
Submitted:
12 October 2023
Posted:
12 October 2023
You are already at the latest version
Abstract
Acid promoted amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with anilines have been studied using hydrochloric acid. Four alcoholic solvents and water were evaluated as reaction medium, and the highest rate was observed in water. Although the initial rate increased by using higher concentration of acid, this leads to more solvolysis of the pyrrolopyrimidine, reducing the potential yield. The substrate scope of the amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine in water using 0.1 equivalent of hydrochloric acid was evaluated using 20 aniline derivatives with variance in basicity and steric bulk. Preparative useful reactions were seen for 14 of the 20 derivatives. Unsuited anilines are ortho substituted anilines with a pKa below 1. Aliphatic and benzylic amines react poorly under these conditions, but such aminations can proceed well in water without acid. Pyrrolopyrimidines with a low water solubility, also reacted slowly in water, however they could easily be aminated in 2-propanol.
Keywords:
acid catalysed amination
; anilines
; pyrrolopyrimidine
; nucleophilic aromatic substitution
; pKa .
1. Introduction
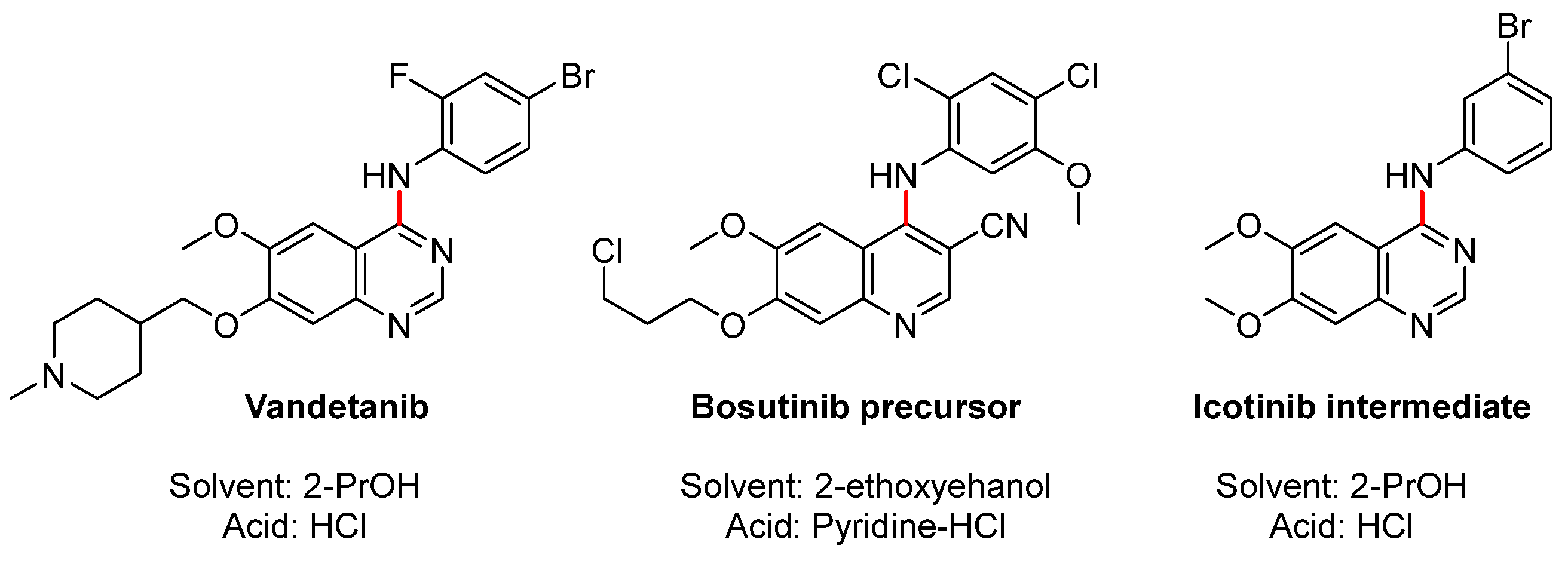
Nucleophilic aromatic substitution with amines is highly useful reactions in pharmaceutical chemistry.[1] Depending on the structure and electronic properties of the coupling partners, the transformations can be conducted in different ways. The use of weakly basic/thermal conditions on electron deficient aryls is suitable for benzyl and alkyl amines, but requires an excess of amine or a co-base to quench the generated acid.[2] Pyrimidines has also been aminated in water with 1 equivalent of amine using potassium fluoride as base.[3] N-Arylation of amines on less activated aromatics can be initiated by NH-deprotonation with strong bases,[4,5] but is restricted to substrates with no labile groups and safety aspects can set limitations. With less activated aromatics palladium catalyzed Buchwald-Hartwig aminations[6,7,8] are highly efficient. Challenges include regioselectivity when multiple halides are present, when there is possibility for racemization,[9] and sometimes strictly water free conditions are needed.[10] Further, the use of palladium should be minimized due to cost and the risk of contaminating the final product. Acid catalyzed amination represent an alternative for aromatic heterocycles, and a review on kinase inhibitor drugs shows that many processes included an amination step where acid is either added or generated as a by-product,[11] some structures are shown in Figure 1. Moreover, Kurup et al. [12] efficiently prepared a series of aniline substituted pyrrolopyrimdidines employing HCl in 2-propanol (2-PrOH). Other acid catalyzed investigated include acetic acid, [13] p-toluenesulfonic acid,[14] silver triflate,[15,16] InCl3 [17,18] and Zn(NO2)2.[19]
Although acid induced amination is frequently employed for anilines, we noticed that the published works are mainly of preparative nature [12,17] and that more in-dept studies on acid catalyzed amination on heteroaryls are lacking. Naturally, such reactions are a balancing act: how can you activate the aryl halide without deactivating the nucleophile. Herein we report our study of effect of solvent and acid amount on the amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine. By testing 20 different anilines, three pyrrolopyrimidines and four representative aliphatic amines, we show the substrate scope of this transformation, and highlight the benefits and limitations of water as reaction medium.
2. Results and Discussion
2.1. Initial reactions in EtOH
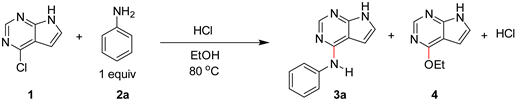
Our interest in the acid catalyzed amination of heteroaromatic chlorides were triggered by failed amination of some heterocycles with aniline under slightly basic conditions. Whereas, amination with 4-chloroquinazoline worked nicely, the corresponding reaction with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine was extremely slow. In contrast, the reaction proceeded much faster in EtOH with addition of a few drops of HCl. We assumed the reaction to proceed as shown in Scheme 1.
The pyrrolopyrimidine is not very basic and should be only transiently activated by protonation or hydrogen bonding, lowering the energy barrier for reaction at C-4. The neutral nucleophilic aniline (2) will be in equilibrium with the non-nucleophilic anilinium ion (2-HCl). The position of this equilibrium depends on the amount of acid added at start, the degree of conversion and the pKa of the aniline. The amination generates the product 3 and one mole equivalent of HCl. The product 3 is more basic than the starting material and will act as a buffer by forming the corresponding hydrochloride salt (3-HCl).
To gain better understanding of the process we started by performing reactions with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1) and aniline (2a) with varying amounts of HCl (0-5 equiv.) in EtOH. The aminations were followed by 1H NMR spectroscopy for 6 h, and integration of the H-2 protons, gave the mole ratio of the starting material 1, the product 3a and the side-product 4. Table 1 shows the conversion (%) after 1 h and the mole % of compounds after 6 h.
The degree of conversion after 1 h is indicative of the initial rate of the reaction. Without acid (entry 1) the product was not detected after 1 h, showing that the amination is only slowly catalyzed by EtOH hydrogen bonding. However, as small amount of 3a and HCl is produced, the rate of this reaction is elevated. Overall, the initial rate increases with the amount of acid used, and clearly, HCl has a catalytic effect on the amination. However, at elevated levels of HCl (0.5-5 equiv., entries 3-6), the side product 4 was also produced, consuming starting material. This is explained by excess acid deactivating the aniline by protonation, which allows EtOH to be a competitive nucleophile.
The levels of the side-product 4 as a function of HCl equiv. and reaction time is plotted in Figure 2. The highest concentrations were noted early in the process using 1-5 equiv. of HCl. On progression the amount of 4 decreases, showing that 4 is not just a side-product, but also a slow reacting substrate. Thus, the amount of acid should be kept low to minimize side-product formation, and in our model system 0.1 equiv. was suitable (Table 1, entry 2).
2.2. Effect of other protic solvents on the reaction
2-Propanol (2-PrOH) has been the most popular solvent for these reactions,[12,20,22] which might be reasonable from a solubility standpoint. We were curious as to how different solvents affected rate and product formation, since the choice of solvent can modulate the relative basicity of the reacting components,[23] or stabilize the transition state and have an impact on the cost profile of the process. The same model reaction was therefore conducted in four different alcohols and water at 60 °C using 0.1 equiv. of HCl. The reaction progress is shown in Figure 3.
From a practical viewpoint, the highest amount of product was obtained with water, MeOH or EtOH as solvent. However, reactions in MeOH gave some solvolysis, and 4-methoxy-7H-pyrrolo[2,3-d]pyrimidine was observed as a side-product (5% after 22 h). The apparent higher rate for the more polar solvents, could be explained by better ability to stabilize a polar transition state. However, the initial rate in 2-PrOH compared to in MeOH/EtOH was rather similar. An alternative explanation is that that the most polar solvents have a better ability to hydrogen bond the released HCl, leading to a more favorable aniline-anilinium ion equilibrium position.
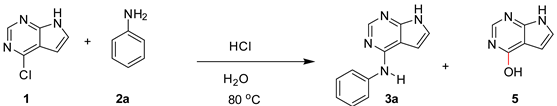
As water was the most promising solvent in the aniline arylation, we proceeded with testing the effect of the HCl amount in the reaction. To increase the rate, the reaction temperature was raised from 60 to 80 °C. The results are shown in Table 2. The initial rate as measured after 20 min was dependent on the amount of acid. The reaction with 0.1 equiv. of HCl (Table 2, entry 1) had a slower onset than the reaction with 1.0 equiv. of HCl (entry 5) which after 20 min had a conversion >50%. Anyhow, all reactions reached full conversion after 6 h. Low levels of the solvolysis side-product 5 was detected in all cases. However, compound 5 has good water solubility, and the actual levels are likely to be higher than that detected due to the extractive work-up performed on the analytical NMR samples.
To minimize the use of chemicals, an amination process with no acid is preferable, but a slow nonreliable reaction onset, can be problematic. A compromise is the use of 0.1 equiv. of HCl. To show the applicability of this procedure a reaction was performed on a 500 mg scale giving 91% of the product 3a.
