
Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
Food allergies are a growing public health concern worldwide, especially in children and young adults. Allergen-specific IgE plays a central role in the pathogenesis of food allergies, but their titers poorly correlate with allergy development. Host immune systems yield allergen-specific immunoglobulin (Ig)A, IgE and IgG subclasses with low or high affinities and differential Fc N-glycosylation patterns that can affect the allergic reaction to food in multiple ways. High affinity IgE is required to induce strong mast cell activation eventually leading to allergic anaphylaxis while low affinity IgE can even inhibit the development of clinically relevant allergic symptoms. IgA and IgG antibodies can inhibit IgE-mediated mast cell activation through various mechanisms, thereby protecting IgE-positive individuals from allergy development. The production of IgE and IgG with differential allergenic potential seems to be affected by the signaling strength of individual B cell receptors, and by cytokines from T cells. This review provides an overview of the diversity of the B cell response and the diverse roles of antibodies in food allergy.
Keywords:
IgE
; B cell response
; allergy
1. Introduction
Food allergies are becoming an increasingly global health concern, especially in urbanized areas, with the prevalence of food allergies rising by about 1.7% between 1997 to 2011 [1]. Allergic reactions to a food antigen are thought to be mainly driven by IgE-mediated activation of mast cells and basophils. They account for most allergies, resulting in symptoms ranging from mild reactions such as hives and itching to more lethal outcomes like anaphylaxis, a severe life-threatening immune reaction [2]. The most common allergenic foods include egg, milk, soy, peanut, and seafood [3]. Allergy management broadly involves avoiding food allergens, undergoing allergen- immunotherapy or taking emergency medications during allergic reactions [4]. Comprehensive studies suggest an age bias in food allergies with infants [5],[6] having more allergies than adults [7]. Also, there is higher prevalence of allergies in women than men after adolescence [8]. Most allergies to food are primed early during childhood between infancy to age 3 when much of the allergen-specific-affinity matured antibodies are accumulated [9]. This is associated with the infant's higher intestinal permeability, increased expression of peptide and sugar transporters at birth that declines during fetal life and adulthood [10],[11],[12]. This may allow increased entry of allergens to the small intestine compared to adults. The developing immune system of infants may also promote sensitization to the ingested food allergens [13].
Nevertheless, very early exposure to egg and peanut allergen, i.e., between 4 to 11 months can enhance allergenic tolerance and decrease the risk of allergies [14]. Conversely, exposure to allergens at an older age may increase the risk of developing allergies [15]. Discrepancies in risk of allergies with early or late exposure to the allergen may result from environmental, genetic factors and timing of allergen exposure. Early susceptibility to a food allergen could promote a short-term or life-long predisposition to the allergic disease. For unknown reasons, some food allergies, like those to milk or egg allergens are often outgrown after childhood [16], while those to peanuts and tree nuts often persist for life [17]. This could be linked to genetic factors, the nature of the allergen and the immune response to the allergen [18],[19],[20],[21],[22],[23]. Some allergens may be good generators of persistent immune memory, induce strong activation of T helper cells and promote/suppress regulatory immune cells [24],[25],[26]. How these factors influence the persistence of an allergy is still unknown.
In the context of type 2 immunity, B cells can undergo differentiation into Be2 cells that secrete IL4 and other cytokines [27]. IL-4 from Be2 cells, basophils or other non-T cells is required to promote the differentiation of naïve T cells into T helper (Th) 2 cells [28],[29],[30]. Through the production of IL-4, Th2 cells eventually promote immunoglobulin class switch to IgE [31]. B cells expressing IgE can rapidly differentiate into plasma cells [32].
Class switch-recombination (CSR) is the rearrangement of the genes coding for the constant regions of antibodies while retaining their antigen-binding region. CSR enables antibodies to change their effector functions and serum half-lifes [36]. This process is associated with the deletion of the gene segments coding for the antibody classes up-stream of the target class. Therefore, CSR is non-reversible. Human IgM+/IgD+, IgA+ and IgG4+ B cells and murine IgM+/IgD+ and IgG1 B cells can class switch to IgE, respectively, but not vice versa. Class-switch from IgM/IgD to IgE is called direct while class switch from human IgA, IgG4 and from murine IgG1 to IgE is called sequential class switch. There is evidence that most IgE in mice is the product of sequential class switch of IgG1+ progenitor cells [33],[34]. CSR to IgE may occur not only in secondary lymphoid tissues but also locally in the mucosa of the nasal cavity, stomach and duodenum [35],[36]. The induced antibody composition may dictate the induction and inhibition of an allergy, which may partly explain the complexity of the clinical findings. Below, we will discuss the roles of various antibody composition features, and how the generation of these antibodies is controlled.
2. The role of low and high-affinity IgE in food allergy
IgE is the least abundant among all antibody isotypes in serum [37]. Even after repeated and long-lasting allergen challenges, it represents only a minor fraction of the total allergen-specific antibodies in serum, suggesting that IgE levels are highly regulated [38]. Its low abundance can be attributed to its comparably short serum half-life of approximately 3 days, and the nature of IgE B cells, which exhibit high rates of apoptosis and little proliferation [39],[40],[41]. Nevertheless, IgE is crucial for the development of type 1 allergic reactions, such as food allergy. Even small quantities of serum IgE efficiently bind to FcεRI receptors on mast cells and basophils. In contrast to its short half-life in serum, IgE bound on mast cells is retained throughout the life cycle of the cell [42]. The FcεRI receptor binds to the Fc region of the IgE molecule with an affinity of 1010 M−1 [43]. In consequence, IgE arms mast cells and basophils with an antigen-specific receptor, which after ingestion of food allergens meditates cross-linking of the FcεRI receptors, eventually leading to the release of histamine and other mediators of the acute allergic reaction [44]. However, the allergenicity of IgE depends on its affinity [45],[46]. Mast cell-bound IgE antibodies with high affinity for their antigens can be cross-linked by trace amounts of antigen. In consequence, the mast cells are activated and may cause allergic anaphylaxis. In contrast, allergen does not efficiently crosslink low-affinity IgE and was shown to prevent anaphylaxis. In addition, low affinity IgE can/may prevent anaphylaxis through competing with high-affinity IgE for binding to FcεRI receptors [47],[48].Though, both high- and low-affinity stimuli of FcεRI elicit similar receptor phosphorylation; the receptor cluster size, mobility, and distribution on mast cells and the down-stream signaling cascade mediated are different, eventually resulting in an altered mast cell response [46]. In accordance with their differential capabilities for mast cell activation, the levels of high-affinity IgE, but of low-affinity IgE correlate with allergic symptoms such as eczema, allergic asthma and anaphylaxis [49],[50],[51].
The physiological role of IgE is poorly understood. It contributes to but is not required for an efficient defense against parasites [52]. Recent data indicate that it is also crucial as a senor of food quality [53],[54]. Natural food consists of a huge number of different substances. In order to minimize uptake of food containing potentially harmful or even toxic molecules, the body has several molecular sensors in the gut that can recognize certain dangerous molecules to provide an early warning to the nervous system, leading to avoidance of this type of food in the future. However, these classical sensors of food quality could recognize only a limited number of harmful substances, leading to nausea and subsequent avoidance behaviour. Now, there is evidence that IgE is another sensor of food quality, which potentially can recognize an unlimited number of potentially harmful substances to promote allergen avoidance behaviour [53],[54],[55]. Of note, food quality sensing requires less IgE-mediated mast cell activation than the development of clinically relevant allergic symptoms and seems to be a feature of the early IgE response. It is possible that early, low-affinity IgE is sufficient as a sensor of food quality while high-affinity IgE formed only later after repeated allergen exposure and appropriate B cell activation leads to allergy. However, this issue needs further investigation [55].
3. The role of antibody isotypes, subclasses and of antibody Fc glycosylation subclasses
Antibodies can exhibit highly diverse functions, ranging from highly inflammatory to anti-inflammatory, and from allergenic to anti-allergenic [37],[56],[57],[58],[59]. The distinct and partly opposing functions of antibodies are based on the high level of heterogeneity antibodies have with respect to isotype subclass, antigen-affinity and FcN-glycosylation pattern. These properties determine their effector functions, i.e. neutralization, opsonization, activation of particular effector cells, complement activation, tissue localization, and eventually their pro- or anti-allergenic capacities [56],[58],[59],[60],[61],[62],[63]. The immune reaction to food antigens generates antibodies of various subclasses and affinities, and temporally changes in the relative ratios of allergenic and protective antibodies seem to have a significant impact on the course of allergy development.
3.1. IgG
While IgE is of major importance for allergy development, in the presence of high allergen doses, IgG-mediated anaphylaxis was also observed in murine models [59]. Murine IgG1, IgG2a and IgGG2b have been shown to promote anaphylaxis through activation of the activating Fcγ receptors (FcγR) FcγRI, FcγRIII, and FcγRIV [64]. IgG-dependent allergic reactions are mediated through secretion of platelet-activating factor (PAF) by neutrophils, monocytes, macrophages and basophils [65]. However, this process requires much higher antigen doses than IgE-mediated anaphylaxis [66]. Whether IgG-mediated anaphylaxis is relevant in patients is a matter of debate [67]. In this context, the food quality sensing function of IgE could be relevant. Food sensing is mediated through low-level mast cell activation, which precedes allergic inflammation and promotes a behavior of allergen avoidance. It has been shown that in the absence of IgE or mast cells, allergen uptake is not avoided, eventually leading to gut inflammation mediated by immune effector mechanisms and antibodies other than IgE [54]. Hence, a low-level, subclinical IgE response may help to avoid the uptake of large allergen quantities required for IgG-mediated anaphylaxis.
