
Submitted:
12 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
The optimal regime of three-photon resonant excitation of a helium atom by a femtosecond ul-traviolet (UV) pulse was discovered and numerically studied, at which the maximum power of the third harmonic of the UV field is achieved in the spectrum of dipole acceleration (the second time derivative of the induced dipole moment) of the atom. It is shown that the optimal frequency of the UV field nearly coincides with the frequency of the three-photon transition 1s2 – 1s2p, taking into account its shift as a result of the dynamic Stark effect, and the intensity of the UV field is dictated by the condition of maximizing the product of the populations of the 1s2 and 1s2p states, averaged over the time interval during which the UV field is non-zero. For the considered UV field durations, from 10 to 100 cycles of the carrier frequency (from units to tens of femtoseconds), the optimal intensity lies in the range from 10^14 W/cm^2 to several units of 10^14 W/cm^2. It is shown that with an optimal choice of the frequency and intensity of the UV field, the dynamics of excitation of bound and continuum states, as well as the shape of the time envelope of the dipole acceleration of the atom, weakly depend on the duration of the UV field envelope; only their time scale changes significantly. In addition, under the optimal conditions the average power of the third harmonic signal in the dipole acceleration spectrum is practically independent of the duration of the UV field envelope.
Keywords:
harmonic generation
; resonant multiphoton processes
; atomic excitation and ionization
; nonperturbative interaction
; vacuum ultraviolet
1. Introduction
The generation of coherent radiation in the vacuum ultraviolet (VUV) and X-ray ranges is one of the topical problems of modern optics. On the one hand, interest in this problem is due to the short period of field oscillations and the possibility of generating radiation pulses of the shortest (attosecond) duration achievable today. On the other hand, the high frequency of radiation, corresponding to the photon energy comparable to or exceeding the ionization potential of an atom, makes it possible to implement regimes of light-matter interaction that are unattainable in the optical range, such as single-photon excitation or ionization of an atom from both the valence and core electronic shells, triggering Auger decay or single-photon excitation of autoionization states [1,2,3,4,5,6,7,8,9,10]. At the same time, the short wavelength of the radiation allows it to be focused into a spot with dimensions significantly smaller than the wavelength of the field in the visible/infrared (IR) range and thereby achieve record spatial resolution [11].
One of the main methods for generating coherent VUV/X-ray radiation is high-order harmonic generation (HHG) of an optical laser field in gases or solids [12,13]. Initially, HHG was considered primarily as a method for generating a wide-band spectrum of harmonics with photon energies significantly exceeding the ionization potential of an atom. In this case, HHG occurs in the regime of tunneling ionization of the atom and is the result of a three-step process, including the release of an active electron, its acceleration in the laser field, and subsequent recombination with the parent ion [14,15]. However, in recent years, the generation of harmonics of moderate orders with photon energies lower than or on the order of the ionization potential of the atom has also attracted considerable interest [16,17,18,19,20,21,22,23,24]. In this case, HHG occurs in the process of multiphoton excitation and multiphoton ionization of the atom and is largely due to transitions between bound states. The advantages of this regime are the high (compared to the tunnel ionization regime) efficiency of harmonic generation and less stringent requirements for the intensity of the laser field. In this case, as a rule, the driver for harmonic generation is the field of the near or mid-IR range, and the order of multiphoton resonances (if any) is somewhere around ten.
As in the above-mentioned studies, this work considers the generation of harmonics under conditions of multiphoton excitation and multiphoton ionization of the atom. The main attention is paid to the properties of the lowest-order (and most intense) harmonic of the external field, namely the third harmonic. It is assumed that the fundamental frequency lies in the ultraviolet range, and the multiphoton resonance is of low order (third). This regime has significant advantages over the harmonic generation driven by the visible/IR range field. Firstly, the role of multiphoton resonances increases and the maximum achievable efficiency of generation of below-threshold and near-threshold harmonics (with photon energies lower than or on the order of the ionization potential of the atom) increases as well. Secondly, with an increase in the fundamental frequency, the phase matching of the driving field with the radiation of its harmonics improves, which makes it possible to achieve a higher efficiency of harmonic generation in an extended medium (note, however, that the analysis of macroscopic effects is beyond the scope of this article).
In the 1970s - 1990s, a number of theoretical [25,26,27,28,29] and experimental [30,31,32,33,34,35,36,37] studies were published on the generation of low-order (primarily third) harmonics of ultraviolet radiation in gases. The theoretical models used in these studies were based on (a) a phenomenological description of the nonlinear response of the medium (nonlinear susceptibility formalism), (b) solution of the Schrödinger equation or equations for the elements of the density matrix for a two-, three-, or four-level system, and (c) on the perturbation theory corresponding to the weak field approximation, as well as a combination of these approaches. In the spectrum of the field and excitation amplitudes of stationary states (or elements of the density matrix), only the lowest- order, often the 1st, 2nd and 3rd harmonics of the fundamental frequency were taken into account.
In this work, the response of an atom to an external field is calculated without using the above approximations: the non-perturbative regime of laser-matter interaction is considered, and all possible harmonics of the fundamental frequency are taken into account in the spectrum of the dipole response of the atom. The harmonic generation process is analyzed using the example of helium atoms by solving the time-dependent Schrödinger equation (TDSE) in a given femtosecond UV field and calculating the induced dipole moment of the atom. Two approaches are used. The first of them involves the expansion of the wave function in terms of a sufficiently large number of bound states of an unperturbed atom, as well as continuum states into which electric dipole transitions from the bound states taken into account are allowed. The second approach is to numerically solve the TDSE from first principles in a two-dimensional model using the effective one-electron potential.
The article is organized as follows. The description of the theoretical models used and an analysis of the results obtained are given in Section 2 and Section 3, respectively. In Section 4 the conclusions are given. A significant part of the analytics, as well as the parameters of the models used, are included in Appendices.
2. Theoretical model
The consideration in this article is based on the TDSE solution for a helium atom in a given external field. In the electric dipole approximation, the TDSE has the form
where is the wave function, and are the radius vectors of electrons in a helium atom, is the Hamiltonian of an atom in the absence of a field, is the dipole moment operator, and is the external (in this case, UV) field. Here and below, atomic units are used. In what follows, we will assume that the UV field is linearly polarized along the z axis and has the form
where is the unit vector along the z axis, and are the amplitude and frequency of the UV field, respectively, and is the trapezoidal envelope:
where is the UV field oscillation period. In accordance with Equations (2) and (3), the UV field pulse has a constant amplitude in a time interval lasting Nosc field cycles, while turning on and off the UV field is characterized by a linear increase (decrease) in its amplitude over 3 field cycles. In what follows, the values Nosc = 10, 30, 50, 70, and 100 are considered (most of the figures are given for Nosc = 30). Note that the absolute duration of the UV field envelope is inversely proportional to its carrier frequency Ω.
As mentioned in the Introduction, the results of the calculations presented in this article were obtained using two approaches to solving the TDSE for a helium atom irradiated with a femtosecond pulse of an ultraviolet field.
The first approach is based on the expansion of the atomic wave function over a limited (sufficiently large) basis of eigenfunctions corresponding to bound states and continuum states into which electric dipole transitions from the considered bound states are allowed. This expansion has the form
where is the spatial part of the wave function of the kth stationary state, is the temporal factor in the same wave function, which we will further call the excitation amplitude of the state , , and is the excitation amplitude of the continuum state with energy and orbital momentum l, . In further calculations, the expansion of the wave function takes into account the first 10 bound states of the helium atom (Kmax=10), corresponding to the values of the principal quantum number n = 1,2,3,4 and the projection of the orbital momentum of the atom onto the field polarization direction m = 0 (electric dipole transitions from ground state into states with m ≠ 0 in a linearly polarized field are prohibited by selection rules). Namely, states , , , , , , , , , and are taken into account (for brevity, here and below, the value of the projection of the orbital momentum of the atom m = 0 is not given in the designation of states). Among the continuum states, those with orbital momentum from 0 to Lmax = 4 (s-, p-, d-, f-, and g-waves) are taken into account. This set of states makes it possible to quite accurately take into account one-, two-, three-, four-, and five-photon transitions from the ground to lower bound states and (transitions to higher-lying states are described with less accuracy). Note that the time dependences take into account oscillations of the phase of the wave function at the natural frequencies of stationary states, which in the atomic system of units are equal to , where is the energy of the kth stationary state in the absence of a field (state energies and other constants used are given in Appendix A). Accordingly, depending on the ratio of the values of and , even in a weak UV field limit, the excitation amplitudes can be fast or slow functions of time on the scale of the UV field cycle.
Further, the excitation amplitudes of bound states are decomposed into harmonics of the UV field:
where is the Fourier component of the excitation amplitude of the kth bound stationary state.
To take into account the ionization of an atom from the bound states under consideration, the approximation of adiabatic exclusion of continuum states is used [38], which, primarily, corresponds to not taking into account transitions between continuum states. In addition, reverse transitions from continuum states to bound states are also not taken into account. These approximations make it possible to express the excitation amplitudes of continuum states through the Fourier components of the excitation amplitudes of bound states and, ultimately, to write a closed system of linear differential equations for , which takes into account the decrease in absolute values of as a result of ionization. These equations have the form
where is the projection of the dipole moment of the transition from state |s〉 to state |k〉 on the z axis, and is the ionization rate of the nth Fourier component of the kth state. Using the residue theory, the following expression can be obtained for :
where is the z-projection of the dipole moment operator, and is the Heaviside unit step function. The derivation of Equations (6) and (7) is given in Appendix B.
Equations (6), (7) were solved numerically using the 4th order one-step explicit Runge-Kutta method using initial conditions
which imply that before exposure to the UV field the atom is in the ground state with energy .
This approach is a simplified and less resource-intensive implementation of the SSEA (state-specific expansion approach) method [39,40]. It makes it possible to quite accurately describe the component of the dipole response of an atom to an external field that dominates in the considered spectral range of below-threshold and near-threshold harmonics (see, for example, [41]) and is caused by transitions between bound states,
In formula (9), the index bb of the induced dipole moment of an atom indicates transitions between bound states, the indices k and s number the stationary states taken into account, and the indices n and m number their Fourier components. Note that within the framework of this approach, the response of the atom due to transitions between continuum states, as well as from continuum to bound states, is not taken into account.
As usual in strong-field physics, the response of an atom to an external field will be characterized by dipole acceleration,
This approach allows one to analyze the contributions of certain stationary states to the generation of harmonics and take into account the properties of a real multielectron atom (through the energies of stationary states and the dipole moments of transitions between them) by choosing the correct basis functions. For this purpose, in this work we used the spectroscopic characteristics of a three-dimensional two-electron helium atom, calculated by the multi-configuration Hartree-Fock method (see Appendix A), the error of which does not exceed several percent relative to experimental data [42].
The second approach used in this article is to solve the TDSE from first principles in a two-dimensional model of a one-electron atom with an effective potential , reproducing the binding energies of ground state and lowest excited bound states and :
where is the vector potential of the UV field, c is the speed of light in vacuum, and
The energies of the lowest stationary states and the dipole moments of transitions between them calculated in this model are given in Appendix A.
This approach has less accuracy in describing the dynamics of the localized part of the atomic wave function, but takes into account recombination and all possible transitions between states of the continuum. Accordingly, the dipole response of an atom to an external field, in addition to the contribution from transitions between bound states, contains components due to transitions from continuum to bound states (recombination) and transitions between continuum states. The dipole acceleration of an atom is calculated using Ehrenfest’s theorem:
where integration is carried out over the entire computational domain.
3. Calculation results
As was shown in [43], the approaches to solving the TDSE used in this article give similar time dependences of the probability of ionization of a helium atom in a UV field with an intensity IUV = 1014 W/cm2. In the first approach, based on the expansion of the wave function in terms of the basis of stationary states, the probability of ionization is determined as
whereas within the framework of the second approach, based on solving a two-dimensional one-electron TDSE from first principles, it has the form
where the integration is carried out inside a square with side A = 100 a.u., in the center of which the core of the He atom is located. In this case, the wave functions of bound states with principal quantum numbers n = 1,2,3,4 are completely contained within the integration region.