2.3. Substrate scope for amination in water
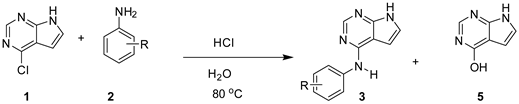
In terms of both reactivity and sustainability, the use of 0.1 equiv. of HCl in water is very attractive. Therefore, we went on to evaluate the substrate scope of the amination by testing 19 more anilines having different pKa and substitution patterns. The reactions were initially monitored on a 100 mg scale. This was followed by preparative reactions with 500 mg of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1). Conversion data for the 100 mg reactions, isolated yields, and experimental/estimated pKa of the anilines are shown in Table 3.
For para and meta substituted anilines (entries 1-10) all aminations reached full conversion within 6 h. Anilines with pKa between 5.2 - 2.7 seemed to react somewhat faster than outside this range. However, 4-nitroaniline (pKa = 1.02, entry 10) was also well suited as substrate, though more of the hydrolytic side-product 5 was formed. This is since water at lower pH becomes a competitive nucleophile. Certain functional groups can be labile under acidic conditions. Using 0.1 equiv. of HCl the pH went from 4.7 at start of the reaction to 2.0 at the end, which are mild conditions and debenzylation of 3f and hydrolysis of the alkyne in 3g were not observed. A comparison of the reaction profiles for 2a (R = H), the most basic aniline 2b (R = 4-OEt) and 4-nitroaniline (2j) is shown in Figure 4 where conversion is plotted vs. reaction time.
To evaluate steric effects, we also performed amination with 10 additional aniline derivatives (entries 11-20). N-Alkylation of anilines increase both their basicity and steric bulk. 4-Fluoro-N-methylaniline (2k, entry 11) reacted more slowly than aniline (2a), but regardless proved to be a good substrate, indicating that some added bulk is allowed for. 2-Hydroxyaniline (2l, entry 12) showed very good reactivity (entry 12). Somewhat slower reaction progress was seen for the bulky 2-iodoaniline (2n, entry 14) and the deiodinated side product 3a was also formed (8-10%). Similar deiodination have been previously observed.[28,29,30] A few control experiments were performed to identify the reason for the deiodination. To test for radical type dehalogenation, degassing of solvent, and protection from day light was done, but this had no effect on level of side-product 3a. Next, palladium contamination could be expected to cause reduction, however the palladium level of 1 and 2n were measure to only 6 µg/Kg and 17 µg/Kg, respectively. The aniline 2n contained ca 19 mg/Kg of iron, which combined with HCl could generate hydrogen gas, but only in minute amounts. Further, when purified product 3n was submitted to heating in water with 1.5 equivalent of HCl no deiodination took place in 22 h. In contrast, 2-iodoaniline (2n) was found to be unstable and provide aniline (2a). When we treated 2n in the absence of 1 with more HCl (1.5 equiv.) for 22 h also 2,4-diiodoaniline and 2,6-diiodoaniline were formed, indicating that disproportionation is occurring under acidic conditions as seen by other.[31,32]
.2,4-Dichloroaniline (2o) with a lower pKa had reactivity in line with that of 2-iodoaniline (2n). Figure 5 shows conversion vs. time for reactions towards 3l, 3k, 3n and 3o compared with that of the parent compound 3a.
Introducing two ortho-substituent or combining ortho substituents with a low pKa (entries 13 and 16-20), prevents efficient amination and minimal production of product was seen. For these anilines with low nucleophilicity, water becomes a competing nucleophile giving higher amounts of the side-product 5.
Preparative reactions were performed with 500 mg of 1 and 1.1 equiv. of the anilines 2a-l, 2n and 2o. The corresponding products were purified by silica-gel column chromatography giving 80-94% isolated yield (average 87%). The moderate yield for the 2-iodo analogue 3n is due to side-product formation (comp. 3a), while the reaction to form the 2,4-dichloro analogue 3o produced more of the hydrolytic side-product 5. To conclude, amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1) proceeds well with meta and para substituted anilines with pKa ranging from 5.3-1.0 Steric hindrance introduced at the nucleophilic nitrogen, and ortho-chloro and ortho-iodo substitution leads to lower rate, but acceptable reactions if the pKa of the aniline is not too low. In addition to cost savings, the use of water instead of 2-PrOH, prevents the formation of 2-chloropropane.[33,34] Some of these reactions, might also proceed well with lower amount of acid, although with a somewhat slow onset. For instance, the parent compound 3a (R= H) can be formed without acid added, while the 2-iodo analogue 3n was not formed without acid.
2.4. Other limitations to the use of water and HCl in amination
Highly crystalline compound with limited water solubility generally represents a challenge for reactions in water. We tested two such pyrrolopyrimidines 6 (mp. 245-247 °C) and 7 (mp. > 300 °C) in acid promoted amination to the corresponding products 8 and 9, see Scheme 2. In water, the pyrrolopyrimidines 6 and 7 were completely insoluble and the amination proceeded very slowly. After 3 days 60% conversion was observed for the least crystalline 6, while no product formation could be detected using the bromo containing 7. For these substrates a change to 2-PrOH, resulted in full conversion after 22 h, giving compound 8 and 9 in 82% and 87% isolated yield, respectively.
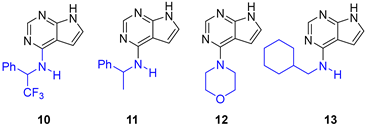
Highly basic amines should be unsuited for amination under acidic conditions as they would be protonated by the acid catalyst. Thus, for comparison with the aniline aminations we also performed a few experiments under acidic conditions with 2,2,2-trifluoro-1-phenylethylamine (pKa = 6.1[35]), morpholine (pKa = 8.33[36] ), 1-phenylethylamine (pKa = 9.45[36] ) and cyclohexylmethanamine (pKa = 10.49[37] ). The results are summarized in Table 4, entries 1-4.
Reaction with 2,2,2-trifluoroethyl-1-phenylamine in water and 0.1 equiv. of HCl gave a low conversion after 22 h (entry 1). Multiple products were formed, however their identity was not confirmed. Possibly, halogen exchange is occurring alongside amination, [38] and this amine is concluded to be an unsuited reactant in acid catalyzed amination. 1-Phenylethylamine (entry 2) and cyclohexylmethanamine (entry 4) were as expected poor nucleophiles under these conditions. The reaction with morpholine reached a higher 61% conversion (entry 3), which is due to its lower basicity.
The morpholine and cyclohexylmethylamine derivatives 12 and 13 have previously been synthesized by Jesumoroti et al. [39]. 4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (1) was aminated in 2-PrOH using 3 equiv. of amine and conc. HCl (3 drops). We repeated these experiment, and weighting of conc. HCl indicated that the 3 drops corresponded to ca 1 equiv. of HCl in this case, which in essence means the reaction is run under basic conditions. The aminations worked (entries 5 and 8), but proceeded with higher rate if the acid was omitted, and the reactions are conducted in water (entries 6-7 and 9). Thus, acid should not be used in aromatic substitutions with aliphatic and benzylic amines. However, water can be a suitable solvent in basic amination of pyrrolopyrimidine, and the products 12 and 13 were isolated in 88 and 92% yield.
3. Materials and Methods
3.1. Chemicals and Analysis
All solvents and most reagents used in the project were purchased from VWR and Merck. 4-Chloro-7H-pyrrolo[2,3-d]pyrimidine was obtained from 1 Click Chem. Compound 6 and 7 were prepared as previously described.[40] The identity of compound 5 was confirmed by comparison a commercial reference from Merck, while 11 was confirmed by in-house made material.[2] Silica-gel chromatography was performed using silica-gel 60A purchased from VWR with a pore size 40-63 um. 1H- and 13C-NMR spectra were recorded using a Bruker Advance III HD NMR spectrometer from Nanaobay electronics with a Smartprobe 5 mm probe head, operating at 400 MHz and 600 MHz for proton, and carbon spectra at 100 MHz and 150 MHz, respectively. All 19F NMR chemical shifts are relative to internal hexafluorobenzene in DMSO at δ = -163.0 ppm. Samples were mainly analyzed in DMSO-d6. 1H and 13C NNR chemical shifts are in ppm relative to the DMSO solvent peak at 2.50 ppm and 39.5 ppm, respectively. High resolution mass spectroscopy (HRMS) was performed using a WaterTM's Synapt G2-S Q-TOF instrument. Samples were ionized by Electrospray Ionization (ESI/70eV) and analyzed using an Atmospheric Solids Analysis Probe (ASAP). Calculated exact mass and spectra processing was done by WatersTM Software (Masslynx V4.1 SCN871).
3.2. General Synthetic Methods
3.2.1. General Procedure A: Test Amination (100 mg scale)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (1, 100 mg, 0.651 mmol, 1.0 equiv.) was mixed with the appropriate aniline (1.0 equiv.), and in the specified solvent (5 mL) and HCl (0-5 equiv. ) were added. The reaction mixtures were stirred for up to 22 h at 60 or 80 °C, with 4-5 samples taken out for 1H NMR analysis. The samples withdrawn was diluted with EtOAc (1-2 mL) and added aq. NaHCO3 (1-2 mL). After phase separation, drying and concentration, the residue was dissolved in DMSO-d6 and analysed by 1H-NMR spectroscopy. Integration of the pyrrolopyrimidine H-2 protons were used to estimate levels of substrate, product and side-product. After cooling to room temperature, the reaction mixtures were suspended in sat. Na2CO3 (aq., 2 mL) and vacuum filtered, washed with water and dried. The compounds were purified by silica-gel column chromatography to confirm the identity of the product.
3.2.2. General Procedure B: 500 mg Scale Amination in Water
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (500 mg, 3.26 mmol 1. equiv.) and the appropriate anilines (1.1 equiv.) were mixed with H2O (25 mL) and HCl (0.61 M, 0.1 equiv.). The reaction mixtures were stirred at 80 °C for 3 - 22 h. After cooling to room temperature, the reaction mixtures were suspended in sat. Na2CO3 (aq. 10 mL) and the formed solid was isolated by filtration. To recover more material the filtrates were extracted with EtOAc (4 × 30 mL). The combined organic phases were dried with brine (2 × 20 mL) and anhydrous Na2SO4 followed by filtration and concentration in vacuo. The filtrate and precipitate were combined and dried in vacuo. The crude product was immobilized on celite and purified using silica-gel flash chromatography as specified for each compound.