Though allergen-specific IgG could be potentially harmful, it often seems to be beneficial for allergic patients. Accordingly, increasing levels of allergen-specific IgGs are associated with the natural resolution of food allergies [67]. Likewise, a positive response to allergen-specific immunotherapy is associated with increased allergen-specific serum IgG [68]. Serum IgG4 is elevated in patients who undergo allergen immunotherapy and has been associated with increased clinical tolerance to specific allergens [69]. Depletion of serum IgG4 from peanut-tolerant patients has been shown to promote stronger mast cell degranulation [68].
The protective effect of IgG is mediated by multiple mechanisms. IgG can block IgE-mediated allergies via allergen neutralization and FCγ-RIIb-mediated inhibition via the IgG inhibitory receptor FcץRIIb [70],[71],[72]. In allergen neutralization, IgG competes with IgE for binding to the allergenic proteins eventually preventing their interaction with IgE. This is an important mechanism by which antibodies provide clinical tolerance to allergic diseases [73]. IgG4 (in humans) and IgG1 (in mice) are clonally related to IgE. Therefore, their antigen-binding sites share the same fine specificity which might be important for efficient competition for allergen binding. In the body fluids, IgG antibodies are present at very higher levels, typically exceeding that of IgE approximately 100-fold or more [33]. The unique blocking properties of IgG4 are associated with their ability to form a Fab arm exchange which allows bispecific antigen recognition thereby interrupting the crosslinking of identical antigens and preventing the formation of immune complexes [74]. Also, IgG4 is unable to activate complement C1q [75]. Accordingly, administration of blocking murine IgG1 against allergen has also been shown to inhibit IgE-mediated anaphylaxis in mice [72].
In addition to its neutralizing activity, IgG can also interact with the FcץRIIb receptor on mast cells and basophils, thereby inhibiting the allergen-IgE-FCεRI activation cascade. Both allergen neutralization and FcץRIIb cross-linking seem to be relevant for IgG-mediated inhibition of IgE-mediated anaphylaxis [72].
3.2. Mucosal IgA
Allergen-specific IgA is also capable of allergen neutralization and is relevant to block IgE-mediated activation of mast cells and basophils [71]. As shown in human samples, a considerable proportion of mucosal IgE is clonally related to IgA [76], indicating that IgA and IgE antibodies share the same antigen binding regions. Mucosal IgA is mostly produced as a dimer that is actively transported to the extracorporeal surface of mucosal epithelial cells [37]. As shown in a murine model of oral immunotherapy, IgA in the mucosal sites binds to allergens and prevents them from penetrating the epithelial barrier and triggering an immune response [71]. Therefore, mucosal IgA can bind to food allergens before they can reach cell-bound IgE and prevents mast cell and basophil degradation in an allergen-specific manner [71]. Since the induction of the most severe and potentially lethal consequence of allergy, systemic anaphylaxis, requires systemic absorption of the ingested allergen [77], the capability of mucosal IgA for allergen neutralization prior to its ingestion, might be of particular relevance for protection from severe allergic reactions.
4. Antibody Ig-Fc glycosylation
Differential Fc N glycosylation of IgG antibodies modulate their binding to activating and inhibiting Fc receptors and inconsequence their impact on the activation of inhibition of innate effector cells, including mast cells [78]. In inflammatory (auto)immune diseases, IgG antibodies with low levels of galactose and sialic acid have been shown to correlate with disease severity. Consistent with the fact that protein glycosylation is an ancient evolutionary development and that sialylated proteins are more likely to be associated with tolerance, non-galactosylated forms of IgA antibodies have also been shown to be associated with inflammatory processes [79].
Allergen-specific IgG subclass glycosylation may also play a role in the inhibition of IgE responses via cross-linking with the IgG inhibitory receptor FcץRIIb or, in the presence of high allergen concentrations, in the induction of IgG-mediated allergic reactions via activating FcץRs [80].
In addition, overall (total) IgG Fc glycosylation may play an important role in the control of IgE- and IgG-mediated allergic reactions. An increase in individuals with higher baseline inflammatory immune states (e.g., obesity, unhealthy diet, altered metabolome and microbiome), which is characterized by low levels of galactosylation and sialylation, may be responsible for more frequent shifts to allergic inflammatory phenotypes. The level of Fc galactosylation and sialylation of the overall (total) serum IgG acts as a vast immunological buffer system by regulating the expression of activating and inhibitory FcץRs and can be controlled, for example, by pregnancy and IVIg treatment [56],[58],[60],[81],[82],[83].
Since IgE is a highly glycosylated antibody isotype, it is very likely that the action of IgE is regulated by its type of glycosylation. In contrast to IgG antibodies, which have one conserved N-glycosylation site at Asn 297 in the Fc portion, murine IgE has nine and human IgE has seven potential N-glycosylation sites [84]. One site does not appear to be coupled by a glycan. The other sites occupied and the coupled glycans are characterized by a conserved pentasaccharide structure of 4 N-acethylglucosamines (GlucNAcs) and three mannoses. One of these glycans is of the high mannose type, which is important for IgE binding to the FcƐRI [85]. The other core glycans are of the complex type and can be further modified with a core fucose, a bisecting N-acetylglucosamine (GlcNAc), as well as one or two galactose residues, each of which can be further capped by a sialic acid [85],[86].
Instead, a recent study claimed that IgE antibodies with high levels of galactosylation and sialylation are associated with allergic severity [87]. In contrast, our work has shown that non-sialylated IgE antibodies have a greater potential to activate mast cells and basophils [88]. Further work is needed to show how IgE glycosylation evolves over the course of allergy severity or after therapy and how differentially glycosylated IgE antibodies function [Figure 1]. Also, the interaction of differentially glycosylated IgE antibodies with FcƐRI(a) and also with soluble or membrane-bound glycan-binding molecules such as IgE binding protein (galectin-3) and CD33, which may be differentially expressed in different inflammatory conditions, needs to be further investigated [89],[90].
5. Development of antibodies in food allergy
5.1. Allergen-specific to T cell activation
Antibody responses to proteins, such as food allergens, are strictly T-dependent [91]. While B cells recognize three-dimensional epitopes Th cells are specific for small peptides presented in MHCII molecules. The initial entrance of the allergen leading to specific sensitization may occur through the skin, gastrointestinal tract, airway or damaged epidermal barrier [92],[93],[94],[95]. In allergic individuals, sensitization results in the formation of Th2 and T follicular helper (Tfh) 13 cells and the formation of allergenic IgE [50]. Subsequently, allergenic re-encounter through food ingestion via the gastrointestinal tract leads to IgE-mediated activation of mast cells and basophils, eventually driving acute allergic symptoms.
Food allergens can access a dysfunctional epithelial barrier and trigger the production of alarmins such as Interleukin 33 (IL33), Interleukin 25 (IL25) and Thymic stromal lymphopoietin (TSLP). These alarmins mediate type 1 hypersensitivity [96] by skewing the T cell response towards the Th2 axis and activating mast cells, dendritic cells, innate lymphoid cells and eosinophils [97],[98]. B cells may also support the generation of Th2 cells and allergy development through the production of IL4, as recently shown in a murine model for allergic asthma [28].
5.2. Production of unmutated, low affinity IgE
During a response to a protein antigen, such as food allergens, activated B cells can follow multiple differentiation pathways. Initially, the extrafollicular pathway of B cell differentiation yields short-lived plasma cells that produce antibodies of relatively low affinity. B cells following this pathway do not introduce much hypermutations into their antigen-binding regions nor do they differentiate into memory cells [99],[100]. Though extrafollicular B cell differentiation might be mainly important during the initial response, it seems to persist for longer periods at least on low level. In a murine model to food allergy, approximately 5% of IgE clones with a considerable expansion rate (more than 50 copies per clone) did not show signs of hypermutation, even after repeated and long-lasting allergen challenge [33]. Hence indicating that an extrafollicular response could continue on a low level during established allergy. To which extent this unmutated, low-affinity IgE may contributes to the inhibition of allergic symptoms has been described above but remains to be further elucidated. At least, low-affinity IgE seems not to correlate with allergic symptoms [49],[50].
5.3. Production of mutated, high affinity IgE
Expressed in a membrane-bound form on the cell surface, antibodies serve as antigen-specific B cell receptor (BCR) which determine the cellular fate at all stages of development [101]. Hypermutated, high-affinity antibodies are the product of the follicular pathway. B cells following the follicular pathway transform primary B cell follicles within secondary lymphoid tissues into germinal centers (GC), where B cells undergo hypermutation, affinity maturation and differentiation into long-lived plasma cells and memory B cells [102],[103] [Figure 2].
GC development requires help from Tfh cells, which provide stimuli such as IL-21 and CD40 essential for induction of hypermutation and positive selection of B cells that have acquired BCR of higher affinity [104],[105]. While Th-derived IL4 is sufficient to induce class switch to IgE and the formation of low-affinity IgE, the generation of high-affinity IgE with anaphylactic properties depends on help from Tfh13 cells which additionally produce high levels of IL-13 and IL-5 together with some IL-21 [50]. Tfh13 cells regulate germinal center responses in type 2 immune reactions and appears to be important for the generation of hypermutated high-affinity IgE and the development of asthma [50]. Genetically modified mice lacking Tfh13 cells show only very low levels of anaphylactic, high-affinity IgE. Tfh13 cells are found in allergic mice and humans with high-affinity IgE to allergens and are further characterized by the expression of the transcription factors BCL6 and GATA3. These cells may represent an interesting target for future therapies of food allergy [106].
A key property of the GC reaction is the generation of memory, mediated by memory B cells and long-lived plasma cells [107]. The pool of long-lived plasma cells secretes antibodies of very high affinity but consist of only a low number of distinct clones. Therefore, they have a limited antigen-binding repertoire [108]. In contrast, memory B cells do not secrete antibodies but provide a backup. Upon antigenic re-stimulation, they can undergo rapid differentiation into antibody-secreting plasma cells. Though the affinities of their antibodies are lower compared to that of long-lived plasma cells, they consist of a higher number of clones that cover a large antigen-binding repertoire [108].
