
Submitted:
12 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
Sirtuins, especially SIRT1, play a significant role in the regulation of inflammatory response, autophagy, and cell response to oxidative stress. Since their discovery, sirtuins have been regarded as anti-aging and longevity-promoting enzymes. Sirtuin-regulated processes seem to take part in most prevalent placental pathologies, such as preeclampsia. Furthermore, more and more research studies indicate that SIRT1 may prevent pre-eclampsia development, or at least alleviate its manifestations. Having taken this into consideration, we have made a review of recent studies on the role of sirtuins, especially SIRT1, in the regulation of processes determining normal or abnormal development and functioning of the placenta.
Keywords:
sirtuin-1
; SIRT1
; sirtuins
; human placenta
; placental physiology
; placental pathophysiology
; preeclampsia
; peroxisome proliferator-activated receptor γ
; PPARγ
; SIRT1/PPARγ signaling
1. Introduction
Placenta is a unique organ occurring during intrauterine life, playing a significant role in the regulation of fetal growth and development [1,2]. It consists of trophectoderm-derived epithelial cells, outer layer of blastocyst, and extraembrional mesodermal cells that derive from inner cellular mass – a group of cells which give rise to embryo proper [3]. In the course of placentation, this combination of cells gives rise to a complex organ which anchors the fetus in the uterine cavity and provides delivery of oxygen, nutrients and hormones required for fetal growth, as well as the excretion of carbon dioxide and other end products of metabolism. While extraembryonal mesodermal cells give rise to mesenchymal parts of the placenta, including fetal circulatory system, trophectoderm-derived epithelial cells differentiate to form two main layers of the trophoblast: villous trophoblast (called labirynthine layer in mice) and extravillous trophoblast (called junctional zone in mice). While villous trophoblast takes part in gas and nutrient exchange, extravillous trophoblast anchors the placenta in the uterine wall and remodels maternal spiral arteries to provide sufficient perfusion of fetoplacental unit [4]. Impaired placental development or function may have significant consequences both for the mother and for the fetus, resulting in complications such as pregnancy-induced hypertension/preeclampsia, intrauterine growth retardation, gestational diabetes, macrosomia, or even accelerate the termination of a pathological pregnancy through miscarriage, stillbirth, and preterm birth [5]. Furthermore, some research studies suggest that gestational complications, especially those resulting in the intrauterine growth retardation, can have a long-term effects even in postnatal life, contributing to metabolic programming which can increase the risk of obesity, diabetes, and cardiovascular disease later in life [6]. This is why there is a need of deeper understanding of placental development, especially in reference to some signaling pathways that can affect fetal growth.
Sirtuins, which are a highly conserved group of epigenetic proteins, play an important role in the comprehensive regulation of metabolic processes at the cellular level. A detailed understanding of their physiological and altered expression in the human placenta may provide a lot of valuable information about the physiology of the placenta and the mechanisms of development of placental pathologies. Much of the latest placental research, the results of which we cite and discuss in this review, concerns sirtuin-1 (silent information regulator 2 homolog 1 or SIRT1), a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase [1,7,8].
2. SIRT1 in the regulation of trophoblast function
2.1. Effects towards placental development and differentiation
SIRT1 is very important for proper trophoblast differentiation and placental development [1,5,7,9,10]. In placentas and serum samples collected from patients with preeclampsia (PE), SIRT1 activity is reduced and can be observed mainly in cell nuclei within cytotrophoblast and syncytiotrophoblast [11,12]. SIRT1 probably takes part in trophoblast maintenance and differentiation through modulating small mothers against decapentaplegic proteins 2/3 (SMAD2/3), signal transducer and activator of transcription (STAT), and peroxisome proliferator-activated receptor γ (PPARγ)-dependent signaling pathways [13,14,15,16]. Placentas collected from SIRT1 KO pregnant mice are small and show visible abnormalities both within labirynthine layer and junctional zone [11]. In addition, trophoblast stem cells (TSC) collected from SIRT1 knockout (KO) mice show impaired differentiation.. In such cells, PPARγ expression is reduced, along with the expression of such proteins as SMAD2, SMAD3, and signal transducer and activator of transcription 3 (STAT3). STAT3 protein is correlated with differentiation of trophoblast giant cells, while its molecules can be deacetylated by SIRT1, which represses their activity [14,17,18]. Moreover, a possible role of PPARγ in placentation and trophoblast differentiation is emphasized [13,19], and PPARγ activity may be regulated by SIRT1, both through deacetylation and through recruitment of cofactors such as nuclear receptor co-repressor-1 (NCoR1), silencing mediator of retinoid and thyroid hormone receptors (SMRT), and positive regulatory domain containing 16 (Prdm16) [20,21]. Results of research studies made so far suggest that labirynthine layer of trophoblast is poorly developed in SIRT1+/- mice, while trophoblast invasive capability is also reduced, being even more reduced in SIRT1 KO mice [22], which may indicate that SIRT1 plays an important role in placental development and differentiation.
2.2. Effects of SIRT1 on autophagy within trophoblast
Autophagy is a cell homeostasis-promoting process directing damaged proteins and organelles to lysosomal degradation [23,24,25,26,27]. Autophagy protects the placenta from pathogens and stress. In PE patients, autophagy in trophoblast is impaired, and accumulation of abnormal proteins within the placenta is increased [28]. Several research studies have shown that SIRT1 prevents H2O2-induced oxidative stress and apoptosis through promoting autophagy within trophoblast [29]. From the mechanistic standpoint, some studies on autophagy-mediating proteins have shown that SIRT1 may regulate autophagy through NAD+-dependent deacetylation of some of them, e.g. transcription factor EB (TFEB), LC3-II – a membrane-bound (lapidated) form of microtubule-associated protein 1 light chain 3 (LC3), protein that contains a Bcl-2 homology-3 domain (Beclin-1), ubiquitin-binding scaffold protein (p62, also known as sequestosome 1, SQSTM1), autophagy related-proteins 5, 7 and 8 (ATG5, ATG7 and ATG8, respectively) [30,31]. Formation of lysosomes taking part in autophagy is strictly regulated by TFEB which can be deacetylated by SIRT1, subsequently activating the expression of several downstream autophagy-related genes, such as lysosomal associated membrane proteins 1 and 2 (LAMP1, LAMP2), and cathepsin D (CTSD) [32,33]. Furthermore, concentrations of protein markers typical for the initial stage of autophagy activation, such as LC-II, Beclin-1 and SQSTM1 [34,35,36] have been also significantly altered in the course of PE and can be regulated by SIRT1 [37,38,39]. These results taken together suggest that SIRT1 may regulate autophagy within the trophoblast through deacetylation of its specific target proteins taking part in the process of autophagy.
2.3. Effects on cell senescence phenotype occurrence within the placenta
Premature senescence of the placenta is a trait typical for PE. It manifests with increased occurrence of cell senescence phenotype (CSP), senescence-associated secretory phenotype (SASP) and enhanced expression of such cell senescence markers as protein encoded by the TP53 tumor suppressor gene (p53) and cyclin-dependent kinase inhibitor p21 (p21). Loss of SIRT1 activity is also a specific marker of cell senescence, and SIRT1 deficiency results in a premature senescence within placentas during their formation [40,41,42,43]. Interestingly, Xiong et al. have found that reduced SIRT1 activity promotes p53 acetylation and p21 expression, as well as impairs trophoblast cell migration and invasion in case of advanced maternal age, which suggests that SIRT1 deficiency can take part in the pathogenesis of PE through inducing CSP within the placenta.
2.4. Effects of other sirtuins towards trophoblast
There are seven sirtuins in mammals (SIRT1 – SIRT7). All of them deacetylate specific target proteins, using NAD+ as a co-substrate, as well as take part in the regulation of such processes as oxidative stress response, energy metabolism, inflammatory response etc. [44]. Several research studies have revealed that sirtuins play a significant role in trophoblast development and differentiation. This does not only apply to SIRT1. For example, SIRT2 is normally expressed in placental syncytiotrophoblast, and its expression is reduced in PE patients. SIRT2 may induce trophoblast cell necrosis while inhibiting trophoblast cell proliferation [45,46]. SIRT3 is reported to affect trophoblast cell migration, invasion, and tube formation, as well as to take part in the pathogenesis of PE [47]. SIRT4 can also induce trophoblast cell senescence [48,49,50]. These results may additionally confirm the hypothesis that SIRT1 deficiency can take part in the pathogenesis of PE through regulating trophoblast cell invasion, migration and proliferation.
3. SIRT1 and PPARγ
Peroxisome proliferator-activated receptor γ (PPARγ) belongs to the family of nuclear hormonal ligand-activated receptors. It can also act as transcription factor, widely known because of its crucial role in glucose and lipid metabolism, as well as in adipocyte differentiation. After dimerization with retinoid X-receptor (RXR), PPARγ binds specific DNA sequences defined as PPARγ-reactive elements (PPRE) and subsequently induces genes involved in fatty acid assimilation and accumulation, which results in lipid accumulation and adipogenesis. PPARγ is actually necessary for formation of both white and brown adipose tissue, with white adipose tissue being the site of energy storage and hormone secretion, while brown adipose tissue being the site of energy expenditure and thermogenesis. PPARγ may be activated by thiazolidinediones (TZD) – synthetic activators that are sometimes used in the treatment of type 2 diabetes mellitus [51,52].
Sirtuin 1 (SIRT1) is a member of NAD+-dependent protein deacetylases, at the same time functioning as a sensor of cell nutritional status. Its orthologue has been initially discovered in budding yeast, Saccharomyces cerevisiae as a longevity-promoting enzyme. SIRT1 has been first identified as a histone deacetylase, promoting chromatin compaction and silencing transcription of some genes in case of undernutrition [53]. However, more recent studies have identified numerous non-histone substrates of SIRT1, including p53, forkhead O class box transcription factors (FoxOs), and PPARγ. Through deacetylating PPARγ, SIRT1 renders its inactivation, thus inhibiting adipogenesis and promoting fat mobilization at the same time. On the other hand, inhibition of SIRT1 expression with small interfering RNA (siRNA) promotes adipogenesis and inhibits lipolysis [54]. In addition to its effects on metabolism, SIRT1 regulates many other signaling pathways, including those involved in cell proliferation, apoptosis, autophagy, and inflammatory response [55,56]. SIRT1 can be activated both by a naturally occurring compound – resveratrol which has been identified as anti-inflammatory and anti-oxidative agent, and by small molecule synthetic activators [57].
Although SIRT1 inhibitory action towards PPARγ has been studied quite well, the interaction between these two proteins is not so simple. It has been found that PPARγ deacetylation by SIRT1 results in the recruitment of positive regulatory (PR) domain zinc finger protein 1 (PRDM1) coactivator, which selectively activates PPARγ to stimulate conversion of white adipose tissue to brown adipose tissue [58]. Furthermore, PPARγ can be also an upstream inhibitor of SIRT1, both through inhibiting its deacetylase activity and through inhibiting its expression at the level of transcription [59]. Finally, both thiazolidinedione PPARγ activators and SIRT1 activators, such as resveratrol, may exert collateral effects. TZD induce a transient overexpression of SIRT1 [60], while resveratrol binds some nuclear receptors from PPAR family, including PPARγ [61]. Thus, evaluation of cross-talk between signaling pathways dependent on these two proteins requires a thorough analysis of experiment results, especially if activators of both proteins are used.
3.1. Role of SIRT1- and PPARγ-dependent signaling pathways in placental pathology
Impaired trophoblast differentiation and placental development are correlated with many complications of pregnancy, including miscarriage, pre-eclampsia, intrauterine growth retardation, and gestational diabetes [62,63,64]. These complications are related to a suboptimal microenvironment at the maternal side of the placenta, showing signs of hypoxia, oxidative stress, inflammation, and /or hyperglycemia. This is why it should be stated how those alterations in the placental microenvironment may affect SIRT1 and PPARγ-dependent signaling in the placenta.
3.1.1. Effects of hypoxia on PPARγ activity
Oxygen tension is an important parameter within the placenta, both in the course of normal development and specific placental pathologies [65]. During hypoxia, i.e. when oxygen tension is too low, many signaling pathways are activated, subsequently affecting tissue homeostasis. Relatively best known among them is a signaling pathway activated by hypoxia-inducible factor (HIF) which is a complex of two component proteins: hypoxia-inducible factor subunit alpha (HIFα) domain that is stabilized with oxygen, and hypoxia-inducible factor subunit beta (HIFβ) domain that is expressed constitutively [66]. HIF-dependent signaling pathway is necessary for both placental formation and development, especially for trophoblast differentiation to invasive cell line (trophoblast giant cells in mice, corresponding to human extravillous trophoblast) [67,68,69]. It is known that hypoxia affects PPARγ activity through HIF complex, and thus hypoxia inhibits adipocyte differentiation through its effect on HIF-dependent PPARγ2 – an isoform of PPARγ typical for adipose tissue [70]. In mouse trophoblast stem cells, hypoxia inhibits PPARγ activity, but this effect is independent from HIF activity. [71]. In addition, forced PPARγ expression in the course of hypoxia may in part rescue trophoblast cell differentiation into labyrinthine layer in mice [71].
