
Submitted:
12 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
This work is the first part of a series of papers on a comprehensive analysis of the processes of prebiotic self-organization and protophotosynthesis on the surface of semiconductor minerals and in systems of natural dispersed semiconductors. Comprehensive analysis within the framework of the "semiconductor world" concept allows integrating a variety of models on a single physical basis - from ZnS world and FeS world (based on inorganic semiconductors) to the PAH world and aromatic world (including organic semiconductors). Thus, we do not put forward a new alternative hypothesis of chemical evolution - a new "chemical world", but only integrate the evolutionary and geochemical criteria of different "chemical worlds" into a single "physical world", which gives one the opportunity to reconstruct and predict the directions of chemical evolution according to the uniform principles of physical chemistry. In the first part of this series we consider photoinduced self-organization and "photo-controlled" evolution of the early protobiological systems performing solar energy conversion on the surface of dispersed semiconductor minerals capable of (photo)catalyzing and photosensitizing prebiological processes. The latter include a transition from the elementary cycles of photocatalytic reactions on the surface of semiconductors to protophotometabolic cycles and protophotosynthesis, from photophysical processes on the surface of mineral semiconductors to photoinduced membrane potentials, from photocontrolled sorption on the surface of such minerals to the formation of photosensitive protomembranes and selection of photosynthetic structures. The evolutionary approach to the analysis of protobiological mechanisms and protobioenergetics within the framework of the "semiconductor world" concept provides a new approach to the study of the last common point of divergence of protobiological systems, where the emergence principles of different energy supply schemes (like the energy source-specific photoautotrophy and substrate-specific chemoautotrophy) merge at the physicochemical level. The proposed integrating scheme is consistent with most biological, geological, and physicochemical concepts, which ensures its complete cross-checking and internal consistency.
Keywords:
semiconductor minerals
; photosynthesis
; photocatalysis
; origin of life
; protocells
; self-assembly
; self-organization
; photoinduced processes
; protometabolism
; protomembranes.
Preface.
This review is the first introductory part of a series of papers planned to be published by the authors since the 2010s during the period of their work at the geochemical institute and later in the photobiological / photobiomimetic laboratory (engaged in research on photosynthesis and the development of biomimetic photovoltaic cells) at the Institute of Chemical Physics of the Russian Academy of Sciences until its reorganization at the end of the 2010s. At present, the full implementation of this work is impossible within the Russian Federation, due to the merger of institutes and laboratories, and the focus of most research projects on the applied problems rather than on the fundamental science. In this regard, the only opportunity for the authors to continue this work is to publish or deposit as a preprint their main concepts and preliminary results obtained earlier in order to ensure the possibility of verifying their ideas by the independent researchers throughout the world, as well as to establish direct communication and further collaboration with the foreign colleagues interested in the development of the above ideas.
This manuscript was initially written at the invitation of the journal Life in 2020 for the cancelled special issue "Origin and Early Evolution of Photosynthesis". The impossibility of submitting the resulting text containing more than 200 pages (excluding illustrations) required its further revision and division into three parts. Therefore, the second version of this manuscript was prepared at the invitation of the same journal in January 2022 for the Topical Collection "Feature Review Papers for Life" edited by Prof. Michael Russell. However, the submission of the final manuscript, unfortunately, coincided with the beginning of the Russian aggression against Ukraine in February 2022, which prevented the authors from submitting the manuscript on time. Finally, the first part of this manuscript was resubmitted at the repeated invitation from Life to the Topical Collection "Feature Review Papers for Life" in June 2023, and since then it is still under review. Thus, several months have passed since the first submission of this manuscript and the authors decided to deposit its original text as a preprint, since with the increasing delay time of its publication the manuscript may loose certain points of novelty (since 2020, a number of similar ideas have already been expressed or verified by the other authors).
Unfortunately, there is almost no opportunity for the authors to continue their work on this topic in the nearest future both in Russia, where academic science is rapidly degrading, and abroad, due to the political situation (since all the attempts of the authors to join the foreign groups studying the origin of life problem within the EU and US failed due to the sanctions and restrictions on cooperation with the Russian citizens regardless of their civic stance). Therefore, in the current circumstances, we can only convey the main ideas of our concept to the international readership and hope that they will be interesting to someone who can provide their further verification and development. We are also opened for any feedback, suggestions and comments from the origin of life community on the improvement of the ideas described in this and in the forthcoming papers, and we are certainly ready for any type of cooperation with those colleagues who are ready to provide us the opportunity to continue this work within their research groups. Fais ce que dois, advienne, que pourra.
1. Plausibility of the protophotosynthesis models depends on the definition of photosynthesis.
The emergence of primary photosynthesis mechanisms is an integral part of the chemical evolution, and hence, should not be studied separately from the geochemical (or astrochemical) conditions of the corresponding processes of chemical evolution. The direction of natural selection, according to the general principles of chemical evolutionary processes [1,2], depends on the conditions in which it occurs, while the nature of structures participating and winning in this process strictly depends on the medium and the selection criteria. Accordingly, the emergence of photosynthesis as a product of long molecular evolution, which began long before the formation of its modern biochemical forms, should be understood not only from the “reverse engineering of molecular biological machinery” of its modern forms, but, above all, from a specific analysis of natural conditions for the processes of chemical evolution in different periods of geological or space time. The subject of this paper is not the emergence of specific biochemical forms of the photosynthetic process, which is investigated by many reputable research teams, but the physical and chemical reasons for the emergence and the evolutionary path of the physical principle itself, which underlies the assimilation and conversion of the solar energy that generated photosynthesis. Here we do not consider the aspects of natural selection of the specific photosynthetic pathways and their hereditary mechanisms that fix them at the taxonomic level, but we consider photosynthesis as a physical and chemical phenomenon resulting from the development of the solar energy conversion mechanisms in the environmental conditions of the early Earth during chemical evolution. Depending on what stage of chemical evolution we are talking about, on which carrier in the studied / validated model the processes are preceding and what level of maturity of the template synthesis of biomacromolecules is typical for this stage, the specific mechanisms for maintaining and fixation the precursor photosynthetic process can vary. The earlier this stage is, the more reductionist is the model (and the less applicable are the modern hereditary concepts, if we are talking about stabilization and reproduction of its mechanisms from generation to generation).
Depending on which model we select (and what definition of the energy photoconversion process we use as the conventional equivalent of photosynthesis), on what biomimetic criteria we apply to reproduce and interpret the evolutionarily pre-photosynthesis phenomena, not only the answer about the origin of photosynthesis will change but the subject matter as well. At the same time, since metonymy / reification of terminology often occurs during the development of science, and the degree of rigor and concretization of the definitions continously increases, modeling of the processes in accordance with their early general definitions is the most simple (less limited by the strict binding to its current form) and heuristically fruitful way for an evolutionist and any other specialist who considers the essence of the phenomenon in its development (either geological stratigraphy or biological evolution). In the case of modeling photosynthesis, this circumstance is fundamental, due to the differences in specific mechanisms or fundamental aspects of the definitions of photosynthesis for different taxa, different biochemical substrates, different evolutionary branches and different environmental conditions (for example, oxygenic and anoxygenic photosynthesis). With excessive chemical or taxonomically correct concretization of definitions, the possibility of finding a common ground in the evolving mechanisms of photosynthesis, starting from its primary prebiological stages, may be lost, as well as the possibility or expediency of searching for this evolutionary context. Let us illustrate this thesis.
Oxford English Dictionary in its early editions defined photosynthesis as "The process by which carbon dioxide is converted into organic matter in the presence of the chlorophyll of plants under the influence of light, which in all plants... involves the production of oxygen from water" [3] (in place of the ellipsis is the mention of "except some bacteria", although this is a biologically / taxonomically outdated definition). This definition clearly focuses on the process of carbon dioxide assimilation and production of oxygen from water in the presence of chlorophyll as a photocatalyst while organic matter there is referred to in a broad sense, without specifying any particular classes of the organic compounds. This allows to implement a wide range of different photochemical (photoinduced / photocatalytic) processes of organic synthesis while talking about photosynthesis modeling (within the limits of compliance with the above definition). Such a broad understanding of photosynthesis in the early 1980s probably contributed to the development of "artificial photosynthesis" research, providing plausibility to the results obtained despite the variety of the synthesized organic matter (sensu lato), often not correlated with the biological prototype.
Later (in the 1990s) in the photosynthesis definitions in academic dictionaries a certain trend towards narrowing the term meaning was observed. "Oxford Dictionary of Biochemistry and Molecular Biology" (2nd edition,1997) [4] defines photosynthesis as “the synthesis by organisms of organic chemical compounds, especially carbohydrates, from carbon dioxide using energy obtained from light rather than the oxidation of chemical compounds". In this case, the chemistry of the organic products synthesized is not yet limited, but the requirement for oxygen emission is already removed (due to the discovery of anoxygenic photosynthesis) and the photocatalytic aspects of the photopigment application are not obvious for a non-specialist (there are no phrases like “in the presence of the chlorophyll”... or “under the influence of light”, as in the 1980s definition). Besides, a crucial opposition "using energy obtained from light rather than the oxidation of chemical compounds" hinders the search for the common roots of photosynthesis and chemosynthesis in molecular and chemical evolution. However, no taxonomic restrictions are implied in this definition (because it is simply written "synthesis by organisms").
In the 2000s the situation gets worse, since most of the definitions used in the literature at the beginning of the 21st century contain restrictions on both organic products of photosynthesis and taxonomy of the photosynthesizing organisms. At the same time, the bioenergetic context of photosynthesis, as a rule, is beyond the first and the most cited definition. In Concise Medical Dictionary photosynthesis is defined as "the process whereby green plants and some bacteria manufacture carbohydrates from carbon dioxide and water" under irradiation [5], while the Oxford Dictionary of Food and Nutrition defines it as "the synthesis of carbohydrates from carbon dioxide and water by plants in sunlight, with the release of oxygen" [6]. The broader and further away from the problem of photosynthesis the readership of the dictionary, the greater the likelihood of narrowing in the term definition, due to which a non-specialist or a specialist from the related field is unlikely to be inspired by such a definition for a scientific search within an evolutionary / developmental context (at the level of search for the possible non-carbohydrate products and intermediates in photoreactions that preceded the emergence of modern photosynthesis). The most universal and consistent definition is from the Oxford Dictionary of Biology (sixth edition, 2008) [7]: "The chemical process by which green plants and other phototrophs synthesize organic compounds from carbon dioxide and water in the presence of sunlight". However, it is almost tautological ("Sepulka") to the definition of phototrophs in the same dictionary: "Any organism that uses energy derived from the sun to manufacture organic compounds by photosynthesis". Therefore, when trying to find the evolutionary roots of this process, we will anyway be forced to go beyond the internal logic of the topic, limited at the organismic level by the modern forms of photo(auto)trophs.
From the above definitions, one can extract several fundamental issues that make it possible to define photosynthesis, invariant to the level of epistemological accuracy:
- 1)
- light absorption / influence of light;
- 2)
- carbon dioxide assimilation / conversion;
- 3)
- synthesis of organic chemical compounds;
- 4)
- release of oxygen / production of oxygen from water.
In total, this is close to the meaning of the term "photosyntax" introduced for this set of processes by C.R. Barnes in 1893, along with the term "photosynthesis" and replacing the term "assimilation" ("For the process of formation of complex carbon compounds out of simple ones under the influence of light, I propose that the term photosyntax be used" [8]). Nevertheless, as it was believed in those years (cited from H. Gest [9]): "The term 'photosynthetic assimilation' is a perfectly general one, and would include the assimilation of other compounds by the aid of light, should any such processes be discovered in the future [In fact, H. Molisch discovered the photoheterotrophic growth mode of purple bacteria in 1907]. 'nitrogen assimilation' would indicate that carbon could be directly assimilated". (H. Gest refers here to A.J. Ewart's addition to "The Physiology of Plants. A Treatise upon the Metabolism and Sources of Energy in Plants" – an English translation of the "Pflanzenphysiologie. Ein Handbuch der Lehre vom Stoffwechsel und Kraftwechsel in der Pflanze" by W. Pfeffer. However, in the reprint edition of 1902 available to the author, this addition is absent [10], so it is impossible to judge the accuracy of the quote). However, in accordance with the known data of modern bioenergetics and photobiology, photosynthesis schemes are not limited to "carbon dioxide assimilation" even involving the light-activated (bio)synthesis of certain compounds, since dozens of biologically significant and vital membrane-mediated and other mechanisms, as a rule, are ignored in any definition of photosynthesis. However, without their reproduction, it is impossible to simulate the early forms of photosynthesis and to understand the mechanisms of its evolution from the simple spontaneous photosynthetic pigment assemblies or their precursors to its modern complex form.
At the same time, every step in the evolution of understanding the forms or ways of photosynthesis in its specific forms, starting from the 19th century, calls into question the possibility of defining any of its modern forms as an uncontested physical and chemical reality within which it is possible. Let us quote the above work of H. Gest [9]: "The discovery of anoxygenic bacterial photosynthesis made the general definition of 'photosynthesis' in... most other dictionaries incorrect, but this is still not widely recognized". And further: "Ten years before Barnes (1893) coined the term 'photosynthesis,' Theodor Engelmann (1883) reported novel experiments that revealed photosensory behavior of purple sulfur bacteria. gave negative results Later, in 1907, H. Molisch demonstrated convincingly that purple bacteria do not produce O2, and that they have the capacity to use organic compounds as sole carbon sources for growth with energy provided by light". Hence, according to H. Gest, "The metabolic/physiological pattern of the purple bacteria obviously did not satisfy the criteria for photosynthesis as originally defined for green plants, and as a consequence, for several decades the bacteria were not generally accepted as being photosynthetic". Thus, an incorrect or an incomplete definition can lead to a "false trail" and slow down the development of science for a long time. Gest further writes: "The original definition of photosynthesis as an oxygenic process led investigators to continue designing fu-tile experiments to find evidence of O2 production by purple bacteria for some time. The last definitive negative experiments were reported in 1954, 71 years after Engelmann's first report!" So, strictly limiting the definitions of photosynthesis, we will never be able to model and to understand the mechanisms of its origin and the divergence of its evolutionary-biochemical pathways, correlated with the natural (for example, geochemistry, chemistry of the atmosphere or aquatic environment, etc.) conditions for the evolution / selection of protophotosynthetic systems.
The consequences of unifying the definition of photosynthesis were clearly demonstrated by H. Gest in the above paper [9]: "The discovery of photophosphorylation in 1954 revealed a basic 'common denominator' of oxygenic and anoxygenic photosyntheses, and paved the way for redefinition of 'photosynthesis.' In 1963, Martin Kamen suggested a revised definition which would have the effect of including anoxygenic bacterial photosynthesis by (a) avoiding any specification of the carbon source for growth, and (b) not indicating O2 as a photosynthetic product". Therefore, if we talk about the general definition, then Gest, distinguishing between photosynthetic and "quasi-photosynthetic" bacteria, proposes an extension of the classical Kamen's definition [11] ("Photosynthesis is a series of processes in which electromagnetic energy is converted to chemical free energy which can be used for biosynthesis") to a more general form [12]: Photosynthesis is a series of processes in which electromagnetic energy is converted to chemical energy used for biosynthesis of organic cell materials; a photosynthetic organism is one in which a major fraction of the energy required for cellular syntheses is supplied by light. Obviously, it does not contain any restrictions on both chemical composition (molecular oxygen and carbon dioxide, as well as the specific photopigments) and taxonomy of autotrophs, which makes such a definition convenient for modeling and reconstructing the evolution of photosynthesis.
Later in [13] H. Gest explained the accepted level of abstraction of the definition: “Molecular oxygen and carbon dioxide are not included in the “common denominator definition” of photosynthesis because photosynthetic bacteria do not produce oxygen and carbon dioxide is not necessarily their required carbon source. A number of the bacterial species can grow with either carbon dioxide or simple organic compounds such as acetate as the sole carbon source for synthesis of all cell constituents with light as the source of energy". Accordingly, evolution, biochemical phylogenetic taxonomy, physiological diversity of the solar energy utilization mechanisms imply taking into account all the possible states of photophysiological processes and the response of organisms to irradiation, starting from the simplest forms of the existence of living matter, and, in particular, the physiology of photosynthesis: "Comparison of the biochemical patterns of diverse organism requires categorization of physiological types, for example, heterotrophs versus autotrophs, aerobes versus anaerobes. Newly discovered aerobic bacteria that contain the “photopigments” bacteriochlorophyll and carotenoids, but which are incapable of using light as a sole or major source of energy for growth". "A number of investigators persist in referring to such organisms as “aerobic anoxygenic photosynthetic bacteria... (1) such organisms do not manifest photosynthetic metabolism as a major feature, and (2) the term anoxygenic refers to the fact that purple and green photosynthetic bacteria convert light energy to chemical energy (used for biosynthesis) anaerobically and do not produce oxygen". Accordingly, Kamen’s and Gest’s definition seems to be more universal. However, this universality is achieved (for oxygenic and non-oxygenic photosyntheses), by H. Gest, by introducing into consideration [14]: a) photobioenergy criteria; b) the photosynthetic apparatus as a structure (he pointed out that "important experiments were reported which revealed the “common denominator" of oxygenic and non-oxygenic photosyntheses", namely, he emphasized the evidence of light-dependent production of ATP by the photosynthetic apparatus — in other words, the conversion of light energy to the chemical energy). Therefore, in a retrospective theoretical analysis and in the photosynthesis evolution models considered in this review, we have to take into account bioenergetics, as well as the ancestral (often pre-membrane) forms of storage and utilization of the solar energy by the early photosynthetic and protophotosynthetic systems, according to the most general definition of photosynthesis (true for its most functionally early forms as the “common denominator”).
Based on the foregoing, in the course of reification and an increase in the level of concreteness in the area of photosynthesis modeling, several limiting forms of the models should inevitably have arisen, focused on:
- ⮚
- only light-assisted adsorption / assimilation of carbon dioxide (in the "technological" limit - not only CO2, but also of other atmospheric agents, in particular, pollutants);
- ⮚
- only photoinduced redox processes (photocatalytic processes used for photodisinfection, especially those based on dispersed semiconductors usually associated with modeling of the photosynthesis elementary stages);
- ⮚
- only photocatalysis and photo-assisted chemical synthesis;
- ⮚
- only on photoinduced purification and oxygenation of the atmosphere (this is also a common practice, from the terrestrial conditions to various technological models of the "space biospheres" developed since the last quarter of the 20th century);
- ⮚
- only on obtaining energy (this aspect can be clearly seen in the design of the biomimetic solar cells based on the principles of the natural photosynthesis in the understanding of technologists and engineers).
This is often observed in practice and leads to the numerous disputes about the photosynthesis models and their compliance with one or another technologically operationally convenient definition.
If we define the process of photosynthesis and, accordingly, artificial photosynthesis, sensu lato, as the synthesis of organic matter under the influence of radiation, mediated by some photochemical or photoelectrochemical mechanisms, then we come to an absurd paradox when dozens of incompatible (or even conflicting) processes, qualitatively different in mechanisms and products, are positioned as model photosynthesis and artificial photosynthesis. Currently, the proper scientific community faces an unacceptable situation when even the most superficial search, restricted by name, in scholar.google.co.uk in 2023 offers about 1,600 publications on “artificial photosynthesis” (unrestricted search for this phrase through the same search engine generates nearly 51,100 references). However, looking at the first ten pages, we see that this concept, used as a “hot label”, refers to more than two dozen qualitatively different implementations of processes - both homogeneous and heterogeneous. The differential content analysis demonstrates that all these works are incomparable to a single set of criteria (not all mechanisms associated with artificial models of photosynthesis support water decomposition, but those where this function is implemented do not always pass an obligatory criterion of carbon dioxide fixation or organic matter synthesis). Obviously, this kind of discourse, which is the product of arbitrary juggling with concepts, is not conducive to constructive analysis, since the concept of modeling photosynthesis by artificial photosynthesis (the result of using the term) and even the definitions of “photosynthesis” and “artificial photosynthesis” (as defined terms) are not falsifiable. It is therefore impossible to implement a model analysis of photosynthesis processes at their initial evolutionary stage using heuristic tools for modeling photosynthesis and creating “artificial photosynthesis” in an extended (sensu lato) chemical interpretation.
If we define photosynthesis as a biological process of converting the light energy, accumulated in chemical compounds, synthesized by photoautotrophs with pigments that are sensitive to the corresponding spectral range, then it is obvious that no existing complex artificial photosynthesis system can reproduce such a set of processes - not to mention the early models of photosynthesis.
Strictly speaking, any previously defined criterion of photosynthesis, in its modern sense, in modeling entails hypostasis (or, in other words, concreteness / reification), which is incompatible with modeling the phenomenon as a whole, since the complete set of descriptors that determine photosynthesis as a process may characterize only photosynthesis itself (in terms of mathematical logic, the highest degree of isomorphism in modeling is automorphism), and any deviation from this definition, if we are guided by purely chemical criteria, entails the incompatibility effects in the model. For example, oxygenic and anoxygenic photosynthesis, by definition, are incompatible in key descriptors of redox processes that determine the terminological difference, thus making impossible their modeling within the same biochemical approach, sensu stricto. We can only observe the similarity of processes at the physical level; therefore, photosynthesis models starting from the emergent causes of its origin, but equally applicable to the later stages of the photosynthesis evolution, sensu lato, can be constructive for the wide nomenclature of these phenomena united by a single term only when modeling concerns the essence of processes at the physical level.
2. Basic principles of the reliable reconstruction of protophotosynthesis: From physical and geochemical selection criteria to evolutionary consistency.
The key to integrating protophotosynthesis / proto-life models and understanding divergence paths lies purely in physical laws, at the time when divergent chemical evolution and molecular selection processes have not yet “separated the sheep from the goats” (ab haedis segregare oves), and have not led to the specialization of individual molecular structures for specific intracellular processes. At that time, multifunctional simple structures had advantages in terms of static survival among prebiological systems, which were the source for divergent selection of the precursor forms of intracellular molecular biomachinery. The selection criteria in this case do not imply adaptation to the conditions of an abiogenic nature that are not optimal for the implementation of molecular biological processes, but imply the emerging adaptability of the selected structures to each other as agents of the emerging protobiological environment [15].
In such systems, selection takes place not so much under the pressure of “selecting conditions” of the external environment [16,17], but according to the criteria of cooperation (adaptability to joint action), which is replaced at the early stages / in the simplest structures by the multifunctionality of these structures. Therefore, it is constructive to start the analysis not from a certain stage of biological evolution of photosynthesis and divergence of the corresponding pigment structures, but from a search for the physical singular stage at which any functionally equivalent circuit of the physical precursor could give rise to the “shoot of the phylogenetic tree” of chemical evolution, that resulted in modern forms of photosynthesis. The questions of polyphilia or monophilia of the tree at this singular stage cannot be solved outside the framework of analysis of the coordination of "molecular symbiosis" as a predecessor of metabolism [18], equifinal in the physical sense, but diverging in the chemical aspect.
In contrast to artificial models of photosynthesis, that ignore the historical and evolutionary factors in choosing the material basis of artificial photosynthesis (moreover, often deviating from the principles of its organization and focusing on the efficiency growth and other technical criteria), the analysis of photosynthesis precursors and the reasons for their selection in natural environment (up to the choice of the modern pool of “bioorganic machinery” of photosynthetic phenomena), must proceed from the physico-chemical factors of abiogenesis based on geophysical and geochemical conditions [19,20]. The reconstruction of the genesis and evolution of any functions and the related biological structures, starting from the prebiological stages with their simpler physico-chemical mechanisms (compared to modern cellular systems) that proceed in a native geochemical environment under the conditions of an imperative set of environmental factors (not reducible to a single selection target factor to which, sensitivity and resistance are developed during selection), should objectively adhere to at least of the following principles (in hierarchical order):
1. Definitive consistency
2. Physico-chemical consistency
3. Geochemical and geophysical consistency (for a number of other hypotheses - astrophysical and astrochemical one)
4. Emergent consistency
5. Evolutionary consistency
Any solution to the problem of abiogenesis and evolutionary formation of the primary functions can be considered as evidence of hypotheses, and not as an illustration of the solution to the problem of forming biomimetic systems (with some deviation from reality inherent in any modeling process), when the selection of model attributes can take into account not only similarity of functions between the model system and the original, but also the evolutionary consistency of the carrier, directly resulting from the reproduction of the geochemical conditions of abiogenesis / initial evolution of functions in this model. At the moment, no existing model satisfies this condition fully. The condition is partially satisfied only for a limited pool of the simplest systems that reproduce geological conditions of chemical evolution rather than the formation of systems and structures with a prebiological interpretation. It is necessary to remember a simple methodological truth that "the presence of prototypes, in itself, outside the evolutionary and geochemical context, proves nothing." Until the consistent chemical mechanisms that turn into protobiological ones are proven, we can be certain only about physics, that is common to any simulated biological / protobiological process, and we can only approximate the truth to the level of physical chemistry projected onto the geochemical environment.
3. Coupling between the light harvesting, charge separation and catalysis in a minimal singular model of protophotosynthetic machinery.
According to comparative photobiology and biomimetic photochemistry, light harvesting is not considered to be the conversion of solar energy during photosynthesis, if the other key components - charge separation and catalysis - are missing [21]. Effective charge separation in modern photosynthetic systems is usually associated with microheterogeneous phase-separated dispersed structures within the cell (reaction centers that are supramolecular associates [22] with reversible electrostatic polarization [23,24]) and gradients of charge carriers at biomembranes [25,26]. Single acts of the polarization process can be mapped / tracked using standard photovoltage techniques [27]. It has been demonstrated by methods of quantum chemistry and molecular dynamics that many electrostatic fluctuations are dynamically “frozen” on the timeline of primary charge separation, however, in general, the processes of primary charge transfer in reaction centers are simulated by three bound electronic states corresponding to the pairs of a donor (photoexcited chlorophyll) and an acceptor, as well as a reduced bridge interacting with the dissipative medium of proteins, corresponding to the degrees of freedom of the solvent [28,29]. If we disregard the subtleties of modern electron transfer paths, recorded by picosecond optical measurements and the Stark spectroscopy method [30], we can illustrate (using a self-consistent procedure for assessing the effects of induced dipoles in the protein and the surrounding membrane) that all such processes are electrostatically controlled at the supramolecular level of organization (ab initio) [31].
According to the above requirement, if we adopt this approach at the level of physics, one of the simulated processes is the charge transfer (along the electron transfer chains or using a proton pump - it is not indifferent to photochemistry, but of no concern to electrophysics). The non-strict determinancy of the nature of the reducing and oxidizing agents in high-level electrochemical schemes, along with the above arguments, allows one to descend to the level of evolutionary uncertainty of the reducing and oxidizing agents (since the first photosynthetic systems used reducing agents other than water, such as hydrogen, hydrogen sulfide, metal sulfides, and ferrous ions as electron sources, and some modern simple organisms replace water with quite exotic agents from the standpoint of modern biochemistry, for example, by oxidizing arsenite to arsenate [32], or using other inorganic ions), and also rise to the formal abstraction level that is optimal for the physical models of photosynthesis that do not appeal to a certain biochemistry.
Indeed, an increasing number of works on the molecular design of bionic schemes of non-chlorophyll-based artificial photosynthesis reproduce photoinduced charge separation [33], accumulative charge separation and / or transport [34,35] (the goal of optimizing such bionic schemes is often to achieve long-lived charge separation [36]), coming to the idea of combining the conversion of light radiation and electrochemical gradients into electrical energy, which is prompted by a photosynthetic prototype [37]. The biological analogy may explain the need to combine electrochemical gradients and photosynthetic machinery: it is well known that in prokaryotic phototrophs the plasmalemma and endomembrane systems perform the function of photosynthetic membranes, and in prochlorophytes, blue-green algae and eukaryotic chloroplasts, this function is performed by thylakoid membranes.
It is not surprising that the most physically adequate protocell models implement the principles of organizing oriented reaction centers generating a potential gradient (proton gradient, charge gradient) on the membrane [38], while progressive versions of artificial photosynthesis with inorganic membranes use “molecular wires” for charge transfer [39]. Outside special cases with exotic transfer scheme / electrophysical properties of the carrier (such as, for example, assumptions about the adequate reproduction of some aspects of photosynthesis with asymmetric charge transfer in ferroelectric media [40], which implicitly correlates with the elegant but insufficiently proven position on the role of such media and ferroelectric effects in membrane physics [41,42,43,44,45,46]), a rather simple system of ideas about the optimal organization of a simple photosynthesis model emerges. It contains a photocatalyst for oxidation-reduction reactions, charge separation by manipulating the physical chemistry of the surface of the membrane-mimetic carrier (which, following the first works of the founder of membrane-mimetic chemistry, J.H. Fendler, include artificial photosynthesis media with charge transfer or separation [47], for example - based on colloidal semiconductors with catalytic nanocoating) or bulk nanophases / clusters (simulating reaction centers), a photoabsorbing part (antenna) and feedback loops for the regulation of processes in this complex system [48].
4. Integration of minimal protophotosynthetic functions in a single structure as a criterion for the unity of their emergence.
From the point of view of the similarity and modeling theories, which postulate the need to reduce variables and operators containing no information, when their presumed function can be carried out in a simpler way, for the case of the most simply organized photosynthetic model (reproducing the early evolutionary stages that took place long before modern molecular biochemical methods and photosynthesis implementation in cellular structures were developed), it is advisable to raise the question about the simplest forms of processes that combine most functions of the model in a single carrier or in a minimal number of carriers. This corresponds to a number of obvious evolutionary considerations, according to which the emergence of a single functional structure or population of structures is usually more probable than the emergence of a coherent complex, made of a small number of structures or populations of structures adapted to each other (compare ideas from [49,50,51,52] and some works on the stochasticity of the medium, which at the prebiological stage may include potential components that form the resulting structure [53,54,55]). This reasoning reminds the reductionist principles of ARIZ / TRIZ that preserve the functionality of biomodels. According to the above principles, an ideal solution suggests a scheme where the maximum number of links is removed while their functions are fully preserved. In other words, where the same links are capable of performing several physical / chemical functions [56,57,58,59,60]. In other words, the model problem in the case of photosynthesis can be formulated as: it is necessary to create a simple (geologically and geochemically relevant to the pre-cellular stage of chemical evolution) system that would combine photosensitivity, key electrophysical features and electrochemical gradients, basic catalytic functions in a given medium but that would consist of one or more simple components physically equivalent to each other and, as a consequence, equifinal in physical criteria that would allow to perform phylogenetic reconstruction of modern principles or earlier biochemical prototypes of photosynthesis.
This principle is intuitively implemented even by the specialists who are not using evolutionary discourses and ARIZ / TRIZ methods of resolving contradictions and who are trying to simulate fully functional photosynthesis by small investments. From 1980s, attempts have been made to integrate charge transfer and separation by the membrane structures or membrane mimetics such as cationic and anionic surfactants [61,62] (including redox-active [63,64] surfactants - see Figure 1 from Fendler's article "Aspects of Artificial Photosynthesis: The Role of Potential Gradients in Promoting Charge Separation in the Presence of Surfactant Vesicles" [61]) in a single system of model photosynthesis, while from 2010s, the trend has been gaining strength to combine within the framework of artificial photosynthesis not only absorption of radiation and charge transfer, but also multi-electron catalysis / photo-redox catalysis [65,66,67,68,69]. However, in some cases, the installations that simulate this process become unacceptably complex, since they ignore the principles of analysis and decomposition of the model with the minimum number of multifunctional units. When trying to directly integrate photosynthetic and other cell functions into a single nanostructure with a size similar to the electron mean free path during photo-induced charge transfer, we come up with an irreducible complexity (not in the sense of “Irreducible complexity” according to Michael Behe, but in that physical sense, when it is impossible to further reduce / simplify the system without losing its key properties. This complexity is usually associated with the minimal protocellular structure, rather than with the machinery of the photosynthetic apparatus (for example, nanoscale protocell with an integrated metabolic, genetic, and container system by S. Rasmussen from Los-Alamos National Lab [70,71]; see Figure 2 and Figure 3), that contains a light sensitizer for energy supply, like in phototrophs. This confirms the previously expressed ideas about the integration of self-assembly of protophotosynthetic systems and the simplest protobiological "minimal protocells".
Such small ultrasystems (≈ 5 nm) containing a photosensitizer [72,73] and a minimal amount of fatty acid (due to which they fit well with the abiogenetic “fatty acid world” model [74]), are typically characterized by the quantum self-assembly (including photo-induced one and photo-induced electron tunneling [75,76,77]. The small size of such structures relative to the mean free path of the charge carriers explains the possibility of spintronics control over photoinduced transport phenomena in them, and therefore over the processes of elementary photosynthesis [78]. This, in turn, is complicated by the possible effects of quantum entanglement [79] (observed, due to the small size of structures, not only on the scale of a single minimal protocell, but also on two or more contacting and interacting protocells [80]). As follows from the quantum calculations, the processes involving quasiparticles can be especially effective at this scale.
Basically, the general model involving quantum self-assembly associated with a membrane-mimetic surfactant, photo-induced electron transfer and colloidal dispersion of nanostructures, which causes many dimensional effects, including ones provided by the quasiparticles, seems to be applicable. However, this approach does not point out what could be a precursor of photosynthesis at the stage when neither the main polymer / supramolecular components of the protocell [81] nor specialized organic photosensitizers were in place, but when energy supply of the chemically evolving photoautotrophic system, unable to function without the energy influx, should have already existed. For this reason, it makes sense to further develop the concept of nanodispersed self-organizing and photosensitive structures with charge transfer and a membrane-mimetic layer with interpolation down the evolutionary and physical geochemical ladder (of molecular and chemical evolution), up to the point of minimal complexity, allowing light harvesting, charge separation and catalysis in a singular model.
Indeed, if the “ideal” model should possess only physical and chemical consistency, then the interpolating hypothesis suitable for describing evolution should also possess geochemical and geophysical consistency (and, therefore, identify chemical agents that implement physical functions that existed in the early stages of prebiological evolution in the natural environment). If the original system possessed the emergent consistency, formed by self-assembly of a minimum of the necessary and sufficient agents, then a model that claims to have abiogenetic value (and the resulting functional biosimilarity) also requires the evolutionary consistency – an explanation of reasons for further complication, based on a comparative analysis of various manifestations of this process at the stages of chemical evolution that are different in terms of material basis, from the period preceding to the one directly following the modeled stage to explain the reasons for the transition to the next stages in the physical conditions of the preceding ones. For the earliest stages, when the mechanisms of biochemical regulation have not yet been launched (and, therefore, their genetic signatures have not been preserved), only physical reductionism in a model study, based on the physical and geochemical conditions of such stages (providing physical and chemical, geophysical and geochemical consistency, the importance of which was discussed above), can serve as a criterion for finding the earliest physical realizations of protophotosynthetic functions in the chemical evolution. Early prebiochemical level of modeling, i.e. orientation on the physical criteria as a methodological technique, disregards specific chemical implementation, which is a typical source of disputes and contradictions between the specialists who favor different models, but who agree to talk about the same mechanism from a physical point of view. Therefore, first of all, it is advisable to find a common ground underlying all the process models implemented at the elementary and undemanding level, and only then consider the probable scenarios for this (inevitable, for physical reasons) developing process, depending on the proposed chemical basis available in the environment, and on the geochemical conditions of the early Earth.
5. What is the minimal set of functions sufficient for (proto)photosynthesis modeling from the standpoint of mathematical biophysics?
So, what could, in conditions of minimal physical complexity, interconnectively perform the above photosynthetic functions (light harvesting, charge separation, catalysis)? To ensure the reliability of modeling, we should treat this issue based on the ideas of mathematical biophysics about the goal of the evolutionary modeling – i.e. reproducing the phenomenology of modern photosynthesis and the functions of its consequents. No doubt, for the early stages of processes simulating it (when the components of the photosystems did not yet exist), most of the kinetic equations of modern photosynthetic processes are not applicable. Therefore, we should confine ourselves to only the most basic physical processes, not to create the illusion of evolutionary predetermination of modern photosynthesis at those stages when the physical question “for what reason” is accurate, but the molecular-selective / genetic question “for what purpose” this or that function or structural motive was selected during the evolution is yet premature.
The further analysis of formal modeling criteria is based on comprehensive publications by prominent specialists in the field of photosynthesis modeling, who developed multiparticle kinetic methods for various photosynthetically relevant structures - A. Rubin and G. Rhiznichenko, who formulated theses on photosynthesis modeling methods, in particular, in their book “Mathematical Biophysics” [82]. In contrast to modeling complex processes with the high level of organization, for which simplifications and qualitative study of the processes are appropriate, and the selection of the details of the simulated processes to be included in the model depends on the aim of the research, the description of basic elementary mechanisms can be achieved in a uniform form both for modern photosynthesis and protophotosynthesis without significant simplifications. As accurately indicated by Rubin and Rhiznichenko [82], “to study the mechanisms of electron transport, energy dissipation, and conformation changes inside photosynthetic reaction centers, detailed models are necessary”. At the same time, “to simulate the main features of the primary photosynthetic processes, organic synthesis and metabolism of a living cell, a simplified dynamic model can be used". The path of reasoning given below is definitive in nature, without claiming to be exhaustive, however, it contains all the minimal components necessary for the construction of a functional model. The analysis of other trends in this area leads to approximately the same list of descriptors by other methods. It may seem tutorial, but without it, it may be impossible to justify the reproduction of the corresponding structures and properties in protophotosynthesis.
5.1. Photochemistry.
The primary property for the receiving element of any photosynthetic system (including protophotosynthesis), by definition, is the ability to absorb light and to transform light quanta into the energy of separated and stabilized charges (followed by conformational changes in the protein structure, which prevent backward electron transport and energy losses, but this is just a consequence, not a cause of the photoinduced rearrangement of the system charge). According to the definition [83], the first stages of the photobiological process, as a rule, are: “absorption of a light quantum by a chromophore group and generation of electronic excited states → transfer of electronic excitation energy → primary photosynthetic act and generation of primary photoproducts → formation of primary stable chemical compounds". However, this definition sensu lato accurately describes the photochemical, but not photosynthetic processes that preceded the emergence of modern complex photosynthesis. In this case, if we exclude from the consideration the “physiologobiochemical processes → final photobiological effect” that are characteristic only of living systems, it is possible to trace the pathways from the primary protophotosynthetic mechanisms towards the contemporal photosynthesis on the principles of describing these acts and their physical actors, not correlated with modern biochemical agents. Based on the premise that in spite of a great diversity of photobiological effects, the initial stages of light energy transformation have common molecular mechanisms [83], it is possible to derive evolutionary-biophysical or evolutionary-physical principles (according to Blumenfeld [84] - the key principles for understanding the formation of those modern biophysical processes that “survived in the course of physical selection”) of physical and chemical divergence of various biological photoprocesses characterized by differences at any of the primary stages or links of the above “conveyor”, which leads to dissimilar results.
5.2. Redox processes.
It is known that in modern photosynthesis under illumination proton flux inside thylakoids leads to alkalization of the chloroplast stroma ..., which in turn leads to an increase in the inward proton fluxes, and the growth of the passive flux inside the cell and growth of proton concentration inside the cell induces membrane depolarization. Thus, both from the standpoint of cell biochemistry and from the standpoint of membrane electrophysiology of photosynthesis, the photoinduced redox mechanism is obligatory, having the illumination of the system at the beginning, and the readjustment of gradients at the system-environment interface at the end, with the final stage of membrane depolarization serving as an example. This also triggers photoinduced reaction-diffusion processes, since the depolarization process is associated with the redistribution of charge carriers, that is, ions. Since hydrogen carriers interchange with electron carriers forming a redox-loop, and hydrogen carriers transfer H+ to the carriers on the opposite side of the membrane, when electrons are transferred transmembraneously from one to the other electron carrier, it is logical to model redox processes on gradients of the carrier concentrations, precursor to membrane processes in osmochemistry, and to reduce all the above processes to the level of transfer of the main differently charged carriers (holes and electrons, as in solid state physics), temporarily neglecting their chemistry and structure (since formal equations do not require this).
5.2.1. The need for redox-catalytic agents for the evolution of redox states of carriers.
The evolution of the redox states of these mobile carriers in the majority of the models so far has been described by means of the mass action law. Therefore, it is advisable to introduce into the membrane structure that maintains the redox state some redox-catalytic agent that satisfies the mass action law and its well-known applications to the dynamics and kinetics of enzymatic processes [85,86,92]. This is logical because of the presence of two different types of photosynthetic electron transport, representing electron tunnels from one electron carrier to the other along the electron path, and hence, electron transfer inside multienzyme complexes (which are embedded in the photosynthetic membrane). This will be described in more detail later in Section 5.3 (Catalysis and Macrokinetics).
5.2.2. The need for electrostatic interactions in redox-evolution of photosynthesis.
Due to the bioelectrochemical mechanisms of redox coupling and dependence of the rate parameters on the transmembrane electric field, it is logical to pay attention to electrostatic interactions in the process of docking and generation of a supercomplex that is a necessary condition of the effective electron transfer. For the earliest stages of evolution this transfer should be interpreted as an electrostatic (non-covalent) interaction, characteristic of many coordination / supramolecular systems, including soft matter ones, as well as of the most phenomena of electrostatic catalysis. For this reason, below we will consider the issues of kinetics and catalysis in photosynthesis.
5.3. Catalysis and macrokinetics.
According to [82], mechanisms of primary photobiological processes (photosynthesis, visual reception), enzyme catalysis in the enzyme active center, and ion transfer through membrane channels are governed by similar physical principles. Therefore, the ideal solution is to combine catalysis, photo-induced charge carrier transfer and photo-electrophysical reception that ensure various links of the kinetics of photo-induced processes on a single structure so that their combination is equifinal and emergently coupled. Since in theoretical biophysics generalized kinetic and physical models of interactions allow to describe different biological phenomena, for the early stages we can postulate “cross-kinetic schemes” and the possibility of implementing the same physical and kinetic mechanisms in the framework of processes that are now identified as independent phases (and sometimes as independent processes), that diverged at later stages of chemical evolution.
5.3.1. Protophotosynthetic catalysis should be photoredox catalysis.
Since any photosynthetic complex contains several components, each of which can be presented in oxidized and reduced forms, and schemes of the states and their transitions for a photosynthetic complex usually include dozens of states, biomimetic catalysis that simulates acts of the photosynthetic process, including the acts of protophotosynthetic catalysis, should, firstly, be a redox catalysis, and, secondly, based on the definition, a photoredox catalysis or a process equivalent to it (with energy pumping in a certain spectral range).
5.3.2. Protophotosynthetic catalysis should be membrane / membrane mimetic catalysis.
Since the main participants of photosynthetic electron transport are embedded in the bilayer lipid membrane (multienzyme complexes) (otherwise they could not provide the path for directed electron transport across the membrane), it is expedient to incorporate photocatalysts / photoredox catalysts of protophotosynthesis into membrane-mimetic surfaces (surface immobilization of the catalyst [87]), for the purpose of interface charge separation, or to use the integration of their own photocatalytic and membrane mimetic properties.
5.3.3. The need for charge separation and reversible charging-discharging cycles in protomenranes.
Since the photo-redox functions and the functions of bioelectrogenesis in this case are related, similarly to photosynthesis models (for example, the scheme of catalytic cycle of photosystem II includes states of the photosystem II complex, which is determined by the redox states of the involved electron carriers), it is logical to take into account the photoelectrochemical states of the catalyst and the double electric layer on its surface / the surface of the integrating layer as a driver of reverse charge-discharge processes and, at the same time, electrochemical / photoelectrochemical oxidation-reduction, and, on the other hand, as a stabilizing buffer capacity, accumulating energy and providing "homeostasis of the periodic steady-states" in the system. This is consistent with modern photosynthesis models, where rapid charge separation in primary photochemical pairs, electron transport from the inner to the outer membrane surfaces, and the following quasi-stabilization of the separated charges at opposite membrane sides correspond to capacitor membrane properties.
5.3.4. Photoelectrocatalytic acceleration of processes in early photosynthesis.
On the other hand, the cyclic discharge of a double layer “capacitor” is, from the standpoint of nonlinear dynamics, an analog model of some oscillatory / self-oscillatory processes inherent in biological processes (according to [82] self-oscillatory biological systems include oscillating metabolic systems and periodic photosynthesis processes), being at the same time, an element of the equivalent circuit for these processes. Thus, if such kinetic phenomena of discharge or gating are observed in an electrochemical or electrocatalytic double layer system, this system is, at the same time, a special case of an oscillatory process, and an equivalent circuit of analog modeling of an “isokinetic” process from any field of nonlinear wave representations and self-oscillating kinetics. Obviously, for the photoelectrochemical system, which is protophotosynthesis, the chemical implementation and its electrochemical equivalent circuit, with high probability, could coincide. Since the rate constants at the electron transfer inside the photosynthetic reaction center are influenced by the electric field [83], the assumption about the integration of catalysis and electrophysical acceleration of processes in early photosynthesis seems quite convincing.
5.3.5. Indifference to the charge carrier nature, similarity of equivalent circuits and the presence of reversible non-covalent interactions.
Since, from the point of view of formal kinetics, the chemistry of reacting substances when they are introduced into the systems of equations does not matter (moreover, the reactions of enzymatic kinetics and ecological schemes describing the coexistence of species are described by equivalent schemes), any ions and charge carriers can participate, depending on environmental conditions. The same “ionic pluralism of vectorial chemistry” led to the evolution of not only the proton pump, but also the schemes involving sodium transport (“sodium world” in the classical [88,89] - see Figure 4 - and modern molecular interpretation [90,91]) at the later stages of the evolutionary process and geochemical selection. It is essential that reversible non-covalent interactions characteristic of enzyme-substrate complexes and enzyme-mimetic complexes were present in the medium. The kinetic feasibility of existence of such complexes is obvious almost ab initio, since the reaction rate equations (for example, classical ones, derived by Henry, Michaelis, Menten, etc. [92,93,94]) were derived on the basis of the postulates about the existence of an enzyme-substrate complex and the rapid establishment of equilibrium between substrates and the latter, in accordance with the law of masses.
5.3.6. The search for enzyme-mimetic catalytic pathways for prebiological photocatalysis.
The situation is simplified (or complicated) by the fact that, according to [82], the reaction centers of photosynthesis can be compared to a special photoactive enzyme, where prosthetic groups of the reaction center carriers play the role of components of the active center. To develop the comparison further, according to [82], the usual low-molecular enzyme substrate here is light quanta, the absorption of which “triggers” the functioning of the reaction center of photosynthesis. Furthermore, the experimental data accumulated recently prove explicitly the principle of conjugation of electron transfers and conformational rearrangements. Indeed, in real enzymes, initial changes in the electronic state of the active center also take very short periods of time (which give an “impetus” to conformational rearrangements in the enzyme protein globule that vary in scale and time). Therefore, light quanta can be introduced into the kinetic scheme as a substrate, which allows us to consider as energy storage in primary photosynthetic systems not only chemical forms of light quanta that are provided by electron transfer and the formation of chemical bonds, but also more direct elementary physical methods of its accumulation, including consumption of the photoelectric effect energy. Indeed, in the case of models of photosynthetic electron transport, which specifically deal with electron transfer in a multienzyme complex, electron transport processes are the basis of the primary photosynthetic light stage where the transformation of solar energy into the energy of chemical bonds takes place.
Therefore, given that the kinetic models of photosynthetic electron transfer between components within multienzyme complexes evolutionally are the most advanced, it is rational to look for less complex functionally equivalent catalytic transfer ways for simplified implementation in conditions of the prebiological physics. This is not surprising, since enzyme mimetics / protein mimetics are used in artificial photosynthesis, in particular, replacing the functions of protein components of antenna structures [95] with their spectrochemical properties, and the possibility of modeling enzymatic catalysis on the simplified (especially inorganic) substances that began precisely with the evolutionary formulation of the question “what existed before the enzymes?” [96], have been under consideration since the 1960s. [97,98].
5.3.7. The need to reproduce the kinetics of multienzyme complexes in models of complex photosynthetic and protophotosynthetic systems.
Due to the fact that modern photosynthesis consider functioning of not single enzymes, but multienzyme complexes as well as electron paths, where electron transport occurs via mobile carriers, it is logical to immediately simulate not a single enzyme-catalytic unit, but an entire multi-enzyme system. This follows not only from the structural similarity (similar power of the multitude of agents), but also from the functional representations. Indeed, the enzyme-mimetic catalysis is increasingly focused on reproducing multi-enzyme systems [99], starting with triple-enzyme mimetic models [100,101] (see Figure 5), as well as on introducing polynuclear complexes [102]. It can be accurately proved that mimetic simulation of enzyme catalysis [103], in some cases, is conceptually associated with the phase of chemical coupling [104], which ensures (similar to cross-catalytic mechanisms with developed feedback loops) the transition of catalysis to a higher stage of evolutionary development. According to the last cited work, this can be directly used in the analysis of biomimetic chemical systems (as close as possible to the conditions of the living systems, since coupled reactions are the basis of biochemical reactions). However, from the point of view of biological expediency, the chemical coupling of reactions in the system is the most effective in the case of colocalization of enzymes and cofactors upon compartmentalization.
Therefore, cytomimetic modeling of chemical coupling in enzymatically-mediated photosynthesis processes implies the application of immobilization of the enzymes (or enzymes and cofactors, or coenzymes, especially in the case of multienzyme complexes with cofactors [105,106] / coenzymes [107,108]; see Figure 6) on a planar carrier imitating biological membranes with the intercalated agents of the indicated type [109,110], which will also ensure the kinetic consistency of the model and its biological prototype [111,112]. The highest form of the model implementation with the participation of immobilized multienzyme complexes is, by definition, a biomimetic scheme based on “artificial cells” with a membrane [113,114,115]. Consequently, the next step in analyzing the criteria of a photosynthesis model with an enzymatic or enzyme-mimetic link should be the consideration of membrane or membrane-mimetic systems (and the processes they implement), respectively.
5.4. Membrane processes.
According to [82], “in more detailed models, we must take into account all processes proceeding in the photosynthetic membrane: for example, … proton transmembrane transfer, transfer of other ions, generation of electric and electrochemical potentials and their effects on electron flows” (the following ATP–synthase activity; the role of buffer groups in the luminal and stromal thylakoid space is not relevant for the stages in which ATP–synthase and the luminal and stromal thylakoid space were absent).
5.4.1. The need for phase separation and compartmentalization for matching the kinetic model of primary photosynthetic processes.
First, any system that simulates photosynthesis completely must have a compartmentalizing interface / membrane to provide the buildup of electric and electrochemical potentials, transmembrane proton fluxes / ion fluxes across the membrane to match the generalized kinetic model of primary photosynthetic processes.
5.4.2. Double electric layer is necessary for reversible charging-discharging and electrochemical / photoelectrochemical oxidation-reduction processes.
Secondly, in the case of dependence of the rate constants on transmembrane electric potential in the model circuit, photosynthetic membrane can be considered as a special capacitor (transmembrane electron and ion transport is accompanied by the formation of a transmembrane electric potential, which in turn affects electron and ion fluxes). In other words, a double electric layer (see Frig. 1) must exist in the structure to ensure reverse charge-discharge and, at the same time, electrochemical / photoelectrochemical oxidation-reduction, as in photosynthesis models, where rapid charge separation in photochemical pairs, electron transport from the inner to the outer membrane surfaces correspond to capacitor membrane properties.
5.4.3. Accounting for the kinetics of membrane processes.
Thirdly, the redox kinetics of photosynthesis should be taken into account – the model structure must inevitably simulate the changes in the redox states of electron carriers, as well as the changes in electric / electrochemical potential values in the photosynthetic membrane.
5.4.4. Accounting for the membrane geometry and diffusion limitations.
Fourth, since it is known that subcellular energy-transforming redox reactions occur in the limited space between membranes of complex geometry, the model should take into account the size effects and the geometry of membrane-mimetic or functionally adequate protobiological structures, at least if we are talking about the processes of restricted diffusion of the mobile electron carriers in photosynthetic membrane, in particular, during regulation of the photosynthetic electron transport by transmembrane electrochemical potential.
5.4.5. The need for a reaction-diffusion approach to the emergence of photosynthesis.
Fifth, since using only differential equations it is difficult to take into account the spatial heterogeneity and complex geometry of interacting macromolecules, as well as the interior of the photosynthetic membrane where these interactions occur, a reaction-diffusion approach is also needed to describe the processes of morphology varying (“morphogenesis”) of structures under irradiation, or N-agent, multiparticle analysis (indifferent to the chemical aspect of their organization, provided that a kinetic similarity of the qualitative solutions is ensured). Since photosynthesis in the boundary conditions created by membranes with different shapes and specific surface is connected with the variability of the geometry, ion strength, pH, and transmembrane proton transfer in the direction opposite that of the concentration gradient is coupled to the electron transport, it seems necessary to use a reaction with diffusion for its modeling, at least for gradient fractionation, which is necessary for functioning of the photochemioosmotic coupling mechanisms observed on the membranes.
5.4.6. Accounting for the chemiosmotic coupling.
Sixth, according to the principle of chemiosmotic coupling (Figure 7), electron transport is coupled with the formation of a transmembrane difference of electrochemical potentials for protons (and the process of proton transport in a photosynthetic system includes at least three proton transfer mechanisms: release of protons, lateral diffusion and passive leakage of protons), while diffusion and electrostatics determine the overall dynamics of the processes. It is well known that transmembrane proton transport / transmembrane transport of other ions, providing the formation of an electrochemical potential, is an important stage of energy transformation in the primary processes of photosynthesis, while spatiotemporal evolution of electrochemical potential ΔμH + in photosynthetic membrane implies integration of electrogenic ion processes and morphogenesis / reaction-diffusion processes involving these ions in the membranes. Mitchell, who applied the principles of chemosmotic coupling to photosynthetic phosphorylation, as well as some of his supporters and opponents, made a decisive contribution to the identification of these mechanisms [116,117,118,119]. However, in that period no one studied the dependence or correlation between the redox parameters in the system, reaction-diffusion processes and bioelectrogenesis with the reaction volume and the boundary conditions (membranes).
