
Submitted:
12 October 2023
Posted:
16 October 2023
You are already at the latest version
Abstract
The type III receptor tyrosine kinase FLT3 is a pivotal kinase for hematopoietic progenitor cell regulation, with significant implications in acute myeloid leukemia (AML) through mutations like internal tandem duplication (ITD). This study delves into the structural intricacies of FLT3, the roles of activation loop mutants, and their interaction with tyrosine kinase inhibitors. Coupled with this, the research leverages molecular contrastive learning and protein language modeling to examine interactions between small molecule inhibitors and FLT3 activation loop mutants. Utilizing the ConPLex platform, over 5.7 million unique FLT3 activation loop mutants-small molecule pairs were analyzed. The binding free energies of three inhibitors were assessed, and cellular apoptotic responses were evaluated under drug treatments. Notably, the introduction of the Xepto50 scoring system provides a nuanced metric for drug efficacy. The findings underscore the modulation of molecular interactions and cellular responses by Y842 mutations in FLT3-KD, highlighting the need for tailored therapeutic approaches in FLT3-ITD-related malignancies.
Keywords:
drug sensitivity scores
; molecular modeling
; molecular docking
; four-parameter logistic curve
Introduction
FLT3, also known as Fms-like tyrosine kinase 3, is a receptor tyrosine kinase that plays a pivotal role in the hematopoietic system. It is predominantly expressed in hematopoietic progenitor cells, acting as a key regulator of their survival, proliferation, and differentiation. This significance of FLT3 has been extensively documented and is reviewed in several studies, one of which is referenced as [1]. In the context of acute myeloid leukemia (AML), FLT3 takes on an even more pronounced role. AML, a malignancy of the myeloid lineage of blood cells, exhibits a variety of genetic anomalies. Notably, about 30% of AML patients carry an activating mutation in the FLT3 gene. This mutation significantly boosts the cell's survival and proliferation capabilities, often leading to aggressive disease progression. The most frequently observed of these mutations is the internal tandem duplication (ITD). This intriguing mutation involves an in-frame duplication of a sequence within the FLT3 gene. This duplication can vary in length, from just a few amino acids to more than a hundred. The result of this mutation is a structural alteration where the juxtamembrane region becomes separated from the kinase domain. Consequently, this change activates the kinase activity, driving the oncogenic properties of the cell. Clinical observations have revealed a grim picture: the presence of an ITD mutation in FLT3 often correlates with poor survival rates and a challenging overall prognosis for AML patients. Diving deeper into the structure of FLT3, within its kinase domain, there lies a conserved tyrosine residue located in what's referred to as the activation loop. This loop is a hallmark of kinase enzymes and is frequently involved in modulating their activity. A wealth of research, including the study referenced as [2], highlights the importance of activation loops across various kinases. However, when it comes to type III receptor tyrosine kinases, a group to which FLT3 belongs, this loop doesn't play the conventional regulatory role. Our previous research efforts have unveiled that this particular tyrosine residue in FLT3, while not crucial for its kinase activity, is indispensable for the transformative capabilities of the FLT3-ITD mutation [3]. Further complicating the clinical landscape, mutations in codon 842, specifically Y842H and Y842C, have emerged as culprits in mediating resistance to tyrosine kinase inhibitors, a common therapeutic strategy for AML [4]. Among these, the Y842C mutation deserves special mention. It has not only been identified as a mechanism of drug resistance but has also been flagged as an activating mutation in AML patients, as detailed in the study referenced as [5].
The extracellular domain of type III Receptor Tyrosine Kinases (RTKs) is architecturally composed of five immunoglobulin-like (Ig-like) domains. Among these, the Ig-like motifs 2 and 3 are specifically involved in ligand binding, providing specificity to the ligand-receptor interaction. In contrast, domains 4 and 5 have the crucial function of mediating receptor dimerization, a fundamental step for the signaling capabilities of these receptors. Anchoring these receptors firmly to the cell membrane is a hydrophobic transmembrane domain. This domain acts as a gateway between the extracellular environment and the cell's interior. Adjacent to the transmembrane domain lies the intricate intracellular region. This region starts with the juxtamembrane region and subsequently houses the bipartite kinase domain, ultimately ending with the carboxyterminal tail. For type III RTKs, the juxtamembrane region is not just a mere structural component. It performs a crucial autoinhibitory function. By strategically binding to the activation loop of the kinase domain, it effectively locks the kinase in an inactive state, ensuring that signaling is tightly regulated [6]. When FLT3 is in this inactive state, it remains unphosphorylated. The activation loop adopts a distinct conformation, often referred to as the 'DFG-out' conformation due to its conserved aspartic acid-phenylalanine-glycine (DFG) sequence. This loop, approximately 27 residues in length, interacts with the alanine-proline-glutamic acid (APE) sequence, a detail that has been elaborated upon in various reviews, including [7]. In a scenario where FLT3 remains unbound to its ligand and thus inactive, the juxtamembrane region interacts with the kinase domain. This interaction maintains the kinase domain in its inhibited state. Interestingly, this DFG-out conformation has been exploited therapeutically. Tyrosine kinase inhibitors that bind to this conformation are termed type II inhibitors. Imatinib, a prototypical tyrosine kinase inhibitor (TKI), is a classic example of this category. Conversely, there are Type I TKIs that differ in their mechanism. Instead of the DFG-out conformation, they interact with the kinase domain when it is in the "DFG-in" configuration, signifying an active state of the kinase. Within the scope of the TKIs discussed here, midostaurin is categorized as a type I inhibitor. In contrast, both sorafenib and quizartinib fall under the type II inhibitors, emphasizing their distinct binding and inhibitory profiles.
