
Submitted:
11 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
Cathepsins (Caths) are lysosomal proteases that participate in various physiological and pathological processes. Accumulating evidence suggests that cathepsins play a multifaceted role in radiotherapy response. Firstly, cathepsins contribute to the remodelling of the tumor microenvironment by degrading extracellular matrix components, promoting angiogenesis, and facilitating tumor invasion and metastasis. Their proteolytic activity influences the tumor's response to radiation by affecting oxygenation, nutrient availability, and immune cell infiltration within the tumor microenvironment. Furthermore, cathepsins have been implicated in DNA damage repair processes, including non-homologous end joining and homologous recombination. Cathepsin-mediated DNA repair mechanisms can promote radioresistance in cancer cells, limiting the efficacy of radiotherapy. Additionally, cathepsins have been associated with the activation of prosurvival signalling pathways, such as PI3K/Akt and NF-κB, which can confer resistance to radiation-induced cell death.
Radiotherapy plays a crucial role in the treatment of various malignancies. However, the effectiveness of radiotherapy can be limited by intrinsic or acquired resistance mechanisms in cancer cells. In recent years, there has been growing interest in understanding the role of cathepsins, a family of proteases, in modulating the response of tumor cells to radiotherapy.
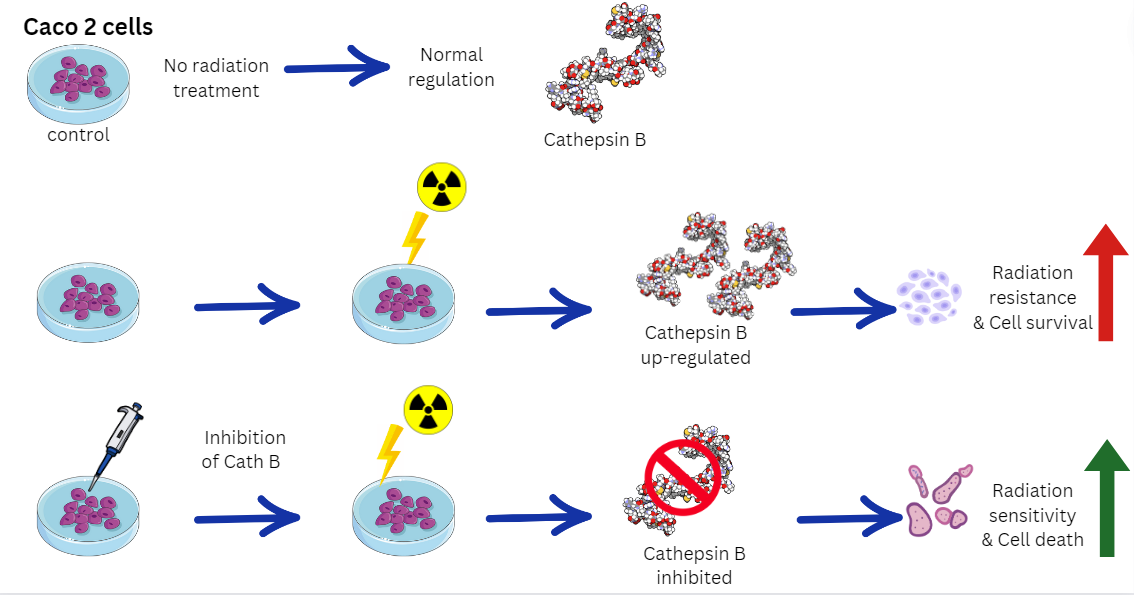
In this study, we shed light on the regulation and expression of cathepsin B in colon carcinoma cell line (Caco-2) before and after exposure to radiation were investigated. Radiation-induced upregulation of cathepsin B in dose-independent manner. Proteolytic inhibition of cathepsin B by cathepsin B specific inhibitor CA074 has increased the cytotoxic effect of ionizing irradiation in caco-2 cells. Similar results were also obtained after cathepsin B knockout by CRISPR CAS9. These results showed that cathepsin B could contribute to ionizing radiation resistance and abolishing of cathepsin B either by inhibition its proteolytic activity or expression has increased in caco-2 cells susceptibility to ionizing irradiation.
Keywords:
colon cancer
; cathepsin B
; radiotherapy
; radioresistance
; cathepsin inhibitors
; CA074
1. Introduction
Colorectal cancer (CRC) is a prevalent malignant tumor globally and ranks as the third major contributor to cancer-related fatalities (following breast and lung cancer) and the fourth highest fatality rate (after lung, liver, and stomach cancer) [1, 2], CRC constitutes approximately 10% of both cancer occurrence and fatality rates [3-5], and the second leading reason for cancer-related fatalities in developed nations [6].
Within the several enzymes housed within lysosomes, cathepsins stand out as a remarkable family of lysosomal proteases, displaying an expansive range of functions. cathepsins play a vital role in intracellular housekeeping, including their involvement in antigen processing during immune responses. Additionally, they contribute to the degradation of various proteins and chemokines, thereby actively involved in maintaining cellular homeostasis (as reviewed in [7-10]).
Cathepsins encompass several types, including serine types (cathepsins A and G), aspartic acid types (cathepsins D and E), and the most abundant cysteine types (cathepsins B, C, F, H, K, L, O, S, V, W, and X) [9, 11, 12]. These diverse cathepsins play crucial roles in numerous cellular activities, such as hormone synthesis and activation, as well as some physiological processes like apoptosis and autophagy.
Regarding cancer development, secreted Caths play a role in the degradation and remodeling of the tumor extracellular matrix (ECM), while intracellular cathepsins serve as crucial components of signaling pathways that can promote cell growth and inflammation [12, 13]. Furthermore, Caths are involved in response to anticancer therapy within the tumor microenvironment and can play pivotal roles in the development of resistance to therapeutic interventions [14-18].
Cathepsin B serves as a cysteine proteinase with a primary role as an endopeptidase in endolysosomal compartments of regular cells. Nonetheless, in the context of tumor growth, cathepsin B's control can undergo modifications at various stages, leading to its increased expression and extracellular release. This phenomenon potentially indicates cathepsin B's involvement in changes that contribute to the advancement of cancer [19, 20].
Alternative splicing and miRNA expression are noteworthy factors. Within the realm of proteases, Cathepsin B holds significant importance owing to its pivotal role in pathological mechanisms. As a vital component of the lysosome cascade, Cathepsin B operates as a cysteine protease, contributing to the control of metalloproteinases [21].
Cathepsin B's influence extends to intracellular communications, autophagy induction, and immune resistance. Its involvement in cell survival varies significantly across different conditions, ranging from caspase-dependent apoptosis to aiding tumor neovascularization and metastasis. In this review, we explore recent research that delves into Cathepsin B's role in pathological mechanisms, particularly within the context of cancer [22-24].
Cancer drug resistance is closely linked to the biology of lysosomes, as many conventional chemotherapeutic agents are sequestered within the acidic pH environment of these organelles [25]. This sequestration limits their interaction with their intended targets, reducing their effectiveness. Studies have demonstrated that the level of drug accumulation within lysosomes is directly correlated with cellular tolerance to their cytotoxic effects [26, 27].
Cancer cells secrete various proteases, including Caths, into the extracellular space, thereby promoting tumor invasiveness [28]. Interestingly, a study has shown that oral administration of a pH-buffering agent, which neutralizes the acidic pH within the tumor microenvironment, can reduce cancer cell metastasis. This effect is accompanied by a decrease in cath B activity [29].
Moreover, cathepsins play a role in angiogenesis by releasing pro-angiogenic factors and stimulating endothelial cell proliferation [21]. Another previous study has identified cathepsins within the tumor microenvironment as potential contributors for resistance during radiotherapy [30].
Given that the primary mechanism of action of radiotherapy is the selective damage to cancer cell DNA, evaluating the involvement of cathepsins in modulating radiation-induced DNA damage and repair mechanisms is of great significance in predicting the efficacy of radiotherapy. The expression and activity of cathepsins can be influenced by various factors, including radiation-induced stress responses and the tumor microenvironment. Exposure to radiation itself can trigger the upregulation and activation of cathepsins [31].
Under stress, Cathepsin B releases from lysosomes, relocating to the cytoplasm and potentially triggering a sequence of events culminating in toxicity. The gradual generation of reactive oxygen species (ROS) not only compromises lysosomal stability but also activates and releases Cathepsin B and L. Given that ROS can initiate both cytoprotective and cytotoxic autophagy, Cathepsin B's participation in either response, contingent on the stimulus, is plausible [32].
Cathepsins have demonstrated their ability to regulate tumor cell survival and apoptosis following radiation exposure. Cathepsin-mediated proteolysis can activate or deactivate specific apoptotic pathways, thus influencing the fate of irradiated cells [33]. Studies have shown that knockdown of cathepsin L using external inhibitors or siRNA-mediated silencing resulted in increased radiosensitivity of the p53-mutant glioma cell line U251. This effect could be attributed to G2/M phase cell cycle arrest or the malfunction of DNA damage repair processes [34].
