
Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
The aim of the study is to develop a population pharmacokinetic (PopPK) model and to investigate the influence of CYP3A5/CYP3A4 and ABCB1 single nucleotide polymorphisms (SNPs) on the Tacrolimus PK parameters after LCP-Tac formulation in stable adult renal transplant patients. The model was developed using NONMEM v7.5, from full PK profiles from a clinical study (n=30) and trough concentrations (C0) from patient follow-up (n=68). The PK profile of the LCP-Tac formulation was best described by a two-compartment model with linear elimination, parameterised in elimination (CL/F) and distributional (CLD/F) clearances and central compartment (Vc/F) and peripheral compartment (Vp/F) distribution volumes. A time lagged first order absorption process was characterized using transit compartment models. According to the structural part of the base model, the LCP-Tac showed an absorption profile characterized by 2 transit compartments and a mean transit time of 3.02 hours. Inter-individual variability was associated with CL/F, Vc/F and Vp/F. Adding inter-occasion variability (IOV) on CL/F caused a statistically significant reduction in the model minimum objective function MOFV (p<0.001). Genetic polymorphism of CYP3A5 and cluster of CYP3A4/A5 SNPs statistically significantly influenced Tac CL/F. In conclusion a PopPK model was successfully developed for LCP-Tac formulation in stable renal transplant patients. CYP3A4/A5 SNPs as a combined cluster including three different phenotypes (high, intermediate, and poor metabolizers) was the most powerful covariate to describe part of inter-individual variability associated with apparent elimination clearance. Incorporating this covariate in the initial dose calculation and during the therapeutic drug monitoring (TDM) would probably optimize Tac exposure attainments.
Keywords:
Tacrolimus
; LCP-Tac
; Population Pharmacokinetics
; CYP3A5
; CYP3A4
; ABCB1
; Renal transplantation
; stable adult patients
; Immunosuppression
1. Introduction
Tacrolimus (Tac) is a major immunosuppressant drug prescribed for over 30 years in kidney transplant patiefnts to avoid graft rejection1–3. High inter- and intra-individual variabilities in Tac exposure have been reported4,5. Among factors, hematocrit levels, time after transplantation, hepatic dysfunction, protein plasma concentrations as well as patients’ age, ethnicity, sex, and CYP3A4/A5 polymorphisms (SNPs), have been described as the main responsible 4,6–11. These factors and the Tac narrow therapeutic index make a challenge to establish the optimal dose. Indeed, Tac might be associated with several dose-dependent side effects such as neurotoxicity, nephrotoxicity, and post-transplant diabetes mellitus12–14, that can be reduced by avoiding too high Tac pre-dose concentration (C0), in the long-term. Conversely, Tac under-dosing resulting in too low C0 should not be attempted because it increases the risk of acute rejection and immunologic sensitization15. In this regard, the dosage of the Tac formulation can play an important role. Several formulations of Tac have been developed, demonstrating a very good efficacy in preventing acute rejection episode. Thus, Tac was originally formulated as immediate release IR-Tac (Prograf®) administered twice daily, then the first once-daily prolonged-release formulation, (Advagraf®) was developed and lastly the once-daily extended-release formulation LCP-Tac was commercialized using a MeltDose® delivery technology that provides a slower release/absorption rate resulting in delayed Tmax and more decreased Cmax and fluctuation profile. Additionally, compared to IR-Tac, reduction of Tac to the smallest possible particle size in LCP-Tac, allows a better dissolution and absorption, increasing its bioavailablity16–20. Other factors such as the varying distribution of CYP3A enzymes along the gut may contribute to the increasing bioavailability of LCP-Tac compared to IR-Tac21. Part of Tac from the LCP-Tac formulation is absorbed in distal parts of the small intestine or even the colon, where the CYP3A enzyme exists to a lesser extent compared to the proximal parts where most of the IR-Tac is absorbed.
The exposure and starting dose of Tac is significantly influenced by genes encoding Tac metabolizing enzymes cytochromes P450 3A4/5 (CYP3A4/5). CYP3A5 expresser (*1 allele carrier) require higher Tac dose in comparison to non-expresser (*3/*3) to achieve similar exposure22–25. While it is suggested that lower dose is required for patients with CYP3A4*22 SNP26,27, as it leads to lower Tac oral clearance. In addition, ABCB1 is also thought to be a contributing factor to the low oral bioavailability of Tac, however the impact ABCB1 SNPs on Tac pharmacokinetics has not been conclusively determined28.
The influence of predictive factors of Tac exposure has been widely studied after IR-Tac administration using modeling approaches4,29–34. While scarce studies describe the influence of these factors for LCP-Tac, among them, only two have been carried out in kidney transplant patients35–37. This study aims at investigating the influence of CYP3A5, CYP3A4 and ABCB1 SNPs on the Tac exposure upon LCP-Tac administration, in stable adult renal transplant patients, using a population modeling approach.
2. Methods
2.1. Study Design
Thirty patients were from an open-label, prospective, non-randomized investigator-driven single-center clinical trial (clinicalTrials.gov NCT02961608), while the rest of the patients were from routine check-ups at the hospital with Tac C0. All the patients were treated with an immunosuppressive drug regimen of oral twice-daily IR-Tac (Prograf; Astellas Pharma Europe Ltd, Staines, UK), for at least six months before conversion to once-daily LCP-Tac oral (Envarsus; Chiesi Farmaceutici, Parma, Italy) with a dose conversion ratio of 0.7. All patients received triple Immunosuppression therapy combining Tac, mycophenolate mofetil and prednisone.
A total of 98 stable renal transplant patients, that underwent a kidney transplant at least six months prior to the study were included. Patients with current infections, hepatitis B or C, severe gastrointestinal disorders, and patients receiving concomitant drugs that could interact with CYP3A enzyme were excluded from this study. The study was conducted in compliance with the Declaration of Helsinki and approved by the ethics committee of the Bellvitge University Hospital (Barcelona).
Patients from the clinical trial had an extensive sampling profile (10-18 sampling times over a 24-hour period). Blood samples were taken at pre-dose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 12.5, 13, 13.5, 14-, 15-, 20-, and 24-hours post-dosing under steady-state conditions. For the remaining patients, from one to five C0 samples per patient were obtained, depending on the date of conversion.
2.2. Tacrolimus Measurement and Data Recording
Tac was measured using a LC-MS/MS method, previously developed and validated38. Chromatographic determination was performed using the Acquity (®) UPLC (®) with a C18 BEH ™ reversed phase column (2.1 × 50mm id, 1.7μm). The limit of quantitation was set at 1.0 ng·mL-1. Tac daily doses and demographic characteristics of the patients were retrieved, from the medical files, at the start of the treatment. Hematocrit (%) and serum creatinine concentrations (µmol·L-1) at each occasion at which concentrations were monitored were also recorded. Clinical outcome variables that were assessed were renal function (eGFR), estimated using the Chronic Kidney Disease Epidemiology Collaboration formula, delayed graft function (DGF) and graft loss.
2.3. Genotyping
Genomic DNA was extracted from a peripheral whole-blood sample using Maxwell RSC® (Promega Corporation, Sydney, Australia) and was stored at −80° C. Genotyping of the CYP3A5*3 G > A (rs776746), CYP3A4*22 C > T (rs35599367) and ABCB1 3435C > T (rs1045642) polymorphisms (SNPs) was carried out using TaqMan SNP Genotyping Assay with the 7900HT Fast Real-time PCR System, Applied Biosystems (Thermo Fisher Scientific, Waltham, MA, United States).
According to the functional defect associated with CYP3A variants, we classified patients into three different clusters of CYP3A metabolizers: Poor metabolizers (PM) (CYP3A4*22 carriers + CYP3A5*3/*3), Intermediate metabolizers (IM) (CYP3A4*22 non-carriers + CYP3A5*3/*3 or CYP3A4*22 carriers + CYP3A5*1 carriers), and High metabolizers (HM) (CYP3A4*22 non-carriers + CYP3A5*1 carriers).
2.4. Statistical Analysis
Unpaired -t-tests were used to evaluate differences in log-transformed values of exposure metrics (C0 normalized by dose and AUC concentration-time curves) between two groups, meanwhile one-way-analysis of variance tests were used for comparisons among three groups. Results with p values of less than 0.05 were considered statistically significant. Statistical analyses were performed with the R program version (4.0.3).