Material and Methods
Preparation of native and mutant FLT3 structures: The native structure of the FLT3 protein, with a resolution of 3.20 Å, was obtained from the Protein Data Bank (PDB). The PDB ID for the dimeric FLT3 structure is 4XUF [8]. For our computational analysis, we utilized only one subunit, specifically Subunit A. The crystallographic structure displayed two missing loops: one between residues Lys649 and Asp651, and the other between Glu708 and Val782. These missing loops were reconstructed using the Modeler plugin within the Chimera software. The co-crystal ligand, quizartinib, was excised from the binding site. Point mutations were then introduced into the native FLT3 protein structure at position Y842 to produce the Y-to-C and Y-to-F mutant proteins. These mutant structures were generated using the Dunbrack rotamer library [9], and among them, structures with the lowest energy and highest probability scores were chosen for subsequent computational analyses. The molecular structures of quizartinib (PubChem CID: 24889392), sorafenib (PubChem CID: 216239), and midostaurin (PubChem CID: 9829523) were sourced from the PubChem database [10].
Molecular Docking: The native and mutant FLT3 protein structures were first prepared by removing water molecules. Subsequently, the structures were converted to the Pdbqt format using AutoDock in preparation for docking. Docking analysis was executed using AutoDock Version 4.2 [11] in conjunction with ADT Tools 1.5.6. Intermediate steps, including energy minimization for protein and ligand structures in the Pdbqt format and grid box generation, were handled using the graphical user interface of AutoDock Tools. AutoDock added polar hydrogens, Kollman atomic charges, solvation parameters, and fragmental volumes to the protein. The prepared structures were saved in Pdbqt format. For grid map file generation, AutoGrid was employed, utilizing a grid box with dimensions set to 60 × 60 × 60 points in x, y, and z, and a grid spacing of 0.375 Å. The grid box center was adjusted based on the position of the co-crystal ligand. AutoDock's iterative local search global optimizer was used to generate protein-ligand poses. Complexes with the lowest binding free energy (greater negative ΔG values) were selected as the starting structures for molecular dynamics (MD) simulations. In total, nine complexes, namely native-quizartinib, Y842C-quizartinib, Y842F-quizartinib, native-sorafenib, Y842C-sorafenib, Y842F-midostaurin, native-midostaurin, Y842C-midostaurin, and Y842F-midostaurin, were chosen as initial structures for MD simulations.
MD simulations: The topologies for both ligand and protein structures were generated using the PRODRG server [12] and the editconf script from the GROMACS software, respectively. The protein topologies were derived using the GROMOS96 43a1 force-field [13]. Subsequently, ligand topologies were combined with protein topologies to create a protein-ligand complex. This complex was situated inside a cubic box populated with the simple point charge (SPC) water model. To neutralize the system, counter ions (Na+ and Cl-) were introduced. The neutralized system then underwent 50,000 steps of energy minimization using the steepest descent algorithm. Position restraints for the ligand and temperature coupling groups were established at this juncture. The energy-minimized systems proceeded to a two-phase equilibration, each spanning 1000 ps. The initial phase operated within an isothermal-isochoric ensemble, ensuring a constant number of particles, volume, and temperature. This step aimed to stabilize the system's temperature. In the subsequent phase, the system's pressure and density were equalized under the isothermal-isobaric ensemble, maintaining a constant number of particles, pressure, and temperature. The temperature and pressure during these ensembles were regulated by the velocity rescaling thermostat [14] and the Parrinello-Rahman barostat [15], respectively. Following equilibration, all position restraints were released, and the systems were subjected to 1000 ns MD simulations. These MD trajectories facilitated the calculation of thermodynamic binding free energies through the Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA) method.
MM-PBSA calculations: We selected the last 50 ns of the most stable trajectories from MD simulations to compute the binding free energies of protein-ligand systems using the g_mmpbsa tool [16]. This tool synergizes binding energy calculations with high-throughput MD simulations, accounting for conformational changes that occur during protein-ligand binding. While the method doesn't compute the entropic terms, it's ideal for comparing the relative binding energies of molecules that interact within the same binding pocket.
The binding free energy for protein-ligand, protein-protein, protein-DNA complexes, or any biomolecular assemblage can be theoretically expressed by the equation:
ΔGbinding = Gcomplex – (Gprotein + Gligand)
Each component in equation (1) can further be defined by:
Gx = (EMM) – TS + (Gsolvation)
In this equation, 'x' can represent Gcomplex, Gprotein, or Gligand. EMM stands for the average molecular mechanics potential energy in a vacuum. The term TS symbolizes the entropic contribution to free energy in a vacuum, with 'T' and 'S' denoting temperature and entropy, respectively. Lastly, Gsolvation refers to the free energy of solvation.
Drug sensitivity assays – The Ba/F3 cell line was procured from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). The cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin, 100µg/mL streptomycin (Corning, Corning, NY, USA), and 10 ng/mL murine IL3 (Thermo Fisher). All inhibitors were sourced from MedChemExpress, Monmouth Junction, NJ, USA. Ba/F3 cells, after being stably transfected with FLT3-ITD and activation loop tyrosine mutants, were maintained in the same medium as the parental Ba/F3 cells. For the drug sensitivity assays, 10,000 cells were seeded in IL3-free medium and exposed to ten distinct drug concentrations, ranging from 5 picomolar to 10 micromolar, for 72 hours. Cell viability was then assessed post the 72-hour period using CellTiter-Glo (Promega, Madison, WI, USA).
Apoptosis assay – Ba/F3 cells stably expressing FLT3-ITD or activation loop mutants were treated with various drug concentrations for 48 hours. Following treatment, the cells were processed, and apoptotic cells were quantified using the FITC-Annexin-V/7-AAD kit (BD Biosciences, Franklin Lakes, NJ, USA) as per the manufacturer’s instructions.
ConPLex analysis: The kinase domain of FLT3 was identified using the NCBI's Conserved Domains Search. For our analysis, we retrieved the Simplified Molecular Input Line Entry System (SMILES) notations of selected small molecules from the ChEMBL Database. To simulate mutations, the Y842 residue in FLT3 was replaced with both Cysteine (C) and Phenylalanine (F). Using a custom Python script, these modified FLT3 sequences were combined with the small molecules' SMILES notations, resulting in more than 5.7 million protein-small molecule pairs. These pairs were then evaluated using the pre-trained ConPLex model to predict interaction scores, providing insight into potential binding affinities between the FLT3 variants and the small molecules.
