
You are currently viewing a beta version of our website. If you spot anything unusual, kindly let us know.
Preprint
Review
Brief Electrical Stimulation Ameliorates Poor Recovery after Surgical Repair of Injured Peripheral Nerves
Altmetrics
Downloads
178
Views
64
Comments
0
A peer-reviewed article of this preprint also exists.

This version is not peer-reviewed
Abstract
Injured peripheral nerves regenerate their axons in contrast to those in the central nervous system. However, functional recovery after surgical repair is often disappointing. The basis for the poor recovery is the progressive deterioration with time and distance, of the growth capacity of the neurons that lose their contact with targets (chronic axotomy) and the growth support of the chronically denervated Schwann cells (SC) in the distal nerve stumps. This is despite the retained capacity of chronically denervated and atrophic muscle to accept reinnervation. Progressive decline in regeneration associated genes in both axotomized neurons and denervated SCs accounts for the decline in regenerative success in association with silencing of neural activity in sensory neurons due to their disconnection from their sense organs and, in motoneurons due to loss of their synaptic contacts in the spinal cord. Whilst exogenous neurotrophic factors promote nerve regeneration, the profuse axonal outgrowth and difficulties in delivery are avoided by promoting their endogenous expression with brief (1 hour) low frequency (20Hz) electrical stimulation (ES) proximal to the injury site. ES accelerates axon outgrowth and in turn, target reinnervation in both animals and human subjects. Applying ES to intact nerve days prior to nerve injury, conditional ES (CES) increases axonal outgrowth and regeneration rate with the potential for application in nerve transfer surgeries and end-to-side neurorrhaphies. However, the additional surgery for applying CES electrodes may be a hurdle. ES is applicable in all surgeries with excellent outcomes.
Keywords:
Subject: Medicine and Pharmacology - Neuroscience and Neurology
1. Introduction
Romon Y Cajal [1] recognized the contrasting capacity of injured nerves in the peripheral nervous system (PNS) and central nervous system (CNS) to regenerate. Yet, recovery of function is generally disappointing despite the contrasting support of the regeneration, the regrowth of the nerve fibres, by the Schwann cells (SCs) of the PNS and lack of support by the oligodendrocytes of the CNS [2,3,4,5,6]. As an example, fewer than 50% of patients regain adequate motor or sensory function after surgical repair of injured median or ulnar nerves [7]. Indeed, only ~10% of the two million Americans who suffer some form of peripheral nerve injury, demonstrate limited functional recovery, many with impaired motor and sensory function, and frequently suffering pain [8]. Functional recovery varies with location and severity. The recovery is the most severe for the more proximal nerve injuries [8], brachial plexus injury being the most disabling with severe functional impairment and poor quality of life [9]. Hand function is rarely restored after these injuries, occurring in only 1.2% of patients with multiple-traumatic injuries with an incidence of 1.64 cases out of 100,000 patients [10]. Patients are typically healthy, young adults who are economically productive [11]. They cannot work for ~21-31 weeks after upper extremity injuries, requiring long periods of rehabilitation [11,12,13]. Many of these patients, being unable to fulfill their former functions, must make changes in their careers [11,14].
The poor functional recovery has been relatively neglected in the scientific literature. Scientists, primarily clinical scientists, have addressed the issue in studies of whether administration of drugs and or various growth factors have a positive effect on nerve regeneration [15,16,17,18,19]. Electrical stimulation (ES) of muscles, used for many years by physiotherapists, is now a standard manipulation with the goals of preventing denervation atrophy and joint fixation [20,21,22]. In contrast, the use of ES for injured nerves has been a relatively recent approach introduced in the 1980s by Nix and Hopf [23] and furthered to promote muscle and sensory reinnervation by Gordon, Brushart, and colleagues in the 2000’s [24,25,26,27,28], their findings, largely confirmed by many investigators [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56]. The efficacy of localized tacrolimus (FK506) treatment demonstrated in in vivo and in vitro animal studies [57,58,59,60,61], and its potential use in human subjects, is considered in detail in a recent review [19] and hence, will not be considered further in this review. This review considers how and why functional recovery is poor after peripheral nerve injury and the efficacy of neurotrophic factors and/or brief low frequency ES for counteracting the negative effects of delayed surgical repair as a prelude for advocating ES to promote nerve regeneration and functional recovery after nerve injuries in patients. Previous reviews have concerned one or more of these issues [6,15,62,63,64,65,66,67,68,69,70].
2. Peripheral Nerve Injury
After crush (axonotmesis) or transection (neurotmesis) nerve injuries, the neurons, and their nerve fibres proximal to the injury are separated from their peripheral targets, a state of axotomy [15,71]. The axotomized neurons, the SCs in the denervated nerve stump distal to the injury, and the denervated muscle fibres and sense organs, undergo growth-associated changes as precursors to nerve regeneration and target reinnervation. These changes are reviewed prior to consideration of their declining capabilities with time after and distance from the nerve injury.
2a. Chromatolysis and Gene Expression in Axotomized Neurons
The classical morphological changes of the movement of the nucleus of axotomized neurons to an eccentric position and the dispersion of the Nissl bodies in the cytoplasm of the cell body, are referred to as chromatolysis [72,73]. The changes reflect the increased neuronal metabolism and protein synthesis [74] as the neurons transition from their normal transmitting state to a growth/regenerative mode [75]. This transition is usually driven by the transcriptome in which transcription factors, discussed in Section 2a. Neuronal molecular signaling of nerve injury, are responsible for coordinating the expression of multiple regeneration associated genes (RAGs) [76]. Genes that translate proteins for neurotransmitter synthesis including choline acetyl transferase in motoneurons, are downregulated whilst the RAGs, including those that transcribe the cytoskeletal proteins, tubulin, and actin, are upregulated (Figure 1A [77,78] reviewed by [75]). The cytoskeletal proteins are essential for the transport of materials from the cell body to the growth cones for elongation of the growing axons [79]. The corresponding downregulation of neurofilament protein that controls the calibre of the axons in motor and sensory nerves [15,77,80,81], accounts for the reduced size of the nerve fibres in the proximal nerve stump of injured nerves, as measured electrophysiologically [15,75,82] and morphologically [15,80,81,83,84]. The expression of neurotrophic factors including BDNF (brain derived neurotrophic factor) and its receptors trkB and p75, in motoneurons, is upregulated (Figure 1A: [15,16,85,86]. The several phenotypes of the heterogeneous DRG sensory neurons and their Ia, Ib, and cutaneous afferent nerve fibres that supply muscle spindles, tendons, and skin, respectively, transition to a more homogenous phenotype after axotomy [87].