By unraveling the specific mechanisms underlying cathepsin B-mediated radiation resistance in Caco-2 cells, this study offers insights into potential therapeutic strategies. Targeting cath B or its downstream pathways may represent a promising approach to sensitize Caco-2 cells to radiation and improve the overall therapeutic outcomes in colon cancer patients. Further investigation is needed to fully elucidate the molecular interactions and regulatory networks involving cath B, with the aim of developing targeted interventions that can enhance the effectiveness of radiotherapy in the clinical setting.
2. Results
2.1. Active-Site Labelling of Cysteine Peptidases with DCG-04 in Cultured Caco-2
Caco-2 cells were subjected to ascending doses of ionizing irradiation followed by treatment with DCG-04, an activity-based probe designed for papain family cysteine cathepsins. DCG-04 contains an epoxide warhead that selectively and irreversibly interacts with the active site cysteine residue of targeted cysteine cathepsins, facilitating the study of their specific activities rather than their abundance. After cell harvesting, SDS-PAGE under reducing conditions was performed to visualize DCG-04 labelled cysteine peptidases using streptavidin-HRP. Four major bands with molecular weights of 70, 40, 30, and 25 KDa were observed (Figure 1).
Considering that lysosomal cathepsins typically ranging in molecular weights between 20 and 45 KDa, the highest 70 KDa band was disregarded. The other three bands co-migrated with different forms of cath B, namely pro-cath B, intermediate form, and mature form. This co-migration pattern strongly indicates that cath B, a key member of the papain family cysteine cathepsins, may be closely involved in the cellular response to ionizing irradiation.
2.2. Pulldown of Cathepsin B
To investigate the potential upregulation of Cath B in response to ionizing irradiation, caco-2 cell lysates were obtained after exposure to different ionizing irradiation doses. Ex-vivo labelled, with DCG-04 before radiation exposure was, followed by incubation with streptavidin agarose beads. The proteins bound to the streptavidin beads were eluted and subjected to SDS-PAGE and immunoblotting using specific antibodies targeting cathepsin B. The results showed that Cath B showed significant upregulation across all tested doses (p < 0.001). At 2 Gy, Cath B exhibited approximately a four-fold upregulation, while at 8 Gy, it was upregulated by over seven-fold. At the highest tested dose of 10 Gy, Cath B showed a nearly nine-fold upregulation (Figure 2).
Notably, although the upregulation generally increased with higher doses, it was not strictly dose-dependent, as the upregulation at 2 Gy was slightly higher than that at 4 Gy.
2.3. Expression of Cath B before and after Exposure to Ionizing Irradiation
A series of experiments were conducted to detect the expression of Cath B before and after exposure to various ionizing irradiation doses. Following the exposure, cells were harvested, and protein extraction was performed. Subsequently, protein electrophoresis and immunoblotting were carried out using specific antibodies targeting cath B, while β-tubulin served as the loading control (Figure 3).
The obtained results demonstrated that the expressions of Cath B at 2 Gy, 4 Gy, 6 Gy, and 8 Gy were more than three times greater than those in non-irradiated cells. Interestingly, at 2 Gy, cath B expression was slightly higher compared to 4 Gy and 6 Gy. However, the most significant upregulation of cath B was observed at 10 Gy, where it exceeded 4.5 times the expression in non-irradiated cells.
Notably, the upregulation of cath B was observed in both its pro-Cath B and processed/mature forms. However, in contrast to the total Cath B levels, the mature cathepsin B was less abundant at the 10 Gy irradiation compared to the smaller doses.
2.4. Implication of Cath B Inhibition on Ionizing Radiation Cytotoxicity
To investigate the potential involvement of cath B in ionizing radiation cytotoxicity, caco-2 cells were exposed to ascending doses of ionizing radiation in the presence and absence of the cath B specific inhibitor CA074.
The MTT assay results demonstrated an escalation in cytotoxicity attributed to elevated ionizing radiation doses. Interestingly, pre-treating Caco-2 cells with a cathepsin B inhibitor further augmented the cytotoxic impact of radiation and correspondingly reduced cell viability. For instance, at 2 Gy, the effect of ionizing radiation on cell viability was relatively mild (around 5%), but it was increased to approximately 18% after cath B inhibition. while at 4 Gy, cell viability was reduced to about 93%, which was further decreased to about 77% after blocking the proteolytic activity of cath B with the specific inhibitor (Figure 4).
Similarly, at 6 Gy, the cytotoxic effect of ionizing radiation was more pronounced, resulting in a cell viability of approximately 64% without cath B inhibitor and 55% after cath B inhibition. At 8 Gy, cell viability was inhibited to 58% in the absence of the cath B inhibitor and 49% in its presence. Finally, at 10 Gy, cell viability was reduced to 40% without cath B inhibition and 36% after cath B inhibition. However, no significant differences were observed between the two control groups.
2.5. Impact of Cath B CRIPSR/CAS9 Mediated Knockout on Ionizing Radiation Cytotoxicity
To validate the previous cytotoxicity findings, we created caco-2 cells with Cath B knockout using CRISPR/Cas9 technology. The expression of Cath B was examined in both the wild-type caco-2 cells and the caco-2 cells with Cath B knockout. In the wild-type caco-2 cells, Cath B was detected, whereas in the caco-2 cells with Cath B knockout, Cath B was not detectable (Figure 5).
Notably, knocking out Cath B alone had no significant effect on cell viability. However, when caco-2 cells were exposed to 2 Gy of radiation, the cell viability decreased to approximately 80% compared to the controls. On the other hand, when Cath B was knocked down, the cell viability was reduced to about 20%. Similarly, with a 4 Gy radiation dose, cell viability decreased to about 66%, while knocking down Cath B resulted in a viability of about 20%. At a 6 Gy radiation dose, cell viability dropped to about 56%, but Cath B knockout further reduced cell viability to approximately 18%. Remarkably, at 8 Gy and 10 Gy radiation doses, there were similar outcomes, with cell viability reaching about 45%, while Cath B knockout in caco-2 cells led to a significant reduction in cell viability to about 10% (Figure 6).
2.6. Impact of Cath B Inhibition on Apoptosis in Ionizing Radiation Exposed Cells
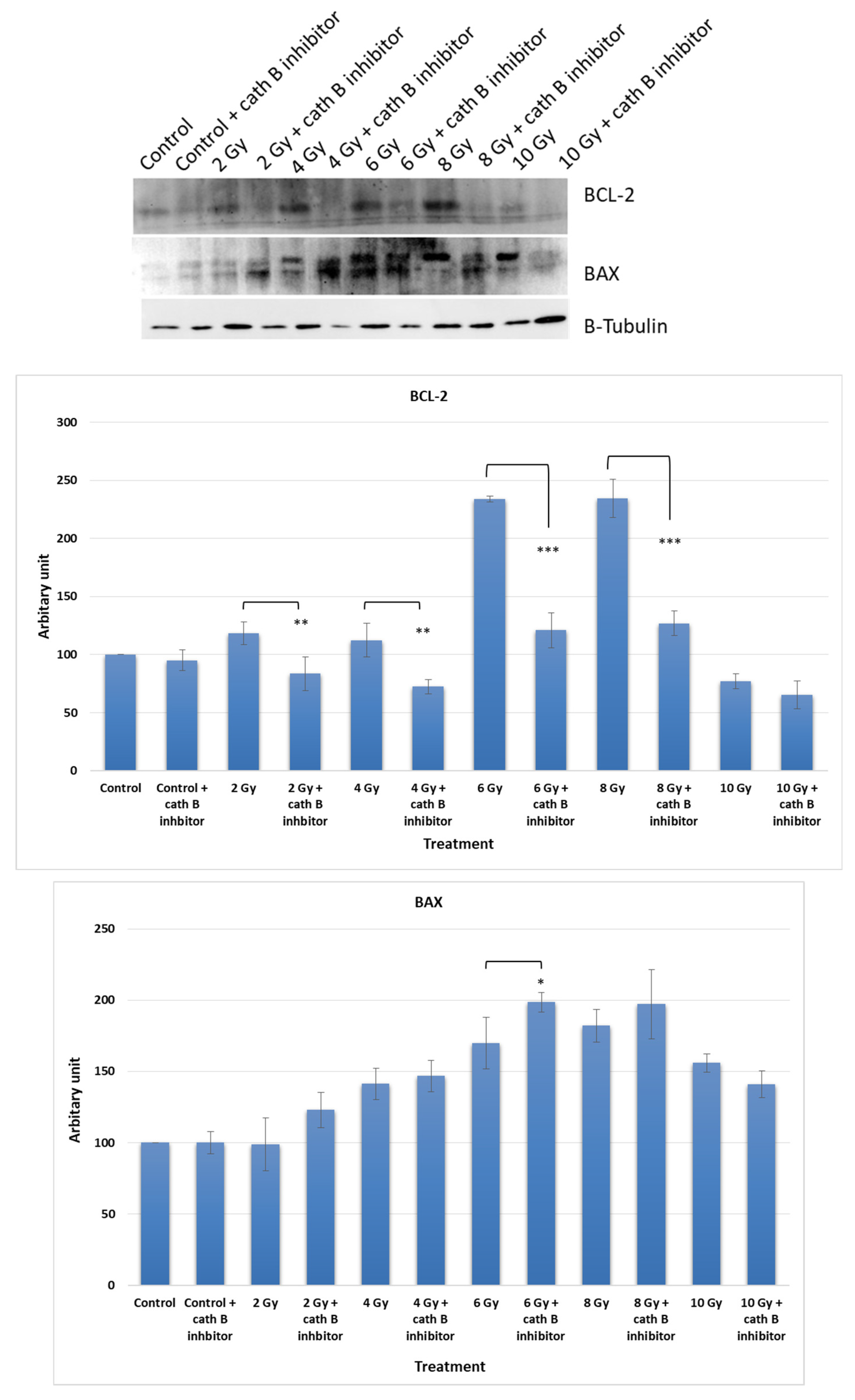
To investigate the potential impact of Cath B inhibition on the expression of apoptotic proteins, caco-2 cells were exposed to increasing doses of ionizing radiation in the presence and absence of the Cath B inhibitor CA074. After exposure, the cells were harvested, and proteins were extracted for analysis. Protein electrophoresis was performed under reducing conditions, followed by immunoblotting on nitrocellulose membranes using specific antibodies targeting the pro-apoptotic protein BAX and the anti-apoptotic protein BCL-2 (Figure 7).