As follows from Figure 1, the frequency dependences of the ionization probability are also qualitatively similar. Figure 1 shows the probability of ionization of an atom at the end of a UV field pulse with a peak intensity IUV = 1014 W/cm2 and a constant-amplitude interval duration equal to 30 UV field cycles (Nosc = 30), as a function of its frequency. The frequency of the UV field is scanned in a range that covers resonances with two-, three-, and four-photon transitions from the ground to excited bound states. The blue curve corresponds to the solution obtained based on expansion (4), and the red curve based on the two-dimensional one-electron model.
Figure 1.
Dependence of the probability of ionization of a helium atom at time t = 50T on the frequency of the UV field with a peak intensity IUV = 1014 W/cm2. The blue solid curve corresponds to the solution obtained by expanding the atomic wave function in stationary states, , and the red dotted curve corresponds to the solution of the two-dimensional one-electron TDSE from first principles, . Vertical dashed lines indicate the frequencies of multiphoton transitions between unperturbed bound states of the atom. In this case, multiphoton resonances of odd order are marked in black, and resonances of even order are marked in green.
Figure 1.
Dependence of the probability of ionization of a helium atom at time t = 50T on the frequency of the UV field with a peak intensity IUV = 1014 W/cm2. The blue solid curve corresponds to the solution obtained by expanding the atomic wave function in stationary states, , and the red dotted curve corresponds to the solution of the two-dimensional one-electron TDSE from first principles, . Vertical dashed lines indicate the frequencies of multiphoton transitions between unperturbed bound states of the atom. In this case, multiphoton resonances of odd order are marked in black, and resonances of even order are marked in green.
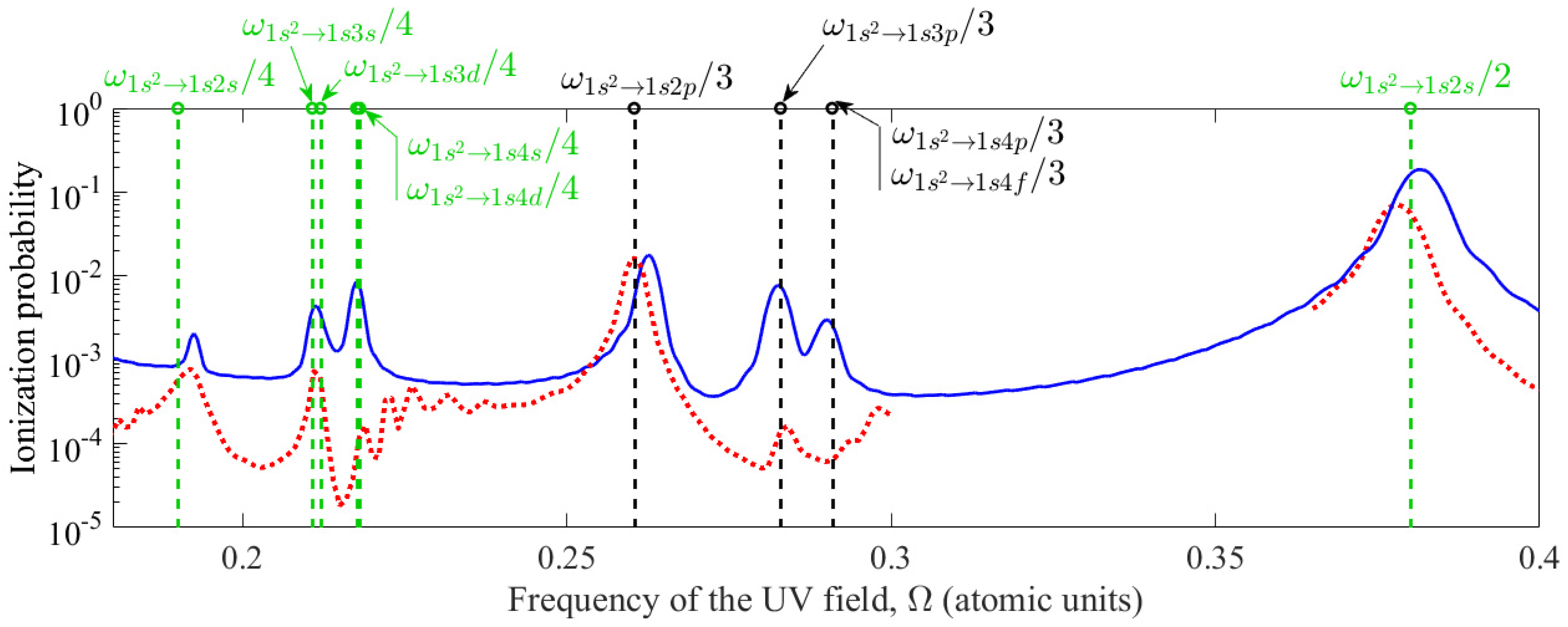
As follows from Figure 1, multiphoton resonances are reproduced in both models, but their frequencies are slightly different due to (a) the neglect of highly excited bound states and the use of the adiabatic continuum exclusion approximation when expanding the wave function in stationary states and (b) the difference in the dipole moments of the transitions, as well as energies of highly excited bound states, in a two-dimensional one-electron model from the corresponding parameters of a real helium atom. The best agreement in the probability of atomic ionization is achieved for a three-photon transition from state to state , as well as two- and four-photon transitions to state , which is explained by the highest accuracy of both models in describing these transitions. The accuracy of the approach based on the expansion of the wave function in stationary states is due to the low probability of transitions from ground state to disregarded intermediate states with n ≥ 5 during three-photon excitation of state or two/four-photon excitation of state in a real helium atom. At the same time, the two-dimensional model of the helium atom is constructed in such a way as to reproduce the energies of states , , and with few percent accuracy. The dipole moments of transitions between them are reproduced with lower, but still reasonable accuracy of tens of percent (see Appendix A). Taken together with the fact that the three-photon transition to state predominantly occurs through state , while the two- and four-photon transitions to state occur through state , this determines the accuracy of the two-dimensional model in describing these transitions.
Let us consider in more detail the region of resonances with three-photon transitions from state to the p- and f-states taken into account. Figure 2 shows the dependence of the probability of ionization of an atom at the end of a UV field pulse on its frequency and peak intensity, calculated by expanding the atomic wave function over stationary states. Similar to Figure 1, this figure is given for UV field pulses with a constant amplitude interval duration equal to 30 carrier cycles (Nosc = 30). Note that the blue curve in Figure 1 in the frequency range 0.25<Ω<0.3 represents a cross-section of Figure 2 for an intensity value of 1014 W/cm2.
Figure 2.
Probability of ionization of a helium atom (in a logarithmic scale) after exposure to a UV field pulse depending on its intensity and frequency in the vicinity of three-photon resonances. The dotted lines indicate the frequencies of three-photon transitions between unperturbed atomic states; here black color corresponds to the transition |1s2〉 - |1s2p〉, green is for the transition |1s2〉 - |1s3p〉, whereas blue is for the transitions |1s2〉 - |1s4p〉 and |1s2〉 - |1s4f〉 (on the scale of the figure they coincide). The solid black curve indicates the frequency of the three-photon transition |1s2〉 - |1s2p〉 in the UV field.
Figure 2.
Probability of ionization of a helium atom (in a logarithmic scale) after exposure to a UV field pulse depending on its intensity and frequency in the vicinity of three-photon resonances. The dotted lines indicate the frequencies of three-photon transitions between unperturbed atomic states; here black color corresponds to the transition |1s2〉 - |1s2p〉, green is for the transition |1s2〉 - |1s3p〉, whereas blue is for the transitions |1s2〉 - |1s4p〉 and |1s2〉 - |1s4f〉 (on the scale of the figure they coincide). The solid black curve indicates the frequency of the three-photon transition |1s2〉 - |1s2p〉 in the UV field.
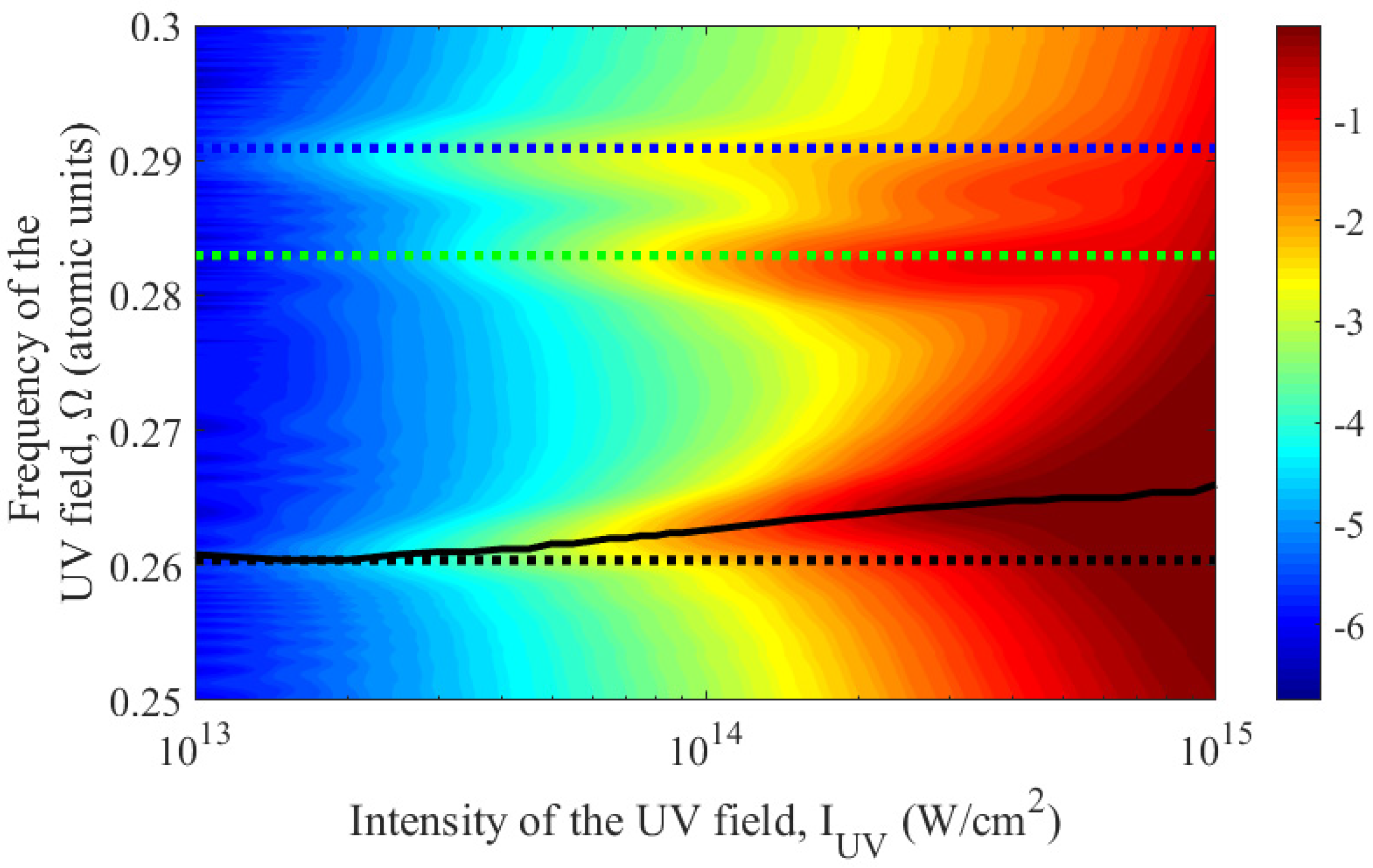
As follows from Figure 2, (i) the highest probability of ionization is achieved in the vicinity of the frequencies of three-photon transitions, (ii) the absolute maximum corresponds to the vicinity of the transition |1s2〉 - |1s2p〉 frequency; with increasing field intensity, (iii) the probability of ionization increases and (iv) the resonances broaden and shift in frequency.
