
Submitted:
13 October 2023
Posted:
16 October 2023
You are already at the latest version
Abstract
Hippocampal neuronal activity generates dendritic and somatic Ca2+ signals, which depending on stimulus intensity, rapidly propagate to the nucleus and induce the expression of transcription factors and genes with crucial roles in cognitive functions. Soluble Amyloid-beta Oligomers (AβOs), the main synaptotoxins engaged in the pathogenesis of Alzheimer's disease, generate aberrant Ca2+ signals in primary hippocampal neurons, increase their oxidative tone and disrupt structural plasticity. Here, we explored the effects of sub-lethal AβOs concentrations on activity-generated nuclear Ca2+ signals and on the Ca2+-dependent expression of neuroprotective genes. To induce neuronal activity, neuron-enriched primary hippocampal cultures were treated with the GABAA receptor blocker gabazine (GBZ), and nuclear Ca2+ signals were measured in AβOs-treated or control neurons transfected with a genetically encoded nuclear Ca2+ sensor. Incubation (6 h) with AβOs significantly reduced the nuclear Ca2+ signals and the enhanced phosphorylation of cyclic AMP response element binding protein (CREB) induced by GBZ. Likewise, incubation (6 h) with AβOs significantly reduced the GBZ-induced increases in the mRNA levels of Neuronal Per Arnt Sim domain protein 4 (Npas4), Brain-derived Neurotrophic Factor (Bdnf), Ryanodine Receptor type-2 (RyR2), and the antioxidant enzyme NADPH-Quinone-Oxidoreductase (Nqo1). Based on these findings we propose that AβOs, by inhibiting the generation of activity induced nuclear Ca2+ signals, disrupt key neuroprotective gene expression pathways required for hippocampal-dependent learning and memory processes.
Keywords:
Alzheimer´s disease
; Neuronal activity
; Synaptotoxicity
; Calcium-dependent signaling pathways
; Calcium-mediated Gene Expression
; Antioxidant Enzyme Expression.
1. Introduction
Alzheimer's disease (AD), the most common form of age-associated dementia worldwide, is a progressive and fatal neurodegenerative illness manifested by severe and progressive deterioration of cognitive and memory processes [1]. Extracellular senile plaques, composed mainly of β-amyloid (Aβ) peptides [2], and intracellular neurofibrillary tangles of hyperphosphorylated microtubule-associated protein (p-Tau) [3,4], are the primary brain histopathological AD markers. At early stages in this disease, Aβ and p-Tau aggregates accumulate in the hippocampus, a brain region with an essential role in learning and memory processes [4], and which is particularly vulnerable to the neurotoxic effects associated with different age-related diseases [5,6,7]. Hippocampal atrophy is an early indication of the conversion from a normal aging process to the development of dementia [8,9]. Therefore, AβOs treatment of primary hippocampal neurons is a suitable experimental in vitro model to study the mechanisms underlying the neuronal dysfunctions promoted by AβOs [10,11,12,13,14,15].
Uncontrolled neuronal activity, as occurs in AD, affects neural network homeostasis and initiates synaptotoxic processes [16]. Current evidence indicates that multiple factors contribute to the development of AD [17]; among them, neuronal oxidative stress and abnormal Ca2+ signaling are central features of this disease [18,19]. Several studies have shown that postmortem AD brains display elevated oxidative stress markers, including oxidized lipids, proteins, and DNA (reviewed in [20]). Even slight imbalances of these molecules can be deleterious to the brain; moreover, elevated neuronal levels of reactive oxygen species (ROS) can cause selective dysfunctions and neurodegeneration [21,22,23]. Memory mechanisms are directly compromised by elevated ROS content, which underscores the importance of establishing how excessive ROS levels may be coupled with other critical aspects of AD pathology. The first link between AβOs and neuronal oxidative stress was established in 2007 [11], and was confirmed by us and other groups [15,24].
Neuronal Ca2+ signals, defined as transient, controlled increments in intracellular Ca2+ concentration, play essential roles in hippocampal synaptic plasticity and memory processes [25,26]. However, uncontrolled Ca2+ signals lead to neuronal dysfunction and eventually cause neuronal death [27]. Accordingly, neurons have developed highly sophisticated mechanisms to control Ca2+ homeostasis and signaling locally in dendritic spines, and in somatic and nuclear compartments [25,28]; these control mechanisms fail to function properly in AD [19,29]. In primary hippocampal cultures, AβOs promote Ca2+ influx through N-methyl-D-aspartate (NMDA) glutamate receptors, which induces Ca2+ release mediated by ryanodine receptor (RyR) Ca2+ channels [12]. The ensuing abnormal Ca2+ signals, which are long-lasting but of low amplitude, engage Ca2+-dependent pathways that hinder BDNF-induced dendritic spine growth and induce excitotoxicity, leading to the loss of synaptic structure and function [12,29,30]. In summary, abnormal intracellular Ca2+ signaling and the related excitable and synaptic dysfunctions are consolidated hallmarks of AD onset and possibly of other neurodegenerative diseases [31,32].
Essentially, all activity-induced, long-term modifications of brain functions require appropriate synapse-to-nucleus communication to precisely control nuclear Ca2+-dependent gene expression [28]. Robust synaptic activity generates Ca2+ signals that, in addition to promoting the activation of cytoplasmic signaling cascades, readily reach and enter the nucleus via passive diffusion through nuclear pores [33,34]. However, nuclear Ca2+ entry requires Ca2+ signals to reach the vicinity of the nucleus, which does not occur by simple Ca2+ diffusion since this process is highly restricted in the cytoplasm [35,36]. In particular, the type-2 (RyR2) RyR isoform plays a major role in the propagation of Ca2+ signals to the nucleus [26]. Once inside the nucleus, Ca2+ signals contribute to activity-dependent transcription by promoting Ca2+-dependent phosphorylation of CREB-binding protein (CBP), which allows the formation of the transcriptionally active CREB-CBP complex [28,37,38].
Blocking GABAA receptor function with gabazine (GBZ) stimulates the activity of rat primary hippocampal neurons [26,39,40], which contain ∼10–15% inhibitory interneurons that are responsible for the tonic inhibition of the neuronal network [41]. The GBZ-induced increase in hippocampal neuronal activity promotes the emergence of oscillatory Ca2+ transients, which propagate from the cytoplasm to the neuronal nucleus, where they promote CREB phosphorylation [26] - a key event in BDNF-mediated synaptic plasticity and hippocampal memory [42,43]. Addition of GBZ also enhances Bdnf, Npas4, and RyR2 expression [26], all of which have central roles in hippocampal synaptic plasticity and spatial memory [43,44,45,46]. In contrast, incubation (6 h) of primary hippocampal neurons with a sub-lethal AβOs concentration eliminates the RyR2 protein increase induced by BDNF and prevents within minutes the dendritic spine remodeling induced by BDNF [12]. Moreover, incubation (6 h) of primary hippocampal neurons with AβOs inhibits the RyR2-mediated generation of mitochondrial ROS, and thus contributes to the synaptic dysfunction caused by AβOs [15]. However, the effects of AβOs on the generation of activity induced nuclear Ca2+ signals, synaptic plasticity, and antioxidant defense mechanisms in hippocampal neurons remain undefined.
In this work, we report that AβOs inhibited the generation of nuclear Ca2+ signals induced by GBZ addition to rat primary hippocampal neurons; AβOs also decreased the GBZ-induced CREB phosphorylation increase, and the expression of genes engaged in memory and neuroprotective processes, such as Bdnf, RyR2 and Npas4. Importantly, AβOs also reduced the expression of NADPH-Quinone-Oxidoreductase-1 (Nqo1), an enzyme with a key role in the neuronal antioxidant response that is significantly impaired in AD [47,48].
2. Materials and Methods
Reagents: The Aβ1-42 peptide was from Bachem Inc. (Torrance, CA, USA), Gabazine SR95531was from Tocris Bioscience (Avonmouth, Bristol, UK); cytoplasmic GCaMP5 and nuclear GCaMP3-NLS were kindly provided by Dr. H. Bading, University of Heidelberg, Germany; DMSO was from Sigma (Darmstadt, Germany), Hexafluoro-2-propanol (HFIP) was from Merck (Darmstadt, Germany). Serum-free Neurobasal Medium, B27 Supplement, GlutaMAXTM, DAPI, Lipofectamine 2000 and TRIzol reagent were from GibcoTM/ Thermofisher Scientific (Waltham, MA, USA). Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix and DAKO mounting medium were from Agilent Technologies (Santa Clara, CA, USA. DNAase Turbo DNA-freeTM kit was from AmbionTM/ Thermofisher Scientific (Waltham, MA, USA). Improm II TM reverse transcriptase was from Promega (Madison, Wisconsin, USA). Protein content was determined with a BCA kit from Thermofisher Scientific (Waltham, MA, USA).
Antibodies: Specific antibodies against p-CREB were from Cell Signaling Technologies (Danvers, MA, USA). Antibodies against MAP2 were from Abcam (Cambridge, UK) and against β-tubulin from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 488 anti-rabbit and Alexa Fluor 635 anti-mouse were from Invitrogen (Carlsbad, CA, USA).
Primary rat hippocampal cultures: Primary hippocampal cultures were prepared from Sprague-Dawley rats at gestation age 18 days, as described (Paula-Lima et al.-, 2011). Briefly, cells were recovered and plated in Petri dishes previously treated with poli-L-lysine (0.1 mg/mL). Then, cultures were maintained in serum-free Neurobasal Medium containing B27 Supplement Glutamax TM, Penicillin (20 U/mL)/ Streptomycin (20 μg / ml) at 37°C under 5% CO2. In all experiments, cultures were used between 12-15 days-in-vitro (DIV). For most of the experiments, hippocampal neurons were pre-incubated with 500 nM AβOs for 6 h before incubation with 5 μM Gabazine for 30 minutes for Ca2+ measurements and CREB immunofluorescence; for determinations by qPCR cultures were incubated for 6 h with 500 nM AβOs before incubation with 5 μM Gabazine for 2 h. Compounds were maintained during the respective incubation period as stated. All experimental protocols used in this work complied with the “Guiding Principles for Research Involving Animals and Human Beings” of the American Physiological Society and were approved by the Bioethics Committee on Animal Research, Faculty of Medicine, Universidad de Chile.
Preparation of AβOs: The Aβ1–42 peptide was prepared as a dried hexafluoro-2-propanol (HIFP) film and stored at -20ºC, as described [12,13,15,24] Before use, the film was resuspended in DMSO, diluted with 100 μM cold phosphate-buffered saline (PBS) and incubated without stirring for 24 h at 4ºC. The resulting Aβ-containing solution was centrifugated at 14,000 g for 10 min at 4°C to remove protofibrils and fibrils (insoluble aggregates). The supernatants that contained Aβ oligomers were transferred to clean tubes and stored at 4ºC; an aliquot was used to determine AβOs content with a BCA kit. Fresh AβOs preparations (up to 48 h) were used in all experiments.
