
Submitted:
16 October 2023
Posted:
17 October 2023
You are already at the latest version
Abstract
Peptide-based opioid ligands are important candidates for the development of novel, safer, and more effective analgesics to treat pain. To develop peptide-based safer analgesics, we synthesized a mixture-based cyclic pentapeptide library containing a total of 24,624 pentapeptides and screened the mixture-based library samples using a 55 °C warm-water tail-withdrawal assay. Using this phenotypic screening approach, we deconvoluted the mixture-based samples to identify a novel cyclic peptide Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt), which produced dose- and time-dependent antinociception with an ED50 (and 95% confidence interval) of 0.70 (0.52-0.97) mg/kg i.p. mediated by the mu-opioid receptor (MOR). Additionally, higher doses (≥ 3 mg/kg, i.p.) of CycloAnt antagonized delta-opioid receptors (DOR) at least 3 hours. Pharmacological characterization of CycloAnt showed the cyclic peptide did not reduce breathing rate in mice at doses up to 15 times the analgesic ED50 value, and dramatically less hyperlocomotion than the MOR agonist, morphine. While chronic administration of CycloAnt resulted in antinociceptive tolerance, it was without opioid-induced hyperalgesia and significantly reduced signs of naloxone-precipitated withdrawal suggestive of reduced physical dependence as compared to morphine. Collectively, the results suggest this dual MOR/DOR multifunctional ligand is an excellent lead for the development of peptide-based safer analgesics.
Keywords:
mixed-based combinatorial library
; structure–activity relationships
; cyclic peptide
; mu-opioid receptor
; delta-opioid receptor
; bifunctional ligand
; antinociception
; opioid antagonist
; opioid liabilities
1. Introduction
Opioid analgesics are effective pain medications for relieving acute, severe pain. However, repeated opioid use results in clinical liabilities such as the development of tolerance and dependence and a potentially lethal respiratory depression [1]. The misuse of opioid analgesics leads to the increasing risk of opioid abuse and the related overdose, which have contributed to the escalating economic burden imposed on society. There is a critical public health need for novel, safer, and more efficacious analgesics to treat pain.
One strategy to address this need focuses on the development of opioid peptides into safer analgesics [2,3]. The opioid system utilizes opioid receptors and the endogenous opioid peptides to modulate pain, yet the endogenous opioid system is not observed to produce respiratory depression and addictive side effects. A recent study using a genetically encoded biosensor revealed the mu-opioid peptide receptor (MOR) and the delta-opioid peptide receptor (DOR) not only function at the cell membrane but also inside the cell at different locations, depending on the chemistry of ligand types [4]. After peptides activated the MOR in plasma membrane, MOR quickly internalized and was detected in active state in endosome for 20 min; while opiate alkaloids penetrated the membrane to produce Golgi-localized receptor activation after MOR was activated in the plasma membrane [4]. The spatiotemporal specificity affects signal duration and pathway selection downstream, contributing to distinct downstream physiological effects [5,6] As the endogenous opioid peptides, not opiate alkaloids, can precisely modulate pain, the different spatiotemporal patterns of opioid receptor activation may link to the severity of adverse effects limiting opiate use in pain management [7].
The endogenous opioid peptides such as enkephalins, endorphins, and dynorphins exert their analgesic effects through the MOR, DOR, and kappa (KOR) opioid receptors. These peptides have varying affinities at the opioid receptors and none of them binds exclusively to one opioid receptor type [8]. For example, Enkephalins bind and activate the DOR and MOR. Endorphins exert their effect primarily through the MOR, but they also have affinity for the DOR. While dynorphins have high affinity for the KOR, they have significant affinity for the MOR and DOR as well. Most importantly, as a whole, the endogenous opioid peptides activate all opioid receptors.
All three opioid receptors mediate opioid-dependent analgesia, while MOR has been the primary target for clinically used opioid analgesics; however, activation of MOR may lead to respiratory depression and physical dependence. Unlike MOR, activation of DOR does not cause respiratory depression and physical dependence [9]. Since DOR is mainly located in the cytoplasm, with a very low level of receptors being associated with the plasma membrane [10], activation of DOR alone produces insufficient analgesia in model of acute pain [9]. However, mounting evidence indicates that activation of DOR produces analgesia in various models of chronic inflammatory and neuropathic pain [11,12].
Importantly, multifunctional ligands that simultaneously modulate MOR and DOR have been demonstrated to produce enhanced antinociception with fewer adverse effects [13,14,15,16,17,18]. For example, co-administration of the DOR agonist SNC80 with the MOR agonist heroin enhanced the antinociceptive effect of heroin while decreasing the sedative and reinforcing effects [19]. Dual agonists effective at both MOR and DOR, such as MMP-2200 [20,21] and SRI-22141 [22], produce high antinociceptive potency as well as decreasing the propensity to produce locomotor stimulation, tolerance, and physical dependence in mouse models of chronic pain. Simultaneous activation of MOR and DOR using a dual agonist LP2 not only significantly ameliorated mechanical allodynia but also produced neuroprotective effects in a rodent model of neuropathic pain [23]. Interestingly, dual functional ligands acting as MOR agonist/DOR antagonist have also been reported to produce enhanced efficacy with reduced tolerance and dependence [13,24,25]. DIPP-NH2[Ψ] [26], SoRi20411 [27], and 14-alkoxy pyridomorphinans [28] are potent DOR antagonists and MOR agonists which produce analgesia with reduced tolerance when administered repeatedly at the ED80 antinociceptive dose supraspinally twice daily for 5−7 days. Ligands simultaneously modulating multiple opioid receptors, such as MOR and DOR, therefore hold a great promise for the development of safe analgesics for pain.
We have previously reported the synthesis of a mixture-based cyclic peptide library consisting of 24,624 anthraniloyl(Ant)-tagged pentapeptides in a positional scanning format (Figure 1) [29]. The fully-defined cyclic peptide Tyr-[D-Lys-Dap(Ant)-Tyr-Gly] identified from this library demonstrated high affinity for the MOR in a competition receptor binding assay [29]. As cyclic peptides have improved stability against proteolysis, we hypothesized a cyclic peptide with a desired antinociceptive, safety, and pharmacokinetic profile would be within the library. To identify such a cyclic peptide analgesic from this library, we employed a direct phenotypic in vivo screening using the 55 °C warm-water tail-withdrawal (WWTW) assay to facilitate the discovery of the analgesic lead [30,31]. As the WWTW assay measures the overall antinociception of a sample in mice, the antinociceptive lead compounds discovered should have a favored pharmacokinetic profile without a preconceived bias for the mechanism of action [32]. Using the direct in vivo screening, we identified a fully-defined, novel, potent, low-liability cyclic peptide analgesic from the mixture library. Further target identification characterization using opioid receptor knock-out mice elucidated the analgesic lead produced agonist activity at MOR, while pharmacological testing antagonizing DOR, indicating it acted as a MOR agonist/DOR antagonist in mice.
2. Results
2.1. Mixture-Based Cyclic Peptide Library
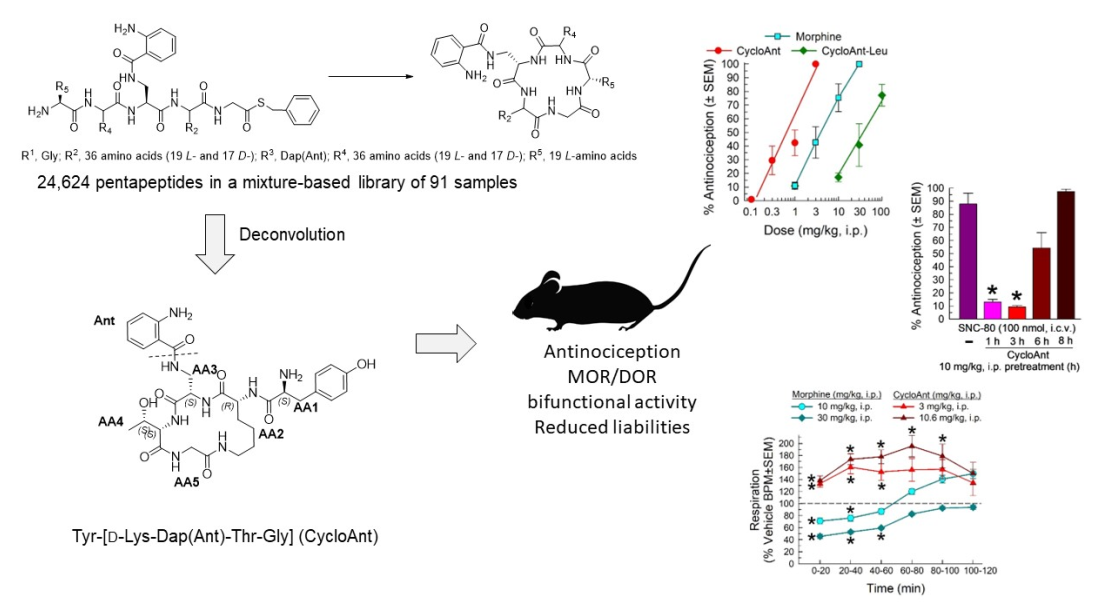
As we reported previously, the cyclic peptides in the library [29] were synthesized from peptide thioesters [33] using a imidazole-promoted cyclization strategy [34]. All the pentapeptides in this library have a Gly fixed at R1 position and a Dap(Ant) at fixed R3 position. The rest of the R2, R4, and R5 positions are made either with a single amino acid (O) or a mixture of amino acids (X) [29]. The library was composed of the R2-, R4-, and R5-sub-libraries (Figure 1). R2-position and R4-position were constituted by a total of 36 amino acids (AAs) including 19 L- and 17 D-amino acids, while R5-position was generated with 19 natural L-amino acids. Thus, there are 36 R2 samples in the R2-sublibrary, 36 R4 samples in the R4-sublibrary, and 19 R5 samples in the R5-sublibrary; each sample is a mixture of pentapeptides. Each sample contains a total of 684 (36 × 19) pentapeptides at the R2- and R4-sublibraries, and 1296 (36 × 36) pentapeptides at the R5-sublibrary. This positional scanning library has a total of 91 (36 + 36 + 19) samples and 24,624 (36 × 36 × 19) pentapeptides in the linear sequences.
Figure 1.
The mixture-based cyclic peptide library and library composition. (Top). General peptide sequence in the library. (Bottom). The defined single amino acid (O) of each sample in the library.
Figure 1.
The mixture-based cyclic peptide library and library composition. (Top). General peptide sequence in the library. (Bottom). The defined single amino acid (O) of each sample in the library.