Despite the high-affinity IgE is derived from GCs, the existence of IgE+ memory B cells and long-lived plasma cells is a matter of debate. At least the majority of IgE cells seem to be excluded from these memory compartments. Because of their very low frequency and the possibility of confusion with B cells that bind IgE via their low-affinity receptor CD23, IgE-expressing B cells are difficult to detect without doubt. In addition, GCs typically stain brightly for IgG, but only some IgE is detectable in these tissue structures. Thus, the production of IgE appears to be tightly regulated [109]. Different antibody classes exhibit qualitatively distinct signaling properties. In a model using forced IgE BCR signaling has been shown to induce apoptosis, independent from antigenic stimulation [41]. Accordingly, in the same study, primary IgE+ cells showed a higher rate of apoptosis than IgG1+ cells. Noteworthy, another study confirmed the finding that IgE expression mediates a tonic, antigen-independent signal. This study could not confirm that IgE directly promotes B cell apoptosis, instead independent of antigen binding, it was found to support terminal differentiation into plasma cells, which involves multiple parts of the IgE BCRs as well as Syk, CD19, BLNK, Btk, and IRF4 [39].
Another study showed that though IgE is formed by reactivation of IgG memory cells, signaling of the membrane IgE BCR, but not of the murine IgG1 BCR is required to yield high IgE levels [110]. IgE BCR expression was still found on plasma cells, which is different to IgG which are not expressed any more on the surface of long-lived plasma cells and IgE signaling on plasma cells was found to be relevant for the production of serum IgE.
Together, IgE BCR expression mediates a tonic signal even in the absence of external stimulation by antigen, if that promotes apoptosis or terminal differentiation might be dependent on the model, and under physiological conditions, on additional factors such as the availability of an anti-apoptotic environment. Some IgE+ B cells seem to survive and signal via their membrane-bound IgE receptor which is a crucial regulator of IgE production.
Comparison of IgE+ and IgG1+ murine B cells by whole-genome CRISPR screening showed that IgE+ B cells have distinct properties [111]. Different form IgG+ cells, IgE+ B cells and IgE+ plasma cells showed chronic calcium signaling eventually resulting in BCL2L11-dependent apoptosis. Moreover, there is evidence that after repeated antigenic stimulation in mice, high-affinity IgE-secreting plasma cells are generated through reactivation and further class-switch recombination of IgG1 memory B cells [34]. Together, these findings indicate that the majority of IgE is formed by short-lived plasma cells generated from IgG+ memory B cells.
Nevertheless, next-generation sequencing of the IgG1 and IgE repertoires in a murine model of food allergy indicates that IgE+ cells may follow individual fates. While most IgE clones showed little clonal expansion, a small proportion of highly hypermutated IgE clones exhibited massive clonal expansion, comparable to that of the most expanded IgG1 clones [33]. These data are in accordance with the view that most IgE+ cells show little proliferation and exhibit a short lifetime. But a few hypermutated, GC-derived IgE+ B cells seem to undergo positive selection and proliferation. If these expanded IgE clones that escape from early apoptosis can survive on the long run to enter the memory compartment remains to be elucidated.
5.4. Regulation of IgG to IgE ratios
Most of the class-switched IgE+ cells are derived from IgG+ B cells that underwent further (sequential) class switch to IgE in a follicular B cell response [31],[33],[115]. The mechanisms controlling the relative IgE to IgG production during the allergen-specific immune response are only partly understood so far. Studies from our laboratory investigating the antibody response to hen´s egg in a murine food allergy model indicate that IgE to IgG ratios are controlled on the level of single B cell clones [33]. Most individual clones containing both IgE and IgG1, showed a several-fold excess of IgG1 compared to IgE, i.e., a high IgG1 to IgE ratio. However, fewer, but still a considerable proportion of clones showed a massive excess of IgE, with ratios above 5-fold more IgE than IgG1. Evidence was provided that the differential IgG1 to IgE ratios are due to individual BCR signaling strength which had two consequences. First, strong BCR signaling inhibited sequential class switch from IgG1 to IgE. Second, BCR crosslinking could optimize help from T follicular helper cells producing IL-21, a cytokine that was found to favor IgG1 over IgE production. Hence, on a clonal level, IgG1 to IgE ratios seem to be strongly affected by the individual antibody affinities [33] [Figure 3]. There is increasing evidence that nutrition and metabolic factors can have a strong impact on B cell activation [116], however, their role on class switch, and the relative ratios of allergenic IgE and anti-allergenic IgG remains to be elucidated.
6. Development of differentially glycosylated antibodies
IgG Fc glycosylation (galactosylation and sialylation) are regulated by 2 glycosyltransferases, β1,4-galactosyltransferase 1 (B4galt1) and α2,6-sialyltransferase 1 (St6gal1), in antibody producing B cells [117]. There is evidence that the expression of these enzymes and hence IgG Fc glycosylation is controlled by Tfh cell-derived cytokines [118]. Within the GC, IL-6/IL-23-dependent IL-17A+ TFH17 cells induce a low IgG Fc sialylation program in B cells. How these mechanisms affect the glycosylation of antibodies in the context of allergies is not known.
Early IgE antibodies from extrafollicular plasma cell responses show low hypermutation rates with correspondingly low affinity [33]. These plasma cells may generate IgE antibodies with high levels of galactosylation and sialylation as shown for early extrafollicular IgG antibodies after immunization [118],[119]. IgG antibodies derived from the germinal center show higher mutation rates and lower levels of galactosylation and sialylation [118]. Depending on the co-stimulation inducing the germinal center response, the derived plasma cells produce IgG antibodies with distinct levels of galactosylation and sialylation, but all lower than the initial extrafollicular level of galactosylation and sialylation [118]. In allergy, germinal center-derived plasma cells may produce different IgE/IgG ratios as well as IgE (and IgG) antibodies with reduced levels of galactosylation and sialylation, which may also depend on the co-stimulation that induce the germinal center response. TFH13 cells have recently been linked to IgE antibodies in asthma [50],[120]. It remains to be investigated which Tfh cell subsets induce which IgE glycosylation profile. Whether inflammatory Tfh13 cells or other Tfh cell subsets, such like Tfh17 cells, which are important for the induction of inflammatory glycosylated IgG antibodies [118],[121], can influence the development of inflammatory IgE glycosylation patterns during the germinal center reaction has to be investigated. Different allergen immunotherapies may also induce different Tfh and GC B cell responses and IgE/IgG(4) ratios, as well as IgE (and IgG) antibodies with different glycosylation levels, depending on the type of adjuvant [80]. Accordingly, anti-cytokine therapies that affect the germinal center response may affect the IgE/IgG ratio and/or the IgE (and IgG) glycosylation pattern.
7. Conclusions
The contribution of the B cell responses and the induced antibody compositions to the development or protection from food allergy is complex. Follicular B cell responses to food allergens yield high-affinity IgE crucial to promote the development of severe allergic symptoms. Extrafollicular B cell responses to food allergens yield low-affinity IgE that might be protective. IL4+ Th2 cells are sufficient to drive the production of low-affinity, potentially protective IgE, but the generation of high allergenic high-affinity IgE requires additionally help from Tfh13 cells. Allergen-specific IgG and IgA antibodies can inhibit IgE-mediatet mast cell activation and seem to limit IgE-mediated allergic symptoms in patients. Recent results imply that Fc-N glycosylation of IgG and possibly IgE may also have a considerable impact on their pro/anti-allergenic properties. Antibody affinities, class switch and antibody Fc-N glycosylation are all controlled within the germinal center reaction.
Acknowledgments
Christopher Udoye and Rudolf Manz were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) MA 2273/16-1. Christopher Udoye was supported by the international Research Training Group 1911. Rudolf Armin Manz was supported by the Excellence Cluster “Inflammation at Interfaces” (EXC 306/2). Rudolf Manz acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany`s Excellence Strategy – EXC 22167-390884018.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jackson, K. D.; Howie, L. D.; Akinbami, O. J. Trends in Allergic Conditions among Children: United States, 1997-2011; US Department of Health and Human Services, Centers for Disease Control and …, 2013.