The results presented above correlate with hypoxia-associated placental pathology, occurring in the course of pre-eclampsia at the maternal site. Abnormal differentiation of syncytiotrophoblast which is an analogue of labirynthine layer of trophoblast in mice, is a typical feature of this condition, regarded as secondary to reduced blood supply to the maternal part of the placenta because of abnormal remodeling of spiral arteries by invasive cells of extravillous trophoblast [5]. Placentas collected from PE patients show a reduced expression of PPARγ, as well as decreased activity of glial cells missing-1 (GCM1) – a main regulator of syncytiotrophoblast formation and probably a target protein for PPARγ. GCM1 can in turn activate protein referred to as syncytin-1 [72,73,74] and trophoblast abnormalities similar to those occurring in the course of PE have been recapitulated in vitro through repressing the level of GCM1 [75]. Therefore, it is presumed that reduced PPARγ activity as a result of hypoxia may inhibit GCM1 and syncytin-1 expression, which in turn negatively affects syncytiotrophoblast differentiation.
Another finding typical for PE is elevated level of anti-angiogenic molecule - soluble vascular endothelial growth factor (VEGF) receptor, also known as soluble fms-like tyrosine kinase 1 (sFlt-1), in maternal blood [76]. Although etiology and origin of increased sFlt-1 release from placentas in PE patients is still a debate subject, several studies have shown a correlation between hypoxia and increased sFlt-1 expression in human trophoblast [77,78,79,80]. Some studies reveal a correlation between sFlt-1 level in syncytiotrophoblast and severity of PE manifestations [81,82]. PPARγ activity has been negatively correlated with increased s-Flt-1 level in the rat model of PE, pregnant female rats show PE symptoms, such as arterial hypertension, proteinuria and fetal growth retardation when treated with PPARγ antagonist. These symptoms are associated with an increased sFlt-1 concentration in the plasma [83]. Quite interestingly, one separate study on mice has shown a correlation between reduced level of GCM1 and raised level of sFlt-1 in the plasma [84]. Combined results of these studies suggest that PPARγ-GCM1 axis can take part in regulating sFlt-1 expression. When evaluating the levels of sFlt-1 mRNA and sFlt-1 release from differentiated mouse trophoblast stem cells (TSC) after their treatment with PPARγ activator, rosiglitazone, reduced levels of both sFlt-1 mRNA and sFlt-1 release can be found. Rosiglitazone does not affect sFlt-1 level in wild-type TSC (WT TSC) exposed to hypoxia and in PPARγ KO TSC exposed to normoxia or hypoxia, which suggests that the observed effect is PPARγ-dependent.
Finally, PE is characterized by increased occurrence of apoptosis in trophoblast cells [85], which is interesting, because PPARγ is one of molecules involved in apoptosis. When trophoblast cells are cultured under hypoxic conditions, their differentiation to form syncytiotrophoblast is impaired, and severe hypoxia leads to apoptosis [86,87]. In similar conditions, treatment of these cells with PPARγ activator – rosiglitazone – promotes their normal differentiation and alleviates apoptotic damage [86]. These results combined may be a premise to regarding PPARγ as a potential target protein in the treatment of placental pathologies, such as PE.
3.1.2. Effects of hypoxia on SIRT1 activity
In comparison to correlations between hypoxia and PPARγ activity, correlation between hypoxia and SIRT1 activity seems to be more complex. In several research studies, SIRT1 has been identified as an upstream regulator of HIFα domains. SIRT1 may deacetylate hypoxia-inducible factor 1 subunit alpha (HIF-1α), which results in blocked recruitment of p300 domains and thus abrogate the expression of HIF-1α effector genes [88]. In addition, SIRT1 selectively stimulates the activity of hypoxia-inducible factor 2 subunit alpha (HIF-2α), thus promoting hypoxia-related signaling dependent on this alternative HIFα domain [89]. Moreover, SIRT1 gene expression can be impaired by hypoxia in a HIF-dependent manner, which suggests a feedback loop between these two proteins [90]. SIRT1 expression within trophoblast under hypoxic conditions has not been evaluated in details. However, one study has shown SIRT1 induction during hypoxia in human trophoblast cells, which results in upregulation of N-myc downstream regulated gene 1 (NDRG1) and reduced expression of p53, thus promoting cell survival [91]. Nevertheless, more detailed studies are thought to be necessary for pinpointing precise and detailed correlations between hypoxia, HIF activity, and SIRT1 activity, both in trophoblast and in placenta.
Similarly to PPARγ, SIRT1 activity has been found to be reduced within syncytiotrophoblast isolated from PE patients placentas [92]. It is hypothesized that this result is related to increased CSP occurrence among trophoblast cells in the course of PE, especially when having taken SIRT1 longevity-promoting function into consideration [92]. In several studies, SIRT1 activity has also been negatively correlated with the severity of PE manifestations. In one of them, SIRT1 activator – resveratrol – has been shown to inhibit sFlt-1 release induced by the treatment of human placentas with tumor necrosis factor alpha (TNF-α) or by exposing them to hypoxia. Resveratrol also reduces sFlt-1 release from placental explants collected from PE patients, although only by 25-30% [93]. In a more recent study, exposal of primary human trophoblast cells to resveratrol has been found to inhibit both sFlt-1 secretion and sFlt-1 mRNA transcription [94]. Also in differentiated mouse WT TSC and SIRT KO TSC, treatment with resveratrol inhibits both sFlt-1 secretion and sFlt-1 mRNA transcription in a SIRT1-dependent manner. Since PPARγ activity also falls with SIRT1 KO [11], there is probably more than one target protein through which SIRT1 regulates basic levels of sFlt-1. Finally, resveratrol has also been reported to alleviate arterial hypertension and proteinuria on rat model of PE [95]. These data suggest that although SIRT1 is required to maintain low expression of sFlt-1, it inhibits sFlt-1 release from cells, and thus SIRT1 activation may be regarded as a therapeutic option in PE patients, along with PPARγ activation.
3.1.3. Effects of SIRT1 and PPARγ action towards placentas exposed to oxidative stress
Increased level of oxidative stress has been also found in placentas collected from PE patients [96]. This means an excess of reactive oxygen species (ROS) which may be secondary to hypoxia, ischemia/reperfusion, or reduced level of antioxidants. Since oxidative stress as a possible effect of hypoxia has been already discussed, this paragraph will be focused on other possible causes of oxidative stress. In rat model of maternal malnutrition, diet deficient in folic acid and cobalamin applied in pregnant females results in the increased level of oxidative stress markers in the plasma, as well as in reduced level of PPARγ mRNA in the placenta. However, it has no consequences for fetuses or mothers unless placental mass is affected [97]. As to SIRT1, oxidative stress induced by the treatment of human placental explants with hypoxanthine/xanthine oxidase reduces both SIRT1 mRNA and protein expression, as well as inhibits the expression of glucose transporter 1 (GLUT1), a glucose transporter responsible for glucose uptake [12]. These phenotypic alterations can be abrogated by resveratrol [12]. Similarly, treatment with resveratrol reduces oxidative stress in the placenta, as well as apoptosis occurrence in rat model of PE induced with L-NG-nitroarginine methyl ester (L-NAME) [95]. RNA profiling in mouse TSC has identified glutathione peroxidase isoform-encoding genes – GPX1 and GPX2 – as included in ten genes most repressed in SIRT1 KO TSC. GPX protein family accounts for cell protection from oxidative stress through catalyzing the reduction of organic hydroperoxides and hydrogen peroxide with glutathione (GSH) [98]. Reduced expression of GPX1 and GPX3 in SIRT1 KO TSC in comparison to WT TSC has been confirmed with quantitative polymerase chain reaction (qPCR) and correlated with increased apoptosis occurrence in SIRT1 KO TSC. It should be a subject of further evaluation whether SIRT1 KO TSC are indeed more susceptible to oxidative stress because of reduced glutathione peroxidase (GPX) expression. A sum of resveratrol actions as a SIRT1 activator is depicted on Figure 1.
Sirtuin-1 (SIRT1) acts primarily by removing acetyl groups from lysine residues within substrate proteins in the presence of nicotinamide adenine diphosphate (NAD+). The NAD+ dependence determines that the levels of NAD+ and SIRT1 activity (deacetylation) are tightly coupled. The acetyl group is transferred to the 2´-OH position of ADP-ribose, ultimately yielding nicotinamide (NAM) and 2´-O-acetyl-ADP-ribose (2-OAADPr). This oxidative stress-induced epigenetic mechanism reveals/increases the expression of genes that counteract preeclampsia by reducing hypertension, oxidative stress, inflammation and apoptosis in the placenta.
The antihypertensive effect of resveratrol through activation of SIRT1 at the placental level in preeclampsia is mainly based on the inhibition of anti-angiogenic factors soluble fms-like tyrosine kinase-1 (sFlt-1) and soluble endoglin (sEng), which are known to cause endothelial and trophoblast dysfunction. In addition, SIRT1 reduces the expression of pro-inflammatory molecules, and increases the expression of anti-oxidant molecules in endothelial cells. Such endothelial antioxidant markers in the preeclamptic placenta are: nuclear factor erythroid 2-related factor 2 (Nrf2), antioxidant response element (ARE), glutathione (GSH), superoxide dismutase (SOD), heme oxygenase-1 (HO-1) and NADPH-quinone oxidoreductase-1 (NQO1). The Nrf2-ARE pathway is an intrinsic mechanism of defense against oxidative stress. Its activation in endothelial cells triggers the transcription of antioxidant genes encoding, among others, catalase (CAT), SOD and glutathione peroxidase (GPX).
Increased SIRT1 activity in the preeclampsia placenta may promote trophoblast cell invasion, migration, and tube formation. This is achieved through activation of epithelial-mesenchymal transition (EMT) and Wnt/β-catenin pathway. Wnt/β-catenin signaling, a highly conserved pathway through evolution, regulates key cellular functions including proliferation, differentiation, migration, genetic stability, apoptosis, and stem cell renewal.
3.1.4. Effects of SIRT1 and PPARγ towards placentas affected by inflammatory response
Inflammatory response within the placenta can occur within the frames of physiology or pathology. An example of physiologic inflammatory response is one observed within the placenta and fetal membranes during a normal delivery. [104]. Such pro-inflammatory conditions at time of delivery have been correlated with unchanged PPARγ expression accompanied by reduced SIRT1 expression both in fetal membranes and in the placenta [12,105]. SIRT1 expression in human placenta is regulated by pro-inflammatory cytokines and has been reported to fall after the exposal on interleukins-1 beta (IL-1β) and TNF-α [12]. Quite interestingly, visfatin/nicotinamide mononucleotide adenyltransferase (Nampt), an adipokine and SIRT1 activator, positively correlates with SIRT1 activity and its level rises in obese women’s placentas just before delivery, which may suggest a possible mechanism preventing SIRT1 activity fall during late pregnancy. It can be sometimes responsible for post-term delivery, commonly observed in obese pregnant women [106].
Pathologic inflammatory response within the placenta is correlated both with PE and maternal obesity [107,108]. In case of microelement deficiency, pronounced inflammation within the placenta has been correlated with reduced expression of PPARγ mRNA in pregnant female rats [109]. On mouse model of lipopolysaccharide (LPS)-induced intrauterine fetal death (IUFD), preliminary treatment of pregnant mice with PPARγ activator – rosiglitazone, reduces IUFD occurrence from 64% to 16% [110]. This effect is related to enhanced nuclear location of PPARγ within placental trophoblast cells, as well as to reduced expression of placental pro-inflammatory mediators, such as interleukin-6 (IL-6) and TNF-α, and abrogating LPS-induced nuclear translocation of PPARγ within labirynthine layer of the trophoblast [110]. Finally, on a rat model of LPS-induced PE, a transplant of human mesenchymal stem cells (MSC) results in a reduced activity of pro-inflammatory mediators, such as IL-6 and TNF-α, as well as increased placental PPARγ activity; this is accompanied by a milder course of arterial hypertension and greater fetal mass in comparison to rats treated with LPS alone [111].
Much less is known about the correlation between obesity-related inflammation within the placenta and SIRT1/PPARγ expression. This type of inflammation characterizes with T lymphocyte and macrophage infiltration within chorionic villi. This type of inflammation occurs twice more often within placentas of female fetuses, although the reason why is unknown [112]. While macrophage infiltration within adipose tissue has been correlated with reduced SIRT1 expression [113], no alterations in SIRT1 expression have been reported in placentas collected from obese mothers. However, decreased placental SIRT1 expression accompanied by increased placental PPARγ expression can be observed on mouse model of high fat diet during pregnancy [11]. It has been correlated with increased activity of placental lipoprotein lipase (LPL), as well as with increased adipose tissue content in fetuses, which suggests that maternal overnutrition affects fetal development through altering the activity of SIRT1 and PPARγ [114]. Dependence of these phenotypic traits on pro-inflammatory mediators has yet to be elucidated. Reduced SIRT1 activity has been reported in mouse WT TSC treated with IL-6 [1], but the way how it is correlated with other markers of trophoblast cell differentiation has not been evaluated.