3.3. Isolated materials
3.3.1. N-Phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3a)
The compound was prepared as described in procedure B, starting with aniline. The reaction time was 6 h The crude material was purified using gradient silica-gel flash chromatography (n-pentane/EtOAc, 1:2, Rf = 0.21, → EtOAc). This yielded 626 mg (2.98 mmol, 91%) as a white powder, mp. 240 – 243 °C (lit. [12] 241 °C). 1H NMR (400 MHz, DMSO-d6) δ 11.74 (s, 1H), 9.28 (s, 1H), 8.27 (s, 1H), 7.93 – 7.85 (m, 2H), 7.38 – 7.28 (m, 2H), 7.23 (dd, J = 3.5, 2.3 Hz, 1H), 7.01 (tt, J = 7.3, 1.2 Hz, 1H), 6.78 (dd, J = 3.5, 1.8 Hz, 1H). The 1H NMR correspond to that found previously.[12]
3.3.2. N-(4-Ethoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3b)
The compound was synthesized on a 500 mg scale as described in procedure B using 4-ethoxyaniline. The reaction time was 9 h. The crude product was immobilized on celite and purified using silica-gel flash chromatography (EtOAc/n-pentane, 2:1, Rf = 0.16). This gave 781 mg (3.07 mmol, 94%) of a white powder, mp. 240 – 242 °C (lit. [12] 241 – 242 °C), 1H NMR (400 MHz, DMSO-d6) δ 11.66 (s, 1H), 9.13 (s, 1H), 8.20 (s, 1H), 7.70 (d, J = 9.0 Hz, 2H), 7.18 (d, J = 3.5 Hz, 1H), 6.91 (d, J = 9.0 Hz, 2H), 6.67 (d, J = 3.4 Hz, 1H), 4.00 (q, J = 6.9 Hz, 2H), 1.32 (t, J = 6.9 Hz, 3H). 1H NMR correspond to those previously described.[12,17]
3.3.3. N-(4-Butylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3c)
The compound was prepared as described in procedure B, starting with 4-butylaniline. The reaction time was 9 h. The crude material was purified using silica-gel flash chromatography (EtOAc/n-pentane, 2:1, Rf = 0.35). This gave 764 mg (2.87 mmol, 88%) of a white powder, mp. 195 – 197 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 9.21 (s, 1H), 8.25 (s, 1H), 7.80 – 7.72 (m, 2H), 7.21 (dd, J = 3.5, 2.2 Hz, 1H), 7.18 – 7.11 (m, 2H), 6.76 (dd, J = 3.5, 1.8 Hz, 1H), 2.55 (t, J = 7.7 Hz, 2H), 1.62 – 1.50 (m, 2H), 1.39 – 1.25 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.1, 151.3, 151.3, 138.4, 136.5, 128.7 (2C), 122.4, 121.0 (2C), 104.0, 99.3, 34.7, 33.8, 22.2, 14.3; HRMS (ES+, m/z): found 267.1614, calcd. for C16H19N4, [M+H]+, 267.1610.
3.3.4. N-(Benzo[d][1,3]dioxol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3d)
The compound was synthesized on a 500 mg scale as described in procedure B, using benzo[d][1,3]dioxol-5-amine. The reaction time was 22 h. The crude product was purified using flash silica-gel chromatography (EtOAc/n-pentane, 4:1, Rf = 0.20, → 100% EtOH, 100% → EtOAc/MeOH, 10:1). This yielded 701 mg (2.77 mmol, 85%) of a light red powder, mp. 282 °C (decomp.) (lit. [17] 282 – 283 °C), 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 9.18 (s, 1H), 8.23 (s, 1H), 7.59 (d, J = 2.2 Hz, 1H), 7.23 – 7.19 (m, 2H), 6.89 (d, J = 8.4 Hz, 1H), 6.71 (dd, J = 3.5, 1.9 Hz, 1H), 6.00 (s, 2H). The spectroscopic data correspond to those previously reported.[17]
3.3.5. N-(4-Fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3e)
The compound was prepared as described in procedure B, starting with 4-fluoroaniline. The reaction time was 6 h. The crude material was purified using silica-gel flash chromatography (EtOAc/n-pentane, 4:1, Rf = 0.23, → EtOAc). This gave 683 mg (3.00 mmol, 92%) of a white powder, mp. 251 – 253 °C (Lit. [39] 253 °C). 1H NMR (400 MHz, DMSO-d6) δ 11.76 (s, 1H), 9.34 (s, 1H), 8.27 (s, 1H), 7.92 – 7.86 (m, 2H), 7.23 (d, J = 3.5 Hz, 1H), 7.20 – 7.13 (m, 2H), 6.76 (d, J = 3.4 Hz, 1H). The 1H NMR correspond that found in the literature.[39]
3.3.6. N-(3-(Benzyloxy)phenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3f)
The compound was synthesized on a 500 mg scale as described in procedure B, using 3-benzyloxyaniline. The reaction time was 6 h. The crude product was immobilized on celite and purified using flash chromatography (EtOAc/n-pentane, 4:1, Rf = 0.25, → EtOAc). This yielded 896 mg (2.83 mmol, 87%) of a light brown powder, mp. 194 – 196 ºC, 1H NMR (400 MHz, DMSO-d6) δ 11.77 (s, 1H), 9.27 (s, 1H), 8.30 (s, 1H), 7.79 (t, J = 2.3 Hz, 1H), 7.52 –7.29 (m, 6H), 7.27 – 7.18 (m, 2H), 6.81 (dd, J = 3.5, 1.9 Hz, 1H), 6.67 (dd, J = 8.2, 2.6, 1H), 5.11 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.1, 153.9, 151.3, 151.2, 142.2, 137.7, 129.6, 128.9 (2C), 128.3, 128.2 (2C), 122.7, 113.1, 108.5, 107.4, 104.3, 99.2, 69.6; HRMS (ES+, m/z): found 317.1407, calcd. for C19H17N4O, [M+H]+, 317.1402.
3.3.7. N-(3-Ethynylphenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3g)
The compound was synthesized on a 500 mg scale as described in general procedure B, using 3-ethynylaniline. The reaction time was 6 h. The crude product was purified using flash chromatography (EtOAc/n-pentane, 4:1, Rf = 0.23). This yielded 627 mg (2.77 mmol, 83%) of a white powder, mp. 228 – 230 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 9.40 (s, 1H), 8.32 (s, 1H), 8.16 (t, J = 2.0 Hz, 1H), 7.91 (ddd, J = 8.4, 2.4, 1.0 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.26 (d, J = 3.5 Hz, 1H), 7.11 (dt, J = 7.6, 1.3 Hz, 1H), 6.80 (d, J = 3.5 Hz, 1H), 4.15 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 153.7, 151.4, 151.1, 141.2, 129.4, 125.5, 123.2, 122.9, 122.2, 121.0, 104.3, 99.2, 84.3, 80.7; HRMS (ES+, m/z): found 235.0987, calcd. for C14H11N4, [M+H]+, 235.0984. Reference spectra has not been found.
3.3.8. N-(3-Chlorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3h)
The compound was prepared as described in procedure B, starting with 3-chloroaniline. The reaction time was 6 h. The crude material was purified using silica-gel flash chromatography (EtOAc/n-pentane, 4:1, Rf = 0.27). This gave 696 mg (2.69 mmol, 81%) of a white powder, mp 226 – 227 °C (lit. [39] 227 °C); 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H), 9.46 (s, 1H), 8.35 (s, 1H), 8.22 (t, J = 2.1 Hz, 1H), 7.81 (dd, J = 8.2, 2.4 Hz, 1H), 7.35 (t, J = 8.1 Hz, 1H), 7.28 (d, J = 3.4 Hz, 1H), 7.03 (dd, J = 7.8, 2.4 Hz, 1H), 6.82 (d, J = 3.4 Hz, 1H). The 1H NMR correspond that found in the literature.[39]
3.3.9. N-(4-Bromo-3-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3i)
The compound was synthesized on a 500 mg scale as described in procedure B. The reaction time was 3 h. The crude product was purified using flash silica-gel chromatography (EtOAc/n-pentane, 1:1, Rf = 0.28 → EtOAc). This yielded 908 mg (2.96 mmol, 91%) of a white solid, mp. 308 – 309 °C, 1H NMR (400 MHz, DMSO-d6) δ 11.88 (s, 1H), 9.61 (s, 1H), 8.37 (s, 1H), 8.26 (dd, J = 12.2, 2.4 Hz, 1H), 7.67 – 7.56 (m, 2H), 7.30 (dd, J = 3.6, 2.0 Hz, 1H), 6.82 (dd, J = 3.5, 1.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 156.8 (d, J = 241.0 Hz), 152.9, 151.0, 150.5, 141.9 (d, J= 10.6 Hz), 132.8 (d, J = 1.3 Hz), 122.9, 116.9 (d, J = 21.3 Hz), 107.5 (d, J = 27.5 Hz) 104.1, 98.6 (d, J = 21.3 Hz), 98.57; 19F NMR (376 MHz, DMSO-d6) δ -107.97 (dd, J = 12.2, 6.8 Hz); HRMS (ES+, m/z): found 307.0000, calcd. for C12H9N4FBr [M+H]+, 306.9995.
3.3.10. N-(4-Nitrophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3j)
The compound was prepared as described in procedure B, starting with 4-nitroaniline. The reaction time was 9 h. The crude material was purified using silica-gel flash chromatography (EtOAc/n-pentane, 2:1, Rf = 0.27 → EtOAc/MeOH, 10:1). This yielded 732 mg (2.87 mmol, 88%) of a yellow powder, mp. 335 – 337 °C (Lit. [39] 331 °C). 1H NMR (400 MHz, DMSO-d6) δ 11.99 (s, 1H), 9.99 (s, 1H), 8.44 (s, 1H), 8.25 (s, 4H), 7.37 (dd, J = 3.5, 2.3 Hz, 1H), 6.89 (dd, J = 3.5, 1.9 Hz, 1H). The 1H NMR correspond that found in the literature.[39]
3.3.11. N-(4-Fluorophenyl)-N-methyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3k)
The compound was prepared as described in general procedure B, starting with N-methyl-4-fluoroaniline. The reaction time was 22 h. The crude material was purified using silica-gel flash chromatography (gradient, EtOAc/n-pentane, 4:1, Rf = 0.10, → EtOAc). This gave 694 mg (2.87 mmol, 88%) of a white powder, mp. 250 – 252 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.27 (s, 1H), 7.48 – 7.28 (m, 4H), 6.90 (d, J = 3.5 Hz, 1H), 4.66 (d, J = 3.5 Hz, 1H), 3.50 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.3 (d, J = 244.1 Hz), 156.4, 151.8, 151.2, 142.5 (d, J = 1.0 Hz), 130.6 (d, J = 8.4 Hz, 2C), 121.5, 116.4 (d, J = 22.7 Hz, 2C), 103.3, 100.8, 39.4; 19F NMR (565 MHz, DMSO-d6) δ -114.95 – -115.04 (m); HRMS (ES+, m/z): found 243.1051, calcd. for C13H12N4F, [M+H]+, 243.1046.