Next, we will focus on the resonance with the three-photon transition |1s2〉 - |1s2p〉, which is characterized by the highest probability of atomic ionization and is most accurately described within the framework of the models used. For each fixed intensity of the UV field, we determine the resonance frequency using the method of multiphoton photoionization spectroscopy [44], based on the position of the maximum probability of atomic ionization in the vicinity of the frequency of the three-photon transition between the unperturbed states |1s2〉 and |1s2p〉. The dependence of the transition frequency on the UV field intensity obtained in this way is shown in Figure 2 by a black solid curve.
An increase in the probability of atomic ionization with increasing UV field intensity is accompanied by real and virtual excitation of bound states. As a consequence, the response of the atom to the UV field (dipole acceleration) is enriched in harmonics of the fundamental frequency. Figure 3 shows the dependence of the energy concentrated in the spectral component of the dipole acceleration of an atom (10) at the frequency of the 3rd harmonic of the UV field (the method for calculating this quantity is described in Appendix С) on its frequency and intensity. Calculations were made for the duration of the interval of constant amplitude in the UV field envelope equal to 30 carrier cycles (Nosc = 30).
Figure 3.
Dependence (in a logarithmic scale) of the third harmonic energy in the spectrum of the dipole acceleration of a helium atom on the intensity and frequency of the UV field in the vicinity of three-photon resonances. The dotted lines indicate the frequencies of three-photon transitions between unperturbed states of the atom; here black color corresponds to the |1s2〉 - |1s2p〉 transition, green is for the |1s2〉 - |1s3p〉 transition, whereas blue is for the |1s2〉 - |1s4p〉 and |1s2〉 - |1s4f〉 transitions (on the scale of the figure they coincide). The solid black curve indicates the frequency of the three-photon transition |1s2〉 - |1s2p〉 in the UV field. The cyan dashed curve corresponds to the dependence of the UV field frequency on its intensity, which maximizes the third harmonic energy in the dipole acceleration spectrum.
Figure 3.
Dependence (in a logarithmic scale) of the third harmonic energy in the spectrum of the dipole acceleration of a helium atom on the intensity and frequency of the UV field in the vicinity of three-photon resonances. The dotted lines indicate the frequencies of three-photon transitions between unperturbed states of the atom; here black color corresponds to the |1s2〉 - |1s2p〉 transition, green is for the |1s2〉 - |1s3p〉 transition, whereas blue is for the |1s2〉 - |1s4p〉 and |1s2〉 - |1s4f〉 transitions (on the scale of the figure they coincide). The solid black curve indicates the frequency of the three-photon transition |1s2〉 - |1s2p〉 in the UV field. The cyan dashed curve corresponds to the dependence of the UV field frequency on its intensity, which maximizes the third harmonic energy in the dipole acceleration spectrum.

Two remarks should be made here. The energy (time-integrated square of the amplitude) of the 3rd harmonic in the dipole acceleration spectrum was chosen as the parameter under study, rather than its peak power spectral density or peak intensity, because under certain conditions, the spectral and time dependences of the harmonic signal may have several maxima of comparable amplitude. In particular, this case is observed under conditions of Rabi oscillations between the resonant states of a multiphoton transition [45]. The second remark concerns the free-induction decay. Under conditions of three-photon resonance, the excited state |1s2p〉 of the atom, coupled by a dipole-allowed transition with the ground state |1s2〉, is effectively populated. As a result, after the end of the UV field pulse, the atom continues to emit at the frequency of transition |1s2〉 – |1s2p〉, close to the third harmonic frequency (however, slightly different from it in the absence of a field). This radiation corresponds to free-induction decay. In order to separate it from the third harmonic radiation, the dipole acceleration of the atom, calculated by formula (10) or (13), is multiplied by a temporary mask, which is nonzero only in the presence of a UV field (for more details, see Appendix С).
Similar to Figure 2, the dotted lines in Figure 3 indicate the frequencies of three-photon transitions to unperturbed states of the atom, and the solid black curve indicates the resonance frequency of the three-photon transition |1s2〉 – |1s2p〉 |in the UV field, determined by the maximum probability of ionization. The UV field frequency that maximizes the 3rd harmonic energy in the dipole acceleration spectrum for a given field intensity is plotted in Figure 3 by the dashed cyan curve. As follows from Figure 3, the maximum energy of the 3rd harmonic in the dipole acceleration spectrum is achieved at a UV field intensity IUV = 2×1014 W/cm2 and a frequency Ω = 0.2644 a.u., which is in close proximity to the resonant frequency, Ωres = 0.2638 a.u., of three-photon transition to the |1s2p〉 state in the presence of a UV field of given intensity (in this case, the detuning from resonance is about 1/3 of the half-width of the resonance curve in the frequency dependence of the probability of ionization of an atom). For the specified optimal parameters, the probability of ionization at the end of the UV field pulse is approximately 24%.
As the UV field intensity decreases, the third harmonic energy (Figure 3) and, in particular, the probability of atomic ionization (Figure 2), quickly decrease. In this case, the frequency that maximizes the harmonic energy practically coincides with the frequency of the resonant three-photon transition |1s2〉 – |1s2p〉 in the UV field. At the same time, for a UV field intensity exceeding the optimal value IUV = 2×1014 W/cm2, the value of the maximum achievable energy of the third harmonic decreases, while the detuning of the optimal (for a given intensity) frequency of the UV field from the frequency of the three-photon resonance with the |1s2〉 – |1s2p〉 transition increases. With a further increase in the intensity of the UV field, the frequency that maximizes the energy of the third harmonic in the spectrum of the dipole acceleration of the atom shifts to the region of resonances with the transitions |1s2〉 – |1s3p〉, |1s2〉 – |1s4p〉, and |1s2〉 – |1s4f〉, while the harmonic energy continues to decrease.
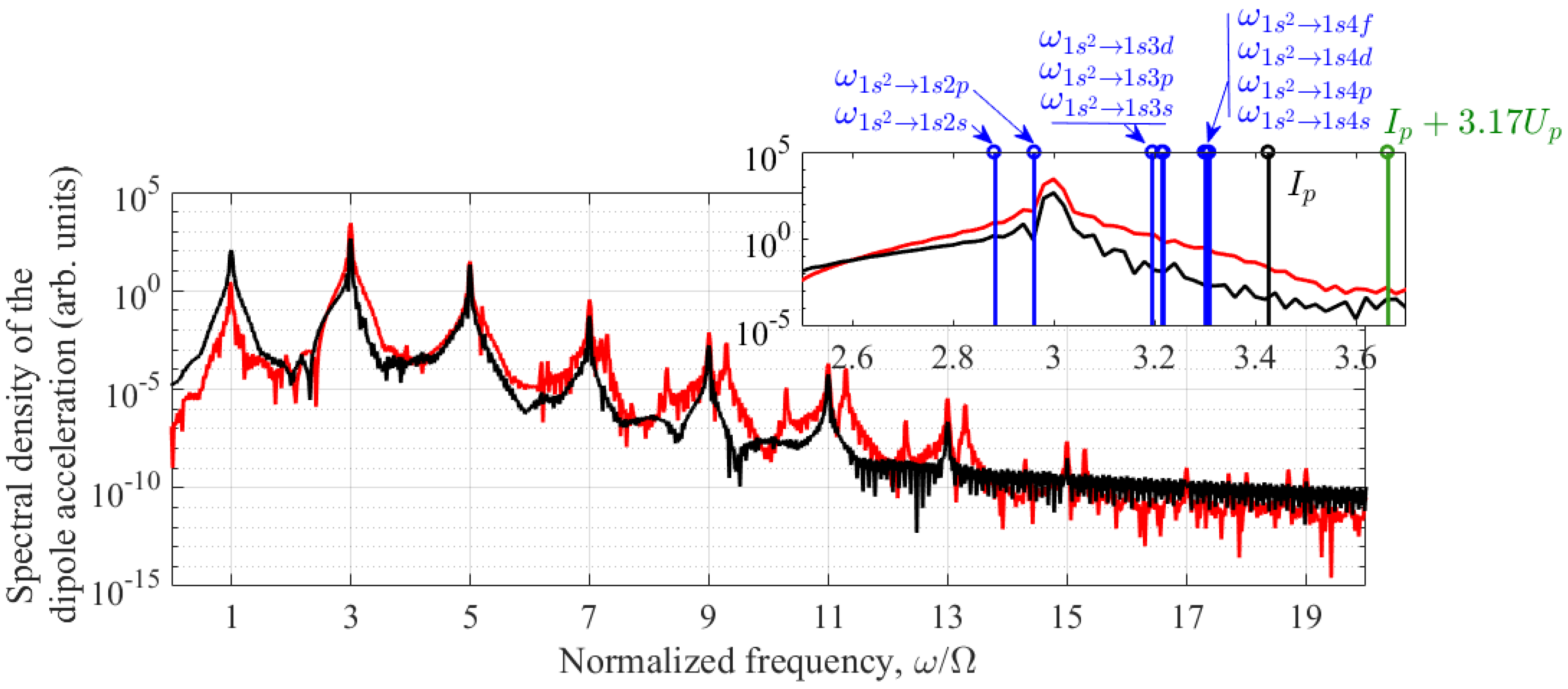
To explain this dependence, let us analyze additional figures. Figure 4 shows the frequency dependence of the power spectral density (the square of the modulus of the spectral amplitude, see Appendix С) of the dipole acceleration of an atom under conditions that maximize the energy of the 3rd harmonic for Nosc = 30. The red curve shows the solution of Equations (6), (7), corresponding to the expansion of the wave function in terms of the basis of stationary states (4). In this case, the third harmonic energy reaches a maximum at IUV = 2×1014 W/cm2 and Ω=0.2644 a.u, see Figure 3. The black curve is obtained based on model (11), (12). In this case, the optimum shifts towards higher UV field intensity and the maximum is achieved at IUV = 4×1014 W/cm2 and Ω = 0.2730 a.u. The increase in the optimal intensity value in the two-dimensional model is most likely due to the lower probability of multiphoton excitation and ionization of the atom, see Figure 1. Similar to Figure 3, in this case the optimal frequency of the UV field, Ω = 0.2730 a.u., turns out to be close to the resonance frequency of the |1s2〉 – |1s2p〉 transition in the UV field, which for IUV = 4×1014 W/cm2 in the two-dimensional model is Ωres = 0.2720 a.u. Several conclusions can be drawn from Figure 4. Firstly, the dipole acceleration of an atom is multi-frequency. In addition to the fundamental frequency of the UV field and its third harmonic, the atomic response contains harmonics of higher orders, up to the 13th one. Secondly, for the chosen UV field parameters, the 3rd harmonic in the dipole acceleration spectrum is dominant. In the solution of Equations (6), (7), it is approximately 90 times more intense than the 5th harmonic, three orders of magnitude more intense than the signal at the fundamental frequency of the UV field, and 4 orders of magnitude more intense than the 7th harmonic. Thus, the contribution of the 3rd harmonic to the spectrum of dipole acceleration is decisive. Third, as shown in the inset of Figure 4, under the conditions considered, the 3rd harmonic is the only below-threshold harmonic; its frequency is comparable to the frequencies of single-photon transitions to all excited bound states of the atom and to the threshold of its photoionization. The fifth and higher harmonics in the case under consideration are above-threshold. Fourthly, as shown in the inset to Figure 4, due to the small ponderomotive energy of a free electron in the UV field, Up ≈ 0.02 a.u. << Ω, the contribution of the Corkum’s mechanism of harmonic generation [14,15] under the conditions under consideration is insignificant. In addition, Ip+Up ≈ Ip (where Ip = 0.9036 a.u. is the ionization potential of an unperturbed atom), and the shift of the energy boundary of the continuum due to the dynamic Stark effect [46] does not play a noticeable role. The fifth and final conclusion from Figure 5 is the qualitative agreement between the dipole acceleration spectra at the 3rd, 5th, and, with less accuracy, the 7th harmonics, calculated (i) using a stationary-state basis and (ii) in a two-dimensional one-electron model of the helium atom, with appropriate (optimal in each model) UV field parameters. The differences in the shape of the spectral lines of higher harmonics are apparently due to the limited basis of the stationary states taken into account in expansion (4).
