
Submitted:
10 October 2023
Posted:
12 October 2023
You are already at the latest version
Abstract
Alzheimer's disease is a multifactorial neurodegenerative disorder, with Acetylcholinesterase, Butyrylcholinesterase, Beta secretase, Glycogen Synthase Kinase beta and Monoamine oxidase B playing central role in it's pathogenesis.
Therefore, this research aims to discover potential curative multitarget drugs derived from African natural products capable of addressing the multifactorial characteristics of the disease.
In silico approaches were used to filter a 880 african natural compounds library based on selected pharmacokinetic properties. Molecular docking against the five aforementioned proteins, followed by Molecular Mechanics with Generalised Born and Surface Area Solvation scoring for the top compounds were used to assess binding. Density Functional Theory studies were used to assess electronic transitions.
Pharmacokinetic filters resulted in 200 compounds, of which only five were selected after molecular docking. Density Functional Theory and Molecular Mechanics with Generalised Born and Surface Area Solvation scoring resulted in 3 potential multitarget compounds. Compound 157 (ZINC000095485950) showed triple-target inhibitory activity against Butyrylcholinesterase, Glycogen Synthase Kinase beta and Beta-secretase, while compounds 159 (ZINC000095485952) and 696 (ZINC000039144622) showed dual-target inhibitory activity against Butyrylcholinesterase and Glycogen Synthase Kinase beta. Molecular dynamics simulation and further experimental assessments are suggested.
Keywords:
Alzheimer's disease
; African natural compounds
; Multitarget-directed ligands
; AChE
; BACE1
; BuChE
; GSK-3β
; MAO-B.
1. Introduction
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative disease affecting behaviour and cognition regions in the brain, primarily the entorhinal cortex [1]. Affecting mainly elderly patients, the disease usually starts with an asymptomatic stage followed by a stage of mild cognitive impairment, and finally, dementia, leading to significant disability and dependency [2,3]. Every 3 seconds, someone develops AD [4], and at least 50 million people live with AD, estimated to reach 152 million by the year 2050 [5]. More than half of the global cases of dementia were reported in low and middle-income countries [6].
AD is characterised by three main pathological hallmarks: Amyloid beta (Aβ) plaque deposition, neurofibrillary tangles (NFTs) of hyperphosphorylated tau protein, and death of neurons and loss of synapses [7]. Recent studies have revealed that several mechanisms, such as the amyloid hypothesis, tau hypothesis, cholinergic hypothesis, oxidative hypothesis and many others, are behind these pathological hallmarks [8], which rely on various essential enzymes namely Amyloid Precursor Protein(APP)-cleaving enzyme 1 (BACE1) [9], MonoAmine Oxidase B (MAO-B) [10], Glycogen Synthase Kinase beta (GSK-3β) [11,12], Acetylcholinesterase (AChE), and Butyrylcholinesterase (BuChE) [13,14,15].
Overall, these findings suggest inhibiting these enzymes could be a potential approach for treating AD. For instance, GSK-3β has emerged as a potential therapeutic target in AD, resulting in many agents with various mechanisms of action, such as lithium, adenosine triphosphate (ATP) and non-ATP competitive inhibitors (e.g. Tideglusib, in Phase II clinical trials) [16,17]. Furthermore, Acetylcholinesterase inhibitors (AChEIs) such as galantamine, donepezil, and rivastigmine, have been the mainstay of AD treatment [18]. By preventing ACh turnover and restoring its synaptic levels, these medications compensate for the loss of cholinergic neurons and provide symptomatic relief [19]. Memantine, a N-methyl-D-aspartate (NMDA) receptor antagonist, is approved for moderate to severe AD by blocking NMDA-mediated calcium ion flux and reducing the effects of elevated glutamate levels [20]. Anti-Aβ immunotherapy; Aducanumab and Lecanemab , was also used yet it was linked to serious amyloid-related imaging abnormalities (ARIA) [21,22].
The complex pathophysiology of AD makes Multitarget-directed ligands have greater potential for being more effective and safer treatments compared to the currently available single-targeted drugs [23]. To date, no multi-target treatment has been approved for AD [24]. Yet, Leuco-methylthioninium, a multitarget compound with inhibitory activity against MAO-B, tau protein aggregation, and Nitric oxide production, is in phase III clinical trial [25].
Several phytochemical compounds, such as alkaloids and Flavonoid-based compounds have been found to be potential natural anti-Alzheimer's agents [26,27].
Africans have depended on ethnomedicine to diagnose, treat, and manage CNS-related illnesses since the early decades [28]. This drives the ongoing search for new lead compounds from this African ethnomedicine [29]. For instance, EGb 761, a natural compound purified from the extract of the African plant Ginkgo biloba was shown to have powerful treatments potential for dementia [30]. Furthermore, plants from 23 families, with the Amaryllidaceae family being the most significant, have been extensively studied for their anti-AD activity in vivo and in vitro. Leaf extracts of Carpobrotus edulis, Terminalia sericea, Tithonia diversifolia, and Boophone disticha have shown neuroprotective properties, inhibiting AChE and BACE1 aggregation [31].
2. Results
2.1. Pharmacokinetics and toxicity prediction
Two hundred compounds out of 880 were found to have no risk of mutagenicity, hepatotoxicity and cardiotoxicity with good bioavailability profile (S1).
2.2. Molecular docking
During the docking procedure, the 200 compounds that successfully passed the Pharmacokinetic filter, were subjected to HTVS, SP, and XP modes of Glide against the five enzymes. Thirty two compounds displayed higher scores compared to the reference for BuChE, compound 27 (ZINC000095485893) for MAO-B, and compound 442 (ZINC000028109109) for AChE. No compound demonstrated docking scores greater than the standard ligand for BACE1 and GSK-3β as shown in (S1). Analysis of the obtained docking results for the promising compounds against the five targets revealed five potential multitarget compounds. (Table 1) shows the docking scores and pharmacokinetic properties of these five compounds with their chemical structures illustrated in (Figure 1).
2.3. Visualization and analysis
Visualization of the intermolecular two-dimensional (2D) and three-dimensional (3D) interactions of the five compounds and the standard ligands with each enzyme are shown in (Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6).
All compounds are shown in green sticks, and the amino acids of the enzyme are shown in gray sticks. Nitrogen and oxygen atoms are shown in blue and red, respectively.
2.4. Free Binding Energy Calculations (MM-GBSA):
The free binding energies of the 5 multitarget compounds are calculated (Table 2).
2.5. Density Functional Theory
3. Discussion
Despite the extensive research in AD pathophysiology, the complete picture of its molecular mechanism is unclear [8]. Therefore, until now no drug can cure, prevent or delay the disease progression [8]. The discovery of several hypotheses for AD pathogenesis and potential therapeutic targets, such as AChE and BuChE, BACE1, GSK-3β and MAO-B, opened the door for novel treatment approaches [8]. As numerous studies have demonstrated the importance of using a multitarget approach for multifactorial diseases such as AD [8], all of the five proteins mentioned above were selected as potential targets in this study [8].
African natural products have always been a rich source of promising candidates that were used in drug discovery of various multifactorial diseases [34]. The Afrodb library, used as the screening library in this study, contains 880 selected compounds from African medicinal plants [33].
The pharmacodynamic activity of these natural products on AD is governed by their pharmacokinetic profile [35]. Since the intended pharmacological activity of these natural compounds is the brain, they must have good predicted permeability across the BBB. The brain permeability of the compounds was measured using (logPS) and (logBB) provided by the pkCSM online server [36]. LogPS is estimated by in situ brain perfusions of a substance administered straight into the carotid artery. At the same time, logBB, is used to quantify blood-brain permeability in living animal models [36]. The pkCSM online server was trained with experimentally measured logBB & logPS values [36]. Furthermore, natural compounds are known for their questionable safety profile [36]. Hence, using the same server, the library was subjected to toxicity prediction using the AMES test for mutagenesis, hERG for cardiotoxicity (by predicting if the compound is likely to inhibit the potassium channels encoded by hERG I & II so leading to QT interval prolongation), and hepatotoxicity [36].
Out of the 880 compounds in the library, 200 passed the cut-off criteria and had predicted logBB > -1, logPS > -3, negative AMES, No hERG I & II inhibition, and no hepatotoxicity. Hence these 200 compounds were predicted to be non-toxic and traverse the CNS and blood-brain barrier easily and were thus included further in the study [36].
The selected 200 compounds were docked against the five enzymes AChE, BuChE, BACE1, GSK-3β and MAO-B using the three glide docking modes HTVS, SP, and XP which differ in accuracy, speed and scoring function [37]. HTVS enables rapid compound screening while minimizing the number of intermediate conformations and reducing final torsion refinement and sampling [37]. Glide SP performs exhaustive sampling and is the recommended balance between speed and accuracy, requiring 10 seconds/compound [37]. XP mode eliminates false positives and penalizes molecules with low binding affinity to the receptor [37]. The molecular docking results were evaluated based on the values of binding affinity and visual inspection of the chemical structure, as well as the interactions with important amino acid residues using the standard ligands of each protein as a control.
For AChE, only one compound (compound 442) had a binding affinity (-12.23 kcal/mol) greater than the standard ligand GNT (binding affinity of -12.11 kcal/mol) (S1). Compound 159 showed a binding affinity value of −7.78 kcal/mol, followed by compound 147 with −7.57 kcal/mol and compound 157 with −7.53 kcal/mol (Table 1). Being a serine hydrolase, AChE creates a tetrahedral intermediate through acid-base reactions with a catalytic triad (Ser203, His447, Glu334 ) [38]. The standard GNT was shown to exhibit van der Waals interaction with His447, pi(π) - pi stacking interaction with Gly121 and π–alkyl interaction with Tyr337 and Trp86. Residues such as Trp86, Glu202, and Tyr337 appear to be involved in the process of hydrophobic interactions [39]. Re-docking of GNT in our study (Figure 2a), revealed that the interactions are favored mainly by the stacking formed against Glu202, where GNT was the Hydrogen bond (H-bond) donor and the oxygen atom of the amino acid Glu202 was the H-bond acceptor. H-bonding was also observed with Gly122 and Glu202. The NH+ group in GNT also acts as H-bond donor with Tyr337 where Aβ joins AChE through hydrophobic stacking interaction which is the same binding site of GNT in AChE. Compound 159 (Figure 2d) exhibited carbon-hydrogen bonding interactions with His447, Tyr337 and Trp86 and conventional H-bonding with Glu202 as H-bond donor, in addition to π–alkyl interaction with Tyr337 and Trp86. Residues such as Trp86 and Tyr337 were shown to be important for the hydrophobic interactions that make a good anchoring effect of the ligand with AChE [39]. Compound 147 (Figure 2b) showed H-bonding with Phe295 and Tyr337 as amino acids being H-bond acceptor. Moreover, it showed higher van der Waals and hydrophobic interactions through the aromatic and furan rings of the ligand with Phe338, Phe295, and Trp236. Finally, Compound 157 (Figure 2c) exhibited only hydrophobic interaction with Trp86 as its single interaction with the enzyme.