3.3.12. 2-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)phenol (3l)
The compound was isolated on a 500 mg scale as described in procedure B, using 2-aminophenol. The reaction time was 22 h. The crude product was immobilized on celite and purified using flash silica-gel chromatography (gradient, EtOAc/n-pentane, 4:1, Rf = 0.21 → EtOAc). This yielded 650 mg (2.90 mmol, 89%) of a light-yellow powder, mp. 232 – 234 °C (Lit.[17] 233 – 235 °C); 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 10.60 (s, 1H), 8.89 (s, 1H), 8.21 (s, 1H), 7.56 (dd, J = 7.9, 1.7 Hz, 1H), 7.21 (dd, J = 3.5, 2.2 Hz, 1H), 7.02 (td, J = 7.3, 1.7 Hz, 1H), 6.92 (dd, J = 8.1, 1.6 Hz, 1H), 6.84 (td, J = 7.5, 1.6 Hz, 1H), 6.70 (dd, J = 3.5, 1.6 Hz, 1H). The 1H NMR correspond to that previously reported.[17]
3.3.13. N-(2-Iodophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amin (3n)
The compound was prepared as described in procedure B, starting with 2-iodoaniline. The reaction time was 22 h. The crude product was immobilized on celite and purified 3 times using flash silica-gel chromatography (CH2Cl2/acetone, 4:1, Rf = 0.20, → CH2Cl2/acetone, 1:1). This gave 865 mg (2.57 mmol, 79%) of an off-white solid, mp. 216–218 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.69 (s, 1H), 9.03 (s, 1H), 8.12 (s, 1H), 7.94 (dd, J = 8.0, 1.5 Hz, 1H), 7.54 (dd, J = 7.9, 1.6 Hz, 1H), 7.43 (td, J = 7.6, 1.5 Hz, 1H), 7.16 (dd, J = 3.5, 2.1 Hz, 1H), 7.03 (td, J = 7.6, 1.6 Hz, 1H), 6.35 (dd, J = 3.4, 1.6 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 154.6, 151.1, 151.0, 141.2, 138.9, 128.8, 128.7, 127.6, 121.9, 102.9, 99.5.0, 98.8; HRMS (ES+, m/z): found 336.9956, calcd. for C12H10N4I, [M+H]+, 336.9950.
3.3.14. N-(2,4-Dichlorophenyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (3o)
The compound was prepared as described in procedure B, starting with 2,4-dichloroaniline. The reaction time was 16 h. The crude material was purified using silica-gel flash chromatography (EtOAc/n-pentane, 1:1, Rf = 0.33 → EtOAc/MeOH, 10:1). This gave 728 mg (2.61 mmol, 80%) of a white powder. 1H NMR (400 MHz, DMSO-d6) δ 11.76 (s, 1H), 9.10 (s, 1H), 8.15 (s, 1H), 7.75 (d, J = 8.7 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.45 (dd, J = 8.6, 2.4 Hz, 1H), 7.22 (dd, J = 3.5, 2.3 Hz, 1H), 6.60 (dd, J = 3.5, 2.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 153.6, 151.5, 151.1, 142.6, 133.3, 130.5, 123.1, 121.8, 119.6, 118.6, 104.4, 99.1; HRMS (ES+, m/z): found 279.0208, calcd. for C12H9N4Cl2, [M+H]+, 279.0204
3.3.15. 4-Ethoxy-7H-pyrrolo[2,3-d]pyrimidine (4)
The material was isolated following an amination of 1 with aniline in EtOH according to method A. Silica-gel flash chromatography (EtOAc/n-pentane, 2:1, Rf = 0.21) gave 5 mg (3.06 mmol) of a solid. 1H NMR (400 MHz, DMSO-d6) δ 11.99 (s, 1H), 8.34 (s, 1H), 7.33 (dd, J = 3.5, 2.3 Hz, 1H), 6.45 (dd, J = 3.4, 1.8 Hz, 1H), 4.52 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.8, 152.5, 150.3, 124.0, 104.4, 97.8, 61.5, 14.5; HRMS (ASAP+, m/z): found 164.0827, calcd for for C8H10N3, (M+H)+, 164.0824
3.3.16. 6-(4-Fluorophenyl)-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (8)
4-Chloro-6-(4-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidine (100 mg, 0.404 mmol, 1.0 equiv.) was mixed with aniline (1.1 equiv.), and 2-PrOH (5 mL) and HCl (0.61 M, 0.1 equiv.) were added. The reaction mixture was stirred for 22 h at 80 °C. After cooling to room temperature, the reaction mixture was suspended in sat. Na2CO3 (aq., 2 mL) and vacuum filtered. To recover more material the filtrate was extracted with EtOAc (4 × 10 mL). The combined organic phases were dried with brine (2 × 5 mL) and anhydrous Na2SO4 followed by filtration and concentration in vacuo. Both filtrate and precipitate were combined and dried in vacuo. The crude product was immobilized on celite and purified using silica-gel flash chromatography (CH2Cl2/acetone, 4:1, Rf = 0.17, → CH2Cl2/acetone, 1:1). This gave 101 mg (0.331 mmol, 82%) of a white solid, mp. 323-326 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.30 (s, 1H), 9.38 (s, 1H), 8.30 (s, 1H), 7.94 – 7.89 (m, 2H), 7.89 – 7.83 (m, 2H), 7.38 – 7.29 (m, 4H), 7.16 (s, 1H), 7.02 (tt, J = 7.3, 1.2 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 161.6 (d, J = 244.9 Hz), 153.2, 152.2, 151.2, 140.3, 133.7, 128.5 (2C), 128.2 (d, J = 3.1 Hz), 126.8 (d, J = 8.2 Hz, 2C), 122.0, 120.1 (2C), 116.0 (d, J = 21.8 Hz, 2C), 105.0, 95.8; 19F NMR (565 MHz, DMSO-d6) δ -114.5 – -114.6 (m); HRMS (ASAP+, m/z): found 305.1206, calcd for C18H14N4F, (M+H)+, 305.1202.
3.3.17. 6-(4-Bromophenyl)-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine (9)
4-Chloro-6-(4-bromophenyl)-7H-pyrrolo[2,3-d]pyrimidine (100 mg, 0.324 mmol, 1.0 equiv.) was mixed with aniline (1.1 equiv.), and 2-PrOH (5 mL) and HCl (0.61 M, 0.1 equiv.) were added. The reaction mixture was stirred for 22 h at 80 °C. After cooling to room temperature, the reaction mixture was suspended in sat. Na2CO3 (aq., 2 mL) and vacuum filtered. To recover more material the filtrate was extracted with EtOAc (4 × 10 mL). The combined organic phases were dried with brine (2 × 5 mL) and anhydrous Na2SO4 followed by filtration and concentration in vacuo. Both filtrate and precipitate were combined and dried in vacuo. The crude product was immobilized on celite and purified using silica-gel flash chromatography (CH2Cl2/acetone, 4:1, Rf = 0.31, → CH2Cl2/acetone, 1:1). This gave 103 mg (0.282 mmol, 87%) of a white solid, mp. 329 - 332 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.35 (s, 1H), 9.43 (s, 1H), 8.31 (s, 1H), 7.91 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 8.2 Hz, 2H), 7.35 (t, J = 7.7 Hz, 2H), 7.25 (s, 1H), 7.03 (t, J = 7.3 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 153.3, 152.2, 151.5, 140.3, 133.4, 132.0 (2C), 130.8, 128.5 (2C), 126.7 (2C), 122.1, 120.6, 120.2 (2C), 105.0, 96.6 ; HRMS (ASAP+, m/z): found 365.0403 calcd for C18H14N4Br (M+H)+, 365.0402.
3.3.18. 4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)morpholine (12)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (500 mg, 3.26 mmol, 1.0 equiv.) and morpholine (3 equiv.) were mixed with H2O (25 mL). The reaction mixture was stirred at 80 °C for 2.5 h. After cooling to room temperature, the reaction mixture was suspended in sat. Na2CO3 (aq. 10 mL) and a solid formed, which was isolated by filtration. The filtrate was extracted with EtOAc (3 × 30 mL). The combined organic phases were washed with brine (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The material from the filtrate and the precipitate were combined and dried in vacuo to afford the title compound 586 mg (2.88 mmol, 88%) as an off-white solid, mp. 212 - 213 °C, (Lit.[39] 207 °C); 1H NMR (400 MHz, DMSO-d6) δ 11.73 (br s, 1H), 8.16 (s, 1H), 7.20 (dd, J = 3.6, 2.4 Hz, 1H), 6.62 (dd, J = 3.6, 1.8 Hz, 1H), 3.84 – 3.82 (m, 4H), 3.73–3.70 (m, 4H). The 1H NMR was in agreement with that reported.[39]
3.3.19. N-(Cyclohexylmethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (13)
The compound was prepared as described for preparation of 12 but using 1-cyclohexylmethanamine and reacting for 8 h. After cooling to room temperature, the reaction mixtures were suspended in sat. Na2CO3 (aq. 10 mL) and vacuum filtered. The filter cake was washed with water and dried in vacuo to give 690 mg (2.99 mmol, 92%) as an off-white solid, mp. 180.5 – 181.5 °C, (Lit. [39] 179 °C); 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 8.06 (s, 1H), 7.34 (t, J = 5.9 Hz, 1H), 7.03 (dd, J = 3.5, 1.7 Hz, 1H), 6.56 (dd, J = 3.5, 1.3 Hz, 1H), 3.30 (t, 5.6 Hz, 2H), 1.79 – 1.54 (m, 6H), 1.26 – 1.09 (m, 3H), 1.00 - 0.86 (m, 2H). The 1H NMR was in agreement with that reported, [39] but we have reported the most down filed shift as multiplet.
5. Conclusions
Nucleophilic aromatic substitution on heterocyclic chlorides with anilines is an im-portant reaction in medicinal chemistry research, and for these substrates the use of acid promoted reactions appear attractive both in terms of simplicity, yield and sustainability. In these aminations the amount of acid used should be kept low to minimize the competing solvolysis. Thus, in reaction between 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1) and aniline 0.1 equivalent of HCl is sufficient, and the reaction proceeds with higher rate in water than in short chain alcohols. The substrate scope of the amination in water was evaluated with 20 aniline derivatives. The amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1) proceeds well with meta and para substituted anilines with pKa ranging from 1.0 - 5.3. Steric hindrance introduced at the nucleophilic nitrogen, and ortho-chloro and iodo substitution leads to lower rate, but still acceptable reactions. No decomposition of benzyl ethers or ethyne substituents were observed, however an unusual deiodination occurred for compound 3n. Considerable cost savings can be achieved by the use of water and HCl instead of organic solvents and more complicated promotors or catalysts. Limitations to the use of water/HCl amination includes 2-substituted anilines with pKa below 1 and 2,6-disubstituted anilines. Also, aliphatic and benzylic amines due to their high basicity cannot be reacted with aryl halides under acidic conditions. Finally, for aminations of pyrrolopyrimidines with very low water solubility, these reactions are best performed with 2-propanol as solvent.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: references to the pKa values, Figure S1-S18: 1H and 13C NMR spectra of new compounds.