2b. Neuronal Molecular Signaling of Nerve Injury
There are two distinct phases of the neuronal signalling of the nerve injury: (i) a rapid phase dictated by a retrograde calcium wave to the neuronal soma and (ii) a later slow signaling phase characterized by the retrograde transport of signaling molecules by motor proteins [88,89,90].
In the first rapid phase, the disruption of the axonal membrane at the site of the nerve crush or transection, exposes the axonal cytoplasm to external ionic concentrations. Within seconds of the injury, calcium ions enter the proximal nerve stump from the external fluid and, as membrane depolarization activates voltage-gated calcium channels, the intracellular calcium concentrations rise and trigger rapid sealing of the axon membrane at the injury site [91,92,93,94,95]. Local protein translation proceeds, long-range retrograde signaling is activated, and the local cytoskeleton contributes to the formation of the growth cone [96,97,98,99]. The growth cone is composed primarily of a microtubule cytoskeleton and F-actin (filamentous actin) with the central domain containing microtubules, organelles, and vesicles, and the P domain, composed of dynamic microtubules and F-actin [100,101]. The F-actin bundles comprise the finger-like projections of the filopodia.
The axonal calcium activates the nucleotide cAMP (cyclic adenosine monophosphate), via the calcium-dependent adenylyl cyclase enzyme which, in turn, activates the pro-regenerative kinase DLK (dual leucine zipper-bearing kinase) via PKA (protein kinase A), and the transcription factor, CREB1 (cAMP-responsive element-binding protein 1), amongst others [102,103]. DLK is a key sensor of local injury that informs the soma about the injury [102,103]. JNK (JUN N-terminal kinase) downstream of DLK signaling, is also transported to the soma where it activates STAT3 (signal transducer and activator of transcription factor 3) and c-Jun {an immediate early gene of the AP-1 (activator protein-1 transcription factor) transcription complex of Jun, c-Fos, and the ATF/CREB families [105]}, to promote nerve regeneration [106,107]. A wavefront of the calcium ions propagates anterograde to the cell body to reach millimolar concentrations in the soma. Calcium is also raised throughout the stump by additional calcium entry that follows the reversal of the sodium-calcium exchange pump by the calcium load [92]. The accumulating calcium ions activate local intracellular calpain (a serine-threonine protein) and oxidative species, resulting in axon swelling, rapid granular disintegration of the axonal cytoskeleton [108,109,110,111,112,113], and dieback to the first node of Ranvier [114,115,116]. The local calpain at the sealed end of the stump cleaves the submembranous spectrin complex, restructures the cytoskeleton by microtubule and actin depolymerization, and, in turn, allows the elaboration of the growth cone [95,117,118,119].
The local calcium activates many other signaling pathways. These include CaMK (Ca2+/calmodulin-dependent protein kinase) that phosphorylates CREB in the nucleus. The phosphorylated CREB later influences gene expression directly. It does so by mediating cAMP-induced transcription [120,121] via PKA and MEK/Erk (extracellular signal-regulated kinase) pathways [122], and the translation of the transcription factor, HIF-1a (hypoxia-inducible factor), that, in turn, activates HIF-1a responsive genes for axonal regeneration [123].
There is an initial and local translational burst of mTOR (mammalian target of rapamycin) that controls ~250 localized mRNAs. These in turn, transcribe several proteins that are transported to the cell body via retrograde axonal transport. The transport is facilitated by the local increase in tyrosinated α-tubulin in the microtubular cytoskeleton [124]. The transported proteins include importin-β1, the adaptor protein that transports cytoplasmic proteins via dynein, the retrograde motor on the microtubules [125], vimentin (the intermediate filament protein), STAT3, ZBP-1 (Z-DNA-binding protein 1), RanBP1 (Ran binding protein 1), Ran being the Ras-related nuclear protein ligand-activated nuclear receptor, and PPARƔ, the peroxisome proliferator-activated receptor ([126,127,128], reviewed by [129]). Luman/ATF3, an endoplasmic reticulum (ER) transmembrane basic leucine zipper transcription factor, is also transported in an importin-dependent manner to the cell body where it is critical regulator of sensory axon regeneration, linking the unfolded protein response and the ensuing endoplasmic stress response to axon repair [130,131]. There is an interesting coordination of the temporal phases of the luman protein levels. They are co-ordinated with the three phases of the growth responsive response of sensory neurons, (1) the stress response phase immediately after injury, (2) the pre-regenerative phase, 9 to 24 hours thereafter when transcription factor activity regulates DNA replication and transcription, and (3) the regenerative phase at 4 days [132].
The transported proteins activate pro-regenerative pathways in the second slow signaling phase of nerve injury. These pathways include the translation and activation of transcription factors, specific epigenetic modifiers, and additional signaling molecules [129].
Transcription factors There are more than 1500 transcription factors in the genome [133] of which c-Jun is markedly increased in axotomized motor and sensory neurons [134]. The genes that coordinate the transcription of many other genes, often the transcription factors, are referred to as Hub genes [102,135,136]. The transcription factors bind to selective DNA promotor regions [137,138,139,140] to increase or repress the transcription of specific target genes, thereby coordinating the expression of multiple RAGs [76,129]. The transcription factor c-JUN was the first to be identified in a RAG network [134,141). CREB serves to coordinate the transcription of many RAGs because the CREB protein not only regulates transcription of BDNF and arginase 1, amongst other genes, but also drives the transcription of both the AP1 and the ATF3 hub genes, being a highly connected node [101]. One study distilled the RAG network to ~40 transcription factors downstream of multiple parallel signaling pathways [142], of which the calcium-dependent cAMP activates only a fraction of injury-induced genes, at least in sensory neurons [143].