2.2. Library Deconvolution Using Direct In vivo Screening
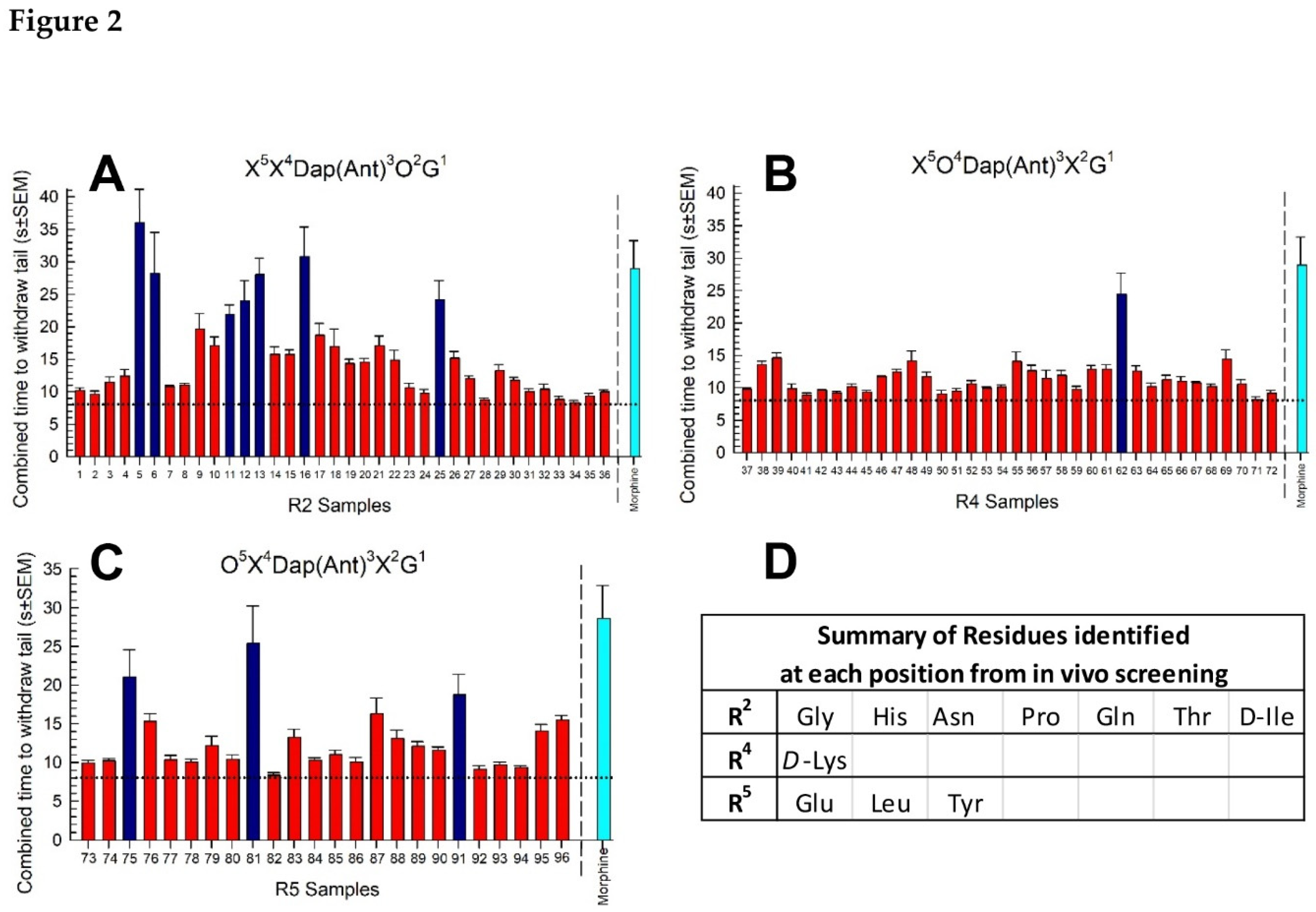
Utilizing a positioning scanning format, the library was screened in vivo via antinociception assessed with the 55 °C WWTW assay as we previously described [30,31]. Each partially-defined, mixture-based sample was administered intraperitonially (i.p.) at a dose of 5 mg/kg. Additional mice received vehicle alone (10%DMSO/90% saline, i.p.) as a negative control, or morphine (10 mg/kg, i.p.) as a positive control. The combined time mice demonstrated to withdraw their tail was summed over six time points examined for each sample tested and is reported by substitution position (Figure 2a–c). Analysis of the responses with one-way ANOVA detected significant global responses in the R2- (F(37,266) = 12.1, p < 0.0001; Figure 2a), R4- (F(37,266) = 13.1, p < 0.0001; Figure 2b) and R5-sublibraries (F(25,182) = 9.50, p < 0.001; Figure 2c). Post hoc analysis (Dunnett’s) identified samples from the R2-, R4- and R5-sublibraries producing a combined average response time to tail-withdrawal that was both significantly different from the vehicle response (p < 0.05 vs dotted line, Figure 2a–c), but NOT different from the morphine response (p > 0.05 vs cyan bar, Figure 2a–c). Meeting these conditions for the R2- position were Samples 5 (Gly), 6 (His), 11 (Asn), 12 (Pro), 13 (Gln), 16 (Thr) and 25 (D-Ile), whereas for the R4 position, only Sample 62 (D-Lys) showed this response, while the R5 position demonstrated three Samples meeting the criteria: 75 (Glu), 81 (Leu) and 91 (Tyr).
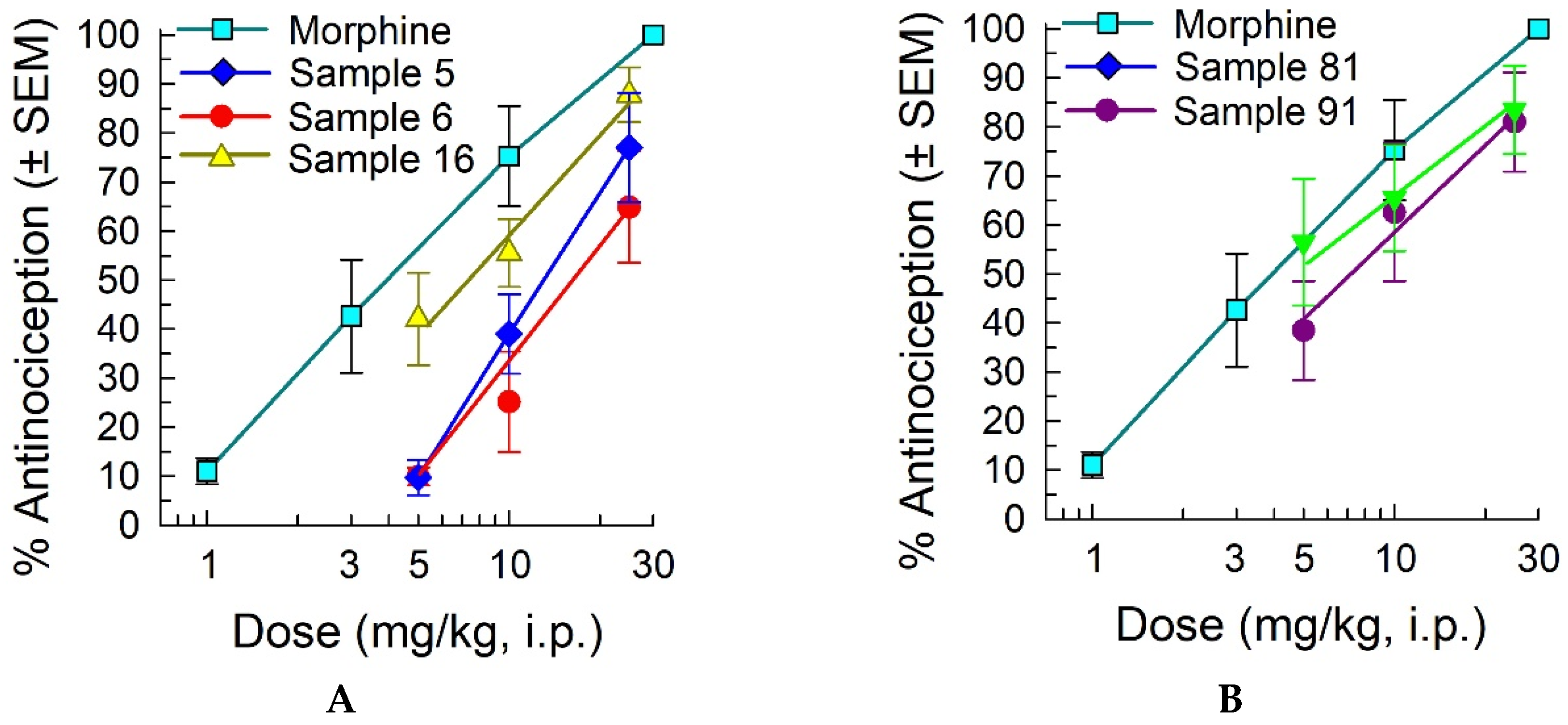
To validate the antinociceptive effects of the five strongest partially-defined samples and determine a potential dose-response relationship, additional doses of 10 mg/kg or 25 mg/kg, i.p. of selected samples were administered to mice and tested in vivo using the 55 °C warm-water tail-withdrawal assay (Figure 3). Samples 5, 6 and 16 defined at the R2-position produced dose-dependent antinociception that peaked at 60 min post administration (Figure 3A). Estimated from the limited doses testing, antinociceptive ED50 (and 95% confidence intervals) values were near that of morphine (4.12 (2.73-5.82) mg/kg, i.p.), at 13.0 (10.2-17.8) mg/kg, i.p. (sample 5), 17.5 (12.5-33.2) mg/kg, i.p. (sample 6) and 7.15 (3.91-9.96) mg/kg, i.p. (sample 16). Similarly, mixture-based samples 81 and 91 defined at the R5-position produced antinociceptive ED50 (and 95% confidence intervals) values of 3.62 (0.00-8.58) mg/kg, i.p. (sample 81) and 7.15 (0.52-12.7) mg/kg, i.p. (sample 91) at their peak effect of 120 min post administration (Figure 3B). While limited to estimates, this data collectively confirms the antinociceptive effects of the partially-defined, mixture-based samples, validating further deconvolution of the library. Taken together, Thr (16) at R2, D-Lys (62) at R4, and Tyr (91) and Leu (81) at R5 were selected as the most active residues at each position to deconvolute the mixture-based library for the identification of the cyclic peptide that produced the most significant antinociceptive activity in mice with the 55 °C WWTW assay.
Figure 3.
Antinociception produced by the top five mixture-based samples in the mouse 55 °C warm-water tail-withdrawal assay is dose-dependent. (a) Dose-response of mixture-based samples 5, 6 and 16 defined at the R2 position, 60 min after administration; (b) Dose-response of mixture-based samples 81 and 91 defined at the R5 position, 120 min after administration. All samples were administered by the i.p. route. Morphine is included as a positive control, with antinociception measured at the 30 min peak effect. All points = 8 mice (7-11 mice for morphine).
Figure 3.
Antinociception produced by the top five mixture-based samples in the mouse 55 °C warm-water tail-withdrawal assay is dose-dependent. (a) Dose-response of mixture-based samples 5, 6 and 16 defined at the R2 position, 60 min after administration; (b) Dose-response of mixture-based samples 81 and 91 defined at the R5 position, 120 min after administration. All samples were administered by the i.p. route. Morphine is included as a positive control, with antinociception measured at the 30 min peak effect. All points = 8 mice (7-11 mice for morphine).
2.3. Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu) for the Deconvolution of the Library.
Two cyclic peptides, Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu), were then synthesized to complete the deconvolution of the library. Using mercaptopropyl isobutyl polyhedral oligomeric silsesquioxane (POSS-SH) as a soluble support [35], Boc-protected amino acids including Gly (R1), Thr(tBu) (R2), Dap[Ant(Boc)] (R3), D-Lys(Alloc) (R4), and Tyr(tBu)/Leu (R5) were subsequentially coupled to form the peptide POSS-thioesters, Boc-Tyr(tBu)-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS and Boc-Leu-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS, respectively (Scheme 1). The peptide POSS-thioesters were then treated with Pd(PPh3)4 in the presence of PhSiH3 in DCM to remove the Alloc protective group, followed by adding imidazole to carry out a one-pot, in situ cyclization to form the final side-to-tail cyclic peptides, CycloAnt and CycloAnt-Leu, respectively [36].
The cyclic peptides were then treated with 55% TFA to remove the protective groups and the resulting compounds were purified by RP-HPLC and structurally characterized by NMR experiments before characterization of their pharmacological profile.
2.4. Screening of the Cyclic Peptide CycloAnt for Protein Interaction
Potential interaction of CycloAnt with a panel of 47 protein targets including GPCRs, kinases, nuclear hormone receptors, ion channels, and enzymes was assessed by assays included in the SAFETYscan service from Eurofins DiscoverX. CycloAnt was tested in both agonist and antagonist modes for each representative GPCRs (see Supplemental Material S9-11). Tested in a cAMP assay utilizing cells overexpressing each receptor, CycloAnt demonstrated MOR and DOR agonism, with EC50 values of 14.0 and 3.36 nM, respectively (Supplemental Table S1). CycloAnt did not produce agonist or antagonist activity at any other target tested in this panel.
