
Submitted:
17 October 2023
Posted:
17 October 2023
You are already at the latest version
Abstract
Brown-Spotted Pit viper (Protobothrops mucrosquamatus), also known as the Chinese habu, is a widespread and highly venomous snake distributed from from northeastern India to eastern China. Genomics research can help provide much insight in understanding venom components and natural selection in vipers. Here, we collected, sequenced and assembled the genome of a male P. mucrosquamatus individual from China, producing a highly continuous reference genome, with the length of 1.53 Gb and 41.18% repeat element content. From this 24,799 genes were identified, and 97.97% genes could be annotated. Nuclear genome single-copy genes phylogenetic tree including 6 species verified the validity of our genome assembly and annotation process. This research will contribute to further study on Protobothrops biology and the genetic basis of the snake venom.
Keywords:
Genetics and Genomics
; Evolutionary Biology
; Zoology
1. Introduction
Protobothrops mucrosquamatus belongs to the Viperidae (viper) family of snakes Commonly known as the brown spotted pit viper or Chinese habu, it is widely distributed in northern Vietnam, Laos, northern Myanmar and northeastern India as well as southwestern and eastern China (Figure 1) [1]. P. mucrosquamatus is a venomous snake with tubular venom-conducting fangs and loreal pit, poisoning of their prey manifested by functional impairment of the blood circulation system [2]. Compared with other terrestrial vipers, the maximum amount of single discharging venom of P. mucrosquamatus is higher than Trimeresurus stejnegeri, Gloydius blomhoffii and Bungarus multicinctus [3]. It’s toxicity per unit dose is also higher than that in Deinagkistrodon acutus and T. stejnegeri [3]. Snake venom, while it may contribute to health damage in organisms [1,2,4,5,6], can also play a role in biomedicine [5,7,8,9], such as snake antivenom development, disease treatment and many other fields [10]. High-quality reference genomes and transcriptomes are required to detect venom genes, insight toxin-manufacturing mechanism and design safe and effective antivenoms and other drugs [11,12]. Moreover, rapid evolution of venom protein generally occurs under environmental stress [13,14]. For instance, predation needs, making the study of proteinaceous-venoms coding genes an excellent model system for the adaptation and nature selection [15].
2. Main Content
Context
While snake venoms represent a danger to human health, they are also a potential gold mine of bioactive proteins that can be harnessed for drug discovery purposes [16]. Snake genomics has huge potential for studying venom evolution and toxinology. Here, we assembled a highly contiguous genome of a male P. mucrosquamatus individual collected from Guilin, Guangxi, China using single-tube long fragment read (stLFR) [17] and Whole Genome Sequencing (WGS) technologies. The total size of the genome is 1.53G, containing 41.18% repeat element content, which supply new evidence for further research on Protobothrops genome and the genetic basis of the snake venom.
Methods
Detailed stepwise protocols are gathered together in a protocols.io collection, with some minor adaptations outlined below [18].
Sample collection and sequencing
The male P. mucrosquamatus sample was captured in Guilin, Guangxi, China. After collection and identification, the specimen was quickly frozen in -80°C drikold dry ice during storage and transport in order to maintain high quality DNA and RNA for further use. Samples from 4 organs, including the heart, stomach, liver, and kidney were utilized for RNA sequencing. The muscle sample was used for stLFR and WGS sequencing. DNA extraction, library construction and sequencing are outlined in the protocols.io protocols [18].
The Institutional Review Board of BGI (BGI-IRB E22017) granted approval for sample collection, experiments, and research design in this study. Throughout this research, strict adherence to the guidelines set forth by BGI-IRB was ensured during all procedures.
Genome assembly, annotation and assessment
Supernova software (v2.1.1) was employed to assemble the stLFR sequencing data. To address any gaps and eliminate redundancies in this assembly, the WGS data was subjected to gap filling and redundancy removal using GapCloser [19] (v1.12-r6) and redundans (v0.14a) tools, respectively.
In order to identify known repeat elements in genome sequences, a combination of tools was utilized: Repeat Finder (TRF) [20] (v. 4.09), LTR_FINDER [21], RepeatModeler [22] (v1.0.8), RepeatMasker [23] (v. 3.3.0) and RepeatProteinMask (v. 3.3.0) [24] were employed for the search. For the prediction of protein-coding genes, multiple approaches were employed. De novo gene prediction was performed using Augustus [25] (v3.0.3). The RNA-seq data underwent filtration with Trimmomatic [26] (v0.30), followed by transcript assembly using Trinity [27] (v2.13.2) based on clean RNA-seq data. Alignment of transcripts against the genome to obtain gene structures was accomplished using Programto Assemble Spliced Alignments (PASA) [28] (v2.0.2). Homology-based prediction involved mapping protein sequences from the UniProt database (release-2020_05), Pseudonaja textilis, Thamnophis elegans and Notechis scutatus to the genome using the Blastall (v2.2.26) [29] with an E-value cut-off of 1e-5. Gene models were predicted by analyzing the alignment results with GeneWise [30] (v2.4.1). Integration of RNA-seq, homology, and de novo predicted genes was achieved using the MAKER pipeline (v3.01.03) [31] to generate the final gene set.