Figure 4.
Frequency dependence of the power spectral density of dipole acceleration under conditions that maximize the third harmonic energy. The red curve corresponds to the solution of the system of Equations (6), (7) at Nosc = 30, IUV = 2×1014 W/cm2, and Ω = 0.2644 a.u. The black curve was obtained based on model (11), (12) with Nosc = 30, IUV = 4×1014 W/cm2, and Ω = 0.2730 a.u. The inset shows the vicinity of the third harmonic frequency; the vertical blue lines indicate the frequencies of single-photon (both dipole-allowed and dipole-forbidden) transitions into the unperturbed bound states taken into account, the vertical black line indicates the ionization potential Ip of the atom in the absence of a UV field, and the vertical green line indicates the plateau boundary in the spectrum of harmonics due to the Corkum’s mechanism, Ip + 3.17×Up, where Up is the ponderomotive energy of the electron (see [14,15]). The frequency values in the inset to the figure correspond to model (6), (7) (see Appendix A, formula (A1)).
Figure 4.
Frequency dependence of the power spectral density of dipole acceleration under conditions that maximize the third harmonic energy. The red curve corresponds to the solution of the system of Equations (6), (7) at Nosc = 30, IUV = 2×1014 W/cm2, and Ω = 0.2644 a.u. The black curve was obtained based on model (11), (12) with Nosc = 30, IUV = 4×1014 W/cm2, and Ω = 0.2730 a.u. The inset shows the vicinity of the third harmonic frequency; the vertical blue lines indicate the frequencies of single-photon (both dipole-allowed and dipole-forbidden) transitions into the unperturbed bound states taken into account, the vertical black line indicates the ionization potential Ip of the atom in the absence of a UV field, and the vertical green line indicates the plateau boundary in the spectrum of harmonics due to the Corkum’s mechanism, Ip + 3.17×Up, where Up is the ponderomotive energy of the electron (see [14,15]). The frequency values in the inset to the figure correspond to model (6), (7) (see Appendix A, formula (A1)).
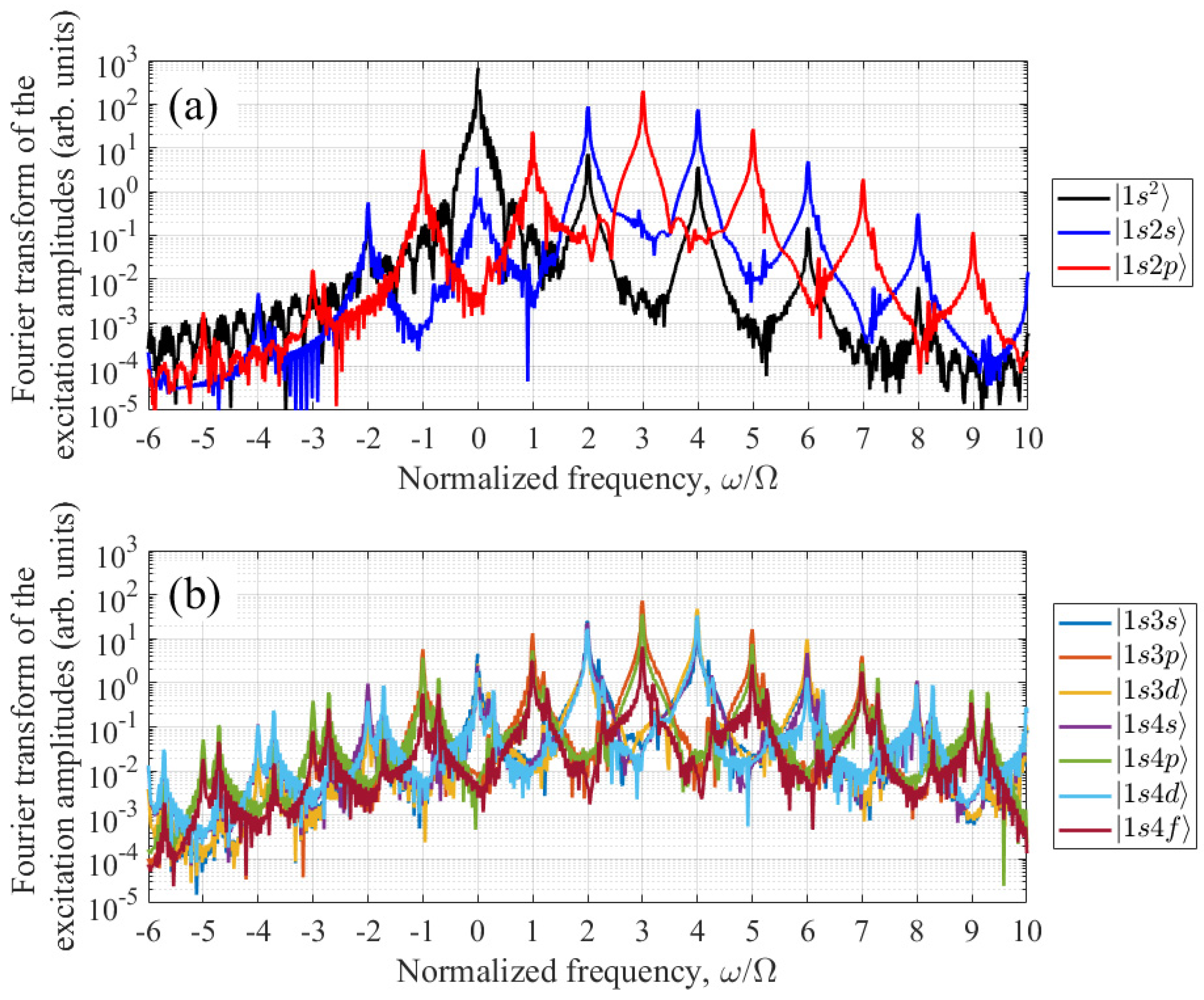
Next, we will analyze the role of various stationary states in the generation of harmonics. When using the expansion of the wave function in stationary states and the adiabatic continuum exclusion approximation, the dipole acceleration of the atom is determined by the excitation amplitudes of bound states (9), (10). The excitation amplitude spectra of the states taken into account for Nosc = 30 (see (3)) and optimal UV field parameters, IUV = 2×1014 W/cm2 and Ω = 0.2644 a.u., are shown in Figure 5. The horizontal axis shows the excitation frequency of the atom, measured from the unperturbed frequency of the ground state |1s2〉. As follows from Figure 5, the states |1s2〉, |1s2s〉, and |1s2p〉, shown in Figure 5a, have the largest excitation amplitudes, while the remaining states, Figure 5b, are excited noticeably weaker. In accordance with Equation (9), the complex amplitude of the dipole moment of an atom at the 3rd harmonic frequency is determined by the sum of the products over all considered states s and k of opposite parity and over the Fourier components of the excitation amplitudes of these states n and n+3. As can be seen from Figure 5a, in the conditions under consideration, of the even parity states, the 0th Fourier component of the excitation amplitude of the state |1s2〉, , has the largest amplitude, and of the odd parity states, this is the case for the 3rd Fourier component of the excitation amplitude of the state |1s2p〉, , oscillating at a frequency close to the frequency of the single-photon transition |1s2〉 - |1s2p〉. Accordingly, the dominant contribution to the amplitude of the 3rd harmonic of the UV field frequency in the spectrum of the induced dipole moment of the atom is made by the product . In this case, the dipole acceleration of the atom at the third harmonic frequency is proportional to , while its energy turns out to be proportional to , where integration is carried out over the time interval at which the UV field is different from zero (see Appendix С). On the other hand, due to the dominant contribution of the Fourier components a1,0 and a3,3 to the excitation amplitudes of the states |1s2〉 and |1s2p〉, respectively, the approximate equality is valid: , where denotes the time-averaged product of the populations of the states |1s2〉 and |1s2p〉, while is the duration of the UV field pulse. The resulting equality is easy to interpret, namely, the energy of the third harmonic reaches its maximum under the conditions of the most effective excitation of the coherent superposition of states |1s2〉 and |1s2p〉.
Figure 5.
Absolute values of the Fourier transform of the excitation amplitudes of bound states in a UV field with Nosc = 30, IUV = 2×1014 W/cm2, and Ω = 0.2644 a.u. In (a), the black, blue, and red curves show the excitation amplitudes of the |1s2〉, |1s2s〉, and |1s2p〉 states, respectively. In (b), the excitation amplitudes of the remaining states taken into account, |1s3s〉, |1s3p〉, |1s3d〉, |1s4s〉, |1s4p〉, |1s4d〉, and |1s4f〉, are shown in different colors. All values are calculated for a time interval within which the UV field is nonzero (similar to formula (C3) in Appendix C).
Figure 5.
Absolute values of the Fourier transform of the excitation amplitudes of bound states in a UV field with Nosc = 30, IUV = 2×1014 W/cm2, and Ω = 0.2644 a.u. In (a), the black, blue, and red curves show the excitation amplitudes of the |1s2〉, |1s2s〉, and |1s2p〉 states, respectively. In (b), the excitation amplitudes of the remaining states taken into account, |1s3s〉, |1s3p〉, |1s3d〉, |1s4s〉, |1s4p〉, |1s4d〉, and |1s4f〉, are shown in different colors. All values are calculated for a time interval within which the UV field is nonzero (similar to formula (C3) in Appendix C).
This statement is illustrated by Figure 6. The upper part of the figure (Figure 6a) shows the time-averaged populations of bound states, , and the total population of continuum states, , as a function of the UV field intensity. In Figure 6, Nosc = 30 (see (3)), and the frequency of the UV field for each intensity is chosen equal to the frequency of the three-photon transition |1s2〉 – |1s2p〉 in the field (taking into account the dynamic Stark effect, see the black solid curve in Figure 2). Lower part of the figure (Figure 6b), shows (i) the time-averaged product of the populations of the states |1s2〉 and |1s2p〉, , and (ii) the energy of the third harmonic in the spectrum of the dipole acceleration of the atom as a function of the intensity of the UV field. As can be seen from Figure 6b, up to the dimension factor, these two dependences practically coincide. Thus, to maximize the third harmonic energy, it is necessary to most effectively excite the |1s2p〉 state without excessively depleting the |1s2〉 state.
Figure 6.
(a) Dependences of the populations of the states |1s2〉 (black curve), |1s2s〉 (blue curve), |1s2p〉 (red curve), the total population of the remaining bound states taken into account with n = 3 and n = 4 (green curve), as well as the total population of continuum states (cyan curve), averaged over the duration of the UV field pulse, on the intensity of the UV field under conditions of resonant three-photon excitation of the |1s2p〉 state. (b) Left axis, blue color: dependence of the product of the populations of the |1s2〉 and |1s2p〉 states averaged over the duration of the UV field pulse on the intensity of the UV field under conditions of three-photon excitation of the |1s2p〉 state; right axis, red color: similar dependence of the energy of the third harmonic of the UV field in the dipole acceleration spectrum. The asterisks indicate the calculated values, the dashed lines are obtained by interpolating them with a spline.
Figure 6.