5.4.7. Accounting for the kinetics of the membrane potential formation.
However, the direct modeling of the cyclic electron transport demonstrates that the experimentally observed redox kinetics of the electron carriers is determined not only by concentrations, redox states, and conformations of the proteins, but also by the spatial distribution of the mobile carriers and the configuration of the reaction volume. It is well known that electrogenic changes in the physiological state leading to different pH and salt concentrations result in changes in the geometry of the system, while changes in the geometrical size change the reaction rate greatly, and thus serve in photosynthetic systems as an effective regulatory mechanism. Moreover, the observed rate constant of the reaction is known to depend on the geometrical size of the reaction volume. At the same time, osmochemistry and the problems with volume regulation in modern cells are coupled through signaling and the electrochemical membrane potential [120,121,122] (including, for the free volume of the membrane in which pores appear [123]), the importance of which in the early photosynthesis was mentioned above, and which makes the model self-consistent.
5.5. Chemical synthesiss.
In the primary photosynthetic processes in subcellular systems the absorbed light energy is transformed into the free energy of chemical bonds. If we talk about the complete reproduction of photosynthesis, it is necessary to take into account its ultimate stages preceding the indirect physiological effect: generation of primary photoproducts → formation of the primary stable chemical compounds [83]. Modern photosynthesis results in formation of numerous secondary (indirect) products, such as amino acids [124], proteins [125], and lipids [126] in addition to the main (primary) product – carbohydrates due to the developed ultrastructure and molecular machinery of the cell [127]. At the same time the absence of complex metabolic pathways and cycles in the early photosystems that preceded modern forms of photosynthesis resulted in synthesis of a wide vaiety of the primary photoproducts on the semiconductor mineral surface with the electron donors depending on the plausible geochemical conditions. However, in this work we do not focus on the particular products of photochemical reactions occurring during protophotosynthesis, since it is the subject of our forthcoming papers.
6. A unified geochemical basis of prebiotic photo- and chemosynthesis: The possible native mineral constituents of protophotosynthesis.
Let us consider the formation of the above mentioned functions from the standpoint of geochemical evolution and take into account the stages preceding the emergence of protophotosynthetic processes. Indeed, to analyze genesis of photosynthetic processes from the level that already requires their implementation for metabolic purposes is tantamount to tailoring a solution to the question “What for?” in biology, but failing to provide the answer about the geochemical prerequisites for the emergence of these processes (that is, to answer the question “Why such a substitution of functions occurred in evolutionary chemistry?”)
In connection with the well-known catalytic role of mineral structures in the process of abiogenic synthesis of organic substances at prebiological stages [128,129], it is logical to start by considering models of the energy supply of primary metabolic processes involving these structures. Consequently, it is necessary to consider the prototypes of both photoautotrophic (using mineral components as photocatalysts, and using light as the primary energy source), and chemoautotrophic systems (using mineral structures as sources of free chemical energy and as redox catalysts). A mineral substance itself without an external light source is the basis for chemotrophy (chemoautotrophy), so this primordial mechanism of the energy supply for the earliest protometabolic processes will be considered first.
G. Wächtershäuser, the originator of the chemoautotrophic hypothesis of life origin, which subsequently formed the basis of the “FeS-world” concept, suggested that enzyme-free synthesis of organic substances during CO2 reduction could occur in cyclic redox reactions on the surface of catalytically active sulfide minerals widespread on the early Earth (FeS / NiS) [130,131,132]. The energy source for the above redox processes is the reaction of pyrite formation during the interaction of FeS with H2S in volcanic systems or deep-sea geothermal vents [133]. As a result of these processes, a layer of chemisorbed (mainly due to electrostatic interaction) organic molecules is formed on the mineral substrate, which can participate in the “surface metabolism” [134], forming autocatalytic reaction cycles similar to the reverse Krebs cycle observed in the currently living ancient organisms (Figure 8). Being good ligands, these organic compounds could form complexes with Fe2+ ions, which possess higher catalytic activity compared to the mineral surfaces [135]. This facilitated transition from the surface two-dimensional mineral template-dependent catalysis to the three-dimensional redox catalysis with the participation of transition metal complexes.
At the same time, certain “structural motifs” of the active centers of ancient redox catalysts could be “inherited” by living organisms from their mineral predecessors. An example of such "ancient traces" in modern biochemical systems are Fe-S clusters - cofactors of ferredoxin electron transport proteins that are similar in structure to iron-sulfur minerals such as greigite (Fe3S4) and mackinawite. Since the active centers of some other ancient redox enzymes have a similar structure, M. Russell and colleagues suggested that such minerals could play a leading role in the formation of protometabolic redox processes on the early Earth [136]. It should be noted that the mineral greigite (Fe3S4) is a close structural analogue of magnetite (Fe3O4) with a reverse spinel structure and also possesses ferrimagnetic and semiconductor properties [137] (in fact, analogous to magnetite in a reducing atmosphere).
However, even the most detailed scheme of the emergence and development of protometabolism cannot pretend to be a complete model of abiogenesis without the associated mechanism of coevolution of protocellular structures, since the effective maintenance of the concentration gradients and metabolic pathways, by definition, requires compartmentalization of the system. M. Russell et al. proposed a scheme for the emergence of the simplest protocell in microscopic cellular mineral structures based on sedimentary colloidal membranes [138,139]. These “cells”, coated with a layer of iron sulfide, could simultaneously ensure the occurrence of the most important protometabolic reactions and create a local environment for autocatalytic cycles involving the above reactions [140] (Figure 9). According to Russell’s hypothesis, serpentinization process (hydration of ultrabasic rocks under the influence of hydrothermal solutions at high temperature), accompanied by the formation of magnetite due to the partial oxidation of Fe2+ ions and the release of free electrons, could serve as a source of energy and electrons for the reduction of carbon dioxide to biologically-relevant organic substances. (“Serpentinization of ultramafic crust would have continuously supplied hydrogen, methane, ammonia, as well as transition metal ions”[141]). Under the conditions of the early Earth, underwater alkaline hydrothermal vents provided the spontaneous formation of a geochemical proton gradient over dynamic mineral membranes formed at the interface between the solutions with different acidity [142]. These membranes consisted predominantly of iron and nickel sulfides and also could contain catalytically active regions, allowing the possibility of reducing the water-dissolved CO2 on their surface, leading to the synthesis of more complex organic substances [143,144]. At the interface of two solutions with different acidity, capable of interacting and forming colloidal membranes from insoluble sediments, a potential gradient appears and ensures directed proton transfer to create conditions for redox processes on the membrane surface (similar to the chemiosmotic mechanism in Mitchell's theory) [145]. The subsequent sorption and coordination fixation of the organics obtained resulted in the enhancement of the catalytic activity of protometalloenzymes in the redox reactions of the primary metabolism. This hypothesis is also supported by the significant similarity in the structure of some iron-sulfur minerals and the structure of active centers of the ancient metalloenzymes containing iron-sulfur clusters.
The advantages of this hypothesis include the non-equilibrium nature of the processes occurring on dynamic inorganic membranes (from solid state physics to soft matter physics), as well as the presence of compartmentalization in the system of the coupled protometabolic processes, which limits diffusion of reagents and products and increases their local concentration to create optimal conditions for the development of the autocatalytic cycles. However, this model is limited by the use of hydrothermal heat as an energy source and describes only the likely mechanism for the emergence of thermophilic chemoautotrophy, despite the fact that most of the minerals involved in the submarine alkaline hydrothermal vent hypothesis possess semiconductor properties, and hence, are capable of photocatalytic performance when exposed to the sunlight. In recent works by M. Russell the crucial role of another redox-active mineral fougerite (a hydrated layered double Fe2+/Fe3+ hydroxide) in the emergence of life is suggested [146,147]. Considering that this naturally occurring mineral is also a semiconductor, it could participate both in catalytic and photocatalytic redox reactions providing the chemical basis for the prebiotic metabolic pathways.
Another example of abiogenically relevant colloidal systems is inorganic coacervates based on mineral deposits that contain semiconductors. In sedimentary rocks and coals, framboidal pyrite is quite common as the spherical mineral structures 10–50 μm in diameter [148,149] formed by the tiny pyrite crystals 0.5–1.5 μm in size (Figure 10 c,d) . Such structures are believed to be pseudomorphs of ancient microorganisms, although there is still no convincing evidence of their biogenic origin [150,151]. Another hypothesis considers these structures as the products of physicochemical processes on the early Earth. In particular, in the works of L. Minaeva and L. Kizilshtein [148,149] it was experimentally demonstrated that similar structures could be formed as a result of diffusion of H2S into coacervate droplets of Fe(OH)2 (Figure 10 a,b). Such colloidal particles [152] are formed in an aqueous solution of Fe2+ at pH above 5.5 and at low values of Eh (reducing medium) favoring the formation of the colloidal precipitate Fe(OH)2. At the same time, in the presence of natural stabilizers (potentially, abiogenically synthesized organic substances), which impede the rapid deposition of colloidal particles; the suspension can be stabilized at the stage of coacervate droplets. Similar processes could occur in hydrothermal vents rich in iron salts and hydrogen sulfide [153]. Semipermeable pyrite-based inorganic membrane (at the first stage composed of hydrotroilite - FeS·nH2O) could likely combine the functions of sorption, catalysis, and energy or electron source for the protometabolic processes.
Despite the geochemical plausibility of the above hypotheses, they do not provide the possibility for participation of semiconductor iron-containing minerals in the processes of photocatalytic conversion of the solar energy. Meanwhile, the second possible option for the energy supply at the early stages of protocell development is photoautotrophy, which is, in fact, a precursor of photosynthesis.
One of the earliest models of photosynthesis is photovoltaic mineral cell, proposed by S. Granick in 1957 [154] and illustrating the possible mechanisms of processes related to respiration and photosynthesis, based on the simple mineral compounds abundant on the early Earth. This model is based on the interaction of simple mineral or organic components of the primordial soup and catalytically active mineral substrates, in particular magnetite (Fe3O4) with impurities of sulfur atoms. The structure of its spinel-type crystal lattice, consisting of iron atoms with varying degree of oxidation, allows it to function both as an electron donor and acceptor, while its semiconductor properties and photosensitivity at the near-UV region provide the possibility of photoinduced charge generation on the mineral surface. If the oxidized forms of compounds are adsorbed on a magnetite surface, they can be photoreduced, and in that case of adsorption of the reduced species, they can readily undergo photooxidation. According to the author, such physicochemical system could well serve as an energy supplier at an early stage in the development of life and could ensure the ordered operation of the redox processes in primary metabolism [155] (see Figure 11).
Obviously, this primitive model of abiogenic photosynthesis, instead of water, could use a more accessible electron donor, and instead of magnetite, any other abiogenically relevant semiconductor mineral (ZnS, FeS2, etc.). It should be noted that the abundant critique of the Granick’s model in modern literature, in fact, does not refer to his photovoltaic semiconductor cell, but to his general theory about the recapitulation of the stages of the evolutionary development in pigment systems via pigment biosynthesis [155,156]. And if we ignore specific chemistry, then in prebiological conditions at the stage when different photochemical systems were developing ways to convert solar energy, this model seems quite plausible.
Another model of the photochemical conversion of the solar energy with the participation of the simplest mineral compounds was developed by S. Schnoll. He proposed a model inorganic system of the coupled photochemical redox reactions involving Fe(II) and Fe(III) compounds that occur in a bicarbonate buffer solution and provide storage of light energy in the form of buffer capacity [157]. The author claims that such a system of energy conversion reproduces the individual stages of photosynthesis, namely, water photosplitting to form oxygen and reduction of the hydrogen acceptors (Figure 12).
This model is a simplified “biomimetic” scheme that does not pretend to meet the conditions and requirements of abiogenetic modeling. However, in spite of the simplified model nature and non-optimal source of electrons for the early stages of abiotic photosynthesis, it demonstrates the importance of the kinetic coupling between the redox and ion reactions in reversible metabolic cycles, as well as the need for the feedback loops to regulate the processes of energy conversion in the system. A significant drawback of this model is its completely homogeneous nature, which does not provide for the spatial separation of the coupled redox processes and the emergence of concentration gradients between individual compartments. In this regard, it is logical to extrapolate the described mechanisms of primitive photochemical redox transformations in the buffer solution (taking into account the availability of the corresponding electron donor) to heterophase, in particular, microheterogeneous colloidal systems.
7. Mineral photosensitive catalytic semiconductors as the basic actors and components of abiotic protophotosynthesis.
It is known that inorganic semiconductors, like organic dye molecules, are capable to absorb light and photosensitize redox reactions [158]. The absorption of photons with energies exceeding the semiconductor band gap leads to the generation of electron-hole pairs capable of participating in redox reactions with components of the medium. Holes formed in the semiconductor valence band oxidize exogenous electron donors, and electrons generated in the conduction band reduce electron acceptors adsorbed on the semiconductor surface. Moreover, the smaller the particles or the thinner the semiconductor layer, the easier it is for charge carriers to reach the surface of the mineral and provide a high yield of heterophase heterogeneous photocatalytic reactions. Particle size also affects the semiconductor band gap and determines its photosensitivity region: the smaller the particle size, the greater the band gap and, as a consequence, from the standpoint of physics of size effects, there is a possibility to shift the spectral parameters that can adapt the system to radiation of a wide spectral range (and in the presence of particles of different sizes, this can lead to the presence of spectral selection of particles according to their efficiency in converting light of certain wavelengths) [159]. Conductivity of semiconductors depends on temperature and illumination in a certain spectral range, as well as on the presence of impurities, which makes their position in the evolution of solids highly diversified under various geophysical and geochemical conditions. In fact, even when geochemical catalysis by the metal micro- / nanoparticles occurs, as a rule, semiconductor particles / coatings also participate. According to F. Vol’kenshtein, one of the founders of photoelectrochemistry of semiconductors, “most catalysts are semiconductors” [160], moreover, “even in cases when metal is used as a catalyst, the process actually occurs on the surface of semiconductor ... because most metals are covered with a semiconductor coating; and (ibid.): "the electrons and holes of the semiconductor, controlling the electrical, magnetic, optical properties, are at the same time ... the main actors in catalysis." Given the combination of these factors, abiogenic photocatalytic processes should most efficiently proceed on dispersed particles of semiconductors.
Heterogeneous photocatalysis on dispersed semiconductors could be the main mechanism of abiogenic synthesis of organic compounds both in interstellar space and on the surface of the cosmic bodies, in particular, on primitive Earth [161]. Photochemical redox processes involving dispersed photocatalysts could occur in atmosphere of the primitive Earth [162] and on the surface of natural colloidal particles of semiconductor minerals [163], facilitating abiogenic synthesis of organic compounds on their surface and providing conditions for the emergence and development of the early metabolic pathways (Figure 13). In the above cited works of V.N. Parmon it is pointed out that such photocatalytic processes could play an extremely important role in prebiotic evolution.
Under prebiological conditions, undoubtedly, there existed semiconductor minerals capable of photocatalysis, including visible-light-driven photocatalysis [164]. Dispersed inorganic semiconductors of biogenic origin are also known to exhibit photochemical activity in biological systems [165]. Thus, for instance, microparticles of hydrated iron oxide with the structure of ferrihydrite mineral, which are a part of the ferritin protein, can act as a catalyst in redox reactions and provide photosensitized reduction or oxidation of various organic substrates [166]. In addition, the direct photoinduced electron transfer from semiconductor CdS [167] and TiO2 [168] nanoparticles to individual enzymes (hydrogenases), as well as to Clostridium butyricum bacterial cells, with hydrogen being evolved as a result of intra- and extracellular activity of hydrogenases [169], is possible. Integration of the template functions (sorption, local concentration and orientation of the reagents in the adsorption layer), catalysis (spatial colocalization and activation of the reagents on the surface), and the energy source (electron donation) provides dispersed semiconductor particles with a key role in the early development of metabolic pathways.
Iron, zinc and titanium compounds possessing semiconductor properties, as well as their mixtures (i.e. paragenesis) obtained in the form of microparticles and consecutive nanolayers, functioning as semiconductor heterostructures, can be used as photocatalytic mineral carriers [170]. Classical examples of photocatalytic semiconductor mineral structures based on iron compounds are magnetite [171,172] (often coupled with ZnS [173,174,175]), ZnO [176,177,178], SiO2 [179], and TiO2 [180,181,182] (in particular, within osmotic membranes [183]). ZnS and ZnO nanoparticles are also used as photocatalysts, both individually and together [184,185,186]. Apart from the potential of pure sulfide mineral semiconductors, it was also shown that mixed heterostructures based on ZnS and ZnO may be formed at high temperatures in an anoxic and weakly reduced atmosphere of the early Earth, and would possess a higher abiogenic photocatalytic activity in the CO2 reduction [187]. Pyrites [188,189] are another example of mineral structures with semiconductor and photocatalytic properties (including non-iron compounds - cattierite CoS2 [190,191], vaesit NiS2 [192,193] and others, often localized in paragenesis, in contact with autochthonous metals in the deposit [194,195]). Pyrite and its structural analogs, semiconductors, are known to form heterostructures with natural minerals with which they exist in paragenesis. This mechanism of interaction is reproduced in the chemical synthesis of multilayer particles and simple nanoheterostructures (such as FeS2 / Fe2O3, Co:FeS2 / CoS2, NiS2@MoS2, etc. [196,197,198,199]).The contact between pyrite-like semiconductors with other catalytically-active semiconductors, such as TiO2, also results in photocatalytic structures. This has been convincingly shown for FeS2 [200,201], NiS2 [202], CoS2 [203,204], as well as for a number of naturally enriched mineral structures with impurities / admixtures [205].
It is possible that the interactions between many microelements and ultramicroelements in a modern cell are a consequence of their coexistence in geochemical, in particular, in paragenetic conditions of the origin of the corresponding structures and the location of substrates with the corresponding composition, that is, “emitters” of the certain ions. Their chemical coupling is in fact the coupling of their physicochemical functions in the cell, and the separation of roles represents their specific physicochemical roles at the divergence stage: one substrate could perform functions that another could not, for example, assimilation of the light energy using one “heir” of a mineral semiconductor substrate could be coupled with the electron transfer mediated by the “heir” of another mineral substrate.
To date, the most complete and consistent concept of autotrophic life origin, describing the possibility of inorganic photosynthesis based on semiconductor minerals, was proposed by A. Mulkidjanian [206,207]. In this concept, the key role is assigned to the porous mineral sediments with semiconductor properties (zinc and manganese sulfides) that serve as abiogenic photocatalysts for CO2 reduction in the synthesis of primary organic compounds (Figure 14). In addition to this function, ZnS and MnS films deposited on a clay / zeolite matrix could protect organic substances from photodissociation under the influence of harmful UV radiation, and also serve as the templates during their polycondensation. Despite the fact that colloidal suspensions of mineral semiconductors, including ZnS, are known to produce reactive oxygen species under illumination, particularly hydroxyl radicals and peroxides, which are powerful oxidizing agents for most organic compounds (this is commonly used in disinfection and wastewater treatment [208,209,210,211,212]), the absence of oxygen in the early Archean atmosphere prevented prebiotic organic compounds from catalytic photooxidation.
Apart from layers, zinc and manganese sulfides could be deposited in the form of colloidal particles of photocatalysts with a large specific surface area. Such ZnS-enriched sediments could be formed near continental geothermal vents, where the water enriched with transition metal cations (Zn2+, Mn2+, etc.) was in contact with hydrogen sulfide, which simultaneously formed poorly soluble sulfides and allowed to compensate the electron deficiency during photooxidation of the semiconductor [213,214]. An additional argument in favor of this concept is the widespread occurrence of zinc ions in the structure of the most evolutionary ancient proteins [215]. The effectiveness of dispersed ZnS as a photocatalyst for prebiologically relevant syntheses has been confirmed by numerous experimental works [216,217,218,219]. In particular, it has been shown that single stages of the ancient reverse Krebs cycle which provides synthesis of organic metabolites from carbon dioxide, could readily proceed under illumination in the absence of specific enzymes with the participation of colloidal ZnS as a photocatalyst [220,221,222,223]. The likely presence of semiconducting colloidal particles in the oceans of the prebiotic Earth suggests that photochemical reactions on their surfaces could have played a significant role in the synthesis of organic molecules [224,225]. Doping of ZnS with copper ions makes it possible to shift its absorption band to the lower energies and significantly increase light utilization efficiency of the photocatalyst [225,226].
The absence of similar experimental studies with iron sulfides as photocatalysts in the literature is apparently due not so much to their limited photochemical activity as to the impossibility of its manifestation in modern oxygen-rich atmosphere due to the rapid redox transformation of the photocatalyst. However, on the early Earth, photochemical processes with iron sulfides could undoubtedly proceed by analogy with the heterogeneous photocatalytic syntheses considered above. Combination of photosensitivity and redox activity for these minerals could significantly expand their catalytic properties and ensure coevolution of the photodependent and dark redox processes of the early metabolism. In this case, the difference in the band gap for zinc and iron sulfides, which ensures absorption in different spectral regions, made it possible for both types of semiconductors to coexist as a part of a common “mineral community”. Thus, different minerals in a primary “semiconductor world” could simultaneously carry out abiogenic photosynthesis and redox catalysis with the further divergence upon specialization to the corresponding functions.
8. Geochemical consistency and "semiconductor worlds" in abiogenesis.
Thus, it is obvious that colloidal semiconductor particles could perform photochemical (pre-photosynthetic) functions within the simplest prebiological or other compartmentalized (pre-membrane) nonequilibrium systems, providing them with active reducing agents, that is, photocatalytic reactions on the surfaces of dispersed semiconductors with adsorbed proto-enzymes could form the basis of protometabolism [165]. In this regard, it is logical to compare their capabilities with the list of requirements for the photosynthetic systems [82,83] described in Section 5.
Strictly speaking, below we discuss not only geochemical consistency, but rather a more specific photogeochemical consistency [227]. The latter includes not only the presence of certain carriers and photostability of the medium under irradiation, but also the ability to perform photocatalysis, or photoredox behavior under given environmental conditions, as well as the ability to provide photochemical coupling [104] of the reactions between various adsorbates. The final aim of such formulation of the problem, including correlating with the particular criteria for “photosynthetic” processes, phenomena and mechanisms, is a logical transition from light to life [228]. According to the last cited work, the subject of this transition is evolution of the structural motifs responsible for redox reactions (the biological “transistors”) across the tree of life, and the photogeochemical reactions on minerals that ultimately came to be the driving force for these biological reactions.
At the same time, the bottom up and top down approaches are equally well suited as arguments for this kind of problem statement. Bottom up concepts often come from the photochemistry of the simple compounds (starting from the earliest and most classical works from the 1930s [229,230] and further [231], since this area has been exhaustively explored over the past 80 years) involving mineral surfaces. This approach answers the question “how photochemical and geothermal energy produces electron donors and acceptors through geophysical processes / photogeochemical reactions, if allowed to persist over geological time scale” [232]). The top down approach proceeds from the paths of electronic transport in modern microorganisms, which inevitably comply with the principles of geochemical and evolutionary consistency. Because of this, the evolutionary development of electron transport pathways in a natural geochemical environment is now considered as the contribution of electron transfer in the coevolution of the biosphere and geosphere [233]. The top down approach also is known to appeal to the “bottom up prototypes” on the basis of validation / verification logic. It is often noted (see the last cited work) that “photogeochemical, abiotic formation of high-energy chemical products is sacrificial rather than catalytic” and “however, these reactions potentially provided prebiotic substrates for the subsequent evolution of biological catalysis.”
When analyzing primary photosynthetic organisms that did not have complex biomacromolecular systems for converting solar energy into the energy of chemical bonds, it is logical to start from the most primitive photosensitive agents widely available in the environment that can ensure photoinduced redox processes. One of the most likely candidates for the role of such agents in conditions of the early Earth are minerals with semiconductor properties. These include, first of all, sulfide minerals such as FeS2 (pyrite), ZnS (sphalerite), Fe1-xS (pyrrhotite), MnS (alabandite), MoS2 (molybdenite), PbS (galena), Cu2S (chalcocyte), CuS (covellite), CuFeS2 (chalcopyrite), FeAsS (arsenopyrite) and some others, as well as, under more oxidizing conditions, such oxide minerals as Fe3O4 (magnetite), α-Fe2O3 (hematite), MnO2 (pyrolusite), TiO2 ( rutile), ZnO (zincite), Cu2O (cuprite), CuO (tenorite), SnO2 (cassiterite), etc. [234]. The advantage of inorganic semiconductors in comparison with π-conjugated organic molecules is higher stability and the possibility of multi-electron photoprocesses.
Under native conditions the existence of absolutely pure minerals of a single stoichiometric composition is an exception rather than a rule. The naturally forming surfaces most often have a mixed composition (especially manifested in the case of layer-by-layer deposition under unstable natural conditions characterized by nonequilibrium in the distribution of chemical agents). Formation of pure semiconductor photocatalytic particles of uniform composition in primary conditions was unlikely. The problem of the essence of the mineral species for such structures still remains unsolved [235,236]. At earlier stages in the development of life on Earth (up to the so-called “oxygen explosion”) in a neutral atmosphere and in the presence of hydrogen sulfide as a product of volcanic activity, the transition metal compounds most likely occurred in the form of sulfide minerals. Among them, iron sulfides were one of the most common salts.
Pure pyrite (FeS2) is a promising earth-abundant semiconductor for thin-film inorganic solar cells and photocatalytic applications. It has a relatively large absorption coefficient of α > 105 cm−1 for photon energies larger than about 1.3 eV, such that a thin layer of less than 1 Å should effectively absorb the visible part of the solar spectrum. The bandgap of pyrite of 0.95 eV is on the borderline of what might still be considered acceptable for a high-efficiency single-junction device, it shows both n- and p-type conduction [237,238]. It is actively used to create inorganic photovoltaic devices [239,240,241,242] and nanodispersed heterogeneous photocatalysts [243,244,245]. It is well known that pyrite crystals, along with crystals of other mineral semiconductors, were used in the design of the simplest detector radio receiver as a detector diode, due to the property of the pyrite-metal contact, the current can be transmitted mainly in one direction. Prior to pyrite, galena, another semiconductor with a small band gap of 0.4 eV, was used for similar purposes [246,247,248,249,250]. The electrogenic properties of pyrite as a high-energy material are evident from its ability to produce sparks upon impact, which explains the Greek name “pyrite”. The electrical conductivity of pyrite is largely determined by the presence of impurities [251] and the chemical composition of the surface [252], which is easily oxidized in a modern oxygen atmosphere. Nevertheless, it is physically obvious that in conditions of the early Earth, a non-oxidizing atmosphere ensured the resistance of thin films and colloidal dispersions of pyrite to oxidation. This fact allows us to consider them as abiogenically relevant potential photocatalysts in prebiotic syntheses. In addition to the photocatalytic reduction of CO2, the ability to abiogenically fix atmospheric nitrogen under irradiation to form ammonium salts may be characteristic of pyrite in the early Earth atmospheric conditions. This process could be essential for the synthesis of nitrogen-containing organic substances, including amino acids and nitrogenous bases, under abiogenic conditions in the absence of specialized enzymatic systems for nitrogen fixation and could be a predecestor of the nitrogen fixation system [253].
Among iron oxides, hematite, wustite, maghemite and magnetite are also semiconductors, which are characterized by emergence of photoconductivity upon generation of electron-hole pairs with subsequent separation of charges or their recombination [254,255]. An important condition for this process is contact with an electrolyte solution, so photoinduced redox reactions could occur only on the surface of mineral particles wetted by the waters of the Primary Ocean or land saltwater reservoirs. The presence of impurity ions changes the band gap and the type of conductivity of the semiconductor; therefore, the presence of natural impurities in mineral structures significantly expands their spectral sensitivity range and possible redox potentials as compared to the ideal crystals of pure laboratory-grown substances.
All of the above minerals, both in pure form and in paragenesis with each other and with other ore-forming minerals, can be natural sources of electrons for photoinduced redox processes occurring on their surface (in the adsorption layer). At the same time, the film thickness of the semiconductor mineral, optimal for photoinduced processes, is quite small (50-100 nm) and is limited by the depth of radiation penetration [240]. Therefore, surface photosensitive layers may form due to the interaction of bulk mineral rocks with components of the atmosphere and hydrosphere (an oxide film in a modern atmosphere and a sulfide film in the case of a reducing atmosphere on the early Earth enriched with volcanic gas components).
In addition to changes in the chemical composition of the surface of the initial minerals under the influence of environmental factors, photosensitive layers or films based on inorganic semiconductors can easily be obtained by precipitation of sulfide minerals from a solution rich in transition metal salts when interacting with water-dissolved sulfides (similar to semipermeable dynamic colloidal membranes in chemobrionics or the so-called "chemical gardens") [256,257]. Such laboratory models have indicative prototypes in the geological nature (Figure 15).
Light sensitivity and semiconductor properties are also typical of organic semiconductors of natural geochemical origin. Thus, polycyclic aromatic hydrocarbons (PAHs), widely distributed in the Universe and found in many astronomical bodies [258,259], as well as a number of other π-conjugated organic compounds, in particular tetrapyrroles, exhibit semiconductor properties under the influence of radiation. Unlike inorganic semiconductors with a band gap of 1-2 eV, molecular crystals or amorphous films of organic semiconductors have a wider bandgap of 2-4 eV and acquire electronic or hole conductivity when irradiated with visible light. In this case, initially photoexcitation of molecules leads to the generation of Frenkel excitons, which then decay to form free charge carriers. Polycyclic aromatic hydrocarbons (PAHs) with an expanded π-electronic system (tetracene, pentacene and their isomers and structural analogues) in the crystalline state are typical organic semiconductors [260,261,262] and have a high absorption coefficient in the UV spectral range. On their basis, OFETs are often made in the form of a thin surface film on quartz glass [263,264,265,266], which leads to comparisons with template phenomena in the evolution of abiogenic organic matter on quartz substrates [266,267,268].
Based on the PAHs / aromatic world hypothesis [269,270], these compounds could be not only structural precursors (in particular, the initial form of organic matter for RNA synthesis and assembly), but also functional precursors of photosensitive pigments and, along with tetrapyrroles, they could support primary processes of solar energy conversion on the early Earth. In this case, the ionizing radiation of the Sun could initiate gradual hydrophilization of RAH molecules by replacing some hydrogen atoms by hydroxy and other functional groups. Such hydrophilization increases their solubility in water, and also gives the original molecule amphiphilic properties, the ability to self-assemble in a polar aqueous medium, to form supramolecular assemblies (micellar systems, vesicles and bilayers) [271] and to be incorporated into simple membrane structures. This process also led to a gradual shift of the absorption maxima of the compounds to the visible region of the spectrum, which made it possible to significantly expand the range of radiation effectively absorbed by the photosensitizing molecules.
In other words, firstly, the mineral semiconductors in conditions of the early Earth were abundant, and secondly, the classical inorganic substances of many “worlds” fully correspond to these semiconductors or their natural geochemical derivatives produced under the existing environmental conditions with high probability (in some cases - almost inevitably).
The question of how they should have been organized (not only topographically, but also from the standpoint of their structural kinetics) in order to integrate into biological functions during abiogenesis, must be solved with the "reverse engineering" of the products of evolution - processes and topographic forms of compartmentalization of the early and modern types of photosynthesis, sensu lato. This is needed to provide comparative analysis between the basic criteria of the latter (according to the above list of papers [82,83]) and the physicochemical capabilities for their development and reproduction (in particular, laboratory modeling) based on semiconductors.
9. Self-organization in “semiconductor worlds” under the solar energy pumping: Energy supply for the processes of protophotosynthesis and abiogenesis.
The problem of the emergence of primary photosynthesis, or the appearance of the first mechanisms of energy consumption necessary for the operation of prebiological systems, cannot be considered in isolation from the problem of the energy supply of abiogenesis on natural surfaces that absorb light energy. Therefore, we can say that, by modeling processes in natural protophotosynthetic structures during the initial formation of their function, we can answer some questions about self-organization of autotrophic protobionts as such. Since many attempts at artificial synthesis of protocells have failed without arriving at a functional result (necessary and sufficient - sensu stricto), it is advisable to give nature the opportunity to reproduce in laboratory conditions its own typical forms and functional systems, according to the principles of physical chemistry or chemical physics independent of the researcher, by ensuring energy input in the form used by nature for self-organization (photochemistry) and / or for lowering the barrier to certain chemical reactions (catalysis). The quantum self-assembly of functional protocell models based on PNA and photosensitizer [75,76,77] (Figure 16) allows us to hope that this is possible. At the same time, from the point of view of reconstruction of the first stages of photosynthesis based on recapitulation by the metabolism of its origin [272], it is desirable to reproduce its origin from those self-assembly mechanisms that directly follow from the physics and chemistry of this “nonequilibrium process with optical pumping”.
It is well known that in “artificial photosynthesis” the principles of self-organization or self-assembly [273,274,275], as well as catalytic assemblies of nanodispersed carriers (vesicles and micells - see Figure 17), which under natural conditions (due to size effects) are usually formed by self-assembly [276], are widely used. Deviation from the biological prototype often leads to the focus on fashionable nanostructures that did not participate in the native photosynthesis [277]. However, it is obvious from the first principles (ab initio) that nothing prevents the use of this principle in an emergent and evolutionary context. It means that only one step remains before the validation of the recapitulation / emergent mechanism using this principle to prove its consistency (when the principle of "assembly and photoactivation recapitulate evolution" [278] is interpolated to the early stages of the photosynthesis origin). In this case, we will need many manifestations of self-organization and self-assembly of substances, considered as probable primary executors of the protophotosynthetic function (or the preceding photocatalytic processes), in particular, semiconductors.
Self-organization will be considered a correct model of the structure formation only in the case of equifinality - the similarity of the resulting and photosynthetic structures (the same forms of self-organization that are used for semiconductors in artificial photosynthesis are too sophisticated and often cannot be correlated with a natural prototype - photosynthesis [279,280]). Since we are talking about the photoinduced processes, both photochemical and photophysical approaches to the energy pumping seem to be quite rational. In the case of semiconductors, it is logical to analyze energy pumping and self-organization in semiconductor systems. Based on these provisions, we will begin a comparative analysis of the above requirements for the processes of elementary photosynthesis with the conditions and the possibility of their reproduction on mineral (photo)semiconductors. In this case, it will become possible to establish correspondence between the protophotosynthetic processes occuring on mineral semiconductors with the processes of natural photosynthesis listed in Section 5, according to [82,83].
10. From semiconductor-based artificial photosynthesis to abiotic photosynthesis / protophotosynthesis modeling.
The first biophysically necessary characteristics of photosynthesis, according to [82,83], is the ability to absorb light and to transform light quanta into the energy of separated and stabilized charges. The ability of semiconductors to implement primary photoprocesses in the chain “absorption of a light quantum ... and generation of electronic excited states → transfer of electronic excitation energy → ... generation of primary photoproducts” (sensu lato), as a rule, does not raise objections. Artificial photosynthesis, starting with the first research by M. Graetzel [281], has been associated with semiconductors, providing light energy harvesting and conversion (or by spectral sensitization, when dyes were used to mediate the process). This process usually occurs on semiconductor – liquid interfaces and junctions [282], and ensures interaction with the natural environment (necessary in the case of reproduction of the stages: “generation of primary photoproducts → formation of primary stable chemical compounds”) including, often, the reaction-diffusion nature of the process. Thus, the following processes are performed on semiconductors: water splitting [283,284] and carbon dioxide reduction / photoelectrochemical conversion [285,286], as well as photocatalytic fixation of CO2 to afford higher organic compounds [287].
The above processes are photochemical, and not only photophysical in nature, although the early concepts on the initial stages of photosynthesis considered only the physical - electronic / semiconductor aspect [288]. For this reason the simplified models of artificial photosynthesis followed along the same path. Meanwhile, there is a direct connection between the photophysical and photochemical sides of the issue. It is well known that even in the early works of Graetzel on artificial photosynthesis [289], the analysis moves from purely photophysical processes (such as, light-induced charge carrier generation and further charge separation) to their coupling with photochemical processes (photoredox reactions involving suitable electron donor / acceptor chemical transformation), that close the energy conversion cycle in thesystems simulating photosynthetic processes within the framework of microheterogeneous photochemistry and redox catalysis (see Figure 18, Figure 19). M. Graetzel et al. proposed to use mineral colloidal semiconductor particles as light absorbing units, in particular, Fe2O3 particles in [290,291], and in other works - various combinations of ZnO [292,293,294,295] or ZnS [296] particles. Thus, this is directly related to semiconductor mineral media corresponding to the conditions of the primary forms of abiogenic photosynthesis indicated above.
On the other hand, the biomimetic nature of the early works of M. Graetzel, directly associated with artificial photosynthesis, clearly correlates with the evolutionary development of primary photoprocesses (for example, compare Figure 18, Figure 19). and selection (not in a strict modern biological interpretation of this term, but in a broader interpretation, probably characteristic of the early prebiological evolutionary stage [297], despite the inconsistency of these simplified constructions for the biological evolution of the modern type [298,299]), since the final stage of development is the modern forms of photosynthesis using metalloporphyrins.
The primary idea underlying the work on artificial Graetzel-style photosynthesis was the bionic reproduction of photobioelectrogenesis using chlorophyll in the late 1960s and early 1970s [300,301]. Initially, the bottom up biomimetic approach in these studies coexisted with the top down approach (when chlorophyll isolated from biological objects such as spinach was used). Currently, this approach is generally preserved, but porphyrins [302] are used instead (including zinc-containing metalloporphyrins [303,304,305,306,307]), especially popular in the 1980s, and phthalocyanines (including zinc-containing ones [308,309,310,311]) more popular in the 21st century, including those applied to photoelectrodes from substrates of the above-mentioned mineral composition.
In this case, observing both the geochemical sequence (bottom up) and the evolutionary (top down) biomimetic strategy allows us to consider this approach as highly relevant for reproducing primary photoprocesses.
11. Photoinduced redox processes and chemical gradients in the coevolution of protorespiration and prothotosynthesis: Is it possible to model redox catalysis in photosynthesis without multienzyme complexes?
The second biophysically necessary property of photosynthesis pointed out in Section 5, according to the criteria listed in [82,83], is redox processes localized on the (proto)membrane, leading to the emergence of a charge gradient as a bioenergetic prerequisite.
The photoinduced redox mechanism, which has the illumination of the system at the beginning and, at the end, the rearrangement of gradients at the system-media interface (with its special case: the final stage of membrane depolarization in photosynthesis), can be implemented on the gradients of the charge-carrier concentration using membrane-like structures and redox-catalytic agent that replaces the functions of multienzyme complexes in photosynthetic electron transport.
In accordance with the well-known consistency criterion for co-evolution of respiration and photosynthesis [312,313], we must proceed, firstly, from the fact that the water splitting system of the ordinary photosynthesis makes it possible to use water as an electron donor — a compound with a high redox potential that is not prone to act in the role of a reducing agent, and secondly, from the "polyphyletic" origin of many photosystems that make it possible to use compounds with lower redox potential (including hydrogen, sulfides, hydrogen sulfide, sulfur, ferrous ions, nitrites) as an electron donor or the cyclic electron flow around the photosystem.
In other words, unlike engineering biomimetics, which comes, as a rule, from the most efficient modern systems, we should not be tied to the concrete implementation of the principles of photosynthesis and high-efficiency redox catalysis, but we can consider the “strictly physical” aspect of redox processes, that is the presence of universal electron donors and their transfer paths in one or another protosystem. In electron transport, an agent with a more negative redox potential serves as a reducing agent (electron donor), and with a more positive redox potential, it serves as an oxidizing agent (electron acceptor).
The presence of a catalytic agent, required in Section 5.2.1, can be achieved using redox catalysis on semiconductors, in particular, photoredox catalysis [314,315,316,317,318,319]. When modeling the catalytic functions of multi-enzyme complexes using semiconductors coexisting with other mineral media that are relevant to mineral photosynthesis (for example, in the processes of mineral self-organization of iron ores [320]), especially, during the initial compartmentalization of processes and the presence of newly-forming physicochemical gradients in the system [321], it is possible to predict the presence of exotic forms of catalytic kinetics (including heterogeneous catalytic kinetics) in such systems, including indirect cross-catalysis and a huge range of feedback loops - not only of a purely reaction type, but also of reaction-diffusion type (characteristic of distributed systems with self-organization of complex forms of charge transfer [322,323]).
Modern physical approaches allow for not only combined biomimetic enzymatic catalysis and photocatalysis in photoinduced synthesis of organic substances and in carbon dioxide conversion [324,325] (including using artificial enzymes and artificial-enzyme-based structures [326,327], which is equivalent not to photocatalyst – enzyme coupled artificial photosynthesis [328], but to biomimetic photocatalysis / biomimetic redox-catalysis [329,330] or enzyme-mimetic catalysis [331]), but also for “protobiomimetic / proto-enzyme mimetic” catalysis using mineral carriers both to contact with several enzymes and to form a peculiar multi-enzymatic complex, and to independently perform “enzymatic” functions (for example, MoS2 [332] as a peroxidase enzyme and visible-light-induced photocatalyst).
The progress of this industry is moving from the simplest techniques requiring the use of biogenic enzymes [333] to biomimetic catalysis with a growing role of non-enzymatic components, which are increasingly moving away from the bionic prototype. (From molecularly imprinted polymeric carriers [334] to functional inorganic ones and, as a rule, having mineral prototypes, to systems that often perform a number of complementary functions, such as photoconductor sensitivity and magnetic charge deflection [335], photoinjection of which under prebiological conditions could be achieved by the photoeffect [336], without any developed biochemical machinery). Thus, the development trend in this industry recapitulates the top down direction of photocatalytic / photosynthetic systems along the evolutionary route. At the same time, as indicated above, at the initial stage there are no restrictions on specialization, which justifies the divergent development of those processes that later transform into conjugated catalytic chains, starting with the most elementary forms of the catalytic process on semiconductors that are not identical in composition, due to natural conditions of their formation.
The need for bioelectrochemical redox coupling mechanisms, the dependence of the rate parameters on the transmembrane electric field and the emphasis on electrostatic interactions for effective electron transfer, which may even lead to electrostatic (non-covalent) interaction, in this case has direct parallels, provided by energy or entropy configurations of supramolecular systems interacting with the medium, like enzymes, as well as with semiconductor matrices, which in this case are of equal importance as actors in the catalytic process, not limited to enzymatic catalysis. This is manifested in well-known, and, in many cases, generally accepted interpretations of integral semiconductor and enzymatic catalysis as a bioelectrochemical and electrocatalytic process with the participation of an active center [337] proceeding at the interfaces, that is, heterojunctions at the photocatalyst / photoenzyme boundary [338].
It is worth noting that organomineral interfaces are used as the most energy-efficient common heterojunctions, including compositions known as media components where mineral photosynthesis could potentially develop. The very fact of photoinduced prebiological organic synthesis and early chemical evolution on such surfaces can be considered as photosynthesis sensu lato (taking into account the synthesis of various primary and secondary organic products - see Section 1 of this article, where the definitions of photosynthesis are considered). The latter could comprise metal compounds found in the natural environment, such as Fe2O3 [339], ZnS and ZnO (this property is used in ChemFET / EnzymeFET [340]), and TiO2 (relevant not only for abiogenesis on the Earth, but also for other astronomical bodies, for example, Mars [341,342,343]). From the point of view of further evolution of the enzyme-free (or pre-enzymatic) synthesis, it is worth noting that porphyrins, which were considered earlier during the analysis of Graetzel's works as the next step in the evolution of biomimetic photosynthesis and photoelectric conversion, are used as components of the energy-efficient heterojunctions in the enzyme-free (in fact, enzyme-asymmetric) coordination substrate. The same principle is used in the non-enzymatic sensing of some biomacromolecules (although the use of more stable phthalocyanines [344] is more effective in this case), and the photosensitivity and photoactivity at the contact - heterojunction - are of vital importance. Interfacial phenomena at heterojunctions during molecular evolution could provide combination of photocatalysis, photoinduced charge carrier transfer, and the emergence of a gradient on mineral "protomembranes".
12. Inseparable complex of photoinduced phenomena on a semiconductor protophotosynthetic interface as a set of reaction-diffusion processes.
It is well known that the emergent combination of catalysis, photoinduced transport of the charge carriers and photoelectrophysical reception, providing various kinetics of photoinduced processes on a single microscopic structure, was an evolutionary prerequisite at the stage when functions that subsequently formed modern photosynthesis were under development. (Since in the absence of integration and spatial coupling between them, photosynthesis could not become a coordinated network of processes both in space and in time, but would evolve similar to autonomously evolved single processes that preceded it). In this case, the kinetics and catalysis at the early stages of the developing precursor photosynthesis must be interpreted based on the possibility of different processes with a single physical basis (including conflicting and spatially separated, reversible and influencing each other indirectly through feedback loops) developing on a single precursor structure – up to the evolutionary separation of the functions of synthesis, photosensitivity and transfer (of charge, particles and mass transfer in the framework of the reaction-diffusion photosynthesis [345,346] - including its early forms [347]). Based on the above, let us consider the requirements to photocatalysis.
The fact that biomimetic catalysis, which models single stages of protophotosynthetic catalysis, should serve as redox catalysis, photo-redox catalysis (with photo-pumping), which has been demonstrated in Section 5.3. This imposes obvious electrochemical / photoelectrochemical requirements on the substance of these catalytic mechanisms in order to reproduce the phenomena of photosynthesis on semiconductors and to satisfy, by definition, the IUPAC terminological recommendations on photoelectrochemical energy conversion and semiconductor electrochemistry [348].
According to the above mentioned recommendations, in photoelectrochemical structures one should take into account the energy states and their densities in electrolytes containing redox systems, as well as diffusion length and mobility of electrons and holes in semiconductor, that is, the redox conditions are obviously set to satisfy the reaction-diffusion dynamics of the charge carrier transport, involved in their formation. At the same time, changes in the redox potential of the electrolyte are identified with Fermi levels (the equivalent of the chemical potential of a system in its basic state at the absolute zero), and for the concentration of nonequilibrium charge carriers - with the quasi Fermi level. During semiconductor illumination for photoelectrochemical energy conversion quasi Fermi levels should be considered, since the quasilevels of electrons and holes do not coincide under such conditions, and the concentrations of nonequilibrium charge carriers in the semiconductor under illumination are non-stationary. These changes can, in general, be measured by the standard voltmetric equipment for electrochemical measurements, revealing complex kinetics (up to autowaves and chemical self-oscillations), since potentiometric methods reveal the Fermi level difference, known as electrochemical potential, rather than pure electric potential (known in electrophysics as Galvani potential). Changes in Fermi level differences in space and time on compartmentalized interfaces where charge and ion fluxes are separated (which also occurs in membrane protocells) being deviations from equilibrium as the overpotential, are definitively correlated with changes in redox potential (regardless of the specific carrier or the presence / absence of bioorganic, in particular bilipid membranes, which are usually associated with the emergence of differently charged ionic gradients and redox gradients at the cell membrane or a protocell / artificial cell boundary). During light excitation the minor carrier density is strongly increased compared to its equilibrium value [348], therefore, at different distances from the semiconductor interface, the activity of photocatalysis and the manifestation of its charge effects, associated with a particular type of carrier, will be different. However, here the regulation of the photo-redox effect as a correlate of the distance dependence of the quasi-Fermi level as a whole is determined by the diffusion length and the penetration depth of light.
Firstly, within the framework of such approach to photoelectrochemical energy conversion, photocatalysis is a photoinduced reaction-diffusion process (speaking about diffusion of the charge carrier of a specific chemical nature). It can be added that in a distributed system this process is position-sensitive (since with any form of the light distribution, the uniform distribution of diffusion lengths and quasi-Fermi levels in all axes cannot be achieved). This affects an analogue or a physical evolutionary prototype of the kinetic integration of photosynthesis and photomorphogenesis in autotrophic organisms (this has been known in plants since the 1970s, and is associated with redox states, cell enzyme activity, bioenergetics, specific endogenous rhythms or oscillations, mediated mainly by the redox enzymatic machinery [349,350]).
Secondly, the entire micro- / nanoenvironment gets involved in the redox processes, corresponding to the effective length of the distance dependence of the quasi-Fermi level. This allows to reconstruct pre-photosynthetic photoredox processes on catalytic semiconductor structures, as well as to talk about the optimal implementation in the boundary conditions determined by the space charge layer, with the separation of the contribution of surface electron density and volume density of the energy states. Redox regulation in modern biochemical pathways significantly contributes to the catalytic processes, including those realized in photosystems, using macroheterocyclic compounds similar to those used in photosynthesis pathways [351,352,353,354], which allows us to speak about the biomimetic nature of the modeling approach. The criterion for the presence of a space charge layer and charge separation directly appeals to the well-known mechanisms of redox regulation of membrane transport processes [355,356,357] (including processes implemented in the modern photosynthesis machinery by various agents, such as plastoquinone, cytochrome P450, chloroplast membrane proteins, etc. [358,359,360,361,362]), and implies the transition to the next step of evolutionary biomimesis – incorporation of the photosensitizer into the interface.
The idea to incorporate photocatalysts / photo-redox catalysts of protophotosynthesis into membrane-mimetic surfaces for interface charge separation, or to use the integration of their own photocatalytic and membrane-mimetic properties, directly follows from the bionic prototype reproduced in the applied artificial photosynthesis systems [363] (much closer to the bionic target, than the homogeneous modifications [364]). Such a way of complexation of the photosynthetic structure with a simultaneous improvement in functionality could be used during the transition from purely geological photocatalysis and ancestral forms of abiotic photosynthesis on mineral surfaces to the early forms of photosynthetic protobionts (Figure 20). The incorporation of (photo)redox machinery in the soft matter interface originates in “direct biomimetics”, which operates with direct equivalents of natural compounds and often synthesizes them using nature-mimicking synthetic pathways. (This is one of the oldest ways of chemical biomimetics [365,366,367], unlike many others, that does not raise either doubts in biosimilarity criteria or negative assessments of the similarity between the prototype and the model due to their equivalence). Thus, in the simplest adequate models of biological photophysical and photochemical processes based on chlorophyll and carotenoids (as components of “artificial photosynthesis”) the latter were immobilized into micellar surfactant and vesicular systems [368] with micron and submicron sizes. At a later time, discussions were held on the prospects of using nanostructured forms of interfaces for water splitting and water oxidation in artificial photosynthesis [369]. But, from a physical point of view, chemical composition is not the determining criterion for similarity when it comes to phase separation and interface transfer. Thus, separation of charges, as well as ensuring biomimetic kinetic effects (not excluding the facts that are usually associated with charge-chemical selectivity), are also proceed at interfaces of simpler composition, and many authors highlight the correspondence of the structure to physicochemical requirements, which, in the reductionist approach, are limited by the requirements of soft mater physics, which includes colloids, polymers, liquid crystals and mesophases [370,371,372,373].
From the point of view of physics, the boundary conditions can be arbitrary without prejudice to the final biomimetic effect. Therefore, the idea of using supramolecular porous microspheres [374] and mesoporous spheres [375] in artificial photosynthesis, and even the earlier versions base on solid-state structures, if they are capable to functionally simulating heterogeneous-catalytic and selective-diffusion functions, do not cause general discussion.
The aforementioned nature of the interphase contact - heterojunction - helps different photocatalysts / photoconductors on this interface to interact, to simulate functions of not one but several enzyme mimetics or, more accurately, to describe the physics of the phenomenon in terms of the photosynthetic prototype, multienzyme complex modeling. This makes sense not only in the aspect of the similarity of the physical result (which is quite sufficient for technical bionics, but not enough for the evolutionary trend in biomimetics emerging from geological semiconductor catalytic prototypes [376]), but also in the aspect of the early evolutionary mechanisms when the contact with a mineral surface was a natural necessity, and the phases of the substance, formed at the contact with the semiconductor catalyst, as a result of the particle integration by heteroepitaxy mechanisms, often remained in adhesion with the catalytic substrate and were forced to function together with it, being selected as a whole. It should be noted that inorganic chemobryonic membranes shown in Figure 8, Figure 11 and Figure 16 could readily act as inorganic active surfaces or protomembranes, since they are good prototypes for geomimetic catalysis (see Figure 21). (Perhaps the scenario resembled the one for multienzyme complexes at a later stage, taking into account the microenvironment or, if we are speaking of the above membrane scenario, it proceeded in a single "multicatalytic" compartmentalization, ensuring that the processes occurring with its various components were inseparable [377,378]).
13. Towards the formation of precursors of the photosensitive systems and photoenzymes at mineral heterojunctions: From membrane mimetic interfaces in “semiconductor worlds” to “heteroepitaxial worlds”.
Phase interfaces between semiconductors of various compositions [379] are an area of joint implementation of processes that would be impossible within the bulk phase of any one of them. (For example, adaptation of the photoconverting properties of structures, whose components are not sensitive in the accessible spectral range, by controlling the band gap at the boundaries of heterojunctions or mobility and effective masses of charge carriers on them). We can assume that in the prebiological evolution of photosynthesis, adaptation to the spectral properties of the accessible radiation proceeded by similar heterojunction-like mechanisms. This is in good agreement with the requirement for the integration of photoredox functions and electrogenesis functions modulating the photoelectrochemical states of the protophotosynthetic catalyst and the electric double layer on its surface, following from similar requirements for photosynthesis [82,83] (Section 5.3.3, 5.4.2).
Since the 1970s, we have known about the contribution of the double electric layer to the modulation, adsorption and scattering of radiation on membranes / films (membrane-mimetic materials) and other surfaces [380,381,382,383] (which is the basis for dynamic light scattering / methods of photon correlation spectroscopy [384,385]), including the spectrum shift (for example, the electrochromic shift [386], which also occurs in chloroplasts [387,388], in photosynthetic pigment systems [389,390]) and not only for the target structure, but also for the molecules adsorbed on it [391]. Thus, in the case of specific sorption, the spectrochemical “effect of the sorbent on the sorbate” will be identical to the spectral shift effects observed in particularcases of templating, partly involved in Cairns-Smith’s “crystals as genes” mechanisms [392] (reducing it to the level of fitting the geometry of bonds and effective atomic / ionic radii, mutually comparable in terms of conformational and spectrochemical properties, similar to the known physical metaphor [393], which is not associated with genes or the nature of substance). That is, regardless of how the modern mechanisms of inheritance formed, such mechanisms could participate in the prebiological adaptation of microsystems sensitive to the particular spectral ranges, due to the shift of the spectroelectrochemical characteristics mediated by the charge mechanisms.
