
Submitted:
11 October 2023
Posted:
12 October 2023
You are already at the latest version
Abstract
Breast cancer (BCa) is the most frequently diagnosed malignant tumor in women and is also one of the leading causes of cancer-related death. Most breast tumors are hormone-dependent and estrogen signaling plays a critical role in promoting the survival and malignant behaviors of these cells. Estrogen signaling involves ligand-activated cytoplasmic estrogen receptors that translocate to the nucleus with various co-regulators such as steroid receptor co-activator (SRC) family members, and bind to the promoters of target genes and regulate their expression. SRC-3 is a member of this family that interacts with, and enhances the transcriptional activity of the ligand activated estrogen receptor. Although SRC-3 has important roles in normal homeostasis and developmental processes, it has been shown to be amplified and overexpressed in breast cancer and to promote malignancy. The malignancy-promoting potential of SRC-3 is diverse and involves both promoting malignant behavior of tumor cells and creating a tumor microenvironment that has an immunosuppressive phenotype. SRC-3 also inhibits the recruitment of tumor-infiltrating lymphocytes with effector function and promotes stemness. Furthermore, SRC-3 is also involved in the development of resistance to hormone therapy and immunotherapy during breast cancer treatment. The versatility of SRC-3 in promoting breast cancer malignancy in this way makes it a good target, and methodical targeting of SRC-3 probably will be important for the success of breast cancer treatment.
Keywords:
breast cancer
; SRC-3
; estrogen signaling
; tumor microenvironment
; tumor infiltrating cells
1. Introduction
Breast cancer (BCa) is one of the most commonly diagnosed malignancies in women and is also a major cause of cancer death [1,2]. BCas are highly heterogeneous tumors and the most commonly used system for classifying BCa is based on the status of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) [3,4]. Furthermore, the status of these receptors has a direct relationship with the prognosis of the disease [5].
Estrogen signaling is a critical mechanism in the normal physiology of the breast, but it is also involved in the pathogenesis of hormone-dependent BCa, which is responsible for two thirds of all cases of BCa [6]. Estrogens transmit the signals by acting on target cells via ERs, which are located on the cell membrane and in the cytoplasm [7]. Estrogen-activated cytoplasmic ERs translocate to the nucleus with various co-regulators including steroid receptor co-activators (SRCs) and regulate the transcription of ER target genes and consequently alter many cellular mechanisms [7,8]. SRC is a family of three transcriptional regulators that enhance the activity of steroid receptors, including ER [9]. SRC-3 (also known as AIB1, NCoA-3, RAC3, ACTR, TRAM1, and p/CIP) is a member of this family and is involved in tissue homeostasis and development in normal physiology [10,11]. The activity of SRC-3 is generally regulated at the post-translational level and a large number of modification sites have been identified on the primary structure of the SRC-3 protein, including phosphorylation, methylation, acetylation, ubiquitination and SUMOylation [12,13].
SRC-3 has also been shown to be overexpressed and amplified in BCa and have critical roles in the development and progression of the disease [14,15]. In fact, detailed studies have shown that SRC-3 is involved in BCa pathogenesis in multiple ways, including promoting proliferation, migration, invasion and metastasis in BCa cells [16,17]. SRC-3 is also involved in the creation of an immunosuppressive tumor microenvironment and promotes stemness [18,19]. In regard to cancer treatment, SRC-3 is involved in the development of resistance to both endocrine therapy and immunotherapy in patients with BCa [20].
Given the mutiple critical roles of SRC-3 in the development and progression of BCa, the potential of SRC-3 as a possible therapeutic target is high. Therefore, various natural and lab-made artificial molecules have been designed to inhibit SRC-3 activity or reduce its levels in cells, as explained in the following parts. The results of both in vitro and in vivo experiments showed that a decrease in SRC-3 levels or activity both inhibited the malignant behavior of BCa cells and changed the tumor microenvironment to a tumor-suppressing phenotype. Overall, the results of current studies on the inhibition of SRC-3 levels or activity are very strong and promising. Therefore, the development of new therapeutic approaches targeting SRC-3 for the treatment of BCa in the clinic in the near future is a strong possibility. However, in order to achieve this, it will be essential to overcome the current limitations of molecules targeting SRC-3 and to design/develop new nanocarrier systems that will enable the delivery of these molecules to cells such as cancer stem cells (CSCs).
2. BCa and Estrogen Action
BCa is the most frequently diagnosed malignant tumor in females and is one of the leading causes of cancer related death in women [1,2]. Although BCa is highly curable in patients with early stages, it is generally incurable in patients with advanced stages who have distant metastases [21]. In fact, BCa are highly heterogeneous tumors with many subtypes, which are generally classified into four groups according to the status of ER, PR, and HER2 status, which are Luminal A, Luminal B, HER2(+), and Triple(-) (TNBC) [3,4]. The Luminal A class of BCa are ER(+) and/ or PR(+) and mostly show a low proliferation rate, in a correlation to low Ki-67 levels. The Luminal B class of breast tumors express ER and/ or PR, similar to Luminal A tumors, but their proliferation rates are high in concordance with higher Ki-67 level. HER2(+) tumors are ER and PR deficient but have HER2. In TNBC all three hormone receptors, (ER, PR and HER2) are deficient. The BCa subclasses show different malignant behaviors, often dependent on the status of hormone receptors, and therefore the absence or presence of these receptors is also associated with BCa prognosis. For example, although ER(-) tumors are considered to be associated with early recurrence risk, ER(+) tumors are generally associated with late recurrence risk [22,23]. Furthermore, although patients with hormone receptor positive Luminal A and B tumors generally have a favorable prognosis, patients with hormone receptor negative tumor sub-classes have been shown to have a poor prognosis [24].
Estrogens are steroid hormones that have crucial functions in the development of secondary sexual characteristics in females. However, they are also involved in the pathogenesis of BCa in multiple ways. Estrogens are mainly produced and secreted by the ovaries under the control of the luteinizing hormone (LH)/ follicle-stimulating hormone (FSH) axis [25] and act on the target cells through ERs which are called ER-α and ER-β that are encoded by ESR1 and ESR2, respectively [26]. ERs are the members of the nuclear hormone receptor (NHR) superfamily and have diverse functional domains to carry out their functions [27]. In the absence of estrogen, ERs are located in the cytoplasm inactively bound with various HSP proteins including HSP70 and HSP90, in a complex form [28]. Estrogen activated ER may act in a genomic or in a non-genomic manner in the cells (Figure 1).
In the genomic mode of action, estrogen binding causes conformational changes in the ER that convert it to an active form and dissociate it from HSPs [29]. Consequently, dimerized ER is translocated to the nucleus, generally with some co-regulators including steroid receptor activators (SRCs), and binds the estrogen response elements (EREs) on the regulatory regions of target genes to regulate their expression [30]. Activated ER may also be indirectly involved in the transcriptional regulation of some genes through interactions with transcription factors such as SP1 and Cyclin G2 [31,32,33]. In both mechanisms, the active ER interacts with co-regulators such as SRCs and other molecules to regulate the transcription of target genes [34,35]. In the non-genomic mode of action, membrane located ER, activated through estrogen binding, leads to many changes in the cytoplasm and affects a variety of signaling pathways including NFκB, MAPK, and PI3K/ AKT [8]. In addition, GPR30 (also known as GPER1), a G protein-coupled receptor, on the cell membrane has been identified as a membrane estrogen receptor [36,37]. Although the affinity of estrogen to GPR30 isn’t as high as compared to classical intracellular ERs, it triggers a rapid response and then conveys the signal to many intracellular signaling cascades through second messenger - dependent or independent mechanisms [38,39].
Although both of the intracellular ERs are expressed in normal breast tissues, the ER-β level is higher making it the dominant receptor in estrogen/ ER signaling [40]. However, in the ER(+) BCa cells the level of ER-β is decreased and ER-α is increased [41,42]. Similarly, increased ER-α to ER-β ratio dependent on the decrease of ER-β expression has been shown also in both uterine myomas and mouse skin tumors and cell lines [43,44]. In this context, although increased ER-α to ER-β ratio seems to be a general mechanism in both malignant and benign hyperproliferative tissues, it is the major source for promoting of malignant behaviors in ER(+) BCa cells. Indeed, it has been shown that the changed expression of ER-α is associated with the development and progression of BCa [45]. Many ER-α target genes involved in cell proliferation have been identified including the genes encoding c-Myc, Cyclin D1, FoxM1, and Greb1 [46,47,48]. Estrogen/ ER-α signaling has been shown also involved in the BCa cell migration but this effect is complicated and may act in both inhibitory and activatory roles depending on the co-regulators and downstream mechanisms. Indeed, estrogen suppresses the E-cadherin level through direct binding of ER-α together with several co-repressors to the promoter of Cdh1 which is the E-cadherin encoding gene [49]. On the other hand, metastasis-associated protein 3 (MTA3), another direct target of ER-α, has been shown to repress the transcription of Snail 1 which is a well-known repressor of Cdh1 [50]. Consequently, increased MTA3 level depending on the estrogen/ ER-α axis represses Snail 1 expression and therefore Cdh1 transcription is restored. Another MTA family member, MTA2, has also been shown to interact with SRC-3, to establish an inhibitory complex, thereby inducing epithelial-mesenchymal transition (EMT) by repressing Cdh1 expression in ER(+) luminal breast tumors [51]. Therefore, it is plausible to postulate the formation of an inhibitory SRC-3/ MTA2 interaction complex under estrogen absence conditions, since SRC-3 expression is negatively regulated by estrogen [52]. Maintaining or restoring of the E-cadherin level on the BCa cells is important in both the maintenance of tissue stability and consequently inhibiting migrative and invasive abilities of cancer cells, and also for targeting tumor cells by immune cells, as described in the next sections.
3. BCa Tumor Microenvironment
The tumor microenvironment is a dynamic complex network that composes of cellular and non-cellular components and has crucial roles in the behaviors of tumor cells including invasion, metastasis, and therapy resistance [53]. Indeed, the effects of the tumor microenvironment on both promoting the malignant behaviors and metastasis processes of tumor cells have been known for a long time. Although the tumor microenvironment is highly heterogeneous across cancer types, the cellular content of the microenvironment is tumor infiltrating cells (TICs), and has been composed of multiple cell groups including T-cells, leukocytes, monocytes, and tumor associated macrophages (TAMs) [54,55,56]. Tumor infiltrating lymphocytes (TILs) are important components of TICs which affect tumor cell metabolism directly and determine the nature and phenotype of the microenvironment. However, they are not found uniformly in all BCa subtypes. Although TILs are found at a relatively low rate in the Luminal A and Luminal B types of tumors, they are found increased in TNBC and HER2(+) breast tumors [57]. The non-cellular components which surround cellular components are basically composed of growth factors, cytokines, chemokines, extracellular vesicles, and extracellular matrix proteins [54,56]. In fact, the tumor microenvironment as a whole, with both cellular and non-cellular components, is a specialized ecosystem and often acts independently, at least partly, from the rest of the organism. For example, it has a low oxygen concentration and this causes recruitment of regulatory T-cells (Tregs) to the microenvironment and inhibition of effector T-cell differentiation, which would otherwise combat cancer cells [58]. In addition, cancer cells secrete fibroblast growth factor (FGF), which causes the recruitment of cancer associated fibroblasts (CAFs) to the tumor microenvironment, where CAFs both change the extracellular matrix and secrete various immunosuppressive cytokines that promote angiogenesis as well as the growth of tumors [53,59].
Although breast tumors generally have an immunosuppressive microenvironment, both the effect of estrogen and the absence/ presence of ER have a prominent impact on the composition of infiltrated TILs [60]. In fact, estrogens have multiple effects on the immune system cells including proliferation, differentiation, and regulation of cytokine production [61,62,63,64,65,66]. For example, estrogen inhibits both the proliferation of CD4+ T-cells and inhibits the activities of natural killer (NK) and cytotoxic T lymphocytes (CTLs) to contribute promoting an immunosuppressive tumor microenvironment [67,68]. However, an immunosuppressive tumor microenvironment is created by the collective actions of many cells, including the tumor cells and some immune system cells. In this context, secretion of some immunosuppressive molecules such as TGF-β and IDO-1 by tumor cells and recruitment of some immunosuppressive immune system cells, such as Tregs, into the tumor microenvironment are the lead causes in the creation of an immunosuppressive phenotype [69]. Indeed, the amplification and immunosuppressive activities of Tregs were shown to be promoted by estrogen [70,71,72]. The increase in the estrogen-dependent immunosuppressive activities of Tregs is provided by the increase in FoxP3 and PD-1 levels [70,71]. Estrogen also induces the production and secretion of TGF-β and IL-10 in Tregs [73,74]. In concordance, the treatment of Tregs with ICI-182780, an ER antagonist, was shown to inhibit both FoxP3 expression and TGF-β production and secretion [75].
Tregs are crucial cells in immune homeostasis and act as the suppressors against excess immune response to prevent autoimmune diseases; they are highly increased in the tumors of many types of solid cancers including BCa [76]. Tumor infiltrating Tregs have a different phenotype from normal tissue Tregs and it has been suggested that the interaction of Tregs with the tumor microenvironment probably drives this distinct Treg phenotype [77,78]. Indeed, it has been shown that the immunosuppressive roles of Tregs are associated with both their presence in the tumor microenvironment and with an increase in the inhibitory receptors [79]. Tregs found in the tumor microenvironments are generally associated with poor prognosis since they both suppress effector T-cells and also inhibit the efficacy of chemotherapy and radiotherapy [80]. In concordance, it has been shown that selective depletion of Tregs results in augmentation of the anti-tumor immune response [81].