The expression of BCL-2 was significantly reduced in CA074-treated cells compared to cells exposed only to ionizing radiation, except for the 10 Gy dose where the effect was less pronounced. At 4 Gy, BCL-2 expression decreased by approximately 40% in CA074-treated cells compared to those exposed only to ionizing radiation. The reduction was more pronounced at 6 Gy and 8 Gy doses, reaching about 50% in CA074-treated cells compared to cells exposed only to ionizing radiation.
Regarding the pro-apoptotic protein BAX, its expression increased by about 25% at 2 Gy and 6 Gy doses. However, the effect was less pronounced at 4 Gy and 8 Gy doses, although the difference was still statistically significant. Remarkably, at 10 Gy, BAX expression was slightly reduced in cells treated with Cath B inhibitor in comparison with cells exposed only to ionizing radiation.
These findings provide evidence of a potential role of cath B in caco-2 cells' resistance to ionizing radiation treatment and suggest a possible mechanism involving the suppression of certain apoptotic events.
3. Discussion
Colon cancer primarily impacts the elderly population, yet it is not limited to a specific age group. Its onset commonly originates from tiny cell clusters known as polyps that develop within the colon. Although most polyps are non-cancerous, certain ones may transform into colon cancer as time progresses. Typically, polyps remain asymptomatic [35]. Colon cancer is regarded as one of the most perilous and life-threatening forms of cancer. When detected at an early, localized stage, the survival rate stands at 91%. However, if the cancer has advanced to involve nearby tissues, organs, and/or regional lymph nodes, the 5-year relative survival rate drops to 72%. The situation is most critical when colon cancer spreads to distant parts of the body, resulting in a mere 13% 5-year relative survival rate [36]. Due to these factors, researchers are actively focusing their efforts on targeting this particular type of cancer to discover optimal strategies for controlling and managing the disease.
Radiation therapy is employed as a treatment method to effectively manage colon cancer. Several single-institution retrospective studies have demonstrated enhanced local control and potentially improved survival rates by incorporating external irradiation and/or intraoperative radiation in the treatment approach [37].
Cathepsins constitute a wide-ranging group of peptidases primarily active within endosomes and lysosomes, crucial components of the lysosomal death pathway [38]. Central to this pathway is lysosomal membrane permeation (LMP), wherein the integrity of the lysosomal membrane is compromised. This results in the release of luminal contents, including cathepsins, into the cytoplasm. The unleashed cathepsins then initiate a sequence of events leading to organelle impairment and triggering various forms of cell demise – encompassing apoptosis, pyroptosis, or necroptosis [39, 40]
Recent investigations have also underlined the connection between LMP and radiosensitivity. The induction of lysosomal biogenesis by irradiation amplifies the discharge of lysosomal hydrolysates into the cytoplasm, consequently intensifying cell mortality and achieving radiosensitization [41]. Importantly, the influence of cathepsins on cell demise hinges on whether their cleavage of substrates is activating or deactivating in nature.
Previous studies have observed an increase in lysosomal cathepsins as a consequence of radiotherapy, which has been associated with a crucial role in promoting resistance to this form of treatment [31, 42, 43]. Specifically, in glioblastoma cancer, upregulation of Cath B has been linked to radiotherapy resistance [42, 43]. This was in agreement with our results when the expression of all bands shown in Figure 2 significantly increased upon exposure to any of the tested irradiation doses, indicating upregulation of many cysteine peptidases in response to ionizing irradiation. This finding suggests that ionizing irradiation may play a role in altering cellular processes related to these peptidases, potentially impacting cellular functions. Interestingly, our own investigation discovered a dose-independent increase in Cath B expression in caco-2 cells after ionizing radiation exposure, with some doses leading to a tenfold rise in expression (Figure 3). These data are supporting the previous results that many of cysteine peptidases are upregulated as a consequence of exposure to ionizing irradiation. These findings suggest a complex regulatory response of cathepsin B to ionizing irradiation.
To examine the specific activities and abundance of Cystine cathepsin with Active site labelling we found that, the expression of all bands significantly increased upon exposure to any of the tested irradiation doses, indicating upregulation of many cysteine peptidases in response to ionizing irradiation. This finding suggests that ionizing irradiation may play a role in altering cellular processes related to these peptidases, potentially impacting cellular functions. Further research is warranted to explore the specific mechanisms through which ionizing irradiation influence the regulation of cysteine peptidases and its implications on cellular behaviour.
Considering that lysosomal cathepsins typically range in molecular weights between 20 and 45 KDa, the highest 70 KDa band was disregarded. The other three bands co-migrated with different forms of Cath B, namely pro-Cath B, intermediate form, and mature form. This co-migration pattern strongly indicates that Cath B, a key member of the papain family cysteine cathepsins, may be closely involved in the cellular response to ionizing irradiation [17]
In order to explore the potential increase in cathepsin B levels in reaction to ionizing irradiation, lysates from caco-2 cells were collected subsequent to exposure to varying doses of ionizing radiation. It was observed that cathepsin B demonstrated substantial upregulation across all doses tested. These data are supporting the previous results that many of cysteine peptidases are upregulated as a consequence of exposure to ionizing irradiation [43], and suggest a complex regulatory response of cathepsin B to ionizing irradiation. Further investigations are needed to understand the underlying mechanisms responsible for this non-linear upregulation pattern.
To verify whether the increase in activity is attributed to higher expression levels of Cath B, Significantly, the increase in Cath B expression was detected in both its pro-Cath B and processed/mature variants. Yet, in contradiction to the overall Cath B levels, the mature cathepsin B exhibited lower abundance at the 10 Gy irradiation dose compared to the smaller doses. Our findings suggest a correlation between increased Cath B activity and its expression, indicating that exposing caco-2 cells to irradiation leads to elevated Cath B expression indicating that the upregulation of Cath B was not strictly dose dependent and these results in agree with previous study [44].
To validate the pivotal role of Cath B in irradiation-induced cytotoxicity and its contribution to the resistance of caco-2 cells against ionizing radiation, we utilized CRISPR/CAS9 to completely abolish Cath B expression or employed the Cath B-specific inhibitor CA07, the sensitivity of the caco-2 cells to ionizing radiation significantly increased. Notably, the impact was more pronounced in caco-2 cells with Cath B knocked down compared to cells with Cath B inhibited. This could be attributed to CRISPR/CAS9 fully eliminating Cath B expression, whereas CA074 only partially inhibits its activity within the cells.
To assess the potential influence of Cath B inhibition on the expression of apoptotic proteins, caco-2 cells were subjected to escalating doses of ionizing radiation under the presence and absence of the Cath B inhibitor CA074. These findings provide evidence of a potential role of Cath B in caco-2 cells' resistance to ionizing radiation treatment and suggest a possible mechanism involving the suppression of certain apoptotic events.
Moreover, in the Cath B inhibited caco-2 cells, the pro-apoptotic protein BAX showed significant increase, while the expression of the anti-apoptotic protein BCL-2 decreased as mentioned in our results. This suggests that apoptosis is more likely to occur when cells are exposed to weak or moderate radiation doses, while necrosis is triggered at higher radiation doses [45].
It is important to note that Cath B is not the sole factor contributing to cell resistance to radiotherapy. Other investigations have highlighted the role of Cath S, for instance, as an important player in promoting resistance to radiotherapy in certain tumors [31].
In conclusion, these data are supporting the previous results that many of cysteine peptidases are upregulated as a consequence of exposure to ionizing irradiation. These findings suggest a complex regulatory response of cathepsin B to ionizing irradiation. Further investigations are needed to understand the underlying mechanisms responsible for this non-linear upregulation pattern. Targeting lysosomal cathepsins shows promise as a potential strategy for enhancing the efficacy of radiotherapy and overcoming radiotherapy resistance in cancer treatment. However, further research is warranted to explore the specific mechanisms through which ionizing irradiation influence the regulation of cysteine peptidases and its implications on cellular behaviour. Further investigations are needed to understand the underlying mechanisms responsible for this non-linear upregulation pattern.