In the subsequent evolutionary history of modern photosynthesis, this is recapitulated on a more complex biochemical base as an adaptation to natural light conditions in catalytic efficiency of photoenzymes. (It should be noted that these adaptation mechanisms aroused a discussion a few years ago [394,395,396] and some issues have not as yet been resolved, if we set aside discussions about model experiments that illustrate the arguments of the warring parties adaptation than proving anything).
Meanwhile, the formation of precursor photosensitive systems and photoenzymes on mineral heterojunctions could be very plausible and energetically justified, given modern studies of photocatalyst / enzyme heterojunctions fabricated for high-efficiency photoenzyme synergic actions in water media [338,397] (Figure 22) and multielectron redox catalysis based on artificial photoenzymes for abiotic oxidase reaction cascades [398]. (The multielectron redox catalysis, described in the last cited work, can be coupled with multielectron transfer in heterojunctions, well known in electrochemical and radiochemical solid state physics [399]). An electric double layer plays a decisive role in the interaction between the enzyme (in the laboratory conditions - an enzyme mimetic or enzyme model) and the interface of the substrate that controls the photoelectrochemical and spectrochemical behavior of the photoenzyme, as well as in the interaction between the enzyme and the electrolyte medium.
If we consider a control electrochemical substrate with photoinjection of a charge (a mineral precursor of the photosynthetic apparatus), in accordance with the rules of electrochemistry, as an electrode, then in the photocatalytic and photoenzymatic case the interpretation of electrostatic interactions between an enzyme (or precursor / enzyme-mimetic catalysts) and electrode will be equally adequate as a direct electron transfer-type bioelectrocatalysis in the electric double layer [400,401].
From the position of plasmonics, it is logical to expand the boundaries of the interpretation of the double layer as the driving force of the photo (electro) chemical or spectrochemical evolution of protophotocatalytic units and in the case of templating on microparticles of natural metal-bearing rocks, since the double electric layer shifts the spectral bands of surface plasmons at the boundaries of metals and electrolytes [402]. Moreover, as in the case of the “bare” layer considered above, in most cases, such shift can be implemented by changing the redox potential or ionic strength of the medium (as a bathochromic or hypsochrome shift in the case of semisynthetic photoenzymes with tetrapyrrole fragments [403]). This means that in the presence of light-color regulation of redox potentials, the foundations are established for chromoselective photocatalysis [404], which preceded photoenzymes and modern spectrozonal forms of photosynthesis during the propagation / catalytic progress for simpler products of prebiological synthesis of organic matter reversibly adsorbed on the mineral substrates.
14. Similarity between the dynamics of processes in the electric double layer of membrane mimetic semiconductor surfaces and photoinduced membrane potential oscillations in biological cells.
In electrochemistry, the sorption effect on the structure of the catalytic double layer and electrochemical / electrocatalytic kinetics has been the subject of experimental research since the time of A. Frumkin [405,406] and has been well studied during the past 70 years. However, the investigation was limited only to forced modes with galvanostatic or potentiostatic for polarographic or other analytical and chemical purposes, in particular, on installations for the study of half-waves of oxidation-reduction. However, in reality, the kinetics of the double electric layer as a capacitor under native conditions “automatically” implies oscillations or pulsations of the charging-discharging (in the presence of sources of constant charge injection, in particular photo-injection, sources and sinks) [407].
Photoinduced processes (polarization-depolarization) in the dynamics of the double electric layer are well studied. The studies demonstrated not only the effect of the double layer on the transfer of particles, in particular, electrons (including photoinjected ones), through membranes and model membrane-mimetic films and layers (such as Langmuir films in various modifications [408]), but also the ability to control layer oscillations by means of laser radiation, including optically polarized radiation [409,410] (Figure 23). The experiments with dispersed semiconductor particles - quantum dots - demonstrated the presence of a photoinduced nonlinear electrical response [411]. Thus, there is a fairly large pool of data on the photoinduced and photosynchronized dynamics of the double layer, on its use in photocatalysis on surfaces that simulate biological membrane surfaces (for which non-equilibrium mechanics works well – non-equilibrium statistical mechanics gives adequate results of calculating the surface conductivity of the double layer of the living cells [412]) and enzymatic surfaces, including those directly related to photocatalysis (which, ab initio, is demonstrated not only for photoenzymes, but also for photoabzymes [413]) [414].
Moreover, a dynamic non-equilibrium double layer (as defined by the Encyclopedia of Surface and Colloid Science [415]) is associated with the aggregative stability of biocolloids and geocolloids, including those capable of aggregation with the heterointerface formation during the emergence and evolution of photosynthesis and photocatalytic semiconductor heterojunction-assisted phenomena [416]. The last argument is supported, in particular, by the fact that the interaction of metal particles and ions with bacterial cells is satisfactorily interpreted and approximated using non-equilibrium thermodynamic surface models with electric double layer [417], being a particular case of this set of non-equilibrium thermodynamic models [418]. The above case also includes other phenomena of electrochemical kinetics, aggregation equilibrium shift in dispersed systems and limitation of their particle size by zeta-potential values determined by the electric double layer [419,420,421]).
A non-equilibrium electric double layer makes a significant contribution to the ion adsorption dynamics and to charge injection and transport in redox polymer films / membrane mimetic media [422,423]. Obviously, this also applies to the creation of ion gradients and transmembrane transport of charge carriers / ions, which is fundamental for modeling the evolution of photosynthesis. In this regard, it is advisable to point out the contribution of a non-equilibrium electric double layer to the mechanism of electroosmosis and ion-exchange processes on membranes [424,425]. Modeling of the reverse osmotic processes implies methods based on the analysis of electrostatic double layer interaction [426] are commonly used. Attractive osmotic pressure in the double layer is characteristic of polyelectrolytes (which include many biological compounds [427,428]) [429] and near-membrane layers, or the microenvironment of surfactant bilayers of membrane-mimetic structures [430]. Modern continuum models allow to analyze the osmotic pressure taking into account the ionic composition of the medium [431], which differentiates them from the theoretical concepts of the 1940s-1950s. [432,433]. In this regard, a qualitatively new approach to the integration of the transfer effects of the specific ions / charge carriers in reaction-diffusion osmotic systems, characterized by the propagation of the ion flows in space under the action of an external field (for example, in electroosmosis [434,435]), seems justified.
The above approach is sufficient to describe transient processes in the double layer leading to electrochemical oscillations or pulsations controlled by an external field (see e.g. [436]), as well as electroosmotic waves in a nonlinear double layer flowing in space [437], such as solitons and some forms of oscillations in chemical self-oscillating systems, similar to the Belousov-Zhabotinsky reaction and self-oscillating biological processes. Some of these processes are observed at different time scales in photosynthesis [438,439], characterizing carbon assimilation and oxygen emission [440,441], charge separation and transfer [442], compartment-interdependent pH changes, regulated by the electron flows [443], ion sorption, biochemical effects and cytotoxicity of differently charged ions [444], phosphate turnover regulated by the membrane electrophysiology-assisted bioenergetic pathways [445], etc.), including those caused by photobiological machinery [446,447].
The ultimate evolutionary-specific case of a prototype for modeling double-layer photoinduced electrophysical phenomena in nature is photosynthesis itself, which has been known since the second half of the 20th century. [448,449]. Unfortunately, there are few studies focused on the analysis of the double layer on photocatalytic semiconductor particles and its contribution to the quantum yield and the photoelectrochemical kinetics [450,451], which, along with significantly different physicochemical focus of these works, often prevents their comparative analysis and integration. Therefore, we are forced to “sketch a picture in broad strokes”, describing only qualitatively possible paths, and not quantitative models of phenomena that can be implemented only if formulated as a “multiphysical” (photoelectrochemical / photoenzymological / photoelectrophysiological or photomembranological [452,453]) multiparametric task [454,455,456].
However, from the biomimetic point of view, it is clear that it is the electric double layer that can cause photoelectrogenic reaction-diffusion processes that reproduce the redistribution of mass (mass transfer) during photomorphogenesis modeled in photobionic systems, since consolidation and aggregation on a surface with a double layer [457,458], as well as hydrodynamic instabilities, and capillary (more precisely, for such a case, electrocapillary [459,460,461,462]) waves [463,464], leading to the formation of chemo-hydrodynamic patterns [465], as well as the accompanying Derjaguin non-equilibrium electrokinetic phenomena [466], are due to the presence of a double (usually non-equilibrium) layer and cannot proceed in its absence.
Thus, we can say that in this system a charging-discharging electric double layer plays a pacemaker or synchronizer role, not only by spreading excitation when an optical "signal" is applied, but also by organizing mass transfer and adaptive (in accordance with the excitation parameters) formation of chemo-hydrodynamic / reaction-diffusion patterns, similar to morphogenesis in biomembranes or physical systems modeling them [467,468].
15. Similarity between the equivalent circuits of semiconductor protophotosynthetic photocatalytic interfaces and photosynthetic membranes.
In accordance with the criteria of Section 5.3.4, “…if such kinetic phenomena of discharge or gating are observed in an electrochemical or electrocatalytic system with a double layer, this system is ... an analog equivalent circuit of the ‘isokinetic’ process” [82,83].
If we consider, in the framework of nonlinear analysis, electric double layer of the surfaces or membranes involved in the occurrence of protometabolic processes, as a capacitor (where the discharge can act as a synchronizing pulse for equivalent circuits of various biomimetic processes at the surface interface) [469,470], then we can construct an equivalent electrochemical transfer circuit for any surface or complex nanostructure involved in pattern formation. Note that finding formal mathematical models is possible for any geometry of the systems containing electric double layer (such as double-layer cylinders [471,472], two parallel plates [473], etc.) including porous ones [474,475]. In this case, both for biochemical membrane carriers of the electric double layer, and for their mineral predecessors, it is possible to find out how the capacity and efficiency of electrokinetic transfer depend on the adsorption of differently charged ions on their surface [476,477].
This allows us to speak about the control role of sorption in synchronization / desynchronization of "electrophysiological processes" in the prototypes of biological membranes in photosynthetic systems at the stage of their functional formation. This mechanism undoubtedly influenced both the reaction-diffusion morphogenesis (through absorption controlled diffusive processes, according to the Landauer equation [478,479]) and catalytic processes (for example, during adsorption-controlled diffusion in catalytic redox-processes [480], from simple inorganic carrier-assisted ones to biopolymer-assisted ones, such as adsorption-controlled redox activity based on nonspecific electrostatic sorption of proteins or aminoacids on inorganic electrode-like surfaces [481,482]). The influence of adsorption is also seen for heterogeneous catalysis [483], photocatalysis (for example, adsorption-controlled selective photocatalytic transformations on semiconductor mineral surfaces [484]) and for the mobility of protocellular structures when they assimilate the mechanisms of surface sorption control / determination of physiological processes (both for coacervate droplet-like structures and surfactant-bearing protocell-like drops with membrane-like surfaces [485,486]). It is possible to consider such complex regulation in a natural electrolyte environment as a consequence of the operation of the electyric double layers on the semiconductor-electrolyte-semiconductor heterojunctions [487]. Even for the most elementary semiconductors of geomineral origin, this statement agrees well with their photovoltaic function, which may precede photosynthesis, since heterojunction interface double layers are typical even for photovoltaic cells based on ZnS-containing structures [488].
It would be extremely attractive (though inaccurate) and simple to conclude that all wave phenomena can be associated with the same oscillation mechanism in the electric double layer, thereby reducing the entire variety of processes to one elementary photosynthetic equivalent circuit (and then to provide adaptive frequency sampling with different oscillation hierarchies, but not to rearrange the whole scheme [489]). For example, for simplicity’ sake, it would be possible to reduce the entire variety of oscillations to models of catalytic oscillations, for which the elementary Langmuir-type kinetics [490] are described in the isothermal version (since the Michaelis-Menten kinetics is derived from the Langmuir-Hinshelwood kinetics). However, according to well-known later works that caused a discussion in Physical Review E [491,492,493], this approach is too simplistic. In fact, during heterogeneous catalysis the initial reaction surface of the catalyst is not retained, and its passivation is not a gradual uniform process, but is characterized by a stage of chaos, after which the local electrochemical properties of the surface (hence, equivalent circuits of the corresponding regions) change, invariably depending on the adsorbate-induced surface restructuring [494] (see Figure 24). In the case of dispersed particles, for example, an oxide layer formation [495,496,497] is observed, which “automatically” transforms the metal particles into semiconductor ones, transferring the reaction mechanism from catalysis on metals to catalysis on semiconductors. At the same time, since there is no strict regularity of (self-)oscillations in heterogeneous catalytic reactions, surface restructuring is also not homogeneous [498]. This results in the integral surface of the oscillating catalytic structure being a perturbed pseudo-stochastic set of points that alternately participate in the opposite electrochemical processes. Thus, in self-oscillatory protobiological catalytic processes there is a number of oscillation scales that differ from the typical spatial and temporal scales of pulsations of the electric double layer on the single photocatalytic particles.
On the other hand, the sorption regulation of oscillatory processes associated with the electric double layer indicates the possible role of adsorbed ions in the overall (more specifically, electrochemical) kinetics of the process. The effect of oscillations of adsorptive concentrations in adsorbent grain surfaces on the adsorption process in mixed kinetics [499] is well-known. At the same time, locally existing random thermodynamic disturbances on the surface (as a rule, in the classical adsorption-kinetic experiment, thermally induced, or sometimes photo-induced) can affect adsorption kinetics [500]. Thus, one more set of feedback / regulation loops can be distinguished, which must correspond to separate sets of kinetic modes (and oscillation frequencies of the corresponding parameters), stable and metastable states. This assumption is well confirmed experimentally in dozens of works, which showed a variety of differences in oscillations and sequences of dynamical states caused by anion adsorption in the redox-electrochemical processes [501]. In some cases, similar sequences of dynamical states may correspond to the models of multistable (bistable in the simple case of a minimum of interacting agents / attainable states) and multimodal periodic regimes of oscillations in chemical kinetics [502,503], which introduced similar concepts into biochemical (as a rule, enzymatic) kinetics [504,505].
An inexperienced evolutionist, may use the "evolutionary enzymological" or "evolutionary biophysical / kinetic" paradigm to compare simplified ab initio models of some kinetic and adsorption processes with the action models of elementary enzymes and conclude that some chemical prototype found in geochemical conditions could be the predecessor of a particular enzyme in its function, based on just catalimetry and kinetic similarity. However, this conclusion, in any case, would be a simplification, since from the point of view of chemical kinetics, many processes can be derived from the same precursor, which are kinetically similar to the processes observed in biology, but adequate to biological structures with different functions and different characteristic periods or frequencies.
When modeling the photosynthesis phenomena on membrane-mimetic prebiological carriers, using only kinetic similarity and frequency separation, it is impossible to accurately assert which function is an "evolutionary predecessor" of which group of oscillations with a known spectral composition, since it is impossible to achieve effective separation of these functions within the frequency domain. In fact, this is also typical for biology where the need for synchronization and capture / assimilation of frequencies has led to the fact that though it is possible to conduct separate analysis of frequency-domain enzyme kinetics [506] for specific ionic oscillations such as calcium waves and frequency domain studies of gating currents for ion channels of membranes that contribute to these oscillations (or, in other cases, pulsations) [507], a clear selection and identification / attribution of oscillations of specific roles, without taking into account the mechanisms of their occurrence, is impossible or extremely difficult. Moreover, in most cases there are also oscillations of a smaller scale, which together provide energy supply or pumping of oscillations of a more general scale (as the activity of ion channels ensures the emergence of a membrane potential, so is the regularity and power spectrum density of electrophysiological burstings made up of the additive action potentials), due to which it is impossible to eliminate some frequencies without prejudice to others generated by them, just as it is impossible to ignore the contribution of the former to the overall energy and kinetics of the process.
So, it is necessary to exercise caution when identifying all oscillations in protophotosynthetic systems with one type of oscillations in a semiconductor medium or caused by charging-discharging of an electric double layer. One should keep in mind that double layers of different origin may display oscillations of a more fundamental level than additive and even cooperative oscillations of the double layer as a continuum, for example, spatiotemporal oscillations of the electron density [508,509] or Friedel oscillations [510,511] that result from screening of the charged impurities (including chemisorbed impurities) in nonspherical symmetries determined by the symmetry of the point group of a given crystal (which can be different from point to point if samples of natural origin exhibit non-stoichiometric composition and contain natural polymorphic modifications). Obviously, during redox reactions or adsorption mediated by an electric double layer (transformed from physical sorption to chemisorption) on an inorganic mineral (membrane-mimetic or protophotosynthetic) surface, the emergence of impurities and changes in symmetry, and even in the coordination parameters of the substrate is usually observed, which makes Friedel oscillations inevitable. The minimum requirement then is to distinguish between the inducing and induced oscillations: for example, photoredox processes can, depending on the electrochemical nature of their products, lead to the accumulation of charge or discharge from the double layer (thus, controlling it). Alternatively, the kinetics of the electric double layer at the membrane mimetic interface can synchronize electrochemical redox processes on semiconductor particles (by controlling them) [512,513]. Thus, in terms of control theory and regulation theory, there is a rearrangement of the feedback system during the reaction or reaction-diffusion processes, which corresponds to the rearrangement of the equivalent circuits (at least, due to the doping that changes with the changing electrochemical conditions of sorption, and thus affects the function of the element formed).
The last statement forces us to interpret consequent-antecedent chains in Markovian chain macrokinetic models not as a predetermination of diffusion-controlled reactions and adsorption processes in the system, but as a dependence of its arbitrary next state on the previous trajectory [514], which can push it to another pathway (by changing flow tracing in equivalent circuits or by changing the nature of elements in them). This approach suggests a comparison with decoding metabolic pathways (especially in evolutionary biochemistry), when by the end of the 20th century, instead of a harmonious and deterministic set of "averaged normal metabolism" lines, a complicated network of processes emerged that was flexibly rearranged from one biochemical pathway to another, as in physiological processes (from permanent adaptation to impulse and trigger rearrangements as a result of cellular signaling), as well as with external physical and chemical signals in a form mediated by the physiological mechanisms. Accordingly, there are no fundamental obstacles to comparing transitions between linear, graph and network (including three-dimensional) kinetic models of biochemical oscillations [515,516] and similar transitions between linear, graph, network (including three-dimensional) kinetic models of other (non-biochemical and, as a rule, preceding and more general in relation to biophysical models [517,518]) kinetic oscillations.
16. The independence of formal kinetics of oscillations from the substrate as a prerequisite of the possible evolutionary transition from mineral semiconductor kinetics / dynamics to photosynthetic kinetics / dynamics.
Considering oscillations in the system as kinetic ones (in particular, kinetic physicochemical oscillations, including dependence on many factors [519], ignored by the simple, minimal kinetic models [520]), we are justified in viewing the isomorphism of processes as a necessary, and in some cases necessary and sufficient, condition to consider them as model systems for biological prototypes (i.e. biomimetic systems). For this reason, we are not limited by the substrate restrictions, but consider physically defined and equivalently mathematically approximated processes with the known interrelationship between them, modeling or replacing the same processes (related in a similar way) in a simulated biological object. Therefore, ab initio, we can choose an equivalent to most types of the wave phenomena in biological kinetics - both kinetic and diffusion-limited and diffusion-controlled [521,522,523,524,525].
The emergence of most oscillations of this kind is determined exclusively by nonlinearity and can initially occur even near equilibrium [526], which allows one to speak about the genesis of the corresponding processes (evolutionarily arising from them as from a natural geological prototype) that are subsequently implemented in biological systems, since the precursor conditions for the emergence of such systems in the natural mineral environment did not always deviate significantly from equilibrium. Nonlinear chemical kinetics in such cases explains instabilities and associated oscillations, but cannot clarify the origin of thermodynamic conditions for them [527] (although in the same systems the conditions can be selected where both the kinetic and thermodynamic aspects coexist and act co-directionally [528]).
In nonlinear physics, variables involved in modeling are usually of minor importance: same equations can describe processes in completely different areas - from plasma physics and mechanics to population dynamics. As it is accurately indicated in the preface to the book [529], it is possible “to demonstrate a real physical analogue of practically any physical process from another area of physics”. Therefore, there is no point in considering the specific chemical basis of protophotosynthetic processes sensu stricto, if the observed nonlinear oscillatory dynamics is physical and common not for the chemistry of carriers of a certain composition, but for the variety of processes described by the similar equations. Such systems may explain, from a chemical point of view, the absurd introduction of light quanta as a substrate into the kinetic scheme of (proto-)photosynthesis, following from the reasoning of Riznichenko and Rubin [82,83]. This becomes possible due to the presence of particles and quasiparticles (for example, "holes") in the kinetic scheme.
Generally speaking, reactions as such are replaced there by reaction-diffusion generation-recombination processes in semiconductors, leading to nonlinear regular oscillatory and chaotic dynamics, as is brilliantly demonstrated by E. Schöll [530,531,532] (Figure 25). In his early works this author has theoretically proved that threshold switching in such systems can be interpreted as generation-recombination induced non-equilibrium phase transition [531,532], and the formation of dissipative structures in semiconductor systems can be associated with recombination instability [533,534].
Obviously, for photosensitive semiconductor structures involved in protophotosynthesis, it is advisable to consider photoinduced generation-recombination processes (well known since the 1960s [535] and that lead to the creation of efficient semiconductor photodetectors, including those beyond the visible spectral range [536,537,538]), which also reproduce some oscillation modes in (bio)chemical systems, including those related to photosynthesis and the corresponding transfer processes. Such processes were investigated for both native photosynthesis and for “subcellular explants” in vitro since 1980s in different forms [539,540,541,542,543,544,545,546,547], not only in tissues of the higher plants but also in purple photosynthetic bacteria pigmented with bacteriochlorophyll a or b, together with various carotenoids, for obtaining the light energy for photosynthesis [548,549]). Developed since the late 1960s, the ideas on kinetic oscillations in semiconductors [550,551,552] agree well with the equivalent concepts for photosynthesis developed in the last quarter of the twentieth century. Currently, the hierarchy of oscillatory processes in semiconductors has been analyzed - from self-sustained and self-regulated current oscillations in the kinetic theory of semiconductor superlattices [553] (lattice oscillations associated with the transport of charge carriers have been known since the 1960s [554]) to additive oscillations of photoconductivity in relaxation kinetics of amorphous semiconductors and thin semiconductor films (for example, semiconductor amorphous hydrogenated silicon used for design of thin-film solar cells and thin-film transistors [555]) and kinetic oscillations in transistors and resistors under the influence of high currents [556]. Various hierarchically interpreted levels of oscillations in semiconductors can be compared with oscillations in models of photosynthesis, which differ both in scale and in the method of registration - from oscillations at the level of charge carriers and fluorescence oscillations to additive membrane current oscillations and cooperative oscillations of ion transport in a plant or plant’s part in general [557,558,559,560,561,562,563,564,565,566,567].
17. From the formal kinetics of elementary excitations in semiconductors to quasiparticle-assisted biophysics of the primary forms of (proto)photosynthesis.
As follows from the above, to discuss the probable prothotosynthesis mechanisms, it is necessary to descend beyond the atomic level of biophysical kinetics associated with the chemical composition [568], namely, to the level of the kinetics of excitation transfer in the system, which includes the transfer of energy by quasiparticles, independent of the chemical composition. Indeed, the concepts of energy transfer by quasiparticles in photosynthesis date back to the early 1960s, when first the electron-hole representations [569] and then exciton representations [570] were assimilated from the solid state physics and during the 1970s - 1980s were compared with experimental data [571,572,573,574]. All this gradually led to the prevailing theoretical understanding of the role of excitons in photosynthesis. Fast computerization (and the consequent automation of data collection) in the 1990s accelerated the adoption of concepts that the overwhelming majority of researchers could verify empirically and calculate using high-speed clusters and supercomputers in remote access (compare the works of the 1990s in the progression [575,576,577,578,579,580]. Therefore, the XXI century physics of photosynthesis can be justifiably called "biophysics of quasiparticles" and, in particular, "excitonic biophysics": this fact is confirmed by the significant increase in the technically and methodologically superb articles published in leading scientific journals [581,582,583,584,585,586,587].
It is known that energy transfer in photosynthetic complexes includes excitons and many quasiparticles differing in their role: polarons [588,589,590,591], polaritons [592,593,594,595], phonons [596,597,598,599,600] and even such specific (for electron transfer in bacterial photosynthesis) quasi-particles, as hystons [601,602,603]. In compartmentalized systems it is necessary to take into account other forms of excitation or quenching of excitation, which, in particular, depend on the microenvironment parameters and its realistic reproduction / consideration in the particular experiment conditions [604]. Therefore, it is not surprising that experiments in the field of artificial photosynthesis, including those with semiconductor structures, rely on the same quasiparticle-based concepts or even the same quasiparticles as in natural photosynthesis: excitons [605,606,607,608], polarons (even on “fashionable" LiNbO3 and hybrid perovskite systems although they little correlate with the biological source of the name "photosynthesis") [609,610,611,612,613,614], polaritons [615], phonons (also without reference to the biological prototype, which is logical, since quasiparticle-based concepts were introduced from physics, but not from biology) [616,617,618,619]. In some cases, plasmonic / nanoplasmonic concepts are added to create "plasmonic photosynthesis" [620,621,622,623,624], "plasmon-induced", "plasmon-assisted" or "plasmon-enhanced" artificial photosynthesis [625,626,627]. However, such synthetic processes are interpreted sensu lato, since no structures supporting plasmonics have been discovered in the phytoanatomy of photosynthesis. Nevertheless, if we discount the “vociferous” extreme radical approaches, then it is clear from the above that practically all energy transfer processes in photosynthesis, and, accordingly, in protophotosynthesis and its model (in particular, semiconductor-based) reconstructions, can be translated into the language of quasiparticle kinetics, well developed for solid state and semiconductor physics by the early 1980s [628,629,630,631]. In other words, it is proposed to model protophotosynthesis (as well as earlier for the design of artificial photosynthesis [632,633]) based on the kinetics of charge transfer and carrier mobility, by analyzing interactions between particles / quasiparticles, regardless of the chemical composition. This thesis can be indirectly confirmed from the standpoint of bioenergetics: if we consider the photosynthetic reaction center as a quantum heat engine, then it is quite obvious that, like the Carnot heat engine, no (bio)chemical requirements can be imposed on “fuel” [634]. This logic well accommodates mathematical concepts of Rubin and Riznichennko [82,83] about light quanta as a reaction substrate at the light stage of photosynthesis.
However, when considering catalysis with the participation of a light inductor or a substrate, it is essential that the above listed concepts fit the logic of the catalytic act on a semiconductor / mineral surface simulating protophotosynthesis. First of all, one should turn to the physics of catalysis, in particular, solid-state catalysis, to identify the role and interplay of quasiparticles.
In the 1970s-1980s. one of the leading researchers in the field of physical and chemical heterogeneous catalysis, A.M. Dobrotvorskii, (co-author of the monograph of the same name [635], who for the first time formulated the problem of prediction / analysis of heterogeneous catalysis mechanisms by pattern recognition methods [636] as it is still implemented in chemoinformatics) proposed a quasi-fermion approach to electronic structure (and dynamics) of solids in the chemisorption and heterogeneous catalysis [637,638], which correlates well with the mechanisms of interatomic interactions in solids. In the 1980s the theoretical foundations of the influence of excitations of the electronic and phonon subsystems of semiconductors on adsorption and catalytic processes on them were laid [639]. Later, the book "Electronic phenomena in adsorption and catalysis on semiconductors and dielectrics" [640] was published (which has not lost its relevance until now and was reprinted in 2012), where, in addition to the sections on "Electron processes in semiconductor adsorbents and catalysts", we find Chapter 8 on "The phonon and shock mechanisms of charge-carrier capture in adsorption and catalysis”, with detailed development of this concept. At this moment in the development of chemical physics and, in particular, "catalytic physics", extending in historical development from the solid state physics (metals, semiconductors) and surface physics [641,642,643] to physics-based enzyme design (based on QSAR / QSPR similarity criteria) and from self-catalytic colloids to living cells [644,645] (which, obviously, becomes fully possible also with the help of QSAR / QSPR similarity criteria), it becomes possible to integrate qualitatively different ideas about particular cases of catalytic acts and associated energy transfer using quasiparticles in the framework of generalized concepts of physics of quasiparticles in condensed (but not necessarily solid state) matter [646]. Indeed, soft matter processes involve mostly the same quasiparticles as in semiconductors and, in particular, in catalytic artificial photosynthesis: phonons [647], polarons [648], polaritons [649] or plasmon-polaritons, as well as plasmons [650,651] (as in "plasmonic photosynthesis"). At the same time, soft matter physics is characterized by the same forces and scales as enzymatic catalysis and its models (the same Van der Waals forces and non-covalent interactions that are involved in the processes of coordination / supramolecular fixation), including those in amphiphilic polymers and polyelectrolytes [652,653]. This makes it possible to draw an evolutionary line from the mineral solid media that participated in prebiological catalysis / photocatalysis to the formation of biopolymer structures (in particular, protocells) with the most optimal mechanisms for the implementation of photosynthetic and, in particular, catalytic processes in biopolymer soft matter conditions. At the same time, the main arena for the developing catalysis and charge transfer events continues to be the interphase boundary - interface (membrane precursor, biomembrane, or membrane-mimetic material simulating it in the experiment) / active surface, along which excitation propagates and through which it energetically amplifies (in particular, with the help of quasiparticles, for example, plasmons, as in SERS - Surface-Enhanced Raman Spectroscopy [654,655] and in methods based on the SPR principle - Surface Plasmon Resonance [656,657,658]). Most functions of surfaces and interfaces in modern photosynthesis are delegated to membranes and catalytic active centers.
18. A fundamental role of the phase boundaries / interfaces in the evolution of protophotosynthesis (charge carrier transport, surface reaction kinetics, etc.)
Before discussing membranes, it is necessary to justify the need for phase separation and the presence of active surfaces. We have to comply with the requirements of evolutionary and emergent consistency asserting that spontaneous synthesis and survivability of structures are impossible if in no way related to environmental conditions and not leading, according to the results of their evolutionary (biophysical and physiological) functionality, to the adaptation of their inherent physical mechanisms under (proto)biological functions. We are therefore forced to look for “evolutionarily selected” functions and functional mechanisms of prebiological active surfaces as “precursors” of the late and more evolutionary progressive functions realized in membrane cytological / ultrastructural systems of modern photosynthesis. The list of basic requirements for model and reductionist evolutionary-chemical prototypes of photosynthesis, according to Rubin and Riznichenko [82,83] (Section 5.4), indicates that the role of membrane structures in these prototypes is reduced to transfer of charge carriers (including ions), to generation of electric and electrochemical potentials and their effects on the charge carrier flow (partly - electron flows). Accordingly, the active membrane model should include sources and sinks, as well as mechanisms for accumulating charge (capacitor-containing circuit). In addition, due to membrane localization of the catalysis, which involves the use of functionally different enzymes in chloroplasts or in the whole cells of photosynthetic organisms [659,660,661,662,663], it is necessary to ensure that the design of the active surface simulating the membrane is compatible with the requirements for the implementation of the catalytic act corresponding to the same act of its living prototype (in the case of artificial photosynthesis it should be photocatalysis). Therefore, in the definitions of "active surfaces" and "active interfaces" we can choose precisely those descriptors and criteria that favor their functional activation in a form that supports the physiological and biophysical progress of photosynthesis (coupled membrane functions) and accompanying biocatalysis or biophotocatalysis (which, despite the differences in methods, is almost synonymous [664,665,666,667], although not all forms of biophotocatalysis / photobiocatalysis can occur in native biological systems and become the products of biological evolution).
The principles of the models of photosynthesis listed in the works of Rubin and Riznichenko [82,83], obviously, determine the following requirements to the surfaces where photosynthesis occurs:
18.1. Photophysical and photochemical activity of surfaces and interfaces
Examples can be given both for pure and dye-doped semiconductor surfaces or interfaces [668,669,670,671]. In a number of such cases, the question about the mechanism of light-induced charge transfer using dyes in active interfaces inevitably arises: is “photoredox interaction or semiconductor junction” the cause of the observed effect? By virtue of the previously outlined principle of feedbacks on photochemical effects in protophotosynthesis on the active surfaces, the reversible and oscillating effects of irradiation on wetting and other surface properties of such systems, in particular, photoinduced high hydrophilicity / high hydrophobicity [672], should also be taken into account. Thus, irradiation performs the function similar to photolithography in catalytic nanotechnologies, when, as a result of irradiation (and etching), active surfaces are created [673]. In other cases, photocatalytically active surfaces favor the interaction with organic matter, including bioorganic systems such as enzymes (even those that regulate genetic functions [674]).
18.2. Electrophysical and electrochemical activity
Electroactive surfaces and interfaces can be implemented on semiconductors with defects [675,676] associated simultaneously with phase boundaries and with quasiparticles. The latter produce electrical instabilities (both in inorganic metal oxide and organic semiconductors [677,678,679], including under optical pumping [680,681]), interphase transport and redox passivation [682,683] (reversible or irreversible, if reversible, then in some cases - oscillatory). However, in other cases, electronically active interfaces can be based on qualitatively different structures, such as metal-organic framework films [684], which satisfies well the requirements of reproducing biophysics of photosynthesis, since metal-organic frameworks (mainly redox-active ones) are capable of carrying out processes of collecting solar energy in artificial photosynthesis combined with electrocatalytic functions [685,686,687]. Nevertheless, no precedents of using classical semiconductor structures associated with early prebiological photosynthesis have been found in the list of such surfaces. The response of semiconducting metal oxides to water as a result of chemical transformations on catalytically active surfaces [688] and roles of active interfaces in the electrochemical water splitting reactions [689] are well studied. Nonlinear electrical responses have not been investigated for ZnS due to the technical futility of the material; however, in the related ZnO used in varistors (the resistance of which varies nonlinearly as the applied voltage changes) electrically active interfaces have been studied repeatedly [690,691,692,693]. Recently, it has become popular to integrate electrochemically active surfaces and electric capacitors [694] in a single structure, which maximally likens such systems to active biological membranes capable of storing and conducting excitation resulting from a set of bioelectrochemical acts.
18.3. (Chemi-)sorption and catalytic activity.
The emergence of catalytic activity is always associated with chemisorption on catalytically active surfaces [695]; and many methods for determining catalytically active surfaces are based on the principles of chemisorption (starting with the classical works of Roginsky and others [696,697,698]). Enthalpies of adsorption on catalytically active surfaces have been determined [699]. There are many methods to characterize catalytically active surfaces, including operando-spectroscopy of electron paramagnetic resonance and in situ electron microscopy (both were introduced in the 1970s-1980s of the XX century [700,701]). At present, methods of molecular design of the active surfaces of catalysts are known that optimize their chemical and biochemical reactivity [702,703,704] both for solid-state active surfaces and for soft matter ones. Starting with the classic works of the first half of the twentieth century (Lewis and others), the correlation between the catalytic and electrophysical properties of active surfaces is indisputable [705,706], which automatically equates the sets of “catalytically active” and “electrically / electrophysically / electronically active surfaces”.
18.4. Surface kinetic activity, in particular, catalytic chemical oscillations.
This principle logically follows from the previous one. Active surfaces in semiconductor structures capable of supporting chemical oscillations are poorly studied (except for kinetic, but not chemical-catalytic oscillations during generation-recombination processes described in the above cited work of Schöll [532]). However, if we limit ourselves to the requirement of similarity within the kinetic scheme, then, to illustrate this thesis, we can consider a system very well studied in the framework of catalytically active surfaces: CO oxidation on Pt surface [707,708,709], on which, since the 1980s, chemical self-oscillations that are not inferior in clarity and identification of the components of kinetic regimes to the chemical oscillations observed in Belousov-Zhabotinsky reaction, were repeatedly reported [710,711,712,713].
18.5. Redox activity.
The above examples of redox activity do not explain the transition from solid-state structures to soft matter in the evolutionary context. Therefore, it is necessary to provide examples (which do not change, in general, the essence of the mechanisms of mediated electron transfer at redox active surfaces [714]), demonstrating the role of redox-active surfaces in polymer, membrane-mimetic systems [715,716,717]. As can be seen from the references cited, micellar redox-active structures with biomimetic and model protobiological properties can be obtained, considered as protocells and artificial cell models [718,719].
18.6. Reaction-diffusion activity.
Surface reactions and chemical transport in conductive systems with electrochemically-active surfaces are well-known [720], but such results are not applicable for semiconductors due to the multiplicity of carriers in the active surfaces of semiconductors. As a result, most of the works on transport in active surfaces are limited to the liquid-phase and gas-phase substances and aerosols for chemical engineering, as well as on soft matter [721,722,723,724,725]. At the same time, the reaction-diffusion activity is characteristic of semiconductor surfaces in various forms [726,727,728,729]. Moreover, in different atmospheres, the response of interfaces will be different [730,731] (which is used for the development of gas sensors). By varying the defect structures on semiconductor active surfaces, it is possible to achieve qualitatively different chemical results [732]. This confirms that atmospheric or liquid conversion of the medium (as in photosynthesis) with energy pumping on the defect structures (as a rule, photochemical or corpuscular one [733,734,735]) can be implemented. Since the concepts of reaction with diffusion and self-organization are correlated, we can formulate the last requirement to the surface activity - non-equilibrium processes and self-organization.
18.7. Structure-forming surface activity.
Self-organized shape kinetics and dynamics of the active surfaces [736] and self-organization processes at the active interfaces [737] (see Figure 26) are well-known phenomena. Reactions on the self-organizing surface produce kinetic roughening [738] (in accordance with the Kardar-Parisi-Zhang equation), which can promote cross-catalytic processes. Such self-organization can be considered as “natural nanotechnology”, which replaces artificial methods of designing an active surface by adapting it to the conditions of the environment (either liquid or gaseous) [739,740]. Feedbacks help to regulate self-organization of active surfaces, due to which self-organization and utilization of the medium components occur synergistically. Only in this context one can describe self-organization on the surface as morphogenesis, that is, a reaction-diffusion process involving an activator and an inhibitor [741]. The reversible nature of structure formation in the system is associated with its functions and is the main criterion of its reactivity, that is, self-organization of the active surface dependent on the environmental conditions is also a tool for functionalization of its activity. Design of the active interfaces using responsive molecular components leads to the synthesis of responsive interfaces that demonstrate a reversible change in physical properties in response to the external stimuli [742].
At this point the discussion is no longer limited to the physically active surface, but extends to potentially prebiological or biomimetic interpretation of the active surface. At the same time, although strictly biomimetic and membrane-mimetic surfaces are associated with soft matter (from biomimetic chromatophores compatible with photosynthesis on soft active surfaces [743] to electrochemically active surfaces for creating membrane models, as well as active surfaces for engineering of templated artificial cells [744,745]), their evolutionary properties begin to manifest themselves long before the moment when a sufficient set of functions for realization of prebiological activity is accumulated. Therefore, the only way to study the mechanisms of transition from mineral active surfaces to biologically relevant ones is to analyze the activity of these surfaces in an evolutionary context, taking into account the geochemical conditions of their operation.
19. Evolution of the active surface as a way of transition from chemical to biological evolution.
The fundamental issue is the evolution of the active surface, i.e. how from a mineral inorganic surface, which can be considered within the competence of solid state physics, a transition was made to an active (according to the same functional requirements for developing photosynthesis with a membrane) surface, which can be described in terms of soft matter physics. We should proceed from what these two systems have in common and consider this set of features not as a way of reducing more complex biochemical realizations to simpler physicochemical models. (The works of Komissarov et al. can serve as an example of incorrect modeling that extrapolates technically accurate solutions of simplified applied problems to biological mechanisms and evolutionary trends. Due to the above methodological error they equalized the conditions of natural photosynthesis and many incompatible photochemical processes on glasses, polymers, crystals, etc. Such false analogies lead to "new ideas about the evolution of the Earth's atmosphere and photosynthesis", which postulated that the source of oxygen in modern photosynthesis is not water, but exo- and endogenous hydrogen peroxide, which decomposes as a result of photoelectrochemical and thermal decomposition during heating of the chloroplast - reactor [746,747,748,749]).
In evolutionary biology, phylogenetic analysis is conducted by searching for the most ancient common ancestor. Therefore, an accurate analysis of physicochemical evolution must proceed ab initio from the first principles common to the product of evolution and its abiogenetic predecessor. In future it will be possible to "screen" the intersection areas of multiple descriptors in order to implement data driven mathematical identification [750] of the first-principle models [751] of prebiological physical and biophysical processes (evolved from physical processes) [752] including nonlinear dynamic systems [753], prerequisitely approximable / qualitatively analyzed using differential equations / partial differential equations [754,755] (such as photosynthesis itself [82,83,756,757]). Here we outline the ways to implement this approach. Considering that photochemical and electrochemical systems were studied using data-driven identification (including oscillatory systems that meet the requirements of the Voltera series approach) [758,759], the proposed approach to identification of protophotosynthetic and membrane-electrochemical processes based on the same principles seems logically correct. At the same time evolution of the kinetics of photosynthesis and other fundamental physiological processes in accordance with the changing environmental conditions led to the substitution of the mechanisms of the corresponding kinetic processes with conservation of their physical nature and formal approximation. However, the approximability of the certain processes with the same equations as the assumed kinetic prototype can indicate only kinetic similarity, but not the similarity in the physical or chemical nature of the acting agents. The opposite, in the case of photosynthesis, leads to physically absurd errors, reminiscent of the notion that the analysis of non-linear Arrhenius plots in photosynthesis (the rates of photosynthesis have temperature dependences characterized by curved Arrhenius plots, for which the logarithm of the activation energy is a linear function of temperature) establishes its similarity with many processes in animal cells and qualitatively different inorganic and polymer materials - from ferromagnetic materials and semiconductors to ferroelectrics [760,761,762]. In this regard, in spite of the existence of the particular aspects of the reductionist similarity between biological and physical systems, in order to incorporate them into protobiological / evolutionary biophysical models, it is necessary to take into account at least the following considerations:
- a)
- which variable or which physiological agent demonstrates this or that kinetic curve (it is inadmissible to identify mechanisms with the equivalent kinetics of different variables or objects that do not correspond to each other);
- b)
- is a function / kinetics-preserving transition possible between the carriers of two formally close kinetic curves (it is obvious that if in one case we are talking about magnetic hysteresis, in the other case - about phenomena specific to ferroelectrics, in the third case - about the physics of semiconductors, and in the fourth case - about photosynthesis, as in a number of just cited works [760,761,762], it is impossible to draw a line of substitution between them);
- c)
- what evolutionary reasons cause the introduction of this or that structure (or one or another member of the equation or an agent formally involved in kinetics) into photosynthesis or another physiological process: if physical introduction does not result in the assimilation, or is rejected by natural selection or is not inherited as “ exotic new acquisition", physically unsupported by the environment, then it is hardly rational to talk about its interpretation in the framework of evolutionary (proto)biology.
In spite of existence of the universal approaches for active interfaces, the common between active interfaces is revealed only at the general physical level. According to [763], “The principle of universality, according to which large scales and long times screen a system intimate details, provides a mean to systematise such knowledge: many growing interfaces, for instance, are described by the same equation - the Kardar-Parisi-Zhang (KPZ) equation”. At the specific / local level, differences inevitably come to light. With the blurring of boundaries and criteria of similarity, statements about the role of surfaces, in particular, active ones, in self-organization and evolution of protophotosynthetic or protobiocatalytic systems as a whole are, rather, intuitive a posteriori categories borrowed from experience, than formalized physical statements. Even in the liquid phase at the gas-liquid interface total polarization vanishes, provided that there is no net flux through the boundaries, and at any planar wall polarization is determined by the magnitude of the bulk current and total interface polarization is rigorously determined by the current difference, that precludes the influence of the total interface polarization on the active bulk coexistence and questions the proposed coupling of the interface to the bulk [764]. Therefore, categories that do not specify the physical requirements for an object (in this case, a surface) may turn out to be non-constructive, even if mathematically accurate.
In fact, although ‘active surface’ and ‘heterogeneous surface’ are, from the standpoint of energetic concepts, intuitively close terms, and statistical (KPZ) models predicts the shapes of cellular membranes by accounting for the active feedbacks between the membranes and attached proteins (including catalytic-active enzymes) [765], concretization of the activity of specific sites within the framework of the Kardar-Parisi-Zhang equation cannot be obtained for reasons correlating with the universality of this concept, as noted by many authors [766,767,768,769,770]. Universality is the observation that there are properties for a large class of systems that are independent of the dynamical details: systems display universality in a scaling limit; universality class is a manifold of mathematical models which share a single scale invariant limit under the process of renormalization group flow. Thus, Kardar-Parisi-Zhang equation, within the framework of the above considered requirements to protomembranes characterized by kinetic surface roughening and dynamic texturing [771,772], self-organization and patter formation [773,774], can postulate the role of diffusion [775,776], but cannot specify the requirements to diffusants; can indicate the role of the formation medium (its noise or turbulence [777,778]), but cannot characterize and predict the emergent contribution of microroughness and the medium noise that generates it to the activity of the resulting surface or its section; can describe growth and development of the surface [779], but cannot limit the class of materials in which they are generated - neither by charge nor by chemical principles (including polymers [780,781] and crystals [782], solids [783] and liquids, as well as liquid crystals [784,785,786], both semiconductors and metals [787,788] into the field of applicability); can explain wave processes during the interface growth [789] and synchronization of processes [790], but cannot select processes that are inherent to the above carriers, or provide synchronization that directs the process to some specific state of surface activity with a given nature of the substrate [791].
20. The inevitability of the membrane participation in the development of protophotosynthesis.
It is obvious that membranes are necessary for the functioning of biosystems and biogeochemical cycling and exchange processes (if we consider the membranes of organisms participating in geochemical exchange, as the active interfaces between biosphere and “geochemical exchange reservoir”, from which they are formed and into which they release biomass during postmortal fossilization and mineralization, different from the vital biomineralization, due to the lack of its regulation / mediation by the membrane mechanisms [792]). The transition from protocells to LUCA would not have been feasible if there were no physicochemical mechanisms which, during the period before the formation of complex molecular biological membranes, enabled the substance transport in protocells, transmembrane transport and separation of components (from ions up to fractionation of isotopes [793], observed in the “geochemical time scale” and well confirmed by mass spectrometry, in vivo or in situ, both on the cells and on a number of model protocells, which do not always contain full-fledged ion channels [794,795,796]; for example, see Figure 27). Membranes in prothotosynthesis, as well as in artificial photosynthesis (conductive bipolar membrane interfaces, capable of initiating water disassociation within the interfacial region, which can perform water splitting for renewable energy in the presence of a pH gradient possible under the electric field conditions [797]), are necessarily associated with the charge separation and charge transfer, therefore, solving the problems of the evolutionary genesis of photosynthesis is always inevitably associated with the problem of the genesis and evolution of the corresponding membranes, and hence, the formation of a concentration gradient and charge transfer / carrier migration. Therefore, the ways of conjugation of the above functions were intuitively outlined even back in the 1980s.
For instance, in 1986 Hyman Hartmann (co-author of A.G. Cairns-Smith in the book “Clay Minerals and the Origins of Life”, co-editor of the well-known collections “Search for the Universal Ancestors”, “The Origin and Evolution of the Cell”) in the paper “Speculations on the Origin and Evolution of Photosynthesis and the membrane” integrated the approaches of membranologists and photosynthetists to the formation of the simplest inorganic precursor of photosynthesis [798]. In this paper “proposal for the origin and evolution of photosynthesis is postulated which involves carbon dioxide, nitrogen, sulfur and self-replicating iron-rich clays”, and “formation of Fe2S2 and Fe4S4 cores” resulting in the reduction of nitrogen by MoFeS compounds which led to the formation of pyrrole, flavin, nicotinamide, phycobilins (tetrapyrroles), heme and chlorophyll. The above list suggests that development of the redox functions under mineral conditions during the evolution of protometabolism did not proceed only in the direction of photosynthesis, but implied divergent evolution and selection for different (photochemical and non-photochemical) conditions of the emerging chemical processes. The key thesis of this hypothesis is the idea that “the electron transport chain evolved from ferrous ion through the Fe2S2 and Fe4S4 cores to the hemes”. Indeed, the origin of photosynthesis (as a physicochemical process combining the light quanta absorption and energy storage in the form of an electrochemical H+ gradient on biomembranes, mediated by the ETC [Electron Transport Chain], and associated with the transfer of protons against the gradient of the electrochemical potential), should be “topographically” associated with the formation of the pathways for photoinduced electron transport, transport of protons or other charge carriers against the potential gradient. That is why the prototypes of this phenomenon should have operated long before the formation of the modern photosynthetic membranes.
This statement is in good agreement with our “semiconductor world” concept. Therefore, in the next articles of this series, we will consider the formation of protomembranes, from inorganic semiconductor surfaces to the conventional phospholipid membranes typical for most types of the protocell models [799,800].
Acknowledgments
The authors are grateful to Dr. Mikhail Nikulichev for translation of the paper.
References
- Kuhn, H. Model consideration for the origin of life. Environmental structure as stimulus for the evolution of chemical systems. Naturwissenschaften 1976, 2, 68–80, [https://pubmed.ncbi.nlm.nih.gov/934343/]. [Google Scholar] [CrossRef]
- Bartsev, S.I.; Mezhevikin, V.V. Natural selection in a flow as a universal mechanism of evolution of prebiological autocatalytic systems. Doklady Biochemistry and Biophysics 2003, 388, No. 1, 35–38, [https://pubmed.ncbi.nlm.nih.gov/12741130/]. [Google Scholar] [CrossRef]
- Oxford English Dictionary, 2nd ed. Clarendon Press: Oxford, 1989.
- Oxford Dictionary of Biochemistry and Molecular Biology. Oxford University Press: Oxford, 1997; 508 p. [ISBN 978-0-19-854768-6]. Oxford Dictionary of Biochemistry and Molecular Biology (2nd ed.) Oxford University Press: Oxford, 2006. [https://www.oxfordreference.com/display/10.1093/acref/9780198529170.001.0001/acref-9780198529170-e-15515].
- Concise Medical Dictionary (8th ed.) Oxford University Press: Oxford, 2010. [https://www.oxfordreference.com/display/10.1093/acref/9780199557141.001.0001/acref-9780199557141-e-7777].
- A Dictionary of Food and Nutrition (3rd ed.) Oxford University Press: Oxford, 2009. [https://www.oxfordreference.com/display/10.1093/acref/9780199234875.001.0001/acref-9780199234875-e-4170].
- Oxford Dictionary of Biology (6th edition). Oxford University Press: Oxford - New York, 2008. [https://www.oxfordreference.com/display/10.1093/acref/9780199204625.001.0001/acref-9780199204625-e-3399].
- Barnes, C.R. On the food of green plants. Bot. Gaz. 1893, 18, 403–411, [http://www.jstor.org/stable/2464454]. [Google Scholar] [CrossRef]
- Gest, H. History of the word photosynthesis and evolution of its definition. Photosynthesis Research, 2002, 73, 7–10. [Google Scholar] [CrossRef]
- Pfeffer, W. The physiology of plants; a treatise upon the metabolism and sources of energy in plants (Ed. and Transl. by A.J. Ewart.) Clarendon Press: Oxford, 1900. [CrossRef]
- Kamen, M.D. Primary Processes in Photosynthesis. Academic Press: New York, 1963. [https://shop.elsevier.com/books/primary-processes-in-photosynthesis/kamen/978-1-4832-2959-1].
- Gest, H. Photosynthetic and quasi-photosynthetic bacteria. FEMS Microbiol. Lett. 1993, 112, 1–6. [Google Scholar] [CrossRef]
- Gest, H. Evolution of knowledge encapsulated in scientific definitions. Persp. Biol. Med. 2001, 44, 556–564. [Google Scholar] [CrossRef]
- Gest, H. History of concepts of the comparative biochemistry of oxygenic and anoxygenic photosyntheses. Photosyn. Res. 1993, 35, 87–96. [Google Scholar] [CrossRef]
- Badyaev, A.V. Evolution despite natural selection? Emergence theory and the ever elusive link between adaptation and adaptability. Acta Biotheoretica 2008, 56, 249–255. [Google Scholar] [CrossRef]
- Van der Steen, W. J. Methodological problems in evolutionary biology. X. Natural selection without selective agents. Acta Biotheoretica 1998, 46, 99–107. [Google Scholar] [CrossRef]
- Sharma, V.; Annila, A. Natural process–Natural selection. Biophysical Chemistry 2007, 127, 123–128, [https://pubmed.ncbi.nlm.nih.gov/17289252/]. [Google Scholar] [CrossRef] [PubMed]
- Oro, J.; Armangue, G.; Mar, A. The principle of cooperation and life's origin and evolution. NASA, Washington Second Symposium on Chemical Evolution and the Origin and Evolution of Life; 1986, p. 78.
- Shnoll, S. Physico-chemical factors of biological evolution. In Soviet Scientific Reviews Supplement Series. Physicochemical Biology (Revised Edition); Routledge, 1981; Volume 1, 280 p.