Author Contributions
Conceptualization, B.H.H.; methodology, H.S, B.H.H; validation, S.Y., B.H.H; formal analysis, H.S, B.H.H.; investigation, H.S, Y.S..; writing—original draft preparation, B.H.H; writing—review and editing, B.H.H., C.E.O, H.S., Y.S.; visualization, H.S, B.H.H.; supervision, B.H.H, C.E.O.; project administration, B.H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Additional data will be made available on request.
Acknowledgments
This research was funded by NTNU (no grant number). The support from the Norwegian NMR Platform (project number 226244/F50) is highly appreciated. So is the help from the Mass Spectrometry Lab at the NV Faculty at NTNU. Roger Aarvik is thanked for technical support.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of most compounds can be provided by the authors.
References
- Roughley, S. D.; Jordan, A. M., The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451-3479, . [CrossRef]
- Aarhus, T. I.; Bjørnstad, F.; Wolowczyk, C.; Larsen, K. U.; Rognstad, L.; Leithaug, T.; Unger, A.; Habenberger, P.; Wolf, A.; Bjørkøy, G.; Pridans, C.; Eickhoff, J.; Klebl, B.; Hoff, B. H.; Sundby, E., Synthesis and Development of Highly Selective Pyrrolo[2,3-d]pyrimidine CSF1R Inhibitors Targeting the Autoinhibited Form. J. Med. Chem. 2023, 66, 6959-6980, . [CrossRef]
- Walsh, K.; Sneddon, H. F.; Moody, C. J., Amination of Heteroaryl Chlorides: Palladium Catalysis or SNAr in Green Solvents? ChemSusChem 2013, 6, 1455-1460, . [CrossRef]
- Lin, Y.; Li, M.; Ji, X.; Wu, J.; Cao, S., n-Butyllithium-mediated synthesis of N-aryl tertiary amines by reactions of fluoroarenes with secondary amines at room temperature. Tetrahedron 2017, 73, 1466-1472, . [CrossRef]
- Borch Jacobsen, C.; Meldal, M.; Diness, F., Mechanism and Scope of Base-Controlled Catalyst-Free N-Arylation of Amines with Unactivated Fluorobenzenes. Chem. - Eur. J. 2017, 23, 846-851, . [CrossRef]
- Dorel, R.; Grugel, C. P.; Haydl, A. M., The Buchwald–Hartwig Amination After 25 Years. Angew. Chem. Int. Ed. 2019, 58, 17118-17129, . [CrossRef]
- Surry, D. S.; Buchwald, S. L., Dialkylbiaryl phosphines in Pd-catalyzed amination: a user's guide. Chem. Sci. 2011, 2, 27-50, . [CrossRef]
- Hartwig, J. F., Evolution of a fourth generation catalyst for the amination and thioetherification of aryl halides. Acc. Chem. Res. 2008, 41, 1534-1544, . [CrossRef]
- Wagaw, S.; Rennels, R. A.; Buchwald, S. L., Palladium-Catalyzed Coupling of Optically Active Amines with Aryl Bromides. J. Am. Chem. Soc 1997, 119, 8451-8458, . [CrossRef]
- Yin, J.; Buchwald, S. L., Pd-Catalyzed Intermolecular Amidation of Aryl Halides: The Discovery that Xantphos Can Be Trans-Chelating in a Palladium Complex. J. Am. Chem. Soc. 2002, 124, 6043-6048, . [CrossRef]
- Ayala-Aguilera, C. C.; Valero, T.; Lorente-Macías, Á.; Baillache, D. J.; Croke, S.; Unciti-Broceta, A., Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2022, 65, 1047-1131, . [CrossRef]
- Kurup, S.; McAllister, B.; Liskova, P.; Mistry, T.; Fanizza, A.; Stanford, D.; Slawska, J.; Keller, U.; Hoellein, A., Design, synthesis and biological activity of N4-phenylsubstituted-7H-pyrrolo[2,3-d]pyrimidin-4-amines as dual inhibitors of aurora kinase A and epidermal growth factor receptor kinase. J. Enzyme Inhib. Med. Chem. 2018, 33, 74-84, . [CrossRef]
- Guo, C.; Dong, L.; Marakovits, J.; Kephart, S., A novel method to enable SNAr reaction of aminopyrrolopyrazoles. Tetrahedron Lett. 2011, 52, 1692-1696, . [CrossRef]
- Wu, Y.; Wang, B.; Wang, J.; Qi, S.; Zou, F.; Qi, Z.; Liu, F.; Liu, Q.; Chen, C.; Hu, C.; Hu, Z.; Wang, A.; Wang, L.; Wang, W.; Ren, T.; Cai, Y.; Bai, M.; Liu, Q.; Liu, J., Discovery of 2-(4-Chloro-3-(trifluoromethyl)phenyl)-N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)acetamide (CHMFL-KIT-64) as a Novel Orally Available Potent Inhibitor against Broad-Spectrum Mutants of c-KIT Kinase for Gastrointestinal Stromal Tumors. J. Med. Chem. 2019, 62, 6083-6101, . [CrossRef]
- Lawhorn, B. G.; Philp, J.; Zhao, Y.; Louer, C.; Hammond, M.; Cheung, M.; Fries, H.; Graves, A. P.; Shewchuk, L.; Wang, L.; Cottom, J. E.; Qi, H.; Zhao, H.; Totoritis, R.; Zhang, G.; Schwartz, B.; Li, H.; Sweitzer, S.; Holt, D. A.; Gatto, G. J., Jr.; Kallander, L. S., Identification of Purines and 7-Deazapurines as Potent and Selective Type I Inhibitors of Troponin I-Interacting Kinase (TNNI3K). J. Med. Chem. 2015, 58, 7431-7448, . [CrossRef]
- Sun, L.; Cui, J.; Liang, C.; Zhou, Y.; Nematalla, A.; Wang, X.; Chen, H.; Tang, C.; Wei, J., Rational design of 4,5-disubstituted-5,7-dihydro-pyrrolo[2,3-d]pyrimidin-6-ones as a novel class of inhibitors of epidermal growth factor receptor (EGF-R) and Her2(p185erbB) tyrosine kinases. Bioorg. Med. Chem. Lett. 2002, 12, 2153-2157, . [CrossRef]
- Nozal, V.; Martínez-González, L.; Gomez-Almeria, M.; Gonzalo-Consuegra, C.; Santana, P.; Chaikuad, A.; Pérez-Cuevas, E.; Knapp, S.; Lietha, D.; Ramírez, D.; Petralla, S.; Monti, B.; Gil, C.; Martín-Requero, A.; Palomo, V.; de Lago, E.; Martinez, A., TDP-43 Modulation by Tau-Tubulin Kinase 1 Inhibitors: A New Avenue for Future Amyotrophic Lateral Sclerosis Therapy. J. Med. Chem. 2022, 65, 1585-1607, . [CrossRef]
- Staderini, M.; Bolognesi, M. L.; Menendez, J. C., Lewis Acid-Catalyzed Generation of C-C and C-N Bonds on π-Deficient Heterocyclic Substrates. Adv. Synth. Catal. 2015, 357, 185-195, . [CrossRef]
- Abou-Shehada, S.; Teasdale, M. C.; Bull, S. D.; Wade, C. E.; Williams, J. M. J., Lewis Acid Activation of Pyridines for Nucleophilic Aromatic Substitution and Conjugate Addition. ChemSusChem 2015, 8, 1083-1087, . [CrossRef]
- Hennequin, L. F.; Stokes, E. S. E.; Thomas, A. P.; Johnstone, C.; Plé, P. A.; Ogilvie, D. J.; Dukes, M.; Wedge, S. R.; Kendrew, J.; Curwen, J. O., Novel 4-Anilinoquinazolines with C-7 Basic Side Chains: Design and Structure Activity Relationship of a Series of Potent, Orally Active, VEGF Receptor Tyrosine Kinase Inhibitors. J. Med. Chem. 2002, 45, 1300-1312, . [CrossRef]
- Boschelli, D. H.; Ye, F.; Wang, Y. D.; Dutia, M.; Johnson, S. L.; Wu, B.; Miller, K.; Powell, D. W.; Yaczko, D.; Young, M.; Tischler, M.; Arndt, K.; Discafani, C.; Etienne, C.; Gibbons, J.; Grod, J.; Lucas, J.; Weber, J. M.; Boschelli, F., Optimization of 4-Phenylamino-3-quinolinecarbonitriles as Potent Inhibitors of Src Kinase Activity. J. Med. Chem. 2001, 44, 3965-3977, . [CrossRef]
- Zhang, D.; Xie, G.; Davis, C.; Cheng, Z.; Chen, H.; Wang, Y.; Kamal, M. Novel fused quinazoline derivatives useful as tyrosine kinase inhibitors. WO2003/82830, 2003.