Tregs may perform their immune suppressor role by several different mechanisms in the tumor microenvironment. Tregs secrete anti-inflammatory cytokines including TGF-β, IL-10, and IL-35 into the microenvironment and thereby suppress the activity of effector immune cells and consequently anti-tumor immunity [82,83,84,85]. Secretion of these cytokines is important in the creating and sustaining of an immunosuppressive tumor microenvironment. For example, IL-10 secreted by Tregs increases the expression of co-inhibitory receptors and thereby induces immunosuppression functions of Tregs [85,86]. IL-35 generally acts jointly with IL-10 in the generation and sustaining of the immunosuppressive tumor microenvironment [85]. Although most of TGF-β in the tumor microenvironment is produced by tumor cells and tumor associated fibroblasts, it also can be produced and secreted by Tregs [87]. TGF-β may affect the tumor microenvironment in multiple ways. For example, TGF-β can induce Treg differentiation from the naive CD4+ T-cells [88]. Tregs, also directly can kill CD8+ T-cells and NK cells through secreting granzyme B and perforin [89,90]. On the other hand, cell to cell interaction is another important mechanism in Treg biology and Tregs commonly express some co-regulatory receptors to modulate their actions. In this context, ICOS, OX40, and GITR function as co-stimulatory receptors, whereas CTLA-4, Lag-3, Tim-3, TIGIT, PD-1, and KLRG1 function as co-inhibitory receptors [91]. The functional consequences of the interaction between many of these receptors and their ligands (usually found in antigen presenting cells, tumor, or epithelial cells) have been revealed, at least partially.
4. SRC-3 Has Multiple Roles in BCa Pathogenesis
SRC-3 is a member of the p160 steroid receptor co-activator family and has critical roles in the regulation of normal cell physiology [9]. The SRC-3 is mainly regulated functionally at the post-translational level and many modifications have been identified on the SRC-3 protein [12,13,92]. Indeed, it has been shown that the changes in functional post-translational modifications of SRC-3 cause systemic changes in growth and metabolism [93]. In particular, phosphorylated SRC-3 interacts with ligand activated nuclear hormone receptors and then recruits various proteins including histone acetyltransferases through its different domains (Figure 2) [94]. The established complex alters chromatin dynamics and consequently enhances the transcriptional activities of the NHRs [94].
SRC-3 is essentially involved in the regulation of metabolic homeostasis similar to other SRC family members. SRC-3 deleted or overexpressed mouse models have been developed by several research groups to elucidate the roles of SRC-3 in normal cellular physiology and crucial data have been derived. For example, SRC-3 is involved in the regulation of adipocyte differentiation, and white adipose differentiation is impaired in SRC-3 deleted mice [95]. SRC-3 is also involved in the regulation of energy metabolism, in preventing obesity, and increasing glucose tolerance, by augmentation of mitochondrial activity [96]. Furthermore, the observations on SRC-3 deleted mice show a pleiotropic phenotype including abnormalities in the mammary gland development and a reduction in reproductive function [11].
4.1. SRC-3 Affects the Tumor Microenvironment and Promotes Stemness
SRC-3 may affect the tumor microenvironment in multiple ways by affecting many immune system cells, as well as tumor cells. Although these effects generally result in an immunosuppressive phenotype, they also may contribute to the creation of an inflammatory environment. Tregs have a special place in the SRC-3 induced immunosuppressive tumor microenvironment. Indeed, Tregs have a strong SRC-3 expression and a high SRC-3 level is required for their immunosuppressive functions as described above [18]. In this context, it can be speculated that SRC-3 has a crucial role in establishing an immunosuppressive microenvironment in BCa. Although the factors that induce SRC-3 expression in Tregs are not yet understood, it is possible that this regulation may be dependent on some common mechanisms including the estrogen, retinoic acid (RA) and TGF-β signaling mechanisms. Silencing or inhibition of SRC-3 in Tregs has been shown causes a decrease in the expression levels of both FoxP3 and PD-1 encode genes [18]. FoxP3 and PD-1 levels are directly related with the immunosuppressive abilities of Tregs and has been shown that estrogen up-regulates the expressions of the genes encode these proteins [70,71]. However, estrogen has the opposite roles on SRC-3 transcription and activity and has been shown that although estrogen inhibits SRC-3 transcription, it phosphorylates and consequently activates SRC-3 protein [13,52]. Consequently, the phosphorylated SRC-3 binds to ERs to potentiate its genomic or non-genomic functions and thereby contribute to the estrogen action. RA and TGF-β has important roles in the generation and differentiation of Tregs and has been shown that RA induces FoxP3 expression in a TGF-β dependent manner and consequently promote generation of Tregs [97,98]. Furthermore, it has been shown that RA and TGF-β increases SRC-3 transcription [13]. Therefore, it seems reasonable to hypothesize that SRC-3 may play a role in RA- and TGF-β-mediated generation and/or differentiation of Tregs. NFκB has been shown to be bind to the SRC-3 promoter and up-regulate its expression, in response to TNF-α [99]. Although NFκB signaling is generally considered to be the mechanism that induces the differentiation of effector T-cells, it also promotes FoxP3 expression and has a role in the generation of Tregs [100,101,102]. SRC-3 expression has been shown to decrease in an AKT/mTOR-dependent manner in hypoxia conditions in preeclampsia, a complication of pregnancy [103]. Although it is unknown whether AKT/ mTOR-dependent regulation of SRC-3 expression is a general mechanism in cells, including Tregs, it is possible that to be a general mechanism in the regulation of SRC-3 expression, since has the versatile effects.
Furthermore, the effects of SRC-3 in the induction of an anti-inflammatory environment have also been shown. Chen et al. have shown that SRC-3 inhibits inflammation, and deletion of SRC-3 in mice results in increased production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6 and consequently increased inflammation in the colon [104]. In concordance, induction of SRC-3 activity through the small molecular MCB-613 results in the enrichment of anti-inflammatory macrophages in mice [105]. The anti-inflammatory effect of SRC-3 has been also observed in vitro. In addition, stimulation of SRC-3 through MCB-613 in the RAW 264.7 macrophages results in decreasing expression of pro-inflammatory cytokine mRNAs including TNF-α, IL-1β, and IL-6 [105]. In concordance, it has been shown that LPS treatment leads to increased secretion of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, in SRC-3 deleted macrophages compared to wild type macrophages [106]. Interestingly, the transcription of pro-inflammatory cytokines is nearly unchanged in SRC-3 deleted macrophages compared to wild type, but the translational efficiency of these cytokine mRNA's has been increased [106]. It has been shown that SRC-3 dependent regulation of this effect occurs at the post-transcriptional level and SRC-3 exerts this effect by promoting binding of some translational repressors to the 3' UTR region of TNF-α mRNA, to inhibit its translation. Furthermore, macrophages from SRC-3 deleted mice produce a high level of TNF-α protein in response to LPS stimulation without changing the TNF-α mRNA level [107].
SRC-3 may also affect the phagocytosis abilities of macrophages. It has been shown that the levels of the scavenger receptor A and catalase are lower in SRC-3 deficient macrophages, compared to wild type macrophages [107]. In this context, SRC-3 directly contributes to the regulation of catalase transcription and SRC-3 deficiency results in a decrease in catalase expression [107]. Catalase is an important enzyme in the regulation of reactive oxygen species and is responsible for the conversion of H2O2 to H2O [108]. It was shown that both ROS level and apoptotic index are higher in SRC-3 deleted macrophages compared to wild types [107]. Indeed, other studies also confirmed an inhibitory role of SRC-3 in both intrinsic and extrinsic apoptotic pathways [109,110].
On the other hand, it seems that SRC-3 is involved in both the activation and recruitment of neutrophils through the regulation of CXCL-2, in a NFκB dependent manner, and thereby SRC-3 may contribute to the creation and regulation of an inflammatory environment, at least in the neutrophil context [111]. Consistently, SRC-3 was shown to be an NFκB co-regulator that promotes NFκB-mediated transcriptional activity, and this activity is regulated by phosphorylation by IκB kinase [112,113]. The role of SRC-3 in regulation of NFκB was further supported by demonstration of a direct interaction between SRC-3 and Rel-A [113]. NFκB signaling is known to inhibit apoptosis and therefore SRC-3 dependent inhibition of apoptosis may be related to activation of NFκB, at least partly. Moreover, SRC-3 is not only a co-activator for NFκB but is also a direct target, and inflammatory cytokines induce SRC-3 expression via direct binding of NFκB to the SRC-3 promoter [99]. Probably a feedback loop operates between SRC-3 and NFκB because SRC-3 also represses the translational efficiency of pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β, and this effect is abolished in SRC-3 deficient mice, as described above [106].
SRC-3 has been shown to drive the CSC phenotype, in concordance with its EMT promoting roles (Figure 3) [19]. In this context, cytoplasmic PELP1/ SRC-3 complexes were shown to mediate the expansion of breast CSCs [114]. Indeed, it was shown that PELP1/ SRC-3 complexes regulate CSCs through modulating metabolic adaptation associated gene expression programs [115]. Furthermore, SRC-3 is required for maintenance and induction of the CSC phenotype; consequently, treatment of BCa cells with an SRC-3 inhibitor decreases SRC-3-induced CSCs in BCa [19,116]. The SRC-3 level is positively associated with ALDH+ CSCs in BCa [19]. ALDH1+ CSCs are especially important since they are associated with both tamoxifen resistance and early recurrence after anti-estrogen therapy in breast tumors [117,118]. Although use of disulfiram, an ALDH inhibitor, results in activating T cell immunity and consequently in clearance of breast CSCs, it is not yet known whether this effect is associated with effects on SRC-3 [119,120]. Furthermore, SRC-3 interacts with SOX-2 and promotes its transcriptional activity [121]. SOX-2 expression increases during development of tamoxifen resistance and high SOX-2 level is important to maintain CSCs in BCa [122,123]. SRC-3 interacts with estrogen-related receptor β (ESRRB) and functions as a co-activator to the induction and sustaining of embryonic stem cell (ESC) renewal and pluripotency [124,125,126]. Indeed, SRC-3 was shown to induce the expression of self-renewal and pluripotency related genes, including KLF-4, in ESCs [127]. Furthermore, SRC-3 is involved in the regulation of Hematopoietic stem cells (HSCs) by modulating their mitochondrial metabolism [128].
4.2. SRC-3 Promotes Malignant Behaviors of Tumor Cells
The implication of SRC-3 in the development and progression of many types of cancers have been reported [17,129]. Indeed, many reports have shown that SRC-3 is involved in carcinogenic processes by multiple pathways (Figure 3). However, SRC-3 has been most extensively studied in BCa. The story of a relationship between SRC-3 and BCa started about 20 years ago and SRC-3 is considered a proto-oncogene since its overexpression leads to BCa in mice [130]. Although both overexpression and amplification of SRC-3 are reported in BCa, its overexpression is much more common compared to amplification [14,131,132]. Furthermore, it has been shown that SRC-3 levels are higher in the advanced stages of the disease and that higher SRC-3 levels are associated with poor prognosis in ER(+) BCa [14,133,134,135,136,137]. The effects of SRC-3 in BCa pathogenesis were shown in mice in which SRC-3 was deleted or overexpressed. It was demonstrated that elevating SRC-3 abundance results in hyperplasia and consequently breast adenocarcinoma in mice [130,138,139]. Interestingly, even moderate overexpression of SRC-3 causes pre-malignant transformation in the mammary epithelium [140]. Conversely, SRC-3 deficiency inhibits both v-Ha-ras and chemical carcinogen induced BCa [141,142]. Furthermore, SRC-3 directly interacts with ER-α in the presence of estrogen, recruits other co-regulators and consequently increases the transcriptional activity of ER-α to promote cell proliferation [143,144,145]. Thereby SRC-3 is involved in the pathogenesis of ER(+) BCa and promotes the malignant behavior of overexpressing cells. In this model, SRC-3 is the primary co-regulator for ER-α activity and its binding allows the sequential binding of secondary co-regulators which are p300/CBP and CARM1 [146]. However, SRC-3 may interact with the mutant estrogen receptor, which is activated in a ligand-independent manner (in the absence of estrogen) [147,148].
It was shown that SRC-3 is also involved in the production and secretion of growth factors and thereby involved in the regulation of growth factor signaling. For example, SRC-3 overexpression results in an increase in the IGF-I mRNA and protein levels, as well as the components of the IGF-I signaling mechanism, such as IGF-I receptor β (IGF-IRβ) [11,130,149]. In addition, the SRC-3 expression level is positively correlated with HER2, and this event is associated with tamoxifen resistance [136,150]. In concordance, breast tumorigenesis induced by HER2 was completely inhibited in SRC-3 deficient mice [151]. SRC-3 was implicated in the migration, invasion and metastasis processes: it was namely shown that SRC-3 overexpression results in an increase in the MMP-7 and MMP-10 levels and thereby promotes metastasis [152]. SRC-3 promotes FAK activation, and also functions as an adapter molecule between EGFR and FAK and consequently promotes cell migration in BCa [153,154]. SRC-3 also promotes EMT in cancer cells through the classical cadherin switching mechanism, by which E-cadherin changes to N-cadherin [19]. This transition mechanism is crucial for tumor cells to gain migrative abilities and is considered as one of the initial steps in the invasion and metastasis processes of cancer cells [155].
Rohira et al., have shown that SRC-3 overexpression induces Snail 1 and Snail 2 expressions and thereby decreases E-cadherin level, and in concordance, Vimentin and N-cadherin levels increase in SRC-3 overexpressing cancer cells [19]. E-cadherin is a glycoprotein on the epithelial cells and is crucial for the establishment of adherens junctions between neighboring cells [156]. Each E-cadherin molecule has a large extracellular region, a transmembrane region, and a short cytoplasmic domain [157]. The extracellular region consists of 5 extracellular cadherin domains and interacts with the extracellular region of cadherin in neighboring cells. The cytoplasmic domain of E-cadherin interacts with the cytoskeleton through catenin proteins. E-cadherin loss is observed in the advanced stages of many cancers, including BCa and this event is a strong marker of EMT [158]. Cadherin switching in advanced stages of cancers is generally associated with a gain in the invasive and metastatic potential of cancer cells, and this event may be a result of various mechanisms that are triggered by genetic or epigenetic alterations [158,159]. Furthermore, it also may be a result of therapeutic approaches, such as androgen deprivation therapy in prostate cancer [160].