Xepto50: Xepto50 is designed to handle data ranging from a single experiment to multiple experiments, encompassing various cell lines and drugs, all within a single Excel file. The software intelligently detects the number of response columns. When there are two or more response columns, Xepto50 calculates using the mean response for subsequent analyses. If there are three or more response columns, the software not only plots the Standard Error of the Mean (SEM) but also provides functionality to compute and remove outliers. Xepto50 is versatile in its data input capabilities; it can accept response data in the form of viability or inhibition, whether presented as a ratio or a percentage. However, for consistency and ease of analysis, it internally converts all input responses to a format that represents inhibition in percentage terms. For curve fitting and analysis, Xepto50 applies a four-parameter logistic regression function.
Xepto50 offers an integrated solution for analyzing drug response experiments. Initially, the tool employs the curve_fit function from scipy.optimize to fit the data. To further refine this fit, the lmfit model is subsequently utilized. In terms of metrics, Xepto50 is equipped to calculate traditional IC50, interpolated IC50, and Area Under the Curve (AUC). Additionally, it determines Drug Sensitivity Scores, DSS1, DSS2, and DSS3. Of note is the unique "Xepto50 score" introduced by the software. This score is derived by determining the AUC between the interpolated IC50 and the sum of the interpolated IC50 and a constant value. The baseline response value used for this calculation is 50. The result is then normalized by dividing it by the total area spanning between the IC50 and the aforementioned sum of the interpolated IC50 and the constant value.
Ensuring data quality and reliability is of utmost importance. To that end, Xepto50 offers a comprehensive suite of quality scores, including R² Score, Adjusted R² Score, Standard Error of the Estimate (Sy.x), Root Mean Squared Error (RMSE), Shapiro-Wilk Normality Test P-value, Explained Variance Score, Maximum Residual Error, Root Mean Absolute Error (RMAE), and Mean Absolute Percentage Error (MAPE), among others. For user accessibility, Xepto50 features a user-friendly Graphical User Interface (GUI). This ensures a seamless experience even for individuals who may not be versed in programming. The tool is also designed for easy setup within a conda environment. Installation is straightforward: pip install xepto50. Once installed, users can initiate the software by simply entering the command xepto50.
Results
- Identification of FLT3 interacting small molecules using molecular contrastive learning and protein language
Molecular contrastive learning is an emerging technique that has garnered significant attention due to its ability to leverage vast datasets of small molecules for probing molecular interactions. A novel integration of this methodology with protein language modeling was observed in a recent publication [17]. For our analysis, we harnessed an expansive dataset of more than 1.9 million small molecules sourced from the ChEMBL database. These were juxtaposed with the FLT3 kinase domain (FLT3-KD) and its mutants, Y842C and Y842F. Consequently, the analysis involved over 5.7 million unique FLT3-KD-small molecule pairs. Employing a pretrained model within the ConPLex platform, we discerned that 938 small molecules manifested interactions with the FLT3-KD, contingent on a ConPLex interaction score threshold of >0.8. Adopting an identical score threshold, we identified interactions of 930 small molecules with FLT3-KD-Y842C and 923 molecules with FLT3-KD-Y842F (Supplementary Table S1 and Figure 1A). Interestingly, while the interaction scores exhibited no significant statistical divergence between the wild-type FLT3-KD and its mutants, an observable trend emerged. The interaction scores consistently descended in the order of FLT3-KD > FLT3-KD-Y842C > FLT3-KD-Y842F (Figure 1B). This trend insinuates that mutations within the activation loop could potentially modulate the interaction dynamics between inhibitors and the FLT3 kinase domain. Furthermore, the specific characteristics of these mutations may influence the nature of these interactions in distinct ways. Notably, established FLT3 inhibitors like quizartinib, ponatinib, and sorafenib all had interaction scores surpassing 0.8. However, midostaurin had a score of less than 0.6, as presented in Figure 1C.
- Binding Free Energy Analysis of Native and Mutant FLT3 Structures with Drug Molecules Using the MM-PBSA Approach
As we observed a trend in ConPLex interaction scores where mutants displayed slightly compromised interactions, we wanted to use structure-based approaches to measure the effect of point mutations. We have selected three inhibitors: quizartinib, sorafenib, and midostaurin, due to their wide use in FLT3 research. We utilized the MM-PBSA method to compute thermodynamic binding free energies for both native and mutant FLT3 structures interacting with various drug molecules. The native FLT3 protein structure was sourced from the PDB database [8]. We introduced point mutations at position Y842 to create models of the Y842C and Y842F mutant structures. The kinase domain of the native experimental structure, in complex with the inhibitor quizartinib, was chosen as the binding site for our free energy calculations. We docked the molecules quizartinib, sorafenib, and midostaurin onto the specified binding pocket of the native and mutant FLT3 structures. The docked complexes exhibiting the most stable conformations underwent MD simulations, followed by thermodynamic binding free energy calculations (Table 1). Over time, the MM-PBSA method has gained traction and is now a recognized approach for predicting and comparing the binding free energies of various biomolecular structures [18,19,20,21]. Binding free energy inversely relates to the affinity between proteins and ligands. Our analyses revealed that mutations in FLT3 structures influenced the binding free energies. Specifically, the binding free energy dropped for both mutant FLT3 proteins when interacting with sorafenib, compared to the native FLT3-sorafenib complex. In contrast, with midostaurin, the binding free energy for mutant structures was higher than for the native protein complex. Intriguingly, quizartinib presented intermediate binding energy levels in both native and mutant structures. The van der Waals energy was the most significant contributor to overall binding free energy. However, with midostaurin, electrostatic energy had a more favorable contribution in both mutant structures compared to the van der Waals energy. The polar solvation energy component contributions were generally unfavorable for the total binding free energy across all protein-ligand complexes.
