
Submitted:
13 October 2023
Posted:
16 October 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Severe COVID-19 is frequently associated with thromboembolic complications. Increased platelet activation and platelet-leukocyte aggregate formation can amplify thrombotic responses by inducing tissue factor (TF) expression on leukocytes. Here, we characterized TF-positive extracellular vesicles (EVs) and their cellular origin in 12 patients suffering from severe COVID-19 (time course, 134 samples overall) and 25 healthy controls. EVs exposing phosphatidylserine (PS) were characterized by flow cytometry. Their cellular origin was determined by staining with anti-CD41, anti-CD45, anti-CD235a, and anti-CD105 as platelet, leukocyte, red blood cell, and endothelial markers. We further investigated the association of EVs with TF, platelet factor 4 (PF4), C-reactive protein (CRP) and high mobility group box-1 protein (HMGB-1). COVID-19 patients showed higher levels of PS-exposing EVs compared to controls. The majority of these EVs originated from platelets. A higher amount of EVs in patient samples were associated with CRP, HMGB-1, PF4, and TF, as compared to EVs from healthy donors. In COVID-19 samples, 16.5% of all CD41+ EVs displayed the leukocyte marker CD45, and 55.5% of all EV aggregates (CD41+CD45+) co-expressed TF, which reflects the interaction of platelets and leukocytes in COVID-19 on an EV level.
Keywords:
platelet-leukocyte aggregates
; extracellular vesicles
; tissue factor
; COVID-19
; immunothrombosis
1. Introduction
Thrombotic events and endothelial dysfunction are key pathomechanisms of coronavirus disease-19 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1,2]. In severe COVID-19, deregulated complement activation enhances the recruitment of neutrophils to the infected lungs and promotes tissue factor (TF) expression on neutrophils, monocytes, and endothelial cells, resulting in microvascular thrombosis and endothelial dysfunction [1,3,4]. These events are best described as immunothrombosis, a simultaneous overactivation of the innate immune response and the coagulation system [5]. Activated platelets are main effectors of immunothrombosis and interact with leukocytes to promote pro-inflammatory and pro-coagulant responses [4]. Platelets from COVID-19 patients show a hyperreactive phenotype characterized by the release of platelet factor 4 (PF4), platelet-derived growth factor (PDGF) and thromboxane A2, as well as increased P-selectin expression, which mediates platelet-leukocyte aggregate formation via P-selectin glycoprotein ligand (PSGL)-1 [6,7,8,9,10]. The enhanced aggregate formation of platelets with monocytes or neutrophils in COVID-19 has been correlated with poor prognosis [7,9,11].
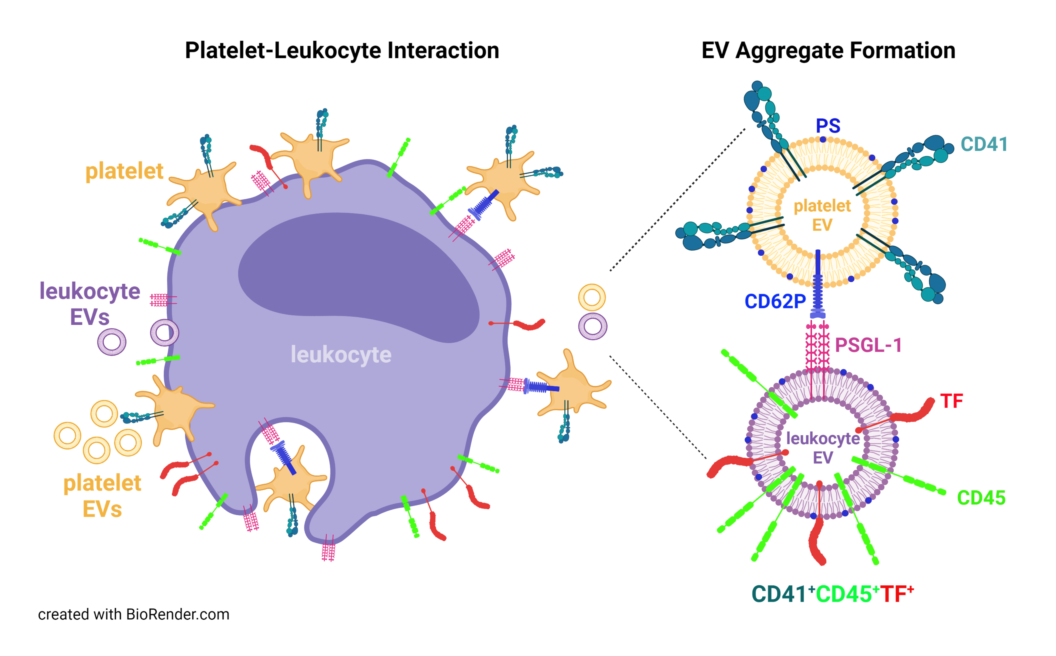
The platelet-leukocyte crosstalk can boost the dysregulated cytokine and chemokine response seen in COVID-19 and triggers TF expression on monocytes, neutrophils, as well as endothelial cells [6]. Moreover, aggregate formation of platelets with monocytes drives TF expression on monocytes via P-selectin and integrin αIIb/β3-dependent signaling and is amplified by TNF-α and IL-1β [12]. Likewise, platelets can induce TF expression on neutrophils and promote the release of neutrophil extracellular traps (NETs), representing a scaffold of expelled nuclear DNA and histones that captures and kills pathogens by antimicrobial proteins and proteolytic enzymes [13]. NETs support the pro-thrombotic phenotype by their ability to initiate coagulation, either by presenting TF [14] or by triggering the contact-dependent pathway via factor XII [15]. NETs promote platelet binding and the formation of platelet-neutrophil aggregates, which fuel further NET release and thus exacerbate endothelial damage and thrombotic complications [6,16,17]. Upon activation, platelets as well as neutrophils and monocytes release extracellular vesicles (EVs) into the circulation, which are important mediators of thrombosis due to their exposure of phosphatidylserine (PS) [18,19,20] and TF [21,22,23,24,25]. Furthermore, EVs exert immunomodulatory functions by transferring regulatory and pro-inflammatory molecules, such as nucleic acids, C-reactive protein (CRP) or high mobility group box-1 protein (HMGB-1) [26,27,28].
Here, we characterized the cellular origin of PS-exposing EVs in plasma from COVID-19 patients, as well as their association with inflammation- and coagulation-related parameters, including CRP, HMGB-1, PF4, and TF. We identified TF-bearing EVs co-expressing platelet (CD41) and leukocyte (CD45) markers, reflecting the recently described platelet-leukocyte aggregate formation in severe COVID-19 on the EV level.
2. Results
2.1. Patient Characteristics
In total, 12 critically ill COVID-19 patients receiving mechanical ventilation were included in this study. Twenty-five healthy individuals served as controls. Baseline characteristics and comorbidities are summarized in Table 1. The medication regimens for individual patients are detailed in Supplementary Table S1. The mean age of COVID-19 patients was 79.5 [77.0–82.9] years and the majority of patients were males (83 %). Secondary bacterial (E. coli) and/or fungal (Aspergillus fumigatus) infections were confirmed in 58 % of all patients. The overall mortality was 58 %. The time points of sample collection for individual patients are shown in Figure 1.
2.2. Cellular Origin of Extracellular Vesicles
COVID-19 patients showed significantly increased levels of PS-exposing circulating EVs compared to healthy donors (204,000 [122,375–450,000] vs. 53,500 EVs/µL, Figure 2). These EVs were derived from platelets (CD41+; 59,465 [25,980–112,155] vs. 9000 [5850–10,913] EVs/µL), red blood cells (CD235a+; 12,385 [7266–57,816] vs. 5400 [4193–7985] EVs/µL), leukocytes (CD45+; 11,958 [7359–22,659] vs. 2450 [1830–3378] EVs/µL), and endothelial cells (CD105+; 9598 [5843–22,898] vs. 2400 [1788–3335] EVs/µL; see also Supplementary Figures S1 and S2 for the time course of EV concentrations). Regarding the relative abundance of PS-exposing EVs in COVID-19 patients, 26.0 [16.0–33.0] % originated from platelets, 7.0 [4.0–21.3] % from red blood cells, 6.0 [4.0–8.0] % from leukocytes, and 5.0 [4.0–5.0] % from endothelial cells. CD36, which has been linked to platelet hyperactivation [28,29], was expressed on 12 [9.0–14.0] % of all EVs.