- Simons, F. E. R.; Ardusso, L. R. F.; Bilo, M. B.; Dimov, V.; Ebisawa, M.; El-Gamal, Y. M.; Ledford, D. K.; Lockey, R. F.; Ring, J.; Sanchez-Borges, M. 2012 Update: World Allergy Organization Guidelines for the Assessment and Management of Anaphylaxis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Poowutikul, P.; Pansare, M.; Kamat, D. Food Allergy: A Review. Pediatr. Ann. 2020, 49, e50–e58. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A. T.; Meize-Grochowski, R. Epinephrine Auto-Injectors for Anaphylaxis Treatment in the School Setting: A Discussion Paper. SAGE Open Nurs. 2019, 5, 2377960819845246. [Google Scholar]
- Gupta, R. S.; Springston, E. E.; Warrier, M. R.; Smith, B.; Kumar, R.; Pongracic, J.; Holl, J. L. The Prevalence, Severity, and Distribution of Childhood Food Allergy in the United States. Pediatrics 2011, 128, e9–e17. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R. S.; Warren, C. M.; Smith, B. M.; Blumenstock, J. A.; Jiang, J.; Davis, M. M.; Nadeau, K. C. The Public Health Impact of Parent-Reported Childhood Food Allergies in the United States. Pediatrics 2018, 142. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R. S.; Warren, C. M.; Smith, B. M.; Jiang, J.; Blumenstock, J. A.; Davis, M. M.; Schleimer, R. P.; Nadeau, K. C. Prevalence and Severity of Food Allergies among US Adults. JAMA Netw. Open 2019, 2, e185630–e185630. [Google Scholar] [CrossRef]
- Kurukulaaratchy, R. J.; Karmaus, W.; Arshad, S. H. Gender and Atopy Influences on the Natural History of Rhinitis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 7. [Google Scholar] [CrossRef]
- Nielsen, S. C. A.; Roskin, K. M.; Jackson, K. J. L.; Joshi, S. A.; Nejad, P.; Lee, J.-Y.; Wagar, L. E.; Pham, T. D.; Hoh, R. A.; Nguyen, K. D. Shaping of Infant B Cell Receptor Repertoires by Environmental Factors and Infectious Disease. Sci. Transl. Med. 2019, 11, eaat2004. [Google Scholar] [CrossRef]
- (10) Younoszai, M. K.; Lynch, A. In Vivo D-Glucose Absorption in the Developing Rat Small Intestine. Pediatr. Res. 1975, 9, 130–133. [Google Scholar] [CrossRef]
- Drozdowski, L. A.; Clandinin, T.; Thomson, A. B. R. Ontogeny, Growth and Development of the Small Intestine: Understanding Pediatric Gastroenterology. World J. Gastroenterol. WJG 2010, 16, 787. [Google Scholar]
- Davidson, N. O.; Hausman, A. M.; Ifkovits, C. A.; Buse, J. B.; Gould, G. W.; Burant, C. F.; Bell, G. I. Human Intestinal Glucose Transporter Expression and Localization of GLUT5. Am. J. Physiol.-Cell Physiol. 1992, 262, C795–C800. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.; Haverson, K.; Inman, C.; Harris, C.; Jones, P.; Corfield, G.; Miller, B.; Stokes, C. The Development of the Mucosal Immune System Pre-and Post-Weaning: Balancing Regulatory and Effector Function. Proc. Nutr. Soc. 2005, 64, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Ierodiakonou, D.; Garcia-Larsen, V.; Logan, A.; Groome, A.; Cunha, S.; Chivinge, J.; Robinson, Z.; Geoghegan, N.; Jarrold, K.; Reeves, T. Timing of Allergenic Food Introduction to the Infant Diet and Risk of Allergic or Autoimmune Disease: A Systematic Review and Meta-Analysis. Jama 2016, 316, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.; Barman, M.; Brekke, H. K.; Hesselmar, B.; Johansen, S.; Sandberg, A.-S.; Wold, A. Late Introduction of Fish and Eggs Is Associated with Increased Risk of Allergy Development–Results from the FARMFLORA Birth Cohort. Food Nutr. Res. 2017, 61, 1393306. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.; Sicherer, S.; Wood, R. The Natural History of Food Allergy. J. Allergy Clin. Immunol. Pract. 2016, 4, 196–203. [Google Scholar] [CrossRef]
- Sicherer, S. H.; Urisu, A. Natural History and Prevention of Food Allergy. Food Allergy E-Book Expert Consult Basic 2011, 251. [Google Scholar]
- Chatchatee, P.; Järvinen, K.-M.; Bardina, L.; Beyer, K.; Sampson, H. A. Identification of IgE-and IgG-Binding Epitopes on As1-Casein: Differences in Patients with Persistent and Transient Cow’s Milk Allergy. J. Allergy Clin. Immunol. 2001, 107, 379–383. [Google Scholar] [CrossRef]
- Järvinen, K.-M.; Beyer, K.; Vila, L.; Bardina, L.; Mishoe, M.; Sampson, H. A. Specificity of IgE Antibodies to Sequential Epitopes of Hen’s Egg Ovomucoid as a Marker for Persistence of Egg Allergy. Allergy 2007, 62, 758–765. [Google Scholar] [CrossRef]
- Vila, L.; Beyer, K.; Järvinen, K.-M.; Chatchatee, P.; Bardina, L.; Sampson, H. A. Role of Conformational and Linear Epitopes in the Achievement of Tolerance in Cow’s Milk Allergy. Clin. Exp. Allergy 2001, 31, 1599–1606. [Google Scholar] [CrossRef]
- Sicherer, S. H.; Wood, R. A.; Vickery, B. P.; Jones, S. M.; Liu, A. H.; Fleischer, D. M.; Dawson, P.; Mayer, L.; Burks, A. W.; Grishin, A. The Natural History of Egg Allergy in an Observational Cohort. J. Allergy Clin. Immunol. 2014, 133, 492–499. [Google Scholar] [CrossRef]
- Kostara, M.; Chondrou, V.; Sgourou, A.; Douros, K.; Tsabouri, S. HLA Polymorphisms and Food Allergy Predisposition. J. Pediatr. Genet. 2020, 9, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Grotenboer, N. S.; Ketelaar, M. E.; Koppelman, G. H.; Nawijn, M. C. Decoding Asthma: Translating Genetic Variation in IL33 and IL1RL1 into Disease Pathophysiology. J. Allergy Clin. Immunol. 2013, 131, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Berin, M. C. Mechanisms That Define Transient versus Persistent Food Allergy. J. Allergy Clin. Immunol. 2019, 143, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, A. B.; Qamar, N.; Erickson, K. A.; Kwasny, M. J.; Cai, M.; Szychlinski, C.; Singh, A. M.; Fuleihan, R. L. Cytokine Responses to Egg Protein in Previously Allergic Children Who Developed Tolerance Naturally. Ann. Allergy. Asthma. Immunol. 2014, 113, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Frischmeyer-Guerrerio, P. A.; Guerrerio, A. L.; Chichester, K. L.; Bieneman, A. P.; Hamilton, R. A.; Wood, R. A.; Schroeder, J. T. Dendritic Cell and T Cell Responses in Children with Food Allergy. Clin. Exp. Allergy 2011, 41, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Harris, D. P.; Haynes, L.; Sayles, P. C.; Duso, D. K.; Eaton, S. M.; Lepak, N. M.; Johnson, L. L.; Swain, S. L.; Lund, F. E. Reciprocal Regulation of Polarized Cytokine Production by Effector B and T Cells. Nat. Immunol. 2000, 1, 475–482. [Google Scholar] [CrossRef]
- Song, Z.; Yuan, W.; Zheng, L.; Wang, X.; Kuchroo, V. K.; Mohib, K.; Rothstein, D. M. B Cell IL-4 Drives Th2 Responses in Vivo, Ameliorates Allograft Rejection, and Promotes Allergic Airway Disease. Front. Immunol. 2022, 13, 762390. [Google Scholar] [CrossRef]
- Hurdayal, R.; Ndlovu, H. H.; Revaz-Breton, M.; Parihar, S. P.; Nono, J. K.; Govender, M.; Brombacher, F. IL-4–Producing B Cells Regulate T Helper Cell Dichotomy in Type 1-and Type 2-Controlled Diseases. Proc. Natl. Acad. Sci. 2017, 114, E8430–E8439. [Google Scholar] [CrossRef]
- Hammad, H.; Plantinga, M.; Deswarte, K.; Pouliot, P.; Willart, M. A. M.; Kool, M.; Muskens, F.; Lambrecht, B. N. Inflammatory Dendritic Cells—Not Basophils—Are Necessary and Sufficient for Induction of Th2 Immunity to Inhaled House Dust Mite Allergen. J. Exp. Med. 2010, 207, 2097–2111. [Google Scholar] [CrossRef]
