
Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
Breast cancer (BC) is the most common malignancy among women worldwide. In recent years, significant progress has been made in BC therapy. However, serious side effects resulting from the use of standard chemotherapeutic drugs, as well as the phenomenon of multidrug resistance (MDR), limit the effectiveness of approved therapies. Advanced research in the BC area is necessary to create more effective and safer forms of therapy to improve the outlook for individuals diagnosed with this aggressive neoplasm. For decades, plants and natural products with anti-cancer properties have been successfully utilized in treating various medical conditions. Anthracene derivatives are tricyclic secondary metabolites of natural origin that have been identified in plants, lichens, and fungi. They represent a few botanical families, e.g., Rhamnaceae, Rubia-ceae, Fabaceae, Polygonaceae, and others. The review comprehensively covers and analyzes the most recent advances in the anticancer activity of anthracene derivatives (emodin, aloe-emodin, hypericin, chrysophanol, rhein, and physcion) applied both individually, or in combination with other chemotherapeutic agents in in vitro and in vivo BC models.
Keywords:
anthracene derivatives
; breast cancer
; natural products
; biological activity
; emodin
; aloe-emodin
; hypericin
; chrysophanol
; rhein
; physcion
1. Introduction
Cancer is one of the most common causes of death in the world [1]. This diverse disease is a multi-stage process that is initiated by genetic changes occurring in normal cells, leading to their transformation into cancer cells. Transformed cancer cells are characterized by uncontrolled growth, the ability to invade nearby tissues, and to create distant metastases [2]. Despite significant progress in cancer treatment, including the emergence of new molecule-based therapies, treatment options for cancer patients remain limited due to serious adverse effects and the phenomenon of multidrug resistance (MDR) [3]. Bioactive substances of natural origin may interfere with the process of carcinogenesis by modifying the behavior of neoplastic cells and affecting signaling pathways that have been improperly activated or inhibited [2].
Anthracene and its derivatives have been the subject of extensive research over the years due to their interesting photochemical and bio-activities [4]. They are present in Liliaceae, Fabaceae, Rubiaceae, Polygonaceae, Rhamnaceae, and Scrophulariaceae families. Within the various chemical classes of natural products, anthraquinones are characterized by their extensive structural diversity, significant biological activity, and relatively low toxicity. It has been demonstrated that anthracene derivatives display anti-inflammatory, immunomodulatory, or antineoplastic activities. Anthraquinones inhibited growth of breast, lung, colon, prostate, cervix, or leukemia cancer cells [5].
The review comprehensively summarizes the most recent advances in the anticancer activity of anthracene derivatives (emodin, aloe-emodin, hypericin, chrysophanol, rhein, and physcion) used both individually, or in combination with other chemotherapeutic agents in in vitro and in vivo breast cancer (BC) models.
2. Molecular Basis of Breast Cancer
Breast cancer (BC) is the most common malignancy among women worldwide with approximately 2.3 million new cases detected in 2020 (GLOBOCAN 2020). Unfortunately, the rates of BC incidence and mortality are still rising [6,7].
Classical immunohistochemistry markers, such as estrogen (ER), progesterone (PR), and human epidermal growth factor (HER2) receptor expression, along with clinicopathological factors like tumor size, grade, and nodal involvement, have traditionally been used for therapy selection and predicting disease progression. However, the widespread use of high-throughput gene expression analysis techniques has revealed that the response of cancer cells to treatment is not solely determined by anatomical prognostic factors but rather by the internal molecular characteristics of BC. Therefore, five distinct molecular subtypes of BC have been identified, including luminal A, luminal B, HER2-overexpressed, triple-negative breast cancer (TNBC), and normal-like (Figure 1) [8].
BCs classified as luminal A exhibit expression of ERs and/or PRs, lack of HER2 expression and low value of Ki-67 cell proliferation index. This subtype of BC depicts a positive response to endocrine therapy and generally has the best prognosis [9]. In turn, luminal B BC (ER+ and/or PR+, HER2+/-) is associated with a less favorable prognosis than the luminal A subtype and presents a higher proliferating index value. Hormonal therapy along with chemotherapy can be beneficial for the treatment of patients suffering from these tumors [10,11]. The HER2-positive subtype accounts for approximately 10-15% of BCs and is characterized by HER2 overexpression alongside the absence of ER and PR. These tumors tend to grow more rapidly than the luminal subtypes, but the prognosis has significantly improved with the introduction of HER2-targeted therapies [11]. TNBC is defined as a type of BC with a lack of ER, PR, and HER2 protein expression. TNBC represents the most aggressive subtype of BC and is associated with a poor prognosis. Currently, treatment options for TNBC are primarily restricted to surgical intervention, adjuvant chemotherapy, and radiotherapy [12].
One of the most important challenges associated with conventional treatments are the serious side effects and MDR phenomenon. Advanced research in the BC area is necessary to create more effective and safer forms of therapy to improve the outlook for individuals diagnosed with this aggressive neoplasm [12]. Numerous research efforts are now exclusively dedicated to discovering alternative forms of treatment (including natural products) for BC [13]. Promising compounds with proven anti-cancer properties are anthracene derivatives.
3. Characteristics of Anthracene Derivatives
Anthracene derivatives are tricyclic secondary metabolites of natural origin which have been identified in plants, lichens and funghi [14]. They were known since the Antiquity as natural pigments. More than 700 structures of these coloured molecules have been described so far, with around one-third in different organs of plants: flowers, fruits, roots or rhizomes [15] representing a few botanical families, like Rhamnaceae, Rubiaceae, Fabaceae, Polygonaceae [5] and others (Table 1).
Anthracene derivatives may occur in a reduced or oxidized form. The former are less diverse and are known to be present in fresh plant material as anthrons or anthranols. In turn, oxidized forms of anthracene derivatives, namely anthraquinones form a very diverse group of metabolites that are also milder in action. They can be present in fresh plant material, but also, they are formed during drying at 105 C degrees or storage of bioactive plant material for 1-2 years from the reduced forms. Anthraquinones – the largest group of anthracene derivatives - in their chemical structure contain an anthracene moiety that is substituted with two ketone groups at the positions 9 and 10 forming a 9,10-dioxoanthracene or 9,10-anthracenedione (Figure 2a). Additionally, some structures may contain two hydroxyl groups at C-1 and C-8 carbon atoms whose presence induces laxative effects of the compounds. Due to the solubility issues, these metabolites are often present in glycosylated forms in plants to be better dissolved in aqueous solutions of plant cells [16].
Two metabolic pathways are proposed for the biosynthesis of anthraquinones. One of them includes the cyclization of octa-β-ketoacyl-CoA that is produced by the addition of acetyl-coenzyme A to three groups of malonyl-coenzyme A [17]. The other is related to the shikimic acid pathway where an addition of succinoylbenzoic acid to mevalonic acid occurs [18].
In the scientific literature, the 1,8-dixydroxyanthraquinones (1,8-OH-AQ) that were proven to exhibit laxative effects and used in traditional medicine are the most recognizable compounds that belong to anthracene derivatives. Differently substituted aglycons of 1,8-OH-AQ can be divided into a few subgroups. So far, the derivatives of emodin, aloe-emodin, physcion, chrysophanol, and rhein have been described in the most thorough manner (Figure 3) [17,18].
An interesting example of a polymeric 1,8-OH-AQ is hypericin. This compound contains two anthracene moieties that are stacked at each other forming a structure of naftodianthrone by means of oxidative coupling, with additional substituents like hydroxyl or ketone groups. Hypericin and their derivatives, like pseudohypericin or isohypericin are strong photosensitizers and play a pivotal role in anticancer photodynamic therapy (Figure 2b) [19].
Because of these characteristics, anthraquinones as interesting molecules have attracted the attention of chemists who elaborated the total synthesis of many derivatives of naturally occurring structures. Many of them can be now used as chemotherapeutics in the treatment of cancer. Among the approved drugs with elaborated synthetic production, we can list doxorubicin, daunorubicin, idarubicin, epirubicin, valrubicin, mitoxantrone, pixantrone, and others [19].
Considering the structural differences between the anthracene derivatives and the number of publications on their anticancer activity, the aim of this review is to put together the information on 1,8-dihydroxyanthraquinones as potential drug candidates for the treatment of BC, their mechanisms of action, and potential synergistic effects with other commercially available chemotherapeutics. This subgroup of anthracene derivatives is well-studied and certainly deserves attention in terms of its anticancer properties.
Further below a detailed biological potential of emodin, aloe-emodin, hypericin, chrysophanol, rhein, and physcion was presented.
4. Anticancer Activity of Anthracene Derivatives in Breast Cancer In Vitro Models
4.1. Emodin
Of all anthraquinones, the use of emodin in BC therapy has been most extensively described in the scientific literature. In vitro studies confirmed anticancer properties of emodin towards several types of human BC cell lines, including BCap-37 [20], MCF7 [21], MDA-MB-453 [22], MDA-MB-231 [23], and GILM2 human BC cells obtained from lung metastasis [24]. The use of different types of cell models helped uncover the molecular mechanisms behind the therapeutic potential of emodin in the treatment of BC.
Emodin has been characterized as a strong proapoptotic agent. It has been showing that the treatment of the human BCaP-37 BC cell line with emodin at 20 and 50 µM for 48 h induced morphological characteristics for apoptosis, decreased in Bcl-2/Bax ratio, and increased cytosolic cytochrome c concentration. These changes indicate the involvement of the mitochondrial signaling pathway in the emodin-induced apoptosis [20]. Subsequent studies by the same researchers revealed that emodin regulates the expression of about 30 specific genes in BCap-37 cells, including insulin-like growth factor 2 (IGF-2, downregulated by emodin) and protein p21 (upregulated by emodin) [25]. In BCap-37 and ZR-75-30 BC cell lines emodin reduced the expression of Bcl-2 (B-cell CLL/lymphoma 2) and increased levels of cleaved caspase-3, PARP (poly (ADP-ribose) polymerase), p53 and Bax, resulting in dose- and time-dependent proapoptotic effects [26].
In MDA-MB-231 cells emodin was significantly cytotoxic at concentrations of 10-80 µM following 24, 48 and 72h treatment. Emodin has also been shown to dose-dependently inhibit the migration and invasion of MDA-MB-231 cells and thus may be considered effective in the prevention of BC metastasis. Enzyme-linked immunosorbent assay (ELISA) and Western blot analyses revealed that the anti-metastatic potential of emodin results from the downregulation of proteolytic enzymes involved in the degradation of extracellular matrix components, triggering metastasis. Emodin at 40 and 80 µM decreased the production of matrix metalloprotease (MMP) 2 and 9, uPA and uPAR in MDA-MB-231 cells and reduced the levels of p38 and ERK kinases [23].
Emodin showed also significant growth inhibitory effects on MCF-7 cells with IC₅₀ = 7.22 µg/ml (∼30 μM) and exerted a concentration-dependent inhibitory effect on the colony-forming ability of MCF-7 cells with IC₅₀ = 7.60 µg/ml (∼30 µM). Single-strand DNA breakage and DNA fragmentation, considered as hallmarks of apoptosis, were observed in emodin-treated MCF-7 cells. Proapoptotic effect of emodin is more likely mediated through modulation of the expression of apoptosis-related genes, such as Fas ligand (FasL), myeloid cell leukemia sequence 1 (MCL1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Bcl2-associated X protein (Bax), cyclin D1 (CCND1) and v-myc myelocytomatosis viral oncogene homolog (C-MYC). The 72-h emodin treatment significantly upregulated the expression of FASL and down-regulated the expression of MCL1, CCND1 and C-MYC genes as compared with untreated control cells [27]. Anticancer properties of emodin are strongly dependent on its tyrosine kinase inhibitory potential. Zhang et al. described that emodin could act as a tyrosine kinase inhibitor, decrease the activity of HER-2/neu tyrosine kinase in MDA-MB-453 cells, inhibit the growth of cancer cells, induce the production of lipid droplets, and promote the mature differentiation of BC cells. One of the emodin derivatives 10-(4-acetamidobenzylidene)-9-anthrone (DK-V-47) was more effective than emodin in repressing the tyrosine phosphorylation of p185neu and in inhibiting the proliferation and transformation of HER-2/neu-overexpressing human BC cells. DK-V-47 was also more potent than emodin in suppressing transformation phenotypes of activated HER-2/neu transformed 3T3 fibroblasts, including anchorage-dependent and -independent growth and metastasis-associated properties. These results clearly indicated that the inhibition of p185neu tyrosine kinase by both emodin and DK-V-47 suppressed the HER-2/neu associated phenotype of BC cells, including their ability to metastasize [28]. The same group has reported that the combination of emodin (20 µM) and paclitaxel (1 µM) synergistically inhibited the anchorage-dependent and -independent growth of HER-2/neu-overexpressing MDA-MB-361, BT-474, MDA-MB-231 and MDA-MB-435 BC cells in vitro. The mechanism is related to the reduction of tyrosine phosphorylation of HER-2/neu, suggesting that HER-2/neu inhibition is one of the important approaches of emodin in BC treatment [29].
Emodin also sensitizes HER2/neu-overexpressing cancer cells to chemotherapeutic agents, including cisplatin, doxorubicin, etoposide, and paclitaxel [30]. Emodin also effectively inhibited the growth of MDA-MB-435 cells with low HER-2/neu expression by decreasing tyrosine kinase [31]. Recently it has also been confirmed that emodin significantly reduced the phosphorylation levels of ERK1/2 and AKT but not p38 MAPK in MDA-MB-231 cancer cells. Emodin inhibited BC cell proliferation and invasion through the serine/threonine kinase (AKT) signaling and extracellular-regulated protein kinase (ERK) pathways [32].
Using virtual screening, Zhang et al. found that emodin is an effective aromatic hydrocarbon receptor (AhR) agonist. Subsequent in vitro experiments also found that the expression levels of AhR and cytochrome P450 1A1 (CYP1A1) in MCF-7 cells were significantly upregulated by emodin treatment, suggesting that the antitumor effects of emodin against BC might be related also to the AhR-CYP1A1 signaling pathway [33].
Emodin has been implicated in the regulation of estrogen signaling in BC cells. Sui et al. found that emodin inhibited estrogen-induced proliferation of MCF-7 and MDA-MB-231 cells, promoted apoptosis and arrested the cell cycle in the G0/G1 phase by downregulating the expression of cyclin D1 Bcl-2 and estrogen receptor (ER) α proteins [34]. In addition, emodin induced breast cell apoptosis and proliferation through ERα inhibition [26,35].
