Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
There are compelling evidences that the immune system is to confer protection not only against foreign pathogens but also against cancer. But if the immune system can’t detect every malignant cell, and at the same time a suitable tumor microenvironment (TME) is formed by inflammatory processes, a carcinogenesis is not longer to be blocked. Once the tumor cells have found their niche, the tumor cells are trained by immune cells turned inside out in how to deal with the immune system "out there". The TME is a training camp and protected space for tumor cells. The Corona crisis has revived a therapeutic approach that was thought to be almost dead: RNA vaccines. Despite decades of research and many clinical studies, no registered RNA cancer vaccine exists today. But the SARS-CoV-2 RNA vaccines against COVID-19 disease have demonstrated their dangerous weaknesses. RNA vaccines manipulate the innate immunity and make the body susceptible to any viral infection. There is also the possibility of integration into the genome. The few approved therapeutic cancer vaccines show little effect. Despite some successes, immunotherapies remain ineffective for most patients with cancer. Powerful therapeutic cancer vaccines remain an unfulfilled dream, a misconception.
Keywords:
Cancer biology
; tumor immunology
; neoantigens
; T cell exhaustion
; immunosenescence
; cancer vaccines
; RNA vaccines
1. Introduction
“Take home the feeling of revolution - groundbreaking - breakthrough - game-changing - vaccination on demand “… With such slogans biotech companies promote their cancer vaccines. But very few things are actually groundbreaking or even a revolution. Nevertheless, the SARS-CoV-2 crisis has virtually created a new hype on cancer vaccines. Especially RNA vaccines are seen as a therapeutic wonder weapon against various cancer types. Let’s have a critical look at the status quo of vaccine developments, the complex biology behind cancer with emphasis of known natural limitations for immune interventions. Is a vaccine able to circumvent the main barriers and to cure this disease?
2. Basic discoveries
The first evidence of immunotherapy against cancer comes from the Egyptian physician Imhotep in 2600 BC [1]. He infected a disclosed breast cancer ulcer with a bacteria-contaminated poultice. Of course, immunology was completely unknown at that time. Imhotep observed something different: the infection causes fever and the fever kills the tumor cells. Fever is a form of hyperthermia. In Greek mythology, heat had sacral significance and treatment by fever was equated with the life-giving power of the sun. Many healers of that time believed that if they could control body temperature, they could cure all diseases, including cancer. The Greek scholar Parmenides (540-470 BC) claimed, "Give me the power to produce fever and I will cure all disease." No less than Hippocrates (460-370 BC) also believed in the healing power of heat [2].
In 1863 Rudolf Virchow discovered active leucocytes in tumor tissues and it was Paul Ehrlich in 1909 who postulated a “body’s own protection system” against tumor cells. “The immune surveillance hypothesis” was born and elaborated by Thomas and Burnet in the 1950s and ‘60s: T-cell-mediated immunity evolved as a specific defense against cancer cells and that T cells constantly patrol the body, searching for abnormal body cells. Until today there are many clinical data demonstrating the correlation between the immune system/ surveillance and the development of tumors: spontaneous remission of colon carcinoma, acute myeloid leukemia, or remission of lung and liver metastases of lung cancer, non-small cell lung cancer (NSCLC). As long as the tumor load is controlled by the immune system, as long as this balance between disease and innate and acquired immunity works, the life is not threatened. The same is for infectious diseases [3].
In the 1890s William B. Coley injected streptococcal organisms in patients with solid tumors (“Coley’s Toxins”) to activate the immune system. Coley (1862-1936) was an American bone surgeon and pioneer of cancer immunotherapy. He was convinced that post-surgical infections had helped patients to recover better from their cancer by provoking an immune response. Because of severe adverse effects due to the living streptococcal organisms, he switched to using dead bacteria. But Coley’s published results were difficult to interpret with confidence. "More research would be needed to determine what benefit, if any, this therapy might have for people with cancer" (American Cancer Society). Nevertheless, Coley is known as the "Father of Immunotherapy" [4].
There are compelling evidences that the immune system is to confer protection not only against foreign pathogens but also against cancer. If this were not so, the body would collapse within a short time. Our live would be a short episode only. Every day abnormal cells are produced somewhere in the body, are recognized by the immune system and eliminated.
But if the immune system can’t detect every malignant cell, and at the same time a suitable tumor microenvironment (TME) is formed by inflammatory processes, a carcinogenesis is not longer to be blocked. The immune surveillance fails.
3. The biology of cancer cells
Let's start with a brief and incomplete overview of the complex biology of cancer cells. Completeness is beyond the scope of this review and would fill a textbook. This overview is important to identify therapeutic targets. Only some essential stages in carcinogenesis can be discussed here, which I have summarized in seven molecular hallmarks:
3.1. Uncontrolled growth by manipulation of the cell cycle
Most cells are renewed throughout life. Maintaining the actual state of the whole organism is a prerequisite for health. Each healthy cell of the body has a predetermined number of cell divisions, then automatic cell death, apoptosis, occurs. Cell division takes place according to a predetermined program, called cell cycle. The cell cycle is monitored at three checkpoints by different proteins to ensure DNA integrity. The first checkpoint is the G1 checkpoint at the transition from G1 to the S stage. The second checkpoint is the G2 checkpoint at the transition of G2 into mitosis. The third and final checkpoint is the spindle checkpoint into mitosis.
The main gatekeeper of the G1 checkpoint is the retinoblastoma protein (pRb). Probably the best known checkpoint protein is p53, a tumor suppressor protein, G1 and G2 checkpoint. P53 is known as the guardian of the genome. Who controls the cell cycle determines the weal or woe of the body. In 50-60 % of all cancers, the gene for p53 is mutated. P53 is described as "the most frequently mutated gene in human cancer" and was voted "Molecule of the Year" in 1993 [5]. Like p53, pRb can mutate, and cancer cells pass unhindered through the G1 checkpoint. The pRb was the first tumor suppressor protein to be discovered. The name is derived from retinoblastoma, a malignant tumor in the retina of the eye.
3.2. Apoptosis is switched off in cancer cells
The two most powerful weapons for suppressing cancer cells are stopping the cell cycle for repair and apoptosis. After successful repair, the cell returns to the cycle and divides. If the DNA damage is irreparable, apoptosis occurs.
For this purpose, genes are started by p53 (transcription factor), which initiate programmed cell death. At the same time, p53 suppresses the formation of new blood vessels, which tumor cells additionally need due to their increased metabolism. If p53 itself is damaged, faulty repairs may occur or damaged cells that should be eliminated escape apoptosis.
Cellular suicide is programmed in each cell by genes. The signal for suicide can come from outside or inside the cell. If the signal comes from outside, it is picked up by receptors on the surface. These receptors are called death receptors because they trigger a chain of signals that mean the death of the cell.
The external signals, the ligands, that bind at the death receptors are molecules from a protein family called tumor necrosis factor (TNF) such as TNF-alpha. Large amounts of TNF-alpha are released by immune cells in response to infection. Cancer cells not only checkmate the two main cell cycle guards p53 and the retinoblastoma protein, they also manipulate the suicide program to turn off apoptosis. Bcl-2 molecules are the antagonist of apoptosis. If too many Bcl-2 proteins are in the cell, suicide does not occur. In cancer cells, the gene for Bcl-2 is mutated. The result is an overproduction of the Bcl-2 protein. Too much BcL-2 suppresses apoptosis. The resistance of tumor cells to radiation and chemotherapy stems from the absence of apoptosis [6,7].
3.3. Telomerase - the key to immortality
Telomeres were discovered as early as the early 1930s and were thought to be protective caps for the ends of chromosomes. Telomeres are DNA sequences of the type TTAGGG-TTAGGG-TTAGGG-TTAGGG repeated thousands of times and located at the end of chromosomes. They help control cell division, with a piece of telomere lost with each division. When the telomere has reached a critical minimum length, the cell stops dividing - the aging process has begun. With each duplication of chromosomes, hundreds of TTAGGG's are lost. A newborn starts with 100% telomere length. At 35 years of age, this length has already decreased by 25% and a person of 65 years of age has only 50% of the original length.
The telomere is built during embryogenesis by telomerase. After that, the enzyme is active only in immature germ cells and some stem cells, otherwise it is dormant until the cell degenerates. In many tumor cells, telomerase is active, preventing, among other things, growth suppression and helping to turn off apoptosis. The telomere is constantly replenished, allowing the tumor cell to undergo an infinite number of cell divisions [8,9,10]. The immortality of the cancer cell is preprogrammed. It should be emphasized that the manipulation of the cell cycle without the manipulation of telomerase would almost come to nothing. Only the unhindered activity of telomerase enables tumor growth.
3.4. The unstable tumor genome
Proto-oncogenes are weak spots in our genome. They are widely distributed on different cell types and can transform into an oncogene at any time. As an oncogene, they cause a mutated protein or a significant overproduction of their protein. Both cause a cell to degenerate. If the overproduction affects receptors, the subsequent signal chains run at full speed and permanently activate or deactivate proteins and genes. The switching on and off of genes, their regulation, is still only partially understood. Even less do we understand the deregulation of genes that precedes every cancer event. We can only superficially describe regulation and deregulation.