2.3. Association of Extracellular Vesicles with CRP, PF4, and HMGB-1
Next to the characterization of their cellular origin, we assessed the association of EVs with CRP, PF4, and HMGB-1, which are mediators of immunothrombosis (Figure 3). COVID-19 patients showed significantly increased levels of CRP+ EVs, which represented 43.0 [19.0–57.0] % of the total count of PS-exposing EVs, vs. 4.0 [3.0–7.5] % in healthy controls.
Albeit only a small percentage of EVs was associated with PF4 (COVID-19, 3.0 [3.0–4.0] %; healthy controls, 2.0 [0.5–3.0] %), PF4+ EVs were significantly elevated in COVID-19 patients compared to healthy donors (7525 [3878–16,385] vs. 1050 [170–2280] EVs/µL).
HMGB-1 was also detected on EVs. Compared to healthy controls, COVID-19 patients had significantly higher concentrations of HMGB-1+ EVs (15,443 [8548–36,240] vs. 1980 [1500–2688] EVs/µL).
2.4. Tissue Factor Expression on Platelet- and Leukocyte-Derived Extracellular Vesicles
In COVID-19 patients, 16.5 [9.0–22.5] % of all PS-exposing platelet EVs were CD41+ and CD45+, providing evidence for aggregate formation between platelet- and leukocyte-derived EVs, whereas only 3.4 [1.6–8.5] % of CD41+CD45+ EVs were detected in healthy donors (Figure 4). Remarkably, 55.5 [42.9–69.2] % of all EV aggregates (CD41+CD45+) in COVID-19 patients co-expressed TF, while the percentage of TF+ EV aggregates remained below the detection limit in healthy donors. For individual patients, the percentages of CD41+CD45+TF+ EVs ranged from 25.0–76.5%. EVs from COVID-19 patients expressed TF to a significantly higher extent than EVs from healthy donors (18,925 [10,053,33,028] vs. 1880 [1335–3945] EVs/µL, respectively). In the COVID-19 group, 8.0 [6.0–11.0] % of all EVs were associated with TF as compared to 4.0 [2.0–5.0] % in healthy donors.
2.5. Time Course of Coagulation- and Inflammation-Related Mediators
Statistical modelling of the time effect on coagulation- and inflammation-related mediators revealed a decrease over time in leukocytes, neutrophils, platelets, PF4, EV counts, CRP+ EVs, TF+ EVs, as well as IP-10 in COVID-19 patients with a probability of >95 % (Table 2; see also Supplementary Figures S3 and S4 for the time course of individual patients). IL-8, IL-1β, G-CSF, nucleosomes, TNF-α, D-dimer, IL-10, and HMGB-1 levels increased over time with a probability in the range of 100.0–92.7 %.
3. Discussion
Immunothrombosis, the joint over-activation of the innate immune response and coagulation, is a central pathomechanism in sepsis and severe COVID-19. Complement activation and cytokine release, platelet hyperactivity, as well as coagulopathy play critical roles in this complex scenario [31,32]. In particular, the crosstalk of platelets and leukocytes has been shown to amplify inflammatory effector functions, as discussed in more detail below [9,17].
Increased levels of circulating EVs released from activated blood cells are well documented in sepsis and severe COVID-19 [33]. EVs released from the plasma membrane expose PS and support coagulation by catalyzing the formation of the tenase and prothrombinase complexes of the coagulation cascade [19]. Therefore, we focused on the characterization of this EV population in our study. Our data confirm the presence of increased levels of circulating PS-exposing EVs in patients with severe COVID-19. In line with previous reports [10,24,34,35], the majority of these EVs originated from platelets.
While flow cytometry is versatile and well established to characterize EVs in plasma samples, the limitations of this approach have to be taken into account. There is evidence that Anx5, which we used to detect PS-exposing EVs in this study, also labels apolipoprotein B-containing lipoproteins, such as low-density lipoprotein [36], challenging the use of Anx5 to uniquely identify EVs in lipoprotein-containing samples. This is why we combined the PS-based approach for EV detection with additional membrane-bound markers (CD41, CD45, CD235a, CD105), which are not exposed on lipoproteins. Moreover, the fact that we compared TF expression on EVs in patient samples to samples from healthy donors greatly limits a potential bias in our results, since lipoproteins would have been present in the control samples, as well.
We found that EVs from COVID-19 patients carry CRP, PF4, and HMGB-1 on their surface, which may further fuel immunothrombosis. Elevated levels of CRP+ EVs have been described in sepsis, but also in myocardial infarction, and several studies have highlighted the pro-inflammatory characteristics of these CRP-bearing EVs [26,37,38]. Sustained platelet activation in COVID-19 boosts the release of PF4 and HMGB-1, inducing neutrophil activation and the release of NETs, which strongly promote coagulation [4,10,17,39]. Platelet-derived HMGB-1+ EVs have been reported as markers of platelet activation and are associated with a poor prognosis in COVID-19 patients [40]. Furthermore, we found increased levels of CD36+ EVs in COVID-19 patients. Platelet glycoprotein CD36 acts as a receptor for membrane-derived EVs, as it binds to PS on their surface [30], resulting in platelet activation, aggregation, and thrombus formation [29]. Accordingly, increased CD36 expression indicates a higher risk of venous and arterial thromboembolism [41].
Next to characterizing PS-exposing EVs, which propagate coagulation as discussed above, we focused on the expression of TF on EVs, which is the main initiator of coagulation. Monocytes represent the predominant source of blood-borne TF, which can be passed on to EVs originating from these cells [42,43]. While we were not able to obtain samples for all time points from each COVID-19 patient due to extubation or death, our data show that each patient had increased levels of TF+ EVs at all available time points in comparison to healthy controls. While several previous studies have also reported increased levels of TF-expressing EVs in COVID-19 patients, which correlated with disease severity [21,24,43], others failed to detect differences in TF+ EVs between COVID-19 patients and healthy controls [35]. These conflicting results may at least in part be attributed to different antibody clones used for the detection of TF or to different fluorochrome-to-protein ratios of antibody-fluorochrome conjugates used for the flow cytometric detection of TF+ EVs.
Enhanced platelet-leukocyte aggregate formation is known to occur in various pathological conditions, including sepsis and COVID-19, where increased platelet-monocyte interaction has been linked to disease severity [39]. Our data suggest that the enhanced interaction of platelets and leukocytes in COVID-19 is also reflected on the EV level. About 17 % of all platelet-derived EVs displayed the leukocyte marker CD45, suggesting aggregate formation between platelet-derived and leukocyte-derived EVs. Notably, 56 % of all CD41+CD45+ EVs expressed TF, likely of monocyte or neutrophil origin. It is well established that activated platelets express P-selectin and interact with monocytes through PSGL-1 [44], a mechanism which may also mediate aggregate formation between platelet- and leukocyte-derived EVs. Platelets induce TF expression on monocytes through P-selectin and integrin αIIb/β3 signaling [39] and stimulate the release of TF-conveying NETs by neutrophils [45]. In turn, increased TF expression has been associated with an upregulation of CD16 on monocytes, inducing a shift from CD16– classical monocytes, which are mainly phagocytic, towards inflammatory CD16+ intermediate and non-classical monocytes [12].