The effects of emodin on MDA-MB-231 and MDA-MB-453 human TNBC cell lines alone or co-cultured with human adipocytes were investigated. The results showed that emodin inhibited TNBC proliferation and invasion more efficiently when co-cultured with adipocytes by downregulating the level of CC-chemokine ligand 5 (CCL5) in adipocyte supernatants; inhibiting the expression level of protein kinase B (AKT); and activating glycogen synthase kinase-3i (GSK3) and β-catenin. This led to the suppressed expression of EMT- and invasion-associated markers, including vimentin, snail, matrix metalloproteinase (MMP)-2 and MMP-9, and upregulation of E-cadherin, contributing to the inhibition of invasion [36].
The anticancer properties of an emodin azide methyl anthraquinone derivative (AMAD), extracted from the nature giant knotweed rhizome of traditional Chinese herbs were investigated. The IC50 of AMAD was 9.06 ± 0.95 μmol/L for MDA-MB-453 cells, whereas for normal mouse fibroblast NIH3T3 cells the IC50 was >100 μmol/L. Apoptotic induction was associated with a collapse of the mitochondrial membrane potential and activated caspase cascade involving caspase-8, caspase-9, caspase-3, and PARP cleavage in a concentration-dependent manner. AMAD also effectively increased the cleavage of Bid, a BH3 domain-containing proapoptotic Bcl-2 family member and induced the subsequent release of cytochrome c from mitochondria into the cytosol [22]. In another study, Yan et al. showed that in cancer cells overexpressing HER2/neu-over treatment with AMAD inhibited MAPK and PI3K/AKT-dependent signaling pathways, leading to growth inhibition and induction of apoptosis. It was shown for the first time that emodin treatment impairs the binding of HER2/neu to Hsp90, intracellular redistribution, enhanced ubiquitinylation and subsequent proteasomal degradation of HER2/neu, which may represent a novel approach for the targeted therapy of HER2/neu-overexpressing cancers [37].
The proapoptotic potential of emodin was demonstrated in vitro at concentrations of >10 µM. At lower concentrations (<10µM) emodin had no or very mild effect on the viability of invasive BC MDA-MB-468 and MDA-MB-435 cell lines in vitro but it was significantly influencing their invasive potential by specifically antagonizing the adenosine 5'-triphosphate (ATP)-gated Ca(2+)-permeable channel P2 × 7 receptor (P2X7R) [38]. P2X7R is highly expressed in many tumors and cancer cells and has been found to play an important role in the migration and invasion of metastatic tumor cells [39]. Studies by Jelassi et al. showed that an increase in gelatinolytic activity, in cancer cell invasiveness in vitro and cell morphology changes induced by ATP are prevented by 1 µM emodin [38].
Emodin has been also shown to prevent the tumor-promoting interactions between cancer cells and tumor-associated macrophages (TAMs). TAMs are the most abundant leucocytes in the tumor microenvironment (TME), responsible for remodeling of TME in response to various signals including those from cancer cells [21]. Emodin reduced the recruitment of macrophages to the tumor and their subsequent M2-like polarization, and thus ameliorated the immunosuppressive state of TME [40,41,42]. Emodin suppressed TGF-β1 production in BC cells and macrophages and attenuated TGF-β1 or macrophage-induced epithelial mesenchymal-transition (EMT) and cancer stem cell (CSC) formation of BC cells [43].
The influence of emodin on the ETM transition induced by fibroblasts isolated from the tissues of TNBC patients was investigated. Using an in vitro co-culture model, interface zone fibroblasts (INFs) or cancer-associated fibroblasts (CAFs) induced EMT and promoted cancer cell migration in epithelial BT20 cells. Interestingly, we found that emodin inhibited EMT programming and phenotype in epithelial BT20 cells induced with INFs and CAFs conditioned medium [44]. Lee et al. showed that emodin suppressed TNF-a induced MMP-1 expression in cultured human dermal fibroblasts through the inhibition of the activator protein-1 (AP-1) signaling pathway.
Emodin is also a promising anti-angiogenic factor. Kwak et al. demonstrated that emodin inhibited VEGF-A-induced proliferation, migration, invasion and tube formation of Human Umbilical Vein Endothelial Cells (HUVEC) in vitro. Moreover, emodin also impaired basic fibroblast growth factor-induced proliferation and migration of HUVECs and VEGF-A-induced tube formation of human dermal microvascular endothelial cells. Emodin arrested growth of VEGF-A-stimulated HUVECs at the G0/G1 phase of the cell cycle through down-regulation of cyclin D1 and E. Emodin blocked VEGF-A-induced tyrosine phosphorylation of VEGF receptor KDR/Flk-1 and downstream signaling molecules including FAK, ERK1/2, p38 MAPK, AKT, and endothelial nitric oxide synthase, showing its potential anti-angiogenic activity [45]. Emodin also attenuated cancer cell metastasis and angiogenesis in vitro via MMPs and vascular endothelial growth factor receptor 2 (VEGFR2) inhibition, which may be associated with the downregulation of the Runx2 [46], ], a transcription factor which is one of members in Runx gene family encoding proteins homologous to Drosophila Runt and a potential target for inhibition of metastatic growth of BC cells [47]. Studies have demonstrated that atypical expression and function of Runx2 are associated with the formation of bone metastasis in BC [46].
Zou et al. found that emodin increased the expression of SerRS, which is a strong transcriptional inhibitor of VEGFA in TNBC cells. In addition, a direct target of emodin - nuclear receptor corepressor 2 (NCOR2), has been identified. When NCOR2 binds to emodin, it is released from the SerRS promoter, resulting in the activation of SerRS and inhibition of VEGFA transcription [46].
Emodin might be also effective in the prevention of multidrug resistance of BC cells and the treatment of drug-resistant BC. Fu et al. reported that emodin at 10 μg/mL downregulated the expression of DNA excision repair protein ERCC-1 and inhibited doxorubicin (DOX)-cisplatin resistance in MCF-7 cells [48]. Zu and co-workers showed that emodin (20 μM) increased the sensitivity of MCF-7 BC cells to chemotherapy and promoted 5’fluorouracil-induced apoptosis and cellular senescence. The mechanism was related to the inhibition of NRARP, and silencing NRARP blocked the effect of emodin on MCF-7 cells [49]. Li et al. reported that DOX combined with emodin can improve the sensitivity of MDA-MB-231 and MCF-7 cells to chemotherapy, and the mechanism is closely related to increasing γH2A in cancer cells and regulating AKT1-mediated DNA damage [49]. These data are particularly promising as DOX is a commonly used chemical drug against BC, but the rapid emergence of drug resistance is a major culprit limiting its clinical use [50].
The other approach important for emodin application in BC therapies is the delivery of the drug to BC cells. Wang et al. used the high-pressure homogenization method to produce emodin-loaded solid lipid nanoparticles (E-SLNs), characterized by stable particle size of 28.6 ± 3.1 nm, good drug entrapment efficiency and relatively good over long time (4 months) storage. In vitro cytotoxicity of E-SLNs (5-30 µM) towards human BC cell lines MCF-7 and MB-MDA-231 was significantly higher than free emodin whereas unloaded SLNs were not cytotoxic for both cell lines [51]. Liu et al. investigated the therapeutic potential of polymer lipid hybrid nanoparticles loaded with emodin (E-PLNs), obtained by the nanoprecipitation method. The average particle size of the E-PLNs was 122.7±1.79 nm, and the encapsulation rate was 72.8%. Compared with free emodin E-PLNs showed greater toxicity towards MCF-7 cells by promoting emodin uptake and inducing apoptosis [52]. Emodin and emodin-containing scaffolds were also shown as cytotoxic toward the GILM2 human BC cell line obtained from lung metastasis [24].
Several in vitro studies on BC models are not limited to emodin alone but also include its combination with other natural compounds: berberine, thymoquinone, daunorubicin and curcumin. These combined treatments seem to achieve better antitumor effects, which may become an effective strategy for BC therapies [53,54,55,56].
4.2. Aloe-emodin
The effect of aloe-emodin (AE) on human BC cell proliferation and survival has also been investigated using various in vitro models. AE in the concentration range of 10 to 50 μM showed estrogen-like activity, by increasing the proliferation of human ER-positive MCF-7 cells was investigated. The observed effects were attenuated by co-treatment with an ER antagonist - fulvestrant. Using another experimental condition AE showed significant cytotoxicity towards human ER-positive MCF-7 and ER-negative MDA-MB-231 BC cells by inducing mitochondria-independent apoptosis by activating the caspase-8 pathway [57]. The cytotoxic effect of AE was also observed using MDA-MB-231 cells [58].
Aloe-emodin was proposed as one of the components of Aloe vera extract responsible for its cytotoxic activity for MCF-7 human BC cell line due to its particularly strong calculated binding affinity toward estrogen receptor alpha (Erα) (-8.8 kcal/mol) as compared to standard drug Tamoxifen (-6.4 kcal/mol) [59].
Effective application of aloe-emodin as an anti-cancer drug is limited due to its poor water solubility and low bioavailability. These difficulties may be overcome by loading aloe-emodin onto various carriers. Freag et al. designed and produced surface-functionalized polyethylene glycol liquid crystalline nanoparticles (PEG-LCNPs) of aloe-emodin (AE) to enhance its water solubility and increase clinical relevance. AE-PEG-LCNPs were characterized by particle size of 190 nm and zeta potential of -49.9 and increased serum stability. Moreover, nanoparticles were also stable following sterilization by autoclaving and γ-radiation. In vitro studies using human breast adenocarcinoma cell line (MCF-7) showed that following 48h of incubation the half maximal inhibitory concentration of AE-PEG-LCNPs was 3.6-fold lower than free AE and their cellular uptake was 3-fold higher than free AE following 24-hour treatment [60]. Chen and co-workers synthesized AE-loaded solid lipid nanoparticles (AE-SLNs) characterized by stable particle size at 88.9 ± 5.2 nm, promising drug entrapment efficiency (EE) of 97.71 ± 0.5% and good stability regard to zeta-potential as high as -42.8 mV. In vitro cytotoxicity studies showed that AE-SLNs are more cytotoxic for MCF-7 BC cells than free AE solution and have no significant toxicity towards non-cancerous human mammary epithelial MCF-10A cells. Incubation of MCF-7 cancer cells with AE-SLNs increased also the number of apoptotic cells and AE uptake when compared with free AE treatment [61].
4.3. Aloin A & B
Aloin might be involved in the suppression of BC cell growth in vitro by DNA intercalating activity, as predicted using computational screening [62]. Esmat et al. investigated the influence of aloin treatment (20 and 60 µg/mL) on the nuclear DNA ploidy of estrogen receptor (ER) and prostaglandin receptor (PgR) positive breast tumor cell line T47D [63]. Change in DNA content (ploidy) has been described in a variety of tumors and it might be related to the patient survival rate and response to the treatment [64]. Aloin treatment has not changed the DNA ploidy but caused a significantly dose-dependent increase of the BC cells in the S phase and the appearance of cells cycling at a higher ploidy level (>G2M). These results suggest that aloin does not inhibit the initiation of DNA synthesis and that cells replicated a full complement of DNA but had difficulty in the M phase. The mechanism of aloin cytotoxic action might involve inhibition of topoisomerase II, an enzyme known for its ability to alter DNA supercoiling, essential for the survival of eukaryotic cells [65].
The cytotoxic potential of aloin using two human BC cell lines MCF-7 and SKBR-3, characterized by the lack or presence of erbB-2-topoIIα gene co-amplification, respectively, was described. MCF-7 cell line was more sensitive to aloin treatment than the SKBR-3 cell line, as demonstrated by MTT and clonogenic assays. Aloin at higher concentrations induced apoptosis, inhibited topo IIα protein expression and downregulated cyclin B1 protein expression in the MCF-7 cell line, whereas erbB-2 protein expression was not affected. Topo IIα protein expression was only mildly decreased in SKBR-3 cell line, only at higher concentrations. It suggests that the aloin cytotoxic effect is regulated by more than one mechanism, depending on the dose level and phenotype of breast tumor cells [66].
Aloin was also shown to significantly decrease the mammosphere formation at 50, 100 and 200 µM, indicating the cytotoxic effect of these compounds on the subpopulation of cancer stem cells (CSC) [67].
4.4. Physcion
Anti-breast cancer properties of physcion were demonstrated in MCF-7 cells due to an influence on the mitochondrial apoptotic pathway mostly by impact on oxidative stress [68]. Physcion was observed to stimulate apoptosis via activation of caspase pathways. Moreover, it caused the accumulation of ROS which disturbed the proper functioning of mitochondria [69]. Other studies showed that physcion influences on the level of pro- and antiapoptotic proteins. The expression of Bax protein was increased but Bcl-xL and Bcl2 were diminished. The creation of the Bcl-2/Bax heterodimer demonstrates a pro-apoptotic impact by inhibiting Bcl-2, leading to the production of ROS and the modulation of MMP levels [70]. The excess of arising ROS leads to the stimulation of the caspase cascade which is involved with apoptosis activation [71]. Intracellular ROS play the role of second messengers for certain growth factors to have an impact on cytokines and hormones, resulting in mitochondrial oxidative harm and programmed cell death through the Nrf2 signaling pathway, subsequently causing additional ROS release from the mitochondria [70].
The current research findings indicate that physcion suppressed the growth of MDA-MB-231 cells in a manner that depended on the dosage used in MDA-MB-231 human BC cells. In cells exposed to physcion, there was a reduction in the number of cells in the S phase, while there was an increase in the number of cells in the G0/G1 phase compared to untreated cells. These findings suggest that physcion prompted cell cycle advancement by causing G0/G1 phase arrest [72]. The advancement of the cell cycle involves a step-by-step activation of CDKs, which require interaction with their respective regulatory cyclins for activation. Specifically, the cyclin D/CDK4 complex plays a crucial role in controlling the G0/G1 phase, while the cyclin E/CDK2 and cyclin A/CDK2 complexes are linked to the G1/S transition and the onset of the S phase, respectively [73].
Consequently, the inhibition of cyclin D, cyclin E, cyclin A, CDK2, and CDK4 by physcion contributed to the arrest of the cell cycle at both the G0/G1 and G1/S phases. The CDK inhibitor p21 can hinder cell cycle progression by suppressing the kinase activities of CDKs/cyclin complexes. Moreover, cyclin-CDK complexes deactivate the tumor suppressor Rb by phosphorylating Rb at serine sites. In the case of MDA-MB-231 cells, the expression of p21 was induced by physcion, while pRb was suppressed. These findings suggest that physcion is involved in causing cell cycle arrest at the G0/G1 phase [72].