5. SRC-3 May Contribute to Therapy Resistance in Multiple Ways in BCa
5.1. SRC-3 May Contributes to Hormone Therapy Resistance
Considering that more than 70% of BCa patients have ER(+) tumors and ER signaling plays a crucial role in the development and progression of BCa, clinically targeting this mechanism for the treatment of BCa is quite reasonable, and indeed this therapeutic approach has proven successful in most patients [161]. However, although the patients are mostly successfully treated with these approaches, resistance often emerges after long term exposure [162,163]. One of the mechanisms of acquired resistance is the acquisition of mutations of ER-LBD which may cause ligand independent activation of the receptor [164,165]. A role of SRC-3 in ligand-independent activation of the ER has been demonstrated: it was shown that SRC-3 interacts more potently with the ER-LBDmut compared to ER-LBDwild-type under hormone-deprived culture conditions, created to mimic estrogen deprivation therapy [166]. SRC-3 may also be involved in endocrine resistance via interacting with estrogen-related receptor α (ERR-α), and ERR-α /SRC-3 complexes may control the expression of estrogen regulated genes, in a hormone independent manner [167].
The current therapies that target ER signaling for treatment of BCa are selective ER modulators (SERMs), selective ER downregulators (SERDs) and aromatase inhibitors (AIs) [168,169]. SERMs are anti-estrogen molecules that compete with estrogen for binding to ER-α and thereby inhibit ER-α dependent signaling mechanisms [170]. SERDs are antagonists of ERs and their affinities to ER are stronger, compared to SERMs. SERD agents generally inhibit the transcriptional activity of ER-α more strongly, compared to SERM agents and their inhibition mechanisms include promoting proteosomal degradation of ER-α, and disrupting its dimerization/ nuclear translocation [171,172,173]. The aromatase enzyme catalyzes the last step of the mechanism of conversion of androgen to estrogen, and therefore AIs are used to inhibit estrogen induced cell proliferation through blocking this biochemical pathway in postmenopausal women with ER(+) BCa [174].
Tamoxifen is the best known SERM agent used in the treatment of ER(+) BCa, and the effects of SRC-3 on tamoxifen in the treatment of BCa have been relatively well studied, compared to all other SERM, SERD and AI group molecules. It was shown that elevated SRC-3, as observed in most BCa patients, inhibits tamoxifen activity, and thereby renders anti-estrogen treatment inefficient [136]. However, SERM group molecules including tamoxifen, 4-Hydroxytamoxifen, and Raloxifene increase SRC-3 stability, interestingly [52,175]. Although tamoxifen increases the SRC-3 level indirectly through induction of TGF-β activity, increased SRC-3 further interacts with ER-α and this event is linked to tamoxifen resistance in ER(+) BCa [52,136]. In concordance, silencing of SRC-3 results in re-sensitization of ER(+) BCa cells to tamoxifen and thereby in treatment success [176]. The role of SRC-3 in this mechanism has been shown, mechanistically. It was specifically shown that tamoxifen/ ER complexes directly bind to the promoter of Erbb2, which is the HER2 encoding gene. However, PAX-2 must also be present in the complex to repress and thereby limit HER2 expression [177]. Nevertheless, SRC-3 competes with PAX-2 for participation to these complexes and if the SRC-3 level is high, as observed in BCa, the complex includes SRC-3 instead of PAX-2, causing an increase in HER2 levels and consequently tamoxifen resistance [177]. Indeed, increased HER2 expression and activity has been associated with hormone therapy resistance, further metastatic potential and overall poor prognosis, in BCa [178]. In this context, a positive correlation has been observed between SRC-3 mRNA level and HER2 status/ activity, in BCa [179]. Furthermore, HER2 may also be involved in the regulation of co-activator function of SRC-3 by phosphorylating it, and thereby increasing the activity of SRC-3 [180]. Indeed it was shown that tamoxifen treatment results in binding of SRC-3 to the promoter of the PS2 gene which is a direct target of ER, in HER2 overexpressing tamoxifen resistant cells [181]. Finally, a model was proposed to explain tamoxifen resistance in ER(+) BCa cells, based on the increased levels of SRC-3 and HER2, dependent on tamoxifen treatment. In the proposed model, tamoxifen acts as agonist on ER in the ER(+) BCa cells that have high SRC-3 and HER2 expressions and therefore these cells develop de novo tamoxifen resistance [136,181]. Elevated SRC-3 is also associated with herceptin resistance in HER2 overexpressing BCa cells. Lahusen et al., have shown that SRC-3 regulates EGFR phosphorylation on multiple sites including autophosphorylation sites and silencing of SRC-3 results in a decrease in the total tyrosine phosphorylation on the EGFR, and also in the EGF induced HER2 activation [182].
5.2. SRC-3 May Contribute to Immunotherapy Resistance
SRC-3 has immunomodulatory activities that contribute to establishing a tumor-promoting immunosuppressive microenvironment. Indeed, it was shown that SRC-3 may contribute to immunotherapy resistance through regulation of immunosuppressive functions of Tregs which are pivotal cells in the creation of an immunosuppressive tumor microenvironment, as described above. SRC-3 expression is high in Tregs and it has critical roles in regulating the gene expression of these cells [18]. Inhibition of SRC-3 in breast cancer was shown to weaken the immunosuppressive functions of Tregs, consequently leading to the establishment of a tumor suppressive microenvironment [18]. In concordance, permanent eradication of an aggressive breast cancer model was demonstrated in Treg-cell-specific SRC-3 deleted mice [183]. Furthermore, deletion of SRC-3 in immune intact mice or inhibition through a chemical inhibitor results in an anti-tumor microenvironment, and consequently suppresses BCa progression [184]. On the other hand, CXCL-9, Mip-1α, and IFN-γ levels significantly increase in SRC-3 deficient mice [183]. CXCL-9 is an IFN-γ inducible chemokine that attracts various CXCR-3 expressing effector immune cells including CD8+ and CD4+ T-cells and also NK cells and thereby changes the tumor microenvironment to an anti-tumor phenotype [185,186,187]. Indeed, CXCL-9 overexpression leads to recruitment of T-cells as well as inhibition of tumor growth and metastasis, in an animal experiment [188]. In concordance, high CXCL-9 levels correlated with an increase in the infiltrating anti-tumor immune cells and also with a better response to chemotherapy in BCa patients [189,190].
SRC-3 decreases E-cadherin expression and increases N-cadherin expression, as discussed above [19]. Although this event has been discussed for the epithelial cancer cells in the previous section, it is also important for the success of TIL related immunotherapy. Indeed, the current literature suggests that presence or absence of E-cadherin on tumor cells may be important in the regulation of immunomodulatory mechanisms, at least in terms of the anti-tumor immune response activities of TILs. This mechanism is based on the fact that E-cadherin is the interaction partner of CD103 which is expressed in some immune system cells, such as NK and effector T-cells, and the interaction between CD103 and E-cadherin activates the cytotoxic functions of these cells (Figure 4).
CD103 is a heterodimeric transmembrane protein expressed on several immune system cells including CTLs, tissue resident T lymphocytes (TRMs) and Tregs [191,192]. Tregs have an immunosuppressive function as discussed above, whereas CTLs attack the tumor cells and perform crucial functions in the anti-tumor response. TRMs are a special population of CTLs and are involved in the protection of epithelial tissues against viruses [193]. CD103 interacts with E-cadherin expressed in epithelial cells and has roles in the retention of immune system cells within epithelial tissues [194,195]. Indeed, CD103 is differentially expressed in TILs, and targeting CD103 or E-cadherin by antibodies or genetic approaches inhibits TCR-mediated killing of tumor cells [196]. A heterophilic interaction was shown between the MIDAS motif of CD103 in domain I and the EC-1 domain of E-cadherin [197]. Therefore, in the case of loss of E-cadherin in tumor cells as in advanced stages of cancers, the tumor infiltrating immune cells are also decreased, as expected [198,199]. CD103 expression is induced by TGF-β and this induction is stronger in CD8+ T-cells such as CTL and TRMs, compared to CD4+ Tregs [200,201]. It was shown that TGF-β regulates CD103 expression at the transcriptional level. Mechanistically, TGF-β, which is abundantly present in the tumor microenvironment, activates the classical TGF-β /Smad pathway in cells that express CD103, and consequently, transcription of the CD103 encoding gene, itgae, increases [202]. In this way, increased CD103 abundance on infiltrating T-cells results in stronger binding of these cells to E-cadherin found on tumor cells [203]. However, TGF-β, produced by tumor cells acts as an immunosuppressive factor that helps cancer cells escape from the immune response by inhibiting the expression of molecules involved in the CTL-mediated tumor cytotoxicity such as perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ [204]. Nevertheless, it is also involved in the migration of T-cells towards epithelial tumors and in promoting the anti-tumor activities of tumor-infiltrating CD8+ T-cells [205,206]. Moreover, TGF-β increases both CD103 expression and its affinity to interact with E-cadherin in an ILK phosphorylation-dependent manner [205].
Another receptor that binds to E-cadherin is killer cell lectin-like receptor G1 (KLRG1), a membrane-spanning glycoprotein expressed on some subsets of NK and T cells [207]. KLRG1 has an extracellular C-type lectin-like domain that can interact with N-cadherin and R-cadherin in addition to E-cadherin [208,209]. KLRG1 is an MHC-independent inhibitory receptor that, when interacting with cadherin molecules on target cells, inhibits TCR signaling and consequently the effector functions of NK and CD8+ T-cells [208,209,210,211,212,213]. Although CD103 and KLRG1 share the same ligand, they have opposite effects on effector T cells. The expression of KLRG1 is also controlled by TGF-β, like CD103, but this regulation results in the repression of KLRG1 expression, in contrast to CD103 [214]. Therefore, it can be speculated that KLRG1(+)cells should be underrepresented in the tumor microenvironment due to the high TGF-β concentration in the milieu. Indeed, the proportion of CD8+ TILs expressing KLRG1 was shown to be significantly lower in melanoma and renal cell carcinoma [215,216].
Furthermore, a negative association has been shown between N-cadherin level and success of TIL-related tumor immunotherapy. It was demonstrated that N-cadherin increases PD-L1 and IDO-1 levels in an IFN-γ-R1/Jack/Stat signaling dependent manner in TILs [217]. PD-L1 and IDO-1 induce apoptosis in T-cells and therefore it was suggested that their inhibition may be useful to increase the success of TIL-related tumor immunotherapy [218,219]. Indeed, Sun et al., have shown that N-cadherin deficiency converts the tumor microenvironment from immunotherapy resistant to responsive through decreasing of PD-L1 and IDO-1 levels, and by inhibition of effector Treg production [217]. Similar results were reported by Kolijn et al., namely that EMT causes an increase in the Treg numbers and also increased IDO-1 levels [220]. In concordance, gene expression analyses from various cancer datasets have shown that the EMT signature is positively associated with immunosuppression signatures, but is negatively correlated with the signature of CD8+ TILs [221]. Taken together, these data suggest that high SRC-3 causes EMT, leading to the recruitment of Tregs into the tumor microenvironment and to an increase in IDO-1 expression, thereby contributing to the development of an immunosuppressive microenvironment and thereby to failure of immunotherapy in BCa.
6. SRC-3 Is a Promising Target to Overcome Therapy Resistance in BCa
All the data summarized here suggest that SRC-3 is a proto-oncogene that is involved in BCa pathogenesis by multiple pathways. Therefore, SRC-3 has been suggested as a promising target to overcome therapy resistance in BCa [222,223].
The cellular level of SRC-3 protein is mainly regulated by both ubiquitin dependent and independent proteasome degradation mechanisms; however it can be also degraded by non-proteasome dependent mechanisms [175,224]. In this context, the phosphorylation of SRC-3 on S505 and S509 residues by GSK3 and then ubiquitination by SCFFbw7α was shown [225]. Moreover, SRC-3 is phosphorylated by cell treatment with RA on S860, then ubiquitinated by the CUL-3-based E3 ligase, and consequently degraded [226]. In this regard, it was suggested that molecules such as Gambogic acid and Thevebioside that promote SRC-3 degradation can be used to increase treatment success in cancers, in addition to standard therapy [227,228]. Verrucarin A is another SRC-3 degradation promoting molecule [229]. Verrucarin A probably controls the upstream mechanisms that promote SRC-3 degradation, as discussed above for other molecules, since there isn't a direct interaction between Verrucarin A and SRC-3 [229]. Although, Gossypol has been identified to directly bind to SRC-3 and to lead to its degradation in a proteasome independent manner, it seems that it isn't a specific inhibitor for SRC-3 [230]. The cardiac glycoside bufalin is another inhibitor that directly binds to SRC-3 and promotes its degradation. It was shown that treatment with Bufalin results in the degradation of SRC-3 and also inhibits growth of cancer cells, at very low concentrations [231]. However, Bufalin is non-specific for SRC-3, like Gossypol. All the present literature demonstrates that decreasing of SRC-3 protein levels by degradation mechanisms may be beneficial in cancer treatment. However, the molecules used for this purpose generally target the entire degradation network, as described above, and are not specific to SRC-3 degradation. Therefore, an approach that can specifically promote degradation of the SRC-3 protein would be of great benefit. PROTAC is a novel small molecule technology to induce ubiquitination and degradation of target proteins [232]. Various degraders have been designed and successfully used to specifically degrade many proteins, including PD-L1, using PROTAC technology [233,234,235]. In this point, development of degraders that specifically target SRC-3 and promote its proteasomal degradation will be a promising approach.