3.1.5. Correlations between hyperglycemia and placental SIRT1/PPARγ activity
Although no studies have been conducted on alterations of placental SIRT1 activity in case of maternal diabetes mellitus, similar studies referring to placental PPARγ activity have shown interesting results. PPARγ activity has been increased both in human primary trophoblast cells exposed to hyperglycemia [115], and in the placentas of pregnant female mice with streptozotocin-induced diabetes mellitus [116]. On the other hand, many other studies have observed reduced placental PPARγ expression in case of gestational diabetes mellitus [117,118,119,120], and one of those studies has revealed reduced expression of this protein both in syncytiotrophoblast and in extravillous trophoblast [119]. Further studies are necessary to precisely evaluate the correlation between gestational diabetes mellitus and PPARγ activity within trophoblast and placenta, taking into consideration both management of maternal hyperglycemia and related effects towards fetal growth.
4. SIRT1-dependent prevention of preeclampsia
4.1. SIRT1 protective actions towards vascular endothelial cells
Endothelial cell dysfunction is one of typical traits of pre-eclampsia, resulting from several factors, including oxidative stress, inflammatory response, autophagy, etc. SIRT1 counteracts oxidative stress, as well as exerts some anti-inflammatory and anti-aging effects. Several research studies have shown that SIRT1 activity is reduced in serum samples collected from PE patients, as well as in human umbilical vein endothelial cells (HUVEC) incubated with such serum [121]. SIRT1 may protect HUVEC from necrosis in PE patients, thus blocking PE development [122]. From the mechanistic point of view, SIRT1 protects endothelial cells from oxidative stress, inflammatory response and cell senescence phenotype through numerous mechanisms, depicted on Figure 2.
SIRT1 (silent information regulator 2 homolog 1) is a crucial cellular survival protein especially in oxidative stress environments. SIRT1 activity depends on oxidized form of nicotinamide adenine dinucleotide (NAD+), which is generated from its precursor – nicotinamide mononucleotide (NMN) – by enzyme nicotinamide-(mono)nucleotide adenylyltransferase (NMNAT). Three NMNAT isoforms have been discovered and they show distinct subcellular localizations: NMNAT1 (nucleus), NMNAT2 (cytosol) and NMNAT3 (mitochondria), which suggests a localization-component to NAD+ synthesis in response to metabolic signals [123,124]. Similarly, although the main site of SIRT1 synthesis is the nucleus, its activity is also observed in the cytoplasm and mitochondria [123,125]. The level of NAD+ is determined by NAD+ synthesis from the salvage pathway or NAD+ /reduced form (NADH) ratio. Mitochondrial redox metabolism within the electron transport chain (ETC) is crucial for SIRT1 level, because NAD+/NADH and AMP/ATP metabolism result from the tricarboxylic acid (TCA) cycle and β-oxidation or oxidative phosphorylation, respectively [126]. NAD+ is required in the SIRT1-mediated deacetylase reaction. This reaction also generates nicotinamide (NAM), which then enters the salvage pathway. Nicotinamide mononucleotide adenyltransferase (Nampt), catalyzing the conversion from NAM to NMN, is the rate limiting enzyme in this pathway. NMN is thereby converted to NAD+ by NMNAT.
The increases in NAD+/NADH ratio and AMP/ATP ratio observed during caloric restriction are well-known inducers of SIRT1.
SIRT1 attenuates oxidative stress and inflammation to regulate vascular endothelial functions through several important signal mediators, such as AMP-activated protein kinase (AMPK), nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox), endothelial nitric oxide synthase (eNOS), and forkhead transcription factors of the O class (FOXOs) [7,127]. SIRT1 can stimulate AMPK via the modulation of upstream AMPK kinase such as liver kinase B1(LKB1), suppressing the production of reactive oxygen species (ROS) and inflammatory response in human umbilical vein endothelial cells (HUVECs), while AMPK influences SIRT1 deacetylation activity by increasing cellular NAD+ levels or directly phosphorylating (P) SIRT1. Increased AMP/ATP ratio induces endothelial AMPK, which in turn suppresses Nox expression and Nox-induced ROS production [128]. AMPK-dependent phosphorylation and SIRT1-dependent deacetylation of eNOS lead to an increase in local nitric oxide (NO) concentration. Moreover, SIRT1 deacetylates FoxOs proteins, and thus stimulates FoxO-dependent antioxidative enzymes, such as catalase (CAT), manganese superoxide dismutase (MnSOD), and thioredoxin (TRX), eliminating ROS from endothelial cells and thus preventing endothelial dysfunction [127,129,130]. SIRT1 protects endothelial cells from senescence through regulating signaling pathways dependent on tumor protein p53 (p53), eNOS, transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) and FOXO3. Expression of these proteins can be in turn regulated at the level of translation by several micro-RNA molecules, such as mi-R217, mi-R34a, mi-R155 and mi-R22 [131,132,133,134,135,136,137]. Optimization of NO concentration and genome stability extend the average lifespan of endothelial cells.
4.1.1. SIRT1 and the protection of endothelial cells against oxidative stress and inflammatory response
Oxidative stress and inflammatory response are mutually related pathophysiologic processes taking part in the pathogenesis of PE. Oxidative stress consists in raised concentration of ROS, which results with inflammatory response and – in case of endothelial cells – with their damage and dysfunction [7]. Mitochondrial function is impaired in the course of PE, which results in increased ROS generation, mainly in the form of superoxide anions, causing oxidative stress and systemic inflammation [138,139,140,141]. SIRT1 inhibition abrogates the activity of endogenous antioxidative systems on in vitro models of PE. Furthermore, SIRT1 is necessary for counteracting oxidative stress and inflammation in the course of diabetic angiopathy [142,143,144], while the same two phenomena (i.e. oxidative stress and inflammation) play a crucial role in the pathogenesis of PE. SIRT1 inhibition in hyperglycemic conditions results in endothelial cell dysfunction, while SIRT1 activation alleviates endothelial aging induced by oxidative stress in diabetic mice [145,146]. Quite interestingly, SIRT1 alleviates oxidative stress and inflammatory response through regulating endothelial cell functions via several signaling pathways dependent on adenosine monophosphate(AMP)-activated protein kinase (AMPK), nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox), endothelial nitric oxide synthase (eNOS) and FoxOs [147]. There is a complex network of interactions between AMPK and SIRT1. SIRT1 may activate AMPK through modulation of liver kinase B1 (LKB1) – an upstream regulatory enzyme, modulating AMPK activity [147,148], which inhibits ROS production and inflammatory response in HUVEC. AMPK also affects SIRT1 deacetylase activity through regulation of intracellular NAD+ concentration, or directly phosphorylating SIRT1 molecules. In addition, increased activity of NADPH oxidase (Nox) can also increase intracellular NAD+ concentration, which stimulates SIRT1 activity in endothelial cells [127]. Moreover, SIRT1 deacetylates FoxO proteins, and thus stimulates FoxO-dependent antioxidative enzymes, such as catalase (CAT), manganese superoxide dismutase (MnSOD), and thioredoxin (TRX), eliminating ROS from endothelial cells and thus preventing endothelial dysfunction [127,128,129,130]. SIRT1 has been reported to stimulate c-Myc expression through promoting forkhead box protein O1 (FoxO1) degradation, which prevents hyperglycemia-induced endothelial cell dysfunction and angiogenesis [149]. eNOs as a nitric oxide synthase (NOS) family protein is expressed in vascular smooth muscle cells and plays a crucial role in the pathogenesis of PE by catalyzing nitric oxide (NO) biosynthesis and at the same time inhibiting ROS production [150]. SIRT1 may directly deacetylate eNOs, or stimulate eNOs activity indirectly, through affecting FoxO proteins and AMPK-dependent signaling pathways [151], which can take part in PE pathogenesis. This evidence suggests that SIRT1 may protect endothelial cells from oxidative stress and inflammatory response through interacting with other enzymes, which can take part in the pathogenesis of PE.
SIRT1 may protect endothelial cells through autophagy regulation
In endothelial cells, autophagy is regulated mainly by SIRT1-dependent and FoxO-dependent signaling pathways, which may take part in the pathogenesis of PE [152]. Research studies have found that SIRT1 activates FoxO1, thus protecting endothelial cells through autophagy regulation [153]. To be more precise, SIRT can deacetylate, and thus activate FoxO1, while activated FoxO1 may stimulate SIRT1 expression [154]. FoxO1 is strictly related to autophagy, since it modulates the expression of such proteins taking part in autophagy as a small GTPase Rab7, LC3, ATG-5, and Beclin-1 [155]. These results suggest that SIRT1 exerts a protective effect on endothelial cells, analogously to the trophoblast (see section 2.2. Effects of SIRT1 on autophagy within trophoblast), also by regulating autophagy via multiple signaling pathways, as shown in Figure 3.
Sirtuin-1 (SIRT1) acts as an energy and redox sensor, because is activated by nicotinamide adenine dinucleotide (NAD+), an important substrate in energy and oxidation reactions. As a result of NAD+-dependent deacetylation of the respective protein substrates, acetyl groups are transferred to the 2´-OH position of ADP-ribose, ultimately yielding nicotinamide (NAM) and 2´-O-acetyl-ADP-ribose (2-OAADPr) [123,124]. There is an increase in the level of gene expression and, consequently, proteins responsible for the induction and promotion of autophagy, such as forkhead box protein O1 (FoxO1), sequestosome 1 (SQSTM1, also known as ubiquitin-binding scaffold protein p62), and – a key regulator of the autophagy/lysosomal-to-nucleus signaling pathway – transcription factor EB (TFEB) [34,35,36,37]. Moreover, formation of lysosomes is strictly regulated by TFEB via activation of several downstream autophagy-related genes, such as lysosomal associated membrane protein 1 and 2 (LAMP1, LAMP2), and cathepsin D (CTSD) [32,33].
Sirt1-deacetylated FOXO1 stimulates the expression of RAB7, encoding a small GTPase that is a crucial factor in the maturation of autophagosomes and endosomes [156]. Other autophagy-related genes activated directly by SIRT1 or via FoxO1 are those encoding membrane-bound, lipidated form of LC3 (LC3-II), a protein containing a Bcl-2 homology-3 domain (BECN1), and autophagy related-proteins 5 and 7 (ATG5, ATG7) [30,31]. In addition, FoxO1 directly activates SIRT1, thus creating an autofeedback loop regulating SIRT1 expression [40]. Autophagy is also induced by SIRT1 inhibiting the mammalian target of rapamycin (mTOR)-related signaling pathway [157,158]. Cell autolysis as a result of autophagy is preceded by the formation of: phagophore, autophagosome and – after fusion of the autophagosome and a lysosome – autolysosome [23,24,25,26,27].
4.1.2. SIRT1 and possible protection of endothelial cells against senescence
CSP occurrence in vascular endothelial cells is a direct cause of most dangerous complications of cardiovascular diseases, and thus most frequent cause of death [159,160]. Quite interestingly, in PE patients CSP has been observed in endothelial progenitor cells, which is related to endothelial dysfunction [161,162]. SIRT1 protects endothelial cells from CSP through regulating some signaling pathways dependent on p53, eNOs, Nrf2, forkhead box protein O3 (FoxO3), as well as p21/p16. Expression of these proteins can be in turn regulated at the level of translation by several micro-RNA molecules, such as mi-R217, mi-R34a, mi-R155 and mi-R22 [131,132,133,134,135,136,137]. Although additional research studies may be required to precisely pinpoint SIRT1 role in endothelial cell senescence phenotype regulation, hitherto performed studies suggest that SIRT1 protects endothelial cells from oxidative stress, inflammatory response, CSP and autophagy through deacetylation of its specific target proteins, which may take part in the pathogenesis of PE.