Like in AChE, none of the 200 compounds had a greater binding affinity to BACE1 compared to the co-crystallized standard ligand (binding affinity of -7.35 kcal/mol). Compound 27 had the best binding affinity of -6.77 kcal/mol, followed by compound 157 with a binding affinity of -6.24 kcal/mol (Table 1). BACE1 is a member of the aspartyl protease family of enzymes, with its active site containing several subsites; S1' at the center of the active site and contains the two catalytic Aspartate residues; Asp32 and Asp228, S2′ subsite containing mostly hydrophobic and amphipathic residues like Ser35, Val69, Tyr71, Ile126, and Tyr198, S3′ and S4′ exposed to the solvent and consisting of residues Pro70, Thr72, Glu125, Arg128, Arg195, and Trp197, S1 and S3 subsites composing the hydrophobic pocket and containing Leu30, Phe108, Ile110, Ile118, and Trp115, S2 and S4 are the hydrophilic subsites exposed to the solvent involving residues like Lys9, Ser10, Thr72, Gln73, Thr231, Thr232, Arg235, Arg307, and Lys321 [40]. The co-crystalized ligand (Figure 4a) had strong interactions with the two catalytic Aspartate of BACE1, Asp32 and Asp228 via the ligand’s 1,3-thiadiazine ring which acts as H-bond donor and also forms salt bridges. Furthermore, the N3 of the 1,3-thiadiazine ring interacts with Tyr71 through ππ-cation bond, conferring a better anchor for the ligand to the binding site. The methyl-substituent of the pyrazole ring of the standard ligand also occupies the lipophilic cleft of Ser35/Tyr198/Ile126 and is suggested to account for its potency [41]. When it comes to compound 27 (Figure 4b), it was shown to occupy the S1' pocket of the active site of BACE-1 without interacting with the catalytic aspartic acid residues (Asp32 and Asp228). Yet, it still possesses activity as it interacts with other key amino acid residues in the active site [42]. Furthermore, the dihydro-chromene benzene ring in compound 27 interacts with flip-flap Tyr 71 amino acid residue in S2' pocket of the active site via π-π stacking. The phenolic OH of compound 27 forms a H-bond with the carbonyl oxygen of Phe108 of the S1 hydrophobic pocket of the active site leading to further stabilization of the inhibitor binding with BACE1 [40,42]. Moreover, the dimethyl substituents of the heteroaryl ring occupy the hydrophobic cleft in the same manner as the methyl of the pyrazole ring in the reference compound, which suggests that the potency of compound 27 could be similar to the standard ligand [41]. When it comes to the interactions of compound 157 with BACE1 (Figure 4c), the docking results showed that it occupies S1' pocket via a weak H-bond linking the Asp32 oxygen with the phenolic OH of the compound. H-bonding between the non phenolic OH of compound 157 and the Thr231 of the hydrophilic S2 pocket, also contributes to its inhibitory activity on the enzyme [40,43].
Five compounds, compound 27,147, 157, 159, and 696, had greater binding affinity to the third enzyme, BuChE, compared to the co-crystalized ligand (-6.05 kcal/mol). Compound 27 had the highest binding affinity of -8.31 kcal/mol followed by compound 147 and 159 with binding affinities of -7.18 kcal/mol and -7.1 kcal/mol, respectively. Compounds 157 and 696 both had a binding energy of -7.0 kcal/mol (Table 1).
The active site of BuChE consists of a catalytic triad, a choline binding pocket, and an acyl binding pocket, which are immersed in a 20 Å deep gorge [44,45]. Three important residues, Ser198, His438, and Glu325, make up the catalytic triad, Gln119, Val288, Leu286 and Ala328 form the acyl binding pocket [44,45,46]. The residues, Phe329 and Trp332 of BuChE help pull ligands toward the inner gorge [45]. BuChE also has Asp70 and Trp82 residues in the peripheral anionic pocket that also support ligand binding [45].
The major interactions between the re-docked standard ligand tacrine and BuChE are the aromatic stacking with Trp82 (figure.3a), which helps to attract tacrine to the deep gorge. Also, the aromatic nitrogen N7 of the standard forms a H-bond with the main chain carbonyl of His438, which is in the catalytic triad pocket [44,47]. Compound 27 showed very good interactions with BuChE (Figure 3b). It formed 3 H-bonds, acting as a H-bond donor with the carbonyl oxygen of Leu286, which is part of the acyl pocket. The other two H-bonds were formed with a water molecule in the active site. Moreover, multiple important π–π stacking hydrophobic bonds were noticed between compound 27 aromatic system, with the aromatic amino acids His438 (which is part of the catalytic triad), Trp82 (found in the peripheral anionic pocket and supports ligand binding), and Phe329 (which help to attract the compound toward the deep gorge and the catalytic triad) [45]. Compound 147 (Figure 3c) formed T shaped π–π stacking hydrophobic with the aromatic amino acids Phe329 and Trp231 which are key residues and major acyl loop components that help in the interaction. There are three H-bonds between three water molecules inside the active site and the carbonyl group of compound 147 [45]. The interaction of compound 157 (Figure.3d) involved mainly π–π stacking hydrophobic of its aromatic ring with His438 (one of the important aromatic catalytic triad amino acids). While the methoxy hydrogen of the compound formed H-bond (as hydrogen bond donor) with the carbonyl group (as hydrogen bond acceptor) of His438. Compound 159 (Figure 3e) had a very good interaction with the catalytic triad amino acids Ser198 and His438 via H-bonding and π–π stacking hydrophobic , respectively. Additionally, what made the interaction better is the oxygen in compound 159 hydroxyl group of the aromatic ring which acts as hydrogen bond acceptor with one water molecule inside the active site. Finally, Compound 696 (Figure 3f) formed a π–π stacking hydrophobic bond with the aromatic amino acid Phe329, which helps to attract the substrate toward the deep gorge. The hydroxyl hydrogen of the compound 696 acts as a H-bond donor with a water molecule from the active site [45].
The fourth enzyme GSK-3β had a strong binding affinity to its co-crystallized ligand of -9.72 kcal/mol. Only compounds 157, 159, and 696 showed noticeable binding affinities of -8.55, -8.6 and -8.77 kcal/mol, respectively (Table 1). GSK-3β is a member of the serine/threonine kinase family to which ATP is a natural ligand [48]. It has 3 binding sites: an ATP binding site, consisting of Leu132, Tyr134, Val135, Pro136, and Arg141( catalytic domain consist of glycine rich loop and hinge region, which connect C and N terminal domain, involving an ATP-binding site at their interface) [49], an Axin binding site, consisting of Lys85, Asp133, Val135, Lys183, and Asp200, and finally a Priming site, consisting of Arg96, Arg180, Ser203, Lys205, and, Val214 [50]. Studies have shown that amino acids Leu130, Val131, Leu132, Asp133, Tyr134, Val135, Pro136 and Glu137 are essential for ATP competitive inhibitors’ activity, while the amino acids Lys85, Glu97, Arg141, Gln185 and Asp200 are essential for activity and selectivity, and finally the Asp133 and Val135 are crucial for all affinity but not selectivity [48]. The co-crystallized ligand with GSK-3β is a selective indazole-based GSK-3β inhibitor with high hERG affinity. The indazole carboxamide of the ligand form triad hydrogen bonds, 2 with Asp133 and the third with Val135 in the hinge region, which is one of GSK-3β domains.While the piperidine moiety of the ligand is oriented to Arg141, and the two fluorine atoms directed to catalytic Lys85 (Figure 5a) [51]. The docking result of Compound 696 (Figure 5d), shows that the hydroxyl group attached to the Chromone nucleus forms H-bonds with Val135 and Asp200. Also it interacts with Leu132 through bad interactions. Despite lacking many crucial bonds compared to the standard ligand, compound 696 has multiple non-covalent interactions with GSK-3β, indicating that it could play an important role as a GSK-3β inhibitor [48]. As for compound 159, also a chromenone derivative, molecular docking (Figure 5c) revealed that it has hydrophobic interactions with Val70, Ala83, Cys199, Leu132, Tyr134 and Leu188. Moreover, the chromone moiety forms three hydrogen bonds, 2 with Val135 and the third Ile62. Although Ile62 is usually involved in hydrophobic interactions, some compounds like compound cpd22 (Which is a GSK-3β inhibitor) form H-bonds with Ile62 [52]. Compounds 159 forms a fourth H-bond with Asp200 while also interacting with the catalytic amino acid Lys85 through bad interaction (Lys85, Glu97 are highly conserved catalytic residues that enhance ATP interactions) [53]. Again, all of the observed interactions suggest that compound 159 could be a potential GSK-3β inhibitor [48,54]. Compound 157 on the other hand (Figure 5b), acted as a H-bond acceptor as well as a donor with Val135 forming two H-bonds. It also formed a third H-bond with Gln185 and it showed bad interaction with Leu132 .The 3 hydrogen bonds contribute greatly to its binding affinity [55].
The fifth and final enzyme MAO-B showed a binding affinity only to compound 27 with a value of -12.55 kcal/mol which is greater than the binding energy of the co-crystalized ligand of -11.6 kcal/mol (Table 1). MAO-B is a flavoprotein that contains 520 amino acids forming two cavity structures, an entry cavity and reactive site cavity for substrate binding [56]. Studies showed that the catalytic activity of MAO-B is donated by amino acids Lys296, Trp388 (which may play an important role in the attachment of FAD to MAO-B non-covalently) and Tyr398, Tyr435 (which form an aromatic sandwich within the substrate binding site) [57]. MAO-B’s catalytic site also contains an Ile199 residue which can take two conformations, closed or open, depending on the property of the binding ligand thus playing an important role in determining the plasticity of the catalytic site [57]. The docking results of the standard ligand, a coumarin derivative, (Figure 6a) showed that its hydroxymethyl group at C4 form an aromatic sandwich with Tyr435 . It also shows hydrophilic interactions through water bridges of the flavin ring of FAD group and the coumarin lactone group of the standard both with the amino acid Lys296. The bis-N-benzylamine moiety of the standard also assumes a hook conformation when it is wedged into the pocket of the entry cavity leading to structural adjustments and conformational changes in the amino acids lining the active site [58]. The docking results of compound 27 (Figure 6b) showed that it forms π-π and hydrophobic interactions with Tyr326 ; This interaction holds the phenyl system of compound 27 in the entrance cavity of MAO-B thus probably contributing to the increase in the binding affinity of compound 27 compared to the standard.
When comparing the docking scores for the top compounds for each of the five enzymes, it could be seen that there are five potential multitarget compounds which were 27, 147, 157, 159, and 696 (Figure 1). Of these, compounds 27 showed triple-target inhibitory activity against BuChE, BACE1 and MAO-B. Compound 147 showed dual-target inhibitory activity against AChE and BACE1. Compound 157 showed triple-target inhibitory activity against BuChE, GSK-3β and BACE1. Finally, compounds 159 and 696 both showed dual-target inhibitory activity against BuChE and GSK-3β.