- Looney, T. J.; Lee, J.-Y.; Roskin, K. M.; Hoh, R. A.; King, J.; Glanville, J.; Liu, Y.; Pham, T. D.; Dekker, C. L.; Davis, M. M. Human B-Cell Isotype Switching Origins of IgE. J. Allergy Clin. Immunol. 2016, 137, 579–586. [Google Scholar] [CrossRef]
- He, J.-S.; Narayanan, S.; Subramaniam, S.; Ho, W. Q.; Lafaille, J. J.; Lafaille, M. A. C. de. Biology of IgE Production: IgE Cell Differentiation and the Memory of IgE Responses. IgE Antibodies Gener. Funct. 2015, 1–19. [Google Scholar]
- Udoye, C. C.; Rau, C. N.; Freye, S. M.; Almeida, L. N.; Vera-Cruz, S.; Othmer, K.; Korkmaz, R. Ü.; Clauder, A.-K.; Lindemann, T.; Niebuhr, M. B-Cell Receptor Physical Properties Affect Relative IgG1 and IgE Responses in Mouse Egg Allergy. Mucosal Immunol. 2022, 15, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- He, J.-S.; Subramaniam, S.; Narang, V.; Srinivasan, K.; Saunders, S. P.; Carbajo, D.; Wen-Shan, T.; Hidayah Hamadee, N.; Lum, J.; Lee, A. IgG1 Memory B Cells Keep the Memory of IgE Responses. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Takhar, P.; Smurthwaite, L.; Coker, H. A.; Fear, D. J.; Banfield, G. K.; Carr, V. A.; Durham, S. R.; Gould, H. J. Allergen Drives Class Switching to IgE in the Nasal Mucosa in Allergic Rhinitis. J. Immunol. 2005, 174, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Hoh, R. A.; Joshi, S. A.; Lee, J.-Y.; Martin, B. A.; Varma, S.; Kwok, S.; Nielsen, S. C. A.; Nejad, P.; Haraguchi, E.; Dixit, P. S. Origins and Clonal Convergence of Gastrointestinal IgE+ B Cells in Human Peanut Allergy. Sci. Immunol. 2020, 5, eaay4209–eaay4209. [Google Scholar] [CrossRef]
- Manz, R. A.; Hauser, A. E.; Hiepe, F.; Radbruch, A. Maintenance of Serum Antibody Levels. Annu Rev Immunol 2005, 23. [Google Scholar] [CrossRef]
- Geha, R. S.; Jabara, H. H.; Brodeur, S. R. The Regulation of Immunoglobulin E Class-Switch Recombination. Nat. Rev. Immunol. 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Yang, Z.; Robinson, M. J.; Chen, X.; Smith, G. A.; Taunton, J.; Liu, W.; Allen, C. D. C. Regulation of B Cell Fate by Chronic Activity of the IgE B Cell Receptor. Elife 2016, 5. [Google Scholar] [CrossRef]
- Croote, D.; Darmanis, S.; Nadeau, K. C.; Quake, S. R. High-Affinity Allergen-Specific Human Antibodies Cloned from Single IgE B Cell Transcriptomes. Science 2018, 362, 1306–1309. [Google Scholar] [CrossRef]
- Laffleur, B.; Duchez, S.; Tarte, K.; Denis-Lagache, N.; Péron, S.; Carrion, C.; Denizot, Y.; Cogné, M. Self-Restrained B Cells Arise Following Membrane IgE Expression. Cell Rep. 2015, 10, 900–909. [Google Scholar] [CrossRef]
- Jr, D. M. IgE Receptor and Signal Transduction in Mast Cells and Basophils. Curr. Opin. Immunol. 2008, 20, 717–723. [Google Scholar]
- Metzger, H. The Receptor with High Affinity for IgE. Immunol. Rev. 1992, 125, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Galli, S. J.; Tsai, M. IgE and Mast Cells in Allergic Disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Mita, H.; Yasueda, H.; Akiyama, K. Affinity of IgE Antibody to Antigen Influences Allergen-induced Histamine Release. Clin. Exp. Allergy 2000, 30, 1583–1589. [Google Scholar] [CrossRef]
- Suzuki, R.; Leach, S.; Liu, W.; Ralston, E.; Scheffel, J.; Zhang, W.; Lowell, C. A.; Rivera, J. Molecular Editing of Cellular Responses by the High-Affinity Receptor for IgE. Science 2014, 343, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Chang, X. Low-Affinity but High-Avidity Interactions May Offer an Explanation for IgE-Mediated Allergen Cross-Reactivity. Allergy 2021, 76. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Dolpady, J.; Wabl, M.; Curotto de Lafaille, M. A.; Lafaille, J. J. Sequential Class Switching Is Required for the Generation of High Affinity IgE Antibodies. J. Exp. Med. 2012, 209, 353–364. [Google Scholar] [CrossRef]
- Kelleher, M. M.; Phillips, R.; Brown, S. J.; Cro, S.; Cornelius, V.; Carlsen, K. C. L.; Skjerven, H. O.; Rehbinder, E. M.; Lowe, A. J.; Dissanayake, E. Skin Care Interventions in Infants for Preventing Eczema and Food Allergy. Cochrane Database Syst. Rev. 2022. [Google Scholar]
- Gowthaman, U.; Chen, J. S.; Zhang, B.; Flynn, W. F.; Lu, Y.; Song, W.; Joseph, J.; Gertie, J. A.; Xu, L.; Collet, M. A. Identification of a T Follicular Helper Cell Subset That Drives Anaphylactic IgE. Science 2019, 365, eaaw6433. [Google Scholar] [CrossRef]
- Burrows, B.; Martinez, F. D.; Halonen, M.; Barbee, R. A.; Cline, M. G. Association of Asthma with Serum IgE Levels and Skin-Test Reactivity to Allergens. N. Engl. J. Med. 1989, 320, 271–277. [Google Scholar] [CrossRef]
- Fitzsimmons, C. M.; Falcone, F. H.; Dunne, D. W. Helminth Allergens, Parasite-Specific IgE, and Its Protective Role in Human Immunity. Front. Immunol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Plum, T.; Binzberger, R.; Thiele, R.; Shang, F.; Postrach, D.; Fung, C.; Fortea, M.; Stakenborg, N.; Wang, Z.; Tappe-Theodor, A. Mast Cells Link Immune Sensing to Antigen-Avoidance Behaviour. Nature 2023, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Florsheim, E. B.; Bachtel, N. D.; Cullen, J.; Lima, B. G. C.; Godazgar, M.; Carvalho, F.; Chatain, C. P.; Zimmer, M. R.; Zhang, C.; Gautier, G. Immune Sensing of Food Allergens Promotes Avoidance Behaviour. Nature 2023, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Udoye, C. C.; Manz, R. A. IgE-Mast Cell Mediated Allergy a Sensor of Food Quality. Signal Transduct. Target. Ther. 2023. [Google Scholar]
- Karsten, C. M.; Pandey, M. K.; Figge, J.; Kilchenstein, R.; Taylor, P. R.; Rosas, M.; McDonald, J. U.; Orr, S. J.; Berger, M.; Petzold, D. Anti-Inflammatory Activity of IgG1 Mediated by Fc Galactosylation and Association of FcγRIIB and Dectin-1. Nat. Med. 2012, 18, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, Z. Y. Induction and Suppression of Allergic Diarrhea and Systemic Anaphylaxis in a Murine Model of Food Allergy. J Allergy Clin Immunol 2012, 129. [Google Scholar] [CrossRef] [PubMed]
- Petry, J.; Rahmöller, J.; Dühring, L.; Lilienthal, G.-M.; Lehrian, S.; Buhre, J. S.; Bartsch, Y. C.; Epp, A.; Lunding, H. B.; Moremen, K. W. Enriched Blood IgG Sialylation Attenuates IgG-Mediated and IgG-Controlled-IgE-Mediated Allergic Reactions. J. Allergy Clin. Immunol. 2021, 147, 763–767. [Google Scholar] [CrossRef]
- Strait, R. T.; Posgai, M. T.; Mahler, A.; Barasa, N.; Jacob, C. O.; Köhl, J.; Ehlers, M.; Stringer, K.; Shanmukhappa, S. K.; Witte, D. IgG1 Protects against Renal Disease in a Mouse Model of Cryoglobulinaemia. Nature 2015, 517, 501–504. [Google Scholar] [CrossRef]
- Bieber, K.; Hundt, J. E.; Yu, X.; Ehlers, M.; Petersen, F.; Karsten, C. M.; Köhl, J.; Kridin, K.; Kalies, K.; Kasprick, A. Autoimmune Pre-Disease. Autoimmun. Rev. 2022, 103236. [Google Scholar] [CrossRef]
- Hess, C.; Winkler, A.; Lorenz, A. K.; Holecska, V.; Blanchard, V.; Eiglmeier, S.; Schoen, A.-L.; Bitterling, J.; Stoehr, A. D.; Petzold, D. T Cell–Independent B Cell Activation Induces Immunosuppressive Sialylated IgG Antibodies. J. Clin. Invest. 2013, 123, 3788–3796. [Google Scholar] [CrossRef]
- Lamprecht, P.; Kerstein, A.; Klapa, S.; Schinke, S.; Karsten, C. M.; Yu, X.; Ehlers, M.; Epplen, J. T.; Holl-Ulrich, K.; Wiech, T. Pathogenetic and Clinical Aspects of Anti-Neutrophil Cytoplasmic Autoantibody-Associated Vasculitides. Front. Immunol. 2018, 9, 680. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Marques, O.; Riemekasten, G. Functional Autoantibodies Directed against Cell Surface Receptors in Systemic Sclerosis. J. Scleroderma Relat. Disord. 2017, 2, 160–168. [Google Scholar] [CrossRef]