4.2. Anti-inflammatory action of SIRT1 within the placenta in the context of preeclampsia
SIRT1 plays an essential role in alleviating inflammatory response and oxidative stress in several physiologic and pathologic conditions [163]. Reduced placental SIRT1 expression means that anti-inflammatory and anti-oxidative protection has been compromised. In addition, SIRT1 inhibits high mobility group box 1 (HMGB1) release from cells to the extracellular space, through deacetylation of this non-histone protein molecule. Although in most cases, HMGB1 binds to DNA and promotes transcription of its specific target genes, it is not sensu stricto nuclear protein, and its molecules can be translocated to other organelles, or even actively released from cells, which is usually induced by exposal to specific factors (e.g. hypoxia). HMGB1 molecules can also be passively released from cells following cell necrosis [164]. Extracellular HMGB1 activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) through interactions with receptor for advanced glycation end-products (RAGE) and toll-like receptor 4 (TLR4), as well as promotes secretion of pro-inflammatory cytokines, such as TNF-α, IL-1, IL-6, and interleukin-8 (IL-8) [164]. Therefore, HMGB1 release to the extracellular environment activates both innate and adaptive immunity. Moreover, some recent research studies suggest that HMGB1 released from hypoxic trophoblast may increase endothelial permeability through signaling pathways dependent on TLR4 and caveolin-1 [165]. Increased permeability of endothelium is the main cause of proteinuria and generalized edemas in the course of PE [165]. HMGB1 concentration in HUVEC-containing medium rises after HUVEC treatment with IL-6 or with serum collected from PE patients, despite reduced HMGB1 concentration in these cells, which suggests that HMGB1 is released from the cells in such conditions. Furthermore, experiments comprising SIRT1 inhibition or activation have shown that SIRT1 may block HMGB1 release from cells on a mouse model of PE, which suggests that SIRT1 can abrogate pro-inflammatory actions of HMGB1 in the course of PE [122]. In addition, SIRT1 inhibits 70-kDa heat shock proteins (HSP70) release from HUVEC after their exposal to IL-6 or serum collected from PE patients. In cells unexposed to stress, HSP70 undergoes a constitutive expression and plays many significant physiologic roles in almost each organelle, including cytoplasm, endoplasmic reticulum, mitochondria, and cell nucleus [166]. HSP70 expression is induced by several kinds of stress and initially, it has been thought that it helps the cells counteract the stress [166]. However, more recent studies have revealed that HSP70 released to the extracellular environment may bind to many specific signaling receptors, such as lectin-like oxidized low-density lipoprotein-1 (LOX-1), toll-like receptor 2 (TLR2), TLR4, 50-kDa integral membrane protein of the tumor necrosis factor receptor (TNF-R) family (CD40), scavenger receptor expressed by endothelial cell-1 (SREC-1), and link domain-containing scavenger receptor-1 (FEEL-1), which implicates ambiguous effects of HSP70 in some conditions [166]. It has been confirmed that HSP70 activates human monocytes, inhibiting the release of some pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6 and interleukin-10 (IL-10). However, another study has revealed that in patients suffering from early-onset PE, HSP70 concentration in the plasma is positively correlated with concentration of TNF-α, soluble type 1 receptor for TNF-α, IL-1β, interleukin-12 (IL-12), glutamicoxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), lactate dehydrogenase (LDH) and uric acid, which in turn suggests that raised level of HSP70 is related to increased negative effect towards maternal and fetal well-being [166,167]. Molvarec et al. [168] have delivered more evidence suggesting that HSP70 may contribute to systemic inflammatory response in PE patients. They have found serum HSP70 level to be positively correlated with increased levels of such proteins as interleukin-12 subunit beta p40 (IL-12p40), monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1). They have also shown that raised levels of HSP70 and sFlt-1/placenta growth factor (PlGF) are independent risk factors of PE development. Although pathogenic functions of HSP70 in PE remain controversial, HUVEC release HSP70 to the extracellular environment in response to stimulation with IL-6 or serum collected from PE patients, which suggests that they perceive such stimulation as a kind of stress. Forced expression of SIRT1 inhibits HSP70 release from cells, which indicates that it may inhibit HUVEC response to stress in the course of PE. One recent research study has shown that SIRT1 indeed affects HSP70 expression in cells. Inducible HSP70 has been shown to be upregulated in spinal cord of mice chronically overexpressing SIRT1 in the central nervous system [169]. Studies on SIRT1 mechanisms of action indicate that it deacetylates heat shock transcription factor 1 (HSF-1) transcription factor that is an essential regulator of heat shock proteins (HSPs) expression, which results in enhanced expression of inducible HSP70 [169]. Few studies, however, deals with SIRT1 role in modulating HSP70 release from cells. Those studies which have explored this issue may deliver a first piece of evidence that SIRT1 can inhibit HSP70 release from HUVEC cells, thus counteracting the effects of exposal to IL-6 or serum collected from PE patients. However, further studies are necessary to pinpoint the mechanisms through which SIRT1 modulates HSP70 release from cells. In the course of PE, excessive inflammatory response and oxidative stress result in endothelial cell damage and death. Necrotic cells may release HMGB1, which can further enhance inflammatory response, resulting in a vicious circle. SIRT1 protects HUVEC from necrosis resulting from their exposal to IL-6 or serum collected from PE patients. This protective effect of SIRT1 is probably related to its anti-inflammatory and anti-oxidative actions, but also to its anti-apoptotic functions. Many studies have confirmed that SIRT1 can deacetylate p53, thus abrogating its proapoptotic actions [170]. Furthermore, SIRT1 has been found to be downregulated in the placentas of PE model mice, while HMGB1 and HSP70 serum concentrations are markedly elevated in such mice. SIRT1 inhibits HMGB1 and HSP70 release from HUVEC exposed to IL-6 or serum collected from PE patients and protects the cells from necrosis. All these findings indicate that SIRT1 can play a protective role in PE, alleviating its manifestations.
SIRT1 alleviates PE course on animal models of PE
SIRT1 activity is reduced both in PE patients’ placentas and sera and in placentas and sera collected from mice used as an animal model of PE [171]. It has been found that SIRT1 inhibition in SIRT1+/- mice induces typical manifestations of PE, such as arterial hypertension, proteinuria, intrauterine growth retardation, renal damage, as well as labirynthine layer atrophy. Moreover, all of these manifestations can be alleviated by the treatment with experimental drug SRT2104, a potent SIRT1 inducer [22]. It has also been reported that SIRT1 KO mice placentas and fetuses show abnormalities both within labirynthine layer and in junctional zone. In addition, SIRT1 KO mice develop numerous abnormalities, from increased prenatal mortality to fetal growth impairment resulting in greater postnatal mortality [172,173,174,175]. Furthermore, placentas collected from SIRT1 KO mice show increased occurrence of cell senescence phenotype, as well as other morphologic abnormalities [176], which is strictly correlated with PE development.
SIRT1 induction alleviates PE manifestations
In reduced uterine perfusion pressure (RUPP) rats, constituting an animal model of PE, supplementation with recombined SIRT1 protein alleviates PE manifestations, such as arterial hypertension, impaired placental angiogenesis, inflammatory response, and unfavorable pregnancy outcome [177]. Similar effect may be achieved through treatment with SIRT1 inducer – SRT2104 [22]. However, more animal studies and clinical trials are needed to precisely determine SIRT1 role in PE.
5. Conclusion
Recently, there have been more and more publications on the activity of sirtuins in placental tissue in normal and complicated pregnancies. This applies in particular to SIRT1 in placental vascular endothelial cells and trophoblast cells. [8,178,179,180,181]. Directly involved in many key intracellular reactions, SIRT1 builds up the connection between epigenetics and metabolism at the placental level [182]. As an NAD+-dependent deacetylase, SIRT1 regulates many aspects of chromatin biology, such as transcription, recombination, and genome stability, by modifying histones, transcription factors, and epigenetic enzymes, and in connection with the above, it directly affects placental homeostasis by modifying a diverse set of metabolic enzymes, both in the cytosol and in the mitochondria [1,183]. The beneficial effects of SIRT1 in the human placenta known so far relate primarily to modulating the activity of factors responsible for the course of the inflammatory response, oxidative stress, autophagy, and cell senescence [7,29,155,184,185,186,187].
Therefore, there are well-documented reasons to treat the activation or inhibition of SIRT1 as a potential therapeutic target, especially in hypertensive disorders complicating pregnancy, in which the main pathomechanism is based on endothelial dysfunction [5,7,8,188]. This is even more important as the incidence of PE is increasing, possibly as a result of increased prevalence of predisposing disorders, such as chronic hypertension, diabetes, and obesity [189,190].
Author Contributions
Conceptualization, M.W. and, D.S.; Writing – Original Draft Preparation, M.W. and, G.S.; Writing – Review & Editing, D.S. and, M.W.; Visualization, D.S.; Supervision, D.S. and, G.S.; Software, G.S. and, D.S.; Funding Acquisition, D.S.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. This review is based on already published data listed in the references.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 2-OAADPr | 2´-O-acetyl-adenosine diphosphate(ADP)-ribose. |
| AMPK | adenosine monophosphate(AMP)-activated protein kinase. |
| ARE | antioxidant response element. |
| ATG5 | ATG7, ATG8 – autophagy related-proteins 5, 7, 8. |
| Beclin-1 | the mammalian ortholog of yeast Atg6/Vps30, an essential autophagy protein that contains a Bcl-2 homology-3 domain. |
| BECN1 | Beclin-1 gene. |
| CAT | CAT – catalase gene, catalase (antioxidant enzyme), respectively. |
| CD40 | 50-kDa integral membrane protein of the tumor necrosis factor receptor (TNF-R) family. |
