
Submitted:
10 October 2023
Posted:
11 October 2023
You are already at the latest version
Abstract
Non-alcoholic fatty liver disease (NAFLD) is defined as the accumulation of lipids in the form of lipid droplets in more than 5% of hepatocytes. It is regarded as a range of diverse pathologies, including simple steatosis and steatohepatitis. The structural characteristics of lipid droplets have been implicated in the etiology of the disease, along with their protein composition, mainly perilipins. These proteins have garnered increasing attention as a pivotal regulator, since their levels and distinct expression appear to be associated with the progression from simple steatosis to steatohepatitis. Perilipins are target proteins of chaperone-mediated autophagy, and their degradation is a prerequisite for lipolysis and lipophagy to access the lipid core. Both lipophagy and chaperone-mediated autophagy have significant implications in the development of the disease, as evidenced by their upregulation during the initial phases of simple steatosis, and their subsequent downregulation once steatosis is established. On the contrary, during steatohepatitis, the process of chaperone-mediated autophagy is enhanced, although lipophagy remains suppressed. Evidently, the reduced levels of autophagic pathways observed in simple steatosis serve as a defensive mechanism against lipotoxicity. Conversely, in steatohepatitis chaperone mediated autophagy fails to compensate for the continuous generation of small lipid droplets and thus cannot protect hepatocytes from lipotoxicity.
Keywords:
Non-alcoholic fatty liver disease
; lipophagy
; chaperone-mediated autophagy
; perilipins
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disorder worldwide, with a global prevalence of 25%, [1]. NAFLD encompasses a spectrum of distinct pathologies characterized by the accumulation of predominantly triglycerides (TGs) within lipid droplets (LDs) in more than 5% of hepatocytes, with concurrent exclusion of alternate etiologies of hepatic lipid infiltration, including excessive alcohol consumption [2,3,4]. Non-alcoholic fatty liver (NAFL) refers to simple steatosis, as the first, benign and reversible stage, while 10-20% of cases are progressing to a more severe and inflammatory, condition known as non-alcoholic steatohepatitis (NASH) [1]. Fibrosis may be present in around 30% of NASH cases and can evolve to an irreversible cirrhotic stage and finally to hepatocellular carcinoma (HCC)[5].
NAFLD strongly correlates with the metabolic syndrome, as the pathogenesis of NAFLD is associated with various manifestations of the syndrome, such as insulin resistance, obesity and dyslipidemia [6]. In accordance with this observation, in the early 2020, the term “Metabolic-dysfunction associated liver disease-MAFLD” was suggested to replace the term “NAFLD” [7]. Nevertheless, for the purpose of this manuscript, we shall adhere to the utilization of the term “NAFLD”.
NAFLD not only has an increasing incidence worldwide, but also constitutes an important economic burden in the health system, as it is estimated to account for enormous medical costs in both European countries and United States [1]. Up to date, there are no approved pharmacological treatments for NAFLD, thus a better understanding of the pathogenetic mechanisms is more than urgent[8].
Various studies have elucidated the role of autophagy in the development of NAFLD[8,9,10]. Autophagy is a conserved catabolic process, which includes the degradation of dysfunctional proteins and organelles in the lysosomal lumen [11]. Autophagy consists of three main pathways: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA)[11]. Macroautophagy facilitates the engulfment of certain autophagic cargo, such as cytosolic organelles and proteins in autophagosomes, which will finally fuse with lysosomes and result in the degradation of the cargo. A selective form of macroautophagy, the so-called lipophagy has been extensively studied, as a pivotal modulator in the progression of NAFLD[2]. Lipophagy involves the sequestration of LDs into autophagosomes, and their subsequent degradation mediated by lysosomal lipases[12]. On the other hand, microautophagy and CMA do not involve autophagosomes. Microautophagy directly drives the fusion of the dysfunctional proteins into the lysosomes, while CMA is a more specific process, as it targets certain cytosolic proteins, which contain the pentapeptide KFERQ motif into the lysosomes for degradation [13]. Macroautophagy and CMA are both constitutively active in cells and they could be upregulated under stressful conditions [14].
Concerning the liver, both macroautophagy (hereafter referred to as “lipophagy”) and CMA have been associated with various physiologic and pathologic conditions, thus these two processes will be the focus of this review. More specifically we will discuss the pathogenesis of NAFLD and provide an up-to-date review on the role of lipophagy and CMA in the development of the disease, mainly through the regulation of LDs.
2. Pathogenesis of NAFLD
The main hallmark of NAFLD is the accumulation of LDs in more than 5% of hepatocytes, known as steatosis [15]. NAFL is characterized by increased lipid accumulation, while NASH is presented with steatosis, inflammation, hepatocyte injury and possibly fibrosis [16].
Emerging evidence suggests that hepatic steatosis is a consequence of altered lipid metabolic processes due to increased accumulation of free fatty acids (FFAs) in the liver [17]. Insulin resistance seems to play a key pathophysiological role in the development of NAFLD. Under these conditions, insulin is no longer available to suppress lipolysis in adipose tissue thus increased circulation of FFAs results in enhanced efflux of lipids towards the liver [2]. Esterification of FFAs into TGs, which are enclosed in LDs and stored into hepatocytes, constitutes an adaptive mechanism due to lipid overload through transformation of potentially toxic FFAs into neutral TGs [18]. Studies have shown that adipose tissue derived fatty acids are the dominant source of hepatic TG accumulation in NAFLD [19,20]. In addition, as NAFLD has been associated with obesity and sedentary lifestyle, intestinal absorption of dietary lipids and subsequent accumulation in the liver in the form of LDs, account for 16% of total lipid flux to the liver [20]. Another source of FFAs is de novo lipogenesis (DNL), a process through which the liver synthesizes fatty acids de novo, which are subsequently esterified into TGs. Although DNL is not abundant in the healthy liver, it contributes up to 26% of lipid accumulation in patients with NAFLD [20].