4. Materials and Methods
4.1. Caco-2 Cell Culture
Caco-2 cells derived from human colon carcinoma were used in this study (Given by Dr. Rosa Lemmens-Gruber, Vienna university). These cells were cultured in Dulbecco’s Eagle’s medium (DMEM, Gibco, ThermoFisher Scientific, Waltham, MA, USA), containing 4 mM glutamine. Additionally, 100 units/mL of penicillin and 100 μg/mL of streptomycin (ThermoFisher Scientific, Waltham, MA, USA) were supplemented as antibiotics, and the cells were incubated at 37 °C in an environment with 5% CO2.
4.2. Radiation Treatment
Two groups of Caco-2 cells, treated and untreated with cathepsin B specific inhibitor CA074 (Merck, Rockville, MD, USA) were irradiated using a 6 MV photon beam of a medical linear accelerator (LINAC) (Varian Medical Systems, Palo Alto, USA) with different doses (2, 4, 6, 8 and 10 Gy, with constant dose rate of 3 Gy/min). For assessment of delivered dose the Eclipse (Varian) treatment planning system (TPS) was used. Another two groups of Caco-2 cells, treated and untreated with cathepsin B inhibitor were not exposed to radiation and served as negative control. Caco-2 cell lines (~107) were treated for 24h at 37 °C in a complete medium containing 10 μM CA074 before radiation. Dimethylsulphoxide solvent (final concentration 0.1%) was added to the untreated groups.
4.3. MTT Assay, Cell Viability Cytotoxicity Test
Cells were placed into 96-well plates, with 2000 cells per well along with 100 μL of medium, and then incubated for a duration of 24 h. Subsequently, the cells were treated with inhibitors (CA074) individually, using concentrations of 10 μM. The viability of the cells after 24 h of treatment was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2 and -5 diphenyltetrazolium bromide (MTT)-based viability assay called EZ4U (Biomedica, Vienna, Austria). For this assay, 20 μL of EZ4U solution was introduced into each well, followed by a 2-hour incubation at 37 °C. The absorbance was then measured at 450 nm using a microplate reader (Infinite F200, Tecan, Männedorf, Switzerland), with 620 nm serving as a reference for unspecific background values. This entire experimental procedure was replicated three times, with each repetition consisting of triplicate samples [46-48].
4.4. Western Blotting
The experiments were carried out following the procedures outlined in prior references [49-53]. Initially, cells were cultured after radiation in 100 mM cell culture dishes within a 5% CO2 incubator, using DMEM medium supplemented with 5% fetal bovine serum (ThermoFisher Scientific, Waltham, MA, USA). Afterward, the medium was aspirated, and the cells were washed twice with PBS. Following this, cells were gently detached using lysis buffer (comprising 200 mM sodium acetate, 150 mM NaCL, pH 5.5, and supplemented with 40 μM E-64), and subsequently transferred to 1.5 μL centrifuge tubes.
For homogenization, the cells were treated with ultrasonication (10sec then 30sec on ice/ 3 times) while maintained on ice, followed by the addition of 0.1% Triton X-100. The homogenized cell mixture was then incubated on ice for 30 min. Subsequently, the samples were cleared by centrifugation at 15,000× g for 10 min. The separated proteins were subjected to 12.5% SDS-PAGE under reducing conditions. These proteins were subsequently transferred onto nitrocellulose membranes (obtained from Santa Cruz Biotechnology, Dallas, TX, USA) using semi-dry blotting at 25 V for 30 min.
To prevent non-specific binding, the membrane was treated with a blocking solution (3% BSA in PBS) for duration of 3 h. Following this, the membrane was exposed to primary antibodies, namely cathepsin B (ThermoFisher Scientific, Waltham, MA, USA) (1:2000), BAX and Bcl-2 (Cell Signaling, Graz, Austria) (1:1000), and β-tubulin (Sigma Aldrich, St. Louis, MO, USA) (1:3000), for a period of 90 min. Subsequently, the membrane was washed five times with PBST and then incubated for an additional 90 min with the corresponding secondary antibodies. This was followed by three washes with PBST and one wash with PBS.
For visualization, enhanced chemiluminescence (Amersham ECL plus Western blotting detection reagent, GE Healthcare, Vienna, Austria) was employed. The membranes were exposed to X-ray films (Amersham Hyper film ECL, GE Healthcare, Vienna, Austria), and the resulting experimental films were scanned and quantified using ImageJ software (NIH, Bethesda, MD, USA).
4.5. Active Site Labelling of Cysteine Cathepsins
In order to label cysteine cathepsins within cultured cells, the utilization of the activity-based probe DCG04 (Medkoo, Morrisville, NC, USA) has proven effective. This probe is essentially a biotinylated variant of the general cysteine peptidase inhibitor E-64, selectively designed for targeting cysteine peptidases. Notably, DCG04 has the unique capacity to selectively bind to active cysteine peptidases present within complex protein mixtures [54].
To label the cells after radiation treatment, Caco-2 cells were incubated for 72 h at 37 °C with 10 μM DCG04. Following this incubation, cellular protein extract was meticulously prepared. Subsequently, 30 μg of this extract underwent separation through 12.5% SDS polyacrylamide gels. The separated proteins were then transferred onto a nitrocellulose membrane sourced from Santa Cruz Biotechnology (Dallas, TX, USA). To ensure specificity and minimize non-specific binding, the transferred proteins were subjected to a blocking step using a solution of 3% bovine serum albumin (BSA) from ThermoFisher Scientific (Waltham, MA, USA) in PBS. The membrane was then exposed to streptavidin-horseradish peroxidase (0.125 μg/mL in PBST) from BioLegend (San Diego, CA, USA). This crucial step preceded the application of enhanced chemiluminescence detection methods, allowing for the visualization of the labeled cysteine cathepsins.
4.6. Pull-Down of DCG04 Labelled Cysteine Cathepsins
For the purpose of isolating in vitro labelled cysteine cathepsins, the following procedure was executed: Initially, 250 μL (equivalent to approximately 400 μg) of cellular protein extracts were quantified utilizing the Bradford method as outlined by Bradford in 1976 [55, 56].
Subsequently, the cellular extracts previously labelled with DCG04 were appropriately diluted by combining them with 750 μL of a binding buffer comprising 20 mM sodium acetate (pH 5.5), 150 mM sodium chloride, 0.1% triton X-100, 10 μg/mL E-64, 10 μg/mL leupeptin sourced from Sigma Aldrich (St. Louis, MO, USA), and 1 mM PMSF from Abcam. This mixture was subjected to centrifugation at 14,000× g for duration of five min, with the resulting supernatant incubated overnight along with 40 μL of settled streptavidin beads at 4 °C.
To facilitate further processing, the beads were collected via centrifugation lasting 5 min at 3000 rpm. Subsequently, a sequence of washes was carried out: five washes employing 20 mM sodium acetate (pH 5.5), 150 mM sodium chloride, and 0.1% triton X-100, followed by two additional washes using 10 mM Tris-HCL at pH 6.8 [34].
The settled beads were then mixed with 40 μL of a 2X sample buffer and heated for a period of five min at 95 °C [35, 36]. The ensuing supernatant underwent separation through SDS-PAGE, followed by blotting onto a nitrocellulose membrane. This membrane was subsequently subjected to immunoblotting utilizing antibodies (1:2000 dilution) obtained from ThermoFisher Scientific (Waltham, MA, USA) and specifically directed against cathepsin B.
4.7. Cathepsin B Knockout Experiment
Twenty-four h post-seeding and cell confluency reached 70–90 %, Caco-2 cells underwent transfection with either the CRISPR/Cas9 plasmid targeting cathepsin B (Santa Cruz sc-400360) or the corresponding control plasmid (Santa Cruz sc-418922). The transfection procedure was done using the X-tremeGENE ™ HP DNA transfection reagent sourced from Roche Diagnostics (catalog Nr. 6366244001, Mannheim, Germany) and strictly adhering to the guidelines stipulated by the manufacturer as follow: X-tremeGENE™ HP DNA transfection reagent, plasmid DNA and diluent were left to warm with gentile vortex. The diluent and plasmid DNA were added and gently mixed in a sterile tube and then X-tremeGENE™ HP DNA transfection reagent was added later to the diluted DNA. The final Mixture was incubated for 15 min at 25 °C and then was added to Caco-2 cells in a dropwise manner with Gentile shaking for proper distribution. The cells were incubated with the mixture cells for 72 h before the exposure to different doses of ionizing radiation [53, 57].
4.8. Statistical Analysis
Statistical analysis was conducted employing a non-parametric t-test to compare two groups. For scenarios involving more than two groups, either one-way ANOVA or two-way ANOVA was employed, based on the number of independent variables. While a significant result from an analysis of variance (ANOVA) F-test provides overall evidence of group differences, it doesn't specify which pairs of means exhibit divergence. To discern specific differences among three or more group means, post hoc tests were employed. Following the ANOVA, Dunnett’s multiple comparison test was utilized to pinpoint pairs with significant differences. This approach compares means from multiple experimental groups against a single control group mean to identify any disparities.