To annotate the genes function of P. mucrosquamatus, a comprehensive analysis was conducted. BLAST searches were executed against multiple databases, including SwissProt, TrEMBL, and Kyoto Encyclopedia of Genes and Genomes (KEGG), with an E-value cut-off of 1e-5. To predict motifs and domains, InterProScan [26] (v5.52-86.0) as well as Gene ontology (GO) were employed. The results of this analysis further enriched our understanding of the genes' roles and their involvement in biological processes.
The completeness of the genome was evaluated using sets of Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.2) with genome mode and lineage data from vertebrata_odb10 [32]. To reconstruct the phylogenetic tree, we used OrthoFinder(v2.3.7) (RRID:SCR_017118) [33] to search for single-copy orthologs among the protein sequences of Anolis carolinensis (GCA_000090745.2), Chelonia mydas (GCA_015237465.2), Danio rerio (GCA_000002035.4), Deinagkistrodon acutus (http://gigadb.org/dataset/100196), Gallus gallus (GCA_016699485.1), Homo sapiens (GCA_000001405.29), Mus musculus (GCA_000001635.9), Ophiophagus hannah (GCA_000516915.1), Python bivittatus (GCA_000186305.2), Xenopus tropicalis (GCA_000004195.4) and Alligator mississippiensis (GCA_000281125.4).
Results
In this snake genomics study, 224.27 Gb linked-reads data was obtained after stLFR sequencing, and 96.93 Gb short reads data was obtained after WGS sequencing, coming to a grand total of 321.20Gb (Table 1).
We produced a high-continuity P. mucrosquamatus genome assembly, with 1.53Gb total genome size, 39.86% GC content and 362.40kb scaffold N50 length (Table 2). The P. mucrosquamatus genome assembly, of which maximal scaffold length reaches 5.31 M, has 149173 scaffolds over 500bp, with 1.51Gb total length, occupying 98.82% in genome total length. That will become effective resource to provide new perspectives on the study of viper genomics.
In the aggregate, we identify 41.18% repetitive element in P. mucrosquamatus genome, among which 32.33% LINEs become the highest proportion of this assembly, accounting for 471.99M, which is very similar to repetitive element content in the previously sequenced Thamnophis elegans genome (42.02%) (accession No. PRJNA561996) and Crotalus tigris genomes (42.31%) [35], indicating plausible values. The other dominant examples of transposable elements, LTR, DNA transposons and SINE, were 11.50%, 4.94%, and 0.80% respectively (Figure 2, Table 3 and Table 4).
After homology-based, De-novo and RNA-sequencing annotation methods, 24,799 protein-coding gene have been identified in our P. mucrosquamatus genome assembly. The average length of P. mucrosquamatus gene is 1.53 bp, containing 8.96 exon for each gene. Additionally, 387 miRNAs, 319 tRNAs, 289 snRNAs were predicted in P. mucrosquamatus genome. (Table 6)
Through comparisons with public datasets , including InterPro [36], Kyoto Encyclopedia of Genes and Genomes (KEGG) [37], SwissProt [38], TrEMBL [38] and Gene ontology terms), 24296 expanded gene family were identified, and 97.97% genes can be annotated based on function.(Table 5)
According to KEGG enrichment analysis consequences, Environmental Information Processing, Organismal Systems and Metabolism pathways took up a great proportion of these, among which Signal transduction pathways took up the largest propotion. Genes associated with Immune system (2445) and Endocrine system (2033) accounted for the largest number of Organismal Systems pathways (Figure 3a). Based on the GO analysis results, there are 7900 genes related to binding and 7740 gene related to cellular processes (Figure 3b).
Data validation and quality control
Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.2.2 was used to evaluate the completeness and quality of our assembly [39]. BUSCO analysis results indicating this genome assembly has up to 83.6% by using the vertebrata_odb10 database. (Figure 4)
For the purpose of checking the quality of our assembly, 7 other kinds of amphibians and reptiles (Anolis carolinensis, Chelonia mydas, Deinagkistrodon acutus, Ophiophagus hannah, Python bivittatus, Xenopus tropicalis and Alligator mississippiensis) , Gallus gallus, Homo sapiens, Mus musculus, Danio rerio protein sequences download from NCBI and CNGB were used to construct a phylogenetic tree. The relationship among all the species reflected by phylogenetic tree conformed to previous research, demonstrating our data can screening related species(Figure 5). A total of 1177 single-copy loci were found.