2.5. Evaluation of the Cyclic Peptides for Their Antinociceptive Effects in Wild-Type Mice.
We tested the antinociceptive effects of the cyclic peptides using the 55 °C WWTW assay in C57BL/6J male mice (Figure 4). Both peptides produced dose- (Figure 4A) and time-dependent antinociception (F(16,152) = 38.5; p < 0.0001; two-way ANOVA; Figure 4B). Peak antinociception was produced by both peptides within 20 min of administration, with a significant duration of 70 min for CycloAnt (3 mg/kg, i.p.) and 50 min for the CycloAnt-Leu analog (100 mg/kg, i.p.) compared to vehicle (p < 0.05, Dunnett’s post hoc test; Figure 4B). The two cyclic peptides produced full antinociception, but with significant differences in potency. CycloAnt-Leu produced antinociception with an ED50 (and 95% C.I.) value of 38.3 (18.7-127.1) mg/kg, i.p., 9.3-fold less potent than the control opioid morphine (ED50 (and 95% C.I.) value = 4.12 (2.73-5.82) mg/kg, i.p.; Figure 4A). In contrast, CycloAnt displayed an ED50 (and 95% C.I.) value of 0.70 (0.52-0.97) mg/kg, i.p., 5.9-fold more potent than morphine. Collectively, this data proves CycloAnt a potent antinociceptive “hit” identified through the direct phenotypic in vivo screening and deconvolution of this mixture-based cyclic peptide library.
2.6. Evaluation of Opioid Receptor Involvement in CycloAnt Antinociception
To assess the contributions of the individual opioid receptors to CycloAnt-mediated antinociception, the cyclic peptide (3 mg/kg, i.p.) was administered to mice lacking the mu- (MOR KO), kappa- (KOR KO) or delta-opioid receptor (DOR KO). Compared to the response in wild-type C57BL/6J mice, CycloAnt-mediated antinociception was significantly reduced 80% only in MOR KO mice (F(3,28) = 216.3, p < 0.0001, one-way ANOVA with Dunnett’s post hoc test), and not KOR KO or DOR KO mice (Figure 5).
2.7. Evaluation of Opioid-Receptor-Selective Antagonism Mediated by CycloAnt
Given the MOR and DOR activity identified in the in vitro screening, and the singular MOR agonist activity displayed in mice, CycloAnt was evaluated for antagonist activity against the KOR-selective agonist U50,488 (10 mg/kg, i.p.), and the DOR-selective agonist SNC-80 (100 nmol, i.c.v.; Figure 6A). MOR KO mice were used to preclude complications of mu-opioid receptor mediated antinociception. U50,488 antinociception was not significantly antagonized by pretreatment with CycloAnt (3 or 10 mg/kg, i.p.; F(2,21) = 0.9, p = 0.42, one-way ANOVA; Figure 6A left bars). However, surprisingly, CycloAnt pretreatment significantly antagonized SNC-80 antinociception in a dose-dependent manner (F(2,21) = 22.3, p < 0.0001, one-way ANOVA with Dunnett’s post-hoc testing; Figure 6A, right bars). Examined at the dose of maximal DOR antagonism (10 mg/kg, i.p.), CycloAnt-mediated DOR antagonism was time dependent (F(4,43) = 16.2, p < 0.0001, one-way ANOVA with Dunnett’s post-hoc testing; Figure 6B), with significant reduction of SNC-80 antinociception for 1 or 3 hours (p < 0.0001), but not 6 hours (p = 0.07) or 8 hours (p = 0.96). This data suggests CycloAnt acts in vivo as a multifunctional MOR agonist and DOR antagonist.
2.8. Evaluation of CycloAnt for Respiratory and Hyperlocomotor Effects
Respiratory depression is a major life-threatening effect of clinically used opioid analgesics. To evaluate whether the cyclic peptide-based MOR/DOR multifunctional ligand can dissociate this side effect from antinociception, we examined CycloAnt for its potential to depress breathing rate over a 120 min period using the automated, computer-controlled Comprehensive Lab Animal Monitoring System (CLAMS) apparatus in male C57BL/6J mice [32]. As expected, the positive control morphine (10 or 30 mg/kg, i.p.) produced significant dose and time-dependent respiratory depression compared to vehicle F(20,360) = 6.67, p < 0.001, two-way RM ANOVA with Tukey’s multiple comparison post hoc test; Figure 7A). In contrast, CycloAnt did not significantly impair breathing when administrated i.p. at either the peak antinociceptive dose (3.0 mg/kg) or a higher dose 15 times the antinociceptive ED50 value (10.6 mg/kg), and in fact statistically increased it in a time and dose-dependent manner up to 100 min compared to vehicle (p < 0.05, Tukey’s test; Figure 7A).
As a MOR agonist, morphine induces psychostimulation demonstrated by hyperlocomotion in mice. Consistent with this, treatment significantly increased ambulation (factor: time × treatment, F(20,360) = 36.7, p < 0.0001, two-way RM ANOVA; Figure 7B), primarily due to morphine (factor: treatment, F(10,250) = 34.8, p < 0.001, two-way RM ANOVA). In contrast, CycloAnt induced significant hyperlocomotor activity (factor: treatment, F(10,205) = 3.19, p = 0.0008, two-way RM ANOVA with Tukey’s test; Figure 7B), but CycloAnt-induced ambulations were significantly less than those induced by morphine after 20 min to the end of the testing period (†p < 0.05, Tukey’s test; Figure 7B).
2.9. Evaluation of CycloAnt for Potential of Opioid-Induced Hyperalgesia, Tolerance and Dependence Development.
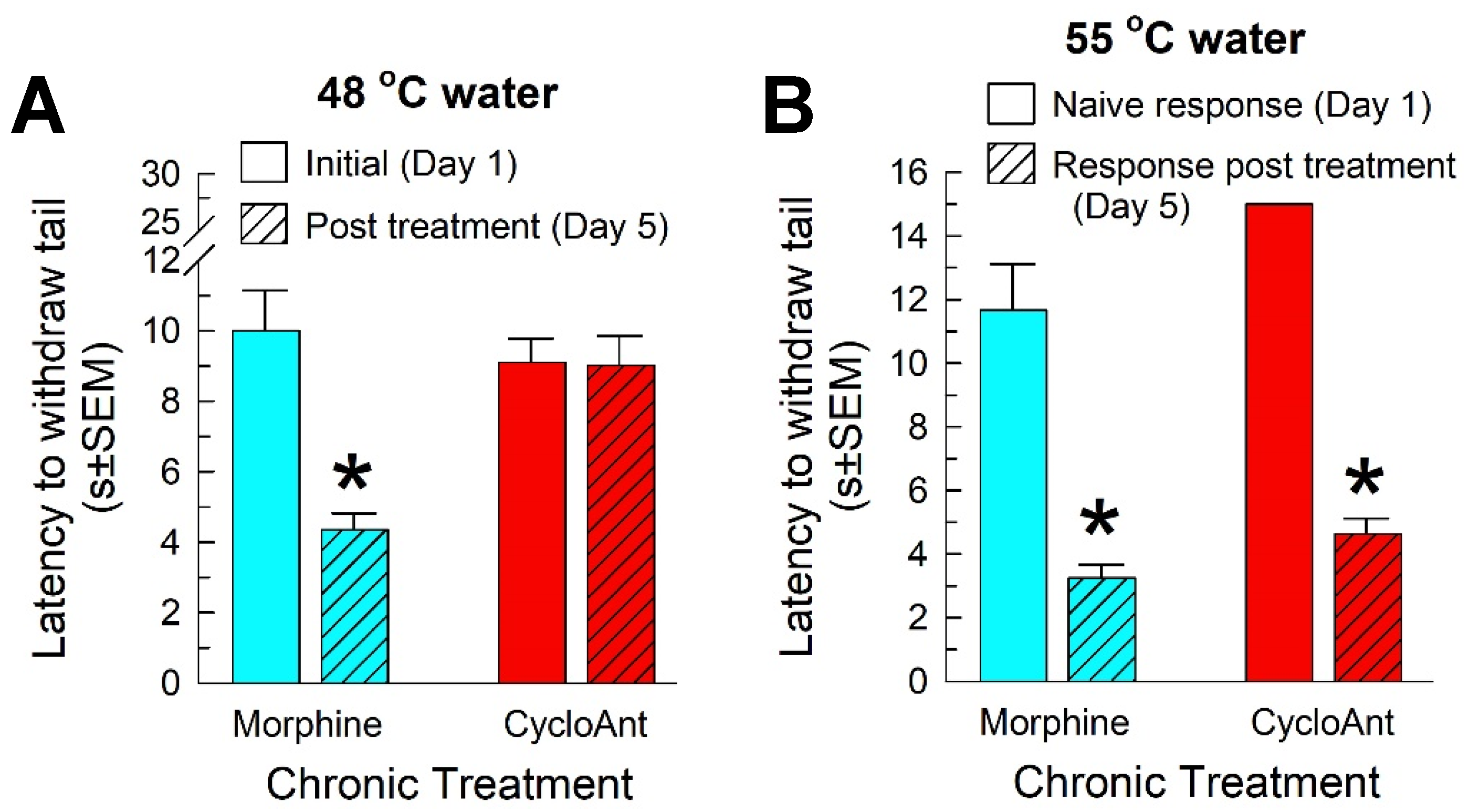
Chronic use of opioid analgesics may cause opioid-induced hyperalgesia, the paradoxical response where a patient becomes more sensitive to painful stimuli, as well as antinociceptive tolerance and physical dependence. To assess this, mice were treated twice daily for four days with the morphine (10 mg/kg, i.p.) or CycloAnt (3 mg/kg, i.p.), and once more on the 5th day before WWTW testing. Tail-withdrawal latencies were collected both prior to the start of treatment (on day 1), and again on the morning of day 5 prior to drug treatment. Each group of mice showed equivalent initial responses on day 1 to a 48 °C warm-water stimulus (morphine, 10 ± 1.14 s vs CycloAnt, 9.1 ± 0.68 s; p = 0.52, Student’s t-test; Figure 8A) or 55 °C warm-water stimulus (morphine, 1.43 ± 0.05 s vs CycloAnt, 1.56 ± 0.08 s; p = 0.20, Student’s t-test). Whereas chronic morphine administration induced significant hyperalgesia (F(1,17) = 14.3, p = 0.002; two-way RM ANOVA) against a 48 °C warm-water stimulus on day 5 (with a tail withdrawal latency of 4.35 ± 0.47 s; p < 0.0001, Sidak’s post hoc test; Figure 8A), chronic CycloAnt treatment did not induce significant OIH (day 5 tail withdrawal latency of 9.02 ± 0.83 s; p = 0.997, Sidak’s post hoc test; Figure 8A). However, chronic administration of either morphine or CycloAnt repeatedly over 4 days produced significant reductions in the tail-withdrawal latency of the compound on day 5 (F(1,18) = 5.75, p = 0.03; two-way REML ANOVA with Sidak’s post hoc test; Figure 8B), indicating opioid-induced tolerance.
We then examined the potential development of physical dependence after chronic administration of morphine or CycloAnt as above, followed by precipitation of withdrawal with naloxone. Repeated treatment with escalating doses of morphine induced physical dependence, demonstrated by administration of naloxone (Table 1). Morphine produced significant increases in the frequency of forepaw tremor, presence of diarrhea, and frequency of jumping and teeth chattering, along with decreases in rearing and forepaw licking frequency characteristic of opioid physical dependence (see one-way ANOVA values in Table 1). In contrast, while CycloAnt showed significant increases in diarrhea and reduced rearing frequency compared to saline control mice, most other signs of withdrawal, such as jumping frequency and teeth chattering, significantly differed from those of morphine or were completely absent (Table 1). Collectively, these results indicate chronic CycloAnt treatment induced much less physical dependence than as seen with morphine.

Table 1.
Assessment of physical dependence in mice treated chronically with CycloAnt, Saline or Morphine and subjected to naloxone-precipitated withdrawal. ‡Saline values reproduced from Wilson et al., 2021 [37]. Average and Standard Error of the Mean reported for n = 9-10 mice/treatment. *p < 0.05 versus Saline control, †p < 0.05 versus Morphine control (Tukey’s post-hoc test).
Table 1.