Until recently, the initial step in the progression of NAFLD has been considered the accumulation of fat in the liver (steatosis). This has been proposed as the “two hit” hypothesis, formulated by Day and James in 1998 [21]. Hepatic accumulation of lipids is considered as the first hit, which sensitizes the liver to further metabolic stressors. These metabolic stressors, such as mitochondrial dysfunction, endoplasmic reticulum (ER) stress, inflammatory signals from circulating cytokines and adipokines represent the second hit, which is a prerequisite for the progression to NASH [4].
However, this pathogenetic theory is considered today outdated and too simplistic to delineate the development of NAFLD, thus a more complex hypothesis has been widely accepted named as “multiple hit hypothesis”[22]. According to this, multiple parallel factors act synergistically in genetically predisposed individuals to promote NAFLD development [23].This leads to dysfunction of mitochondria with increased production of reactive oxygen species (ROS), presence of elevated levels of ER stress and activation of inflammatory cascades [24]. In addition, other metabolic factors, such as altered gut microbiota, which result in increased permeability and absorption of FFAs, contribute to enhanced circulation of toxic compounds and therefore to the release of proinflammatory cytokines [25]. Lipotoxicity, along with the activation of inflammatory pathways, leads to hepatocyte death, a crucial determinant that distinguishes NASH from NAFL [26]. Thereby, dead hepatocytes release factors that induce wound healing responses, such as the activation of resident immune cells (Kupffer cells) and hepatic stellate cells, leading intimately to the replacement of injured hepatocytes by fibrotic cells [27].
Both two hit and multiple hit hypotheses consider NAFLD development as a progressive spectrum of pathological features, with steatosis being the primary and reversible stage, followed by steatohepatitis, a more severe inflammatory condition[22]. However, evidence suggest that NAFL and NASH may represent two independent conditions, without steatosis being a prerequisite stage for the progression to NASH [28]. In fact, some studies have documented that steatosis seems to occur in parallel with lipotoxicity, thus NASH could potentially constitute the initial liver lesion, without an earlier stage of steatosis [4]. In the vast majority of patients with NAFLD, simple steatosis seems to be stable over time [29], with the histological progression to NASH being rare. Thus, some studies propose that NAFLD does not constitute a consecutive linear progression of different stages, but a complex metabolic liver disease with two distinct clinical entities; NAFL and NASH [4].
Although steatosis has been considered as a totally benign situation, it seems that various disruptions that happen during increased accumulation of lipids in hepatocytes and contribute to the long-term development of lipotoxic conditions. More specifically, during steatosis, TGs constitute the main type of lipid enclosed in LDs. Studies have shown that TGs per se, although they are correlated with the severity of steatosis, they do not harm hepatocytes[30]. However, among TGs, other types of lipids, such as fatty acids, cholesterol, diacylglycerol and phospholipids are accumulated in the liver and can injure hepatocytes[31]. The chronic accumulation of these lipids could promote lipotoxic conditions and therefore to the progression of simple steatosis to steatohepatitis [32].
3. Role of LDs in NAFLD progression
3.1. Biogenesis of LDs
In order to protect against elevated FFAs flux towards the liver, originated from diet, adipose tissue and DNL, hepatocytes esterify FFAs in neutral lipids and store them in highly dynamic organelles, known as LDs [33]. Excess FFAs are primarily stored in the ER, where specific enzymes catalyze their esterification mainly into TGs, the most common neutral lipid found in hepatic steatosis and also in cholesterol esters [17]. Once a critical concentration of neutral lipids is achieved, they are beginning to deform ER bilayer and they are finally stored in the cytoplasm [34]. Thus, LDs are composed of a hydrophobic core of neutral lipids, comprised predominantly of TGs and cholesterol esters, which are coated by a phospholipid monolayer along with specific proteins responsible for lipid metabolism [35].
3.2. Structural changes of LDs affecting NAFLD progression
Structural characteristics of LDs, such as changes in lipid composition have been associated with different stages of fatty liver disease. In fact, cholesterol esters in LDs have been found in liver biopsies of patients with NASH, but not in patients with NAFL, thereby pointing out a potential role of cholesterol esters in the promotion of inflammation and fibrosis [36].
Apart from lipid core composition, LDs size also adapts to metabolic changes. Under normal conditions where the presence of LDs does not exceed over 5% of hepatocytes, LDs appear small with a diameter of 100-200nm [37]. However, histopathological studies from liver biopsies have shown that NAFL is commonly observed in the form of macrovesicular steatosis, which entails the accumulation of large droplets of fat within hepatocytes causing the nuclei to be displaced towards the periphery of the cytoplasm [38].These large LDs are associated with higher quantities of TGs in the lipid core, and morphological characteristics, such as low surface to volume ratio, functioning in favor of lipid storage rather than lipolysis, thus protecting against cytotoxic effects of FFAs[37,38]. On the other hand, studies from more severe steatohepatic stages have shown that NASH steatosis is usually seen as a coexistence of macrovesicular steatosis along with patches of microvesicular steatosis, referring to distended hepatocytes with a foamy, vacuolated cytoplasm, known as balloon cells [38]. In contrast to macrovesicular steatosis where the nucleus is displaced peripherally, in microvesicular steatosis the nucleus is usually centrally positioned[38]. Macrovesicular steatosis alone is considered a good long term prognostic factor, as these patients rarely develop fibrosis or cirrhosis[38]. On the contrary, microvesicular steatosis has been found to co-exist with higher frequencies of severe macrovesicular steatosis, inflammation, cellular injury, and hepatocyte ballooning, features that are commonly found in NASH livers [39]. Thus, microvesicular steatosis could be considered as a significant prognostic factor of the transition of simple steatosis to steatohepatitis [40].
3.3. Perilipins-the gatekeeper of LDs surface and their role in NAFLD
The surface of LDs is coated by numerous proteins, such as the adipose triglyceride lipase 1 (ATGL1), the hormone sensitive lipase (HSL) and the perilipins (PLINs) [41]. PLINs, the best characterized and the most abundant LD associated proteins, consist of five members; PLIN1 to PLIN5[41]. PLIN1 is found primarily in mature adipocytes, PLIN2 and PLIN3 are found ubiquitously,PLIN4 is mainly restricted to adipocytes), while PLIN5 is found in oxidative tissues, such as those of the heart, muscle and liver [33,41].