Transcription factors that were identified independently, include ATF3 (cAMP-dependent transcription factor 3) [144,145,146,147,148], STAT3 [149,150], SOX11 {(SRY-Box Transcription Factor 11) [151,152,153,154]}, SMAD1 (Suppressor of Mothers against Decapentaplegic family member 1) [155,156,157,158,159], C/EBPβ (CCAAT/enhancer binding protein β) [160,161], p53 (tumor protein p53) [162,162], GSK3β (glycogen synthase kinase 3β [163]), c-Myc [151,152,153,154], and KLF4 (Krüppel-like factor 4) [163,164]. ATF-3, a cAMP-dependent transcription factor, is upregulated rapidly in injured motor and sensory neurons following JNK signalling [87,146,147,148]. The factor is used frequently and reliably as a biomarker of axotomy [27;87,145]. Both Jun and ATF3 mediate peripheral nerve regeneration in vitro [151] and in vivo [152]. Phosphorylated STAT3 stimulates growth initiation but does not perpetuate axonal growth [150]. SOX11 is also elevated in injured nerves and promotes their regeneration [151,153]. It does so via the activation of the transcription factors, ATF3 and c-Jun, and the RAGs, Arpc3 and Sprr1a [151,153], and by increasing the responsiveness of neurotrophic factors [153]. Elevated SMADs are phosphorylated and accumulate in the nuclei of injured DRGs and motoneurons, SMAD1 in the sensory neurons [155,156,157,158] and SMADs 1,2, and 4 in the motoneurons [159]. They act as modulators of activated GSK3β (glycogen synthase kinase 3) downstream of P13K (phophoinositide-3-kinase)/Akt pathway, by interacting with the transcriptional coregulator p300 HAT (histone acetyltransferase p300), to promote expression of several pro-regenerative target genes and in turn, nerve regeneration [155,156,158]. The transcription factor C/EBP (CCAAT/enhancer binding protein) that is induced in injured neurons, is also an essential transcription factor for nerve regeneration. It binds to the promoters of Tα1-tubulin of the microtubules and the growth cone protein, GAP-43 [161]. It was reported that p53 stimulates regeneration of transected sciatic nerve fibres with reduced reinnervation of target muscle fibres after facial nerve injury in p53 knockout mice [162,163]. The c-Myc transcription factor stimulates peripheral nerve regeneration by elevating expression of the RAGs arpc3 and Sprr1a and the transcription factors c-Jun and Atf3, in the nucleus of axotomized DRG neurons, and by increasing the responsiveness of the neurons to neurotrophic factors [151,152,153,154]. In contrast to the other factors, KLF4 is downregulated after injury, thereby negating its inhibitory effect on nerve regeneration [164,165].
Epigenetic modifications, or “tags,” regulate patterns of gene expression by altering DNA accessibility and chromatin structure without altering the DNA sequence [166]. They include the miRNA (microRNA), and the non-coding RNAs, namely the long chain and circular RNAs. They can affect nerve regeneration by altering the access of transcription factors to DNA by DNA methylation, post-translational modification of the histone protein that wraps around nuclear DNA, or by controlling ncRNAs (noncoding RNA) that silence genes [101,130,167].
DNA methylation generally, is associated with repression of transcription [129]. There are >200 identified histone modifications [168]. An important example is the p300/CBP-associated factor (PCAF)-dependent acetylation of H3K9ac (histone 3 lysine 9) that is paralleled by the reduction in methylation of H3K9 (H3K9me2). This reduced methylation relaxes the chromatin environment surrounding the promoters of several pro-regenerative genes. In DRG sensory neurons, this modification results in the expression of the growth-associated genes GAP-43, galanin, and BDNF [169]. The acetylation requires retrograde ERK signaling. Also, PCAF overexpression promotes axonal regeneration of the central axons of the DRG neurons across injured spinal cord.
MicroRNAs (miRNA), of which lncRNA (long chain non-coding RNAs) account for 60 to 80% of the mammalian genome transcriptome [170], are differentially expressed after peripheral nerve injury [101,171]. They target specific mRNAs with resulting repression or degradation of their translation. For example, miR-21 is upregulated after sciatic nerve injury and by targeting Sprouty2 (a specific inhibitor of the Ras/Raf/Erk pathway), promotes axonal growth from adult DRG neurons [172,173]. A second example is the nerve regeneration that results from miR-26a that specifically targets GSK3β to rescue axon regeneration, the miR26a-GSK3β pathway regulating axon regeneration at the neuronal soma by controlling the expression of the regeneration-associated transcription factor, SMAD1 [160].
2c. Wallerian Degeneration
The peripheral nerve fibres distal to the site of crush or transection, are isolated from their neuronal cell bodies and, in turn are deprived, for all intents and purposes, of their source of synthesis of proteins, lipids, glycoproteins, and carbohydrates [15,72,109]. They undergo the self-destructive process of Wallerian degeneration of nerve fibres and their myelin, including the axons that die back to the first node of Ranvier in the proximal nerve stump that prevents the scarring that occurs in CNS injury [114,115,116,174]. Ramon Y Cajal [1], using his silver staining technique, elucidated the degeneration of the axons distal to the nerve injury, myelin breakdown, and the proliferation of the remaining SCs. The calcium ions that enter the nerve via calcium channels, activate calpain proximal and distal to the injury site, that mediates proteolysis with degeneration of axon segments several hundred micrometers from the injury site [175,176]. The expression of SARM1 (sterile alpha TIR motif-containing protein 1), the essential protein for axon degeneration in the distal nerve stump, is elevated due to the loss of the anterograde transport by the calcium-dependent proteolysis of the cytoskeletal structures in the stump, followed by a rapid depletion of NMNAT2 (NAD-synthesizing enzyme, nicotinamide mononucleotide acetyltransferase 2; reviewed by [18]). The downstream steps to axon degeneration remain to be determined. Meanwhile, the remaining fast axonal transport allows for continued propagation of action potentials in the distal stump for hours and even days [108,177,178,179,180].
Ras/raf/ERK signaling in SCs is evident immediately after injury with ERK levels returning to lower levels just prior to SC proliferation [181]. Activation of Raf-kinase drives SC dedifferentiation as well as inducing much of the inflammatory response important for nerve repair, including breakdown of the blood nerve barrier and the delayed recruitment of macrophages into the denervated nerve stump [182]. SCs break their myelin sheaths down through autophagy [183,184,185] with ~50% of the total myelin thought to be broken down by the SCs [186]. The SC expression of proinflammatory cytokines and chemokines within 3 to 5 hours of nerve injury, contribute to myelin and axon breakdown and the phagocytosis of their debris within the first 3 days of injury [187]. The cytokines, including IL-1α (interleukin 1α), and LIF (leukemia inhibitory factor), their receptors IL-6R and gp130 respectively, and tumor necrosis factor α (TNF-α), stimulate the expression of the two cystolic forms of PLA2 (phospholipase enzyme-A2) [188]. These remain high for two weeks [187]. TNF-α hydrolyses the phosphatidylcholines in the myelin membranes, thereby releasing the potent myelinolytic agent, lysophosphotidylcholine. Lysophosphotidylcholine also feeds back to sustain cytokine expression and the expression of the chemokines, including MIP-1 (macrophages inflammatory protein-1), MMP-1α or CCL2 (monocyte chemoattractant protein-1α) and IL-lβ [17,189,190,191,192,193,194,195,196]. Within a day of the nerve injury, IL-6 and TNF-α induce the expression of MMP-9 (matrix metallopeptidase 9) that contributes to myelinolysis [197,198].