Cytoplasmic Ca2+ Signals: To detect cytoplasmic Ca2+ signals, primary hippocampal cultures were transfected with the genetically encoded cytoplasmic Ca2+ sensor GCaMP5. Transfected neurons were recorded in Tyrode's solution (mM: 129 NaCl, 5 KCl, 25 HEPES, pH 7.3, 30 glucose, 2 CaCl2, 1 MgCl2). As an additional strategy to detect neuronal Ca2+ signals, primary cultures were loaded for 15 minutes at 37° C with the Ca2+ probe Fluo 4-AM, prepared in Tyrode extracellular medium. Cells were placed in the microscopy stage of a wide field Zeiss Cell Observer epifluorescence microscope (Zeiss, Oberkochen, Germany), using as objectives Plan-Neofluar 20x/0.4 or Plan Apochromat, 40x/1.3 water, with light source 470 nm Colibri 2 LED-based module and a digital camera EMCCD Evolve 512 delta (Teledyne Photometrics Tucson, AZ). All settings were adjusted to minimize bleaching and maximize acquisition frequency. After recording a stable baseline, 10 μM gabazine was added to the culture; Ionomycin (3 μM) was added at the end of the experiment to visualize the maximum fluorescence signals.
To determine basal Ca2+ levels, cultures were incubated at 37°C for 20 minutes in the dark with 2 µM Fura2-AM. Subsequently, cells were washed three times with Tyrode’s solution (mM: 129 NaCl, 5 KCl, 25 Hepes, pH 7.3, 30 glucose, 2 CaCl2, 1 MgCl2). Cells were placed in the microscopy stage of a Spinning Disc IX81 microscope (Olympus), using a xenon lamp as excitation source, with excitation filters for 340/402 nm wavelengths, and 40x objective. The first minutes of the record yielded the fluorescence levels corresponding to the resting Ca2+ concentration. Then, ionomycin was added to saturate the Ca2+ probe. For data analysis, the background fluorescence was subtracted and the ratio of 340 nm and 380 nm fluorescence intensity (ratio; F340/F380) was calculated.
Nuclear Ca2+ Signals: To detect nuclear Ca2+ signals, primary hippocampal cultures (14-16 DIV) were transfected with the genetically encoded Ca2+ sensor GCaMP3-NLS, which has a sequence of nuclear destination and thus accumulates in the neuronal nucleus. The GCaMP3 transfected neurons were recorded in Tyrode’s solution (mM: 129 NaCl, 5 KCl, 25 Hepes, pH 7.3, 30 glucose, 2 CaCl2, 1 MgCl2). To assess the direct effect of AβOs on nuclear Ca2+ signals, cultures were incubated at the microscope stage with 500 nM AβOs and acquired by image time lapse for 15 min. To determine the effect of AβOs on activity induced nuclear Ca2+ signals, neurons were incubated for 6 h with 500 nM AβOs before addition of 5 μM GBZ at the microscope stage. A wide-field Zeiss Cell Observer epifluorescence microscope (Zeiss, Jena, Germany), using Plan-Neofluar 20Å~/0.4 or Plan Apochromat as objectives, 40Å~/1.3 water, with a light source, 470-nm Colibri 2 light-emitting diode (LED)-based module, and a digital camera, electron-multiplying charge-coupled device (EMCCD) Evolve 512 delta (Teledyne Photometrics, Tucson, AZ, USA) was used. All settings were adjusted to minimize bleaching and maximize acquisition frequency. After recording a stable baseline, 5 μM GBZ was added to the culture; 3 μM ionomycin was added at the end of the experiment to visualize the maximum fluorescence signals. The analysis of Ca2+ signals was performed as described [26]. Briefly, images were segmented by hand to generate binary masks suitable for morphological and functional analysis using the Fiji distribution of ImageJ software. All fluorescence signals, recorded at the region of stimulation, are expressed as (Fmax-F0/F0) or as F/F0, where Fmax and F represent, respectively, the maximal recorded fluorescence intensity and the recorded fluorescence intensity; F0 corresponds to the intensity at the initial time (mean intensity of 20 to 50 frames recorded before the stimulus).
CREB phosphorylation and nuclear staining: To determine whether AβOs interrupt the GBZ-induced increase in synaptic activity and CREB phosphorylation, hippocampal cultures were pre-incubated for 6 h with 500 nM AβOs before the addition of 5 μM GBZ for 30 minutes. Cells were then fixed and CREB phosphorylation was detected by immunofluorescence, using an antibody against the phosphorylated form of CREB (Ser-133 p-CREB). In addition, cultures were probed with an antibody against the microtubule-associated protein MAP2 to identify neuronal cells and with DAPI to detect all cell nuclei. All images were recorded with a 40x objective in the z-axis of all confocal planes acquired by Spinning-Disk microscopy (Olympus XI 81 Spinning Disk super zoom, Tokyo, Japan). Quantification of the relative levels of p-CREB fluorescence was detected in different conditions by background subtraction (50 pixels ball radius) and segmentation by Otsu threshold, using DAPI staining as total nuclear staining reference. Mean fluorescence intensity was quantified only in neuronal cells (MAP2 positive) and expressed as fold change relative to control cells.
RNA Extraction and qPCR: Total RNA was isolated using TRIzol reagent following the manufacturer’s recommendations; to remove any contamination with genomic DNA, a digestion step with DNAase (Turbo DNA-freeTM kit, Ambion, Invitrogen Carlsbad, CA) was included to remove residual contaminating DNA. The RNA purity was determined by the 260/280 absorbance ratio in nanodrop (Nano-400 Nucleic Acid Analyzer from Allsheng (Hangzhou, China). Complementary DNA (cDNA) was synthesized with Improm II TM reverse transcriptase using 2 μg of total RNA. The mRNA levels of RyR2, BDNF exon IV, Npas4, NqoI were amplified using specific primers (Table I) from 2 μg of cDNA in 20 μL final volume and were performed in an Mx3000P qPCR System (Stratagene, Santa Clara, CA, USA) using the Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix. The mRNA levels were quantified with the 2−ΔΔCT method [98] using β-actin as housekeeping gene. All samples were run in triplicate and included controls. To verify the purity of the products, the dissociation curves were analyzed in all cases.
Statistical and Data Analysis: Results are expressed as mean ± SE. Statistical analysis between groups was performed with One-way ANOVA followed by the Bonferroni test, as indicated in the respective figure legends. Comparison between two groups was performed by two-tailed Student’s t-test. All statistical analyses were performed using SigmaPlot version 12.0.
3. Results
3.1. AβOs disrupt the nuclear Ca2+ transients induced by Gabazine
Previous reports have described that addition of AβOs elicits long-lasting and low amplitude cytoplasmic Ca2+ signals in primary hippocampal neurons [12,13]. Following AβOs addition (500 nM), cytoplasmic Ca2+ signals, detected with Fluo-4 (Figure 1A), exhibited a similar response pattern (Figure 1B). To evaluate if AβOs also induce nuclear Ca2+ signals, AβOs (500 nM) were added at the microscope stage to hippocampal cultures transfected with the nuclear Ca2+ sensor GCaMP3-NLS (Figure 1C). This treatment induced a slow, sustained, and low intensity increase in nuclear Ca2+ levels (Figure 1D). This nuclear response was slower than that displayed by the cytoplasmic Ca2+ signals following AβOs addition to primary hippocampal neurons (Figure 1B) [12,15,24].
To test the effects of AβOs on the Ca2+ signals induced by neuronal activity, primary cultures preincubated (6 h) with AβOs were treated with the GABAA receptor antagonist GBZ, which by decreasing the inhibitory GABAergic tone produces an excitatory/inhibitory imbalance and a rapid increase in neuronal activity [39,49]. As reported previously, treatment of primary hippocampal cultures with GBZ induced the emergence of oscillatory and synchronized cytoplasmic (Figure 2A,B) and nuclear Ca2+ signals (Figure 2C,D), measured as described [26]. The GBZ-induced nuclear Ca2+ transients, recorded in primary neurons transfected with the genetically encoded nuclear Ca2+ sensor GCaMP3-NLS (Figure 2C), displayed frequencies, intensities, and durations that exhibited some experimental variability (Figure 2D). In contrast, primary neurons pre-treated (6 h) with 500 nM AβOs before GBZ addition displayed a conspicuous decrease in the emergence of the oscillatory and synchronic nuclear Ca2+ signals elicited by GBZ (Supplementary video 1). Quantification of different nuclear Ca2+ signals parameters revealed that AβOs significantly decreased the maximum amplitude (Figure 3A) and the number of detected peaks (Figure 3B) induced by GBZ addition. These results show that incubation (6 h) with 500 nM AβOs, which is a non-lethal concentration [12,50], disrupts the oscillatory and synchronic nuclear Ca2+ signals caused by the increase in neuronal activity induced by GBZ. Of note, treatment with 500 nM AβOs (6 h) did not modify the basal cytoplasmic Ca2+ levels (Supplementary Figure 1).
3.2. AβOs prevent the CREB phosphorylation increase induced by Gabazine
The increase in hippocampal neuronal activity induced by GBZ triggers the early induction of gene expression changes commanded initially, among other factors, by the phosphorylation of the transcription factor CREB and of its binding protein CBP by different nuclear kinases [26,28]. To determine whether treatment with AβOs disrupts the CREB phosphorylation increase induced by GBZ, primary cultures were incubated for 6 h with 500 nM AβOs prior to GBZ addition. Phosphorylated CREB (p-CREB) levels were detected by immunofluorescence 30 minutes after GBZ addition, using an antibody against CREB phosphorylated in serine 133 (green in Figure 4). An antibody against the microtubule-associated protein MAP2 was used to identify neurons (red in Figure 4) and DAPI was applied to detect all cell nuclei (blue in Figure 4). Treatment with GBZ for 30 minutes produced a significant increase in CREB phosphorylation in primary neurons (Figure 4A,B), as described [26]. In contrast, neurons preincubated for 6 h with 500 nM AβOs and then incubated with 5 µM GBZ for 30 minutes showed significantly lower p-CREB levels, which were not different from those displayed by the untreated controls (Figure 4B). These results show that incubation for 6 h with AβOs abolished the subsequent GBZ-induced CREB phosphorylation increase.