The expression of PLINs on the LDs surface varies depending on the LD size, with small LDs expressing PLIN3, PLIN4 and PLIN5, medium LDs expressing PLIN2 and large LDs expressing PLIN1 [42]. PLIN1, is a key regulator of lipolysis and mediates exchange of lipids between LDs, thus contributing to the formation and stabilization of large LDs [43]. PLIN1 is exclusively expressed in the steatotic liver, while on the contrary it is absent in normal healthy hepatocytes [44]. In fact, during adipocyte differentiation, the initially expressed PLIN2 in premature adipocytes, is replaced by PLIN1 in mature adipocytes [41]. In line with this notion, PLIN1 is considered a marker of chronic steatosis, a condition characterized by LD maturation and the sequential expression of PLIN3, PLIN5, PLIN2 and finally PLIN1 on LD surface during this gradual process [45]. In contrast to PLIN1, PLIN2 has been detected in few hepatocellular LDs of the normal liver, along with PLIN3 [44]. PLIN2 is the major hepatic LD protein as it is responsible for lipid accumulation [46] and plays a significant role in the accessibility of lipolytic mechanisms to LDs. More specifically, under energy deprivation, phosphorylation of PLIN2 in response to AMP-activated protein kinase (AMPK) activation acts as a recognition marker by Heat shock cognate 71 kDa protein (HSC70), thereby contributing to the degradation of PLIN2 by CMA[47]. This modification allows cytosolic lipases, such as ATGL1 and autophagy related proteins, to begin lipolytic processes [47]. In line with this notion, PLIN2, is the most upregulated PLIN in the fatty liver [44], and it has been proposed to be the most important marker of hepatic LDs accumulation [48]. Straub et al have not detected any significant correlation between the levels of PLIN1 and PLIN2 in steatosis compared to steatohepatitis [44]. PLIN3 is ubiquitously expressed and is mainly associated with LDs biogenesis, while it has also antilipolytic properties and, similar to PLIN2, is degraded by CMA after its phosphorylation by AMPK [49]. Furthermore, PLIN5 is the most dynamic protein and is highly expressed in fasted hepatocytes [50]. During resting conditions or when cells are overloaded with lipids, PLIN5 acts as a gatekeeper, since it inhibits ATGL-mediated lipolysis and mitochondrial beta- oxidation, serving as a protective factor against hepatic lipotoxicity [51]. On the contrary, during fasting conditions, PLIN5 upregulates lipolysis and mitochondrial beta-oxidation, to cover energy demands [52]. Considering NAFLD, studies have shown significantly increased levels of PLIN5 in severely steatotic liver [53], as it has been shown to be required for the adaptation to lipid overload and it has been proposed as a crucial regulator of LD metabolism, hepatic inflammation and mitochondrial function[50]. Apart from simple steatosis, a limited number of studies has investigated the expression pattern of PLINs in NASH livers. Notably, researchers have pointed out that during steatohepatitis, PLIN2 exhibits a major expression in small LDs, especially around ballooned hepatocytes, with the levels of expression being correlated with the severity of the inflammation [54]. Apart from PLIN2, levels of PLIN3 and PLIN5 on the surface of small LDs are also elevated, as markers of acute hepatocellular injury, while, PLIN1 has not been detected since its expression is observed in chronic steatotic conditions[45] (Figure 1).
3.4. Breakdown of lipid droplets
LDs undergo a biogenesis and degradation cycle which contributes to LD homeostasis. Dysregulation of LD homeostasis may result in increased intracellular lipid accumulation and thus development of NAFLD [55]. Catabolism of LDs into FFAs and glycerol is induced under energy deprivation conditions, thus LDs constitute a significant energy storage pool. Consequently the tight regulation of LD metabolism is crucial, especially for energy dependent organs, such as the liver [35]. Lipolytic pathways include lipolysis, which is mediated through cytosolic lipases, such as the ATGL, and lipophagy, a selective breakdown process of LDs through macroautophagy components [56].
3.4.1. Degradation of PLINs via CMA
Degradation of LDs is mediated through two major lipolytic pathways- lipolysis by neutral lipases and lipophagy by acid lysosomal lipases [57]. As mentioned above, numerous proteins harbor on the LD surface, playing major roles in LD homeostasis and in communication with other organelles [58]. Along with this notion, studies have focused on PLINs and their role in LD degradation, with PLIN2, PLIN3 and PLIN5 acting as negative regulators of lipolytic mechanisms, therefore the dissociation of these proteins from LD membrane is a prerequisite for the accessibility of lipolytic mechanisms[59,60]. Further studies have shown the presence of the characteristic KFERQ-motif in PLIN2, PLIN3, PLIN5 which have been identified as CMA substrates[53,61]. CMA constitutes a selective form of autophagy, by targeting specifically proteins containing the KFERQ motif and delivering this cargo directly to the lysosomes by the interaction of HSC70 with the lysosomal associated membrane protein 2A (LAMP2A). HSC70 recognizes the characteristic motif in the PLINs, and together they interact with the cytosolic tail of LAMP2A, thus mediating the translocation of PLINs into the lysosomal lumen [62]. CMA constitutes a significant metabolic regulator in the liver as it is activated under various stressful conditions, such as starvation or in response to lipid overload [63]. However, during chronic lipid dysregulation, as it is observed in NAFL, CMA is downregulated, mostly due to changes in the lipid composition of the lysosomal membrane[64].
3.4.2. Degradation of lipid core via lipolysis and lipophagy
After the removal of PLINs, LDs are accessible to lipolytic mechanisms. Lipolysis occurs in the cytoplasm by neutral lipases that directly act on the lipid core of LDs, to produce FFAs, a substrate for mitochondrial beta-oxidation [65].