Author Contributions
Huan Liu designed and initiated the project. Anhui Normal University collected the samples. Haorong Lu, Yajie Zhou and Minhui Shi performed the DNA extraction, library construction. Xiaotong Niu and Shiqing Wang performed data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) [40] of China National GeneBank DataBase (CNGBdb) [41] with accession number CNP0004048.
Acknowledgments
Our project was supported by the China National GeneBank (CNGB) and the the Guangdong Provincial Key Laboratory of Genome Read and Write (grant no. 2017B030301011). This work was also supported by BGI-Shenzhen.
Conflicts of Interest
The authors declare no conflict financial interests.
Reuse Potential
This genomic data will provide new resources for further study of viper biology and evolution, alongside the genetic basis of viper snake venom.
References
- Liu C-C, Wu C-J, Hsiao Y-C, Yang Y-H, Liu K-L, Huang G-J, et al.. Snake venom proteome of Protobothrops mucrosquamatus in Taiwan: Delaying venom-induced lethality in a rodent model by inhibition of phospholipase A2 activity with varespladib. Journal of Proteomics. 2021. [CrossRef]
- Valenta J, Stach Z, Otahal M. Protobothrops mangshanensis bite: First clinical report of envenoming and its treatment. Biomedical papers of the Medical Faculty of the University Palacký, Olomouc, Czechoslovakia. 2012. [CrossRef]
- He Y, Sun W-Q, Li J-L, Zhang S, Li J, You H-J, et al.. Yuanmaotoufu Shedu Duxing Ji Huifu Guilv[Toxicity and Recovery of Protobothrops Mucrosquamatus Venoms]. Xiandai Shengwu Yixue Jinzhan. 2016; 0:1623–62016;
- Mao Y-C, Liu P-Y, Chiang L-C, Lee C-H, Lai C-S, Lai K-L, et al.. Clinical manifestations and treatments of Protobothrops mucrosquamatus bite and associated factors for wound necrosis and subsequent debridement and finger or toe amputation surgery. Clinical Toxicology. Taylor & Francis; 2021. [CrossRef]
- Zeng F, Chen C, Chen X, Zhang L, Liu M. Small Incisions Combined with Negative-Pressure Wound Therapy for Treatment of Protobothrops Mucrosquamatus Bite Envenomation: A New Treatment Strategy. Med Sci Monit. 2019. [CrossRef]
- Lin C-C, Chen Y-C, Goh ZNL, Seak C-K, Seak JC-Y, Shi-Ying G, et al.. Wound Infections of Snakebites from the Venomous Protobothrops mucrosquamatus and Viridovipera stejnegeri in Taiwan: Bacteriology, Antibiotic Susceptibility, and Predicting the Need for Antibiotics—A BITE Study. Toxins. Multidisciplinary Digital Publishing Institute; 2020. [CrossRef]
- Offor BC, Muller B, Piater LA. A Review of the Proteomic Profiling of African Viperidae and Elapidae Snake Venoms and Their Antivenom Neutralisation. Toxins. Multidisciplinary Digital Publishing Institute; 2022. [CrossRef]
- Whitaker R, Whitaker S. Venom, antivenom production and the medically important snakes of India. Current Science. Temporary Publisher; 103:635–432012;
- Chippaux J-P, Goyffon M. Venoms, antivenoms and immunotherapy. Toxicon. 1998. [CrossRef]
- Vogel G, Pan H, Chettri B, Yang D, Jiang K, Wang K, et al.. A New Species of the Genus Protobothrops (Squamata: Viperidae) from Southern Tibet, China and Sikkim, India. Asian Herpetological Research. 2013. [CrossRef]
- Xin H, Tao P, Demin H, Liang Z, Yinxu H, Lei Y, et al.. A New Species of the Genus Protobothrops (Squamata: Viperidae: Crotalinae) from the Dabie Mountains, Anhui, China: A New Species of the Genus Protobothrops (Squamata: Viperidae: Crotalinae) from the Dabie Mountains, Anhui, China. Asian Herpetological Research. 2012. [CrossRef]
- Tan CH. Snake Venomics: Fundamentals, Recent Updates, and a Look to the Next Decade. Toxins. Multidisciplinary Digital Publishing Institute; 2022. [CrossRef]
- Aird SD, Aggarwal S, Villar-Briones A, Tin MM-Y, Terada K, Mikheyev AS. Snake venoms are integrated systems, but abundant venom proteins evolve more rapidly. BMC Genomics. 2015. [CrossRef]
- Casewell NR, Wüster W, Vonk FJ, Harrison RA, Fry BG. Complex cocktails: the evolutionary novelty of venoms. Trends in Ecology & Evolution. 2013. [CrossRef]