The recruited macrophages also express these cytokines and chemokines and are responsible for the bulk of the myelin and axonal phagocytosis over the following ~3 weeks [116,199,200,201,202]. They play the major role in removing inhibitory molecules, including MAGs (myelin associated glycoproteins), from the degenerating axons [202]. Their release of nitric oxide has been implicated in the myelin breakdown [203] and their phagocytosis of the myelin debris includes the myelin that is opsonized by complement components binding to the complement receptor type 3 on the macrophages [204]. Antibodies to non-opsonized myelin are phagocytosed via the macrophage Fc receptor [205]. The third macrophage receptor used is the scavenger receptor-AI,II [206]. The spectrum of macrophages has been separated into two “polarizing” phenotypes, M1 and M2 with the M1 macrophages associated with pro-inflammatory and neurodegenerative functions and the M2 macrophages broadly viewed as anti-inflammatory and promoting cellular repair [207]. Although an early increase in M1 macrophages 1-2 days after injury with M2 macrophages replacing these from days 3 to 7, the majority of the accumulating M2 macrophages may be of a mixed phenotype because these mixed type macrophages were not included in their analysis [207,208]. In addition to their essential role in phagocytosis of degenerating axons and their myelin, the macrophages sense hypoxic conditions and stimulate angiogenesis for a polarized vasculature that guides SCs and elongating axons [209,210].
2d. Schwann Cell Response to Nerve Injury
Once the SCs lose their myelin, they re-enter the cell cycle, undergo mitosis, and differentiate toward a state supportive of nerve regeneration [5,15,75,108,139,211]. The transient expression of Cdc2 (cyclin-dependent kinase, cell cycle division 2) by the denervated SCs, possibly induced by c-Jun [212], is involved in their proliferation and migration [213]. The SC genes that transcribe myelin proteins, including MAP (myelin associated protein), MBP (myelin basic protein), P0 (myelin protein zero), and PLP (proteolipid protein), are downregulated in the denervated distal stump with the RAGs associated with nerve regeneration, upregulated as the SCs dedifferentiate and acquire the ability to survive without axonal interactions (Figure 1A; [214]). This change in SC transcription occurs with their transition from their myelinating state to the growth supportive state [75]. The transcription factor, c-Jun, programs the SCs to generate the repair cells that are essential for nerve regeneration, c-Jun accelerating the downregulation of myelin genes, promoting myelin breakdown, and amplifying the upregulation of a broad spectrum of repair-supportive features, including the expression of trophic factors ([137,215]; reviewed by Jessen and Arthur-Farraj [216]). As early as 1991, the upregulation of hundreds of growth-supportive RAGs was reported in denervated SCs [217] with more genes differentially expressed in the SCs than in sensory neurons [218].
IL-6, synthesized in the SCs within 24 hours [190,192,219,220], signals the expression of RAGs via its receptor [191]. The SCs express NRG-1 (neuregulin-1), a member of the family of glial growth factors, and its ErbB2/3 receptor [221,222,223,224]. The NRG-1 levels remain elevated for at least 30 days [225,226,227]. NRG-1 strongly inhibits the expression of genes involved in myelination and in glial cell differentiation, suggesting that it might be involved in the dedifferentiation process of SCs from the myelinating to the repair phenotype [228]. In addition, NRG-1 likely mediates, at least in part, the second phase of SC proliferation that is stimulated when regenerating axons contact the SCs in the Bands of Büngner [229,230,231,232,233,234,235]. The scaffolding oncoprotein Gab2 (Grb2-associated binder-2) is required for SC proliferation after nerve injury, its activation leading to the migration of the SCs [236], possibly through actin modulation [237]. In the model based on their findings, autocrine/paracrine activation by NRG of its erbB2 receptor on the SCs, promotes the SC migration by leading to transcriptional Gab2 expression via the Rac-JNK-cJun pathway, and the phosphorylation of Gab2 via the paracrine HGF (hepatocyte growth factor) from fibroblasts [236].
The non-coding RNAs, namely the long chain and circular RNAs, play important roles in SC proliferation and migration [101]. Examples of the long chain RNAs include NEAT1 (nuclear enriched abundant transcript 1) that promotes SC proliferation and migration [101], MALAT1 (metastasis associated lung adenocarcinoma transcript 1) that elevates BDNF [238], and Loc680254 that promotes nerve regeneration by inducing SC proliferation [239]. BC088259 which showed the most significant upregulation after sciatic nerve injury, interacts with vimentin to regulate SC migration [240,241]. Downregulation of some long chain RNAs also enhances SC proliferation and migration. An example is MEG-3 (maternally expressed gene 3) that increases SC proliferation and migration and facilitates nerve regeneration through the PTEN/P13K/ADT pathway [242]. Some circular RNAs are also upregulated in the denervated nerve stumps and are associated with SC proliferation. For example, cirRNA-Spidr targets P13K-Akt to promote nerve regeneration after rat sciatic nerve crush injury [243].
Within the first week of nerve injury, the denervated SC’s express several neurotrophic factors and their receptors, their levels peaking within a month. The factors include the neurotrophins, NGF (nerve growth factor), BDNF, and their p75 receptor, NT-3 and NT-4/5 (neurotrophin-3 and 4/5) and the TrkC receptor, the GDNF (glial derived neurotrophic factor) family and their receptors, GDFRα1 and ret, and other factors including IGF-1 and IGF-II (insulin-like neurotrophic factor I and II), VEGF (vascular endothelial growth factor), HGF (hepatocyte growth factor), platelet-rederived growth factor-BB, FGF (fibroblast growth factor), TFG-β (transforming growth factor β), and their receptors, and pleiotrophin [16,244,245,246,247,248,249] and Xu et al 2023 unpublished data. The expression of GDNF and pleiotrophin is specific for the denervated SCs in the motor pathways of the quadriceps nerve branch of the femoral nerve, whereas the remaining neurotrophic factors, HGF, BDNF, NGF, and IGF I and II, are more specific for the denervated SCs located in the sensory pathways of the saphenous nerve branch [245,246,247]. The time course of expression varies for different trophic factors. NGF rises rapidly, then declines prior to a 5-fold upregulation possibly in response to macrophage release of IL-1β and persisting for at least 3 weeks [250,251]. The upregulation of BDNF is much slower, detectable at 7 days and increasing up to 28 days after nerve injury to levels much higher than NGF [16,251]. On the other hand, GDNF and its GDFRα1, but not its coreceptor Ret, are upregulated and reach a peak within 7 days [16,252]. The expression is not sustained, declining with time when the denervation period of the distal nerve stump is prolonged for more than a month [16,244,245,247,252]. NT-3 upregulates c-Jun [215] and regulates the levels of the upregulated p75 receptor in the SCs [182,254,255,256]. The transcription factor, Notch, is also upregulated in the SCs [137,138,139,140].