About 100 proto-oncogenes, mostly growth genes, are known today. Different proteins contribute to the growth of a cell, like the different growth factors. Many growth factors are directly involved in the cell cycle. Cells that are dormant in the G0 region are stimulated by growth factors to return to the G1 stage. Other growth factors are necessary for cells to enter the S stadium from G1. Still other growth factors are mitogens that trigger mitosis.
In addition to growth factors, their receptors are also proto-oncogenes. Growth factor and receptor are spatially separated from each other. One cell produces the factor, and the other cell has the receptor for it. This is another control mechanism of the cells. It prevents a cell from stimulating itself to grow on a whim. The spatial separation only applies to healthy cells. Since tumor cells proliferate continuously, they have established their own self-sufficient supply of growth factors.
One very well studied factor is EGF, epidermal growth factor, which is produced by cells of the brain, salivary glands, kidneys and other organs. EGF is the signal to the cell to initiate mitosis. In many tumors, the receptor for the growth factor EGF is mutated. The proto-oncogene has become an oncogene.
EGFRvIII is the abbreviation for the mutated EGF receptor (EGFR), which is frequently found in brain tumors, prostate carcinomas, ovarian carcinomas and small-cell bronchial carcinomas. The abbreviation refers to a crippled form of receptor that is no longer able to bind a ligand on the cell surface. This would actually be the end of the matter with the growth factor: no ligand, no stimulus, no growth. However, the modified receptor part that reaches into the cell interior is highly active and no longer needs an external stimulus. EGFRvIII sends non-stop growth signals into the cell. This phenomenon is called "autocrine loop", in which a cell secretes the signal for growth in order to receive it again itself. The tumor cell controls its own growth and is independent of external factors. There is a small glimpse of light to break this growth autonomy. EGFRvIII is found exclusively on tumor cells and not on healthy ones. This is where the therapeutic approach lies: novel drugs aim to specifically switch off EGFRvIII. They block the receptor and suppress its signals within the cell.
EGFRvIII is a special form of the EGF receptor. Even the pathological overproduction of the normal EGF receptor is sufficient to turn a healthy cell into a tumor cell. A healthy cell contains between 20,000 and 200,000 EGF receptors. A tumor cell can contain up to 2,000,000 receptors. Of these, up to 60% of the overproduced receptors are EGFRvIII cripple receptors. EGF receptor overproduction is associated with resistance to chemotherapy and poor prognosis [11,12,13].
3.5. Tumor microenvironment (TME)
Pre-cancerous cells need a niche in which they can thrive and prosper to cancer cells. This niche is the TME and protects the malignant cells against the immune system. Apart from malignant cells, immune cells, fibroblasts, tube-forming endothelial cells and their surrounding pericytes (for generation of the tumor vasculature and lymphatics), and other cells form the TME. The non-cancerous cell mass can comprise > 50% of the tumor mass. These non-cancerous cells are suggested to support cancer development and progression. The tumor and its microenvironment are closely related with an intensive intercellular communication. Without the support of normal tissue and stromal cells cancerous cells would not evolve and would be unable to manifest the disease.
The niche is a space that is separated from the surrounding healthy tissue by its own cell barrier. In the niche, the tumor cells are protected from attack by the immune system. A dedicated blood supply brings nourishment to the cancer cells. The tumor cells live together peacefully as in a commune with other cells, exchanging information and supporting each other. Even more: the other non-cancer cells become willing service providers for the few cancer cells, see Figure 1.
Non-cancerous cells in the commune include stromal cells that form the connective tissue. They are fibroblasts that organize the extracellular tissue. The extracellular tissue is mainly composed of various fibers that store water. The extracellular tissue is a kind of buffer between the cells, balancing the cell movements. In the niche, the healthy fibroblasts become cancer-associated fibroblasts (CAF - cancer-associated fibroblast) with new tasks. The CAFs secrete various enzymes whose task is to destroy the extracellular tissue, also called extracellular matrix. This makes room for new tumor cells. At the same time, the CAFs use messenger substances to stimulate the formation of new vessels in the niche [14,15,16,17].
3.6. The immune surveillance fails
Timely detection and elimination of cancer cells is the essence of immune surveillance. Once the tumor cells have found their niche, the immune system has a hard time. Because in the niche, the tumor cells are trained by immune cells turned inside out in how to deal with the immune system "out there". In the niche, only one sets the tone, the tumor cell. All other cells are submissive service providers. The training within the niche is called tumor editing.
In 2002 Robert Schreiber et al. proposed the „tumor immunoediting“ [18]: tumor elimination, equilibrium with the immune system, and escape from control. It’s a process between immune surveillance and tumor formation in three phases. Tumor editing is training on the job for tumor cells. This is a model of how the transition from immune surveillance to tumor formation is imagined. Reality may differ from it. But the model offers a plausible explanation and is supported by animal experimental data.
Tumor Editing Phase 1: elimination of tumor cells
As long as isolated tumor cells form somewhere in the body, immune cells are on the spot. For example, the natural killer cells (NK cells). They play an important role in the innate defense against microbial invaders and in recognizing and killing tumor cells. Killer cells scan every body cell in their area of operation for the presence of MHC I, the recognition proteins for "self." Macrophages also hunt down tumor cells because there is no protective niche yet. With a delay, cytotoxic T lymphocytes (CTL) intervene in the fight against tumor cells.
Tumor Editing Phase 2: equilibrium
A niche does not yet exist. But there is a stalemate between tumor cells and immune cells. The stalemate can be explained by the fact that tumor cells that have escaped elimination have learned and are dressing themselves in an immunologically inconspicuous manner. There is nothing on their surface to indicate a degenerate cell. MHC I has disappeared in many tumor cells. Interferon gamma (IFN-γ) is responsible for this. IFN-γ exerts a selection pressure on the tumor cells, which leads to the fact that only those tumor cells survive which are immunologically inconspicuous. Under the influence of IFN-γ, the CTLs cells are on a brake, called PD-1 and CTLA-4, which we will discuss later. The braked killer cells do not attack tumor cells. The tumor cells remain in a dormant state. This can last weeks, months, perhaps even years. The tumor cells neither grow nor shrink during this phase. They are stuck in stage G0 of the cell cycle. The immune defense has nothing to do. The few tumor cells that drop their mask and reveal themselves as cancer are immediately destroyed by CTLs. But some of them manage to escape once again.
Tumor editing phase 3: escape
Tumor cells also mutate during the equilibrium phase. They simply have to mutate because they do not have a stable genome like a healthy cell and are constantly exposed to the selection pressure of the immune system. The resistant tumor cells eventually prevail and break the blockade of the immune defense. Tumor cells secrete growth factors and other soluble molecules into their environment. They create a niche for themselves and recruit cells as roommates for their cancer commune. Only now have the tumor cells arrived in the niche. And only now do the laws of the niche, such as oxygen deficit and hypoxia-inducible factors (HIF) activation, take effect. Tumor-associated macrophages (TAM) appear, transcription factors like NF-kappaB go haywire and, and, and [19].
The weapon of microorganisms in the fight against the immune system is their strong reproductive activity. The weapons of tumor cells against the immune system are their many tricks to escape surveillance. Escape from immunosurveillance is considered a cancer hallmark.
3.7. Contact inhibition
Among the many tricks a tumor cell uses to evade immune surveillance is to disrupt cell-to-cell contact. Cell-to-cell contact is necessary, among other things, to stop growth as soon as cells physically touch each other. This growth arrest is called contact inhibition. Contact inhibition is mediated by direct cell-to-cell contact. Various molecules on the surface of the cells are involved in this, which are grouped together as Contact Adhesion Molecules (CAM). Five different CAMs exist, all of which appear simultaneously on the cell surface. The contacted cell A has the same five CAMs as the contact-seeking cell B.
The CAMs of cell A combine with the CAMs of cell B. Already the loss of only one contact molecule from the group of five leads to the loss of the entire contact inhibition. No sooner have the tumor cells arrived in the niche than they rid themselves of at least one of the five CAM contact molecules. One of the CAMs is a protein called E-cadherin (E-cad). At the top of the hit list is E-Cad because of its proximity to the EGF receptor. The EGF receptor ensures growth and is needed by the tumor cell. Once the contact inhibition is resolved, the tumor cells can invade the surrounding tissue [20,21].
4. Short interim summary
The evolutionary mandate of life means to adapt to the environment, to permanently improve the genetic configuration, everything to do for survival only. The multidrug resistance pump system (MDR) in many cancer cells underlines this survival strategy, and immunediting is only another mechanism of the evolutionary survival strategy.
From what has been said so far, it is clear that the transformation of a healthy cell into a tumor cell is made possible by a multitude of mutations that accumulate. On the one hand, mechanisms within the cell that are designed for growth are affected; on the other hand, the tumor cell develops strategies to escape destruction by immune cells. The changes affect signaling pathways, metabolism, receptors, growth factors, adhesion molecules, to name a few. Which of these changes can actually be influenced by vaccines? Interaction with a vaccine always requires an antigenic structure on the tumor cell surface.