2.5. Population Pharmacokinetic Analysis
- Base model development
The popPK analysis was performed with the non-linear mixed-effects model approach using NONMEM® version 7.5 (ICON Development Solutions, Hanover, MD, USA). Perl-Speaks-NONMEM (PsN) version 5.2.6, R package version 4.0.3, Pirana Modeling Workbench version 3.0 (Certara L.P. (Pharsight), St. Louis, MO), and Xpose 4.7.2 were used as support tools for model evaluation. The first-order conditional estimation (FOCEI) method with interaction was used throughout the modeling process.
One- and two-compartment open models with linear elimination and first order absorption were tested. The standard lag-model and transit compartment models in which the optimal amount of transit compartments is estimated39, were used to model the absorption delay. Inter-individual variability (IIVs) and inter-occasion variability (IOVs)40 were tested in all PK parameters assuming log-normal distributions. Additive, proportional, and combined error models were tested to describe the residual error (RE) variability. To compare the different nested models statistically, the likelihood ratio test, based on the reduction of the minimum objective function value (MOFV) at a significance level of p < 0.005 (change in MOFV [∆MOFV] = –7.879 for 1 degree of freedom) was considered. For non-hierarchical models, the most parsimonious model according to the Akaike information criterion (AIC) was chosen41. The decrease in MOFV, parameter precision expressed as percentage of relative standard error (RSE%), reductions in IIV associated with parameters, η- and ε-shrinkage values42, model completion status, and visual inspection of goodness-of-fit plots were also considered for model selection.
- Covariate Model
The effect of factors that could be physiologically and clinically meaningful on the PK parameters was investigated. Specifically, the influence of patient age, gender, total body weight, body mass index and hematocrit were evaluated. The effect of CYP3A4/A5 SNPs, cluster phenotypes and ABCB1 genotypes were also tested on Tac CL/F.
Firstly, correlations between continuous covariates were explored. Then, univariate analysis, forward inclusion and backward elimination stepwise procedures were carried out to explore the covariates43. Significance levels of 5% (ΔMOFV = –3.841 units, p<0.05) and 0.1% (ΔMOFV = 10.8 units, p<0.001) were considered during the forward addition and backward elimination steps, respectively. Only covariates providing a reduction of IIV associated with parameters of at least 10% were considered clinically relevant and were retained in the model.
2.6. Model Evaluation and Internal Validation
Goodness-of-fit plots were analysed throughout the modelling process. The predictive capability was evaluated using prediction-corrected visual predictive checks (pcVPC) based on 1000 simulations44. The median and 2.5th and 97.5th percentiles of the simulated data and their respective 95% prediction intervals were calculated and visually compared with the same percentiles obtained from the original raw data. Also Npde45 (Normalized prediction distribution errors) diagnostics were performed. Model adequacy was evaluated by checking the evenly distribution of predicted discrepancies and comparing the shape, location and variance of distribution parameters to the theoretical normal distribution.
3. Results
3.1. Patient Characteristics and Datasets
Blood Tac concentration-time data (n=655) from 98 renal transplant recipients were simultaneously analyzed. From this data, 480 out of 655 were obtained from the rich AUC sampled group (n=30) at least six months after transplantation and 175 were obtained from C0 values (n=68).
The median total daily administered dose was 2 mg with a wide variation between patients ranging from 0.5 mg to 12 mg. Figure 1 shows the overlaid individual full concentration-time and normalized by dose concentration -time profiles. In both cases, a large variability was observed, especially in the absorption phase, with a wide range of variation in Tmax among patients and more than one peak in some cases.
Demographic, biochemical, and genetic characteristics of the patient population are summarized in Table 1. Patient characteristics were similar when compared both groups (Clinical and follow-up study). Similar values in the hematocrit, serum creatinine and renal function values between groups were observed. Table 2 displays a comparative C0 and AUC values sorted by the CYP3A4/A5, ABCB1 SNPs groups and cluster phenotypes (Table 2). Statistically significant (p<0.001) differences were found when comparing normalized by dose AUC values between CYP3A5 *1 expressers vs. non-expressers. Similarly, statistically significant (p<0.001) differences were found when comparing C0 normalized by dose of CYP3A5 *1 expressers vs. non-expressers.
Regarding CYP3A4 no significant difference (p>0.05) was found when comparing the normalized by dose AUC values between CYP3A4*22 carriers vs. CYP3A4*22 non-carriers. By contrast, CYP3A4*22 carriers vs. CYP3A4*22 non-carriers C0 normalized by dose showed significant differences when compared (p<0.01).
When cluster combinations were considered, the C0 normalized by dose were significantly different among all phenotypes (p<0.01) (Table 2). For AUC/D values statistically significant differences were found among all phenotypes excepting between IM and PM (Table 2).
By contrast, no statistically significant differences were found when AUCs values were compared normalized by dose between ABCB1 *C carriers, (high pumper) vs. non-carriers, (low pumper) (p>0.05).
3.2. Population PK Analysis
The PK profile of the Tac whole-blood concentration versus time data was best described by a two-compartment model with linear elimination, parameterized in terms of apparent elimination and distributional clearances (CL/F and CLD/F, respectively) and apparent central compartment (Vc/F) and peripheral compartment (Vp/F) distribution volumes. A time lagged first order absorption process was characterized using transit compartment models and the number of absorption compartments were fixed to 2. According to the base model, the MeltDose® Tac shows a mean absorption transit time of 2.91 hours. Inter-individual variability could be associated with CL/F, Vc/F and Vp/F. Adding IOV on CL/F caused statistically significant reduction in the model MOFV (∆MOFV= -232 units, p<0.005) and was therefore retained in the final model. Further inclusion of the IOV on other parameters did not improve the model. A proportional error model best described the RE distribution. When covariates were entered univariately on distribution Vc/F and Vp/F volumes or clearances, neither body weight nor body mass index or age provided a significant drop in the MOFV (p > 0.05). Allometric inclusion of body weigh on these PK parameters worsened the model. No improvement of the model was observed when residual error associated with concentrations was standardized by hematocrit values of 45%.
Genetic polymorphism in CYP3A5 (*1 expressers vs non expressers) statistically significantly influenced Tac CL/F, resulting in an MOFV drop of 25 points (p<0.001), and a reduction in unexplained IIV associated with CL/F of 19%. Two different clearance values were observed for each group, i.e. 20.4 L·h-1 for expressers vs. 10.1 L·h-1 for non-expressers. In contrast, when CYP3A4 genotypes were tested on CL/F (*22 carriers vs. non-carriers), no statistically significant difference was found and no reduction of the IIV associated with CL/F was observed. A cluster of CYP3A4 and CYP3A5 polymorphism was created combining both SNPs. The different cluster phenotype groups resulted in significantly different means of C0 normalized by dose (Table 2), 1.23, 2.94 and 4.04 ng.mL-1 for HM, IM and PM groups (p<0.001) , respectively. The inclusion of cluster as a covariate significantly decreased the MOFV with respect to the base model, resulting in an MOFV drop by 37.5 points (p<0.001). Three different CL/F values were identified one per each cluster phenotype, i.e. CL/F values were of 19.6 L·h-1, 10.6 L·h-1, and 7.37 L·h-1, for high, intermediate, and poor metabolizers, respectively. After inclusion of this covariate a reduction of 22.5% was observed in IIV associated with CL/F.
On the other hand, the ABCB1 *C/*C SNP failed to influence the model significantly. The unexplained IIV associated with the CL/F was reduced from 49% to 38% from base to final covariate model. The final model parameters were estimated with good precision, the RSE% of all the parameters being lower than 35%. The eta shrinkage for CL/F in the final model was 13%. Residual unexplained variability (RUV) associated with the final model was 9.67% and the corresponding shrinkage was 23.5%. The final population pharmacokinetic parameter values are displayed in Table 3.
The descriptive capability of the model was confirmed by the goodness of fit plots. Good correlations between the observed and population and individual predicted concentrations were found with a random distribution around the identity line. Individual weighted residuals (IWRES) did not show any trend when plotted against individual predicted concentrations, this confirming that residual error was adequately modelled. Similarly, conditional weighted residuals (CWRES) indicated that the structural part of the model adequately described the data (Figure 2). The scatter plots of NPDE vs time and individual predicted concentrations (Figure 3) showed a random distribution around null line with most of predicted npde values within the 95% confidence interval of the theoretical normal distribution, this proving the descriptive capability of the model.