- Volkenstein, M.Y. Physical Approaches to Biological Evolution (reprint of the original 1st ed. 1994); Springer: Berlin, Heidelberg, 2011; 420 p. [Google Scholar] [CrossRef]
- McConnell, I.; Li, G.; Brudvig, G.W. Energy conversion in natural and artificial photosynthesis. Chem. Biol. 2010, 17, 434–447, [https://pubmed.ncbi.nlm.nih.gov/20534342/]. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Shkuropatov, A.Y.; Shuvalov, V.A. Femtosecond nuclear oscillations under charge separation in reaction centers of photosynthesis. Biochemistry (Moscow) 2003, 68, 541–550, [https://pubmed.ncbi.nlm.nih.gov/12882636/]. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Shuvalov, V.A. Modeling of reversible charge separation in reaction centers of photosynthesis: an incoherent approach. J. Theor. Biol. 2014, 343, 92–101, [https://pubmed.ncbi.nlm.nih.gov/24270095/]. [Google Scholar] [CrossRef]
- Yakovlev, A.G.; Shuvalov, V.A. Reversible charge separation in reaction centers of photosynthesis: a classical model. Doklady Biochemistry and Biophysics 2013, 450, 143–146, [https://pubmed.ncbi.nlm.nih.gov/23824456/]. [Google Scholar] [CrossRef]
- Slovacek, R.E.; Hind, G. Correlation between photosynthesis and the transthylakoid proton gradient. Biochim. Biophys. Acta 1981, 635, 393–404, [https://pubmed.ncbi.nlm.nih.gov/7236671/]. [Google Scholar] [CrossRef]
- Enser, U.; Heber, U. Metabolic regulation by pH gradients. Inhibition of photosynthesis by indirect proton transfer across the chloroplast envelope. Biochim. Biophys. Acta 1980, 592, 577–591, [https://pubmed.ncbi.nlm.nih.gov/6251871/]. [Google Scholar] [CrossRef]
- Geacintov, N.E. Tracing charge separation events in photosynthesis: anomalous photovoltage polarity events explained. Biophys. J. 1993, 65, 11–12, [https://pubmed.ncbi.nlm.nih.gov/8369419/]. [Google Scholar] [CrossRef] [PubMed]
- LeBard, D.N.; Kapko, V.; Matyushov, D.V. Energetics and kinetics of primary charge separation in bacterial photosynthesis. J. Phys. Chem. B. 2008, 112, 10322–10342, [https://pubmed.ncbi.nlm.nih.gov/18636767/]. [Google Scholar] [CrossRef] [PubMed]
- Makri, N.; Sim, E.; Makarov, D.E.; Topaler, M. Long-time quantum simulation of the primary charge separation in bacterial photosynthesis. Proc. Nat. Acad. Sci. USA 1996, 93, 3926–3931, [https://pubmed.ncbi.nlm.nih.gov/8632991/]. [Google Scholar] [CrossRef] [PubMed]
- Fajer, J.; Brune, D.C.; Davis, M.S.; Forman, A.; Spaulding, L.D. Primary charge separation in bacterial photosynthesis: oxidized chlorophylls and reduced pheophytin (reduced bacteriopheophytin / transient electron acceptor). Proc. Nat. Acad. Sci. USA 1975, 72, 4956–4960, [https://pubmed.ncbi.nlm.nih.gov/174084/]. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.W.; Chu, Z.T.; Warshel, A. Electrostatic control of charge separation in bacterial photosynthesis. Biochim. Biophys. Acta 1990, 1017, 251–272, [https://pubmed.ncbi.nlm.nih.gov/2196939/]. [Google Scholar] [CrossRef]
- Oremland, R.S.; Stolz, J.F. Arsenic microbes and contaminated aquifers. Trends Microbiol. 2005, 13, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Wadas, T.J.; Hester, H.; Schmehl, R.; Eisenberg, R. Platinum chromophore-based systems for photoinduced charge separation: a molecular design approach for artificial photosynthesis. Inorg. Chem. 2005, 44, 6865–6878. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Boixel, J.; Pellegrin, Y.; Blart, E.; Becker, H.C.; Odobel, F.; Hammarström, L. Accumulative charge separation inspired by photosynthesis. J. Am. Chem. Soc. 2010, 132, 17977–17979. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Boixel, J.; Pellegrin, Y.; Blart, E.; Becker, H.C.; Odobel, F.; Hammarström, L. Accumulative electron transfer: multiple charge separation in artificial photosynthesis. Faraday Discuss. 2012, 155, 233–252, [https://pubmed.ncbi.nlm.nih.gov/22470977/]. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Ohkubo, K.; Suenobu, T. Long-lived charge separation and applications in artificial photosynthesis. Acc. Chem. Res. 2014, 47, 1455–1464, [https://pubmed.ncbi.nlm.nih.gov/24793793/]. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Xiao, T.; Zhang, Q.; Liu, Z. Photosynthesis-inspired bifunctional energy-harvesting devices that convert light and salinity gradients into electricity. Chem. Commun. 2018, 54, 12310–12313, [https://pubmed.ncbi.nlm.nih.gov/30272063/]. [Google Scholar] [CrossRef] [PubMed]
- Altamura, E.; Milano, F.; Tangorra, R.R.; Trotta, M.; Omar, O.H.; Stano, P.; Mavelli, F. Highly oriented photosynthetic reaction centers generate a proton gradient in synthetic protocells. Proc. Nat. Acad. Sci. 2017, 114, 3837–3842, [https://pubmed.ncbi.nlm.nih.gov/28320948/]. [Google Scholar] [CrossRef]
- Katsoukis, G.; Frei, H. Heterobinuclear light absorber coupled to molecular wire for charge transport across ultrathin silica membrane for artificial photosynthesis. ACS Appl. Mater. Interfaces 2018, 10, 31422–31432, [https://pubmed.ncbi.nlm.nih.gov/30146876/]. [Google Scholar] [CrossRef]
- Matyushov, D.V. Reorganization asymmetry of electron transfer in ferroelectric media and principles of artificial photosynthesis. J. Phys. Chem. B. 2006, 110, 10095–10104, [https://pubmed.ncbi.nlm.nih.gov/16706471/]. [Google Scholar] [CrossRef]
- Leuchtag, H.R. Indications of the existence of ferroelectric units in excitable-membrane channels. J. Theor. Biol.; 1987, 127, 321–340, [https://pubmed.ncbi.nlm.nih.gov/2448549/]. [Google Scholar] [CrossRef] [PubMed]
- Leuchtag, H.R. Phase transitions and ion currents in a model ferroelectric channel unit. J. Theor. Biol.; 1987, 127, 341–359, [https://pubmed.ncbi.nlm.nih.gov/2448550/]. [Google Scholar] [CrossRef] [PubMed]
- Leuchtag, H.R. A proposed physical explanation of the activation of sodium channels. Ferroelectrics 1988, 86, 105–113. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Leuchtag, H.R. Bioferroelectricity: Modeling the transitions of the sodium channel. Ferroelectrics 1994, 155, 19–24. [Google Scholar] [CrossRef]
- Leuchtag, H.R. Fit of the dielectric anomaly of squid axon membrane near heat-block temperature to the ferroelectric Curie-Weiss law. Biophysical Chemistry 1995, 53, 197–205, [https://pubmed.ncbi.nlm.nih.gov/17020847/]. [Google Scholar] [CrossRef] [PubMed]
- Leuchtag, H.R.; Bystrov, V.S. Theoretical models of conformational transitions and ion conduction in voltage-dependent ion channels: Bioferroelectricity and superionic conduction. Ferroelectrics 1999, 220, 157–204. [Google Scholar] [CrossRef]
- Fendler, J.H. Artificial photosynthesis–an example of membrane mimetic chemistry. BioEssays 1984, 1, 165–167. [Google Scholar] [CrossRef]
- Brown, K.A.; King, P.W. Coupling biology to synthetic nanomaterials for semi-artificial photosynthesis. Photosynth. Res. 2020, 143, 193–203, [https://pubmed.ncbi.nlm.nih.gov/31641988/]. [Google Scholar] [CrossRef]
- Hafizi, B. Nonlinear evolution equations, recurrence and stochasticity. The Physics of Fluids 1981, 24, 1791–1798. [Google Scholar] [CrossRef]
- Saccone, C.; Preparata, G.; Lanave, C.; Quagliariello, E.; Bernardi, G.; Ullmann, A. Chance, stochasticity and evolution: the Markov clock. In Enzyme Adaptation to Natural Philosophy: Heritage from Jacques Monod.; Elsevier Science Publishers BV (Biomedical Division): Amsterdam – New York, Netherlands – USA, 1987; pp. 159–172. [Google Scholar]
- Lenormand, T.; Roze, D.; Rousset, F. Stochasticity in evolution. Trends in Ecology and Evolution 2009, 24, 157–165, [https://pubmed.ncbi.nlm.nih.gov/19178980/]. [Google Scholar] [CrossRef]
- Rosenfeld, S. Mathematical descriptions of biochemical networks: stability, stochasticity, evolution. Progress in Biophysics and Molecular Biology 2011, 106, 400–409, [https://pubmed.ncbi.nlm.nih.gov/21419158/]. [Google Scholar] [CrossRef]
- Pradas, M.; Schmuck, M.; Pavliotis, G.; Kalliadasis, S. Understanding the evolution of complex multiscale systems: Dynamic renormalization, non-equilibrium entropy and stochasticity. In Bulletin of the American Physical Society (66th Annual Meeting of the APS Division of Fluid Dynamics), 2013, 58, H35.00008.
- Egel, R. “Parabiotic Evolution”: From Stochasticity in Geochemical and Subsequent Processes to Genes, Genomes and Modular Cells 2017, MDPI Preprint. [CrossRef]
- Danino, M.; Kessler, D.A.; Shnerb, N.M. Environmental stochasticity and the speed of evolution. Journal of Statistical Physics 2018, 172, 126–142. [Google Scholar] [CrossRef]
- Hyun, J.S.; Park, C.J. Classification of contradiction relations and their solving dimensions based on the butterfly model for contradiction solving for physical contradiction of TRIZ. Knowledge Management Research 2014, 15, 15–34. [Google Scholar] [CrossRef]
- Choi, S.W. Review and application of creative problem-solving processes for technical and physical contradictions using cause-and-effect contradiction tree and integrated principles of TRIZ. Journal of the Korea Safety Management and Science 2015, 17, 215–228. [Google Scholar] [CrossRef]
- Zhang, X.P.; Qiu, M.; Pi, Y.M. Discussion on applying the ideal final result of TRIZ in the simplified physical problems. Journal of Heihe University 2010, 2, 10. [Google Scholar]
- Kim, J.; Kim, J.; Lee, Y.; Lim, W.; Moon, I. Application of TRIZ creativity intensification approach to chemical process safety. Journal of Loss Prevention in the Process Industries 2009, 22, 1039–1043. [Google Scholar] [CrossRef]
- Li, Y.L.; Zhao, H.Y.; Jiang, T.; Zhang, Q.M.; Huang, Y.B. The inventing principles of 40 TRIZ reflected in the chemical industry. Guangzhou Chemical Industry, 2010, 30, 8. [Google Scholar]
- Tunuli, M.S.; Fendler, J.H. Aspects of artificial photosynthesis. Photosensitized electron transfer across bilayers, charge separation, and hydrogen production in anionic surfactant vesicles. J. Amer. Chem. Soc. 1981, 103, 2507–2513. [Google Scholar] [CrossRef]
- Infelta, P.P.; Graetzel, M.; Fendler, J.H. Aspects of artificial photosynthesis. Photosensitized electron transfer and charge separation in cationic surfactant vesicles. J. Amer. Chem. Soc. 1980, 102, 1479–1483. [Google Scholar] [CrossRef]
- Kurihara, K.; Tundo, P.; Fendler, J.H. Aspects of artificial photosynthesis. Photosensitized electron transfer and charge separation in redox active surfactant aggregates. J. Phys. Chem. 1983, 87, 3777–3782. [Google Scholar] [CrossRef]
- Tunuli, M.S.; Fendler, J.H. Aspects of artificial photosynthesis: the role of potential gradients in promoting charge separation in the presence of surfactant vesicles. In Inorganic Reactions in Organized Media; American Chemical Society, Washington, D.C., USA, 1982; Volume 177, pp. 53-70. [CrossRef]
- Macnaughtan, M.L.; Frei, H.M. Synthesis of heterobimetallic charge transfer chromophores and coupled oxidation catalysts for artificial photosynthesis. In Preprints of Symposia – Division of Fuel Chemistry, American Chemical Society, CD ROM Edition; American Chemical Society, Washington, D.C., USA, 2011; Volume 56 (Issue 1), 149.
- Lee, S.H.; Kim, J.H.; Park, C.B. Coupling photocatalysis and redox biocatalysis toward biocatalyzed artificial photosynthesis. Chem. Eur. J. 2013, 19, 4392–4406, [https://pubmed.ncbi.nlm.nih.gov/23436280/]. [Google Scholar] [CrossRef]
- Đokić, M.; Soo, H.S. Artificial photosynthesis by light absorption, charge separation, and multielectron catalysis. Chem. Commun. 2018, 54, 6554–6572. [Google Scholar] [CrossRef]
- Pannwitz, A.; Wenger, O.S. Proton-coupled multi-electron transfer and its relevance for artificial photosynthesis and photoredox catalysis. Chem. Commun. 2019, 55, 4004–4014, [https://pubmed.ncbi.nlm.nih.gov/30810148/]. [Google Scholar] [CrossRef] [PubMed]
- Moberg, S. Artificial photosynthesis-4-aminobenzoic acids effect on charge transfer in a photo catalytic system. PhD Thesis, Uppsala University (Department of Physics and Astronomy), Uppsala, 2019. [Google Scholar]
- Rasmussen, S.; Chen, L.; Deamer, D.; Krakauer, D.C.; Packard, N.H.; Stadler, P.F.; Bedau, M.A. Transitions from nonliving to living matter. Science 2009, 303, 963–965, [https://pubmed.ncbi.nlm.nih.gov/14963315/]. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.; Chen, L.; Nilsson, M.; Abe, S. Bridging nonliving and living matter. Artificial Life 2003, 9, 269–316, [https://pubmed.ncbi.nlm.nih.gov/14556688/]. [Google Scholar] [CrossRef] [PubMed]
- Tamulis, A.; Grigalavicius, M.; Krisciukaitis, S.; Medzevicius, G. Quantum processes in 8-oxo-guanine-Ru (bipyridine)32+ photosynthetic systems of artificial minimal cells. Open Physics 2011, 9, 775–791. [Google Scholar] [CrossRef]
- Tamulis, A. Quantum mechanical investigations of photosynthetic systems of artificial minimal cells based on 8-oxo-guanine-Ru (bipyridine) 2+3. Journal of Computational and Theoretical Nanoscience 2011, 8, 624–636. [Google Scholar] [CrossRef]
- Tamulis, A.; Grigalavicius, M. The emergence and evolution of life in a “fatty acid world” based on quantum mechanics. Origins Life Evol. Biospheres 2011, 41, 51–71, [https://pubmed.ncbi.nlm.nih.gov/20443139/]. [Google Scholar] [CrossRef] [PubMed]
- Tamulis, A.; Tamulis, V. Question 9: Quantum self-assembly and photoinduced electron tunneling in photosynthetic systems of artificial minimal living cells. Origins Life Evol. Biospheres 2007, 37, 473–476, [https://pubmed.ncbi.nlm.nih.gov/17610046/]. [Google Scholar] [CrossRef] [PubMed]
- Tamulis, A.; Tamulis, V. Quantum self-assembly and photoinduced electron tunneling in photosynthetic system of minimal living cell. Viva Origino 2007, 35, 66–72. [Google Scholar] [CrossRef]
- Tamulis, A.; Berteska, L.; Grigalavicius, M.; Baltrusaitis, J. Quantum dynamics of self-assembly of minimal photosynthetic cells. Quantum Matter 2016, 5, 5–18. [Google Scholar] [CrossRef]
- Tamulis, A.; Grigalavicius, M. Molecular spintronics control of photosynthesis in artificial cell. J. Comput. Theor. Nanosci. 2013, 10, 989–995. [Google Scholar] [CrossRef]
- Tamulis, A.; Grigalavicius, M. Quantum entanglement in photoactive prebiotic systems. Systems and Synthetic Biology 2014, 8, 117–140, [https://pubmed.ncbi.nlm.nih.gov/24799958/]. [Google Scholar] [CrossRef]
- Tamulis, A.; Grigalavicius, M.; Baltrusaitis, J. Phenomenon of quantum entanglement in a system composed of two minimal protocells. Origins Life Evol. Biospheres 2013, 43, 49–66, [https://pubmed.ncbi.nlm.nih.gov/23242832/]. [Google Scholar] [CrossRef] [PubMed]
- Tamulis, A.; Grigalavicius, M.; Serbenta, J.; Plausinaitis, K. Quantum entangled single bioorganic supramolecules as light absorbing and light emitting logical devices. J. Comput. Theor. Nanosci. 2015, 12, 1827–1840. [Google Scholar] [CrossRef]
- Rubin, A.; Riznichenko, G. Mathematical Biophysics; Springer: New York, Heidelberg, Dordrecht, London, 2014; 274 p. [Google Scholar]
- Rubin, A.B. Fundamentals of Biophysics; John Wiley & Sons, Inc.: Hoboken, New Jersey, and Scrivener Publishing LLC: Salem, Massachusetts, 2014; 212 p. [Google Scholar]
- Blumenfeld, L.A. Problems of Biological Physics; Springer: Berlin, Heidelberg, 1981; 224 p. [Google Scholar]
- Armstrong, E.F. Studies on enzyme action. II.--The rate of the change, conditioned by sucroclastic enzymes, and its bearing on the law of mass action. Proceedings of the Royal Society of London 1904, 73, 500–516. [Google Scholar] [CrossRef]
- Armstrong, E.F.; Hilditch, T.P. A study of catalytic actions at solid surfaces. V.—The rate of change conditioned by a nickel catalyst and its bearing on the law of mass action. Proceedings of the Royal Society of London, Series A 1920, 98, 27–40. [Google Scholar] [CrossRef]
- Pomogailo, A.D. Catalysis by polymer-immobilized metal complexes. Gordon Breach Sci. Publ.: Amsterdam, 1998; 424 p. [CrossRef]
- Skulachev, V.P. The sodium cycle: a novel type of bacterial energetics. J. Bioenergetics Biomembranes 1989, 21, 635–647, [https://pubmed.ncbi.nlm.nih.gov/2687258/]. [Google Scholar] [CrossRef]
- Skulachev, V.P. The latest news from the sodium world. Biochimica et Biophysica Acta – Bioenergetics 1994, 1187, 216–221. [Google Scholar] [CrossRef]
- Dibrova, D.; Mulkidjanian, A. Reconstruction of the primordial “Sodium World”. Biochimica et Biophysica Acta – Bioenergetics 2014, 1837, e84. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Dibrov, P.; Galperin, M.Y. The past and present of sodium energetics: may the sodium-motive force be with you. Biochimica et Biophysica Acta –Bioenergetics 2008, 1777, 985–992. [Google Scholar] [CrossRef]
- Henry, V. Théorie générale de l'action de quelques diastases (présentée par M. Roux.). Comptes rendus hebdomadaires des séances de l'Académie des sciences 1902, 135, 916–919. [Google Scholar]
- Michaelis, L.; Menten, M.L. The kinetics of the inversion effect. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Michaelis, L.; Menten, M.L. The kinetics of invertin action. FEBS Letters 2013, 587, 2712–2720, [https://pubmed.ncbi.nlm.nih.gov/23867202/]. [Google Scholar] [CrossRef]
- Kwak, J.; Kim, M.C.; Lee, S.Y. An enzyme-coupled artificial photosynthesis system prepared from antenna protein-mimetic tyrosyl bolaamphiphile self-assembly. Nanoscale 2016, 8, 15064–15070. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, L.A. The evolution of biocatalysts. Russ. Chem. Rev. 1961, 30, 117–133. [Google Scholar] [CrossRef]
- Nikolaev, L.A. The principles of biocatalyst modeling. Russ. Chem. Rev. 1964, 33, 275–286. [Google Scholar] [CrossRef]
- Purmal, A.P.; Nikolaev, L.A. The modelling of biological catalysts. Russ. Chem. Rev. 1985, 54, 466–475. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, C.; Zhang, Y.; He, F.; Liu, M.; Li, X. Preparation of graphene nano-sheet bonded PDA/MOF microcapsules with immobilized glucose oxidase as a mimetic multi-enzyme system for electrochemical sensing of glucose. J. Mater. Chem. B 2016, 4, 3695–3702. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, L.; Shang, C.; Zhang, Z.; Dong, S. Triple-enzyme mimetic activity of nickel–palladium hollow nanoparticles and their application in colorimetric biosensing of glucose. Chem. Commun. 2016, 52, 5410–5413. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, G.; Sun, F.; Lin, Y. Heterogeneous nanostructure design based on the epitaxial growth of spongy MoSx on 2D Co(OH)2 nanoflakes for triple-enzyme mimetic activity: experimental and density functional theory studies on the dramatic activation mechanism. ACS Appl. Mater. Interfaces 2018, 10, 32567–32578. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, H.; Liu, C.; Bing, W.; Wang, Z.; Qu, X. A multinuclear metal complex based DNase-mimetic artificial enzyme: matrix cleavage for combating bacterial biofilms. Angew. Chem. Int. Ed. 2016, 55, 10732–10736, [https://pubmed.ncbi.nlm.nih.gov/27484616/]. [Google Scholar] [CrossRef] [PubMed]
- Nagiev, T.M. Mimetic simulation of enzyme catalysis. Russ. J. Phys. Chem. 1996, 70, 895–903. [Google Scholar]
- Nagiev, T. The Theory of Conjugate Reactions in the Context of Modern Ideas. Advances in Chemical Engineering and Science 2019, 10, 52–68. [Google Scholar] [CrossRef]
- Chang, T.M.S.; Yu, Y.T.; Grunwald, J. Artificial cell immobilized multienzyme systems and cofactors. In Enzyme Engineering? Vol. 6; Springer: Boston, MA, 1982; pp. 451–456. [Google Scholar] [CrossRef]
- Chang, T.M.S.; Kuntarian, N. Galactose conversion using a microcapsule immobilized multienzyme cofactor recycling system. In Enzyme engineering, Vol. 4; Springer: Boston, MA, 1978; pp. 193–197. [Google Scholar] [CrossRef]
- Campbell, J.; Chang, T.M.S. Immobilized multienzyme systems and coenzyme requirements: perspectives in biomedical applications. Biomedical Applications of Immobilized Enzymes and Proteins 1977, 2, 281–302. [Google Scholar]
- Sun, G.; Shi, J.; Jia, S.; Luo, Y.; Jiang, Z.; Yuan, X. General model for artificial photosynthesis with capsule-immobilized enzyme. AIChE Journal 2022, 68, E17409. [Google Scholar] [CrossRef]
- Sarma, R.; Islam, M.; Running, M.P.; Bhattacharyya, D. Multienzyme immobilized polymeric membrane reactor for the transformation of a lignin model compound. Polymers 2018, 10, 463. [Google Scholar] [CrossRef] [PubMed]
- Yotova, L.; Medhat, N. Optical biosensor with multienzyme system immobilized onto hybrid membrane for pesticides determination. Int. J. Bioautomation 2011, 15, 267–276. [Google Scholar]
- Ho, S.P.; Kostin, M.D. Kinetics of immobilized multienzyme systems. J. Chem. Phys. 1974, 61, 918–920. [Google Scholar] [CrossRef]
- Fernandes, P.M. Mathematical modeling of immobilized multienzyme systems. Doctoral dissertation, Rutgers University, 1977.
- Gu, K.F.; Chang, T.M.S. Conversion of α-ketoglutarate into L-glutamic acid with urea as ammonium source using multienzyme systems and dextran-NAD+ immobilized by microencapsulation within artificial cells in a bioreactor. Biotechnol. Bioeng. 1988, 32, 59–62. [Google Scholar] [CrossRef]
- Chang, T.M.S. Recycling of NAD(P) by multienzyme systems immobilized by microencapsulation in artificial cells. In Methods in Enzymology; Academic Press, 1987; Volume 136, pp. 67-82. [CrossRef]
- Chang, T. Biotechnological approach using artificial cells immobilized multienzyme systems and hepatocytes for bioartificial liver. Biomaterials Artificial Cells and Artificial Organs 1988, 16, 844. [Google Scholar]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biological Reviews 1966, 41, 445–501. [Google Scholar] [CrossRef]
- Slater, E.C. An evaluation of the Mitchell hypothesis of chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Eur. J. Biochem. 1967, 1, 317–326, [https://pubmed.ncbi.nlm.nih.gov/4293928/]. [Google Scholar] [CrossRef]
- Telfer, A.; Evans, M.C.W. Evidence for chemiosmotic coupling of electron transport to ATP synthesis in spinach chloroplasts. Biochimica et Biophysica Acta – Bioenergetics 1972, 256, 625–637, [https://pubmed.ncbi.nlm.nih.gov/5020234/]. [Google Scholar] [CrossRef]
- Hangarter, R.P.; Good, N.E. Energy thresholds for ATP synthesis in chloroplasts. Biochimica et Biophysica Acta – Bioenergetics 1982, 681, 397–404. [Google Scholar] [CrossRef]
- Jakobsson, E. Interactions of cell volume, membrane potential, and membrane transport parameters. American Journal of Physiology-Cell Physiology 1980, 238, C196–C206, [https://pubmed.ncbi.nlm.nih.gov/7377338/]. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Dunham, P.B. Membrane mechanisms and intracellular signalling in cell volume regulation. International Review of Cytology 1995, 161, 173–262, [https://pubmed.ncbi.nlm.nih.gov/7558691/]. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Mills, J.W. Membrane events involved in volume regulation. In Current Topics in Membranes; Academic Press, 1999; Volume 48, pp. 123-196. [CrossRef]
- Marrink, S.J.; Sok, R.M.; Berendsen, H.J.C. Free volume properties of a simulated lipid membrane. Journal of Chemical Physics 1996, 104, 9090–9099. [Google Scholar] [CrossRef]
- Smith, D.C.; Bassham, J.A.; Kirk, M. Dynamics of the photosynthesis of carbon compounds II. Amino acid synthesis. Biochimica et Biophysica Acta 1961, 48, 299–313. [Google Scholar] [CrossRef]
- Heber, U. Protein synthesis in chloroplasts during photosynthesis. Nature 1962, 195, 91–92, [https://pubmed.ncbi.nlm.nih.gov/13905812/]. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Morris, I. Synthesis of lipid during photosynthesis by phytoplankton of the Southern Ocean. Science 1980, 207, 197–199, [https://pubmed.ncbi.nlm.nih.gov/17809104/]. [Google Scholar] [CrossRef]
- Weber, A.P. Synthesis, export and partitioning of the end products of photosynthesis. In The structure and function of plastids; Springer: Dordrecht, 2007; pp. 273–292. [Google Scholar] [CrossRef]
- Hazen, R.M.; Sverjensky, D.A. Mineral surfaces, geochemical complexities, and the origins of life. Cold Spring Harbor perspectives in biology 2010, 2, a002162, [https://pubmed.ncbi.nlm.nih.gov/20452963/]. [Google Scholar] [CrossRef]
- Schoonen, M.; Smirnov, A.; Cohn, C. A perspective on the role of minerals in prebiotic synthesis. AMBIO: A Journal of the Human Environment 2004, 33, 539–551, [https://pubmed.ncbi.nlm.nih.gov/15666687/]. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Evolution of the first metabolic cycles. Proceedings of the National Academy of Sciences 1990, 87, 200–204, [https://pubmed.ncbi.nlm.nih.gov/2296579/]. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Groundworks for an evolutionary biochemistry: the iron-sulfur world. Progress in Biophysics and Molecular Biology 1992, 58, 85–201, [https://pubmed.ncbi.nlm.nih.gov/1509092/]. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. From volcanic origins of chemoautotrophic life to Bacteria, Archaea and Eukarya. Philosophical Transactions of the Royal Society, B, Biological Science, 2006, 361, 1787–1806, [https://pubmed.ncbi.nlm.nih.gov/17008219/]. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Pyrite formation, the first energy source for life: a hypothesis. Systematic and Applied Microbiology 1988, 10, 207–210. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Before enzymes and templates: theory of surface metabolism. Microbiological reviews 1988, 52, 452. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. α-Hydroxy and α-Amino Acids Under Possible Hadean, Volcanic Origin-of-Life Conditions. Science 2006, 314, 630–632, [https://pubmed.ncbi.nlm.nih.gov/17068257/]. [Google Scholar] [CrossRef]
- White, L.M.; Bhartia, R.; Stucky, G.D.; Kanik, I.; Russell, M.J. Mackinawite and greigite in ancient alkaline hydrothermal chimneys: identifying potential key catalysts for emergent life. Earth and Planetary Science Letters 2015, 430, 105–114. [Google Scholar] [CrossRef]
- Wu, M.; John, S.T.; Pan, Y. Electronic structures of greigite (Fe3S4): A hybrid functional study and prediction for a Verwey transition. Scientific Reports 2016, 6, 21637. [Google Scholar] [CrossRef]
- Mielke, R.E.; Robinson, K.J.; White, L.M.; McGlynn, S.E.; McEachern, K.; Bhartia, R.; Kanik, I,; Russell, M. J. Iron-sulfide-bearing chimneys as potential catalytic energy traps at life's emergence. Astrobiology 2011, 11, 933–950, [https://pubmed.ncbi.nlm.nih.gov/22111762/]. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origins of cells: a hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 2003, 358, 59–85, [https://pubmed.ncbi.nlm.nih.gov/12594918/]. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philosophical Transactions of the Royal Society B: Biological Sciences 2007, 362, 1887–1926, [https://pubmed.ncbi.nlm.nih.gov/17255002/]. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Hall, A.J.; Martin, W. Serpentinization as a source of energy at the origin of life. Geobiology 2010, 8, 355–371, [https://pubmed.ncbi.nlm.nih.gov/20572872/]. [Google Scholar] [CrossRef] [PubMed]
- Macleod, G.; McKeown; C. ; Hall; A. J.; Russell, M.J. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Origins of Life and Evolution of the Biosphere 1994, 24, 19–41, [https://pubmed.ncbi.nlm.nih.gov/11536657/]. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Daniel, R.M.; Hall, A.J. On the emergence of life via catalytic iron-sulphide membranes. Terra Nova 1993, 5, 343–347. [Google Scholar] [CrossRef]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends in Biochemical Sciences 2004, 29, 358–363, [https://pubmed.ncbi.nlm.nih.gov/15236743/]. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. Journal of the Geological Society 1997, 154, 377–402, [https://pubmed.ncbi.nlm.nih.gov/11541234/]. [Google Scholar] [CrossRef]
- Russell, M.J. Green rust: The simple organizing ‘seed’of all life? Life 2018, 8, 35. [Google Scholar] [CrossRef]
- Russell, M.J.; Ponce, A. Six ‘must-have’minerals for life’s emergence: Olivine, pyrrhotite, bridgmanite, serpentine, fougerite and mackinawite. Life 2020, 10, 291. [Google Scholar] [CrossRef]
- Kizilstein, L.Ya. Framboidal pyrite involved in the revival of life on The Earth? Priroda 2007, 1, 49–54. (in Russian). [Google Scholar]
- Kizilshtein, L.Ya.; Minaeva, L.G. Origin of the framboidal pyrite. Dokl. Akad. Nauk SSSR 1972, 206, 1187–1189. [Google Scholar]
- Liu, A.G. Framboidal pyrite shroud confirms the ‘death mask'model for moldic preservation of ediacaran soft-bodied organismsediacaran taphonomy. Palaios 2016, 31, 259–274. [Google Scholar] [CrossRef]
- Retallack, G.J. Comment to Liu. Framboidal pyrite shroud confirms the ‘death mask'model for moldic preservation of ediacaran soft-bodied organisms. Palaios 2017, 32, 195–196. [Google Scholar] [CrossRef]
- Kalliokoski, J. Framboids—macrocrystals of colloidal pyrite. Econ. Geol. 1965, 60, 1562. [Google Scholar]
- Sawłowicz, Z. Framboids: from their origin to application. Prace Mineralogiczne (Mineralogical Transactions, Polska Akademia Nauk - Komisja Nauk Mineralogicznych) 2000, 88, 3–58. [Google Scholar]
- Granick, S. Speculations on the origins and evolution of photosynthesis. Annals of the New York Academy of Sciences 1957, 69, 292–308, [https://pubmed.ncbi.nlm.nih.gov/13479007/]. [Google Scholar] [CrossRef]
- Granick, S. Evolution of heme and chlorophyll. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, N. Y.; 1965; pp. 67–88. [Google Scholar]
- Mauzerall, D. Light, iron, Sam Granick and the origin of life. Photosynthesis research 1992, 33, 163–170, [https://pubmed.ncbi.nlm.nih.gov/24408576/]. [Google Scholar] [CrossRef] [PubMed]
- Shnoll, S. Physico-chemical factors of biological evolution; Nauka: Moscow, USSR, 1979; 263 p. (in Russian) [Google Scholar]
- Grätzel, M. Energy resources through photochemistry and catalysis. Acad. Press: New York; 1983, 632 p. [CrossRef]
- Ferreira, D.L.; Sousa, J.C.L.; Maronesi, R.N.; Bettini, J.; Schiavon, M.A.; Teixeira, A.V.; Silva, A.G. Size-dependent bandgap and particle size distribution of colloidal semiconductor nanocrystals. Journal of Chemical Physics 2017, 147, 154102. [Google Scholar] [CrossRef]
- Volkenstein, F.F. Semiconductors as catalysts for chemical reactions; Publishing House of Moscow State University: Moscow, 1968; pp. 1–40. [Google Scholar]
- Emeline, A.V.; Otroshchenko, V.A.; Ryabchuk, V.K.; Serpone, N. Abiogenesis and photostimulated heterogeneous reactions in the interstellar medium and on primitive earth: relevance to the genesis of life. Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2003, 3, 203–224. [Google Scholar] [CrossRef]
- Parmon, V.N.; Zakharenko, V.S. Photocatalysis and photosorption in the Earth's atmosphere. Cattech, 2001 5, 96-115. [CrossRef]
- Parmon, V.N. Abiogenic catalysis in Nature. Colloids and Surfaces A: Physicochemical and Engineering Aspects 1999, 151, 351–365. [Google Scholar] [CrossRef]
- Xia, D.; Wang, W.; Wong, P.K. Visible-light-driven photocatalytic treatment by environmental minerals. In Advances in Photocatalytic Disinfection; Springer: Berlin, Heidelberg, 2017; pp. 41–61. [Google Scholar] [CrossRef]
- Nikandrov, V.V. Inorganic semiconductors as photosensitizers in biochemical redox reactions. Membr. Cell Biol. 1998, 12, 755–769. [Google Scholar]
- Nikandrov, V.V.; Grätzel, C.K.; Moser, J.E.; Grätzel, M. Light induced redox reactions involving mammalian ferritin as photocatalyst. Journal of Photochemistry and Photobiology B: Biology 1997, 41, 83–89, [https://pubmed.ncbi.nlm.nih.gov/9440316/]. [Google Scholar] [CrossRef]
- Shumilin, I.A.; Nikandrov, V.V.; Popov, V.O.; Krasnovsky, A.A. Photogeneration of NADH under coupled action of CdS semiconductor and hydrogenase from Alcaligenes eutrophus without exogenous mediators. FEBS letters 1992, 306, 125–128, [https://pubmed.ncbi.nlm.nih.gov/1633866/]. [Google Scholar] [CrossRef]
- Nikandrov, V.V.; Shlyk, M.A.; Zorin, N.A.; Gogotov, I.N.; Krasnovsky, A.A. Efficient photoinduced electron transfer from inorganic semiconductor TiO2 to bacterial hydrogenase. FEBS Letters 1988, 234, 111–114. [Google Scholar] [CrossRef]
- Krasnovsky, A.A.; Nikandrov, V.V. The photobiocatalytic system: Inorganic semiconductors coupled to bacterial cells. FEBS letters 1987, 219, 93–96. [Google Scholar] [CrossRef]
- Wang, D.; Han, D.; Shi, Z.; Wang, J.; Yang, J.; Li, X.; Song, H. Optimized design of three-dimensional multi-shell Fe3O4/SiO2/ZnO/ZnSe microspheres with type II heterostructure for photocatalytic applications. Applied Catalysis B: Environmental 2018, 227, 61–69. [Google Scholar] [CrossRef]
- Bagheri, S.; Julkapli, N.M. Magnetite hybrid photocatalysis: advance environmental remediation. Reviews in Inorganic Chemistry 2016, 36, 135–151. [Google Scholar] [CrossRef]
- Fakhri, A.; Naji, M.; Nejad, P.A. Adsorption and photocatalysis efficiency of magnetite quantum dots anchored tin dioxide nanofibers for removal of mutagenic compound: toxicity evaluation and antibacterial activity. Journal of Photochemistry and Photobiology B: Biology 2017, 173, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Lee, Z.; Wei, M.; Chang, C.C.; Chu, K.W. Photocatalytic hydrogen production by magnetically separable Fe3O4@ZnS and NiCo2O4@ZnS core–shell nanoparticles. International Journal of Hydrogen Energy 2015, 40, 11436–11443. [Google Scholar] [CrossRef]
- Preethi, V.; Kanmani, S. Photocatalytic hydrogen production using Fe2O3-based core shell nanoparticles with ZnS and CdS. International Journal of Hydrogen Energy 2014, 39, 1613–1622. [Google Scholar] [CrossRef]
- Roychowdhury, A.; Pati, S.P.; Kumar, S.; Das, D. Effects of magnetite nanoparticles on optical properties of zinc sulfide in fluorescent-magnetic Fe3O4/ZnS nanocomposites. Powder technology 2014, 254, 583–590. [Google Scholar] [CrossRef]
- Atla, S.B.; Lin, W.R.; Chien, T.C.; Tseng, M.J.; Shu, J.C.; Chen, C.C.; Chen, C.Y. Fabrication of Fe3O4/ZnO magnetite core shell and its application in photocatalysis using sunlight. Materials Chemistry and Physics 2018, 216, 380–386. [Google Scholar] [CrossRef]
- Akkari, M.; Aranda, P.; Mayoral, A.; García-Hernández, M.; Amara, A.B.H.; Ruiz-Hitzky, E. Sepiolite nanoplatform for the simultaneous assembly of magnetite and zinc oxide nanoparticles as photocatalyst for improving removal of organic pollutants. Journal of Hazardous Materials 2017, 340, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, J.; Li, X.; Wang, D.; Wei, B.; Song, H.; Li, X.; Fu, S. Preparation and photocatalytic properties of magnetically reusable Fe3O4@ZnO core/shell nanoparticles. Physica E: Low-dimensional Systems and Nanostructures 2016, 75, 66–71. [Google Scholar] [CrossRef]
- Huang, S.; Gu, L.; Zhu, N.; Feng, K.; Yuan, H.; Lou, Z.; Li, Y.; Shan, A. Heavy metal recovery from electroplating wastewater by synthesis of mixed-Fe3O4@SiO2/metal oxide magnetite photocatalysts. Green Chemistry 2014, 16(5), 2696–2705. [Google Scholar] [CrossRef]
- Beydoun, D.; Amal, R.; Low, G.K.C.; McEvoy, S. Novel photocatalyst: titania-coated magnetite. Activity and photodissolution. The Journal of Physical Chemistry B 2000, 104, 4387–4396. [Google Scholar] [CrossRef]
- Xu, J.; Ao, Y.; Fu, D.; Yuan, C. Low-temperature preparation of anatase titania-coated magnetite. Journal of Physics and Chemistry of Solids 2008, 69, 1980–1984. [Google Scholar] [CrossRef]
- Yan, X.; Yuan, K.; Lu, N.; Xu, H.; Zhang, S.; Takeuchi, N.; Kobayashi, H.; Li, R. The interplay of sulfur doping and surface hydroxyl in band gap engineering: Mesoporous sulfur-doped TiO2 coupled with magnetite as a recyclable, efficient, visible light active photocatalyst for water purification. Applied Catalysis B: Environmental 2017, 218, 20–31. [Google Scholar] [CrossRef]
- Darabi, R.R.; Jahanshahi, M.; Peyravi, M. A support assisted by photocatalytic Fe3O4/ZnO nanocomposite for thin-film forward osmosis membrane. Chemical Engineering Research and Design 2018, 133, 11–25. [Google Scholar] [CrossRef]
- Hu, J.S.; Ren, L.L.; Guo, Y.G.; Liang, H.P.; Cao, A.M.; Wan, L.J.; Bai, C.L. Mass production and high photocatalytic activity of ZnS nanoporous nanoparticles. Angewandte Chemie International Edition 2005, 44, 1269–1273, [https://pubmed.ncbi.nlm.nih.gov/15651014/]. [Google Scholar] [CrossRef]
- Nasi, L.; Calestani, D.; Besagni, T.; Ferro, P.; Fabbri, F.; Licci, F.; Mosca, R. ZnS and ZnO nanosheets from ZnS(en)0.5 precursor: nanoscale structure and photocatalytic properties. The Journal of Physical Chemistry C 2012, 116, 6960–6965. [Google Scholar] [CrossRef]
- Hitkari, G.; Singh, S.; Pandey, G. Structural, optical and photocatalytic study of ZnO and ZnO–ZnS synthesized by chemical method. Nano-Structures Nano-Objects 2017, 12, 1–9. [Google Scholar] [CrossRef]
- Park, J.M.; Oh, S.H.; Kim, Y. ZnS–ZnO heterostructure nanorings grown under a possible early Earth atmosphere. Crystal Growth Design 2020, 20, 1196–1202. [Google Scholar] [CrossRef]
- Liu, S.; Li, M.; Li, S.; Li, H.; Yan, L. Synthesis and adsorption/photocatalysis performance of pyrite FeS2. Applied surface science 2013, 268, 213–217. [Google Scholar] [CrossRef]
- Morales-Gallardo, M.V.; Ayala, A.M.; Pal, M.; Jacome, M.C.; Antonio, J.T.; Mathews, N.R. Synthesis of pyrite FeS2 nanorods by simple hydrothermal method and its photocatalytic activity. Chemical Physics Letters 2016, 660, 93–98. [Google Scholar] [CrossRef]
- Liu, L.; Kankam, I.; Zhuang, H.L. Single-layer antiferromagnetic semiconductor CoS2 with pentagonal structure. Physical Review B 2018, 98, 205425. [Google Scholar] [CrossRef]
- Faber, M.S.; Park, K.; Caban-Acevedo, M.; Santra, P.K.; Jin, S. Earth-abundant cobalt pyrite (CoS2) thin film on glass as a robust, high-performance counter electrode for quantum dot-sensitized solar cells. Journal Of Physical Chemistry Letters 2013, 4, 1843–1849. [Google Scholar] [CrossRef]
- Anand, J.S.; Rajan, R.K.; Zaidan, A.A.M. Electrosynthesized NiS2 thin films and their optical and semiconductor studies. Reports in Electrochemistry 2013, 3, 25–29. [Google Scholar] [CrossRef]
- Saeed, S.; Rashid, N. Growth and characterization of semiconducting nickel sulfide nanocrystals from air-stable single-source metal organic precursors. Cogent Chemistry 2015, 1, 1030195. [Google Scholar] [CrossRef]
- Mitsui, T.; Môri, N.; Yomo, S.; Ogawa, S. Semiconductor-metal phase diagram of Co-doped NiS2. Solid State Communications 1974, 15, 917–920. [Google Scholar] [CrossRef]
- Jarrett, H.S.; Bouchard, R.J.; Gillson, J.L.; Jones, G.A.; Marcus, S.M.; Weiher, J.F. The métal-semiconductor phase diagram for NiS2− xSex. Materials Research Bulletin 1973, 8, 877–882. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, J.; Lu, Z.; Xia, H. Hierarchical FeS2 nanosheet@Fe2O3 nanosphere heterostructure as promising electrode material for supercapacitors. Materials Letters 2016, 166, 223–226. [Google Scholar] [CrossRef]
- Yan, S.; Wang, K.; Zhou, F.; Lin, S.; Song, H.; Shi, Y.; Yao, J. Ultrafine Co:FeS2/CoS2 heterostructure nanowires for highly efficient hydrogen evolution reaction. ACS Applied Energy Materials 2020, 3, 514–520. [Google Scholar] [CrossRef]
- Wang, K.; Song, H.; Lin, Z.; Gao, Y.; Wu, H.; Yan, S.; Wang, J.; Shi, Y. Improving hydrogen evolution performance of Co:FeS2/CoS2 nano-heterostructure at elevated temperatures. Materials Express 2019, 9, 786–791. [Google Scholar] [CrossRef]
- Zhang, Z.; Lv, X.; Chen, Y.; Zhang, P.; Sui, M.; Liu, H.; Sun, X. NiS2@MoS2 nanospheres anchored on reduced graphene oxide: a novel ternary heterostructure with enhanced electromagnetic absorption property. Nanomaterials 2019, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Kang, M. Physicochemical properties of core/shell structured pyrite FeS2/anatase TiO2 composites and their photocatalytic hydrogen production performances. Current Applied Physics 2013, 13, 1482–1489. [Google Scholar] [CrossRef]
- Rashid, J.; Saleem, S.; Awan, S.U.; Iqbal, A.; Kumar, R.; Barakat, M.A.; Arshad, M.; Zaheer, M.; Rafique, M.; Awad, M. Stabilized fabrication of anatase-TiO2/FeS2 (pyrite) semiconductor composite nanocrystals for enhanced solar light-mediated photocatalytic degradation of methylene blue. RSC Advances 2018, 8, 11935–11945. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Meng, Z.D.; Ghosh, T.; Oh, W.C. Enhanced photocatalytic efficiency of nanoscale NiS2/TiO2 catalysts synthesized by hydrothermal and sol-gel method. Journal of the Korean Ceramic Society 2012, 49, 135–141. [Google Scholar] [CrossRef]
- Zeda, M.E.N.G.; Wonchun, O.H. Photodegradation of organic dye by CoS2 and carbon (C60, Graphene, CNT)/TiO2 composite sensitizer. Chinese Journal of Catalysis 2012, 33, 1495–1501. [Google Scholar] [CrossRef]
- Zhu, L.; Jo, S.B.; Ye, S.; Ullah, K.; Meng, Z.D.; Oh, W.C. A green and direct synthesis of photosensitized CoS2–graphene/TiO2 hybrid with high photocatalytic performance. Journal of Industrial and Engineering Chemistry 2015, 22, 264–271. [Google Scholar] [CrossRef]
- Zhang, G.; Yan, Y.; Hu, Z.; Xiao, B. Investigation on preparation of pyrite tailings-based mineral admixture with photocatalytic activity. Construction and Building Materials 2017, 138, 26–34. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y. On the origin of life in the zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth. Biology Direct 2009, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y.; Galperin, M.Y. On the origin of life in the zinc world. 2. Validation of the hypothesis on the photosynthesizing zinc sulfide edifices as cradles of life on Earth. Biology direct 2009, 4, 27. [Google Scholar] [CrossRef]
- Rao, H.; Lu, Z.; Liu, X.; Ge, H.; Zhang, Z.; Zou, P.; He, H.; Wang, Y. Visible light-driven photocatalytic degradation performance for methylene blue with different multi-morphological features of ZnS. RSC Advances 2016, 6, 46299–46307. [Google Scholar] [CrossRef]
- Ye, Z.; Kong, L.; Chen, F.; Chen, Z.; Lin, Y.; Liu, C. A comparative study of photocatalytic activity of ZnS photocatalyst for degradation of various dyes. Optik 2018, 164, 345–354. [Google Scholar] [CrossRef]
- Sharma, M.; Jain, T.; Singh, S.; Pandey, O.P. Photocatalytic degradation of organic dyes under UV–Visible light using capped ZnS nanoparticles. Solar Energy 2012, 86, 626–633. [Google Scholar] [CrossRef]
- Mani, S.K.; Saroja, M.; Venkatachalam, M.; Rajamanickam, T. Antimicrobial activity and photocatalytic degradation properties of zinc sulfide nanoparticles synthesized by using plant extracts. Journal of Nanostructures 2018, 8, 107–118. [Google Scholar] [CrossRef]
- Rafiq, A.; Imran, M.; Ikram, M.; Naz, M.; Aqeel, M.; Majeed, H.; Hussain, S.G.; Ali, S. Photocatalytic and catalytic degradation of organic dye by uncapped and capped ZnS quantum dots. Materials Research Express 2019, 6, 055801. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of first cells at terrestrial, anoxic geothermal fields. Proceedings of the National Academy of Sciences 2012, 109, E821–E830, [https://pubmed.ncbi.nlm.nih.gov/22331915/]. [Google Scholar] [CrossRef]
- Guzman, M.I. Abiotic photosynthesis: from prebiotic chemistry to metabolism. In Origins of Life: The Primal Self-Organization; Springer: Berlin, Heidelberg; 2011; pp. 85–105. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Galperin, M.Y. On the abundance of zinc in the evolutionarily old protein domains. Proceedings of the National Academy of Sciences 2010, 107, E137–E137, [https://pubmed.ncbi.nlm.nih.gov/20693418/]. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Yang, B.; Liu, X.; Yang, Y.; Su, W. Photocatalytic reversible amination of α-keto acids on a ZnS surface: implications for the prebiotic metabolism. Chemical Communications 2012, 48, 2146–2148, [https://pubmed.ncbi.nlm.nih.gov/22237955/]. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Liu, X.; Yang, Y.; Su, W. Enhanced photocatalytic performance of ZnS for reversible amination of α-oxo acids by hydrothermal treatment. Origins of Life and Evolution of Biospheres 2012, 42, 263–273, [https://pubmed.ncbi.nlm.nih.gov/22638837/]. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Guzman, M.I. CO2 reduction under periodic illumination of ZnS. Journal of Physical Chemistry C 2014, 118, 11649–11656. [Google Scholar] [CrossRef]
- Zhou, R.; Guzman, M.I. Photocatalytic reduction of fumarate to succinate on ZnS mineral surfaces. Journal of Physical Chemistry C 2016, 120, 7349–7357. [Google Scholar] [CrossRef]
- Zhang, X.V.; Martin, S.T. Driving parts of Krebs cycle in reverse through mineral photochemistry. Journal of the American Chemical Society 2006, 128, 16032–16033. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Martin, S.T. Prebiotic metabolism: production by mineral photoelectrochemistry of α-ketocarboxylic acids in the reductive tricarboxylic acid cycle. Astrobiology 2009, 9, 833–842, [https://pubmed.ncbi.nlm.nih.gov/19968461/]. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Martin, S.T. Photo-production of lactate from glyoxylate: how minerals can facilitate energy storage in a prebiotic world. Chemical Communications 2010, 46, 2265–2267. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Martin, S.T. Oxaloacetate-to-malate conversion by mineral photoelectrochemistry: implications for the viability of the reductive tricarboxylic acid cycle in prebiotic chemistry. International Journal of Astrobiology 2008, 7, 271–278. [Google Scholar] [CrossRef]
- Zhang, X.V.; Ellery, S.P.; Friend, C.M.; Holland, H.D.; Michel, F.M.; Schoonen, M.A.; Martin, S.T. Photodriven reduction and oxidation reactions on colloidal semiconductor particles: Implications for prebiotic synthesis. Journal of Photochemistry and Photobiology A: Chemistry 2007, 185, 301–311. [Google Scholar] [CrossRef]
- Mamajanov, I.; Caudan, M.; Jia, T.Z. Protoenzymes: The case of hyperbranched polymer-scaffolded ZnS nanocrystals. Life 2020, 10, 150, [https://pubmed.ncbi.nlm.nih.gov/32823487/]. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Influence of Fe, Ni, and Cu doping on the photocatalytic efficiency of ZnS: implications for prebiotic chemistry, 2016, arXiv preprint arXiv:1610.00859.
- Doane, T.A. A survey of photogeochemistry. Geochemical transactions 2017, 18, 1, [https://pubmed.ncbi.nlm.nih.gov/28246525/]. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G. From light to life. Origins of Life and Evolution of Biospheres 2015, 45, 347–350, [https://pubmed.ncbi.nlm.nih.gov/26105723/]. [Google Scholar] [CrossRef] [PubMed]
- Dhar, N.R. Denitrification in sunlight. Nature 1934, 134, 572–573. [Google Scholar] [CrossRef]
- Rao, G.G.; Varadanam, C.I. Photo-ammonification of organic nitrogenous compounds in the soil. Nature 1938, 142, 618. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; Strampach, N.; Hui, L.N.; Palmer, M.R.; Salehi, J. Nitrogen photoreduction on desert sands under sterile conditions. Proceedings of the National Academy of Sciences 1983, 80, 3873–3876, [https://pubmed.ncbi.nlm.nih.gov/16593330/]. [Google Scholar] [CrossRef]
- Kim, J.D. The evolution of biological geochemical electron transfer reactions. PhD Thesis, State University of New Jersey, New Brunswick, 2013. [Google Scholar]
- Jelen, B.I.; Giovannelli, D.; Falkowski, P.G. The role of microbial electron transfer in the coevolution of the biosphere and geosphere. Annual Review of Microbiology 2016, 70, 45–62, [https://pubmed.ncbi.nlm.nih.gov/27297124/]. [Google Scholar] [CrossRef]
- Shuey, R.T. Semiconducting ore minerals; Elsevier Scientific Publishing Company: Amsterdam – Oxford – New York; 1975; pp. 1-415. [CrossRef]
- Borutzky, B.Y. Essays on Fundamental and Genetic Mineralogy: 1. What is the Mineral and Mineral Species? New Data on Minerals 2005, 40, 159–166. [Google Scholar]
- Borutzky, B.Y. Essays on Fundamental and Genetic Mineralogy: 2. The practive of working out “natural genetic” systematics of minerals. New Data on Minerals 2006, 41, 162-171.