4.4. Rhein
Recent studies demonstrated that modifying the structure of rhein by introducing a benzyloxy group in the place of 1,8-phenolic hydroxyl group changes the properties of this 4F compound. This indicates that derivative 4F may be a selective anthracycline candidate with low toxicity. Based on this, the effect of derivative 4F on the cancer cells was observed in recent experiments. In recent research, we observed that derivative 4F effectively slowed down cell growth, exhibiting a time and dosage-dependent response. Remarkably, it managed to spare normal breast cells from excessive harm while targeting BC cells [74]. Moreover, its impact on curtailing the proliferation and expansion of BC cells surpassed that of rhein and the control, significantly extending the time it takes for cancer cells to double. This indicates a successful modification of the rhein side chain, suggesting that derivative 4F could be a promising, low-toxicity candidate within the anthracycline class [75]. Another protein which has garnered attention as a potential therapeutic target for tumors due to its role in regulating various signaling pathways governing physiological processes is Rac1 protein, a small signal GTPase. In this research molecular docking was tested to assess the binding stability between derivative 4F and Rac1, discovering that derivative 4F exhibited stronger interactions by forming more hydrogen bonds and arene-cation bonds with Rac1, along with increased hydrophobic interactions with amino acid residues compared to Rhein and the control inhibitor. This suggests greater stability of the Rac1-derivative 4F complex. To substantiate these findings, a luciferase reporter gene assay was conducted, revealing that derivative 4F effectively reduced Rac1 promoter activity in BC cells in a dose-dependent manner, accompanied by a dose-dependent down-regulation of Rac1 protein expression. These results indicate that derivative 4F has practical potential in regulating Rac1[74].
There are examples of other rhein derivatives that have promising effects in BC treatment. Researchers observed that rhein lysinate (RHL), an analog of rhein, effectively suppressed the phosphorylation of EGFR, MEK, c-Raf, and ERK while inducing apoptosis in MCF-7, SK-Br-3, and MDA-MB-231 cells. This investigation also revealed that rhein could enhance the sensitivity of BC cells to taxol by reducing phospho-epidermal growth factor receptor (p-EGFR) levels, shedding light on its potential to mitigating drug resistance issues in BC cells [76]. Other reports indicate that rhein exerts inhibitory effects on Akt phosphorylation, promoting the activation of FOXO3a. This activation, in turn, enhances the pro-apoptotic protein Bim's activity, resulting in caspase protein cleavage and the subsequent initiation of apoptosis within MCF-7 cells. Rhein's capabilities extend to the inhibition of NF-κB activation and its downstream targets, namely HIF-1α and VEGF165, in BC cells such as MCF-7 and MDA-MB-435. Furthermore, in vitro studies demonstrated that rhein effectively suppresses HER-2 protein phosphorylation in SK-Br-3 cells, suggesting its potential in the development of therapies tailored for HER-2-positive BC [76].
4.5. Chrysophanol
In the research conducted on BC cell lines, it was discovered that chrysophanol effectively suppressed the growth of BT-474 and MCF-7 cells. This suppression occurred by inducing the production of reactive oxygen species (ROS) and triggering endoplasmic reticulum stress. These effects were mediated through the activation of unfolded protein response (UPR) regulatory proteins such as PERK, eIF2α, GADD153, and IRE1α, involving the Akt and MAPK pathways [77]. Chrysophanol also initiated a non-apoptotic form of cell death via the mitochondrial cell death pathway. Anticancer properties of chrysophanol regarding arresting cancer cells in the S phase of the cell cycle due to reductions in proteins like cyclin D, CDK2, and thymidylate synthase were observed in BC MCF-7 and MDA-MB-231 cells [78]. Specifically, chrysophanol suppressed the proliferation of MCF-7 and MDA-MB-231 cells in a concentration-dependent manner by halting the progression of BC cells at the G1-S cell cycle checkpoint [79]. This was achieved by significantly inhibiting the expression of cyclin family proteins, including cyclin D1 and cyclin E, while increasing P21 levels in both cell lines, as confirmed by Western blot analysis and PCR results [80].
Chryspohanol effectively restrained the proliferation of MCF-7 and MDA-MB-231 cells in a dose-dependent manner. Additionally, it induced cell cycle arrest at the G1-S checkpoint, leading to decreased levels of cyclin D1 and cyclin E proteins. Chr also enhanced the apoptotic effects of paclitaxel (PTX) and reduced the expression of Bcl-2. Notably, Chr was observed to deactivate IκB and p65 phosphorylation, which are crucial components of the NF-κB pathway. To confirm the role of NF-κB in chrysophanol's anti-cancer effects, an NF-κB inhibitor, PDTC, was employed. In PDTC-treated cells, the impact of chrysophanol on Bcl-2 was less pronounced compared to normal MCF-7 and MDA-MB-231 cells, suggesting that chrysophanol exerts its effects by inhibiting NF-κB activity [81]. These findings align with previous reports indicating that chrysophanol hinders cancer cell growth through NF-κB/cyclin signaling modulation. Furthermore, chrysophanol promoted apoptosis, coinciding with decreased levels of Bcl-2 protein and the cleavage of caspase 3 and PARP. Overall, these results suggest that chrysophanol targets NF-κB/Bcl-2 to suppress BC cell proliferation and enhance sensitivity to chemotherapy, potentially serving as a valuable chemotherapeutic agent against BC cells [79].
It was demonstrated that chrysophanol exhibits anti-cancer properties when applied to human BT-474 and MCF-7 BC cells. Chrysophanol effectively inhibited the proliferation of these BC cells and induced apoptosis, while sparing normal breast ductal cells. Its influence on BT-474 and MCF-7 cell fate involved the activation of pro-apoptotic proteins within the mitochondria, the generation of reactive oxygen species (ROS), and the induction of endoplasmic reticulum (ER) stress proteins. Additionally, chrysophanol played a role in regulating signaling proteins related to MAPK and PI3K/AKT pathways. Furthermore, unveiled new findings, including the increased presence of pro-apoptotic proteins (Bax, Bak, and cytochrome c), elevated cytosolic calcium ions, and the loss of mitochondrial membrane potential (MMP) in response to chrysophanol treatment in BT-474 and MCF-7 cells. Moreover, chrysophanol triggered the activation of ER stress regulatory proteins, such as PERK, eIF2α, IRE1α, and GADD153, in a dose-dependent manner [77].
4.6. Hypericin
In recent studies, the impact of hypericin (HYP) encapsulated within Pluronic F127 (F127/HYP) in photodynamic therapy (PDT) on the MDA-MB-231BC cell line (representing TNBC) was explored compared to normal human breast ductal cells (MCF-10A). The spectroscopic properties of HYP when formulated in F127 copolymeric micelles indicated successful solubilization and suggested the potential of this biocompatible copolymer as a drug delivery system for HYP [82]. The in vitro findings revealed that F127/HYP micelles exhibited potent and selective phototoxic effects on BC cells, demonstrating time- and dose-dependent behavior, while sparing normal cells (MCF-10A). These results align with previous research, where HYP in P123 micelles PDT showed a similar selective impact on MCF-7 cells but not on MCF-10A normal cells [83]. This investigation underscores the robust photodynamic activity of F127/HYP, devoid of dark toxicity, making it a promising option for PDT in the context of breast cancer, particularly TNBC [82].
Furthermore, F127/HYP micelles were observed to accumulate in both the endoplasmic reticulum (ER) and mitochondria, leading to cell death via necrosis. The cellular uptake and subcellular distribution of F127/HYP micelles effectively addressed the hydrophobicity issue associated with HYP. Concerning cell death, exposure to F127/HYP PDT induced necrosis in MDA-MB-231 cells. It is known that photosensitizers (PSs) located in mitochondria or the ER typically induce apoptosis, while PSs targeting the plasma membrane or lysosomes can hinder the apoptotic process, potentially leading to necrosis [84]. Overall, these results suggest that F127/HYP micelles hold promise as a valuable platform for the targeted delivery of HYP, offering an effective approach for the treatment of TNBC through PDT.
Photoactivated HYP effectively reduced the mRNA and protein expression of HER2 in both SKBR-3 and MCF-7 cells [85]. It also increased the generation of ROS in MCF-7 and MDA-MB-231 cells. Furthermore, the inhibition of superoxide dismutase-2 (SOD-2) by methoxyestradiol significantly heightened the sensitivity of MCF-7 cells to HYP [86]. In vitro experiments confirmed that HYP and hypericinates could permeate the membrane of MCF-7 cells and accumulate in organelles proximal to the nucleus. Moreover, photodynamic assessments indicated that HYP could impede the formation of cellular colonies, suggesting its potential in preventing tumor recurrence [87,87]. It was demonstrated that HYP at concentrations of ≥50 μg/mL induces rapid cell death in cancer cells. Recent investigations have highlighted the significance of ADAMTS1 activity in BC development and progression [88]. In the recent resarch, HYP at concentrations of ≥5 μg/mL also prompted swift cancer cell death, suggesting that hypericin's inhibitory effect on cell proliferation may be mediated through its tumor-suppressive and cytotoxic properties, primarily involving ADAMTS1 [89].
It has been established that HYP exhibits an antiproliferative impact at lower concentrations, while at higher doses, it induces apoptosis and can disrupt cell mitosis [90]. In a prior study examining the relationship between BC cells and ADAMTS9. Hypericin's potential antitumor effects may be attributed to its interaction with ADAMTS1, ADAMTS3, and ADAMTS9, considering the apoptotic and extracellular effects of ADAMTS9 and the antiangiogenic properties of ADAMTS1 [89].
BC cells release substances that influence pre-osteoclasts, osteoblasts, and bone stromal cells. This stimulation leads to the development of mature osteoclasts that break down bone tissue. Consequently, growth factors are released, further promoting the proliferation of BC cells and perpetuating a harmful cycle of bone degradation [91]. RANKL prompted the formation of many multinucleated osteoclasts initially. However, HYP treatment significantly hindered osteoclast differentiation, leading to a dose-dependent reduction in osteoclast numbers. Notably, the appearance of TRAP-positive osteoclasts occurred after 3 days of RANKL stimulation, with more mature osteoclasts forming and merging over the subsequent 2 days. In contrast, HYP treatment consistently inhibited osteoclast differentiation throughout this process [92]. Importantly, previous research demonstrated that HP, at the same doses that inhibited osteoclast differentiation, had no cytotoxic effects [93] which suggests that HP can effectively suppress osteoclast formation in a dose-dependent manner.
To pinpoint when HYP interferes with osteoclastogenesis, HYP (1.2 μM) was introduced to the culture medium on days 0, 1, 2, 3, or 4 of osteoclast differentiation. The most significant inhibitory effects were observed when HYP was administered alongside RANKL treatment, particularly at the outset. In contrast, exposing precursor cells to HYP at later stages (after 3 days) resulted in less effective suppression. These findings indicate that HYP primarily inhibits early osteoclast differentiation [92]. BC cells can directly influence osteoclast precursor cells to promote osteoclast differentiation. HYP significantly reduced these stimulatory effects induced by BC MDA-MB-231 cells, indicating its inhibitory role in osteoclastogenesis and osteoclast activity driven by these cancer cells [92].
One of the latest studies demonstrated that utilizing gold nanoparticles (AuNPs) as carriers for hydrophobic PSs like HYP enhances the efficacy of PDT, primarily causing apoptosis-mediated cell death. PDT remains an appealing cancer therapy due to its minimally invasive nature and tumor cell selectivity. However, achieving efficient drug delivery to tumor cells is crucial and requires comprehensive exploration. In this research, HYP was attached to AuNPs as carriers to enhance its uptake by MCF-7 BC cells and consequently improve PDT effectiveness. In this investigation, hHYP was physically attached to AuNPs via sonication, forming a compound held together by non-covalent bonds. This approach increased the drug's accumulation within MCF-7 BC cells, consequently elevating PDT efficacy across various concentrations. Therefore, the non-covalent conjugation of HYP with AuNPs holds promise as a strategy for enhancing PDT's effectiveness in delivering hydrophobic PS drugs [93].
5. Anticancer Activity of Anthracene Derivatives in Breast Cancer In Vivo Models
The evidence on the anticancer potential of 1,8-OH-AQ proved by the in vitro experiments encouraged the researchers to plan experiments on living organisms. Certainly, the number of undertaken experiments of this type is growing within recent years.
Among the compounds of interest, emodin is studied to the greatest extent. Ma et al. investigated the impact of emodin on xenograft mice. In their experiment, 30 male BALB/c nu/nu athymic mice of 6-8 weeks old with LS1034 cells injected subcutaneously were used for the in vivo assay that lasted 39 days. From the whole group, 10 animals were receiving intraperitoneally 40 mg/kg of emodin solution next to a control and a fluorouracil (33 mg/kg i.p.) groups. The authors stated that emodin performed a G0/G1 phase cell cycle arrest which resulted in the apoptotic effects in mice suffering from colon cancer. The administration of emodin once every three days caused a significant reduction in the tumor size – both concerning its weight (from 1.041 g to 0.272 g in the control and emodin-treated groups, respectively) and volume (46 % volume reduction) [94].
The induction of apoptosis by emodin was also observed in the study of Wang et al. on xenograft mice bearing gallbladder carcinoma. This carcinoma was a multidrug-resistant phenotype. This anthraquinone (at a dose of 50 mg/kg) was administered intraperitoneally to the 6-week-old BALB/c nu/nu mice with SGC996 cells injected subcutaneously, for the following 18 days, starting from the third day after inoculation. Next to the emodin-treated group, the authors studied the impact of cisplatin (1 mg/kg) and the mixture emodin-cisplatin on the xenograft mice. It was noted that the combined treatment of cancer using cisplatin and emodin brought promising results. Mice exposed to the combinative treatment were characterized by significantly smaller tumors. Together with the stronger cytotoxicity of the drugs’ mixture, the systemic toxicity was not elevated, showing a neutral impact of this treatment on normal cells. Additionally, the co-treatment cisplatin-emodin inhibited the overexpression of MRP1 to a stronger extent than cisplatin itself, leading to an enhanced apoptosis of cancer cells. Emodin increases the sensitivity of gallbladder tumor (cell line SGC996) when administered with cisplatin, oxaliplatin and carboplatin. [95].