SRC-3 inhibitor-2 (SI-2) is an non-natural molecule that has been developed in the Lab as an effort of a multidisciplinary study, and it was shown that SI-2 selectively inhibits SRC-3 expression at both the mRNA and protein levels [223]. Although SI-2 has a short half-life, its low nanomolar activity has made it a promising candidate [17,223]. Song et al., have shown that SI-2 significantly repressed BCa cell proliferation in vitro, and inhibited breast tumor growth in a xenograft model [223]. Furthermore, inhibition of SRC-3 by SI-2 also inhibits immunosuppressive functions of Tregs and their tumor infiltrations, but causes an increase in the CTL and NK cells and consequently changes the tumor microenvironment from immunosuppressive to tumor-suppressive [18,184,236]. SI-2 treatment also targets TIC populations and blocks EMT [19]. Another research group has developed the molecules SI-10 and SI-12 on the basis of the SI-2 scaffold, which have longer half-lives [237]. SI-10 and SI-12 inhibit the malignant behavior of BCa cells and the growth of breast tumors in xenograft models, and suppress the growth of BCa in PDX organoids [237]. It seems that SI-2-based molecules are promising agents for specifically inhibiting SRC-3 in BCa, and we will probably discuss them further in the next years. On the other hand, it was shown that salinomycin directly inhibits SRC-3 transcription, and increases sensitivity of BCa cells to tamoxifen [238]. Although salinomycin inhibits malignant behaviors of BCa cells and kills CSCs, it isn't a specific SRC-3 inhibitor [239]. In this regard, some well-designed nanocarriers can be used to effectively deliver SRC-3 inhibitors into rapidly proliferating cells, such as CSCs. Recent studies have shown that KU-55933, an ATM inhibitor [240], and chloroquine, a lysosome inhibitor [241], can be effectively delivered to breast CSCs using the triphenylphosphonium-functionalized hyperbranched polyethylenimine nanoparticles (PTPP) [242,243]. Similarly, PTPP or PTPP-based nanocarriers could be promising approaches to efectively deliver SRC-3 inhibitors to cells.
There are also nucleic acid-based approaches used to target SRC-3. For example, it has been shown that AY-3, a DNA aptamer, interacts with SRC-3 and abrogates its interaction with p300 [244]. The SRC-3/ p300 association is important for the transcriptional activity of ER-α and disruption of this interaction inhibits the transcriptional activity of ER-α and thereby ER-α-promoted malignant behaviors in BCa cells [146]. Therefore, further investigation for the new aptamers that would disrupt and/or abrogate SRC-3 activity and designing new nanocarrier systems that will effectively deliver these aptamers into CSCs and tumor cells will be important to inhibit/ overcome SRC-3 promoted malignant behaviors in BCa.
7. Conclusions and Future Directions
BCa is one of the most frequently diagnosed malignant tumors in women and is also a leading cause of cancer related death. Although the prognosis for patients diagnosed at an early stage is generally favorable, the prognosis for those diagnosed at an advanced stage is generally poor. SRC-3 has long been considered a proto-oncogene in BCa and its increased expression is associated with poor prognosis in BCa patients. It acts in multiple ways on both tumor cells and the tumor microenvironment, increasing the malignant behavior of the tumor and contributing to therapy resistance. Therefore, targeting SRC-3 during BCa treatment is likely to be crucial both for inhibiting the malignant behavior of tumor cells and for overcoming therapy resistance, ultimately increasing overall treatment success.
Author Contributions
Conceptualization, L.V. and S.V.; methodology, L.V. and S.V.; software, L.V., G.M.D., V.T. and S.V.; validation, L.V., G.M.D., V.T. and S.V.; formal analysis, L.V., G.M.D., V.T. and S.V.; investigation, L.V., G.M.D., V.T. and S.V.; resources, L.V., G.M.D., V.T. and S.V.; data curation, L.V., G.M.D., V.T. and S.V.; writing—original draft preparation, L.V., G.M.D., V.T. and S.V.; writing—review and editing, L.V., G.M.D., V.T. and S.V.; visualization, L.V.; supervision, S.V.; project administration, L.V. and S.V.; funding acquisition, S.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin 2023, 73, 17-48. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209-249. [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nunez, P.; Acuna-Aguilar, L.E.; Gomez-Valles, F.O.; Ramirez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer, Mayrovitz, H.N., Ed.; Brisbane (AU), 2022.
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol 2014, 5, 412-424. [CrossRef]
- Zattarin, E.; Leporati, R.; Ligorio, F.; Lobefaro, R.; Vingiani, A.; Pruneri, G.; Vernieri, C. Hormone Receptor Loss in Breast Cancer: Molecular Mechanisms, Clinical Settings, and Therapeutic Implications. Cells 2020, 9. [CrossRef]
- Faltas, C.L.; LeBron, K.A.; Holz, M.K. Unconventional Estrogen Signaling in Health and Disease. Endocrinology 2020, 161. [CrossRef]
- Clusan, L.; Ferriere, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int J Mol Sci 2023, 24. [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol 2019, 116, 135-170. [CrossRef]
- Xu, J.; Wu, R.C.; O'Malley, B.W. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer 2009, 9, 615-630. [CrossRef]
- Stashi, E.; York, B.; O'Malley, B.W. Steroid receptor coactivators: servants and masters for control of systems metabolism. Trends Endocrinol Metab 2014, 25, 337-347. [CrossRef]
- Xu, J.; Liao, L.; Ning, G.; Yoshida-Komiya, H.; Deng, C.; O'Malley, B.W. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci U S A 2000, 97, 6379-6384. [CrossRef]
- Wu, R.C.; Qin, J.; Yi, P.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell 2004, 15, 937-949. [CrossRef]
- Zheng, F.F.; Wu, R.C.; Smith, C.L.; O'Malley, B.W. Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol Cell Biol 2005, 25, 8273-8284. [CrossRef]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965-968. [CrossRef]
- Guan, X.Y.; Xu, J.; Anzick, S.L.; Zhang, H.; Trent, J.M.; Meltzer, P.S. Hybrid selection of transcribed sequences from microdissected DNA: isolation of genes within amplified region at 20q11-q13.2 in breast cancer. Cancer Res 1996, 56, 3446-3450.
- Gojis, O.; Rudraraju, B.; Gudi, M.; Hogben, K.; Sousha, S.; Coombes, R.C.; Cleator, S.; Palmieri, C. The role of SRC-3 in human breast cancer. Nat Rev Clin Oncol 2010, 7, 83-89. [CrossRef]
- Li, L.; Deng, C.X.; Chen, Q. SRC-3, a Steroid Receptor Coactivator: Implication in Cancer. Int J Mol Sci 2021, 22. [CrossRef]
- Nikolai, B.C.; Jain, P.; Cardenas, D.L.; York, B.; Feng, Q.; McKenna, N.J.; Dasgupta, S.; Lonard, D.M.; O'Malley, B.W. Steroid receptor coactivator 3 (SRC-3/AIB1) is enriched and functional in mouse and human Tregs. Sci Rep 2021, 11, 3441. [CrossRef]
- Rohira, A.D.; Yan, F.; Wang, L.; Wang, J.; Zhou, S.; Lu, A.; Yu, Y.; Xu, J.; Lonard, D.M.; O'Malley, B.W. Targeting SRC Coactivators Blocks the Tumor-Initiating Capacity of Cancer Stem-like Cells. Cancer Res 2017, 77, 4293-4304. [CrossRef]
- Kiliti, A.J.; Sharif, G.M.; Martin, M.B.; Wellstein, A.; Riegel, A.T. AIB1/SRC-3/NCOA3 function in estrogen receptor alpha positive breast cancer. Frontiers in Endocrinology 2023, 14. [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat Rev Dis Primers 2019, 5, 66. [CrossRef]
- Kennecke, H.; McArthur, H.; Olivotto, I.A.; Speers, C.; Bajdik, C.; Chia, S.K.; Ellard, S.; Norris, B.; Hayes, M.; Barnett, J.; et al. Risk of early recurrence among postmenopausal women with estrogen receptor-positive early breast cancer treated with adjuvant tamoxifen. Cancer 2008, 112, 1437-1444. [CrossRef]
- Kennecke, H.F.; Olivotto, I.A.; Speers, C.; Norris, B.; Chia, S.K.; Bryce, C.; Gelmon, K.A. Late risk of relapse and mortality among postmenopausal women with estrogen responsive early breast cancer after 5 years of tamoxifen. Ann Oncol 2007, 18, 45-51. [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010, 28, 3271-3277. [CrossRef]
- Auchus, M.L.; Auchus, R.J. Human steroid biosynthesis for the oncologist. J Investig Med 2012, 60, 495-503. [CrossRef]
- Jensen, E.V.; Jacobson, H.I.; Walf, A.A.; Frye, C.A. Estrogen action: a historic perspective on the implications of considering alternative approaches. Physiol Behav 2010, 99, 151-162. [CrossRef]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol 2017, 16, 4-20. [CrossRef]
- Echeverria, P.C.; Picard, D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta 2010, 1803, 641-649. [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 2006, 7, 497-508. [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 2001, 29, 2905-2919. [CrossRef]
- Li, C.; Briggs, M.R.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology 2001, 142, 1546-1553. [CrossRef]
- Safe, S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm 2001, 62, 231-252. [CrossRef]
- Stossi, F.; Likhite, V.S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem 2006, 281, 16272-16278. [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 2000, 14, 121-141.
- McKenna, N.J.; O'Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465-474. [CrossRef]
- Rae, J.M.; Johnson, M.D. What does an orphan G-protein-coupled receptor have to do with estrogen? Breast Cancer Res 2005, 7, 243-244. [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625-1630. [CrossRef]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953-2962. [CrossRef]
- Prossnitz, E.R.; Barton, M. Estrogen biology: new insights into GPER function and clinical opportunities. Mol Cell Endocrinol 2014, 389, 71-83. [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERalpha) and beta (ERbeta): subtype-selective ligands and clinical potential. Steroids 2014, 90, 13-29. [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab 2015, 29, 557-568. [CrossRef]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors alpha and beta in cancer. Crit Rev Oncol Hematol 2004, 50, 3-22. [CrossRef]
- Bakas, P.; Liapis, A.; Vlahopoulos, S.; Giner, M.; Logotheti, S.; Creatsas, G.; Meligova, A.K.; Alexis, M.N.; Zoumpourlis, V. Estrogen receptor alpha and beta in uterine fibroids: a basis for altered estrogen responsiveness. Fertil Steril 2008, 90, 1878-1885. [CrossRef]
- Logotheti, S.; Papaevangeliou, D.; Michalopoulos, I.; Sideridou, M.; Tsimaratou, K.; Christodoulou, I.; Pyrillou, K.; Gorgoulis, V.; Vlahopoulos, S.; Zoumpourlis, V. Progression of mouse skin carcinogenesis is associated with increased ERalpha levels and is repressed by a dominant negative form of ERalpha. PLoS One 2012, 7, e41957. [CrossRef]
- Welboren, W.J.; Sweep, F.C.; Span, P.N.; Stunnenberg, H.G. Genomic actions of estrogen receptor alpha: what are the targets and how are they regulated? Endocr Relat Cancer 2009, 16, 1073-1089. [CrossRef]
- Dubik, D.; Dembinski, T.C.; Shiu, R.P. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res 1987, 47, 6517-6521.
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.S.; Myatt, S.S.; Kwok, J.M.; Sivanandan, K.; Coombes, R.C.; Medema, R.H.; Hartman, J.; et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983-2995. [CrossRef]
- Sabbah, M.; Courilleau, D.; Mester, J.; Redeuilh, G. Estrogen induction of the cyclin D1 promoter: involvement of a cAMP response-like element. Proc Natl Acad Sci U S A 1999, 96, 11217-11222. [CrossRef]
- Oesterreich, S.; Deng, W.; Jiang, S.; Cui, X.; Ivanova, M.; Schiff, R.; Kang, K.; Hadsell, D.L.; Behrens, J.; Lee, A.V. Estrogen-mediated down-regulation of E-cadherin in breast cancer cells. Cancer Res 2003, 63, 5203-5208.
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207-219. [CrossRef]
- Vareslija, D.; Ward, E.; Purcell, S.P.; Cosgrove, N.S.; Cocchiglia, S.; O'Halloran, P.J.; Charmsaz, S.; Bane, F.T.; Brett, F.M.; Farrell, M.; et al. Comparative analysis of the AIB1 interactome in breast cancer reveals MTA2 as a repressive partner which silences E-Cadherin to promote EMT and associates with a pro-metastatic phenotype. Oncogene 2021, 40, 1318-1331. [CrossRef]
- Lauritsen, K.J.; List, H.J.; Reiter, R.; Wellstein, A.; Riegel, A.T. A role for TGF-beta in estrogen and retinoid mediated regulation of the nuclear receptor coactivator AIB1 in MCF-7 breast cancer cells. Oncogene 2002, 21, 7147-7155. [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J Cancer 2017, 8, 761-773. [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr Biol 2020, 30, R921-R925. [CrossRef]
- Ben-Baruch, A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor-microenvironment interactions. Breast Cancer Res 2003, 5, 31-36. [CrossRef]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal Cell Pathol (Amst) 2020, 2020, 6283796. [CrossRef]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer-Updates on Therapeutic Implications and Pathologic Assessment. Cancers (Basel) 2021, 13. [CrossRef]
- De Guillebon, E.; Dardenne, A.; Saldmann, A.; Seguier, S.; Tran, T.; Paolini, L.; Lebbe, C.; Tartour, E. Beyond the concept of cold and hot tumors for the development of novel predictive biomarkers and the rational design of immunotherapy combination. Int J Cancer 2020, 147, 1509-1518. [CrossRef]
- Farc, O.; Cristea, V. An overview of the tumor microenvironment, from cells to complex networks (Review). Exp Ther Med 2021, 21, 96. [CrossRef]
- Dannenfelser, R.; Nome, M.; Tahiri, A.; Ursini-Siegel, J.; Vollan, H.K.M.; Haakensen, V.D.; Helland, A.; Naume, B.; Caldas, C.; Borresen-Dale, A.L.; et al. Data-driven analysis of immune infiltrate in a large cohort of breast cancer and its association with disease progression, ER activity, and genomic complexity. Oncotarget 2017, 8, 57121-57133. [CrossRef]
- Cunningham, M.; Gilkeson, G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol 2011, 40, 66-73. [CrossRef]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front Immunol 2015, 6, 635. [CrossRef]
- Okasha, S.A.; Ryu, S.; Do, Y.; McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Evidence for estradiol-induced apoptosis and dysregulated T cell maturation in the thymus. Toxicology 2001, 163, 49-62. [CrossRef]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol 2004, 173, 2227-2230. [CrossRef]
- Staples, J.E.; Gasiewicz, T.A.; Fiore, N.C.; Lubahn, D.B.; Korach, K.S.; Silverstone, A.E. Estrogen receptor alpha is necessary in thymic development and estradiol-induced thymic alterations. J Immunol 1999, 163, 4168-4174.