- Aird SD, Arora J, Barua A, Qiu L, Terada K, Mikheyev AS. Population Genomic Analysis of a Pitviper Reveals Microevolutionary Forces Underlying Venom Chemistry. Genome Biology and Evolution. 2017. [CrossRef]
- Rao W, Kalogeropoulos K, Allentoft ME, Gopalakrishnan S, Zhao W, Workman CT, et al.. The rise of genomics in snake venom research: recent advances and future perspectives. GigaScience. 2022. [CrossRef]
- Wang O, Chin R, Cheng X, Wu MKY, Mao Q, Tang J, et al.. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 2019. [CrossRef]
- Liu B, Cui L, Deng Z, Ma Y, Yang D, Gong Y, et al.. The annotation pipeline for the genome of a snake. 2023. https://www.protocols.io/view/the-annotation-pipeline-for-the-genome-of-a-snake-cnv3ve8n.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al.. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012. [CrossRef]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 1999. [CrossRef]
- Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 2007. [CrossRef]
- Smit AFA, Hubley R, Green P. RepeatModeler Open-1.0. 2008–2015. Seattle, USA: Institute for Systems Biology Available from: httpwww repeatmasker org, Last Accessed May. 1:20182015;
- arailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences Curr Protoc Bioinformatics. 2009. Chapter.
- Tempel S. Using and understanding RepeatMasker. Mobile genetic elements: protocols and genomic applications. Springer; :29–51 2012.
- Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research. 2004. [CrossRef]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014. [CrossRef]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al.. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. Nature Publishing Group; 2013. [CrossRef]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al.. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008. [CrossRef]
- Mount DW. Using the Basic Local Alignment Search Tool (BLAST). Cold Spring Harb Protoc. Cold Spring Harbor Laboratory Press; 2007. [CrossRef]
- Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004. [CrossRef]
- Campbell MS, Holt C, Moore B, Yandell M. Genome Annotation and Curation Using MAKER and MAKER-P. Current Protocols in Bioinformatics. 2014. [CrossRef]
- Wick RR, Holt KE. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Research. F1000 Research Limited London, UK; 8:21382021.
- Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology. 2015. [CrossRef]
- Margres MJ, Rautsaw RM, Strickland JL, Mason AJ, Schramer TD, Hofmann EP, et al.. The Tiger Rattlesnake genome reveals a complex genotype underlying a simple venom phenotype. Proceedings of the National Academy of Sciences. Proceedings of the National Academy of Sciences; 2021. [CrossRef]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al.. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014. [CrossRef]
- Kanehisa M. The KEGG Database. ‘In Silico’ Simulation of Biological Processes. John Wiley & Sons, Ltd.
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 2000. [CrossRef]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015. [CrossRef]
- Guo X, Chen F, Gao F, Li L, Liu K, You L, et al.. CNSA: a data repository for archiving omics data. Database. 2020. [CrossRef]
- Chen FZ, You LJ, Yang F, Wang LN, Guo XQ, Gao F, et al.. CNGBdb: China National GeneBank DataBase. Yi Chuan. 2020. [CrossRef]
Figure 1.
A Brown-Spotted Pit viper (Protobothrops mucrosquamatus) individual, photographed by Diancheng Yang in Guilin, Guangxi Province.
Figure 1.
A Brown-Spotted Pit viper (Protobothrops mucrosquamatus) individual, photographed by Diancheng Yang in Guilin, Guangxi Province.

Figure 2.
Distribution of transposable elements (TEs) in the P. mucrosquamatus genome. The TEs include DNA transposons (DNA) and RNA transposons (i.e. DNAs, LINEs, LTRs, and SINEs). (a) De novo sequence divergence rate distribution. (b) Known sequence divergence rate distribution.
Figure 2.
Distribution of transposable elements (TEs) in the P. mucrosquamatus genome. The TEs include DNA transposons (DNA) and RNA transposons (i.e. DNAs, LINEs, LTRs, and SINEs). (a) De novo sequence divergence rate distribution. (b) Known sequence divergence rate distribution.