Lipophagy on the other hand, takes place inside the lysosomes and involves the autophagic sequestration of LDs into the autophagosomes and the subsequent fusion with lysosomes [57]. LDs are finally degraded by lysosomal lipases and FFAs are released for energy production. Lipolysis and lipophagy seem to be concomitantly activated, however studies have underlined a potential preference of lipolysis to larger LDs, whereas lipophagy targets smaller LDs [66]. Along with this line, ATGL mediated lipolysis has been proposed to act as an upstream pathway in larger LDs in order to produce small newly formed LDs by re-esterification that could be finally targeted by lipophagy [66]. Studies have shown that cytosolic lipases, such as ATGL, interact with key autophagic proteins, such as light chain (LC3B) and p62 to recruit LDs to autophagosomes [67], thus proposing that these two pathways not only work in tandem, but they are also cross regulated.
4. Autophagy modulation during NAFLD
Steatosis, as mentioned above, is a benign and reversible stage of NAFLD, in which toxic FFAs are transformed into neutral TGs, stored finally as LDs in the cytoplasm of hepatocytes[18]. During the past decade autophagy has gained significant attention, as a potential crucial regulator of the pathogenesis of NAFLD, thereby autophagy activators and inhibitors could possibly serve as promising therapeutic targets. It is well-established that autophagy not only regulates the degradation of damaged organelles and cytosolic components, but also various metabolic processes. For instance, lipid balance, , is maintained mainly through lipophagy, while CMA has emerged as a potential co-regulator. Both lipophagy and CMA are constitutively active in hepatocytes, thereby contributing to lipid homeostasis [14].
4.1. Lipophagy modulation during NAFLD
Various studies have suggested the importance of lipophagy in the maintenance of liver homeostasis, as it mediates catabolization of LDs to FFAs [17]. Lipophagy plays a crucial role, not only in the regulation of lipid transportation, but also in the adaptation of cells to several insults, such as lipid imbalance[9]. It is well established that lipophagy activity is impaired in NAFLD [68].
Immunohistochemical studies have demonstrated inhibition of lipophagy in human liver samples diagnosed with NAFLD[69]. In accordance with this observation, increased levels of LC3-II and p62, the most highly studied lipophagy markers, have been found in patients with NAFLD [70]. Under conditions of energy surplus and high-fat diet, mammalian target of rapamycin (mTOR), a widely known negative regulator of lipophagy, is frequently hyperactivated thus inhibiting initiation of lipophagy [71]. However, the long-term inhibition of lipophagy in the steatotic liver has been found to be mediated through downregulation of autophagy related transcription factors, such as forkhead box transcription factors (FOXO) and transcription factor EB (TFEB) [72,73]. Consequently, the steatotic liver is characterized by decreased expression of genes associated with autophagic core mechanism and the formation of autophagosomes[9]. Additionally, a disturbance in lysosome biogenesis and an impediment in autophagosome-lysosome fusion due to alterations in the cellular membrane lipid composition of autophagosomes and lysosomes has been reported [74].
Despite the well-established notion that lipophagy is impaired in NAFLD, there is limited evidence considering the levels of lipophagy through the different stages of NAFLD. A recent study has demonstrated that lipophagy is impaired in the later stages of NAFLD, in both in vivo and in in vitro models [8]. Further investigation of LC3-II and p62 protein levels have shown an increased lipophagy flux in the early stages of NAFL with a gradual decrease during propagation of the disease [75]. Similar findings have been reported in human liver specimens suffering from NASH, as evidenced by elevated levels of LC3-II and p62, thus indicating suppression of lipophagy, with the perpetual accumulation of LC3-II and p62 being positively correlated with the severity of the disease [70].
4.2. CMA involved in LD degradation during NAFLD
Recently, the role of CMA has emerged concerning the metabolic functions of the liver[76]. CMA has been shown to be associated with lipid homeostasis, while mice with defects in liver CMA have been shown to develop hepatosteatosis [10]. CMA substrates involve only proteins, not lipids, however it is strongly associated with LD metabolism. Intriguingly PLIN2, PLIN3 and PLIN5 have been found to contain the pentapeptide KFERQ[53]. Therefore, they interact with HSC70, promoting the delivery of PLINs to lysosomes for selective degradation [61]. In fact, mutation of KFERQ peptitide in PLIN2 resulted in LD accumulation, thereby elucidating a crucial role for CMA as an upstream regulator for both lipolysis and lipophagy [59].
A limited number of studies have identified the role of CMA as a crucial regulator of hepatic metabolism and hepatic adaptation to stressful stimuli, such as energy deprivation and lipid overload [63]. The cellular response to various stressors, including prolonged starvation, oxidative stress, and exposure to factors that result in protein damage, conditions that characterize NASH livers, elicits maximal activation of CMA [64]. Constitutive blockade of CMA has been shown to induce hepatic steatosis, despite that lipophagy machinery has remained intact [63]. LAMP2A has been widely used as a marker of CMA activity. Levels of LAMP2A have been found elevated in mildly steatotic liver compared to normal liver, while these levels were significantly decreased in progressive steatosis, suggesting that CMA activity is inversely correlated with severity of hepatosteatosis [53]. The initial elevated levels of CMA in cases of mild steatosis acted as a compensatory mechanism to lipid overload, however when this lipid overload was prolonged, CMA was dramatically decreased [53]. A study by Rodriguez-Navaro et al., has identified a negative impact of dietary lipid challenges on CMA activity. In fact, they have suggested that this effect is primarily mediated by alterations in the lipid composition of the lysosomal membrane resulting from lipid exposure. Furthermore, they observed a decrease in the stability of lysosomal membrane proteins. Specifically, LAMP2A was found to be particularly susceptible to lipid composition changes, thereby revealing a distinct mechanism that contributes to compromised CMA under lipid overload conditions [64].
On the other hand, there is limited evidence on CMA levels in NASH disease. In fact, it is widely known that metabolic oxidative stress, primarily characterized by oxidatively modified proteins, triggers CMA, augments substrate translocation by HSC70 towards the lysosomal membrane, and elevates LAMP2A levels [77]. Das et al have reported that LAMP2A levels were elevated in an induced mouse model of steatohepatic liver, therefore, this study proposes a pathogenetic model, with NASH being characterized by increased CMA levels [78]. That study reported that a subset of proinflammatory protein receptors, such as purinergic receptors, mainly the P2X7 receptor, which undergoes upregulation in response to metabolic oxidative stress, may modulate the autophagy process by boosting, albeit to a limited extent, the mRNA levels of HSC70 and LAMP-2A and promoting the association of LAMP2A with the lysosomal membrane [78] (Figure 2). However, the exact pathway that activates CMA in the NASH liver remains to be elucidated.