The pcVPC (Figure 4) showed that the model properly describes the mean tendency of the entire data. In general, median, 5th and 95th percentiles of the observations, were within the 95% confidence intervals for the median, the 5th and 95th percentiles of the simulated profiles.
4. Discussion
As the best of our knowledge, this is the first population approach study focused on the influence of genetic polymorphisms of CYP3A5, CYP3A4 and ABCB1 on Tac pharmacokinetics upon LCP-Tac administration in stable kidney transplant patients. Only in a previous study from Woillard et al.37 a similar number of data was analyzed (637 from two full PK profiles vs. 655 in our study) from less patients (47 vs. 98 in our study), but no influence of genetics was explored. In another study, Henin et al.35 included a larger set of data and also addressed the influence of CYP3A5 SNPs but not of CYP3A4 and ABCB1, which are also involved in Tac pharmacokinetics, in “de novo” patients.
Unlike previous studies in kidney transplant patients35,37, the disposition profile of Tac after LCP-Tac administration was best described by a two-compartment model. The delayed first order absorption was described by 2 transit compartments, and a mean transit time of 2.91 hours. The number of transit compartments had to be fixed in the final model to better estimate the central compartment distribution volume and inter-individual variability associated with this parameter. The description of delayed absorption and disposition profile provided by our final model was consistent with that of Martial et al.36 in stable adult hepatic transplant patients (two compartmental model, NN=1.6 and MTT=3.4 hours). By contrast, other studies in renal transplantation35,37 omitted one disposition compartment to better describe the complex Tac delayed absorption from LCP-Tac, hereby characterizing two and three absorption phases with rapid/slow37 or rapid/medium/slow35 absorption kinetics. According to these models, the highest fraction of drug absorbed occurred in the slow (60%)37 and the medium kinetic absorption phases (61%, MTT=4.82 h)35, that probably represent what occurs at the more distal parts of the small intestine as described before for LCP_Tac46. Thus, a scintigraphy study carried out in healthy volunteers revealed that LCP-Tac reached the colon at around 3.8-4.8 hours post administration46. Interestingly, our MTT (2.91 h) was between the fast (1.06 h) and medium (4.82 h) mean times reported by Henin for the absorption process. On the other hand, in our study and previous35, in general, a high inter-individual variability was observed for parameters characterizing the absorption process. Herein, further analyses with more data/patients would be required to confirm previous findings. In any case, all these results confirm the delayed absorption rate of Tac from LCP-Tac, with probably the most fractions being absorbed at the distal part of the small intestine, but also the importance of using Erlang distribution or transit compartment models to describe it.
Our model is less mechanistic than those previously developed35,37, and it did not describe the multiple peaks observed in the absorption profiles (Figure 1), whereas it adequately described the mean trend of the data. The observed peak and C0 were correctly captured by our model as evidenced by the prediction visual predictive check plot (Figure 3). Thus, a feasible estimation of the central compartment distribution volume was found which is crucial for a correct prediction of peak concentrations. Of note, the total distribution volume estimated by our model (Vc/F+Vp/F) was higher than the distribution volumes previously reported by Henin (629 L vs. 452 L35, respectively) but closer to that reported by Martial et al36 in hepatic transplant patients, although these authors fixed the Vp/F value to 500 L.
Among all the covariates tested, the CYP3A5 SNPs expressers vs. non-expressers was statistically significant on CL/F, but no significance was found when the influence of CYP3A4 SNPs (*22 carriers vs. non-carriers) were tested on CL/F. Neither SNP associated with ABCB1 (*C carriers vs. non-carriers) showed influence on CL/F. These findings were consistent with those of the statistical analyses of exposure metrics estimated from raw data. Indeed, statistically significant differences were found in AUC/D and C0/D values (p<0.005) when compared CYP3A5 expressers vs. non-expressers (Table 2) with lower values for expressers that also had higher CL/F values (20.4 L·h-1 vs. 10.1 L·h-1 for expressers vs. non-expressers, respectively). By contrast although a trend to higher exposure (AUC/D) was found for CYP3A4*22 carriers vs. non carriers, these differences did not achieve significance (p=0.2410), but they became significant when a higher sample size was available in the case of C0/D (p<0.005). In any case, the relative magnitude of the differences in exposure metrics between genetic groups was lower for CYP3A4 (from 20 to 27%) than CYP3A5 (from 46 to 60%). This could be explained by a less contribution of CYP3A4 on the Tac metabolism as a consequence of a lower intrinsic clearance47, but also by the low number of patients of the *22 carrier group that lead to the mean AUC/D value estimated in this group.
As previously32, all these findings, led to investigate the effect of a combination of CYP3A4/A5 genotypes as a cluster grouped with three phenotypes (HM, IM, PM). The inclusion of the cluster on CL/F was statistically significant resulting in three different CL/F values (19.6, 10.6 and 7.37 L·h-1 for HM, IM and PM, respectively) with a slightly higher reduction of MOFV. In line with this, statistically significant differences were found among AUC/D values of HM and IM or PM, but not between IM and PM. However, differences between IM and PM were evidenced when sample size was increased in C0/D comparisons (p<0.005). Thus, the significant inclusion of the cluster reinforced the impact of CYP3A4*22 carriers vs. non carriers on Tac metabolism.
After comparing our results with those previously reported by our group32, in Tac-IR formulations, similar values for HM (CL/FHM: 19.7, n=47 and 19.6 L·h-1, n=19 was found for Tac-IR and LCP-Tac respectively. On the other hand, slightly higher values were found for IM (CL/FIM: 12.5 L·h-1, n= 230 and 10.6 L·h-1, n=68 and PM (CL/FPM: 9.1 L·h-1, n=27 and 7.37 L·h-1, n=11) in Tac-IR vs LCP-Tac formulations respectively. In our previous model32, the delayed absorption was not described with transit models, thereby this leading to lower predicted concentrations at the end of the dosing interval than expected with a transit compartment modeling. Furthermore, although differences in the inter-individual variability found in both studies, can bias comparisons, a trend to higher bioavailability (or lower CL/F values) for LCP-Tac formulation than the Tac-IR is evidenced, as it should be expected19. Indeed, taking into account the CYP3A4/A5 expression along the gut, these differences could be attributed to the different release profile of Tac from both formulations. Unlike Tac-IR, according to results of our model, LCP-Tac is not immediately released at the proximal part of the intestine, where due to the higher expression of CYP3A4/A5, higher first pass effect takes place. Regarding the effect of ABCB1 SNP (*T/*T, *C/*T and *C/*C SNPs), no influence on CL/F was detected and differences between exposure metrics of *C carriers vs. non-carriers (Table 2). This is in agreement with previous studies carried out with the Tac-IR formulation exposure7,25.
Our CL/F values were lower in CYP3A5 expressers vs non-expressers (20.4 L·h-1 and 10.1 L·h-1 respectively), than those reported by Henin et al.35 in “de novo” patients. Specifically in this population, considering a mean bodyweight around 70 kg, CL/F of 33.2 and 20 L·h-1 were estimated for CYP3A5 expressers. These differences could be due to co-administered corticosteroids at high doses at the early period post-transplant but also to hematocrit concentrations between “de novo” and stable patient sub-populations. Patients of our study received reduced doses or discontinued corticosteroids, compared to the higher doses given to “de novo” patients, according to standard practice. Higher doses of corticosteroids can lead to greater induction of CYP3A4 metabolizing enzyme and thus leading to higher Tac clearance 48. This finding again supports the role of CYP3A4 on the metabolism of Tac. On the other hand, hematocrit concentrations of our study were higher (from 37.4 to 44.0 % (IQR)) than those reported in “de novo” patients35 (from 22.9 to 37%). As a restrictive clearance drug, Tac is expected to decrease its clearance as the free fraction decreases, as occurs in our stable patients where hematocrit concentrations reach its almost normal values compared with “de novo” patients. This was also the reason why probably this covariate was not predictive of CL/F variability in the final model. All these findings support the development of the current model for LCP-Tac dose adjustments in stable patients. There is still ongoing debate about the inclusion of size metrics in Tac population PK models, particularly when these models are used for dose calculation during the clinical practice.
The major limitation of the current study is the high number of sparse data compared to intensive sampling to better describe the delayed absorption phase. Another limitation is the low prevalent PM patients in Caucasian population. Thus, larger studies with intensive sampling over a dosing interval are required to confirm our results.