The Corona crisis has revived a therapeutic approach that was thought to be almost dead. Companies such as Moderna and BioNTech have launched mRNA vaccines against SARS-CoV-2. The oldest German RNA vaccine company, CureVac, failed to gain marketing approval due to lack of efficacy. All three companies were founded with the goal of developing RNA vaccines against cancer, CureVac in 2000, BioNTech in 2008 and Moderna in 2010. There are NO registered RNA cancer vaccines. On the other hand, there are again current announcements that RNA vaccines against cancer will soon be launched on the market.
Let's turn to cancer vaccine development and critically examine different aspects.
5. The immunogenicity of tumor cells
Like all cells in the body, tumor cells produce different proteins on the cell surface. They are all potential antigens because they can be recognized by antibodies and CTL- killer cells. Therein lies the definition of an antigen: an antigen triggers an immune response.
5.1. Harakiri of tumor cells
As immune surveillance has shown, this works quite well. If the tumor cell undergoes too many mutations, the antigens on the surface also change and immune surveillance lags behind. If these robust tumor cells escape surveillance, they flee to a protected niche and proliferate there, not to mention that immune cells in the niche are reprogrammed and no longer attack the tumor. These immune cells have become tolerant. But not all of them. There are still immune cells that continue to recognize tumor antigens and eliminate the tumor cells. These immune cells can be dangerous to the tumor. From the tumor's point of view, they have to go. For this purpose, the tumor cells reach deep into their bag of tricks: they kill themselves voluntarily - Harakiri!
Harakiri has been foreseen by biology only for exceptional cases like diseased cells. There is another exceptional case: Harakiri of the tumor cell so that other tumor cells can survive. Particularly aggressive cancer types with poor prognosis often contain relatively high levels of constitutively apoptotic cells [22,23].
That sounds paradoxical at first. When a tumor cell dies by apoptosis, cell debris is produced. Among them are the many tumor antigens. The cell debris is quickly cleared away by macrophages. Thus, fragments of tumor antigens enter the antigen presenting cells (APCs), which activate the CTLs. In most cases, however, complete activation does not occur because these tumor antigens have little or no immunogenicity. The semi-activated T cells die. That is, these antigens cannot elicit an immune response. T cell activation requires three signals: the presentation of antigen (signal 1), co-stimulatory molecules B7 and CD28 (signal 2) and pro-inflammatory molecules (signal 3). If only one signal is missing or the antigen is not sufficiently immunogenic (see above), the T cell dies on apoptosis [24].
A normal apoptosis of tumor cells does not usually produce immunogenic molecules. An antigen from a dead tumor cell is therefore inconsequential, whereas the same antigen from a living tumor cell can be recognized by the immune system. Once a tumor antigen is recognized on a living tumor cell, CTLs proliferate rapidly and attack tumor cells with exactly the same antigen. To prevent this, the identified tumor cell immediately commits suicide after being unmasked in order to protect the other tumor cells. Thus, the target antigens for the CTL killer cells are gone. An immune response against dying cancer cell is generally missing. The paradox is solved. This suicide is part of the escape strategy to escape the immune system. Fortunately, medical science has special cytostatic drugs that induce immunogenic apoptosis. Irradiation also leads to the same effect. This creates antigens that are recognized again by the CTL killer cells.
5.2. Tumor antigens
As recently as 50 years ago, it was not certain whether tumor cells expressed specific antigens [25]. The first tumor antigen (TA) was discovered on melanoma cells in 1991, MAGE-1, a milestone in cancer immunology. Today, more than 2000 TAs have been described.
In principle, every abnormally expressed protein on cancer cells can serve as tumor antigen, based on the recognition by cytotoxic T lymphocytes (CTLs), which is crucial for cancer-vaccine development. Four requirements must be fulfilled for TAs:
(1) Isolation of stable human T lymphocyte clones or lines recognizing the peptide.
(2) Identification of the peptide recognized by the T cells.
(3) Identification of the HLA/ MHC presenting molecule.
(4) Evidence that the peptide is processed by antigen presenting cells (APCs) and presented to the specific CTL.
Tumor antigens are broadly classified into two categories, unique and shared antigens. Examples of unique TAs are p53 - (head and neck squamous cell carcinoma -, positive for 14% of patients), bcr-abl, (chronic myeloid leukemia – positive for 16% of patients) and neoantigens. Examples of shared TAs are MAGE-A1 (melanoma, positive for 16% of patients) and PSA (prostate carcinoma, positive for 16% of patients.
The frequency of TAs varies from patient to patient, consequently also the immunotherapy. In addition, the expression level in the patient can also vary, so that one patient responds to immunotherapy while another patient does not, because his tumor cells do not produce sufficient targets.
Another problem is the usually low affinity of TAs. Affinity measures the strength of interaction between an epitope of an antigen and an antibody's antigen binding site. The antigen-antibody interaction is non-covalent and does not rely on shared electrons (strong interaction), but on various electromagnetic interactions such as hydrogen bonds and van der Waals forces (weak interaction).
In 2009 the National Cancer Institute/ USA published a project on prioritization of cancer antigens and defined criteria for the "ideal" cancer antigen [26]. The ranking criteria were based on the likelihood for efficacy in clinical trials and were as follows (Table 1).
75 representative antigens were selected. None of the 75 antigens met all criteria. 46 antigens were immunogenic, and only 20 antigens had a therapeutical function and induced immune responses.
Therapeutic function is considered the most important criteria. This is ignored in many clinical trials with therapeutic vaccines. The induction of a high number of CD8+ T cells is described as success. Whether these CTLs actually do their job is often not questioned. The mere number of CTLs in the tumor infiltrate is not an indication of vaccine efficacy [27]. Responsible for this phenomenon, many CTLs with zero effect, could be the immunosuppressive milieu dominated by cytokines such as IL-4, IL-10, IL-13 or TGF-β. A key role is played by IL-10, which suppresses the effector functions of macrophages, NK cells, mast cells, B cells and T cells. A high IL-10 expression is associated with aggressive clinical manifestations in melanoma and squamous cell carcinoma [28].
The same applies to the level of antibodies in response to a vaccine. The measurement of antibodies in response to a vaccine is presumed to be level of protection and duration. This vaccine immunogenicity is only an indication for a possible protection but definitely not equal to the real factual protection. The antibody titer or the count of CTLs in the infiltrate are only laboratory markers. The only convincing method to measure the efficacy of a tumor vaccine is the overall survival time (OS) as the most meaningful endpoint of a phase III clinical trial. OS is defined by the date of death from any cause.
This endpoint is not included in the revised RECISTS guidelines [29]. These rules called Response Evaluation Criteria in Solid Tumors, or RECIST, determine when tumor size in cancer patients changes during treatment: improvement, complete or partial respond, CR or PR, stable disease (SD) or progressive disease (PD). The majority of all clinical trials evaluating cancer drugs use RECIST. The clinical response of immunotherapeutical agents, however, is quite different to chemical drugs. The kinetics of antitumor effects is much slower. Whereas a response to a chemical drug commonly occurs within some weeks only after initial administration, the response to an immunological drug can be delayed up to six months after initial treatment. In addition, the tumor size can increase and new lesions (progressive disease) can appear after starting the immunotherapy before a regression begins.
Novel criteria for evaluation of antitumor responses with immunotherapeutic agents are strongly required.
5.2.1. Neoantigens
In second place of the ideal tumor vaccine is the immunogenicity of the antigen. The success of any vaccination stands and falls with this. With the classical TAs, the immunogenicity is often very low, as already described. With the discovery of neoantigens, it was believed that a great leap in vaccine development had been made.
Neoantigens are a new class of mutated and patient-specific antigens, present in tumor cells but not in normal cells and with a lack of central tolerance against them. As a reminder, classical TAs encounter both a central and a peripheral tolerance. Targeting TAs may also lead to autoimmune toxicity [30].
Neoantigens are the result of mutations [31]. So far analyzed neoantigens are missense mutations, a type of nonsynonymous substitution (missense neoantigens), in which one DNA nucleotide is switched out with another one in a way that changes the amino acid specified. The so made neo-protein looks quite different to the corresponding wild type and may not even be functional but an ideal antigen for a personalized vaccine because it is recognized by the immune system: MHC class I (in humans, HLA class I) molecules bind intracellular antigens and present them to cytotoxic CD8+ T cells, whereas MHC II bind extracellular antigens and present them to CD4+ T helper cells.
Many perhaps all neoantigen prediction algorithms use predicted binding affinity to MHC class I as a surrogate for immunogenicity. But not each neoantigen is a suitable vaccine candidate and able to induce a T cell response. Schumacher et al. [32] used in silico prediction tools to describe 448 neoantigens detected in a patient with melanoma. Only one strong and one weak neoantigen out of 448 were found.
Like all other antigens, neoantigens are classified into dominant and subdominant. A third category describes cryptic peptides which have low MHC I affinity. Cryptic peptides are presented at the cell surface and are therefore non-immunogenic. Protein antigens typically contain multiple epitopes capable of binding MHC II molecules, or MHC I but T cell responses are limited to only a small number of these determinants in each individual. A vaccine that induces responses against subdominant epitopes will be less protective than a vaccine that induces a robust immunodominant response. Immunodominance is evident for both antibody-mediated immunity and cell-mediated immunity.