Platelet-monocyte aggregates also promote the secretion of inflammatory mediators. Incubation of platelets from COVID-19 patients with monocytes from healthy donors triggers the release of IL-1β, IL-6, IL-8, MIP-1α, MCP-1, TNF-α, PF4, and PDGF [12,39], and elevated levels of these factors were observed in clinical samples from COVID-19 patients [46]. Likewise, our study cohort presented increased levels of inflammatory mediators that dynamically changed during the course of mechanical ventilation, with an increase of IL-8, IL-1β, G-CSF, nucleosomes, TNF-α, D-dimer, IL-10, and HMGB-1 over time. A similar time-dependent increase has been reported for IL-6, IL-8, IL-1β, and TNF-α in COVID-19 [47]. We further observed decreasing platelet, leukocyte, and neutrophil counts in mechanically ventilated COVID-19 patients over time.
To conclude, we report here for the first time that TF-expressing platelet- and leukocyte-derived EV aggregates are present in severely ill COVID-19 patients, and we propose that these aggregates may act as amplifiers of immunothrombosis.
4. Materials and Methods
4.1. Patients and Sample Collection
Twelve patients with PCR-confirmed or suspected SARS-CoV-2 infection, requiring mechanical ventilation were included in this study at the Department of Internal Medicine, Hospital St. Vinzenz, Zams, Austria between November 2020 and January 2021 [32]. Sample collection was approved by the ethics committee of the Medical University of Innsbruck (1144/2020). The study was conducted in accordance with the declaration of Helsinki. Whole blood samples anticoagulated with EDTA (S-Monovette® K3 EDTA, Sarstedt, Nümbrecht, Germany) were obtained during routine blood collection every 24 h. Overall 134 samples were collected. The control group (n=25) consisted of healthy individuals from whom EDTA-anticoagulated blood was obtained after written consent. Platelet-poor plasma was obtained by centrifugation of whole blood at 2000 x g for 15 min at 22°C and stored at –80°C until further analysis. Routine laboratory measurements (C-reactive protein, CRP; procalcitonin, PCT; D-dimer) and blood cell counts were obtained as part of standard medical care. Non-survival of patients was defined as death during mechanical ventilation or within 14 days following extubation.
4.2. Flow Cytometric Characterization of Phosphatidylserine-Exposing Extracellular Vesicles
Due to their pro-coagulant properties, we focused on PS-exposing EVs, which represent a subpopulation of larger EVs derived from the cell membrane (“microvesicles”). These EVs were characterized by flow cytometry using a CytoFLEX LX device (Beckman Coulter, Brea, CA) equipped with 405 nm, 488 nm, 561 nm, and 631 nm lasers. Calibration of the flow cytometer was performed with fluorescent silica beads (1 μm, 0.5 μm, 0.1 μm; excitation/emission 485/510 nm; Kisker Biotech, Steinfurt, Germany). The triggering signal for EVs was set to the violet side scatter (405 nm), and the EV gate was set below the 1 µm bead cloud as previously described [48,49].
For staining, plasma samples were diluted 1:100 in 0.1 µm sterile-filtered Annexin V (Anx5) binding buffer (BD Biosciences, San Jose, CA). The cellular origin of EVs was assessed by staining of the diluted samples (100 μL each; 30 min; room temperature; in the dark) with a combination of 2 µL PC7-conjugated anti-CD41 (Beckman Coulter) as platelet marker, 2 µL FITC-conjugated anti-CD235a (eBioscience, San Diego, CA) as red blood cell marker, 5 µL PE-conjugated anti-CD105 (Becton Dickinson, Franklin Lakes, NJ) as endothelial marker, 2 µL PB-conjugated anti-CD45 (Beckman Coulter) as leukocyte marker, and 2.5 µL AF700-conjugated anti-hCD36 (BioLegend) to detect platelet glycoprotein IV. The association of EVs with coagulation- and inflammation-related mediators was assessed by staining aliquots of the diluted samples (100 µL each; 30 min; room temperature; in the dark) with 1 µL FITC-conjugated anti-CRP (Abcam) to detect CRP, 5 µL AF700-conjugated anti-hHMGB-1 (R&D Systems) to detect HMGB-1, and 2.5 µL PE-conjugated anti-hCXCL4 (R&D Systems) to detect PF4. Calibration and all controls for the flow cytometric characterization of EVs are shown in Supplementary Figure S5.
The co-expression of TF on EVs of platelet and leukocyte origin was assessed by staining with 2 µL FITC-conjugated anti-hTF (Biomedica Diagnostics, Stamford, CT), 2 µL PC7-conjugated anti-CD41, and 2 µL PB-conjugated anti-CD45 for 30 min at 4°C in the dark. APC-conjugated Anx5 (2 µL, BD Biosciences) was used as marker for EVs exposing PS. The gating strategy to define TF+ EVs is shown in Supplementary Figure S6. Prior to use, all fluorochrome conjugates were centrifuged at 18,600 x g for 10 min at 4°C to remove eventual precipitates. All fluorochrome conjugates as well as the respective antibody clones are listed in Table 3.
Prior to analysis, stained samples were diluted 1:5 in 0.1 µm sterile-filtered Anx5 binding buffer. Acquisition was performed for 2 min at a flow rate of 10 µL/min and Anx5 positive events in the EV gate were quantified. Data were analyzed using the Kaluza Software 2.1 (Beckman Coulter). Further details on the flow cytometric characterization of EVs are reported according to the MIFlowCyt-EV framework in Supplementary Table S2.
4.3. Quantification of Cytokines, Chemokines, and Growth Factors
A total of 27 cytokines, chemokines, and growth factors were analyzed using a magnetic bead array assay (Bio-Plex ProTM human cytokine 27-plex; Bio-Rad, Vienna, Austria), including interleukin (IL)-1β, IL-1 receptor antagonist (IL-1ra), IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17A, interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1 alpha and beta (MIP-1α, MIP-1β), regulated on activation, normal T-cell expressed and secreted (RANTES), eosinophil chemotactic protein (eotaxin), interferon-inducible protein 10 (IP-10), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), basic fibroblast growth factor (bFGF), platelet derived growth factor (PDGF), as well as vascular endothelial growth factor (VEGF). Plasma samples were diluted 1:4 with sample diluent and analyzed according to the manufacturer instructions.
4.4. Quantification of PF4, HMGB-1, and Nucleosomes
Plasma concentrations of PF4 and HMGB-1 were quantified by ELISA (R&D Systems, Minneapolis, MN, and IBL International, Hamburg, Germany). The cell death detection ELISA (Roche, Mannheim, Germany) was used to determine nucleosome levels in plasma samples.
4.5. Statistical Analysis
Statistical analysis was carried out using GraphPad Prism version 9.5.1 (La Jolla, CA, USA). Data are represented as median [IQR; interquartile range]. Groups were compared using the Mann-Whitney test. A value of p < 0.05 was considered as statistically significant.
To assess the effect of time on different parameters, we used the Bayesian methods due to advantages over the frequentist framework [50,51]. Hierarchical models were created taking the repeated measures for the individual patients into account. To model the effect of time, we used the individual parameters as outcome variables, as well as time and patient as predictors. To estimate the probability of time being positively or negatively associated with the parameters (indicating an increase or decrease over time, respectively), we used the posterior percentages with positive vs. negative slope values for the predictor time. We used the default priors for this analysis. To estimate whether the change of the parameters was associated with survival, we employed the change of the measure from the first observation as predictor variable. A logistic model with survival as outcome variable was created. Again, the patient was used as grouping variable. Prior for the slope were set to normal with mean = 0 and standard deviation = 3. We used the logit of the slope estimate (transformed to percentages) for the effect of change of the parameters on survival. Analysis was carried out using R (version 4.2.2). The tidyverse [52] package was used for data wrangling and brms [53] to create the statistical models.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table S1: Medication regimens for individual patients; Table S2: MIFlowCyt-EV framework; Figure S1: Individual time course of extracellular vesicle concentrations in COVID-19 patients; Figure S2: Time course of extracellular vesicles in COVID-19 patients; Figure S3: Individual time course of coagulation- and inflammation-related parameters in COVID-19 patients; Figure S4: Time course of inflammatory mediators in mechanically ventilated COVID-19 patients; Figure S5: Calibration and controls for the flow cytometric characterization of phosphatidylserine-exposing extracellular vesicles; Figure S6: Characterization of TF-positive phosphatidylserine-exposing extracellular vesicles in COVID-19 patients.