Assessment of physical dependence in mice treated chronically with CycloAnt, Saline or Morphine and subjected to naloxone-precipitated withdrawal. ‡Saline values reproduced from Wilson et al., 2021 [37]. Average and Standard Error of the Mean reported for n = 9-10 mice/treatment. *p < 0.05 versus Saline control, †p < 0.05 versus Morphine control (Tukey’s post-hoc test).
| Measure | Treatment (drug on day 5, -120 min prior to naloxone) | Outcome | |||||
| CycloAnt (n = 9) | Saline (n = 10)‡ | Morphine (n = 10) | (1-way ANOVA) | ||||
| Ave | SEM | Ave | SEM | Ave | SEM | F(2,26) = | |
| Forepaw Tremor | 23.7† | 6.85 | 20.2 | 80.4 | 66* | 17.0 | 4.77, p = 0.02 |
| Wet Dog Shakes | 2.22 | 0.43 | 0.8 | 0.51 | 1.3 | 0.56 | 1.95, p = 0.16 |
| Straightening | 1.22 | 0.47 | 5.7 | 2.33 | 1.1 | 0.46 | 3.35, p = 0.05 |
| Presence of Diarrhea | 6.67* | 0.91 | 1.1 | 0.41 | 5.4* | 0.78 | 16.6, p < 0.001 |
| Jumping Frequency | 1.89† | 1.31 | 0 | 0 | 41.6* | 2.64 | 188.1, p < 0.001 |
| Rearing Frequency | 17.8* | 5.94 | 55.4 | 6.59 | 22.2* | 2.46 | 15.4, p < 0.0001 |
| Forepaw Licking Frequency | 11.2† | 1.80 | 15.1 | 3.27 | 1.7* | 1.00 | 9.59, p = 0.0008 |
| Teeth Chattering Frequency | 0† | 0 | 0.4 | 0.27 | 9.5* | 1.79 | 25.0, p < 0.0001 |
3. Discussion
From the direct in vivo phenotypic screening of the mixture-based cyclic peptide library using the 55 °C WWTW assay, we have identified a novel hit CycloAnt, Tyr-[D-Lys-Dap(Ant-Thr-Gly], exhibiting time- and dose-dependent antinociception with an ED50 value of 0.7 mg/kg (i.p.) in mice. Notably, this library was screened earlier using a receptor binding assay for the MOR, identifying a hit with a binding affinity of 14 nM [29]. However, that cyclic peptide has a sequence of Tyr-[D-Lys-Dap(ant)-Phe-Gly], differing from the sequence Tyr-[D-Lys-Dap(ant)-Thr-Gly] identified in this study. The differing results may stem from the two screening methods utilized, based on different mechanisms and readouts. First, as the in vivo direct phenotypic screening tests the relative antinociceptive effect of a sample, in terms of the MOR, only agonists can be identified from the assay. While the binding assay at MOR measures the affinity of a sample at the receptor, whether the sample is an agonist or antagonist cannot be differentiated, leaving activity unresolved. Second, the samples presently were administered to mice through the intraperitoneal route for the in vivo direct phenotypic screening. As the mice are complete organisms with systemic physiology and functions such as metabolism, only these samples with a preferred pharmacokinetic profile will succeed in this screening. It seems likely that the Tyr-[D-Lys-Dap(ant)-Phe-Gly] previously identified using the receptor binding assay is unable to adequately activate the MOR under physiological conditions in a complete organism, precluding antinociception in the mouse WWTW assay.
CycloAnt interactions with a broad set of molecular targets were determined with the Safety47 Panel Dose Response, using the SAFETYscan provided by Eurofins DiscoverX. This panel utilizes 78 assays at 47 protein targets, including GPCRs, kinases, nuclear hormone receptors, ion channels, and enzymes. Notably, a number of targets associated with antinociception are included in this panel, including MOR, KOP, and DOR, cyclooxygenase 1 (COX1), cyclooxygenase 2 (COX 2), human serotine receptors 1A (5-HT1A), 1B (5-HT1B), 2A (5-HT2A), and 2B (5-HT2B), human adrenoceptors 3A (5-HT3A), alpha2A (ADRA1), beta1 (ADRB1), and beta2 (ADRB2), voltage gated sodium channel (Nav1.5), and the human glutamate ion channel (NMDAR). CycloAnt only interacted with MOR and DOR in the panel, showing agonist activity at both receptors using the cAMP assay. It did not produce agonist or antagonist activity at any other pain-associated targets in this panel. While the results of MOR testing were confirmed with our in vivo testing presently, they diverged with the surprising demonstration in vivo of DOR antagonism. Notably, the DOR agonist activity in the cAMP assay was made with cells expressing a high level of DOR, whereas the in vivo results utilizing mice with a physiologically-normal level of DOR expression indicated the compound behaved as an antagonist. This discrepancy on the function at DOR may be accounted for receptor reserve [38,39,40], as a very weak partial agonist/antagonist can appear as a full agonist when receptor reserve is present. While the discrepancy is beyond the scope of this initial identification and characterization, future investigations may prove beneficial in better understanding this interaction. Regardless, consistent with the SAFETYscan results, CycloAnt was not found to produce any significant biological activity at any other targets in this study than DOR and MOR.
The combination of DOR antagonism with MOR agonism has been reported to produce antinociception while possessing safety advantages over typical MOR agonists alone [41]. Although antinociceptive tolerance has been shown to be reduced by co-administration of MOR agonists with DOR antagonists [42,43]] or by bifunctional MOR agonist/DOR antagonist compounds [44]], chronic administration of CycloAnt presently produced a reduction of subsequent antinociception consistent with significant tolerance. However, chronic exposure to CycloAnt did not produce the significant opioid-induced hyperalgesia demonstrated by morphine, and displayed reduced signs of opioid physical dependence, consistent with earlier pharmacological studies [42,43] and reports where i.c.v. pretreatment with DOR-selective antisense oligonucleotides decreased morphine dependence in mice [45]]. Importantly, CycloAnt did not suppress breathing rate at high doses up to 15 times of the antinociceptive ED50 value, and in fact increased breathing rate significantly. CycloAnt treatment also induced significantly less hyperlocomotion than less-than-equianalgesic doses of morphine. All of these results are consistent with the recently reported activity of the bifunctional MOR agonist/DOR antagonist [Nal(2’)4]CJ-15,208, which also did not produce hyperlocomotion or respiratory depression; indeed, the bifunctional peptide briefly and significantly increased respiration as well [44]. Given the absence of CycloAnt-mediated activity at off-target proteins, future studies are anticipated to investigate potential mechanisms by which the addition of DOR antagonism may contribute to the improved safety advantages demonstrated by this cyclic peptide.
The potent antinociceptive effect and low liability profile after the i.p. administration suggests this cyclic peptide has a preferred pharmacodynamic and pharmacokinetic profile for systemic use as a potentially safer pain modulator. Therefore, this dual MOR/DOR agonist is an excellent initial lead for the development of peptide-based safer opioid analgesic.
4. Materials and Methods
4.1. General Materials.
Silica gel, 130-270 mesh, 60Å, BET surface area 500m2/g, pore volume 0.75cm3/g, was purchased from Aldrich Chemical Company Inc. Boc amino acid derivatives, benzotriazole-1-yloxy-trispyrrolidino-phosphonium hexafluorophosphate (PyBOP), N,N-diisopropylethylamine (DIEA), and trifluoroacetic acid (TFA) were purchased from Chem-Impex. HF were purchased from Air Products (San Marcos, CA). Polypropylene mesh (74 micro) was utilized to prepare mesh packets. Phenyltrimethoxysilane and p-Chloromethylphenyltrimethoxysilane were purchased from Gelest, Inc. (Tullytown, PA). Mercaptopropyl isobutyl POSS (POSS-SH) was purchased from Hybrid Plastics (Hattiesburg, MS). Pd(PPh3)4 and all solvents were purchased from Fisher Scientific.
LC-MS (APCI and ESI) was recorded on an Agilent LCMS-1260 infinity II at 6120 quadrupole LC/MS mass spectrometer at 214 nm using a Zorbax Eclipse Plus C18 (3.0 × 100 mm, 3.5 μm) column. Preparative RP-HPLC was performed on a Shimadzu preparative HPLC using a Phenomenex Luna 10 μ C18 100Å column (21.2 × 250 mm). NMR spectra were recorded on a Bruker Avance III-HD 400 instrument at 400 MHz for 1H NMR and 100 MHz for 13C NMR. NMR chemical shifts are expressed in ppm relative to internal solvent peak and coupling constant were calculated in hertz.
4.2. Synthesis of the Mixture-Based Cyclic Peptide Library.
The cyclic peptide library was synthesized as we have previously reported [29]. Briefly, all linear peptide thioesters were synthesized using the functionalized mercaptomethylphenyl silica gel as ‘volatilable’ support as we described. Boc-amino acids were coupled on the silica support using PyBOP/DIEA as coupling reagents. X-positions were coupled as mixtures of Nα-Boc protected amino acids using concentration ratios to compensate for the relative reaction rates in competitive couplings. Position 3 was coupled with a Nα-Boc-Nβ-Alloc-diaminoproponic acid. After peptide elongation on resin, the Boc-protected peptides were treated with Pd(PPh3)4 (0.1 equiv.) in the presence of PhSiH3 (20 equiv.) in DCM to remove the Alloc protection group. The resin bound peptides were coupled with 2-nitrobenzoic acid, followed by the treatment of 2 M SnCl2 in DMF overnight to reduce the nitro group to an amino group to generate the Ant-label. After removal of the Boc group with 55% TFA, the resin bound Ant-labeled peptides were treated with anhydrous HF in the presence of 5% anisole at 0 °C for 2 h. After removal of the HF with nitrogen stream and lyophilization, the linear peptide thioesters were cyclized in a mixed solution of 1.5 M imidazole (aq.) and acetonitrile (1:7 v/v) at a concentration of 1 mM for 72 h [34], forming the Ant-labeled cyclic peptides.
4.3. Solution-Phase Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu).
4.3.1. Synthesis of Boc-Dap[Ant(Boc)]-OH.
All Boc-amino acids used for the POSS-thioester synthesis were commercially available, except the 3rd amino acid Boc-Dap[Ant(Boc)], which was synthesized in the lab and directly utilized for the peptide synthesis without further purification. Briefly, Boc-Dap-OH (1 mmol) was dispersed in 2 ml of DMF with the presence of DIEA (1 mmol). In a separated vial, Boc-2-aminobenzoic acid (1 mmol, 0.11 M in DCM) was mixed with PyBOP (1 mmol) and DIEA (1.5 mmol) and activated for 5 min. The solution of Boc-Dap-OH was then added to the activated Boc-2-aminobenzoic acid. The reaction was stirred at r.t and the completion of the reaction was monitored by LC-MS. After 5 hrs the resulting Boc-Dap[Ant(Boc)] was utilized directly for peptide synthesis without purification.
4.3.2. Synthesis of Linear Peptide POSS-thioesters Boc-Tyr(tBu)-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS and Boc-Leu-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS.
The linear peptide thioester was synthesized using POSS-SH as a soluble support as we previously described [35]. Briefly, Boc-Gly-OH (1.05 mmol) preactivated with PyBOP (1.05 mmol) and DIEA (2.1 mmol) was added to POSS-SH (0.10 M in 10 mL DCM). The reaction mixture was stirred at room temperature for 3 hours. After removal of DCM, Boc-Gly-S-POSS was washed 3 times with 75% acetonitrile containing aqueous HCl (0.05 M). The resulting precipitation was collected by centrifugation and freeze-dried. Boc-Gly-S-POSS was treated with 55% TFA for 30 min to remove the Boc protective group. After removal of TFA/DCM in vacuo, the peptide chain was elongated by coupling to the subsequent Boc-amino acid using the method described above. At each of the peptide coupling steps, an extra amount of DIEA was added to the reaction mixture to adjust the pH value to 8. The completion of the amino acid coupling reaction was monitored by a ninhydrin test. The linear peptide POSS-thioesters were obtained at an overall total yield of 81.8%.