Furthermore, to mitigate the risk of false positives, the Bonferroni test was implemented. This adjustment, pioneered by Bonferroni, serves to maintain the integrity of statistical significance assessments. All statistical analyses were executed using GraphPad Prism by GraphPad Software, San Diego, CA, USA, in conjunction with Microsoft Excel 365. Significance was established at a probability level of p < 0.05. Data were presented as mean ± standard error (SE). For detailed statistical parameters relevant to specific experiments, please refer to the appropriate sections or figure legends.
Acknowledgments
We thank Tamer Z. Salem (Zewail City of Science and Technology, Egypt) for assistance with proof reading and for comments that greatly improved the manuscript.
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2019. Geneva, World Health Organization; 2020. Accessed January 06, 2023.
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol. 2014, 20, 923–942. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J Clin 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Schoen, R.E.; Pinsky, P.F.; Weissfeld, J.L.; Yokochi, L.A.; Church, T.; Laiyemo, A.O.; Bresalier, R.; Andriole, G.L.; Buys, S.S.; Crawford, E.D.; et al. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. N. Engl. J. Med. 2012, 366, 2345–2357. [Google Scholar] [CrossRef]
- Zarour, L.R.; Anand, S.; Billingsley, K.G.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, X.; Lu, Z. Local histone acetylation by ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Autophagy 2017, 13, 1790–1791. [Google Scholar] [CrossRef] [PubMed]
- Bright, N.A.; Davis, L.J.; Luzio, J.P. Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr. Biol. 2016, 26, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chou, P.M.; Mirkin, B.L.; Rebbaa, A. Senescence-initiated reversal of drug resistance: Specific role of cathepsin L. Cancer Res. 2004, 64, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Shi, C.; Yan, Z.; Wu, M. Overexpression of Cathepsin L is associated with chemoresistance and invasion of epithelial ovarian cancer. Oncotarget 2016, 7, 45995–46001. [Google Scholar] [CrossRef] [PubMed]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Bestvater, F.; Dallner, C.; Spiess, E. The C-terminal subunit of artificially truncated human cathepsin B mediates its nuclear targeting and contributes to cell viability. BMC Cell Biol. 2005, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Mongrain, S.; Cagnol, S.; Langlois, M.J.; Boulanger, J.; Bernatchez, G.; Carrier, J.C.; Boudreau, F.; Rivard, N. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol. Carcinog. 2016, 55, 671–687. [Google Scholar] [CrossRef]
- Kryczka, J.; Papiewska-Pajak, I.; Kowalska, M.A.; Boncela, J. Cathepsin B Is Upregulated and Mediates ECM Degradation in Colon Adenocarcinoma HT29 Cells Overexpressing Snail. Cells 2019, 8. [Google Scholar] [CrossRef]
- Sigloch, F.C.; Knopf, J.D.; Weißer, J.; Gomez-Auli, A.; Biniossek, M.L.; Petrera, A.; Schilling, O. Proteomic analysis of silenced cathepsin B expression suggests non-proteolytic cathepsin B functionality. Biochim. Biophys. Acta 2016, 1863, 2700–2709. [Google Scholar] [CrossRef]
- Formolo, C.A.; Williams, R.; Gordish-Dressman, H.; MacDonald, T.J.; Lee, N.H.; Hathout, Y. Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 2011, 10, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Hao, S.; Young, P.; Zhang, H. Targeting Cathepsin B for Cancer Therapies. Horiz. Cancer Res. 2015, 56, 23–40. [Google Scholar] [PubMed]
- Mijanović, O.; Branković, A.; Panin, A.N.; Savchuk, S.; Timashev, P.; Ulasov, I.; Lesniak, M.S. Cathepsin B: A sellsword of cancer progression. Cancer Lett. 2019, 449, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Tam, A.; Santi, S.A.; Parissenti, A.M. Role of autophagy and lysosomal drug sequestration in acquired resistance to doxorubicin in MCF-7 cells. BMC Cancer 2016, 16, 762. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, M.R.; Barar, J.; Pourseif, M.M.; Eskandani, M.; Jafari Niya, M.; Mashayekhi, M.R.; Omidi, Y. Molecular machineries of pH dysregulation in tumor microenvironment: Potential targets for cancer therapy. Bioimpacts 2017, 7, 115–133. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, C.; Chen, H.; Ren, M.; Liu, X. Cathepsins Trigger Cell Death and Regulate Radioresistance in Glioblastoma. Cells 2022, 11, 4108. [Google Scholar] [CrossRef]
- Seo, H.R.; Bae, S.; Lee, Y.S. Radiation-induced cathepsin S is involved in radioresistance. International Journal of Cancer 2009, 124, 1794–1801. [Google Scholar] [CrossRef]
- Radogna, F.; Cerella, C.; Gaigneaux, A.; Christov, C.; Dicato, M.; Diederich, M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2016, 35, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Alapati, K.; Gopinath, S.; Malla, R.R.; Dasari, V.R.; Rao, J.S. uPAR and cathepsin B knockdown inhibits radiation-induced PKC integrated integrin signaling to the cytoskeleton of glioma-initiating cells. Int. J. Oncol. 2012, 41, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-q.; Wang, W.-j.; Li, J.; Yang, N.; Chen, G.; Wang, Z.; Liang, Z.-q. Cathepsin L suppression increases the radiosensitivity of human glioma U251 cells via G2/M cell cycle arrest and DNA damage. Acta Pharmacologica Sinica 2015, 36, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Labianca, R.; Beretta, G.D.; Kildani, B.; Milesi, L.; Merlin, F.; Mosconi, S.; Pessi, M.A.; Prochilo, T.; Quadri, A.; Gatta, G.; et al. Colon cancer. Crit. Rev. Oncol. Hematol. 2010, 74, 106–133. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Platell, C.; McCaul, K.; Millward, M.; van Hazel, G.; Bayliss, E.; Trotter, J.; Ransom, D.; Iacopetta, B. Survival rates for stage II colon cancer patients treated with or without chemotherapy in a population-based setting. Int. J. Colorectal Dis. 2007, 22, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Czito, B.G.; Bendell, J.; Willett, C.G. Radiation therapy for resectable colon cancer. Is there a role in the modern chemotherapy era? Oncology 2006, 20, 179–187, discussion 187-8, 192. [Google Scholar]
- Quesnel, A.; Karagiannis, G.S.; Filippou, P.S. Extracellular proteolysis in glioblastoma progression and therapeutics. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188428. [Google Scholar] [CrossRef]
- Oberle, C.; Huai, J.; Reinheckel, T.; Tacke, M.; Rassner, M.; Ekert, P.G.; Buellesbach, J.; Borner, C. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010, 17, 1167–1178. [Google Scholar] [CrossRef]
- Repnik, U.; Hafner Česen, M.; Turk, B. Lysosomal membrane permeabilization in cell death: Concepts and challenges. Mitochondrion 2014, 19 Pt A, 49–57. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, Y.; Zhang, X.; Jiang, Z. Lys05 induces lysosomal membrane permeabilization and increases radiosensitivity in glioblastoma. J. Cell Biochem. 2020, 121, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Xu, S.; Li, X.; Ma, X. Cathepsin B contributes to radioresistance by enhancing homologous recombination in glioblastoma. Biomed. Pharmacother. 2018, 107, 390–396. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, C.; Chen, H.; Ren, M.; Liu, X. Cathepsins Trigger Cell Death and Regulate Radioresistance in Glioblastoma. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Mönch, D.; Maaß, A.; Gromoll, C.; Hehr, T.; Leibold, T.; Schlitt, H.J.; Dahlke, M.H.; Renner, P. Three dimensional cultivation increases chemo- and radioresistance of colorectal cancer cell lines. PLoS ONE 2021, 16, e0244513. [Google Scholar] [CrossRef] [PubMed]
- Przybyszewski, W.M.; Wideł, M.; Szurko, A.; Maniakowski, Z. [Dose rate-dependent cellular and molecular effects of ionizing radiation]. Postepy Hig Med Dosw 2008, 62, 468–477. [Google Scholar]
- Ghasemi, M.; Liang, S.; Luu, Q.M.; Kempson, I. The MTT Assay: A Method for Error Minimization and Interpretation in Measuring Cytotoxicity and Estimating Cell Viability. Methods Mol. Biol. 2023, 2644, 15–33. [Google Scholar] [PubMed]
- Labuda, R.; Bacher, M.; Gratzl, H.; Doppler, M.; Parich, A.; Aufy, M.; Lemmens-Gruber, R.; Schuhmacher, R.; Rychli, K.; Wagner, M.; et al. Luteapyrone, a Novel ƴ-Pyrone Isolated from the Filamentous Fungus Metapochonia lutea. Molecules 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Moreira, V.P.; da Silva Mela, M.F.; Anjos, L.R.; Saraiva, L.F.; Arenas Velásquez, A.M.; Kalaba, P.; Fabisiková, A.; Clementino, L.; Aufy, M.; Studenik, C.; et al. Novel Selective and Low-Toxic Inhibitor of LmCPB2.8ΔCTE (CPB) One Important Cysteine Protease for Leishmania Virulence. Biomolecules 2022, 12, 1903. [Google Scholar]
- Søderstrøm, S.; Lie, K.K.; Lundebye, A.K.; Søfteland, L. Beauvericin (BEA) and enniatin B (ENNB)-induced impairment of mitochondria and lysosomes - Potential sources of intracellular reactive iron triggering ferroptosis in Atlantic salmon primary hepatocytes. Food Chem. Toxicol. 2022, 161, 112819. [Google Scholar] [CrossRef]
- Ahmad, F.; Leake, D.S. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro-inflammatory cytokines in human macrophages [S]. J. Lipid Res. 2019, 60, 98–110. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Aufy, M.; Shabbir, W.; Lemmens-Gruber, R. Identification of phosphorylation sites and binding pockets for modulation of Na(V) 1.5 channel by Fyn tyrosine kinase. Febs j 2018, 285, 2520–2530. [Google Scholar] [CrossRef]
- Willam, A.; Aufy, M.; Tzotzos, S.; Evanzin, H.; Chytracek, S.; Geppert, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Czikora, I.; et al. Restoration of Epithelial Sodium Channel Function by Synthetic Peptides in Pseudohypoaldosteronism Type 1B Mutants. Front. Pharmacol. 2017, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, W.; Topcagic, N.; Aufy, M.; Oz, M. CRISPR/Cas9 Mediated Knock Down of δ-ENaC Blunted the TNF-Induced Activation of ENaC in A549 Cells. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Medzihradszky, K.F.; Burlingame, A.; Bogyo, M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 2000, 7, 569–581. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Aufy, M.; Abdelaziz, R.F.; Hussein, A.M.; Topcagic, N.; Shamroukh, H.; Abdel-Maksoud, M.A.; Salem, T.Z.; Studenik, C.R. Impact of Enniatin B and Beauvericin on Lysosomal Cathepsin B Secretion and Apoptosis Induction. Int. J. Mol. Sci. 2023, 24, 2030. [Google Scholar] [CrossRef] [PubMed]
- Gnanamony, M.; Gondi, C.S. Targeting the Expression of Cathepsin B Using CRISPR/Cas9 System in Mammalian Cancer Cells. Methods Mol. Biol. 2018, 1731, 123–131. [Google Scholar]
Figure 1.