- Differential apoptotic responses in FLT3-ITD expressing Ba/F3 cells harboring Y842 mutations
We next aimed to compare the apoptosis responses among different Y842 mutants. To do this, we generated Ba/F3 cells that stably express FLT3-ITD, FLT3-ITD-Y842F, and FLT3-ITD-Y842C. These cells were cultured in the presence of murine Interleukin 3 (IL3), but we removed IL3 prior to adding drugs for the apoptosis assays. Cells were treated with either 1 nM or 5 nM of Quizartinib, Sorafenib, Midostaurin, or the equivalent volume of DMSO, which was used to prepare the drug solutions. Our observations revealed that while the expression of FLT3-ITD alone was sufficient to support the survival of Ba/F3 cells in the absence of IL3, cells expressing FLT3-ITD alongside Y842F or Y842C mutations had approximately 4 times more apoptotic cells (as shown in Figure 2A). Regardless of the Y842 mutations, the treatment with inhibitors enhanced the apoptosis response. Given that different drug-mutant combinations showed varied binding energies (Figure 2B), we calculated the relative apoptosis by subtracting the number of apoptotic cells in the DMSO-treated samples from the total. A similar trend was observed in the samples treated with 50 nM Midostaurin, whereas an opposite trend was evident in the 5 nM Sorafenib-treated samples (Figure 2C). This finding underscore the role of the Y842 mutations in modulating apoptotic responses in Ba/F3 cells expressing FLT3-ITD. Specifically, cells harboring FLT3-ITD alongside Y842F or Y842C mutations demonstrated a heightened apoptotic response, approximately fourfold greater, in comparison to cells expressing only FLT3-ITD. This suggests that the presence of these mutations may render cells more susceptible to apoptosis in the absence of IL3. Interestingly, while drug treatment amplified apoptosis across the board, different drug-mutant combinations exhibited varied responses. The differential binding energies observed for each drug-mutant pair may offer insights into the mechanistic differences in drug efficacy and specificity. Importantly, while Midostaurin at 50 nM followed the general trend, Sorafenib at 5 nM behaved oppositely. This highlights the nuanced interplay between specific mutations and drug treatments, emphasizing the need for personalized therapeutic strategies in targeting FLT3-ITD associated malignancies.
- Evaluation of drug sensitivity metrics and the introduction of the Xepto50 scoring system for enhanced drug efficacy analysis
As apoptotic responses demonstrated a partial correlation with in silico data, our subsequent objective was to assess cell viability to determine drug sensitivity indices. Initially, we quantified the interpolated IC50 and area under the curve (AUC) employing GraphPad Prism 9. Notably, there were no significant disparities in terms of IC50 (represented as -log10IC50, Figure 3A) or AUC (Figure 3B), with the exception that the Y842C mutant exhibited reduced responsiveness to sorafenib. To further assess various metrics, we introduced Xepto50, a robust tool capable of determining IC50, interpolated IC50, AUC, and drug sensitivity scores (DSS1, DSS2, and DSS3) in batch mode from an Excel file input. Xepto50, a Python-based application with a graphical user interface (GUI), exhibited interpolated IC50 and AUC values consistent with those of GraphPad Prism 9 (Supplementary Figure S1B-C). Additionally, the trends observed in DSS1 (Figure 3C), DSS2 (Supplementary Figure S1D), and DSS3 (Supplementary Figure S1E) paralleled those of IC50 and AUC metrics, implying that these drug sensitivity metrics might not fully encapsulate theoretical observations. The four-parameter logistic regression curve remains a prevalent model for gauging drug sensitivity. A lateral shift in this curve denotes reduced potency (Supplementary Figure S1E), whereas a diminished slope indicates compromised cooperativity (Supplementary Figure S1F). Conversely, a vertical shift of the maximum value alludes to heightened efficacy (Supplementary Figure S1G). Beyond these, multiple other curve manifestations can be discerned (Supplementary Figure S1H-J). Given that a drug's impact is an amalgamation of these factors, deriving conclusions from a singular parameter could obscure true drug efficacy. For instance, drugs with identical IC50 values might display stark differences in cooperativity and efficacy (Supplementary Figure S1G-H). However, a perusal of the logistic regression curve could elucidate these nuances. It is crucial to underline that a drug exhibiting low potency might be highly efficacious at elevated concentrations, a nuance potentially overlooked by prevailing scoring techniques. Thus, we advocate for an alternative metric—the Xepto50 score—that gauges the normalized area under the curve at the 50% interpolated value within a specified range. Distinctly, the Xepto50 score remains unaffected by the logistic regression curve's position but is acutely responsive to its shape, rendering it ideal for discerning drug efficacy. Importantly, our findings revealed that the Xepto50 score better mirrors apoptosis response and theoretical values (Figure 3D).
Discussion
The advancements in molecular modeling, combined with the rise of machine learning in drug discovery, are poised to bring transformative changes to pharmacology. Among these innovations, molecular contrastive learning stands out as a burgeoning technique, demonstrating its aptitude in deciphering vast molecular interactions with remarkable accuracy. In line with findings from prior studies [17], our research capitalizes on the extensive dataset of small molecules sourced from ChEMBL, shedding light on interactions within the FLT3 kinase domain. We observed a distinct trend in interaction scores, descending in the sequence of FLT3-KD > FLT3-KD-Y842C > FLT3-KD-Y842F. This pattern indicates that mutations within the activation loop might be instrumental in altering inhibitor interactions with the FLT3 kinase domain. Additionally, the protein language model discerned variations resulting from amino acid alterations in the protein sequences. Given the established knowledge that protein mutations can profoundly impact therapeutic outcomes [22,23,24], it is crucial to recognize and comprehend these nuanced genetic shifts when considering therapeutic strategies.