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov 2017, 7, 72-85. [CrossRef]
- McMurray, R.W.; Ndebele, K.; Hardy, K.J.; Jenkins, J.K. 17-beta-estradiol suppresses IL-2 and IL-2 receptor. Cytokine 2001, 14, 324-333. [CrossRef]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv Exp Med Biol 2020, 1277, 33-52. [CrossRef]
- Gajewski, T.F.; Meng, Y.; Harlin, H. Immune suppression in the tumor microenvironment. J Immunother 2006, 29, 233-240. [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int Immunol 2007, 19, 337-343. [CrossRef]
- Prieto, G.A.; Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 2006, 118, 58-65. [CrossRef]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol 2008, 214, 456-464. [CrossRef]
- Askenasy, N.; Kaminitz, A.; Yarkoni, S. Mechanisms of T regulatory cell function. Autoimmun Rev 2008, 7, 370-375. [CrossRef]
- Luo, C.Y.; Wang, L.; Sun, C.; Li, D.J. Estrogen enhances the functions of CD4(+)CD25(+)Foxp3(+) regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol 2011, 8, 50-58. [CrossRef]
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-alpha binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci Rep 2017, 7, 17289. [CrossRef]
- Itahashi, K.; Irie, T.; Nishikawa, H. Regulatory T-cell development in the tumor microenvironment. Eur J Immunol 2022, 52, 1216-1227. [CrossRef]
- Kang, M.J.; Kim, K.M.; Bae, J.S.; Park, H.S.; Lee, H.; Chung, M.J.; Moon, W.S.; Lee, D.G.; Jang, K.Y. Tumor-infiltrating PD1-Positive Lymphocytes and FoxP3-Positive Regulatory T Cells Predict Distant Metastatic Relapse and Survival of Clear Cell Renal Cell Carcinoma. Transl Oncol 2013, 6, 282-289. [CrossRef]
- Park, H.J.; Kusnadi, A.; Lee, E.J.; Kim, W.W.; Cho, B.C.; Lee, I.J.; Seong, J.; Ha, S.J. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell Immunol 2012, 278, 76-83. [CrossRef]
- Kim, H.R.; Park, H.J.; Son, J.; Lee, J.G.; Chung, K.Y.; Cho, N.H.; Shim, H.S.; Park, S.; Kim, G.; In Yoon, H.; et al. Tumor microenvironment dictates regulatory T cell phenotype: Upregulated immune checkpoints reinforce suppressive function. J Immunother Cancer 2019, 7, 339. [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol 2019, 16, 356-371. [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A 2013, 110, 17945-17950. [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924-940. [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Menetrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers (Basel) 2021, 13. [CrossRef]
- Pedroza-Pacheco, I.; Madrigal, A.; Saudemont, A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol 2013, 10, 222-229. [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 2019, 20, 724-735. [CrossRef]
- Saraiva, M.; O'Garra, A. The regulation of IL-10 production by immune cells. Nat Rev Immunol 2010, 10, 170-181. [CrossRef]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J Hematol Oncol 2022, 15, 135. [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003, 198, 1875-1886. [CrossRef]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635-646. [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007, 204, 1257-1265. [CrossRef]
- Stockis, J.; Roychoudhuri, R.; Halim, T.Y.F. Regulation of regulatory T cells in cancer. Immunology 2019, 157, 219-231. [CrossRef]
- Li, C.; Liang, Y.Y.; Feng, X.H.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol Cell 2008, 31, 835-849. [CrossRef]
- York, B.; Yu, C.; Sagen, J.V.; Liu, Z.; Nikolai, B.C.; Wu, R.C.; Finegold, M.; Xu, J.; O'Malley, B.W. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc Natl Acad Sci U S A 2010, 107, 11122-11127. [CrossRef]
- Liao, L.; Kuang, S.Q.; Yuan, Y.; Gonzalez, S.M.; O'Malley, B.W.; Xu, J. Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J Steroid Biochem Mol Biol 2002, 83, 3-14. [CrossRef]
- Louet, J.F.; Coste, A.; Amazit, L.; Tannour-Louet, M.; Wu, R.C.; Tsai, S.Y.; Tsai, M.J.; Auwerx, J.; O'Malley, B.W. Oncogenic steroid receptor coactivator-3 is a key regulator of the white adipogenic program. Proc Natl Acad Sci U S A 2006, 103, 17868-17873. [CrossRef]
- Coste, A.; Louet, J.F.; Lagouge, M.; Lerin, C.; Antal, M.C.; Meziane, H.; Schoonjans, K.; Puigserver, P.; O'Malley, B.W.; Auwerx, J. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1alpha. Proc Natl Acad Sci U S A 2008, 105, 17187-17192. [CrossRef]
- Hill, J.A.; Hall, J.A.; Sun, C.M.; Cai, Q.; Ghyselinck, N.; Chambon, P.; Belkaid, Y.; Mathis, D.; Benoist, C. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 2008, 29, 758-770. [CrossRef]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 2008, 181, 2277-2284. [CrossRef]
- Alvarado, C.V.; Rubio, M.F.; Fernandez Larrosa, P.N.; Panelo, L.C.; Azurmendi, P.J.; Ruiz Grecco, M.; Martinez-Noel, G.A.; Costas, M.A. The levels of RAC3 expression are up regulated by TNF in the inflammatory response. FEBS Open Bio 2014, 4, 450-457. [CrossRef]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921-931. [CrossRef]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932-940. [CrossRef]
- Grinberg-Bleyer, Y.; Caron, R.; Seeley, J.J.; De Silva, N.S.; Schindler, C.W.; Hayden, M.S.; Klein, U.; Ghosh, S. The Alternative NF-kappaB Pathway in Regulatory T Cell Homeostasis and Suppressive Function. J Immunol 2018, 200, 2362-2371. [CrossRef]
- He, C.; Shan, N.; Xu, P.; Ge, H.; Yuan, Y.; Liu, Y.; Zhang, P.; Wen, L.; Zhang, F.; Xiong, L.; et al. Hypoxia-induced Downregulation of SRC-3 Suppresses Trophoblastic Invasion and Migration Through Inhibition of the AKT/mTOR Pathway: Implications for the Pathogenesis of Preeclampsia. Sci Rep 2019, 9, 10349. [CrossRef]
- Chen, W.; Zhuo, M.; Lu, X.; Xia, X.; Zhao, Y.; Huang, Z.; Xu, J.; Li, W.; Yu, C. SRC-3 protects intestine from DSS-induced colitis by inhibiting inflammation and promoting goblet cell differentiation through enhancement of KLF4 expression. Int J Biol Sci 2018, 14, 2051-2064. [CrossRef]
- Mullany, L.K.; Rohira, A.D.; Leach, J.P.; Kim, J.H.; Monroe, T.O.; Ortiz, A.R.; Stork, B.; Gaber, M.W.; Sarkar, P.; Sikora, A.G.; et al. A steroid receptor coactivator stimulator (MCB-613) attenuates adverse remodeling after myocardial infarction. Proc Natl Acad Sci U S A 2020, 117, 31353-31364. [CrossRef]
- Yu, C.; York, B.; Wang, S.; Feng, Q.; Xu, J.; O'Malley, B.W. An essential function of the SRC-3 coactivator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell 2007, 25, 765-778. [CrossRef]
- Chen, Q.; Chen, T.; Xu, Y.; Zhu, J.; Jiang, Y.; Zhao, Y.; Xu, J.; Yu, C. Steroid receptor coactivator 3 is required for clearing bacteria and repressing inflammatory response in Escherichia coli-induced septic peritonitis. J Immunol 2010, 185, 5444-5452. [CrossRef]
- Alfonso-Prieto, M.; Biarnes, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J Am Chem Soc 2009, 131, 11751-11761. [CrossRef]
- Colo, G.P.; Rosato, R.R.; Grant, S.; Costas, M.A. RAC3 down-regulation sensitizes human chronic myeloid leukemia cells to TRAIL-induced apoptosis. FEBS Lett 2007, 581, 5075-5081. [CrossRef]
- Colo, G.P.; Rubio, M.F.; Nojek, I.M.; Werbajh, S.E.; Echeverria, P.C.; Alvarado, C.V.; Nahmod, V.E.; Galigniana, M.D.; Costas, M.A. The p160 nuclear receptor co-activator RAC3 exerts an anti-apoptotic role through a cytoplasmatic action. Oncogene 2008, 27, 2430-2444. [CrossRef]
- Chen, W.; Lu, X.; Chen, Y.; Li, M.; Mo, P.; Tong, Z.; Wang, W.; Wan, W.; Su, G.; Xu, J.; et al. Steroid Receptor Coactivator 3 Contributes to Host Defense against Enteric Bacteria by Recruiting Neutrophils via Upregulation of CXCL2 Expression. J Immunol 2017, 198, 1606-1615. [CrossRef]
- Werbajh, S.; Nojek, I.; Lanz, R.; Costas, M.A. RAC-3 is a NF-kappa B coactivator. FEBS Lett 2000, 485, 195-199. [CrossRef]
- Wu, R.C.; Qin, J.; Hashimoto, Y.; Wong, J.; Xu, J.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol 2002, 22, 3549-3561. [CrossRef]
- Truong, T.H.; Hu, H.; Temiz, N.A.; Hagen, K.M.; Girard, B.J.; Brady, N.J.; Schwertfeger, K.L.; Lange, C.A.; Ostrander, J.H. Cancer Stem Cell Phenotypes in ER(+) Breast Cancer Models Are Promoted by PELP1/AIB1 Complexes. Mol Cancer Res 2018, 16, 707-719. [CrossRef]
- Truong, T.H.; Benner, E.A.; Hagen, K.M.; Temiz, N.A.; Kerkvliet, C.P.; Wang, Y.; Cortes-Sanchez, E.; Yang, C.H.; Trousdell, M.C.; Pengo, T.; et al. PELP1/SRC-3-dependent regulation of metabolic PFKFB kinases drives therapy resistant ER(+) breast cancer. Oncogene 2021, 40, 4384-4397. [CrossRef]
- Panelo, L.C.; Machado, M.S.; Rubio, M.F.; Jaworski, F.; Alvarado, C.V.; Paz, L.A.; Urtreger, A.J.; Vazquez, E.; Costas, M.A. High RAC3 expression levels are required for induction and maintaining of cancer cell stemness. Oncotarget 2018, 9, 5848-5860. [CrossRef]
- Dubrovska, A.; Hartung, A.; Bouchez, L.C.; Walker, J.R.; Reddy, V.A.; Cho, C.Y.; Schultz, P.G. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br J Cancer 2012, 107, 43-52. [CrossRef]
- Miyoshi, Y.; Shien, T.; Ogiya, A.; Ishida, N.; Yamazaki, K.; Horii, R.; Horimoto, Y.; Masuda, N.; Yasojima, H.; Inao, T.; et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res 2016, 18, 73. [CrossRef]
- Dancik, G.M.; Voutsas, I.F.; Vlahopoulos, S. Aldehyde Dehydrogenase Enzyme Functions in Acute Leukemia Stem Cells. Front Biosci (Schol Ed) 2022, 14, 8. [CrossRef]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res 2021, 81, 5919-5934. [CrossRef]
- Huang, Y.; Duan, X.; Wang, Z.; Sun, Y.; Guan, Q.; Kang, L.; Zhang, Q.; Fang, L.; Li, J.; Wong, J. An acetylation-enhanced interaction between transcription factor Sox2 and the steroid receptor coactivators facilitates Sox2 transcriptional activity and function. J Biol Chem 2021, 297, 101389. [CrossRef]
- Domenici, G.; Aurrekoetxea-Rodriguez, I.; Simoes, B.M.; Rabano, M.; Lee, S.Y.; Millan, J.S.; Comaills, V.; Oliemuller, E.; Lopez-Ruiz, J.A.; Zabalza, I.; et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151-3169. [CrossRef]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine Therapy of Estrogen Receptor-Positive Breast Cancer Cells: Early Differential Effects on Stem Cell Markers. Front Oncol 2017, 7, 184. [CrossRef]
- Percharde, M.; Azuara, V. Essential roles for the nuclear receptor coactivator Ncoa3 in pluripotency. Cell Cycle 2013, 12, 195-196. [CrossRef]
- Percharde, M.; Lavial, F.; Ng, J.H.; Kumar, V.; Tomaz, R.A.; Martin, N.; Yeo, J.C.; Gil, J.; Prabhakar, S.; Ng, H.H.; et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev 2012, 26, 2286-2298. [CrossRef]
- Wu, Z.; Yang, M.; Liu, H.; Guo, H.; Wang, Y.; Cheng, H.; Chen, L. Role of nuclear receptor coactivator 3 (Ncoa3) in pluripotency maintenance. J Biol Chem 2012, 287, 38295-38304. [CrossRef]
- Chitilian, J.M.; Thillainadesan, G.; Manias, J.L.; Chang, W.Y.; Walker, E.; Isovic, M.; Stanford, W.L.; Torchia, J. Critical components of the pluripotency network are targets for the p300/CBP interacting protein (p/CIP) in embryonic stem cells. Stem Cells 2014, 32, 204-215. [CrossRef]
- Hu, M.; Zeng, H.; Chen, S.; Xu, Y.; Wang, S.; Tang, Y.; Wang, X.; Du, C.; Shen, M.; Chen, F.; et al. SRC-3 is involved in maintaining hematopoietic stem cell quiescence by regulation of mitochondrial metabolism in mice. Blood 2018, 132, 911-923. [CrossRef]
- Ma, G.; Ren, Y.; Wang, K.; He, J. SRC-3 has a role in cancer other than as a nuclear receptor coactivator. Int J Biol Sci 2011, 7, 664-672. [CrossRef]
- Torres-Arzayus, M.I.; Font de Mora, J.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004, 6, 263-274. [CrossRef]
- Reiter, R.; Oh, A.S.; Wellstein, A.; Riegel, A.T. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Delta3 on estrogenic ligands with different intrinsic activity. Oncogene 2004, 23, 403-409. [CrossRef]
- Reiter, R.; Wellstein, A.; Riegel, A.T. An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J Biol Chem 2001, 276, 39736-39741. [CrossRef]
- Bautista, S.; Valles, H.; Walker, R.L.; Anzick, S.; Zeillinger, R.; Meltzer, P.; Theillet, C. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res 1998, 4, 2925-2929.