Regarding the pharmacokinetic profiles of these five candidates, all were predicted to be non-toxic. Compound 147 was predicted to be the most permeable across the BBB, followed by compounds 696 and 27, while compounds 159 and 157 had the least BBB permeability (Table 1).
An important post-docking investigation done in this study was the free binding energy calculations of the five potential multitarget compounds with their respective target proteins compared to the standard ligands using MM-GBSA. These calculations are crucial for ensuring the accuracy of the docking results to avoid any false positive or false negative results. As the negative value of free binding energy increases, the stability of the ligand-protein complex will increase and thus the ability to exert pharmacological action [59]. In these calculations the OPLS3e force field is usually used for energy minimization [59]. The docking results indicated that AChE would potentially be inhibited by compounds 147, 157, and 159. Yet these compounds had positive MM-GBSA binding affinities with AChE, suggesting their unfavorable binding to the enzyme. The five potential multitarget compounds showed comparable docking scores with BuChE and all had negative MM-GBSA binding affinity values to the enzyme. CCompounds 147 and 157 had the highest MM-GBSA binding affinities hence the most favorable binding, followed by compounds 27 and 159 and finally compound 696 had the least affinity with the least favorable binding. As for GSK-3β, the results indicate comparable MM-GBSA scores and docking scores for compounds 157, 159, and 696, with compound 159 showing slightly stronger binding affinity compared to compounds 157 and 696. The MM-GBSA results of MAO-B suggest its highly favorable binding of compound 27. Although docking scores of compound 27 and compound 157 with BACE1 revealed comparable affinities, MM-GBSA results indicate a much stronger binding of compound 157 with the enzyme, as shown in (Table 2).
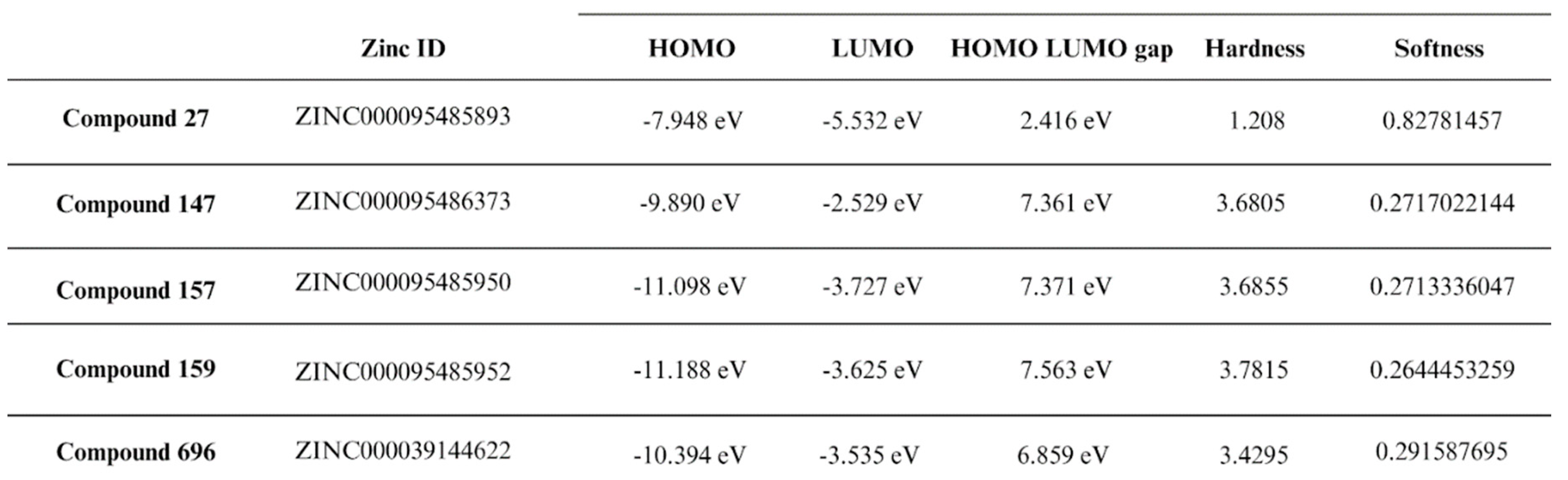
The final step in our study involved performing DFT analysis for the five potential multitarget candidates. In this analysis, the HOMO energy describes the electron-donating ability of the compound or the stable state. In contrast, LUMO energy describes the compound’s ability for electron acceptance or the excited state. HUMO-LUMO gap is an important parameter in determining the compound's tendency to undergo electronic transitions and participate in chemical reactions, a larger energy gap indicates that the compound is less likely to be reactive [60,61]. Compound 159 is thought to be the most stable and unreactive, with compounds 157 and 147 coming next Compound 696 has an intermediate reactivity compared to the other four compounds, while compound 27 shows a smaller energy gap, hence instability and high tendency to undergo electronic transitions. Beside the HUMO-LUMO gap, chemical hardness (η) can also be used to indicate chemical stability. Hard molecules have a wide energy gap, and soft molecules have a narrow energy gap. Soft molecules require less energy for excitation than hard molecules, making them more polarisable [62]. As can be seen from the supplied data (Table 3), compounds 157, 159, and 147 have comparable chemical hardness values, indicating that they have almost identical stability and resistance to chemical reactions. Compound 696 on the other hand, has a somewhat lower chemical hardness value than the other compounds, indicating that it is more reactive. Compound 27 has a significantly lower chemical hardness and higher softness value thus would potentially conduct chemical reactions more readily than the other compounds.
Considering all obtained data, the following generalizations could be made for the five potential multitarget candidates and their interactions with the five selected targets. Compound 27 has a good pharmacokinetic and toxicity profile and good binding affinities to BuChE, BACE1 and MAO-B. Yet, its high chemical instability revealed by DFT made it unqualified for further investigations as a potential triple target compound for AD. Compound 147 also had a good pharmacokinetic and toxicity profile, being the most BBB and CNS permeable, and good chemical stability according to the DFT analysis. Nevertheless, its MM-GBSA revealed unfavorable binding towards AChE, making it potentially acting as a single-target molecule against BuChE. Compound 157 is suggested to be a potential multitarget (triple-target) molecule, with highly favourable binding towards BuChE, BACE1 and GSK-3β, good pharmacokinetic and toxicity profile, and good chemical stability. Finally, compounds 159 and 696 are also considered potential multitarget (dual-target) molecules, with highly favorable binding with BuChE and GSK-3β, good pharmacokinetic and toxicity profiles, and good chemical stability.
Compounds 157, 159, and 696 are all phytochemically classified as chromones. Studies have shown that chromones bearing γ-benzopyrone nucleus have beneficial effects in AD. The core fragment of different flavonoids containing the chromone ring is responsible for inhibiting cholinesterase and Aβ aggregation, has neuroprotective, scavenger and anti-inflammatory activities [63]. Another study on chromone-2-carboxamido-alkyl benzyl amine derivatives has revealed that chromone rings play an important role in treating AD as it shows an antioxidant, anti-Aβ aggregation and neuroprotection activity [63]. Again, pyridinium moiety linked to chromone is crucial as a ChEs inhibitor [63]. The information provided by these studies indicate that the chromone moiety, and thus the compounds 157, 159, and 696 show promising potential in treating AD [63].
4. Materials and Methods
4.1. Data sources
African natural compounds were obtained from the Afrodb library, which contains 880 selected compounds from African medicinal plants. The library was downloaded from the Zinc database. (https://zinc.docking.org/)
4.2. Pharmacokinetics and toxicity prediction:
The pkCSM online server (https://biosig.lab.uq.edu.au/pkcs) was used to predict the pharmacokinetic profile of African natural compounds. The results were viewed and filtered according to Blood Brain Barrier (BBB) permeability (LogBB > -1), CNS permeability (LogPS > -3), hepatotoxicity, mutagenicity and cardiotoxicity.
4.3. In silico molecular docking
4.3.1. Protein and ligand preparation:
The crystallographic structures of the five proteins AChE, BuChE, BACE1, GSK-3β and MAO-B, with Protein Data Bank (PDB) identity (ID) 4EY6, 4BDS, 4XXS, 6TCU and 7P4F respectively, with their co-crystallized small molecule inhibitors (ligands), were obtained from National Center for Biotechnology Information (NCBI) - The structures accessed on 25 September 2022. (https://www.ncbi.nlm.nih.gov/structure/?term=)
Prior to the analysis, the proteins were prepared by the Protein Preparation Wizard tool. Their problems were first assessed and listed as follows: Problems with overlapping atoms, alternate positions, missing atoms, and atom types. These issues were resolved by the addition of hydrogen atoms, positional changes, minimisation, and filling in of missing side chains. Then water molecules beyond 0.5Å were deleted, and the proteins were preprocessed.
Ligands that passed pharmacokinetic analysis were prepared using the ligPrep tool; under the OPLS3e (Optimized Potentials for Liquid Simulations) force field. The library was exposed to energy minimisation. The software was adjusted to generate at most 32 conformers for each ligand, and states were generated at 7 ± 2 pH. Protein reliability reports were generated to ensure the validity of the proteins for further processing.
4.3.2. Molecular docking:
The grid boxes of the five proteins where the molecular docking tooks place were generated using Maestro's Receptor Grid Generation tool. As the protein structures were obtained with co-crystallized ligands, the grid box was generated using the centroid of workspace ligands. Default settings of the van der Waals radius scaling were used; Scaling factor = 1 and partial charge cutoff = 0.25. Then using the ligand docking tool, molecular docking proceeded by firstly screening the 200 filtered compounds of the prepared ligands using High-Throughput Virtual Screening mode (HTVS), then the resulting compounds were further prepared with the generation of only one conformer at most, and then Standard Precision (SP) and Extra Precision (XP) were conducted with ligand preparation step in between.
The same docking process was followed for each standard ligand against its enzyme, and the binding energy readings (glide score) were used as cut-off points for the library compounds. Galantamine (GNT), was used as the standard ligand for the enzyme AChE (PDB ID: 4EY6) as it has high inhibitory potential against the enzyme [39]. While the co-crystalized ligands were used as standard for BACE1 (PDB: 4XXS), BuChE (PDB: 4BDS), GSK-3β (PDB: 6TCU), and MAO-B (PDB:7P4F).
4.4. Free Binding Energy Calculations (MM-GBSA) And Density Functional Theory (DFT):
To determine the free binding energies, MM-GBSA calculations were done by Schrödinger’s Maestro interface [55], [Schrödinger Release 2023-3: Maestro, Schrödinger, LLC, New York, NY, 2023.] using the MM-GBSA panel of the prime module for the compounds with the highest XP docking score. The enzyme was included in the workspace, and the compounds for estimation were selected from the project table, setting (VSGB) as the solvation model and OPLS3e force field.