The current trend in tumor vaccine development is towards the use of neoantigens as personalized vaccines. RNA vaccines or peptide vaccines in particular rely on patient-specific neoantigens. However, neoantigens can also be patient independent, meaning that a sequence identical neoantigen can appear in multiple patients as shared neoantigens [33,34].
There are encouraging reports of successful vaccination of patients with neoantigens. Phase I clinical trials have also been conducted [35]. Quite a few of these trials employ combination therapies, for example vaccine plus immune checkpoint inhibitors such as anti-PD-1 antibody. The crux of such combinations is that therapeutic success, if any, cannot be clearly attributed to either agent. If the neoantigens are so powerful, then they should be vaccinated stand-alone.
The Achilles' heel of neoantigens is the same as for classical TAs: mutations, mutations, mutations. As briefly described in section 3.4, the tumor genome is very unstable. All antigens on a tumor cell are constantly threatened by mutations. In the fight against cancer, it is moving targets that make immunotherapy so difficult. Neoantigens are proteins that have already been mutated. But mutations do not stop; even mutated neoantigens continue to mutate. If a cocktail of neoantigens is prepared for a vaccine regimen of 3-5 vaccinations over a period of several weeks for a patient on day 0, a single batch, which is usually the case, it may well be that the designated neoantigens have continued to mutate in the patient during the vaccination interval. The vaccine antigens from day 0 would no longer match the current antigen situation on day 30 or day 40.. The vaccination would be ineffective.
5.3. Tumor escape mechanisms
Tumor cells have developed a number of ways to evade the immune system. I have already introduced the tumor microenviroment (TME). Once the tumor cells have found their niche, the tumor cells are trained by immune cells turned inside out in how to deal with the immune system "out there". The TME is a training camp and protected space for tumor cells.
All solid tumors contain infiltrates of leukocytes. Among these, T cells (Th1, Th2, Th17, Treg, γδ T cells, CTLs), and tumor-associated macrophages (TAMs). Immune cells stimulate the tumor proliferation and angiogenesis by secretion of mitogenic growth factor such as tumor necrosis factor-α (TNF-α), epidermal growth factor (EGF), transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF); chemokines such as CXCL-12; various cytokines. Tumor cells depend on pro-inflammatory cytokine stimulation. The basis for cytokine production is in the permanent activation of the NF-kappaB pathway. Permanent activation of the NF-kappaB pathway of tumor infiltrating cells represents one mechanism that appears to favor tumor survival.
The NF-kappaB activation is strongly maintained by the hypoxic environment which favors the influx of cells depending on the glycolytic pathway, macrophages and granulocytes. These cells also produce many reactive oxygen species (ROS) [36]. The NF-kappaB pathway is regulated by (mitochondrial) ROS. The sustained NF-kappaB activation is a direct link between inflammation and cancer.
Dendritic cells found in TME are defective and cannot present (tumor) antigens. Moreover, it seems that many DCs suppress T cell activities. Tumor cells themselves can downregulate their MHC I complex to escape recognition by cytotoxicity of CD8+ T-cells. Approximately 40-90% of human tumors are MHC I downregulated [37,38].
Antitumor CTL activity is blocked, NK and T cell mediated killing is blocked – cancer cells can escape, successfully edited by TAMs and others. TAMs are re-programmed to inhibit lymphocyte functions through release of inhibitory cytokines such as IL-10, prostaglandins and reactive oxygen. TAMs also recruit regulatory T cells (Treg) cells via the chemokine CCL22, a further contribution to the immune suppressive milieu. In TME, Treg cells expand downregulate T-cell proliferation and suppress anti-tumor responses of both CD4+ and CD8+ T cells by production of inhibitory cytokines such as TGF-β, IL-10 and IL-35. Treg cells are partly responsible, not only for local, but also for peripheral, systemic immunosuppression
The tumor vascularization is particularly enhanced by TAMs via production of VEGF. The tumor vasculature itself has lost high endothelial venules which support high levels of lymphocyte extravasation. That means, that the influx of healthy CTLs, NK cells, T cells, monocytes from outside the TME into the TME is blocked.
Evasion mechanisms include tumor cells not expressing the co-stimulatory signals necessary for T cell activation. T cell activation requires three signals, the presentation of antigen, co-stimulatory molecules B7 and CD28 and pro-inflammatory molecules. If only one signal is missing or the antigen is not sufficiently immunogenic, the T cell dies on apoptosis.
5.4. State of exhaustion
Tumor editing has taught us that the immune system's fight against cancer is very successful as long as the number of tumor cells remains manageable. After all, immune surveillance protects the body from cancer for years, if not for a lifetime. If the number of tumor cells grows, the immune system still resists. This can go on for many years. An unstable equilibrium prevails. But multiple gene changes in the tumor cells end the equilibrium and the tumor cells break out. The escape succeeds thanks to the mutations in the tumor cell. These mutations also regularly change the proteins on the surface, the tumor antigens. But if the tumor antigens are constantly changing, the immune system can no longer keep up. It lags behind in its response.
Another phenomenon is added: if the killer cells are bombarded with tumor antigens without interruption, the killer cells react by refusing to work. Instead of killing, they lay down their weapons and surrender. Burnout of the killer cells. The tumor antigens can dance around on the nose of the killer cell, and the killer cell watches only. Nothing works anymore. Immunologists speak of a "state of exhaustion" (T cell exhaustion) and of anergy in the complete absence of the immune response. Not only tumor antigens cause burnout. The state of exhaustion also occurs after permanent activation by infectious antigens, including vaccine antigens [39,40,41,42].
We already know this T cell hyporesponsiveness from the niche: immune cells do not attack cancer cells. Exhausted T cells express high levels of inhibitory receptors, including programmed cell death protein 1 and 2 (PD-1/2) and cytotoxic T lymphocyte antigen-4 (CTLA-4) and less effector cytokines such as interleukin-2 (IL-2), tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ). The inhibitory receptors are immune checkpoints and important for the regulation of T cells.
Little is known about the mechanisms behind this. A transcription factor has been identified, TOX (thymocyte selection-associated HMG BOX), which controls a number of genes responsible for CTL burnout. Several studies have shown that TOX is involved in promoting T cell exhaustion [43].
PD-1/2 and CTLA-4 are immune checkpoint proteins and are located on the surface of all T cells (killer cells, helper cells, regulatory cells). The names reveal that the checkpoint proteins are part of programmed cell death. PD-1/2 (protein programmed cell death protein 1/2) and CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) are receptors. The checkpoint proteins monitor natural T cell activity and stop it in time. Activation of PD-1 and 2 receptors leads to a shutdown of the CTL cell.
The same happens upon activation of CTLA-4. Tumor cells form ligands, such as PD-L1/L2 or CD-80, which block PD-1/2 and CTLA-4. Thus, the T cells are inhibited and no longer fulfill their mission. The tumor has induced immunsuppression and evade the immune response [44,45].
T cell dysfunction is attempted to be cured by therapeutic antibodies known as immune checkpoint inhibitors (ICI). However, clinical efficacy is modest. Only a few types of cancer respond to checkpoint inhibition. There is no or low response to antibodies like pembrolizumab in prostate, pancreatic, ovarian cancer. One reason is the low expression level of, e.g., PD-L1 on these tumor cells. Thus, the FDA requires a high level of PD-L1 expression as a criterion for pembrolizumab: Tumor Proportion Score (TPS) ≥ 50%. In non-small cell lung cancer (NSCLC) the expression rate of PD-L1 is 25-30%.
5.5. Immunosenescence
When the healthy immune system of a young person sees an antigen for the first time, it is able to form a robust humoral and cellular response. The antigen is recognized, presented and the immune system responds. B and T memory cells form and are present into old age. Booster vaccinations, e.g. influenza, benefit from this mechanism. From about age 50, the ability of immune cells to respond to new antigens diminish, and at about age of ˃ 65, new antigens are no longer transformed by the immune system into a robust immune response. Therefore, immunizing the elderly after the age of ˃ 65 with a new vaccine that their body has never seen before is not very successful. Immunosenescence causes impaired new antigen response.
The reason is that as we age, the stability of our genes decreases and mutations occur more frequently. Aging is the result of accumulation of somatic damages and genomic rearrangements. The DNA repair mechanisms for these mutations such as mismatch repair or double-strand break repair also decline with age [46,47,48,49]. It seems that all DNA repair systems are affected. This is due to a decrease in expression of the repair enzymes and a decrease in their functionality [50,51]. The decrease in DNA repair mechanisms leads to further mutations. A devil's circle.
The immune system is one function of the body profoundly affected by aging. The reduced efficacy of vaccines in the elderly is generally attributed to immunosenescence [52,53]. It involves both the host's capacity to respond to infections and the development of long-term immune memory, especially by vaccination. The decline in age-associated immune function on molecular, cellular and organismal changes is common to most if not all vertebrates, it's an evolutionary ubiquitious process which run in only one direction - a way of no return. Most of the parameters affected by immunosenescence appear to be under genetic control.
Consequently, older adults are more susceptible to infectious diseases, cancer, autoimmune diseases and increase in low-grade systemic inflammation known as inflammaging [54]. Age-associated immune changes take place in the innate and acquired immune systems and affect not only lymphocytes, but also myeloid cells with a change in pro-inflammatory cytokines.