5. Discussion
NAFLD consists of distinct liver disorders that occur due to excessive accumulation of fat in the liver[23]. NAFLD manifests in various forms, from simple fatty liver to NASH and the resulting fibrosis/ cirrhosis[16]. Approximately one-fifth of NAFLD patients will experience disease progression to NASH, while roughly 20% will experience further progression to cirrhosis or to End-Stage Liver Disease (ESLD), which can result in various complications and confer significant morbidity and mortality [79].
Recently, LDs have emerged as the focal point of studies pertaining to NAFLD pathogenesis. A range of LD characteristics, including their size, concentration of cholesterol esters, and expression of various PLINs in their lipid membranes, appear to contribute to liver steatosis[36,38]. It is noteworthy that different stages of NAFLD are distinguished by varying sizes of LDs and PLINs[42]. In the initial stages of NAFL, small LDs begin to accumulate in the cytoplasm and are primarily engulfed by PLIN2, a marker of liver steatosis[41]. However, in the later stages of NAFL, the size of LDs increases considerably and PLIN2 is replaced by PLIN1, a hallmark of LD maturation and macrovesicular steatosis [45]. Furthermore, studies in NASH liver specimens have shown increased expression of PLIN2, PLIN3 and PLIN5, mainly found in small LDs[45,54]. In line with this notion, histopathological studies of NASH livers have pointed out the existence of a background of macrovesicular steatosis in conjunction with microvesicular steatosis patches, resulting in the foamy appearance of hepatocytes [45,54].
Up to date, various pathogenetic mechanisms have been identified as potential mechanisms that underlie NAFLD[16]. Autophagy pathways have been proposed as crucial regulators of hepatic steatosis, by being implicated in lipid metabolism[9]. Therefore, various studies have pointed out the role of decreased levels of autophagy in the pathogenesis of NAFLD, but only a limited number of studies have investigated the levels of both lipophagy and CMA pathways during NAFLD progression, from simple steatosis to NASH. In the initial stages of simple steatosis, there is an upregulation of lipophagy, which acts as a compensatory mechanism to counter the accumulation of lipids [8]. However, chronic lipid accumulation results in downregulation of lipophagy, mediated by different factors, such as mTOR activation and FOXO/TFEB downregulation[12,71]. Under these conditions, accumulation of LDs is favored, however acting as a protective mechanism against lipotoxicity. Apart from NAFL, a limited number of studies has focused on the levels of lipophagy during NASH. Inflammation, oxidative stress and lipotoxicity result in excessive ER stress that further downregulate lipophagy [70,80].
CMA has also been elucidated as a significant regulator of NAFLD pathogenesis[63]. Early stages of simple steatosis are characterized by increased levels of CMA, which acts in coordination with elevated lipophagy, to compensate for lipid accumulation and restore hepatic homeostasis[53]. However, prolonged lipid overload, results in downregulation of CMA by altering lipid composition of the lysosomal membrane, thereby decreasing the stability of LAMP2A protein[64]. Therefore, we could speculate that advanced stages of simple steatosis are characterized by decreased levels of both lipophagy and CMA, with aiming to protect hepatocytes from oxidative stress, resulting from excessive FFAs production and subsequent lipotoxicity and ER stress.
On the other hand, during NASH development, inflammatory conditions are identified as excessive stressful stimuli that upregulates CMA, as evidenced by elevated levels of LAMP2A and HSC70 [78]. Oxidative stress, one of the key characteristics of NASH progression activates CMA, as a survival pathway in order to get rid of oxidized proteins and protect hepatocytes from apoptosis[81]. Although CMA seems to act as a compensatory mechanism to lipophagy inhibition in NASH liver, it is reasonable to hypothesize that the persistent biogenesis of small LDs coated by PLIN2, PLIN3, and PLIN5 may not be counteracted. Although these PLINs are substrates of CMA, it could be speculated that the levels of LD biogenesis exceed the capacity of CMA to degrade them. Therefore, the inhibition of lipophagy along with the continuous production of small LDs engulfed by PLIN2, PLIN3 and PLIN5 could be a proposed mechanism for NAFLD progression from simple steatosis to NASH. In line with this notion, a study by Asimakopoulou et al. has shown that tumor areas of HCC are characterized by markedly increased levels of PLIN5, thereby pointing out a potential role for the overexpression of this PLIN in the propagation of the disease [82].
Although considerable advancements have been made in the investigation of the pathogenesis of NAFLD, the evidence regarding the levels of lipophagy and mainly CMA during the progression of the disease from NAFL to NASH remains inadequate. Further research endeavors should concentrate on the precise signaling pathways that regulate both the levels of lipophagy and CMA and how these pathways are cross regulated during the different stages of the disease. Furthermore, the histopathological characteristics of NAFLD livers should be further studied in correlation with the autophagic levels, in order to elucidate the exact pathogenetic mechanism of macrovesicular and microvesicular steatosis. Ultimately, delving into the investigation of levels of PLINs along with other structural characteristics of LDs during the different stages of NAFLD could provide a more comprehensive understanding of this complex disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Perilipins and NAFLD; Figure S2: Autophagy modulation during NAFLD.