(a) Dependences of the populations of the states |1s2〉 (black curve), |1s2s〉 (blue curve), |1s2p〉 (red curve), the total population of the remaining bound states taken into account with n = 3 and n = 4 (green curve), as well as the total population of continuum states (cyan curve), averaged over the duration of the UV field pulse, on the intensity of the UV field under conditions of resonant three-photon excitation of the |1s2p〉 state. (b) Left axis, blue color: dependence of the product of the populations of the |1s2〉 and |1s2p〉 states averaged over the duration of the UV field pulse on the intensity of the UV field under conditions of three-photon excitation of the |1s2p〉 state; right axis, red color: similar dependence of the energy of the third harmonic of the UV field in the dipole acceleration spectrum. The asterisks indicate the calculated values, the dashed lines are obtained by interpolating them with a spline.
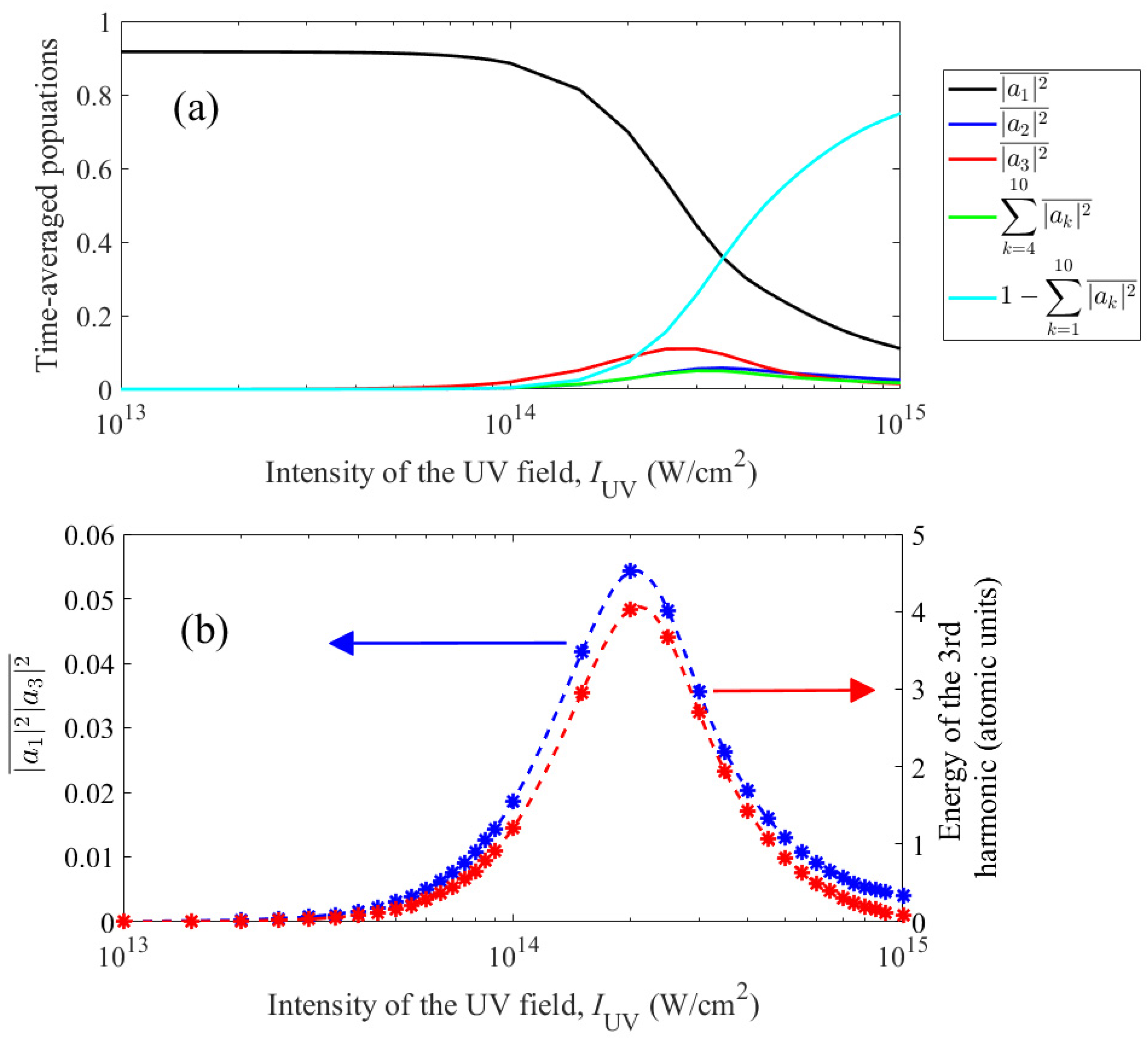
This conclusion allows us to explain the dependence of the frequency that maximizes the energy of the third harmonic in the spectrum of the dipole acceleration of an atom on the intensity of the UV field in Figure 3. In a relatively weak field, the depletion of the |1s2〉 state is not significant (see Figure 6a), and the optimum is achieved at maximum excitation of the |1s2p〉 state, i.e. when adjusting the frequency of the UV field to three-photon resonance. At the optimal UV field intensity, IUV = 2×1014 W/cm2, a balance is achieved between excitation of the |1s2p〉 state and depletion of the |1s2〉 state, in this case the optimal frequency of the UV field remains in the immediate vicinity of the resonant transition frequency. With a further increase in the intensity of the UV field during resonant excitation of the |1s2〉 – |1s2p〉 transition, both the populations of bound states and the third harmonic energy decrease as a result of ionization of the atom (see Figure 6a). Accordingly, the optimal frequency of the UV field in Figure 3 moves away from the resonant one, which makes it possible to reduce the probability of ionization and, thereby, maintain a noticeable population of the |1s2〉 and |1s2p〉 states.
Next, using the expansion of the wave function in terms of the basis of stationary states, we study the dependence of the conditions that maximize the energy of the third harmonic in the spectrum of the dipole acceleration of an atom on the duration of the constant amplitude interval in the UV field envelope (parameter Nosc, see (3)). Figure 7 for three values of Nosc = 10, 30, and 100 shows the dependences of (i) the frequencies of the resonant three-photon transition |1s2〉 – |1s2p〉 and (ii) the frequencies of the UV field that maximize the energy of the third harmonic in the dipole acceleration spectrum on the intensity of the UV field.
Figure 7.
Dependence of the frequency of the three-photon transition |1s2〉 – |1s2p〉 on the intensity of the UV field (solid curves); dependence of the UV field frequency, which maximizes the energy of the third harmonic in the spectrum of the dipole acceleration of an atom, on the field intensity (dashed curves). Black color corresponds to Nosc = 10, red color is for Nosc = 30, and blue color is for Nosc = 100. The black asterisk, red cross, and blue circle indicate combinations of UV field intensity and frequency at which the absolute maximum energy of the third harmonic in the dipole acceleration spectrum is achieved for Nosc = 10, 30, and 100, respectively.
Figure 7.
Dependence of the frequency of the three-photon transition |1s2〉 – |1s2p〉 on the intensity of the UV field (solid curves); dependence of the UV field frequency, which maximizes the energy of the third harmonic in the spectrum of the dipole acceleration of an atom, on the field intensity (dashed curves). Black color corresponds to Nosc = 10, red color is for Nosc = 30, and blue color is for Nosc = 100. The black asterisk, red cross, and blue circle indicate combinations of UV field intensity and frequency at which the absolute maximum energy of the third harmonic in the dipole acceleration spectrum is achieved for Nosc = 10, 30, and 100, respectively.
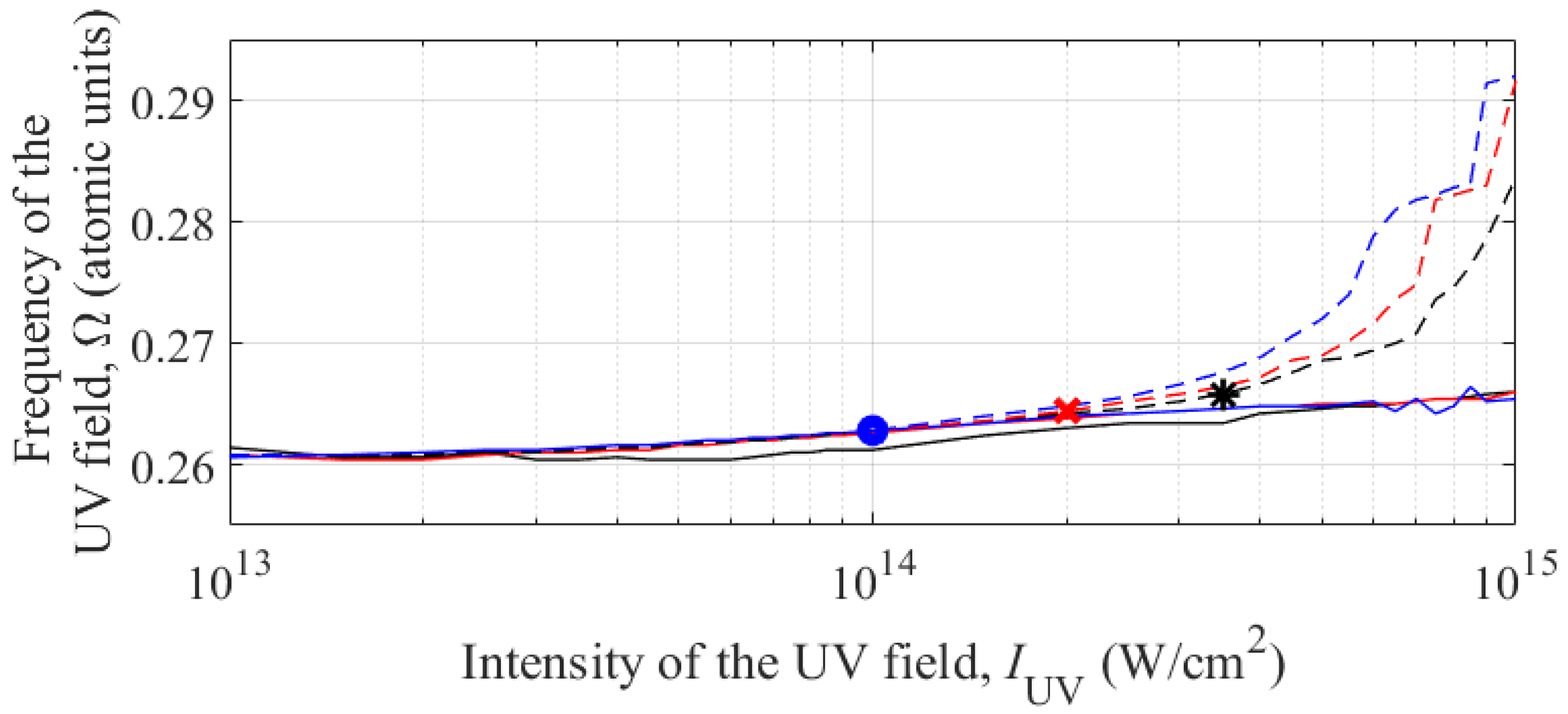
The resonance frequencies of the three-photon transition |1s2〉 – |1s2p〉 for different durations of the UV field envelope practically coincide, which is due to the short establishing time of the dynamic Stark effect [46]. Some difference in the transition frequency is observed for the shortest pulse (Nosc = 10) and is explained by the fact that the total duration of the intervals of turning on and off the UV field (3 + 3 = 6 carrier cycles) in this case is comparable to the duration of the constant amplitude interval (10 carrier cycles). Accordingly, the time-average intensity of the UV field turns out to be noticeably lower than the peak intensity, and the Stark effect at the same peak intensity is weaker than for UV field pulses with a longer duration of the constant amplitude interval. At the same time, jumps in the position of the resonance frequency in the longest pulse (Nosc = 100) with an intensity of the order of 1015 W/cm2 are explained by the extremely high, exceeding 99.99%, probability of single ionization of the atom and the unreliability of the algorithm for determining the resonance frequency under such conditions.