4.3.3. Synthesis of Cyclic Peptides Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) and Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu)
The cyclic peptides were synthesized using the imidazole-promoted cyclization methods as we previously reported [36]. Under N2 protection, the Boc-Tyr(tBu)-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS (1.0 mmol) or the Boc-Leu-D-Lys(Alloc)-Dap(Ant)-Thr-Gly-S-POSS (1.0 mmol) was dissolved in 100 mL of DCM to a concentration of 10 mM. A portion of PhSiH3 (24 mmol) and Pd(PPh3)4 (0.07 mmol) were then added to facilitate the removal of the Alloc protective group. After reacting overnight, imidazole (1.36 g, 20 mmol) was directly added to the reaction mixture to a concentration of 0.2 M and the reaction was carried out for 24 hrs. After removal of the solvent in vacuo, 75% acetonitrile (aq.) was added to dissolve the yielded cyclic peptide while precipitating the POSS-SH released from peptide cyclization. The supernatant was collected and then lyophilized to dry. The resulting powder was washed with water to remove imidazole. The precipitation was collected and re-dissolved in 65% acetonitrile. After centrifugation at 4000 rpm for 10 min, the supernatant was collected and freeze-dried to yield the protected cyclic peptide Boc-Tyr(tBu)-[D-Lys-Dap(Ant)-Thr-Gly] or Boc-Leu-[D-Lys-Dap(Ant)-Thr-Gly]. Following the treatment with 55% TFA for 30 min, the fully unprotected cyclic peptide Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) or Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt) was yielded. The crude compound was purified by reverse-phase HPLC. The cyclic peptide was obtained as a TFA salt after HPLC purification.
Tyr-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt). 1H NMR (400 MHz, DMSO-d6) δ 0.99-1.22 (m, 2H), 1.03 (d, 3H, J = 6 Hz), 1.08-1.18 (m, 1H), 1.29-1.39 (m, 3H), 1.49-1.56 (m, 1H), 2.85-2.87 (m, 1H), 2.90 (m, 1H), 2.96-2.98 (m, 1H), 3.08-3.12 (m, 1H), 3.50 (td, 1H, J = 14, 6 Hz), 3.88 (d, 1H, J = 6.8 Hz), 3.90-3.94 (m, 1H), 3.99-4.02 (m, 2H), 4.08 (t, 1H, J = 6 Hz), 4.2-4.24 (m, 1H), 5.07 (d, 1H, J = 4.8 Hz), 6.34 (br.s, 2H), 6.49-6.52 (dd, 1H, J = 7.2, 1 Hz), 6.54 (d, 1H, J = 2 Hz), 6,72 (d, 3H, J = 8 Hz), 7.03 (d, 2 H, J = 8 Hz), 7.16 (td, 1H , J = 7.6, 1.6 Hz ), 7.33 (t, 1H, J = 5.2 Hz), 7.46 (dd, 1H, J = 8, 1.2 Hz), 7.64 (d, 1H, J = 5.2 Hz), 7.72 (br.s, 1H), 8.13 (br.s, 2H), 8.29 (t, 1H, J = 5.6 Hz), 8.38 (t, 1H, J = 5.6 Hz), 8.43 (d, 1H, J = 7.2 Hz), 8.47 (d, 1H, J = 6.4 Hz), 9.39 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 20.1, 21.2, 28.1, 31.5, 36.8, 38.2, 40.7, 43.8, 53.6, 54.2, 56.4, 60.2, 66.3, 114.9, 115.3, 115.7, 115.8, 116.9, 118.7, 125.2, 128.8, 130.9, 132.5, 149.7, 157.1, 158.4, 158.7, 168.2, 169.2, 170.5(2), 170.6, 172.1. 19F NMR (376 MHz) δ -37.7 (s).
Leu-[D-Lys-Dap(Ant)-Thr-Gly] (CycloAnt-Leu). 1H NMR (400 MHz, DMSO-d6) δ 0.92 (d, 6H, J = 8 Hz), 1.03 (d, 3H, J = 4 Hz), 1.29-1.32 (m, 2H), 1.38-1.39 (m, 2H), 1.57-1.65 (m, 5H), 2.96-2.99 (m, 1H), 3.12-3.16 (m, 1H), 3.36 (dd, 1H, J = 16, 4 Hz), 3.53 (m, 1H), 3.88 (dd, 1H, J = 16, 8 Hz), 3.93-4.00 (m, 1H), 4.02-4.04 (m, 2H), 4.12 (t, 1H, J = 8 Hz), 4.19 (t, 1H, J= 8 Hz), 6.51 (td, 1H, J = 8, 1 Hz), 6.73 (d, 1H, J = 8 Hz), 7.16 (td, 1 H, J = 8, 1 Hz), 7.41 (t, 1H, J = 8 Hz), 7.48 (dd, 1H , J = 7.6, 1.6 Hz ), 7.66 (d, 1H, J = 8 Hz), 8.15 (br.s, 3H), 8.26 (t, 1H, J = 6 Hz), 8.38 (t, 1H, J = 5.6 Hz), 8.57 (d, 1H, J = 8 Hz), 8.83 (d, 1H, J = 6.8 Hz). 13C NMR (100 MHz, DMSO-d6) δ 20.1, 21.7, 22.1, 23.2, 24.1, 28.2, 31.6, 38.2, 40.4, 40.5, 43.8, 51.4, 54.4, 56.4, 60.0, 66.3, 114.7, 115.2, 115.8, 116.9, 118.8, 128.8, 132.5, 149.8, 158.4, 158.8, 169.2, 169.5, 170.5(2), 170.7, 172.4.
4.4. Animals.
Male C57BL/6J mice (19–27 g each, Jackson Laboratories, Bar Harbor, ME) were used for screening and deconvoluting the library. Male mu-opioid receptor gene knockout mice (MOR KO), kappa-opioid receptor gene-knockout mice (KOR KO), and delta-opioid receptor gene-knockout mice (DOR KO) obtained from a breeding colony established at the University of Florida with progenitors from Jackson Labs were used for additional testing of the lead. Four Mice per cage were housed in a room with temperature and 12-hrs light/dark cycle controls with the lights on from 07:00 am to 19:00 pm; food and water were available ad libitum. All procedures with mice were approved by the local IACUC.
4.5. Assessment of Opioid-Induced Hyperalgesia Using the 48 °C Tail-Withdrawal Assay in Mice.
The 48 °C warm-water tail-withdrawal assay was performed as previously described [37], with the latency to withdraw the tail taken as the end point. The water temperature of 48 °C was selected for this work to ensure a moderate tail-withdrawal response, with a measurable decrease in withdrawal time possible but also a significant temperature for hyperalgesic testing. If the mouse failed to display a tail-flick in 30 s, the tail was removed from the water to minimize tissue damage. After determining control latencies on day 1, mice received chronic morphine (10 mg/kg, i.p.) or CycloAnt (3 mg/kg, i.p.) treatment twice a day for 4 days as described below. Dosing of these compounds was based on the demonstration of equianalgesic efficacy (see results). Mice were then tested prior to treatment on day 5 for evidence of induced hyperalgesia. Experimentally induced decreases in tail-withdrawal latencies in this assay indicate hyperalgesic effects [46,47].
4.6. Antinociceptive Test Using the 55 °C Tail-Withdrawal Assay in Mice.
The 55 °C warm-water tail-withdrawal assay was performed as previously described [48] to screen the library [30] (Reilley et al., 2010). Mixed-based samples were dissolved at a concentration of 5 mg/mL in saline containing 10% DMSO. Each sample was given to the mice (n = 8) intraperitoneally (i.p.) at a dose of 5 mg/kg. At 0.5, 1, 2, 3.5, 5, 8 hours post-administration of the samples, the tail-withdrawal latencies were measured for each sample. The average response time to tail-withdrawal at the six time points were then combined. The combined response time to withdrawal tail was used to determine the antinociceptive effect of each sample. A cutoff of 15 s was used to avoid tissue damage; those mice failing to withdraw their tails within this time were assigned a maximal antinociceptive score of 100%.
To determine antinociceptive activity of individual compounds, the tail-withdrawal latency was determined repeatedly every 10 min following administration of the compounds for 1 h or until latencies returned to baseline values. At each time point, antinociception was calculated according to the following formula: % antinociception = 100 x (test latency - control latency)/(15 - control latency). Tail-withdrawal data points are the means of 8–16 mice, unless otherwise indicated, with SEM shown by error bars.
The opioid receptor involvement in the agonist activity of the individual compounds was determined by testing in MOR KO, KOR KO and DOR KO mice as utilized previously [49].
To determine antagonist activity, MOR KO mice were pretreated with CycloAnt 20 min prior to the administration of the KOR-selective agonist U50,488 (10 mg/kg, i.p.), or DOR-selective agonist SNC-80 (100 nmol, i.c.v.); the MOR KO mice do not display antinociceptive activity of CycloAnt. Antinociception produced by the established opioid agonists was then measured 40 min after their administration. Additionally, the duration of DOR antagonism produced by CycloAnt was further determined by evaluating SNC-80-induced antinociception 3, 6 or 8 hours after administration of CycloAnt.
4.7. Respiratory Depression and Hyperlocomotion.
Male C57Bl/6J mice were tested for respiratory effects using automated, computer-controlled Comprehensive Lab Animal Monitoring System (CLAMS) apparatus. Mice (n = 8) were habituated in closed apparatus cages (23.5 × 11.5 × 13 cm3) for 30 min before being administrated the hit i.p. at the experimented high dose of 3.0 mg/kg, and an even higher dose at 15 times of the ED50 value, 10.6 mg/kg, i.p.. Mice were then returned to chambers, and their respiration rate (breaths/min) were measured for 100 min in 30-s intervals using a pressure transducer built into the enclosed, sealed CLAMS cage. The resultant change in pressure during mouse breathing was recorded to reveal the respiration rate (breaths/min).
Infrared beams located in the floor measured locomotion as number of beam breaks. Respiration and locomotive data were averaged over 20 min periods for 120 min post-injection of the test compound. Data are presented as % vehicle response ± SEM, ambulation or breaths per minute.
4.8. Antinociceptive Tolerance and Naloxone-Precipitated Opioid Withdrawal Assay.
Mice were randomly assigned to one of three groups given saline (i.p.), morphine (10 mg/kg, i.p.) or CycloAnt (3 mg/kg, i.p.) twice daily for 4 days, and once more on the morning of the 5th day. Mice were then tested in the 55 °C WWTW assay to assess for the loss of therapeutic efficacy (antinociceptive tolerance; see above). Two hours after this test, naloxone (10 mg/kg, i.p.) was administrated to all mice to precipitate opioid withdrawal [37]. Mouse activity was digitally recorded and evaluated later for an established battery of withdrawal behaviors for 15 min [37]. Immediately upon injection of naloxone, all mice were scored for number of jumps, forepaw tremor, wet dog shakes, straightening, rearing, forepaw licking, teeth chattering, and the presence and number of soft stools.
4.9. Statistical Analysis.
All dose–response lines were analyzed by regression, and ED50 (effective dose producing 50% antinociception) values and 95% C.I. were determined using individual data points from graded dose—response curves with Prism 9.0 software (GraphPad, La Jolla, CA, USA). Percent antinociception was used to determine within-group effects and to allow comparison to baseline latency in tail-withdrawal experiments. The statistical significance of differences between ED50 values was determined by evaluation of the ED50 value shift via nonlinear regression modeling with Prism software. Significant differences in behavioral data were analyzed by ANOVA (one-way or two-way with repeated measures (RM), as appropriate). Significant results were further analyzed with Sidak’s, Tukey’s, or Dunnett’s multiple comparison post hoc tests, as appropriate. All data are presented as mean ± SEM, with significance set at p < 0.05.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR profiles of CycloAnt and CycloAnt-Leu and Safety47 Panel response.
Author Contributions
Conceptualization, R.A.H.; methodology, Y.L. and J.P.M.; formal analysis, J.P.M and Y.L.; investigation, Y.L., M.G.S., S.O.E., A.E. and J.P.M.; writing—original draft preparation, Y.L.; writing—review and editing, J.P.M. and Y.L.; visualization, J.P.M. and Y.L.; supervision, J.P.M. and Y.L.; project administration, J.P.M. and Y.L.; funding acquisition, J.P.M. and Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by grants from the National Institute for Health 5P20 GM109091. Y.L. acknowledges the financial support from the University of South Carolina for and the Aspire I Innovation funding.