- Barawi, M.; Ferrer, I.J.; Flores, E.; Yoda, S.; Ares, J.R.; Sánchez, C. Hydrogen photoassisted generation by visible light and an earth abundant photocatalyst: pyrite (FeS2). Journal of Physical Chemistry C 2016, 120, 9547–9552. [Google Scholar] [CrossRef]
- Mateo-Marti, E.; Galvez-Martinez, S.; Gil-Lozano, C.; Zorzano, M.P. Pyrite-induced UV-photocatalytic abiotic nitrogen fixation: implications for early atmospheres and life. Scientific Reports 2019, 9, 15311. [Google Scholar] [CrossRef]
- Puthussery, J.; Seefeld, S.; Berry, N.; Gibbs, M.; Law, M. Colloidal iron pyrite (FeS2) nanocrystal inks for thin-film photovoltaics. Journal of the American Chemical Society 2011, 133, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Kirkeminde, A.; Gong, M.; Ren, S. The renaissance of iron pyrite photovoltaics: progress, challenges, and perspectives. In Low-cost Nanomaterials; Springer: London; 2014; pp. 137-166. [CrossRef]
- Macpherson, H.A.; Stoldt, C.R. Iron pyrite nanocubes: size and shape considerations for photovoltaic application. Acs Nano 2012, 6, 8940–8949. [Google Scholar] [CrossRef]
- Dasbach, R.; Willeke, G.; Blenk, O. Iron sulfide for photovoltaics. MRS Bulletin 1993, 18, 56–60. [Google Scholar] [CrossRef]
- Li, W.; Döblinger, M.; Vaneski, A.; Rogach, A.L.; Jäckel, F.; Feldmann, J. Pyrite nanocrystals: shape-controlled synthesis and tunable optical properties via reversible self-assembly. Journal of Materials Chemistry 2011, 21, 17946–17952. [Google Scholar] [CrossRef]
- Tian, A.; Xu, Q.; Shi, X.; Yang, H.; Xue, X.; You, J.; Wang, X.; Dong, C.; Yan, X.; Zhou, H. Pyrite nanotube array films as an efficient photocatalyst for degradation of methylene blue and phenol. RSC Advances 2015, 5, 62724–62731. [Google Scholar] [CrossRef]
- Moradi, M.; Kalantary, R.R.; Esrafili, A.; Jafari, A.J.; Gholami, M. Visible light photocatalytic inactivation of Escherichia coli by natural pyrite assisted by oxalate at neutral pH. Journal of Molecular Liquids 2017, 248, 880–889. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Singh, S. Temperature and pressure behaviour of narrow-gap semiconductors including galena. Current Applied Physics 2014, 14, 496–507. [Google Scholar] [CrossRef]
- Schuhmann, D.; Vanel, P.; Talib, A. influence of the semiconductor character of some minerals upon the adsorption of surfactants-application to the galena xanthate system. Journal de Chimie Physique et de Physico-Chimie Biologique 1988, 85, 551–554. [Google Scholar] [CrossRef]
- Thompson, K.C.; Simkovich, G.; Aplan, F.F. Flotation and electrokinetic properties of the semiconductor, galena. Journal of The Electrochemical Society 1984, 131, c99. [Google Scholar] [CrossRef]
- Martínez, M.D.C.L. Influencia del carácter semiconductor de la galena sobre su potencial de electrodo y sobre la adsorción del xantato. Doctoral dissertation, Universidad Complutense de Madrid, Italy, 1975.
- Dimitrova, S.; Moldovanova, M. Semiconductor properties of pure galena crystals. Physica Status Solidi 1965, 8, 173–176. [Google Scholar]
- Steinhagen, C.; Harvey, T.B.; Stolle, C.J.; Harris, J.; Korgel, B.A. Pyrite nanocrystal solar cells: promising, or fool’s gold? The Journal of Physical Chemistry Letters 2012, 3, 2352–2356. [Google Scholar] [CrossRef]
- Bi, Y.; Yuan, Y.; Exstrom, C.L.; Darveau, S.A.; Huang, J. Air stable, photosensitive, phase pure iron pyrite nanocrystal thin films for photovoltaic application. Nano Letters 2011, 11, 4953–4957. [Google Scholar] [CrossRef]
- Du, H.; Yang, C.; Pu, W.; Zeng, L.; Gong, J. Enhanced electrochemical reduction of N2 to ammonia over pyrite FeS2 with excellent selectivity. ACS Sustainable Chemistry and Engineering 2020, 8, 10572–10580. [Google Scholar] [CrossRef]
- Matsumoto, Y. Energy positions of oxide semiconductors and photocatalysis with iron complex oxides. Journal of Solid State Chemistry 1996, 126, 227–234. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The iron oxides: structure, properties, reactions, occurrences and uses. John Wiley & Sons; 2003; 793 p.
- Cartwright, J.H.; García-Ruiz, J.M.; Novella, M.L.; Otálora, F. Formation of chemical gardens. Journal of Colloid and Interface Science 2002, 256, 351–359. [Google Scholar] [CrossRef]
- Barge, L.M.; Cardoso, S.S.; Cartwright, J.H.; Cooper, G.J.; Cronin, L.; De Wit, A.; Doloboff, I.J.; Escribano, B.; Goldstein, R.E.; Haudin, F.; Jones, D.E.; Mackay, A.L.; Maselko, J.; Pagano, J.J.; Pantaleone, J.; Russel, M.J.; Sainz-Díaz, C.I.; Steinbock, O.; Stone, D.A.; Tanimoto, Y.; Thomas, N.L. From chemical gardens to chemobrionics. Chemical reviews 2015, 115, 8652–8703. [Google Scholar] [CrossRef]
- Allamandola, L.J.; Sandford, S.A.; Wopenka, B. Interstellar polycyclic aromatic hydrocarbons and carbon in interplanetary dust particles and meteorites. Science 1987, 237, 56–59, [https://pubmed.ncbi.nlm.nih.gov/17813622/]. [Google Scholar] [CrossRef] [PubMed]
- Lovas, F.J.; McMahon, R.J.; Grabow, J.U.; Schnell, M.; Mack, J.; Scott, L.T.; Kuczkowski, R.L. Interstellar chemistry: a strategy for detecting polycyclic aromatic hydrocarbons in space. Journal of the American Chemical Society 2005, 127, 4345–4349. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Koretsune, T.; Hotta, C.; Takeya, J.; Kimura, T.; Wakabayashi, Y. Large surface relaxation in the organic semiconductor tetracene. Nature Communications 2014, 5, 5400. [Google Scholar] [CrossRef] [PubMed]
- Hepp, A.; Heil, H.; Weise, W.; Ahles, M.; Schmechel, R.; von Seggern, H. Light-emitting field-effect transistor based on a tetracene thin film. Physical Review Letters 2003, 91, 157406. [Google Scholar] [CrossRef] [PubMed]
- Blasberger, A.; Behar, E.; Perets, H.B.; Brosch, N.; Tielens, A.G. Observational evidence linking interstellar UV absorption to PAH molecules. The Astrophysical Journal 2017, 836, 173. [Google Scholar] [CrossRef]
- Koch, N. Organic electronic devices and their functional interfaces. Chem. Phys. Chem 2007, 8, 1438–1455. [Google Scholar] [CrossRef]
- Hasegawa, T.; Takeya, J. Organic field-effect transistors using single crystals. Science and Technology of Advanced Materials 2009, 10, 024314. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y. Organic semiconductors for organic field-effect transistors. Science and Technology of Advanced Materials 2009, 10, 024313. [Google Scholar] [CrossRef]
- Zaia, D.A.M. A review of adsorption of amino acids on minerals: was it important for origin of life? Amino Acids 2004, 27, 113–118, [https://pubmed.ncbi.nlm.nih.gov/15309580/]. [Google Scholar] [CrossRef]
- Klabunovskii, E.I. Can enantiomorphic crystals like quartz play a role in the origin of homochirality on earth? Astrobiology 2001, 1, 127–131, [https://pubmed.ncbi.nlm.nih.gov/12467116/]. [Google Scholar] [CrossRef]
- Fedo, C.M.; Whitehouse, M.J. Metasomatic origin of quartz-pyroxene rock, Akilia, Greenland, and implications for Earth's earliest life. Science 2002, 296, 1448–1452, [https://pubmed.ncbi.nlm.nih.gov/12029129/]. [Google Scholar] [CrossRef]
- Ehrenfreund, P.; Rasmussen, S.; Cleaves, J.; Chen, L. Experimentally tracing the key steps in the origin of life: the aromatic world. Astrobiology 2006, 6, 490–520, [https://pubmed.ncbi.nlm.nih.gov/16805704/]. [Google Scholar] [CrossRef]
- Menor-Salván, C.; Ruiz-Bermejo, M.; Osuna-Esteban, S.; Muñoz-Caro, G.; Veintemillas-Verdaguer, S. Synthesis of polycyclic aromatic hydrocarbons and acetylene polymers in ice: a prebiotic scenario. Chemistry Biodiversity 2008, 5, 2729–2739, [https://pubmed.ncbi.nlm.nih.gov/19089832/]. [Google Scholar] [CrossRef]
- Groen, J.; Deamer, D.W.; Kros, A.; Ehrenfreund, P. Polycyclic aromatic hydrocarbons as plausible prebiotic membrane components. Origins of Life and Evolution of Biospheres 2012, 42, 295–306, [https://pubmed.ncbi.nlm.nih.gov/22798228/]. [Google Scholar] [CrossRef]
- Morowitz, H.J. Beginnings of cellular life: metabolism recapitulates biogenesis. Yale University Press: New Haven – London; 1993; 210 p.
- Girerd, J.J.; Philouze, C.; Anxolabehere-Mallart, E.; Sainton, J.; Blondin, G.; Frapart, Y. Manganese models for photosynthesis: from self-assembly to design. Journal of Inorganic Biochemistry 1995, 59, 610. [Google Scholar] [CrossRef]
- Hansen, M, Troppmann, S, König, B. Artificial photosynthesis at dynamic self-assembled interfaces in water. Chemistry – A European Journal, 2016; 22 , 58-72. [CrossRef]
- Hsin, J.; Chandler, D.E.; Gumbart, J.; Harrison, C.B.; Sener, M.; Strumpfer, J.; Schulten, K. Self-assembly of photosynthetic membranes. Chem. Phys. Chem. 2010, 11, 1154–1159. [Google Scholar] [CrossRef]
- Lee, J.S.; Nam, D.H.; Kuk, S.K.; Park, C.B. Near-infrared-light-driven artificial photosynthesis by nanobiocatalytic assemblies. Chemistry, 2014, 20, 3584–3588, [https://pubmed.ncbi.nlm.nih.gov/24615772/]. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, M.; Lee, J.S.; Park, C.B. Self-assembled light-harvesting peptide nanotubes for mimicking natural photosynthesis. Angewandte Chemie International Edition 2012, 51, 517–520, [https://pubmed.ncbi.nlm.nih.gov/21976303/]. [Google Scholar] [CrossRef] [PubMed]
- Cardona, T. Reconstructing the origin of oxygenic photosynthesis: do assembly and photoactivation recapitulate evolution? Frontiers in Plant Science 2016, 7, 257, [https://pubmed.ncbi.nlm.nih.gov/26973693/]. [Google Scholar] [CrossRef] [PubMed]
- Pang, F.; Zhang, R.; Lan, D.; Ge, J. Synthesis of magnetite–semiconductor–metal trimer nanoparticles through functional modular assembly: a magnetically separable photocatalyst with photothermic enhancement for water reduction. ACS Applied Materials Interfaces 2018, 10, 4929–4936, [https://pubmed.ncbi.nlm.nih.gov/29345458/]. [Google Scholar] [CrossRef] [PubMed]
- Kotov, N.A.; Dékány, I.; Fendler, J.H. Ultrathin graphite oxide–polyelectrolyte composites prepared by self-assembly: Transition between conductive and non-conductive states. Advanced Materials 1996, 8, 637–641. [Google Scholar] [CrossRef]
- Graetzel, M. Artificial photosynthesis, very efficient visible light energy harvesting, and conversion by spectral sensitization of fractal oxide semiconductor films. In Photochemical Energy Conversion (Proc. Int. Conf. Photochem. Convers. Storage Solar Energy), 1989.
- Guijarro, N.; Formal, F.L.; Sivula, K. Artificial photosynthesis with semiconductor–liquid junctions. CHIMIA International Journal for Chemistry 2015, 69, 30–40, [https://pubmed.ncbi.nlm.nih.gov/26507086/]. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Domen, K. Introductory lecture: sunlight-driven water splitting and carbon dioxide reduction by heterogeneous semiconductor systems as key processes in artificial photosynthesis. Faraday Discussions 2017, 198, 11–35, [https://pubmed.ncbi.nlm.nih.gov/28272623/]. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Long, J.; Yang, L.; Chen, W.; Dai, W.; Fu, X.; Wang, X. Organic semiconductor for artificial photosynthesis: water splitting into hydrogen by a bioinspired C3N3S3 polymer under visible light irradiation. Chemical Science 2011, 2, 1826–1830. [Google Scholar] [CrossRef]
- Zhou, H.; Li, P.; Liu, J.; Chen, Z.; Liu, L.; Dontsova, D.; Yan, R.; Fan, T.; Zhang, D.; Ye, J. Biomimetic polymeric semiconductor based hybrid nanosystems for artificial photosynthesis towards solar fuels generation via CO2 reduction. Nano Energy 2016, 25, 128–135. [Google Scholar] [CrossRef]
- Pang, H.; Masuda, T.; Ye, J. Semiconductor-based photoelectrochemical conversion of carbon dioxide: stepping towards artificial photosynthesis. Chemistry–an Asian Journal 2018, 13, 127-142. [CrossRef]
- Hoffmann, M.R.; Moss, J.A.; Baum, M.M. Artificial photosynthesis: semiconductor photocatalytic fixation of CO2 to afford higher organic compounds. Dalton Transactions 2011, 40, 5151–5158, [https://pubmed.ncbi.nlm.nih.gov/21373667/]. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.; Clayton, R.K. The first step in photosynthesis: evidence for its electronic nature. Proceedings of the National Academy of Sciences of the USA 1960, 46, 769–776, [https://pubmed.ncbi.nlm.nih.gov/16590669/]. [Google Scholar] [CrossRef] [PubMed]
- Graetzel, M. Artificial photosynthesis: water cleavage into hydrogen and oxygen by visible light. Accounts of Chemical Research 1981, 14, 376–384. [Google Scholar] [CrossRef]
- Duret, A.; Grätzel, M. Visible light-induced water oxidation on mesoscopic α-Fe2O3 films made by ultrasonic spray pyrolysis. Journal of Physical Chemistry B 2005, 109, 17184–17191. [Google Scholar] [CrossRef]
- Le Formal, F.; Grätzel, M.; Sivula, K. Controlling photoactivity in ultrathin hematite films for solar water-splitting. Advanced Functional Materials 2010, 20, 1099–1107. [Google Scholar] [CrossRef]
- Ravirajan, P.; Peiró, A.M.; Nazeeruddin, M.K.; Graetzel, M.; Bradley, D.D.; Durrant, J.R.; Nelson, J. Hybrid polymer/zinc oxide photovoltaic devices with vertically oriented ZnO nanorods and an amphiphilic molecular interface layer. The Journal of Physical Chemistry B 2006, 110, 7635–7639. [Google Scholar] [CrossRef]
- Mershin, A.; Matsumoto, K.; Kaiser, L.; Yu, D.; Vaughn, M.; Nazeeruddin, M.K.; Bruce, B.D.; Graetzel, M.; Zhang, S. Self-assembled photosystem-I biophotovoltaics on nanostructured TiO2 and ZnO. Scientific Reports 2012, 2, 234. [Google Scholar] [CrossRef]
- Abdi-Jalebi, M.; Chandiran, A.K.; Nazeeruddin, M.; Grätzel, M. Low temperature dye-sensitized solar cells based on conformal thin zinc oxide overlayer on mesoporous insulating template by atomic layer deposition. Scientia Iranica - Transactions on Nanotechnology (F) 2014, 21, 2479–2484. [Google Scholar]
- Kumar, M.H.; Yantara, N.; Dharani, S.; Graetzel, M.; Mhaisalkar, S.; Boix, P.P.; Mathews, N. Flexible, low-temperature, solution processed ZnO-based perovskite solid state solar cells. Chemical Communications 2013, 49, 11089–11091, [https://pubmed.ncbi.nlm.nih.gov/24141601/]. [Google Scholar] [CrossRef]
- Nguyen, M.; Tran, P.D.; Pramana, S.S.; Lee, R.L.; Batabyal, S.K.; Mathews, N.; Wong, L.H.; Graetzel, M. In situ photo-assisted deposition of MoS2 electrocatalyst onto zinc cadmium sulphide nanoparticle surfaces to construct an efficient photocatalyst for hydrogen generation. Nanoscale 2013, 5, 1479–1482. [Google Scholar] [CrossRef]
- Lima-de-Faria, A. Evolution without selection: Form and function by autoevolution. Elsevier: Amsterdam, Netherlands; 1988; 372 p.
- Dyer, B.D; Schuster, P.; Holm, N.G. A. Lima-de-Faria, evolution without selection form and function by autoevolution. Origins of Life and Evolution of Biospheres 1989, 19, 645–652. [Google Scholar] [CrossRef]
- Hughes, A.L. Evolution without selection: form and function in autoevolution. By A. Lima-de-Faria. Molecular Biology and Evolution 1990, 7, 634. [Google Scholar]
- Gerischer, H.; Michel-Beyerle, M.E.; Rebentrost, F.; Tributsch, H. Sensitization of charge injection into semiconductors with large band gap. Electrochimica Acta 1968, 13, 1509–1515. [Google Scholar] [CrossRef]
- Tributsch, H.; Calvin, M. Electrochemistry of excited molecules: photo-electrochemical reactions of chlorophylls. Photochemistry and Photobiology 1971, 14, 95–112. [Google Scholar] [CrossRef]
- Yi, C.; Giordano, F.; Cevey-Ha, N.L.; Tsao, H.N.; Zakeeruddin, S.M.; Grätzel, M. Influence of structural variations in push–pull zinc porphyrins on photovoltaic performance of dye-sensitized solar cells. Chem. Sus. Chem. 2014, 7, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K.; Grätzel, M. Light induced redox reactions of water soluble porphyrins, sensitization of hydrogen generation from water by zinc porphyrin derivatives. Helvetica Chimica Acta 1980, 63, 478–485. [Google Scholar] [CrossRef]
- Pileni, M.P.; Graetzel, M. Zinc porphyrin sensitized reduction of simple and functional quinones in micellar systems. Journal of Physical Chemistry 1980, 84, 1822–1825. [Google Scholar] [CrossRef]
- Hurst, J.K.; Lee, L.Y.; Graetzel, M. Photoredox behavior of zinc(II) porphyrins in vesicle assemblies. Journal of the American Chemical Society 1983, 105, 7048–7056. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Vlachopoulos, N.; Krishnan, V.; Monnier, A.; Graetzel, M. Sensitization of titanium dioxide in the visible light region using zinc porphyrins. Journal of Physical Chemistry 1987, 91, 2342–2347. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Shelnutt, J.A.; Graetzel, M. Sensitization and photoredox reactions of zinc(II) and antimony(V) uroporphyrins in aqueous media. Inorganic Chemistry 1988, 27, 2820–2825. [Google Scholar] [CrossRef]
- Yum, J.H.; Jang, S.R.; Humphry-Baker, R.; Grätzel, M.; Cid, J.J.; Torres, T.; Nazeeruddin, M.K. Effect of coadsorbent on the photovoltaic performance of zinc pthalocyanine-sensitized solar cells. Langmuir 2008, 24, 5636–5640, [https://pubmed.ncbi.nlm.nih.gov/18435553/]. [Google Scholar] [CrossRef] [PubMed]
- Giribabu, L.; Kumar, C.V.; Reddy, P.Y.; Yum, J.H.; Grätzel, M.; Nazeeruddin, M.K. Unsymmetrical extended π-conjugated zinc phthalocyanine for sensitization of nanocrystalline TiO2 films. Journal of Chemical Sciences 2009, 121, 75. [Google Scholar] [CrossRef]
- Ince, M.; Cardinali, F.; Yum, J.H.; Martínez-Díaz, M.V.; Nazeeruddin, M.K.; Grätzel, M.; Torres, T. Convergent synthesis of near-infrared absorbing,“push–pull”, bisthiophene-substituted, zinc(II) phthalocyanines and their application in dye-sensitized solar cells. Chemistry–A European Journal 2012, 18, 6343–6348, [https://pubmed.ncbi.nlm.nih.gov/22473900/]. [Google Scholar] [CrossRef]
- Molina, D.; Ruiz-Preciado, M. A.; Sadegh, F.; Álvaro-Martins, M.J.; Grätzel, M.; Hagfeldt, A.; Sastre-Santos, Á. p-Phenylene-bridged zinc phthalocyanine-dimer as hole-transporting material in perovskite solar cells. Journal of Porphyrins and Phthalocyanines 2019, 23, 546–553. [Google Scholar] [CrossRef]
- Xiong, J.; Bauer, C.E. A cytochrome b origin of photosynthetic reaction centers: an evolutionary link between respiration and photosynthesis. Journal of Molecular Biology 2002, 322, 1025–1037, [https://pubmed.ncbi.nlm.nih.gov/12367526/]. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. A redox switch hypothesis for the origin of two light reactions in photosynthesis. FEBS Letters 2005, 579, 963–968, [https://pubmed.ncbi.nlm.nih.gov/15710376/]. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lin, J.; Wang, X. Semiconductor–redox catalysis promoted by metal–organic frameworks for CO2 reduction. Physical Chemistry Chemical Physics 2014, 16, 14656–14660, [https://pubmed.ncbi.nlm.nih.gov/24921181/]. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, M. Catalysis of redox processes at illuminated semiconductor electrodes. Journal of the Electrochemical Society 1983, 130, C124. [Google Scholar]
- Mei, B.; Han, K.; Mul, G. Driving surface redox reactions in heterogeneous photocatalysis: the active state of illuminated semiconductor-supported nanoparticles during overall water-splitting. ACS Catalysis 2018, 8, 9154–9164. [Google Scholar] [CrossRef]
- Annadhasan, M.; Selvam, K.; Swaminathan, M. A combined-redox synthesis of 2-alkylbenzimidazoles from 2-nitroanilines by semiconductor photocatalysis. Synthetic Communications 2012, 42, 1500–1508. [Google Scholar] [CrossRef]
- Zhou, R. Semiconductor photocatalysis: mechanisms, photocatalytic performances and lifetime of redox carriers. PhD Thesis, University of Kentucky, Lexington, Kentucky, USA, 2017. [Google Scholar]
- Dukovic, G. Excited state processes in semiconductor nanocrystals and their relationships with light-driven multi-electron catalysis. ECS Meeting Abstracts 2019, 41, 1956. [Google Scholar] [CrossRef]
- Hongbo, L. Auto-catalysis and cross-catalysis in mineralization of enriched ore of anshan-type iron deposits. Journal of Northeastern University 1995, P618.310.1 [in Chineese].
- Tóth, J. Gradient systems are cross-catalytic. Reaction Kinetics and Catalysis Letters 1979, 12, 253–257. [Google Scholar] [CrossRef]
- Basza, G.; Beck, M.T. Autocatalysis, cross-catalysis, self-inhibition and crosswise inhibition: Pathways into exotic chemical kinetics. Acta Chim. Hung 1972, 73, 26–37. [Google Scholar]
- Rastogi, R.P.; Mathur, P. Complex Dynamics in Systems Involving Both Cross Catalytic and Autocatalytic Processes. Proceedings of the Indian National Science Academy-Part A: Physical Sciences 2009, 75, 159. [Google Scholar]
- Wu, Y.; Shi, J.; Ding, F.; Zhao, J.; Zou, X.; Wang, M.; Zhang, S.; Tong, Z.; Zhang, S.; Jiang, Z. Integrated enzyme-photocatalysis system for carbon dioxide conversion. Scientia Sinica Chimica 2016, 47, 315–329. [Google Scholar] [CrossRef]
- Ding, X.; Dong, C.L.; Guan, Z.; He, Y.H. Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: direct enantioselective synthesis of 2,2-disubstituted indol-3-ones from 2-arylindoles. Angewandte Chemie 2019, 131, 124–130, [https://pubmed.ncbi.nlm.nih.gov/30421485/]. [Google Scholar] [CrossRef]
- Ju, E.; Dong, K.; Wang, Z.; Zhang, Y.; Cao, F.; Chen, Z.; Pu, F.; Ren, J.; Qu, X. Confinement of reactive oxygen species in an artificial-enzyme-based hollow structure to eliminate adverse effects of photocatalysis on UV filters. Chemistry–A European Journal, 2017, 23, 13518-13524; [https://pubmed.ncbi.nlm.nih.gov/28741846/]. [Google Scholar] [CrossRef]
- Yi, H.; Yan, M.; Huang, D.; Zeng, G.; Lai, C.; Li, M.; Huo, X.; Qin, L.; Liu, S.; Liu, X.; Li, B.; Wang, H.; Shen, M.; Fu, Y.; Guo, X. Synergistic effect of artificial enzyme and 2D nano-structured Bi2WO6 for eco-friendly and efficient biomimetic photocatalysis. Applied Catalysis B: Environmental 2019, 250, 52-62. [CrossRef]
- Yadav, R.K.; Baeg, J.O.; Oh, G.H.; Park, N.J.; Kong, K.J.; Kim, J.; Hwang, D.W.; Biswas, S.K. A photocatalyst–enzyme coupled artificial photosynthesis system for solar energy in production of formic acid from CO2. Journal of the American Chemical Society 2012, 134, 11455–11461, [https://pubmed.ncbi.nlm.nih.gov/22769600/]. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Wang, Y.; Guan, Y.; Deng, K.; Lv, K.; Sun, J.; Li, Z.; Li, M. Photocatalytic properties and electrochemical characteristic of a novel biomimetic oxygenase enzyme photocatalyst iron(II) tetrahydroxymethyltetra(1,4-dithiin)porphyrazine for the degradation of organic pollutants. Journal of Molecular Catalysis A: Chemical 2013, 372, 114–120. [Google Scholar] [CrossRef]
- Li, W.; Pei, X.; Deng, F.; Luo, X.; Li, F.; Xiao, Y. Bio-inspired artificial functional photocatalyst: biomimetic enzyme-like TiO2/reduced graphene oxide nanocomposite with excellent molecular recognition ability. Nanotechnology 2015, 26, 175706. [Google Scholar] [CrossRef] [PubMed]
- Crocker, L.; Koehler, P.; Bernhard, P.; Kerbs, A.; Euser, T.; Fruk, L. Enzyme-inspired flavin–polydopamine as a biocompatible nanoparticle photocatalyst. Nanoscale Horizons 2019, 4, 1318–1325. [Google Scholar] [CrossRef]
- Borthakur, P.; Boruah, P.K.; Das, M.R.; Artemkina, S.B.; Poltarak, P.A.; Fedorov, V.E. Metal free MoS2 2D sheets as a peroxidase enzyme and visible-light-induced photocatalyst towards detection and reduction of Cr(VI) ions. New Journal of Chemistry 2018, 42, 16919–16929. [Google Scholar] [CrossRef]
- Inoue, H. Fixation of carbon-dioxide using photocatalyst and enzyme. Denki Kagaku 1993, 61 (1), 113–114. [Google Scholar]
- Shen, X.; Zhu, L.; Liu, G.; Tang, H.; Liu, S.; Li, W. Photocatalytic removal of pentachlorophenol by means of an enzyme-like molecular imprinted photocatalyst and inhibition of the generation of highly toxic intermediates. New Journal of Chemistry 2009, 33, 2278–2285. [Google Scholar] [CrossRef]
- Habibi, N.; Etemadifari, Z.; Dianati, M. Magnetic nanocomposite thin film photocatalyst and cell extract enzyme biocatalyst in application of nanobiotechnology for development of a photo-bio desulfurization system. Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry 2016, 46, 857–860. [Google Scholar] [CrossRef]
- Lu, A.; Li, Y.; Wang, X.; Ding, H.; Zeng, C.; Yang, X.; Hao, R.; Wang, C.; Santosh, M. Photoelectrons from minerals and microbial world: A perspective on life evolution in the early Earth. Precambrian Research 2013, 231, 401–408. [Google Scholar] [CrossRef]
- Varfolomeev, S.D.; Bachurin, S.O.; Osipov, I.V.; Aliev, K.V.; Berezin, I.V.; Kabanov, V.A. Bioelectro-catalysis-enzyme active-center-semiconductor matrix electron-transfer. Doklady Akademii Nauk SSSR 1978, 239, 348–351. [Google Scholar]
- Zhang, H.; Wu, J.; Han, J.; Wang, L.; Zhang, W.; Dong, H.; Li, C.; Wang, Y. Photocatalyst/enzyme heterojunction fabricated for high-efficiency photoenzyme synergic catalytic degrading Bisphenol A in water. Chemical Engineering Journal 2020, 385, 123764. [Google Scholar] [CrossRef]
- Wang, F.X.; Ye, C.; Mo, S.; Liao, L.L.; Luo, H.Q.; Li, N.B. A novel photoelectrochemical sensing platform based on Fe2O3@Bi2S3 heterojunction for an enzymatic process and enzyme activity inhibition reaction. Sensors and Actuators B: Chemical 2019, 288, 202–209. [Google Scholar] [CrossRef]
- Koike, K.; Takagi, D.; Hashimoto, M.; Hashimoto, T.; Inoue, T.; Ogata, K.I.; Sasa, S.; Inoue, M.; Yano, M. Characteristics of enzyme-based ZnO/Zn0.7Mg0.3O heterojunction field-effect transistor as glucose sensor. Japanese Journal of Applied Physics 2009, 48, 04C081. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kobayashi, K.; Saito, T. (1996). Martian Soil Analysis. Its implication for life on mars.In: Chemical Evolution: Physics of the Origin and Evolution of Life (Proceedings of the Fourth Trieste Conference on Chemical Evolution, Trieste, Italy, 4-8 September 1995); Kluwer Academic Publishers, Dordrecht – Boston – London, Netherlands – USA – UK, 1996; pp. 389-398. [CrossRef]
- Fox, A. C.; Eigenbrode, J. L.; Freeman, K. H. Radiolysis of macromolecular organic material in Mars-relevant mineral matrices. Journal of Geophysical Research: Planetsn 2019, 124, 3257–3266. [Google Scholar] [CrossRef]
- Ochiai, E.I. Inorganic Chemistry of Earliest Sediments: Bioinorganic Chemical Aspects of the Origin and Evolution of Life. In Proceedings of the NATO Advanced Study Institute held at Maratea (Italy, June 1–12 , 1981); Ponnamperuma, C., Ed.; D.Reidel Publishing Company, Dordrecht : Hollanf / Boston : USA / London: England, 1981, 235-276. [CrossRef]
- Dai, H.; Zhang, S.; Xu, G.; Peng, Y.; Gong, L.; Li, X.; Li, Y.; Lin, Y.; Chen, G. Highly photoactive heterojunction based on gC3N4 nanosheets decorated with dendritic zinc (II) phthalocyanine through axial coordination and its ultrasensitive enzyme-free sensing of choline. RSC Advances 2014, 4, 58226–58230. [Google Scholar] [CrossRef]
- Giersch, C. Stationary diffusion gradients associated with photosynthetic carbon flux—a study of compartmental versus diffusion–reaction models. Journal of Theoretical Biology 2003, 224, 385–397, [https://pubmed.ncbi.nlm.nih.gov/12941596/]. [Google Scholar] [CrossRef]
- Wang, H.; Lin, S.; Allen, J.P.; Williams, J.C.; Blankert, S.; Laser, C.; Woodbury, N.W. Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science 2007, 316, 747–750, [https://pubmed.ncbi.nlm.nih.gov/17478721/]. [Google Scholar] [CrossRef] [PubMed]
- Rothman, D.; Petroff, A.P.; Liang, B.; Sim, M.; Bosak, T. Reaction-diffusion, early photosynthesis, and the spatial organization of conical stromatolites. In American Geophysical Union, Fall Meeting 2009, San Francisco, California, 2009; NG42A-04.
- Bard, A.J.; Memming, R.; Miller, B. Terminology in semiconductor electrochemistry and photoelectrochemical energy conversion (Recommendations 1991). Pure and Applied Chemistry 1991, 63, 569–596. [Google Scholar] [CrossRef]
- Wagner, E.; Tetzner, J.; Haertle, U.; Deitzer, G.F. Endogenous rhythmicity and energy transduction VIII. Kinetics in enzyme activity, redox state and energy charge as related to photomorphogenesis in seedlings of Chenopodium rubrum L. 1. Berichte der Deutschen Botanischen Gesellschaft 1974, 87, 291–302. [Google Scholar] [CrossRef]
- Deitzer, G.; Hopkins, D.; Wagner, E. Analysis of ultradian rhythms of enzyme-activity in chenopodium rubrum during photomorphogenesis. Plant Physiology 1976, 57, 19. [Google Scholar]
- Petrov, R.; Popov, V. Regulation of the catalytic properties of NAD-dependent hydrogenase-influence of the redox potential of the medium. Biochemistry-Moscow, 1988, 53, 1466-1470. [Google Scholar]
- Kim, J.Y.; Park, H.S.; Im Kang, S.; Choi, E. J.; Kim, I.Y. Redox regulation of cytosolic glycerol-3-phosphate dehydrogenase: Cys102 is the target of the redox control and essential for the catalytic activity. Biochimica et Biophysica Acta-General Subjects 2002, 1569, 67–74, [https://pubmed.ncbi.nlm.nih.gov/11853959/]. [Google Scholar] [CrossRef]
- Banerjee, R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Current Opinion in Chemical Biology 2017, 37, 115–121, [https://pubmed.ncbi.nlm.nih.gov/28282633/]. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chai, J.; Ou, X.; Li, M.; Liu, Z. Structural insights into substrate selectivity, catalytic mechanism, and redox regulation of rice photosystem II core phosphatase. Molecular Plant 2019, 12, 86–98, [https://pubmed.ncbi.nlm.nih.gov/30453087/]. [Google Scholar] [CrossRef] [PubMed]
- Grabov, A.; Bottger, M. Are redox reactions involved in regulation of K+ channels in the plasma membrane of Limnobium stoloniferum root hairs? Plant Physiology 1994, 105, 927–935, [https://pubmed.ncbi.nlm.nih.gov/12232255/]. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Callegaro, M.T.; Barzon, E.; Benetti, M.; Bindoli, A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radical Biology and Medicine 1998, 24, 370–376, [https://pubmed.ncbi.nlm.nih.gov/9433913/]. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, A.; Rigobello, M.P. Redox regulation of mitochondrial membrane permeability transition. Free Radical Biology and Medicine 2002, 33, S96. [Google Scholar]
- Horton, P.; Allen, J.F.; Black, M.T.; Bennett, J. Regulation of phosphorylation of chloroplast membrane polypeptides by the redox state of plastoquinone. FEBS Letters 1981, 125, 193–196. [Google Scholar] [CrossRef]
- Sies, H.; Dafré, A.L.; Ji, Y.; Akerboom, T.P. Protein S-thiolation and redox regulation of membrane-bound glutathione transferase. Chemico-Biological Iinteractions 1998, 111, 177–185, [https://pubmed.ncbi.nlm.nih.gov/9679553/]. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Li, H.; Grinkova, Y.; Sibhatu, H.; Jamal, J.; Poulos, T.L.; Sligar, S.G. Understanding redox regulation in membrane associated cytochrome P450s and the FMN domain of nitric oxide synthase. Chemical Research in Toxicology 2010, 23, 268–269. [Google Scholar]
- Lu, Y.; Wang, H.R.; Li, H.; Cui, H.R.; Feng, Y.G.; Wang, X.Y. A chloroplast membrane protein LTO1/AtVKOR involving in redox regulation and ROS homeostasis. Plant cell reports 2013, 32, 1427–1440, [https://pubmed.ncbi.nlm.nih.gov/23689258/]. [Google Scholar] [CrossRef]
- Spinello, A.; Ritacco, I.; Magistrato, A. The catalytic mechanism of steroidogenic cytochromes P450 from all-atom simulations: entwinement with membrane environment, redox partners, and post-transcriptional regulation. Catalysts 2019, 9, 81. [Google Scholar] [CrossRef]
- Kornienko, N.; Zhang, J.Z.; Sakimoto, K.K.; Yang, P.; Reisner, E. Interfacing nature's catalytic machinery with synthetic materials for semi-artificial photosynthesis. Nat. Nanotechnol. 2018, 13, 890–899, [https://pubmed.ncbi.nlm.nih.gov/30291349/]. [Google Scholar] [CrossRef]
- McCormick, T.M.; Calitree, B.D.; Orchard, A.; Kraut, N.D.; Bright, F.V.; Detty, M.R.; Eisenberg, R. Reductive side of water splitting in artificial photosynthesis: new homogeneous photosystems of great activity and mechanistic insight. J. Am. Chem. Soc. 2010, 132, 15480–15483, [https://pubmed.ncbi.nlm.nih.gov/20945839/]. [Google Scholar] [CrossRef]
- Botha, J.J.; Ferreira, D.; Roux, D.G. Synthesis of condensed tannins. Part 4. A direct biomimetic approach to [4, 6]-and [4, 8]-biflavanoids. Journal of the Chemical Society, Perkin Transactions 1981, 1, 1235–1245. [Google Scholar] [CrossRef]
- Roux, D.G.; Ferreira, D. The direct biomimetic synthesis, structure and absolute configuration of angular and linear condensed tannins. In Fortschritte der Chemie organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Springer, Vienna, 1982; pp. 47-76. [CrossRef]
- Pelter, A.; Satchwell, P.; Ward, R.S.; Blake, K. Effective, direct biomimetic synthesis of dibenzocyclooctene lignans by hypervalent iodine oxidation of phenolic dibenzylbutyrolactones. Journal of the Chemical Society, Perkin Transactions 1995, 1, 2201–2202. [Google Scholar] [CrossRef]
- Fuchino, Y.; Amao, Y. Photochemical and photophysical properties of carotenoid immobilized on a surfactant micellar medium including chlorophyll as an artificial photosynthesis system. Biophysics 2006, 2, 57–61, [https://pubmed.ncbi.nlm.nih.gov/27857560/]. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Sartorel, A.; Toma, F.M.; Puntoriero, F.; Scandola, F.; Campagna, S.; Prato, M.; Bonchio, M. Artificial photosynthesis challenges: water oxidation at nanostructured interfaces. Top. Curr. Chem. 2011, 303, 121–150, [https://pubmed.ncbi.nlm.nih.gov/21547686/]. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.; Duwe, H. P.; Pfeiffer, W. On the biomembranes as composite lamellae of smectic A liquid crystal and macromolecular network: elastic properties, local and collective dynamics. Physica Scripta 1989, 1989, 107–113. [Google Scholar] [CrossRef]
- Kajiyama, T.; Kumano, A.; Takayanagi, M.; Okahata, Y.; Kunitake, T. Crystal-liquid crystal phase transformation and water permeability of artificial amphiphiles as biomembrane model. Chemistry Letters 1979, 8, 645–648. [Google Scholar] [CrossRef]
- Smieja, J.M.; Benson, E.E.; Kumar, B.; Grice, K.A.; Seu, C.S.; Miller, A.J.; Mayer, J.M.; Kubiak, C.P. Kinetic and structural studies, origins of selectivity, and interfacial charge transfer in the artificial photosynthesis of CO. Proc. Natl. Acad. Sci. U S A 2012, 109, 15646–50, [https://pubmed.ncbi.nlm.nih.gov/22652573/]. [Google Scholar] [CrossRef] [PubMed]
- Schaming, D.; Hatay, I.; Cortez, F.; Olaya, A.; Méendez, M.A.; Ge, P.Y.; Deng, H.; Voyame, P.; Nazemi, Z.; Girault, H. Artificial photosynthesis at soft interfaces. Chimia 2011, 65, 356–359, [https://pubmed.ncbi.nlm.nih.gov/21744694/]. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Xue, B.; Frere, S.; Slutsky, I.; Cao, Y.; Wang, W.; Gazit, E. Multiporous supramolecular microspheres for artificial photosynthesis. Chem. Mater. 2017, 29, 4454–4460, [https://pubmed.ncbi.nlm.nih.gov/28572704/]. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Antonietti, M.; Liu, J. Bio-inspired carbon nitride mesoporous spheres for artificial photosynthesis: photocatalytic cofactor regeneration for sustainable enzymatic synthesis. Journal of Materials Chemistry A 2014, 2, 7686–7693. [Google Scholar] [CrossRef]
- Bacsa, R.R.; de Parseval, P.; Martin, F.; Serp, P. Geomimetic catalysis: From volcanic stones to ultra-selective Fe–Mo/Al2O3–TiO2 catalysts for few-walled carbon nanotube production. Carbon 2013, 64, 219–224. [Google Scholar] [CrossRef]
- Poe, S.L.; Kobašlija, M.; McQuade, D.T. Microcapsule enabled multicatalyst system. Journal of the American Chemical Society 2006, 128, 15586–15587, [https://pubmed.ncbi.nlm.nih.gov/17147357/]. [Google Scholar] [CrossRef] [PubMed]
- Poe, S.L.; Kobašlija, M.; McQuade, D.T. Mechanism and application of a microcapsule enabled multicatalyst reaction. Journal of the American Chemical Society 2007, 129, 9216–9221, [https://pubmed.ncbi.nlm.nih.gov/17602626/]. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, D.; Amal, R.; Low, G.; McEvoy, S. Occurrence and prevention of photodissolution at the phase junction of magnetite and titanium dioxide. Journal of Molecular Catalysis A: Chemical 2002, 180, 193–200. [Google Scholar] [CrossRef]
- Krupp, H.; Schnabel, W. Light-modulated electrostatic double layer adhesion. Journal of Adhesion 1973, 5, 269–277. [Google Scholar] [CrossRef]
- Barker, G.C.; Cloke, G. Electrical double layer perturbation by light absorption at the interface. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1974, 52, 468–473. [Google Scholar] [CrossRef]
- Donners, W.A.B.; Rijnbout, J.B.; Vrij, A. Light scattering from soap films: I. Determination of double-layer repulsion forces. Journal of Colloid and Interface Science 1977, 6, 249–260. [Google Scholar] [CrossRef]
- Plieth, W.J. Light absorption and light scattering in the field of the electrochemical double layer. In “Nonlinear behaviour of molecules, atoms, and ions in electric, magnetic, or electromagnetic fields”: Proceedings of the 31st International Meeting of The Société de Chimie Physique (25-28 September, 1978); Elsevier: Amsterdam – New York, Netherlands – USA, 1979; p. 251. [Google Scholar]
- Joosten, J.G.H. Electrical double layer and London—van der Waals forces in soap films studied by laser light scattering. Berichte der Bunsengesellschaft für Physikalische Chemie 1984, 88, 1153–1161. [Google Scholar] [CrossRef]
- Semenov, S.N. Electrophoresis and field flow fractionation in electric double layer observed by dynamic light scattering as possible analytical instrument. Jpn. J. Electroph. 1999, 43, 8. [Google Scholar]
- Plieth, W.J.; Gruschinske, P.; Hensel, H.J. Electrochromic changes of light absorption by the electric field of the electrolytic double layer. Berichte der Bunsengesellschaft für physikalische Chemie 1978, 82, 615–620. [Google Scholar] [CrossRef]
- De Grooth, B.G.; Van Gorkom, H.J.; Meiburg, R.F. Electrochromic absorbance changes in spinach chloroplasts induced by an external electrical field. Biochimica et Biophysica Acta-Bioenergetics 1980, 589, 299–314. [Google Scholar] [CrossRef]
- Schlodder, E.; Witt, H.T. Electrochromic absorption changes of a chloroplast suspension induced by an external electric field. FEBS Letters 1980, 112, 105–113. [Google Scholar] [CrossRef]
- De Grooth, B.G.; Amesz, J. Electrochromic absorbance changes of photosynthetic pigments in Rhodopseudomonas sphaeroides. I. Stimulation by secondary electron transport at low temperature. Biochimica et Biophysica Acta-Bioenergetics 1977, 462, 237–246. [Google Scholar] [CrossRef] [PubMed]
- De Grooth, B.G.; Amesz, J. Electrochromic absorbance changes of photosynthetic pigments in Rhodopseudomonas sphaeroides II. Analysis of the band shifts of carotenoid and bacteriochlorophyll. Biochimica et Biophysica Acta-Bioenergetics 1977, 462, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Plieth, W. Dependence of light-absorption of adsorbed molecules on the electric-field of the double-layer in the visible and ultraviolet spectral region. Abstracts of Papers of the American Chemical Society 1985, 190, 38. [Google Scholar]
- Bullard, T.; Freudenthal, J.; Avagyan, S.; Kahr, B. Test of Cairns-Smith’s ‘crystals-as-genes’ hypothesis. Faraday Discussions 2007, 136, 231–245, [https://pubmed.ncbi.nlm.nih.gov/17955812/]. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, S.; Jie, J.; Li, S.; Zheng, S.; Weng, M.; Yu, C.; Li, S.; chen, D.; Pan, F. A descriptor of “material genes”: Effective atomic size in structural unit of ionic crystals. Science China Technological Sciences 2019, 62, 849–855. [Google Scholar] [CrossRef]
- Hanf, R.; Fey, S.; Schmitt, M.; Hermann, G.; Dietzek, B.; Popp, J. Catalytic efficiency of a photoenzyme—an adaptation to natural light conditions. Chem. Phys. Chem. 2012, 13, 2013–2015, [https://pubmed.ncbi.nlm.nih.gov/22505323/]. [Google Scholar] [CrossRef] [PubMed]
- Björn, L.O. Comment on “Catalytic efficiency of a photoenzyme—an adaptation to natural light conditions” by J. Popp et al. Chem. Phys. Chem. 2013, 14, 2595–2597, [https://pubmed.ncbi.nlm.nih.gov/23712896/]. [Google Scholar] [CrossRef]
- Hermann, G.; Schmitt, M.; Dietzek, B.; Popp, J. Response to the comments by L.O. Björn on our paper “catalytic efficiency of a photoenzyme—an adaptation to natural light conditions”. Chem. Phys. Chem. 2013, 14, 2598–2600, [https://pubmed.ncbi.nlm.nih.gov/23712948/]. [Google Scholar] [CrossRef]
- Zhang, P.; Hu, J.; Shen, Y.; Yang, X.; Qu, J.; Du, F.; Sun, W.; Li, C.M. Photoenzymatic catalytic cascade system of a pyromellitic diimide/g-C3N4 heterojunction to efficiently regenerate NADH for highly selective CO2 reduction toward formic acid. ACS Applied Materials & Interfaces 2021, 13, 46650–46658. [Google Scholar] [CrossRef]
- Ertl, M.; Reichl, E.; Knör, G. Multielectron redox catalysis with efficient tyrosinase activity based on a visible-light controlled artificial photoenzyme. European Journal of Organic Chemistry 2020, 2020, 3077–3080. [Google Scholar] [CrossRef]
- Sakaushi, K.; Lyalin, A.; Tominaka, S.; Taketsugu, T.; Uosaki, K. Two-dimensional corrugated porous carbon-, nitrogen-framework/metal heterojunction for efficient multielectron transfer processes with controlled kinetics. ACS Nano 2017, 11, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Kitazumi, Y.; Tsujimura, S.; Shirai, O.; Yamamoto, M.; Kano, K. Electrostatic interaction between an enzyme and electrodes in the electric double layer examined in a view of direct electron transfer-type bioelectrocatalysis. Biosensors and Bioelectronics 2015, 63, 138–144, [https://pubmed.ncbi.nlm.nih.gov/25078712/]. [Google Scholar] [CrossRef] [PubMed]
- Higson, S.P.; Vadgama, P. A study of electrical double layer effects in the pretreatment of two-electrode cells for enzyme electrodes. Electroanalysis 1994, 6, 431–436. [Google Scholar] [CrossRef]
- Urbakh, M.; Brodskii, A. Effect of the double-layer structure on surface-plasmon frequencies and light-reflection at a metal-electrolyte interface. Soviet Electrochemistry 1979, 15, 726–731. [Google Scholar]
- Willner, I.; Zahavy, E.; Heleg-Shabtai, V. Eosin-modified reconstituted Co(II) protoporphyrin IX Mmoglobin: a semisynthetic photoenzyme for H2 evolution and hydrogenation. Journal of the American Chemical Society 1995, 117, 542–543. [Google Scholar] [CrossRef]
- Ghosh, I.; König, B. Chromoselective photocatalysis: controlled bond activation through light-color regulation of redox potentials. Angewandte Chemie Int. Ed. 2016, 55, 7676–7679, [https://pubmed.ncbi.nlm.nih.gov/27198967/]. [Google Scholar] [CrossRef]
- Mairanovskii, S.G.E.; Klyukina, L.D.; Frumkin, A.N. The polarographic catalytic surface waves of hydrogen as affected by the structure of the double layer. Doklady Akademii Nauk 1961, 141, 147–150. [Google Scholar]
- Mairanovskii, S.G.; Frumkin, A.N. Effect of the catalyst adsorption and of the double layer structure on the catalytic waves of hydrogen evolution. Review of Polarography 1963, 11, 96–101. [Google Scholar] [CrossRef]
- Pohoaţa, V.; Popa, G.; Schrittwieser, R.; Ionita, C.; Cercek, M. Properties and control of anode double layer oscillations and related phenomena. Physical Review E 2003, 68, 016405, [https://pubmed.ncbi.nlm.nih.gov/12935256/]. [Google Scholar] [CrossRef]
- Kondo, T.; Yanagisawa, M.; Fujihira, M. Effect of electrical double layers on photoinduced electron transfer quenching of an amphiphilic Ru(II)(bpy)2+3 derivative in Langmuir—Blodgett films. Electrochimica Acta 1991, 36, 1793–1798. [Google Scholar] [CrossRef]
- Wu, D.; Zheng, C.Y.; Zhou, C.T.; Yan, X.Q.; Yu, M.Y.; He, X.T. Suppressing longitudinal double-layer oscillations by using elliptically polarized laser pulses in the hole-boring radiation pressure acceleration regime. Physics of Plasmas 2013, 20, 023102. [Google Scholar] [CrossRef]
- Kondo, T. ; Effect of electrical double layers on photoinduced electron transfer in heterogeneous Langmuir-Blodgett films. Doctoral dissertation, PhD Thesis, Tokyo Institute of Technology, 1993.
- Nair, V.; Ananthoju, B.; Mohapatra, J.; Aslam, M. Photon induced non-linear quantized double layer charging in quaternary semiconducting quantum dots. Journal of Colloid and Interface Science 2018, 514, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Kaler, K.V.; Jones, T. A nonequilibrium statistical mechanical calculation of the surface conductance of the electrical double layer of biological cells and its application to dielectrophoresis. Journal of Physical Chemistry 1993, 97, 4745–4755. [Google Scholar] [CrossRef]
- Saphier, S.; Piran, R.; Keinan, E. Photoenzymes and photoabzymes. In Catalytic Antibodies; Keinan, E., Ed.; Wiley-WCH: Weinheim, Germany; 2005; pp. 350–369. [Google Scholar] [CrossRef]
- Pennline, J.A.; Rosenbaum, J.S.; Desimone, J.A.; Mikulecky, D.C. A nonlinear boundary value problem arising in the structure of the double layer at an enzymatic surface. Mathematical Biosciences 1977, 37, 1–17. [Google Scholar] [CrossRef]
- Dukhin, S.S. Non-equilibrium (dynamic) electrical double layer. In Encyclopedia of Surface and Colloid Science; CRC Press: Boca Raton, USA, 2015; pp. 4969–4975. [Google Scholar]
- Wang, H.; Adeleye, A.S.; Huang, Y.; Li, F.; Keller, A.A. Heteroaggregation of nanoparticles with biocolloids and geocolloids. Advances in Colloid and Interface Science 2015, 226, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, E.; Felmy, A.R.; Chilakapati, A. Non-equilibrium thermodynamic simulation of metal uptake in the bacterial electrical double-layer. Colloids and Surfaces B: Biointerfaces 2000, 18, 19–29. [Google Scholar] [CrossRef]
- Dreyer, W.; Guhlke, C.; Müller, R. Modeling of electrochemical double layers in thermodynamic non-equilibrium. Physical Chemistry Chemical Physics 2015, 17, 27176–27194. [Google Scholar] [CrossRef]
- Dukhin, S.S.; Shilov, V.N. Kinetic aspects of electrochemistry of disperse systems. Part II. Induced dipole moment and the non-equilibrium double layer of a colloid particle. Advances in Colloid and Interface Science 1980, 13, 153–195. [Google Scholar] [CrossRef]
- Lyklema, J. Non-equilibrium double layers in connection with colloid stability. In The Structure, Dynamics and Equilibrium Properties of Colloidal Systems; Springer: Dordrecht, 1990; pp. 789–799. [Google Scholar] [CrossRef]
- Baca, J.M.; Hernandez, F.R.; De las Nieves Lopez, F.J.; Hidalgo-Alvarez, R. Calculation of ζ-potential by non-equilibrium double layer theory in positive polystyrene model colloids. Journal of Non-Equilibrium Thermodynamics 1991, 16, 187–199. [Google Scholar] [CrossRef]
- Abarzhi, I.; Malkin, E.; Dukhin, S. Non-equilibrium frontal ion adsorption dynamics in a long bed when the double-layer is taken into account. Colloid Journal of the USSR 1978, 40, 351–356. [Google Scholar]
- Anishchenko, D.V.; Levin, O.V.; Malev, V.V. Double layer structural effects in cyclic voltammetry curves complicated with non-equilibrium injection of charge carriers into redox polymer films. Electrochimica Acta 2017, 241, 375–385. [Google Scholar] [CrossRef]
- Manzanares, J.A.; Murphy, W.D.; Mafe, S.; Reiss, H. Numerical simulation of the nonequilibrium diffuse double layer in ion-exchange membranes. Journal of Physical Chemistry 1993, 97, 8524–8530. [Google Scholar] [CrossRef]
- Haran, S.O. Non-equilibrium electric double layer and electroosmosis at ion-exchange membranes. PhD Thesis, Ben Gurion University, Negev, 2006. [Google Scholar]
- Thom, A. Electrostatic double layer interactions in the transport modelling of reverse osmosis. PhD Thesis, McMaster University, Hamilton, Ontario, 1993. [Google Scholar]
- Rubinstein, M.; Papoian, G.A. Polyelectrolytes in biology and soft matter. Soft Matter 2012, 8, 9265–9267. [Google Scholar] [CrossRef]
- Markley, L.L.; Bixler, H.J.; Cross, R.A. Utilization of polyelectrolyte complexes in biology and medicine. Journal of Biomedical Materials Research 1968, 2, 145–155, [https://pubmed.ncbi.nlm.nih.gov/5708002/]. [Google Scholar] [CrossRef] [PubMed]
- Ennis, J.; Sjöström, L.; Åkesson, T.; Jönsson, B. Attractive osmotic pressure in an electric double layer with grafted polyelectrolytes. Journal of Physical Chemistry B 1998, 102, 2149–2164. [Google Scholar] [CrossRef]
- Parsegian, V.A.; Rand, R.P.; Fuller, N.L. Direct osmotic stress measurements of hydration and electrostatic double-layer forces between bilayers of double-chained ammonium acetate surfactants. Journal of Physical Chemistry 1991, 95, 4777–4782. [Google Scholar] [CrossRef]
- Moon, G.J.; Ahn, M.M.; Kang, I.S. Osmotic pressure of ionic liquids in an electric double layer: Prediction based on a continuum model. Physical Review E 2015, 92, 063020, [https://pubmed.ncbi.nlm.nih.gov/26764817/]. [Google Scholar] [CrossRef]
- Levich, V.G. Theory of the nonequilibrium double layer. Dokl. Akad. Nauk SSSR 1949, 67, 309. [Google Scholar]
- Sparnaay, M.J. Non-equilibrium diffuse double-layer. Transactions of the Faraday Society 1957, 53, 306–314. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, X.J.; Barber, R.W.; Emerson, D.R. Influence of the electric double layer on induced pressure fields and development lengths in electro-osmotic flows. Modern Physics Letters B 2005, 19, 1655–1658. [Google Scholar] [CrossRef]
- Sadr, R.; Yoda, M.; Gnanaprakasam, P.; Conlisk, A.T. Velocity measurements inside the diffuse electric double layer in electro-osmotic flow. Applied Physics Letters 2006, 89, 044103. [Google Scholar] [CrossRef]
- van der Wouden, E.J.; Gardeniers, J.G.; van den Berg, A. Transient charging of the electric double layer in field effect-flow. In Proc. 10th International Conference on Miniaturized Systems for Chemistry and Life Sciences, µTAS – 2006 (5 Nov 2006 – 9 Nov 2006); Kitamori, T., Fujita, H., Hasebe, S., Eds.; Japan Academic Association: Tokyo, Japan, 2006; pp. 83–85. [Google Scholar]
- Ramos, A.; Castellanos, A. Travelling wave electro-osmosis: nonlinear double layer analysis and application to pumping of liquid. In American Physical Society March Meeting (March 21-25, 2005); 2005; N37.004.