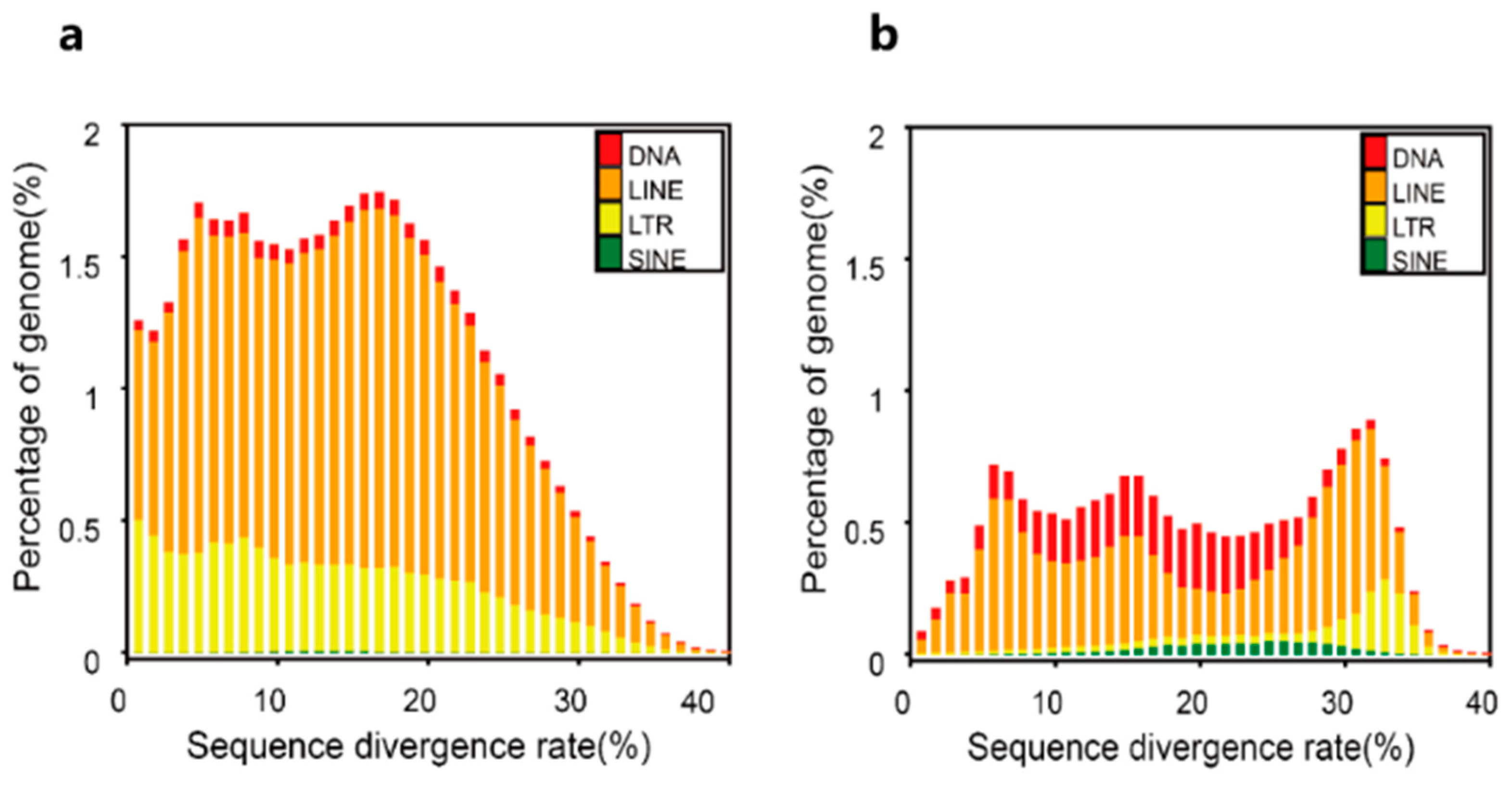
Figure 3.
Gene annotation information of P. mucrosqamatus. (a) KEGG enrichment of P. mucrosquamatus. (b) GO enrichment of P. mucrosquamatus (c) Venn of InterPro, KEGG and Swissport.
Figure 3.
Gene annotation information of P. mucrosqamatus. (a) KEGG enrichment of P. mucrosquamatus. (b) GO enrichment of P. mucrosquamatus (c) Venn of InterPro, KEGG and Swissport.