| CSP | cell senescence phenotype. |
| CTSD | cathepsin D . |
| EMT | epithelial-mesenchymal transition. |
| eNOS | endothelial nitric oxide synthase. |
| ETC | electron transport chain. |
| FEEL-1 | link domain-containing scavenger receptor-1. |
| FoxOs | forkhead O class box transcription factors. |
| FoxO1 | FoxO3 – forkhead box protein O1 and O3, respectively. |
| GCM1 | glial cells missing-1 (transcription factor). |
| GLUT1 | glucose transporter 1. |
| GOT | glutamicoxaloacetic transaminase. |
| GPT | glutamic pyruvic transaminase. |
| GPX | glutathione peroxidase. |
| GPX1 | GPX2, GPX3 – glutathione peroxidase isoforms 1, 2 and 3, respectively. |
| GSH | glutathione. |
| H2O2 | hydrogen peroxide. |
| HIF | hypoxia-inducible factor (HIF). |
| HIF-1α | hypoxia-inducible factor 1 subunit alpha. |
| HIF-2α | hypoxia-inducible factor 2 subunit alpha. |
| HIFα | HIFβ – domains that make up the (hypoxia-inducible factor) HIF molecule domain. |
| HMGB1 | high mobility group box 1 (nonhistone nuclear protein). |
| HO-1 | heme oxygenase-1. |
| HSF1 | heat shock transcription factor 1. |
| HSP70 | 70-kDa heat shock proteins. |
| HSPs | heat shock proteins. |
| HUVEC | human umbilical vein endothelial cells. |
| ICAM-1 | intercellular adhesion molecule 1, also known as CD54 (cluster of differentiation 54). |
| IL-1 | IL-1β, IL-6, IL-8, IL-10, IL-12 –interleukins: 1, 1 beta, 6, 8, 10 and 12. |
| IUFD | intrauterine fetal death. |
| IL-12p40 | interleukin-12 subunit beta (p40). |
| KO | knockout. |
| L-NAME | L-NG-nitroarginine methyl ester. |
| LAMP1 | LAMP2 – lysosomal associated membrane protein 1,2. |
| LC3 | microtubule-associated protein 1 light chain 3 (MAP1LC3), a human homologue of yeast Atg8, an essential component of autophagy. |
| LC3-II | membrane-bound: lipidated form of LC3. |
| LDH | lactate dehydrogenase. |
| LKB1 | liver kinase B1. |
| LOX-1 | lectin-like oxidized low-density lipoprotein-1. |
| LPL | placental lipoprotein lipase. |
| LPS | lipopolysaccharide. |
| MCP-1 | monocyte chemoattractant protein-1. |
| mi-R217 | mi-R34a, mi-R155, mi-R22 – micro-RNA molecules. |
| MnSOD | manganese superoxide dismutase (antioxidant enzyme). |
| mTOR | mammalian target of rapamycin (an ubiquitous serine-threonine protein kinase). |
| NAD | nicotinamide adenine dinucleotide. |
| NAD+ | nicotinamide adenine dinucleotide (oxidized form). |
| NADH | nicotinamide adenine dinucleotide (reduced form, H for hydrogen). |
| NAM | nicotinamide. |
| NCoR1 | nuclear receptor co-repressor-1. |
| NDRG1 | N-myc downstream regulated gene 1 (formerly known as Drg1, Cap43, Rit42, RTP, and PROXY-1). |
| Nampt | nicotinamide mononucleotide adenyltransferase. |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells. |
| NMN | nicotinamide mononucleotide. |
| NMNAT | nicotinamide-(mono)nucleotide adenylyltransferase. |
| NMNAT1 | NMNAT2, NMNAT3 – nicotinamide-(mono)nucleotide adenylyltransferase isoforms: 1,2, and 3. |
| NO | nitric oxide. |
| NOS | nitric oxide synthase. |
| Nox | nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. |
| NQO1 | nicotinamide adenine dinucleotide phosphate (NADPH)-quinone oxidoreductase-1. |
| Nrf2 | nuclear factor erythroid 2-related factor 2. |
| p62 | ubiquitin-binding scaffold protein, also known as sequestosome 1 (SQSTM1). |
| p53 | protein encoded by the TP53 tumor suppressor gene, marker of cell senescence. |
| p21 | cyclin-dependent kinase inhibitor p21, protein marker of cell senescence. |
| PE | preeclampsia. |
| PlGF | placenta growth factor. |
| PPARγ | peroxisome proliferator-activated receptor γ. |
| PPARγ2 | an isoform of PPARγ typical for adipose tissue. |
| PPRE | PPARγ-reactive elements. |
| PRDM1 | positive regulatory (PR) domain zinc finger protein 1, a coactivator selectively activating PPARγ. |
| Prdm16 | positive regulatory domain containing 16. |
| qPCR | quantitative polymerase chain reaction. |
| Rab7 | a small GTPase: member of the Rab family that controls transport to late endocytic compartments such as late endosomes and lysosomes. |
| RAB7 | Rab7 gene. |
| RAGE | receptor for advanced glycation end-products . |
| ROS | reactive oxygen species. |
| RUPP | reduced uterine perfusion pressure. |
| RXR | retinoid X-receptor. |
| SASP | senescence-associated secretory phenotype. |
| sEng | soluble endoglin: the extracellular domain of membrane endoglin. |
| sFlt-1 | soluble fms-like tyrosine kinase 1, also known as soluble vascular endothelial growth factor (VEGF) receptor-1 . |
| siRNA | small interfering RNA. |
| SIRT1 | silent information regulator 2 homolog 1 or sirtuin-1. |
| SIRT1 | SIRT7 – sirtuins 1 to 7 . |
| SMAD2 | SMAD3 – small mothers against decapentaplegic proteins 2 and 3 (transcription factors). |
| SMRT | silencing mediator of retinoid and thyroid hormone receptors. |
| SOD | superoxide dismutase. |
| SQSTM1 | sequestosome 1: also known as ubiquitin-binding scaffold protein p62. |
| SREC-1 | scavenger receptor expressed by endothelial cell-1. |
| SRT2104 | experimental drug, a selective small molecule activator of SIRT1. |
| STAT | signal transducer and activator of transcription (transcription factor). |
| STAT3 | signal transducer and activator of transcription 3 (transcription factor). |
| TCA | tricarboxylic acid cycle, also known as the Krebs cycle or the citric acid cycle. |
| TFEB | transcription factor EB (TFEB), a member of the MiT/TFE family of basic helix-loop-helix leucine zipper transcription factors, a key regulator of the autophagy/lysosomal-to-nucleus signaling pathway. |
| TLR2 | TLR4 – toll-like receptor 2 and 4. |
| TNF-α | tumor necrosis factor alpha. |
| TRX | thioredoxin (antioxidant protein). |
| TSC | trophoblast stem cells. |
| TZD | thiazolidinediones: synthetic activators of PPARγ. |
| WT TSC | wild-type trophoblast stem cells (TSC). |
| VCAM-1 | vascular cell adhesion molecule 1, also known as CD106 (cluster of differentiation 106). |
| VEGF | vascular endothelial growth factor. |
References
- Pham, J.; Arul Nambi Rajan, K.; Li, P.; Parast, M.M. The role of Sirtuin1-PPARγ axis in placental development and function. J Mol Endocrinol. 2018, 60, R201–R212. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. What is the placenta? Am J Obstet Gynecol. 2015, 213(4 Suppl):S6.e1, S6-8. [CrossRef]
- James, J.L.; Carter, A.M.; Chamley, L.W. Human placentation from nidation to 5 weeks of gestation. Part I: What do we know about formative placental development following implantation? Placenta. 2012, 33, 327–34. [Google Scholar] [CrossRef] [PubMed]
- Soncin, F.; Natale, D.; Parast, M.M. Signaling pathways in mouse and human trophoblast differentiation: a comparative review. Cell Mol Life Sci. 2015, 72, 1291–302. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.J. Why is placentation abnormal in preeclampsia? Am J Obstet Gynecol. 2015, 213(4 Suppl):S115-22. [CrossRef]
- Thornburg, K.L.; Marshall, N. The placenta is the center of the chronic disease universe. Am J Obstet Gynecol. 2015, 213(4 Suppl):S14-20. [CrossRef]
- Liu, Z.; Wang, C.; Pei, J.; Li, M.; Gu, W. SIRT1: A Novel Protective Molecule in Pre-eclampsia. Int J Med Sci. 2022, 19, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Kojima, J.; Dai, Y.; Suzuki, T.; Ono, M.; Nishi, H. Sirtuin 1 is a potential therapeutic candidate gene for fetal growth restriction via insulin-like 4. J Matern Fetal Neonatal Med. 2023, 36, 2253486. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta. 2015, 36, 101–14. [Google Scholar] [CrossRef]
- Kwak-Kim, J.; Bao, S.; Lee, S.K.; Kim, J.W.; Gilman-Sachs, A. Immunological modes of pregnancy loss: inflammation, immune effectors, and stress. Am J Reprod Immunol. 2014, 72, 129–40. [Google Scholar] [CrossRef]
- Arul Nambi Rajan, K.; Khater, M.; Soncin, F.; Pizzo, D.; Moretto-Zita, M.; Pham, J.; Stus, O.; Iyer, P.; Tache, V.; Laurent, L.C.; Parast, M.M. Sirtuin1 is required for proper trophoblast differentiation and placental development in mice. Placenta. 2018, 62:1-8. [CrossRef]
- Lappas, M.; Mitton, A.; Lim, R.; Barker, G.; Riley, C.; Permezel, M. SIRT1 is a novel regulator of key pathways of human labor. Biol Reprod. 2011, 84, 167–78. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999, 4, 585–95. [Google Scholar] [CrossRef]
- Borg, A.J.; Yong, H.E.; Lappas, M.; Degrelle, S.A.; Keogh, R.J.; Da Silva-Costa, F.; Fournier, T.; Abumaree, M.; Keelan, J.A.; Kalionis, B.; Murthi, P. Decreased STAT3 in human idiopathic fetal growth restriction contributes to trophoblast dysfunction. Reproduction. 2015, 149, 523–32. [Google Scholar] [CrossRef]
- Erlebacher, A.; Price, K.A.; Glimcher, L.H. Maintenance of mouse trophoblast stem cell proliferation by TGF-beta/activin. Dev Biol. 2004, 275, 158–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Huang, G.; Fan, W.; Chen, Y.; Ward, J.M.; Xu, X.; Xu, Q.; Kang, A.; McBurney, M.W.; Fargo, D.C.; Hu, G.; Baumgart-Vogt, E.; Zhao, Y.; Li, X. SIRT1-mediated deacetylation of CRABPII regulates cellular retinoic acid signaling and modulates embryonic stem cell differentiation. Mol Cell. 2014, 55, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Takahashi, M.; Carpino, N.; Jou, S.T.; Chao, J.R.; Tanaka, S.; Shigeyoshi, Y.; Parganas, E.; Ihle, J.N. Leukemia inhibitory factor regulates trophoblast giant cell differentiation via Janus kinase 1-signal transducer and activator of transcription 3-suppressor of cytokine signaling 3 pathway. Mol Endocrinol. 2008, 22, 1673–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, F.; Xu, Y.; Wei, J.; Zhang, Y.; Yang, H.; Gao, B.; Yu, G.; Fang, D. JAK1-mediated Sirt1 phosphorylation functions as a negative feedback of the JAK1-STAT3 pathway. J Biol Chem. 2018, 293, 11067–11075. [Google Scholar] [CrossRef] [PubMed]
- Tache, V.; Ciric, A.; Moretto-Zita, M.; Li, Y.; Peng, J.; Maltepe, E.; Milstone, D.S.; Parast, M.M. Hypoxia and trophoblast differentiation: a key role for PPARγ. Stem Cells Dev. 2013, 22, 2815–24. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004, 429, 771–6. [Google Scholar] [CrossRef]
- Matsuda, S.; Kobayashi, M.; Kitagishi, Y. Expression and Function of PPARs in Placenta. PPAR Res. 2013, 2013:256508. [CrossRef]
- Pei, J.; Liu, Z.; Wang, C.; Chu, N.; Liu, L.; Tang, Y.; Liu, H.; Xiang, Q.; Cheng, H.; Li, M.; Gu, W. Progesterone Attenuates SIRT1-Deficiency-Mediated Pre-Eclampsia. Biomolecules. 2022, 12, 422. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: the importance of maintaining "clean" cells. Autophagy. 2005, 1, 131–40. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008, 451, 1069–75. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Klionsky, D.J. Autophagy contributes to lysosomal storage disorders. Autophagy. 2012, 8, 715–6. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T. Autophagy: paying Charon's toll. Cell. 2007, 128, 833–6. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Cheng, S.B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y.; Saito, S.; Sharma, S. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy. 2020, 16, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, C.X.; Gao, J.J.; Shi, Y.; Li, H.; Yan, H.; Yan, S.J.; Zhang, Z. Resveratrol induces SIRT1-Dependent autophagy to prevent H2O2-Induced oxidative stress and apoptosis in HTR8/SVneo cells. Placenta. 2020, 91:11-18. [CrossRef]