Chem3D version 22.2.0 software was utilized to perform DFT analysis on the five mutual compounds. First, structures were exported to the workspace; then from the extended Hückel bar of the calculation menu, the calculate surface option was selected. Then in the surface menu, the molecular orbital bar was chosen to obtain Highest Occupied Molecular Orbital (HOMO) and the Lower Unoccupied Molecular Orbital (LUMO) values, which were then used to calculate Hardness (η) and Softness (S), using the following equations (equations(1-2)): [61].
η= [εLUMO−εHOMO] / 2
S =1/η
5. Conclusions
In conclusion, our study intended to identify new promising multitarget compounds from African medicinal plants that are capable of inhibiting two or more enzymes involved in the pathophysiology of Alzheimer’s Disease. Pharmacokinetics and toxicity predictions, molecular docking, MM-GBSA binding affinities, and DFT studies were utilized to screen a library of 880 African natural compounds for their multitarget activities. The above-mentioned investigations resulted in recognising compound 157 as a promising triple-target molecule and compounds 159 and 696 as promising dual-target molecules with good chemical stability, good pharmacokinetic and toxicity profiles, and high docking and MM-GBSA scores together with favourable interactions that are supported by the previous studies.
Based on these findings, we recommend further experimental investigations for compounds 157 as triple-target inhibitor of BuChE, BACE1 and GSK-3β, and compounds 159 and 696 as dual-target inhibitors of BuChE and GSK-3β. These investigations should first include In silico Molecular dynamic simulation in addition to in vitro and in vivo testing to confirm the activity of the selected compounds. We are ultimately hoping that these compounds could be developed as effective and safe drugs for treating Alzheimer’s Disease.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Docking scores and pharmacokinetic properties of the 200 compounds that passed the Pharmacokinetic filters; Table S2: MM-GBSA scores of the top 5 compounds for each enzyme and the 32 ones of BuChE.
Author Contributions
Methodology, E.M.O.A.I.; Validation, A.A.A., A.A.M., L.I.T., R.R.B., and T.A.H.; Formal Analysis, A.A.A. , A.A.M., L.I.T., R.R.B. and T.A.H.; Investigation, A.A.A., A.A.M., L.I.T., R.R.B., and T.A.H.; Data Curation, A.A.A. , A.A.M., L.I.T., R.R.B. and T.A.H. ; Writing – Original Draft Preparation, A.A.A., A.A.M., L.I.T., R.R.B. and T.A.H.; Writing – Review & Editing, E.M.O.A.I. , L.Y.M.E., and S.W.S.; Visualization, A.A.A., A.A.M., L.I.T., R.R.B. and T.A.H.; Supervision, E.M.O.A.I., S.W.S. and L.Y.M.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data are within the paper.
Acknowledgments
The authors extend their appreciation to Sabah Ahmed, Mohammed Bader Almaerif, and Mahir Mohammed for their help and support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- What Are the Signs of Alzheimer’s Disease? Available online: https://www.nia.nih.gov/health/what-are-signs-alzheimers-disease (accessed on 29 September 2023).
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 29 September 2023).
- Alzheimer’s: Facts, Figures & Stats. Available online: https://www.brightfocus.org/alzheimers/article/alzheimers-disease-facts-figures (accessed on 29 September 2023).
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Gong, C.-X.; Dai, C.-L.; Liu, F.; Iqbal, K. Multi-Targets: An Unconventional Drug Development Strategy for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 837649. [Google Scholar] [CrossRef]
- Gong, C.-X.; Dai, C.-L.; Liu, F.; Iqbal, K. Multi-Targets: An Unconventional Drug Development Strategy for Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 837649. [Google Scholar] [CrossRef]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 Inhibitors: Current Status and Future Directions in Treating Alzheimer’s Disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jasiecki, J.; Targońska, M.; Wasąg, B. The Role of Butyrylcholinesterase and Iron in the Regulation of Cholinergic Network and Cognitive Dysfunction in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2033. [Google Scholar] [CrossRef]
- Highly Potent and Selective Aryl-1,2,3-Triazolyl Benzylpiperidine Inhibitors toward Butyrylcholinesterase in Alzheimer’s Disease. Bioorg. Med. Chem. 2019, 27, 931–943. [CrossRef]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10. [Google Scholar] [CrossRef]
- Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1560. [Google Scholar] [CrossRef]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Ferrucci, L.; Kapogiannis, D. Effects of Monoclonal Antibodies against Amyloid-β on Clinical and Biomarker Outcomes and Adverse Event Risks: A Systematic Review and Meta-Analysis of Phase III RCTs in Alzheimer’s Disease. Ageing Res. Rev. 2021, 68, 101339. [Google Scholar] [CrossRef]
- Cheong, S.L.; Tiew, J.K.; Fong, Y.H.; Leong, H.W.; Chan, Y.M.; Chan, Z.L.; Kong, E.W.J. Current Pharmacotherapy and Multi-Target Approaches for Alzheimer’s Disease. Pharmaceuticals 2022, 15, 1560. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer’s Disease: Updated Multi-Targets Therapeutics Are in Clinical and in Progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef]
- Vrabec, R.; Blunden, G.; Cahlíková, L. Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4399. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7. [Google Scholar] [CrossRef]
- Tettevi, E.J.; Maina, M.; Simpong, D.L.; Osei-Atweneboana, M.Y.; Ocloo, A. A Review of African Medicinal Plants and Functional Foods for the Management of Alzheimer’s Disease-Related Phenotypes, Treatment of HSV-1 Infection And/or Improvement of Gut Microbiota. Journal of Evidence-Based Integrative Medicine 2022. [CrossRef]
- Tettevi, E.J.; Maina, M.; Simpong, D.L.; Osei-Atweneboana, M.Y.; Ocloo, A. A Review of African Medicinal Plants and Functional Foods for the Management of Alzheimer’s Disease-Related Phenotypes, Treatment of HSV-1 Infection And/or Improvement of Gut Microbiota. Journal of Evidence-Based Integrative Medicine 2022. [CrossRef]
- Tettevi, E.J.; Maina, M.; Simpong, D.L.; Osei-Atweneboana, M.Y.; Ocloo, A. A Review of African Medicinal Plants and Functional Foods for the Management of Alzheimer’s Disease-Related Phenotypes, Treatment of HSV-1 Infection And/or Improvement of Gut Microbiota. Journal of Evidence-Based Integrative Medicine 2022. [CrossRef]
- Tettevi, E.J.; Maina, M.; Simpong, D.L.; Osei-Atweneboana, M.Y.; Ocloo, A. A Review of African Medicinal Plants and Functional Foods for the Management of Alzheimer’s Disease-Related Phenotypes, Treatment of HSV-1 Infection And/or Improvement of Gut Microbiota. Journal of Evidence-Based Integrative Medicine 2022. [CrossRef]
- Tettevi, E.J.; Maina, M.; Simpong, D.L.; Osei-Atweneboana, M.Y.; Ocloo, A. A Review of African Medicinal Plants and Functional Foods for the Management of Alzheimer’s Disease-Related Phenotypes, Treatment of HSV-1 Infection And/or Improvement of Gut Microbiota. Journal of Evidence-Based Integrative Medicine 2022. [CrossRef]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A Select Highly Potent and Diverse Natural Product Library from African Medicinal Plants. PLoS One 2013, 8, e78085. [Google Scholar] [CrossRef] [PubMed]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [PubMed]