Author Contributions
Conceptualization, A.V.C.; writing—original draft preparation, E.M.M.; A.V.C., designed the figure, E.M.M., writing—review and editing, A.V.C., A.C.G. and P.K; visualization, A.V.C.; supervision, A.V.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 601627. [Google Scholar] [CrossRef]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Yilmaz, Y. Review article: is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment. Pharmacol. Ther. 2012, 36, 815–823. [Google Scholar] [CrossRef]
- Loomba, R.; Adams, L.A. The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis Caused by NASH. Hepatology 2019, 70, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Ge, G.; Tseng, Y.; Ma, Y.; Zhang, J.; Liu, J. Hepatic autophagy fluctuates during the development of non-alcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.; Lin, M.; Rios-Colon, L.; Qi, Q.; Moore, J.T.; Kumar, D. Emerging Roles of Impaired Autophagy in Fatty Liver Disease and Hepatocellular Carcinoma. Int. J. Hepatol. 2021, 2021, 6675762. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am. J. Physiol. Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef]
- Carotti, S.; Aquilano, K.; Zalfa, F.; Ruggiero, S.; Valentini, F.; Zingariello, M.; Francesconi, M.; Perrone, G.; Alletto, F.; Antonelli-Incalzi, R.; et al. Lipophagy Impairment Is Associated With Disease Progression in NAFLD. Front. Physiol. 2020, 11, 850. [Google Scholar] [CrossRef]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 159–166. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Júnior, W.S.S.; Valerio, C.M. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of Different Fatty Acid Sources to Very Low-Density Lipoprotein-Triacylglycerol in the Fasted and Fed States. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited after a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Cusi, K. Role of Insulin Resistance and Lipotoxicity in Non-Alcoholic Steatohepatitis. Clin. Liver Dis. 2009, 13, 545–563. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut–liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef]
- E Feldstein, A.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; Sauver, J.S.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The Natural History of Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Filozof, C.; Goldstein, B.J.; Williams, R.N.; Sanyal, A. Non-Alcoholic Steatohepatitis: Limited Available Treatment Options but Promising Drugs in Development and Recent Progress Towards a Regulatory Approval Pathway. Drugs 2015, 75, 1373–1392. [Google Scholar] [CrossRef]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef]
- Thiam, A.R.; Forêt, L. The physics of lipid droplet nucleation, growth and budding. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2016, 1861, 715–722. [Google Scholar] [CrossRef]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Landis, C.S.; Jin, G.-Y; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated With Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases Associate with Isolated Lipid Droplets (LDs) and Show Altered Content After Ethanol Administration: Potential Role in Alcohol-Impaired LD Metabolism. Alcohol. Clin. Exp. Res. 2013, 38, 327–335. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef]
- Celebi, G.; Cicek, A.F.; Gurel, H.; Genc, H.; Kirik, A.; Ercin, C.N.; Dogru, T. Microvesicular steatosis : a missed item in the management of nonalcoholic fatty liver disease? . 2020, 83, 565–570. [Google Scholar]
- Germano, C.W.; Mega, P.F.; Mattosinho, T.J.A.P.; Dias, L.L.C.; Gestic, M.A.; Utrini, M.P.; Chaim, F.D.M.; Callejas-Neto, F.; Chaim, E.A.; Cazzo, E. Microvesicular Steatosis in Individuals with Obesity: a Histological Marker of Non-alcoholic Fatty Liver Disease Severity. Obes. Surg. 2023, 33, 813–820. [Google Scholar] [CrossRef]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Wolins, N.E. Isolation of Lipid Droplets from Cells by Density Gradient Centrifugation. Curr. Protoc. Cell Biol. 2005, 29, 3–15. [Google Scholar] [CrossRef]