3.3. AβOs inhibit the increase in Npas4, RyR2, Bdnf and Nqo1 mRNA levels induced by Gabazine
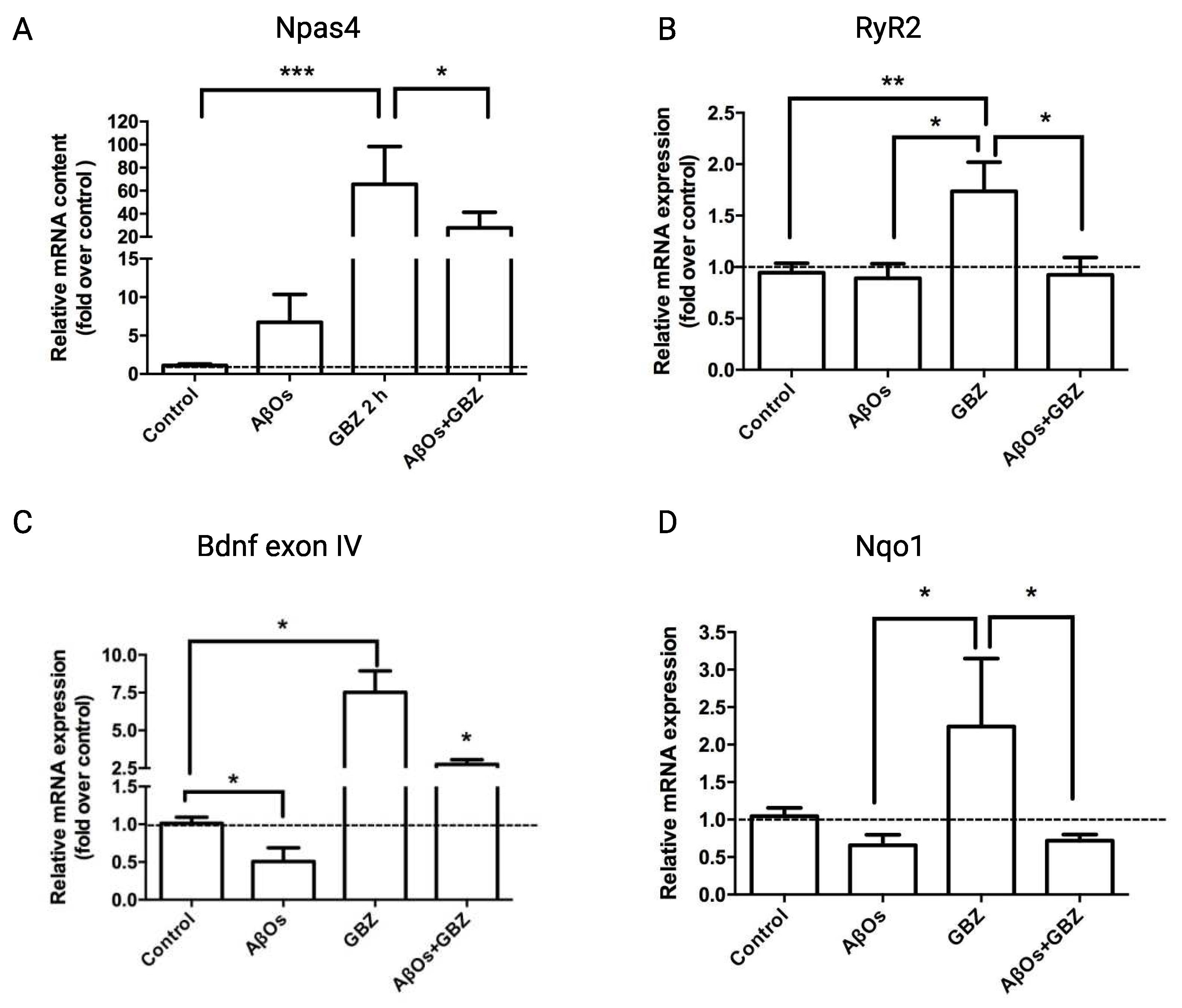
The increase in neuronal activity induced by GBZ, together with promoting CREB phosphorylation also increases the expression of the early response gene Npas4 [26]. In agreement, 2 h treatment of hippocampal cultures with 5 µM GBZ induced a significant increase in the mRNA levels of Npas4 (Figure 5A). Addition of GBZ also increased RyR2 mRNA levels (Figure 5B), and triggered a neuroprotective response, evidenced by significant increases in the mRNA levels of exon IV of the neurotrophic factor Bdnf (Figure 5C). Of note, GBZ also promoted a significant increase in the mRNA levels of the antioxidant enzyme Nqo1 (Figure 5D). However, neurons treated with 500 nM AβOs for 6 h before addition of 5 µM GBZ displayed after 30 minutes significantly lower mRNA levels of Npas4 (Figure 5A) and Bdnf exon IV (Figure 5C), whereas the RyR2 and Nqo1 mRNA levels were reduced to the control values (Figure 5B,D).
Taken together, these results show that incubation with AβOs inhibited the nuclear Ca2+ signals induced by neuronal activity and reduced or abolished the GBZ-induced increases in CREB phosphorylation and of Npas4, RyR2, Bdnf, and Nqo1 mRNA levels (Graphical Abstract).
4. Discussion
Alterations in synapse to nucleus communication, which occur in different central nervous system disorders, contribute to the development of neurodegenerative diseases [32]. The results presented here show that treatment for 6 h of primary hippocampal neurons with AβOs decreased the production of GBZ-induced nuclear Ca2+ signals and the phosphorylation of the transcription factor CREB, and reduced or abolished the enhanced expression of Npas4, Bdnf, RyR2 and Nqo1 induced by GBZ.
Neuronal Ca2+ signals play a wide-range of functions in the activity-dependent synaptic plasticity processes that underlie hippocampal spatial memory [51,52,53]. Among the molecular players controlling neuronal Ca2+ signaling, the endoplasmic reticulum resident inositol 1,4,5-trisphosphate receptors and RyR channels have central roles. Both Ca2+ channel types amplify Ca2+ entry signals via Ca2+-induced Ca2+ release and thus contribute to regulate the function of neuronal Ca2+-dependent signaling pathway [24,54,55,56,57]. Of note, RyR-mediated Ca2+ release plays an essential role in hippocampal synaptic plasticity, and the formation/consolidation of spatial memory [43,44,58,59]. Changes in cellular oxidative state particularly affect RyR function, so that reducing agents impede RyR activation by Ca2+ whereas oxidizing agents have the opposite effects [24,54,55,56,57]. Excessive ROS-induced RyR channel activation has been associated to abnormal Ca2+ release and the functional defects associated to AD and age-related hippocampal dysfunction [12,15,24,44,60,61,62,63].
Our in vitro rat model of AβOs associated synaptotoxicity, which entails acute treatment of primary hippocampal cultures with non-lethal AβOs concentrations [12], adequately represents the deleterious changes in Ca2+ signaling that occur during aging and in response to Aβ peptide toxicity [64]. In agreement with these findings, a recent study reported that iPSC differentiated from neurons of patients with presenilin-1 (PS1) mutations display Ca2+ homeostasis failures and increased β-amyloid and p-Tau levels; negative allosteric modulators of RyR channels reverses these effects, reaffirming the importance of regulating RyR-mediated Ca2+ release in AD affected neurons [65]. Additional studies, performed in a triple transgenic AD model, have confirmed the relevance of controlling RyR-mediated Ca2+ release for neutralizing the AD disease phenotype [66]. Moreover, studies in elderly Rhesus macaques have reported that RyR-mediated Ca2+ release enhances Tau phosphorylation and is associated with reduced neuronal activation and cognitive impairment [60]. Additionally, RyR2 isoform overactivation induces neuronal hyperactivity and Aβ accumulation in a feedback cycle that triggers dendritic spine loss, decreased memory, and death; the negative RyR2 modulator R-Carvedilol and RyR2 mutations aimed at decreasing its activity prevent these processes in humans and mice [61,67].
Adding to these studies, the present results show that acute AβOs treatment inhibited the generation of nuclear Ca2+ signals induced by neuronal activity, a process that engages RyR2-mediated Ca2+ release [26]. Of note, acute AβOs treatment of primary hippocampal neurons induce the generation of sustained, low magnitude cytoplasmic Ca2+ signals that are RyR-mediated [12]. As reported here, the same pattern of Ca2+ signals were generated by AβOs in the neuronal nucleus. Interestingly, basal cytoplasmic Ca2+ levels, as determined by the ratiometric fluorescence indicator Fura2, were not significantly different between control neurons and neurons treated for 6 h with AβOs. Nevertheless, the incubation for 6 h with AβOs significantly reduced the nuclear Ca2+ signal generation induced by GBZ. Hence, neuronal stimulation seems to potentiate the inhibitory effects of AβOs on Ca2+ dynamics, particularly on nuclear Ca2+ signal generation, a result which may have implications for the deleterious effects of AβOs on hippocampal neuronal function during the performance of tasks that entail neuronal activation and engage RyR-mediated Ca2+ release. The development of pharmacological strategies that modulate the activity of RyR channels will continue to attract interest in the development of new drugs to regulate neurodegenerative processes [68].
The inhibitory effects of AβOs on the enhanced expression of Npas4 triggered by GBZ-induced neuronal activity reported here may contribute to understand the novel and central role of Npas4 in the context of AD. The immediate-early gene Npas4 is expressed only in neurons and is considered as a molecular link between neuronal activity and memory [69]. Moreover, Npas4 is among the most rapidly induced early genes and its expression, which requires nuclear Ca2+ signals, is selectively induced by neuronal activity [70,71]. By orchestrating distinct activity-dependent gene programs in different neuronal populations, the transcription factor Npas4 affects synaptic connections in excitatory and inhibitory neurons, neural circuit plasticity, memory formation, reward-related gene expression and behavior and has been involved in neuroprotection [69]. Recent studies have proposed a close relationship between the pathological processes that cause AD and the activation of pathways related to neuronal activity. Among them, Npas4 displays decreased expression in AD patients, which is related to increased aggregation and deposition of Tau protein in neurofibrillary tangles, one of the hallmarks of AD progression [72]. The decreased Npas4 expression reported in AD patients may have additional harmful effects, since Npas4 facilitates autophagy-mediated Tau elimination [73]. In contrast, the Amyloid Precursor Protein (APP), through the generation of an intracellular (AICD) domain, controls Npas4 expression helping to maintain excitatory/inhibitory tone balance [74]. In the same vein, a novel activity-dependent DNA repair mechanism involving the Npas4–NuA4 complex was recently uncovered; this novel mechanism has an important role in maintaining genome stability, a process that is disrupted during pathological aging [75].
Neuroinflammation, closely related to neurodegenerative processes, also seems to play an important role in the regulation of the excitatory/inhibitory tone; infiltration of CD8+ T lymphocytes into the hippocampus of a transgenic AD model (APP-PS1) alters the expression of Npas4 and Arc along with other genes related to synaptic plasticity and Ca2+ signaling [76]. Moreover, HDAC3 maintains basal epigenetic repression of Npas4 and Bdnf transcription, highlighting the role of epigenetic modifications in the control of neuronal activity and induction of immediate early genes [77,78]. These studies position Npas4 and BDNF as outstanding therapeutic targets against AβOs-induced synaptotoxicity and may constitute targets to prevent the development of AD and other neurodegenerative diseases [79,80,81]. In addition, strategies to control intracellular ROS- and Ca2+-mediated signaling might prevent uncontrolled activation of Npas4 and neurodegenerative processes [82].