Author Contributions
Conceptualization, Tanja Eichhorn, René Weiss and Viktoria Weber; Data curation, Tanja Eichhorn, René Weiss, Silke Huber, Ludwig Knabl, Sr., Ludwig Knabl and Reinhard Würzner; Formal analysis, Tanja Eichhorn, René Weiss and Robert Emprechtinger; Funding acquisition, Tanja Eichhorn, René Weiss and Viktoria Weber; Investigation, Tanja Eichhorn, René Weiss, Marie Ebeyer-Masotta and Marwa Mostageer; Methodology, Tanja Eichhorn and René Weiss; Project administration, Tanja Eichhorn, René Weiss and Viktoria Weber; Resources, Silke Huber, Ludwig Knabl, Sr., Ludwig Knabl and Reinhard Würzner; Supervision, Viktoria Weber; Visualization, Tanja Eichhorn, René Weiss and Marwa Mostageer; Writing – original draft, Tanja Eichhorn, René Weiss, Marwa Mostageer and Viktoria Weber; Writing – review & editing, Silke Huber, Marie Ebeyer-Masotta, Robert Emprechtinger, Ludwig Knabl, Sr., Ludwig Knabl and Reinhard Würzner. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Lower Austrian Society for Research Funding (GFF, grant number K3-F-744/005-2019) and by the Technology Fund of Lower Austria (grant number WST3-F-5030664/032-2022). Open Access Funding was provided by the University for Continuing Education Krems.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. The collection of blood samples from patients and from healthy individuals was approved by the Ethics Committee of the Medical University of Innsbruck (1144/2020, date of approval: May 20, 2020 and 1166/2018, date of approval: November 14, 2018, respectively).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are available on request from the authors.
Acknowledgments
The authors are indebted to the nurses and technicians at the intensive care unit and the central laboratory of the Hospital St. Vinzenz, Zams, Austria. The support by the infrastructure provided by the Core Facility Campus Krems is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; Abbate, A. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; Endeman, H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Portier, I.; Campbell, R.A.; Denorme, F. Mechanisms of immunothrombosis in COVID-19. Curr Opin Hematol 2021, 28, 445–453. [Google Scholar] [CrossRef]
- Schrottmaier, W.C.; Mussbacher, M.; Salzmann, M.; Assinger, A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis 2020, 307, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; Fiorelli, S.; Bonomi, A.; Cavalca, V.; Trabattoni, D.; Andreini, D.; Omodeo Sale, E.; Parati, G.; Tremoli, E.; Camera, M. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl Sci 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Hottz, E.D.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Bozza, F.A.; Bozza, P.T. Platelet-leukocyte interactions in the pathogenesis of viral infections. Platelets 2022, 33, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; Weyrich, A.S.; Yost, C.C.; Rondina, M.T.; Campbell, R.A. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Bozza, P.T. Platelet-leukocyte interactions in COVID-19: Contributions to hypercoagulability, inflammation, and disease severity. Res Pract Thromb Haemost 2022, 6, e12709. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; Mahir, W.; Belayachi, L.; Belefquih, B.; Benouda, A.; Cheikh, A.; Langlois, M.A.; Cherrah, Y.; Flamand, L.; Guessous, F.; Boilard, E. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ Res 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Le Joncour, A.; Biard, L.; Vautier, M.; Bugaut, H.; Mekinian, A.; Maalouf, G.; Vieira, M.; Marcelin, A.G.; Rosenzwajg, M.; Klatzmann, D.; Corvol, J.C.; Paccoud, O.; Carillion, A.; Salem, J.E.; Cacoub, P.; Boulaftali, Y.; Saadoun, D. Neutrophil-Platelet and Monocyte-Platelet Aggregates in COVID-19 Patients. Thromb Haemost 2020, 120, 1733–1735. [Google Scholar] [CrossRef]
- Hottz, E.D.; Martins-Goncalves, R.; Palhinha, L.; Azevedo-Quintanilha, I.G.; de Campos, M.M.; Sacramento, C.Q.; Temerozo, J.R.; Soares, V.C.; Dias, S.S.G.; Teixeira, L.; Castro, I.; Righy, C.; Souza, T.M.L.; Kurtz, P.; Andrade, B.B.; Nakaya, H.I.; Monteiro, R.Q.; Bozza, F.A.; Bozza, P.T. Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv 2022, 6, 5085–5099. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; Tektonidou, M.; Konstantinidis, T.; Papagoras, C.; Mitroulis, I.; Germanidis, G.; Lambris, J.D.; Ritis, K. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; Sidiropoulos, P.; Koffa, M.; Boumpas, D.T.; Ritis, K.; Mitroulis, I. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis 2014, 73, 1854–1863. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front Immunol 2022, 13, 838011. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; Cody, M.J.; Manne, B.K.; Portier, I.; Harris, E.S.; Petrey, A.C.; Beswick, E.J.; Caulin, A.F.; Iovino, A.; Abegglen, L.M.; Weyrich, A.S.; Rondina, M.T.; Egeblad, M.; Schiffman, J.D.; Yost, C.C. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Tripisciano, C.; Weiss, R.; Eichhorn, T.; Spittler, A.; Heuser, T.; Fischer, M.B.; Weber, V. Different Potential of Extracellular Vesicles to Support Thrombin Generation: Contributions of Phosphatidylserine, Tissue Factor, and Cellular Origin. Sci Rep 2017, 7, 6522. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ Res 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Jacob, V.; Lambour, A.; Swinyard, B.; Zerbib, Y.; Diouf, M.; Soudet, S.; Brochot, E.; Six, I.; Maizel, J.; Slama, M.; Guillaume, N. Annexin-V positive extracellular vesicles level is increased in severe COVID-19 disease. Front Med (Lausanne) 2023, 10, 1186122. [Google Scholar] [CrossRef]
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thalin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality-Brief Report. Arterioscler Thromb Vasc Biol 2021, 41, 878–882. [Google Scholar] [CrossRef]
- Burrello, J.; Caporali, E.; Gauthier, L.G.; Pianezzi, E.; Balbi, C.; Rigamonti, E.; Bolis, S.; Lazzarini, E.; Biemmi, V.; Burrello, A.; Frigerio, R.; Martinetti, G.; Fusi-Schmidhauser, T.; Vassalli, G.; Ferrari, E.; Moccetti, T.; Gori, A.; Cretich, M.; Melli, G.; Monticone, S.; Barile, L. Risk stratification of patients with SARS-CoV-2 by tissue factor expression in circulating extracellular vesicles. Vascul Pharmacol 2022, 145, 106999. [Google Scholar] [CrossRef]
- Balbi, C.; Burrello, J.; Bolis, S.; Lazzarini, E.; Biemmi, V.; Pianezzi, E.; Burrello, A.; Caporali, E.; Grazioli, L.G.; Martinetti, G.; Fusi-Schmidhauser, T.; Vassalli, G.; Melli, G.; Barile, L. Circulating extracellular vesicles are endowed with enhanced procoagulant activity in SARS-CoV-2 infection. EBioMedicine 2021, 67, 103369. [Google Scholar] [CrossRef]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; Velier, M.; Papazian, L.; Albanese, J.; Kaplanski, G.; Dignat-George, F.; Lacroix, R. Dissemination of extreme levels of extracellular vesicles: tissue factor activity in patients with severe COVID-19. Blood Adv 2021, 5, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Archibald, S.J.; Hisada, Y.; Bae-Jump, V.L.; Mackman, N. Evaluation of a new bead-based assay to measure levels of human tissue factor antigen in extracellular vesicles in plasma. Res Pract Thromb Haemost 2022, 6, e12677. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; Mellett, N.A.; Miles, L.A.; Du, X.J.; Meikle, P.J.; Huber-Lang, M.; Stark, G.B.; Parker, M.W.; Peter, K.; Eisenhardt, S.U. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun 2017, 8, 14188. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; De Lorenzo, R.; Clementi, N.; Antonia Diotti, R.; Criscuolo, E.; Godino, C.; Tresoldi, C.; Angels For Covid-Bio, B.S.G.B.; Bonini, C.; Clementi, M.; Mancini, N.; Ciceri, F.; Rovere-Querini, P.; Manfredi, A.A. Unconventional CD147-dependent platelet activation elicited by SARS-CoV-2 in COVID-19. J Thromb Haemost 2022, 20, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Tahyra, A.S.C.; Calado, R.T.; Almeida, F. The Role of Extracellular Vesicles in COVID-19 Pathology. Cells 2022, 11. [Google Scholar] [CrossRef]