Radiation treatment influence on the active site labelling of cysteine cathepsins by DCG04. (A) Active-site labelling with DCG04. Protein samples were subjected to protein electrophoresis and Western blotting with streptavidin–horseradish peroxidase. (B) Cellular content of 25 KDa and 30 KDa, 40 KDa and 70 KDa bands in caco-2 irradiated and DCG-04 labelled cells were compared to DCG-04 only labelled cells (Control). The data were analysed using two-way ANOVA with Dunnett’s post hoc analysis (* p < 0.05; *** p < 0.001; N = 5). Statistics were calculated using GraphPad Prism.
Figure 1.
Radiation treatment influence on the active site labelling of cysteine cathepsins by DCG04. (A) Active-site labelling with DCG04. Protein samples were subjected to protein electrophoresis and Western blotting with streptavidin–horseradish peroxidase. (B) Cellular content of 25 KDa and 30 KDa, 40 KDa and 70 KDa bands in caco-2 irradiated and DCG-04 labelled cells were compared to DCG-04 only labelled cells (Control). The data were analysed using two-way ANOVA with Dunnett’s post hoc analysis (* p < 0.05; *** p < 0.001; N = 5). Statistics were calculated using GraphPad Prism.
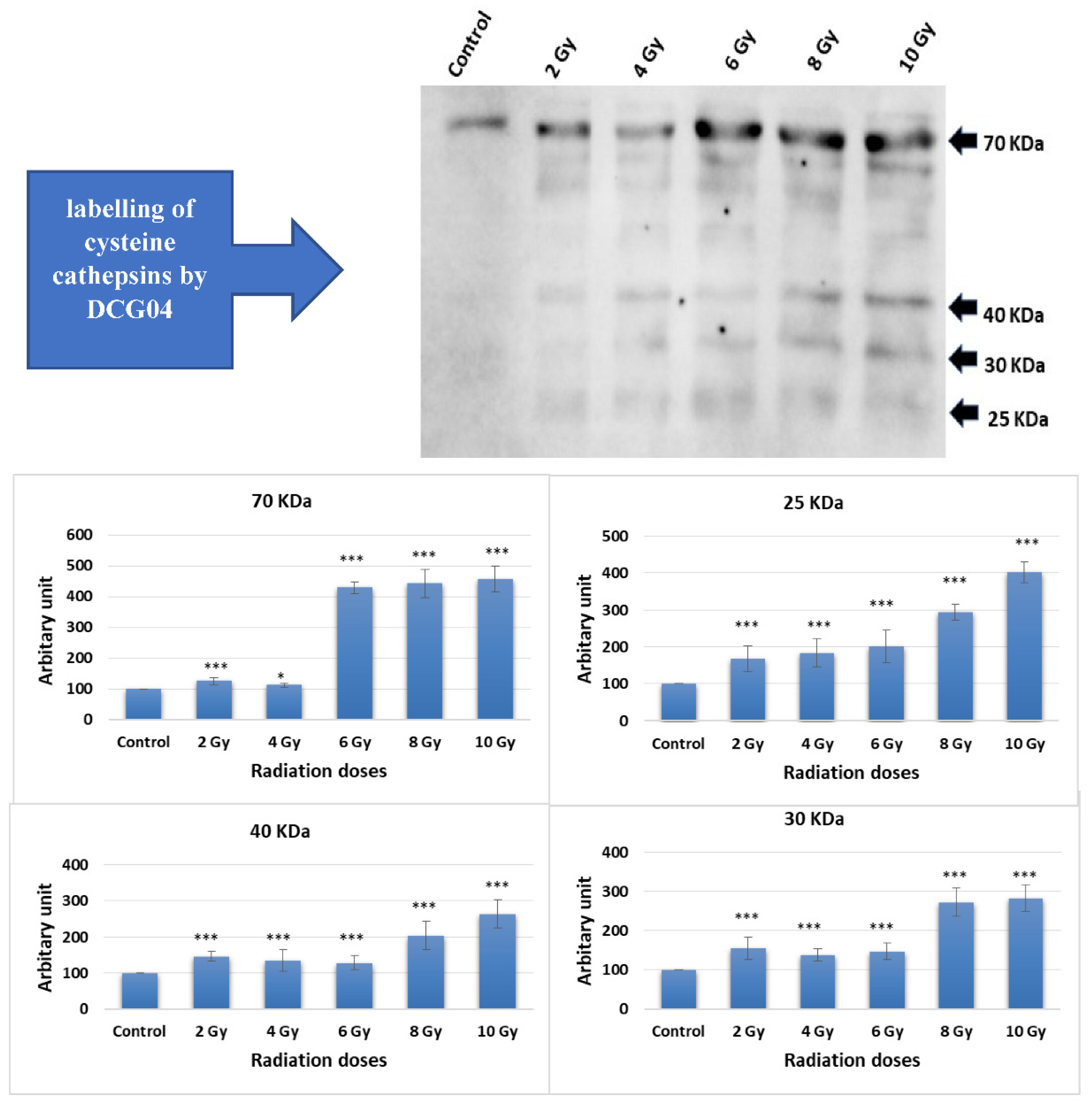
Figure 2.
Avidin pull-down experiment of cysteine cathepsins with and without radiation treatment. (A) Avidin pull-down experiment was performed as described in materials and methods. Conjugated proteins to avidin Sepharose beads were subjected to SDS-PAGE and Western blotting with antibodies specific to human cathepsin B. (B) Ratio of cellular contents of cathepsin B with and without radiation treatment. The data were analyzed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 5.
Figure 2.
Avidin pull-down experiment of cysteine cathepsins with and without radiation treatment. (A) Avidin pull-down experiment was performed as described in materials and methods. Conjugated proteins to avidin Sepharose beads were subjected to SDS-PAGE and Western blotting with antibodies specific to human cathepsin B. (B) Ratio of cellular contents of cathepsin B with and without radiation treatment. The data were analyzed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 5.