- Hudelist, G.; Czerwenka, K.; Kubista, E.; Marton, E.; Pischinger, K.; Singer, C.F. Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat 2003, 78, 193-204. [CrossRef]
- Lee, K.; Lee, A.; Song, B.J.; Kang, C.S. Expression of AIB1 protein as a prognostic factor in breast cancer. World J Surg Oncol 2011, 9, 139. [CrossRef]
- Osborne, C.K.; Bardou, V.; Hopp, T.A.; Chamness, G.C.; Hilsenbeck, S.G.; Fuqua, S.A.; Wong, J.; Allred, D.C.; Clark, G.M.; Schiff, R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 2003, 95, 353-361. [CrossRef]
- Zhao, C.; Yasui, K.; Lee, C.J.; Kurioka, H.; Hosokawa, Y.; Oka, T.; Inazawa, J. Elevated expression levels of NCOA3, TOP1, and TFAP2C in breast tumors as predictors of poor prognosis. Cancer 2003, 98, 18-23. [CrossRef]
- Nakles, R.E.; Shiffert, M.T.; Diaz-Cruz, E.S.; Cabrera, M.C.; Alotaiby, M.; Miermont, A.M.; Riegel, A.T.; Furth, P.A. Altered AIB1 or AIB1Delta3 expression impacts ERalpha effects on mammary gland stromal and epithelial content. Mol Endocrinol 2011, 25, 549-563. [CrossRef]
- Tilli, M.T.; Reiter, R.; Oh, A.S.; Henke, R.T.; McDonnell, K.; Gallicano, G.I.; Furth, P.A.; Riegel, A.T. Overexpression of an N-terminally truncated isoform of the nuclear receptor coactivator amplified in breast cancer 1 leads to altered proliferation of mammary epithelial cells in transgenic mice. Mol Endocrinol 2005, 19, 644-656. [CrossRef]
- Avivar, A.; Garcia-Macias, M.C.; Ascaso, E.; Herrera, G.; O'Connor, J.E.; Font de Mora, J. Moderate overexpression of AIB1 triggers pre-neoplastic changes in mammary epithelium. FEBS Lett 2006, 580, 5222-5226. [CrossRef]
- Kuang, S.Q.; Liao, L.; Wang, S.; Medina, D.; O'Malley, B.W.; Xu, J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res 2005, 65, 7993-8002. [CrossRef]
- Kuang, S.Q.; Liao, L.; Zhang, H.; Lee, A.V.; O'Malley, B.W.; Xu, J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res 2004, 64, 1875-1885. [CrossRef]
- Planas-Silva, M.D.; Shang, Y.; Donaher, J.L.; Brown, M.; Weinberg, R.A. AIB1 enhances estrogen-dependent induction of cyclin D1 expression. Cancer Res 2001, 61, 3858-3862.
- Suen, C.S.; Berrodin, T.J.; Mastroeni, R.; Cheskis, B.J.; Lyttle, C.R.; Frail, D.E. A transcriptional coactivator, steroid receptor coactivator-3, selectively augments steroid receptor transcriptional activity. J Biol Chem 1998, 273, 27645-27653. [CrossRef]
- Tikkanen, M.K.; Carter, D.J.; Harris, A.M.; Le, H.M.; Azorsa, D.O.; Meltzer, P.S.; Murdoch, F.E. Endogenously expressed estrogen receptor and coactivator AIB1 interact in MCF-7 human breast cancer cells. Proc Natl Acad Sci U S A 2000, 97, 12536-12540. [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Chou, C.K.; Pintilie, G.D.; Shen, H.; Foulds, C.E.; Fan, G.; Serysheva, I.; Ludtke, S.J.; et al. Structural and Functional Impacts of ER Coactivator Sequential Recruitment. Mol Cell 2017, 67, 733-743 e734. [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016, 5. [CrossRef]
- Zhao, Y.; Laws, M.J.; Guillen, V.S.; Ziegler, Y.; Min, J.; Sharma, A.; Kim, S.H.; Chu, D.; Park, B.H.; Oesterreich, S.; et al. Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors. Cancer Res 2017, 77, 5602-5613. [CrossRef]
- Wang, Z.; Rose, D.W.; Hermanson, O.; Liu, F.; Herman, T.; Wu, W.; Szeto, D.; Gleiberman, A.; Krones, A.; Pratt, K.; et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci U S A 2000, 97, 13549-13554. [CrossRef]
- Kirkegaard, T.; McGlynn, L.M.; Campbell, F.M.; Muller, S.; Tovey, S.M.; Dunne, B.; Nielsen, K.V.; Cooke, T.G.; Bartlett, J.M. Amplified in breast cancer 1 in human epidermal growth factor receptor - positive tumors of tamoxifen-treated breast cancer patients. Clin Cancer Res 2007, 13, 1405-1411. [CrossRef]
- Fereshteh, M.P.; Tilli, M.T.; Kim, S.E.; Xu, J.; O'Malley, B.W.; Wellstein, A.; Furth, P.A.; Riegel, A.T. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res 2008, 68, 3697-3706. [CrossRef]
- Qin, L.; Liao, L.; Redmond, A.; Young, L.; Yuan, Y.; Chen, H.; O'Malley, B.W.; Xu, J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol 2008, 28, 5937-5950. [CrossRef]
- Long, W.; Yi, P.; Amazit, L.; LaMarca, H.L.; Ashcroft, F.; Kumar, R.; Mancini, M.A.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell 2010, 37, 321-332. [CrossRef]
- Yan, J.; Erdem, H.; Li, R.; Cai, Y.; Ayala, G.; Ittmann, M.; Yu-Lee, L.Y.; Tsai, S.Y.; Tsai, M.J. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res 2008, 68, 5460-5468. [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8. [CrossRef]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int 2003, 3, 17. [CrossRef]
- Oda, H.; Takeichi, M. Evolution: structural and functional diversity of cadherin at the adherens junction. J Cell Biol 2011, 193, 1137-1146. [CrossRef]
- Kaszak, I.; Witkowska-Pilaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Ngosa Toka, F.; Jurka, P. Role of Cadherins in Cancer-A Review. Int J Mol Sci 2020, 21. [CrossRef]
- Varisli, L.; Tolan, V. Increased ROS alters E-/N-cadherin levels and promotes migration in prostate cancer cells. Bratisl Lek Listy 2022, 123, 752-757. [CrossRef]
- Varisli, L.; Tolan, V.; Cen, J.H.; Vlahopoulos, S.; Cen, O. Dissecting the effects of androgen deprivation therapy on cadherin switching in advanced prostate cancer: A molecular perspective. Oncol Res 2022, 30, 137-155. [CrossRef]
- Ferreira Almeida, C.; Oliveira, A.; Joao Ramos, M.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER(+)) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem Pharmacol 2020, 177, 113989. [CrossRef]
- Early Breast Cancer Trialists' Collaborative, G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005, 365, 1687-1717. [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427-438 e426. [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714-3728. [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013, 45, 1446-1451. [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013, 45, 1439-1445. [CrossRef]
- Heck, S.; Rom, J.; Thewes, V.; Becker, N.; Blume, B.; Sinn, H.P.; Deuschle, U.; Sohn, C.; Schneeweiss, A.; Lichter, P. Estrogen-related receptor alpha expression and function is associated with the transcriptional coregulator AIB1 in breast carcinoma. Cancer Res 2009, 69, 5186-5193. [CrossRef]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J Biol Chem 2015, 6, 231-239. [CrossRef]
- Ozyurt, R.; Ozpolat, B. Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers (Basel) 2022, 14. [CrossRef]
- Feng, Q.; O'Malley, B.W. Nuclear receptor modulation--role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 2014, 90, 39-43. [CrossRef]
- Fawell, S.E.; White, R.; Hoare, S.; Sydenham, M.; Page, M.; Parker, M.G. Inhibition of estrogen receptor-DNA binding by the "pure" antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc Natl Acad Sci U S A 1990, 87, 6883-6887. [CrossRef]
- Wijayaratne, A.L.; Nagel, S.C.; Paige, L.A.; Christensen, D.J.; Norris, J.D.; Fowlkes, D.M.; McDonnell, D.P. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology 1999, 140, 5828-5840. [CrossRef]
- Yeh, W.L.; Shioda, K.; Coser, K.R.; Rivizzigno, D.; McSweeney, K.R.; Shioda, T. Fulvestrant-induced cell death and proteasomal degradation of estrogen receptor alpha protein in MCF-7 cells require the CSK c-Src tyrosine kinase. PLoS One 2013, 8, e60889. [CrossRef]
- Boszkiewicz, K.; Piwowar, A.; Petryszyn, P. Aromatase Inhibitors and Risk of Metabolic and Cardiovascular Adverse Effects in Breast Cancer Patients-A Systematic Review and Meta-Analysis. J Clin Med 2022, 11. [CrossRef]
- Lonard, D.M.; Tsai, S.Y.; O'Malley, B.W. Selective estrogen receptor modulators 4-hydroxytamoxifen and raloxifene impact the stability and function of SRC-1 and SRC-3 coactivator proteins. Mol Cell Biol 2004, 24, 14-24. [CrossRef]
- Su, Q.; Hu, S.; Gao, H.; Ma, R.; Yang, Q.; Pan, Z.; Wang, T.; Li, F. Role of AIB1 for tamoxifen resistance in estrogen receptor-positive breast cancer cells. Oncology 2008, 75, 159-168. [CrossRef]
- Hurtado, A.; Holmes, K.A.; Geistlinger, T.R.; Hutcheson, I.R.; Nicholson, R.I.; Brown, M.; Jiang, J.; Howat, W.J.; Ali, S.; Carroll, J.S. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature 2008, 456, 663-666. [CrossRef]
- Lebert, J.; Lilly, E.J. Developments in the Management of Metastatic HER2-Positive Breast Cancer: A Review. Curr Oncol 2022, 29, 2539-2549. [CrossRef]
- Bouras, T.; Southey, M.C.; Venter, D.J. Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res 2001, 61, 903-907.