- Chen, C.; Xia, B.; Tang, L.; Wu, W.; Tang, J.; Liang, Y.; Yang, H.; Zhang, Z.; Lu, Y.; Chen, G.; Yang, Y.; Zhao, Y. Echinacoside protects against MPTP/MPP+-induced neurotoxicity via regulating autophagy pathway mediated by Sirt1. Metab Brain Dis. 2019, 34, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, R.; Wang, W.; Gao, H.; Wang, C. SIRT1/Atg5/autophagy are involved in the antiatherosclerosis effects of ursolic acid. Mol Cell Biochem. 2016, 420(1-2):171-84. [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; Settembre, C.; Wang, W.; Gao, Q.; Xu, H.; Sandri, M.; Rizzuto, R.; De Matteis, M.A.; Ballabio, A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015, 17, 288–99. [Google Scholar] [CrossRef]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018, 9, 3312. [Google Scholar] [CrossRef]
- Akaishi, R.; Yamada, T.; Nakabayashi, K.; Nishihara, H.; Furuta, I.; Kojima, T.; Morikawa, M.; Yamada, T.; Fujita, N.; Minakami, H. Autophagy in the placenta of women with hypertensive disorders in pregnancy. Placenta. 2014, 35, 974–80. [Google Scholar] [CrossRef]
- Gao, L.; Qi, H.B.; Kamana, K.C.; Zhang, X.M.; Zhang, H.; Baker, P.N. Excessive autophagy induces the failure of trophoblast invasion and vasculature: possible relevance to the pathogenesis of preeclampsia. J Hypertens. 2015, 33, 106–17. [Google Scholar] [CrossRef]
- Oh, S.Y.; Choi, S.J.; Kim, K.H.; Cho, E.Y.; Kim, J.H.; Roh, C.R. Autophagy-related proteins, LC3 and Beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod Sci. 2008, 15, 912–20. [Google Scholar] [CrossRef]
- Feng, L.; Chen, M.; Li, Y.; Li, M.; Hu, S.; Zhou, B.; Zhu, L.; Yu, L.; Zhou, Q.; Tan, L.; An, H.; Wang, X.; Jin, H. Sirt1 deacetylates and stabilizes p62 to promote hepato-carcinogenesis. Cell Death Dis. 2021, 12, 405. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; Liu, W. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015, 57, 456–66. [Google Scholar] [CrossRef] [PubMed]
- Pi, Q.Z.; Wang, X.W.; Jian, Z.L.; Chen, D.; Zhang, C.; Wu, Q.C. Melatonin Alleviates Cardiac Dysfunction Via Increasing Sirt1-Mediated Beclin-1 Deacetylation and Autophagy During Sepsis. Inflammation. 2021, 44, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Ye, X.; Chen, Z.; Fu, H.; Li, S.; Xu, P.; Yu, J.; Wen, L.; Gao, R.; Fu, Y.; Qi, H.; Kilby, M.D.; Saffery, R.; Baker, P.N.; Tong, C. Advanced Maternal Age-associated SIRT1 Deficiency Compromises Trophoblast Epithelial-Mesenchymal Transition through an Increase in Vimentin Acetylation. Aging Cell. 2021, 20, e13491. [Google Scholar] [CrossRef] [PubMed]
- Sultana, Z.; Maiti, K.; Dedman, L.; Smith, R. Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am J Obstet Gynecol. 2018, 218(2S):S762-S773. [CrossRef]
- Biron-Shental, T.; Sukenik-Halevy, R.; Sharon, Y.; Goldberg-Bittman, L.; Kidron, D.; Fejgin, M.D.; Amiel, A. Short telomeres may play a role in placental dysfunction in preeclampsia and intrauterine growth restriction. Am J Obstet Gynecol. 2010, 202, 381.e1–7. [Google Scholar] [CrossRef] [PubMed]
- Tasta, O.; Swiader, A.; Grazide, M.H.; Rouahi, M.; Parant, O.; Vayssière, C.; Bujold, E.; Salvayre, R.; Guerby, P.; Negre-Salvayre, A. A role for 4-hydroxy-2-nonenal in premature placental senescence in preeclampsia and intrauterine growth restriction. Free Radic Biol Med. 2021, 164:303-314. [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000, 403, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.J.; Beard, S.; Binder, N.K.; Onda, K.; Kaitu'u-Lino, T.J.; Chen, Q.; Tuohey, L.; De Silva, M.; Tong, S. Key players of the necroptosis pathway RIPK1 and SIRT2 are altered in placenta from preeclampsia and fetal growth restriction. Placenta. 2017, 51: 1-9. [CrossRef]
- Yu, Y.; An, X.; Fan, D. Histone Deacetylase Sirtuin 2 Enhances Viability of Trophoblasts Through p65-Mediated MicroRNA-146a/ACKR2 Axis. Reprod Sci. 2021, 28, 1370–1381. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, Y.; Liu, M.; Liao, L.; Wei, X.; Zhou, R. SIRT3 deficiency affects the migration, invasion, tube formation and necroptosis of trophoblast and is implicated in the pathogenesis of preeclampsia. Placenta. 2022, 120:1-9. [CrossRef]
- Castex, J.; Willmann, D.; Kanouni, T.; Arrigoni, L.; Li, Y.; Friedrich, M.; Schleicher, M.; Wöhrle, S.; Pearson, M.; Kraut, N.; Méret, M.; Manke, T.; Metzger, E.; Schüle, R.; Günther, T. Inactivation of Lsd1 triggers senescence in trophoblast stem cells by induction of Sirt4. Cell Death Dis. 2017, 8, e2631. [Google Scholar] [CrossRef]
- Sandvoß, M.; Potthast, A.B.; von Versen-Höynck, F.; Das, A.M. HELLP Syndrome. Reprod Sci. 2017, 24, 568–574. [Google Scholar] [CrossRef]
- Bartho, L.A.; O'Callaghan, J.L.; Fisher, J.J. ; Cuffe JSM, Kaitu'u-Lino, T.J.; Hannan, N.J.; Clifton, V.L.; Perkins, A.V. Analysis of mitochondrial regulatory transcripts in publicly available datasets with validation in placentae from pre-term, post-term and fetal growth restriction pregnancies. Placenta. 2021, 112:162-171. [CrossRef]
- Park, K.W.; Halperin, D.S.; Tontonoz, P. Before they were fat: adipocyte progenitors. Cell Metab. 2008, 8, 454–7. [Google Scholar] [CrossRef] [PubMed]
- Koppen, A.; Kalkhoven, E. Brown vs white adipocytes: the PPARgamma coregulator story. FEBS Lett. 2010, 584, 3250–9. [Google Scholar] [CrossRef] [PubMed]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–86. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004, 429, 771–6. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.; Milner, J. SIRT1, metabolism and cancer. Curr Opin Oncol. 2012, 24, 68–75. [Google Scholar] [CrossRef]
- Simmons GE Jr, Pruitt, W. M.; Pruitt, K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int J Mol Sci. 2015, 16, 950–65. [CrossRef]
- Farghali, H.; Kutinová Canová, N.; Lekić, N. Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol Res. 2013, 62, 1–13. [Google Scholar] [CrossRef]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; Accili, D. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell. 2012, 150, 620–32. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–71. [Google Scholar] [CrossRef]
- Wei, S.; Kulp, S.K.; Chen, C.S. Energy restriction as an antitumor target of thiazolidinediones. J Biol Chem. 2010, 285, 9780–9791. [Google Scholar] [CrossRef] [PubMed]
- Calleri, E.; Pochetti, G. ; Dossou KSS, Laghezza, A. ; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; Moaddel, R. Resveratrol and its metabolites bind to PPARs. Chembiochem. 2014, 15, 1154–1160. [Google Scholar] [CrossRef]
- Fisher, S.J. Why is placentation abnormal in preeclampsia? Am J Obstet Gynecol. 2015, 213(4 Suppl):S115-22. [CrossRef]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol. 2014, 72, 107–16. [Google Scholar] [CrossRef]
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta. 2015, 36, 101–14. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Wakeland, A.K.; Parast, M.M. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J Endocrinol. 2018, 236, R43–R56. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Adelman, D.M.; Gertsenstein, M.; Nagy, A.; Simon, M.C.; Maltepe, E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000, 14, 3191–203. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Krampitz, G.W.; Okazaki, K.M.; Red-Horse, K.; Mak, W.; Simon, M.C.; Fisher, S.J. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development. 2005, 132, 3393–403. [Google Scholar] [CrossRef] [PubMed]
- Wakeland, A.K.; Soncin, F.; Moretto-Zita, M.; Chang, C.W.; Horii, M.; Pizzo, D.; Nelson, K.K.; Laurent, L.C.; Parast, M.M. Hypoxia Directs Human Extravillous Trophoblast Differentiation in a Hypoxia-Inducible Factor-Dependent Manner. Am J Pathol. 2017, 187, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Maecker, H.L.; Johnson, R.S.; Giaccia, A.J. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell. 2002, 2, 331–41. [Google Scholar] [CrossRef] [PubMed]
- Tache, V.; Ciric, A.; Moretto-Zita, M.; Li, Y.; Peng, J.; Maltepe, E.; Milstone, D.S.; Parast, M.M. Hypoxia and trophoblast differentiation: a key role for PPARγ. Stem Cells Dev. 2013, 22, 2815–24. [Google Scholar] [CrossRef]
- He, P.; Chen, Z.; Sun, Q.; Li, Y.; Gu, H.; Ni, X. Reduced expression of 11β-hydroxysteroid dehydrogenase type 2 in preeclamptic placentas is associated with decreased PPARγ but increased PPARα expression. Endocrinology. 2014, 155, 299–309. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, C.Y.; Yang, Y.C.; Su, T.H.; Chen, H. Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta. 2004, 25, 413–21. [Google Scholar] [CrossRef]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced Syncytin and increased apoptosis in patients with placental dysfunction. Mol Reprod Dev. 2008, 75, 175–83. [Google Scholar] [CrossRef] [PubMed]
- Baczyk, D.; Drewlo, S.; Proctor, L.; Dunk, C.; Lye, S.; Kingdom, J. Glial cell missing-1 transcription factor is required for the differentiation of the human trophoblast. Cell Death Differ. 2009, 16, 719–27. [Google Scholar] [CrossRef] [PubMed]
- Karumanchi, S.A.; Epstein, F.H. Placental ischemia and soluble fms-like tyrosine kinase 1: cause or consequence of preeclampsia? Kidney Int. 2007, 71, 959–61. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: an implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology. 2004, 145, 4838–45. [Google Scholar] [CrossRef]
- Li, H.; Gu, B.; Zhang, Y.; Lewis, D.F.; Wang, Y. Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta. 2005, 26(2-3):210-7. [CrossRef]
- Nevo, O.; Soleymanlou, N.; Wu, Y.; Xu, J.; Kingdom, J.; Many, A.; Zamudio, S.; Caniggia, I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol. 2006, 291, R1085–93. [Google Scholar] [CrossRef] [PubMed]
- Munaut, C.; Lorquet, S.; Pequeux, C.; Blacher, S.; Berndt, S.; Frankenne, F.; Foidart, J.M. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum Reprod. 2008, 23, 1407–15. [Google Scholar] [CrossRef] [PubMed]
- Taché, V.; LaCoursiere, D.Y.; Saleemuddin, A.; Parast, M.M. Placental expression of vascular endothelial growth factor receptor-1/soluble vascular endothelial growth factor receptor-1 correlates with severity of clinical preeclampsia and villous hypermaturity. Hum Pathol. 2011, 42, 1283–8. [Google Scholar] [CrossRef]
- Huppertz, B.; Abe, E.; Murthi, P.; Nagamatsu, T.; Szukiewicz, D.; Salafia, C. Placental angiogenesis, maternal and fetal vessels--a workshop report. Placenta. 2007 Apr;28 Suppl A:S94-6. [CrossRef]