Of note to underline that immunosenescence is not only restricted to aging. During chronic infections certain pathogens, such as CMV and HIV remodel the immune system towards aged T cells [55,56]. Chronic antigen stimulation like in chronic infections is the major trigger of immunosenecsence.
Cancer can be diagnosed at any age. But many cancers show a clear age relationship. 80 percent of cancer cases are diagnosed in patients older than 55 and only 3.7% are detected in people younger than 34 [57]. Most clinical trials with immunotherapeutics involve few or no patients older than 55 years.
T cells are particularly affected. The decreased production of naïve T cells is followed by a diminished response of TCR signalling (NF-kappaB, MAPK) leading to a blunted T cell proliferation to antigen stimulation and reduced T cell differentiation. In contrast to the memory pool of CD4+ and CD8+ T cells, induced at an early age, that can persist for a life-time of an individual. Besides a premature senescence caused by DNA damage, cytokines, mitochondrial dysfunction, there also exist a telomere shortening of senescent CD8+ T cells, called replicative senescence [58,59,60]. The main changes in the T cells repertoire are summarized in Table 2.
The B lymphopoiesis is limited by a loss of naïve follicular B cells. The B cell number in the periphery (blood, spleen, lymph nodes) decline moderately with aging or not. But the peripheral B cell repertoire in elderly is less diverse correlating with poor health status. Antibody responses are decrasing and also the duration of protection after immunization is declining [61]. The deterioration of B cell immunity is accompanied by an increase in pro-inflammatory molecules in the elderly [62,63].
TNF-α, IL-6 and acute C-reactive protein (CRP) are used as indicators of inflammaging [64]. Table 3 summarizes the main changes of aging B cells.
Immunosenescene is responsible for a higher failure rate of immunotherapy, including poor vaccination outcomes.
6. Types of cancer vaccines
When we talk about cancer vaccines, we are talking about therapeutic and not prophylactic ones. Existing vaccines against hepatitis B virus (HBV) (in the context of liver carcinoma) or human papillomavirus (HPV) (in the context of cervical carcinoma) are classical antiviral vaccines and not cancer vaccines. These vaccines may reduce the risk of developing cancer. So far, there are no clinical data demonstrating anti-tumor efficacy of these vaccines. Vaccinating prophylactically against cancer would be a sensation, but the unstable and constantly changing tumor genome does not produce a stable antigen target.
6.1. Cell-based vaccines
Among the first experimental approaches for a cancer vaccine were cell vaccines. The rationale behind this is to present as many tumor antigens as possible to the immune system. The first scientific report on an autologous (personalized) cancer vaccine (whole tumor homogenate mixed with Freund's adjuvant and injected 3x intramuscularly into patient) appeared in 1964.
The author of this review himself produced an autologous whole cell vaccine in 1994 with his laboratory team in Vienna as part of a phase I clinical trial. For this purpose, tumor cells were individually removed from melanoma patients, expanded in the laboratory and transfected with the gene for human interleukin 2. Patients received up to 4 vaccinations within 4-5 weeks. The therapeutic effect failed to materialize and the study was discontinued after phase II [65].
Cell vaccines continued to be developed. In 2010, the first FDA approval for the first autolougus dendritic cell (DC) vaccine against metastatic prostate cancer Sipuleucel-T (Provenge®). Dendritic cells from the patient are primed in vitro with tumor antigens and injected into the patient. As monotherapy, sipuleucel-T provides no additional benefit [66], in combination with androgen receptor-targeting agents (ARTAs), patients have a longer median OS [67].
6.2. BCG vaccine
Bacillus Calmette-Guérin (BCG) is also approved to treat early-stage bladder cancer. BCG contains live-attenuated mycobacterium tuberculosis and is the oldest in wide use across the world. The bacille Calmette-Guérin (BCG) vaccination is more than 100 years old, first used in humans in 1921. Therapy with BCG can lead to very severe side effects such as spinal tuberculosis, pneumonia or hepatitis. To date, the mechanism of action has not been elucidated. It is assumed that the antitumor effect is non-specific [68].
6.3. Oncolytic virus vaccine
Talimogen-Laherparepvec (T-Vec) is also approved by the FDA against malignant melanoma. It is a recombinant herpes simplex virus-1 that infects tumor cells. Massive viral replication in the tumor cell causes it to die. T-Vec is administered as monotherapy or in combination with checkpoint inhibitors. Unfortunately most of the patients still relapse and die of their disease [69].
6.4. Peptide vaccines
6.5. Gene based vaccines
These vaccines consist of either DNA or RNA. Since the Corona crisis, RNA has been the talk of the town as a vaccine. However, the first marketing authorization for a gene-based vaccine was granted to a DNA vaccine in 2010.
The U.S. Department of Agriculture (USDA) approved the marketing of ONCEPT, an agent for dogs against malignant melanoma. It is a xenogeneic DNA vaccine. The plasmid DNA contains a cDNA for the human thyrosinase (huTyr), a tumor antigen (TA). The human tyrosinase protein is at least 85% homologous to canine tyrosinase. Tyrosinase is overexpressed in tumor cells and therefore an ideal target in cancer therapy. After more than 10 years of use, it can be said that the DNA vaccine is biologically safe and leads to low-grade, reversible toxicity only in a small number of patients. The most important question, effectiveness, cannot be answered. This is because, unlike the FDA approval of human drugs, the USDA only reviews the safety of the product and accepts a reasonable expectation of efficacy [72]. DNA vaccines induce a complete immune response that can last for a long time, but the antibody titer does not reach the level achieved e.g. after vaccination with recombinant peptides [73]. Also, DNA vaccines must be heavily adjuvanted.
More figures are available on mRNA vaccines, thanks to Corona. However, to date, the complete FDA approval dossiers for e.g. Comirnaty (BioNTech/Pfizer) and Spikevax (Moderna) vaccines have not been published.
The key issues are again safety and efficacy. The efficacy of Comirnaty was initially reported as 91.3% and a good safety profile [74]. Both have to be negated. Not all data from clinical trials have been evaluated yet, as only a U.S. court in 2022 ordered the FDA to release all relevant papers. An efficacy of only about 50% is discussed [75]. To date, more than 50 serious adverse events have been described [76].
It was the U.S. Securities and Exchange Commission (SEC) that forced BioNTech to disclose the truth about its mRNA Corona vaccine [77]. BioNTech's answer: ... We may not be able to demonstrate sufficient efficacy or safety of our COVID-19 vaccine and/or variant-specific formulations to obtain permanent regulatory approval in the United States, the United Kingdom, the European Union or other countries where the vaccine has been approved for emergency use or granted conditional marketing approval...Serious adverse events may occur during our clinical trials or even after we receive regulatory approval, which could delay or terminate the clinical trials and delay or prevent regulatory approval or market acceptance of any of our product candidates [78].
The RNA vaccine does not produce a stable and sterile immune response. Most importantly, it does not protect against a severe course. A fairy tale that is repeatedly strained. Evidence-based studies are lacking on this. Neither self-protection nor protection from others is given.
I will discuss two cardinal points only, firstly the influence of mRNA on the innate immune system and secondly the danger of integration into DNA.
Ad 1. One of the severe side effects is that the RNA vaccine massively interferes with the innate immune system and provokes serious disorders. 90% of the immune system belongs to the innate immunity, which emphasises the great importance of this defence system. The initial immunological attack against viruses, bacteria, fungi and parasites by innate immunity is very fast and occurs in a few minutes. In contrast, the specific immunological response occurs about 3 weeks after infection.
Interferons are part of the immunological first strike against invading viruses. The mRNA vaccine significantly interferes with innate immunity and reduces the natural interferon response to viruses by manipulating the IFN I signaling pathways [79]. It is an inhibition of the IFN I immune response to viruses, an immune tolerance induced by the vaccine. An increase in other viral infections such as parainfluenza, rhinoviruses and Rous sarcoma virus (RSV) has been reported in 2021. Severe courses of RSV can occur in all age groups.
Another consequence of the reprogramming of innate immunity by the mRNA vaccines is a reduced defense against fungal diseases. Fungal infections will increase among the vaccinated people.
Ad 2. LINE-1, long interspersed nuclear elements (LINEs), is part of a family of autonomous retrotransposons. 17% of the human genome consists of LINE-1 of which most L1 elements are inactive. But the still active L1 elements can modify the genome by insertions, deletions, or rearrangements. LINE-1 is associated with carcinogenesis and may serve as a biomarker for neoplasia [80].
mRNA vaccines become dangerous when they are transcribed by the endogenous enzyme LINE-1 into DNA, which migrates into the nucleus and can be incorporated into the genome. That this possibility is real was demonstrated in cell culture experiments as early as 2021 [81]. In 2022, it was published by another group that the vaccine BNT162b2 mRNA is reverse transcribed intracellularly into DNA in as fast as 6 h upon BNT162b2 exposure [82]. The possible reverse transcription and integration of vaccine RNA is obviously a taboo topic in vaccinology and is not sufficiently addressed and discussed experimentally [83]. Does politics have too much influence on science?