- Wang, S.S.Y.; Chee, K.; Wong, S.W.; Tan, G.B.; Ang, H.; Leung, B.P.; Tan, C.W.; Ramanathan, K.; Dalan, R.; Cheung, C.; Lye, D.C.; Young, B.E.; Yap, E.S.; Chia, Y.W.; Fan, B.E.; Clotting, C.-. .; Bleeding, I. Increased Platelet Activation demonstrated by Elevated CD36 and P-Selectin Expression in 1-Year Post-Recovered COVID-19 Patients. Semin Thromb Hemost 2023, 49, 561–564. [Google Scholar]
- Silverstein, R.L. Type 2 scavenger receptor CD36 in platelet activation: the role of hyperlipemia and oxidative stress. Clin Lipidol 2009, 4, 767. [Google Scholar] [CrossRef]
- Schiavello, M.; Vizio, B.; Bosco, O.; Pivetta, E.; Mariano, F.; Montrucchio, G.; Lupia, E. Extracellular Vesicles: New Players in the Mechanisms of Sepsis- and COVID-19-Related Thromboinflammation. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Huber, S.; Massri, M.; Grasse, M.; Fleischer, V.; Kellnerova, S.; Harpf, V.; Knabl, L.; Knabl, L., Sr.; Heiner, T.; Kummann, M.; Neurauter, M.; Rambach, G.; Speth, C.; Wurzner, R. Systemic Inflammation and Complement Activation Parameters Predict Clinical Outcome of Severe SARS-CoV-2 Infections. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Puhm, F.; Flamand, L.; Boilard, E. Platelet extracellular vesicles in COVID-19: Potential markers and makers. J Leukoc Biol 2022, 111, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; De Vita, N.; Scotti, L.; Vaschetto, R.; Dianzani, U.; Chiocchetti, A. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Traby, L.; Kollars, M.; Kussmann, M.; Karer, M.; Sinkovec, H.; Lobmeyr, E.; Hermann, A.; Staudinger, T.; Schellongowski, P.; Rossler, B.; Burgmann, H.; Kyrle, P.A.; Eichinger, S. Extracellular Vesicles and Citrullinated Histone H3 in Coronavirus Disease 2019 Patients. Thromb Haemost 2022, 122, 113–122. [Google Scholar] [CrossRef]
- Botha, J.; Handberg, A.; Simonsen, J.B. Lipid-based strategies used to identify extracellular vesicles in flow cytometry can be confounded by lipoproteins: Evaluations of annexin V, lactadherin, and detergent lysis. J Extracell Vesicles 2022, 11, e12200. [Google Scholar] [CrossRef]
- Fendl, B.; Weiss, R.; Eichhorn, T.; Linsberger, I.; Afonyushkin, T.; Puhm, F.; Binder, C.J.; Fischer, M.B.; Weber, V. Extracellular vesicles are associated with C-reactive protein in sepsis. Sci Rep 2021, 11, 6996. [Google Scholar] [CrossRef]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; Meredith, I.; Peter, K. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res 2012, 96, 64–72. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; Bozza, F.A.; Bozza, P.T. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D'Angelo, A.; Bianchi, M.E.; Rovere-Querini, P.; Manfredi, A.A. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- McCafferty, C.; Van Den Helm, S.; Letunica, N.; Attard, C.; Karlaftis, V.; Cai, T.; Praporski, S.; Swaney, E.; Burgner, D.; Neeland, M.; Dohle, K.; Crawford, N.W.; Clucas, L.; Tosif, S.; Ignjatovic, V.; Monagle, P. Increased platelet activation in SARS-CoV-2 infected non-hospitalised children and adults, and their household contacts. Br J Haematol 2021, 195, 90–94. [Google Scholar] [CrossRef]
- Osterud, B. Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul Fibrinolysis 1998, 9 (Suppl 1), S9–S14. [Google Scholar]
- Hisada, Y.; Sachetto, A.T.A.; Mackman, N. Circulating tissue factor-positive extracellular vesicles and their association with thrombosis in different diseases. Immunol Rev 2022, 312, 61–75. [Google Scholar] [CrossRef]
- Bournazos, S.; Rennie, J.; Hart, S.P.; Fox, K.A.; Dransfield, I. Monocyte functional responsiveness after PSGL-1-mediated platelet adhesion is dependent on platelet activation status. Arterioscler Thromb Vasc Biol 2008, 28, 1491–1498. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; Ritis, K. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Eichhorn, T.; Huber, S.; Weiss, R.; Ebeyer-Masotta, M.; Laukova, L.; Emprechtinger, R.; Bellmann-Weiler, R.; Lorenz, I.; Martini, J.; Pirklbauer, M.; Orth-Holler, D.; Wurzner, R.; Weber, V. Infection with SARS-CoV-2 Is Associated with Elevated Levels of IP-10, MCP-1, and IL-13 in Sepsis Patients. Diagnostics (Basel) 2023, 13. [Google Scholar] [CrossRef]
- Loftus, T.J.; Ungaro, R.; Dirain, M.; Efron, P.A.; Mazer, M.B.; Remy, K.E.; Hotchkiss, R.S.; Zhong, L.; Bacher, R.; Starostik, P.; Moldawer, L.L.; Brakenridge, S.C. Overlapping but Disparate Inflammatory and Immunosuppressive Responses to SARS-CoV-2 and Bacterial Sepsis: An Immunological Time Course Analysis. Front Immunol 2021, 12, 792448. [Google Scholar] [CrossRef] [PubMed]
- George, S.K.; Laukova, L.; Weiss, R.; Semak, V.; Fendl, B.; Weiss, V.U.; Steinberger, S.; Allmaier, G.; Tripisciano, C.; Weber, V. Comparative Analysis of Platelet-Derived Extracellular Vesicles Using Flow Cytometry and Nanoparticle Tracking Analysis. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Semak, V.; Laukova, L.; Fischer, M.B.; Weber, V. Heparin-Functionalized Adsorbents Eliminate Central Effectors of Immunothrombosis, including Platelet Factor 4, High-Mobility Group Box 1 Protein and Histones. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Kruschke, J.K.; Liddell, T.M. The Bayesian New Statistics: Hypothesis testing, estimation, meta-analysis, and power analysis from a Bayesian perspective. Psychon Bull Rev 2018, 25, 178–206. [Google Scholar] [CrossRef] [PubMed]
- Szucs, D.; Ioannidis, J.P.A. When Null Hypothesis Significance Testing Is Unsuitable for Research: A Reassessment. Front Hum Neurosci 2017, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; D'Agostino McGowan, L.; Francois, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; Kuhn, M.; Lin Pedersen, T.; Miller, E.; Milton Bache, S.; Müller, K.; Ooms, J.; Robinson, D.; Seidel, D.P.; Spinu, V.; Takahashi, K.; Vaughan, D.; Wilke, C.; Woo, K.; Yutani, H. Welcome to the Tidyverse. J Open Soure Softw 2019, 4, 1686. [Google Scholar] [CrossRef]
- Bürkner, P.C. brms: An R Package for Bayesian Multilevel Models Using Stan. J Stat Soft 2017, 80. [Google Scholar] [CrossRef]
Figure 1.
Timeline of sampling. Samples from 12 COVID-19 patients were analyzed in this study (134 samples in total). Sampling was initiated on the first day of mechanical ventilation. Blue frames indicate sampling, (♦) marks extubation, and (†) death of patients.