Institutional Review Board Statement
The animal study protocols governing this work were approved prior to testing by the Institutional Animal Care and Use Committees at the Torrey Pines Institute for Molecular Studies and the University of Florida (protocol #201808990, approved 7/17/2018).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and the supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors upon request.
Abbreviations
Alloc: allyloxycarbonyl; ANOVA: analysis of variance; Ant, anthraniloyl; Boc, tert-butyloxycarbonyl; CI, confidence interval; CycloAnt, Tyr-[D-Lys-Dap(Ant)-Thr-Gly]; CycloAnt-Leu, Leu-[D-Lys-Dap(Ant)-Thr-Gly]; CLAMS; Comprehensive Lab AnimalMonitoring System; DIEA, diisopropylethylamine; DCM, dichloromethane; DMSO, dimethyl sulfoxide; DOR, delta opioid receptor; DOR KO, delta opioid receptor gene-disrupted mice; ED50, dose for 50% maximum effect; HOBt, 1-hydroxybenzotriazole; i.c.v., intracerebroventricular; i.p., intraperitoneal; KOR, kappa opioid receptor; KOR KO, kappa opioid receptor gene-disrupted mice; LC-MS, liquid chromatography-mass spectrometry; MOR, mu opioid receptor; MOR KO, mu opioid receptor gene-disrupted mice; MS, morphine sulfate; NMR, nuclear magnetic resonance; POSS, polyhedral oligomeric silsesquioxane; PyBOP, benzotriazole-1-yloxy-trispyrrolidino-phosphonium hexafluorophosphate; RM, repeated measures; RP-HPLC, reverse-phase high-performance liquid chromatography; SNC-80, (+)-4-[(αR)-α((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide; TFA, trifluoroacetic acid; U50,488,(±)-trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide.
Amino acids are the L-isomer unless otherwise specified; abbreviations for standard amino acids follow IUPAC-IUB Joint Commission of Biochemical Nomenclature [50].
References
- Savage, S.R.; Kirsh, K.L.; Passik, S.D. Challenges in using opioids to treat pain in persons with substance use disorders. Addict. Sci. Clin. Pract. 2008, 4, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W. Opioid peptide-derived analgesics. AAPS J. 2005, 7, E560–E565. [Google Scholar] [CrossRef] [PubMed]
- Webster, L.; Schmidt, W.K. Dilemma of addiction and respiratory depression in the treatment of pain: A prototypical endomorphin as a new approach. Pain Med. 2020, 21, 992–1004. [Google Scholar] [CrossRef]
- Stoeber, M.; Jullié, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; Von Zastrow, M. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 2018, 98, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Tsvetanova, N.G.; Von Zastrow, M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 2014, 10, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.-J. I.; Sergin, I.; Purgert, C.A.; O’Malley, K.L. Location-dependent signaling of the group 1 metabotropic glutamate receptor mGlu5. Mol. Pharmacol. 2014, 86, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacol. 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Sierra, S.; Lueptow, L.; Gupta, A.; Gouty, S.; Margolis, E.B.; Cox, B.M.; Devi, L.A. Biased signaling by endogenous opioid peptides. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 11820–11828. [Google Scholar] [CrossRef]
- Gallantine, E.L.; Meert, T.F. A Comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Clin. Pharmacol. Toxicol. 2005, 97, 39–51. [Google Scholar] [CrossRef]
- Zhang, X.; Arvidsson, L.B.; Elde, U.R.; Hökfelt, T. Localization and regulation of the delta-opioid receptor in dorsal root ganglia and spinal cord of the rat and monkey: evidence for association with the membrane of large dense-core vesicles. Neurosci. 1998, 82, 1225–1242. [Google Scholar] [CrossRef]
- Mas-Orea, X.; Basso, L.; Blanpied, C.; Gaveriaux-Ruff, C.; Cenac, N.; Dietrich, G. Delta opioid receptors on nociceptive sensory neurons mediate peripheral endogenous analgesia in colitis. J. Neuroinflammation. 2022, 19(1), 7. [Google Scholar] [CrossRef]
- Conibear, A. E.; Asghar, J.; Hill, R.; Henderson, G.; Borbely, E.; Tekus, V.; Helyes, Z.; Palandri, J.; Bailey, C.; Starke, I.; et al. A novel G protein–biased agonist at the δ opioid receptor with analgesic efficacy in models of chronic pain. J. Pharmacol. Exp. Ther. 2020, 372, 224–236. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Lei, W.; Ray, A.; Saini, S.K.; Zhang, S.; Molnar, G.; Barlow, D.; Karlage, K.L.; Bilsky, E.J.; Houseknecht, K.L.; et al. Synthesis and structure–activity relationships of 5′-aryl-14-alkoxypyridomorphinans: Identification of a μ opioid receptor agonist/δ opioid receptor antagonist ligand with systemic antinociceptive activity and diminished opioid side effects. J. Med. Chem. 2020, 63, 7663–7694. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, A.; Uprety, R.; Gomes, I.; Massaly, N.; Keresztes, A.I.; Le Rouzic, V.; Gupta, A.; Zhang, T.; Yoon, H.J.; Ansonoff, M.; et al. Synthesis and pharmacology of a novel μ–δ opioid receptor heteromer-selective agonist based on the carfentanyl template. J. Med. Chem. 2020, 63, 13618–13637. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Narita, M.; Muramatsu, N.; Nakayama, T.; Misawa, K.; Kitajima, M.; Tashima, K.; Devi, L.A.; Suzuki, T.; Takayama, H.; et al. Orally active opioid μ/δ dual agonist MGM-16, a derivative of the indole alkaloid mitragynine, exhibits potent antiallodynic effect on neuropathic pain in mice. J. Pharmacol. Exp. Ther. 2014, 348, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, A.T.; Sandweiss, A.; Hu, J.; Bilsky, E.J.; Cain, J.P.; Kumirov, V.K.; Lee, Y.S.; Hruby, V.J.; Vardanyan, R.S.; Vanderah, T.W. Novel fentanyl-based dual μ/δ-opioid agonists for the treatment of acute and chronic pain. Life Sci 2013, 93(25-26), 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Lenard, N.R.; Daniels, D.J.; Portoghese, P.S.; Roerig, S.C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol 2007, 566(1-3), 75–82. [Google Scholar] [CrossRef] [PubMed]
- Dietis, N.; Niwa, H.; Tose, R.; McDonald, J.; Ruggieri, V.; Filaferro, M.; Vitale, G.; Micheli, L.; Ghelardini, C.; Salvadori, S.; et al. In vitro and in vivo characterization of the bifunctional mu and delta opioid receptor ligand UFP-505. Br. J. Pharmacol. 2018, 175, 2881–2896. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.W.; Folk, J.E.; Rice, K.C.; Negus, S.S. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] and heroin. J. Pharmacol. Exp. Ther. 2005, 314, 221–231. [Google Scholar] [PubMed]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef]
- Stevenson, G.W.; Luginbuhl, A.; Dunbar, C.; Lavigne, J.; Dutra, J.; Atherton, P.; Bell, B.; Cone, K.; Giuvelis, D.; Polt, R.; et al. The mixed-action delta/mu opioid agonist MMP-2200 does not produce conditioned place preference but does maintain drug self-administration in rats, and induces in vitro markers of tolerance and dependence. Pharmacol. Biochem. Behav. 2015, 132, 49–55. [Google Scholar] [CrossRef]
- Lei, W.; Vekariya, R.H.; Ananthan, S.; Streicher, J.M. A novel mu-delta opioid agonist demonstrates enhanced efficacy with reduced tolerance and dependence in mouse neuropathic pain models. J. Pain 2020, 21(1-2), 146–160. [Google Scholar] [CrossRef]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous activation of mu and delta opioid receptors reduces allodynia and astrocytic connexin 43 in an animal model of neuropathic pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef]
- Anand, J.P.; Kochan, K.E.; Nastase, A.F.; Montgomery, D.; Griggs, N.W.; Traynor, J.R.; Mosberg, H.I.; Jutkiewicz, E.M. In vivo effects of μ-opioid receptor agonist/δ-opioid receptor antagonist peptidomimetics following acute and repeated administration. Br. J. Pharmacol. 2018, 175, 2013–2027. [Google Scholar] [CrossRef]
- Nastase, A.F.; Anand, J.P.; Bender, A.M.; Montgomery, D.; Griggs, N.W.; Fernandez, T.J.; Jutkiewicz, E.M.; Traynor, J.R.; Mosberg, H.I. Dual pharmacophores explored via structure-activity relationship (SAR) matrix: Insights into potent, bifunctional opioid ligand design. J. Med. Chem. 2019, 62, 4193–4203. [Google Scholar] [CrossRef]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.-D.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid µ agonist/δ antagonist DIPP-NH2[Ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 7. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S. Opioid ligands with mixed μ/δ opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Saini, S.K.; Dersch, C.M.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dooley, C.T.; Misler, J.A.; Debevec, G.; Giulianotti, M.A.; Cazares, M.E.; Maida, L.; Houghten, R.A. Fluorescent mu selective opioid ligands from a mixture based cyclic peptide library. ACS Comb. Sci. 2012, 14, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; McLaughlin, J.P.; Houghten, R.A. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Banerjee, J.; Ganno, M.L.; Reilley, K.J.; Eans, S.O.; Mizrachi, E.; Gyanda, R.; Hoot, M.R.; Houghten, R.A.; McLaughlin, J.P. Discovery of novel antinociceptive α-conotoxin analogues from the direct in vivo screening of a synthetic mixture-based combinatorial library. ACS Comb. Sci. 2013, 15, 153–161. [Google Scholar] [CrossRef]
- Houghten, R.A.; Ganno, M.L.; McLaughlin, J.P.; Dooley, C.T.; Eans, S.O.; Santos, R.G.; Lavoi, T.; Nefzi, A.; Welmaker, G.; Giulianotti, M.A.; Toll, L. Direct phenotypic screening in mice: Identification of individual, novel antinociceptive compounds from a library of 734 821 pyrrolidine bis-piperazines. ACS Comb. Sci. 2016, 18, 51–64. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Y.; Giulianotti, M.; Houghten, R. A. High throughput synthesis of peptide α-thioesters through the use of “volatilizable” support. J. Comb. Chem. 2008, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yongye, A.; Giulianotti, M.; Martinez-Mayorga, K.; Yu, Y.; Houghten, R.A. Synthesis of cyclic peptides through direct aminolysis of peptide thioesters catalyzed by imidazole in aqueous organic solutions. J. Comb. Chem. 2009, 11, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Perry, K.; Sui, B.; Li, Y. Mercapto-functionalized polyhedral oligomeric silsesquioxane as a soluble support for the synthesis of peptide thioesters. Tetrahedron Lett. 2021, 85, 153483. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Giulionatti, M.; Houghten, R.A. Macrolactonization of peptide thioesters catalyzed by imidazole and its application in the synthesis of Kahalalide B and analogues. Org. Lett. 2010, 12, 2250–2253. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.L.; Eans, S.O.; Cirino, T.J.; Stacy, H.M.; Simons, C.A.; Uprety, R.; Majumdar, S.; McLaughlin, J.P. Kratom alkaloids, natural and semi-synthetic, show less physical dependence and ameliorate opioid withdrawal. Cell Mol. Neurobiol. 2021, 41(5), 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017, 170, 414–427. [Google Scholar] [CrossRef]
- Singleton, S.; Bastista-Hon, D.T.; Edelsten, E.; McCaughey, K.S.; Camplisson, E.; Hales, T.G. TRV130 partial agonism and capacity to induce anti-nociceptive tolerance revealed through reducing available μ-opioid receptor number. Br. J. Pharmacol. 2021, 178, 1855–1868. [Google Scholar] [CrossRef]
- Kelly, E. Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010, 86, 598–603. [Google Scholar] [CrossRef]
- Abdelhamid, E.E.; Sultana, M.; Portoghese, P.S.; Takemori, A.E. Selective blockage of delta-opioid receptors prevents the development of morphine-tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 1991, 258, 299–303. [Google Scholar]
- Fundytus, M.E.; Schiller, P.W.; Shapiro, M.; Weltrowska, G.; Coderre, T.J. Attenuation of Morphine Tolerance and Dependence with the Highly Selective Delta-Opioid Receptor Antagonist TIPPY. Eur. J. Pharmacol. 1995, 286, 105–108. [Google Scholar] [CrossRef]
- Scherrer, K.H., Eans, S.O., Medina, J.M., Senadheera, S.N., Khaliq, T., Murray, T.F., McLaughlin, J.P., Aldrich, J.V. Tryptophan substitution in CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) introduces δ-opioid receptor antagonism, preventing antinociceptive tolerance and stress-induced reinstatement of extinguished cocaine-conditioned place preference. Pharmaceuticals 2023, 16(9), 1218–1238. [Google Scholar] [CrossRef]
- Sanchez-Blazquez, P.; Garcia-Espana, A.; Garzon, J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: Role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 1997, 280, 1423–1431. [Google Scholar] [PubMed]
- Crain, S.M.; Shen, K.F. Acute thermal hyperalgesia elicited by low-dose morphine in normal mice is blocked by ultra-low-dose naltrexone, unmasking potent opioid analgesia. Brain Res. 2001, 888(1), 75–82. [Google Scholar] [CrossRef] [PubMed]
- Journigan, V.B.; Mésangeau, C.; Vyas, N.; Eans, S.O.; Cutler, S.J.; McLaughlin, J.P.; Mollereau, C.; McCurdy, C.R. Nonpeptide small molecule agonist and antagonist original leads for neuropeptide FF1 and FF2 receptors. J. Med. Chem. 2014, 57(21), 8903–9827. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. J. Pharmacol. Exp. Ther. 1999, 289, 304. [PubMed]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine-seeking behaviour. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef] [PubMed]
- Nomenclature Committee of IUB (NC-IUB) and IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Eur. J. Biochem. 1984, 138, 9–37.