The mRNA is very unstable by nature. Among other things, the cell regulates protein balance via this instability. Furthermore, RNA receptors of the Toll-like receptor (TLR) family are located within the cell, in this case TLR3,7,8. These receptors are part of the innate immunity and recognize viral RNA, but also the vaccine RNA and give the signal for its destruction. What is desirable for viruses would be a disaster in the case of vaccine RNA: the innate immune system destroys the vaccine RNA before it can take effect.
Karikó et al. [84] found a solution to this problem. They replaced the building block uridine in the vaccine RNA with pseudouridine. The vaccine RNA is no longer recognizable by the internal RNA receptors and the immune response against the RNA vaccine but also against invading viruses is absent. The mRNA vaccines are shooting themselves in the foot because they naturally allow SARS-CoV-2 viruses to pass. Several reinfections in multiple corona vaccinated people is the consequence. What does this mean for a RNA cancer vaccine? The patient, already suffering from a weak immune system due to cancer, runs the risk of becoming severely ill from some viral infection. The title of the Karikó paper: Suppression of RNA Recognition by Toll-like Receptors… This says it all. What a disastrous message.
The vaccine RNA is stabilized once and for all and is untouchable. Pseudouridine instead of uridine in mRNA is not an invention of Karikó et al, rather solid basic research. This is because pseudouridine occurs naturally in the cell, wherever a particular RNA molecule is to be stabilized. Pseudouridine RNA was discovered as early as 1951 [85].
And this is especially the case with cancer cells. A cancer cell gains survival advantages over a healthy cell in many ways. One survival strategy of the cancer cell is to stabilize the tumor RNA by pseudouridine.
No wonder that in recent years, cancer research has increasingly looked at tumor pseudouridine mRNA [86]. Far advanced is the diagnostic development in prostate carcinoma. Pseudouridine mRNA diagnostics would be much more specific than current PSA diagnostics [87].
Karikó et al. have created a vaccine RNA that is extremely stable, like a tumor RNA. But stability comes at a very high price: innate immunity to viruses and other pathogens is virtually eliminated. RNA vaccines are risky, a time bomb.
7. Conclusion and future perspectives
CureVac is the oldest RNA vaccine company, perhaps the oldest in the world, which lost out in the worldwide race for the first mRNA vaccine against SARS-CoV-2. Their study came too late and showed only 48% efficacy. Critics claim that the low dose of 12 µg mRNA (vs 30 µg for BNT162b2) is to blame for the low efficacy. If that were the case, all one would have to do is increase the dose and the problem would be solved. This is not so. Efficacy is not solely a dose issue. In 2017 CureVac prostate cancer vaccine candidate fails phase IIb trial. CureVac's lead mRNA drug was the first RNA vaccine in man. "These results...pave the way for us to advance more potent prophylactic vaccine formulations into the clinic…" said Ingmar Hoerr, former CEO of CureVac. CureVac started the development of a Rabies mRNA vaccine. This is an admission that cancer vaccines don't work.
CureVac/Germany founded in 2000, BioNTech/Germany (2008) or Moderna/USA, (2010), all these companies were originally founded with the goal of developing therapeutic cancer vaccines. These companies have already conducted many clinical trials, all vaccine candidates of all companies have flopped since then [88]. To date, there is no approved mRNA cancer vaccine on the market.
A therapeutic cancer vaccine is confronted with several massive natural barriers as summarized in Table 4.
Based on this knowledge a therapeutic vaccine must fulfil at least three fundamental functions:
- on the anti-inflammatory side - dam up the anti-inflammatory process
- on the pro-inflammatory side - reconstitution of the pro-inflammatory environment
- on the T cell immunity side - reconstitution of the T cell immunity
Cancer patients go to physicians when their tumors have immunosuppressive activities. The correlation of immune dysfunctions and the development of tumors is evident [89]. There is no sense in creating any cancer vaccine while ignoring these clear coherences and the strong influence of the peripheral immunity on the whole system. But this is what currently happens! Behind the hype of RNA cancer vaccines is the same boring attitude as in the last two decades: Take an antigen, put this antigen in a suitable packing, look for an appropiate delivery system and into the patient. And this construct shall work? It did not work in the past and it will not work today or in the future. Old wine in new wineskins.
Despite some successes, immunotherapies remain ineffective for most patients with cancer. This is true for experimental vaccines as well as for therapeutic antibodies, cell vaccines or immune checkpoint inhibitors (ICI). Clinical relevance of a therapeutic vaccination should be evaluated in terms of a therapeutic benefit for the patients, and not as a change of a surrogate marker.
In many studies, combinations of chemical drugs and immunotherapeutics or two immunotherapeutics are used. The rationale behind these combinations is that different therapeutic agents have different mechanisms of action. The combination should avoid the development of resistance. This is the case when various chemical drugs are used which intervene with various signaling pathways. But antibodies, vaccines and also ICIs intervene in the same mechanism: a protein on the cell surface. Above all, if the sufficient efficacy (˃50%) of an immunotherapeutic agent has not been proven as a stand-alone drug, then the agent should not be used. After all, most of the active ingredients have severe side effects.
What remains to be done? As a vaccine developer with 30 years of experience, I act as advocatus diaboli and plead for greater efforts in the development of innovative chemical drugs: new small molecules affecting cancer specific signaling pathways, which regulate the communication between tumor cells. New drugs for treatment-resistant tumors. New drugs for blocking cancer metastasis. Innovative chemical drugs are needed to interrupt the self-sustaining mechanisms of tumor cells, the autocrine loops, which regulate growth and survival. We need drugs that penetrate the TME as a trojan horse and attack tumor cells already in their niche. Or best of all a drug that prevents the formation of TME from the outset. Vaccines have no mission here. The development of such intelligent drugs is highly complex, requires a high degree of intellectual input, it’s time and money consuming and can only be done in a team with different disciplines. This is much more demanding than: take a tumor antigen or a cocktail, a nice formulation, etc. etc. etc.
"We have to learn how to aim chemically." Paul Ehrlich (1854-1915)
This statement has lost none of its meaning to this day. And for me, therapeutic cancer vaccines remain an unfulfilled dream, a misconception. I really hope I am wrong. Research is well on its way to making cancer a chronic and manageable disease.
Funding
This research received no external funding.
Conflicts of Interest
The author declare no conflict of interest.
References
- Felgner, S.; Kocijancic, D.; Frahm, M.; Weiss, S. Bacteria in Cancer Therapy: Renaissance of an Old Concept. Int. J. Microbiol. 2016, 2016, 1–14. [Google Scholar] [CrossRef]
- Gaz, P. Essential Facts on the History of Hyperthermia and their Connections with Electromedicine. Przeglad Elektrotechniczny 2011, 87, 37–40. [Google Scholar]
- Giese, M. Cancer Vaccines. In Introduction to Molecular Vaccinology (Textbook); Giese, M., Ed.; Springer NATURE International Publishing: Switzerland, 2016; pp. 295–333. [Google Scholar] [CrossRef]
- McCarthy, E.F. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Colleen, A.; Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar]
- Arbiser, J.L.; Bonner, M.Y.; Gilbert, L.C. Targeting the duality of cancer. NPJ Precis. Oncol. 2017, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Claire, M.; Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448–458. [Google Scholar] [CrossRef]
- Robinson, N.J.; Schiemann, W.P. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers (Basel). 2022, 14, 808–830. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.R.; Garbe, J.; Levine, G.; Lichtsteiner, S.; Vasserot, A.P.; Yaswen, P. Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor β growth inhibition in p16INK4A(−) human mammary epithelial cells. Proc.Natl.Acad.Sci.USA 2001, 98, 4498–4503. [Google Scholar] [CrossRef]
- Cao, Y.; Li, H.; Deb, S.; Liu, J.P. TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene 2002, 21, 3130–3138. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Levantini, E.; Maroni, G.; Del Re, M.; Tenen, D.G. EGFR signaling pathway as therapeutic target in human cancers. Seminars in Cancer Biology 2022, 85, 253–275. [Google Scholar] [CrossRef]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of Cancer Cells Is Maintained by EGFR Independent of Its Kinase Activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. ; Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Joyce, J.A. . Microenvironmental regulation of therapeutic response in cancer. Trends Cell. Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002, 11, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm. Sin. B. 2020, 10, 2156–2170. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.N. Editorial: Metabolism and Cell Adhesion in Cancer. Front. Cell Dev. Biol. 2022, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C. The paradox of cancer cell apoptosis. Front. Biosci. (Landmark Ed.) 2011, 16, 1759–1767. [Google Scholar] [CrossRef]