5. Conclusion
A new population pharmacokinetic model has been developed in stable renal adult transplant patients upon LCP-Tac administration. The influence of CYP3A4/A5 SNPs as a combined cluster including three different phenotypes has been identified as a powerful covariate to describe part of inter-individual variability associated with the apparent elimination clearance. This suggests the use of this covariate for dose optimization during the routine clinical practice.
Author Contributions
Conceptualion, H.C. and N.L.; Methodology, Z.M.A., M.M., B.F., P.F., A.V-A, and R. R-B.; Software, H.C.; Acquisition of data Z.M.A., M.M., B.F., P.F. and A.V-A , Analysis and interpretation of data Z.M.A., M.M., A.V-A., E.M., JM.C., JM.G., H.C., N.L. Writing–Original Draft Preparation, Z.M.A., M.M., A.V-A, JM.C., JM.G., H.C., N.L.; Writing–Review & Editing, Z.M.A., M.M., A.V-A, JM.C., JM.G., H.C., N.L.; Supervision, H.C. and N.L.; Project Administration, H.C. and N.L.; Funding Acquisition, H.C and N.L. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
We are especially grateful to Gema Cerezo of the nephrology laboratory team for her crucial technical assistance in this study. We thank CERCA Programme/Generalitat de Catalunya for institutional support. The present study was supported Instituto de Salud Carlos III and Ministerio de Sanidad y Consumo (PI18/01740, PI21/00559) (co-founded by FEDER funds/European Regional Development Fund (ERDF), a way to Build Europa, RICORS (kidney disease, RD21/00050021).
References
- Bowman, L.J.; Brennan, D.C.; Facp, D.P. The role of tacrolimus in renal transplantation. Expert Opin. Pharmacother. 2008, 9, 635–643. [Google Scholar] [CrossRef]
- Kamel, M.; Kadian, M.; Srinivas, T.; Taber, D.; Salas, M.A.P. Tacrolimus confers lower acute rejection rates and better renal allograft survival compared to cyclosporine. World J. Transplant. 2016, 6, 697–702. [Google Scholar] [CrossRef]
- Venkataramanan, R.; Swaminathan, A.; Prasad, T.; Jain, A.; Zuckerman, S.; Warty, V.; McMichael, J.; Lever, J.; Burckart, G.; Starzl, T. Clinical Pharmacokinetics of Tacrolimus. Clin. Pharmacokinet. 1995, 29, 404–430. [Google Scholar] [CrossRef]
- E Staatz, C.; E Tett, S. Clinical Pharmacokinetics and Pharmacodynamics of Tacrolimus in Solid Organ Transplantation. Clin. Pharmacokinet. 2004, 43, 623–653. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, D.R.J. Intrapatient Variability of Tacrolimus Exposure in Solid Organ Transplantation: A Novel Marker for Clinical Outcome. Clin. Pharmacol. Ther. 2020, 107, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Neylan, J. Effect of Race and Immunosuppression in Renal Transplantation: Three-Year Survival Results From a US Multicenter, Randomized Trial. Transplant. Proc. 1998, 30, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Staatz, C.E.; Goodman, L.K.; Tett, S.E. Effect of CYP3A and ABCB1 Single Nucleotide Polymorphisms on the Pharmacokinetics and Pharmacodynamics of Calcineurin Inhibitors: Part I. Clin. Pharmacokinet. 2010, 49, 141–175. [Google Scholar] [CrossRef] [PubMed]
- Bekersky, I.; Dressler, D.; Mekki, Q.A.; Bekersky, F.I.; Bs, D.D. Effect of Low- and High-Fat Meals on Tacrolimus Absorption following 5 mg Single Oral Doses to Healthy Human Subjects. J. Clin. Pharmacol. 2001, 41, 176–182. [Google Scholar] [CrossRef]
- Tuteja, S.; Alloway, R.R.; Johnson, J.A.; Gaber, A.O. The effect of gut metabolism on tacrolimus bioavailability in renaltransplant Recipients1,2. Transplantation 2001, 71, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Kim, D.H.; Lee, S.M.; Han, N.Y.; Oh, J.M.; Ha, J.; Kim, Y.S. Pharmacokinetics of tacrolimus according to body composition in recipients of kidney transplants. Kidney Res. Clin. Pr. 2012, 31, 157–162. [Google Scholar] [CrossRef]
- Seegars, M.B.; Tooze, J.; Isom, S.; Anders, B.; Kennedy, L.; Rodriguez, C. Tacrolimus Levels and Correlation to Age and Weight Calculation. Biol. Blood Marrow Transplant. 2018, 24, S337–S337. [Google Scholar] [CrossRef]
- Katari, S.R.; Magnone, M.; Shapiro, R.; Jordan, M.; Scantlebury, V.; Vivas, C.; Gritsch, A.; McCauley, J.; Starzl, T.; Demetris, A.J.; et al. Clinical features of acute reversible tacrolimus (FK 506) nephrotoxicity in kidney transplant recipients. Clin Transplant. 1997, 11, 237. [Google Scholar] [PubMed]
- Weng, L.; Chiang, Y.; Lin, M.; Hsieh, C.; Lin, S.; Wei, T.; Chou, H. Association Between Use of FK506 and Prevalence of Post-Transplantation Diabetes Mellitus in Kidney Transplant Patients. Transplant. Proc. 2014, 46, 529–531. [Google Scholar] [CrossRef]
- Sierra-Hidalgo, F.; Martínez-Salio, A.; Moreno-García, S.; de Pablo-Fernández, E.; Correas-Callero, E.; Ruiz-Morales, J. Akinetic Mutism Induced by Tacrolimus. Clin. Neuropharmacol. 2009, 32, 293–294. [Google Scholar] [CrossRef]
- Wallemacq, P.E.; Furlan, V.; Möller, A.; Schäfer, A.; Stadler, P.; Firdaous, I.; Taburet, A.-M.; Reding, R.; De Clety, S.C.; Goyet, J.D.V.D.; et al. Pharmacokinetics of tacrolimus (FK506) in paediatric liver transplant recipients. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, S.; Nigro, V.; Weinberg, J.; Woodle, E.S.; Alloway, R.R. A Steady-State Head-to-Head Pharmacokinetic Comparison of All FK-506 (Tacrolimus) Formulations (ASTCOFF): An Open-Label, Prospective, Randomized, Two-Arm, Three-Period Crossover Study. Am. J. Transplant. 2017, 17, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Gaber, A.O.; Alloway, R.R.; Bodziak, K.; Kaplan, B.; Bunnapradist, S. Conversion from Twice-Daily Tacrolimus Capsules to Once-Daily Extended-Release Tacrolimus (LCPT). Transplantation 2013, 96, 191–197. [Google Scholar] [CrossRef]
- Trofe-Clark, J.; Brennan, D.C.; West-Thielke, P.; Milone, M.C.; Lim, M.A.; Neubauer, R.; Nigro, V.; Bloom, R.D. Results of ASERTAA, a Randomized Prospective Crossover Pharmacogenetic Study of Immediate-Release Versus Extended-Release Tacrolimus in African American Kidney Transplant Recipients. Am. J. Kidney Dis. 2018, 71, 315–326. [Google Scholar] [CrossRef]
- Staatz, C.E.; Tett, S.E. Clinical Pharmacokinetics of Once-Daily Tacrolimus in Solid-Organ Transplant Patients. Clin. Pharmacokinet. 2015, 54, 993–1025. [Google Scholar] [CrossRef]
- Budde, K.; Bunnapradist, S.; Grinyo, J.M.; Ciechanowski, K.; Denny, J.E.; Silva, H.T.; Rostaing, L.; Envarsus study, group. Novel Once-Daily Extended-Release Tacrolimus (LCPT) Versus Twice-Daily Tacrolimus in De Novo Kidney Transplants: One-Year Results of Phase III, Double-Blind, Randomized Trial. Am. J. Transplant. 2014, 14, 2796–2806. [Google Scholar] [CrossRef]
- Thörn, M.; Finnström, N.; Lundgren, S.; Rane, A.; Lööf, L. Cytochromes P450 and MDR1 mRNA expression along the human gastrointestinal tract. Br. J. Clin. Pharmacol. 2005, 60, 54–60. [Google Scholar] [CrossRef]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Z.; Zheng, J.; Chen, Z.; Tang, Z.; Chen, J.; Li, L. Influence of CYP3A5 and MDR1 polymorphisms on tacrolimus concentration in the early stage after renal transplantation. Clin. Transplant. 2005, 19, 638–643. [Google Scholar] [CrossRef]