Figure 1.
Timeline of sampling. Samples from 12 COVID-19 patients were analyzed in this study (134 samples in total). Sampling was initiated on the first day of mechanical ventilation. Blue frames indicate sampling, (♦) marks extubation, and (†) death of patients.
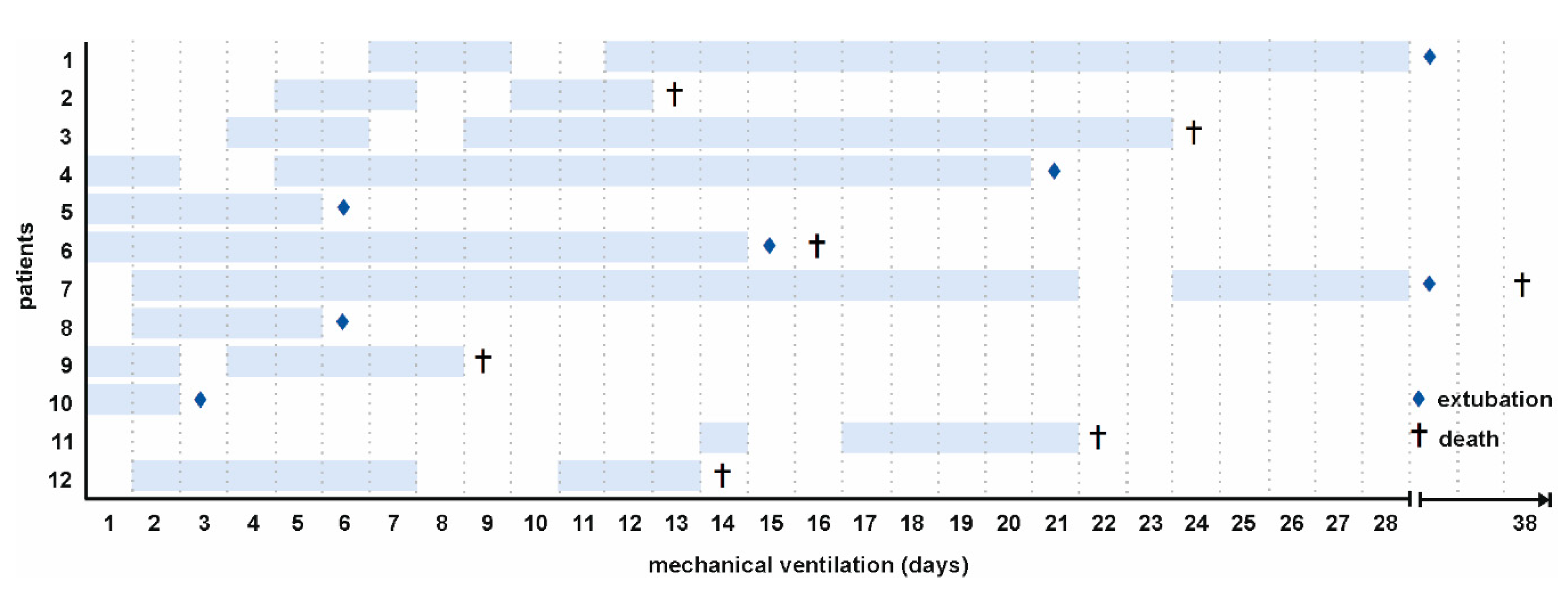
Figure 2.
Cellular origin of extracellular vesicles in COVID-19 patients. Extracellular vesicles in COVID-19 patients (n=134) and healthy controls (n=25) were characterized by flow cytometry and stained by a combination of anti-CD41 as platelet marker, anti-CD235a as red blood cell marker, anti-CD45 as leukocyte marker, anti-CD105 as endothelial marker, and anti-CD36 to detect platelet glycoprotein IV as described in the Methods section. Annexin V (Anx5) was used as marker for EVs exposing phosphatidylserine. Data are given as median [IQR] and were compared using the Mann-Whitney test (** p < 0.01, *** p < 0.001).
Figure 2.
Cellular origin of extracellular vesicles in COVID-19 patients. Extracellular vesicles in COVID-19 patients (n=134) and healthy controls (n=25) were characterized by flow cytometry and stained by a combination of anti-CD41 as platelet marker, anti-CD235a as red blood cell marker, anti-CD45 as leukocyte marker, anti-CD105 as endothelial marker, and anti-CD36 to detect platelet glycoprotein IV as described in the Methods section. Annexin V (Anx5) was used as marker for EVs exposing phosphatidylserine. Data are given as median [IQR] and were compared using the Mann-Whitney test (** p < 0.01, *** p < 0.001).
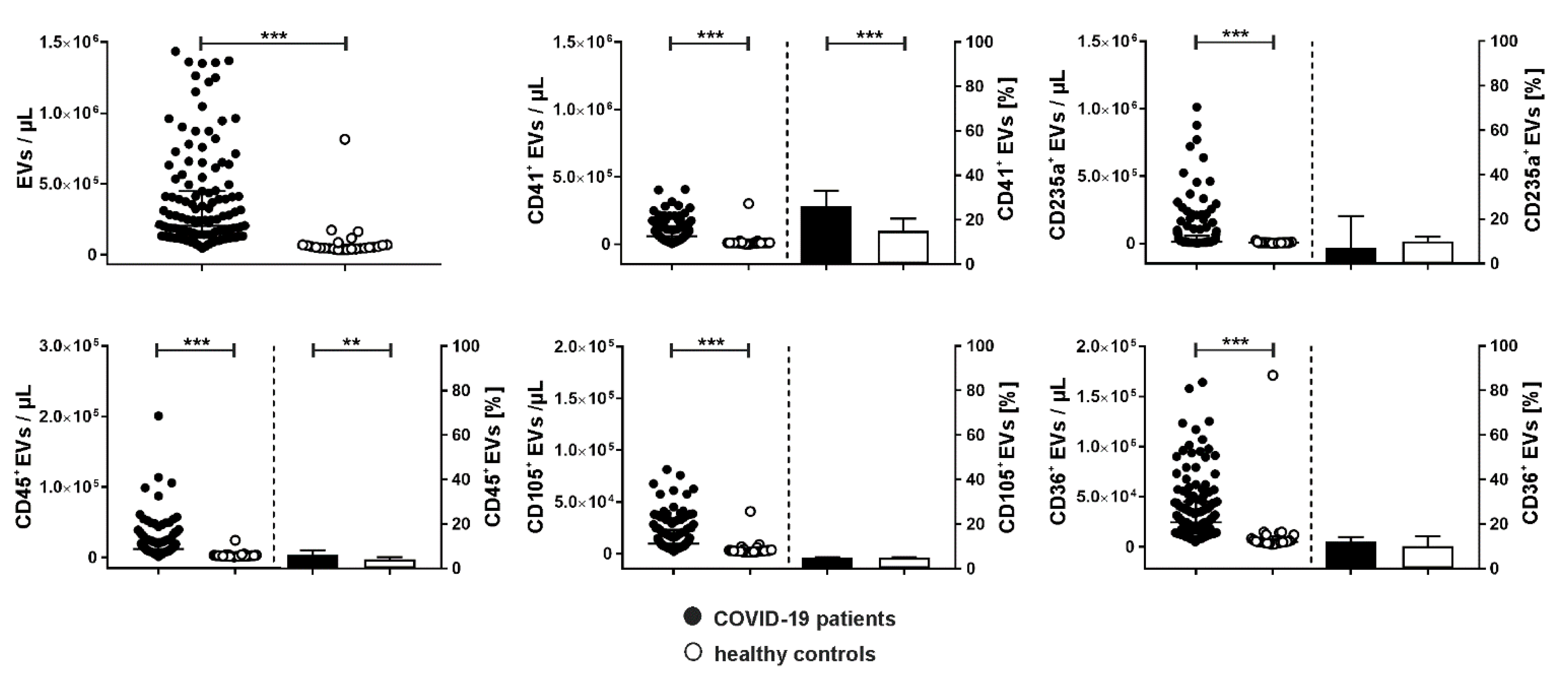
Figure 3.