This tendency was confirmed by other authors. Li and colleagues working with stem-like side population (SP) cells of gallbladder cancer, described a sensitizing effect of emodin to cisplatin. The experiments were performed on 6-week-old BALB/c nu/nu mice with subcutaneously injected SP and non-SP cells (n=6) for 8 days. The drugs were administered every second day: emodin (50 mg/kg), cisplatin (2 mg/kg) and emodin/cisplatin. The population of mice treated with both cisplatin and emodin was found to contain significantly smaller tumors and with smaller weights in the case of both SGC-996 and GBC-SD cells. The non-SP groups delivered no significant difference between the treated and untreated groups. Similarly, to previous results, no impact on the body weight was noted during the experiment which demonstrated a lack of toxicity towards normal cells or organs. Also, the combination of these drugs in the SP group induced the inhibition of the mRNA level of ABCG2.
A combined treatment of emodin with another chemotherapeutic, namely gemcitabine was studied. This chemotherapeutic is currently a successful drug used in the treatment of pancreatic cancer, however, it is bearing several side effects. That is why the search for other molecules that can be used together with gemcitabine and lower its dose is in high demand. Tumor xenografts prepared using SW1990 cells were established by the authors in 4-6 weeks BALB/c female mice (n=12) and treated with 40 mg/kg emodin, 126 mg/kg gemcitabine or 40 mg/kg emodin+80 mg/kg gemcitabine every 6 days for the following 37 days. As a result of the treatment a significant reduction in the tumor weight and volume was observed for every treatment group, however, the strongest effects were noted for the combinational therapy. The basis for the reduction of a tumor was the induced apoptosis. The combination therapy elevated the integrated optical density of a tumor, decreased the Bcl-Bax ratio and promoted the activation of caspase-3 and the release of CytC [96]. Emodin had no effect on VEGFR expression in vivo [97]. On the other hand, the inhibition of MMPs and VEGFR2 receptor which was associated with the downregulation of Runx2 transcriptional activity by emodin was reported in a recent study of Ma et al. [46].
Liu et al. developed the studies on pancreatic cancer in xenograft mice. For this purpose, they used a group of thirty BALB/c nu/nu mice (4-6 weeks old) that were given orally two doses of emodin for 8 weeks: 20 and 40 mg/kg b.w. As a result of the treatment, a 12.7 and 26.4 % decrease in tumor weight was noted for the doses of 20 and 40 mg/kg, respectively. The results obtained for the first dose were insignificant. It is worth noting that emodin suppressed the peritoneal dissemination of the pancreatic tumor, whereas the control group had nodal metastases. In post-mortem studies the group of mice treated with 40 mg/kg p.o. showed large areas of cell debris and loci with destroyed tumors. Also, a significantly larger number of TUNEL-positive cells was presented in the higher emodin dose group. Post-mortem immunohistochemical reactions showed reduced Ki-67, survivin, and MMP-9 levels in the treated groups of animals, in relation to the untreated ones [98].
The impact on the apoptosis-related proteins, like Bcl-2 decrease or Bax increase, was also noted for the emodin-based treatment of blood system cancer (K562-based xenograft models). According to their studies, the weight and the volume of tumors were significantly reduced in all groups treated with emodin. The authors increased the previously reported doses to 100 mg/kg and conducted their studies on three groups of emodin-treated animals with 25, 50 and 100 mg/kg of emodin that was administered intraperitoneally every day for 12 consecutive days. All doses did not cause any changes in the body weight of the tested animals which proves the low toxicity of the drug. The apoptosis of K562 tumor cells was visible in post-mortem studies, in a dose dependent manner. The highest dose showed destroyed cell membranes, karyopyknosis, and margination of the cells that were followed by the signs of phagocytose of the tumor tissue by macrophages [99].
Emodin inhibited breast cancer liver metastasis by regulating lipid synthesis in obese mice (fed with thigh fat diet for 8 weeks prior to the tumor injection) that were administered 400 mg/kg b.w., p.o. for 4 weeks. Interestingly, next to the anticancer effect, the compound exhibited regulatory properties towards the animal's weight – it regulated the abnormal increase in the liver weight by its inhibitory properties towards cholesterol and fatty acids synthesis, fatty acids oxidation, and downregulation of triglycerides synthesis genes (Fasn, Sed1, Gpat1) [100].
Among the group of anthraquinones of interest, emodin is certainly the best-studied compound, however in the scientific literature we can still find a few manuscripts that focus on other derivatives of 1,8-dihyroxyanthraquinone derivatives. Aloe-emodin 3-O-glucoside was described as an effective drug in the treatment of non-small-cell lung cancer. In the mouse xenograft model (BALB/c nu/nu 5-weeks of age female mice) that was based on the cell lines A549, the authors proved that the anthraquinone administered at the doses 13 and 26 mg/kg/day for 14 days i.p. induced cancer cell death by reducing the volume and weight of the tumor. TUNEL staining results showed a reduction in cell nuclei size and an increase in the apoptotic DNA fragmentation in the tumor tissues. Moreover, the levels of caspase 9 and cleaved PARP proteins were increased upon the treatment with the compound, in both groups [101].
The aglycon of the upper compound – aloe-emodin was proved to have sensitizing properties in consolidation with gefitinib in the treatment of the same type of cancer (non-small cell lung cancer PC9-GR) in xenograft mice. Similarly, to the in vitro studies, the level of E-cadherin was increased, whereas the vimetin, Slug and Twist1 proteins were downregulated when the combination of drugs was administered to the laboratory animals [102].
Also, chrysophanol was described as a potential drug candidate that be applied in the treatment of colorectal cancer. Deng et al. in their xenograft model described a visible proapoptotic activity of this anthraquinone visualized by a decreased tumor size and volume after the i.p. administration of two doses of chrysophanol (10 and 20 mg/kg) to 8-week male athymic BALB/c nu/nu mice every day twice a week until the 27th day. The treatment increased the number of positive apoptotic cells in the TUNEL test, upregulated the levels of PUMA, Bax and caspase-3 and decreased the levels of Bcl-2 protein and Ki-67 positive cells. The observed changes indicated a proapoptotic and anti-proliferative action of the compound. Similarly, to other representatives of the same group of metabolites, no significant toxicological changes were observed in soft organs or the blood parameters [103].
The bioavailability of chrysophanol was tested. In the xenograft model using eighty 8-week-old C57BL/6J male mice, the authors proved that nanoparticles with chrysophanol (gold chloride nanoparticles) showed a significantly higher blood plasma level than free chrysophanol. The plasma concentration of this anthraquinone from nanomaterial after the i.p. administration of 50 mg/kg was ca. 10 times higher than the free chrysophanol injected at a dose of 100 mg/kg. No toxicity or histological changes in liver, kidneys or lungs were observed on the tested animals. The antitumor activity of nano-chrysophanol was assessed on the prostate cancer model (LNCap cells) in xenograft nude mice by the same authors. The study lasted 38 days and the animals were given two doses of nanoparticles with chrysophanol: 25 and 50 mg/kg. TUNEL levels of tumor tissue confirmed the strong anticancer potential of the tested formulation. Also, a decreased size and volume of the tumor was observed [104].
Animal experiments were conducted on another anthraquinone derivative – a glucoside of physcion: physcion 8-O-β-D-glucoside. Male nude mice BALB/c-nu were treated twice a week for 21 days with the glucoside at a dose of 60 mg/kg, orally – alone or in combination with paclitaxel (20 mg/kg, i.p.) to study the effects on ovarian cancer. As a result, the elaborated SK-OV-3/PTX xenograft model was sensitive to the combinational treatment of physcion glycoside and paclitaxel. Both drugs, when administered separately, had an insignificant inhibitory effect on tumor growth. It was clear that the derivative of physcion sensitized the tumor to the paclitaxel treatment. The immunohistochemical study showed that the number of P-gp-positive tumor cells was reduced by the anthraquinone glucoside [105].
Physcion was studied in the xenograft mice model of BC. Female BALB/c nude mice that were administered with 30 mg/kg of physcion, i.p. every day for 2 weeks were found to have a suppressed tumor growth starting from the 8th day of treatment with no toxic effects observed on their body weight or organs’ structure. The anticancer activity could be related to the enhanced expression levels of Bax and cleaved caspase-3, but also to the suppressed expression levels of Bcl-xL, Bcl-2, Nrf2, HO-1, SOD-1 and SOD-2 proteins suggesting an antioxidant mode of action. In further studies on immunosuppressive mice, the authors denote that physcion exhibits immunomodulatory action and is able to regulate the levels of interleukins, interferons and tumor necrosis factors in a way to reduce the progression of BC [68].
The treatment with 20 and 40 mg/kg/day i.p. of physcion to xenograft mice with hepatocellular carcinoma resulted in the delay in the development of tumor, an increased number of apoptotic cells in TUNEL assay, increased content of cleaved caspase-3, activation of caspase-12, overexpression of CHOP and a decreased level of Ki-67. All studied parameters confirm proapoptotic properties of this anthraquinone [106].
Similar observations were described by other authors. Physcion was found to be effective in the treatment of human nasopharyngeal carcinoma [107], physcion 8-o-β-glucopyranoside – in the treatment of cervical cancer [108] based on the same modes of action.
An impact on tumor apoptosis was also observed for rhein. Another anthraquinone derivative was extensively studied in both in vitro and in vivo models. Rhein was capable of inducing apoptosis and inhibiting the EGFR and STAT3 pathways in tumor remnants, in combination with EGFR inhibitors. The study was performed on five-week-old athymic BALB/c female xenograft mice with pancreatic cancer treated with 60 mg/kg of rhein i.p., 10 mg/kg i.p. of erlotinib or both drugs. The observed proapoptic activity of the drugs was visible especially when administered in combination, with clear suppression of Bcl-2 level and an increase in the level of BAX [109].
The sensitization of known chemotherapeutics by rhein (10 mg/kg i.p.) was also confirmed by Shen et al. who studied the antiproliferative properties of atezolizumab (10 mg/kg i.p.) within BC treatment. The combination therapy on 4T1 BC xenografts in mice led to a strong reduction in the volume and weight of the tumor, but also to an increase in the CD8+ T cells in the tumor and in the spleen. The serum levels of IL-6 and TNF-α were stimulated by rhein itself and in the rhein and atezolizumab group leading to a conclusion that both rhein and atezolizumab – alone or in combination are potent drugs for the treatment of 4T1 BC [110].
Another proven mechanism of action that is characteristic of rhein is its ability to inhibit the expression of β-catenin which was proved in a mice HepG2 xenograft model together with the inhibition of tumor growth [111].
To sum up, recent publications on the anticancer properties of anthraquinone derivatives list a few interesting studies that were performed on xenograft mice models. All others, even if performed on different cancer types, underline the ability of these plant secondary metabolites to reduce the tumor volume and weight, induce the process of apoptosis, induce the cell cycle arrest, increase the levels of caspase-3, E-cadherin, Bax protein, CD8+ T cells, and serum levels of interleukins and tumor necrosis factors. Additionally, the antiradical mechanisms inside the tumor are disturbed upon the administration of anthraquinones and the level of Bcl-2 protein is decreased. All authors state that the compounds in the tested doses did not show any toxicity to the normal cells or soft organs, which makes the anthraquinone derivatives important drug candidates that can be used in cancer treatment alone, but also, what is important, together with other chemotherapeutics. The major properties of these compounds are set together in Table 2.
6. Sensitizing properties of 1,8-dihydroxyanthraquinones towards known drugs
As mentioned above, the derivatives of anthraquinones are often tested together with other known chemotherapeutics. Anthraquinone derivatives of interest, namely emodin, aloe-emodin, chrysophanol, physcion and rhein were proven to interact with other drugs leading to a more effective therapy. The sensitizing effects are particularly important due to the fact that they can decrease the toxicity and diminish the side effects that often accompany standard therapeutical procedures. As shown in Table 2 and the previous sections, the safety profile of anthraquinone derivatives is promising. The doses as high as 400 mg/kg b.w. did not trigger any toxic effects on the soft organs or body weight of laboratory animals. Previously, scientists proved that the route of administration of an accompanying drug can differ from the main chemotherapeutic agent.
In recent years, combined therapy has become increasingly popular, which involves the simultaneous use of several substances, including compounds of natural origin. Combined therapy can lead to a reduction in toxicity to the patient's body by replacing part of the dose of a conventional anticancer drug with a natural substance with known properties. Many studies emphasize that a combined approach may be more effective than using conventional chemotherapy agents alone.
The scientific literature lists a number of examples that confirm stronger anticancer potential of a joint administration of the selected anthraquinones with standard chemotherapeutics. The selected examples of this approach are listed in the Table 3 below.
7. Discussion
Despite significant progress in the diagnosis and treatment of BC, this disease continues to pose a substantial global health challenge, impacting millions of women worldwide each year. Standard therapeutic options, which are commonly used in BC treatment, like radio- or chemotherapy, can result in a range of undesirable side effects that can significantly impact the quality of life for cancer patients. Additionally, the emergence of MDR in cancer cells can reduce the effectiveness of these therapies over time. Phytochemicals, which are natural compounds found in plants, hold promise in improving BC treatment outcomes. They have the potential to work synergistically with several chemotherapeutic agents, enhancing their effectiveness while minimizing some of the side effects associated with traditional therapeutic regimens. Anthracene derivatives are a group of tricyclic secondary metabolites that occur naturally and have been identified in various organisms, including plants, lichens, and fungi. They are promising compounds with proven anti-cancer properties. Mechanistic studies have established that the natural products derived from anthracene derivatives exert their anti-breast cancer activities through a wide array of molecular targets and mechanisms, including the modulation of angiogenesis, apoptotic pathways, autophagy, synthesis and repair genes expression, damage response, cell cycle regulators, EMT markers, epigenetic mechanisms, heat shock response, inflammation, metastasis-related markers, oxidative status, miRNA, protein synthesis, proliferation or stem-like markers. The evolving insights into the tumor microenvironment and the underlying molecular pathways associated with BC create opportunities for the identification and development of new natural compounds that can be harnessed for their anti-cancer properties. These discoveries not only expand our arsenal of treatment options but also offer avenues for preventive strategies.
8. Conclusions
The review aimed to consolidate the information on 1,8-dihydroxyanthraquinones, including emodin, aloe-emodin, hypericin, chrysophanol, rhein, and physcion, as promising drug candidates for the BC treatment. All these compounds, belonging to a subgroup of anthracene derivatives, have been extensively studied for their anticancer properties. In the manuscript, the potential synergistic interactions between these natural compounds and commercially available chemotherapeutic agents in the in vitro and in vivo settings were also described. The evidence from the literature suggests that 1,8-dihydroxyanthraquinones indeed appear to be a promising group of natural compounds with potential effectiveness in the treatment of BC. This conclusion underscores their potential significance as candidates for further research and development in BC therapy.