- Website Available online: ADMET. [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. 2015. [CrossRef]
- Docking and Scoring. Available online: https://www.schrodinger.com/science-articles/docking-and-scoring (accessed on 20 August 2023).
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls [Internet]; StatPearls Publishing, 2023.
- Silva, L.B.; Ferreira, E.F.B.; Maryam; Espejo-Román, J.M.; Costa, G.V.; Cruz, J.V.; Kimani, N.M.; Costa, J.S.; Bittencourt, J.A.H.M.; Cruz, J.N.; et al. Galantamine Based Novel Acetylcholinesterase Enzyme Inhibitors: A Molecular Modeling Design Approach. Molecules 2023, 28. [CrossRef]
- Mouchlis, V.D.; Melagraki, G.; Zacharia, L.C.; Afantitis, A. Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Brodney, M.A.; Beck, E.M.; Butler, C.R.; Barreiro, G.; Johnson, E.F.; Riddell, D.; Parris, K.; Nolan, C.E.; Fan, Y.; Atchison, K.; et al. Utilizing Structures of CYP2D6 and BACE1 Complexes to Reduce Risk of Drug-Drug Interactions with a Novel Series of Centrally Efficacious BACE1 Inhibitors. J. Med. Chem. 2015, 58, 3223–3252. [Google Scholar] [CrossRef]
- Rombouts, F.J.R.; Alexander, R.; Cleiren, E.; De Groot, A.; Carpentier, M.; Dijkmans, J.; Fierens, K.; Masure, S.; Moechars, D.; Palomino-Schätzlein, M.; et al. Fragment Binding to β-Secretase 1 without Catalytic Aspartate Interactions Identified via Orthogonal Screening Approaches. ACS Omega 2017, 2, 685–697. [Google Scholar] [CrossRef]
- Liu, S.; Fu, R.; Cheng, X.; Chen, S.-P.; Zhou, L.-H. Exploring the Binding of BACE-1 Inhibitors Using Comparative Binding Energy Analysis (COMBINE). BMC Struct. Biol. 2012, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Jamal, Q.M.S.; Alharbi, A.H. Molecular Docking and Dynamics Studies of Cigarette Smoke Carcinogens Interacting with Acetylcholinesterase and Butyrylcholinesterase Enzymes of the Central Nervous System. Environ. Sci. Pollut. Res. 2021, 29, 61972–61992. [Google Scholar] [CrossRef]
- Jamal, Q.M.S.; Khan, M.I.; Alharbi, A.H.; Ahmad, V.; Yadav, B.S. Identification of Natural Compounds of the Apple as Inhibitors against Cholinesterase for the Treatment of Alzheimer’s Disease: An In Silico Molecular Docking Simulation and ADMET Study. Nutrients 2023, 15, 1579. [Google Scholar] [CrossRef]
- Ha, Z.Y.; Mathew, S.; Yeong, K.Y. Butyrylcholinesterase: A Multifaceted Pharmacological Target and Tool. Curr. Protein Pept. Sci. 2020, 21, 99–109. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal Structures of Human Cholinesterases in Complex with Huprine W and Tacrine: Elements of Specificity for Anti-Alzheimer’s Drugs Targeting Acetyl- and Butyryl-Cholinesterase. Biochem. J 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer’s Disease Models. International Journal of Alzheimer’s Disease 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Phukan, S.; Babu, V.S.; Kannoji, A.; Hariharan, R.; Balaji, V.N. GSK3beta: Role in Therapeutic Landscape and Development of Modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, G.J.; Zhang, Y.G.; Mu, W.X.; Zhang, J.Y. [Total residue analysis of Amitraz in the rabbit tissue by gas chromatography]. Hua Xi Yi Ke Da Xue Xue Bao 1988, 19, 419–421. [Google Scholar] [PubMed]
- Zhu, J.; Wu, Y.; Wang, M.; Li, K.; Xu, L.; Chen, Y.; Cai, Y.; Jin, J. Integrating Machine Learning-Based Virtual Screening with Multiple Protein Structures and Bio-Assay Evaluation for Discovery of Novel GSK3β Inhibitors. Front. Pharmacol. 2020, 11, 566058. [Google Scholar] [CrossRef] [PubMed]
- Rippin, I.; Khazanov, N.; Ben Joseph, S.; Kudinov, T.; Berent, E.; Arciniegas Ruiz, S.M.; Marciano, D.; Levy, L.; Gruzman, A.; Senderowitz, H.; et al. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. Int. J. Mol. Sci. 2020, 21, 8709. [Google Scholar] [CrossRef]
- Balboni, B.; Tripathi, S.K.; Veronesi, M.; Russo, D.; Penna, I.; Giabbai, B.; Bandiera, T.; Storici, P.; Girotto, S.; Cavalli, A. Identification of Novel GSK-3β Hits Using Competitive Biophysical Assays. Int. J. Mol. Sci. 2022, 23, 3856. [Google Scholar] [CrossRef]
- Xie, H.; Wen, H.; Zhang, D.; Liu, L.; Liu, B.; Liu, Q.; Jin, Q.; Ke, K.; Hu, M.; Chen, X. Designing of Dual Inhibitors for GSK-3 β and CDK5: Virtual Screening and in Vitro Biological Activities Study. Oncotarget 2017, 8, 18118–18128. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 209773. [Google Scholar] [CrossRef] [PubMed]
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of Conserved Active Site Residues in Monoamine Oxidase A and B and Their Three-Dimensional Molecular Modeling. J. Biol. Chem. 2002, 277, 17209. [Google Scholar] [CrossRef]
- Ekström, F.; Gottinger, A.; Forsgren, N.; Catto, M.; Iacovino, L.G.; Pisani, L.; Binda, C. Dual Reversible Coumarin Inhibitors Mutually Bound to Monoamine Oxidase B and Acetylcholinesterase Crystal Structures. ACS Med. Chem. Lett. 2022, 13, 499–506. [Google Scholar] [CrossRef]
- Targeting SARS-CoV-2 Main Protease by Teicoplanin: A Mechanistic Insight by Docking, MM/GBSA and Molecular Dynamics Simulation. J. Mol. Struct. 2021, 1246, 131124. [CrossRef] [PubMed]
- El-Lateef, H.M.A.; Khalaf, M.M.; Shehata, M.R.; Abu-Dief, A.M. Fabrication, DFT Calculation, and Molecular Docking of Two Fe(III) Imine Chelates as Anti-COVID-19 and Pharmaceutical Drug Candidate. Int. J. Mol. Sci. 2022, 23, 3994. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.M.; Maowa, J.; Alam, A.; Dey, S.; Hosen, A.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. In Silico DFT Study, Molecular Docking, and ADMET Predictions of Cytidine Analogs with Antimicrobial and Anticancer Properties. In Silico Pharmacology 2021, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Adejumo, T.T.; Tzouras, N.V.; Zorba, L.P.; Radanović, D.; Pevec, A.; Grubišić, S.; Mitić, D.; Anđelković, K.K.; Vougioukalakis, G.C.; Čobeljić, B.; et al. Synthesis, Characterization, Catalytic Activity, and DFT Calculations of Zn(II) Hydrazone Complexes. Molecules 2020, 25, 4043. [Google Scholar] [CrossRef] [PubMed]
- Chromone Derivatives Bearing Pyridinium Moiety as Multi-Target-Directed Ligands against Alzheimer’s Disease. Bioorg. Chem. 2021, 110, 104750. [CrossRef]
- [Schrödinger Release 2023-3: Maestro, Schrödinger, LLC, New York, NY, 2023.].
Figure 1.
The chemical structures of the five compounds with potential multitarget activity.
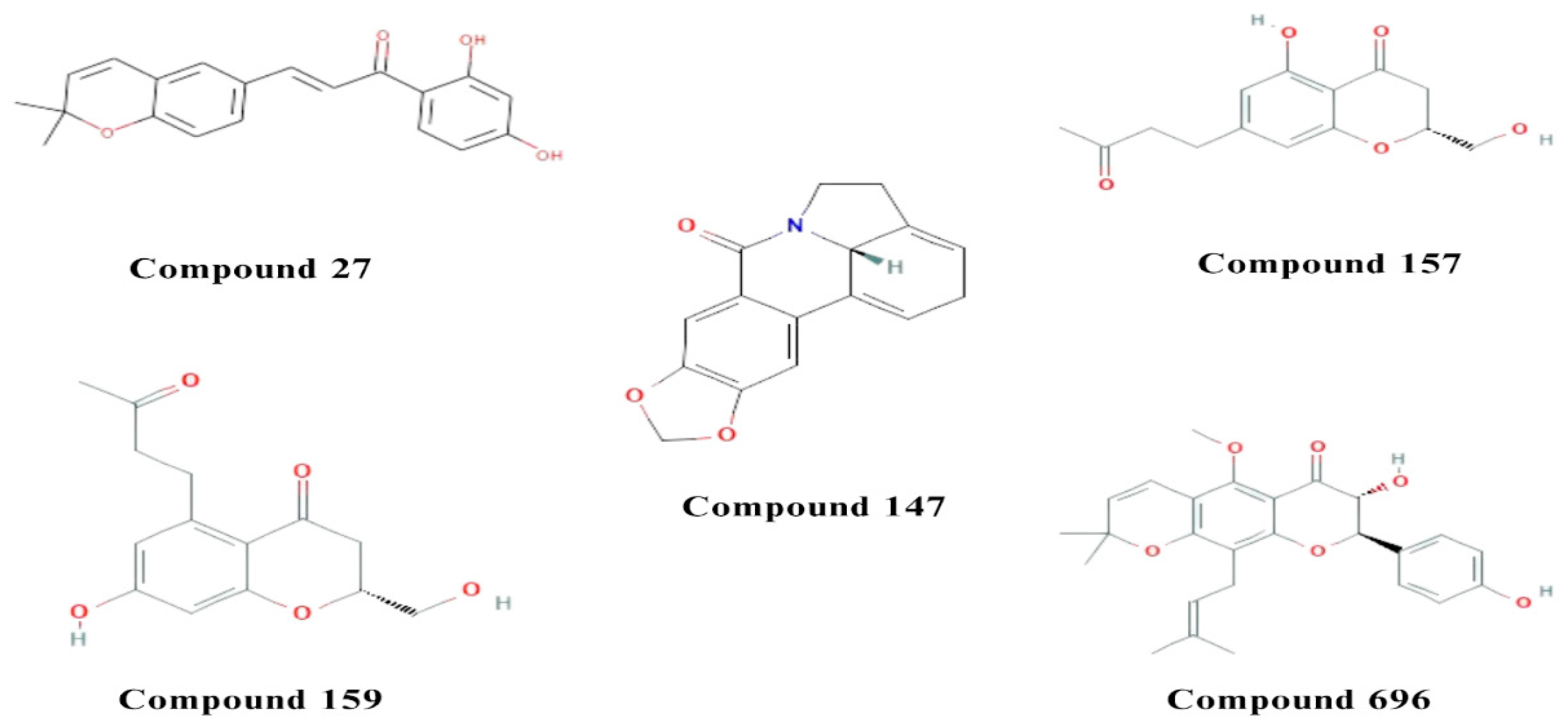
Figure 2.
2D and 3D interactions of the standard and the 3 multitarget compounds in complex with AChE (PDB ID: 4EY6) using XP docking mode of Glide software. (a) Standard Ligand (GNT) ; (b) compound 147; (c) compound 157; (d) compound 159.
Figure 2.
2D and 3D interactions of the standard and the 3 multitarget compounds in complex with AChE (PDB ID: 4EY6) using XP docking mode of Glide software. (a) Standard Ligand (GNT) ; (b) compound 147; (c) compound 157; (d) compound 159.
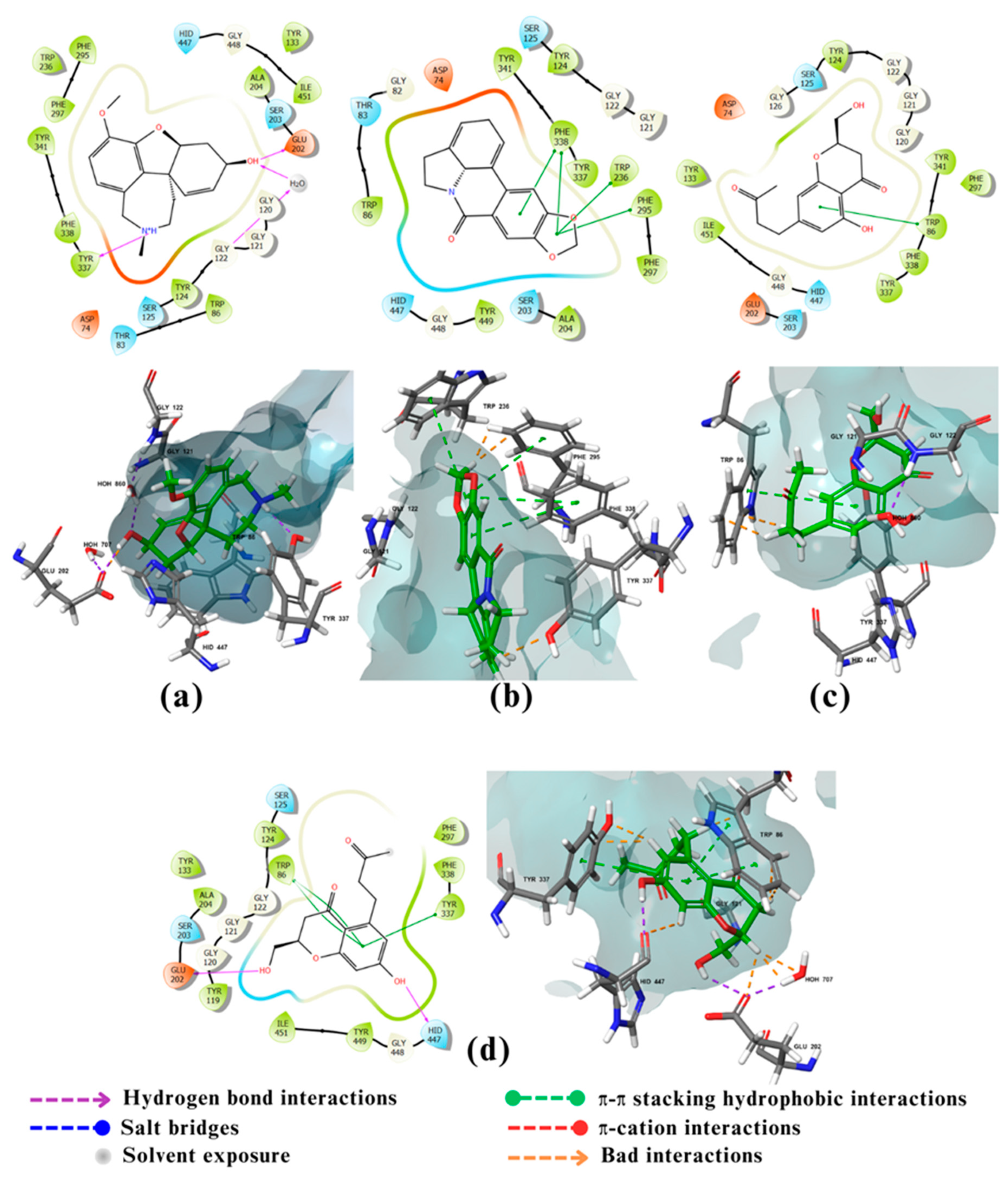
Figure 3.