- de Jonge, H.; de Loor, H.; Verbeke, K.; Vanrenterghem, Y.; Kuypers, D.R. In Vivo CYP3A4 Activity, CYP3A5 Genotype, and Hematocrit Predict Tacrolimus Dose Requirements and Clearance in Renal Transplant Patients. Clin. Pharmacol. Ther. 2012, 92, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Capron, A.; Mourad, M.; De Meyer, M.; De Pauw, L.; Eddour, D.C.; Latinne, D.; Elens, L.; Haufroid, V.; Wallemacq, P. CYP3A5 and ABCB1 polymorphisms influence tacrolimus concentrations in peripheral blood mononuclear cells after renal transplantation. Pharmacogenomics 2010, 11, 703–714. [Google Scholar] [CrossRef]
- Pallet, N.; Jannot, A.-S.; El Bahri, M.; Etienne, I.; Buchler, M.; de Ligny, B.H.; Choukroun, G.; Colosio, C.; Thierry, A.; Vigneau, C.; et al. Kidney Transplant Recipients Carrying the CYP3A4*22 Allelic Variant Have Reduced Tacrolimus Clearance and Often Reach Supratherapeutic Tacrolimus Concentrations. Am. J. Transplant. 2015, 15, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Kahaar, E.; Winter, S.; Tremmel, R.; Schaeffeler, E.; Olbricht, C.J.; Wieland, E.; Schwab, M.; Shipkova, M.; Jaeger, S.U. The Impact of CYP3A4*22 on Tacrolimus Pharmacokinetics and Outcome in Clinical Practice at a Single Kidney Transplant Center. Front. Genet. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Andrews, L.; van Gelder, T.; Shi, Y.; van Schaik, R.; Wang, L.; Hesselink, D. Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: recent developments and ethnic considerations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 555–565. [Google Scholar] [CrossRef]
- Brooks, E.; Tett, S.E.; Isbel, N.M.; Staatz, C.E. Population Pharmacokinetic Modelling and Bayesian Estimation of Tacrolimus Exposure: Is this Clinically Useful for Dosage Prediction Yet? Clin. Pharmacokinet. 2016, 55, 1295–1335. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.-B.; Mourad, M.; Neely, M.; Capron, A.; van Schaik, R.H.; van Gelder, T.; Lloberas, N.; Hesselink, D.A.; Marquet, P.; Haufroid, V.; et al. Tacrolimus Updated Guidelines through popPK Modeling: How to Benefit More from CYP3A Pre-emptive Genotyping Prior to Kidney Transplantation. Front. Pharmacol. 2017, 8, 358–358. [Google Scholar] [CrossRef]
- Andrews, L.M.; Hesselink, D.A.; van Schaik, R.H.N.; van Gelder, T.; de Fijter, J.W.; Lloberas, N.; Elens, L.; Moes, D.J.A.R.; de Winter, B.C.M. A population pharmacokinetic model to predict the individual starting dose of tacrolimus in adult renal transplant recipients. Br. J. Clin. Pharmacol. 2018, 85, 601–615. [Google Scholar] [CrossRef]
- Andreu, F.; Colom, H.; Elens, L.; van Gelder, T.; van Schaik, R.H.N.; Hesselink, D.A.; Bestard, O.; Torras, J.; Cruzado, J.M.; Grinyó, J.M.; et al. A New CYP3A5*3 and CYP3A4*22 Cluster Influencing Tacrolimus Target Concentrations: A Population Approach. Clin. Pharmacokinet. 2017, 56, 963–975. [Google Scholar] [CrossRef]
- Andreu, F.; Colom, H.; Grinyó, J.M.; Torras, J.; Cruzado, J.M.; Lloberas, N. Development of a Population PK Model of Tacrolimus for Adaptive Dosage Control in Stable Kidney Transplant Patients. Ther. Drug Monit. 2015, 37, 246–255. [Google Scholar] [CrossRef]
- Woillard, J.-B.; Saint-Marcoux, F.; Debord, J.; Åsberg, A. Pharmacokinetic models to assist the prescriber in choosing the best tacrolimus dose. Pharmacol. Res. 2018, 130, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Henin, E.; Govoni, M.; Cella, M.; Laveille, C.; Piotti, G. Therapeutic Drug Monitoring Strategies for Envarsus in De Novo Kidney Transplant Patients Using Population Modelling and Simulations. Adv. Ther. 2021, 38, 5317–5332. [Google Scholar] [CrossRef] [PubMed]
- Martial, L.C.; Biewenga, M.; Ruijter, B.N.; Keizer, R.; Swen, J.J.; van Hoek, B.; Moes, D.J.A.R. Population pharmacokinetics and genetics of oral meltdose tacrolimus (Envarsus) in stable adult liver transplant recipients. Br. J. Clin. Pharmacol. 2021, 87, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.-B.; Debord, J.; Monchaud, C.; Saint-Marcoux, F.; Marquet, P. Population Pharmacokinetics and Bayesian Estimators for Refined Dose Adjustment of a New Tacrolimus Formulation in Kidney and Liver Transplant Patients. Clin. Pharmacokinet. 2017, 56, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Rigo-Bonnin, R.; Arbiol-Roca, A.; de Aledo-Castillo, J.M.G.; Alía, P. Simultaneous Measurement of Cyclosporine A, Everolimus, Sirolimus and Tacrolimus Concentrations in Human Blood by UPLC–MS/MS. Chromatographia 2015, 78, 1459–1474. [Google Scholar] [CrossRef]
- Savic, R.M.; Jonker, D.M.; Kerbusch, T.; Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007, 34, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.O.; Sheiner, L.B. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J. Pharmacokinet. Biopharm. 1993, 21, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Nakagawa, T.; Uno, T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 1978, 6, 165–175. [Google Scholar] [CrossRef]
- Savic, R.M.; Karlsson, M.O. Importance of Shrinkage in Empirical Bayes Estimates for Diagnostics: Problems and Solutions. AAPS J. 2009, 11, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, E.N.; Karlsson, M.O. Automated Covariate Model Building Within NONMEM. Pharm. Res. 1998, 15, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-Corrected Visual Predictive Checks for Diagnosing Nonlinear Mixed-Effects Models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Comets, E.; Brendel, K.; Mentré, F. Computing normalised prediction distribution errors to evaluate nonlinear mixed-effect models: The npde add-on package for R. Comput. Methods Programs Biomed. 2008, 90, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Glicklich, A.; Weinberg, J. Improved Bioavailability of MELTDOSE Once-Daily Formulation of Tacrolimus (LCP-Tacro) with Controlled Agglomeration Allows for Consistent Absorption over 24 Hrs: A Scintigraphic and Pharmacokinetic Evaluation. American Journal of Transplants. Published 2013. Available online: https://atcmeetingabstracts.com/abstract/improved-bioavailability-of-meltdose-once-daily-formulation-of-tacrolimus-lcp-tacro-with-controlled-agglomeration-allows-for-consistent-absorption-over-24-hrs-a-scintigraphic-and-pharmacokinetic-ev/ (accessed on 26 October 2022).
- Kamdem, L.K.; Streit, F.; Zanger, U.M.; Brockmöller, J.; Oellerich, M.; Armstrong, V.W.; Wojnowski, L. Contribution of CYP3A5 to the in Vitro Hepatic Clearance of Tacrolimus. Clin. Chem. 2005, 51, 1374–1381. [Google Scholar] [CrossRef]
- Undre, N.A. Pharmacokinetics of tacrolimus-based combination therapies. Nephrol. Dial. Transplant. 2003, 18 (Suppl. S1), i12–i15. [Google Scholar] [CrossRef]
Figure 1.
(A) Overlaid individual steady-state PK profiles of Tac following LCP-Tac administration. (B) Overlaid normalized by dose individual steady-state PK profiles of Tac following LCP-Tac administration.
Figure 1.
(A) Overlaid individual steady-state PK profiles of Tac following LCP-Tac administration. (B) Overlaid normalized by dose individual steady-state PK profiles of Tac following LCP-Tac administration.