Author Contributions
Conceptualization, E.O., W.K.-K., A.W.; investigation, E.O., K.G.-B., A.J., W.K., W.K.-K., A.W.; resources, E.O., K.G.-B., A.J., W.K., W.K.-K., A.W.; writing—original draft preparation E.O., K.G.-B., A.J., W.K., W.K.-K., A.W.; writing—review and editing, A.W.; visualization, W.K.-K., A.W.; supervision, A.W.; project administration, E.O., A.W.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created and analyzed in this manuscript. Data sharing is not applicable.
Acknowledgments
We would like to thank Agnieszka Styczyńska from the Chair and Department of Biochemistry and Molecular Biology, Medical University of Lublin, for her help with editorial assistance and proof-reading.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roy, P.S.; Saikia, B.J. Cancer and cure: A critical analysis. Indian J. Cancer 2016, 53, 441–442. [Google Scholar] [CrossRef]
- Fabiani, R. Antitumoral Properties of Natural Products. Molecules 2020, 25, 650. [Google Scholar] [CrossRef] [PubMed]
- Arrousse, N.; Harras, M.F.; El Kadiri, S.; Haldhar, R.; Ichou, H.; Bousta, D.; Grafov, A.; Rais, Z.; Taleb, M. New anthraquinone drugs and their anticancer activities: Cytotoxicity, DFT, docking and ADMET properties. Results Chem. 2023, 6, 100996. [Google Scholar] [CrossRef]
- Baviera, G.S.; Donate, P.M. Recent advances in the syntheses of anthracene derivatives. Beilstein J. Org. Chem. 17131 2021, 17, 2028–2050. [Google Scholar] [CrossRef] [PubMed]
- Şeker Karatoprak, G.; Küpeli Akkol, E.; Yücel, Ç.; Bahadir Acikara, Ö.; Sobarzo-Sánchez, E. Advances in Understanding the Role of Aloe Emodin and Targeted Drug Delivery Systems in Cancer. Oxid. Med. Cell. Longev. 2022, 7928200. [Google Scholar] [CrossRef] [PubMed]
- Montazeri Aliabadi, H.; Manda, A.; Sidgal, R.; Chung, C. Targeting Breast Cancer: The Familiar, the Emerging, and the Uncharted Territories. Biomolecules 2023, 13, 1306. [Google Scholar] [CrossRef] [PubMed]
- Azadnajafabad, S.; Saeedi Moghaddam, S.; Mohammadi, E.; Delazar, S.; Rashedi, S.; Baradaran, H.R.; Mansourian, M. Patterns of better breast cancer care in countries with higher human development index and healthcare expenditure: Insights from GLOBOCAN 2020. Front. Public Heal. 2023, 11, 1137286. [Google Scholar] [CrossRef]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, Vol. 13, Page 3409 2021, 13, 3409. [Google Scholar] [CrossRef]
- Uchida, N.; Suda, T.; Ishiguro, K. Effect of Chemotherapy for Luminal A Breast Cancer. Yonago Acta Med. 2013, 56, 51. [Google Scholar]
- Creighton, C.J. The molecular profile of luminal B breast cancer. Biol. Targets Ther. 2012, 6, 289–297. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer. Breast Cancer 2022, 31–42. [Google Scholar]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022 151 2022, 15, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Dash, R. Natural Products for the Management and Prevention of Breast Cancer. Evid. Based. Complement. Alternat. Med. 2018, 8324696. [Google Scholar] [CrossRef]
- Thomson, R. Naturally Occurring Quinones. Available online: https://books.google.pl/books?hl=pl&lr=&id=iwFs2rpKOFgC&oi=fnd&pg=PP1&ots=E10gXwxkcL&sig=MxN3Epb0yXu_kfr-mF5weiKnjwg&redir_esc=y#v=onepage&q&f=false.
- Duval, J.; Pecher, V.; Poujol, M.; Lesellier, E. Research advances for the extraction, analysis and uses of anthraquinones: A review. Ind. Crops Prod. 2016, 94, 812–833. [Google Scholar] [CrossRef]
- Farmacognosia Da Planta Ao Medicamento | PDF | Biodiversidade | Biologia de Conservação . Available online: https://www.scribd.com/document/377628211/Farmacognosia-Da-Planta-Ao-Medicamento (accessed on 5 October 2023).
- Han, Y.S.; Van Der Heijden, R.; Verpoorte, R. Improved anthraquinone accumulation in cell cultures of Cinchona “Robusta” by feeding of biosynthetic precursors and inhibitors. Biotechnol. Lett. 2002, 24, 705–710. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Naayer, M.; van Beek, T.A.; Capelle, A.; Haaksman, I.K.; van Doren, H.A.; de Groot, Æ. Chemical and enzymatic hydrolysis of anthraquinone glycosides from madder roots. Phytochem. Anal. 2003, 14, 137–144. [Google Scholar] [CrossRef]
- de Morais, F.A.P.; Balbinot, R.B.; Bakoshi, A.B.K.; Lazarin-Bidoia, D.; da Silva Souza Campanholi, K.; da Silva Junior, R.C.; Gonçalves, R.S.; Ueda-Nakamura, T.; de Oliveira Silva, S.; Caetano, W.; et al. Advanced theranostic nanoplatforms for hypericin delivery in the cancer treatment. J. Photochem. Photobiol. B. 2023, 247, 112782. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, G.; Shi, P. Emodin-induced apoptosis in human breast cancer BCap-37 cells through the mitochondrial signaling pathway. Arch. Pharm. Res. 2008, 31, 742–748. [Google Scholar] [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef]
- Yan, Y.; Su, X.; Liang, Y.; Zhang, J.; Shi, C.; Lu, Y.; Gu, L.; Fu, L. Emodin azide methyl anthraquinone derivative triggers mitochondrial-dependent cell apoptosis involving in caspase-8-mediated Bid cleavage. Mol. Cancer Ther. 2008, 7, 1688–1697. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Zhou, Q.; Lu, Y.; Zhang, H.; Chen, Q.; Zhao, M.; Su, S. Inhibitory effect of emodin on migration, invasion and metastasis of human breast cancer MDA-MB-231 cells in vitro and in vivo. Oncol. Rep. 2015, 33, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Mun, G.H.; Choi, B.; Aseh, A.; Mildred, L.; Patel, A.; Zhang, Q.; Price, J.E.; Chang, D.; Robb, G.; et al. Repair and reconstruction of a resected tumor defect using a composite of tissue flap-nanotherapeutic-silk fibroin and chitosan scaffold. Ann. Biomed. Eng. 2011, 39, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, G.; Shi, P. Effects of emodin on the gene expression profiling of human breast carcinoma cells. Cancer Detect. Prev. 2009, 32, 286–291. [Google Scholar] [CrossRef]
- Zu, C.; Zhang, M.; Xue, H.; Cai, X.; Zhao, L.; He, A.; Qin, G.; Yang, C.; Zheng, X. Emodin induces apoptosis of human breast cancer cells by modulating the expression of apoptosis-related genes. Oncol. Lett. 2015, 10, 2919. [Google Scholar] [CrossRef]
- Li, W.Y.; Chan, R.Y.K.; Yu, P.H.F.; Chan, S.W. Emodin induces cytotoxic effect in human breast carcinoma MCF-7 cell through modulating the expression of apoptosis-related genes. Pharm. Biol. 2013, 51, 1175–1181. [Google Scholar] [CrossRef]
- Zhang, L.; Lau, Y.K.; Xi, L.; Hong, R.L.; Kim, D.S.H.L.; Chen, C.F.; Hortobagyi, G.N.; Chang, C.J.; Hung, M.C. Tyrosine kinase inhibitors, emodin and its derivative repress HER-2/neu-induced cellular transformation and metastasis-associated properties. Oncogene 1998, 16, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Tyrosine Kinase Inhibitor Emodin Suppresses Growth of HER-2/neu-overexpressing Breast Cancer Cells in Athymic Mice and Sensitizes These Cells to the Inhibitory Effect of Paclitaxel1 | Clinical Cancer Research | American Association for Cancer Research . Available online: https://aacrjournals.org/clincancerres/article/5/2/343/199283/Tyrosine-Kinase-Inhibitor-Emodin-Suppresses-Growth (accessed on 5 October 2023).
- Shao-Chun, W.; Lisha, Z.; Hortobagyi, G.N.; Mien-Chie, H. Targeting HER2: Recent developments and future directions for breast cancer patients. Semin. Oncol. 2001, 28, 21–29. [Google Scholar] [CrossRef]
- N, U.; N, K.; M, H. Growth suppression of low HER-2/neu-expressing breast cancer cell line MDA-MB-435 by tyrosine kinase inhibitor emodin. Oncol. Rep. 1996, 3, 509–511. [Google Scholar] [CrossRef]
- Li, F.; Song, X.; Zhou, X.; Chen, L.; Zheng, J. Emodin attenuates high lipid-induced liver metastasis through the AKT and ERK pathways in vitro in breast cancer cells and in a mouse xenograft model. Heliyon 2023, 9, e17052. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, J.; Sheng, A.; Huang, S.; Tang, Y.; Ma, S.; Hong, G. Emodin Inhibits the Proliferation of MCF-7 Human Breast Cancer Cells Through Activation of Aryl Hydrocarbon Receptor (AhR). Front. Pharmacol. 2020, 11, 622046. [Google Scholar] [CrossRef]
- Sui, J.Q.; Xie, K.P.; Zou, W.; Xie, M.J. Emodin inhibits breast cancer cell proliferation through the ERα-MAPK/Akt-cyclin D1/Bcl-2 signaling pathway. Asian Pac. J. Cancer Prev. 2014, 15, 6247–6251. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Huang, C.Y.; Chen, M.C.; Lee, Y.T.; Yue, C.H.; Wang, H.Y.; Lin, H. Emodin and Aloe-Emodin Suppress Breast Cancer Cell Proliferation through ERα Inhibition. Evid. Based. Complement. Alternat. Med. 2013, 2013, 376123. [Google Scholar] [CrossRef]
- Song, X.; Zhou, X.; Qin, Y.; Yang, J.; Wang, Y.; Sun, Z.; Yu, K.; Zhang, S.; Liu, S. Emodin inhibits epithelial-mesenchymal transition and metastasis of triple negative breast cancer via antagonism of CC-chemokine ligand 5 secreted from adipocytes. Int. J. Mol. Med. 2018, 42, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Y.; Zheng, L.S.; Zhang, X.; Chen, L.K.; Singh, S.; Wang, F.; Zhang, J.Y.; Liang, Y.J.; Dai, C.L.; Gu, L.Q.; et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol. Pharm. 2011, 8, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Jelassi, B.; Anchelin, M.; Chamouton, J.; Cayuela, M.L.; Clarysse, L.; Li, J.; Goré, J.; Jiang, L.H.; Roger, S. Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2 × 7 receptors. Carcinogenesis 2013, 34, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2 × 7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Iwanowycz, S.; Wang, J.; Altomare, D.; Hui, Y.; Fan, D. Emodin Bidirectionally Modulates Macrophage Polarization and Epigenetically Regulates Macrophage Memory. J. Biol. Chem. 2016, 291, 11491. [Google Scholar] [CrossRef]
- Iwanowycz, S.; Wang, J.; Hodge, J.; Wang, Y.; Yu, F.; Fan, D. Emodin inhibits breast cancer growth by blocking the tumor-promoting feedforward loop between cancer cells and macrophages. Mol. Cancer Ther. 2016, 15, 1931. [Google Scholar] [CrossRef]
- Jia, X.; Yu, F.; Wang, J.; Iwanowycz, S.; Saaoud, F.; Wang, Y.; Hu, J.; Wang, Q.; Fan, D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res. Treat. 2014, 148, 291–302. [Google Scholar] [CrossRef]
- Liu, Q.; Hodge, J.; Wang, J.; Wang, Y.; Wang, L.; Singh, U.; Li, Y.; Yao, Y.; Wang, D.; Ai, W.; et al. Emodin reduces Breast Cancer Lung Metastasis by suppressing Macrophage-induced Breast Cancer Cell Epithelial-mesenchymal transition and Cancer Stem Cell formation. Theranostics 2020, 10, 8365–8381. [Google Scholar] [CrossRef]
- Hsu, H.C.; Liu, L.C.; Wang, H.Y.; Hung, C.M.; Lin, Y.C.; Ho, C.T.; Way, T. Der Stromal Fibroblasts from the Interface Zone of Triple Negative Breast Carcinomas Induced Epithelial-Mesenchymal Transition and its Inhibition by Emodin. PLoS One 2017, 12, e0164661. [Google Scholar] [CrossRef]
- Kwak, H.J.; Park, M.J.; Park, C.M.; Moon, S.I.; Yoo, D.H.; Lee, H.C.; Lee, S.H.; Kim, M.S.; Lee, H.W.; Shin, W.S.; et al. Emodin inhibits vascular endothelial growth factor-A-induced angiogenesis by blocking receptor-2 (KDR/Flk-1) phosphorylation. Int. J. Cancer 2006, 118, 2711–2720. [Google Scholar] [CrossRef]
- Ma, J.; Lu, H.; Wang, S.; Chen, B.; Liu, Z.; Ke, X.; Liu, T.; Fu, J. The anthraquinone derivative Emodin inhibits angiogenesis and metastasis through downregulating Runx2 activity in breast cancer. Int. J. Oncol. 2015, 46, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Fu, J. min; Zhou, J.; Shi, J.; Xie, J. sheng; Huang, L.; Yip, A.Y.S.; Loo, W.T.Y.; Chow, L.W.C.; Ng, E.L.Y. Emodin affects ERCC1 expression in breast cancer cells. J. Transl. Med. 2012, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zu, C.; Qin, G.; Yang, C.; Liu, N.; He, A.; Zhang, M.; Zheng, X. Low dose Emodin induces tumor senescence for boosting breast cancer chemotherapy via silencing NRARP. Biochem. Biophys. Res. Commun. 2018, 505, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Christowitz, C.; Davis, T.; Isaacs, A.; Van Niekerk, G.; Hattingh, S.; Engelbrecht, A.M. Mechanisms of doxorubicin-induced drug resistance and drug resistant tumour growth in a murine breast tumour model. BMC Cancer 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Wang, S.; Chen, T.; Chen, R.; Hu, Y.; Chen, M.; Wang, Y. Emodin loaded solid lipid nanoparticles: preparation, characterization and antitumor activity studies. Int. J. Pharm. 2012, 430, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhuang, Y.; Wang, P.; Zou, T.; Lan, M.; Li, L.; Liu, F.; Cai, T.; Cai, Y. Polymeric Lipid Hybrid Nanoparticles as a Delivery System Enhance the Antitumor Effect of Emodin in Vitro and in Vivo. J. Pharm. Sci. 2021, 110, 2986–2996. [Google Scholar] [CrossRef]
- Bhattacharjee, M.; Upadhyay, P.; Sarker, S.; Basu, A.; Das, S.; Ghosh, A.; Ghosh, S.; Adhikary, A. Combinatorial therapy of Thymoquinone and Emodin synergistically enhances apoptosis, attenuates cell migration and reduces stemness efficiently in breast cancer. Biochim. Biophys. Acta - Gen. Subj. 2020, 1864, 129695. [Google Scholar] [CrossRef]
- Fu, M.; Tang, W.; Liu, J.J.; Gong, X.Q.; Kong, L.; Yao, X.M.; Jing, M.; Cai, F.Y.; Li, X.T.; Ju, R.J. Combination of targeted daunorubicin liposomes and targeted emodin liposomes for treatment of invasive breast cancer. J. Drug Target. 2020, 28, 245–258. [Google Scholar] [CrossRef]
- Guo, J.; Li, W.; Shi, H.; Xie, X.; Li, L.; Tang, H.; Wu, M.; Kong, Y.; Yang, L.; Gao, J.; et al. Synergistic effects of curcumin with emodin against the proliferation and invasion of breast cancer cells through upregulation of miR-34a. Mol. Cell. Biochem. 2013, 382, 103–111. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Kothandan, G.; Manoharan, R. Berberine and Emodin abrogates breast cancer growth and facilitates apoptosis through inactivation of SIK3-induced mTOR and Akt signaling pathway. Biochim. Biophys. Acta - Mol. Basis Dis. 2020, 1866, 165897. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Park, S.J.; Choi, S.; Kim, S.H.; Kang, K.S. In Vitro Estrogenic and Breast Cancer Inhibitory Activities of Chemical Constituents Isolated from Rheum undulatum L. Mol. 2018, Vol. 23, Page 1215 2018, 23, 1215. [Google Scholar] [CrossRef] [PubMed]
- Effect of Aloe emodin on invasion and metastasis of high metastatic breast cancer MDA-MB-231 cells . Available online: https://pubmed.ncbi.nlm.nih.gov/24620697/ (accessed on 6 October 2023).