2e. Axonal Regeneration
Regenerating axonal sprouts emanate from the first node of Ranvier proximal to the injury site [1,257,258]. The formation of the growth cones occurs without direct support from the cell body and depending on material that is locally available in the axons in the proximal nerve stump that includes the preexisting cytoskeletal elements of actin and tubulin [258,259]. The major source of materials for subsequent axonal elongation comes from the cell body via anterograde axonal transport [79]. As described above in Section 2d. Schwann cell response to nerve injury, the growth cones link to ECM glycoproteins that become organized at the injury site, navigate across the site, and regenerate axons along the bands of Bungner in the denervated distal stumps. A single regenerating axon can give rise to as many of 50-100 branches [260] but, it is an average of 5 daughter axons that regenerate with more regenerating into the distal stump after crush than after transection injuries [261,262,263].
Following the nerve injury, the SCs migrate to form the repair SC layer of the Bands of Bungner on the endothelial laminal sheath [214]. They do so by their sorting through intercellular N-cadherin linkage mediated by EphB-Sox signaling [264]. The growing axons contact the SC basement membrane glycoproteins that are secreted by both SCs and fibroblasts. These glycoproteins include collagen, fibronectin, tenascin C, and laminin ([265,266,268,269]; reviewed by [5,214,267,270,272]). Via adaptor molecules on the SC membranes, including N-cadherin and integrins, the interaction of the axons with the glycoproteins mediates the progression of the growth cones as the axons regenerate [271,273,274,275,276,277,278,279,280].
Contact between the axolemma and SCs initiates remyelination of regenerating axons with the SCs forming myelin layers (lamellae) in proportion to the size of the regenerating axons [281]. Both NRG-III from axons and NRG1-I from SCs play a pivotal role in remyelination [226,282,283] with tyrosine phosphorylation of GAB1 in the denervated SCs, principally regulated by NRG-1, being essential for remyelination [236]. The internodal distances between the formed myelin sheaths are shorter than normal as a result of the 3-fold increase in SC numbers after denervation [284] but they lengthen with time [281]. The regenerating axons increase their diameters in proportion to the size of their parent nerve [285], recovering their normal size if and when they make functional connections [286].
3. Poor Recovery of Function after Peripheral Nerve Injury and Repair
The clinical examinations and overall patient health being considered, decisions to operate after nerve injuries are not as straight forward as the surgeons would like. The simplest classification of nerve injuries is whether the injury is closed or open. Traumatic injuries that are classified as open injuries result in neurotmesis (nerve transection), but the timing of surgical intervention depends on whether the nerve transection is ragged or sharp [287,288]. The primary repair of sharp injuries from knives or razors should be repaired within 3 days of trauma but the repair of ragged nerve transections from blast, gunshot, fracture, or crush injuries is usually delayed for at least 3 weeks to allow the nerve ends to be demarcated [288]. Frequently, an early surgical evaluation is made of whether any of the nerve remains in continuity. When the nerve is transected, the nerve stumps may be sutured to a nearby local structure to prevent their retraction and thereby, to facilitate later surgical repair. Especially when a nerve injury is associated with comorbidities that include vascular problems, the later repair is undertaken once the local inflammation has subsided [287,289]. Irrespective of whether or not surgical repairs are delayed, proximal injuries in particular, result in long delays before regenerating nerve fibres contact distal denervated targets (Figure 1B). Even after early surgical repair after proximal injuries such as the brachial plexus nerve injuries, many axotomized neurons remain chronically axotomized without target contacts for periods of 2 or more years as they regenerate their axons slowly over long distances at the regeneration rate of the estimated 1 mm/day using the Tinel sign that identifies the site at which a tap on the regenerating nerve elicits a tingling sensation in the conscious patient [290,291]. The SCs in the nerve stump distal to the injury are subjected progressively to chronic denervation especially those far distal to the microsurgical suture of the proximal and distal nerve stumps.
Based on morphological evidence, the predominant view of the denervated distal nerve stump is that the SCs progressively die as the endoneurial tubes break down and are replaced by connective tissue that likely is impenetrable to regenerating axons. It was reported in rat and rabbit hindlimb nerves that, over periods of 1 to 26 months, denervated SCs undergo progressive atrophy and loss with disruption of their basement membranes, shrinkage of their columns, and filling of the columns with densely packed longitudinally orientated collagen fibrils [292,293,294,295,296]. Yet, SCs do remain 6 -25 months after denervation of rat and rabbit sciatic distal nerve stumps and intramuscular nerve trunks, although they are reduced in number [224,296,297]. The numbers decline to less than 10% but, the surviving SCs proliferate in response to mitogens, and they myelinate neurites normally (Figure 2A-C; [298,299]). Indeed, these SCs support the full recovery of the size of regenerated nerve fibres in vivo [300,301] The transcription factor STAT3, has been implicated in the long-term maintenance of SCs [302] and c-Jun influences both apoptosis and proliferation of denervated SCs [303,304,305]. The most accepted interpretation of poor functional recovery remains the irreversible atrophy of muscle and sensory targets and their replacement by fat [3,306]. Muscle mass is known to decline rapidly within two weeks after denervation, to decline more gradually thereafter [307,308,309], and to undergo permanent damage after two years [310].