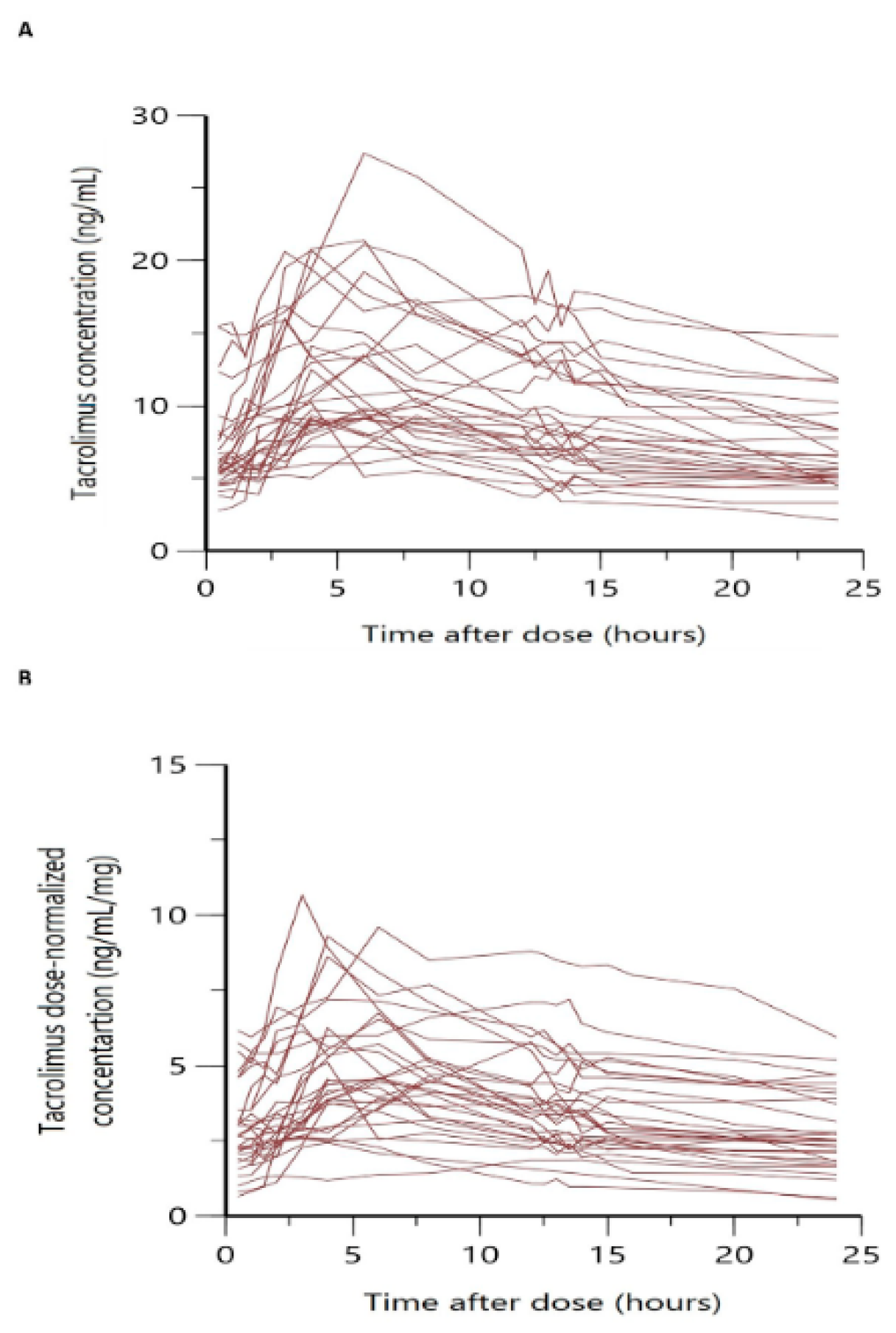
Figure 2.
Goodness-of-fit plots of the final model. (A) Observed concentrations vs. population predicted concentrations. (B) Observed concentrations vs. individual predicted concentrations. (C) Individual weighted residuals (IWRES) vs. individual predicted concentrations. (D) Conditional weighted residuals (CWRES) vs. time from the start of the study.
Figure 2.
Goodness-of-fit plots of the final model. (A) Observed concentrations vs. population predicted concentrations. (B) Observed concentrations vs. individual predicted concentrations. (C) Individual weighted residuals (IWRES) vs. individual predicted concentrations. (D) Conditional weighted residuals (CWRES) vs. time from the start of the study.
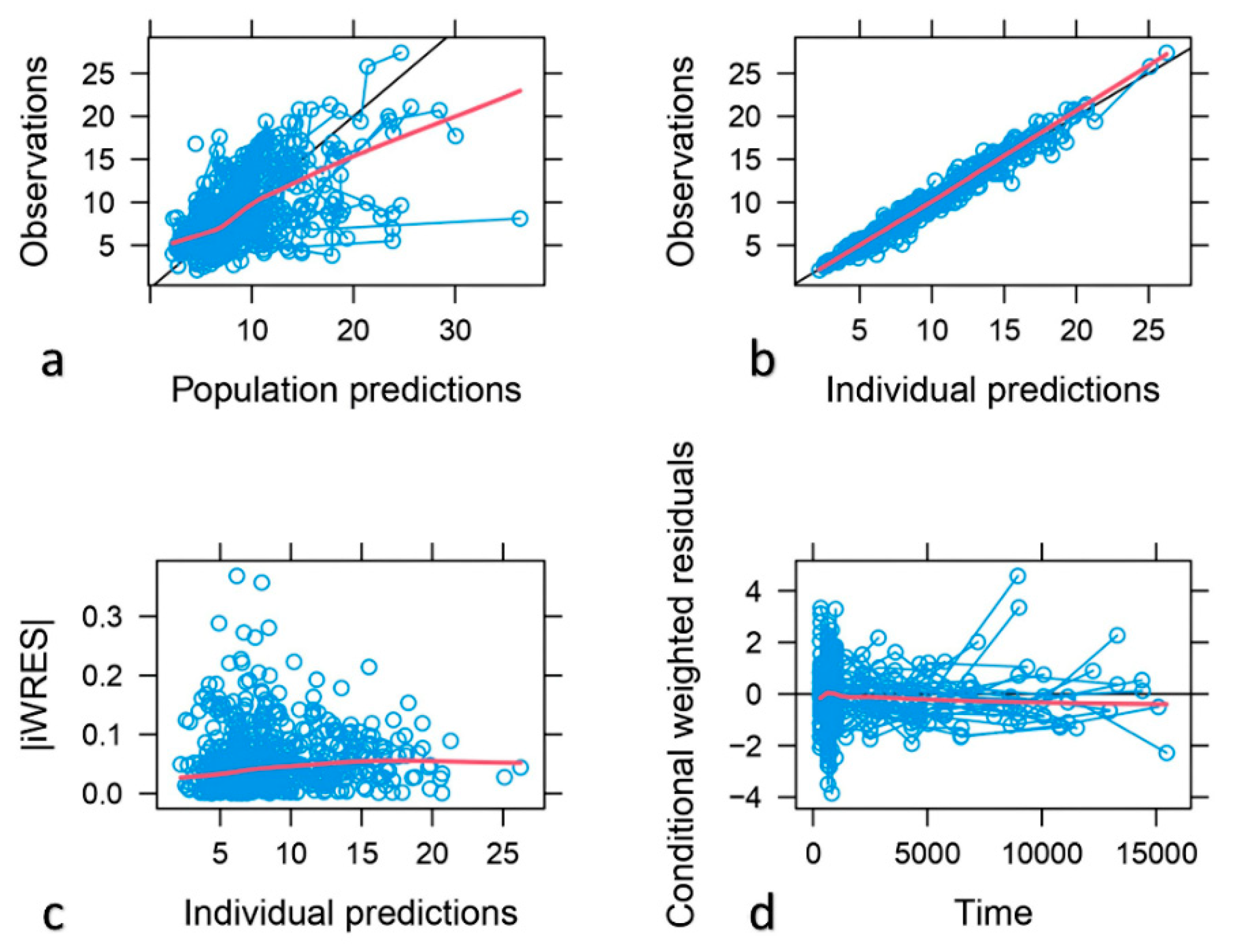
Figure 3.
Normalized prediction distribution errors (NPDE) of the final PopPK model. (A) Scattered plot of NPDE vs. time from the start of the treatment (B) Scattered plot of observed NPDE vs. individual predicted concentrations. The central dashed black line represents the null line, while the dotted and dashed black lines above and below the zero line represent the 95% confidence interval for the distribution NPDEs.
Figure 3.