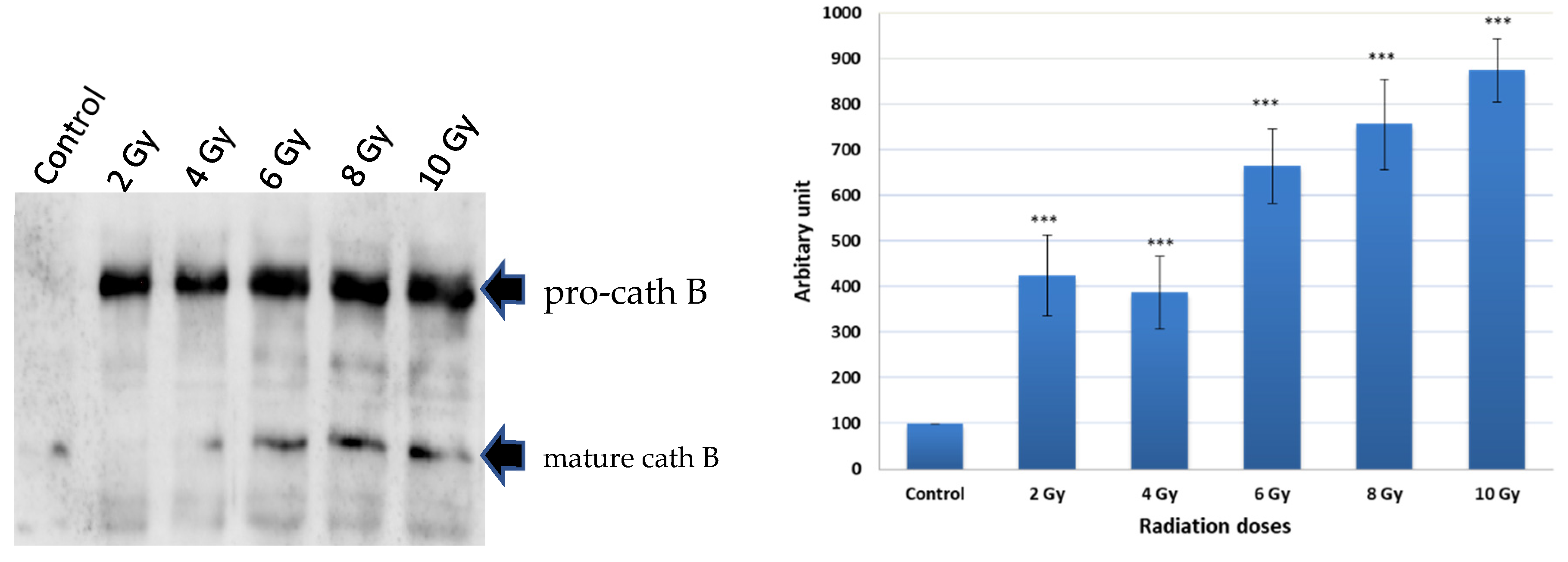
Figure 3.
Cath B expression evaluation with and without different doses of radiation. Western blot analysis of Cath B expression. Ratio of cellular contents of cathepsin B with and without radiation treatment. The data were analyzed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 5.1.
Figure 3.
Cath B expression evaluation with and without different doses of radiation. Western blot analysis of Cath B expression. Ratio of cellular contents of cathepsin B with and without radiation treatment. The data were analyzed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 5.1.
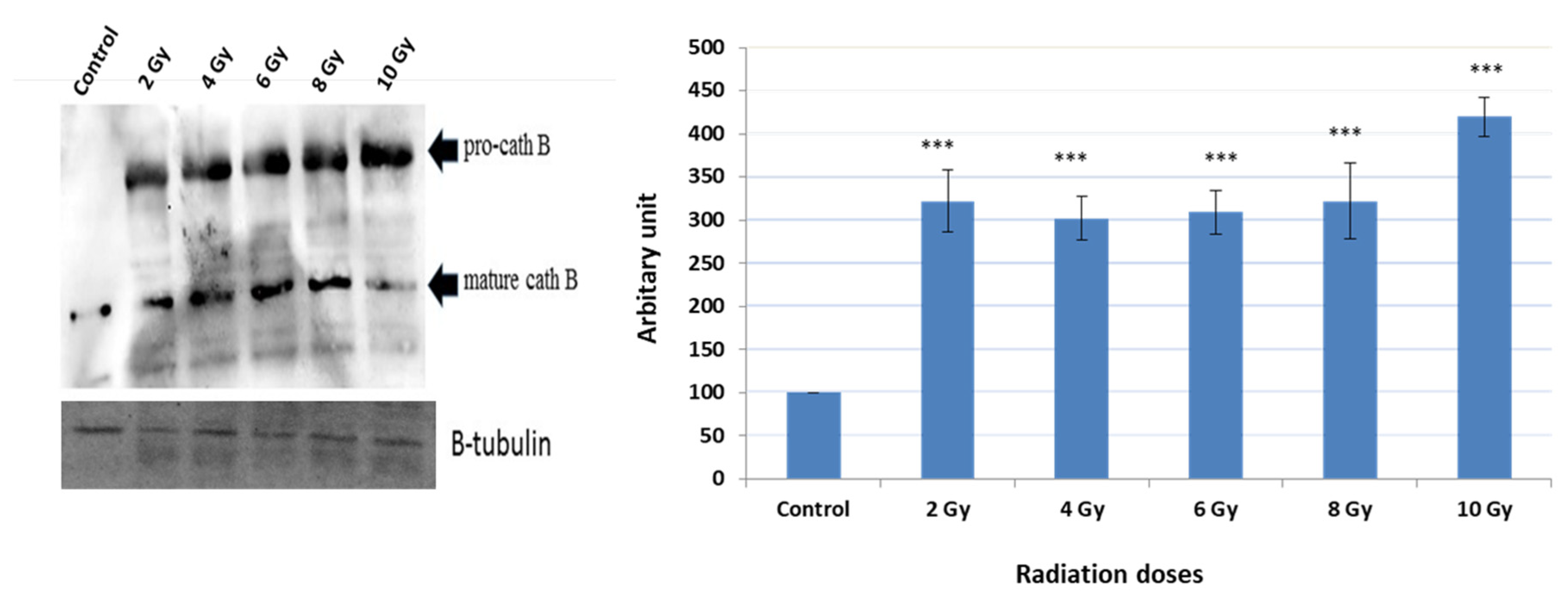
Figure 4.
Cytotoxicity analysis for treated and no treated cells with radiation and cath B inhibitor. The data were analyzed by t-test and statistics using GraphPad Prism. ;** p < 0.01; *** p < 0.001. Graphs are shown as mean ± SE, N = 8.
Figure 4.
Cytotoxicity analysis for treated and no treated cells with radiation and cath B inhibitor. The data were analyzed by t-test and statistics using GraphPad Prism. ;** p < 0.01; *** p < 0.001. Graphs are shown as mean ± SE, N = 8.
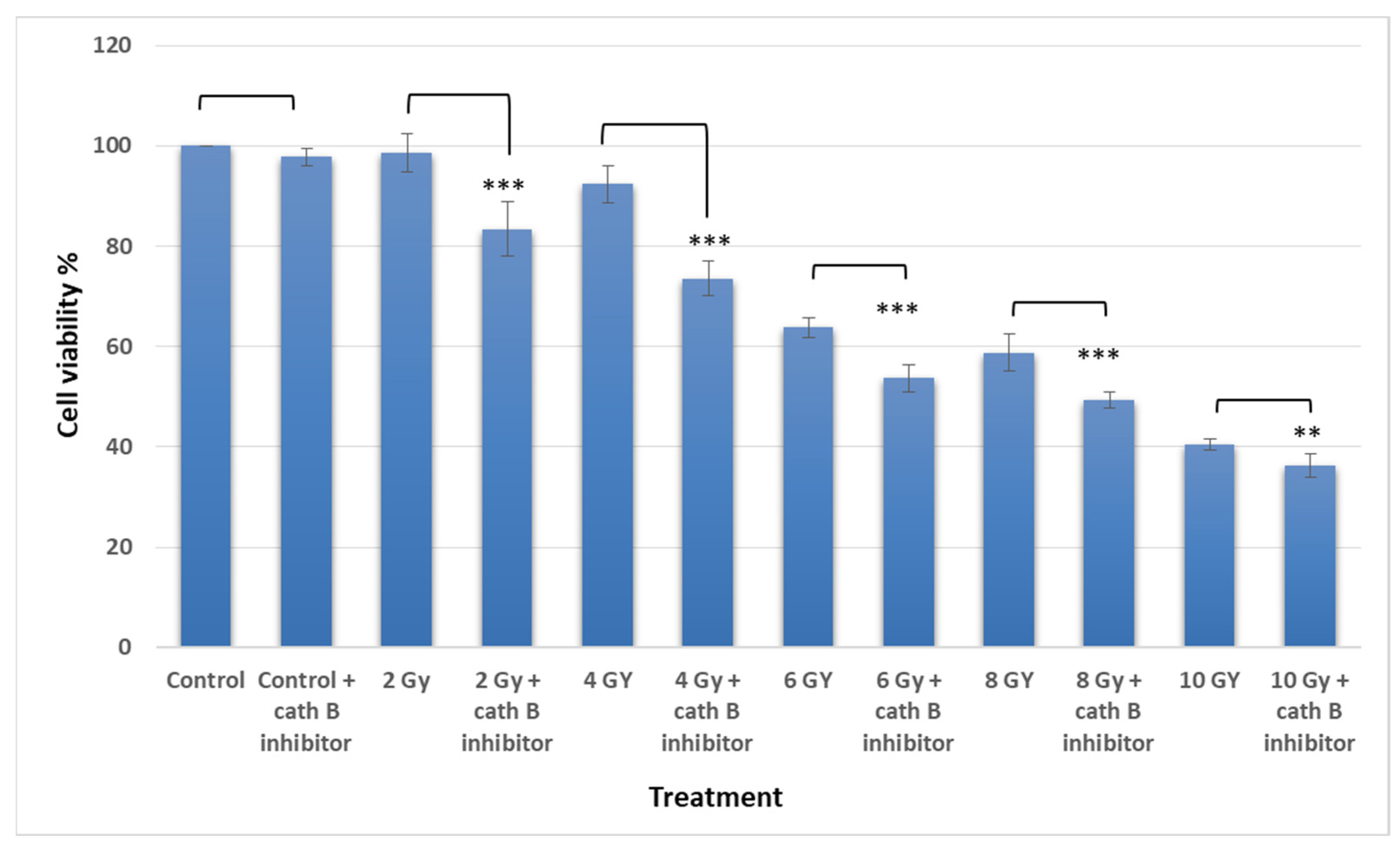
Figure 5.