- Mempel, T.R.; Henrickson, S.E.; von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–153. [Google Scholar] [CrossRef]
- Piessens, W.F. Evidence for human cancer immunity. Cancer 1970, 26, 1212–1220. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; Matrisian, L.M. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Sherry, R.M.; Morton, K.E.; Scharfman, W.J.; Yang, J.C.; Topalian, S.L.; Royal, R.E.; Kammula, U.; Restifo, N.P.; Hughes, M.S.; Schwartzentruber, D.; Berman, D.M.; Schwarz, S.L.; Ngo, L.T.; Mavroukakis, S.A.; White, D.E.; Steinberg, S.M. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 2005, 175, 6169–6176. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Sung, W.W.; Su, T.C.; Chen, M.K.; Wu, P.R.; Yeh, K.T.; Ko, J.L.; Lee, H. High expression of interleukin 10 might predict poor prognosis in early stage oral squamous cell carcinoma patients. Clin. Chim. Acta. 2013, 415, 25–30. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; Rubinstein, L.; Shankar, L.; Dodd, L.; Kaplan, R.; Lacombe, D.; Verweij, J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1. 1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Amos, S.M.; Duong, C.P.; Westwood, J.A.; Ritchie, D.S.; Junghans, R.P.; Darcy, P.K.; Kershaw, M.H. Autoimmunity associated with immunotherapy of cancer. Blood 2011, 118, 499–509. [Google Scholar] [CrossRef]
- Monach, P.A.; Meredith, S.C.; Siegel, C.T.; Schreiber, H. A unique tumor antigen produced by a single amino acid substitution. Immunity 1995, 2, 45–59. [Google Scholar] [CrossRef]
- van Rooij, N.; van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; van Dijk, L.J.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; Nieuwland, M.; Stratton, M.R.; Kerkhoven, R.M.; Kesmir, C.; Haanen, J.B.; Kvistborg, P.; Schumacher, T.N. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013, 31, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Tureci, O. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared neoantigens: ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics 2020, 21, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; Chen, C.; Olive, O.; Carter, T.A.; Li, S.; Lieb, D.J.; Eisenhaure, T.; Gjini, E.; Stevens, J.; Lane, W.J.; Javeri, I.; Nellaiappan, K.; Salazar, A.M.; Daley, H.; Seaman, M.; Buchbinder, E.I.; Yoon, C.H.; Harden, M.; Lennon, N.; Gabriel, S.; Rodig, S.J.; Barouch, D.H.; Aster, J.C.; Getz, G.; Wucherpfennig, K.; Neuberg, D.; Ritz, J.; Lander, E.S.; Fritsch, E.F.; Hacohen, N.; Wu, C.J. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Lluis, J.M.; Buricchi, F.; Chiarugi, P.; Morales, A.; Fernandez-Checa, J.C. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007, 67, 7368–7377. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers (Basel) 2020, 12, 1760–1788. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Baitsch, L.; Fuertes-Marraco, S.A.; Legat, A.; Meyer, C.; Speiser, D.E . The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012, 33, 364–372. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Macián, F.; Im, S.H.; Garcı́a-Cózar, F.J.; Rao, A. T-cell anergy. Curr. Opin. Immunol. 2004, 16, 209–216. [Google Scholar] [CrossRef]
- Huang, S.; Chaofeng Liang, C.; Zhao, Y.; Deng, T.; Jiaxiong Tan, J.; Zha, X.; Li, Y.; Chen, S. Increased TOX expression concurrent with PD-1,Tim-3, and CD244. Asia Pac. J. Clin. Oncol. 2022, 18, 143–149. [Google Scholar] [CrossRef]
- Ding, Z.C.; Lu, X.; Yu, M.; Lemos, H.; Huang, L.; Chandler, P.; Liu, K.; Walters, M.; Krasinski, A.; Mack, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H.; Zhou, G. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014, 74, 3441–3453. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fang, Y.C.; Li, J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol. Lett. 2019, 18, 5399–5407. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Morley, A. Somatic mutation and aging. Ann. N. Y. Acad. Sci. 1998, 854, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.R.; Oda, Y.; de Boer, J.G.; Glickman, B.W. Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice. Genetics 2000, 154, 1291–1300. [Google Scholar] [CrossRef]
- Dolle, M.E.T.; Giese, H.; Hopkins, C.L.; Martus, H.-J.; Hausdorf, J.M.; Vijg, J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat. Genet. 1997, 17, 431–434. [Google Scholar] [CrossRef]
- Seluanov, A.; Danek, J.; Hause, N.; Gorbunova, V. Changes in the level and distribution of Ku proteins during cellular senescence. DNA Repair (Amst) 2007, 6, 1740–1748. [Google Scholar] [CrossRef]
- Goukassian, D.A.; Bagheri, S.; el-Keeb, L.; Eller, M.S.; Gilchrest, B.A. DNA oligonucleotide treatment corrects the age-associated decline in DNA repair capacity. FASEB J. 2002, 16, 754–756. [Google Scholar] [CrossRef]
- McElhaney, J.E.; Effros, R.B. Immunosenescence: what does it mean to health outcomes in older adults? Curr. Opin. Immunol. 2009, 21, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Fastenackels, S.; Katlama, C.; Ait-Mohand, H.; Schneider, L.; Guihot, A.; Keller, M.; Grubeck-Loebenstein, B.; Simon, A.; Lambotte, O.; Hunt, P.W.; Deeks, S.G.; Costagliola, D.; Autran, B.; Sauce, D. Old age and anti-cytomegalovirus immunity are associated with altered T-cell reconstitution in HIV-1-infected patients. AIDS 2011, 25, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.J.; van der Sande, M.; Jeffries, D.; Kaye, S.; Ismaili, J.; Ojuola, O.; Sanneh, M.; Touray, E.S.; Waight, P.; Rowland-Jones, S.; Whittle, H.; Marchant, A. Cytomegalovirus infection in Gambian infants leads to profound CD8 T-cell differentiation. J. Virol. 2007, 81, 5766–5776. [Google Scholar] [CrossRef] [PubMed]
- https://www.cancer.gov/about-cancer/causes-prevention/risk/age.
- Linton, P.J.; Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 2004, 5, 133–139. [Google Scholar] [CrossRef]
- Gillis, S.; Kozak, R.; Durante, M.; Weksler, M.E. Immunological studies of aging. Decreased production of and response to T cell growth factor by lymphocytes from aged humans. J. Clin. Invest. 1981, 67, 937–942. [Google Scholar] [CrossRef]
- Palmer, S.; Albergante, L.; Blackburn, C.; Newman, T. Thymic involution and rising disease incidence with age. Proc. Natl Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Garcia, D.; Blomberg, B.B. B Cell Immunosenescence. Annu. Rev. Cell. Dev. Biol. 2020, 36, 551–574. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Stingl, G.; Brŏcker, E.B.; Mertelsmann, R.; Wolff, K.; Schreiber, S.; Kămpgen, E.; Schneeberger, A.; Dummer, W.; Brennscheid, U.; Veelken, H.; Birnstiel, M.L.; Zatloukal, K.; Schmidt, W.; Maass, G.; Wagner, E.; Baschle, M.; Giese, M.; Kempe, E.R.; Weber, H.A.; Voigt, T. Phase I study to the immunotherapy of metastatic malignant melanoma by a cancer vaccine consisting of autologous cancer cells transfected with the human IL-2 gene. Hum. Gene Ther. 1996, 7, 551–563. [Google Scholar] [CrossRef]
- https://www.iqwig.de/en/presse/press-releases/press-releases-detailpage_10651.html.
- Hafron, J.M.; Wilfehrt, H.M.; Ferro, C.; Harmon, M.; Flanders, S.C.; McKay, R.R. Real-World Effectiveness of Sipuleucel-T on Overall Survival in Men with Advanced Prostate Cancer Treated with Androgen Receptor-Targeting Agents. Adv. Ther. 2022, 39, 2515–2532. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Ryk, C.; Chatzakos, V.; Hallén Grufman, K.; Bavand-Chobot, N.; Flygare, J.; Wiklund, N.P.; de Verdier, P.J. Secondary stimulation from Bacillus Calmette-Guérin induced macrophages induce nitric oxide independent cell-death in bladder cancer cells. Cancer Lett. 2014, 348, 119–125. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers (Basel). 2021, 13, 1383–1397. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; Kendra, K.L.; White, R.L.; Gonzalez, R.; Kuzel, T.M.; Curti, B.; Leming, P.D.; Whitman, E.D.; Balkissoon, J.; Reintgen, D.S.; Kaufman, H.; Marincola, F.M.; Merino, M.J.; Rosenberg, S.A.; Choyke, P.; Vena, D.; Hwu, P. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Malonis, R. J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 6–3210. [Google Scholar] [CrossRef]
- Pellin, M.A. The Use of Oncept Melanoma Vaccine in Veterinary Patients: A Review of the Literature. Vet. Sci. 2022, 9, 597. [Google Scholar] [CrossRef]
- Giese, M.; Bahr, U.; Jakob, N.J.; Kehm, R.; Handermann, M.; Müller, H.; Vahlenkamp, T.H.; Spiess, C.; Schneider, T.H.; Schusser, G.; Darai, G. Stable and long-lasting immune response in horses after DNA vaccination against equine arteritis virus. Virus Genes 2002, 25, 159–467. [Google Scholar] [CrossRef]
- https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-confirm-high-efficacy-and-no-serious.
- Kelly, A. War Room / DailyClout Pfizer Documents Analysis Volunteers’ Reports eBook: Find Out What Pfizer, FDA Tried to Conceal: Investigation Team, Pfizer Documents, DailyClout.