- Rossini, E.; Bochevarov, A. D.; Knapp, E. W., Empirical Conversion of pKa Values between Different Solvents and Interpretation of the Parameters: Application to Water, Acetonitrile, Dimethyl Sulfoxide, and Methanol. ACS Omega 2018, 3, 1653-1662, . [CrossRef]
- Gross, K. C.; Seybold, P. G.; Peralta-Inga, Z.; Murray, J. S.; Politzer, P., Comparison of Quantum Chemical Parameters and Hammett Constants in Correlating pKa Values of Substituted Anilines. J. Org. Chem. 2001, 66, 6919-6925, . [CrossRef]
- Tehan, B. G.; Lloyd, E. J.; Wong, M. G.; Pitt, W. R.; Gancia, E.; Manallack, D. T., Estimation of pKa Using Semiempirical Molecular Orbital Methods. Part 2: Application to Amines, Anilines and Various Nitrogen Containing Heterocyclic Compounds. Quant. Struct.-Act. Relat. 2002, 21, 473-485, . [CrossRef]
- Broderius, S. J.; Kahl, M. D.; Hoglund, M. D., Use of joint toxic response to define the primary mode of toxic action for diverse industrial organic chemicals. Environ. Toxicol. Chemi. 1995, 14, 1591-1605, . [CrossRef]
- Eastes, J. W.; Aldridge, M. H.; Kamlet, M. J., Effects of N-alkylation and N,N-dialkylation of the pKa of anilinium and nitroanilinium ions. J. Chem. Soc. (B) 1969, 922-928, . [CrossRef]
- Pietra, F.; Bartolozzi, M.; Del Cima, F., Competition between reductive dehalogenation and nucleophilic aromatic substitution of nitro-activated iodine by amines. J. Chem. Soc. D: Chem. Commun. 1971, 1232-1232, . [CrossRef]
- Talekar, R. S.; Chen, G. S.; Lai, S.-Y.; Chern, J.-W., Nonreductive Deiodination of ortho-Iodo-Hydroxylated Arenes Using Tertiary Amines. J. Org. Chem. 2005, 70, 8590-8593, . [CrossRef]
- Choguill, H. S.; Ridd, J. H., The mechanism of protodeiodination of p-iodoaniline. J. Chem. Soc. 1961, 822-826, . [CrossRef]
- Choi, H. Y.; Chi, D. Y., A Facile Debromination Reaction: Can Bromide Now Be Used as a Protective Group in Aromatic Systems? J. Am. Chem. Soc. 2001, 123, 9202-9203, . [CrossRef]
- Gilow, H. M.; Burton, D. E., Bromination and chlorination of pyrrole and some reactive 1-substituted pyrroles. J. Org. Chem 1981, 46, 2221-2225, . [CrossRef]
- Teasdale, A.; Fenner, S.; Ray, A.; Ford, A.; Phillips, A., A Tool for the Semiquantitative Assessment of Potentially Genotoxic Impurity (PGI) Carryover into API Using Physicochemical Parameters and Process Conditions. Org. Proc. Res. Dev. 2010, 14, 943-945, . [CrossRef]
- Borukhova, S.; Noël, T.; Hessel, V., Continuous-Flow Multistep Synthesis of Cinnarizine, Cyclizine, and a Buclizine Derivative from Bulk Alcohols. ChemSusChem 2016, 9, 67-74, . [CrossRef]
- ChemBK 2,2,2-Trifluoro-1-phenyl-ethylamine: https://www.chembk.com/en/chem/2,2,2-Trifluoro-1-phenyl-ethylamine. (05.05.2023),.
- Smith, P. J.; Noble, A., A Primary Hydrogen-Deuterium Isotope Effect Study on the Carbonyl Elimination Reaction of 9-Fluorenyl Nitrate with Various Bases. Can. J. Chem. 1975, 53, 263-268, https://cdnsciencepub.com/doi/pdf/10.1139/v75-036.
- Juranic, I., Simple Method for the Estimation of pKa of Amines. Croat. Chem. Acta 2014, 87, 343-347, http://dx.doi.org/10.5562/cca2462.
- Lansbergen, B.; Meister, C. S.; McLeod, M. C., Unexpected rearrangements and a novel synthesis of 1,1-dichloro-1-alkenones from 1,1,1-trifluoroalkanones with aluminium trichloride. Beilstein J. Org. Chem. 2021, 17, 404-409, . [CrossRef]
- Jesumoroti, O. J.; Beteck, R. M.; Jordaan, A.; Warner, D. F.; Legoabe, L. J., Exploration of 4-aminopyrrolo[2,3-d]pyrimidine as antitubercular agents. Mol. Divers. 2023, 27, 753-756, . [CrossRef]
- Kaspersen, S. J.; Sørum, C.; Willassen, V.; Fuglseth, E.; Kjøbli, E.; Bjørkoy, G.; Sundby, E.; Hoff, B. H., Synthesis and in vitro EGFR (ErbB1) tyrosine kinase inhibitory activity of 4-N-substituted 6-aryl-7H-pyrrolo[2,3-d]pyrimidine-4-amines. Eur. J. Med. Chem. 2011, 46, 6002-6014, . [CrossRef]
Figure 1.
Acid assisted amination to medically important compounds. Vandetanib: ref.[20]; Bosutinib precursor ref.[21]; Icotinib intermediate ref. [22].
Scheme 1.
Proposed intermediates and equilibriums for the amination of pyrrolopyrimidine 1. The numbering system for pyrrolopyrimidines is shown for compound 1.
Scheme 1.
Proposed intermediates and equilibriums for the amination of pyrrolopyrimidine 1. The numbering system for pyrrolopyrimidines is shown for compound 1.
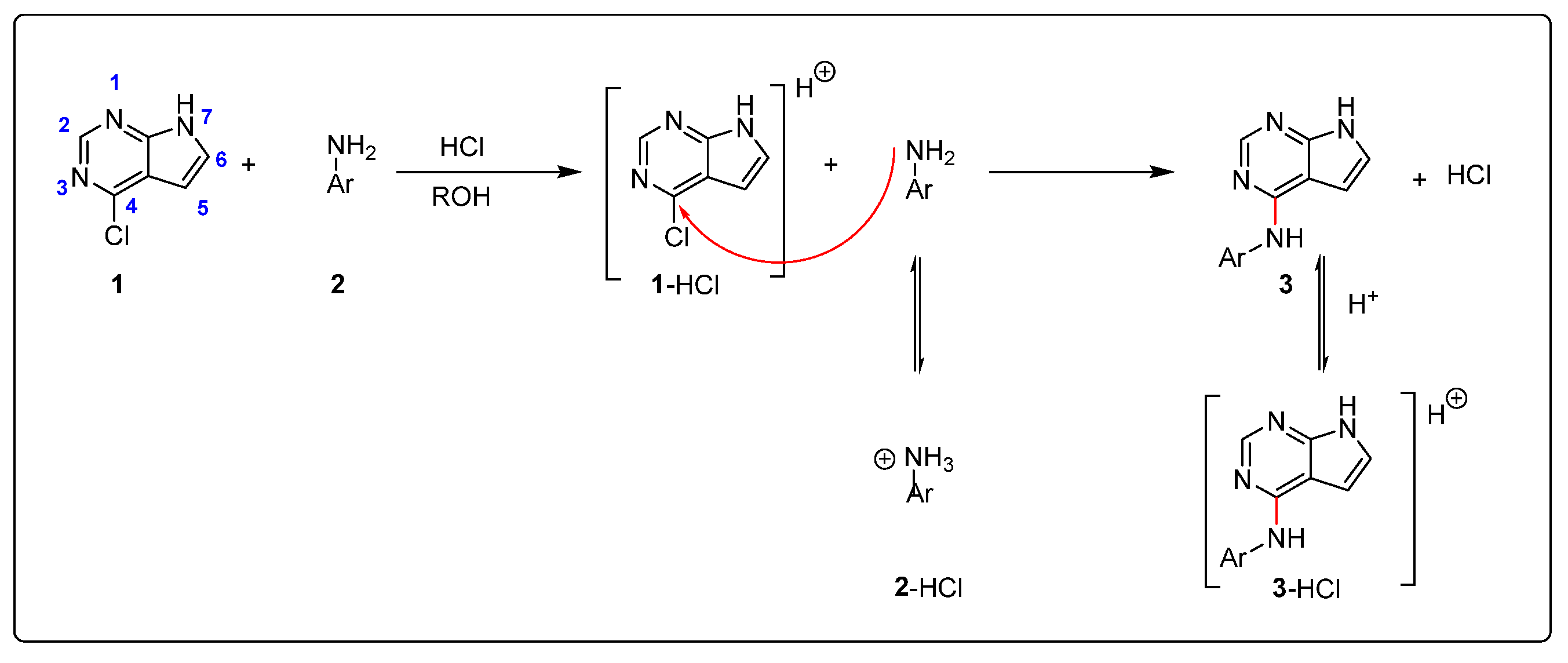
Figure 2.
Effect of HCl equiv. and reaction time on level of the side-product 4.
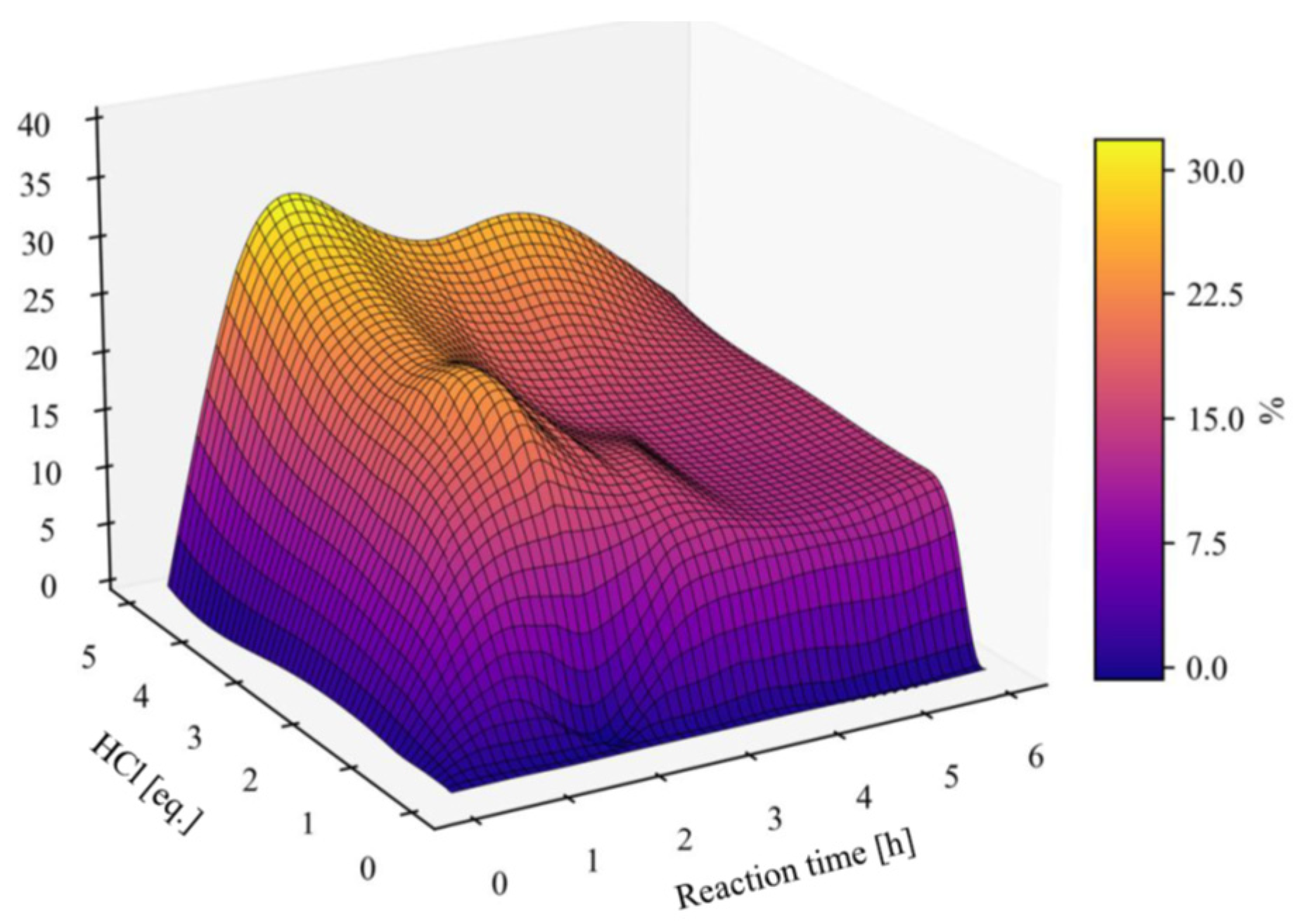
Figure 3.