The dependences of the UV field frequencies that maximize the third harmonic energy differ more significantly for different durations of the UV field envelope. At a fixed intensity, with increasing duration of the UV field pulse, the optimal detuning from the resonance frequency increases. At the same time, the optimal intensity of the UV field decreases with increasing envelope duration, namely, for Nosc =10, 30, and 100 the optimal values are IUV = 3.5×1014 W/cm2, 2×1014 W/cm2, and 1014 W/cm2, respectively. To interpret these data, let us turn to Figure 8, which shows the time dependences of the populations of states |1s2〉, |1s2s〉, and |1s2p〉, as well as the total population of more highly excited bound states and the total population of states of the continuum (a,c) and electron dipole acceleration as a function of time (b,d). Figure 8a,b correspond to the UV field pulse of the shortest duration considered, Nosc = 10, while Figure 8c,d are plotted for the case of Nosc = 100.
Figure 8.
(a,c) Time dependences of the populations of states|1s2〉 (black curve), |1s2s〉 (blue curve), |1s2p〉 (red curve), the total population of the remaining bound states taken into account (green curve), and the total population of continuum states (cyan curve). (b,d) Time dependences of the dipole acceleration (left axis, red curve) and the envelope (in amplitude) of the UV pulse (right axis, black broken curve). Figures (a) and (b) are plotted for Nosc = 10, IUV = 3.5x1014 W/cm2, and Ω = 0.2658, Figures (c) and (d) for Nosc = 100, IUV = 1014W/cm2, and Ω = 0.2628.
Figure 8.
(a,c) Time dependences of the populations of states|1s2〉 (black curve), |1s2s〉 (blue curve), |1s2p〉 (red curve), the total population of the remaining bound states taken into account (green curve), and the total population of continuum states (cyan curve). (b,d) Time dependences of the dipole acceleration (left axis, red curve) and the envelope (in amplitude) of the UV pulse (right axis, black broken curve). Figures (a) and (b) are plotted for Nosc = 10, IUV = 3.5x1014 W/cm2, and Ω = 0.2658, Figures (c) and (d) for Nosc = 100, IUV = 1014W/cm2, and Ω = 0.2628.
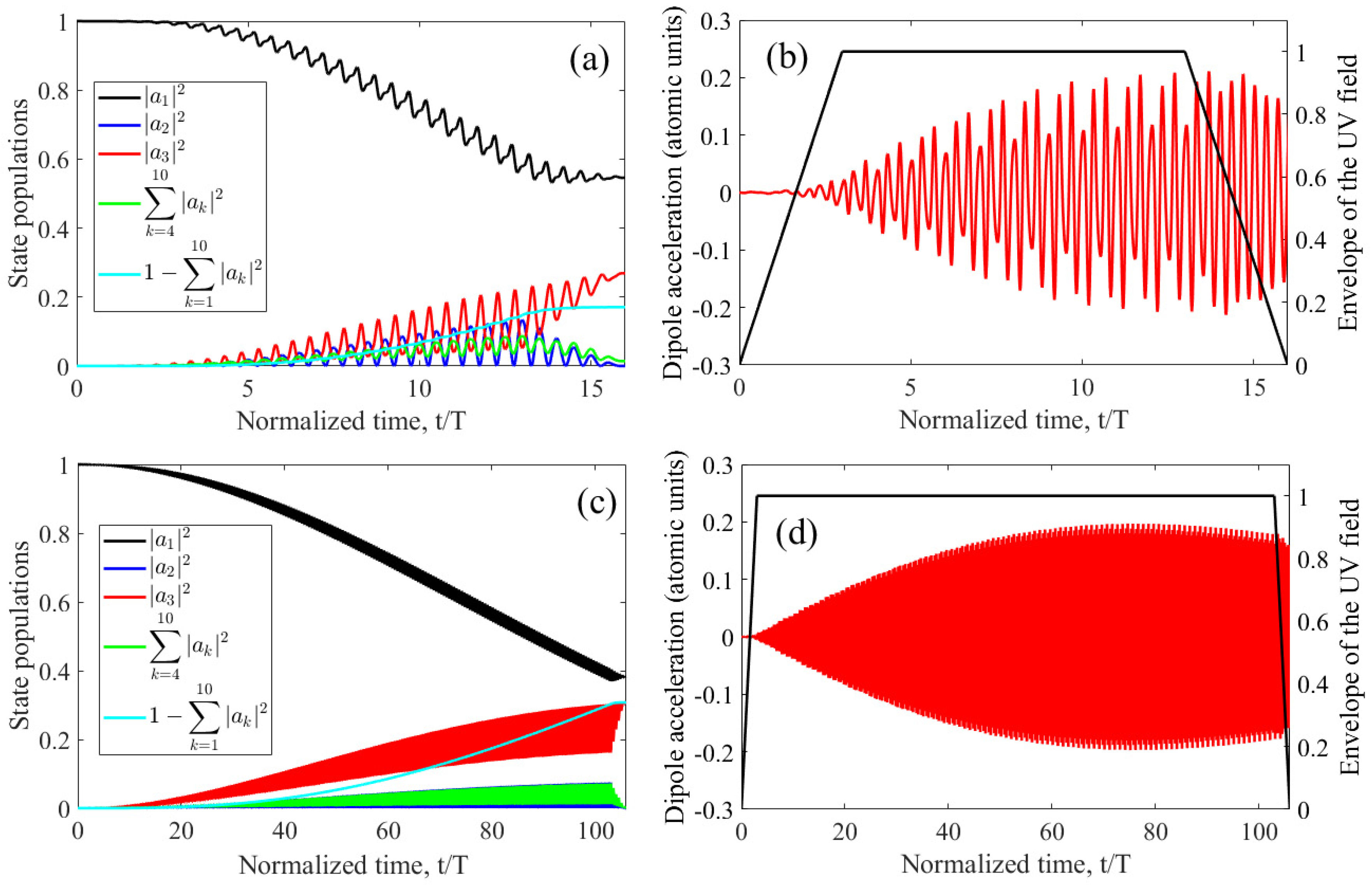
As can be seen from a comparison of Figure 8b and Figure 8d, for the considered UV field parameters, which are optimal for each envelope duration, (i) the time dependences of the dipole acceleration in the interval of constant amplitude of the UV field are similar to each other, and (ii) the peak amplitude of the dipole acceleration is approximately the same. Similar conclusions follow from Figure 8a and Figure 8c, namely, with an optimal choice of UV field parameters, the dynamics of excitation of stationary states weakly depends on the duration of the field envelope, and only the time scale of the process changes significantly. Under optimal conditions, at the end of the UV pulse, the atom goes into the resonant state |1s2p〉 with a noticeable probability of about 20-30%, ionizes with a comparable probability and remains in the ground state |1s2〉 with a probability of about 40-50%. Accordingly, the optimal parameters of the UV field are those that, for different envelope durations, provide the same (optimal) character of excitation of the atom during its interaction with the field. Thus, an increase in the optimal intensity of the UV field with a decrease in the duration of its envelope is caused by the need to provide the same (optimal) degree of excitation of the atom in a shorter interaction time.
At the same time, at a fixed intensity of the UV field, the excitation and ionization of the atom increase or decrease as the frequency of the UV field approaches or moves away from the frequency of the three-photon resonance with the |1s2〉 - |1s2p〉 transition. Accordingly, with increasing duration of the UV field envelope at a fixed intensity, the optimal detuning of the field frequency from resonance increases, and with decreasing duration, vice versa, it decreases. Note that some differences in the degree of excitation and ionization of an atom for different durations of the envelope (and, accordingly, different intensities) of the UV field (see Figure 8a, c) are due to differences in the dependences of (i) excitation probabilities and (ii) probabilities of ionization of an atom on the strength of the UV field.
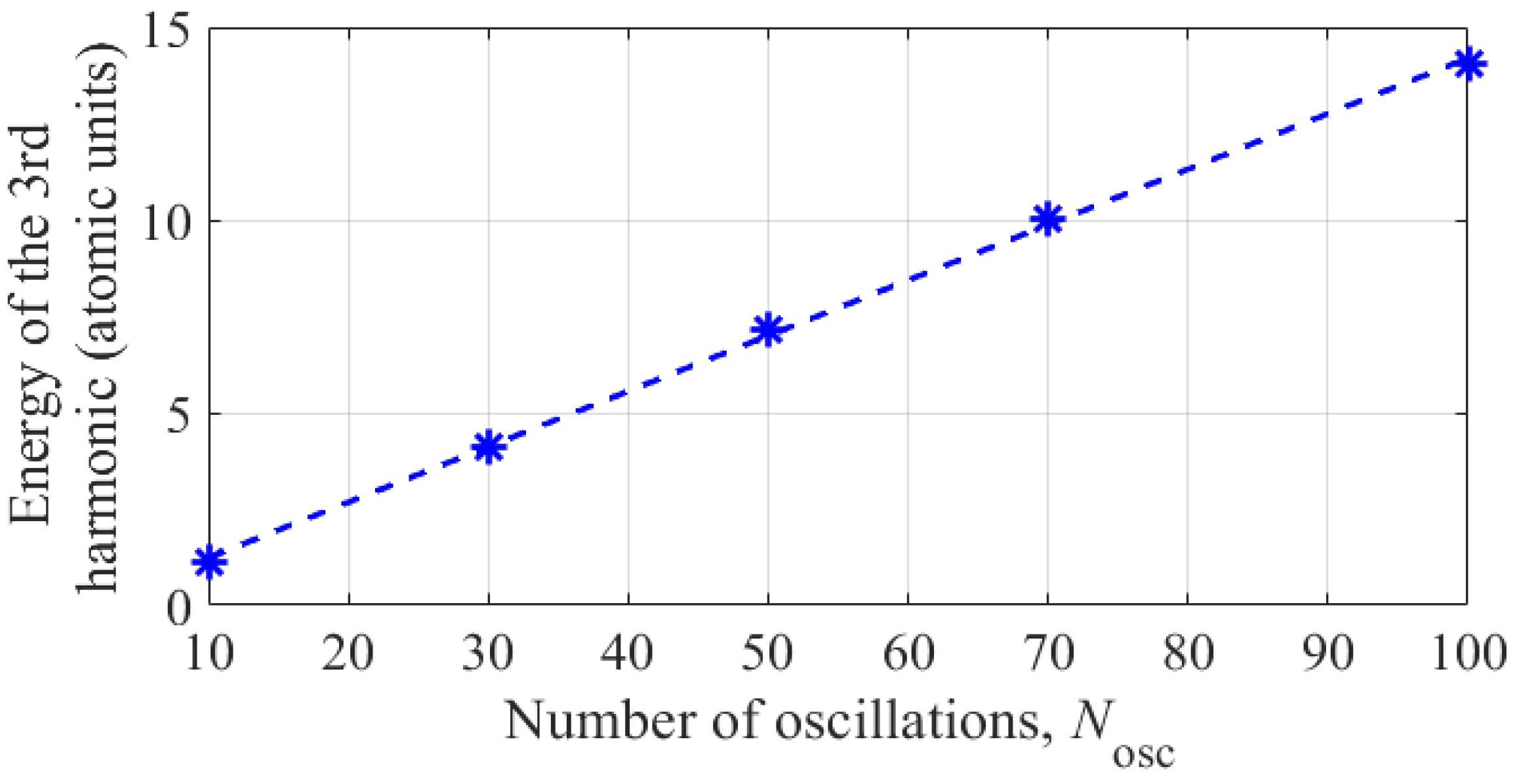
In conclusion of this section, we note that the energy concentrated in the third harmonic of the dipole acceleration of an atom varies approximately in proportion to the duration of the constant amplitude interval in the UV field envelope, see Figure 9. This means that in the conditions under consideration, the average power concentrated in the third harmonic in the spectrum of dipole acceleration of the atom does not depend on the duration of the UV field envelope. Note, however, that with the shortening of the UV field pulse envelope, to achieve the same atomic response power at the third harmonic frequency, an increasing (proportional to the optimal intensity value, see Figure 7) UV radiation power is required.
Figure 9.