2D and 3D interactions of the standard and the 5 multitarget compounds in complex with BuChE (PDB ID: 4BDS) using XP docking mode of Glide software. (a) standard ligand; (b) compound 27; (c) compound 147; (d) compound 157; (e) compound 159; and (f) compound 696.
Figure 3.
2D and 3D interactions of the standard and the 5 multitarget compounds in complex with BuChE (PDB ID: 4BDS) using XP docking mode of Glide software. (a) standard ligand; (b) compound 27; (c) compound 147; (d) compound 157; (e) compound 159; and (f) compound 696.
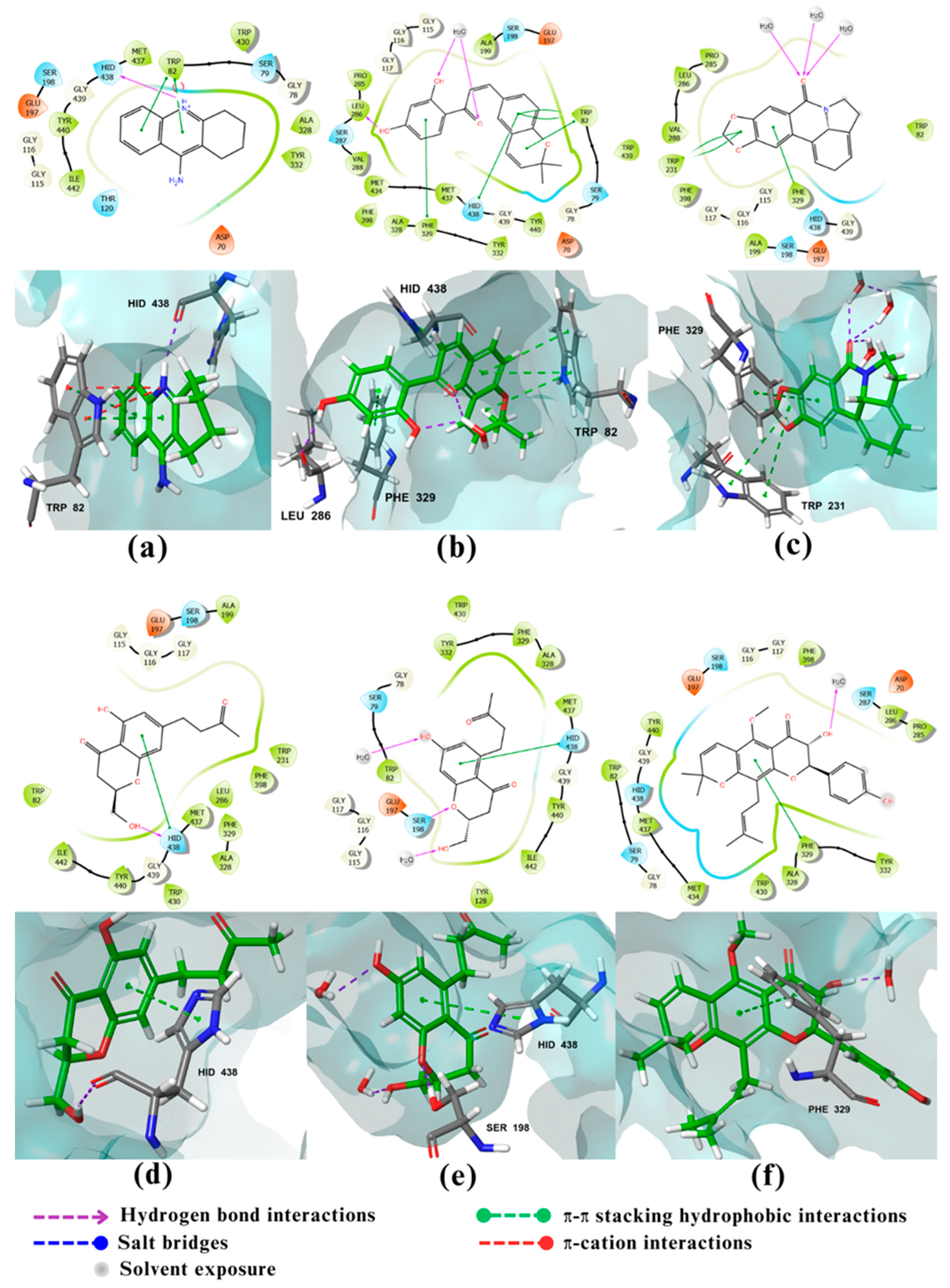
Figure 4.
2D and 3D interactions of the standard and the 2 multitarget compounds in complex with BACE1 (PDB ID: 4XXS) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 27; (c) compound 157. Fluorine and sulfur atoms are presented in neon green and sulfur green, respectively.
Figure 4.
2D and 3D interactions of the standard and the 2 multitarget compounds in complex with BACE1 (PDB ID: 4XXS) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 27; (c) compound 157. Fluorine and sulfur atoms are presented in neon green and sulfur green, respectively.
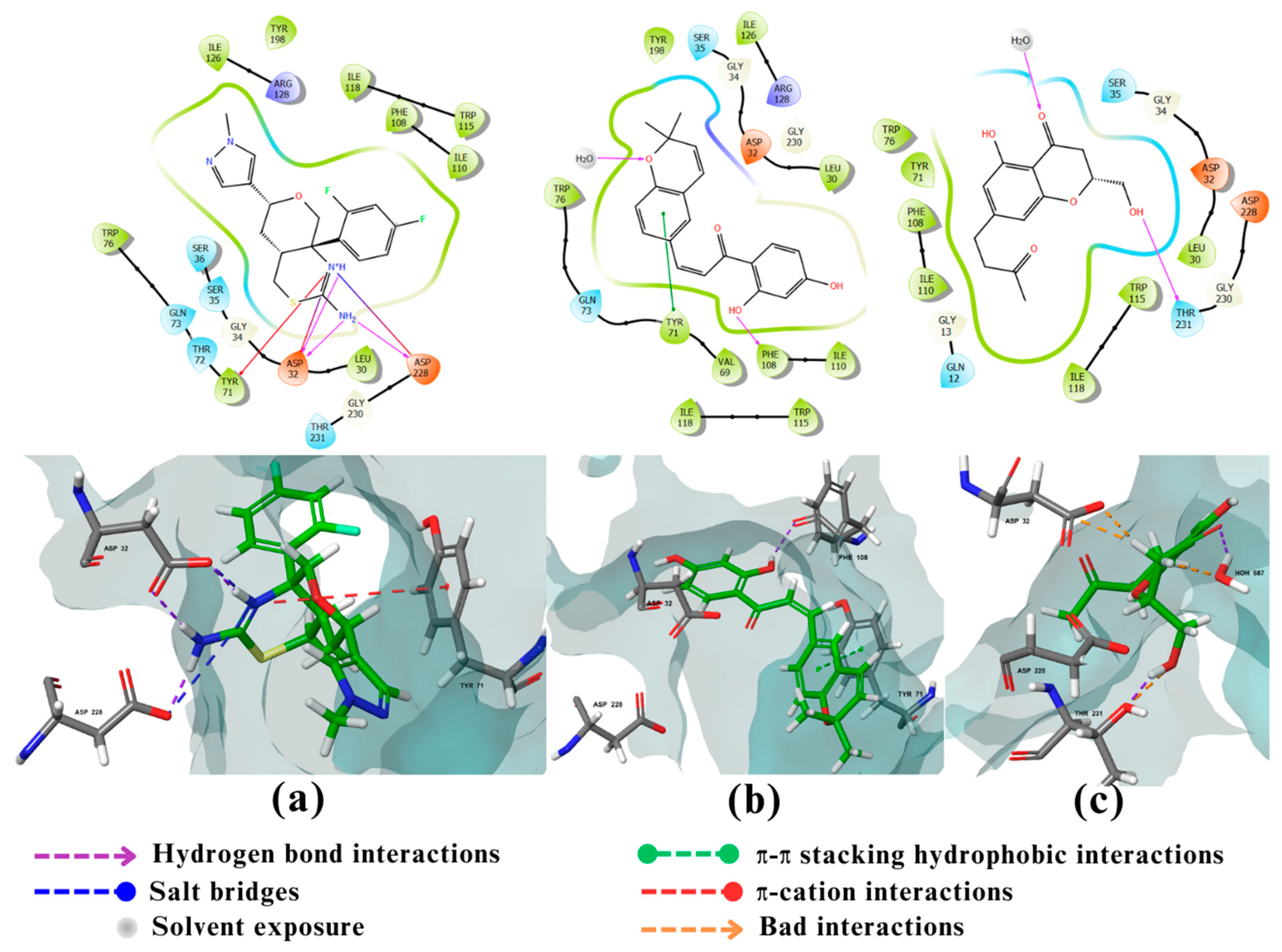
Figure 5.
2D and 3D interactions of the standard and the 3 multitarget compounds in complex with GSK-3β (PDB ID: 6TCU) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 157; (c) compound 159; (d) compound 696. Fluorine atoms are presented in neon green.
Figure 5.
2D and 3D interactions of the standard and the 3 multitarget compounds in complex with GSK-3β (PDB ID: 6TCU) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 157; (c) compound 159; (d) compound 696. Fluorine atoms are presented in neon green.