Figure 2.
Positional scan screening of the cyclic peptide library in vivo using the 55 °C warm-water tail-withdrawal assay in young adult male C57BL/6J mice across an 8-h period. A: library defined at position 2, B: library defined at position 4, C: library defined at position 5. Each sample was given to the mice (n = 8) i.p. at a dose of 5 mg/kg. The tail-withdrawal latencies were measured at six time points post-administration, with combined time to withdraw tails (s; y-axis) calculated by taking the sum of the average tail-withdrawal latencies from each time point. Dotted line = vehicle alone (i.p.); cyan line = morphine control (10 mg/kg, i.p.). D: Summary of key functionality residues identified in the screening. Data represented the combined time to tail withdrawal ± SEM for each sample. Dark blue bars in A, B and C represent significantly different from vehicle (p<0.05) but not morphine (p>0.05); One-way ANOVA with Dunnett’s post hoc tests.
Figure 2.
Positional scan screening of the cyclic peptide library in vivo using the 55 °C warm-water tail-withdrawal assay in young adult male C57BL/6J mice across an 8-h period. A: library defined at position 2, B: library defined at position 4, C: library defined at position 5. Each sample was given to the mice (n = 8) i.p. at a dose of 5 mg/kg. The tail-withdrawal latencies were measured at six time points post-administration, with combined time to withdraw tails (s; y-axis) calculated by taking the sum of the average tail-withdrawal latencies from each time point. Dotted line = vehicle alone (i.p.); cyan line = morphine control (10 mg/kg, i.p.). D: Summary of key functionality residues identified in the screening. Data represented the combined time to tail withdrawal ± SEM for each sample. Dark blue bars in A, B and C represent significantly different from vehicle (p<0.05) but not morphine (p>0.05); One-way ANOVA with Dunnett’s post hoc tests.
Scheme 1.
Solution phase synthesis of the cyclic peptides using POSS-SH as a soluble support.
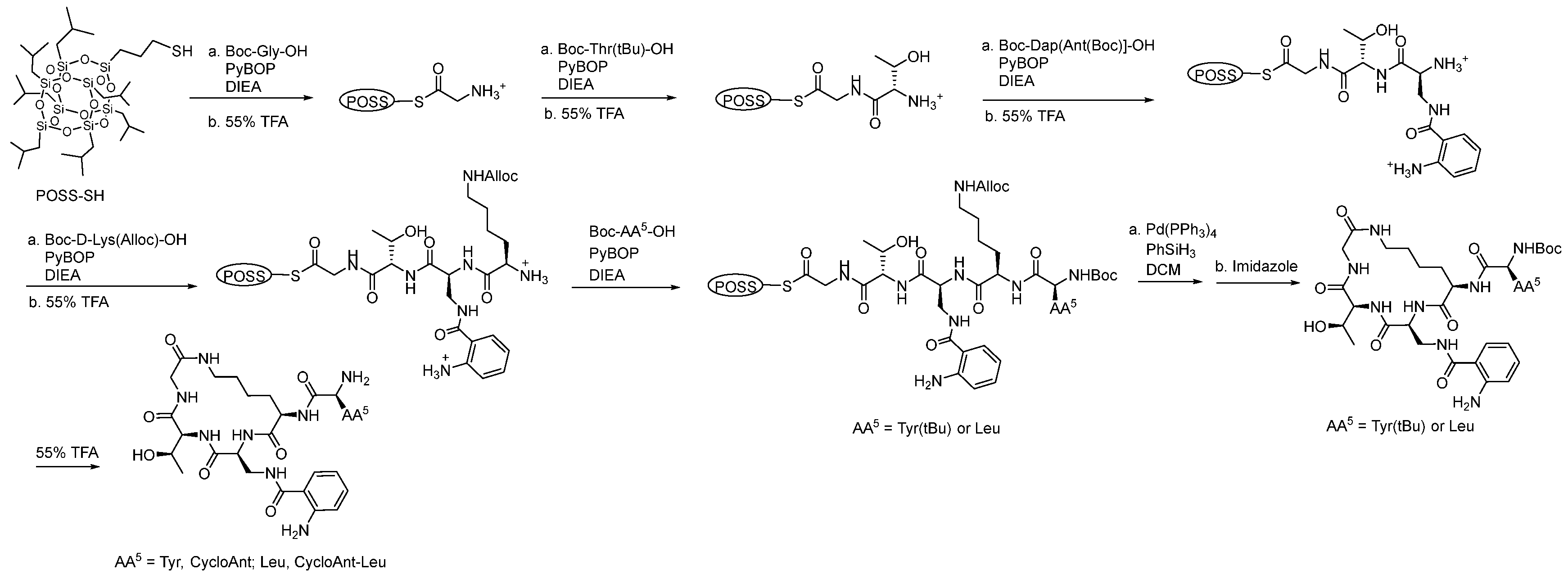
Figure 4.
Acute antinociceptive activity in the 55 °C WWTW assay following i.p. administration in C57BL/6J mice. Shown as A) dose-response and B) time-course of antinociception following a maximally efficacious dose of CycloAnt or CycloAnt-Leu. Morphine and vehicle are included as controls where appropriate. Points represent average % Antinociception ± SEM of 6-11 mice for each compound presented.
Figure 4.
Acute antinociceptive activity in the 55 °C WWTW assay following i.p. administration in C57BL/6J mice. Shown as A) dose-response and B) time-course of antinociception following a maximally efficacious dose of CycloAnt or CycloAnt-Leu. Morphine and vehicle are included as controls where appropriate. Points represent average % Antinociception ± SEM of 6-11 mice for each compound presented.
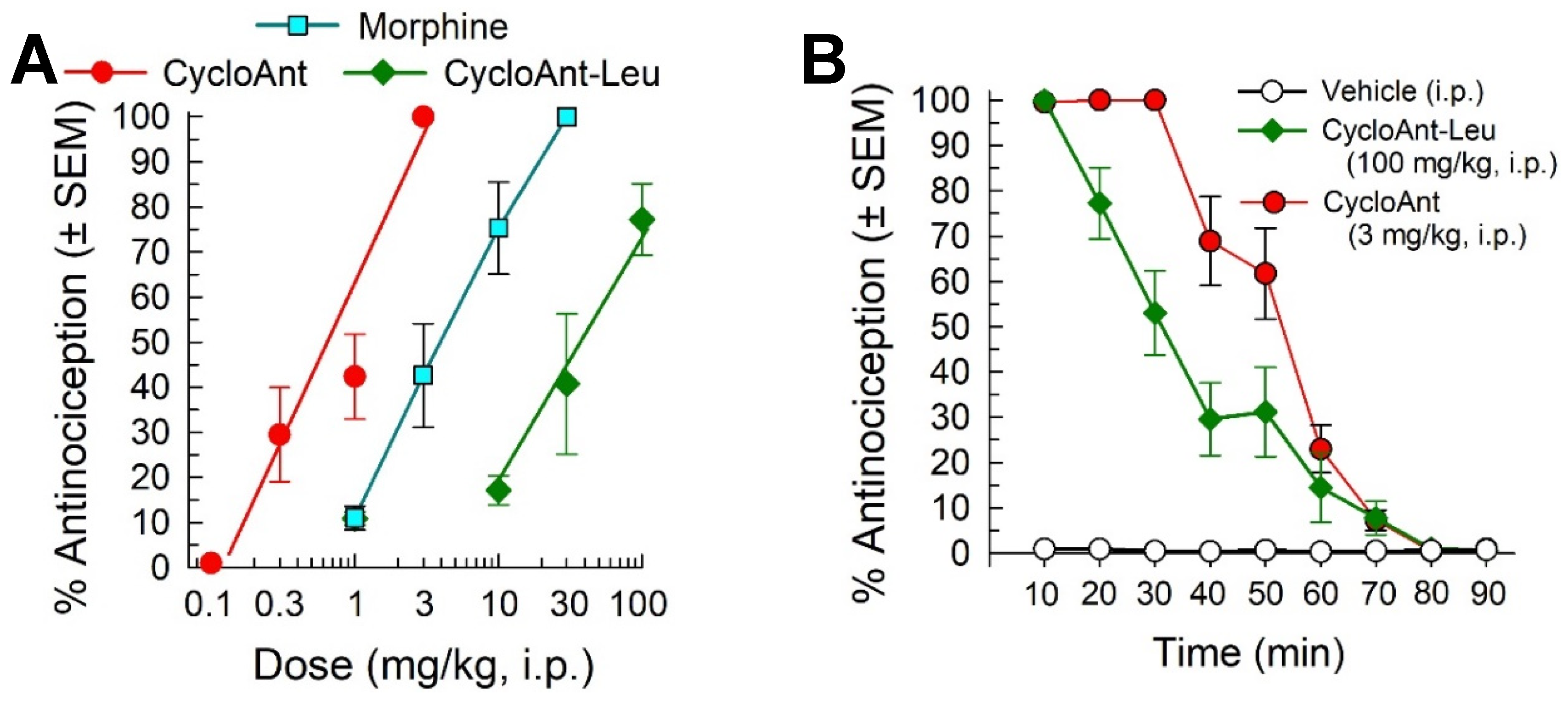
Figure 5.
Evaluation of opioid receptor involvement in the antinociceptive activity of CycloAnt (3 mg/kg, i.p.) 20 min after administration to wild-type (C57BL/6J) or mice where individual opioid receptors have been knocked out. * Significantly different from vehicle-control response (p < 0.05); † Significantly different from wild-type mice response (p < 0.0001); one way ANOVA with Dunnett’s post hoc test. Bars each represent average % Antinociception ± SEM of 8 mice.
Figure 5.
Evaluation of opioid receptor involvement in the antinociceptive activity of CycloAnt (3 mg/kg, i.p.) 20 min after administration to wild-type (C57BL/6J) or mice where individual opioid receptors have been knocked out. * Significantly different from vehicle-control response (p < 0.05); † Significantly different from wild-type mice response (p < 0.0001); one way ANOVA with Dunnett’s post hoc test. Bars each represent average % Antinociception ± SEM of 8 mice.