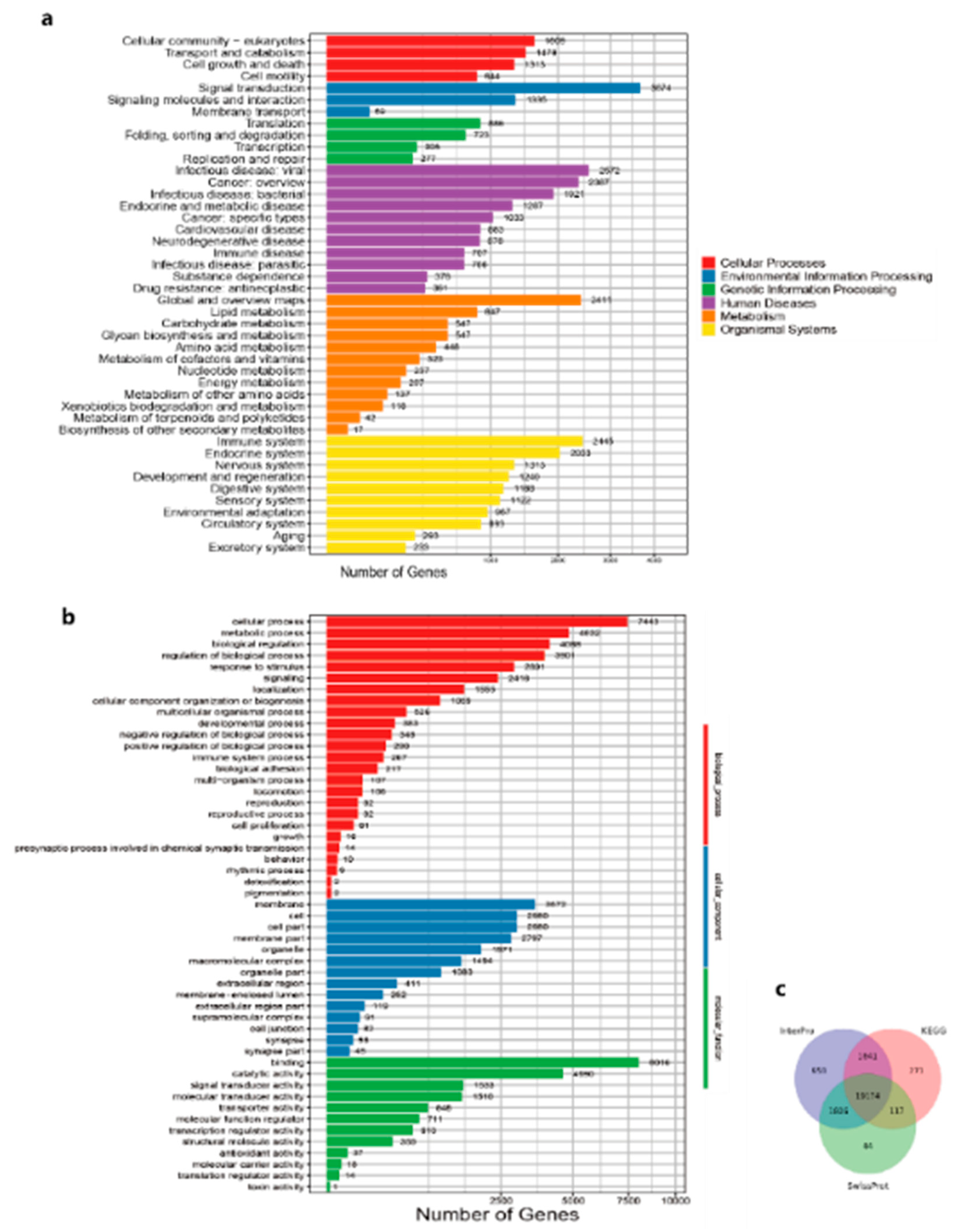
Figure 4.
BUSCO Assessment result of the P. mucrosquamatus genome.
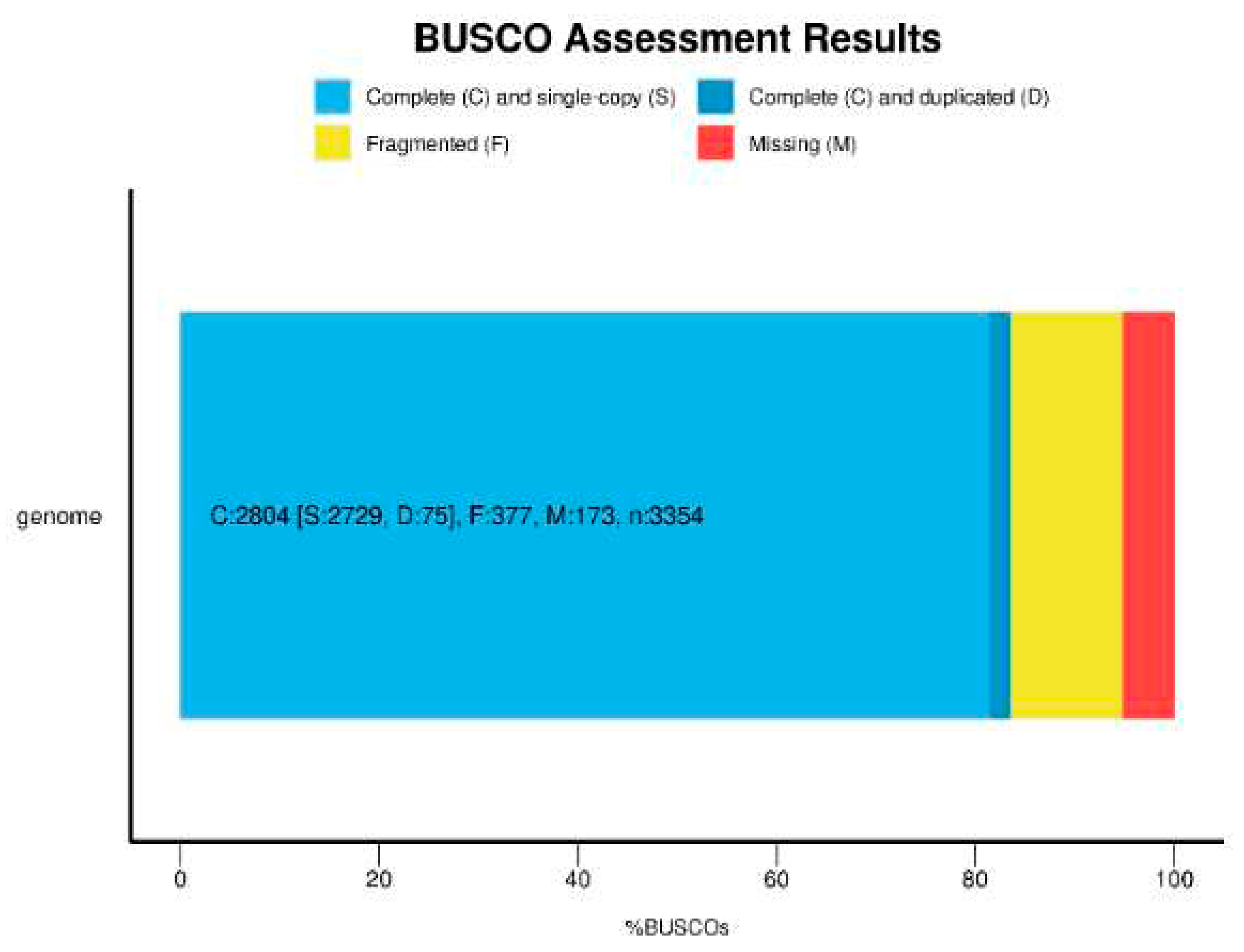
Figure 5.
Phylogenetic tree reconstructed using nuclear genome single-copy genes. The numbers in the branches of the phylogenetic tree