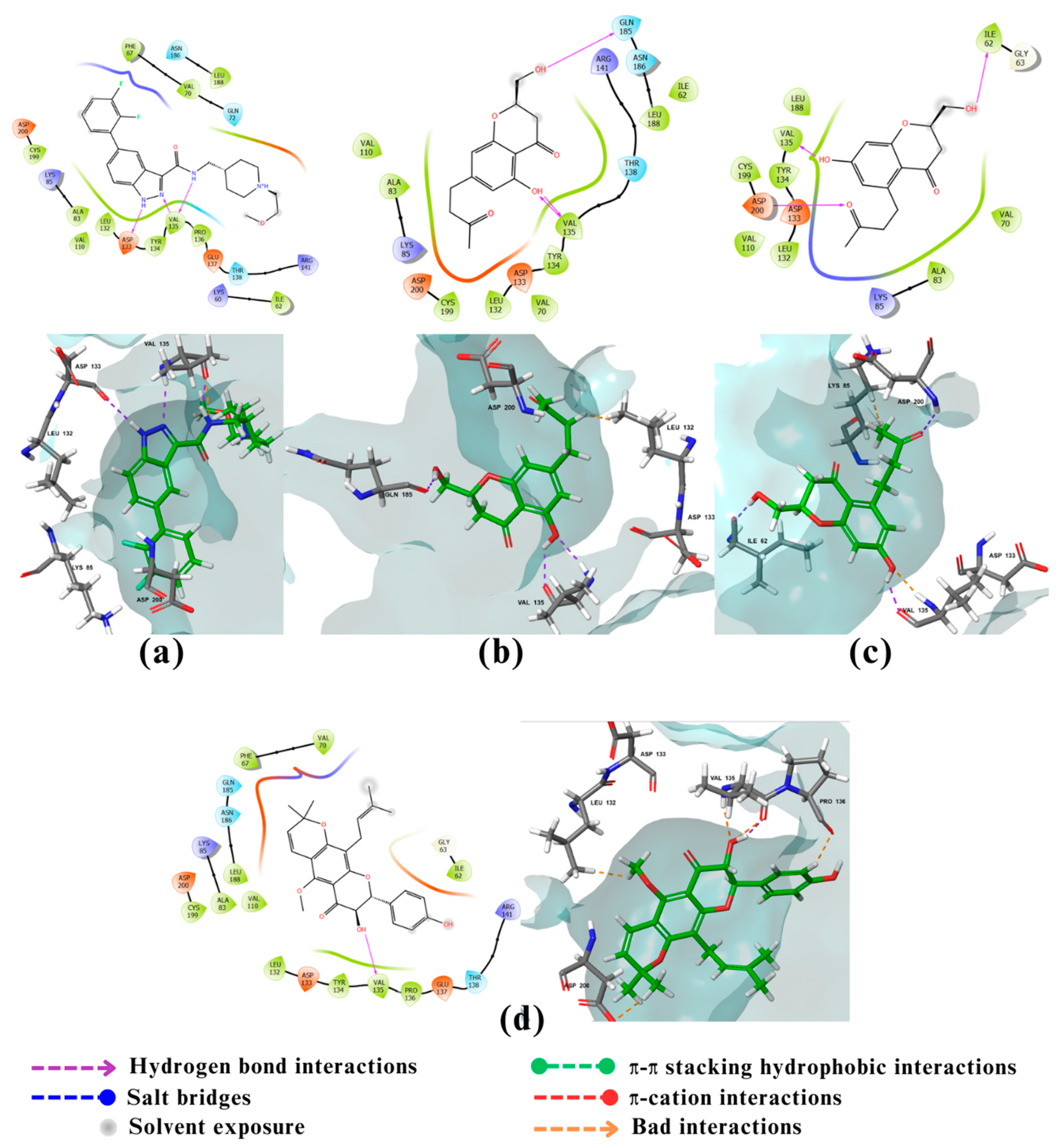
Figure 6.
2D and 3D interactions of the standard and the 2 multitarget compounds in complex with MAO-B (PDB ID: 7P4F) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 27. FAD 601, 5IK602, UNK0 refer to flavin adenine dinucleotide cofactor (FAD), standard compound, and compound 27, respectively. Phosphorus atom presented in pink.
Figure 6.
2D and 3D interactions of the standard and the 2 multitarget compounds in complex with MAO-B (PDB ID: 7P4F) using XP docking mode of Glide software. (a) Standard ligand; (b) compound 27. FAD 601, 5IK602, UNK0 refer to flavin adenine dinucleotide cofactor (FAD), standard compound, and compound 27, respectively. Phosphorus atom presented in pink.
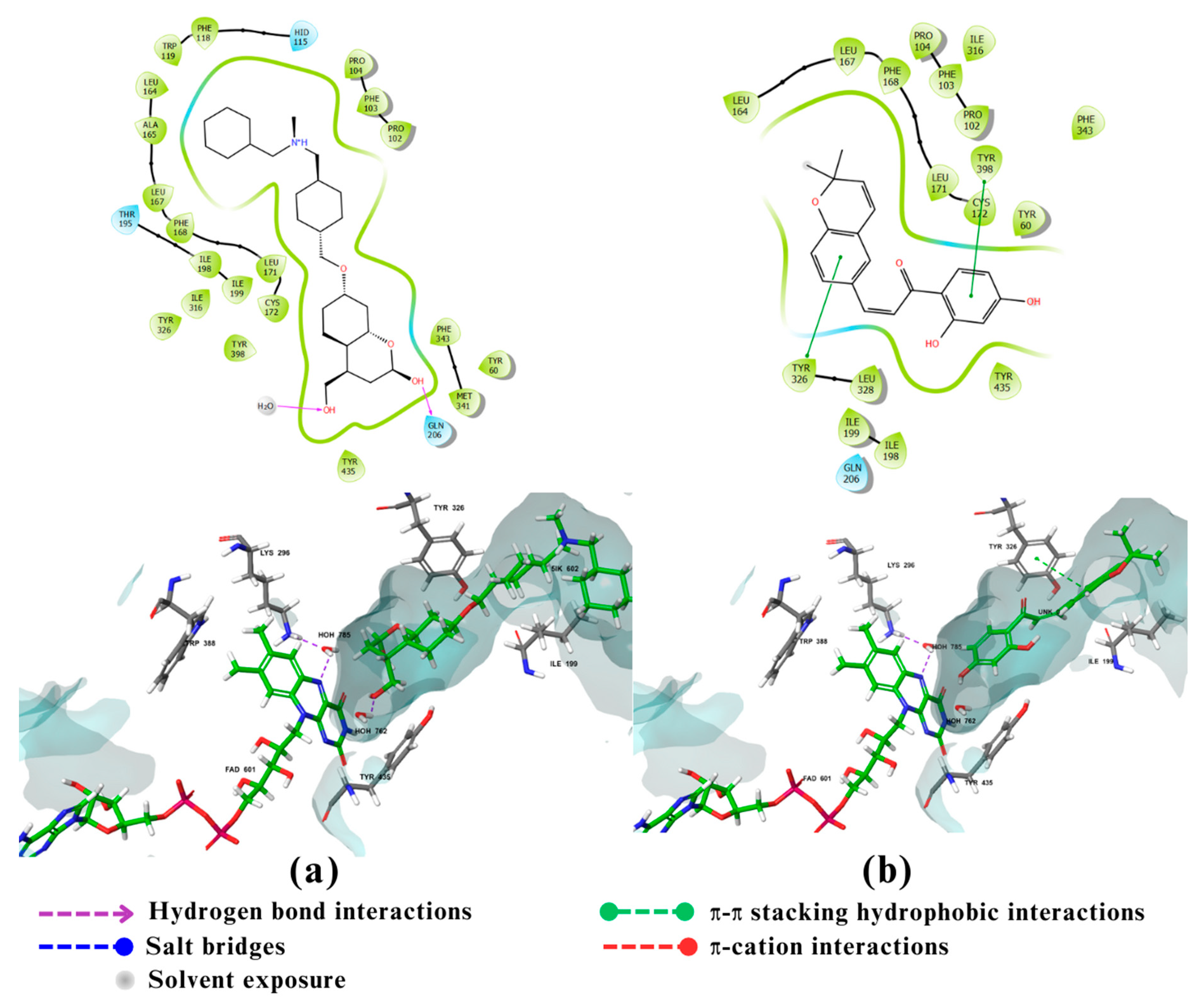
Figure 7.
Molecular orbital distribution plots of HOMO and LUMO of the 5 potential multitarget compounds.
Figure 7.
Molecular orbital distribution plots of HOMO and LUMO of the 5 potential multitarget compounds.
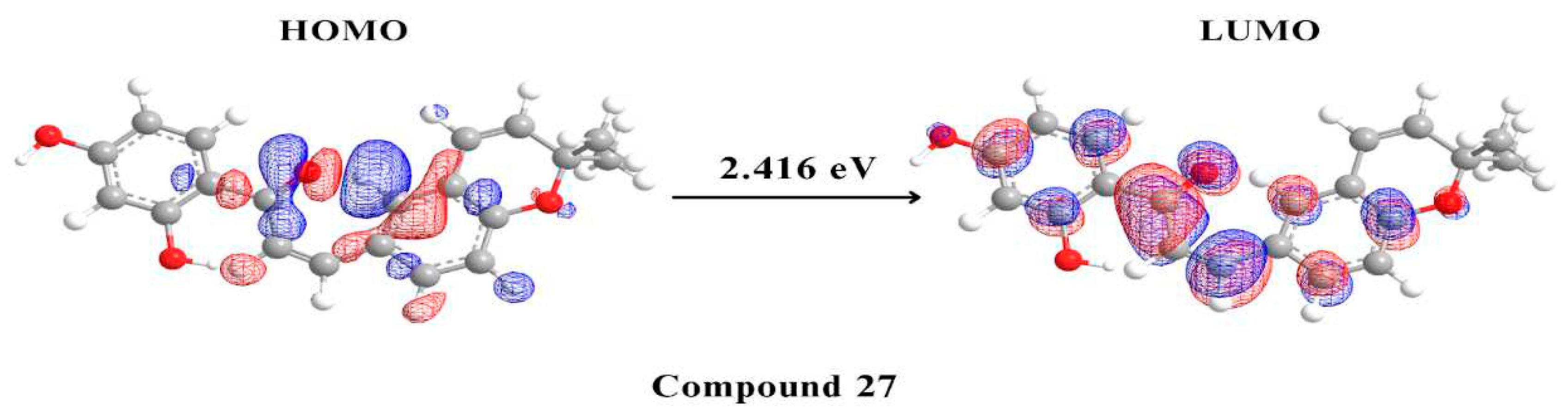
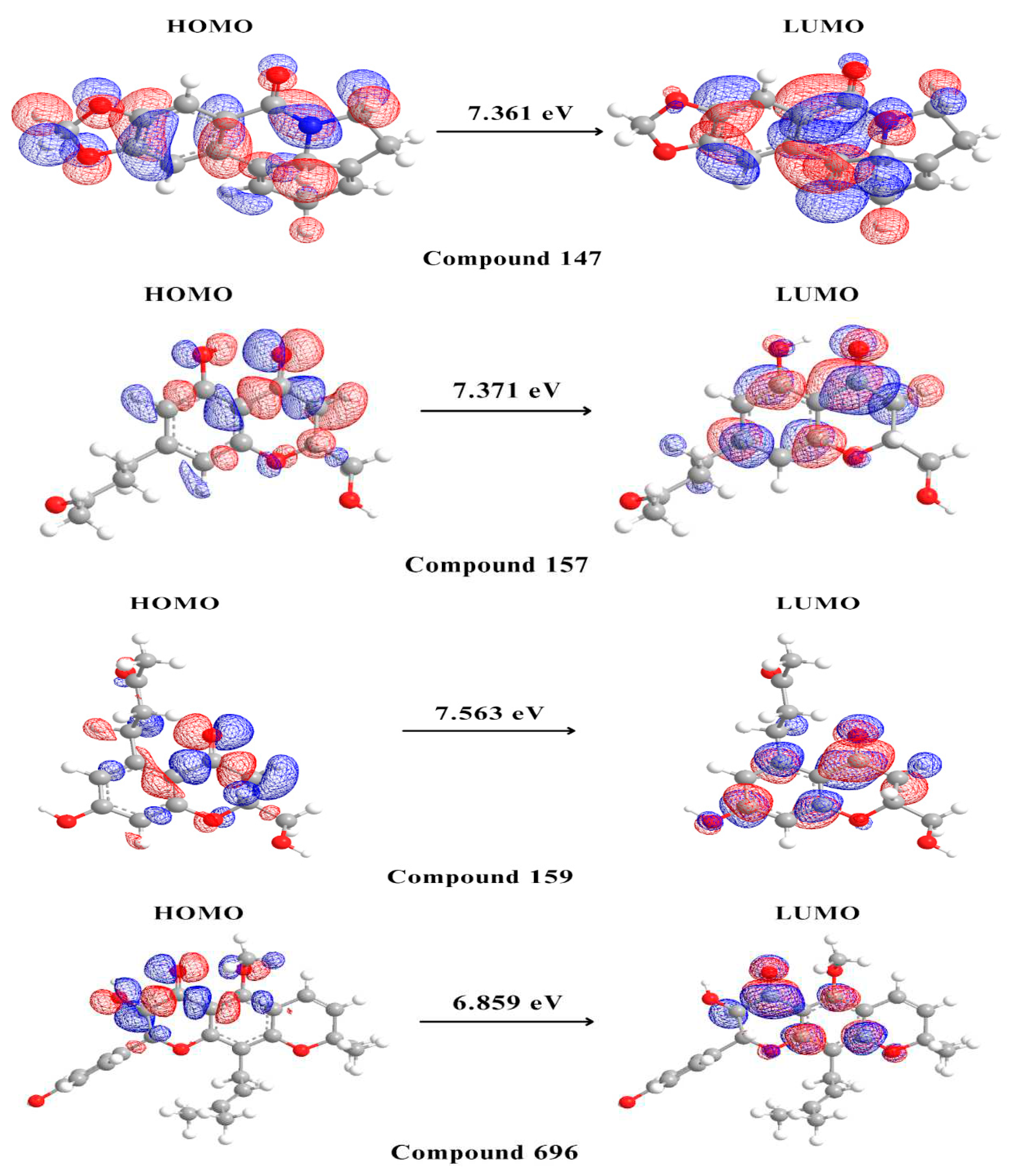

Table 1.