Many studies have established that BDNF, a neurotrophin synthesized and released from neurons in an activity dependent manner [83], mediates the morphological changes entailing the generation and growth of dendritic spines during synaptic plasticity [84,85,86]. Upon binding to TrkB receptors, BDNF stimulates several intracellular signaling cascades that require RyR-mediated Ca2+ release [43], including Ca2+-dependent kinase pathways that contribute to induce and maintain hippocampal long-term potentiation (LTP) [87]. In view of the key roles played by both RyR and BDNF in synaptic plasticity [85,87,88,89,90], our findings that AβOs interrupt GBZ-induced nuclear Ca2+ signals and the ensuing expression of both Bdnf and RyR2 mRNA levels, may add to our current understanding of AD pathology. In particular, the AβOs-induced decrease of Bdnf mRNA levels induced by neuronal activity may compromise BDNF-induced intracellular signaling pathways relevant to hippocampal function [12,50,91,92].
Of note, coupled with an increase in neuronal activity, BDNF induces the nuclear translocation of the transcription factor Nrf2 in neurons [93], a master regulator of antioxidant protein expression that protects brain cells against oxidative damage [94,95,96]. It has been proposed that the key role played by BDNF as an inducer of neuronal antioxidant responses entails crosstalk between RyR-mediated Ca2+ release and ROS [93]. One of the targets of Nrf2 signaling is the expression of Nqo1, an oxidoreductase that catalyzes the 2-electron reduction of quinones to hydroquinones, which is a cellular detoxification process [97]. Here, we show that the activity dependent increase in Bdnf expression mediated by GBZ is accompanied by an increase in the expression of Nqo1. Moreover, pre-treatment of hippocampal neurons with AβOs prevented the nuclear Ca2+-dependent expression of Nqo1 induced by GBZ, indicating that these aggregates disrupt neuroprotective pathways induced by neuronal activity that regulate neuronal oxidative tone.
5. Conclusions
This work provides the first integrative evidence that AβOs disrupt the generation of activity dependent nuclear Ca2+ signals and further reduce the expression of genes engaged in synaptic plasticity and the antioxidant response. It, therefore, adds to the understanding of the toxic effects of AβOs on neuronal function and reveals new possible therapeutic targets to treat neurodegenerative diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Cytoplasmic Ca2+ levels in primary hippocampal neurons.; Table S1: I shows the sequence of primers used to determine the mRNA levels.; Video S1: Treatment with AβOs disrupts the transient nuclear Ca2+ signals induced by GBZ in hippocampal neurons.
Author Contributions
Conceptualization, A.P.-L. and C.H.; methodology, P.L.; I.V-V.; B.B; S.G.; formal analysis, P.L., I.V-V., S.G., J.T.; investigation, A.P.-L., P.L.; resources, A.P.-L.; C.H.; S.H.; writing—original draft preparation, P.L.; writing—review and editing, A.P-L., C.H.; supervision, A.P-L. and C.H.; project administration, A.P.-L. and C.H..; funding acquisition, A.P.-L.; C.H.; S.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Biomedical Neuroscience Institute (BNI) (grant P09-015F), the Chilean Scientific Millennium Initiative, the German Federal Ministry of Education and Research (grant BMBF180051), the Chilean Fondo Nacional de Desarrollo Científico y Tecnológico for the FONDECYT grants 1150736, 1170053, 1190958 and 1211988; the FONDEF 23I10337 grant, the CTI220001 Center, to the National Chilean Agency of Research and Development for the BASAL FB210005 grant and the CONICYT, Ph.D. scholarships (21161086 and 21200346), the Interuniversity Center for Healthy Aging, RED211993 and by the Chilean Fondo de Equipamiento Científico y Tecnológico (FONDEQUIP grants EQM120164, 140119 and 140156).
Institutional Review Board Statement
The animal study protocol was approved by the Bioethics Committee on Animal Research, Faculty of Medicine, Universidad de Chile (protocols code FMUCH #0755 and #1142)” for studies involving animals. All experimental protocols used in this work complied with the “Guiding Principles for Research Involving Animals and Human Beings” of the American Physiological Society.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available upon request from the authors.
Acknowledgments
The excellent technical help of Ms. Nicole Henriquez is gratefully acknowledged. We also thank Hervé Camus for his help in the analysis of confocal microscopy images.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s & Dementia 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proceedings of the National Academy of Sciences 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-Associated Protein Tau. A Component of Alzheimer Paired Helical Filaments. J Biol Chem 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat Rev Mol Cell Biol 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Hibar, D.P.; Adams, H.H.H.; Jahanshad, N.; Chauhan, G.; Stein, J.L.; Hofer, E.; Renteria, M.E.; Bis, J.C.; Arias-Vasquez, A.; Ikram, M.K.; et al. Novel Genetic Loci Associated with Hippocampal Volume. Nat Commun 2017, 8, 13624. [Google Scholar] [CrossRef]
- Fotuhi, M.; Do, D.; Jack, C. Modifiable Factors That Alter the Size of the Hippocampus with Ageing. Nat Rev Neurol 2012, 8, 189–202. [Google Scholar] [CrossRef]
- Younan, D.; Petkus, A.J.; Widaman, K.F.; Wang, X.; Casanova, R.; Espeland, M.A.; Gatz, M.; Henderson, V.W.; Manson, J.E.; Rapp, S.R.; et al. Particulate Matter and Episodic Memory Decline Mediated by Early Neuroanatomic Biomarkers of Alzheimer’s Disease. Brain 2020, 143, 289–302. [Google Scholar] [CrossRef]
- Leal, S.L.; Yassa, M.A. Neurocognitive Aging and the Hippocampus across Species. Trends Neurosci 2015. [CrossRef]
- Adler, D.H.; Wisse, L.E.M.; Ittyerah, R.; Pluta, J.B.; Ding, S.-L.; Xie, L.; Wang, J.; Kadivar, S.; Robinson, J.L.; Schuck, T.; et al. Characterizing the Human Hippocampus in Aging and Alzheimer’s Disease Using a Computational Atlas Derived from Ex Vivo MRI and Histology. Proceedings of the National Academy of Sciences 2018, 115, 4252–4257. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, Nonfibrillar Ligands Derived from Aβ 1–42 Are Potent Central Nervous System Neurotoxins. Proceedings of the National Academy of Sciences 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-D-Aspartate Receptor-Dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. Journal of Biological Chemistry 2007, 282, 11590–11601. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; SanMartin, C.; Sebollela, A.; Hetz, C.; Carrasco, M.A.; Ferreira, S.T.; Hidalgo, C. Amyloid Beta-Peptide Oligomers Stimulate RyR-Mediated Ca2+ Release Inducing Mitochondrial Fragmentation in Hippocampal Neurons and Prevent RyR-Mediated Dendritic Spine Remodeling Produced by BDNF. Antioxid Redox Signal 2011, 14, 1209–1223. [Google Scholar] [CrossRef]
- Lobos, P.; Bruna, B.; Cordova, A.; Barattini, P.; Galáz, J.L.; Adasme, T.; Hidalgo, C.; Muñoz, P.; Paula-Lima, A. Astaxanthin Protects Primary Hippocampal Neurons against Noxious Effects of A β -Oligomers. Neural Plast 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Ortiz-Sanz, C.; Gaminde-Blasco, A.; Valero, J.; Bakota, L.; Brandt, R.; Zugaza, J.L.; Matute, C.; Alberdi, E. Early Effects of Aβ Oligomers on Dendritic Spine Dynamics and Arborization in Hippocampal Neurons. Front Synaptic Neurosci 2020, 12. [Google Scholar] [CrossRef]
- SanMartin, C.D.; Veloso, P.; Adasme, T.; Lobos, P.; Bruna, B.; Galaz, J.; Garcia, A.; Hartel, S.; Hidalgo, C.; Paula-Lima, A.C. RyR2-Mediated Ca(2+) Release and Mitochondrial ROS Generation Partake in the Synaptic Dysfunction Caused by Amyloid Beta Peptide Oligomers. Front Mol Neurosci 2017, 10, 115. [Google Scholar] [CrossRef]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef]
- Tse and Herrup, K. Re-imagining Alzheimer’s Disease – the Diminishing Importance of Amyloid and a Glimpse of What Lies Ahead. ARPN Journal of Engineering and Applied Sciences 2017, 12, 3218–3221. [Google Scholar] [CrossRef]
- Fani, G.; La Torre, C.E.; Cascella, R.; Cecchi, C.; Vendruscolo, M.; Chiti, F. Misfolded Protein Oligomers Induce an Increase of Intracellular Ca2+ Causing an Escalation of Reactive Oxidative Species. Cellular and Molecular Life Sciences 2022, 79, 500. [Google Scholar] [CrossRef]
- Rummel, N.G.; Butterfield, D.A. Altered Metabolism in Alzheimer Disease Brain: Role of Oxidative Stress. Antioxid Redox Signal 2022, 36, 1289–1305. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat Rev Neurosci 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb Perspect Med 2011, 1. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative Stress in the Brain: Novel Cellular Targets That Govern Survival during Neurodegenerative Disease. Prog Neurobiol 2005, 75, 207–246. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid Redox Signal 2021, 34, 591–610. [Google Scholar] [CrossRef]
- SanMartin, C.D.; Adasme, T.; Hidalgo, C.; Paula-Lima, A.C. The Antioxidant N-Acetylcysteine Prevents the Mitochondrial Fragmentation Induced by Soluble Amyloid-Beta Peptide Oligomers. Neurodegener Dis 2012, 10, 34–37. [Google Scholar] [CrossRef]
- Hagenston, A.M.; Bading, H. Calcium Signaling in Synapse-to-Nucleus Communication. Cold Spring Harb Perspect Biol 2011, 3, a004564–a004564. [Google Scholar] [CrossRef]