Dependence of the energy concentrated in the third harmonic of the fundamental frequency in the dipole acceleration spectrum on the duration (in units of the oscillation period) of the constant amplitude interval in the UV field envelope (3). Asterisks are the results of numerical calculation based on system of Equations (6), (7); the energy corresponding to different Nosc is 1.098 a.u. for Nosc = 10, 4.122 a.u. for Nosc = 30, 7.13 a.u. for Nosc = 50, 10.05 a.u. for Nosc = 70, and 14.03 a.u. for Nosc = 100. The dashed line is the result of linear interpolation.
Figure 9.
Dependence of the energy concentrated in the third harmonic of the fundamental frequency in the dipole acceleration spectrum on the duration (in units of the oscillation period) of the constant amplitude interval in the UV field envelope (3). Asterisks are the results of numerical calculation based on system of Equations (6), (7); the energy corresponding to different Nosc is 1.098 a.u. for Nosc = 10, 4.122 a.u. for Nosc = 30, 7.13 a.u. for Nosc = 50, 10.05 a.u. for Nosc = 70, and 14.03 a.u. for Nosc = 100. The dashed line is the result of linear interpolation.
4. Conclusions
This article analyzes the conditions under which the energy of the third harmonic of the UV field in the dipole acceleration spectrum (the second time derivative of the induced dipole moment) for a helium atom reaches a maximum. The frequency range of the UV field is addressed, which includes resonances with three-photon transitions from the ground to excited bound states of the atom. It is shown that the optimal frequency of the UV field practically coincides with the frequency of the three-photon transition |1s2〉 - |1s2p〉 taking into account its shift due to the dynamic Stark effect. In this case, the highest energy of the third harmonic is achieved under conditions of maximizing the time-averaged (over the duration of the UV field pulse) product of the populations of the states |1s2〉 and |1s2p〉. For the considered UV field pulses with a constant amplitude interval duration from 10 to 100 periods of field oscillations (which, for different frequencies of the UV field, corresponds to from 5 to 60 femtoseconds), the optimal UV field intensity ranges from 1014 W/cm2 to several units of 1014 W/cm2. Under optimal conditions, at the end of the UV pulse, the atom is excited to the resonant state |1s2p〉 with a probability of about 20-30%, ionized with a comparable probability, and remains in the ground state |1s2〉 with a probability of about 40-50%. It is shown that with an optimal choice of the frequency and intensity of the UV field, the dynamics of excitation of bound and continuum states, as well as the shape of the time envelope of the dipole acceleration of the atom, weakly depend on the duration of the UV field envelope; only their time scale changes significantly. In addition, the average power of the third harmonic signal in the dipole acceleration spectrum is also practically independent of the duration of the UV field envelope. Thus, the optimal regime of resonant three-photon excitation of the helium atom, which maximizes the energy of the third harmonic of the UV field in its spectrum of dipole acceleration, has been found and numerically investigated.
Author Contributions
Conceptualization, V.A.A., I.R.K. and M.Y.R.; methodology, V.A.A., I.R.K., M.Y.E. and E.V.G.; software, I.R.K., M.Y.E. and M.P.P.; validation, V.A.A., E.V.G. and M.Y.R.; investigation, I.R.K., M.Y.E., M.P.P. and V.A.A.; visualization, I.R.K.; writing—original draft, I.R.K. and V.A.A.; writing—review and editing, I.R.K., V.A.A. and M.Y.R.; supervision, V.A.A., E.V.G. and M.Y.R.; funding acquisition, M.Y.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant No. 22-12-00389).
Data Availability Statement
Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Main parameters of the models used
While solving Equations (6) and (7) we used the spectroscopic characteristics of a three-dimensional two-electron helium atom, calculated by the multiconfiguration Hartree-Fock approach. The energies of the bound states are
The dipole moments of transitions between the states taken into account are given by the matrix
The squared values of dipole moments of transitions from the bound states to the continuum states as functions of energy of the continuum state, ε, are shown in Figure A1.
Figure A1.
The dependencies of squared dipole moments of transitions from the bound states taken into account to the corresponding continuum states as functions of energy of the continuum state, ε. Panel (a) corresponds to the |1s2〉 state, panel (b) corresponds to the bound states with principal quantum number n = 2, panels (c) and (d) correspond to n = 3 and n = 4, respectively.
Figure A1.
The dependencies of squared dipole moments of transitions from the bound states taken into account to the corresponding continuum states as functions of energy of the continuum state, ε. Panel (a) corresponds to the |1s2〉 state, panel (b) corresponds to the bound states with principal quantum number n = 2, panels (c) and (d) correspond to n = 3 and n = 4, respectively.
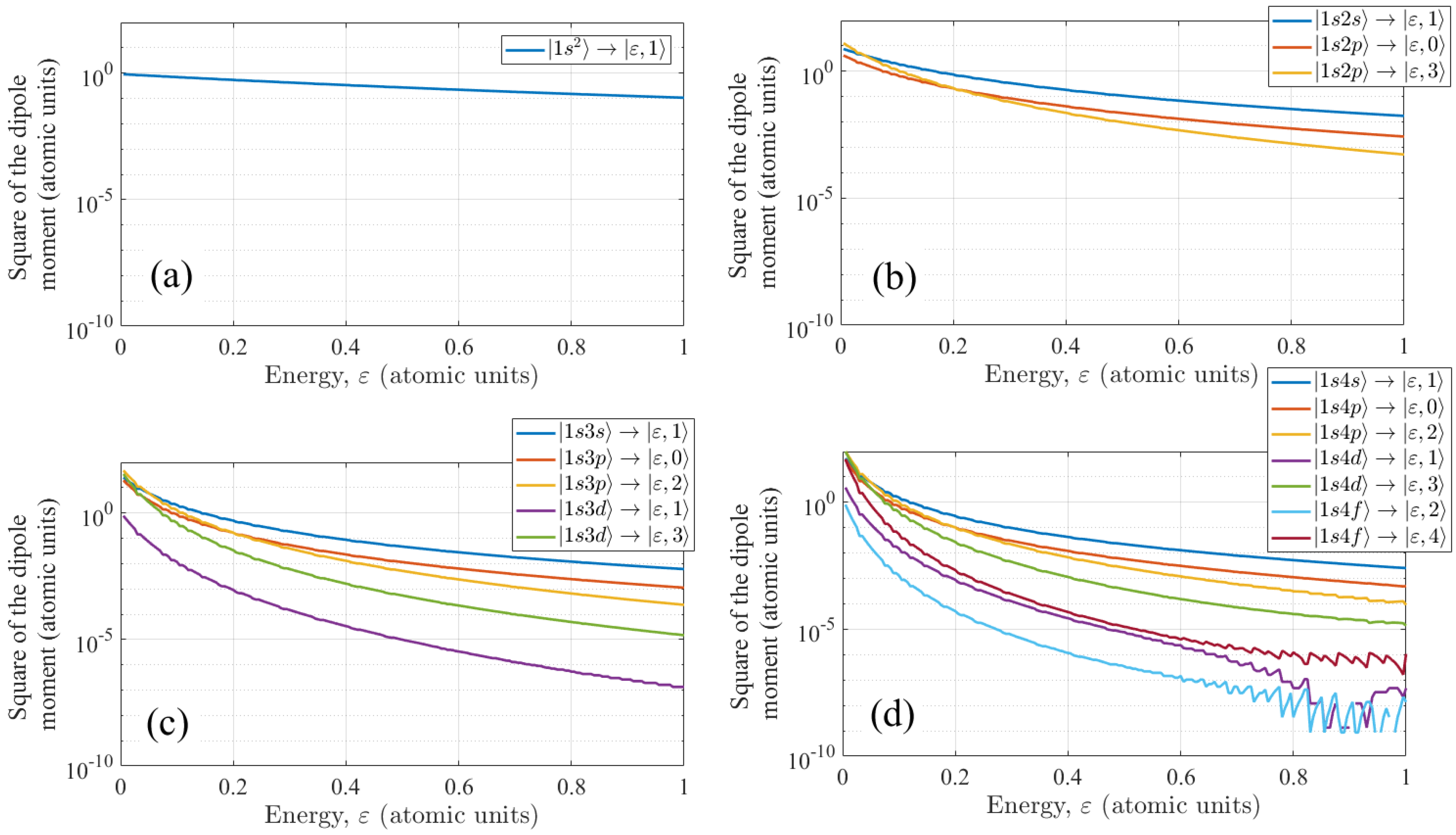
In turn, the single-electron two-dimensional model of a helium atom (11), (12) gives the following energies of the lowest bound states:
The corresponding matrix of dipole moments of transitions between these states is
Appendix B. Solving TDSE by decomposing the atomic wave function into stationary states: basic equations
Substitution of the expansion of the wave function (4) into TDSE (1) and taking the scalar products to the considered stationary states results in the following system of equations:
where the sums over s, l, and p include terms corresponding to dipole-allowed transitions between the considered stationary states of the helium atom, while the influence of transitions between the continuum states on their excitation amplitudes is neglected.
The solution of equations for has a form
where it is taken into account that . Putting (B2) into Equations (B1) for ak(t) and neglecting the transitions between the bound states through the continuum states, one obtains the following system of integro-differential equations for the amplitudes of excitation of the bound states:
Let us consider the integral over the time , included in the third term on the right-hand side of (B3),
Further, let us use the expansion
and suppose that are the slowly varying functions on the time scale of . Then, putting (B5) and (2) in (B4), one obtains
The integral in Equation (B6) can be represented as
Since the product is a slowly varying function of time, we neglect the second term in (B7), see [38]. Thus, Ik(t) takes the form
Substituting (B8) in (B3), after some transformations one obtains
where
The integral over energy ε in (B10) can be calculated via residue theory. Thus, (B10) takes the form
Next, let us use an expansion
where . Substituting (B12) in (B9) and equating the terms with the same n in the exponential functions , we get a system of equations for ak,n(t): where . Substituting (B12) in (B9) and equating the terms with the same n in the exponents , we get a system of equations for ak,n(t):
where (with the substitution of (B11) one obtains Equations (7)). The last two terms in the right-hand side of Equations (B13) correspond to the transitions from the kth bound state through the continuum states to the same state. Neglecting these transitions we finally get the system of Equations (6).
Appendix C. Calculation of the power spectral density of the dipole acceleration of an atom and the energy of the 3rd harmonic in its spectrum
In order to calculate the power spectral density of the dipole acceleration of an atom within the time interval, in which the UV field is nonzero, we used a time mask of the following form
with smooth turning on and off, and the duration of the interval, at which the mask differs from zero, equal to the duration of the UV field pulse. Thus, the frequency dependence of the power spectral density of the dipole acceleration of an atom within the duration of an UV field pulse was calculated using the formula
where
is the dipole acceleration spectrum calculated within the duration of the UV field pulse. The Fourier transforms of the excitation amplitudes of bound states in the UV field, shown in Figure 5, were calculated in the same way (similarly to (C3).