represents branch length obtained in OrthoFinder.
Figure 5.
Phylogenetic tree reconstructed using nuclear genome single-copy genes. The numbers in the branches of the phylogenetic tree represents branch length obtained in OrthoFinder.
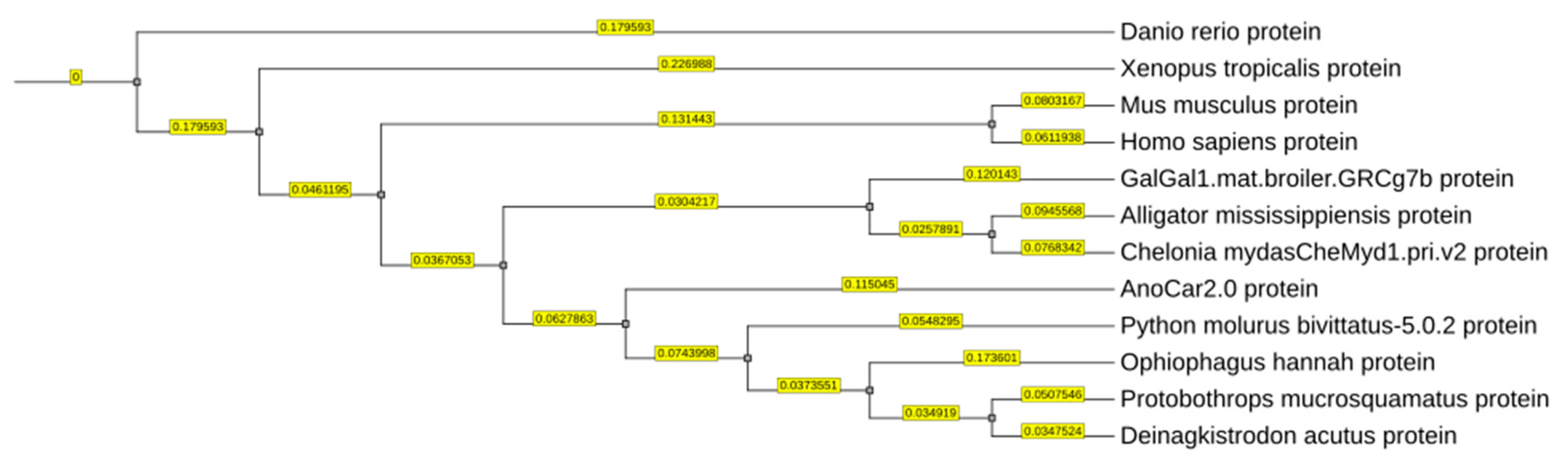

Table 1.