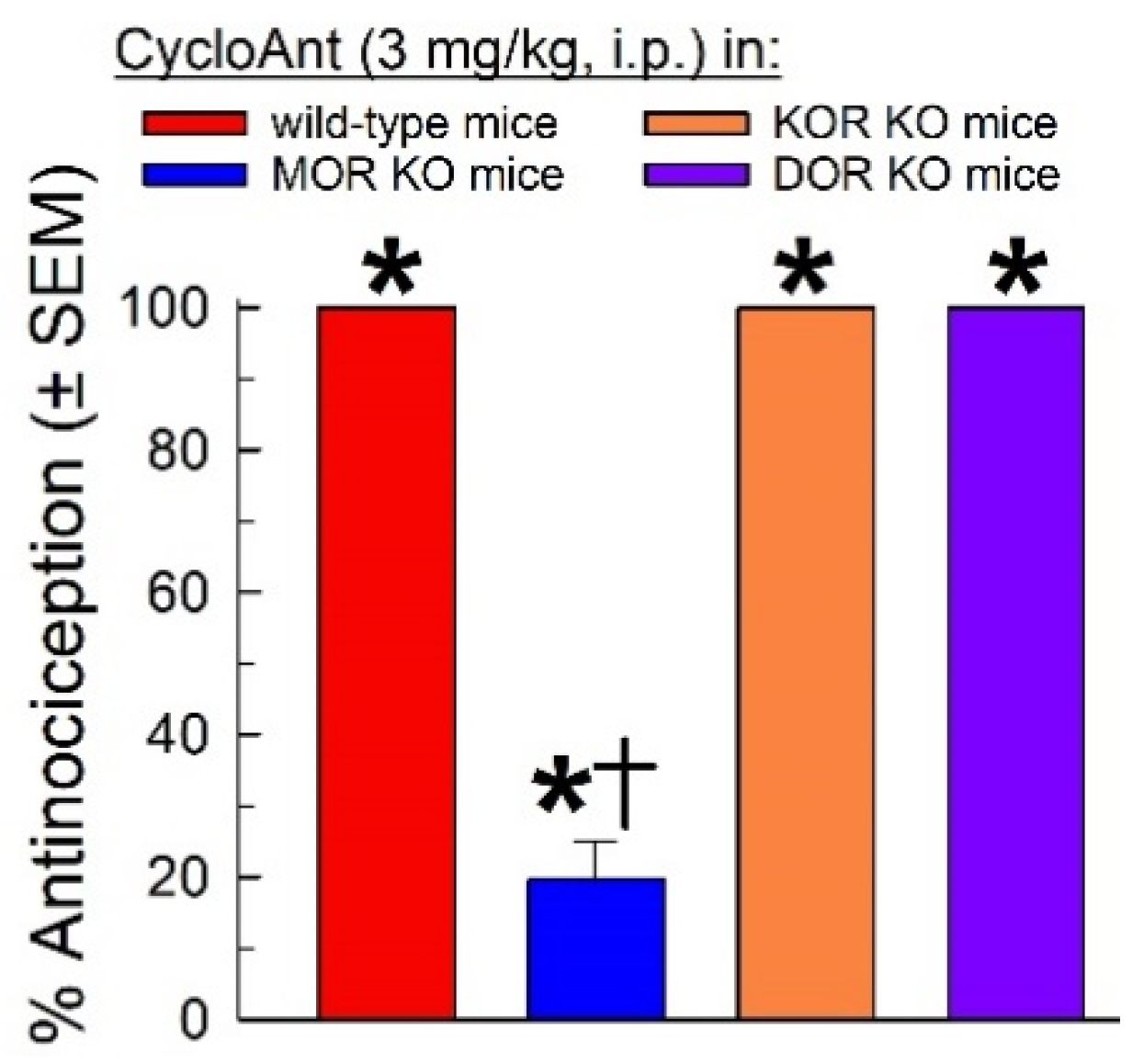
Figure 6.
Evaluation of opioid antagonist activity of CycloAnt in the 55 °C warm-water tail-withdrawal assay. MOR KO mice were tested to avoid confounding antinociception mediated by this receptor. A) A 60-min pretreatment with CycloAnt (3 or 10 mg/kg, i.p.) dose-dependently prevented SNC-80-induced antinociception (100 nmol, i.c.v.), but not that of U50,488 (10 mg/kg, i.p.). B) Time-dependent DOR antagonism by CycloAnt. The antinociceptive effect of SNC-80 (100 nmol, i.c.v.) was determined in mice pretreated for 1, 3, 6 or 8 h with CycloAnt (10 mg/kg, i.p.). Points represent average % antinociception ± SEM from 6 to 12 mice for each bar. *significantly different from response of agonist alone (p < 0.05); one-way ANOVA with Dunnett's post hoc test.
Figure 6.
Evaluation of opioid antagonist activity of CycloAnt in the 55 °C warm-water tail-withdrawal assay. MOR KO mice were tested to avoid confounding antinociception mediated by this receptor. A) A 60-min pretreatment with CycloAnt (3 or 10 mg/kg, i.p.) dose-dependently prevented SNC-80-induced antinociception (100 nmol, i.c.v.), but not that of U50,488 (10 mg/kg, i.p.). B) Time-dependent DOR antagonism by CycloAnt. The antinociceptive effect of SNC-80 (100 nmol, i.c.v.) was determined in mice pretreated for 1, 3, 6 or 8 h with CycloAnt (10 mg/kg, i.p.). Points represent average % antinociception ± SEM from 6 to 12 mice for each bar. *significantly different from response of agonist alone (p < 0.05); one-way ANOVA with Dunnett's post hoc test.
Figure 7.
Effects of the hit on (a) respiration and (b) ambulation in C57BL/6J mice tested in the CLAMS/Oxymax system. Respiratory and ambulation were monitored after i.p. administration of vehicle, morphine (10 or 30 mg/kg, i.p.), CycloAnt (3 or 10.6 mg/kg, i.p.) or vehicle. Data from 12-20 mice presented as % vehicle ± SEM; breaths per minute, BPM (A) or ambulation, XAMB (B). Dashed lines represent normalized vehicle response. * Significantly different from vehicle control response (p < 0.05); † Significantly different from response of morphine at either dose (p < 0.05); two-way RM ANOVA with Tukey’s multiple comparison post hoc test.
Figure 7.
Effects of the hit on (a) respiration and (b) ambulation in C57BL/6J mice tested in the CLAMS/Oxymax system. Respiratory and ambulation were monitored after i.p. administration of vehicle, morphine (10 or 30 mg/kg, i.p.), CycloAnt (3 or 10.6 mg/kg, i.p.) or vehicle. Data from 12-20 mice presented as % vehicle ± SEM; breaths per minute, BPM (A) or ambulation, XAMB (B). Dashed lines represent normalized vehicle response. * Significantly different from vehicle control response (p < 0.05); † Significantly different from response of morphine at either dose (p < 0.05); two-way RM ANOVA with Tukey’s multiple comparison post hoc test.
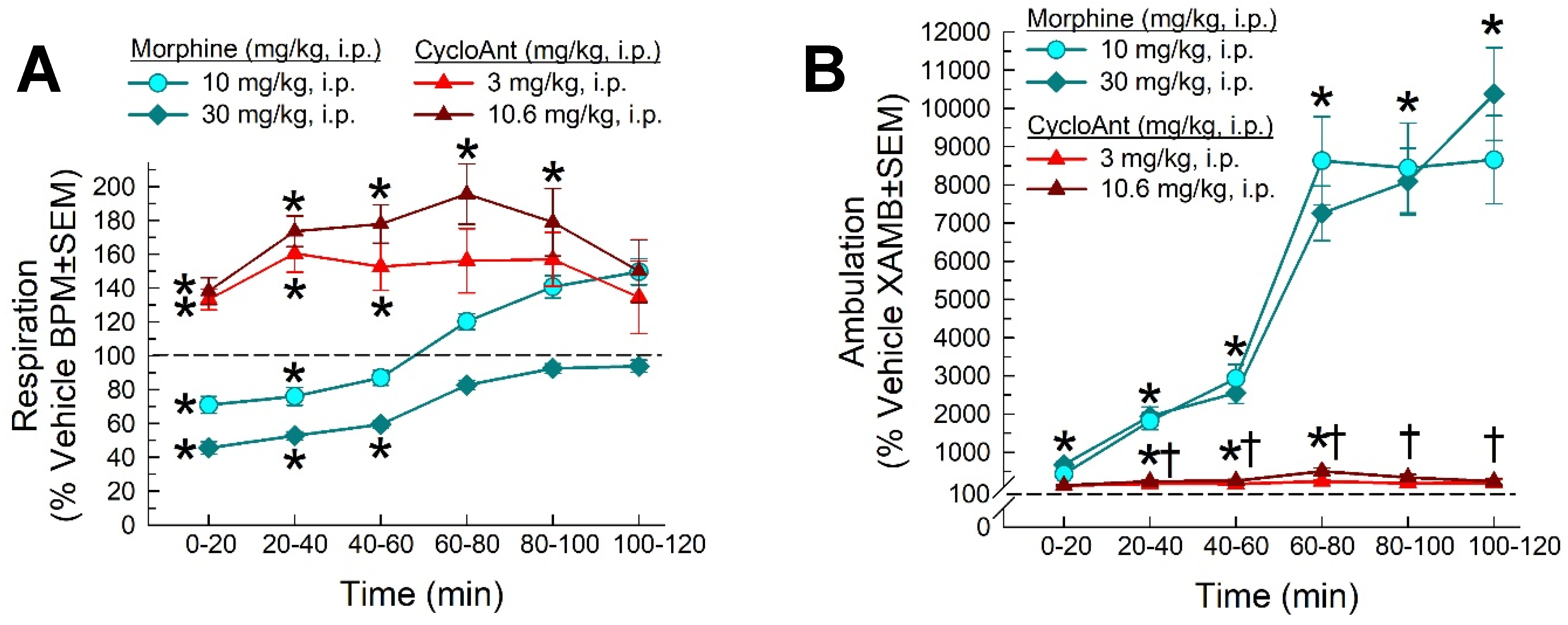
Figure 8.
Assessment of chronic treatment on A) induced hyperalgesia in the 48 °C warm-water tail-withdrawal test or B) antinociceptive tolerance in the 55 °C warm-water tail-withdrawal test. Mice were first tested on day 1 to establish baseline responses prior to any treatment. Mice were then subjected to a dosing regimen with morphine (10 mg/kg, i.p., twice a day; blue bars) or CycloAnt (3 mg/kg, i.p., twice a day; red bars) for 4 days. On day 5, tail-withdrawal latency from 48 °C water was assessed prior to final dosing (A), whereas tail-withdrawal latency from 55 °C water was tested 20 min (for CycloAnt, 3 mg/kg, i.p.) or 30 min (for morphine, 10 mg/kg, i.p.) after drug administration (B). Data are shown as mean ± SEM of A) pre- and post-dosing testing or B) responses to listed drug in naïve and chronically-treated mice. *p < 0.01 versus baseline (A) or response in naïve mice (B), Two-way RM ANOVA with Sidak’s multiple comparisons post hoc test. n = 9-11 mice/treatment.
Figure 8.
Assessment of chronic treatment on A) induced hyperalgesia in the 48 °C warm-water tail-withdrawal test or B) antinociceptive tolerance in the 55 °C warm-water tail-withdrawal test. Mice were first tested on day 1 to establish baseline responses prior to any treatment. Mice were then subjected to a dosing regimen with morphine (10 mg/kg, i.p., twice a day; blue bars) or CycloAnt (3 mg/kg, i.p., twice a day; red bars) for 4 days. On day 5, tail-withdrawal latency from 48 °C water was assessed prior to final dosing (A), whereas tail-withdrawal latency from 55 °C water was tested 20 min (for CycloAnt, 3 mg/kg, i.p.) or 30 min (for morphine, 10 mg/kg, i.p.) after drug administration (B). Data are shown as mean ± SEM of A) pre- and post-dosing testing or B) responses to listed drug in naïve and chronically-treated mice. *p < 0.01 versus baseline (A) or response in naïve mice (B), Two-way RM ANOVA with Sidak’s multiple comparisons post hoc test. n = 9-11 mice/treatment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