Association of extracellular vesicles with CRP, PF4, and HMGB-1 in COVID-19 patients. Characterization of EVs carrying C-reactive protein (CRP+), platelet factor 4 (PF4+), and high mobility group box-1 protein (HMGB-1+) in plasma samples from COVID-19 patients (n=134) in comparison to healthy controls (n=25) using flow cytometry. Data are given as median [IQR] and were compared using the Mann-Whitney test (*** p < 0.001).
Figure 3.
Association of extracellular vesicles with CRP, PF4, and HMGB-1 in COVID-19 patients. Characterization of EVs carrying C-reactive protein (CRP+), platelet factor 4 (PF4+), and high mobility group box-1 protein (HMGB-1+) in plasma samples from COVID-19 patients (n=134) in comparison to healthy controls (n=25) using flow cytometry. Data are given as median [IQR] and were compared using the Mann-Whitney test (*** p < 0.001).
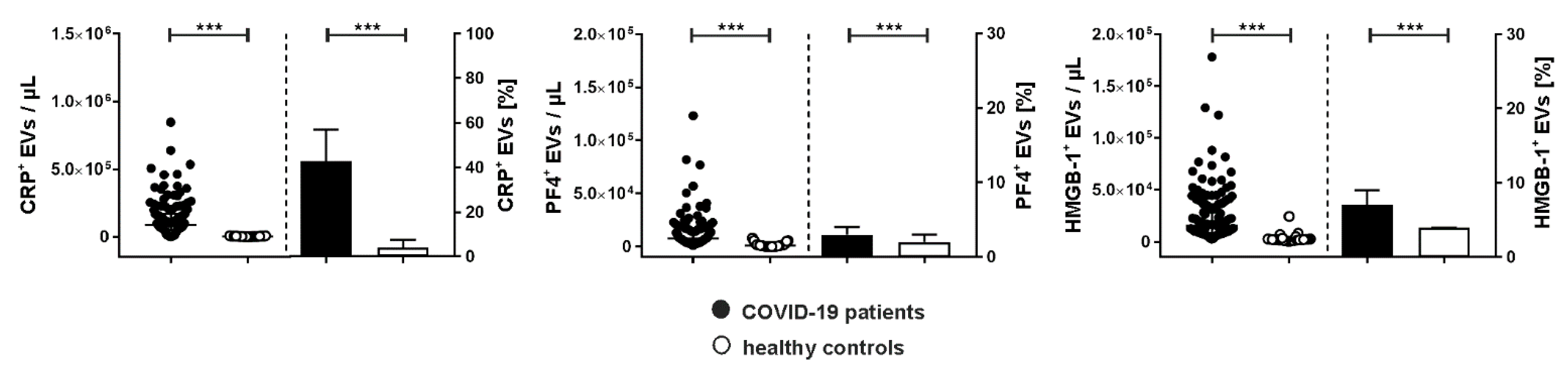
Figure 4.
Aggregate formation between platelet-derived and leukocyte-derived EVs and TF expression on EVs in COVID-19 patients. Aggregates of platelet- and leukocyte-derived EVs were identified as CD41+CD45+ events in the EV gate as described in the Methods section (upper left graph; EV counts/µL and the percentage of platelet EVs carrying CD45 as leukocyte marker are shown). Overall, the presence of CD41+CD45+ EVs correlated with significantly enhanced expression of TF in COVID-19 patients (upper right and lower left graph). For individual patients, TF+ EV aggregates (CD41+CD45+) are shown in the lower right graph. Data are given as median [IQR] and were compared using the Mann-Whitney test (*** p < 0.001). b.d.; below detection.
Figure 4.
Aggregate formation between platelet-derived and leukocyte-derived EVs and TF expression on EVs in COVID-19 patients. Aggregates of platelet- and leukocyte-derived EVs were identified as CD41+CD45+ events in the EV gate as described in the Methods section (upper left graph; EV counts/µL and the percentage of platelet EVs carrying CD45 as leukocyte marker are shown). Overall, the presence of CD41+CD45+ EVs correlated with significantly enhanced expression of TF in COVID-19 patients (upper right and lower left graph). For individual patients, TF+ EV aggregates (CD41+CD45+) are shown in the lower right graph. Data are given as median [IQR] and were compared using the Mann-Whitney test (*** p < 0.001). b.d.; below detection.
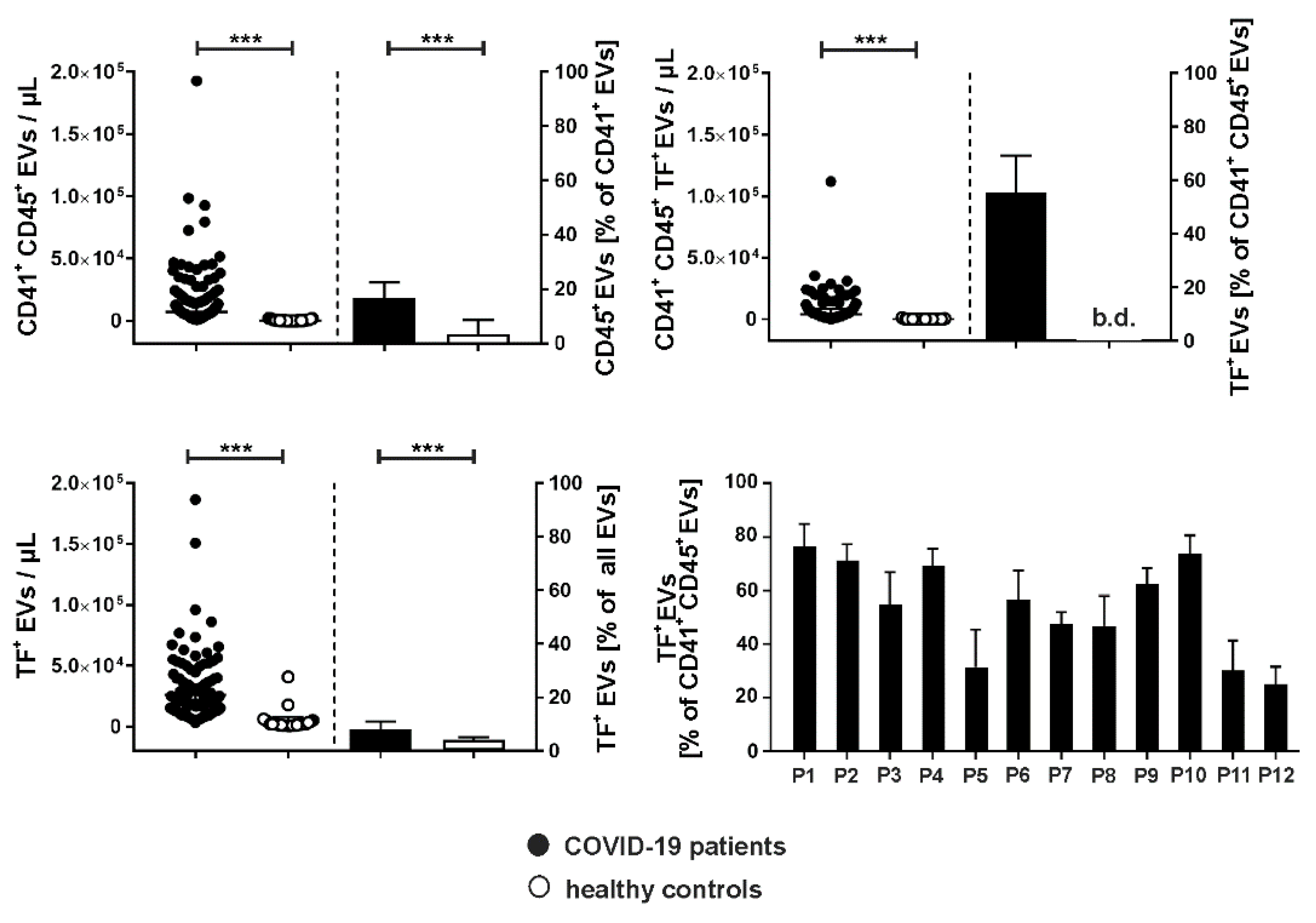

Table 1.