- Majumder, R.; Parida, P.; Paul, S.; Basak, P. In vitro and in silico study of Aloe vera leaf extract against human breast cancer. Nat. Prod. Res. 2020, 34, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Freag, M.S.; Elnaggar, Y.S.R.; Abdelmonsif, D.A.; Abdallah, O.Y. Stealth, biocompatible monoolein-based lyotropic liquid crystalline nanoparticles for enhanced aloe-emodin delivery to breast cancer cells: in vitro and in vivo studies. Int. J. Nanomedicine 2016, 11, 4799–4818. [Google Scholar] [CrossRef]
- Chen, R.; Wang, S.; Zhang, J.; Chen, M.; Wang, Y. Aloe-emodin loaded solid lipid nanoparticles: formulation design and in vitro anti-cancer study. Drug Deliv. 2015, 22, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Majumder, R.; Das, C.K.; Banerjee, I.; Chandra Jena, B.; Mandal, A.; Das, P.; Pradhan, A.K.; Das, S.; Basak, P.; Das, S.K.; et al. Screening of the Prime bioactive compounds from Aloe vera as potential anti-proliferative agents targeting DNA. Comput. Biol. Med. 2022, 141, 105052. [Google Scholar] [CrossRef]
- Esmat, A.Y.; El-Gerzawy, S.M.; Rafaat, A. DNA ploidy and S phase fraction of breast and ovarian tumor cells treated with a natural anthracycline analog (aloin). Cancer Biol. Ther. 2005, 4, 115–119. [Google Scholar] [CrossRef]
- Beerman, H.; Kluin, P.M.; Hermans, J.; Van de Velde, C.J.H.; Cornelisse, C.J. Prognostic significance of DNA-ploidy in a series of 690 primary breast cancer patients. Int. J. cancer 1990, 45, 34–39. [Google Scholar] [CrossRef]
- Järvinen, T.A.H.; Tanner, M.; Rantanen, V.; Bärlund, M.; Borg, Å.; Grénman, S.; Isola, J. Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am. J. Pathol. 2000, 156, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Esmat, A.Y.; Tomasetto, C.; Rio, M.C. Cytotoxicity of a natural anthraquinone (Aloin) against human breast cancer cell lines with and without ErbB-2: topoisomerase IIalpha coamplification. Cancer Biol. Ther. 2006, 5, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Identification of natural products and FDA-approved drugs for targeting cancer stem cell (CSC) propagation. Aging (Albany. NY). 2022, 14, 9466–9483. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, R.; Wang, Y.; Wang, L.; Zhou, T.; Jia, D.; Meng, Z. The anti-breast cancer property of physcion via oxidative stress-mediated mitochondrial apoptosis and immune response. Pharm. Biol. 2021, 59, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.H.; Zhao, S.S.; Liu, D.Y.; Wang, Z.; Niu, L.L.; Hou, L.H.; Wang, C.L. ROS-Ca(2+) is associated with mitochondria permeability transition pore involved in surfactin-induced MCF-7 cells apoptosis. Chem. Biol. Interact. 2011, 190, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.J.F.; Low, I.C.C.; Pervaiz, S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion 2014, 19, 39–48. [Google Scholar] [CrossRef]
- Kim, B.M.; Chung, H.W. Hypoxia/reoxygenation induces apoptosis through a ROS-mediated caspase-8/Bid/Bax pathway in human lymphocytes. Biochem. Biophys. Res. Commun. 2007, 363, 745–750. [Google Scholar] [CrossRef]
- Hong, J.-Y.; Chung, H.-J.; Bae, S.Y.; Trung, T.N.; Bae, K.; Lee, S.K. Induction of Cell Cycle Arrest and Apoptosis by Physcion, an Anthraquinone Isolated From Rhubarb (Rhizomes of Rheum tanguticum), in MDA-MB-231 Human Breast Cancer Cells. J. cancer Prev. 2014, 19, 273–278. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Zhao, Y.; Tian, W.; Zhai, L.; Pang, H.; Kang, J.; Hou, H.; Chen, Y.; Li, D. Rhein Derivative 4F Inhibits the Malignant Phenotype of Breast Cancer by Downregulating Rac1 Protein. Front. Pharmacol. 2020, 11, 754. [Google Scholar] [CrossRef]
- Tian, W.; Li, J.; Su, Z.; Lan, F.; Li, Z.; Liang, D.; Wang, C.; Li, D.; Hou, H. Novel Anthraquinone Compounds Induce Cancer Cell Death through Paraptosis. ACS Med. Chem. Lett. 2019, 10, 732–736. [Google Scholar] [CrossRef] [PubMed]
- Henamayee, S.; Banik, K.; Sailo, B.L.; Shabnam, B.; Harsha, C.; Srilakshmi, S.; Naidu, V.G.M.; Baek, S.H.; Ahn, K.S.; Kunnumakkara, A.B. Therapeutic emergence of rhein as a potential anticancer drug: A review of its molecular targets and anticancer properties. Molecules 2020, 25, 2278. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, W.; Song, G. Chrysophanol selectively represses breast cancer cell growth by inducing reactive oxygen species production and endoplasmic reticulum stress via AKT and mitogen-activated protein kinase signal pathways. Toxicol. Appl. Pharmacol. 2018, 360, 201–211. [Google Scholar] [CrossRef]
- Ni, C.H.; Chen, P.Y.; Lu, H.F.; Yang, J.S.; Huang, H.Y.; Wu, S.H.; Ip, S.W.; Wu, C.T.; Chiang, S.Y.; Lin, J.G.; et al. Chrysophanol-induced necrotic-like cell death through an impaired mitochondrial ATP synthesis in Hep3B human liver cancer cells. Arch. Pharm. Res. 2012, 35, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Li, Z.; Dai, C.; Zhao, D.; Wang, Y.; Ma, C.; Liu, C. Chrysophanol inhibits proliferation and induces apoptosis through NF-κB/cyclin D1 and NF-κB/Bcl-2 signaling cascade in breast cancer cell lines. Mol. Med. Rep. 2018, 17, 4376. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wu, J.; Gao, Y.; Luo, Y.; Yang, D.; Wang, P. The pharmacological properties of chrysophanol, the recent advances. Biomed. Pharmacother. 2020, 125, 110002. [Google Scholar] [CrossRef]
- Ren, L.; Li, Z.; Dai, C.; Zhao, D.; Wang, Y.; Ma, C.; Liu, C. Chrysophanol inhibits proliferation and induces apoptosis through NF-κB/cyclin D1 and NF-κB/Bcl-2 signaling cascade in breast cancer cell lines. Mol. Med. Rep. 2018, 17, 4376–4382. [Google Scholar] [CrossRef]
- de Souza, M.V.F.; Shinobu-Mesquita, C.S.; Meirelles, L.E.F.; Mari, N.L.; César, G.B.; Gonçalves, R.S.; Caetano, W.; Damke, E.; Silva, V.R.S.; Damke, G.M.Z.F.; et al. Effects of hypericin encapsulated on Pluronic F127 photodynamic therapy against triple negative breast cancer. Asian Pac. J. Cancer Prev. 2022, 23, 1741–1751. [Google Scholar] [CrossRef]
- Damke, G.M.Z.F.; Damke, E.; de Souza Bonfim-Mendonça, P.; Ratti, B.A.; de Freitas Meirelles, L.E.; da Silva, V.R.S.; Gonçalves, R.S.; César, G.B.; de Oliveira Silva, S.; Caetano, W.; et al. Selective photodynamic effects on cervical cancer cells provided by P123 Pluronic®-based nanoparticles modulating hypericin delivery. Life Sci. 2020, 255, 117858. [Google Scholar] [CrossRef]
- Diwu, Z.; William Lown, J. Photosensitization with anticancer agents 17. EPR studies of photodynamic action of hypericin: Formation of semiquinone radical and activated oxygen species on illumination. Free Radic. Biol. Med. 1993, 14, 209–215. [Google Scholar] [CrossRef]
- Solár, P.; Ferenc, P.; Koval, J.; Mikeš, J.; Solárova, Z.; Hrčková, G.; Fulton, B.L.; Fedoročko, P. Photoactivated hypericin induces downregulation of HER2 gene expression. Radiat. Res. 2011, 175, 51–56. [Google Scholar] [CrossRef]
- Kimáková, P.; Solár, P.; Fecková, B.; Sačková, V.; Solárová, Z.; Ilkovičová, L.; Kello, M. Photoactivated hypericin increases the expression of SOD-2 and makes MCF-7 cells resistant to photodynamic therapy. Biomed. Pharmacother. 2017, 85, 749–755. [Google Scholar] [CrossRef]
- de Andrade, G.P.; Manieri, T.M.; Nunes, E.A.; Viana, G.M.; Cerchiaro, G.; Ribeiro, A.O. Comparative in vitro study of photodynamic activity of hypericin and hypericinates in MCF-7 cells. J. Photochem. Photobiol. B Biol. 2017, 175, 89–98. [Google Scholar] [CrossRef]
- Freitas, V.M.; do Amaral, J.B.; Silva, T.A.; Santos, E.S.; Mangone, F.R.; Pinheiro, J. de J.; Jaeger, R.G.; Nagai, M.A.; Machado-Santelli, G.M. Decreased expression of ADAMTS-1 in human breast tumors stimulates migration and invasion. Mol. Cancer 2013, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- The effects of hypericin on ADAMTS and p53 gene expression in MCF-7 breast cancer cells . Available online: https://pubmed.ncbi.nlm.nih.gov/25261644/ (accessed on 6 October 2023).