Moreover, the application of the MM-PBSA method, a widely acknowledged technique, reaffirmed the impact of point mutations on binding free energies [25,26]. The variable free energy readings between native and mutant structures, in the presence of different inhibitors, might elucidate some mechanistic underpinnings of the observed efficacy differences. This could help inform inhibitor selections based on specific mutation profiles. Furthermore, our empirical findings in Ba/F3 cells highlighted the functional implications of the Y842 mutations. Their increased apoptotic responses, especially in the absence of IL3, suggest that these mutations might render the cells more vulnerable to therapeutic interventions. These data further advocate for the development of personalized therapeutic regimes. Drug-specific responses, especially the contrasting behavior of Midostaurin and Sorafenib, serve as an important reminder of the intricate and multifaceted interactions between drugs and their molecular targets.
Apart from the established theoretical values, our exploration into comparing drug sensitivity both at apoptosis and viability levels unveiled some inconsistencies with theoretical predictions. Specifically, while Quizartinib and Midostaurin exhibited higher congruence with theoretical values, the cellular response to Sorafenib did not align with its predicted theoretical binding energy. This disparity may either highlight the limitations of our theoretical models or suggest that Sorafenib interacts at different sites within the kinase domain, especially given that we utilized the Quizartinib association site for docking.
Moreover, our findings indicate that traditional drug sensitivity metrics might not consistently represent real-world outcomes. The assessment of drug sensitivity metrics, punctuated by the introduction of the Xepto50 scoring system, has addressed a longstanding challenge in drug discovery. Although widely used metrics like IC50 provide invaluable perspectives, they occasionally miss capturing the entire spectrum of drug efficacy. This gap becomes pronounced in situations where drugs have similar IC50 values but divergent mechanisms of action. Given the Xepto50 score's emphasis on curve shapes rather than mere positions, it promises a more comprehensive insight into drug mechanisms. By leveraging such advanced metrics, the drug development process could be refined, paving the way for therapies that are both potent and adaptive to diverse mechanisms of action.
In conclusion, our findings underscore the potential of leveraging advanced molecular modeling techniques, reinforced with empirical validations, to enhance our understanding of drug-target interactions. The discerning insights obtained from such analyses, when combined with innovative metrics like Xepto50, can pave the way for more informed and effective therapeutic strategies. Future studies could further delve into the mechanistic intricacies of these interactions, potentially revealing novel therapeutic targets or strategies to combat FLT3-ITD-associated malignancies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Drug Sensitivity Scores. (A) Four-parameter logistic regression curves plotted using GraphPad Prism 9. (B-E) Drug sensitivity metrics including interpolated IC50 (B), area under the curve (C), as well as drug sensitivity scores DSS2 (D) and DSS3 (E), were determined using the Xepto50 application. (F-K) Sample plots were produced using Python scripts
Author Contributions
J.U.K, L.A.A., R.P., and L.R. designed and performed the experiments, collected and analyzed the data, and J.U.K., R.P. and L.R. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Crafoord Foundation (J.U.K), the Swedish Cancer Society (J.U.K. and L.R.), the Swedish Research Council (L.R.), the Swedish Childhood Cancer Foundation (J.U.K.), and SUS Stiftelser och Donationer (L.R.), Governmental Funding of Clinical Research within the National Health Service (ALF) (L.R.).
Data and code availability
The Python library, Xepto50, can be obtained from the Python Package Index (PyPI) at www.pypi.org/project/xepto50/, or from the GitHub repository at www.github.com/kazilab/xepto50.
Acknowledgments
We thank the Lund University Library for their generous assistance in covering the costs associated with open-access publication.
References
- J.U. Kazi, L. Rönnstrand, FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications, Physiol Rev, 99 (2019) 1433-1466.
- L.R. Klug, J.D. Kent, M.C. Heinrich, Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases, Pharmacol Ther, 191 (2018) 123-134. [CrossRef]
- J.U. Kazi, R.A. Chougule, T. Li, X. Su, S.A. Moharram, K. Rupar, A. Marhall, M. Gazi, J. Sun, H. Zhao, L. Rönnstrand, Tyrosine 842 in the activation loop is required for full transformation by the oncogenic mutant FLT3-ITD, Cell Mol Life Sci, 74 (2017) 2679-2688. [CrossRef]
- C.C. Smith, Q. Wang, C.S. Chin, S. Salerno, L.E. Damon, M.J. Levis, A.E. Perl, K.J. Travers, S. Wang, J.P. Hunt, P.P. Zarrinkar, E.E. Schadt, A. Kasarskis, J. Kuriyan, N.P. Shah, Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia, Nature, 485 (2012) 260-263. [CrossRef]
- T. Kindler, F. Breitenbuecher, S. Kasper, E. Estey, F. Giles, E. Feldman, G. Ehninger, G. Schiller, V. Klimek, S.D. Nimer, A. Gratwohl, C.R. Choudhary, C. Mueller-Tidow, H. Serve, H. Gschaidmeier, P.S. Cohen, C. Huber, T. Fischer, Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML), Blood, 105 (2005) 335-340. [CrossRef]
- J. Griffith, J. Black, C. Faerman, L. Swenson, M. Wynn, F. Lu, J. Lippke, K. Saxena, The structural basis for autoinhibition of FLT3 by the juxtamembrane domain, Mol Cell, 13 (2004) 169-178. [CrossRef]
- N.J. Dibb, S.M. Dilworth, C.D. Mol, Switching on kinases: oncogenic activation of BRAF and the PDGFR family, Nat Rev Cancer, 4 (2004) 718-727. [CrossRef]
- J.A. Zorn, Q. Wang, E. Fujimura, T. Barros, J. Kuriyan, Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220), PLoS One, 10 (2015) e0121177. [CrossRef]
- M.V. Shapovalov, R.L. Dunbrack, Jr., A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions, Structure, 19 (2011) 844-858. [CrossRef]
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B.A. Shoemaker, P.A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang, E.E. Bolton, PubChem in 2021: new data content and improved web interfaces, Nucleic Acids Res, 49 (2021) D1388-D1395. [CrossRef]
- G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell, A.J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J Comput Chem, 30 (2009) 2785-2791. [CrossRef]
- A.W. Schuttelkopf, D.M. van Aalten, PRODRG: a tool for high-throughput crystallography of protein-ligand complexes, Acta Crystallogr D Biol Crystallogr, 60 (2004) 1355-1363.