Docking scores and pharmacokinetic properties of the five multitarget compounds and the standard ligands for the five enzymes.
Table 1.
Docking scores and pharmacokinetic properties of the five multitarget compounds and the standard ligands for the five enzymes.
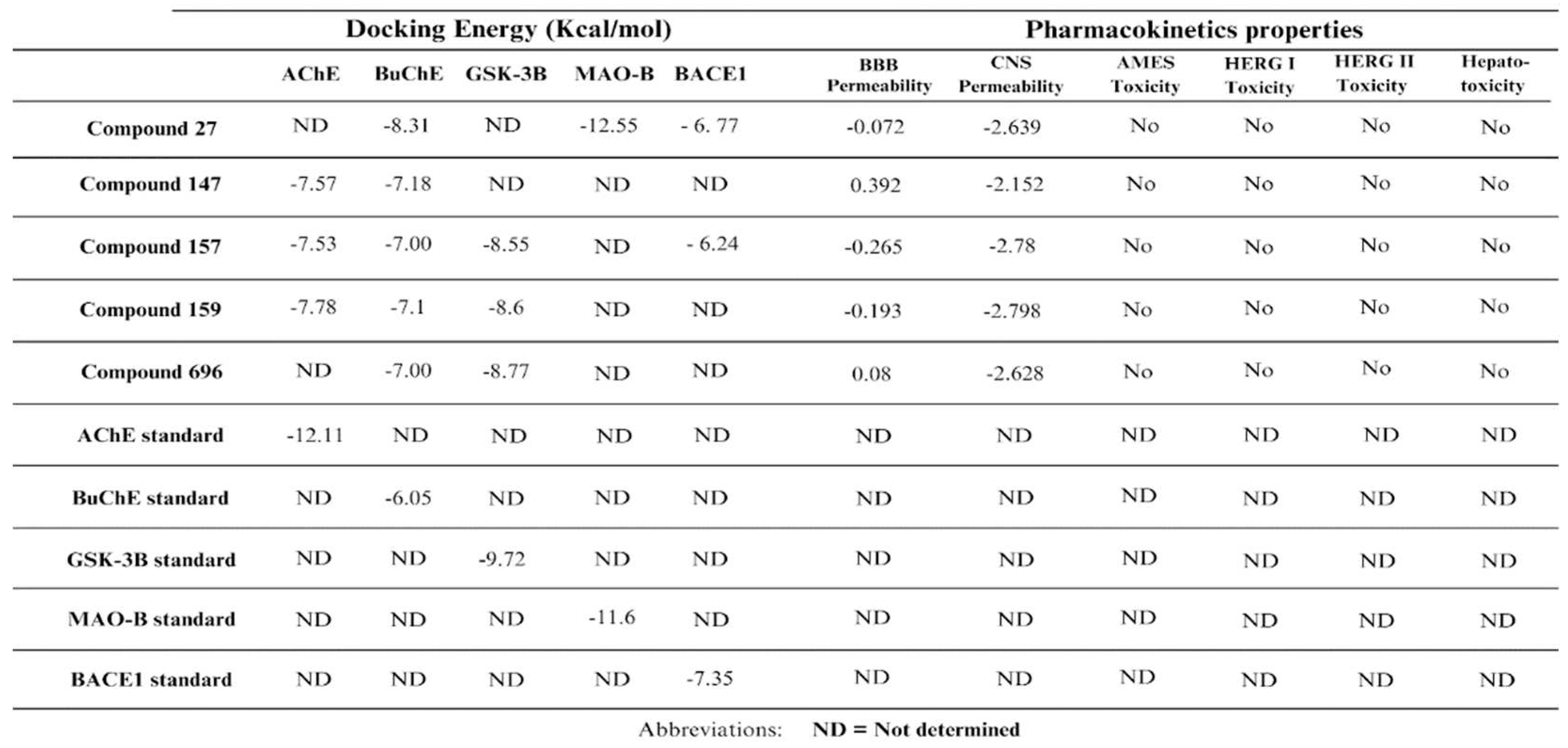
Table 2.
MM-GBSA binding affinity results for the five potential multitarget compounds and standard ligands against the five enzymes.
Table 2.
MM-GBSA binding affinity results for the five potential multitarget compounds and standard ligands against the five enzymes.
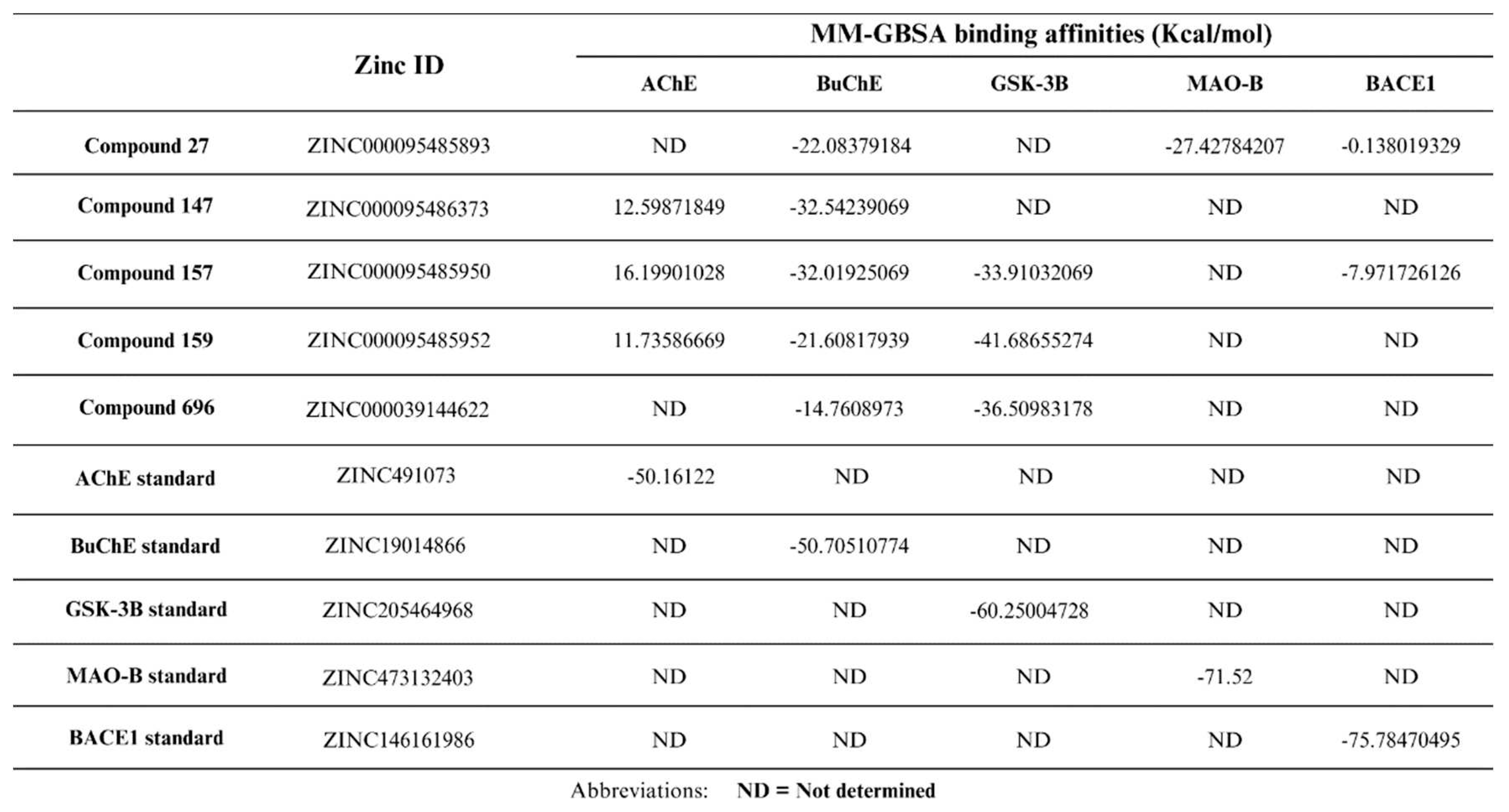
Table 3.
DFT parameters for the five potential multitarget compounds.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