- Lobos, P.; Córdova, A.; Vega-Vásquez, I.; Ramírez, O.A.; Adasme, T.; Toledo, J.; Cerda, M.; Härtel, S.; Paula-Lima, A.; Hidalgo, C. RyR-Mediated Ca2+ Release Elicited by Neuronal Activity Induces Nuclear Ca2+ Signals, CREB Phosphorylation, and Npas4/RyR2 Expression. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat Rev Mol Cell Biol 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bading, H. Nuclear Calcium Signalling in the Regulation of Brain Function. Nat Rev Neurosci 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Brito-Moreira, J.; Ferreira, S.T. Deregulation of Excitatory Neurotransmission Underlying Synapse Failure in Alzheimer’s Disease. J Neurochem 2013, 126, 191–202. [Google Scholar] [CrossRef]
- Brito-Moreira, J.; Paula-Lima, A.C.; Bomfim, T.R.; Oliveira, F.F.; Sepúlveda, F.J.; de Mello, F.G.; Aguayo, L.G.; Panizzutti, R.; Ferreira, S.T. Aβ Oligomers Induce Glutamate Release from Hippocampal Neurons. Curr Alzheimer Res 2011, 8. [Google Scholar] [CrossRef]
- Chiantia, G.; Hidisoglu, E.; Marcantoni, A. The Role of Ryanodine Receptors in Regulating Neuronal Activity and Its Connection to the Development of Alzheimer’s Disease. Cells 2023, 12, 1236. [Google Scholar] [CrossRef]
- Marcello, E.; Di Luca, M.; Gardoni, F. Synapse-to-Nucleus Communication: From Developmental Disorders to Alzheimer’s Disease. Curr Opin Neurobiol 2018, 48, 160–166. [Google Scholar] [CrossRef]
- Brini, M.; Murgia, M.; Pasti, L.; Picard, D.; Pozzan, T.; Rizzuto, R. Nuclear Ca2+ Concentration Measured with Specifically Targeted Recombinant Aequorin. EMBO J 1993, 12, 4813–4819. [Google Scholar] [CrossRef]
- Eder, A.; Bading, H. Calcium Signals Can Freely Cross the Nuclear Envelope in Hippocampal Neurons: Somatic Calcium Increases Generate Nuclear Calcium Transients. BMC Neurosci 2007, 8, 57. [Google Scholar] [CrossRef]
- Allbritton, N.L.; Meyer, T.; Stryer, L. Range of Messenger Action of Calcium Ion and Inositol 1,4,5-Trisphosphate. Science (1979) 1992, 258, 1812–1815. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Chawla, S.; Hardingham, G.E.; Quinn, D.R.; Bading, H. CBP: A Signal-Regulated Transcriptional Coactivator Controlled by Nuclear Calcium and CaM Kinase IV. Science (1979) 1998, 281, 1505–1509. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Arnold, F.J.L.; Bading, H. Nuclear Calcium Signaling Controls CREB-Mediated Gene Expression Triggered by Synaptic Activity. Nat Neurosci 2001, 4, 261–267. [Google Scholar] [CrossRef]
- Pegoraro, S.; Broccard, F.D.; Ruaro, M.E.; Bianchini, D.; Avossa, D.; Pastore, G.; Bisson, G.; Altafini, C.; Torre, V. Sequential Steps Underlying Neuronal Plasticity Induced by a Transient Exposure to Gabazine. J Cell Physiol 2009, n/a-n/a. [CrossRef]
- Sokal, D.M.; Mason, R.; Parker, T.L. Multi-Neuronal Recordings Reveal a Differential Effect of Thapsigargin on Bicuculline- or Gabazine-Induced Epileptiform Excitability in Rat Hippocampal Neuronal Networks. Neuropharmacology 2000, 39, 2408–2417. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol Rev 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Esvald, E.-E.; Tuvikene, J.; Sirp, A.; Patil, S.; Bramham, C.R.; Timmusk, T. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. The Journal of Neuroscience 2020, 40, 1405–1426. [Google Scholar] [CrossRef]
- Adasme, T.; Haeger, P.; Paula-Lima, A.C.; Espinoza, I.; Casas-Alarcón, M.M.; Carrasco, M.A.; Hidalgo, C. Involvement of Ryanodine Receptors in Neurotrophin-Induced Hippocampal Synaptic Plasticity and Spatial Memory Formation. Proceedings of the National Academy of Sciences 2011, 108, 3029–3034. [Google Scholar] [CrossRef]
- More, J.Y.; Bruna, B.A.; Lobos, P.E.; Galaz, J.L.; Figueroa, P.L.; Namias, S.; Sánchez, G.L.; Barrientos, G.C.; Valdés, J.L.; Paula-Lima, A.C.; et al. Calcium Release Mediated by Redox-Sensitive RyR2 Channels Has a Central Role in Hippocampal Structural Plasticity and Spatial Memory. Antioxid Redox Signal 2018, 29, 1125–1146. [Google Scholar] [CrossRef]
- Weng, F.-J.; Garcia, R.I.; Lutzu, S.; Alviña, K.; Zhang, Y.; Dushko, M.; Ku, T.; Zemoura, K.; Rich, D.; Garcia-Dominguez, D.; et al. Npas4 Is a Critical Regulator of Learning-Induced Plasticity at Mossy Fiber-CA3 Synapses during Contextual Memory Formation. Neuron 2018, 97, 1137–1152.e5. [Google Scholar] [CrossRef]
- Nicastri, C.M.; McFeeley, B.M.; Simon, S.S.; Ledreux, A.; Håkansson, K.; Granholm, A.; Mohammed, A.H.; Daffner, K.R. BDNF Mediates Improvement in Cognitive Performance after Computerized Cognitive Training in Healthy Older Adults. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2022, 8. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The Diverse Functionality of NQO1 and Its Roles in Redox Control. Redox Biol 2021, 41, 101950. [Google Scholar] [CrossRef]
- Sharma, V.; Kaur, A.; Singh, T.G. Counteracting Role of Nuclear Factor Erythroid 2-Related Factor 2 Pathway in Alzheimer’s Disease. Biomedicine & Pharmacotherapy 2020, 129, 110373. [Google Scholar] [CrossRef]
- Ueno, S.; Bracamontes, J.; Zorumski, C.; Weiss, D.S.; Steinbach, J.H. Bicuculline and Gabazine Are Allosteric Inhibitors of Channel Opening of the GABA A Receptor. The Journal of Neuroscience 1997, 17, 625–634. [Google Scholar] [CrossRef]
- Tong, L.; Balazs, R.; Thornton, P.L.; Cotman, C.W. β-Amyloid Peptide at Sublethal Concentrations Downregulates Brain-Derived Neurotrophic Factor Functions in Cultured Cortical Neurons. The Journal of Neuroscience 2004, 24, 6799–6809. [Google Scholar] [CrossRef]
- Ross, W.N. Understanding Calcium Waves and Sparks in Central Neurons. Nat Rev Neurosci 2012, 13, 157–168. [Google Scholar] [CrossRef]
- Mozolewski, P.; Jeziorek, M.; Schuster, C.M.; Bading, H.; Frost, B.; Dobrowolski, R. The Role of Nuclear Ca2+ in Maintaining Neuronal Homeostasis and Brain Health. J Cell Sci 2021, 134. [Google Scholar] [CrossRef]
- O’Hare, J.K.; Gonzalez, K.C.; Herrlinger, S.A.; Hirabayashi, Y.; Hewitt, V.L.; Blockus, H.; Szoboszlay, M.; Rolotti, S.V.; Geiller, T.C.; Negrean, A.; et al. Compartment-Specific Tuning of Dendritic Feature Selectivity by Intracellular Ca2+ Release. Science (1979) 2022, 375. [Google Scholar] [CrossRef]
- Marengo, J.J.; Bull, R.; Hidalgo, C. Calcium Dependence of Ryanodine-Sensitive Calcium Channels from Brain Cortex Endoplasmic Reticulum. FEBS Lett 1996, 383, 59–62. [Google Scholar] [CrossRef]
- Marengo, J.J.; Hidalgo, C.; Bull, R. Sulfhydryl Oxidation Modifies the Calcium Dependence of Ryanodine-Sensitive Calcium Channels of Excitable Cells. Biophys J 1998, 74, 1263–1277. [Google Scholar] [CrossRef]
- Bull, R.; Marengo, J.J.; Finkelstein, J.P.; Behrens, M.I.; Alvarez, O. SH Oxidation Coordinates Subunits of Rat Brain Ryanodine Receptor Channels Activated by Calcium and ATP. Am J Physiol Cell Physiol 2003, 285, C119–C128. [Google Scholar] [CrossRef]
- Riquelme, D.; Alvarez, A.; Leal, N.; Adasme, T.; Espinoza, I.; Valdes, J.A.; Troncoso, N.; Hartel, S.; Hidalgo, J.; Hidalgo, C.; et al. High-Frequency Field Stimulation of Primary Neurons Enhances Ryanodine Receptor-Mediated Ca2+ Release and Generates Hydrogen Peroxide, Which Jointly Stimulate NF-KappaB Activity. Antioxid Redox Signal 2011, 14, 1245–1259. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; Hidalgo, C. Contribution of Ca2+ Release Channels to Hippocampal Synaptic Plasticity and Spatial Memory: Potential Redox Modulation. Antioxid Redox Signal 2014, 21, 892–914. [Google Scholar] [CrossRef]
- Valdés-Undurraga, I.; Lobos, P.; Sánchez-Robledo, V.; Arias-Cavieres, A.; SanMartín, C.D.; Barrientos, G.; More, J.; Muñoz, P.; Paula-Lima, A.C.; Hidalgo, C.; et al. Long-Term Potentiation and Spatial Memory Training Stimulate the Hippocampal Expression of RyR2 Calcium Release Channels. Front Cell Neurosci 2023, 17. [Google Scholar] [CrossRef]
- Datta, D.; Leslie, S.N.; Wang, M.; Morozov, Y.M.; Yang, S.; Mentone, S.; Zeiss, C.; Duque, A.; Rakic, P.; Horvath, T.L.; et al. Age-related Calcium Dysregulation Linked with Tau Pathology and Impaired Cognition in Non-human Primates. Alzheimer’s & Dementia 2021, 17, 920–932. [Google Scholar] [CrossRef]
- Yao, J.; Liu, Y.; Sun, B.; Zhan, X.; Estillore, J.P.; Turner, R.W.; Chen, S.R.W. Increased RyR2 Open Probability Induces Neuronal Hyperactivity and Memory Loss with or without Alzheimer’s Disease–Causing Gene Mutations. Alzheimer’s & Dementia 2022, 18, 2088–2098. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M.F. An Endoplasmic-Reticulum-Specific Apoptotic Pathway Is Involved in Prion and Amyloid-Beta Peptides Neurotoxicity. Neurobiol Dis 2006, 23, 669–678. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-Translational Remodeling of Ryanodine Receptor Induces Calcium Leak Leading to Alzheimer’s Disease-like Pathologies and Cognitive Deficits. Acta Neuropathol 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Kharitonova, E.K.; Bacskai, B.J. Therapeutic Strategies to Target Calcium Dysregulation in Alzheimer’s Disease. Cells 2020, 9, 2513. [Google Scholar] [CrossRef]
- Schrank, S.; McDaid, J.; Briggs, C.A.; Mustaly-Kalimi, S.; Brinks, D.; Houcek, A.; Singer, O.; Bottero, V.; Marr, R.A.; Stutzmann, G.E. Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology. Int J Mol Sci 2020, 21, 1030. [Google Scholar] [CrossRef] [PubMed]
- Aloni, E.; Oni-Biton, E.; Tsoory, M.; Moallem, D.H.; Segal, M. Synaptopodin Deficiency Ameliorates Symptoms in the 3xTg Mouse Model of Alzheimer’s Disease. The Journal of Neuroscience 2019, 39, 3983–3992. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Liu, Y.; Reiken, S.; Liu, X.; Argyrousi, E.K.; Yuan, Q.; Miotto, M.C.; Sittenfeld, L.; Meddar, A.; Soni, R.K.; et al. Heart Failure-Induced Cognitive Dysfunction Is Mediated by Intracellular Ca2+ Leak through Ryanodine Receptor Type 2. Nat Neurosci 2023, 26, 1365–1378. [Google Scholar] [CrossRef]