3a. Time- and Distance-Related Decline in Nerve Regenerative Capacity
A cross-suture technique was pioneered by Holmes and Young in 1942 to examine their hypothesis that chronic motoneuron axotomy accounts for the poor functional recovery after surgical repair [311]. Their finding that reinnervated muscle contractile force was restored to normal levels negated their hypothesis. leading them to conclude that indeed, denervated targets are replaced by fat. The conclusion of fat replacement of denervated muscle accounting for poor functional recovery after nerve injury remained the accepted view for the next 50 years. More recently, we systematically examined each of the contributions of chronic axotomy, chronic denervation of the distal nerve stump, and chronic denervation of target musculature, to poor recovery after nerve transection (Figure 2; [28,71,300,312,313,314,315,316,317,318]; reviewed by [6,15,16,62,64,69,248,319,320,321,322]. Our surgical technique, illustrated in Figure 3A,B, was to prolong the axotomy of TIB (tibial) motoneurons or to prolong denervation of the CP (common peroneal) distal nerve stump prior to the cross-suture of TIB and CP proximal and distal nerve stumps, respectively. After at least 5 months, we determined how many motoneurons (1) reinnervated tibialis anterior (TA) muscle fibres by estimating the number of reinnervated motor units (MUs) by the ratio of the whole muscle to mean MU isometric contractile forces; Figure 3C) and (2) regenerated their nerve fibers into the CP nerve stump by applying retrograde dyes of either FG (fluorogold) or FR (fluororuby) to the regenerated nerves in the CP nerve stump. Either dye was used in the experiments that followed because the motoneuron counts using either dye, were not significantly different (Figure 3D,E).
Contrary to the conclusions made in 1942 [311], chronic axotomy of motoneurons had a pronounced negative effect on the regeneration of their nerve fibres: both the numbers of (MN) motoneurons that regenerated their axons into the freshly denervated nerve stumps and the numbers of MUs, the motoneurons whose nerves reinnervated muscle fibres, declined exponentially with a time constant (Ƭ) of 40 days to reach a plateau of 33% of the numbers after immediate nerve repair (Figure 4A,C; [71,312,313]). It was the capacity of regenerating motor nerve fibres to reinnervate 3-5 times as many denervated fibres as they normally do [323,324], the same capacity as that of intact nerve fibres sprouting in partially denervated muscles [325,326]. This 3- 5- fold increase in MU size resulted in the reinnervation of all the denervated muscle fibres and, with their full recovery from muscle atrophy, the full recovery of the reinnervated muscle contractile force [71]. Complete recovery of muscle force by reinnervation by chronically axotomized motoneurons, was the basis for the faulty conclusion made previously in 1942 [311] and is the reason why the clinical assessments of reinnervated skeletal muscle strength by the Medical Research Council (MRC) 5-point scale [327] or by revised scales [328]. are frequently misleading. These clinical assessments continue to underestimate the deleterious effects of chronic axotomy as well as chronic denervation of distal nerve stumps and muscles on nerve regeneration and muscle reinnervation. It is interesting, as an aside, that the size of the nerve supplying the enlarged MUs does not increase [329], the regenerated nerve fibres recovering their normal size once they made functional connection with denervated muscle fibres [300].
Chronic denervation of the distal nerve stump and the muscles that the intact nerve normally supplies, is even more deleterious to nerve regeneration than chronic motoneuron axotomy (Figure 4B; D;300,312.318]. The exponential decline in numbers of MUs and freshly axotomized motoneurons that regenerate their axons into the chronically denervated nerve stumps plateaued at ~5% at 180 days of chronic denervation (Figure 4B). To determine whether the poor regeneration was due to chronic SC denervation within the distal nerve stump and/or prolonged muscle denervation, a chronically denervated autograft was inserted between a freshly cut TIB proximal nerve stump and a chronically denervated CP distal nerve stump [318]. The experiments demonstrated that it is the chronic denervation of the SCs and not the chronic denervation of the TA muscle that was the causative factor of the poor nerve regeneration through chronically denervated distal nerve stumps [318]. It is noteworthy that the chronically denervated SCs that do survive support myelination in vitro [298] and in vivo [318].
The reduced numbers of nerve fibres that regenerated through the chronically denervated nerve stumps reinnervated as many as 3-times as many TA muscle fibres as normal [312]. However, this capacity of the nerves to expand the size of the reinnervated MUs was insufficient to reinnervate all the remaining denervated muscle fibres. In addition, the reinnervate muscle fibres did not recover their normal size [312]. As a result, the partially reinnervated muscles failed to recover their normal contractile force after their chronic denervation, [312].
Chronically denervated muscle fibres undergo extreme atrophy within a year of denervation, declining rapidly within the first two weeks and falling more gradually thereafter [306,307,308,309]. Mitochondrial dysfunction with upregulation of the miRNA, miR142a-5p and subsequent lysosomal degradation, accompany this muscle denervation atrophy [309]. Nonetheless, the fibres survive with 98% reduction in their size after two years of chronic denervation [330]. They also conserve their fascicular arrangement and their functionally important proteins, including acetylcholine receptors and neural-cell adhesion molecules [330]. Although the few freshly axotomized motoneurons (~5% of the normal number) that regenerated their axons through chronically denervated distal nerve stumps, did reinnervate chronically denervated muscle fibres, the reinnervated muscle fibres failed to recover their former size [312]. We suggested that this incomplete recovery was the result of declining numbers of their resident satellite cells able to provide nuclei to the fibres [312]. This was demonstrated in morphological studies [331,332] and later evidence of apoptosis of the cells [332]. The satellite cells are a population of heterogenous stem cells that lie between the sarcolemma of the myofibres and the ensheathing basal lamina [333]. Normally, they are mitotically quiescent and express the paired box protein, Pax7 [334,335,336]. They are activated upon nerve and/or muscle injury [333]. They divide by asymmetrical division to produce myogenic progenitor cells, and by symmetrical division, they produce multiple satellite cells that contribute nuclei to the atrophic muscle fibres driving reinnervated muscle fibres to increase their size [334,335,336]. As the activated satellite cells differentiate into myocytes, they co-express Pax7 and the muscle-specific basic helix-loop-helix protein, MyoD (myoblast determination protein1) of the MRF (myogenic regulatory factor) family of transcription factors [334,335,336]. A linear regression line was fitted to the decline in the numbers of MyD positive cells in chronically denervated biceps muscle of patients as a function of the time to the surgical repair of their brachial plexus injury [337]. Examination of their data, however, indicates that the data are more readily fitted by an exponential rather than a linear decline with the MyD expressing cells declining to a low plateau rather than to zero. Earlier morphological studies of chronically denervated frog muscles, suggested that the proliferative capacity of the satellite cell pool is exhaustible [338] and evidence of repeated cycles of muscle fibre degeneration and regeneration [339] indicated but did not prove that the ultimate fate of chronically denervated muscle fibres is their replacement by fatty and connective tissues [340]. The question remains as to whether the explanation for incomplete recovery of reinnervated muscle fibre diameters after chronic denervation, is that the satellite population of the denervated muscles is depleted with time and/or that the cells fail to divide and express MyD.