- Ogawa, T. Simple oscillations in photosynthesis of higher plants. Biochimica et Biophysica Acta -Bioenergetics 1982, 681, 103–109. [Google Scholar] [CrossRef]
- Slvak, M.N.; Walker, D.A. Oscillations in photosynthesis. In Hungarian-USA Binational Symposium on Photosynthesis: a conference held at Salve Regina College, Newport, Rhode Island (August 15-18, 1986); Salve Regina College: Newport, Rhode Island, 1986; p. 105. [Google Scholar]
- Zvalinskii, V.I.; Litvin, F.F. Modelling the oscillations of the evolution of oxygen during photosynthesis. Biophysics 1990; 35, 288-293.
- Hennessey, T.L. , Field, C.B. Circadian rhythms in photosynthesis: oscillations in carbon assimilation and stomatal conductance under constant conditions. Plant Physiology 1991, 96(3), 831–836, [https://pubmed.ncbi.nlm.nih.gov/16668261/]. [Google Scholar] [CrossRef]
- Lakhno, V.D. Oscillations in the primary charge separation in bacterial photosynthesis. Physical Chemistry Chemical Physics 2002, 4, 2246–2250. [Google Scholar] [CrossRef]
- Siebke, K.; Yin, Z.H.; Raghavendra, A.S.; Heber, U. Vacuolar pH oscillations in mesophyll cells accompany oscillations of photosynthesis in leaves: Interdependence of cellular compartments, and regulation of electron flow in photosynthesis. Planta 1992, 186, 526–531, [https://pubmed.ncbi.nlm.nih.gov/24186782/]. [Google Scholar] [CrossRef]
- Anjum, S.A. , Ashraf, U.; Khan, I.; Tanveer, M.; Saleem, M.F.; Wang, L. Aluminum and chromium toxicity in maize: implications for agronomic attributes, net photosynthesis, physio-biochemical oscillations, and metal accumulation in different plant parts. Water, Air, and Soil Pollution 2016, 227, 326. [Google Scholar] [CrossRef]
- Laisk, A.; Walker, D.A. Control of phosphate turnover as a rate-limiting factor and possible cause of oscillations in photosynthesis: a mathematical model. Proceedings of the Royal society of London. Series B. Biological Sciences 1986, 227, 281–302. [Google Scholar] [CrossRef]
- Barber, J.; Laisk, A.; Schreiber, U. Towards understanding oscillations: A mathematical model of the biochemistry of photosynthesis: Discussion. Philosophical Transactions of the Royal Society of London Series B 1989, 323, 383–384. [Google Scholar] [CrossRef]
- Dubinsky, A.Y.; Ivlev, A.A.; Igamberdiev, A.U. Theoretical analysis of the possibility of existence of oscillations in photosynthesis. Biophysics 2010, 55, 55–58. [Google Scholar] [CrossRef]
- Barber, J.; Mills, J.; Love, A. Electrical diffuse layers and their influence on photosynthetic processes. FEBS Letters 1977, 74, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Barber, J. Membrane surface charges and potentials in relation to photosynthesis. Biochimica et Biophysica Acta-Reviews on Bioenergetics 1980, 594, 253–308, [https://pubmed.ncbi.nlm.nih.gov/7018576/]. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, F.B.; Gu, G.B.; Wang, L.Y.; Zheng, S.J.; Zhang, Q. Photocatalytic reaction kinetics model based on electrical double layer theory. II. Infrared spectroscopic characterization of methyl orange adsorption on TiO2 surface. Transactions of the Nonferrous Metals Society of China 2002, 12, 1187–1190. [Google Scholar]
- Venkatesh, D.; Pavalamalar, S.; Anbalagan, K. Photocatalytic, gas-sensing and double layer capacitance properties of nanoscale SnO2 obtained from template free solution phase synthesis. Journal of Materials Science: Materials in Electronics 2019, 30, 9245–9258. [Google Scholar] [CrossRef]
- Szechyńska-Hebda, M.; Kruk, J.; Górecka, M.; Karpińska, B.; Karpiński, S. Evidence for light wavelength-specific photoelectrophysiological signaling and memory of excess light episodes in Arabidopsis. The Plant Cell 2010, 22, 2201–2218, [https://pubmed.ncbi.nlm.nih.gov/20639446/]. [Google Scholar] [CrossRef] [PubMed]
- Engel, G.S. Quantum coherence in photosynthesis. Procedia Chemistry 2011, 3, 222–231. [Google Scholar] [CrossRef]
- Dickinson, E.J.; Ekström, H.; Fontes, E. COMSOL Multiphysics®: Finite element software for electrochemical analysis. A mini-review. Electrochemistry communications 2017, 40, 71–74. [Google Scholar] [CrossRef]
- Kaffash, A.; Rostami, K.; Zare, H.R. Modeling of an electrochemical nanobiosensor in COMSOL Multiphysics to determine phenol in the presence of horseradish peroxidase enzyme. Enzyme and Microbial Technology 2019, 121, 23–28. [Google Scholar] [CrossRef]
- Li, A.; Lin, Z.J. Efficient mass transport and electrochemistry coupling scheme for reliable multiphysics modeling of planar solid oxide fuel cell stack. Chinese Journal of Chemical Physics 2017, 30, 139. [Google Scholar] [CrossRef]
- Sugimoto, T.; Kobayashi, M.; Adachi, Y. Orthokinetic aggregation of charged colloidal particles in the presence of repulsive double layer force: a trajectory analysis with the solution of non-linear Poisson–Boltzmann equation. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2015, 483, 321–327. [Google Scholar] [CrossRef]
- Tawari, S. L.; Koch, D. L.; Cohen, C. Electrical double-layer effects on the Brownian diffusivity and aggregation rate of Laponite clay particles. Journal of Colloid and Interface Science 2001, 240, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Chiew, Y.C.; Valentini, J.E. The study of surface dilational properties of nonionic surfactant solutions by propagation of electrocapillary waves. Journal of Colloid and Interface Science 1993, 155, 8–15. [Google Scholar] [CrossRef]
- Zubarev, N.M. A nonlinear dispersion relationship for electrocapillary waves on the charged surface of a dielectric liquid. Technical Physics Letters 2001, 27, 689–691. [Google Scholar] [CrossRef]
- Belonozhko, D.F.; Grigor’ev, A.I. Nonlinear electrocapillary waves on a charged surface of the ideal liquid. Technical Physics Letters 2003, 29, 768–770. [Google Scholar] [CrossRef]
- Zubarev, N.M.; Zubareva, O.V. Nonlinear dispersion relation for electrocapillary waves on the surface of a dielectric liquid. Technical Physics Letters 2006, 32, 1027–1029. [Google Scholar] [CrossRef]
- Daikhin, L.I.; Kornyshev, A.A.; Urbakh, M. Effect of capillary waves on the double layer capacitance of the interface between two immiscible electrolytes. Electrochimica Acta 1999, 45, 685–690. [Google Scholar] [CrossRef]
- Budroni, M.A. Cross-diffusion-driven hydrodynamic instabilities in a double-layer system: General classification and nonlinear simulations. Physical Review E 2015, 92, 063007, [https://pubmed.ncbi.nlm.nih.gov/26764804/]. [Google Scholar] [CrossRef]
- Budroni, M.A.; De Wit, A. Dissipative structures: From reaction-diffusion to chemo-hydrodynamic patterns. Chaos: An Interdisciplinary Journal of Nonlinear Science 2017, 27, 104617. [Google Scholar] [CrossRef] [PubMed]
- Derjaguin, B.V.; Dukhin, S.S.; Matijevic, E. Nonequilibrium double layer and electrokinetic phenomena. Surface and Colloid Science 1974, 7, 273–335. [Google Scholar] [CrossRef]
- Paquin-Lefebvre, F.; Xu, B.; DiPietro, K.L.; Lindsay, A.E.; Jilkine, A. Pattern formation in a coupled membrane-bulk reaction-diffusion model for intracellular polarization and oscillations. Journal of Theoretical Biology 2020, 110242. [Google Scholar] [CrossRef]
- Yochelis, A. Catalytic membrane reactor model as a laboratory for pattern emergence in reaction-diffusion-advection media. Israel Journal of Chemistry 2018, 58, 722–732. [Google Scholar] [CrossRef]
- Xu, N.; Riley, J. Nonlinear analysis of a classical system: The double-layer capacitor. Electrochemistry Communications 2011, 13, 1077–1081. [Google Scholar] [CrossRef]
- Maenhout, G.; Schulenberg, T. Linear and non-linear interface model based on the electric double layer theory. Wissenschaftliche Berichte FZKA 2002, 6669, 3–54. [Google Scholar]
- Stigter, D. The charged colloidal cylinder with a Gouy double layer. Journal of Colloid and Interface Science 1975, 53, 296–306. [Google Scholar] [CrossRef]
- Hsu, L. Y.; Keh, H. J. Diffusioosmosis of electrolyte solutions around a circular cylinder at arbitrary zeta potential and double-layer thickness. Industrial & Engineering Chemistry Research 2009, 48, 2443-2450. [CrossRef]
- Ohshima, H. Diffuse double layer interaction between two parallel plates with constant surface charge density in an electrolyte solution IV. Numerical calculation of the interaction between similar plates using the non-linear Poisson-Boltzmann equation. Colloid and Polymer Science 1976, 254, 484–491. [Google Scholar] [CrossRef]
- Posey, F.A.; Morozumi, T. Theory of potentiostatic and galvanostatic charging of the double layer in porous electrodes. Journal of the Electrochemical Society 1966, 113, 176. [Google Scholar] [CrossRef]
- Chang, N.; Zhang, H.; Shi, M.S.; Li, J.; Yin, C.J.; Wang, H.T.; Wang, L. Regulation of the adsorption affinity of metal-organic framework MIL-101 via a TiO2 coating strategy for high capacity adsorption and efficient photocatalysis. Microporous and Mesoporous Materials 2018, 266, 47–55. [Google Scholar] [CrossRef]
- Vorotyntsev, M.A.; Izotov, V.Y.; Kornyshev, A.A. Differential capacitance of the electric double-layer in dilute-solutions of surface-inactive electrolytes and upon the specific adsorption of ions-nonlocal and non-linear effects. Soviet Electrochemistry 1983, 19, 364–368. [Google Scholar]
- Hurwitz, H.; Botte, P.; Mulenga, M. Status of ion adsorption investigations at the Hg electrode-related aspects of double-layer phenomena at biomembrane surfaces. Abstracts of Papers of the American Chemical Society 1984, 187, 120. [Google Scholar]
- Godoy, S.; García-Colín, L.S.; Micenmacher, V. Generalized Landauer equation: Absorption-controlled diffusion processes. Physical Review E 1999, 59, 6180. [Google Scholar] [CrossRef]
- Godoy, S.; Garcı́a-Colı́n, L.S.; Micenmacher, V. Multiple-scattering coefficients and absorption controlled diffusive processes. Journal of Chemical Physics 1999, 111, 9389–9392. [Google Scholar] [CrossRef]
- Shichi, A.; Satsuma, A.; Hattori, T. Adsorption-controlled diffusion in catalytic reduction of NO with hydrocarbons over zeolite catalysts. Catalysis Today 2004, 93, 777–781. [Google Scholar] [CrossRef]
- Wantanabe, T.; Maeda, H. Adsorption controlled redox activity. Surface enhanced Raman investigation of cystine versus cystein on silver electrodes. J. Phys. Chem. 1989, 93, 3258–3260. [Google Scholar] [CrossRef]
- Rouxhet, P.G. Lysozyme on apatites: a model of protein adsorption controlled by electrostatic interactions. Colloids and Surfaces 1989, 37, 339–355. [Google Scholar] [CrossRef]
- Dunwell, M.; Yan, Y.; Xu, B. Understanding the influence of the electrochemical double-layer on heterogeneous electrochemical reactions. Current Opinion in Chemical Engineering 2018, 20, 151–158. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Saito, N.; Hirai, T. Adsorption-driven photocatalytic activity of mesoporous titanium dioxide. Journal of the American Chemical Society 2005, 127, 12820–12822. [Google Scholar] [CrossRef]
- Jinnan, C.; Jiong, P. Velocity of droplet with sorption-controlled surfactant in electrolyte solution. Journal of Chemical Industry and Engineering (China) 2000, 51, 120–125. [Google Scholar]
- Chan, K.Y.; Borhan, A. Spontaneous spreading of surfactant-bearing drops in the sorption-controlled limit. Journal of Colloid and Interface Science 2006, 302, 374–377, [https://pubmed.ncbi.nlm.nih.gov/16860811/]. [Google Scholar] [CrossRef]
- Miklavcic, S.J.; Said, E. Electrostatic potential and double layer force in a semiconductor–electrolyte-semiconductor heterojunction. Physical Review E 2006, 74, 061606. [Google Scholar] [CrossRef]
- Böer, K.W. Heterojunction interface double layer and consequences for photovoltaic cells, specifically CdS(z)ZnS(1-z)S/Cu2S. Physica Status Solidi. A, Applied Research 1978, 49, 455–462. [Google Scholar] [CrossRef]
- Antonini, G.; Deschrijver, D.; Dhaene, T. Broadband rational macromodeling based on the adaptive frequency sampling algorithm and the partial element equivalent circuit method. IEEE Transactions on Electromagnetic Compatibility 2008, 50, 128–137. [Google Scholar] [CrossRef]
- Li, R.S.; Liu, Q.H. Sustained oscillations in isothermal, heterogeneously catalyzed reactions with the simplest Langmuir-type kinetics. Chemical Engineering Science 1992, 47, 3156–3158. [Google Scholar] [CrossRef]
- Zhdanov, V.P.; Kasemo, B. Surface Restructuring, Kinetic Oscillations, and Chaos in Heterogeneous Catalytic Reactions. Journal of Statistical Physics 2000, 101, 631–647. [Google Scholar] [CrossRef]
- Kuzovkov, V.N.; Kortlüke, O.; von Niessen, W. Comment on “Surface restructuring, kinetic oscillations, and chaos in heterogeneous catalytic reactions”. Physical Review E 2001, 63, 023101, [https://pubmed.ncbi.nlm.nih.gov/11308525/]. [Google Scholar] [CrossRef]
- Zhdanov, V.P. Reply to “Comment on ‘Surface restructuring, kinetic oscillations, and chaos in heterogeneous catalytic reactions’”. Physical Review E 2001, 63, 023102, [https://pubmed.ncbi.nlm.nih.gov/11308526/]. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, V.P. Simulation of kinetic oscillations in catalytic reactions accompanied by adsorbate-induced surface restructuring. Surface Science 1999, 426, 345–357. [Google Scholar] [CrossRef]
- Zhdanov, V.P. Simulation of kinetic oscillations in catalytic reactions accompanied by oxide formation. Surface Review and Letters 1999, 6, 347–353. [Google Scholar] [CrossRef]
- Zhdanov, V.P.; Kasemo, B. Kinetic oscillations on nm-sized catalyst particles: oxide model. Surface Science 2002, 511, 23–33. [Google Scholar] [CrossRef]
- Zhdanov, V.P.; Kasemo, B. Fluctuations in kinetic oscillations on nm-sized catalyst particles. Surface Science 2005, 588, L220–L226. [Google Scholar] [CrossRef]
- Zhdanov, V.P. Surface restructuring and aperiodic kinetic oscillations in heterogeneous catalytic reactions. Physica D: Nonlinear Phenomena 2000, 144, 87–96, [https://pubmed.ncbi.nlm.nih.gov/11970707/]. [Google Scholar] [CrossRef]
- Zolotarev, P.P.; Starov, V.M. Effect of oscillations of adsorptive concentration in adsorbent grain surface on adsorption process in case of nonlinear isotherm and mixed kinetics. Zhurnal Fizicheskoi Khimii 1975, 49, 2437–2439. [Google Scholar]
- Zolotarev, P.P.; Starov, V. Effect of random temperature oscillations on physical adsorption-kinetics. Zhurnal Fizicheskoi Khimii 1974, 48, 2598–2600. [Google Scholar]
- Chen, S.; Noles, T.; Schell, M. Differences in oscillations and sequences of dynamical states caused by anion adsorption in the electrochemical oxidation of formic acid. Journal of Physical Chemistry A 2000, 104, 6791–6798. [Google Scholar] [CrossRef]
- Wang, X.J.; Gaspard, P.; Gray, P.; Nicolis, G.; Baras, F.; Borckmans, P.; Scott, S. Homoclinicity and multimodal periodic or chaotic oscillations in chemical kinetics. In Spatial Inhomogeneities and Transient Behavior in Chemical Kinetics; Gray, P., Ed.; Manchester University Press: Manchester, England, 1990; pp. 687–690. [Google Scholar]
- Kulginov, D.; Zhdanov, V.P.; Kasemo, B. Oscillatory surface reaction kinetics due to coupling of bistability and diffusion limitations. Journal of Chemical Physics 1997, 106, 3117–3128. [Google Scholar] [CrossRef]
- Thames Jr, H.D.; Elster, A.D. Equilibrium states and oscillations for localized two-enzyme kinetics: A model for circadian rhythms. Journal of Theoretical Biology 1976, 59, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Selegny, E.; Vincent, J. Chemical oscillations in homogeneous Michaelian multi-enzyme systems. 1. Analytical kinetic treatment. Journal de Chimie Physique et de Physico-Chimie Biologique 1980, 77, 1083–1091. [Google Scholar] [CrossRef]
- Hau, S.S. Frequency-domain enzyme kinetics in the context of artificial calcium oscillations. PhD Thesis, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA, 1996. [Google Scholar]
- Fernández, J.M.; Bezanilla, F.; Taylor, R.E. Distribution and kinetics of membrane dielectric polarization. II. Frequency domain studies of gating currents. The Journal of General Physiology 1982, 79, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Gokhshtein, A.Y. Electron density oscillations in the double-layer field. Doklady Physical Chemistry 1996, 351, 292–295. [Google Scholar]
- Gokhshtein, A.Y. Oscillations of the electron density in the double layer field. Doklady Akademii Nauk 1996, 351, 59–63. [Google Scholar]
- Evstigneev, A.; Sachenko, A. Effect of Friedel oscillations on the capacity of a double electric layer. Fizika Tverdogo Tela 1992, 34, 2287–2290. [Google Scholar]
- Evstigneev, A.M.; Sachenko, A.V. Influence of Friedel oscillations on the capacitance of a double electrical layer. Soviet Physics. Solid State 1992, 34, 1224–1225. [Google Scholar]
- Gradov, O.V.; Gradova, M.A. Photoinduced spatiotemporal oscillations and self-organization of dissipative structures in polymer-immobilized dispersed semiconductors. Journal of Nano- and Electronic Physics 2018, 10, 04022-1–04022-8. [Google Scholar] [CrossRef] [PubMed]
- Gradov, O.V.; Gradova, M.A. Synchronization of photochemical processes and photoinduced self-organization in dispersed semiconductors under optical pumping. In Abstract Book of the 3rd International Symposium “Molecular Photonics", St. Petersburg – Repino (June 24- 29 2012); 2012; p. 156. [CrossRef]
- Schmidtnaake, G.; Pippel, W. Modeling of diffusion controled reaction and adsorption processes by means of Markov-chains. Chemische Technik 1984, 36, 411–415. [Google Scholar]
- Goldstein, B.N.; Aksirov, A.M.; Zakrjevskaya, D.T. A new kinetic model for biochemical oscillations: Graph-theoretical analysis. Biophysical Chemistry 2009, 145, 111–115, [https://pubmed.ncbi.nlm.nih.gov/19837504/]. [Google Scholar] [CrossRef]
- Mincheva, M. Oscillations in non-mass action kinetics models of biochemical reaction networks arising from pairs of subnetworks. Journal of Mathematical Chemistry 2012, 50, 1111–1125. [Google Scholar] [CrossRef]
- Peusner, L.; Mikulecky, D.C.; Bunow, B.; Caplan, S.R. A network thermodynamic approach to Hill and King–Altman reaction–diffusion kinetics. Journal of Chemical Physics 1985, 83, 5559–5566. [Google Scholar] [CrossRef]
- Rieckmann, C.; Keil, F.J. Multicomponent diffusion and reaction in three-dimensional networks: General kinetics. Industrial and Engineering Chemistry Research 1997, 36, 3275–3281. [Google Scholar] [CrossRef]
- Fuchs, O. Kinetik physikalisch-chemischer oszillationen. Colloid and Polymer Science 1980, 258, 985–986. [Google Scholar] [CrossRef]
- Skorobogatov, G. A minimal kinetic scheme providing the effect of chemical oscillations. Doklady Akademii Nauk SSSR 1986, 290, 403–409. [Google Scholar]
- Kolthoff, I.M.; Yamashita, K.; Hie, T.B.; Kanbe, A. Characteristics of polarographic catalytic waves observed with bovine-serum albumin: kinetic or diffusion control. Proceedings of the National Academy of Sciences 1973, 70, 2020–2024. [Google Scholar] [CrossRef] [PubMed]
- Mattern, K.; Felderhof, B.U. Self-consistent cluster expansion for wave propagation and diffusion-controlled reactions in a random medium. Physica A: Statistical Mechanics and its Applications 1987, 143, 21–39. [Google Scholar] [CrossRef]
- Assel, M.; Höfer, T.; Laubereau, A.; Kaiser, W. Diffusion-controlled intermolecular electron transfer studied by transient absorption and degenerate four-wave mixing measurements. Chemical Physics Letters 1995, 234, 151–158. [Google Scholar] [CrossRef]
- Lebiedz, D.; Brandt-Pollmann, U. Manipulation of self-aggregation patterns and waves in a reaction-diffusion system by optimal boundary control strategies. Physical Review Letters 2003, 91, 208301, [https://pubmed.ncbi.nlm.nih.gov/14683405/]. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.H.; Nirdosh, I.; Sedahmed, G.H. Intensification of the rate of electropolishing and diffusion controlled electrochemical machining by workpiece oscillation. Journal of the Taiwan Institute of Chemical Engineers 2014, 45, 840–845. [Google Scholar] [CrossRef]
- Walz, D.; Caplan, S.R. Chemical oscillations arise solely from kinetic nonlinearity and hence can occur near equilibrium. Biophysical Journal 1995, 69, 1698–1707, [https://pubmed.ncbi.nlm.nih.gov/8580313/]. [Google Scholar] [CrossRef] [PubMed]
- Romanovsky, Y.M. Chemical oscillations and instabilities. Non-linear chemical kinetics. Zeitschrift für Physikalische Chemie 1995, 192, 138. [Google Scholar] [CrossRef]
- Barragán, D. Essentials of kinetics and thermodynamics for understanding chemical oscillations. Foundations of Chemistry 2015, 17, 93–106. [Google Scholar] [CrossRef]
- Sagdeev R., Z.; Usikov D., A.; Zaslavsky G., M. Nonlinear Physics: From the Pendulum to Turbulence and Chaos. Harwood Academic Publishers (Gordon and Breach): New York, USA, 1988; 656 p.
- Schöll, E. Theoretical approaches to nonlinear and chaotic dynamics of generation-recombination processes in semiconductors. Applied Physics A 1989, 48, 95–106. [Google Scholar] [CrossRef]
- Landsberg, P.T.; Robbins, D.J.; Schöll, E. Threshold switching as a generation-recombination induced non-equilibrium phase transition. Physica Status Solidi A 1978, 50, 423–426. [Google Scholar] [CrossRef]
- Schöll, E. Nonequilibrium Phase Transitions in Semiconductors: Self-Organization Induced by Generation and Recombination Processes; Haken, H., Ed.; Springer-Verlag: Springer, Berlin, Heidelberg, 1987; 313 p. [Google Scholar] [CrossRef]
- Schöll, E. Continuous bifurcation and dissipative structures associated with a soft mode recombination instability in semiconductors. In Dynamical System and Chaos – Proceedings of the Sitges Conference on Statistical Mechanics Sitges, Barcelona, Spain (September 5 – 11, 1982); Garrido, L., Ed.; Springer: Berlin, Heidelberg, 1983; pp. 204–211. [Google Scholar] [CrossRef]
- Schöll, E. Stability of generation-recombination induced dissipative structures in semiconductors. Zeitschrift für Physik B Condensed Matter 1983, 52, 321–334. [Google Scholar] [CrossRef]
- Klaassen, F.M.; Van Vliet, K.M.; Fassett, J.R. Generation-recombination noise in various photoconductive semiconductors. Journal of Physics and Chemistry of Solids 1961, 22, 391–399. [Google Scholar] [CrossRef]
- Long, D. Generation-recombination noise limited detectivities of impurity and intrinsic photoconductive 8–14μ infrared detectors. Infrared Physics 1967, 7, 121–128. [Google Scholar] [CrossRef]
- Beck, W.A. Photoconductive gain and generation-recombination noise in multiple-quantum-well infrared detectors. Applied Physics Letters 1993, 63, 3589–3591. [Google Scholar] [CrossRef]
- Shadrin, V.D.; Mitin, V.V.; Kochelap, V.A.; Choi, K.K. Photoconductive gain and generation-recombination noise in quantum well infrared photodetectors. Journal of Applied Physics 1995, 77, 1771–1775. [Google Scholar] [CrossRef]
- Rudenko, T.; Gerz, S.; Nikitenko, V.; Makarov, A. Frequency-spectrum of chloroplast cross-section oscillations. Biofizika 1983, 28, 445–450. [Google Scholar]
- Sivak, M.N.; Walker, D.A. Oscillations and other symptoms of limitation of in vivo photosynthesis by inadequate phosphate supply to the chloroplast. Plant Physiology and Biochemistry 1987, 25, 635–648. [Google Scholar]
- Sayeed, S.A.; Mohanty, P. Oscillations in wheat chloroplast photochemical activity: Effect of uncouplers. In Membrane Receptors, Dynamics, and Energetics; Wirtz, K.W.A., Ed.; Springer: Boston, 1987; pp. 311–318. [Google Scholar] [CrossRef]
- Sayeed, S.A.; Mohanty, P. Rhythmic oscillations in wheat chloroplast photochemical activity. I. Oscillations in whole chain, photosystem II and photosystem I electron transport activities. Proceedings: Plant Sciences 1988, 98, 157–174. [Google Scholar] [CrossRef]
- Sayeed, S.A.; Mohanty, P. Oscillations in wheat chloroplast photochemical activity: Part III--Characterization of the possible oscillators in electron transport chain. Indian Journal of Biochemistry and Biophysics 1988, 25, 625–630. [Google Scholar]
- Sayeed, S.A.; Mohanty, P. Rhythmic oscillations in wheat chloroplast photochemical activity. II. Further characterization of the rhythm in photosystem II photoelectron transport activity. Proceedings: Plant Sciences 1988, 98, 175–181. [Google Scholar] [CrossRef]
- Kocks, P.; Ross, J. Kinetic model for (damped) oscillations of transthylakoid pH in plants. Journal of Physical Chemistry 1995, 99, 16490–16497. [Google Scholar] [CrossRef]
- Fritz, L.; Stringher, C.G.; Colepicolo, P. Imaging oscillations in Gonyaulax: a chloroplast rhythm of nitrate reductase visualized by immunocytochemistry. Brazilian journal of medical and biological research 1996, 29, 111–117. [Google Scholar]
- Smrčinová, M.; Sørensen, P.G.; Krempasky, J.; Ballo, P. Chaotic oscillations in a chloroplast system under constant illumination. International Journal of Bifurcation and Chaos 1998, 8, 2467–2470. [Google Scholar] [CrossRef]
- Shchepetov, D.S.; Chernavsky, D.S.; Gorokhov, V.V.; Grishanova, N.P.; Pashchenko, V.Z.; Rubin, A.B. The nature of oscillations in the kinetics of electron transfer in the reaction center of purple bacteria. Doklady. Biochemistry and Biophysics 2009, 425, 87–90, [https://pubmed.ncbi.nlm.nih.gov/19496329/]. [Google Scholar] [CrossRef] [PubMed]
- Shchepetov, D.S.; Chernavsky, D.S.; Gorokhov, V.V.; Paschenko, V.Z.; Rubin, A.B. Application of the standard theory of electronic transitions to the description of oscillations in the kinetics of electron transfer in reaction centers of purple bacteria. Biophysics 2009, 54, 691–698, [https://pubmed.ncbi.nlm.nih.gov/20067182/]. [Google Scholar] [CrossRef]
- Karageorgiy, P.M.; Leiderman, A.Y. Theory of kinetic oscillations in semiconductors. Fizika Tverdogo Tela 1967, 9, 2151–2156. [Google Scholar]
- Karageorgiy, P.M.; Leiderman, A.Y. Forced kinetic oscillations in semiconductor diode structures Soviet Physics: Semiconductors 1967, 1, 617. 1.
- Parfenev, R. V.; Sologub, V. V.; Goltsman, B. M. (1968). Quantum oscillations of the kinetic and photo- electric coefficients of n-type bismuth telluride. Fizika Tverdogo Tela 1968, 10, 3087. [Google Scholar]
- Cebrián, E.; Bonilla, L.L.; Carpio, A. Self-sustained current oscillations in the kinetic theory of semiconductor superlattices. Journal of Computational Physics 2009, 228, 7689–7705. [Google Scholar] [CrossRef]
- Firsov, I.; Lang, I. Kinetic theory of semiconductors with low mobility developed for strong coupling between the current carriers and lattice oscillations. Soviet Physics-JETP 1963, 16, 1301–1312. [Google Scholar]
- Budagyan, B.G.; Aivazov, A.A.; Stanovov, O.N. Oscillations of the photoconductivity and characteristic features of the relaxation kinetics of a-Si:H. Semiconductors 1993, 27, 822–825. [Google Scholar]
- Pavlyuk, S.P.; Kushnyirenko, V.V. The kinetics of onset of oscillations in n+-n-n+ transistors and resistors under influence of high density pulses of current. Vyisnyk Kyivskogo Universytetu. Fyiziko-Matematichnyi Nauki 2007, 280-283.
- Walker, D.A. Concerning oscillations. Photosynthesis research 1992, 34, 387–395, [https://pubmed.ncbi.nlm.nih.gov/24408834/]. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, M.; Gimmler, H.; Schreiber, U.; Heber, U. Relative insensitivity of photosynthesis to the dissipation of a transthylakoid proton gradient in intact chloroplasts. Physiologie Végétale 1985, 23, 801–812. [Google Scholar]
- Peterson, R.B.; Sivak, M.N.; Walker, D.A. Carbon dioxide-induced oscillations in fluorescence and photosynthesis: role of thylakoid membrane energization in regulation of photosystem II activity. Plant Physiology 1988, 88, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Vanselow, K.H.; Kolbowski, J.; Hansen, U.P. Further evidence for the relationship between light-induced changes of plasmalemma transport and transthylakoid proton uptake. Journal of Experimental Botany 1989, 40, 239–245. [Google Scholar] [CrossRef]
- Laasch, H.; Ihle, C.; Günther, G. Detecting localized proton currents in photophosphorylation by procaine inhibition of the transthylakoid pH-gradient. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1993, 1140, 251–261. [Google Scholar] [CrossRef]
- Cruz, J.A.; Sacksteder, C.A.; Kanazawa, A.; Kramer, D.M. Contribution of electric field (Δψ) to steady-state transthylakoid proton motive force (pmf) in vitro and in vivo. Control of pmf parsing into Δψ and ΔpH by ionic strength. Biochemistry 2001, 40, 1226–1237, [https://pubmed.ncbi.nlm.nih.gov/11170448/]. [Google Scholar] [CrossRef]
- Cruz, J.A.; Kanazawa, A.; Treff, N.; Kramer, D.M. Storage of light-driven transthylakoid proton motive force as an electric field (Δψ) under steady-state conditions in intact cells of Chlamydomonas Reinhardtii. Photosynthesis Research 2005, 85, 221–233, [https://pubmed.ncbi.nlm.nih.gov/16075322/]. [Google Scholar] [CrossRef]
- Radenović, Č.; Jeremić, M.; Maksimov, G.; Beljanski, M.; Filipović, M.; Čamdžija, Z. Mechanisms and parameters of transients and oscillations of delayed chlorophyll fluorescence induction processes in the excited thylakoid membrane of the maize intact leaf. Journal of Scientific Agricultural Research 2008, 69, 5–21. [Google Scholar]
- Radenović, Č.N.; Jeremić, M.G.; Maximov, G.V.; Beljanski, M.V.; Radojčić, A.R. Mechanisms and parameters of transients and oscillations of delayed chlorophyll fluorescence in the thylakoid membrane of the intact maize leaf. Russian Journal of Physical Chemistry A 2009, 83, 1582–1591. [Google Scholar] [CrossRef]
- Radenović, Č.; Marković, K.; Radojčić, A.; Anđelković, V.; Kalauzi, A.J. Interdependence between oscillations and transients of delayed fluorescence induction processes in the thylakoid membrane of the intact maize leaf: Responses to effects of increased temperatures and drought. Zbornik Matice Srpske za Prirodne Nauke 2010, 118, 7–26. [Google Scholar] [CrossRef]
- Maynard, S.N. Determining the origin of a Ca2+ Wave released in Arabidopsis Thaliana upon photostimulation of the ER-Chloroplast Nexus. PhD Thesis, Texas A&M University, College Station, Texas, 2017. [Google Scholar]
- Elber, R. ; A new paradigm for atomically detailed simulations of kinetics in biophysical systems. Quarterly reviews of biophysics 2017, 50, e8, [https://pubmed.ncbi.nlm.nih.gov/29233220/]. [Google Scholar] [CrossRef]
- Arnold, W. ; An Electron-Hole Picture of Photosynthesis. The Journal of physical chemistry 1965, 69, 788–791, [https://pubmed.ncbi.nlm.nih.gov/14296949/]. [Google Scholar] [CrossRef]
- Pearlstein, R.M. ; Photosynthetic exciton theory in the 1960s. Photosynthesis Research 2002, 73, 119–126, [https://pubmed.ncbi.nlm.nih.gov/16245112/]. [Google Scholar] [CrossRef]
- Campillo, A.J.; Shapiro, S.L.; Kollman, V.H.; Winn, K.R.; Hyer, R.C. Picosecond exciton annihilation in photosynthetic systems. Biophysical journal 1976, 16, 93–97, [https://pubmed.ncbi.nlm.nih.gov/1244893/]. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, R. ; Exciton percolation in mixed molecular crystals and aggregates: from naphthalene to photosynthesis. The Journal of Physical Chemistry 1976, 80, 2191–2195. [Google Scholar] [CrossRef]
- Pearlstein, R.M. ; Exciton migration and trapping in photosynthesis. Photochemistry and Photobiology 1982, 35, 835–844. [Google Scholar] [CrossRef]
- Pearlstein, R.M. Structure and exciton effects in photosynthesis. New Comprehensive Biochemistry 1987, 15, 299–317. [Google Scholar] [CrossRef]
- Lavergne, J.; Trissl, H.W. Theory of fluorescence induction in photosystem II: derivation of analytical expressions in a model including exciton-radical-pair equilibrium and restricted energy transfer between photosynthetic units. Biophysical journal 1995, 68, 2474–2492, [https://pubmed.ncbi.nlm.nih.gov/7647250/]. [Google Scholar] [CrossRef]
- Novoderezhkin, V.I.; Razjivin, A.P. ; Exciton dynamics in circular aggregates: application to antenna of photosynthetic purple bacteria. Biophysical journal 1995, 68, 1089–1100, [https://pubmed.ncbi.nlm.nih.gov/7756528/]. [Google Scholar] [CrossRef]
- Monshouwer, R.; Abrahamsson, M.; Van Mourik, F.; Van Grondelle, R. Superradiance and exciton delocalization in bacterial photosynthetic light-harvesting systems. The Journal of Physical Chemistry B 1997, 101, 7241–7248. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Kühn, O.; Pullerits, T.; Sundström, V. ; Excitons in photosynthetic purple bacteria: wavelike motion or incoherent hopping? The Journal of Physical Chemistry B 1997, 101, 7275–7283. [Google Scholar] [CrossRef]
- Renger, T.; May, V. ; Multiple exciton effects in molecular aggregates: Application to a photosynthetic antenna complex. Physical review letters 1997, 78, 3406. [Google Scholar] [CrossRef]
- Renger, T.; May, V. ; Ultrafast exciton motion in photosynthetic antenna systems: the FMO-complex. The Journal of Physical Chemistry A 1998, 102, 4381–4391. [Google Scholar] [CrossRef]
- Lee, H.; Cheng, Y.C.; Fleming, G.R. ; Coherence dynamics in photosynthesis: protein protection of excitonic coherence. Science 2007, 316, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Abramavicius, D.; Mukamel, S. ; Quantum oscillatory exciton migration in photosynthetic reaction centers. The Journal of chemical physics 2010, 133, 08B603, [https://pubmed.ncbi.nlm.nih.gov/20707578/]. [Google Scholar] [CrossRef] [PubMed]
- Abramavicius, D.; Voronine, D.V.; Mukamel, S. ; Double-quantum resonances and exciton-scattering in coherent 2D spectroscopy of photosynthetic complexes. Proceedings of the National Academy of Sciences 2008, 105, 8525-8530, [https://pubmed.ncbi.nlm.nih.gov/18562293/]. [Google Scholar] [CrossRef]
- Bode, S.; Quentmeier, C.C.; Liao, P.N.; Hafi, N.; Barros, T.; Wilk, L.; Bittner, F.; Walla, P.J. ; On the regulation of photosynthesis by excitonic interactions between carotenoids and chlorophylls. Proceedings of the National Academy of Sciences 2009, 106, 12311-12316, [https://pubmed.ncbi.nlm.nih.gov/19617542/]. [Google Scholar] [CrossRef]
- Mostame, S.; Rebentrost, P.; Eisfeld, A.; Kerman, A.J.; Tsomokos, D.I.; Aspuru-Guzik, A. ; Quantum simulator of an open quantum system using superconducting qubits: exciton transport in photosynthetic complexes. New Journal of Physics 2012, 14, 105013. [Google Scholar] [CrossRef]
- Westenhoff, S.; Palecek, D.; Edlund, P.; Smith, P.; Zigmantas, D. ; Coherent picosecond exciton dynamics in a photosynthetic reaction center. Journal of the American Chemical Society 2012, 134, 16484–16487, [https://pubmed.ncbi.nlm.nih.gov/23009768/]. [Google Scholar] [CrossRef]
- Scholes, G.D.; Smyth, C. ; Perspective: detecting and measuring exciton delocalization in photosynthetic light harvesting. The Journal of Chemical Physics 2014, 140, 03B201_1, [https://pubmed.ncbi.nlm.nih.gov/24655162/]. [Google Scholar] [CrossRef]
- Warshel, A.; Chu, Z.T.; Parson, W.W. ; Dispersed polaron simulations of electron transfer in photosynthetic reaction centers. Science 1989, 246, 112–116, [https://pubmed.ncbi.nlm.nih.gov/2675313/]. [Google Scholar] [CrossRef] [PubMed]
- Damjanović, A.; Kosztin, I.; Kleinekathöfer, U.; Schulten, K. ; Excitons in a photosynthetic light-harvesting system: a combined molecular dynamics, quantum chemistry, and polaron model study. Physical Review E 2002, 65, 031919, [https://pubmed.ncbi.nlm.nih.gov/11909121/]. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, A.; Rätsep, M.; Timpmann, K.; Trinkunas, G. ; Excitonic polarons in quasi-one-dimensional LH1 and LH2 bacteriochlorophyll a antenna aggregates from photosynthetic bacteria: A wavelength-dependent selective spectroscopy study. Chemical Physics 2009, 357, 102–112. [Google Scholar] [CrossRef]
- Qin, M.; Shen, H.Z.; Zhao, X.L.; Yi, X.X. ; Effects of system-bath coupling on a photosynthetic heat engine: A polaron master-equation approach. Physical Review A 2017, 96, 012125. [Google Scholar] [CrossRef]
- Zhang, Z.; Saurabh, P.; Dorfman, K.E.; Debnath, A.; Mukamel, S. ; Monitoring polariton dynamics in the LHCII photosynthetic antenna in a microcavity by two-photon coincidence counting. The Journal of chemical physics 2018, 148, 074302, [https://pubmed.ncbi.nlm.nih.gov/29471638/]. [Google Scholar] [CrossRef]
- Coles, D.; Flatten, L.C.; Sydney, T.; Hounslow, E.; Saikin, S.K.; Aspuru-Guzik, A.; Vedral, V.; Tang, J.K.H.; Taylor, R.A.; Smith, J.M.; Lidzey, D.G. ; A nanophotonic structure containing living photosynthetic bacteria. Small 2017, 13, 1701777. [Google Scholar] [CrossRef]
- Coles, D.M.; Flatten, L.C.; Sydney, T.; Hounslow, E.; Saikin, S.K.; Aspuru-Guzik, A.; Vedral, V.; Tang, J.K.H.; Taylor, R.A.; Smith, J.M.; Lidzey, D.G. Polaritons in living systems: modifying energy landscapes in photosynthetic organisms using a photonic structure. arXiv preprint arXiv:1702.01705, 2017.
- Squire, R.; March, N.; Ingles, J. Coherent exciton-polariton model for photosynthetic energy transfer. Bulletin of the American Physical Society 2015, 60, BB1–000019. [Google Scholar]
- Hayes, J.M.; Small, G.J. ; Photochemical hole burning and strong electron-phonon coupling: primary donor states of reaction centers of photosynthetic bacteria. The Journal of Physical Chemistry 1986, 90, 4928–4931. [Google Scholar] [CrossRef]
- Jankowiak, R.; Reppert, M.; Zazubovich, V.; Pieper, J.; Reinot, T. Site selective and single complex laser-based spectroscopies: A window on excited state electronic structure, excitation energy transfer, and electron–phonon coupling of selected photosynthetic complexes. Chem. Rev. 2011, 111, 4546–4598, [https://pubmed.ncbi.nlm.nih.gov/21595428/]. [Google Scholar] [CrossRef]
- Kell, A.; Feng, X.; Reppert, M.; Jankowiak, R. On the shape of the phonon spectral density in photosynthetic complexes. J. Phys. Chem. B 2013, 117, 7317–7323, [https://pubmed.ncbi.nlm.nih.gov/23718713/]. [Google Scholar] [CrossRef]
- Pajusalu, M.; Rätsep, M.; Freiberg, A. Temperature dependent electron–phonon coupling in chlorin-doped impurity glass and in photosynthetic FMO protein containing bacteriochlorophyll a. J.Luminescence 2014, 152, 79–83. [Google Scholar] [CrossRef]
- Chen, H.; Wang, X.; Fang, A.P.; Li, H.R. Phonon-assisted excitation energy transfer in photosynthetic systems. Chinese Physics B 2014, 25(9), 098201. [Google Scholar] [CrossRef]
- Pavlovich, V.S. Model for primary electron transfer and coupling of electronic states at reaction centers of purple bacteria. Journal of Applied Spectroscopy 2006, 73, 328–339. [Google Scholar] [CrossRef]
- Pavlovich, V.S. Hystons, new quasi-particles, and electron transfer in bacterial photosynthesis. Physica E: Low-dimensional Systems and Nanostructures 2002, 14, 282–288. [Google Scholar] [CrossRef]
- Pavlovich, V.S. Conception of hystons in bacterial photosynthesis: spectra, exciton dynamics, and electron transfer. Proc SPIE 2007, 6727, 67271T. [Google Scholar] [CrossRef]
- Lee, M.K.; Huo, P.; Coker, D.F. Semiclassical path integral dynamics: Photosynthetic energy transfer with realistic environment interactions. Ann. Rev. Phys. Chem. 2016, 67, 639–668, [https://pubmed.ncbi.nlm.nih.gov/27090842/]. [Google Scholar] [CrossRef]
- Onizhuk, M.; Sohoni, S.; Galli, G.; Engel, G.S. Spatial Patterns of Light-Harvesting Antenna Complex Arrangements Tune the Transfer-to-Trap Efficiency of Excitons in Purple Bacteria. The Journal of Physical Chemistry Letters 2021, 12, 6967–6973. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Liu, Z.; Miyatake, T.; Tamiaki, H.; Kobayashi, T.; Zhang, Z.; Du, J.; Leng, Y. Ultrafast dynamics of multi-exciton state coupled to coherent vibration in zinc chlorin aggregates for artificial photosynthesis. Optics Express 2017, 25, 29667–29675, [https://pubmed.ncbi.nlm.nih.gov/29221004/]. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.; Chen, X.; Lin, L.; Fang, Y.; Wang, X. Biomimetic donor–acceptor motifs in conjugated polymers for promoting exciton splitting and charge separation. Angew. Chem. Int. Ed. 2018, 57, 8729–8733, [https://pubmed.ncbi.nlm.nih.gov/29797759/]. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.S.; Lloyd, L.T.; Sohail, S.H.; Allodi, M.A.; Otto, J.P.; Saer, R.G.; Wood, R.E.; Massey, S.C.; Ting, P.C.; Blankenship, R.E.; Engel, G.S. Photosynthesis tunes quantum-mechanical mixing of electronic and vibrational states to steer exciton energy transfer. Proceedings of the National Academy of Sciences 2021, 118, E2018240118, [https://pubmed.ncbi.nlm.nih.gov/33688046/]. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Z.; Brennaman, M.K.; Concepcion, J.J.; Patrocinio, A.O.T.; Iha, N.Y.M.; Meyer, T.J. Making solar fuels by artificial photosynthesis. Pure and Applied Chemistry 2011, 83, 749–768. [Google Scholar] [CrossRef]
- Stock, M.; Dunn, S. LiNbO3 - A new material for artificial photosynthesis. IEEE Transactions on Ultrasonics, Ferroelectrics, and Frequency Control 2011, 58, 1988–1993, [https://pubmed.ncbi.nlm.nih.gov/21937336/]. [Google Scholar] [CrossRef] [PubMed]
- Nath, R.K.; Zain, M.M.; Kadhum, A.A.H. Artificial photosynthesis using LiNbO3 as photocatalyst for sustainable and environmental friendly construction and reduction of global warming: A review. Catalysis Reviews 2014, 56, 175–186. [Google Scholar] [CrossRef]
- Nath, R.K.; Zain, M.F.M. Artificial photosynthesis in concrete surface by using LiNbO3. Advances in Environmental Biology 2015, 9, 1–9, [https://pubmed.ncbi.nlm.nih.gov/24376384/]. [Google Scholar] [CrossRef]
- Cortecchia, D.; Yin, j.; Bruno, A.; Lo, S.-Z.A.; Gurzadyan, G.G.; Mhaisalkar, S.; Brédas, J.-L.; Soci, C. Polaron self-localization in white-light emitting hybrid perovskites. J. Mat. Chem. C 2017, 5, 2771–2780. [Google Scholar] [CrossRef]
- Zheng, F.; Wang, L.W. Large polaron formation and its effect on electron transport in hybrid perovskites. Energy & Environmental Science 2019, 12, 1219–1230. [Google Scholar] [CrossRef]
- Chen, Y.C.; Song, B.; Leggett, A.J.; Ao, P.; Zhu, X. Resonant confinement of an excitonic polariton and ultraefficient light harvest in artificial photosynthesis. Phys. Rev. Lett. 2019, 122, 257402, [https://pubmed.ncbi.nlm.nih.gov/31347870/]. [Google Scholar] [CrossRef]
- Liu, S.; Weng, B.; Tang, Z.R.; Xu, Y.J. Constructing one-dimensional silver nanowire-doped reduced graphene oxide integrated with CdS nanowire network hybrid structures toward artificial photosynthesis. Nanoscale 2015, 7, 861–866. [Google Scholar] [CrossRef]
- Liu, S.Q.; Zhou, S.S.; Chen, Z.G.; Liu, C.B.; Chen, F.; Wu, Z.Y. An artificial photosynthesis system based on CeO2 as light harvester and N-doped graphene Cu(II) complex as artificial metalloenzyme for CO2 reduction to methanol fuel. Cat. Commun. 2016, 73, 7–11. [Google Scholar] [CrossRef]
- Guiglion, P.; Berardo, E.; Butchosa, C.; Wobbe, M.C.; Zwijnenburg, M.A. Modelling materials for solar fuel synthesis by artificial photosynthesis; predicting the optical, electronic and redox properties of photocatalysts. Journal of Physics: Condensed Matter 2016, 28, 074001, [https://pubmed.ncbi.nlm.nih.gov/26808228/]. [Google Scholar] [CrossRef]
- Jena, N.; Rawat, A.; De Sarkar, A. Strain and pH facilitated artificial photosynthesis in monolayer MoS2 nanosheets. J. Mat. Chem. A 2017, 5, 22265–22276. [Google Scholar] [CrossRef]
- Jain, P.K. Plasmonic Photosynthesis. ECS Meeting Abstracts 2019, 41, 1949. [Google Scholar] [CrossRef]
- Yu, S.; Jain, P.K. Plasmonic photosynthesis of C1–C3 hydrocarbons from carbon dioxide assisted by an ionic liquid. Nature Commun. 2019, 10, 1–7, [https://pubmed.ncbi.nlm.nih.gov/31043604/]. [Google Scholar] [CrossRef]
- Yu, S.; Jain, P.K. Selective branching of plasmonic photosynthesis into hydrocarbon production and hydrogen generation. ACS Energy Letters 2019, 4, 2295–2300. [Google Scholar] [CrossRef]
- Yu, S.; Jain, P.K. Isotope effects in plasmonic photosynthesis. Angew. Chem. 2020, 132, 22666–22669, [https://pubmed.ncbi.nlm.nih.gov/32898311/]. [Google Scholar] [CrossRef]
- Yu, S.; Jain, P.K. Plasmonic catalysis, photoredox chemistry, and photosynthesis. Plasmonic Catalysis: From Fundamentals to Applications 2021 (in press). [CrossRef]
- Ueno, K.; Oshikiri, T.; Shi, X.; Zhong, Y.; Misawa, H. Plasmon-induced artificial photosynthesis. Interface Focus 2015, 5, 20140082, [https://pubmed.ncbi.nlm.nih.gov/26052419/]. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Ueno, K.; Mori, Y.; Shi, X.; Oshikiri, T.; Murakoshi, K.; Inoue, H.; Misawa, H. Plasmon-assisted water splitting using two sides of the same SrTiO3 single-crystal substrate: conversion of visible light to chemical energy. Angew. Chem. Int. Ed. 2014, 53, 10350–10354, [https://pubmed.ncbi.nlm.nih.gov/24988943/]. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Kim, D.; Rungtaweevoranit, B.; Trickett, C.A.; Barmanbek, J.T.D.; Alshammari, A.S.; Yang, P.; Yaghi, O.M. Plasmon-enhanced photocatalytic CO2 conversion within metal–organic frameworks under visible light. J. Am. Chem. Soc. 2017, 139, 356–362, [https://pubmed.ncbi.nlm.nih.gov/28004911/]. [Google Scholar] [CrossRef]
- Glushko, E.Y. Nonuniform kinetics of quasiparticles in a steady-state PACKET representation. Ukrainskii Fizicheskii Zhurnal 1981, 26, 2037–2043. [Google Scholar]
- Seminozhenko, V.P. Kinetics of interacting quasiparticles in strong external fields. Physics Reports 1982, 91, 103–182. [Google Scholar] [CrossRef]
- Sinisky, I.; Golosov, A.; Men, A. The kinetics of the reactions of solid-phases in the crystalline quasiparticles method. Berichte der Bunsen-Gesellschaft-Physical Chemistry Chemical Physics 1982, 86, 482–482. [Google Scholar]
- Glushko, E.Ya. Nonhomogeneous kinetics of quasiparticles in steady-state packet representation. Physica Status Solidi B 1982, 114, 685–694. [Google Scholar] [CrossRef]
- Rawat, A.; Jena, N.; De Sarkar, A. A comprehensive study on carrier mobility and artificial photosynthetic properties in group VI B transition metal dichalcogenide monolayers. J. Mater. Chem. A 2018, 6, 8693–8704. [Google Scholar] [CrossRef]
- Wang, H.; Liu, W.; Jin, S.; Zhang, X.; Xie, Y. Low-dimensional semiconductors in artificial photosynthesis: An outlook for the interactions between particles/quasiparticles. ACS Central Science 2020, 6, 1058–1069, [https://pubmed.ncbi.nlm.nih.gov/32724841/]. [Google Scholar] [CrossRef]
- Dorfman, K.E.; Voronine, D.V.; Mukamel, S.; Scully, M.O. Photosynthetic reaction center as a quantum heat engine. Proceedings of the National Academy of Sciences 2013, 110, 2746–2751, [https://pubmed.ncbi.nlm.nih.gov/23365138/]. [Google Scholar] [CrossRef]
- Ioffe I., I.; Reshetov, V.A.; Dobrotvorsky, A.M. Heterogeneous catalysis: Physical and chemical bases; Chemistry: Leningrad, 1985; 224 p. [Google Scholar]
- Ioffe, I.I.; Dobrotvorskii, A.M.; Belozerskikh, V.A. Prediction and analysis of heterogeneous catalysis mechanisms by pattern recognition methods with a computer. Russ. Chem. Rev. 1983, 52, 229–241. [Google Scholar] [CrossRef]
- Dobrotvorskii, A.M. A quasi-fermion model of electronic-structure and its applications in the chemosorption and heterogeneous catalysis. Doklady Akademii Nauk SSSR 1984, 279, 915–919. [Google Scholar]
- Dobrotvorskii, A.M. , Afanasjeva, O.V. A quasifermion approach to modelling interatomic interactions in solids. Journal of Physics: Condensed Matter 1993, 5, 8839–8848. [Google Scholar]
- Kiselev, V.F.; Plotnikov, G.S.; Bespalov, V.A.; Zoteev, A.V.; Fomin, Y.D. Elementary excitations in a semiconductor-adsorbed-molecule system. Kinetics and Catalysis 1987, 28, 14–27. [Google Scholar]
- Kiselev, V.F.; Krylov, O.V. Electronic phenomena in adsorption and catalysis on semiconductors and dielectrics., 2nd ed.; Springer: Berlin, 2012; 287 p. [Google Scholar]
- McCarroll, J.J. Surface physics and catalysis. Surface Science 1975, 53, 297–316. [Google Scholar] [CrossRef]
- Lundström, I.; Armgarth, M.; Petersson, L.G. Physics with catalytic metal gate chemical sensors. Critical Reviews in Solid State and Material Sciences 1989, 15, 201–278. [Google Scholar] [CrossRef]
- Zamaraev, K.I. Chemical physics and catalysis. Pure and Applied Chemistry 1997, 69, 865–876. [Google Scholar] [CrossRef]
- Sirin, S.; Pearlman, D.A.; Sherman, W. Physics-based enzyme design: Predicting binding affinity and catalytic activity. Proteins: Structure, Function, and Bioinformatics 2014, 82, 3397–3409, [https://pubmed.ncbi.nlm.nih.gov/25243583/]. [Google Scholar] [CrossRef] [PubMed]
- Fodor, É.; Marchetti, M.C. The statistical physics of active matter: From self-catalytic colloids to living cells. Physica A: Statistical Mechanics and its Applications 2018, 504, 106–120. [Google Scholar] [CrossRef]
- Brandt N., B.; Kulbachinsky, V.A. Quasiparticles in condensed matter physics, 3rd ed.; Fizmatlit: Moscow, 2016. [Google Scholar]
- Fan, T.Y.; Sun, J.J. Four-phonon model of soft-matter quasicrystals for studying thermodynamics. Philosophical Magazine Letters 2014, 94, 112–117. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Sharma, P. Geometrically nonlinear deformation and the emergent behavior of polarons in soft matter. Soft Matter 2015, 11, 8042–8047, [https://pubmed.ncbi.nlm.nih.gov/26345397/]. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.; Wecker, A.; Yuan, J. Advanced optical materials in the new decade: From metamaterials, smart soft matter, and lasers to terahertz, polaritons, and AIE. Advanced Optical Materials 2020, 8, 1901916. [Google Scholar] [CrossRef]
- Yuan, H. Single molecules in soft matter: A study of biomolecular conformation, heterogeneity and plasmon enhanced fluorescence. PhD Thesis, Leiden Institute of Physics, Leiden University, 2013. [Google Scholar]
- Matsko, N.; Letofsky-Papst, I.; Mittal, V. What is hidden in the volume plasmon? EFTEM plasmon to carbon map for soft matter characterization. Imaging & Microscopy 2014, 16, 2–4. [Google Scholar]
- Miller, T. Multiscale dynamics in soft-matter systems: Enzyme catalysis, sec-facilitated protein translocation, and ion-conduction in polymers. Bulletin of the American Physical Society 2016, 61, S22–009. [Google Scholar]
- Khokhlov, A.R. Water solutions of amphiphilic polymers: Nanostructure formation and possibilities for catalysis (soft matter as structured materials). Physical Characteristics Research 2005, 84, 832. [Google Scholar]
- Dornhaus, R.; Benner, R.E.; Chang, R.K.; Chabay, I. Surface plasmon contribution to SERS. Surface Science 1980, 101, 367–373. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, J.; Wu, Y.; Xu, Y.; Su, Y.; Zhang, L.; Qi, Y.; Wen, X.; Yang, H. Hybrid surface plasmon effect and SERS characterization in a heterogeneous composite structure of Au nano-array and Ag film. Results in Physics 2020, 17, 103175. [Google Scholar] [CrossRef]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors. Sensors and Actuators B: Chemical 1999, 54, 3–15. [Google Scholar] [CrossRef]
- Yamamoto, M. Surface plasmon resonance (SPR) theory: Tutorial. Review of Polarography 2002, 48, 209–237. [Google Scholar] [CrossRef]
- Piliarik, M.; Homola, J. Surface plasmon resonance (SPR) sensors: Approaching their limits? Optics Express 2009, 17, 16505–16517, [https://pubmed.ncbi.nlm.nih.gov/19770865/]. [Google Scholar] [CrossRef]
- Herrmann, F.H.; Börner, T.; Hagemann, R. Biosynthesis of thylakoids and the membrane-bound enzyme systems of photosynthesis. Results and Problems in Cell Differentiation 1980, 10, 147–177, [https://pubmed.ncbi.nlm.nih.gov/6999569/]. [Google Scholar] [CrossRef] [PubMed]
- Latzko, E.; Kelly, G.J. Photosynthesis control of carbon metabolism through enzyme regulation and membrane-mediated metabolite transport. In Thirty Years of Photosynthesis 1974–2004; Springer: Berlin, Heidelberg, 2006; pp. 33–52. [Google Scholar] [CrossRef]
- Miller, K.; Staeheli, L.A. Direct identification of photosynthetic enzymes on membrane surfaces revealed by deep-etching. Journal of Cell Biology 1973, 59, A226–A226. [Google Scholar]
- Anderson, L.E.; Avron, M. Light modulation of enzyme activity in chloroplasts: Generation of membrane-bound vicinal-dithiol groups by photosynthetic electron transport. Plant Physiology 1976, 57, 209–213, [https://pubmed.ncbi.nlm.nih.gov/16659452/]. [Google Scholar] [CrossRef] [PubMed]
- Krogmann, D.W. The organization of photosynthetic enzymes on the chloroplast membrane. In The Enzymes of Biological Membranes; Martonosi, A., Ed.; Springer: Boston, MA, 1976; pp. 143-162. [Google Scholar] [CrossRef]
- Cuendet, P.; Gratzel, M. Biophotocatalysis based on semiconducting powders. Experientia 1984, 40, 604–604. [Google Scholar]
- López-Vidal, M.G.; Gamboa, G.; Oksdath-Mansilla, G.; Bisogno, F.R. Photobiocatalysis. In Biocatalysis for Practitioners: Techniques, Reactions and Applications 2021, 317-359. [CrossRef]
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew. Chem. Int. Ed. 2018, 57, 7958–7985, [https://pubmed.ncbi.nlm.nih.gov/29194901/]. [Google Scholar] [CrossRef] [PubMed]
- Maciá Agulló, J.A.; Corma Canós, A.; García Gómez, H. Photobiocatalysis: The power of combining photocatalysis and enzymes. Chemistry-a European Journal 2015, 21, 10940–10959, [https://pubmed.ncbi.nlm.nih.gov/26014675/]. [Google Scholar] [CrossRef] [PubMed]
- Drbohlavová, J. Preparation of photocatalytically active surfaces. Ph. D. Thesis, FCH VUT – IRCELYON, Brno – Lyon, France, 2008.