- Webber, E.K.; Fivaz, M.; Stutzmann, G.E.; Griffioen, G. Cytosolic Calcium: Judge, Jury and Executioner of Neurodegeneration in Alzheimer’s Disease and Beyond. Alzheimer’s & Dementia 2023, 19, 3701–3717. [Google Scholar] [CrossRef]
- Sun, X.; Lin, Y. Npas4: Linking Neuronal Activity to Memory. Trends Neurosci 2016, 39, 264–275. [Google Scholar] [CrossRef]
- Brigidi, G.S.; Hayes, M.G.B.; Delos Santos, N.P.; Hartzell, A.L.; Texari, L.; Lin, P.-A.; Bartlett, A.; Ecker, J.R.; Benner, C.; Heinz, S.; et al. Genomic Decoding of Neuronal Depolarization by Stimulus-Specific NPAS4 Heterodimers. Cell 2019, 179, 373–391.e27. [Google Scholar] [CrossRef]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.-K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-Dependent Regulation of Inhibitory Synapse Development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef]
- Miyashita, A.; Hatsuta, H.; Kikuchi, M.; Nakaya, A.; Saito, Y.; Tsukie, T.; Hara, N.; Ogishima, S.; Kitamura, N.; Akazawa, K.; et al. Genes Associated with the Progression of Neurofibrillary Tangles in Alzheimer’s Disease. Transl Psychiatry 2014, 4, e396. [Google Scholar] [CrossRef]
- Fan, W.; Long, Y.; Lai, Y.; Wang, X.; Chen, G.; Zhu, B. NPAS4 Facilitates the Autophagic Clearance of Endogenous Tau in Rat Cortical Neurons. Journal of Molecular Neuroscience 2016, 58, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Opsomer, R.; Contino, S.; Perrin, F.; Gualdani, R.; Tasiaux, B.; Doyen, P.; Vergouts, M.; Vrancx, C.; Doshina, A.; Pierrot, N.; et al. Amyloid Precursor Protein (APP) Controls the Expression of the Transcriptional Activator Neuronal PAS Domain Protein 4 (NPAS4) and Synaptic GABA Release. eNeuro 2020, 7, ENEURO–0322. [Google Scholar] [CrossRef]
- Pollina, E.A.; Gilliam, D.T.; Landau, A.T.; Lin, C.; Pajarillo, N.; Davis, C.P.; Harmin, D.A.; Yap, E.-L.; Vogel, I.R.; Griffith, E.C.; et al. A NPAS4–NuA4 Complex Couples Synaptic Activity to DNA Repair. Nature 2023, 614, 732–741. [Google Scholar] [CrossRef]
- Unger, M.S.; Li, E.; Scharnagl, L.; Poupardin, R.; Altendorfer, B.; Mrowetz, H.; Hutter-Paier, B.; Weiger, T.M.; Heneka, M.T.; Attems, J.; et al. CD8+ T-Cells Infiltrate Alzheimer’s Disease Brains and Regulate Neuronal- and Synapse-Related Gene Expression in APP-PS1 Transgenic Mice. Brain Behav Immun 2020, 89, 67–86. [Google Scholar] [CrossRef]
- Louis Sam Titus, A.S.C.; Sharma, D.; Kim, M.S.; D’Mello, S.R. The Bdnf and Npas4 Genes Are Targets of HDAC3-Mediated Transcriptional Repression. BMC Neurosci 2019, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Herre, M.; Korb, E. The Chromatin Landscape of Neuronal Plasticity. Curr Opin Neurobiol 2019, 59, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The Emerging Field of Epigenetics in Neurodegeneration and Neuroprotection. Nat Rev Neurosci 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.-H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 Reverses Alzheimer’s Disease-Related Pathologies in Vitro and in the 3xTg-AD Mouse Model. Proceedings of the National Academy of Sciences 2018, 115. [Google Scholar] [CrossRef]
- Mousa, H.H.; Sharawy, M.H.; Nader, M.A. Empagliflozin Enhances Neuroplasticity in Rotenone-Induced Parkinsonism: Role of BDNF, CREB and Npas4. Life Sci 2023, 312, 121258. [Google Scholar] [CrossRef]
- Burnside, S.W.; Hardingham, G.E. Transcriptional Regulators of Redox Balance and Other Homeostatic Processes with the Potential to Alter Neurodegenerative Disease Trajectory. Biochem Soc Trans 2017, 45, 1295–1303. [Google Scholar] [CrossRef]
- Kuczewski, N.; Porcher, C.; Lessmann, V.; Medina, I.; Gaiarsa, J.-L. Activity-Dependent Dendritic Release of BDNF and Biological Consequences. Mol Neurobiol 2009, 39, 37–49. [Google Scholar] [CrossRef]
- Danzer, S.C.; Crooks, K.R.C.; Lo, D.C.; McNamara, J.O. Increased Expression of Brain-Derived Neurotrophic Factor Induces Formation of Basal Dendrites and Axonal Branching in Dentate Granule Cells in Hippocampal Explant Cultures. The Journal of Neuroscience 2002, 22, 9754–9763. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Horiike, Y.; Matsuzaki, M.; Miyazaki, T.; Ellis-Davies, G.C.R.; Kasai, H. Protein Synthesis and Neurotrophin-Dependent Structural Plasticity of Single Dendritic Spines. Science (1979) 2008, 319, 1683–1687. [Google Scholar] [CrossRef] [PubMed]
- Kitanishi, T.; Ikegaya, Y.; Matsuki, N.; Yamada, M.K. Experience-Dependent, Rapid Structural Changes in Hippocampal Pyramidal Cell Spines. Cerebral Cortex 2009, 19, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Poo, M. Neurotrophins as Synaptic Modulators. Nat Rev Neurosci 2001, 2, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Balschun, D. Deletion of the Ryanodine Receptor Type 3 (RyR3) Impairs Forms of Synaptic Plasticity and Spatial Learning. EMBO J 1999, 18, 5264–5273. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, N.; Quattrone, A.; Vivoli, E.; Norcini, M.; Bartolini, A.; Ghelardini, C. Different Involvement of Type 1, 2, and 3 Ryanodine Receptors in Memory Processes. Learning & Memory 2008, 15, 315–323. [Google Scholar] [CrossRef]
- Kang, H.; Welcher, A.A.; Shelton, D.; Schuman, E.M. Neurotrophins and Time: Different Roles for TrkB Signaling in Hippocampal Long-Term Potentiation. Neuron 1997, 19, 653–664. [Google Scholar] [CrossRef]
- Tong, L.; Thornton, P.L.; Balazs, R.; Cotman, C.W. β-Amyloid-(1–42) Impairs Activity-Dependent CAMP-Response Element-Binding Protein Signaling in Neurons at Concentrations in Which Cell Survival Is Not Compromised. Journal of Biological Chemistry 2001, 276, 17301–17306. [Google Scholar] [CrossRef]
- Garzon, D.J.; Fahnestock, M. Oligomeric Amyloid Decreases Basal Levels of Brain-Derived Neurotrophic Factor (BDNF) MRNA via Specific Downregulation of BDNF Transcripts IV and V in Differentiated Human Neuroblastoma Cells. The Journal of Neuroscience 2007, 27, 2628–2635. [Google Scholar] [CrossRef] [PubMed]
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The Signaling Pathways Underlying BDNF-Induced Nrf2 Hippocampal Nuclear Translocation Involve ROS, RyR-Mediated Ca2+ Signals, ERK and PI3K. Biochem Biophys Res Commun 2018, 505, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol Rev 2018, 70, 348–383. [Google Scholar] [CrossRef]
- Qiu, J.; Dando, O.; Febery, J.A.; Fowler, J.H.; Chandran, S.; Hardingham, G.E. Neuronal Activity and Its Role in Controlling Antioxidant Genes. Int J Mol Sci 2020, 21, 1933. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive Regulation of the Brain’s Antioxidant Defences by Neurons and Astrocytes. Free Radic Biol Med 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) Localizes to the Mitotic Spindle in Human Cells. PLoS One 2012, 7, e44861. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
AβOs addition induces a slow and sustained increase in nuclear Ca2+ levels in pyramidal hippocampal neurons. Treatment with AβOs induces a slow and sustained increase in nuclear Ca2+ signals in pyramidal hippocampal neurons. (A) Representative image showing the cytoplasmic fluorescence signals emitted by a hippocampal neuron loaded with Fluo-4 after treatment with 500 nM AβOs, at the maximum level of intensity recorded. Scale bar, 15 μm. A rainbow scale was used to show the fluorescence intensity of the Ca2+ indicator (black: low values; red: high values). (B) Representative trace of changes over time in cytoplasmic Ca2+ levels recorded in a neuron loaded with Fluo-4, before (baseline) and after the addition of 500 nM AβOs at the indicated time (arrow). (C) Representative image showing the nuclear fluorescence signals emitted by the nucleus of a neuron transfected with GCaMP3-NLS after the addition of 500 nM AβOs to primary hippocampal cultures, at the maximum level of intensity recorded. Scale bar, 20 μm. A rainbow scale was used to show the fluorescence intensity of the Ca2+ indicator (blue: low values; red: high values). (D) Representative trace of changes over time in nuclear Ca2+ levels recorded in a neuron transfected with GCaMP3-NLS before (baseline) and after the addition of 500 nM AβOs at the indicated time (arrow).
Figure 1.
AβOs addition induces a slow and sustained increase in nuclear Ca2+ levels in pyramidal hippocampal neurons. Treatment with AβOs induces a slow and sustained increase in nuclear Ca2+ signals in pyramidal hippocampal neurons. (A) Representative image showing the cytoplasmic fluorescence signals emitted by a hippocampal neuron loaded with Fluo-4 after treatment with 500 nM AβOs, at the maximum level of intensity recorded. Scale bar, 15 μm. A rainbow scale was used to show the fluorescence intensity of the Ca2+ indicator (black: low values; red: high values). (B) Representative trace of changes over time in cytoplasmic Ca2+ levels recorded in a neuron loaded with Fluo-4, before (baseline) and after the addition of 500 nM AβOs at the indicated time (arrow). (C) Representative image showing the nuclear fluorescence signals emitted by the nucleus of a neuron transfected with GCaMP3-NLS after the addition of 500 nM AβOs to primary hippocampal cultures, at the maximum level of intensity recorded. Scale bar, 20 μm. A rainbow scale was used to show the fluorescence intensity of the Ca2+ indicator (blue: low values; red: high values). (D) Representative trace of changes over time in nuclear Ca2+ levels recorded in a neuron transfected with GCaMP3-NLS before (baseline) and after the addition of 500 nM AβOs at the indicated time (arrow).