Normalized prediction distribution errors (NPDE) of the final PopPK model. (A) Scattered plot of NPDE vs. time from the start of the treatment (B) Scattered plot of observed NPDE vs. individual predicted concentrations. The central dashed black line represents the null line, while the dotted and dashed black lines above and below the zero line represent the 95% confidence interval for the distribution NPDEs.
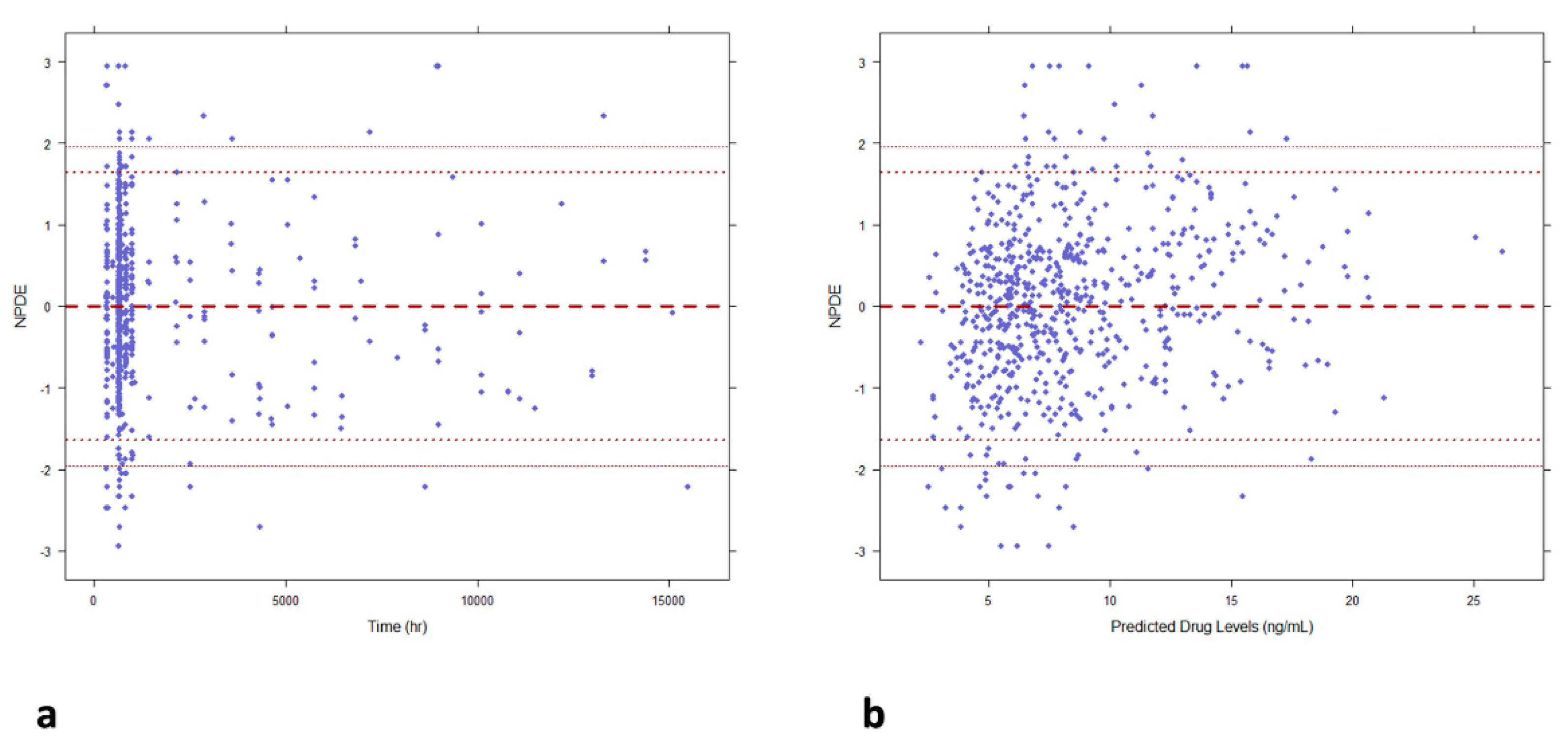
Figure 4.
Prediction-corrected visual predictive check (pcVPC) for the final model. Tac concentrations given in ng·mL-1, time after dose given in hours. The solid line represents the median observed whole blood concentrations (ng·mL-1); prediction-corrected plasma concentration in the pcVPC to the right, and the red band represents a simulation-based 95% confidence interval for the median. The observed 5% and 95% percentiles are presented with dashed red lines, and the 95% confidence intervals for the corresponding model predicted percentiles are shown as blue bands. The observed whole blood concentrations (prediction corrected in the pcVPC) are represented by blue circles.
Figure 4.
Prediction-corrected visual predictive check (pcVPC) for the final model. Tac concentrations given in ng·mL-1, time after dose given in hours. The solid line represents the median observed whole blood concentrations (ng·mL-1); prediction-corrected plasma concentration in the pcVPC to the right, and the red band represents a simulation-based 95% confidence interval for the median. The observed 5% and 95% percentiles are presented with dashed red lines, and the 95% confidence intervals for the corresponding model predicted percentiles are shown as blue bands. The observed whole blood concentrations (prediction corrected in the pcVPC) are represented by blue circles.
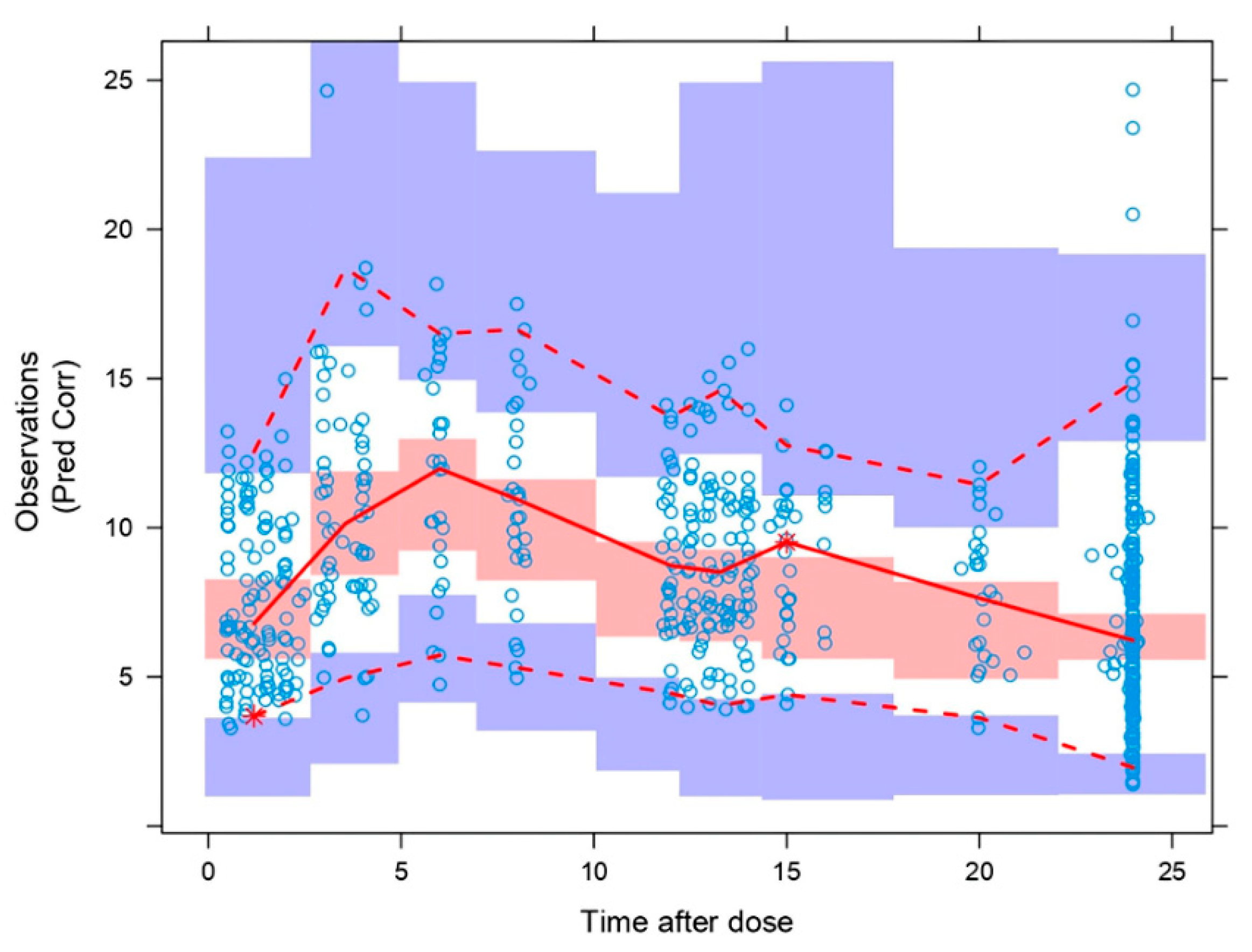

Table 1.