Effect of solvent type on formation of 3a using 0.1 equiv. of HCl. The reactions were performed at 60 °C.
Figure 3.
Effect of solvent type on formation of 3a using 0.1 equiv. of HCl. The reactions were performed at 60 °C.
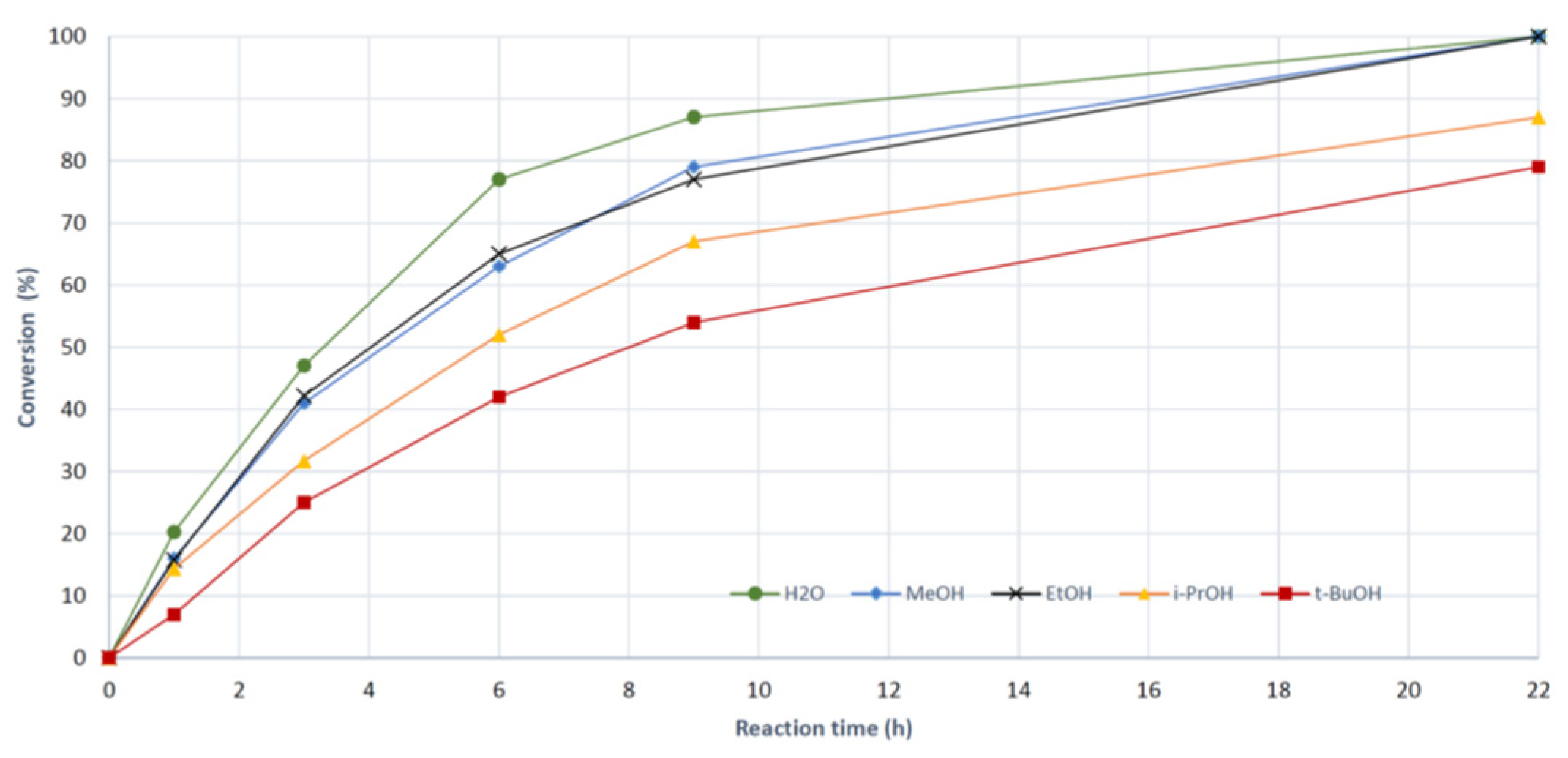
Figure 4.
Comparison of reaction profiles for preparation of 3a (black circles), 3b (green squares) and 3j (red triangles). Conv. (%) = 100 × (3 + 5)/ (1+3+5).
Figure 4.
Comparison of reaction profiles for preparation of 3a (black circles), 3b (green squares) and 3j (red triangles). Conv. (%) = 100 × (3 + 5)/ (1+3+5).
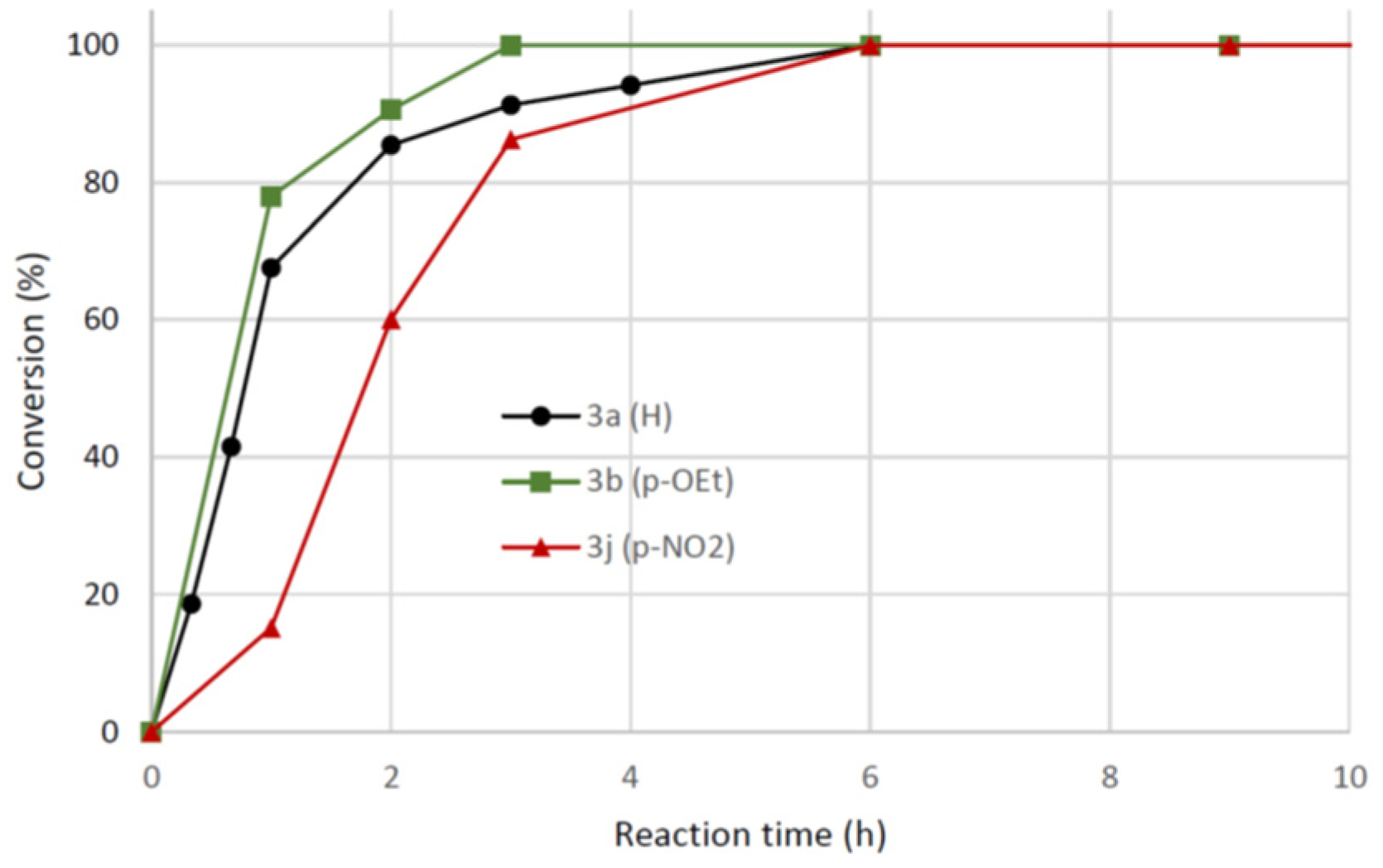
Figure 5.
Comparison of reaction profiles for preparation of 3a (black circles), 3l (green squares), 3k, (blue triangles), 3n (pink tilted squares) and 3o (red cross). Conv. (%) = 100 × (3 + 5)/ (1+3+5).
Figure 5.
Comparison of reaction profiles for preparation of 3a (black circles), 3l (green squares), 3k, (blue triangles), 3n (pink tilted squares) and 3o (red cross). Conv. (%) = 100 × (3 + 5)/ (1+3+5).