- Khodoun, M. V.; Kucuk, Z. Y.; Strait, R. T.; Krishnamurthy, D.; Janek, K.; Clay, C. D.; Morris, S. C.; Finkelman, F. D. Rapid Desensitization of Mice with Anti-FcγRIIb/FcγRIII MAb Safely Prevents IgG-Mediated Anaphylaxis. J. Allergy Clin. Immunol. 2013, 132, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Beutier, H.; Gillis, C. M.; Iannascoli, B.; Godon, O.; England, P.; Sibilano, R.; Reber, L. L.; Galli, S. J.; Cragg, M. S.; Rooijen, N. V. IgG Subclasses Determine Pathways of Anaphylaxis in Mice. J. Allergy Clin. Immunol. 2017, 139, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Strait, R. T.; Morris, S. C.; Yang, M.; Qu, X.-W.; Finkelman, F. D. Pathways of Anaphylaxis in the Mouse. J. Allergy Clin. Immunol. 2002, 109, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Kanagaratham, C.; Ansari, Y. S. E.; Lewis, O. L.; Oettgen, H. C. IgE and IgG Antibodies as Regulators of Mast Cell and Basophil Functions in Food Allergy. Front. Immunol. 2020, 11, 3000. [Google Scholar] [CrossRef]
- Santos, A. F.; James, L. K.; Bahnson, H. T.; Shamji, M. H.; Couto-Francisco, N. C.; Islam, S.; Houghton, S.; Clark, A. T.; Stephens, A.; Turcanu, V. IgG4 Inhibits Peanut-Induced Basophil and Mast Cell Activation in Peanut-Tolerant Children Sensitized to Peanut Major Allergens. J. Allergy Clin. Immunol. 2015, 135, 1249–1256. [Google Scholar] [CrossRef]
- Akdis, C. A.; Akdis, M. Mechanisms of Allergen-Specific Immunotherapy and Immune Tolerance to Allergens. World Allergy Organ. J. 2015, 8, 17. [Google Scholar] [CrossRef]
- Shamji, M. H.; Valenta, R.; Jardetzky, T.; Verhasselt, V.; Durham, S. R.; Würtzen, P. A.; van Neerven, R. J. J. The Role of Allergen-specific IgE, IgG and IgA in Allergic Disease. Allergy 2021, 76, 3627–3641. [Google Scholar] [CrossRef]
- Ansari, Y. S. E.; Kanagaratham, C.; Burton, O. T.; Santos, J. V.; Hollister, B.-M. A.; Lewis, O. L.; Renz, H.; Oettgen, H. C. Allergen-Specific IgA Antibodies Block IgE-Mediated Activation of Mast Cells and Basophils. Front. Immunol. 2022, 13, 881655. [Google Scholar] [CrossRef]
- Strait, R. T.; Morris, S. C.; Finkelman, F. D. IgG-Blocking Antibodies Inhibit IgE-Mediated Anaphylaxis in Vivo through Both Antigen Interception and FcγRIIb Cross-Linking. J. Clin. Invest. 2006, 116, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Vickery, B. P.; Lin, J.; Kulis, M.; Fu, Z.; Steele, P. H.; Jones, S. M.; Scurlock, A. M.; Gimenez, G.; Bardina, L.; Sampson, H. A. Peanut Oral Immunotherapy Modifies IgE and IgG4 Responses to Major Peanut Allergens. J. Allergy Clin. Immunol. 2013, 131, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, R.; Karagiannis, S. N.; Jordakieva, G.; Jensen-Jarolim, E. The Role of IgG4 in the Fine Tuning of Tolerance in IgE-Mediated Allergy and Cancer. Int. J. Mol. Sci. 2020, 21, 5017. [Google Scholar] [CrossRef] [PubMed]
- Lilienthal, G.-M.; Rahmöller, J.; Petry, J.; Bartsch, Y. C.; Leliavski, A.; Ehlers, M. Potential of Murine IgG1 and Human IgG4 to Inhibit the Classical Complement and Fcγ Receptor Activation Pathways. Front. Immunol. 2018, 9, 958. [Google Scholar] [CrossRef]
- Coker, H. A.; Durham, S. R.; Gould, H. J. Local Somatic Hypermutation and Class Switch Recombination in the Nasal Mucosa of Allergic Rhinitis Patients. J. Immunol. 2003, 171, 5602–5610. [Google Scholar] [CrossRef]
- Strait, R. T.; Mahler, A.; Hogan, S.; Khodoun, M.; Shibuya, A.; Finkelman, F. D. Ingested Allergens Must Be Absorbed Systemically to Induce Systemic Anaphylaxis. J. Allergy Clin. Immunol. 2011, 127, 982–989. [Google Scholar] [CrossRef]
- Li, T.; DiLillo, D. J.; Bournazos, S.; Giddens, J. P.; Ravetch, J. V.; Wang, L.-X. Modulating IgG Effector Function by Fc Glycan Engineering. Proc. Natl. Acad. Sci. 2017, 114, 3485–3490. [Google Scholar] [CrossRef]
- Yeo, S. C.; Cheung, C. K.; Barratt, J. New Insights into the Pathogenesis of IgA Nephropathy. Pediatr. Nephrol. 2018, 33, 763–777. [Google Scholar] [CrossRef]
- Epp, A.; Hobusch, J.; Bartsch, Y. C.; Petry, J.; Lilienthal, G.-M.; Koeleman, C. A. M.; Eschweiler, S.; Möbs, C.; Hall, A.; Morris, S. C. Sialylation of IgG Antibodies Inhibits IgG-Mediated Allergic Reactions. J. Allergy Clin. Immunol. 2018, 141, 399–402. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J. V. Anti-Inflammatory Activity of Immunoglobulin G Resulting from Fc Sialylation. science 2006, 313, 670–673. [Google Scholar] [CrossRef]
- Buhre, J. S.; Becker, M.; Ehlers, M. IgG Subclass and Fc Glycosylation Shifts Are Linked to the Transition from Pre-to Inflammatory Autoimmune Conditions. Front. Immunol. 2022, 13, 1006939. [Google Scholar] [CrossRef] [PubMed]
- Seeling, M.; Pöhnl, M.; Kara, S.; Horstmann, N.; Riemer, C.; Wöhner, M.; Liang, C.; Brückner, C.; Eiring, P.; Werner, A. Immunoglobulin G-Dependent Inhibition of Inflammatory Bone Remodeling Requires Pattern Recognition Receptor Dectin-1. Immunity 2023, 56, 1046–1063. [Google Scholar] [CrossRef] [PubMed]
- Plomp, R.; Hensbergen, P. J.; Rombouts, Y.; Zauner, G.; Dragan, I.; Koeleman, C. A. M.; Deelder, A. M.; Wuhrer, M. Site-Specific N-Glycosylation Analysis of Human Immunoglobulin e. J. Proteome Res. 2014, 13, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.-T. C.; Platzer, B.; Washburn, N.; Mani, V.; Bartsch, Y. C.; Conroy, M.; Pagan, J. D.; Bosques, C.; Mempel, T. R.; Fiebiger, E. A Single Glycan on IgE Is Indispensable for Initiation of Anaphylaxis. J. Exp. Med. 2015, 212, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Seeling, M.; Brückner, C.; Nimmerjahn, F. Differential Antibody Glycosylation in Autoimmunity: Sweet Biomarker or Modulator of Disease Activity? Nat. Rev. Rheumatol. 2017, 13, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.-T. C.; Conroy, M. E.; Washburn, N.; Kitaoka, M.; Huynh, D. J.; Laprise, E.; Patil, S. U.; Shreffler, W. G.; Anthony, R. M. Sialylation of Immunoglobulin E Is a Determinant of Allergic Pathogenicity. Nature 2020, 582, 265–270. [Google Scholar] [CrossRef]
- Dühring, L.; Petry, J.; Lilienthal, G.-M.; Bartsch, Y. C.; Kubiak, M.; Pfeufer, C.; Lehrian, S.; Buhre, J. S.; Lunding, H. B.; Kern, C. Sialylation of IgE Reduces FcεRIα Interaction and Mast Cell and Basophil Activation in Vitro and Increases IgE Half-life in Vivo. Allergy 2023. [Google Scholar] [CrossRef]
- Gao, P.; Simpson, J. L.; Zhang, J.; Gibson, P. G. Galectin-3: Its Role in Asthma and Potential as an Anti-Inflammatory Target. Respir. Res. 2013, 14, 1–9. [Google Scholar] [CrossRef]
- Duan, S.; Koziol-White, C. J.; Jester, W. F.; Smith, S. A.; Nycholat, C. M.; Macauley, M. S.; Panettieri, R. A.; Paulson, J. C. CD33 Recruitment Inhibits IgE-Mediated Anaphylaxis and Desensitizes Mast Cells to Allergen. J. Clin. Invest. 2021, 129. [Google Scholar] [CrossRef]
- Adler, L. N.; Jiang, W.; Bhamidipati, K.; Millican, M.; Macaubas, C.; Hung, S.; Mellins, E. D. The Other Function: Class II-Restricted Antigen Presentation by B Cells. Front. Immunol. 2017, 8, 319. [Google Scholar] [CrossRef]
- Izadi, N.; Luu, M.; Ong, P. Y.; Tam, J. S. The Role of Skin Barrier in the Pathogenesis of Food Allergy. Children 2015, 2, 382–402. [Google Scholar] [CrossRef] [PubMed]