- Schmidt, H.K.; Akarsu, M.; Naumann, M.; Müller, T.S. Doped nanoparticles for photocatalytically active surfaces. Transactions of the Materials Research Society of Japan 2004, 29, 2717–2724. [Google Scholar] [CrossRef]
- Böttger, M.; Graumann, T.; Boughaled, R.; Neumann, F.; Kowalsky, W.; Johannes, H.H. Development of a new qualification method for photocatalytically active surfaces based on a solid state luminescent dye. J. Photochem. Photobiol. A: Chem. 2013, 253, 7–15. [Google Scholar] [CrossRef]
- Schlettwein, D. Light-induced charge transfer using phthalocyanines in active interfaces: Photoredox interaction or semiconductor junction? J.f Porphyrins Phthalocyanines 2008, 12, 337. [Google Scholar]
- Irie, H.; Hashimoto, K. Photocatalytic active surfaces and photo-induced high hydrophilicity/high hydrophobicity. In Environmental Photochemistry, Part II; Boule, P., Bahnemann, D. W., Robertson, P. K. J., Eds.; Springer: Berlin, Heidelberg, 2005; pp. 425–450. [Google Scholar] [CrossRef]
- Prins, R.; Schildenberger, M.; Bonetti, Y.C.; Gobrecht, J. Nanotechnology and model catalysis: The use of photolithography for creating active surfaces. CHIMIA International Journal for Chemistry 2000, 54, 63–65. [Google Scholar] [CrossRef]
- Kurz, J.; Eberle, F.; Graumann, T.; Kaschel, M.-E.; Sähr, A.; Neumann, F.; Dalpke, A.H.; Erdinger, L. Inactivation of LPS and RNase A on photocatalytically active surfaces. Chemosphere 2011, 84, 1188–1193, [https://pubmed.ncbi.nlm.nih.gov/21762949/]. [Google Scholar] [CrossRef]
- Hamedani Golshan, N. Understanding electrically active interface formation on wide bandgap semiconductors through molecular beam epitaxy using Fe₃O₄ for spintronics as a base case. PhD Thsesis, Northeastern University, Boston, 2017.
- Dimoulas, A. Electrically active interface and bulk semiconductor defects in high-k/Germanium structures. In Defects in High-k Gate Dielectric Stacks; Gusev, E., Ed.; Springer: Dordrecht, 2006; pp. 237-248. [Google Scholar] [CrossRef]
- Raynaud, C.; Autran, J.L.; Balland, B.; Guillot, G.; Jaussaud, C.; Billon, T. Electrical characterization of instabilities in 6H silicon carbide metal-oxide-semiconductor capacitors. Journal of Applied Physics 1994, 76, 993–997. [Google Scholar] [CrossRef]
- Gomes, H.L.; Stallinga, P.; Cölle, M.; De Leeuw, D.M.; Biscarini, F. Electrical instabilities in organic semiconductors caused by trapped supercooled water. Applied Physics Letters 2006, 88, 082101. [Google Scholar] [CrossRef]
- Di Pietro, R.; Sirringhaus, H. High resolution optical spectroscopy of air-induced electrical instabilities in n-type polymer semiconductors. Advanced Materials 2012, 24, 3367–3372, https://pubmed.ncbi.nlm.nih.gov/22605674/. [Google Scholar] [CrossRef]
- Jones, B.L.; Beaudet, P.R. Negative photoconductivity and electrical instabilities in semiconductors. Canadian Journal of Physics 1967, 45, 4091–4101. [Google Scholar] [CrossRef]
- Hajto, J.P. Optical and electrical instabilities in amorphous semiconductors. PhD Thesis, University of Edinburgh, Edinburgh, 1993. [Google Scholar]
- Hurley, P.K.; Cherkaoui, K.; Groenland, A. Electrically active interface defects in the (100) Si/SiOx/HfO2/TiN system: Origin, instabilities and passivation. ECS Transactions 2006, 3, 97. [Google Scholar] [CrossRef]
- Djara, V.; O’Regan, T.P.; Cherkaoui, K.; Schmidt, M.; Monaghan, S.; O’Connor, E.; Povey, I.M.; O’Connell, D.; Pemble, M.E.; Hurley, P.K. Electrically active interface defects in the In0.53Ga0.47As MOS system. Microelectronic Engineering 2013, 109, 182–188. [Google Scholar] [CrossRef]
- Rubio-Gimenez, V.; Tatay, S.; Volatron, F.; Martinez-Casado, F.J.; Martí-Gastaldo, C.; Coronado, E. High-quality metal–organic framework ultrathin films for electronically active interfaces. Journal of the American Chemical Society, 2016, 138, 2576–2584, [https://pubmed.ncbi.nlm.nih.gov/26847507/]. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Y.S.; Mulfort, K.L. Redox-active MOF with bio-mimetic cobalt nodes: Toward artificial photosynthesis in framework architectures. In Abstracts of Papers of The American Chemical Society 2012, 244. [Google Scholar]
- Xu, J.; He, S.; Zhang, H.; Huang, J.; Lin, H.; Wang, X.; Long, J. Layered metal–organic framework/graphene nanoarchitectures for organic photosynthesis under visible light. J. Mater. Chem. A 2012, 3, 24261–24271. [Google Scholar] [CrossRef]
- Heidary, N.; Harris, T.G.; Ly, K.H.; Kornienko, N. Artificial photosynthesis with metal and covalent organic frameworks (MOFs and COFs): Challenges and prospects in fuel-forming electrocatalysis. Physiologia Plantarum 2019, 166, 460–471, [https://pubmed.ncbi.nlm.nih.gov/30706497/]. [Google Scholar] [CrossRef]
- Malyshev, V.V. Response of semiconducting metal oxides to water vapor as a result of water molecules chemical transformations on catalytically active surfaces. Russian Journal of Physical Chemistry A, Focus on Chemistry, 2008, 82, 2329–2339. [Google Scholar] [CrossRef]
- O’Mullane, A.P. Creating active interfaces as a strategy to improve electrochemical water splitting reactions. Journal of Physics: Energy 2020, 2, 041001. [Google Scholar] [CrossRef]
- Greuter, F. Electrically active interfaces in ZnO varistors. Solid State Ionics 1995, 75, 67–78. [Google Scholar] [CrossRef]
- Ling, Z.; Russell, J.D.; Leach, C. The effect of variations in sintering temperature on the structure of electrically active interfaces in zinc oxide varistors. Key Engineering Materials 1997, 132, 1305–1308. [Google Scholar] [CrossRef]
- Leach, C.; Ling, Z.; Freer, R. The effect of sintering temperature variations on the development of electrically active interfaces in zinc oxide based varistors. Journal of the European Ceramic Society 2000, 20, 2759–2765. [Google Scholar] [CrossRef]
- Elfwing, M.; Olsson, E. Electron holography study of active interfaces in zinc oxide varistor materials. Journal of Applied Physics 2002, 92, 5272–5280. [Google Scholar] [CrossRef]
- Cho, K.G.; Kim, H.S.; Jang, S.S.; Kyung, H.; Kang, M.S.; Lee, K.H.; Yoo, W.C. Optimizing electrochemically active surfaces of carbonaceous electrodes for ionogel based supercapacitors. Advanced Functional Materials 2020, 30, 2002053. [Google Scholar] [CrossRef]
- Zhukov, V.; Winkler, A.; Rendulic, K. The energetics of coadsorbate interaction on catalytically active surfaces. In Proc. ÖPG 93, Österreichischen Physikalischen Gesellschaft, Graz, Austria (20 Sep 1993 – 24 Sep 1993).
- Roginskii, S.Z. Isotopic methods for studying the heterogeneity of active surfaces and interactions in the adsorption layer. Zhurnal Fizicheskoi Khimii 1958, 32, 737–745. [Google Scholar]
- Keier, N.P.; Roginskii, S.Z. Investigation of the nonhomogeneity of active surfaces by the differential isotopic method. I. Active surfaces of metallic nickel and of zinc oxide. Izvest. Akad. Nauk SSSR 1950, (1), 51. [Google Scholar]
- Charcosset, H.; Barthome. D.; Nicolova, R. ; Revillon, A. ; Tournaya. L.; Trambouz. Y. Method for determining active surfaces of some catalytic systems by chemisorption. Bulletin De La Societe Chimique De France 1967, 12, 4555. [Google Scholar]
- Flosdorf, E.W.; Kistiakowsky, G.B. Heats of adsorption on catalytically active surfaces. J. Phys. Chem. 2002, 34, 1907–1918. [Google Scholar] [CrossRef]
- Chapaeva, A.; Chokaev, K.K.; Loginov, A. The formation of the active surfaces of modified lanthanide catalysts. I, Paramagnetic centres of chromium-containing yttrium and scandium oxides. Russian Journal of Physical Chemistry 1990, 64, 1034–1036. [Google Scholar]
- Niklewsk, J.B.; Sis, L.; Wirtz, G. Characterization of catalitically active surfaces with scanning electron microscope. American Ceramic Society Bulletin 1971, 50, 381. [Google Scholar]
- Iwasawa, Y.; Shido, T.; Fukui, K. Molecular design and characterization of active surfaces for molecular-level understanding and development of catalysis. Abstracts of Papers of the American Chemical Society 2001, 221, U310. [Google Scholar]
- Tada, M.; Iwasawa, Y. Chemical design and in situ characterization of active surfaces for selective catalysis. Annu. Rev. Mater. Res. 2005, 35, 397–426. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Palivan, C.G.; Gademann, K.; Meier, W.; Calame, M.; Mikhalevich, V.; Zhang, X.; Piel, E.; Szponarski, M.; Wiesler, A.; Lanzilotto, A.; Constable, E.C.; Fanget, A.; Stoop, S.L. “Active surfaces” as possible functional systems in detection and chemical (bio) reactivity. CHIMIA International Journal for Chemistry 2016, 70, 402–412, [https://pubmed.ncbi.nlm.nih.gov/27363368/]. [Google Scholar] [CrossRef]
- Lewis, D.R. Electrical condition of catalytically active surfaces. Thesis, University of Manchester, Manchester, 1926.
- Bowden, F.P.; Rideal, E.K. On the electrolytic behaviour of thin films. Part II. The areas of catalytically active surfaces. Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character 1928, 120, 80–89. [Google Scholar] [CrossRef]
- Chen, M.; Wang, X.V.; Zhang, L.; Tang, Z.; Wan, H. Active surfaces for CO oxidation on palladium in the hyperactive state. Langmuir 2010, 26, 18113–18118, [https://pubmed.ncbi.nlm.nih.gov/21053982/]. [Google Scholar] [CrossRef]
- Chen, M.; Zheng, Y.; Wan, H. Kinetics and active surfaces for CO oxidation on Pt-group metals under oxygen rich conditions. Topics in Catalysis 2013, 56, 1299–1313. [Google Scholar] [CrossRef]
- Kondoh, H.; Toyoshima, R.; Monya, Y.; Yoshida, M.; Mase, K.; Amemiya, K.; Mun, B.S. In situ analysis of catalytically active Pd surfaces for CO oxidation with near ambient pressure XPS. Catalysis Today 2016, 260, 14–20. [Google Scholar] [CrossRef]
- Ertl, G.; Norton, P.R.; Rüstig, J. Kinetic oscillations in the platinum-catalyzed oxidation of CO. Physical Review Letters 1982, 49, 177. [Google Scholar] [CrossRef]
- Imbihl, R.; Cox, M.P.; Ertl, G.; Müller, H.; Brenig, W. Kinetic oscillations in the catalytic CO oxidation on Pt (100): Theory. J. Chem. Phys. 1985, 83, 1578–1587. [Google Scholar] [CrossRef]
- Eiswirth, M.; Ertl, G. Kinetic oscillations in the catalytic CO oxidation on a Pt (110) surface. Surface Science 1986, 177, 90–100. [Google Scholar] [CrossRef]
- Imbihl, R.; Cox, M.P.; Ertl, G. Kinetic oscillations in the catalytic CO oxidation on Pt (100): Experiments. J. Chem. Phys. 1986, 84, 3519–3534. [Google Scholar] [CrossRef]
- Lyons, M.E. The mechanism of mediated electron transfer at redox active surfaces. Electroanalysis 2015, 27, 992–1009. [Google Scholar] [CrossRef]
- Lan, Y.M.; Cheng, K.J.; Luk, Y.Y. Development of redox-active surfaces and micelles for biocompatible systems: Caging ferrocene in cyclic oligosaccharides. In Abstracts of Papers of the American Chemical Society 2005, 230, U1110–U1111. [Google Scholar]
- Yzambart, G.; Fabre, B.; Camerel, F.; Roisnel, T.; Lorcy, D. Controlled grafting of tetrathiafulvalene (TTF) containing diacetylenic units on hydrogen-terminated silicon surfaces: from redox-active TTF monolayer to polymer films. J. Phys. Chem. C 2012, 116, 12093–12102. [Google Scholar] [CrossRef]
- Joy, S.; Pal, P.; Mondal, T.K.; Talapatra, G.B.; Goswami, S. Synthesis of amphiphilic azo-anion-radical complexes of chromium(III) and the development of ultrathin redox-active surfaces by the Langmuir–Schaefer technique. Chemistry–A European Journal 2012, 18, 1761–1771, [https://pubmed.ncbi.nlm.nih.gov/22237915/]. [Google Scholar] [CrossRef] [PubMed]
- Fellermann, H. Micelles as containers for protocells. Beiträge des Instituts für Umweltsystemforschung der Universität Osnabrück, 2005; 33, 1-83. [Google Scholar]
- Chang, G.G.; Huang, T.M.; Hung, H.C. Reverse micelles as life-mimicking systems. Proceedings of the National Science Council, Republic of China. Part B, Life Sciences 2000, 24, 89–100. [Google Scholar]
- Ciucci, F.; Chueh, W.C.; Goodwin, D.G.; Haile, S.M. Surface reaction and transport in mixed conductors with electrochemically-active surfaces: a 2D numerical study of ceria. Physical Chemistry Chemical Physics 2011, 13, 2121–2135. [Google Scholar] [CrossRef]
- Suwono, A.; Daguenet, M.; Bodiot, D. Theoretical study of the diffusion or conduction fluxes on a finite number of active surfaces in interaction, one with another, separated by inert zones, in a Newtonian or non-Newtonian viscous fluid in laminar or turbulent flow. International Journal of Heat and Mass Transfer 1976, 19, 239–244. [Google Scholar] [CrossRef]
- Beringuier, H.; Suwono, A.; Delmas, A.; Daguenet, M.; Spinner, B.; Bodiot, D. Experimental study of diffusion flows on a finite number of interacting active surfaces divided each other by inert zones into a newtonian or not viscous-fluid in a laminar or turbulent-flow. Journal de Chimie Physique et de Physico-Chimie Biologique 1976, 73, 868–871. [Google Scholar] [CrossRef]
- Suwono, A.; Daguenet, M. Theoretical study of diffusion or conduction interaction between active surfaces in laminar-flow of a fluid with small schmidt or prandtl numbers. Journal de Chimie Physique et de Physico-Chimie Biologique 1977, 74, 681–684. [Google Scholar] [CrossRef]
- Vurdelja, A. A study on the selective transport in the emulsions containing droplets with active surfaces [Badania transportu selektywnego w środowisku emulsji z czynnymi powierzchniami kropel]. PhD Thesis, Warsaw University of Technology, Faculty of Chemical and Process Engineering, Department of Process Kinetics and Thermodynamics, Warsaw, 2017. [Google Scholar]
- Amelin, A.G.; Kabanov, A.N.; Shchukin, E.R.; Shulimanova, Z.L. Features of the motion of aerosol particles near catalytically active surfaces. Kinet. Catal. 1985, 26, 93–101. [Google Scholar]
- Parisi, J.; Peinke, J.; Röhricht, B.; Rau, U.; Klein, M.; Rössler, O.E. Comparison between a generic reaction-diffusion model and a synergetic semiconductor system. Zeitschrift für Naturforschung A 1987, 42, 655–656. [Google Scholar] [CrossRef]
- Merz, W. Strong solutions for reaction-drift-diffusion problems in semiconductor technology. Journal of Applied Mathematics and Mechanics 2001, 81, 623–635. [Google Scholar] [CrossRef]
- Justin, M.; Betchewe, G.; Doka, S.Y.; Crepin, K.T. Exact solutions of a semiconductor nonlinear reaction diffusion equation through factorization method. Applied Mathematics and Computation 2012, 219, 2917–2922. [Google Scholar] [CrossRef]
- Justin, M.; Marcel, G.; Betchewe, G.; Doka, S.Y.; Crepin, K.T. New exact solutions for a semiconductor nonlinear reaction-diffusion equation: the combination of the factorization method to the projective Riccati equation method. Electronic Journal of Mathematical Analysis and Applications 2017, 5, 271–288. [Google Scholar]
- Gardner, J.W. A non-linear diffusion-reaction model of electrical conduction in semiconductor gas sensors. Sensors and Actuators B: Chemical 1990, 1, 166–170. [Google Scholar] [CrossRef]
- Lavine, I.S.; Levinson, J.A.; Glogovsky, K.G. Modeling and simulation of hydrogen diffusion and reaction in semiconductor photonic materials. In Abstracts of Papers of the American Chemical Society 2014, 247. [Google Scholar]
- Peirce, A.P. Mathematical analysis of chemical systems: The effect of defect structures on chemically active surfaces; Optimal control of quantum molecular systems. PhD Thesis, Princeton University, Princeton, New Jersey, 1987. [Google Scholar]
- Glitzky, A. An electronic model for solar cells including active interfaces and energy resolved defect densities. SIAM Journal on Mathematical Analysis 2012, 44, 3874–3900. [Google Scholar] [CrossRef]
- Masuduzzaman, M.; Weir, B.; Alam, M.A. Probing bulk defect energy bands using generalized charge pumping method. Journal of Applied Physics 2012, 111, 074501. [Google Scholar] [CrossRef]
- Kumar, N.T.; Pinto, M.A.D.C.; Shmavonyan, G. Reaction–diffusion cellular automata framework-based understanding of radiation-induced effects from alpha-particles on the performances of microprocessors/FPGAs/other electronic devices using higher order logic (HOL) System and CAVA library in the R&D of semiconductor industry. International Journal of Applied Research on Information Technology and Computing 2018, 9, 39–49. [Google Scholar] [CrossRef]
- Mietke, A.; Jülicher, F.; Sbalzarini, I.F. Self-organized shape dynamics of active surfaces. Proceedings of the National Academy of Sciences 2019, 116, 29–34, [https://pubmed.ncbi.nlm.nih.gov/30567977/]. [Google Scholar] [CrossRef]
- Alonso, S.; Chen, H.Y.; Bär, M.; Mikhailov, A.S. Self-organization processes at active interfaces. European Physical Journal Special Topics 2010, 191, 131–145. [Google Scholar] [CrossRef]
- Cagnetta, F.; Evans, M.R.; Marenduzzo, D. Kinetic roughening in active interfaces. EPJ Web of Conferences 2020, 230, 00001. [Google Scholar] [CrossRef]
- Rubino, M. Developing active surfaces through the implementation of nanotechnology. Abstracts of Papers of the American Chemical Society 2017, 254. [Google Scholar]
- Neretina, S.; Hughes, R. Nanostructure synthesis at the liquid-substrate interface: A new strategy for obtaining plasmonic and chemically active surfaces. Abstracts of Papers of the American Chemical Society 2017, 254. [Google Scholar]
- Hoffmann, L.; Giomi, L. Active surfaces and defect-mediated morphogenesis. Bulletin of the American Physical Society 2021, 66, C05.00001. [Google Scholar]
- Buten, C.; Kortekaas, L.; Ravoo, B.J. Design of active interfaces using responsive molecular components. Advanced Materials 2020, 32, 1904957, [https://pubmed.ncbi.nlm.nih.gov/31573115/]. [Google Scholar] [CrossRef]
- Rossiter, J.; Yap, B.; Conn, A. Biomimetic chromatophores for camouflage and soft active surfaces. Bioinspiration & Biomimetics 2012, 7, 036009, [https://pubmed.ncbi.nlm.nih.gov/22549047/]. [Google Scholar] [CrossRef]
- Krysiński, P.; Blanchard, G.J. Synthesis and characterization of amphiphilic biomimetic assemblies at electrochemically active surfaces. Langmuir 2003, 19, 3875–3882. [Google Scholar] [CrossRef]
- Garni, M.; Wehr, R.; Avsar, S.Y.; John, C.; Palivan, C.; Meier, W. Polymer membranes as templates for bio-applications ranging from artificial cells to active surfaces. European Polymer Journal 2019, 112, 346–364. [Google Scholar] [CrossRef]
- Komissarov, G.G. A new concept of photosynthesis. In: Process Advancement in Chemistry and Chemical Engineering Research. CRC, Waretown, 2016. p. 303-327.
- Komissarov, G.G.; Lobanov, A.V. Photoinduced processes of hydrogen peroxide formation and decomposition and their role in photosynthesis and biosphere origin. Geochemistry International 2014, 52, 1239–1251. [Google Scholar] [CrossRef]
- Lobanov, A.V.; Komissarov, G.G. Hydrogen peroxide in artificial photosynthesizing systems. Biophysics 2014, 59, 169–182. [Google Scholar] [CrossRef]
- Komissarov, G.G.; Lobanov, A.V.; Nevrova, O.V.; Popov, I.A.; Kononikhin, A.S.; Pekov, S.I.; Nikolaev, E.N. New step towards artificial photosynthesis: photogeneration of organic compounds in the inorganic carbon-hydrogen peroxide-phthalocyanine system. Doklady Physical Chemistry 2013, 453, 275–278. [Google Scholar] [CrossRef]
- Sammaknejad, N.; Zhao, Y.; Huang, B. A review of the expectation maximization algorithm in data-driven process identification. Journal of Process Control 2019, 73, 123–136. [Google Scholar] [CrossRef]
- Czop, P.; Kost, G.; Sławik, D.; Wszołek, G. Formulation and identification of first-principle data-driven models. Journal of Achievements in Materials and Manufacturing Engineering 2011, 44, 179–186. [Google Scholar]
- Chang, H.; Zhang, D. Identification of physical processes via combined data-driven and data-assimilation methods. Journal of Computational Physics 2019, 393, 337–350. [Google Scholar] [CrossRef]
- Brewick, P.T.; Masri, S.F. An evaluation of data-driven identification strategies for complex nonlinear dynamic systems. Nonlinear Dynamics 2016, 85, 1297–1318. [Google Scholar] [CrossRef]
- Meidani, K.; Farimani, A.B. Data-driven identification of 2D partial differential equations using extracted physical features. Computer Methods in Applied Mechanics and Engineering 2021, 381, 113831. [Google Scholar] [CrossRef]
- Rudy, S.; Alla, A.; Brunton, S.L.; Kutz, J.N. Data-driven identification of parametric partial differential equations. SIAM Journal on Applied Dynamical Systems 2019, 18, 643–660. [Google Scholar] [CrossRef]
- Laisk, A.; Eichelmann, H. Towards understanding oscillations: A mathematical model of the biochemistry of photosynthesis. Philosophical Transactions of the Royal Society of London. B, Biological Sciences 1989, 323, 369–384. [Google Scholar] [CrossRef]
- Nedbal, L.; Červený, J.; Rascher, U.; Schmidt, H. E-photosynthesis: A comprehensive modeling approach to understand chlorophyll fluorescence transients and other complex dynamic features of photosynthesis in fluctuating light. Photosynthesis Research 2007, 93, 223–234, [https://pubmed.ncbi.nlm.nih.gov/17492490/]. [Google Scholar] [CrossRef]
- Gebhardt, R.S.; Du, P.; Wodo, O.; Ganapathysubramanian, B. A data-driven identification of morphological features influencing the fill factor and efficiency of organic photovoltaic devices. Computational Materials Science 2017, 129, 220–225. [Google Scholar] [CrossRef]
- Ritzberger, D.; Jakubek, S. Nonlinear data-driven identification of polymer electrolyte membrane fuel cells for diagnostic purposes: A Volterra series approach. Journal of Power Sources 2017, 361, 144–152. [Google Scholar] [CrossRef]
- Cope, F.W. Cooperative interactions in nerve membrane potential and in photosynthesis, evidenced by non-linear Arrhenius plots and critical exponents. Physiol. Chern. Phys. 1977, 9, 247-258. [https://pubmed.ncbi.nlm.nih.gov/594192/].
- Cope, F.W. Sigmoid biological time curves for muscle, nerve, growth, firefly, and infrared phosphorescence of green leaves, melanin, and cytochrome c. Physiol. Chern. Phys. 1977, 9, 443–459. [Google Scholar]
- Cope, F.W. Critical exponent analysis of activation energies of nonlinear Arrhenius plots as a test for cooperative interactions in amorphous semiconductors and in biological systems. Physiol. Chern. Phys. 1977, 9, 329–335. [Google Scholar]
- Cagnetta, F. Active interfaces, a universal approach. PhD Thesis, The University of Edinburgh, Edinburgh, 2020. [Google Scholar]
- Hermann, S.; Schmidt, M. Active interface polarization as a state function. Physical Review Research 2020, 2, 022003. [Google Scholar] [CrossRef]
- Hannezo, E. A toy model for active interfaces. Physics 2018, 11, 61. [Google Scholar] [CrossRef]
- Al Hammal, O.; De Los Santos, F.; Munoz, M.A. A non-order parameter Langevin equation for a bounded Kardar–Parisi–Zhang universality class. Journal of Statistical Mechanics: Theory and Experiment, 2005; 10, P10013. [Google Scholar] [CrossRef]
- Sasamoto, T.; Spohn, H. One-dimensional Kardar-Parisi-Zhang equation: an exact solution and its universality. Phys. Rev. Lett. 2010, 104, 230602, [https://pubmed.ncbi.nlm.nih.gov/20867222/]. [Google Scholar] [CrossRef]
- Corwin, I. The Kardar–Parisi–Zhang equation and universality class. Random Matrices: Theory and Applications 2012, 1, 1130001. [Google Scholar] [CrossRef]
- Sasamoto, T. The 1D Kardar–Parisi–Zhang equation: height distribution and universality. Progress of Theoretical and Experimental Physics 2016, 2, 022A01. [Google Scholar] [CrossRef]
- Mukherjee, S. Conserved Kardar-Parisi-Zhang equation: Role of quenched disorder in determining universality. Phy.l Rev. E 2021, 103, 042102. [Google Scholar] [CrossRef] [PubMed]
- Nattermann, T.; Tang, L.H. Kinetic surface roughening. I. The Kardar-Parisi-Zhang equation in the weak-coupling regime. Phys. Rev. A 1992, 45, 7156. [Google Scholar] [CrossRef] [PubMed]
- Fogedby, H.C. Localized growth modes, dynamic textures, and upper critical dimension for the Kardar-Parisi-Zhang Equation in the weak-noise limit. Phys. Rev. Lett. 2005, 94, 195702. [Google Scholar] [CrossRef] [PubMed]
- Szabó, G.; Alava, M.; Kertész, J. Self-organized criticality in the Kardar-Parisi-Zhang equation. Europhysics Letters (EPL) 2002, 57, 665–671. [Google Scholar] [CrossRef]
- Fogedby, H.C. Patterns in the Kardar-Parisi-Zhang equation. Pramana 2008, 71, 253–262. [Google Scholar] [CrossRef]
- Katzav, E. Growing surfaces with anomalous diffusion: Results for the fractal Kardar-Parisi-Zhang equation. Phys. Rev. E 2003, 68, 031607. [Google Scholar] [CrossRef]
- Le Doussal, P.; Thiery, T. Diffusion in time-dependent random media and the Kardar-Parisi-Zhang equation. Phys. Rev. E 2017, 96, 010102. [Google Scholar] [CrossRef]
- Antonov, N.V.; Gulitskiy, N.M.; Kakin, P.I.; Kostenko, M.M. Effects of turbulent environment on the surface roughening: The Kardar-Parisi-Zhang model coupled to the stochastic Navier–Stokes equation. Physica Scripta 2020, 95, 084009. [Google Scholar] [CrossRef]
- Sayfidinov, O.; Bognár, G.V. Numerical solutions of the Kardar-Parisi-Zhang interface growing equation with different noise terms. In Vehicle and Automotive Engineering 2020, 3, 302–311. [Google Scholar] [CrossRef]
- Kechagia, P.; Yortsos, Y.C.; Lichtner, P. Nonlocal Kardar-Parisi-Zhang equation to model interface growth. Phys. Rev. E 2001, 64, 016315, [https://pubmed.ncbi.nlm.nih.gov/11461399/]. [Google Scholar] [CrossRef]
- Hartmann, A.K.; Krajenbrink, A.; Le Doussal, P. Probing large deviations of the Kardar-Parisi-Zhang equation at short times with an importance sampling of directed polymers in random media. Phys. Rev. E 2020, 101, 012134. [Google Scholar] [CrossRef]
- Santalla, S.N.; Rodríguez-Laguna, J.; Cuerno, R. Circular Kardar-Parisi-Zhang equation as an inflating, self-avoiding ring polymer. Phys. Re. E 2014, 89, 010401, [https://pubmed.ncbi.nlm.nih.gov/24580156/]. [Google Scholar] [CrossRef]
- Balibar, S.; Bouchaud, J.P. Kardar-Parisi-Zhang equation and the dynamic roughening of crystal surfaces. Phys. Rev. Lett. 1992, 69, 862. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kahng, B. Exact derivation of the Kardar-Parisi-Zhang equation for the restricted solid-on-solid model. Phys. Rev. E 1995, 51, 796–798, [https://pubmed.ncbi.nlm.nih.gov/9962709/]. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.A.; Sano, M. Evidence for geometry-dependent universal fluctuations of the Kardar-Parisi-Zhang interfaces in liquid-crystal turbulence. J. Stat. Phys. 2012, 147, 853–890. [Google Scholar] [CrossRef]
- Golubović, L.; Wang, Z.G. Kardar-Parisi-Zhang model and anomalous elasticity of two-and three-dimensional smectic-A liquid crystals. Phys. Rev. 1994, 49, 2567–2578, [https://pubmed.ncbi.nlm.nih.gov/9961517/]. [Google Scholar] [CrossRef]
- Golubović, L.; Wang, Z.G. Erratum: Kardar-Parisi-Zhang model and anomalous elasticity of two-and three-dimensional smectic-A liquid crystals [Phys. Rev. E 49, 2567 (1994)]. Phys. Rev. E 1994, 50, 4265, [https://pubmed.ncbi.nlm.nih.gov/9962490/]. [Google Scholar] [CrossRef]
- Schilardi, P.L.; Azzaroni, O.; Salvarezza, R.C.; Arvia, A.J. Validity of the Kardar-Parisi-Zhang equation in the asymptotic limit of metal electrodeposition. Physical Review-Section B-Condensed Matter 1999, 59, 4638–4641. [Google Scholar] [CrossRef]
- Lütt, M.; Schlomka, J.P.; Tolan, M.; Stettner, J.; Seeck, O.H.; Press, W. Kardar-Parisi-Zhang growth of amorphous silicon on Si/SiO2. Phys. Rev. B 1997, 56, 4085–4091. [Google Scholar] [CrossRef]
- Barna, I.F.; Bognár, G.; Mátyás, L.; Guedda, M.; Hriczó, K. Travelling-wave solutions of the Kardar-Parisi-Zhang interface growing equation with different kind of noise terms. In AIP Conference Proceedings 2020, 2293, 280005. [Google Scholar] [CrossRef]
- Lauter, R.; Mitra, A.; Marquardt, F. From Kardar-Parisi-Zhang scaling to explosive desynchronization in arrays of limit-cycle oscillators. Phys. Rev. E 2017, 96, 012220. [Google Scholar] [CrossRef] [PubMed]
- Lassig, M.; Kinzelbach, H. Phase transition of the Kardar-Parisi-Zhang equation in four substrate dimensions-Reply. Phys. Rev. Lett. 1998, 80, 889–889. [Google Scholar]
- Rieß, W. In situ measurements of respiration and mineralisation processes. Interaction between fauna and geochemical fluxes at active interfaces. PhD Thesis, University of Bremen, Bremen, Germany, 1999. [Google Scholar]
- Melkikh, A.V.; Sutormina, M. Protocells and LUCA: Transport of substances from first physicochemical principles. Progress in Biophysics and Molecular Biology 2019, 145, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Gradov, O.V.; Gradova, M.A. "MS-patch-clamp" or the possibility of mass spectrometry hybridization with patch-clamp setups for single cell metabolomics and channelomics. Advances in Biochemistry 2015, 3, 66–71. [Google Scholar] [CrossRef]
- Zhang, L.; Vertes, A. Einzelzell-massenspektrometrie zur untersuchung zellulärer heterogenität. Angewandte Chemie, 2018, 130, 4554–4566. [Google Scholar] [CrossRef]
- Zhang, L.; Vertes, A. Single-cell mass spectrometry approaches to explore cellular heterogeneity. Angewandte Chemie International Edition 2018, 57, 4466–4477, [https://pubmed.ncbi.nlm.nih.gov/29218763/]. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.B.; Freund, M.S.; Hammond, P.T. Catalytic, conductive bipolar membrane interfaces through layer-by-layer deposition for the design of membrane-integrated artificial photosynthesis systems. Chem. Sus. Chem. 2017, 10, 4599–4609. [Google Scholar] [CrossRef]
- Hartman, H. Speculations on the origin and evolution of photosynthesis and the membrane. Origins of Life 1986, 16, 384. [Google Scholar] [CrossRef]
- Nakamura, H. Origin of the proto-cell membrane - great importance of phospholipid bilayer. In Exobiology: Matter, Energy, and Information in the Origin and Evolution of Life in the Universe (Proceedings of the Fifth Trieste Conference on Chemical Evolution: An Abdus Salam Memorial Trieste, Italy, 22–26 September 1997); Chela-Flores, J., Raulin, F., Eds.; Springer, Dordrecht, 1998; pp. 191-194. [CrossRef]
- Kundu, N.; Mondal, D.; Sarkar, N. Dynamics of the vesicles composed of fatty acids and other amphiphile mixtures: unveiling the role of fatty acids as a model protocell membrane. Biophysical Reviews 2020, 12, 1117–1131, https://pubmed.ncbi.nlm.nih.gov/32926295/. [Google Scholar] [CrossRef]
Figure 1.
Potential profile at the interface and across the bilayer of a positively charged surfactant vesicle used for artificial photosynthesis. "Surface potential, charge separation potential, diffusion potential and Donnan potential are exploited for enhanced energy and electron transfer on charged vesicle surfaces, for the utilization of field effects for charge separation, for partitioning between the inner and outer compartments of radicals expelled from vesicle bilayers and for facilitating electron transfer across bilayers" [64]. Reproduced with permission from [64] published by the American Chemical Society.
Figure 1.
Potential profile at the interface and across the bilayer of a positively charged surfactant vesicle used for artificial photosynthesis. "Surface potential, charge separation potential, diffusion potential and Donnan potential are exploited for enhanced energy and electron transfer on charged vesicle surfaces, for the utilization of field effects for charge separation, for partitioning between the inner and outer compartments of radicals expelled from vesicle bilayers and for facilitating electron transfer across bilayers" [64]. Reproduced with permission from [64] published by the American Chemical Society.
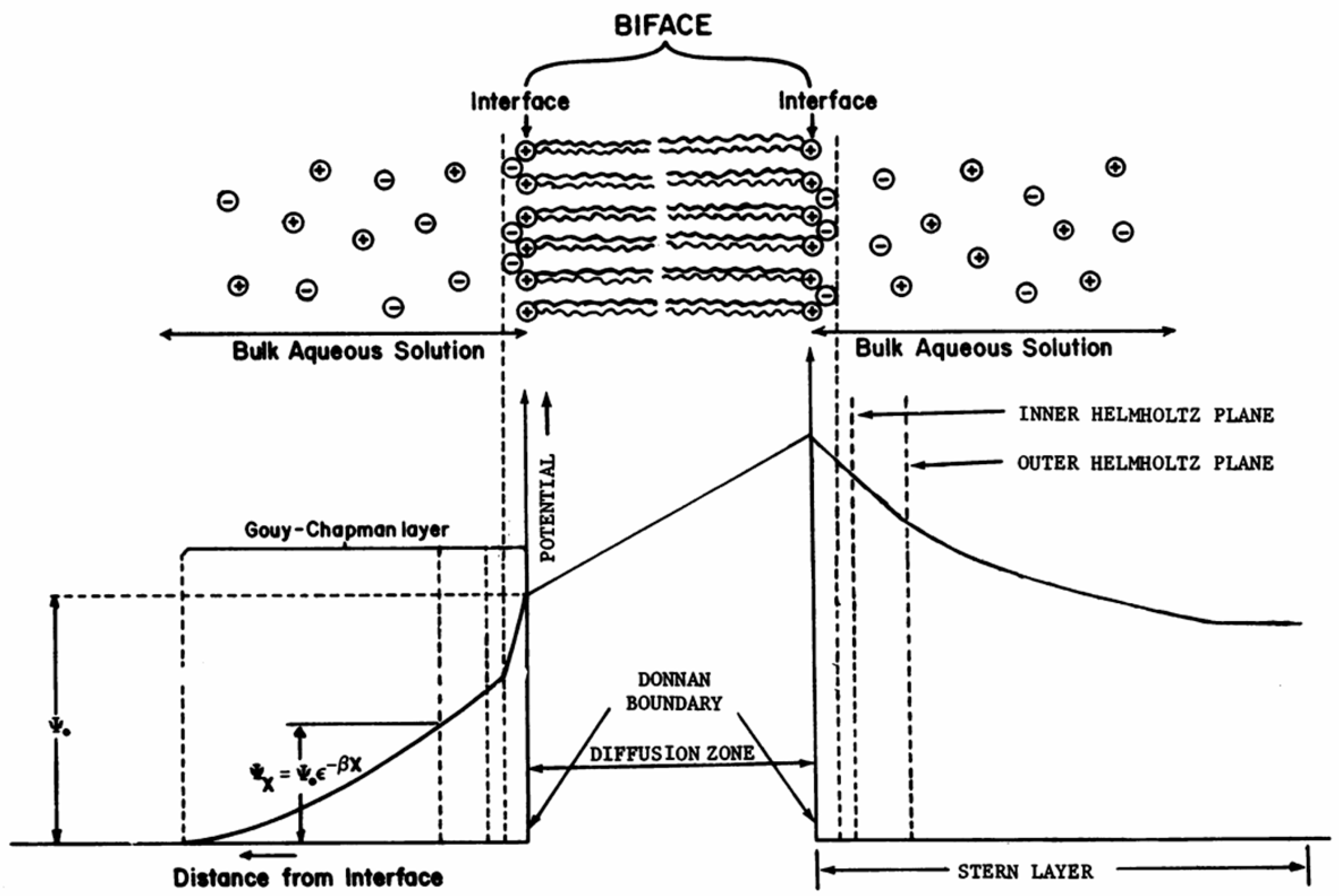
Figure 2.
"A model of a dividing protocell, with an integrated metabolic, genetic, and container system... In the Chen-Rasmussen protocell, light-driven metabolic processes synthesize lipids and PNA, with the PNA acting as both an information molecule and as an electron-relay chain" [70]. "A proto-metabolic process (redox- or photo-driven) is implemented, which enables the structure to grow and reproduce: As more lipid molecules are produced, the aggregate grows and eventually divides, which constitutes self-reproduction... Photo-driven metabolism requires a particular electron relay chain to function properly, and this electron relay is implemented within the templating polymers" [71]. Reproduced with permission from [70] published by the American Association for the Advancement of Science.
Figure 2.
"A model of a dividing protocell, with an integrated metabolic, genetic, and container system... In the Chen-Rasmussen protocell, light-driven metabolic processes synthesize lipids and PNA, with the PNA acting as both an information molecule and as an electron-relay chain" [70]. "A proto-metabolic process (redox- or photo-driven) is implemented, which enables the structure to grow and reproduce: As more lipid molecules are produced, the aggregate grows and eventually divides, which constitutes self-reproduction... Photo-driven metabolism requires a particular electron relay chain to function properly, and this electron relay is implemented within the templating polymers" [71]. Reproduced with permission from [70] published by the American Association for the Advancement of Science.
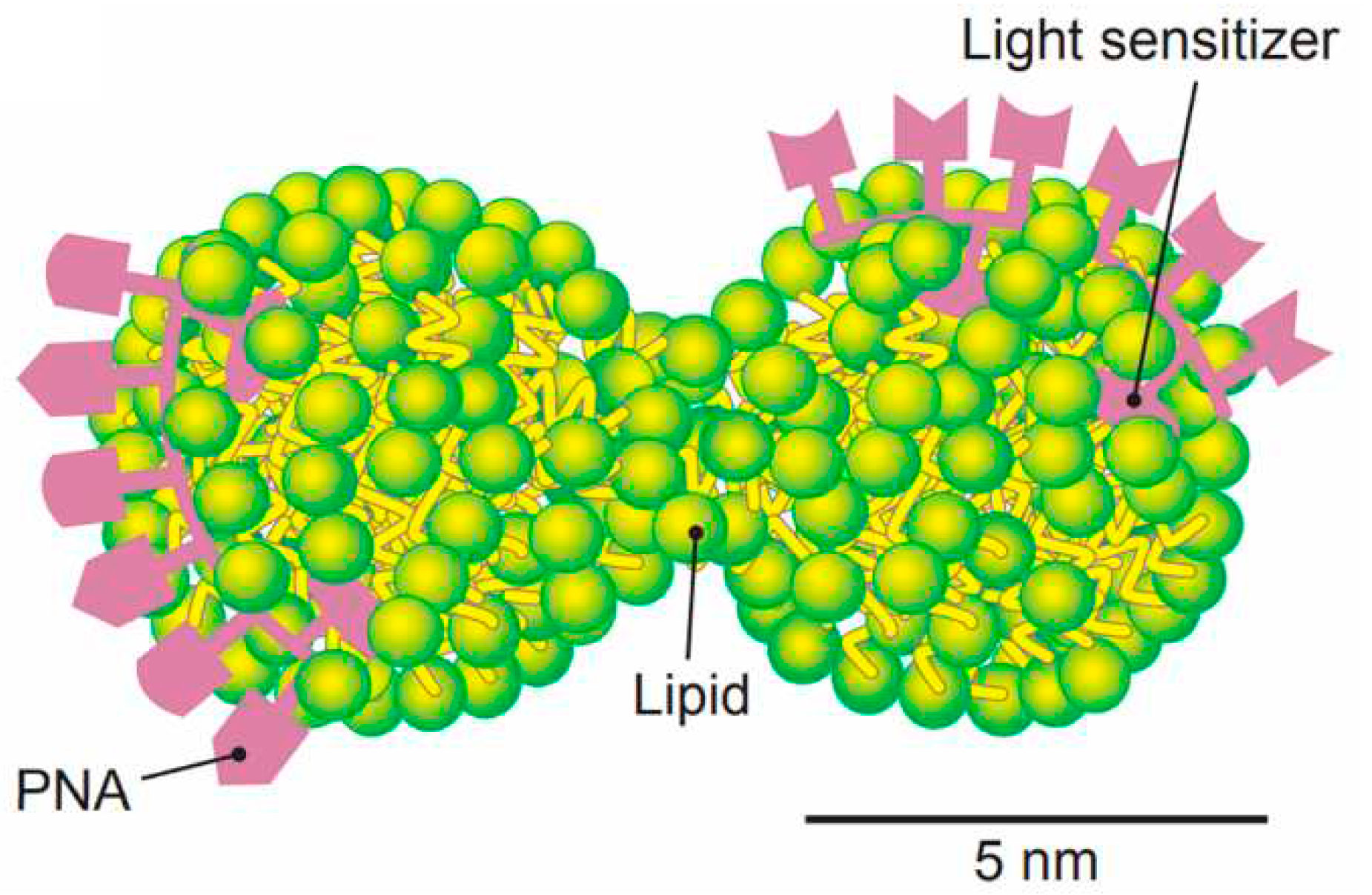
Figure 3.
Causal structure of the full proto-organism model: "three aggregates with associated production cycles represented by light dotted circle arrows: container reproduction (I), metabolic cycle (II), and gene replication (III). The light solid arrows indicate production, and the heavy dashed arrows indicate catalysis. The (gray) precursor molecules constitute the food resources, and the whole system is fueled either by inorganic chemical or light energy" [71]. Reproduced with permission from [71] published by the MIT press.
Figure 3.
Causal structure of the full proto-organism model: "three aggregates with associated production cycles represented by light dotted circle arrows: container reproduction (I), metabolic cycle (II), and gene replication (III). The light solid arrows indicate production, and the heavy dashed arrows indicate catalysis. The (gray) precursor molecules constitute the food resources, and the whole system is fueled either by inorganic chemical or light energy" [71]. Reproduced with permission from [71] published by the MIT press.