3b. Transient Expression of Regeneration Associated Genes
We addressed the question of why the regenerative capacity of the motoneurons and the growth support of the SCs decline with time and distance. We reasoned that elevation of RAG expression by axotomized motoneurons and/or denervated SCs declines with time after chronic nerve injury, a hypothesis that proved to be correct. The parallel decline in expression of RAGs that likely accounts at least in part, for poor functional recovery, is illustrated by the exponential decline of the expression of tubulin in chronically axotomized motoneurons (Figure 5), and of c-jun (Figure 6), p75 (Figure 6,Figure 7), and GNDF (Figure 7), in chronically denervated SCs.
Chronic axotomy After section and ligation of the sciatic nerve in rats (Figure 2A,B and A), the mRNA of cytoskeletal proteins, tubulin and actin, and of GAP-43, was detected and quantified in the axotomized sciatic motoneurons by in situ hybridization [341]. The mRNA levels in the chronically axotomized motoneurons were expressed relative to the high levels of the proteins that were detected at a week after axotomy of the contralateral sciatic nerve (Figure 5B-J) They declined exponentially to the low levels of intact motoneurons by 6 months (Figure 5B-G). Yet, a refreshment injury of a second sciatic nerve section after 1,3,4, and 6 months of chronic axotomy resulted in upregulation of the genes to the high levels detected after the first axotomy (Figure 5D,H-J). This elevation however, decayed more rapidly than after the first axotomy of the motoneurons (Figure 5D). Similar findings were reported for facial motoneurons [342].
Chronic denervation Gene expression and protein translation in denervated SCs also declines progressively after chronic denervation of the distal nerve stump [16,253,342,343,344]. The transcription factor c-Jun which is central to reprograming SCs to the repair phenotype and is the major determinant of effective repair [215], is upregulated in mouse denervated SCs within a week of injury but, the protein levels decline progressively after chronic denervation (Figure 6A,B; [344]). The location of c-Jun in the denervated SCs was verified using mice in which c-Jun was inactivated selectively in SCs in c-Jun knockout mice (Figure 6C,D; [215]). The p75 receptor (p75NTR) protein declined with time in chronically denervated nerves (Figure 6E in mice and Figure 7C,D. in rats; [253]) in parallel with the decline in its mRNA (Figure 7B; [253]). These in vivo findings were replicated in SC cultures where both c-Jun and p75NTR proteins were reduced in the 9th passage of the SCs (Figure 6F-H; [344]). Downstream of c-Jun, the expression of GDNF in the chronically denervated nerve stumps declines exponentially (Figure 7E-G; [339]). The decline is specific for the neurotrophic factors and not other proteins in the distal stump, including NTN (Neurturin), PSP (Persiphin), and ART (Artemin) (Figure 7E-F). The transient nature of gene expression of neurotrophins and their receptors in motoneurons is summarized in Figure 2 of the extensive review of Boyd and Gordon [16].
Shh, a gene that is not expressed in either developing SCs or in intact nerves, is upregulated strongly in SCs after injury [214,345,346,347] and improves nerve regeneration in several settings [347,348,349,350]. Applied Shh elevated c-Jun in cultured SCs and the expression of both Shh and c-Jun declined during chronic denervation [344]. Furthermore, the activation of c-Jun is reduced in the SCs of injured nerves together with reduced expression of its target p75NTR when the Shh gene was knocked out conditionally in the SCs [344]. Inhibiting Shh signaling also reduces SC expression of BDNF, motoneuron survival after injury, and axon regeneration [345,347].
The identified gene set of Aqp5, Gpr37L1, Igfbp2, and Olig1, regulated by c-Jun in injured nerves [214], is highly enriched amongst the genes affected by chronic denervation as well as in aging and it is positively correlated with both c-Jun levels and regeneration [344]. Igfbp2 for example, promotes phosphorylation of Akt, a pathway that is linked to SC proliferation and differentiation (reviewed in several publications [215,351,353]). Gpr37L1 is a receptor for prosaposin and prosapeptide [353] that are secreted after nerve injury and facilitate regeneration [354]. In experiments on chronic denervation, this gene group encompasses Cxcl5, Egfl8, Gas2I3, Megf10, and Pcdh20, all of which are upregulated in SCs after injury ([355,356,357] reviewed by [358,359]). Cxcl5 activates STAT3 [355], the transcription factor shown to be important for maintaining repair cells during chronic denervation [302]. Gas2I3 has a role in the cell cycle, and Megf10 in phagocytosis [360,361].
4. Neurotrophic Factors to Sustain Their Levels
4a. Exogenous Application of Neurotrophic Factors
The demonstration of dramatic neurite outgrowth from sympathetic and DRG (dorsal root ganglion) neurons in response to NGF [362]) resulted in many in vitro and in vivo studies which advocated that growth factors, including BDNF and GDNF, promote nerve regeneration [62,67,248,301,320]. Prior to 2000, the evaluations of the effects of exogenous neurotrophic factors on peripheral nerve regeneration relied on several outcome measures. These included axon counts distal to the site of injury, the ‘pinch test’ that evaluates sensory nerve regeneration in animals by identifying the most distal point of the regenerating nerves at which a nerve pinch with fine forceps elicits an intake of breath in the lightly anesthetized animal [84,363,364,365,366,367,368,369], and walking track analysis to evaluate motor nerve regeneration [15,370]. While these evaluations may have clinical relevance, their relevance for nerve regeneration is confounded by their failure to consider the outgrowth of several axonal sprouts from the proximal stump of injured nerves in both the PNS [15,67,261,262,372] and the CNS [371]. As described in 2e. Axonal regeneration, each nerve fibre in the proximal stump regenerates an average of 5 or more axons into the denervated distal nerve stump [261,262,372]. As a result, the effects of neurotrophic factors on the numbers of axotomized neurons that (1) regenerate their axons, (2) reinnervate denervated targets, and (3) result in functional recovery, have been overestimated. This led us to adopt our retrograde labelling technique to examine how many neurons regenerate their axons into the distal nerve stump in response to exogenous application of neurotrophic factors in addition to counting the number of regenerated axons in the distal stump.