- Jakwerth, C. A.; Ordovas-Montanes, J.; Blank, S.; Schmidt-Weber, C. B.; Zissler, U. M. Role of Respiratory Epithelial Cells in Allergic Diseases. Cells 2022, 11, 1387. [Google Scholar] [CrossRef] [PubMed]
- Splunter, M. van; Liu, L.; Neerven, R. J. J. van; Wichers, H. J.; Hettinga, K. A.; Jong, N. W. D. Mechanisms Underlying the Skin-Gut Cross Talk in the Development of IgE-Mediated Food Allergy. Nutrients 2020, 12, 3830. [Google Scholar] [CrossRef] [PubMed]
- Asero, R.; Antonicelli, L. Does Sensitization to Foods in Adults Occur Always in the Gut? Int. Arch. Allergy Immunol. 2010, 154, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Khodoun, M. V.; Tomar, S.; Tocker, J. E.; Wang, Y. H.; Finkelman, F. D. Prevention of Food Allergy Development and Suppression of Established Food Allergy by Neutralization of Thymic Stromal Lymphopoietin, IL-25, and IL-33. J. Allergy Clin. Immunol. 2018, 141, 171–179. [Google Scholar] [CrossRef]
- Divekar, R.; Kita, H. Recent Advances in Epithelium-Derived Cytokines (IL-33, IL-25 and TSLP) and Allergic Inflammation. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, C. E.; Ranjbar, M.; Omer, H.; Cusack, R. P.; Gauvreau, G. M. The Role of Airway Epithelial Cell Alarmins in Asthma. Cells 2022, 11, 1105. [Google Scholar] [CrossRef]
- MacLennan, I. C. M.; Toellner, K.-M.; Cunningham, A. F.; Serre, K.; Sze, D. M.-Y.; Zúñiga, E.; Cook, M. C.; Vinuesa, C. G. Extrafollicular Antibody Responses. Immunol. Rev. 2003, 194, 8–18. [Google Scholar] [CrossRef]
- Elsner, R. A.; Shlomchik, M. J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef]
- Mackay, F.; Figgett, W. A.; Saulep, D.; Lepage, M.; Hibbs, M. L. B-cell Stage and Context-dependent Requirements for Survival Signals from BAFF and the B-cell Receptor. Immunol. Rev. 2010, 237, 205–225. [Google Scholar] [CrossRef]
- Finney, J.; Yeh, C.-H.; Kelsoe, G.; Kuraoka, M. Germinal Center Responses to Complex Antigens. Immunol. Rev. 2018, 284, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M. J.; Weisel, F. Germinal Center Selection and the Development of Memory B and Plasma Cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M. J.; Vries, V. C. D.; Wasiuk, A.; Guo, Y.; Noelle, R. J. Molecular Mechanism and Function of CD40/CD40L Engagement in the Immune System. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular Helper CD4 T Cells (Tfh). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Berin, M. C. Targeting Type 2 Immunity and the Future of Food Allergy Treatment. J. Exp. Med. 2023, 220, e20221104. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, L. M.; Tarlinton, D. M. Regulation of Germinal Center Responses, Memory B Cells and Plasma Cell Formation—an Update. Curr. Opin. Immunol. 2016, 39, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Chong, A. S.; Ansari, M. J. Heterogeneity of Memory B Cells. Am. J. Transplant. 2018, 18, 779–784. [Google Scholar] [CrossRef]
- Wade-Vallance, A. K.; Allen, C. D. C. Intrinsic and Extrinsic Regulation of IgE B Cell Responses. Curr Opin Immunol 2021, 72. [Google Scholar] [CrossRef]
- Schmitt, M. E. R. The B-Cell Antigen Receptor of IgE-Switched Plasma Cells Regulates Memory IgE Responses. J Allergy Clin Immunol 2020, 146. [Google Scholar] [CrossRef]
- Newman, R.; Tolar, P. Chronic Calcium Signaling in IgE+ B Cells Limits Plasma Cell Differentiation and Survival. Immunity 2021, 54, 2756–2771. [Google Scholar] [CrossRef]
- Asrat, S.; Kaur, N.; Liu, X.; Ben, L.-H.; Kajimura, D.; Murphy, A. J.; Sleeman, M. A.; Limnander, A.; Orengo, J. M. Chronic Allergen Exposure Drives Accumulation of Long-Lived IgE Plasma Cells in the Bone Marrow, Giving Rise to Serological Memory. Sci. Immunol. 2020, 5, eaav8402. [Google Scholar] [CrossRef] [PubMed]
- Luger, E. O. Induction of Long-Lived Allergen-Specific Plasma Cells by Mucosal Allergen Challenge. J Allergy Clin Immunol 2009, 124. [Google Scholar] [CrossRef] [PubMed]
- Wu, L. C.; Scheerens, H. Targeting IgE Production in Mice and Humans. Curr Opin Immunol 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Panetta, V.; Cappella, A.; Hofmaier, S.; Hatzler, L.; Rohrbach, A.; Tsilochristou, O.; Bauer, C.-P.; Hoffmann, U.; Forster, J. IgG and IgG4 to 91 Allergenic Molecules in Early Childhood by Route of Exposure and Current and Future IgE Sensitization: Results from the Multicentre Allergy Study Birth Cohort. J. Allergy Clin. Immunol. 2016, 138, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Jellusova, J. Metabolic Control of B Cell Immune Responses. Curr. Opin. Immunol. 2020, 63, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ise, W.; Harazono, A.; Takakura, D.; Fukuyama, H.; Baba, Y.; Narazaki, M.; Shoda, H.; Takahashi, N.; Ohkawa, Y. Sialylation Converts Arthritogenic IgG into Inhibitors of Collagen-Induced Arthritis. Nat. Commun. 2016, 7, 11205. [Google Scholar] [CrossRef]
- Bartsch, Y. C.; Eschweiler, S.; Leliavski, A.; Lunding, H. B.; Wagt, S.; Petry, J.; Lilienthal, G.-M.; Rahmöller, J.; Haan, N. de; Hölscher, A. IgG Fc Sialylation Is Regulated during the Germinal Center Reaction Following Immunization with Different Adjuvants. J. Allergy Clin. Immunol. 2020, 146, 652–666. [Google Scholar] [CrossRef]
- Buhre, J. S.; Pongracz, T.; Künsting, I.; Lixenfeld, A. S.; Wang, W.; Nouta, J.; Lehrian, S.; Schmelter, F.; Lunding, H. B.; Dühring, L. MRNA Vaccines against SARS-CoV-2 Induce Comparably Low Long-Term IgG Fc Galactosylation and Sialylation Levels but Increasing Long-Term IgG4 Responses Compared to an Adenovirus-Based Vaccine. Front. Immunol. 2023, 13. [Google Scholar] [CrossRef]
- Tong, X.; Guan, C.; Ji, T.; Cao, C.; Jiang, J.; Liu, M.; Guo, Q.; Zhou, P.; Gong, F. Increased Circulating T Follicular Helper 13 Subset Correlates with High IgE Levels in Pediatric Allergic Asthma. Eur. J. Immunol. 2022, 52, 2010–2012. [Google Scholar] [CrossRef]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H. U.; Culemann, S.; Harre, U.; Ackermann, J. A.; Seefried, M.; Kleyer, A.; Uderhardt, S. Regulation of Autoantibody Activity by the IL-23–TH17 Axis Determines the Onset of Autoimmune Disease. Nat. Immunol. 2017, 18, 104–113. [Google Scholar] [CrossRef]
Figure 1.
Role of subclasses and antibody Fc glycosylation in allergy: Allergen-specific IgG with low levels of sialylation and galactosylation promote IgG-driven allergy via the FcγRs on Neutrophils, Basophils, Macrophages or inhibit allergy via the FcγRIIb receptor. High level of IgG sialylation and galactosylation promote upregulation of inhibitory immune receptors FcγRIIb. The function of glycosylated IgE in allergy is still under debate. IgE: Immunoglobulin E, IgG: Immunoglobulin G, MC: Mast cells, NEU: Neutrophils, BAS: Basophils, FcεRI: Fc epsilon RI, FCγ-RIIb: Fc-gamma RII-b, FCγ: Fc-gamma.
Figure 1.
Role of subclasses and antibody Fc glycosylation in allergy: Allergen-specific IgG with low levels of sialylation and galactosylation promote IgG-driven allergy via the FcγRs on Neutrophils, Basophils, Macrophages or inhibit allergy via the FcγRIIb receptor. High level of IgG sialylation and galactosylation promote upregulation of inhibitory immune receptors FcγRIIb. The function of glycosylated IgE in allergy is still under debate. IgE: Immunoglobulin E, IgG: Immunoglobulin G, MC: Mast cells, NEU: Neutrophils, BAS: Basophils, FcεRI: Fc epsilon RI, FCγ-RIIb: Fc-gamma RII-b, FCγ: Fc-gamma.
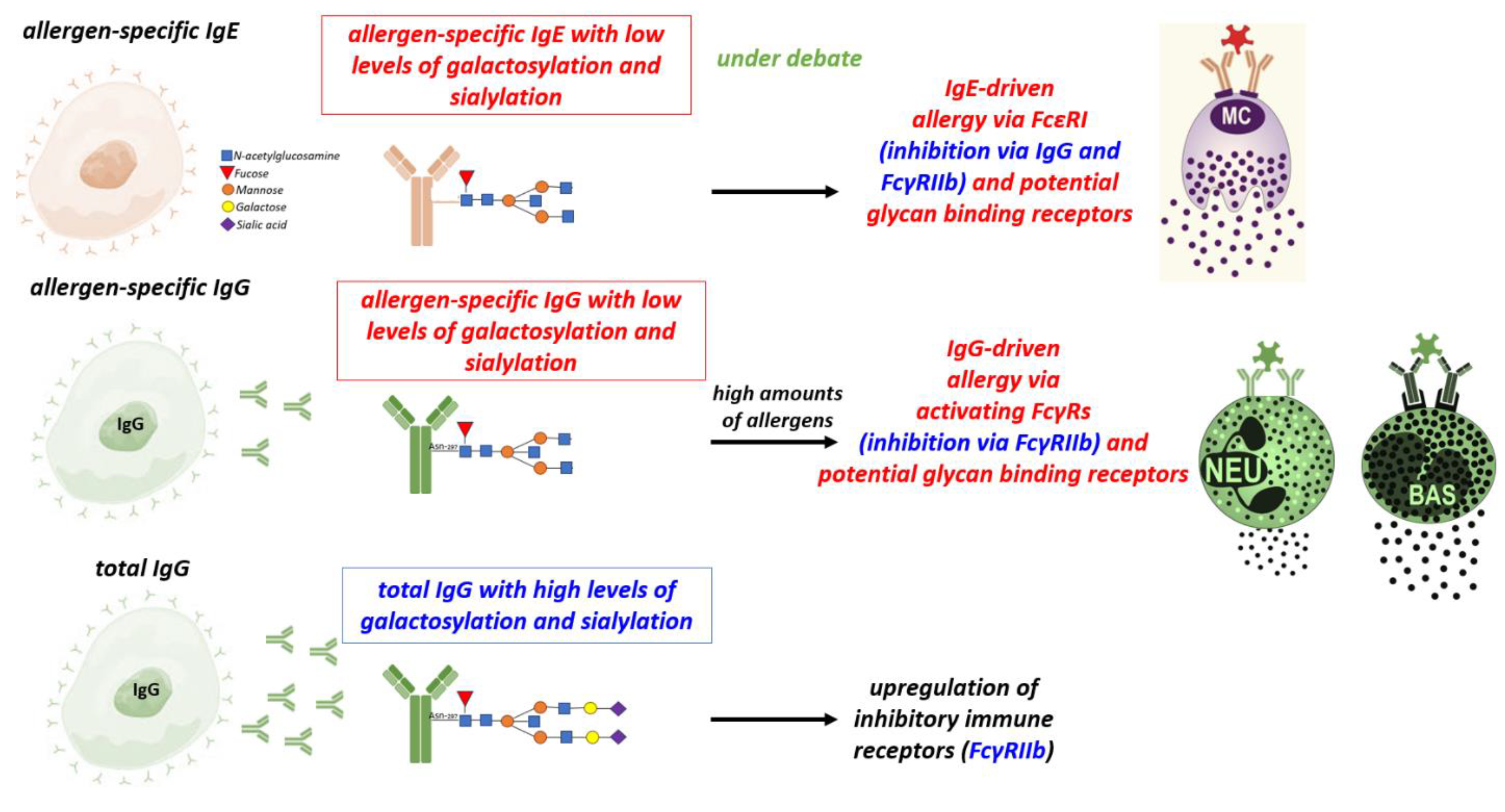
Figure 2.
