DURING OSTEOCLAST DIFFERENTIATION*. Journal of Biological Chemistry 2003, 278, 22023-22030. https://doi.org/.
- Tran, M.T.; Okusha, Y.; Feng, Y.; Morimatsu, M.; Wei, P.; Sogawa, C.; Eguchi, T.; Kadowaki, T.; Sakai, E.; Okamura, H.; et al. The Inhibitory Role of Rab11b in Osteoclastogenesis through Triggering Lysosome-Induced Degradation of c-Fms and RANK Surface Receptors. International Journal of Molecular Sciences 2020, 21, 9352.
- Okusha, Y.; Tran, M.T.; Itagaki, M.; Sogawa, C.; Eguchi, T.; Okui, T.; Kadowaki, T.; Sakai, E.; Tsukuba, T.; Okamoto, K. Rab11A Functions as a Negative Regulator of Osteoclastogenesis through Dictating Lysosome-Induced Proteolysis of c-fms and RANK Surface Receptors. Cells 2020, 9, 2384.
- Feng, Y.; Tran, M.T.; Lu, Y.; Htike, K.; Okusha, Y.; Sogawa, C.; Eguchi, T.; Kadowaki, T.; Sakai, E.; Tsukuba, T.; et al. Rab34 plays a critical role as a bidirectional regulator of osteoclastogenesis. Cell Biochem Funct 2022, 40, 263-277. [CrossRef]
Figure 1.
The graphical abstract for the role of Rab11a and Rab11b in regulating lysosomal degradation of c-fms and RANK surface receptor through facilitating the vesicular transport of these receptors to lysosomes via the axis of early endosomes (EEs)- late endosomes (LEs)- lysosomes (L) in OCs, following the binding of ligands M-CSF and RANKL to c-fms and RANK receptors, respectively. It triggers an elevation of lysosomal enzymatic activity via increasing the expression of LAMP1, CTSB and CTSD, thereby debilitating the secretion of lysosomal proteases such as TRAF, CTSK and MMP9 into BME, abolishing the bone-resorbing activity of OCs, and eventually stabilizing the resting state and maintaining bone homeostasis.
Figure 1.
The graphical abstract for the role of Rab11a and Rab11b in regulating lysosomal degradation of c-fms and RANK surface receptor through facilitating the vesicular transport of these receptors to lysosomes via the axis of early endosomes (EEs)- late endosomes (LEs)- lysosomes (L) in OCs, following the binding of ligands M-CSF and RANKL to c-fms and RANK receptors, respectively. It triggers an elevation of lysosomal enzymatic activity via increasing the expression of LAMP1, CTSB and CTSD, thereby debilitating the secretion of lysosomal proteases such as TRAF, CTSK and MMP9 into BME, abolishing the bone-resorbing activity of OCs, and eventually stabilizing the resting state and maintaining bone homeostasis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