Summary statistics of P. mucrosquamatus sequenced reads.
| Base Number | GC content(%) | Q20(%) | Q30(%) | ||
| WGS | fq1 | 52036970400 | 40.30 | 97.58 | 92.48 |
| fq2 | 52036970400 | 40.23 | 97.98 | 92.71 | |
| stLFR | fq1 | 104698910600 | 38.89 | 96.9 | 90.75 |
| fq2 | 136108583780 | 41.72 | 97.79 | 91.85 | |
Table 2.
Summary of the features of the P. mucrosquamatus genome.
| Statistical level | Original | Scaffold >(500)bp | ||||
| scaffold | contig | contig>(500) | scaffold | contig | ||
| Total number (>) | 203555 | 287462 | 192124 | 149173 | 232200 | |
| Total length of (bp) | 1530648812 | 1481196605 | 1457896424 | 1512499815 | 1463075630 | |
| Average length (bp) | 7519.58 | 5152.67 | 7588.31 | 10139.23 | 6300.93 | |
| N50 Length (bp) | 380005 | 36547 | 37585 | 390274 | 37334 | |
| N90 Length (bp) | 2960 | 2304 | 2773 | 3453 | 2667 | |
| Maximum length (bp) | 5566463 | 488153 | 488153 | 5566463 | 488153 | |
| GC content (%) | 39.86 | 39.86 | 39.79 | 39.8 | 39.8 | |
Table 3.
Statistics for repetitive sequences identified in the P. mucrosquamatus genome.
| Type | Repeat Size | % of genome |
| Trf | 48630912 | 3.177144 |
| Repeatmasker | 248960159 | 16.265008 |
| Proteinmask | 178699911 | 11.674782 |
| De novo | 591205406 | 38.624497 |
| Total | 630311866 | 41.179391 |
Table 4.
Summary of transposable elements (TEs) in the P. mucrosquamatus genome.
| Type | Repbase TEs | TE protiens | De novo | Combined TEs | ||||
| Length (Bp) | % in genome | Length (Bp) | % in genome | Length (Bp) | % in genome | Length (Bp) | % in genome | |
| DNA | 54802686 | 3.580357 | 2721607 | 0.177807 | 23812202 | 1.555693 | 75566775 | 4.936911 |
| LINE | 173499745 | 11.335046 | 145892994 | 9.531448 | 446008208 | 29.138507 | 494919112 | 32.333943 |
| SINE | 11128833 | 0.727066 | 0 | 0 | 1414004 | 0.092379 | 12299674 | 0.80356 |
| LTR | 27382417 | 1.788942 | 30199813 | 1.973007 | 165177572 | 10.791344 | 175979322 | 11.497041 |
| Other | 95860 | 0.006263 | 0 | 0 | 0 | 0 | 95860 | 0.006263 |
| Total | 248960159 | 16.265008 | 178699911 | 11.674782 | 588493585 | 38.447329 | 618611286 | 40.414972 |
Table 6.
Statistics for miRNA, tRNA, rRNA and snRNA discerned in the P. mucrosquamatus genome.
| Type | Copy(w) | Average length(bp) | Total length(bp) | % of genome | |
| miRNA | 387 | 115.3540052 | 44642 | 0.002917 | |
| tRNA | 319 | 76.38244514 | 24366 | 0.001592 | |
| rRNA | rRNA | 75 | 111.8266667 | 8387 | 0.000548 |
| 18S | 18 | 141.5555556 | 2548 | 0.000166 | |
| 28S | 52 | 104.3269231 | 5425 | 0.000354 | |
| snRNA | snRNA | 289 | 115.6955017 | 33436 | 0.002184 |
| CD-box | 110 | 90.2 | 9922 | 0.000648 | |
| HACA-box | 66 | 144.7575758 | 9554 | 0.000624 | |
| splicing | 98 | 112.1734694 | 10993 | 0.000718 |
Table 5.
Consequences of gene functional annotation.
| Values | Total | Swissprot-Annotated | KEGG-Annotated | TrEMBL-Annotated | Interpro-Annotated | GO-Annotated | Overall |
| Number | 24,799 | 21,141 | 21,203 | 23,741 | 23,579 | 15,322 | 24,296 |
| Percentage | 100% | 85.25% | 85.50% | 95.73% | 95.08% | 61.78% | 97.97% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