- S.W. Chiu, S.A. Pandit, H.L. Scott, E. Jakobsson, An improved united atom force field for simulation of mixed lipid bilayers, J Phys Chem B, 113 (2009) 2748-2763. [CrossRef]
- G. Bussi, D. Donadio, M. Parrinello, Canonical sampling through velocity rescaling, J Chem Phys, 126 (2007) 014101. [CrossRef]
- M. Parrinello, A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, Journal of Applied Physics, 52 (1981) 7182-7190. [CrossRef]
- R. Kumari, R. Kumar, C. Open Source Drug Discovery, A. Lynn, g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations, J Chem Inf Model, 54 (2014) 1951-1962.
- R. Singh, S. Sledzieski, B. Bryson, L. Cowen, B. Berger, Contrastive learning in protein language space predicts interactions between drugs and protein targets, Proc Natl Acad Sci U S A, 120 (2023) e2220778120. [CrossRef]
- A.R. Brice, B.N. Dominy, Analyzing the robustness of the MM/PBSA free energy calculation method: application to DNA conformational transitions, J Comput Chem, 32 (2011) 1431-1440. [CrossRef]
- S.P. Brown, S.W. Muchmore, Large-scale application of high-throughput molecular mechanics with Poisson-Boltzmann surface area for routine physics-based scoring of protein-ligand complexes, J Med Chem, 52 (2009) 3159-3165. [CrossRef]
- V.K. Bhardwaj, R. Purohit, A lesson for the maestro of the replication fork: Targeting the protein-binding interface of proliferating cell nuclear antigen for anticancer therapy, J Cell Biochem, 123 (2022) 1091-1102. [CrossRef]
- V.K. Bhardwaj, A. Oakley, R. Purohit, Mechanistic behavior and subtle key events during DNA clamp opening and closing in T4 bacteriophage, Int J Biol Macromol, 208 (2022) 11-19. [CrossRef]
- O. Lindblad, E., Cordero, A. Puissant, L. Macaulay, A. Ramos, N.N. Kabir, J. Sun, J. Vallon-Christersson, K. Haraldsson, M.T. Hemann, A. Borg, F. Levander, K. Stegmaier, K. Pietras, L. Rönnstrand, J.U. Kazi, Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML, Oncogene, 35 (2016) 5119-5131. [CrossRef]
- J.U. Kazi, R.A. Chougule, T. Li, X. Su, S.A. Moharram, K. Rupar, A. Marhall, M. Gazi, J. Sun, H. Zhao, L. Rönnstrand, Tyrosine 842 in the activation loop is required for full transformation by the oncogenic mutant FLT3-ITD, Cell Mol Life Sci, 74 (2017) 2679-2688. [CrossRef]
- J.U. Kazi, L. Rönnstrand, FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications, Physiol Rev, 99 (2019) 1433-1466.
- G. Poli, C. Granchi, F. Rizzolio, T. Tuccinardi, Application of MM-PBSA Methods in Virtual Screening, Molecules, 25 (2020). [CrossRef]
- C. Wang, D. Greene, L. Xiao, R. Qi, R. Luo, Recent Developments and Applications of the MMPBSA Method, Front Mol Biosci, 4 (2017) 87. [CrossRef]
Figure 1.
Interaction dynamics of FLT3 kinase domain and its mutants with small molecules. A) Distribution of interaction scores for over 5.7 million unique FLT3-KD-small molecule pairs sourced from the ChEMBL database. ConPLex platform with a threshold score of >0.8 was used to identify interactions between small molecules and FLT3 kinase domains. B) Trend analysis of interaction scores, revealing a descending order from FLT3-KD > FLT3-KD-Y842C > FLT3-KD-Y842F. C) Specific interaction scores for FLT3 inhibitors.
Figure 1.
Interaction dynamics of FLT3 kinase domain and its mutants with small molecules. A) Distribution of interaction scores for over 5.7 million unique FLT3-KD-small molecule pairs sourced from the ChEMBL database. ConPLex platform with a threshold score of >0.8 was used to identify interactions between small molecules and FLT3 kinase domains. B) Trend analysis of interaction scores, revealing a descending order from FLT3-KD > FLT3-KD-Y842C > FLT3-KD-Y842F. C) Specific interaction scores for FLT3 inhibitors.