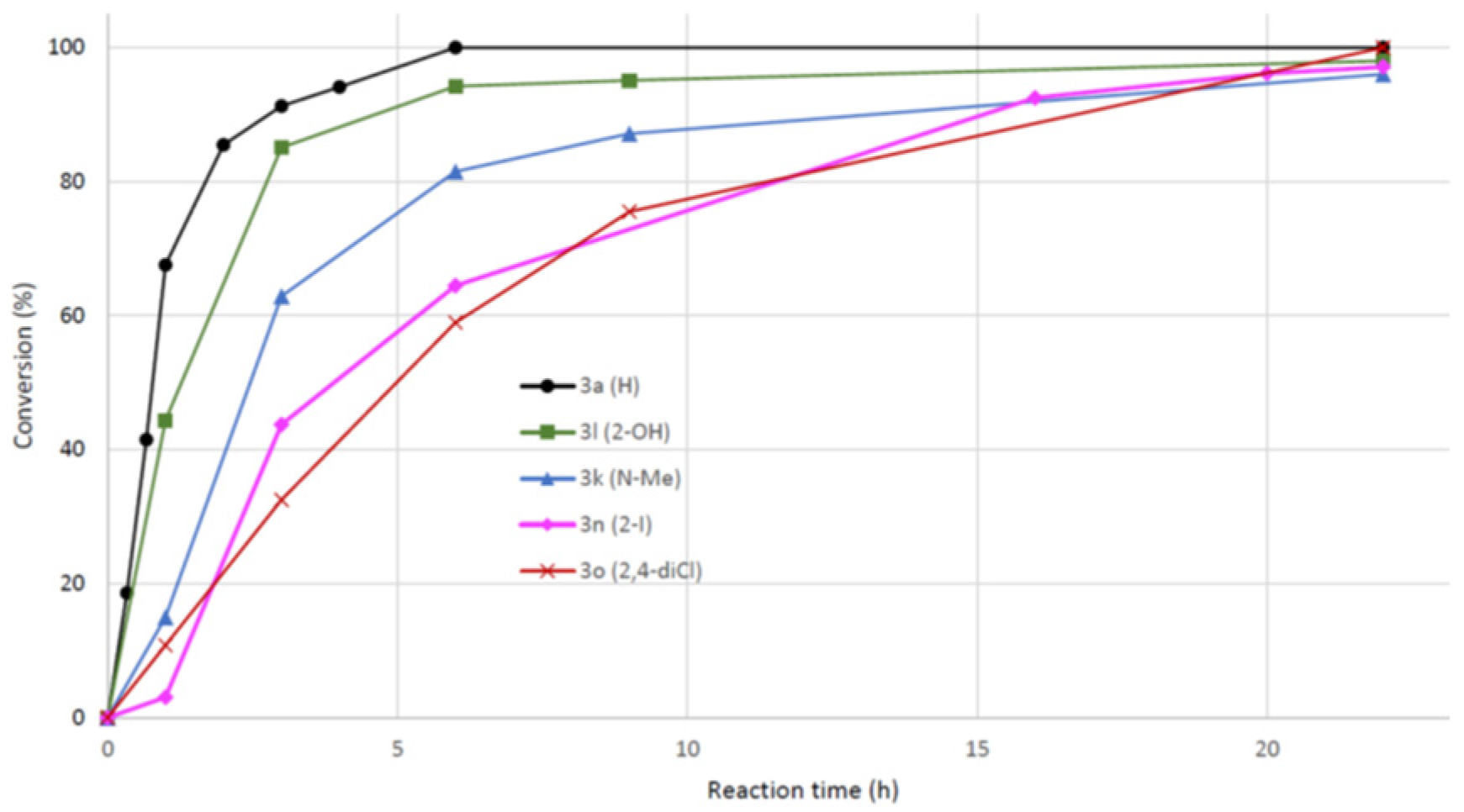
Scheme 2.
Substrates 6 and 7 used to test the limitation of acid catalyzed amination in water. The reaction failed in water, but proceeded well in 2-PrOH.
Scheme 2.
Substrates 6 and 7 used to test the limitation of acid catalyzed amination in water. The reaction failed in water, but proceeded well in 2-PrOH.
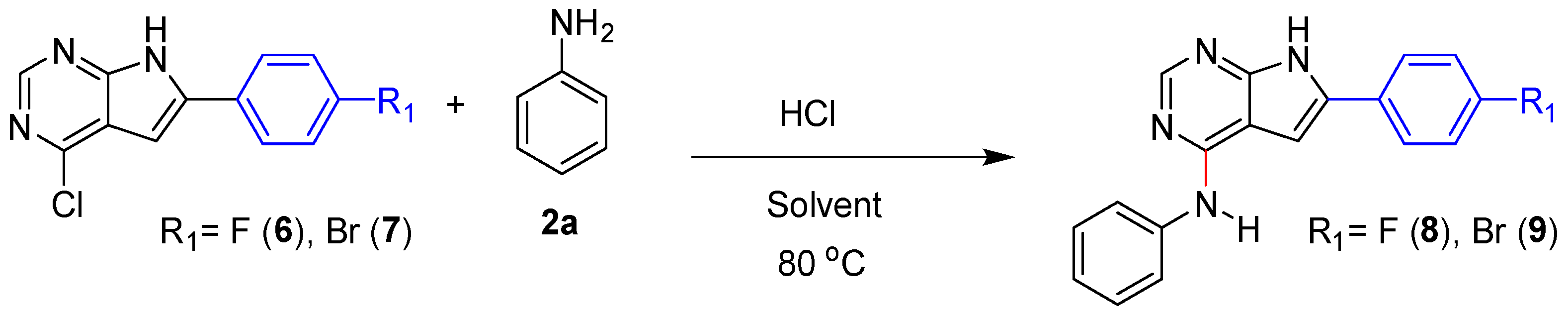

Table 1.
Effect of HCl amount on reaction progress and formation 3a and the side-product 4.
| Mole % after 6 h. 2 | |||||
|---|---|---|---|---|---|
| Entry | HCl (equiv) | Conv. 1 h (%) 1 | 1a | 3a | 4 |
| 1 | 0 | < 1 | 31 | 69 | <1 |
| 2 | 0.1 | 51 | < 1 | >98 | < 1 |
| 3 | 0.5 | 78 | < 1 | 90 | 10 |
| 4 | 1.0 | 82 | < 1 | 86 | 14 |
| 5 | 3 | 82 | < 1 | 85 | 15 |
| 6 | 5 | 86 | < 1 | 83 | 17 |
1 Conversion was measured by 1H NMR, conv (%) = 100×[3a+4]/[1+3a+4]. 2 Mole % of 1, 3a and 4. Values denoted as < 1 means not detected.
Table 2.
Effect of HCl amount on reaction progress in water and formation 3a and the side-product 5.
Table 2.
Effect of HCl amount on reaction progress in water and formation 3a and the side-product 5.
| Mole % after 6 h. 2 | |||||
|---|---|---|---|---|---|
| Entry | HCl (equiv) | Conv. 0.33 h (%) 1 | 1a | 3a | 5 3 |
| 1 | 0.1 | 19 | < 1 | >98 | <1 |
| 2 | 0.2 | 28 | < 1 | >98 | <1 |
| 3 | 0.5 | 48 | < 1 | 98 | 1 |
| 4 | 0.8 | 59 | < 1 | 98 | 1 |
| 5 | 1.0 | 54 | < 1 | 98 | 1 |
1 Conversion was measured by 1H NMR, conv (%) = 100×[3a+5]/[1+3a+5]. 2 Mole % of 1, 3a and 5, < denoted an undetected compound. 3 Detected levels are likely to be lower than actual levels due to extractive work-up.
Table 3.
Substrate scope in HCl promoted amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with anilines. Conversion data and mole (%) data is from 100 mg reactions, while isolated yields are from 500 mg reactions.
Table 3.
Substrate scope in HCl promoted amination of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with anilines. Conversion data and mole (%) data is from 100 mg reactions, while isolated yields are from 500 mg reactions.
| . | Mole (%)3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Aniline | pKa1 | Conv 1 h (%)2 |
Reaction time (h) |
1 | 3 | 5 | Yield (%) | Prod. |
| 1 | 4-OEt (2b) | 5.19 | 78 | 3 | < 1 | >98 | < 1 | 94 | 3b |
| 2 | 4-Bu (2c) | 4.95 | 66 | 6 | < 1 | >98 | < 1 | 88 | 3c |
| 3 | 3,4-methylene- dioxy (2d) |
4.46 | 74 | 3 | < 1 | >98 | < 1 | 85 | 3d |
| 4 | 4-F (2e) | 4.65 | 77 | 6 | < 1 | >98 | < 1 | 92 | 3e |
| 5 | H (2a) | 4.58 | 68 | 6 | < 1 | >98 | < 1 | 91 | 3a |
| 6 | 3-OBn (2f) | ca 4.2 | 86 | 6 | < 1 | >98 | < 1 | 87 | 3f |
| 7 | 3-ethyne (2g) | 3.82 | 81 | 6 | < 1 | >98 | < 1 | 83 | 3g |
| 8 | 3-Cl (2h) | 3.34 | 80 | 6 | < 1 | >98 | < 1 | 81 | 3h |
| 9 | 4-Br-3-F (2i) | 2.73 | 84 | 3 | < 1 | >98 | < 1 | 91 | 3i |
| 10 | 4-NO2 (2j) | 1.02 | 15 | 6 | < 1 | 97 | 3 | 88 | 3j |
| 11 | N-Me-4-F (2k) | ca 4.9 | 15 | 22 | 4 | 96 | < 1 | 88 | 3k |
| 12 | 2-OH (2l) | 4.84 | 44 | 22 | 2 | 96 | 2 | 89 | 3l |
| 13 | 2,6-(i-Pr)2 (2m) | 4.51 | 0 | 22 | 83 | 0 | 17 | - | 3m |
| 14 | 2-I (2n) | 2.6 | 3 | 22 | 3 | 94 | 3 | 79 | 3n |
| 15 | 2,4-Cl (2o) | 2.0 | 11 | 22 | < 1 | 85 | 15 | 80 | 3o |
| 16 | 2,4,5-Cl (2p) | 1.09 | 0 | 22 | 73 | 15 | 12 | - | 3p |
| 17 | 2,6-Cl (2q) | 0.42 | 0 | 22 | 83 | 0 | 17 | - | 3q |
| 18 | 2-NO2 (2r) | -0.31 | 0 | 22 | 78 | 5 | 17 | - | 3r |
| 19 | 2-CF3, 4-NO2 (2s) | < 0 | 0 | 22 | 85 | 0 | 15 | - | 3s |
| 20 | 2,3,4,5,6-F (2t) | -0.28 | 0 | 22 | 82 | 3 | 15 | - | 3t |
1 The specific sources for the experimental and calculated pKa values,[24,25,26,27] are given in the Supporting Information File. 2 Conversion of the amination after 1 h measure by 1H NMR: Conv. = 100 ×[3+5]/[1+3+5], using signal from H-2. 3 Mole % of 1, 3 and 5 at the termination point measured by 1H NMR. Values denoted as < 1 means not detected. The levels of 5 is underestimated by the analysis.
Table 4.
Amination of non-aromatic amines under different conditions.
| Entry | Amine (equiv.) |
HCl (equiv.) |
Solvent | Reaction time (h) |
Conv. (%) | Product |
|---|---|---|---|---|---|---|
| 1 | 1.1 | 0.1 | H2O | 22 | low | 10 1 |
| 2 | 1.1 | 0.1 | H2O | 22 | 13 | 11 |
| 3 | 1.1 | 0.1 | H2O | 22 | 61 | 12 |
| 4 | 1.1 | 0.1 | H2O | 22 | 24 | 13 |
| 5 | 3 | 3 drops 2 | 2-PrOH | 6 | >98 | 12 |
| 6 | 3 | None | H2O | 2.5 | >98 | 12 |
| 7 | 1.5 | None | H2O | 22 | 86 | 12 |
| 8 | 3 | 3 drops 2 | 2-PrOH | 22 | 93 | 13 |
| 9 | 3 | None | H2O | 8 | >98 | 13 |
1 The identity of the product could not be confirmed. 2 HCl 3 drops corresponds to approximately 1 equiv. of HCl in this setting.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