- Sun, Z.; Gong, J.; Wu, H.; Xu, W.; Wu, L.; Xu, D.; Gao, J.; Wu, J.-W.; Yang, H.; Yang, M.; et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 2013, 4, 1594. [Google Scholar] [CrossRef]
- Straub, B.K.; Stoeffel, P.; Heid, H.; Zimbelmann, R.; Schirmacher, P. Differential pattern of lipid droplet-associated proteins andde novoperilipin expression in hepatocyte steatogenesis. Hepatology 2008, 47, 1936–1946. [Google Scholar] [CrossRef]
- Pawella, L.M.; Hashani, M.; Eiteneuer, E.; Renner, M.; Bartenschlager, R.; Schirmacher, P.; Straub, B.K. Perilipin discerns chronic from acute hepatocellular steatosis. J. Hepatol. 2013, 60, 633–642. [Google Scholar] [CrossRef]
- McManaman, J.L.; Bales, E.S.; Orlicky, D.J.; Jackman, M.; MacLean, P.S.; Cain, S.; Crunk, A.E.; Mansur, A.; Graham, C.E.; Bowman, T.; et al. Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. J. Lipid Res. 2013, 54, 1346–1359. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.K.; Gyoengyoesi, B.; Koenig, M.; Hashani, M.; Pawella, L.M.; Herpel, E.; Mueller, W.; Macher-Goeppinger, S.; Heid, H.; Schirmacher, P. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology 2012, 62, 617–631. [Google Scholar] [CrossRef]
- Najt, C.P.; Devarajan, M.; Mashek, D.G. Perilipins at a glance. J. Cell Sci. 2022, 135. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.B.M.; Krizanac, M.; Weiskirchen, R.; Asimakopoulos, A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. Int. J. Mol. Sci. 2021, 22, 5284. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2014, 61, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.-M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.Y.; Sun, K.; Zhang, M.; Zhou, X.; Zheng, X.; Tian, S.; Liu, Y.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of Perilipin and Adipophilin in Nonalcoholic Fatty Liver Disease; Relevance to Oxidative Injury and Hepatocyte Ballooning. J. Atheroscler. Thromb. 2009, 16, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2020, 50, 101115. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2019, 9, 778. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.-P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with Adipose Triglyceride Lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.K.; Strub, M.-P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient Chaperone-Mediated Autophagy in Liver Leads to Metabolic Dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall'Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. 2012, 109, E705–14. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS - Lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.-M. Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease. Liver Res. 2018, 2, 112–119. [Google Scholar] [CrossRef]
- Kashima, J.; Shintani-Ishida, K.; Nakajima, M.; Maeda, H.; Unuma, K.; Uchiyama, Y.; Yoshida, K. Immunohistochemical study of the autophagy marker microtubule-associated protein 1 light chain 3 in normal and steatotic human livers. Hepatol. Res. 2013, 44, 779–787. [Google Scholar] [CrossRef]
- González-Rodríguez, Á.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, T.; Yang, L.; Wang, H.-Y. Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK. Int. J. Mol. Sci. 2017, 18, 1063. [Google Scholar] [CrossRef]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of Carbohydrate Metabolism by the Farnesoid X Receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef]
- Oberkofler, H.; Pfeifenberger, A.; Soyal, S.; Felder, T.; Hahne, P.; Miller, K.; Krempler, F.; Patsch, W. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans. Diabetologia 2010, 53, 1971–1975. [Google Scholar] [CrossRef]