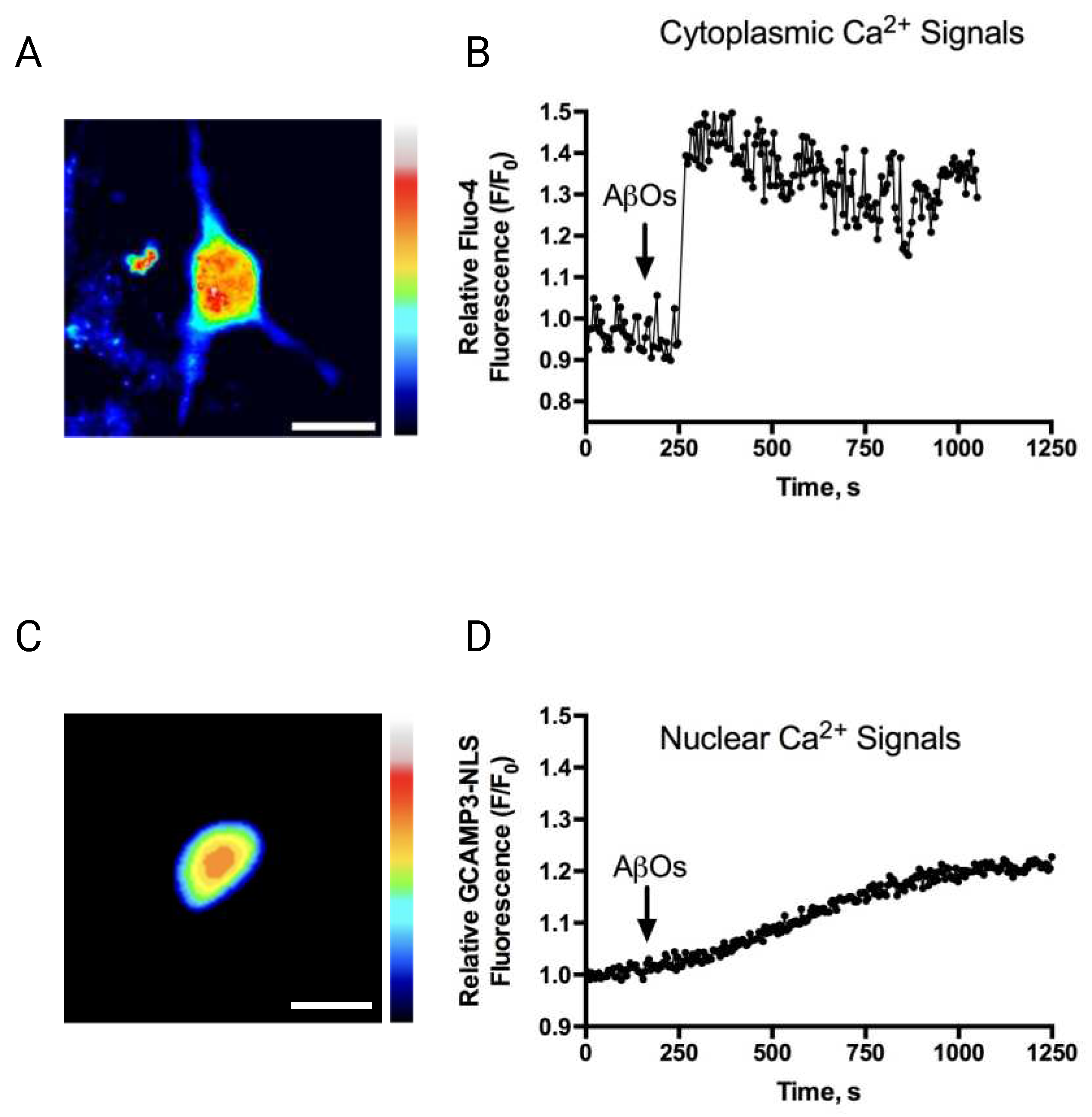
Figure 2.
Gabazine addition to primary hippocampal neurons induces synchronized cytoplasmic (A,B) and nuclear Ca2+ signals (C,D). Gabazine induces synchronized cytoplasmic and nuclear Ca2+ signals in primary hippocampal neurons. (A) Representative image showing the fluorescence signals emitted by the cytoplasmic Ca2+ sensor GCaMP5 (green). (B) Representative trace showing the relative changes of the GCaMP5 sensor fluorescence signals recorded from the dendrites of neurons before (baseline) and after the addition of 5 μM GBZ to the culture, which caused synchronous oscillatory Ca2+ transients. (C) Representative image showing the fluorescence signals emitted by the nuclear Ca2+ sensor GCaMP3-NLS (green) recorded in basal condition; two nuclei of hippocampal neurons are observed. (D) Traces of recordings of the relative changes of the signal intensity of GCaMP3-NLS, recorded in the nuclei illustrated in D before (baseline) and after the addition of 5 μM GBZ to a control culture at the indicated time (arrow).
Figure 2.
Gabazine addition to primary hippocampal neurons induces synchronized cytoplasmic (A,B) and nuclear Ca2+ signals (C,D). Gabazine induces synchronized cytoplasmic and nuclear Ca2+ signals in primary hippocampal neurons. (A) Representative image showing the fluorescence signals emitted by the cytoplasmic Ca2+ sensor GCaMP5 (green). (B) Representative trace showing the relative changes of the GCaMP5 sensor fluorescence signals recorded from the dendrites of neurons before (baseline) and after the addition of 5 μM GBZ to the culture, which caused synchronous oscillatory Ca2+ transients. (C) Representative image showing the fluorescence signals emitted by the nuclear Ca2+ sensor GCaMP3-NLS (green) recorded in basal condition; two nuclei of hippocampal neurons are observed. (D) Traces of recordings of the relative changes of the signal intensity of GCaMP3-NLS, recorded in the nuclei illustrated in D before (baseline) and after the addition of 5 μM GBZ to a control culture at the indicated time (arrow).
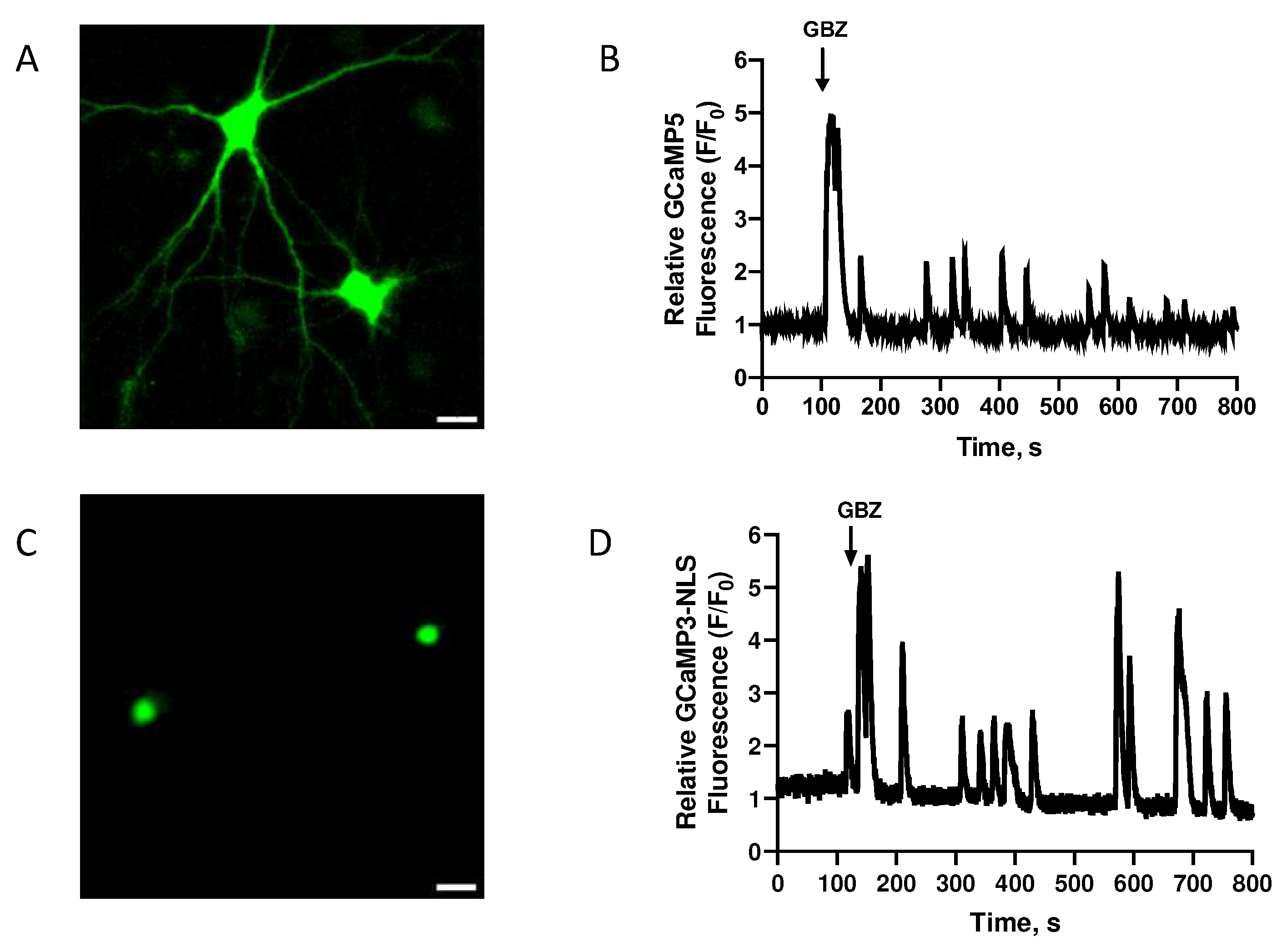
Figure 3.
Treatment with AβOs disrupts the transient nuclear Ca2+ signals induced by GBZ in pyramidal hippocampal neurons. Neurons were incubated for 6 h with AβOs (500 nM) before GBZ addition and the relative change in fluorescence with time values (Fmax-F0/F0) was recorded in the nucleus of neurons expressing GCaMP3-NLS. A) The maximum amplitude and B) the frequency of the detected peaks were quantified; the average values obtained from five experiments performed in independent cultures show significant differences between GBZ addition to controls or to neurons preincubated with AβOs. Data are expressed as Mean ± SE; n = 5. Statistical analysis was performed with Student's t-test. *: p < 0.05.
Figure 3.
Treatment with AβOs disrupts the transient nuclear Ca2+ signals induced by GBZ in pyramidal hippocampal neurons. Neurons were incubated for 6 h with AβOs (500 nM) before GBZ addition and the relative change in fluorescence with time values (Fmax-F0/F0) was recorded in the nucleus of neurons expressing GCaMP3-NLS. A) The maximum amplitude and B) the frequency of the detected peaks were quantified; the average values obtained from five experiments performed in independent cultures show significant differences between GBZ addition to controls or to neurons preincubated with AβOs. Data are expressed as Mean ± SE; n = 5. Statistical analysis was performed with Student's t-test. *: p < 0.05.