Demographic, biochemical and clinical characteristics of the patients included in the study.
Table 1.
Demographic, biochemical and clinical characteristics of the patients included in the study.
| Characteristics | All Patients | Full PK Profile | Sparse sampling (C0) |
|---|---|---|---|
| No. of Patients | 98 | 30 | 68 |
| No. of Sampling | 655 | 480 | 175 |
| Gender (Male/Female) | 68/30 | 20/10 | 45/23 |
| Weight (Kg) | 74.73 (65-81.13) | 72.82 (63.75-80) | 75.36 (65.5-88) |
| Age (Years) | 56 (46-68) | 57 (48-67) | 56 (45-68) |
| BMI (Kg.m-2) | 26.33 (22.94-28.94) | 26.44 (23.44-29.70) | 26.28 (22.89-28.64) |
| HTC ( %) | 40.53 (37.4-44.0) | 39.7 (37.1-41.9) | 40.64 (37.42 -44.0) |
| GF (mL.min-1) | 47.72 (36-58) | 54.86 (45.5 - 67) | 46.7 (36-57) |
| CR (μmol.L-1) | 146.9 (116-163) | 130.47 (106.5-149) | 149 (118-164) |
| CYP3A5 Genotype | |||
| *1/*1 | 4 (4.1%) | 1 (3.3%) | 3 (4.5%) |
| *1/*3 | 17 (17.3%) | 9 (30%) | 8 (11.5%) |
| *3/*3 | 77 (78.6%) | 20 (66.7%) | 57 (84%) |
| CYP3A4 Genotype | |||
| *1/*1 | 86 (86.7%) | 28 (93.3%) | 58 (84%) |
| *1/*22 | 12 (13.3%) | 2 (6.7%) | 10(16%) |
| Cluster | |||
| HM | 19 (19.4%) | 10 (33%) | 9 (13.25%) |
| IM | 68 (69.4%) | 18 (60%) | 50 (73.5%) |
| PM | 11 (11.2%) | 2 (7%) | 9 (13.25%) |
| ABCB1 Genotype | |||
| *T/*T | 21 (21%) | 9 (30%) | 12 (18%) |
| *C/*T | 46 (47%) | 12 (40%) | 34 (50%) |
| *C/*C | 31 (32%) | 9 (30%) | 22 (32%) |
Values are given as arithmetic mean (interquartile range) for continuous variables, and as count (percentage) for categorical variables BMI, body mass index; HTC, hematocrit; GF, glomerular filtration rate estimated by the CKD-EPI formula; Cr, Serum creatinine; HM, High metabolizer; IM, Intermediate metabolizer; PM, poor metabolizer.
Table 2.
Comparative normalized by dose C0 and AUC values sorted by the CYP3A5, CYP3A4, ABCB1 SNPs groups and by the three different cluster phenotypes.
Table 2.
Comparative normalized by dose C0 and AUC values sorted by the CYP3A5, CYP3A4, ABCB1 SNPs groups and by the three different cluster phenotypes.
| Genotype Group | Dose (mg.day-1) |
C0 | N | AUC | N | C0 /D | p-value* | AUC/D | p-value z | |||
| (ng.mL-1) | (ng.h.mL-1) | |||||||||||
| CYP3A5 | ||||||||||||
| CYP3A5 *1/*1, *1/*3 | 5 (2-12) | 6.14 (5.47-6.90) | 46 | 252(180-353) | 10 | 1.22 (1.07-1.42) | <0.001 | 51 (41-64) | <0.001 | |||
| CYP3A5 *3/*3 | 2 (0.5-8) | 6.4 (6.06-6.75) | 187 | 191 (165-223) | 20 | 3.08 (2.94-3.44) | 95 (81-110) | |||||
| CYP3A4 | ||||||||||||
| CYP3A4 *1/*1 | 2.5 (0.75-12) | 6.19 (5.86-6.54) | 180 | 207 (178-241) | 28 | 2.42 (2.23-2.67) | <0.05 | 76 (64-91) | 0.2410 | |||
| CYP3A4 *1/*22 | 1.75 (0.5-8) | 7.05 (5.71-7.20) | 53 | 198 (99-398) | 2 | 3.01 (2.80-4.15) | 122 (76-194) | |||||
| CLUSTER | ||||||||||||
| High-Metabolizer | 3 (2-12) | 6.16 (5.47-6.93) | 46 | 252(180-353) | 10 | 1.23 (1.06-1.42) | <0.001 # | 51 (41-64) | <0.001 # | |||
| <0.001 & | <0.05 & | |||||||||||
| Intermediate-Metabolizer | 2 (0.75-8) | 6.16 (5.80-6.54) | 141 | 190 (161-224) | 18 | 2.91 (2.67-3.18) | <0.05 $ | 92 (80-106) | 0.5756 $ | |||
| Poor-Metabolizer | 1.75 (0.5-8) | 7.3 (6.60-8.10) | 46 | 198 (99-398) | 2 | 4.04 (0.48-4.1) | 122 (76-194) | |||||
| ABCB1 | ||||||||||||
| ABCB1 *C/*T, *C/*C | 2 (0.5-12) | 6.25 (5.94-6.63) | 181 | 203 (170-242) | 21 | 2.58 (2.33-2.85) | 0.3073 | 78 (64-95) | 0.4537 | |||
| ABCB1 *T/*T | 2.125 (1-8) | 6.4 (5.79-7.30) | 52 | 213 (163-279) | 9 | 2.81 (2.48-3.19) | 83 (62-112) | |||||
| Values are reported as geometric means (95% CI). Abbreviations: AUC, area under the whole-blood concentration-time curve; Ctrough: trough whole blood concentrations, Ctrough/D and AUC/D normalized by dose Ctrough and AUC values. N=number of occasions data analyzed is the same for Ctrough and Ctrough/D. Similarly, N is the same for AUC and AUC/D. Dose expressed as median (range) | ||||||||||||
| *p-values for Ctrough/D mean values statistical comparisons | ||||||||||||
| z p-values for AUC/D mean values statistical comparisons | ||||||||||||
| # differences between HM and IM | ||||||||||||
| &differences between HM and PM | ||||||||||||
| $differences between IM and PM | ||||||||||||
Table 3.
Pharmacokinetic parameter estimates of the final model. Relative standard errors are given in parenthesis.
Table 3.
Pharmacokinetic parameter estimates of the final model. Relative standard errors are given in parenthesis.
| Parameter | Value (RSE %) | IIV % (RSE %) |
|---|---|---|
| CL/FHM (L.h-1) | 19.6 (10) | 37.9 (17.9) |
| CL/FIM (L.h-1) | 10.6 (5.2) | - |
| CL/FPM (L.h-1) | 7.37 (11.9) | - |
| Vc/F (L) | 169 (17.2) | 70 (41.4) |
| CLD/F (L.h-1) | 37.6 (13.5) | - |
| Ka(h-1) | 0.72 (33.2) | - |
| Vp (L) | 460 (27.8) | 75 (44.3) |
| MTT (h) | 2.91 (15.5) | 54.6 (37.1) |
| NN | 2 FIX | - |
| IOV (%) | 44.8 (27.4) | - |
| RE (%) | 9.67 (8) | - |
RSE: Relative Standard Error; IIV: Inter-Individual Variability; CL/FHM: Elimination clearance (High metabolizer); CL/FIM (L.h-1) : Elimination clearance (Intermediate metabolizer); CL/FPM (L.h-1): Elimination clearance (Poor metabolizer); Vc/F (L): Central compartment distribution volume; CLD/F (L.h-1) : Distributional clearance; Ka(h-1): Absorption rate constant; Vp (L): Peripheral compartment distribution volume; MTT (h): Mean transit time; NN: Number of transit compartments; IOV: Inter-occasion variability; RE: Residual error.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