To calculate the energy of the third (N = 3) harmonic in the dipole acceleration spectrum within the UV field pulse, the spectrum was multiplied by a spectral filter of the form
Then the time dependence of the complex amplitude of the dipole acceleration at the frequency of the third (N = 3) harmonic of the UV field was calculated via the inverse Fourier transform of the product . Finally, the energy contained in the third harmonic of the dipole acceleration within the duration of the UV field pulse was calculated using the formula
References
- Hentschel, M.; Kienberger, R.; Spielmann, Ch.; Reider, G.A.; Milosevic, N.; Brabec, T.; Corkum, P.; Heinzmann, U.; Drescher, M.; Krausz, F. Attosecond metrology. Nature 2001, 414, 509–513. [Google Scholar] [CrossRef]
- Drescher, M.; Hentschel, M.; Kienberger, R.; Uiberacker, M.; Yakovlev, V.; Scrinzi, A.; Westerwalbesloh, Th.; Kleineberg, U.; Heinzmann, U.; Krausz, F. Time-resolved atomic inner-shell spectroscopy. Nature 2002, 419, 803–807. [Google Scholar] [CrossRef]
- Gagnon, E.; Ranitovic, P.; Tong, X.-M.; Cocke, C.L.; Murnane, M.M.; Kapteyn, H.C.; Sandhu, A.S. Soft x-ray-driven femtosecond molecular dynamics. Science 2007, 317, 1374–1378. [Google Scholar] [CrossRef]
- Cavalieri, A.L.; Müller, N.; Uphues, Th.; Yakovlev, V.S.; Baltuška, A.; Horvath, B.; Schmidt, B.; Blümel, L.; Holzwarth, R.; Hendel, S.; Drescher, M.; Kleineberg, U.; Echenique, P.M.; Kienberger, R.; Krausz, F.; Heinzmann, U. Attosecond spectroscopy in condensed matter. Nature 2007, 449, 1029–1032. [Google Scholar] [CrossRef]
- Chini, M.; Zhao, B.; Wang, H.; Cheng, Y.; Hu, S.X.; Chang, Z. Subcycle ac Stark shift of helium excited states probed with isolated attosecond pulses. Phys. Rev. Lett. 2012, 109, 073601. [Google Scholar] [CrossRef]
- Ott, C.; Kaldun, A.; Raith, P.; Meyer, K.; Laux, M.; Evers, J.; Keitel, C.H.; Greene, C.H.; Pfeifer, T. Lorentz meets Fano in spectral line shapes: A universal phase and its laser control. Science 2013, 340, 716–720. [Google Scholar] [CrossRef]
- Ott, C.; Kaldun, A.; Argenti, L.; Raith, P.; Meyer, K.; Laux, M.; Zhang, Y.; Blättermann, A.; Hagstotz, S.; Ding, T.; Heck, R.; Madroñero, J.; Martín, F.; Pfeifer, T. Reconstruction and control of a time-dependent two-electron wave packet. Nature 2014, 516, 374–378. [Google Scholar] [CrossRef]
- Kuleff, A.I.; Kryzhevoi, N.V.; Pernpointner, M.; Cederbaum, L.S. Core ionization initiates subfemtosecond charge migration in the valence shell of molecules. Phys. Rev. Lett. 2016, 117, 093002. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, S.; Camp, S.; Schafer, K.J.; Gaarde, M.B. Theory of strong-field attosecond transient absorption. J. Phys. B: At. Mol. Opt. Phys. 2016, 49, 062003. [Google Scholar] [CrossRef]
- Young, L.; Ueda, K.; Gühr, M.; Bucksbaum, P.H.; Simon, M.; Mukamel, S.; Rohringer, N.; Prince, K.C.; Masciovecchio, C.; Meyer, M.; et al. Roadmap of ultrafast x-ray atomic and molecular physics. J. Phys. B: At. Mol. Opt. Phys. 2018, 51, 032003. [Google Scholar] [CrossRef]
- Yamauchi, K.; Mimura, H.; Kimura, T.; Yumoto, H.; Handa, S.; Matsuyama, S.; Arima, K.; Sano, Y.; Yamamura, K.; Inagaki, K. Single-nanometer focusing of hard x-rays by Kirkpatrick–Baez mirrors. J. Phys.: Condens. Matter 2011, 23, 394206. [Google Scholar] [CrossRef]
- Schoenlein, R.; Elsaesser, T.; Holldack, K.; Huang, Z.; Kapteyn, H.; Murnane, M.; Woerner, M. Recent advances in ultrafast X-ray sources. Philos. Trans. R. Soc. A 2019, 377, 20180384. [Google Scholar] [CrossRef]
- Li, J.; Lu, J.; Chew, A.; Han, S.; Li, J.; Wu, Y.; Wang, H.; Ghimire, S.; Chang, Z. Attosecond science based on high harmonic generation from gases and solids. Nat. Commun. 2020, 11, 2748. [Google Scholar] [CrossRef] [PubMed]
- Corkum, P.B. Plasma perspective on strong field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Lewenstein, M.; Balcou, Ph.; Ivanov, M.Yu.; L’Huillier, A.; Corkum, P.B. Theory of high-harmonic generation by low-frequency laser fields. Phys. Rev. A 1994, 49, 2117–2132. [Google Scholar] [CrossRef] [PubMed]
- Yost, D.C.; Schibli, T.R.; Ye, J.; Tate, J.L.; Hostetter, J.; Gaarde, M.B.; Schafer, K.J. Vacuum-ultraviolet frequency combs from below-threshold harmonics. Nat. Phys. 2009, 5, 815–820. [Google Scholar] [CrossRef]
- Chini, M.; Wang, X.; Cheng, Y.; Wang, H.; Wu, Y.; Cunningham, E.; Li, P.-C.; Heslar, J.; Telnov, D.A.; Chu, S.-I. , Chang, Z. Coherent phase-matched VUV generation by field-controlled bound states. Nat. Photonics 2014, 8, 437–441. [Google Scholar] [CrossRef]
- Xiong, W.-H.; Geng, J.-W.; Tang, J.-Y.; Peng, L.-Y.; Gong, Q. Mechanisms of below-threshold harmonic generation in atoms. Phys. Rev. Lett. 2014, 112, 233001. [Google Scholar] [CrossRef] [PubMed]
- Spott, A.; Becker, A.; Jaroń-Becker, A. Transition from perturbative to nonperturbative interaction in low-order-harmonic generation. Phys. Rev. A 2015, 91, 023402. [Google Scholar] [CrossRef]
- Beaulieu, S.; Camp, S.; Descamps, D.; Comby, A.; Wanie, V.; Petit, S.; Légaré, F.; Schafer, M. B. Gaarde, F. Catoire, and Y. Mairesse, Role of excited states in high-order harmonic generation. Phys. Rev. Lett. 2016, 117, 203001. [Google Scholar] [CrossRef]
- Xiong, W.-H.; Peng, L.-Y.; Gong, Q. Recent progress of below-threshold harmonic generation. J. Phys. B: At. Mol. Opt. Phys. 2017, 50, 032001. [Google Scholar] [CrossRef]
- Guo, Q.-L.; Li, P.-C.; Zhou, X.-X.; Chu, S.-I. Efficient enhancement of below-threshold harmonic generation by laser-driven excited states of Cs atom. Opt. Commun. 2018, 410, 262–268. [Google Scholar] [CrossRef]
- Felicio Zuffi, A.V.; Vieira Junior, N.D.; Samad, R.E. Below-threshold-harmonics-generation limitation due to laser-induced ionization in noble gases. Phys. Rev. A 2022, 105, 023112. [Google Scholar] [CrossRef]
- Schönberg, A.; Salman, H.S.; Tajalli, A.; Kumar, S.; Hartl, I.; Heyl, C.M. Below-threshold harmonic generation in gas-jets for Th-229 nuclear spectroscopy. Opt. Express 2023, 31, 12880–12893. [Google Scholar] [CrossRef]
- Stappaerts, E.A. Harmonic generation at high field strengths. Frequency shifts and saturation phenomena. Phys. Rev. A 1975, 11, 1664–1667. [Google Scholar] [CrossRef]
- Georges, A.T.; Lambropoulos, P.; Marburger, J.H. Theory of third-harmonic generation in metal vapors under two-photon resonance conditions. Phys. Rev. A 1977, 15, 300–307. [Google Scholar] [CrossRef]
- Diels, J.-C.; Georges, A.T. Coherent two-photon resonant third- and fifth-harmonic vacuum-ultraviolet generation in metal vapors. Phys. Rev. A 1979, 19, 1589–1606. [Google Scholar] [CrossRef]
- Poirier, M. Competition between resonant multiphoton ionization and third-harmonic generation: A mean-field model. Phys. Rev. A 1983, 27, 934–943. [Google Scholar] [CrossRef]
- Agarwal, G.S.; Tewari, S.P. Theory of four-photon resonant vacuum-ultraviolet generation with reabsorption via resonant two-photon process. J. Opt. Soc. Am. B 1985, 2, 1409–1416. [Google Scholar] [CrossRef]
- Slabko, V.V.; Popov, A.K.; Lukinykh, V.F. Generation of coherent radiation at 89.6 nm through two-photon resonant phase-matched tripling of fourth-harmonic Nd:glass laser radiation in Hg vapors. Appl. Phys. 1977, 15, 239–241. [Google Scholar] [CrossRef]
- Timmermann, A.; Wallenstein, R. Generation of tunable single-frequency continuous-wave coherent vacuum-ultraviolet radiation. Opt. Lett. 1983, 8, 517–519. [Google Scholar] [CrossRef]
- Bokor, J.; Bucksbaum, P.H.; Freeman, R.R. Generation of 35.5-nm coherent radiation. Opt. Lett. 1983, 8, 217–219. [Google Scholar] [CrossRef]
- Hilber, G.; Lago, A.; Wallenstein, R. Broadly tunable vacuum-ultraviolet/extreme-ultraviolet radiation generated by resonant third-order frequency conversion in krypton. J. Opt. Soc. Am. B 1987, 4, 1753–1764. [Google Scholar] [CrossRef]
- Miyazaki, K.; Sakai, H.; Sato, T. Two-photon resonances in Xe and Kr for the generation of tunable coherent extreme UV radiation. Appl. Opt. 1989, 28, 699–702. [Google Scholar] [CrossRef]
- Tünnermann, A.; Mossavi, K.; Wellegehausen, B. Nonlinear-optical processes in the near-resonant two-photon excitation of xenon by femtosecond KrF-excimer-laser pulses. Phys. Rev. A 1992, 46, 2707–2717. [Google Scholar] [CrossRef]
- Le Blanc, S.P.; Qi, Z.; Sauerbrey, R. Generation of femtosecond vacuum-ultraviolet pulses. Appl. Phys. B 1995, 61, 439–449. [Google Scholar] [CrossRef]
- Ganeev, R.A.; Usmanov, T. Frequency conversion of picosecond radiation in ultraviolet (338–366 nm) and vacuum ultraviolet (113.5–117.0 nm) ranges. J. Opt. A: Pure Appl. Opt. 2000, 2, 550–556. [Google Scholar] [CrossRef]
- Parker, J.; Stroud Jr., C. R. Population trapping in short-pulse laser ionization. Phys. Rev. A 1990, 41, 1602–1608. [Google Scholar] [CrossRef]
- Mercouris, Th.; Komninos, Y.; Dionissopoulou, S.; Nicolaides, C.A. Computation of strong-field multiphoton processes in polyelectronic atoms: State-specific method and applications to H and Li–. Phys. Rev. A 1994, 50, 4109–4121. [Google Scholar] [CrossRef]
- Mercouris, Th.; Komninos, Y.; Nicolaides, C.A. The state-specific expansion approach to the solution of the polyelectronic time-dependent Schrödinger equation for atoms and molecules in unstable states. Adv. Quantum Chem. 2010, 60, 333–405. [Google Scholar]
- de Morisson Faria, C.F.; Dörr, M.; Sandner, W. Importance of excited bound states in harmonic generation. Phys. Rev. A 1998, 58, 2990–2999. [Google Scholar] [CrossRef]
- https://www.nist.gov/pml/atomic-spectra-database.
- Khairulin, I.R.; Antonov, V.A.; Emelin, M.Yu.; Popova, M.M.; Gryzlova, E.V.; Ryabikin, M.Yu. Multilevel model of multiphoton processes in a helium atom in a strong laser field: ionization description. Opt. Spectrosc. 2023, 131, 128–132. [Google Scholar]
- Letokhov, V.S. Laser Photoionization Spectroscopy; Academic Press, 1987. [Google Scholar] [CrossRef]
- Avanaki, K.N.; Telnov, D.A.; Chu, S.-I. Harmonic generation of Li atoms in one- and two-photon Rabi-flopping regimes. Phys. Rev. A 2016, 94, 053410. [Google Scholar] [CrossRef]
- Delone, N.B.; Krainov, V.P. AC Stark shift of atomic energy levels. Phys.-Usp. 1999, 42, 669–687. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