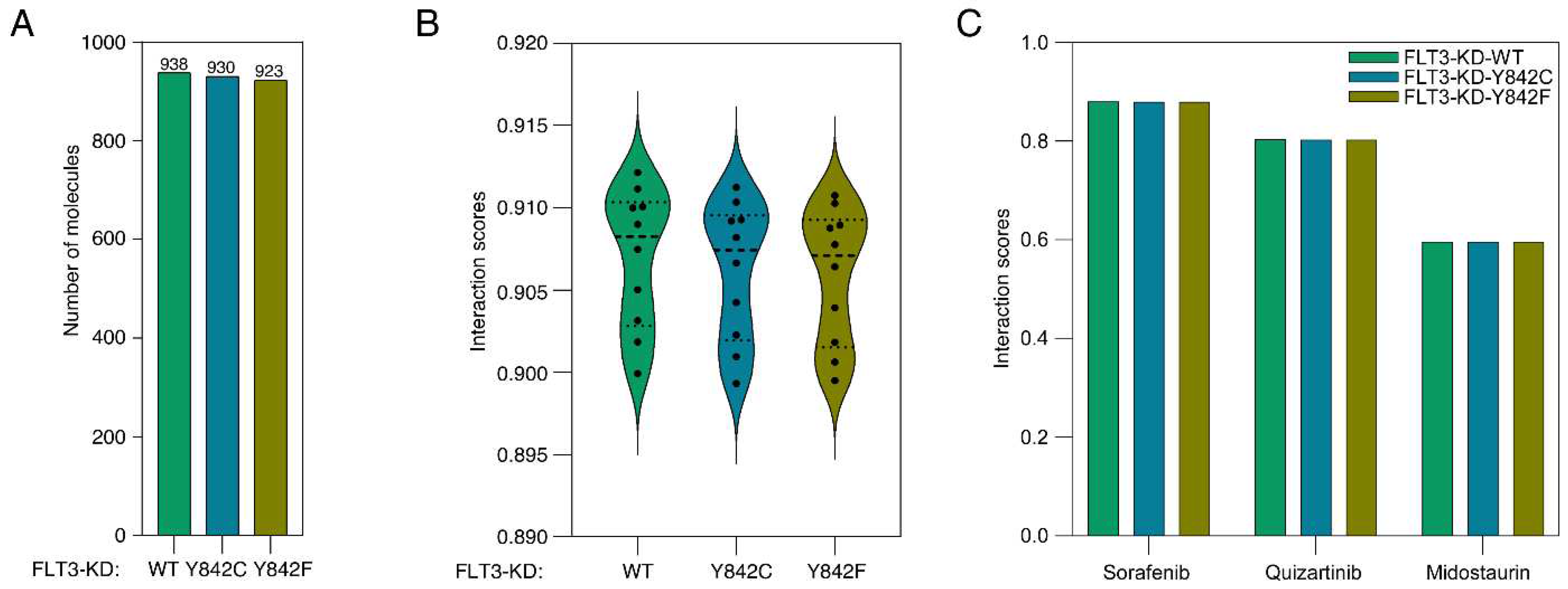
Figure 2.
Differential apoptotic responses and binding energy analyses of FLT3-ITD Y842 mutants under drug treatments. (A) Measurement of apoptotic cells using the annexin V-7-AAD kit after treating cells with specific inhibitors for 48 hours prior to processing and analysis. (B) Binding energy, represented as negative values, is plotted against various drug-mutant pairs. (C) Calculation of relative apoptotic cells by subtracting the number of apoptotic cells observed in DMSO-treated controls from those treated with specific inhibitors.
Figure 2.
Differential apoptotic responses and binding energy analyses of FLT3-ITD Y842 mutants under drug treatments. (A) Measurement of apoptotic cells using the annexin V-7-AAD kit after treating cells with specific inhibitors for 48 hours prior to processing and analysis. (B) Binding energy, represented as negative values, is plotted against various drug-mutant pairs. (C) Calculation of relative apoptotic cells by subtracting the number of apoptotic cells observed in DMSO-treated controls from those treated with specific inhibitors.
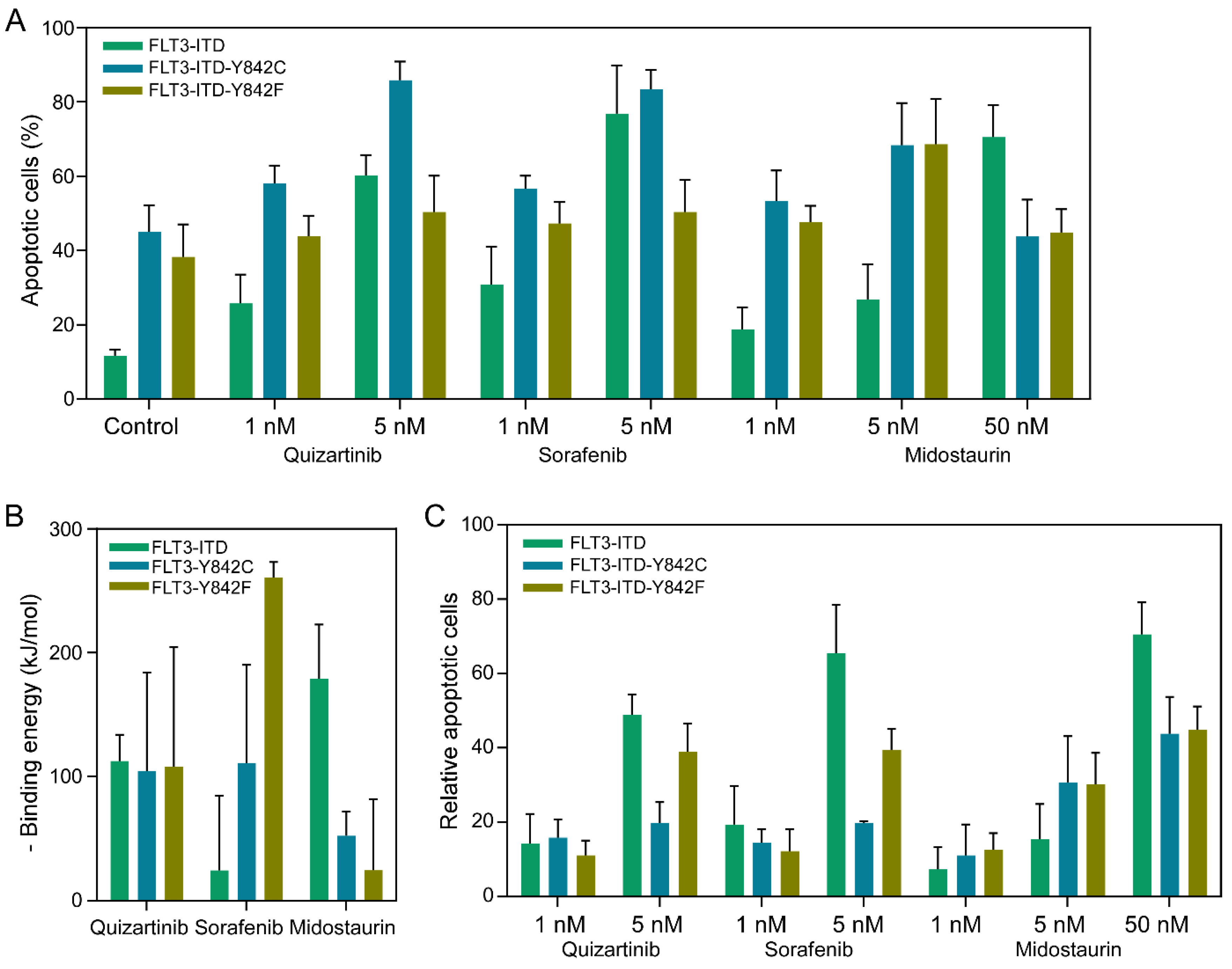
Figure 3.