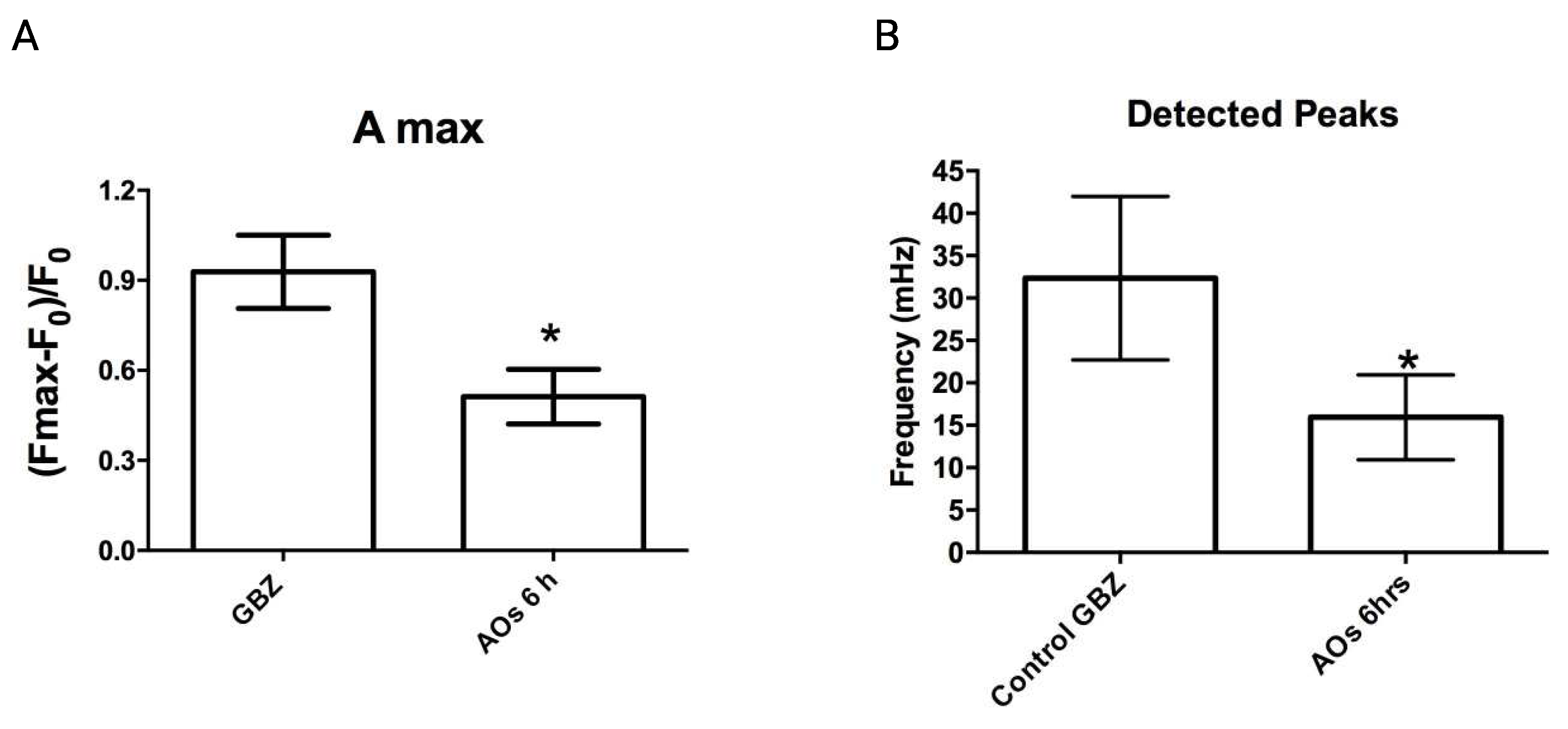
Figure 4.
Treatment with AβOs prevents the CREB phosphorylation increase induced by GBZ in hippocampal neurons. (A) Representative images of control hippocampal cultures and/or cultures pre-incubated for 6 h with 500 nM AβOs before the addition of GBZ. The images show the signals originated from incubation with antibodies against phosphorylated CREB (p-CREB; green) and the neuronal marker MAP2 (red); cell nuclei were labeled with DAPI (blue). All images illustrate the sum of the fluorescence recorded in the z-axis of all confocal planes acquired by Spinning-Disk microscopy, with a 40x objective. Scale bar, 20 μm (B) Quantification of the relative levels of p-CREB fluorescence detected in different conditions. Bars represent Mean ± SE, n = 3. Statistically significant differences were evaluated by One-way ANOVA followed by Bonferroni post-test for multiple comparisons. **: p < 0.01.
Figure 4.
Treatment with AβOs prevents the CREB phosphorylation increase induced by GBZ in hippocampal neurons. (A) Representative images of control hippocampal cultures and/or cultures pre-incubated for 6 h with 500 nM AβOs before the addition of GBZ. The images show the signals originated from incubation with antibodies against phosphorylated CREB (p-CREB; green) and the neuronal marker MAP2 (red); cell nuclei were labeled with DAPI (blue). All images illustrate the sum of the fluorescence recorded in the z-axis of all confocal planes acquired by Spinning-Disk microscopy, with a 40x objective. Scale bar, 20 μm (B) Quantification of the relative levels of p-CREB fluorescence detected in different conditions. Bars represent Mean ± SE, n = 3. Statistically significant differences were evaluated by One-way ANOVA followed by Bonferroni post-test for multiple comparisons. **: p < 0.01.
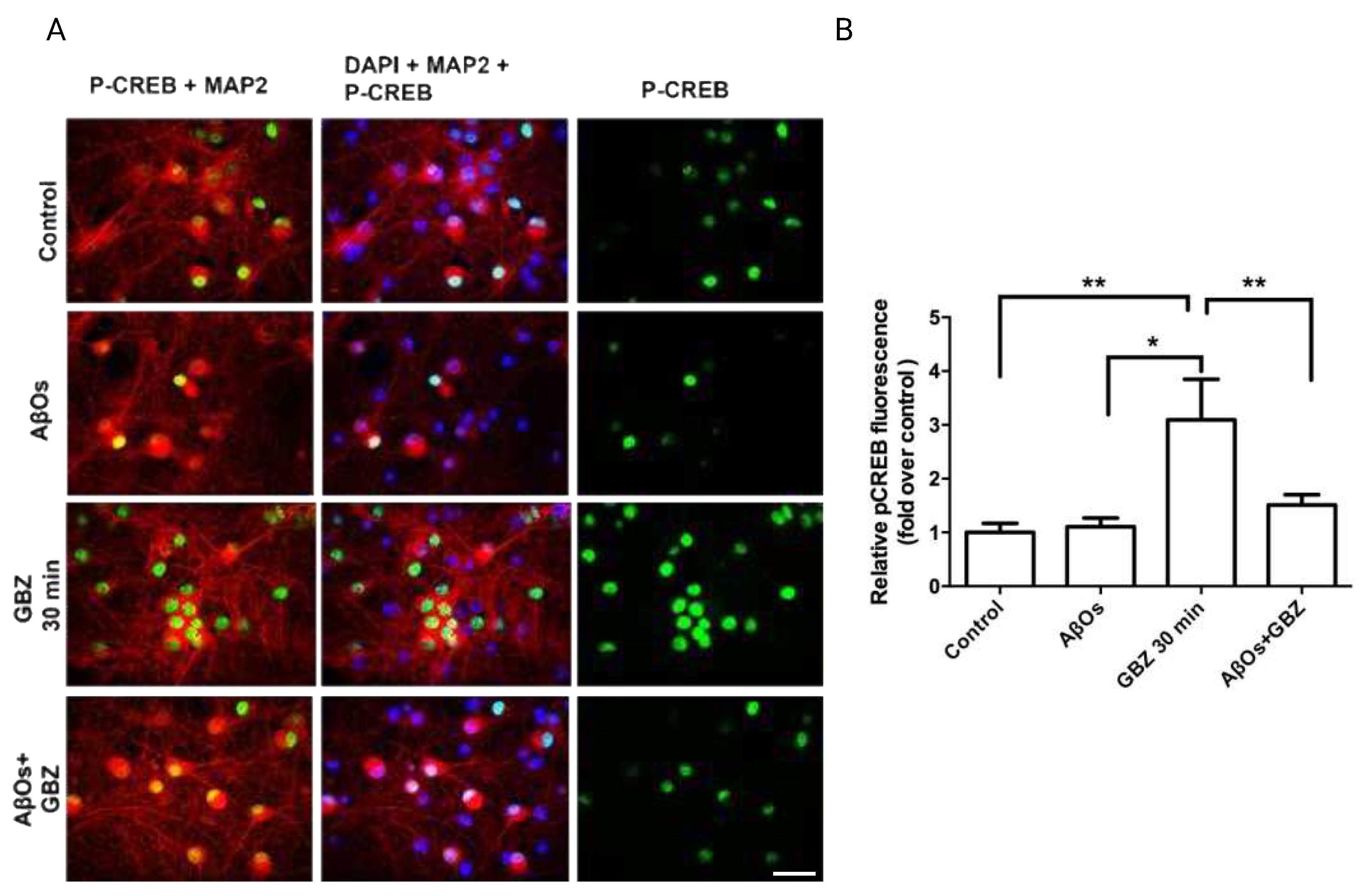
Figure 5.
Treatment with AβOs decrease or prevent the Gabazine-induced increase in the mRNA levels of Npas4, RyR2, Bdnf exon IV and the antioxidant enzyme Nqo1. Treatment with AβOs decreases or prevent the Gabazine-induced increase in the mRNA levels of Npas4, RyR2, Bdnf exon IV and the antioxidant enzyme Nqo1. The relative mRNA levels of Npas4 (A), RyR2 (B), Bdnf exon IV (C) and Nqo1 (D) were determined by qPCR of primary neuronal cultures treated with GBZ for 2 h and incubated next for 6 h with 500 nM AβOs or saline. Values, normalized for β-actin mRNA levels, are expressed as fold over the values displayed by control cultures.
Figure 5.
Treatment with AβOs decrease or prevent the Gabazine-induced increase in the mRNA levels of Npas4, RyR2, Bdnf exon IV and the antioxidant enzyme Nqo1. Treatment with AβOs decreases or prevent the Gabazine-induced increase in the mRNA levels of Npas4, RyR2, Bdnf exon IV and the antioxidant enzyme Nqo1. The relative mRNA levels of Npas4 (A), RyR2 (B), Bdnf exon IV (C) and Nqo1 (D) were determined by qPCR of primary neuronal cultures treated with GBZ for 2 h and incubated next for 6 h with 500 nM AβOs or saline. Values, normalized for β-actin mRNA levels, are expressed as fold over the values displayed by control cultures.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