- McCarthy, F.P.; Drewlo, S.; English, F.A.; Kingdom, J.; Johns, E.J.; Kenny, L.C.; Walsh, S.K. Evidence implicating peroxisome proliferator-activated receptor-γ in the pathogenesis of preeclampsia. Hypertension. 2011, 58, 882–7. [Google Scholar] [CrossRef]
- Bainbridge, S.A.; Minhas, A.; Whiteley, K.J.; Qu, D.; Sled, J.G.; Kingdom, J.C.; Adamson, S.L. Effects of reduced Gcm1 expression on trophoblast morphology, fetoplacental vascularity, and pregnancy outcomes in mice. Hypertension. 2012, 59, 732–9. [Google Scholar] [CrossRef]
- Leung, D.N.; Smith, S.C.; To, K.F.; Sahota, D.S.; Baker, P.N. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol. 2001, 184, 1249–50. [Google Scholar] [CrossRef]
- Nelson, D.M.; Johnson, R.D.; Smith, S.D.; Anteby, E.Y.; Sadovsky, Y. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am J Obstet Gynecol. 1999, 180, 896–902. [Google Scholar] [CrossRef]
- Elchalal, U.; Humphrey, R.G.; Smith, S.D.; Hu, C.; Sadovsky, Y.; Nelson, D.M. Troglitazone attenuates hypoxia-induced injury in cultured term human trophoblasts. Am J Obstet Gynecol. 2004, 191, 2154–9. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010, 38, 864–78. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009, 324, 1289–93. [Google Scholar] [CrossRef]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem. 2011, 286, 13869–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Nelson, D.M.; Sadovsky, Y. N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J Biol Chem. 2006, 281, 2764–72. [Google Scholar] [CrossRef]
- Broady, A.J.; Loichinger, M.H.; Ahn, H.J.; Davy, P.M.; Allsopp, R.C.; Bryant-Greenwood, G.D. Protective proteins and telomere length in placentas from patients with pre-eclampsia in the last trimester of gestation. Placenta. 2017, 50:44-52. [CrossRef]
- Cudmore, M.J.; Ramma, W.; Cai, M.; Fujisawa, T.; Ahmad, S.; Al-Ani, B.; Ahmed, A. Resveratrol inhibits the release of soluble fms-like tyrosine kinase (sFlt-1) from human placenta. Am J Obstet Gynecol. 2012, 206, 253–e10. [Google Scholar] [CrossRef]
- Hannan, N.J.; Brownfoot, F.C.; Cannon, P.; Deo, M.; Beard, S.; Nguyen, T.V.; Palmer, K.R.; Tong, S.; Kaitu'u-Lino, T.J. Resveratrol inhibits release of soluble fms-like tyrosine kinase (sFlt-1) and soluble endoglin and improves vascular dysfunction - implications as a preeclampsia treatment. Sci Rep. 2017, 7, 1819. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zuo, Q.; Huang, S.; Yu, X.; Jiang, Z.; Zou, S.; Fan, M.; Sun, L. Resveratrol inhibits trophoblast apoptosis through oxidative stress in preeclampsia-model rats. Molecules. 2014, 19, 20570–9. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009, 30, 473–82. [Google Scholar] [CrossRef]
- Meher, A.P.; Joshi, A.A.; Joshi, S.R. Maternal micronutrients, omega-3 fatty acids, and placental PPARγ expression. Appl Physiol Nutr Metab. 2014, 39, 793–800. [Google Scholar] [CrossRef]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci. 2015, 16, 4600–14. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.R.; Tong, S.; Kaitu'u-Lino, T.J. Placental-specific sFLT-1: role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol Hum Reprod. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int J Mol Sci. 2022, 23, 15105. [Google Scholar] [CrossRef]
- Bokuda, K.; Ichihara, A. Preeclampsia up to date-What's going on? Hypertens Res. 2023, 46, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Yagel, S.; Verlohren, S. Role of placenta in development of pre-eclampsia: revisited. Ultrasound Obstet Gynecol. 2020, 56, 803–808. [Google Scholar] [CrossRef]
- Hu, M.; Li, J.; Baker, P.N.; Tong, C. Revisiting preeclampsia: a metabolic disorder of the placenta. FEBS J. 2022, 289, 336–354. [Google Scholar] [CrossRef]
- Hadley, E.E.; Richardson, L.S.; Torloni, M.R.; Menon, R. Gestational tissue inflammatory biomarkers at term labor: A systematic review of literature. Am J Reprod Immunol. 2018, 79, 10–1111. [Google Scholar] [CrossRef]
- Kim, S.H.; Shim, S.H.; Sung, S.R.; Lee, K.A.; Shim, J.Y.; Cha, D.H.; Lee, K.J. Gene expression analysis of the microdissected trophoblast layer of human placenta after the spontaneous onset of labor. PLoS One. 2013, 8, e77648. [Google Scholar] [CrossRef]
- Tsai, P.J.; Davis, J.; Thompson, K.; Bryant-Greenwood, G. Visfatin/Nampt and SIRT1: Roles in Postterm Delivery in Pregnancies Associated With Obesity. Reprod Sci. 2015, 22, 1028–36. [Google Scholar] [CrossRef]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L. ; Cunningham MW Jr, Wallace, K. ; LaMarca, B. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond). 2016, 130, 409–19. [Google Scholar] [CrossRef]
- Doshani, A.; Konje, J.C. Placental dysfunction in obese women and antenatal surveillance. Best Pract Res Clin Obstet Gynaecol. 2023, 91:102407. [CrossRef]
- Meher, A.P.; Joshi, A.A.; Joshi, S.R. Maternal micronutrients, omega-3 fatty acids, and placental PPARγ expression. Appl Physiol Nutr Metab. 2014, 39, 793–800. [Google Scholar] [CrossRef]
- Bo, Q.L.; Chen, Y.H.; Yu, Z.; Fu, L.; Zhou, Y.; Zhang, G.B.; Wang, H.; Zhang, Z.H.; Xu, D.X. Rosiglitazone pretreatment protects against lipopolysaccharide-induced fetal demise through inhibiting placental inflammation. Mol Cell Endocrinol. 2016, 423:51-9. [CrossRef]
- Wang, L.L.; Yu, Y.; Guan, H.B.; Qiao, C. Effect of Human Umbilical Cord Mesenchymal Stem Cell Transplantation in a Rat Model of Preeclampsia. Reprod Sci. 2016, 23, 1058–70. [Google Scholar] [CrossRef]
- Leon-Garcia, S.M.; Roeder, H.A.; Nelson, K.K.; Liao, X.; Pizzo, D.P.; Laurent, L.C.; Parast, M.M.; LaCoursiere, D.Y. Maternal obesity and sex-specific differences in placental pathology. Placenta. 2016, 38:33-40. [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; Banks, A.S.; Qiang, L.; Bhanot, S.; Olefsky, J.M.; Sears, D.D.; Caprio, S.; Shulman, G.I. SirT1 regulates adipose tissue inflammation. Diabetes. 2011, 60, 3235–45. [Google Scholar] [CrossRef]
- Qiao, L.; Guo, Z.; Bosco, C.; Guidotti, S.; Wang, Y.; Wang, M.; Parast, M.; Schaack, J. ; Hay WW Jr, Moore, T. R.; Shao, J. Maternal High-Fat Feeding Increases Placental Lipoprotein Lipase Activity by Reducing SIRT1 Expression in Mice. Diabetes. 2015, 64, 3111–20. [Google Scholar] [CrossRef]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am J Obstet Gynecol. 2014, 211, 541–e1. [Google Scholar] [CrossRef]
- Suwaki, N.; Masuyama, H.; Masumoto, A.; Takamoto, N.; Hiramatsu, Y. Expression and potential role of peroxisome proliferator-activated receptor gamma in the placenta of diabetic pregnancy. Placenta. 2007, 28, 315–23. [Google Scholar] [CrossRef]
- Jawerbaum, A.; Capobianco, E.; Pustovrh, C.; White, V.; Baier, M.; Salzberg, S.; Pesaresi, M.; Gonzalez, E. Influence of peroxisome proliferator-activated receptor gamma activation by its endogenous ligand 15-deoxy Delta12,14 prostaglandin J2 on nitric oxide production in term placental tissues from diabetic women. Mol Hum Reprod. 2004, 10, 671–6. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth-Carson, S.J.; Lim, R.; Mitton, A.; Whitehead, C.; Rice, G.E.; Permezel, M.; Lappas, M. Peroxisome proliferator-activated receptors are altered in pathologies of the human placenta: gestational diabetes mellitus, intrauterine growth restriction and preeclampsia. Placenta. 2010, 31, 222–9. [Google Scholar] [CrossRef] [PubMed]
- Knabl, J.; Hüttenbrenner, R.; Hutter, S.; Günthner-Biller, M.; Vrekoussis, T.; Karl, K.; Friese, K.; Kainer, F.; Jeschke, U. Peroxisome proliferator-activated receptor-gamma (PPARγ) is down regulated in trophoblast cells of gestational diabetes mellitus (GDM) and in trophoblast tumour cells BeWo in vitro after stimulation with PPARγ agonists. J Perinat Med. 2014, 42, 179–87. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; Fornes, D.; Linenberg, I.; Powell, T.L.; Jansson, T.; Jawerbaum, A. A novel rat model of gestational diabetes induced by intrauterine programming is associated with alterations in placental signaling and fetal overgrowth. Mol Cell Endocrinol. 2016, 422:221-232. [CrossRef]
- Viana-Mattioli, S.; Nunes, P.; Cavalli, R.; Sandrim, V. Analysis of SIRT1 Expression in Plasma and in an In Vitro Model of Preeclampsia. Oxid Med Cell Longev. 2020, 2020:4561083. [CrossRef]
- Yin, Y.; Feng, Y.; Zhao, H.; Zhao, Z.; Yua, H.; Xu, J.; Che, H. SIRT1 inhibits releases of HMGB1 and HSP70 from human umbilical vein endothelial cells caused by IL-6 and the serum from a preeclampsia patient and protects the cells from death. Biomed Pharmacother. 2017, 88:449-458. [CrossRef]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; Hamidane, H.B.; Elrayess, M.A.; Rafii, A.; Graumann, J.; Schmidt, F.; Mazloum, N.A. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci Rep. 2021 ;11, 8177. [CrossRef]
- Hopp, A.K.; Grüter, P.; Hottiger, M.O. Erratum: Hopp, A. K., et al. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890, Erratum for: Cells. 2019, 8(8)11. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Baker, J.R.; Vuppusetty, C.; Koga, T.; Colley, T.; Fenwick, P.; Donnelly, L.E.; Barnes, P.J.; Ito, K. The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS One. 2018, 13, e0193921. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants (Basel). 2021, 10, 1838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid Med Cell Longev. 2017, 2017:7543973. [CrossRef]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic Biol Med. 2021, 166:238-254. [CrossRef]
- Giannakou, M.E.; Partridge, L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004, 14, 408–12. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int J Mol Med. 2005, 16, 237–43. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhang, H.; Zhang, B.; Zhang, E.; Wu, Y. Tumor Necrosis Factor-alpha (TNF-α) Enhances miR-155-Mediated Endothelial Senescence by Targeting Sirtuin1 (SIRT1). Med Sci Monit. 2019, 25:8820-8835. [CrossRef]
- Guo, Y.; Chao, L.; Chao, J. Kallistatin attenuates endothelial senescence by modulating Let-7g-mediated miR-34a-SIRT1-eNOS pathway. J Cell Mol Med. 2018, 22, 4387–4398. [Google Scholar] [CrossRef]
- Ming, G.F.; Wu, K.; Hu, K.; Chen, Y.; Xiao, J. NAMPT regulates senescence, proliferation, and migration of endothelial progenitor cells through the SIRT1 AS lncRNA/miR-22/SIRT1 pathway. Biochem Biophys Res Commun. 2016, 478, 1382–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, D.; Zhang, N.; Yuan, T.; Tao, H. MiR-217 promotes endothelial cell senescence through the SIRT1/p53 signaling pathway. J Mol Histol. 2021, 52, 257–267. [Google Scholar] [CrossRef]
- Chen, L.; Holder, R.; Porter, C.; Shah, Z. Vitamin D3 attenuates doxorubicin-induced senescence of human aortic endothelial cells by upregulation of IL-10 via the pAMPKα/Sirt1/Foxo3a signaling pathway. PLoS One. 2021, 16, e0252816. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, J.; Fu, M.; Dong, R.; Yang, Y.; Luo, J.; Hu, S.; Li, W.; Xu, X.; Tu, L. Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem Pharmacol. 2020, 177:113951. [CrossRef]