The B cell response to allergens and its impact on mast cell activation. Immune response to food allergens induces follicular and extrafollicular B cell response which yield plasma cells secreting hypermutated (high-affinity) and unmutated (low affinity) IgE, and IgG with differential impact on the activation of mast cells and disease development. Ig: Immunoglobulin, IgE: Immunoglobulin E, IgG: Immunoglobulin G, IgM: Immunoglobulin M, IgD: Immunoglobulin D, FcεRI: Fc epsilon RI, FCγ-RIIb: Fc-gamma RIIb.
Figure 2.
The B cell response to allergens and its impact on mast cell activation. Immune response to food allergens induces follicular and extrafollicular B cell response which yield plasma cells secreting hypermutated (high-affinity) and unmutated (low affinity) IgE, and IgG with differential impact on the activation of mast cells and disease development. Ig: Immunoglobulin, IgE: Immunoglobulin E, IgG: Immunoglobulin G, IgM: Immunoglobulin M, IgD: Immunoglobulin D, FcεRI: Fc epsilon RI, FCγ-RIIb: Fc-gamma RIIb.
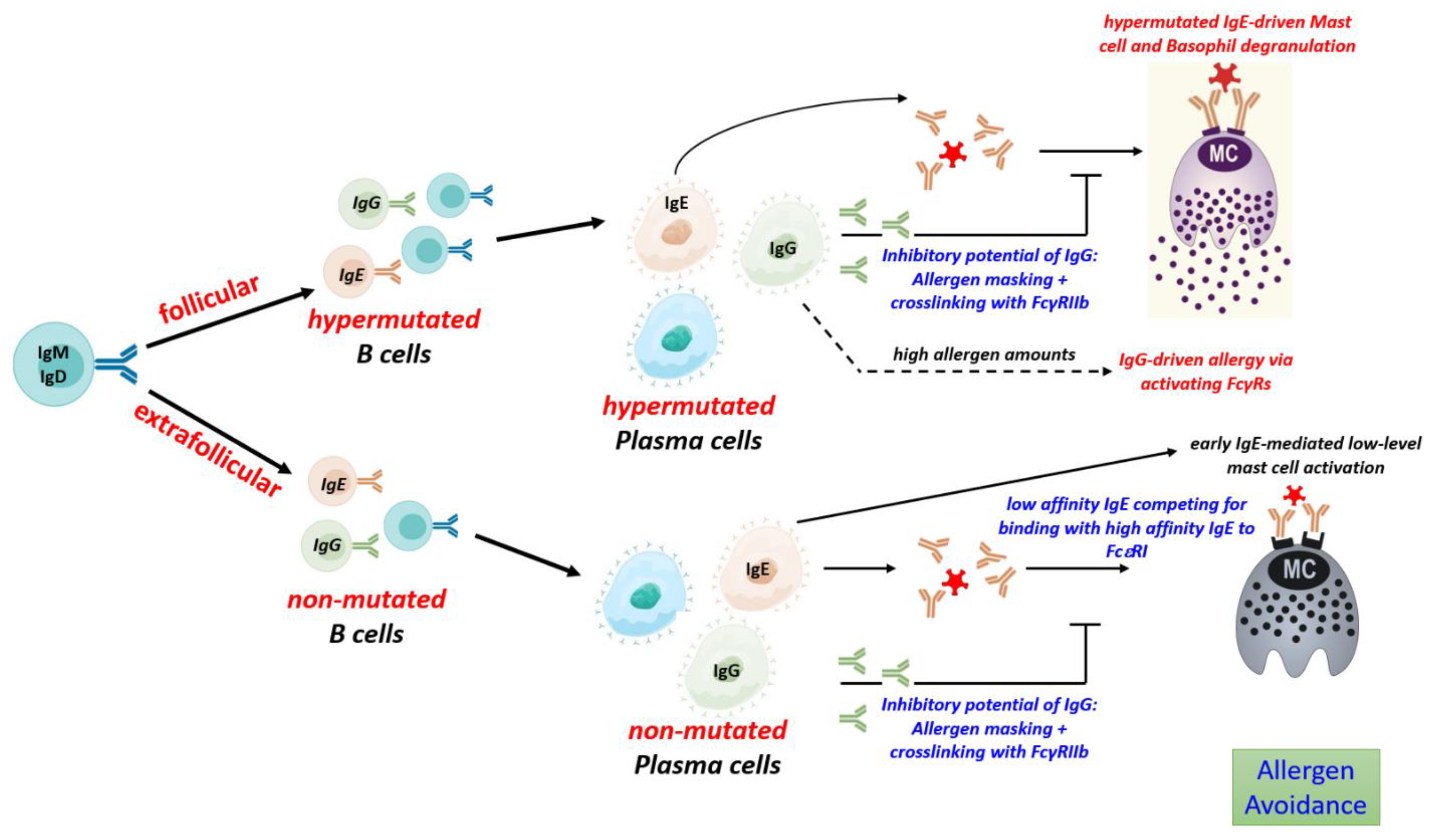
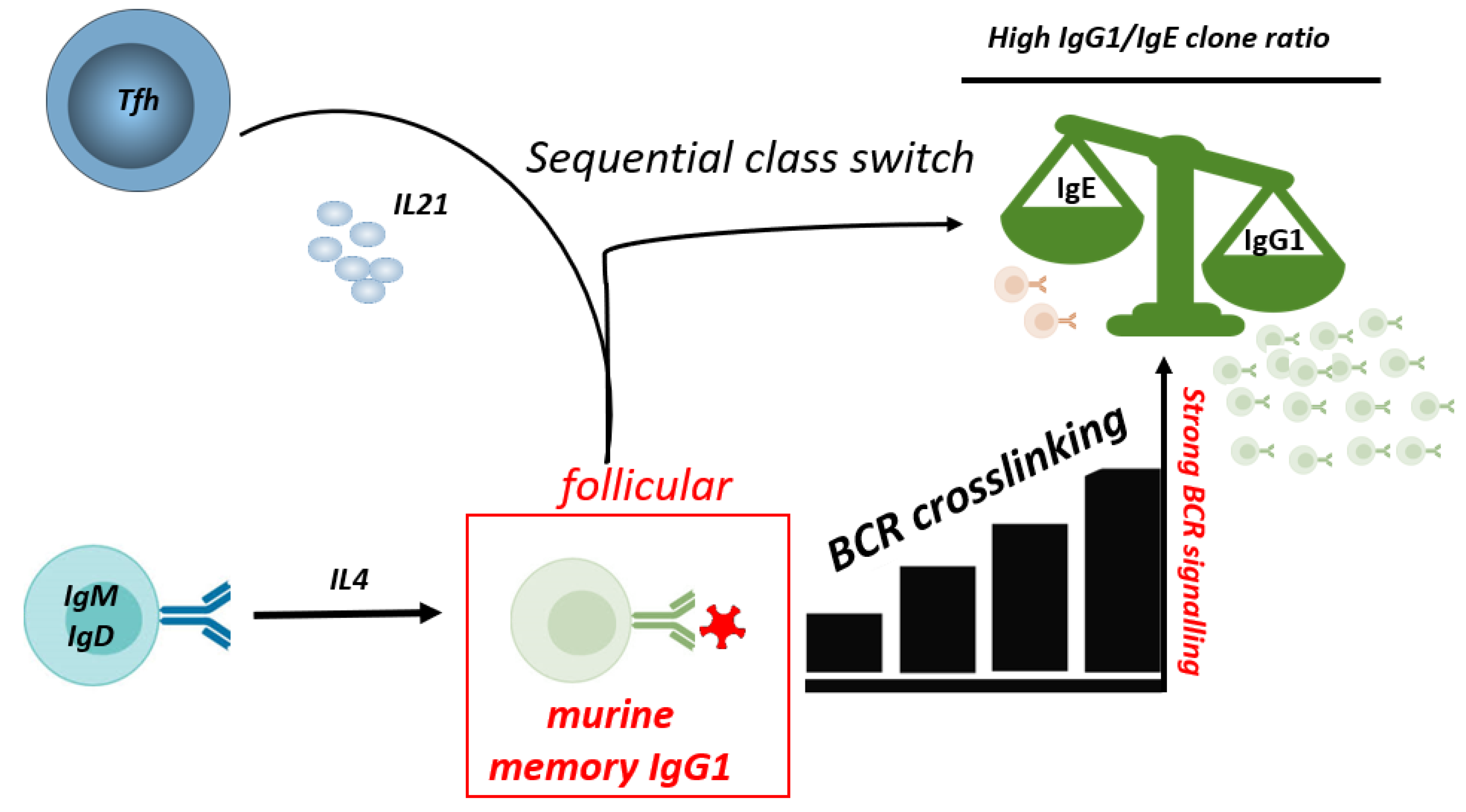
Figure 3.
BCR signaling strength and IL-21 affect IgE/IgG1 ratios. Strong BCR signaling of mouse IgG1 B cells and IL-21 from Tfh cells constraints sequential class-switch to IgE, thereby reducing the IgE/IgG1 ratio. IgE: Immunoglobulin E, IgG1: Immunoglobulin G1, IgM: Immunoglobulin M, IgD: Immunoglobulin D, BCR: B cell receptor, IL-4: Interleukin 4, IL-21: Interleukin 21.
Figure 3.
BCR signaling strength and IL-21 affect IgE/IgG1 ratios. Strong BCR signaling of mouse IgG1 B cells and IL-21 from Tfh cells constraints sequential class-switch to IgE, thereby reducing the IgE/IgG1 ratio. IgE: Immunoglobulin E, IgG1: Immunoglobulin G1, IgM: Immunoglobulin M, IgD: Immunoglobulin D, BCR: B cell receptor, IL-4: Interleukin 4, IL-21: Interleukin 21.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