Cathepsin B knockout impact on non irradiated and radiated Caco-2 cells.
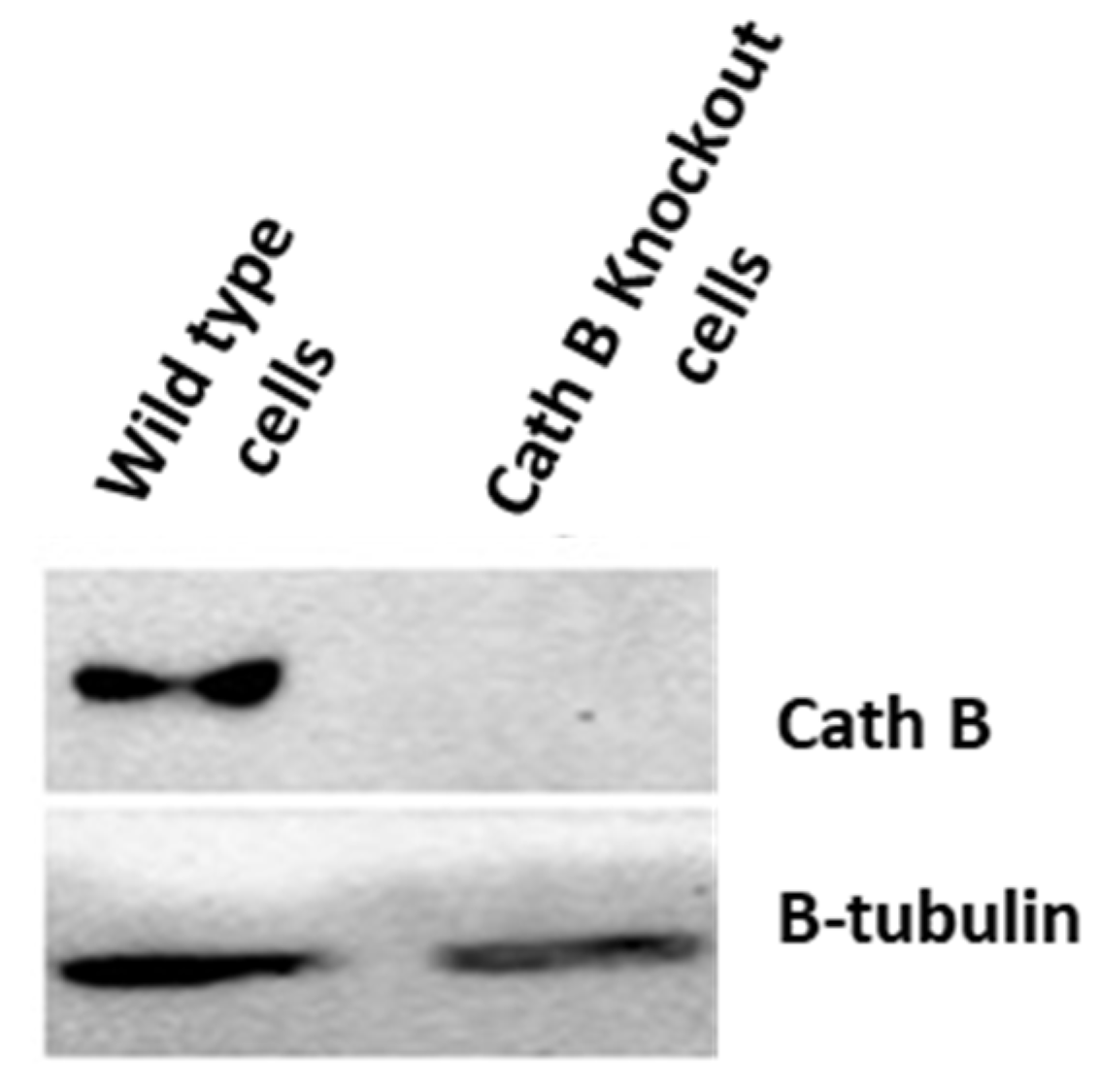
Figure 6.
Cathepsin B knockout impact on non irradiated and radiated cells toxicity. The data were analysed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 8.
Figure 6.
Cathepsin B knockout impact on non irradiated and radiated cells toxicity. The data were analysed by t-test and statistics using GraphPad Prism. *** p < 0.001. Graphs are shown as mean ± SE, N = 8.
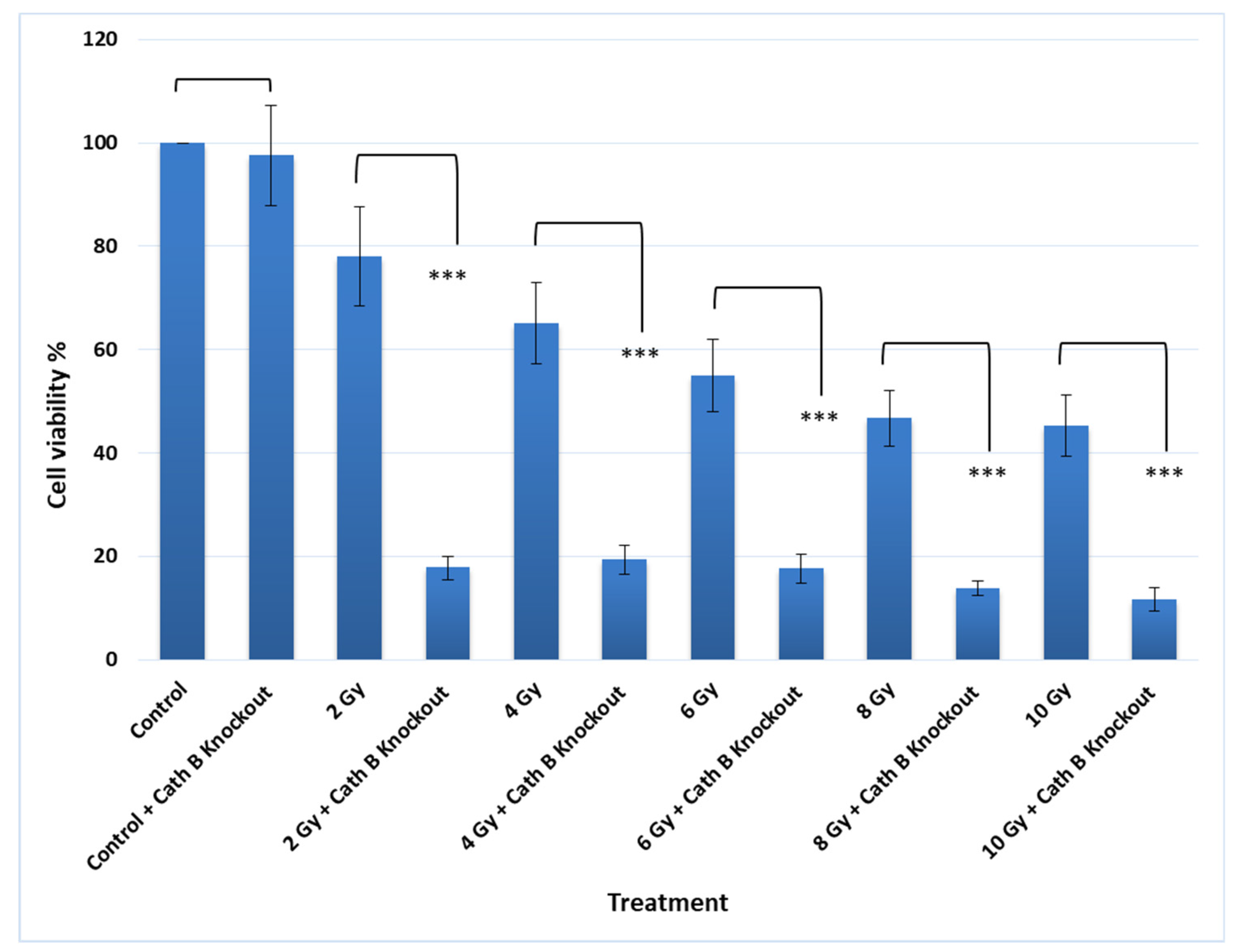
Figure 7.
Cath B inhibition impact on apoptosis rate in treated and non-treated cells with ionizing. The data were analysed using two-way ANOVA with Dunnett’s post hoc analysis (* p < 0.05;** p < 0.01; *** p < 0.001; N = 3). Statistics were calculated using GraphPad Prism.
Figure 7.
Cath B inhibition impact on apoptosis rate in treated and non-treated cells with ionizing. The data were analysed using two-way ANOVA with Dunnett’s post hoc analysis (* p < 0.05;** p < 0.01; *** p < 0.001; N = 3). Statistics were calculated using GraphPad Prism.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