- Stojanovic, G.; Dordevic, A.; Smelcerovic, A. Do other Hypericum species have medical potential as St. John’s wort (Hypericum perforatum)? Curr. Med. Chem. 2013, 20, 2273–2295. [Google Scholar] [CrossRef] [PubMed]
- Shemanko, C.S.; Cong, Y.; Forsyth, A. What Is Breast in the Bone? Int. J. Mol. Sci. 2016, 17, 1764. [Google Scholar] [CrossRef]
- Ouyang, Z.; Guo, X.; Chen, X.; Liu, B.; Zhang, Q.; Yin, Z.; Zhai, Z.; Qu, X.; Liu, X.; Peng, D.; et al. Hypericin targets osteoclast and prevents breast cancer-induced bone metastasis via NFATc1 signaling pathway. Oncotarget 2018, 9, 1868. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Zhai, Z.; Li, H.; Liu, X.; Qu, X.; Li, X.; Fan, Q.; Tang, T.; Qin, A.; Dai, K. Hypericin suppresses osteoclast formation and wear particle-induced osteolysis via modulating ERK signalling pathway. Biochem. Pharmacol. 2014, 90, 276–287. [Google Scholar] [CrossRef]
- Ma, Y.S.; Weng, S.W.; Lin, M.W.; Lu, C.C.; Chiang, J.H.; Yang, J.S.; Lai, K.C.; Lin, J.P.; Tang, N.Y.; Lin, J.G.; et al. Antitumor effects of emodin on LS1034 human colon cancer cells in vitro and in vivo: Roles of apoptotic cell death and LS1034 tumor xenografts model. Food Chem. Toxicol. 2012, 50, 1271–1278. [Google Scholar] [CrossRef]
- Wang, W.; Sun, Y. ping; Huang, X. zhi; He, M.; Chen, Y. ying; Shi, G. ying; Li, H.; Yi, J.; Wang, J. Emodin enhances sensitivity of gallbladder cancer cells to platinum drugs via glutathion depletion and MRP1 downregulation. Biochem. Pharmacol. 2010, 79, 1134–1140. [Google Scholar] [CrossRef]
- Chen, H.; Wei, W.; Guo, Y.; Liu, A.; Tong, H.; Wang, Z.; Tan, W.; Liu, J.; Lin, S. Enhanced effect of gemcitabine by emodin against pancreatic cancer in vivo via cytochrome C-regulated apoptosis. Oncol. Rep. 2011, 25, 1253–1261. [Google Scholar] [CrossRef]
- Lin, S.Z.; Wei, W.T.; Chen, H.; Chen, K.J.; Tong, H.F.; Wang, Z.H.; Ni, Z.L.; Liu, H. Bin; Guo, H.C.; Liu, D.L. Antitumor Activity of Emodin against Pancreatic Cancer Depends on Its Dual Role: Promotion of Apoptosis and Suppression of Angiogenesis. PLoS One 2012, 7, e42146. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, H.; Wei, W.; Ye, S.; Liao, W.; Gong, J.; Jiang, Z.; Wang, L.; Lin, S. Antiproliferative and antimetastatic effects of emodin on human pancreatic cancer. Oncol. Rep. 2011, 26, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Chun-Guang, W.; Jun-Qing, Y.; Bei-Zhong, L.; Dan-Ting, J.; Chong, W.; Liang, Z.; Dan, Z.; Yan, W. Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur. J. Pharmacol. 2010, 627, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, X.; Zhou, X.; Chen, L.; Zheng, J. Emodin attenuates high lipid-induced liver metastasis through the AKT and ERK pathways in vitro in breast cancer cells and in a mouse xenograft model. Heliyon 2023, 9. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, J.W.; Ree, J.; Lim, J.S.; Lee, J.; Kim, J. Il; Thapa, S.B.; Sohng, J.K.; Park, Y. Il Aloe emodin 3-O-glucoside inhibits cell growth and migration and induces apoptosis of non-small-cell lung cancer cells via suppressing MEK/ERK and Akt signalling pathways. Life Sci. 2022, 300, 120495. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Zheng, Z.; Chen, S.; Fang, L.; Feng, R.; Zhang, L.; Tang, Q.; Liu, X. Sensitization of Non-Small Cell Lung Cancer Cells to Gefitinib and Reversal of Epithelial–Mesenchymal Transition by Aloe-Emodin Via PI3K/Akt/TWIS1 Signal Blockage. Front. Oncol. 2022, 12, 908031. [Google Scholar] [CrossRef]
- Deng, M.; Xue, Y.; Xu, L.; Wang, Q.; Wei, J.; Ke, X.; Wang, J.; Chen, X. Chrysophanol exhibits inhibitory activities against colorectal cancer by targeting decorin. Cell Biochem. Funct. 2020, 38, 47–57. [Google Scholar] [CrossRef]
- Lu, L.; Li, K.; Mao, Y.H.; Qu, H.; Yao, B.; Zhong, W.W.; Ma, B.; Wang, Z.Y. Gold-chrysophanol nanoparticles suppress human prostate cancer progression through inactivating AKT expression and inducing apoptosis and ROS generation in vitro and in vivo. Int. J. Oncol. 2017, 51, 1089–1103. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Wei, L.; Zhang, J.; Li, X.; Cui, W.; Zhang, S. Physcion-8-O-β-d-glucoside interferes with the nuclear factor-κB pathway and downregulates P-glycoprotein expression to reduce paclitaxel resistance in ovarian cancer cells. J. Pharm. Pharmacol. 2021, 73, 545–552. [Google Scholar] [CrossRef]
- Pan, X. ping; Wang, C.; Li, Y.; Huang, L. hua Physcion induces apoptosis through triggering endoplasmic reticulum stress in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 99, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.J.; Yang, Z.; Zhang, X.L.; Liu, Z.F.; Fan, J.; Zhang, H.Y. Physcion, a naturally occurring anthraquinone derivative, induces apoptosis and autophagy in human nasopharyngeal carcinoma. Acta Pharmacol. Sin. 2016 3712 2016, 37, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Q.; Liu, H.N.; Li, X.X.; Wang, G.Y. Antitumor activity of physcion 8-o-β-glucopyranoside against cervical cancer by induction of apoptosis. Trop. J. Pharm. Res. 2016, 15, 1145–1150. [Google Scholar] [CrossRef]
- Yang, L.; Lin, S.; Kang, Y.; Xiang, Y.; Xu, L.; Li, J.; Dai, X.; Liang, G.; Huang, X.; Zhao, C. Rhein sensitizes human pancreatic cancer cells to EGFR inhibitors by inhibiting STAT3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef]
- Shen, Z.; Shen, Z.; Li, J.; Qin, L. Rhein Augments Antiproliferative Effects of Atezolizumab Based on Breast Cancer (4T1) Regression. Planta Med. 2019, 85, 1143–1149. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Shao, T.; Song, P.; Kong, Q.; Hua, H.; Luo, T.; Jiang, Y. The natural agent rhein induces β-catenin degradation and tumour growth arrest. J. Cell. Mol. Med. 2018, 22, 589–599. [Google Scholar] [CrossRef]
- Li, X.X.; Dong, Y.; Wang, W.; Wang, H.L.; Chen, Y.Y.; Shi, G.Y.; Yi, J.; Wang, J. Emodin as an effective agent in targeting cancer stem-like side population cells of gallbladder carcinoma. Stem Cells Dev. 2013, 22, 554–566. [Google Scholar] [CrossRef]
- Ma, L.; Cheng, Q. Inhibiting 6-phosphogluconate dehydrogenase reverses doxorubicin resistance in anaplastic thyroid cancer via inhibiting NADPH-dependent metabolic reprogramming. Biochem. Biophys. Res. Commun. 2018, 498, 912–917. [Google Scholar] [CrossRef]
- Li, B.; Zhao, X.; Zhang, L.; Cheng, W. Emodin Interferes With AKT1-Mediated DNA Damage and Decreases Resistance of Breast Cancer Cells to Doxorubicin. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Guo, H.; Xiang, Z.; Zhang, Y.; Sun, D. Inhibiting 6-phosphogluconate dehydrogenase enhances chemotherapy efficacy in cervical cancer via AMPK-independent inhibition of RhoA and Rac1. Clin. Transl. Oncol. 2019, 21, 404–411. [Google Scholar] [CrossRef]
- Chen, H.; Wu, D.; Bao, L.; Yin, T.; Lei, D.; Yu, J.; Tong, X. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed. Pharmacother. 2019, 111, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Fenig, E.; Nordenberg, J.; Beery, E.; Sulkes, J.; Wasserman, L. Combined effect of aloe-emodin and chemotherapeutic agents on the proliferation of an adherent variant cell line of Merkel cell carcinoma. Oncol. Rep. 2004, 11, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Savenkova, D. V.; Havrysh, V.; Skripova, V.S.; Ionova, N.E.; Nurgalieva, A.K.; Minigulova, L.F.; Bogdanov, M. V.; Kiyamova, R.G. Physcion Enhances Sensitivity of Pancreatic Adenocarcinoma and Lung Carcinoma Cell Lines to Cisplatin. Bionanoscience 2020, 10, 549–553. [Google Scholar] [CrossRef]
Figure 1.
Molecular subtypes of breast cancer (HR - hormone receptor, ER - estrogen receptor, PR - progesterone receptor, HER2 – human epidermal growth factor receptor 2). The figure was created with BioRender.com (Toronto, Canada) (accessed on 28th of September, 2023).
Figure 1.
Molecular subtypes of breast cancer (HR - hormone receptor, ER - estrogen receptor, PR - progesterone receptor, HER2 – human epidermal growth factor receptor 2). The figure was created with BioRender.com (Toronto, Canada) (accessed on 28th of September, 2023).
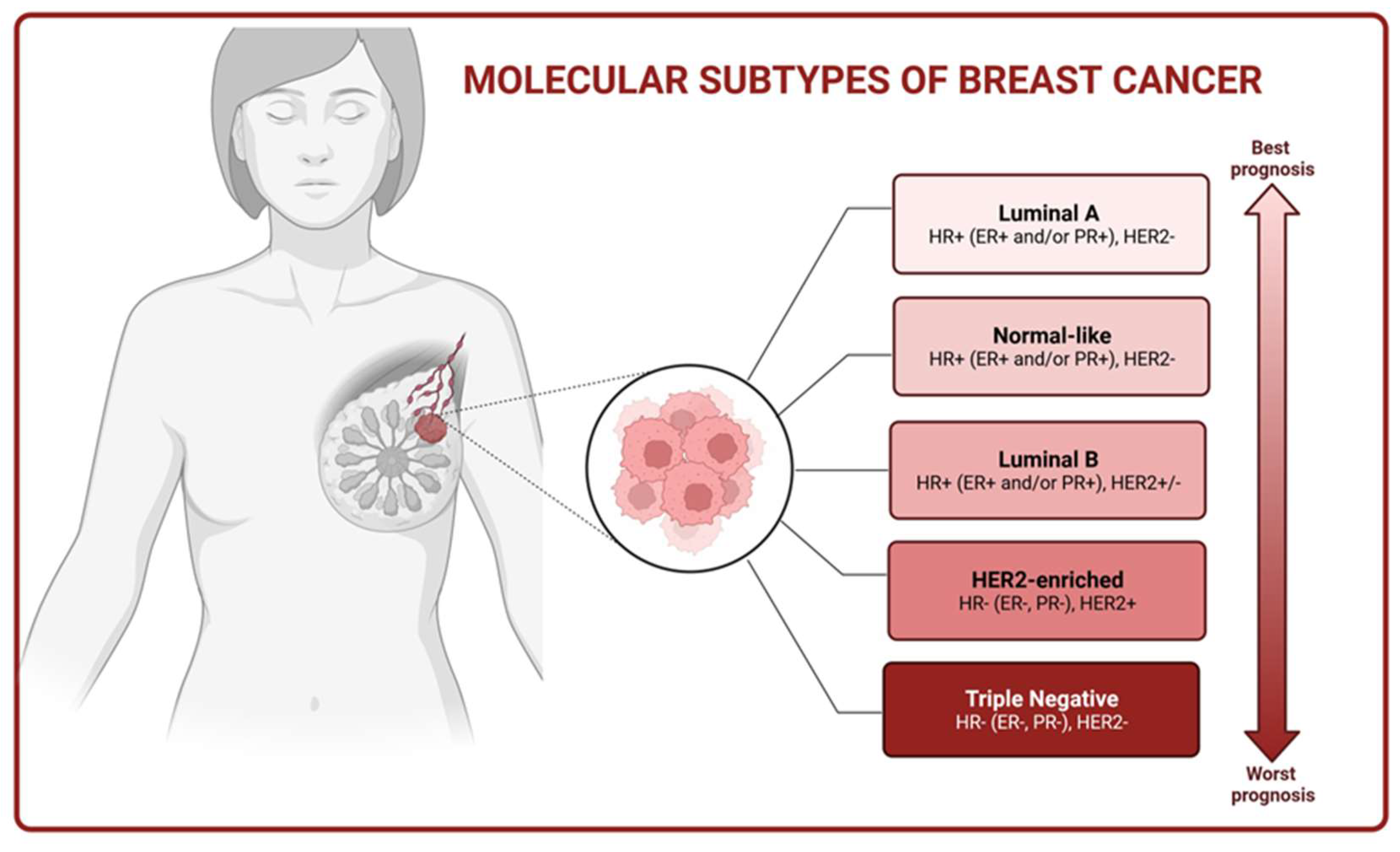
Figure 2.
The chemical structures of an anthraquinone (a) and hypericin (b).
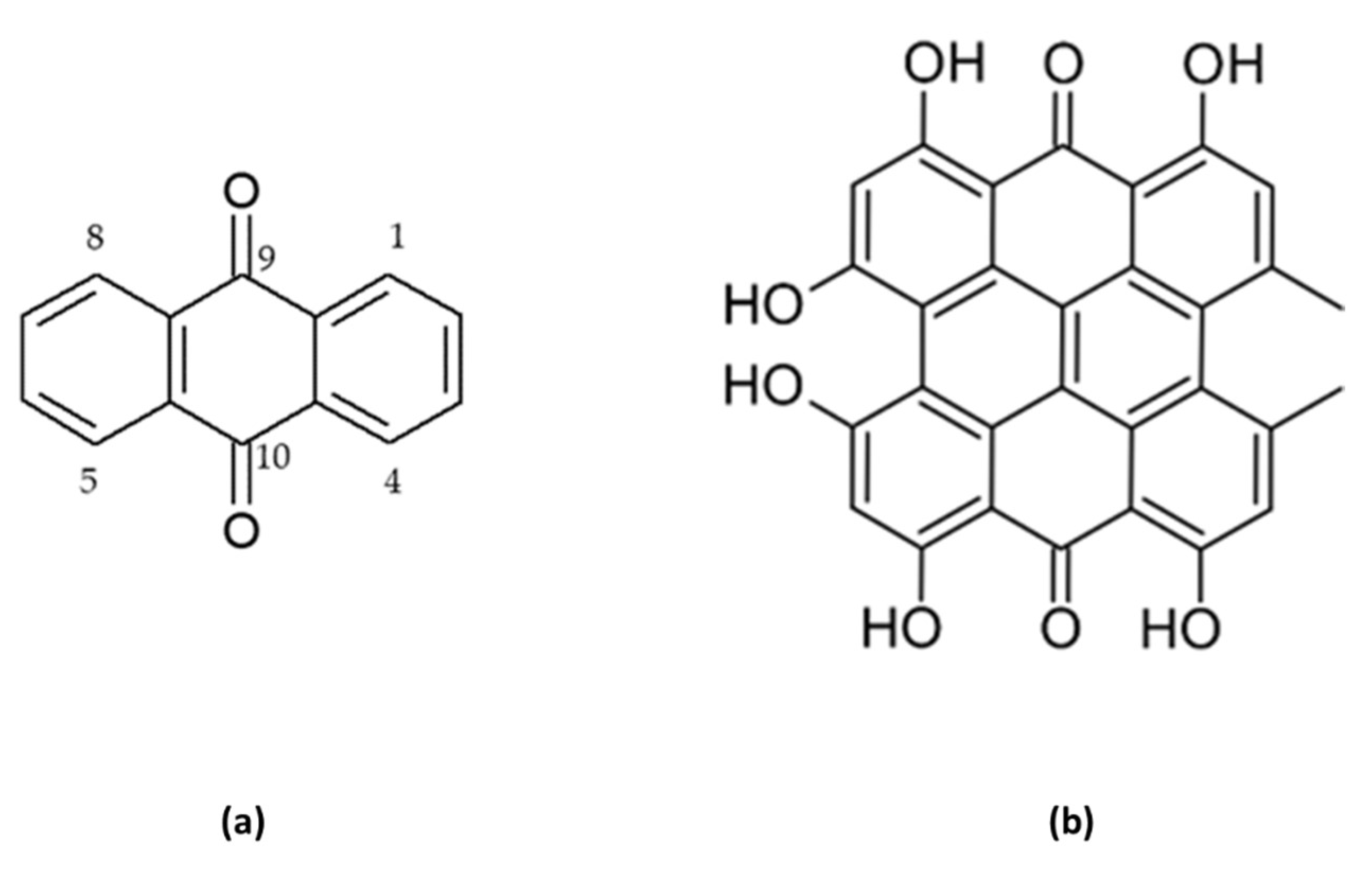
Figure 3.
The chemical structures of the main subgroups of anthraquinones.