- Font de Mora, J.; Brown, M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol 2000, 20, 5041-5047. [CrossRef]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 2004, 96, 926-935. [CrossRef]
- Lahusen, T.; Fereshteh, M.; Oh, A.; Wellstein, A.; Riegel, A.T. Epidermal growth factor receptor tyrosine phosphorylation and signaling controlled by a nuclear receptor coactivator, amplified in breast cancer 1. Cancer Res 2007, 67, 7256-7265. [CrossRef]
- Han, S.J.; Jain, P.; Gilad, Y.; Xia, Y.; Sung, N.; Park, M.J.; Dean, A.M.; Lanz, R.B.; Xu, J.; Dacso, C.C.; et al. Steroid receptor coactivator 3 is a key modulator of regulatory T cell-mediated tumor evasion. Proc Natl Acad Sci U S A 2023, 120, e2221707120. [CrossRef]
- Han, S.J.; Sung, N.; Wang, J.; O'Malley, B.W.; Lonard, D.M. Steroid receptor coactivator-3 inhibition generates breast cancer antitumor immune microenvironment. Breast Cancer Res 2022, 24, 73. [CrossRef]
- Kunz, M.; Toksoy, A.; Goebeler, M.; Engelhardt, E.; Brocker, E.; Gillitzer, R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol 1999, 189, 552-558. [CrossRef]
- Neo, S.Y.; Lundqvist, A. The Multifaceted Roles of CXCL9 Within the Tumor Microenvironment. Adv Exp Med Biol 2020, 1231, 45-51. [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859-874. [CrossRef]
- Walser, T.C.; Ma, X.; Kundu, N.; Dorsey, R.; Goloubeva, O.; Fulton, A.M. Immune-mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J Immunother 2007, 30, 490-498. [CrossRef]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 2010, 28, 105-113. [CrossRef]
- Specht, K.; Harbeck, N.; Smida, J.; Annecke, K.; Reich, U.; Naehrig, J.; Langer, R.; Mages, J.; Busch, R.; Kruse, E.; et al. Expression profiling identifies genes that predict recurrence of breast cancer after adjuvant CMF-based chemotherapy. Breast Cancer Res Treat 2009, 118, 45-56. [CrossRef]
- Hardenberg, J.B.; Braun, A.; Schon, M.P. A Yin and Yang in Epithelial Immunology: The Roles of the alpha(E)(CD103)beta(7) Integrin in T Cells. J Invest Dermatol 2018, 138, 23-31. [CrossRef]
- Hoffmann, J.C.; Schon, M.P. Integrin alpha(E)(CD103)beta(7) in Epithelial Cancer. Cancers (Basel) 2021, 13. [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 2013, 14, 1294-1301. [CrossRef]
- Cepek, K.L.; Shaw, S.K.; Parker, C.M.; Russell, G.J.; Morrow, J.S.; Rimm, D.L.; Brenner, M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature 1994, 372, 190-193. [CrossRef]
- Karecla, P.I.; Bowden, S.J.; Green, S.J.; Kilshaw, P.J. Recognition of E-cadherin on epithelial cells by the mucosal T cell integrin alpha M290 beta 7 (alpha E beta 7). Eur J Immunol 1995, 25, 852-856. [CrossRef]
- Le Floc'h, A.; Jalil, A.; Vergnon, I.; Le Maux Chansac, B.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 2007, 204, 559-570. [CrossRef]
- Karecla, P.I.; Green, S.J.; Bowden, S.J.; Coadwell, J.; Kilshaw, P.J. Identification of a binding site for integrin alphaEbeta7 in the N-terminal domain of E-cadherin. J Biol Chem 1996, 271, 30909-30915. [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res 2019, 79, 1113-1123. [CrossRef]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J Urol 2015, 194, 556-562. [CrossRef]
- Austrup, F.; Rebstock, S.; Kilshaw, P.J.; Hamann, A. Transforming growth factor-beta 1-induced expression of the mucosa-related integrin alpha E on lymphocytes is not associated with mucosa-specific homing. Eur J Immunol 1995, 25, 1487-1491. [CrossRef]
- Pauls, K.; Schon, M.; Kubitza, R.C.; Homey, B.; Wiesenborn, A.; Lehmann, P.; Ruzicka, T.; Parker, C.M.; Schon, M.P. Role of integrin alphaE(CD103)beta7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol 2001, 117, 569-575. [CrossRef]
- Robinson, P.W.; Green, S.J.; Carter, C.; Coadwell, J.; Kilshaw, P.J. Studies on transcriptional regulation of the mucosal T-cell integrin alphaEbeta7 (CD103). Immunology 2001, 103, 146-154. [CrossRef]
- Franciszkiewicz, K.; Le Floc'h, A.; Boutet, M.; Vergnon, I.; Schmitt, A.; Mami-Chouaib, F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res 2013, 73, 617-628. [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369-380. [CrossRef]
- Boutet, M.; Gauthier, L.; Leclerc, M.; Gros, G.; de Montpreville, V.; Theret, N.; Donnadieu, E.; Mami-Chouaib, F. TGFbeta Signaling Intersects with CD103 Integrin Signaling to Promote T-Lymphocyte Accumulation and Antitumor Activity in the Lung Tumor Microenvironment. Cancer Res 2016, 76, 1757-1769. [CrossRef]
- Gebhardt, T.; Mackay, L.K. Local immunity by tissue-resident CD8(+) memory T cells. Front Immunol 2012, 3, 340. [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age (Dordr) 2009, 31, 285-291. [CrossRef]
- Grundemann, C.; Bauer, M.; Schweier, O.; von Oppen, N.; Lassing, U.; Saudan, P.; Becker, K.F.; Karp, K.; Hanke, T.; Bachmann, M.F.; et al. Cutting edge: identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1. J Immunol 2006, 176, 1311-1315. [CrossRef]
- Ito, M.; Maruyama, T.; Saito, N.; Koganei, S.; Yamamoto, K.; Matsumoto, N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med 2006, 203, 289-295. [CrossRef]
- Li, L.; Wan, S.; Tao, K.; Wang, G.; Zhao, E. KLRG1 restricts memory T cell antitumor immunity. Oncotarget 2016, 7, 61670-61678. [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 2015, 15, 243-254. [CrossRef]
- Meinicke, H.; Bremser, A.; Brack, M.; Schrenk, K.; Pircher, H.; Izcue, A. KLRG1 impairs regulatory T-cell competitive fitness in the gut. Immunology 2017, 152, 65-73. [CrossRef]
- Tessmer, M.S.; Fugere, C.; Stevenaert, F.; Naidenko, O.V.; Chong, H.J.; Leclercq, G.; Brossay, L. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol 2007, 19, 391-400. [CrossRef]
- Schwartzkopff, S.; Woyciechowski, S.; Aichele, U.; Flecken, T.; Zhang, N.; Thimme, R.; Pircher, H. TGF-beta downregulates KLRG1 expression in mouse and human CD8(+) T cells. Eur J Immunol 2015, 45, 2212-2217. [CrossRef]
- Attig, S.; Hennenlotter, J.; Pawelec, G.; Klein, G.; Koch, S.D.; Pircher, H.; Feyerabend, S.; Wernet, D.; Stenzl, A.; Rammensee, H.G.; et al. Simultaneous infiltration of polyfunctional effector and suppressor T cells into renal cell carcinomas. Cancer Res 2009, 69, 8412-8419. [CrossRef]
- Baitsch, L.; Legat, A.; Barba, L.; Fuertes Marraco, S.A.; Rivals, J.P.; Baumgaertner, P.; Christiansen-Jucht, C.; Bouzourene, H.; Rimoldi, D.; Pircher, H.; et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One 2012, 7, e30852. [CrossRef]
- Sun, Y.; Jing, J.; Xu, H.; Xu, L.; Hu, H.; Tang, C.; Liu, S.; Wei, Q.; Duan, R.; Guo, J.; et al. N-cadherin inhibitor creates a microenvironment that protect TILs from immune checkpoints and Treg cells. J Immunother Cancer 2021, 9. [CrossRef]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med 2011, 3, 111ra120. [CrossRef]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633-642. [CrossRef]
- Kolijn, K.; Verhoef, E.I.; Smid, M.; Bottcher, R.; Jenster, G.W.; Debets, R.; van Leenders, G. Epithelial-Mesenchymal Transition in Human Prostate Cancer Demonstrates Enhanced Immune Evasion Marked by IDO1 Expression. Cancer Res 2018, 78, 4671-4679. [CrossRef]
- Gu, Y.; Zhang, Z.; Ten Dijke, P. Harnessing epithelial-mesenchymal plasticity to boost cancer immunotherapy. Cell Mol Immunol 2023, 20, 318-340. [CrossRef]
- Raj, G.V.; Sareddy, G.R.; Ma, S.; Lee, T.K.; Viswanadhapalli, S.; Li, R.; Liu, X.; Murakami, S.; Chen, C.C.; Lee, W.R.; et al. Estrogen receptor coregulator binding modulators (ERXs) effectively target estrogen receptor positive human breast cancers. Elife 2017, 6. [CrossRef]
- Song, X.; Chen, J.; Zhao, M.; Zhang, C.; Yu, Y.; Lonard, D.M.; Chow, D.C.; Palzkill, T.; Xu, J.; O'Malley, B.W.; et al. Development of potent small-molecule inhibitors to drug the undruggable steroid receptor coactivator-3. Proc Natl Acad Sci U S A 2016, 113, 4970-4975. [CrossRef]
- Li, X.; Lonard, D.M.; Jung, S.Y.; Malovannaya, A.; Feng, Q.; Qin, J.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 2006, 124, 381-392. [CrossRef]
- Wu, R.C.; Feng, Q.; Lonard, D.M.; O'Malley, B.W. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell 2007, 129, 1125-1140. [CrossRef]
- Ferry, C.; Gaouar, S.; Fischer, B.; Boeglin, M.; Paul, N.; Samarut, E.; Piskunov, A.; Pankotai-Bodo, G.; Brino, L.; Rochette-Egly, C. Cullin 3 mediates SRC-3 ubiquitination and degradation to control the retinoic acid response. Proc Natl Acad Sci U S A 2011, 108, 20603-20608. [CrossRef]
- Yao, C.; Su, L.; Zhang, F.; Zhu, X.; Zhu, Y.; Wei, L.; Jiao, X.; Hou, Y.; Chen, X.; Wang, W.; et al. Thevebioside, the active ingredient of traditional Chinese medicine, promotes ubiquitin-mediated SRC-3 degradation to induce NSCLC cells apoptosis. Cancer Lett 2020, 493, 167-177. [CrossRef]
- Zhao, Z.; Zhang, X.; Wen, L.; Yi, S.; Hu, J.; Ruan, J.; Zhao, F.; Cui, G.; Fang, J.; Chen, Y. Steroid receptor coactivator-3 is a pivotal target of gambogic acid in B-cell Non-Hodgkin lymphoma and an inducer of histone H3 deacetylation. Eur J Pharmacol 2016, 789, 46-59. [CrossRef]
- Yan, F.; Yu, Y.; Chow, D.C.; Palzkill, T.; Madoux, F.; Hodder, P.; Chase, P.; Griffin, P.R.; O'Malley, B.W.; Lonard, D.M. Identification of verrucarin a as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. PLoS One 2014, 9, e95243. [CrossRef]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.C.; Palzkill, T.G.; O'Malley, B.W. Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol 2011, 25, 2041-2053. [CrossRef]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.C.; Palzkill, T.G.; Wang, J.; Qi, R.; Matzuk, A.J.; Song, X.; Madoux, F.; et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res 2014, 74, 1506-1517. [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001, 98, 8554-8559. [CrossRef]
- Cheng, B.; Ren, Y.; Cao, H.; Chen, J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem 2020, 199, 112377. [CrossRef]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct Target Ther 2022, 7, 181. [CrossRef]
- Wang, Y.; Zhou, Y.; Cao, S.; Sun, Y.; Dong, Z.; Li, C.; Wang, H.; Yao, Y.; Yu, H.; Song, X.; et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem 2021, 111, 104833. [CrossRef]
- Han, S.J.; Jain, P.; Gilad, Y.; Xia, Y.; Sung, N.; Park, M.J.; Dean, A.M.; Lanz, R.B.; Xu, J.; Dacso, C.C.; et al. Steroid Receptor Coactivator-3 is a Key Modulator of Regulatory T Cell-Mediated Tumor Evasion. bioRxiv 2023. [CrossRef]
- Qin, L.; Chen, J.; Lu, D.; Jain, P.; Yu, Y.; Cardenas, D.; Peng, X.; Yu, X.; Xu, J.; Wang, J.; et al. Development of improved SRC-3 inhibitors as breast cancer therapeutic agents. Endocr Relat Cancer 2021, 28, 657-670. [CrossRef]
- Manmuan, S.; Sakunrangsit, N.; Ketchart, W. Salinomycin overcomes acquired tamoxifen resistance through AIB1 and inhibits cancer cell invasion in endocrine resistant breast cancer. Clin Exp Pharmacol Physiol 2017, 44, 1042-1052. [CrossRef]
- Wang, H.; Zhang, H.; Zhu, Y.; Wu, Z.; Cui, C.; Cai, F. Anticancer Mechanisms of Salinomycin in Breast Cancer and Its Clinical Applications. Front Oncol 2021, 11, 654428. [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004, 64, 9152-9159. [CrossRef]
- Varisli, L.; Cen, O.; Vlahopoulos, S. Dissecting pharmacological effects of chloroquine in cancer treatment: interference with inflammatory signaling pathways. Immunology 2020, 159, 257-278. [CrossRef]
- Stagni, V.; Kaminari, A.; Contadini, C.; Barila, D.; Sessa, R.L.; Sideratou, Z.; Vlahopoulos, S.A.; Tsiourvas, D. A Triphenylphosphonium-Functionalized Delivery System for an ATM Kinase Inhibitor That Ameliorates Doxorubicin Resistance in Breast Carcinoma Mammospheres. Cancers (Basel) 2023, 15. [CrossRef]
- Stagni, V.; Kaminari, A.; Sideratou, Z.; Sakellis, E.; Vlahopoulos, S.A.; Tsiourvas, D. Targeting breast cancer stem-like cells using chloroquine encapsulated by a triphenylphosphonium-functionalized hyperbranched polymer. Int J Pharm 2020, 585, 119465. [CrossRef]
- An, Y.; Wu, J.; Yang, B.; Zhu, Z.; Gao, M.; Yu, C.; Yang, C.J. Selection and Application of DNA Aptamer Against Oncogene Amplified in Breast Cancer 1. J Mol Evol 2015, 81, 179-185. [CrossRef]
Figure 1.
The estrogen signaling is conveyed through ERs in cells. ERs may act in as genomic or non-genomic manner, upon estrogen induction. In genomic action, ERs located in the cytoplasm leave from HSPs upon estrogen induction, translocate to the nucleus with some co-regulators, including the SRC family members, and consequently bind to the promoter of estrogen-sensitive genes and directly regulate their expression. In non-genomic action, ERs located in the cytoplasm leave HSPs upon estrogen induction and are involved in the regulation of activity of various intracellular signaling pathways. GPR30, an ER located to the cell membrane, is involved in non-genomic actions in an intracellular second messengers dependent or independent manner and regulate the activity of many signaling pathways. Although non genomic actions of ERs generally results in the regulation of expression of various genes, ERs do not bind directly to the promoter of target genes.
Figure 1.
The estrogen signaling is conveyed through ERs in cells. ERs may act in as genomic or non-genomic manner, upon estrogen induction. In genomic action, ERs located in the cytoplasm leave from HSPs upon estrogen induction, translocate to the nucleus with some co-regulators, including the SRC family members, and consequently bind to the promoter of estrogen-sensitive genes and directly regulate their expression. In non-genomic action, ERs located in the cytoplasm leave HSPs upon estrogen induction and are involved in the regulation of activity of various intracellular signaling pathways. GPR30, an ER located to the cell membrane, is involved in non-genomic actions in an intracellular second messengers dependent or independent manner and regulate the activity of many signaling pathways. Although non genomic actions of ERs generally results in the regulation of expression of various genes, ERs do not bind directly to the promoter of target genes.