- Duan, J.L.; Ruan, B.; Song, P.; Fang, Z.Q.; Yue, Z.S.; Liu, J.J.; Dou, G.R.; Han, H.; Wang, L. Shear stress-induced cellular senescence blunts liver regeneration through Notch-sirtuin 1-P21/P16 axis. Hepatology. 2022, 75, 584–599. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim Biophys Acta. 2014, 1840, 2709–29. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front Cardiovasc Med. 2020, 7:12. [CrossRef]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R. ; de Bittencourt PI Jr, Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxid Med Cell Longev. 2015, 2015:181643. [CrossRef]
- Volpe CMO, Villar-Delfino, P. H.; Dos Anjos PMF, Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Kowluru, R.A. Sirt1: A Guardian of the Development of Diabetic Retinopathy. Diabetes. 2018, 67, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic retinopathy. Biomed Pharmacother. 2018, 97:190-194. [CrossRef]
- Kume, S.; Uzu, T.; Kashiwagi, A.; Koya, D. SIRT1, a calorie restriction mimetic, in a new therapeutic approach for type 2 diabetes mellitus and diabetic vascular complications. Endocr Metab Immune Disord Drug Targets. 2010, 10, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2009, 29, 889–94. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Eto, M.; Kano, M.R.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler Thromb Vasc Biol. 2010, 30, 2205–11. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose reductase regulates hyperglycemia-induced HUVEC death via SIRT1/AMPK-α1/mTOR pathway. J Mol Endocrinol. 2019, 63, 11–25. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, J.; Chen, G.; Niu, C.; Wang, Y.; Zhao, C.; Sun, J.; Huang, H.; Huang, S.; Liang, Y.; Shen, Y.; Cong, W.; Jin, L.; Zhu, Z. Resveratrol Promotes Diabetic Wound Healing via SIRT1-FOXO1-c-Myc Signaling Pathway-Mediated Angiogenesis. Front Pharmacol. 2019, 10:421. [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur Heart, J. 2012, 33, 829–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Peng, I.C.; Sun, W.; Su, M.I.; Hsu, P.H.; Fu, Y.; Zhu, Y.; DeFea, K.; Pan, S.; Tsai, M.D.; Shyy, J.Y. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ Res. 2009, 104, 496–505. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Wallace, K. Autophagy in preeclampsia: A new target? EBioMedicine. 2020, 57:102864. [CrossRef]
- Wu, Q.; Hu, Y.; Jiang, M.; Wang, F.; Gong, G. Effect of Autophagy Regulated by Sirt1/FoxO1 Pathway on the Release of Factors Promoting Thrombosis from Vascular Endothelial Cells. Int J Mol Sci. 2019, 20, 4132. [Google Scholar] [CrossRef]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004, 101, 10042–7. [Google Scholar] [CrossRef]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 Functions in Mammalian Autophagy: Rab7 Knockout Studies. Cells. 2018, 7, 215. [Google Scholar] [CrossRef]
- Takeda-Watanabe, A.; Kitada, M.; Kanasaki, K.; Koya, D. SIRT1 inactivation induces inflammation through the dysregulation of autophagy in human THP-1 cells. Biochem Biophys Res Commun. 2012, 427, 191–6. [Google Scholar] [CrossRef]
- Ma, Y.N.; Jiang, X.; Tang, W.; Song, P. Influence of intermittent fasting on autophagy in the liver. Biosci Trends. 2023. [CrossRef] [PubMed]
- Heo, J.I.; Kim, K.I.; Woo, S.K.; Kim, J.S.; Choi, K.J.; Lee, H.J.; Kim, K.S. Stromal Cell-Derived Factor 1 Protects Brain Vascular Endothelial Cells from Radiation-Induced Brain Damage. Cells. 2019, 8, 1230. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics. 2014, 41, 485–95. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Maeng, Y.S.; Park, Y.W.; Koos, B.J.; Kwon, Y.G.; Kim, Y.H. Increased senescence and reduced functional ability of fetal endothelial progenitor cells in pregnancies complicated by preeclampsia without intrauterine growth restriction. Am J Obstet Gynecol. 2008, 199, 259–e1. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, J.; Mitsui-Saito, M.; Hayashi, C.; Hoshiai, T.; Senoo, M.; Chisaka, H.; Yaegashi, N.; Okamura, K. Decrease and senescence of endothelial progenitor cells in patients with preeclampsia. J Clin Endocrinol Metab. 2005, 90, 5329–32. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci Rep. 2015, 5:15971. [CrossRef]
- Holmlund, U.; Wähämaa, H.; Bachmayer, N.; Bremme, K.; Sverremark-Ekström, E.; Palmblad, K. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology. 2007, 122, 430–7. [Google Scholar] [CrossRef]
- Jiang, R.; Cai, J.; Zhu, Z.; Chen, D.; Wang, J.; Wang, Q.; Teng, Y.; Huang, Y.; Tao, M.; Xia, A.; Xue, M.; Zhou, S.; Chen, A.F. Hypoxic trophoblast HMGB1 induces endothelial cell hyperpermeability via the TRL-4/caveolin-1 pathway. J Immunol. 2014, 193, 5000–12. [Google Scholar] [CrossRef]
- Lai, H.; Nie, L.; Zeng, X.; Xin, S.; Wu, M.; Yang, B.; Luo, Y.; Liu, B.; Zheng, J.; Liu, H. Enhancement of heat shock protein 70 attenuates inducible nitric oxide synthase in preeclampsia complicated with fetal growth restriction. J Matern Fetal Neonatal Med. 2022, 35, 2555–2563. [Google Scholar] [CrossRef]
- Peraçoli, J.C.; Bannwart-Castro, C.F.; Romao, M.; Weel, I.C.; Ribeiro, V.R.; Borges, V.T.; Rudge, M.V.; Witkin, S.S.; Peraçoli, M.T. High levels of heat shock protein 70 are associated with pro-inflammatory cytokines and may differentiate early- from late-onset preeclampsia. J Reprod Immunol. 2013, 100, 129–34. [Google Scholar] [CrossRef]
- Molvarec, A.; Szarka, A.; Walentin, S.; Beko, G.; Karádi, I.; Prohászka, Z.; Rigó J, Jr. Serum heat shock protein 70 levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in women with preeclampsia. Clin Chim Acta. 2011, 412(21-22):1957-62. [CrossRef]
- Watanabe, S.; Ageta-Ishihara, N.; Nagatsu, S.; Takao, K.; Komine, O.; Endo, F.; Miyakawa, T.; Misawa, H.; Takahashi, R.; Kinoshita, M.; Yamanaka, K. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol Brain. 2014, 7:62. [CrossRef]
- Gu, X.; Gu, B.; Lv, X.; Yu, Z.; Wang, R.; Zhou, X.; Qiao, W.; Mao, Z.; Zuo, G.; Li, Q.; Miao, D.; Jin, J. 1, 25-dihydroxy-vitamin D3 with tumor necrosis factor-alpha protects against rheumatoid arthritis by promoting p53 acetylation-mediated apoptosis via Sirt1 in synoviocytes. Cell Death Dis. 2016, 7, e2423. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, X.D.; Li, H. Protective role of SIRT1-mediated Sonic Hedgehog signaling pathway in the preeclampsia rat models. J Assist Reprod Genet. 2021, 38, 1843–1851. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin, No. 202: Gestational Hypertension and Preeclampsia. Obstet Gynecol. 2019, 133, 1. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003, 100, 10794–9. [Google Scholar] [CrossRef]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003, 23, 38–54. [Google Scholar] [CrossRef]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; Jia, R.; Zheng, Z.M.; Appella, E.; Wang, X.W.; Ried, T.; Deng, C.X. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008, 14, 312–23. [Google Scholar] [CrossRef]
- Xiong, L.; Ye, X.; Chen, Z.; Fu, H.; Li, S.; Xu, P.; Yu, J.; Wen, L.; Gao, R.; Fu, Y.; Qi, H.; Kilby, M.D.; Saffery, R.; Baker, P.N.; Tong, C. Advanced Maternal Age-associated SIRT1 Deficiency Compromises Trophoblast Epithelial-Mesenchymal Transition through an Increase in Vimentin Acetylation. Aging Cell. 2021, 20, e13491. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech Ageing Dev. 2020, 187:111215. [CrossRef]
- Taskin, I.I.; Gurbuz, S.; Icen, M.S.; Derin, D.C.; Findik, F.M. Expression of sirtuin 2 and 7 in placenta accreta spectrum. Rev Assoc Med Bras (1992). 2023, 69, e20230360. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, X.; Xu, X.; Yan, J. Placenta-derived exosomal miR-135a-5p promotes gestational diabetes mellitus pathogenesis by activating PI3K/AKT signalling pathway via SIRT1. J Cell Mol Med. 2023. [CrossRef]
- Su, S.; Zhong, L.; Huang, S.; Deng, L.; Pang, L. MiRNA-494 induces trophoblast senescence by targeting SIRT1. Hypertens Pregnancy. 2023, 42, 2219774. [Google Scholar] [CrossRef]
- Liu, Z.; Pei, J.; Zhang, X.; Wang, C.; Tang, Y.; Liu, H.; Yu, Y.; Luo, S.; Gu, W. CD74 deficiency reduces trophoblast invasion and proliferation mediated by SIRT1 in preeclampsia. Reproduction. 2023:REP-23-0202. [CrossRef]
- Deodati, A.; Inzaghi, E.; Cianfarani, S. Epigenetics and In Utero Acquired Predisposition to Metabolic Disease. Front Genet. 2020, 10:1270. [CrossRef]
- Zhang, T.; Kraus, W.L. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. 2010 ;1804, 1666-75. [CrossRef]
- Fortuny, L.; Sebastián, C. Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function. Genes (Basel). 2021, 12, 1698. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front Immunol. 2022, 13:831168. [CrossRef]
- Tamura, N.; Heidari, N. ; Faragher RGA, Smith RKW, Dudhia, J. Effects of resveratrol and its analogues on the cell cycle of equine mesenchymal stem/stromal cells. J Equine Sci. 2023, 34, 67–72. [Google Scholar] [CrossRef]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front Physiol. 2021, 12:724506. [CrossRef]
- Aksu, K.; Golal, E.; Aslan, M.A.; Ustunel, I.; Acar, N. The investigation of the role of sirtuin-1 on embryo implantation in oxidative stress-induced mice. J Assist Reprod Genet. 2021, 38, 2349–2361. [Google Scholar] [CrossRef]
- Sande, A.K.; Dalen, I.; Torkildsen, E.A.; Sande, R.K.; Morken, N.H. Pregestational maternal risk factors for preterm and term preeclampsia: A population-based cohort study. Acta Obstet Gynecol Scand. 2023. [CrossRef]
- Wang, W.; Xie, X.; Yuan, T.; Wang, Y.; Zhao, F.; Zhou, Z.; Zhang, H. Epidemiological trends of maternal hypertensive disorders of pregnancy at the global, regional, and national levels: a population-based study. BMC Pregnancy Childbirth. 2021, 21, 364. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of the beneficial effect of the natural sirtuin-1 activator, resveratrol, at the level of the endothelium and trophoblast under oxidative stress (e.g. accompanying preeclampsia) [7,99,100,101,102,103].

Figure 2.
Beneficial effects of SIRT1 on vascular endothelial cells in preeclampsia: limiting oxidative stress, inflammatory response and cellular aging.
Figure 2.
Beneficial effects of SIRT1 on vascular endothelial cells in preeclampsia: limiting oxidative stress, inflammatory response and cellular aging.
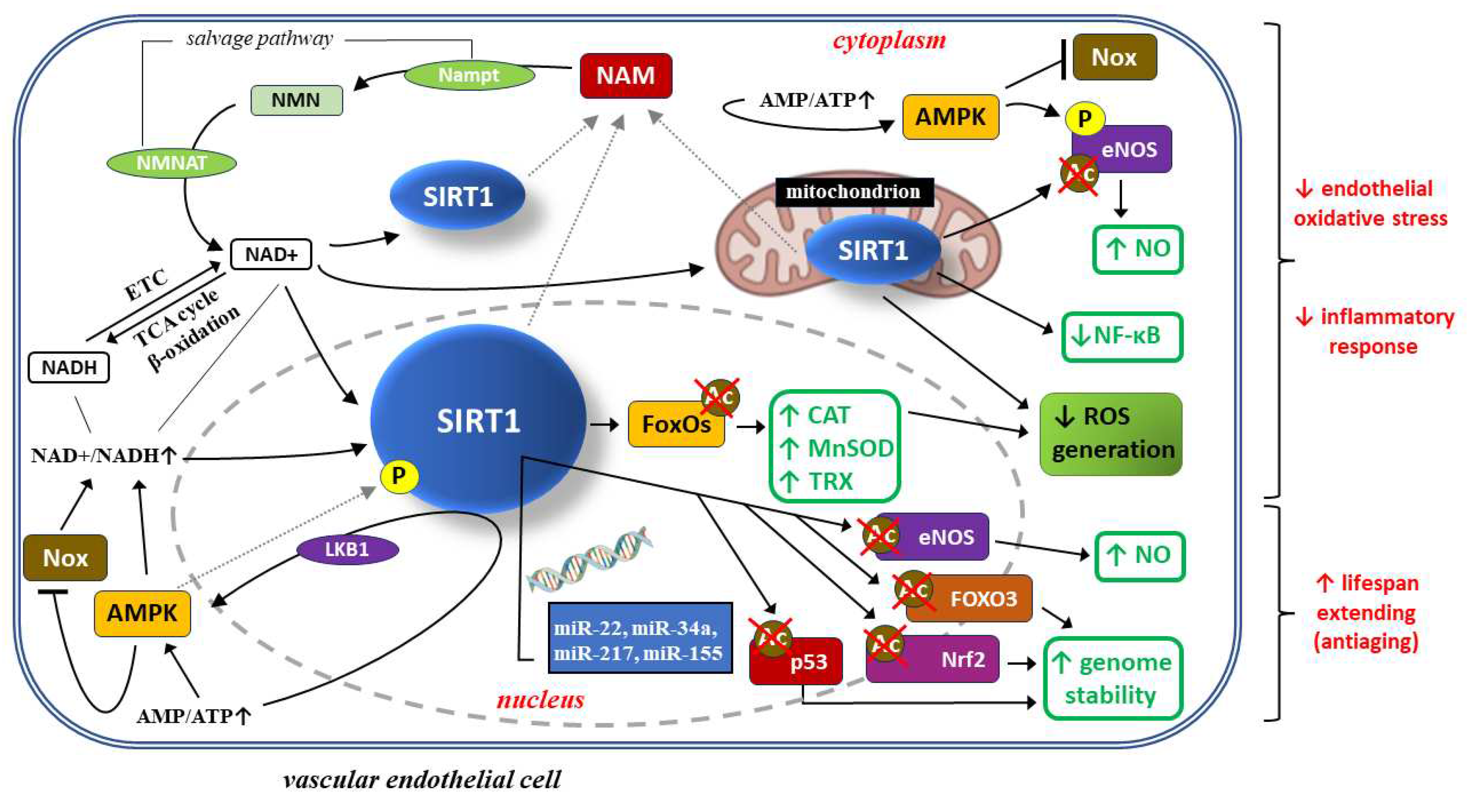
Figure 3.
Mechanisms of action of SIRT1 towards maintaining the homeostasis of both endothelial cells and trophoblast by promoting autophagy.
Figure 3.
Mechanisms of action of SIRT1 towards maintaining the homeostasis of both endothelial cells and trophoblast by promoting autophagy.
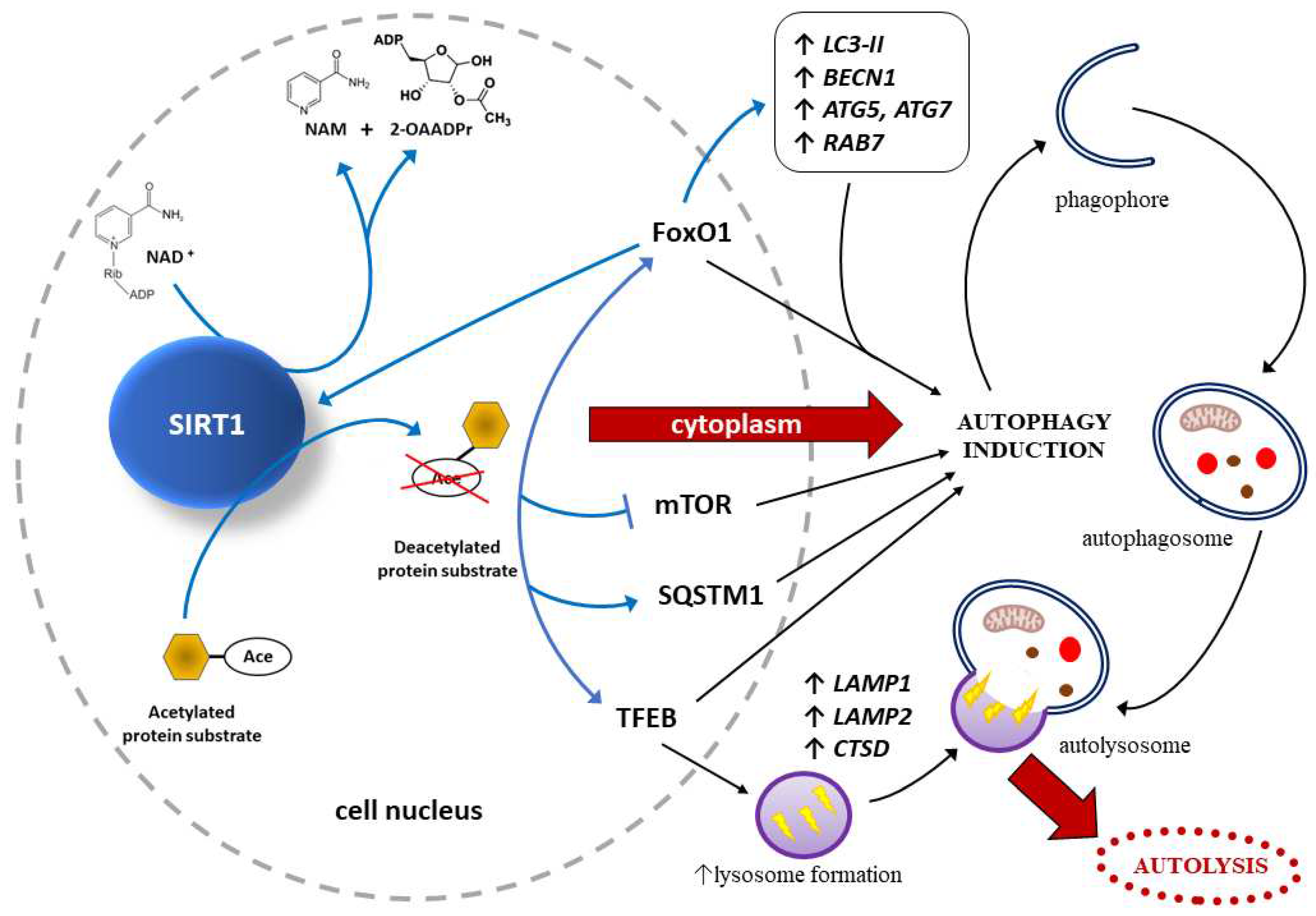
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