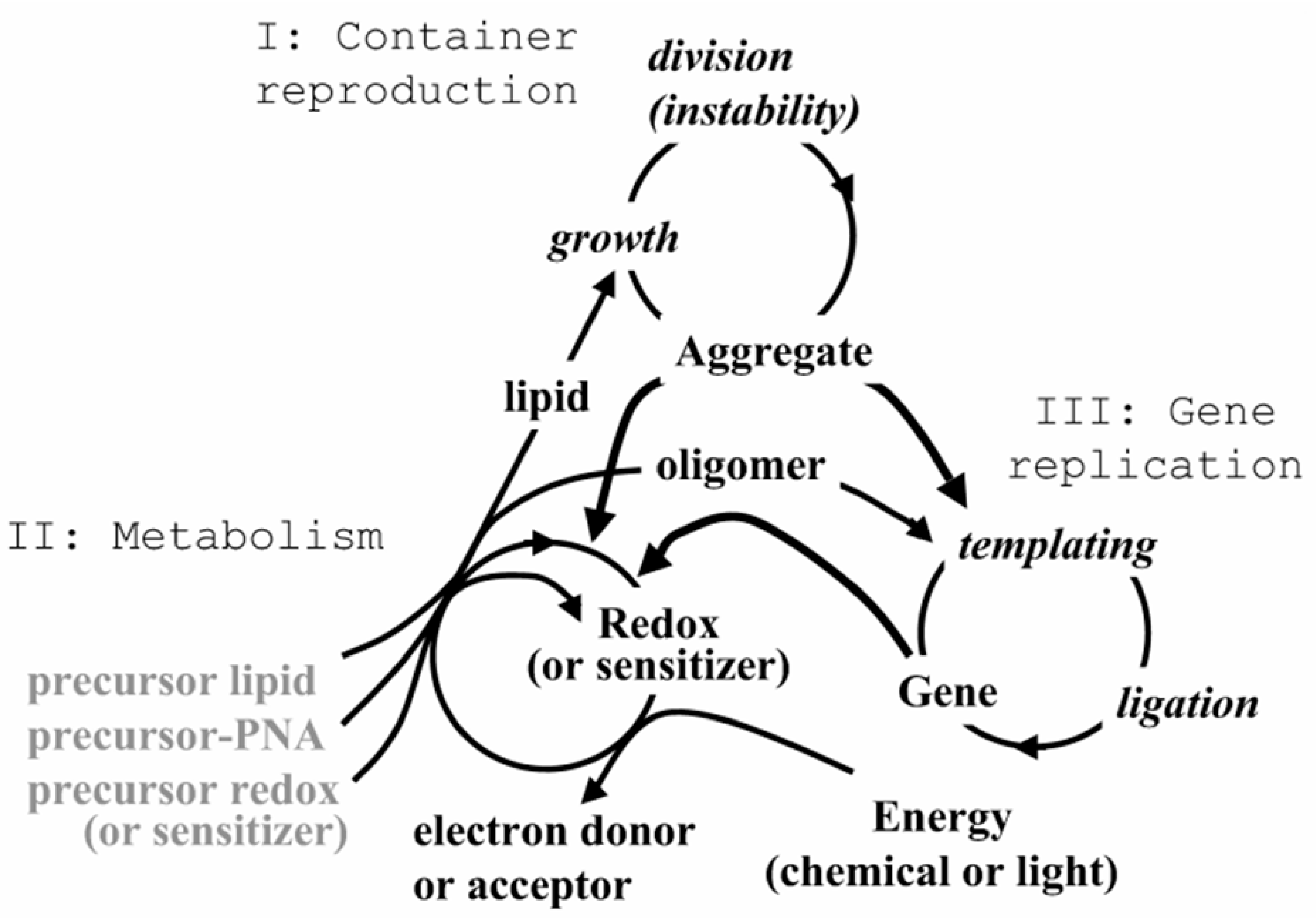
Figure 4.
Na+- and H+-motive respiratory chain phosphorylations (a "minimal" scheme). (1) Na+-motive NADH-quinone reductase. (2) H+-motive redox reaction(s) in the middle and/or terminal segments of the respiratory chain. (3) Na+ (H+)-driven ATP-synthase which translocates Na+ or H+ at high or low [Na+]0/[H+]0 ratios, respectively [88]. Reproduced with permission from [88] published by Springer Nature BV.
Figure 4.
Na+- and H+-motive respiratory chain phosphorylations (a "minimal" scheme). (1) Na+-motive NADH-quinone reductase. (2) H+-motive redox reaction(s) in the middle and/or terminal segments of the respiratory chain. (3) Na+ (H+)-driven ATP-synthase which translocates Na+ or H+ at high or low [Na+]0/[H+]0 ratios, respectively [88]. Reproduced with permission from [88] published by Springer Nature BV.
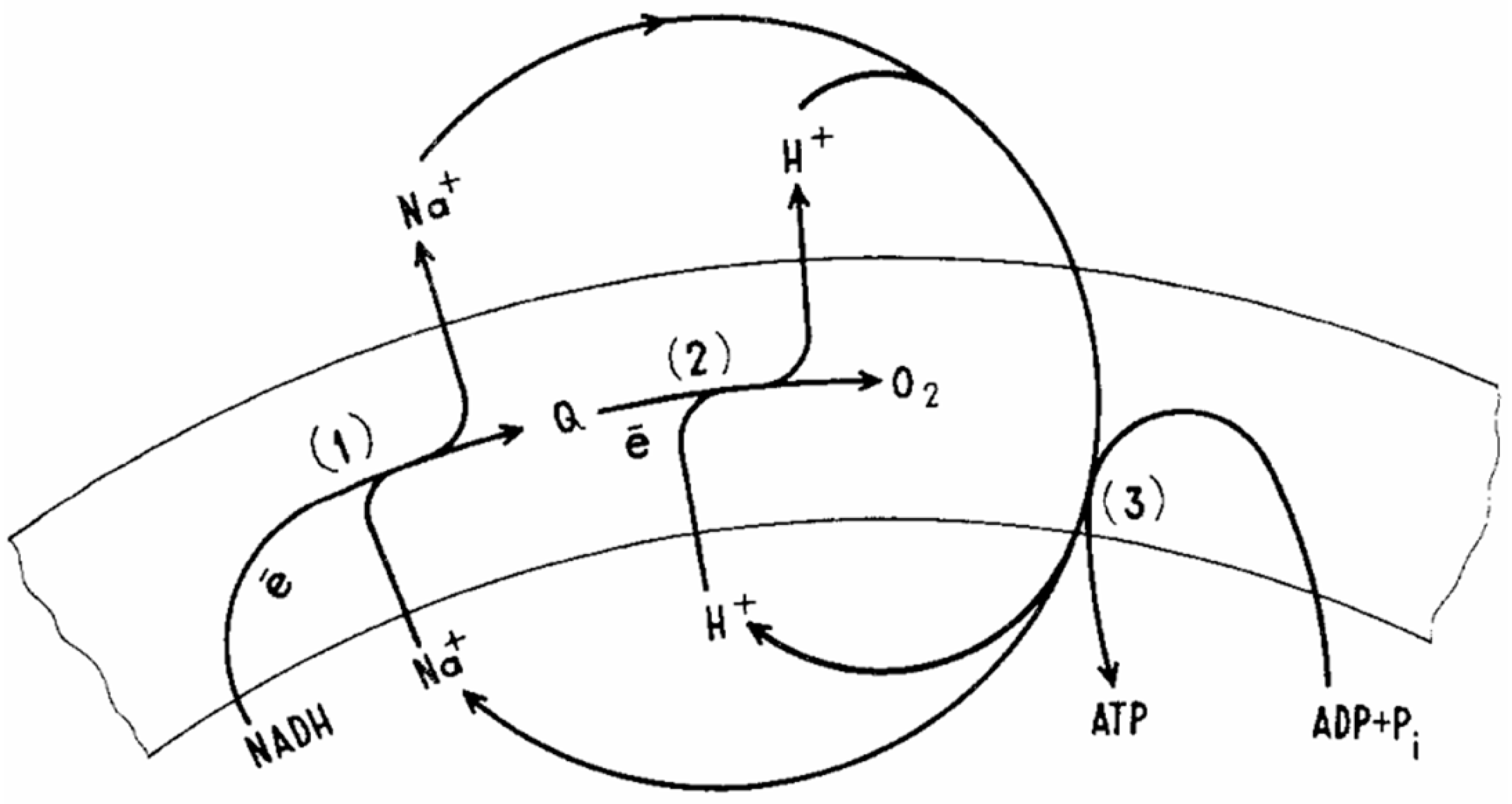
Figure 5.
Schematic presentation of triple-enzyme mimetic activity on an inorganic substrate [101]. Reproduced with permission from [101] published by the American Chemical Society.
Figure 6.
“Two enzyme scheme (a) and three enzyme scheme (b) of mmobilizat immobilization in artificial cells”. Elementary and earliest (1978) examples of multienzyme scheme mmobilization in artificial cells [106]. Reproduced with permission from [106] published by Springer Nature.
Figure 7.
Earliest (1966) Mitchell's schematic presentations for the proton-translocating respiratory chain in mitochondria (a) and for the proton-translocating light-driven oxido-reduction systems for photophosphorylation for noncyclic light-driven oxido-reduction in chloroplasts (b) [116]. Reproduced with permission from [116] published by John Wiley and Sons.
Figure 7.
Earliest (1966) Mitchell's schematic presentations for the proton-translocating respiratory chain in mitochondria (a) and for the proton-translocating light-driven oxido-reduction systems for photophosphorylation for noncyclic light-driven oxido-reduction in chloroplasts (b) [116]. Reproduced with permission from [116] published by John Wiley and Sons.
Figure 8.
Schematic representation of protometabolism on the FeS mineral surface (a) and hypothetic mineral-catalyzed reaction pathway with an autocatalytic feedback [132]. Reproduced with permission from [132] published by the Royal Society.
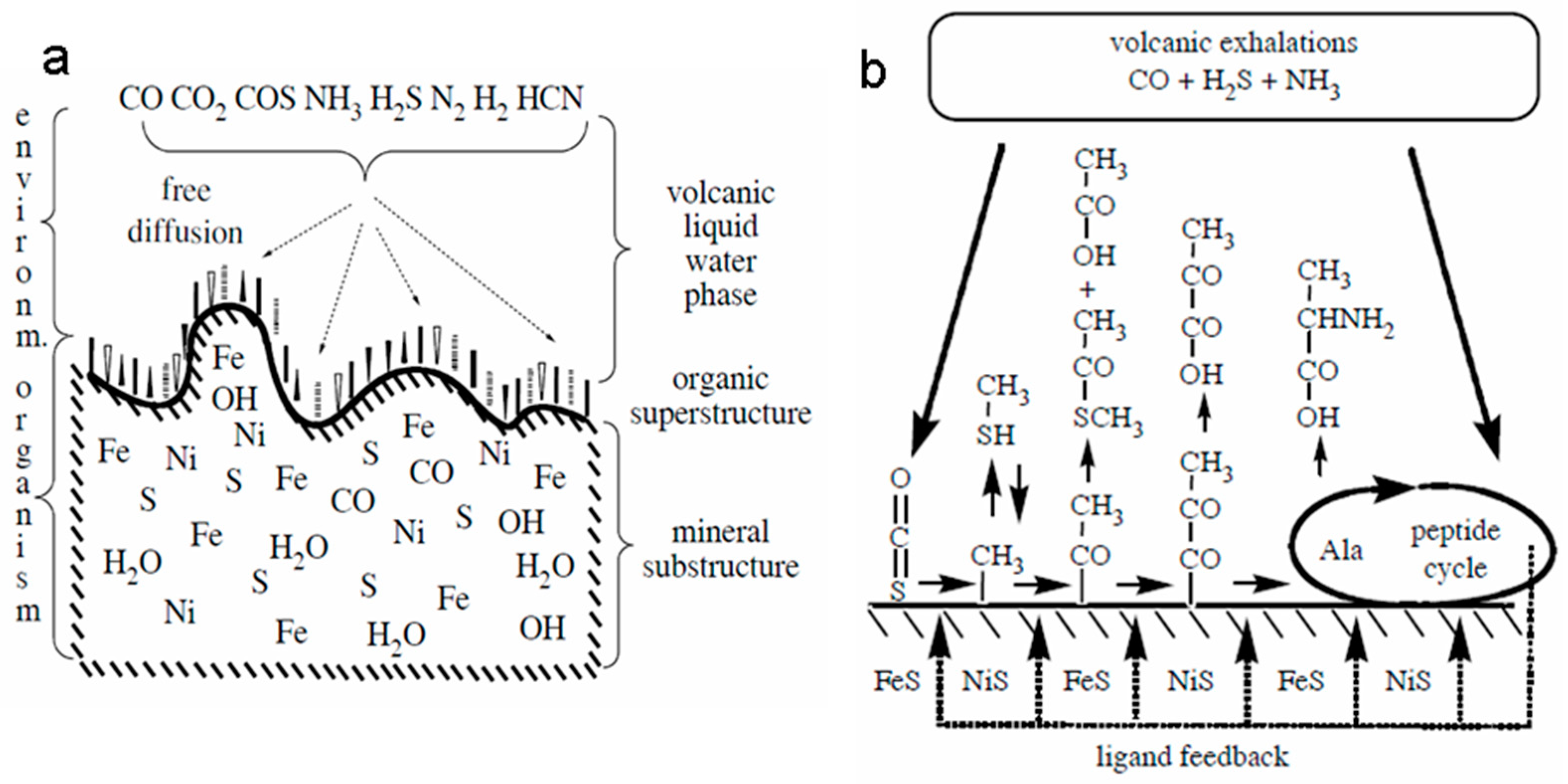
Figure 9.
Schematic representation of the natural prebiotic hydrothermal reactor with catalytically-active inorganic membranes operating under the thermal, redox and pH gradients [144]. Reproduced with permission from [144] published by Elsevier.
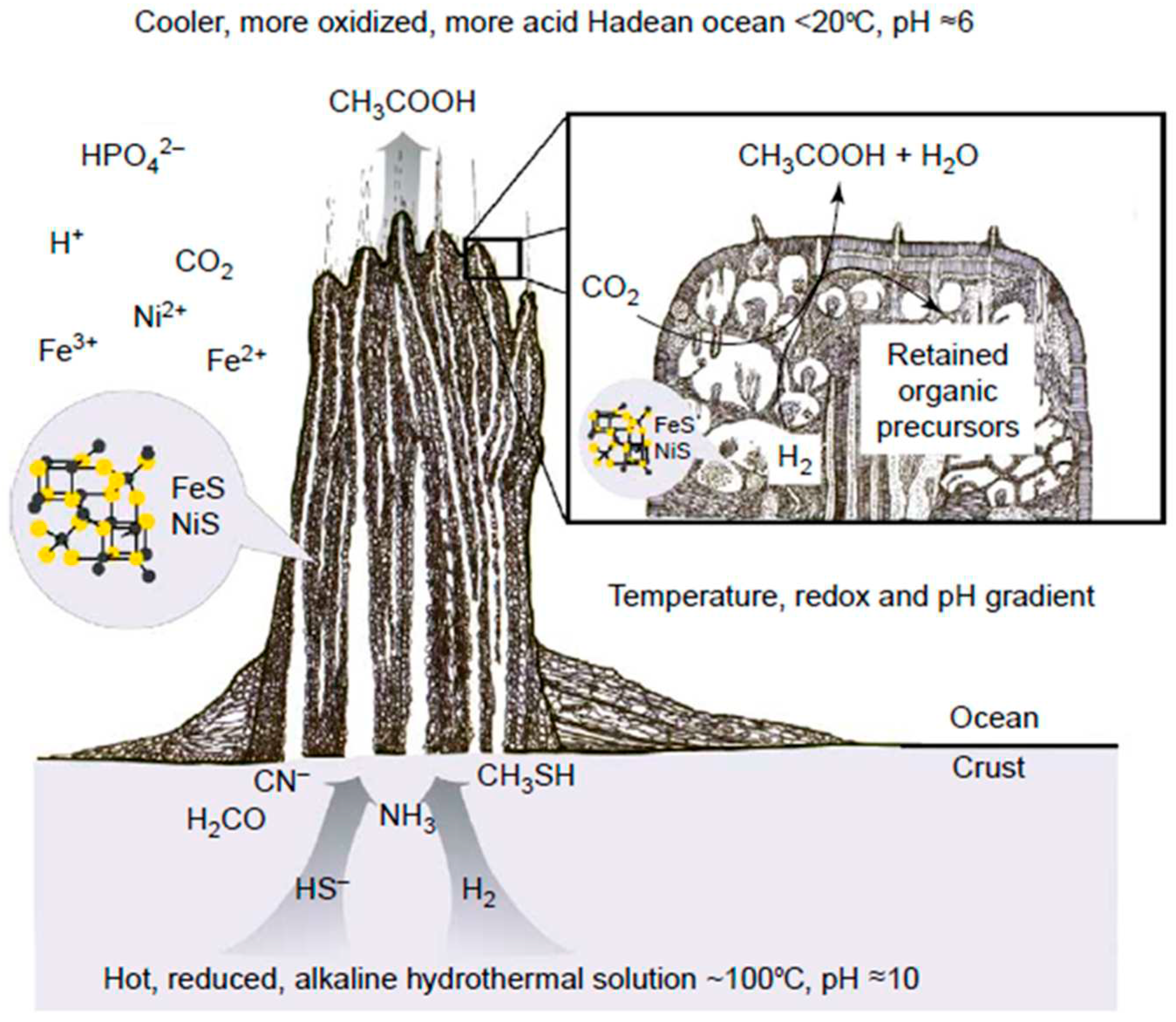
Figure 10.
Inorganic coacervate droplets composed of ferrous hydroxyde / oxohydroxyde (a) and the same droplets substituted with hydrotroilite (b); framboidal pyrite (c-d) [148]. Reproduced with permission from [148] published by Nauka.
Figure 11.
Comparison of the principal schemes of natural photosynthesis (left) and its simple photovoltaic model based on the catalytically active magnetite substrate [156]. Reproduced with permission from [156] published by Springer Nature.
Figure 12.
A simplified scheme of the coupled photochemical redox reactions involving ferrous and ferric ions in a bicarbonate buffer solution providing solar energy conversion (a) and its autocatalytic water splitting unit with a unified hydrogen acceptor A (b) [157]. Reproduced with permission from [157] published by Nauka Press.
Figure 12.
A simplified scheme of the coupled photochemical redox reactions involving ferrous and ferric ions in a bicarbonate buffer solution providing solar energy conversion (a) and its autocatalytic water splitting unit with a unified hydrogen acceptor A (b) [157]. Reproduced with permission from [157] published by Nauka Press.
Figure 13.
A principle scheme of a photocatalytic reaction on a semiconductor particle (a) and a possible scheme of the surface photosensitization of the metal oxides (b) [162]. Reproduced with permission from [162] published by Springer Nature.
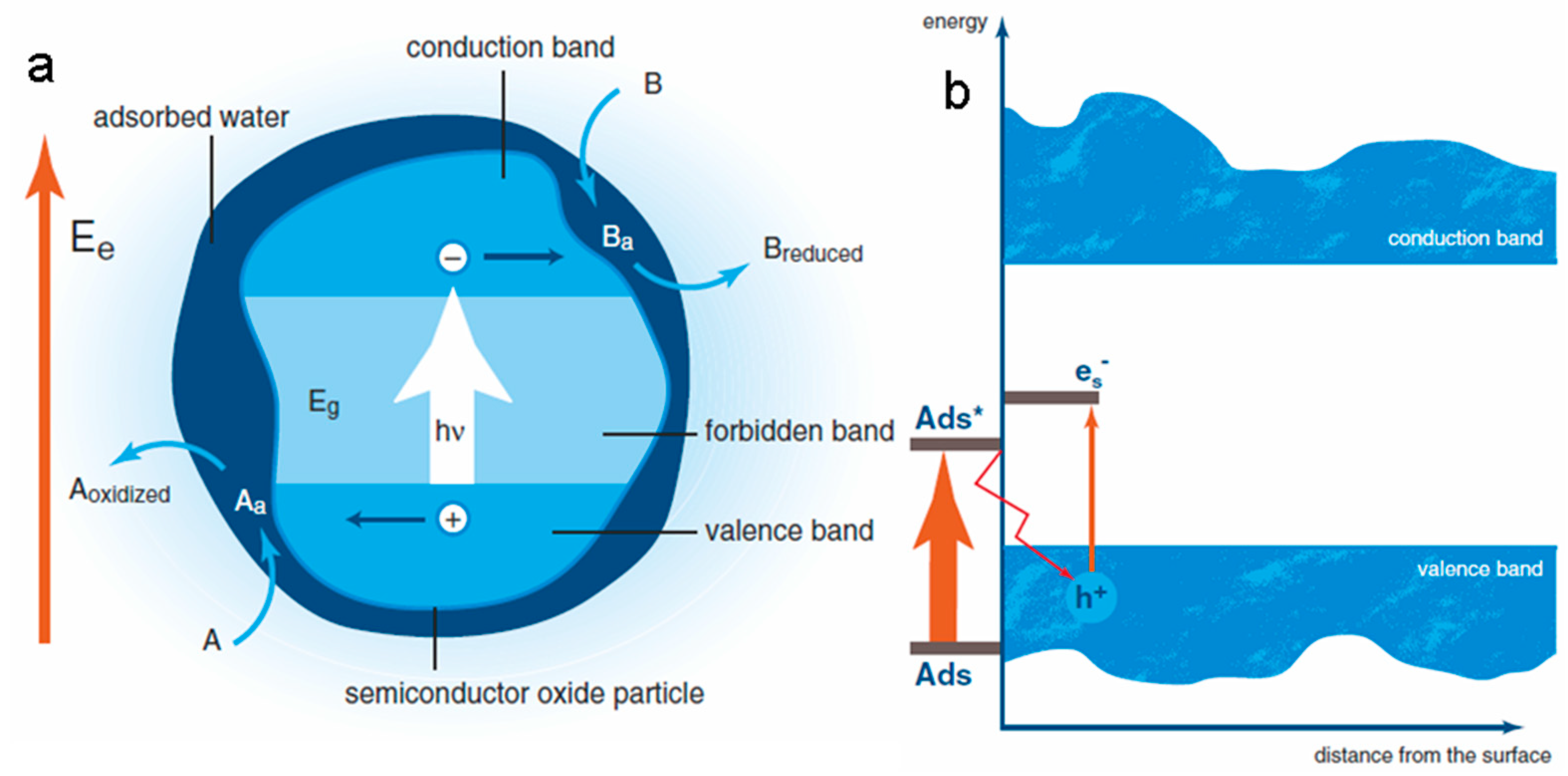
Figure 14.
a) Energy diagrams for different metal sulfides as potential photo-excited electron donors and for the biologically relevant electron acceptors; b) Comparison of the energy diagrams for a single ZnS nanoparticle (left) and a bacterial photochemical reaction center (right) [207].
Figure 14.
a) Energy diagrams for different metal sulfides as potential photo-excited electron donors and for the biologically relevant electron acceptors; b) Comparison of the energy diagrams for a single ZnS nanoparticle (left) and a bacterial photochemical reaction center (right) [207].
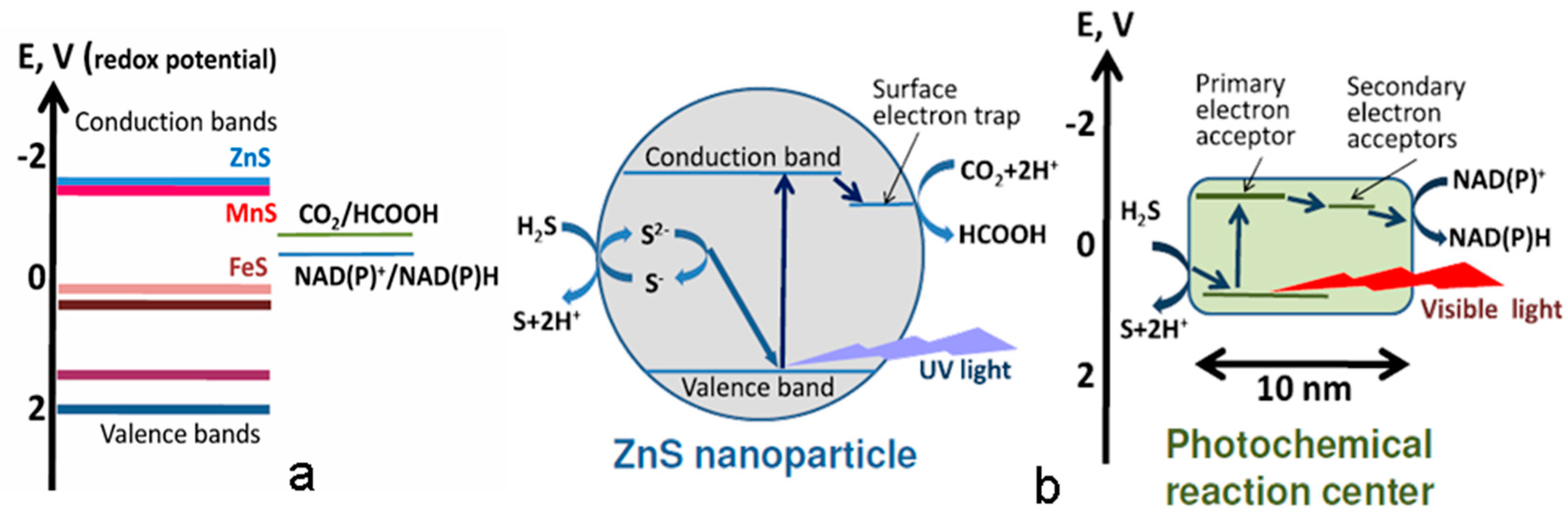
Figure 15.
Natural iron sulfide garden (Tynagh mine, Ireland), now composed of pyrite embedded in barite, produced as a somewhat acidic, iron(III)-bearing buoyant hydrothermal solution invaded a mildly alkaline sulfanide (bisulfide)-bearing brine occupying a void below the carboniferous seafloor ∼350 million years ago; (b, c) hollow pyrite botryoids produced as for (a) but perhaps more slowly; (d) reverse gardens found at Navan, also in Ireland, whereby a dense sulfanide-bearing surface brine seeped down into an open horizontal joint occupied by iron-bearing mineralizing solution ∼350 million years ago (full image box from [257]).
Figure 15.
Natural iron sulfide garden (Tynagh mine, Ireland), now composed of pyrite embedded in barite, produced as a somewhat acidic, iron(III)-bearing buoyant hydrothermal solution invaded a mildly alkaline sulfanide (bisulfide)-bearing brine occupying a void below the carboniferous seafloor ∼350 million years ago; (b, c) hollow pyrite botryoids produced as for (a) but perhaps more slowly; (d) reverse gardens found at Navan, also in Ireland, whereby a dense sulfanide-bearing surface brine seeped down into an open horizontal joint occupied by iron-bearing mineralizing solution ∼350 million years ago (full image box from [257]).

Figure 16.
A full life cycle of the proto-organism (proposed by S. Rasmussen), for which the quantum self-assembly models have been developed. 1, 2: Self-assembly of the proto-organism components— lipids, PNA, and the sensitizer molecules. 3: Feeding the proto-organism with PNA and lipid precursors and more photosentisizer molecules. The proto-organism swells up in particular as it is loaded with many precursor lipids. 4: As light is provided, the phenyl group is first fragmented from the precursor PNA, forming PNA oligomers that can ligate, once they are aligned by the template. 5: As the precursor lipids are turned into lipids (surfactants), the large aggregate becomes unstable and starts breaking up. A thermodynamic balance between hybridized and non-hybridized (double- and single-stranded) PNA has to be established to allow a reasonable partition of the proto-genes between the two aggregates. 6: The life cycle is complete, as the proto-organism has generated a copy of itself. The overall rate limiting steps are believed to be the PNA template directed ligation process and the balanced lipid-PNA and PNA hybridization kinetics [71]. Reproduced with permission from [71] published by MIT press.
Figure 16.
A full life cycle of the proto-organism (proposed by S. Rasmussen), for which the quantum self-assembly models have been developed. 1, 2: Self-assembly of the proto-organism components— lipids, PNA, and the sensitizer molecules. 3: Feeding the proto-organism with PNA and lipid precursors and more photosentisizer molecules. The proto-organism swells up in particular as it is loaded with many precursor lipids. 4: As light is provided, the phenyl group is first fragmented from the precursor PNA, forming PNA oligomers that can ligate, once they are aligned by the template. 5: As the precursor lipids are turned into lipids (surfactants), the large aggregate becomes unstable and starts breaking up. A thermodynamic balance between hybridized and non-hybridized (double- and single-stranded) PNA has to be established to allow a reasonable partition of the proto-genes between the two aggregates. 6: The life cycle is complete, as the proto-organism has generated a copy of itself. The overall rate limiting steps are believed to be the PNA template directed ligation process and the balanced lipid-PNA and PNA hybridization kinetics [71]. Reproduced with permission from [71] published by MIT press.
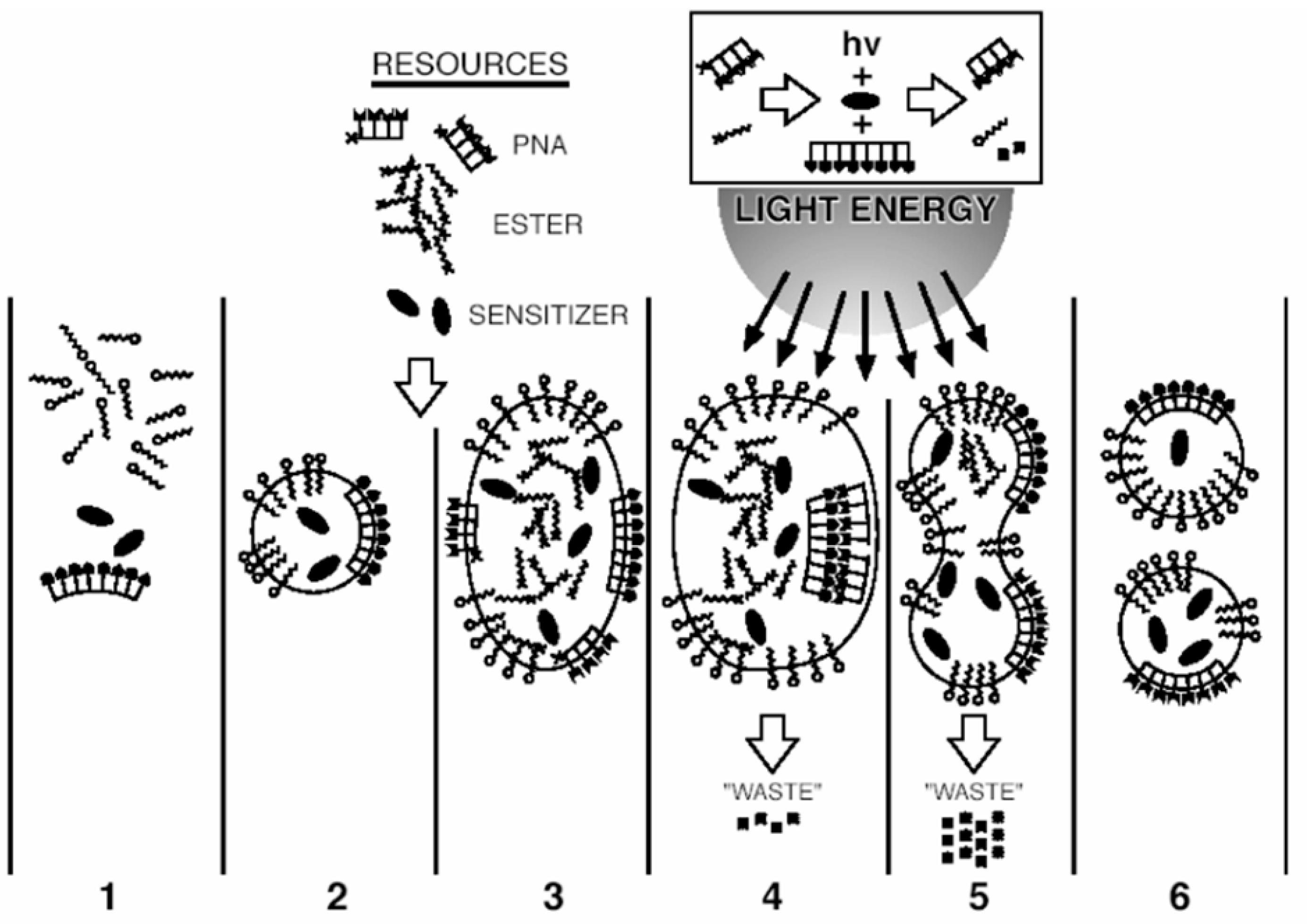
Figure 17.
Representations of a) a vesicle formed from cylindrically shaped amphiphiles and b) a micelle (spatially comparable with Chen-Rasmussen’s "minimal cell" model - see. Figure 2) formed from conical surfactants applicable in model / artificial photosynthesis at dynamic self-assembled interfaces in water [274]. Reproduced with permission from [274] published by John Wiley and Sons.
Figure 17.
Representations of a) a vesicle formed from cylindrically shaped amphiphiles and b) a micelle (spatially comparable with Chen-Rasmussen’s "minimal cell" model - see. Figure 2) formed from conical surfactants applicable in model / artificial photosynthesis at dynamic self-assembled interfaces in water [274]. Reproduced with permission from [274] published by John Wiley and Sons.
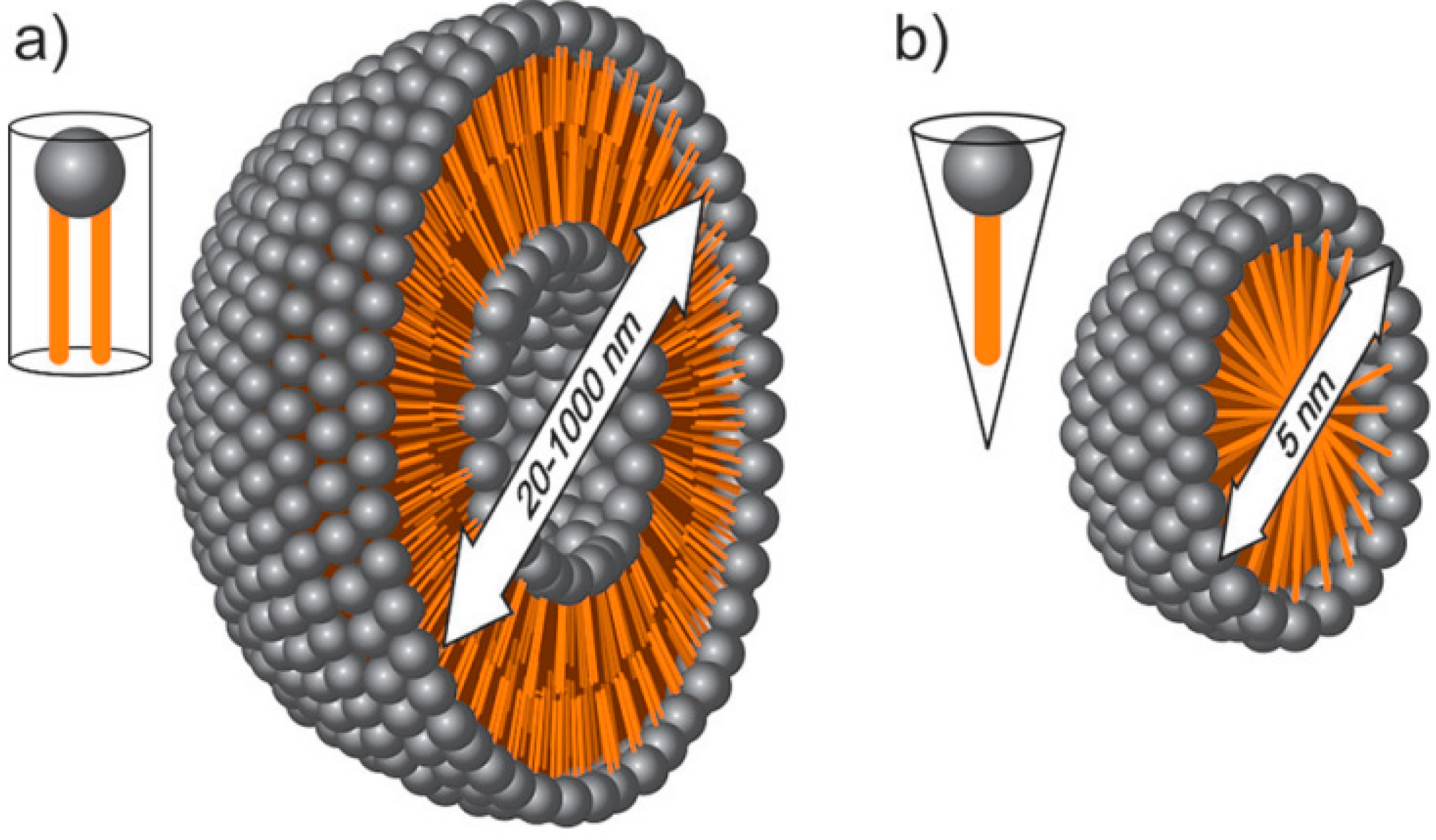
Figure 18.
Light-induced charge separation in functional micelles [289]. This scheme is similar to that of light-induced charge separation in primary "membrane mimetic" systems, corresponding in size to Rasmussen-Tamulis minimal cell models (Fig, 2). If we ignore the specific chemical substrate indicated in [289], one can speak about the participation of such light-induced charge separation schemes in protobiological and prebiological analogues (or prototypes) of the photosynthetic process. Reproduced with permission from [289] published by the American Chemical Society.
Figure 18.
Light-induced charge separation in functional micelles [289]. This scheme is similar to that of light-induced charge separation in primary "membrane mimetic" systems, corresponding in size to Rasmussen-Tamulis minimal cell models (Fig, 2). If we ignore the specific chemical substrate indicated in [289], one can speak about the participation of such light-induced charge separation schemes in protobiological and prebiological analogues (or prototypes) of the photosynthetic process. Reproduced with permission from [289] published by the American Chemical Society.
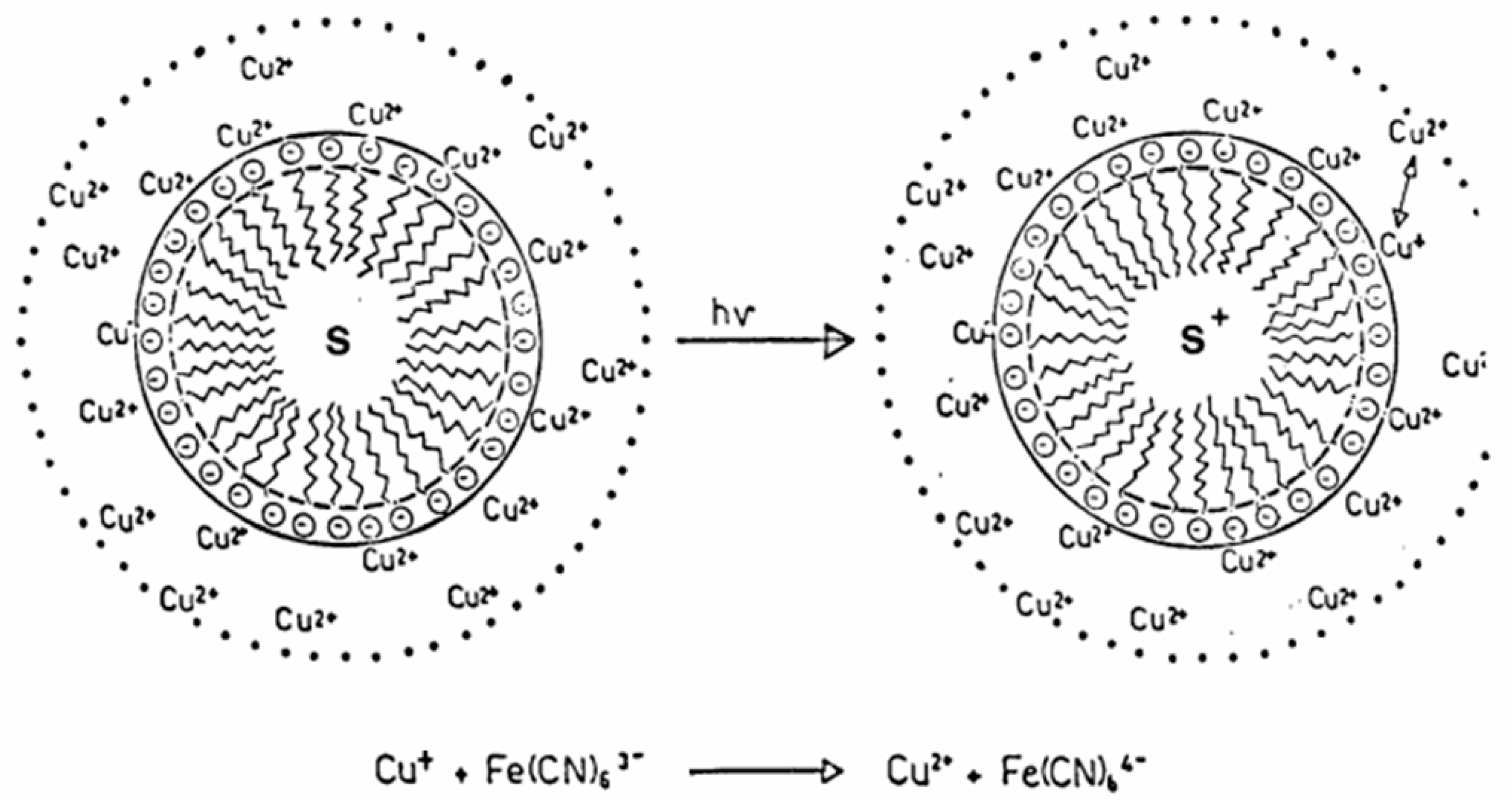
Figure 19.
Schematic illustration of the light-harvesting units used in photochemical water splitting devices [289]. A scheme based on dispersed semiconductors that may have occurred in geological systems in the prebiological era is a good geochemical precursor to more complex light-harvesting and photochemical water splitting schemes involving micelles / surfactant /membrane. It seems to be an evolutionarily plausible prototype of photosynthesis, since it does not require organic matter. Reproduced with permission from [289] published by the American Chemical Society.
Figure 19.
Schematic illustration of the light-harvesting units used in photochemical water splitting devices [289]. A scheme based on dispersed semiconductors that may have occurred in geological systems in the prebiological era is a good geochemical precursor to more complex light-harvesting and photochemical water splitting schemes involving micelles / surfactant /membrane. It seems to be an evolutionarily plausible prototype of photosynthesis, since it does not require organic matter. Reproduced with permission from [289] published by the American Chemical Society.
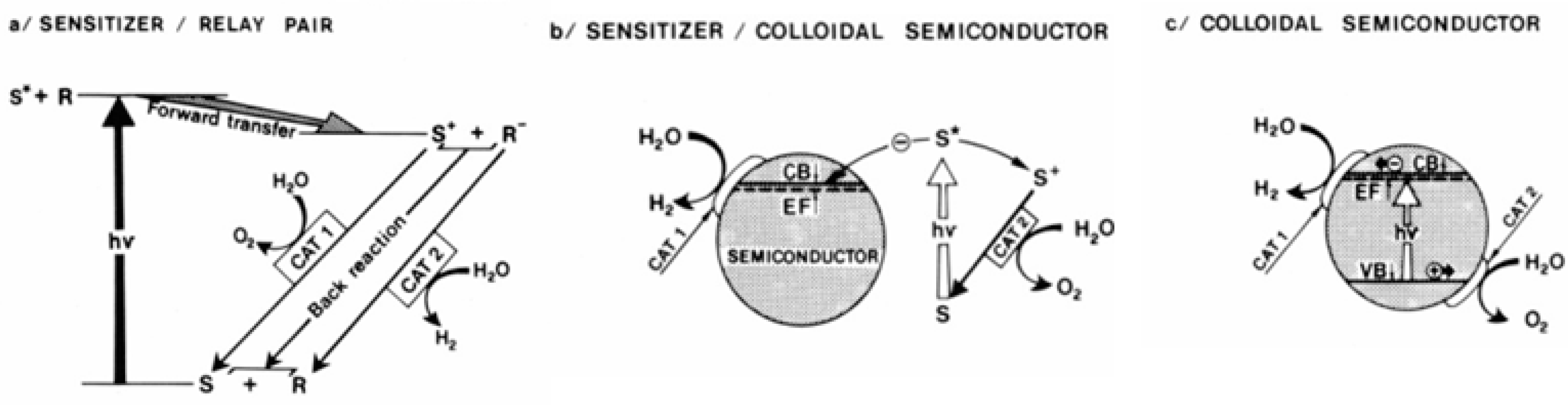
Figure 20.
"Semi-artificial photoanodes" based on photosystem II (a) and on cyanobacterium (b) as light-absorbing and catalytic components of water oxidation photoanodes. Modes of interfacing between the inorganic and biological components vary depending on complexity of the system, with PSII charge transfer pathways... Ambiguity regarding charge and energy flow increases correspondingly with increasing levels of hierarchical organization within the biological unit [363]. . Reproduced with permission from [363] published by Springer Nature.
Figure 20.
"Semi-artificial photoanodes" based on photosystem II (a) and on cyanobacterium (b) as light-absorbing and catalytic components of water oxidation photoanodes. Modes of interfacing between the inorganic and biological components vary depending on complexity of the system, with PSII charge transfer pathways... Ambiguity regarding charge and energy flow increases correspondingly with increasing levels of hierarchical organization within the biological unit [363]. . Reproduced with permission from [363] published by Springer Nature.
Figure 21.
Examples of the native mineral structured catalysts - prototypes of geomimetic catalysts from the paper [376]: SEM images of (a) the surface of the red volcanic rock after the reaction; (c) TEM images of geologica nanotubes produced from the red stone. Such structures are very similar to geological chemical gardens – the subject of chemobryonics (see Figure 10, Figure 15). There are similar mineral structures with photocatalytic functions, which in ancient geological (prebiological) conditions could be not only geomimetic but also (proto)biomimetic and protophotosynthetic. . Reproduced with permission from [376] published by Elsevier.
Figure 21.
Examples of the native mineral structured catalysts - prototypes of geomimetic catalysts from the paper [376]: SEM images of (a) the surface of the red volcanic rock after the reaction; (c) TEM images of geologica nanotubes produced from the red stone. Such structures are very similar to geological chemical gardens – the subject of chemobryonics (see Figure 10, Figure 15). There are similar mineral structures with photocatalytic functions, which in ancient geological (prebiological) conditions could be not only geomimetic but also (proto)biomimetic and protophotosynthetic. . Reproduced with permission from [376] published by Elsevier.

Figure 22.
A possible photoenzyme synergic catalytic mechanism based on photocatalyst-enzyme heterojunction in water. Reproduced with permission from [338] published by Elsevier.
Figure 22.
A possible photoenzyme synergic catalytic mechanism based on photocatalyst-enzyme heterojunction in water. Reproduced with permission from [338] published by Elsevier.
Figure 23.
The structure of a heterogeneous membrane mimetic Langmuir-Blodgett film and the electrical double layer (EDL) created between the monolayers. (a) Side view of the heterogeneous membrane mimetic Langmuir-Blodgett film on the mineral surface (quartz plate), (b) schematic diagram of the electrical double layer (EDL) created in the S/AFc pair with counter cation representation; and (c) EDL in the S/CFc pair with counter anion representation. The contribution of counter cations and counter anions, as well as the corresponding contribution of the substrate to the structure of the photoactivated electric double layer is obvious. Reproduced with permission from [408] published by Elsevier.
Figure 23.
The structure of a heterogeneous membrane mimetic Langmuir-Blodgett film and the electrical double layer (EDL) created between the monolayers. (a) Side view of the heterogeneous membrane mimetic Langmuir-Blodgett film on the mineral surface (quartz plate), (b) schematic diagram of the electrical double layer (EDL) created in the S/AFc pair with counter cation representation; and (c) EDL in the S/CFc pair with counter anion representation. The contribution of counter cations and counter anions, as well as the corresponding contribution of the substrate to the structure of the photoactivated electric double layer is obvious. Reproduced with permission from [408] published by Elsevier.
Figure 24.
Example of dissipative structure formation (a,b) and kinetic oscillations (c) in heterogeneous catalytic reactions with the surface restructuring [491]. Example of the oscillatory reaction kinetics can be compared with Lotka–Volterra (predator–prey) models well-known in biophysics or with classical biochemical oscillators with first-order phase transitions and trsnsitions through the metastable state. The results were obtained on the lattice with periodic boundary conditions. Reproduced with permission from [491] published by Springer Nature.
Figure 24.
Example of dissipative structure formation (a,b) and kinetic oscillations (c) in heterogeneous catalytic reactions with the surface restructuring [491]. Example of the oscillatory reaction kinetics can be compared with Lotka–Volterra (predator–prey) models well-known in biophysics or with classical biochemical oscillators with first-order phase transitions and trsnsitions through the metastable state. The results were obtained on the lattice with periodic boundary conditions. Reproduced with permission from [491] published by Springer Nature.
Figure 25.
Examples of the phase spaces of the charge carrier densities in semiconductors including a model with dielectric relaxation [530]. They are given for comparison with the phase spaces known in nonlinear dynamics of biochemical oscillations observed in enzymatic kinetics, membrane potential oscillations, photosynthesis, etc. Considering the concepts indicated below about the role of quasiparticles in biological photosynthesis and the corresponding transfer processes, the possibility of comparing them with the kinetics of the charge carriers or quasiparticles becomes a very important element of the concept argumentation. Reproduced with permission from [530] published by Springer Nature.
Figure 25.
Examples of the phase spaces of the charge carrier densities in semiconductors including a model with dielectric relaxation [530]. They are given for comparison with the phase spaces known in nonlinear dynamics of biochemical oscillations observed in enzymatic kinetics, membrane potential oscillations, photosynthesis, etc. Considering the concepts indicated below about the role of quasiparticles in biological photosynthesis and the corresponding transfer processes, the possibility of comparing them with the kinetics of the charge carriers or quasiparticles becomes a very important element of the concept argumentation. Reproduced with permission from [530] published by Springer Nature.
Figure 26.
Towards self-organization processes at active interfaces and membrane-structure templating: Evolution of the membrane-bound fraction (a), radius of the domain (b) and growing velocity of the domain (c) for three simulations with different initial conditions (d–f). Reproduced with permission from [737] published by Springer Nature.
Figure 26.
Towards self-organization processes at active interfaces and membrane-structure templating: Evolution of the membrane-bound fraction (a), radius of the domain (b) and growing velocity of the domain (c) for three simulations with different initial conditions (d–f). Reproduced with permission from [737] published by Springer Nature.
Figure 27.
How the analysis of ion transport functions in various cells helps to reconstruct the possible ion transport pathways in protocells and in minimal cells: a) scheme of the membrane transport of ions in cells of archaea Halobacterium; b) transport processes in the model Diatom algae cell; c) analogically modeled / reconstructed scheme of the ion transport in a possible "minimal cell". Reproduced with permission from [793] published by Elsevier.
Figure 27.
How the analysis of ion transport functions in various cells helps to reconstruct the possible ion transport pathways in protocells and in minimal cells: a) scheme of the membrane transport of ions in cells of archaea Halobacterium; b) transport processes in the model Diatom algae cell; c) analogically modeled / reconstructed scheme of the ion transport in a possible "minimal cell". Reproduced with permission from [793] published by Elsevier.
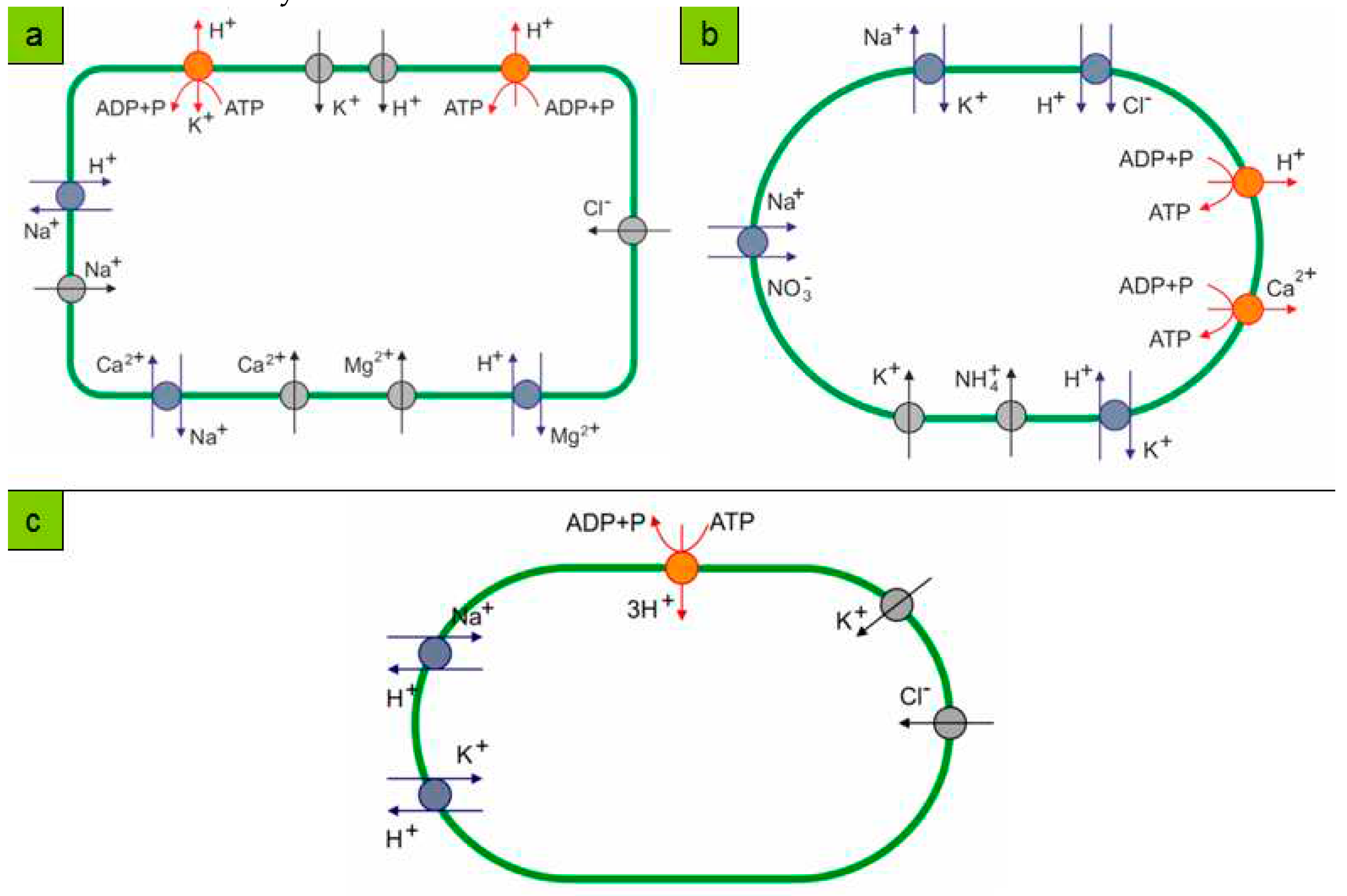
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