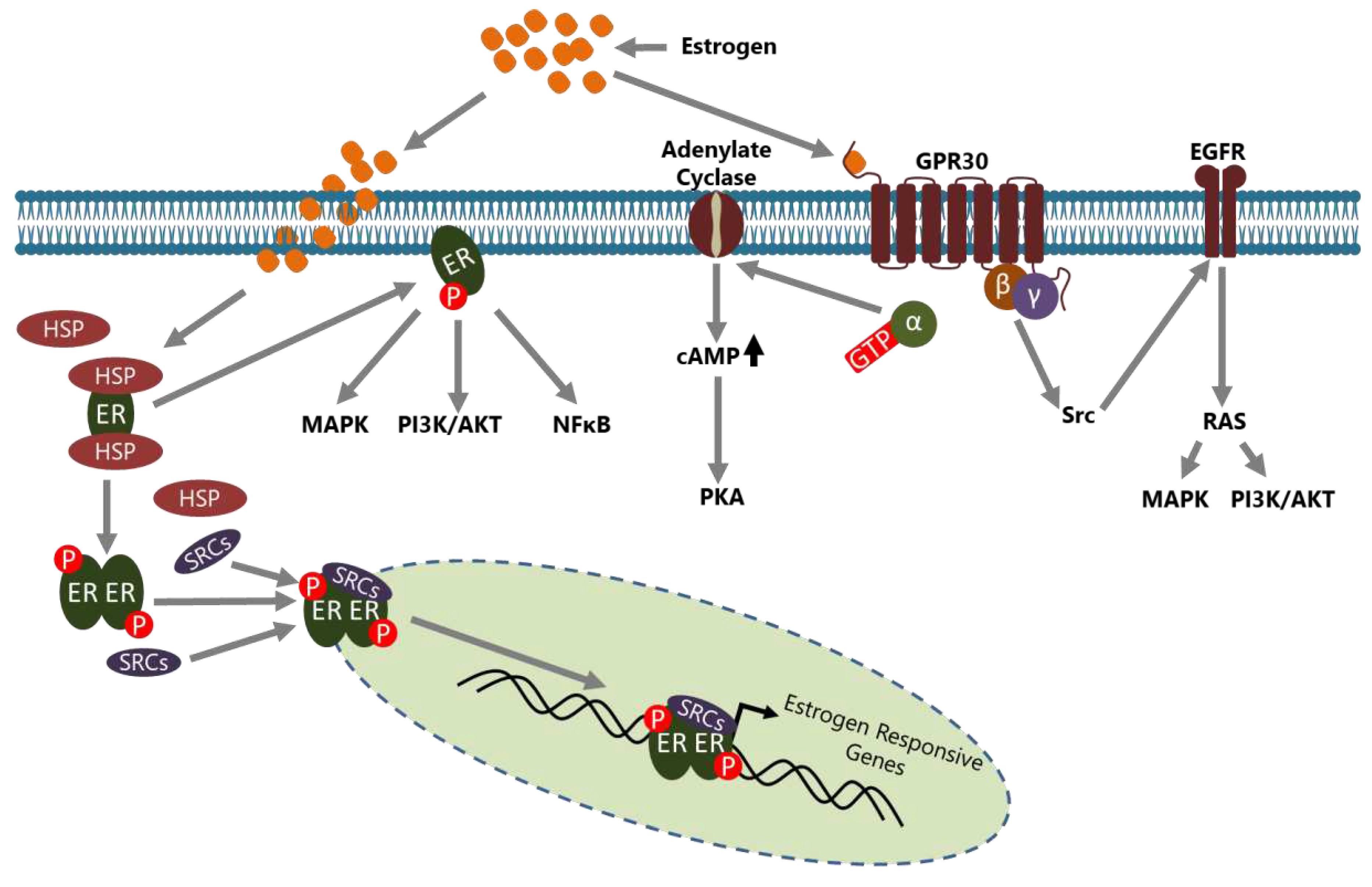
Figure 2.
Primary structure and functional sites of SRC-3 protein. The figure has been constructed on the data presented in refs [9], [17], and [94].
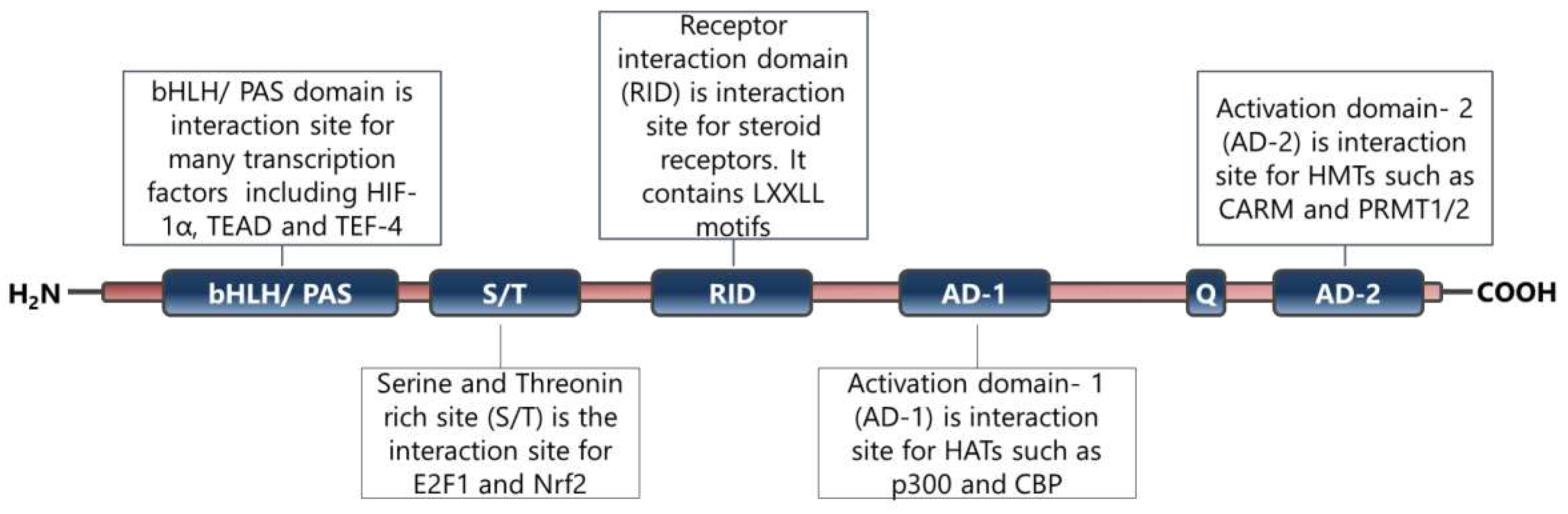
Figure 3.
SRC-3 promotes malignancy in BCa, in multiple ways. SRC-3 not only promotes the malignant behavior of tumor cells, but also changes the tumor microenvironment to a immunosuppressive phenotype which support CSCs and contributes therapy resistance.
Figure 3.
SRC-3 promotes malignancy in BCa, in multiple ways. SRC-3 not only promotes the malignant behavior of tumor cells, but also changes the tumor microenvironment to a immunosuppressive phenotype which support CSCs and contributes therapy resistance.
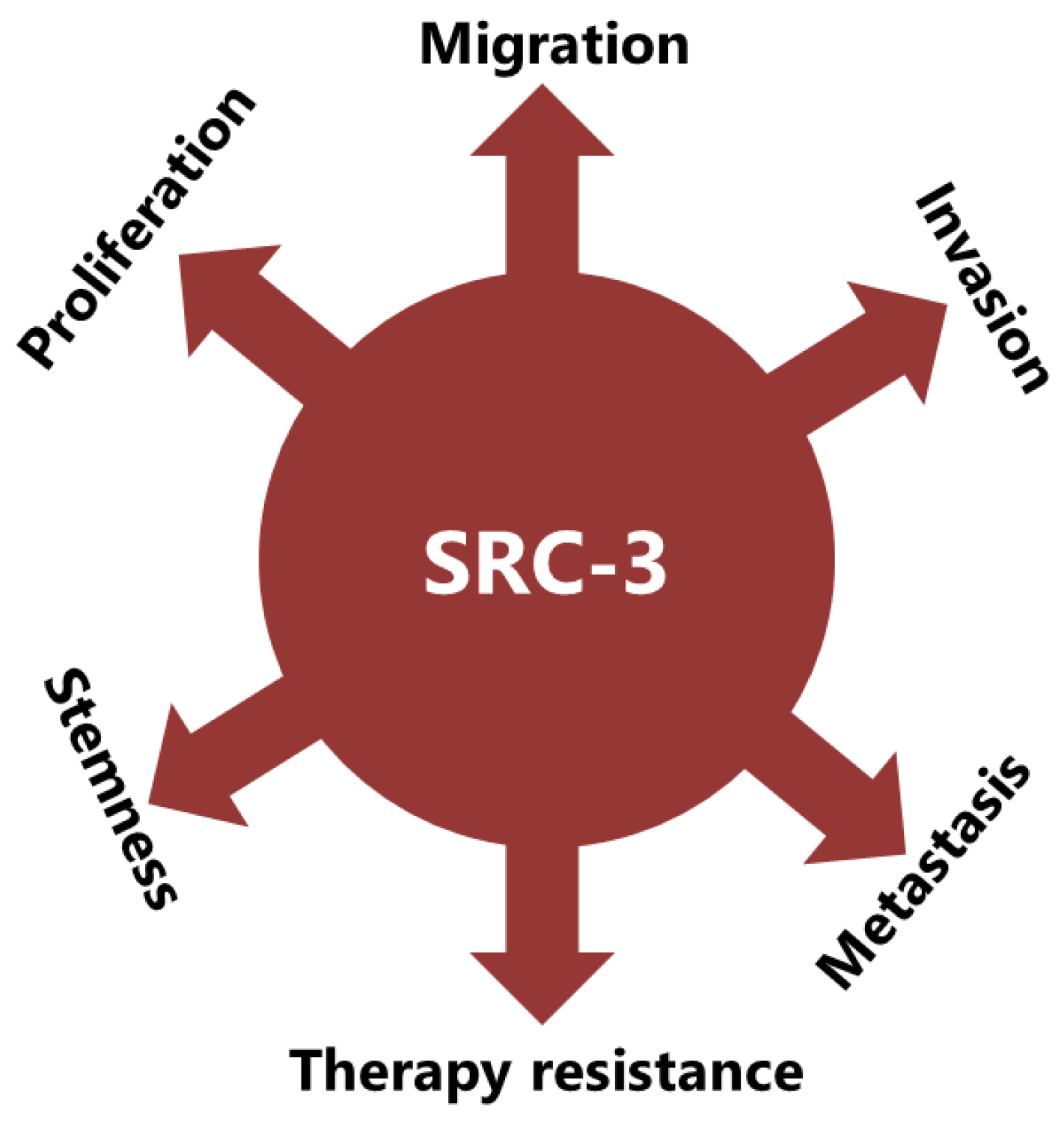
Figure 4.
Up-regulated SRC-3 expression inhibits immunotherapy in BCa, and this effect is directly associated with the SRC-3 dependent repression of E-cadherin expression in tumor cells. Increased SRC-3 level dependent decrease of E-cadherin in tumor cells results abrogation the interaction between CD103 on tumor infiltrated CD8+ T-cells and E-cadherin on target (tumor) cells. Consequently, disruption of an interaction between CD103 and E-cadherin, blocks production and release of cytotoxic granules by CD8+ T-cells to destroy tumor cells, and the tumor cells survive.
Figure 4.
Up-regulated SRC-3 expression inhibits immunotherapy in BCa, and this effect is directly associated with the SRC-3 dependent repression of E-cadherin expression in tumor cells. Increased SRC-3 level dependent decrease of E-cadherin in tumor cells results abrogation the interaction between CD103 on tumor infiltrated CD8+ T-cells and E-cadherin on target (tumor) cells. Consequently, disruption of an interaction between CD103 and E-cadherin, blocks production and release of cytotoxic granules by CD8+ T-cells to destroy tumor cells, and the tumor cells survive.
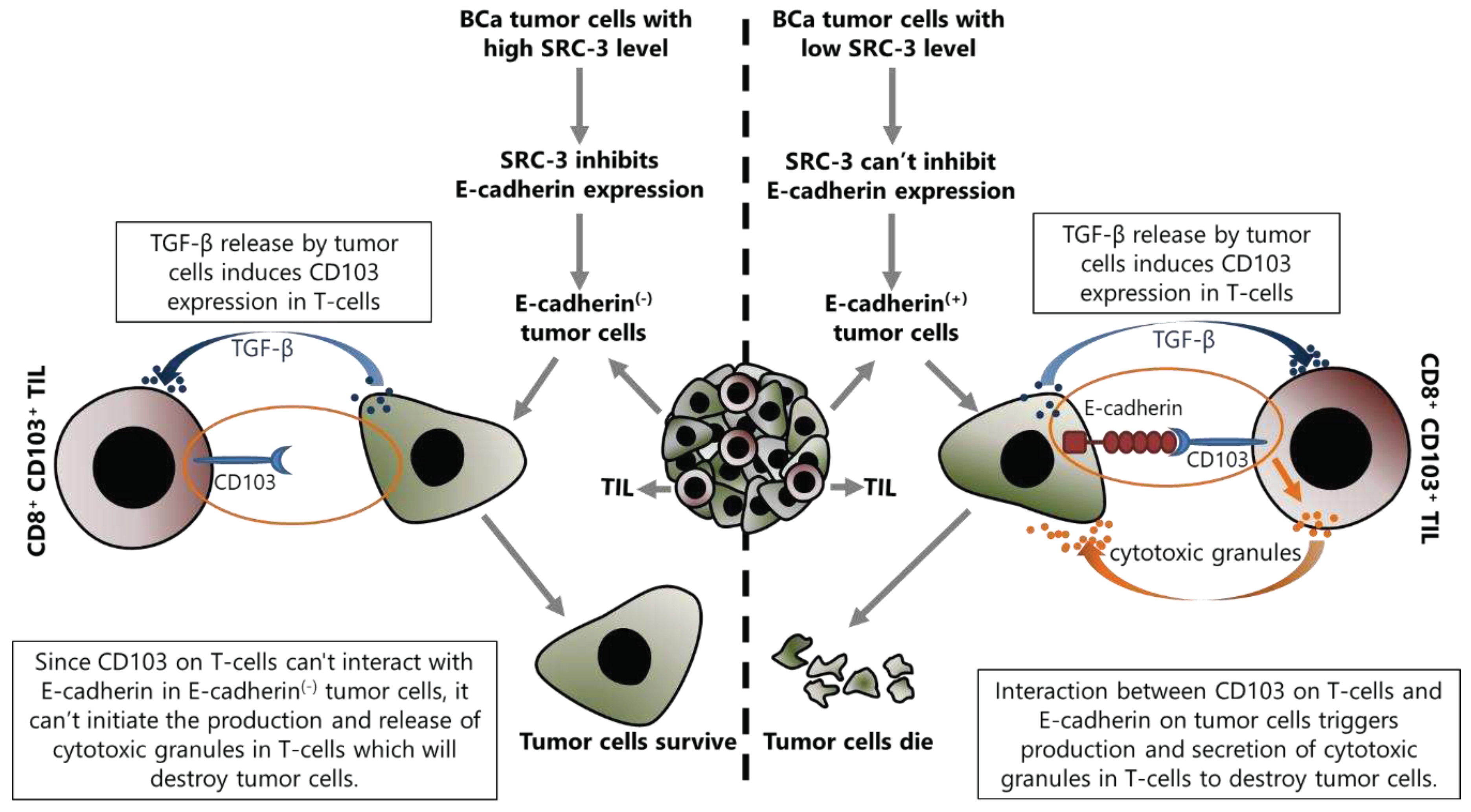
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