- Fraiman, J.; Erviti, J.; Jones, M.; Greenland, S.; Whelan, P.; Kaplan, R.M.; Doshi, P. Serious adverse events of special interest following mRNA COVID-19 vaccination in randomized trials in adults. Vaccine 2022, 40, 5798–5805. [Google Scholar] [CrossRef] [PubMed]
- https://www.sec.gov/Archives/edgar/data/1776985/000156459021016723/bntx-20f_20201231.htm#ITEM_3_D.
- https://investors.biontech.de/static-files/50d0cafc-b2c1-4392-a495-d252f84be105 ( page 8: Risk Factors).
- Seneff, S.; Nigh, G.; Kyriakopoulos, A.M.; McCullough, P.A. Innate immune suppression by SARS-CoV-2 mRNA vaccinations: The role of G-quadruplexes, exosomes, and MicroRNAs. Food Chem. Toxicol. 2022, 164, 113008. [Google Scholar] [CrossRef] [PubMed]
- Ardeljan, D.; Taylor, M.S.; Ting, D.T.; Burns, K.H. The Human Long Interspersed Element-1 Retrotransposon: An Emerging Biomarker of Neoplasia. Clin. Chem. 2017, 63, 816–822. [Google Scholar] [CrossRef]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Hughes, S.H.; Young, R.A.; Jaenisch, R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2105968118. [Google Scholar] [CrossRef] [PubMed]
- Aldén, M.; Olofsson Falla, F.; Yang, D.; Barghouth, M.; Luan, C.; Rasmussen, M.; De Marinis, Y. Intracellular Reverse Transcription of Pfizer BioNTech COVID-19 mRNA Vaccine BNT162b2 In Vitro in Human Liver Cell Line. Curr. Issues Mol. Biol. 2022, 44, 1115–1126. [Google Scholar] [CrossRef]
- Domazet-Lošo, T. mRNA Vaccines: Why Is the Biology of Retroposition Ignored? Genes 2022, 13, 719. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity, 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Cohn, W.E. Pseudouridine, a Carbon-Carbon Linked Ribonucleoside in Ribonucleic Acids: Isolation, Structure, and Chemical Characteristics. J. Biol. Chem. 1960, 235, 1488–1498. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Deng, X.; Chen, J. RNA Modifications in Cancer: Functions, Mechanisms, and Therapeutic Implications. Annual Review of Cancer Biology 2020, 4, 221–240. [Google Scholar] [CrossRef]
- Stockert, J.A.; Weil, R.; Yadav, K.K.; Kyprianou, N.; Tewari, A.K. Pseudouridine as a novel biomarker in prostate cancer. Uro.l Oncol. 2021, 39, 63–71. [Google Scholar] [CrossRef]
- https://clinicaltrials.gov/.
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Tumor and its microenvironment are closely related with an intensive intercellular communication. Without the support of normal tissue and stromal cells cancerous cells would not evolve and would be unable to manifest the disease. Apart from malignant cells, immune cells, fibroblasts, tube-forming endothelial cells and their surrounding pericytes and other cells form the TME. The non-cancerous cell mass can comprise > 50% of the tumor mass [3].
Figure 1.
Tumor and its microenvironment are closely related with an intensive intercellular communication. Without the support of normal tissue and stromal cells cancerous cells would not evolve and would be unable to manifest the disease. Apart from malignant cells, immune cells, fibroblasts, tube-forming endothelial cells and their surrounding pericytes and other cells form the TME. The non-cancerous cell mass can comprise > 50% of the tumor mass [3].
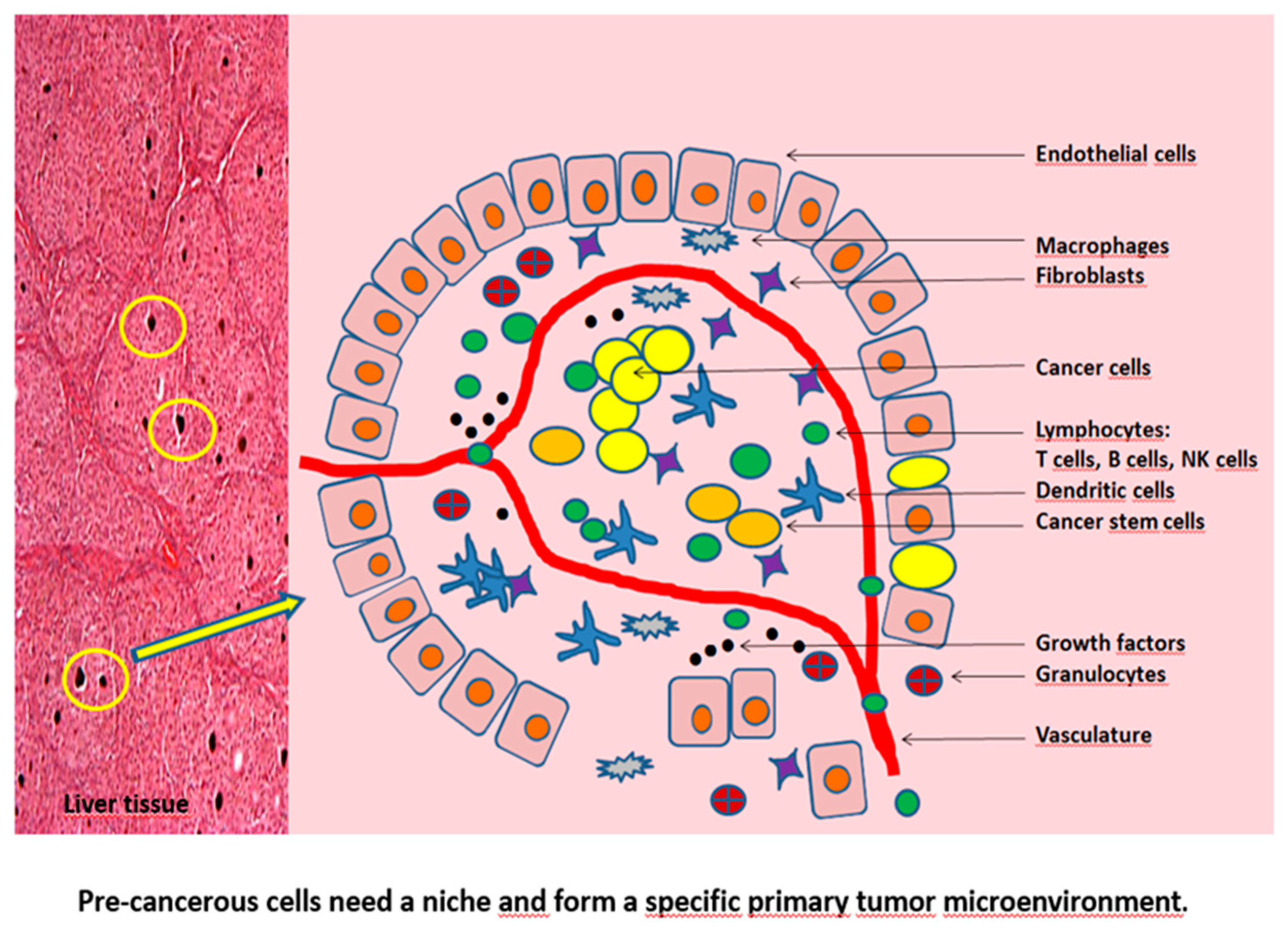

Table 1.
Prioritization of cancer antigens.
| Criteria for cancer antigen | Defined by | Weighting 100% |
|---|---|---|
| 1. Therapeutic function | Clinical trial data showing a vaccine induced response | 32 |
| 2. Immunogenicity | T cell and/ or antibody elicited in clinical trials | 17 |
| 3. Oncogenicity | Associated with oncogenic processes: “self” antigens, viral antigens; tissue differentiation | 15 |
| 4. Specificity | Unique or shared antigens; location of expression | 15 |
| 5. Expression level | Highly expressed to lower level of expression | 7 |
| 6. Stem cell expression | Evidence for expression on putative cancer stem cells | 5 |
| 7. Number of patient with antigen-positive cancer | High – to low level expression in n patients | 4 |
| 8. Number of antigenic epitopes | Multiple epitopes vs. one/ few epitopes | 4 |
| 9. Cellular localization of antigen expression | Cell surface/ circulating antigen | 1 |
Table 2.
The age-related changes in the T cell life.
| Aging T Cell | ||
|---|---|---|
| Decrease | Increase | Unchanged |
| • naïve T cell (TC) number |
• memory pool of CD4+ and CD8+ T cells |
• overall number of T cells in young and elderly people are comparable becauseof increase in memory T cells |
| •TC receptor repertoire | • senescence marker:PD-1CD57,KLRG-1AID ( CD8+ T cell) | |
| • TC receptor signaling | ||
| • TC differentiation | ||
|
• CD8+ TC replicative senescence |
||
|
• CD28, CD27 for T cell activation |
||
Table 3.
The age-related changes in the B cell life.
| Aging B Cell | ||
|---|---|---|
| Decrease | Increase | Unchanged |
|
• naïve follicular B cells ● immunglobulin diversity ● high affinity antibodies ● memory B cells ● plasma cells ● hematopoietic stem cells quantitative as qualitative changes with functional modification |
• low-affinity antibodies |
• B cell number in the periphery decline moderately or not |
Table 4.
Natural barriers for cancer vaccines.
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