- W. K. K. Wu, L. Zhang, and M. T. V. Chan, "Autophagy, NAFLD and NAFLD-Related HCC," (in eng), Adv Exp Med Biol, vol. 1061, pp. 127-138, 2018 2018. [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-Mediated Autophagy in the Liver:Good or Bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am. J. Physiol. Liver Physiol. 2013, 305, G950–63. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; Fu, X.; Pang, M.; Zhou, Y.; Yin, G.; Zhang, J.; Fan, D. The Antioxidative Role of Chaperone-Mediated Autophagy as a Downstream Regulator of Oxidative Stress in Human Diseases. Technol. Cancer Res. Treat. 2022, 21, 15330338221114178. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 Expression in Hepatocellular Carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Perilipins and NAFLD. Under normal conditions PLIN2 has been detected in a few hepatocellular LDs along with PLIN3 (a). During the early stages of steatosis, small LDs are beginning to accumulate in the hepatocytes, coated by PLIN2, PLIN3 and PLIN5. Subsequently, under chronic lipid overload conditions, these PLINs are substituted by PLIN1, as LDs are becoming larger, a hallmark of LD maturation and macrovesicular steatosis (b). On the contrary, during the development of NASH, levels of PLIN2, PLIN3 and PLIN5 on the surface of small LDs are elevated, as markers of acute hepatocellular injury (c).
Figure 1.
Perilipins and NAFLD. Under normal conditions PLIN2 has been detected in a few hepatocellular LDs along with PLIN3 (a). During the early stages of steatosis, small LDs are beginning to accumulate in the hepatocytes, coated by PLIN2, PLIN3 and PLIN5. Subsequently, under chronic lipid overload conditions, these PLINs are substituted by PLIN1, as LDs are becoming larger, a hallmark of LD maturation and macrovesicular steatosis (b). On the contrary, during the development of NASH, levels of PLIN2, PLIN3 and PLIN5 on the surface of small LDs are elevated, as markers of acute hepatocellular injury (c).
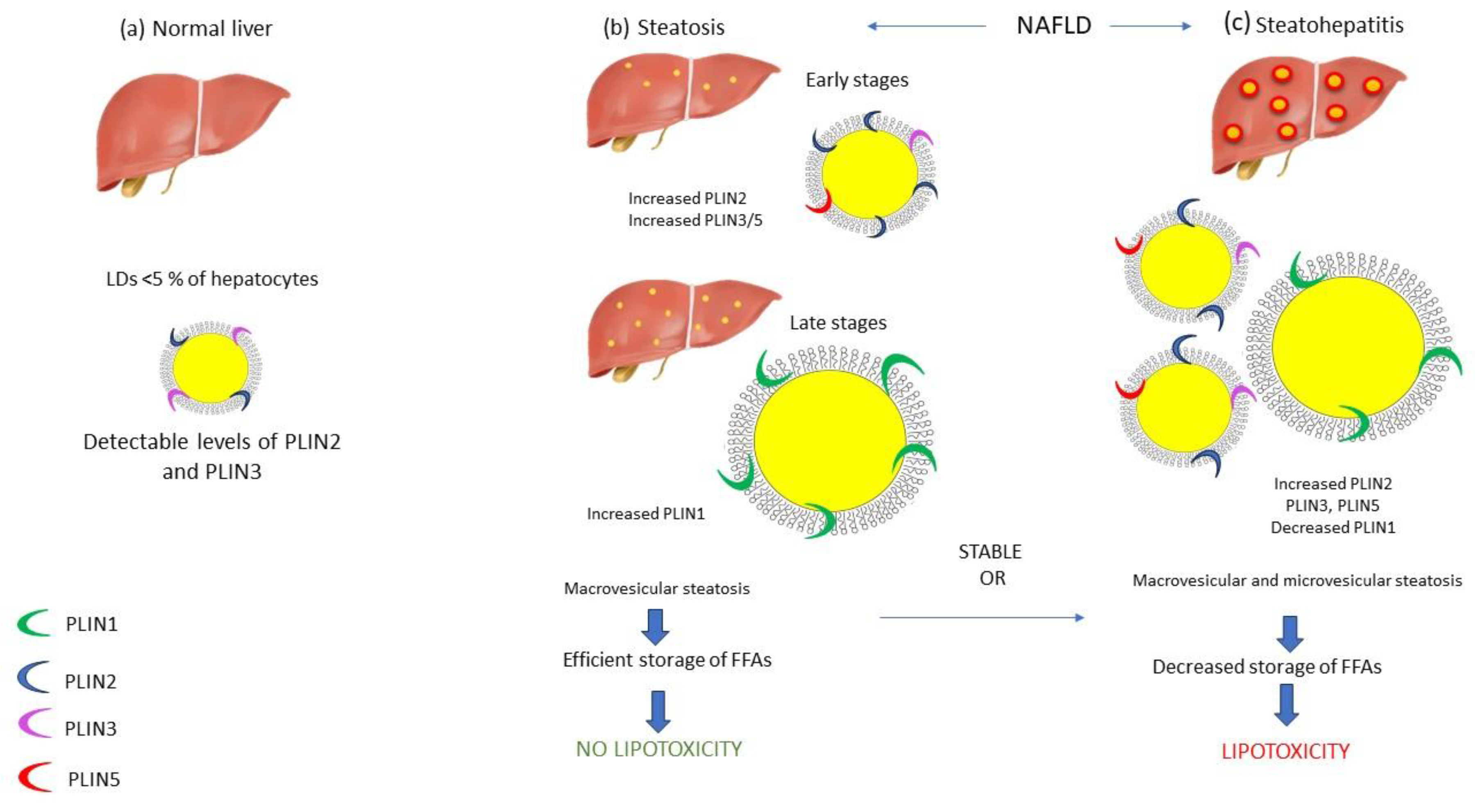
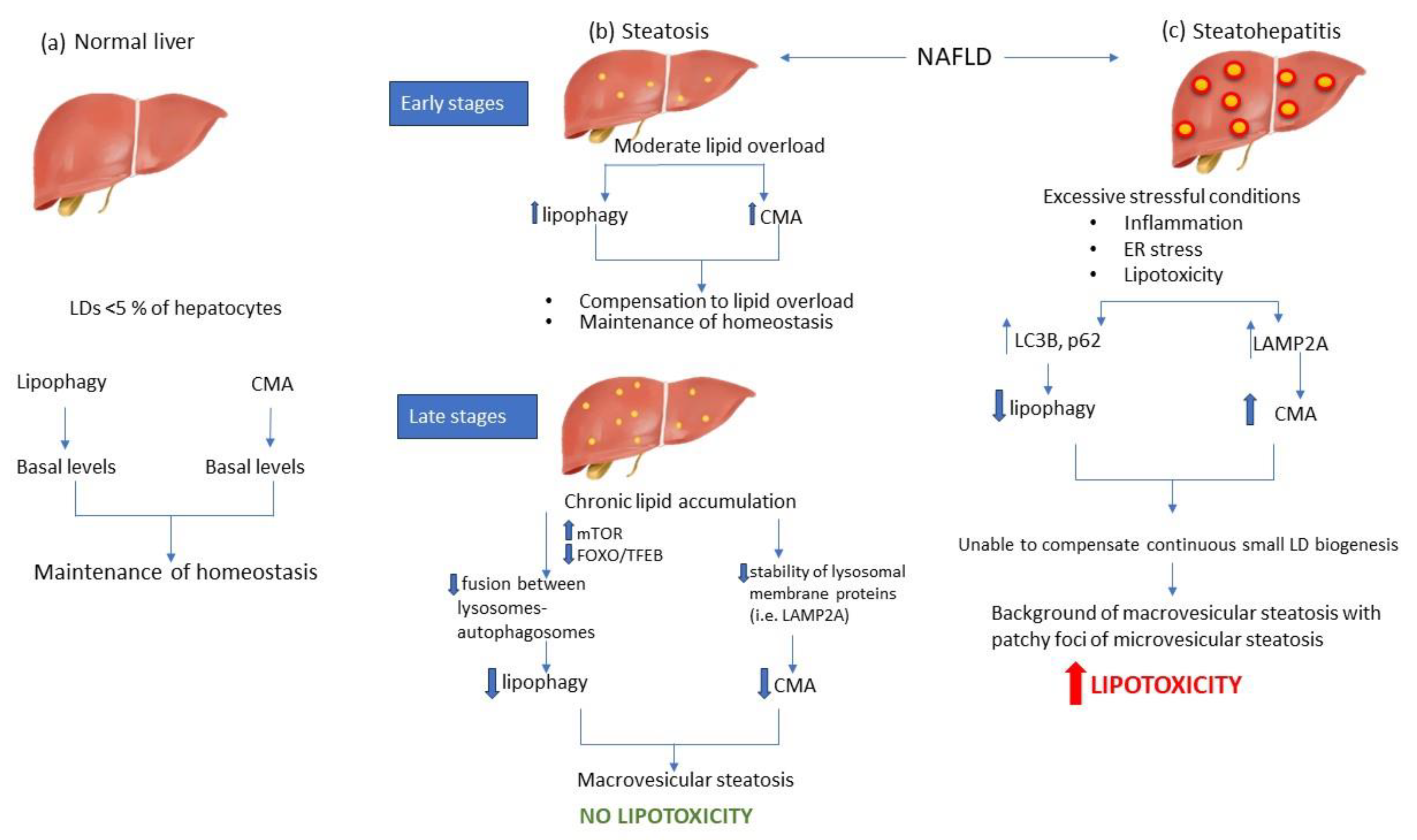
Figure 2.
Autophagy modulation during NAFLD. In the normal liver, both lipophagy and CMA exist on basal levels to maintain lipid balance (a). During the early stages of steatosis, lipophagy and CMA are upregulated to compensate increased lipid flux, and therefore to inhibit LD accumulation and maintain lipid homeostasis. However, livers with advanced steatosis are characterized by decreased levels of lipophagy and CMA, mainly due to alterations in lysosomal lipid composition. Inhibition of autophagic mechanisms acts as a protective mechanism against lipotoxicity (b). On the other hand, NASH livers are distinguished by the presence of inflammation, oxidative stress and lipotoxicity, which act as excessive stressful stimuli, thereby inhibiting lipophagy, while on the same time CMA is activated to alleviate the liver from the proteotoxic stress of oxidized proteins. However, the constant biogenesis of small lipid droplets could not be counteracted by CMA levels, thereby leading in uncontrollable lipotoxic environment (c). .
Figure 2.
Autophagy modulation during NAFLD. In the normal liver, both lipophagy and CMA exist on basal levels to maintain lipid balance (a). During the early stages of steatosis, lipophagy and CMA are upregulated to compensate increased lipid flux, and therefore to inhibit LD accumulation and maintain lipid homeostasis. However, livers with advanced steatosis are characterized by decreased levels of lipophagy and CMA, mainly due to alterations in lysosomal lipid composition. Inhibition of autophagic mechanisms acts as a protective mechanism against lipotoxicity (b). On the other hand, NASH livers are distinguished by the presence of inflammation, oxidative stress and lipotoxicity, which act as excessive stressful stimuli, thereby inhibiting lipophagy, while on the same time CMA is activated to alleviate the liver from the proteotoxic stress of oxidized proteins. However, the constant biogenesis of small lipid droplets could not be counteracted by CMA levels, thereby leading in uncontrollable lipotoxic environment (c). .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