Patient characteristics at baseline.
| COVID-19 (n=12) |
Survivors (n=5) |
Non-survivors(n=7) | Controls (n=25) |
|
|---|---|---|---|---|
| Age, years | 79.5 [77.0–82.9] | 78.1 [70.6–83.9] | 80.6 [77.8–81.5] | 29.1 [27.4–42.9] |
| Female | 2 (17 %) | 1 (20 %) | 1 (14 %) | 15 (60 %) |
| Male | 10 (83 %) | 4 (80 %) | 6 (86 %) | 10 (40 %) |
| Mortality | 7 (58 %) | |||
| Secondary infections | 7 (58 %) | |||
| Comorbidities, n (%) | ||||
| Cardiovascular | 10 (83) | 4 (80) | 6 (86) | n.a. |
| Renal/genitourinary | 1 (8) | 0 (0) | 1 (14) | n.a. |
| Endocrine | 2 (17) | 1 (20) | 1 (14) | n.a. |
| Metabolic | 6 (50) | 2 (40) | 4 (57) | n.a. |
| Musculoskeletal | 1 (8) | 1 (20) | 0 (0) | n.a. |
| Psychiatric/mental | 2 (17) | 2 (40) | 0 (0) | n.a. |
| Autoimmune/immunological | 1 (8) | 0 (0) | 1 (14) | n.a. |
| Cancer | 1 (8) | 0 (0) | 1 (14) | n.a. |
Data are represented as median [interquartile range] and counts (percentage). n.a., not applicable.
Table 2.
Effect of time on inflammation- and coagulation-related parameters. Probability (P) increase and decrease indicate the probability that a certain parameter will increase or decrease, respectively, depending on time according to the statistical model. The Estimate refers to the effect size, as an increase of time by one will change the parameter by the Estimate value. The lower and upper 95% credibility intervals (CI) are denoted as CIlower and CIupper. As a measure for the model fit Rhat as well as the Bulk and Tail effective sample size (ESS) are given, confirming acceptable model fit.
Table 2.
Effect of time on inflammation- and coagulation-related parameters. Probability (P) increase and decrease indicate the probability that a certain parameter will increase or decrease, respectively, depending on time according to the statistical model. The Estimate refers to the effect size, as an increase of time by one will change the parameter by the Estimate value. The lower and upper 95% credibility intervals (CI) are denoted as CIlower and CIupper. As a measure for the model fit Rhat as well as the Bulk and Tail effective sample size (ESS) are given, confirming acceptable model fit.
| Parameter | Pincrease | Pdecrease | Estimate | Estimate Error | CIlower | CIupper | Rhat | Bulk ESS | Tail ESS |
|---|---|---|---|---|---|---|---|---|---|
| Leukocytes [cells/µL] | 0.07 | 99.92 | -153.67 | 48.1 | -246.91 | -58.22 | 1 | 2344.63 | 2722.01 |
| Neutrophils [cells/µL] | 0 | 100 | -213.65 | 47.33 | -308.98 | -118.91 | 1.01 | 3150.99 | 2606.26 |
| Platelets [cells/µL] | 0.9 | 99.1 | -7024.9 | 2941.64 | -12,645.01 | -1393.28 | 1 | 6704.61 | 2862.77 |
| PF4 [ng/mL] | 0 | 100 | -53.12 | 9.13 | -70.53 | -35.17 | 1 | 3299.33 | 3140.46 |
| D-dimer [ng/L] | 95.97 | 4.03 | 0.1 | 0.06 | -0.02 | 0.2 | 1 | 2889.52 | 2843.01 |
| Nucleosomes [AU] | 99.22 | 0.78 | 0.01 | 0 | 0 | 0.01 | 1 | 3642.96 | 2814.7 |
| HMGB-1 [ng/mL] | 92.67 | 7.33 | 0.42 | 0.28 | -0.15 | 0.96 | 1 | 3536.52 | 2048.85 |
| EVs [events/µL] | 4.65 | 95.35 | -10,038.83 | 5802.75 | -21,576.23 | 1638.06 | 1 | 6796.11 | 3067.23 |
| TF+ EVs [events/µL] | 0 | 100 | -1543.5 | 479.62 | -2716.92 | -813.98 | 1.38 | 8.97 | 13.71 |
| CRP+ EVs [events/µL] | 0.07 | 99.92 | -6998.63 | 2129.1 | -11,234.85 | -2893.5 | 1 | 6375.93 | 3141 |
| CRP [mg/L] | 75.8 | 24.2 | 0.65 | 0.93 | -1.14 | 2.52 | 1 | 3608.36 | 2938.3 |
| PCT [ng/mL] | 53.42 | 46.58 | 0 | 0.03 | -0.06 | 0.06 | 1 | 2454.24 | 2441.22 |
| IL-1β [pg/mL] | 99.98 | 0.02 | 0.1 | 0.03 | 0.04 | 0.16 | 1 | 2596.99 | 2760.37 |
| IL-6 [pg/mL] | 40.95 | 59.05 | -1.7 | 7.5 | -16.63 | 13.11 | 1 | 4165.65 | 3158.01 |
| IL-8 [pg/mL] | 100 | 0 | 2.84 | 0.51 | 1.85 | 3.83 | 1 | 3460.46 | 2930.64 |
| IL-10 [pg/mL] | 95.95 | 4.05 | 0.15 | 0.09 | -0.02 | 0.33 | 1 | 3720.19 | 3263.86 |
| IFN-γ [pg/mL] | 71.25 | 28.75 | 0.13 | 0.22 | -0.31 | 0.56 | 1 | 2979.59 | 2620.35 |
| TNF-α [pg/mL] | 97.97 | 2.03 | 0.71 | 0.35 | 0.04 | 1.39 | 1 | 3549.91 | 2730.32 |
| IP-10 [pg/mL] | 0 | 100 | -171.03 | 29.29 | -228.73 | -112.8 | 1 | 1797.06 | 2200.2 |
| MCP-1 [pg/mL] | 69.5 | 30.5 | 2.08 | 4.1 | -6.18 | 10 | 1 | 2603.32 | 2691.08 |
| G-CSF [pg/mL] | 99.72 | 0.28 | 6.87 | 2.6 | 1.69 | 11.77 | 1 | 3776.26 | 3357.83 |
Table 3.
Antibodies and fluorochrome conjugates used for flow cytometry.
| Flow cytometry | ||||||
|---|---|---|---|---|---|---|
| Antigen | Origin | Clone | Marker for | Fluorochrome | Abbreviation | Supplier |
| CD41 | mouse | P2 | platelets | PhycoerythrinCyanin 7 | PC7 | Beckman Coulter |
| CD235a | mouse | HIR2 (GA-R2) | red blood cells | Fluorescein Isothiocyanate |
FITC | eBioscience |
| CD105 | mouse | 266 | endothelial cells | Phycoerythrin | PE | Becton Dickinson |
| CD45 | mouse | J33 | leukocytes | Pacific Blue | PB | Beckman Coulter |
| CD36 | mouse | 5-271 | platelet glycoprotein IV | Alexa Fluor 700 | AF700 | BioLegend |
| PF4 | mouse | # 170138 | platelet factor 4 | Phycoerythrin | PE | R&D Systems |
| CRP | goat | - | C-reactive protein | Fluorescein Isothiocyanate |
FITC | Abcam |
| TF | mouse | VD8 | tissue factor | Fluorescein Isothiocyanate |
FITC | BioMedica Diagnostics |
| HMGB-1 | mouse | 115603 | high mobility group box-1 protein | Alexa Fluor 700 | AF700 | R&D Systems |
| Anx5 | - | - | phosphatidylserine | Allophyco-cyanin | APC | BD Biosciences |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