Assessment of various drug sensitivity metrics. (A) IC50 values were determined using GraphPad Prism 9, derived from interpolated values at 50 and subsequently transformed to a negative log10 scale. (B) The area under the curve (AUC) was computed from the same dataset, with a baseline response set at 10. (C) Drug Sensitivity Score 1 (DSS1) was determined using the Xepto50 software. (D) The Xepto50 score was derived from the normalized AUC at a specific interval on a logarithmic concentration axis.
Figure 3.
Assessment of various drug sensitivity metrics. (A) IC50 values were determined using GraphPad Prism 9, derived from interpolated values at 50 and subsequently transformed to a negative log10 scale. (B) The area under the curve (AUC) was computed from the same dataset, with a baseline response set at 10. (C) Drug Sensitivity Score 1 (DSS1) was determined using the Xepto50 software. (D) The Xepto50 score was derived from the normalized AUC at a specific interval on a logarithmic concentration axis.
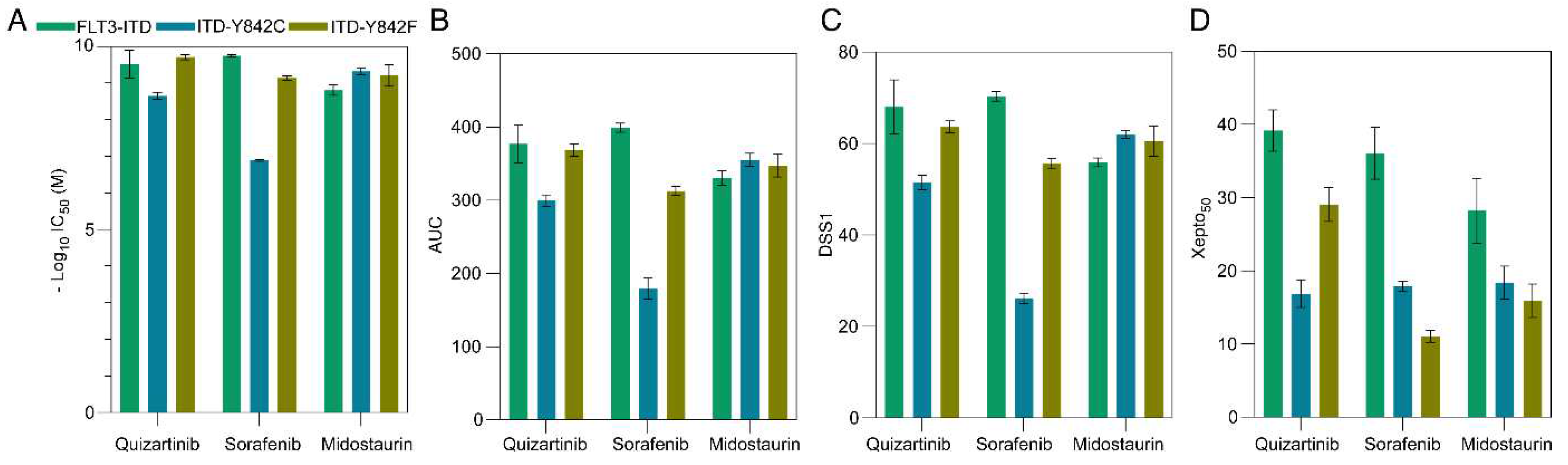

Table 1.
The thermodynamic binding free energy and its constituents calculated by the MM-PBSA approach.
Table 1.
The thermodynamic binding free energy and its constituents calculated by the MM-PBSA approach.
| Native FLT3 Protein | FLT3 Inhibitors | van der Waal energy (kJ/mol) | Electrostatic energy (kJ/mol) | Polar solvation energy (kJ/mol) | SASA energy (kJ/mol) | Binding energy (kJ/mol) |
| Native | Quizartinib | -190.70± 15.48 | -110.75 ± 24.56 | 206.74 ± 34.49 | -17.72± 1.28 | -112.45± 21.08 |
| Sorafenib | - 42.12± 78.17 | - 7.15± 14.10 | 28.66± 48.37 | - 3.65± 7.00 | - 24.27± 60.15 | |
| Midostaurin | -326.70± 49.07 | -199.75± 40.48 | 372.96± 58.41 | -25.37± 3.19 | -178.85± 43.74 | |
| Mutant I (Y842C) |
Quizartinib | -258.69± 74.61 | - 12.69± 29.22 | 186.68± 26.86 | -19.79± 3.54 | -104.49± 79.30 |
| Sorafenib | -140.86±107.91 | - 13.04± 16.37 | 54.82± 53.67 | -11.69± 6.83 | -110.76± 77.65 | |
| Midostaurin | -116.65± 66.32 | -132.48±106.74 | 209.11±177.76 | -12.02± 6.89 | - 52.04± 19.61 | |
| Mutant II (Y842F) |
Quizartinib | -179.77±134.54 | - 53.30± 43.73 | 139.41±109.23 | -14.19± 9.53 | -107.85± 96.60 |
| Sorafenib | -329.85± 12.03 | - 70.41± 9.66 | 162.84± 11.38 | -23.24± 0.87 | -260.67± 12.46 | |
| Midostaurin | -120.79±113.89 | -121.86±115.33 | 228.13±196.64 | -10.12± 9.78 | -24.643± 57.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