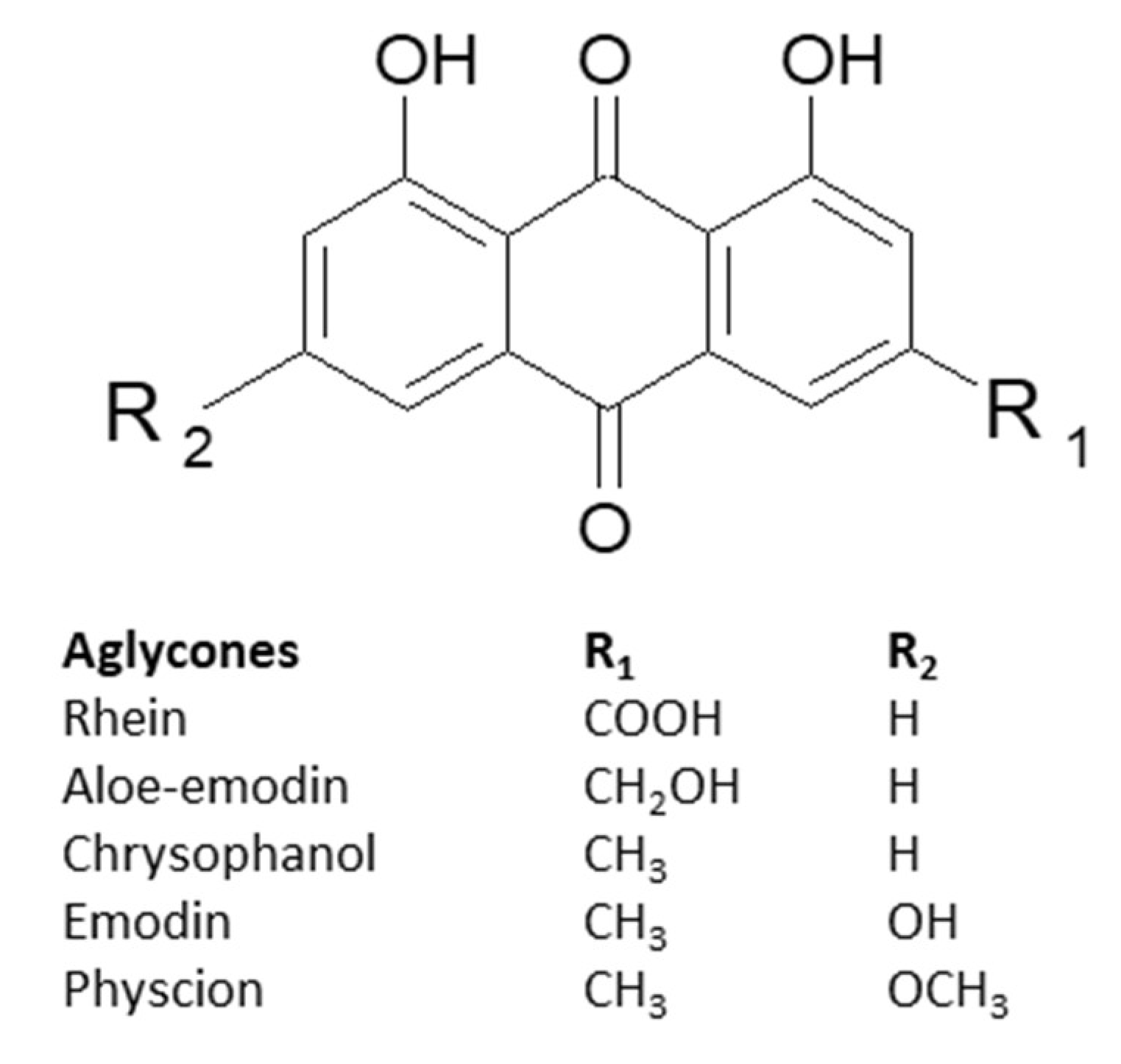

Table 1.
The selected plant species containing anthracene derivatives.
| Botanical family | Gender name | Selected species |
|---|---|---|
| Rhamnaceae |
Rhamnus, Ventilago |
Rhamnus purshiana, Rhamnus frangula, Rhamnus catartica, Ventilago maderaspatana |
| Fabaceae | Cassia | Cassia angustifolia, Cassia senna, Cassia obtusifolia |
| Polygonaceae |
Rheum, Rumex |
Rheum palmatum, Rheum officinale, Rheum rabarbarum Rumex conglumeratus, Rumex pulcher, Rumex alpinus, Rumex maritimus, Rumex obtusifolius |
| Liliaceae |
Aloe, Bulbine |
Aloe ferox, Aloe vera, Bulbine capitate |
| Guttiferae | Hypericum | Hypericum perforatum |
| Rubiaceae |
Morinda, Rubia, Galium, Heterophyllea |
Morinda citrifolia, Morinda officinalis Rubia tinctorum, Galium sinaicum, Galium verum, Heterophyllea pustulata |
Table 2.
Anticancer properties of 1,8-dihydroxyanthraquinones in in vivo xenograft mice models (+ - increase/stimulation, - - decrease/inhibition).
Table 2.
Anticancer properties of 1,8-dihydroxyanthraquinones in in vivo xenograft mice models (+ - increase/stimulation, - - decrease/inhibition).
| Compound | Cancer type | Model | Dosing | Effect | References |
|---|---|---|---|---|---|
| Emodin | colon cancer LS1034 cells |
6-8-weeks male BALB/c nu/nu athymic mice | 40 mg/kg i.p., once every three days for 39 days | G0/G1 phase cell cycle arrest + apoptosis - tumour size (weight and volume) |
[94] |
| Emodin | multidrug resistant gallbladder carcinoma SGC996 cells |
6-weeks BALB/c nu/nu mice | 50 mg/kg i.p. for 15 days | - tumor size + apoptosis - overexpression of MRP1 Sensitizing cisplatin, oxaliplatin and carboplatin |
[95] |
| Emodin | gallbladder cancer SGC-996 GBC-SD cells |
6-weeks BALB/c nu/nu mice | emodin (50 mg/kg i.p.), cisplatin (2 mg/kg i.p.) and emodin/cisplatin every 2 days for 8 days | sensitising effect of emodin to cisplatin - mRNA level of ABCG2 |
[112] |
| Emodin | pancreatic cancer SW1990 cells | 4-6 weeks female BALB/c mice | 40 mg/kg emodin, 126 mg/kg gemcitabine or 40 mg/kg emodin+80 mg/kg gemcitabine every 6 days for 37 days | - tumour size (weight and volume) + tumor integrity + apoptosis -Bcl-Bax ratio + caspase-3 and CytC no effect on VEGFR - MMPs and VEGFR2 receptor expression |
[46] |
| Emodin | pancreatic cancer | 4-6 weeks BALB/c nu/nu mice | 20 and 40 mg/kg p.o. for 8 weeks | - tumour size (weight and volume) - peritoneal dissemination + apoptosis + TUNEL-positive cells - Ki-67, survivin, MMP-9 expression |
[98] |
| Emodin | Blood system cancer K562 cells |
4–6 weeks male BALB/c nude mice | 25, 50, and 100 mg/kg i.p. daily for 12 days | + apoptosis No toxicity - Ki-67 |
[99] |
| Emodin | breast cancer MDA-MB-231 cells |
nude BALB/c nu/nu mice fed a high-fat diet | 400 mg/kg, p. o., q. d. for 4 weeks | Regulation of body weight - cholesterol and fatty acids synthesis - fatty acids oxidation - Fasn, Sed1, Gpat1 genes |
[32] |
| Aloe-emodin 3-O-glucoside | non-small-cell lung cancer A549 cells |
5-6 weeks female BALB/c nude mice | 13 and 26 mg/kg/day for 14 days i.p. | - tumour size (weight and volume) - cell nuclei size + the apoptic DNA fragmentation + caspase 9 and cleaved PARP |
[101] |
| Aloe-emodin | non-small cell lung cancer PC9-GR cells | 4-5 weeks female BALB/c nude mice | 30 mg/kg aloe-emodin, 30 mg/kg gefitinib and their combination | Sensitising gefitinib + E-cadherin -vimetin, Slug and Twist1 proteins |
[102] |
| Chrysophanol | prostate cancer LNCap cells |
4-6 weeks female BALB/c athymic nude mice (nu/nu) | 25 and 50 mg/kg i.p. a day in the form of nanoparticlesfor 28 days | + TUNEL positive cells - tumour size (weight and volume) |
[103] |
| Physcion 8-O-β-D-glucoside | ovarian cancer SK-OV-3/PTX cells |
4-6 weeks male nude mice BALB/c-n | 60 mg/kg, orally in combination with paclitaxel (20 mg/kg, i.p.), twice a week for 21 days | In combination: - P-gp positive tumor cells - tumour size (weight and volume) |
[105] |
| Physcion | breast cancer | 8 weeks male BALB/c nude mice | 30 mg/kg/day i.p. for 2 weeks | + Bax and cleaved caspase-3, - Bcl-xL, Bcl-2, Nrf2, HO-1, SOD-1 and SOD-2 proteins expression in tumor tissue Immunomodulatory action |
[106] |
| Physcion | hepatocellular carcinoma Huh7 cells |
4-6 weeks male BALB/c nude mice | 20 and 40 mg/kg/day i.p. | + TUNEL positive cells - tumour size (weight and volume) + cleaved caspase-3, + caspase-12, overexpression of CHOP - Ki-67 |
[106] |
| Rhein | pancreatic cancer | 5 weeks female athymic BALB/c mice | 60 mg/kg of rhein i.p., 10 mg/kg i.p. of erlotinib | With erlotinib + apoptosis - EGFR and STAT3 pathways - Bcl-2 level + Bax level |
[109] |
| Rhein | breast cancer 4T1 cells |
6–8 weeks female BALB/c mice | 10 mg/kg i.p. rhein every 3 days for a total of three times alone or with 10 mg/kg i.p. atezolizumab once every 2 days for a total of three times | With and without atezolizumab - tumour size (weight and volume) + CD8+ T cells production in the tumor and in the spleen + serum levels of IL-6 and TNF-α |
[110] |
| Rhein | hepatocellular carcinoma HepG2 cells |
5 weeks female nude mice BALB/c-nu | 100 mg/kg/0.2 ml, once a day, i.p. for 3 weeks | - the expression of β-catenin | [111] |
Table 3.
Combinatorial effect of anthraquinones with chemotherapeutic in breast cancer settings (+ - increase/stimulation, - - decrease/inhibition).
Table 3.
Combinatorial effect of anthraquinones with chemotherapeutic in breast cancer settings (+ - increase/stimulation, - - decrease/inhibition).
| Type of cancer | Anthraquinone | Chemotherapeutic | Effect | Ref. |
|---|---|---|---|---|
| Anaplastic thyroid cancer | Physcion 20 µM |
Doxorubicin 0.5 µM |
The inhibition of 6PGD by physcion in combination with doxorubicin sensitizes ATC cells to the treatment | [113] |
| Breast cancer | Rhein 10 mg/kg i.p. every 3 days, 3 times |
Atezolizumab 10 mg/kg i.p. every 2 days, 3 times |
- tumour size + CD8+ T cells production in the tumor and in the spleen + serum levels of IL-6 and TNF-α |
[110] |
| Breast cancer | Emodin 110 µM |
Doxorubicin 5µM |
Sensitisation to doxorubicin + apoptosis + DNA damage in cancer cells + γH2Ax expression - drug resistance to doxorubicin (PI3K-AKT pathway) |
[114] |
| Breast cancer | Emodin 20μM |
5-Fluorouracyl 40 μM |
Sensitisation to 5-fluorouracyl +apoptosis + ROS generation +cyclin dependant kinase inhibitors expression - E2F1expression - notch-regulated ankyrin repeat protein expression |
[49] |
| Breast cancer | Physcion 5 mg/kg, i.p. |
Paclitaxel 5 mg/kg, i.p. |
Inhibition of 6PGD by physcion that results in the sensitization of breast cancer cells to paclitaxelpaclitaxel | [113] |
| Cervical cancer | Physcion 10 mg/kg, i.p. |
Paclitaxel 1 mg/kg, i.p. |
Physcion sensitizes paclitaxel-treated cervical cancer by the inhibition of 6PGD | [115] |
| Gallbladder cancer | Emodin 50 mg/kg i.p., every 2 days for 8 days |
Cisplatin (2 mg/kg i.p. every 2 days for 8 days |
sensitising effect of emodin to cisplatin - mRNA level of ABCG2 no toxicity |
[112] |
| Gallbladder cancer | Emodin 50 µM |
cisplatin 2 µM |
Downregulation of MRP1 protein expression Glutathione depletion Enhanced chemosensitivity + apoptosis |
[95] |
| Hepatocellular carcinoma | Physcion 20 and 40 µM |
Cisplatin 100 nM Paclitaxel 500 nM Doxorubicin 10 nM |
Inhibited growth, induced apoptosis and annexin V level in the combination of physcion with other chemotherapeutics | [116] |
| Merkel cell carcinoma | Aloe-emodin From 5 µM |
Abiplatin > 0.25 µg/mL Adriablastin > 0.025 µg/mL 5-Fluorouracyl > 0.1 µg/mL |
Additive effect with abiplatin, adriablastin, 5-fluorouracyl, - cell proliferation No toxicity |
[117] |
| Non-small cell lung cancer | Aloe-emodin 30 mg/kg i.p. |
Gefitinib 30 mg/kg i.p. |
Sensitising gefitinib + E-adherin - vimetin, Slug and Twist1 proteins |
[102] |
| Lung cancer | Physcion 5-15 µM |
Cisplatin 0-128 µM |
+ ROS level Decreased IC50 of cisplatin by 1.6-2.2 times |
[118] |
| Ovarian cancer | Physcion 8-O-β-D-glucoside 60 mg/kg, orally twice a week for 21 days |
Paclitaxel 20 mg/kg, i.p., twice a week for 21 days |
In combination: - P-gp positive tumor cells - tumour size (weight and volume) |
[105] |
| Pancreatic cancer | Rhein 60 mg/kg i.p. |
Erlotinib 10 mg/kg i.p. |
+ apoptosis - EGFR and STAT3 pathways - Bcl-2 level + Bax level |
[109] |
| Pancreatic cancer | Emodin 40 mg/kg emodin every 6 days for 37 days |
Gemcitabine 80 mg/kg every 6 days for 37 days |
- tumour size (weight and volume) + tumor integrity + apoptosis no toxicity -Bcl/Bax ratio + caspase-3 and CytC expression no effect on VEGFR - MMPs and VEGFR2 receptor expression |
[96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