BDNF and/or GDNF When an Alzet mini-osmotic pump was implanted to deliver BDNF and/or GDNF to the site of TIB-CP nerve cross-suture in rats, either immediately or after a 2 month period of chronic axotomy of TIB neurons, retrograde labeling revealed that the reduced numbers of chronically axotomized TIB motoneurons regenerating their axons into the freshly denervated CP distal nerve stump were elevated significantly by the exogenous growth factors (Figure 8A-E; [314]). BDNF was effective only in low doses and not at high doses due to the binding of this neurotrophic factor to both the trkB and p75NTR receptors on the motor nerve membranes, the former receptor mediating a positive effect on nerve regeneration and the latter, an inhibitory effect. Further details of the receptors and the dose-dependent effects of BDNF can be found elsewhere [313]. GDNF on the other hand, was effective at all doses, acting via two receptors ret and GFR-α1, with only positive effects [314]. GDNF administration to the cross-sutured TIB-CP nerves after immediate repair did not promote nerve regeneration as it did after prolonged axotomy [314]. However, a 2- and/or 4- week delivery of GDNF encased in polymeric microspheres and placed on the suture sites of a 10 mm long ANA (acellular nerve graft) between CP nerve stumps (Figure 8F), was effective in elevating the numbers of CP motor and sensory neurons that regenerated their axons to normal levels by 8 weeks (Figure 8H,I; [373]).
Unfortunately, the regeneration enhancing effects of GDNF and BDNF were confounded by increased axonal sprouting from the site of the TIB-CP coaptation [314]. Transmitting electron micrographs of SCs and their regenerated axons in the distal nerve stump revealed a regenerated SC unit of a SC with four daughter axons when saline was administered to the suture site (Figure 8I-B) rather than the one-to-one relationship of SCs and myelinated axons in the intact nerve (Figure 8I-A; [314]). BDNF alone increased the number of daughter axons (Figure 8I-C) and the combination of BDNF and GDNF increased the number even more (Figure 8I-D). The numbers increased to as high as nine axons in each endothelial sheath (Figure 8K-A,B,C,D), This increased axonal outgrowth led us to consider increasing endogenous rather than exogenous neurotrophic factors to promote nerve regeneration.
4b. Endogenous Neurotrophic Factors
The slow upregulation of neurotrophins and cytoskeletal proteins after axotomy, described above in Section 2a. Chromatolysis and gene expression in axotomized neurons, was dramatically accelerated and amplified by a 1-hour period of low frequency (20Hz) electrical stimulation (ES; Figure 9A-G [86]). The accelerated RAG upregulation of tubulin and GAP-43 (Figure 9F,G) and the decline in expression of neurofilament (Figure 9H) are preceded by the striking accelerated expression of BDNF and its trkB receptor (Figure 9D,E). That ES increases and accelerates RAG expression after elevating BDNF and trkB expression, suggests causation between neurotrophic factor upregulation, RAG expression, and accelerated nerve regeneration as discussed below in Section 5.
5. Neuronal Activity and Nerve Regeneration
5a. Reduced Activity and Synapse Withdrawal
We addressed the question of whether the decline in regenerative capacity of chronically axotomized neurons over time/distance (described above in Section POOR RECOVERY OF FUNCTION AFTER PERIPHERAL NERVE INJURY AND REPAIR) is due to reduced neuronal activity and, in turn, to their reduced interaction with denervated SCs in the distal nerve stump.
When hindlimb motor and sensory neural activity after ligation, nerve-nerve or nerve-muscle anastomoses of transected sciatic nerve branches, was recorded on implanted electrodes during cat treadmill locomotion and separated by cross-correlation (Figure 10A,B; [374]), the motor potentials were found to decline within the first month and the sensory potentials to decline to levels below resolution (Figure 10B; [374]). The latter decline was due to the disconnection of the sensory neurons from their sense organs. The reported synaptic depression in axotomized motoneurons [375,376,377,378,379] and the decline in their motor activity levels were accounted for by the withdrawal of synapses from the neurons, the excitatory VGLUT1 glutamatergic synapses declining by ~50% and the coverage by inhibitory GAD67 GABAergic synapses declining by ~35% (Figure 10C-F; [380,381,382,383,384,385,386,387,388]). The activation of astrocytes after motoneuron axotomy likely is responsible, at least in part, for the displacement of the synaptic boutons [381,389]. The loss of synapses was reversed by daily treadmill exercise implemented 3 days after nerve transection and surgical repair, the reversal depending on slow, continuous exercise in males and interval exercise (short high-speed sprints followed by rest periods) in female mice (Figure 10G, H; [387,388]). The effectiveness of the exercise was reduced when the exercise program was delayed [388]. These findings elucidated (1) the profound effect of synapse withdrawal on neural activity during the period when the expression of RAGs, including the cytoskeletal proteins and neurotrophic factors, decline after axotomy and (2) how appropriate patterned treadmill exercise prevents this withdrawal. The exercise effect likely depends on BDNF because the synaptic contacts on intact motoneurons were reduced in conditional BDNF knock-out transgenic mice [390].
5b. Staggered Nerve Regeneration
In 1987, Brushart and Seiler [391] introduced the rat femoral nerve as a model to study peripheral nerve regeneration. It was used later in studies of Brushart and Gordon that used retrograde labeling of the motoneurons that regenerated their axons to demonstrate (1) ‘staggering’ of regenerating axons across the site of femoral nerve transection and microsurgical repair [24], and (2) preferential reinnervation of the quadiceps motor branch by motor nerves and of the cutaneous sensory branch by sensory nerves [24,26,392]. The staggering became evident when only ~220 of a total of 350 femoral motoneurons were found to regenerate axons the distance of 25 mm into the distal nerve stump (Figure 11A,B; [24]). At the accepted regeneration rate of 3 mm/day in animals [393], measured primarily with the pinch test (as described in 4a. Exogenous application of neurotrophic factors; [86,363,364,365,366,367,368,369], and including estimates of latencies of ~1-2 days prior to axonal outgrowth [81,381,382,383,384,385,386,387,388,389], all 350 motoneurons would have been expected to regenerate their axons over the distance of 25 mm. Yet, the time for all the axotomized femoral motoneurons to regenerate their axons to the site of application of the fluorescent dye(s) was 8-10 weeks (Figure 11B; [24]) The regeneration rates have been measured with the widely used pinch test in animals [86,363,364,365,366,367,368,369].
Our explanation for the protracted period of regeneration after nerve transection and repair was that growth cones ‘stagger’ across the suture site prior to entering into the denervated nerve stump [24]. That longitudinal alignment of laminin and the SCs across the suture site was observed ~10 days after nerve transection and surgical repair (Figure 11C; [394]) supported this explanation. More directly, the results of experiments in which the retrograde dye FR was applied just distal to the suture to determine when regenerating axons cross the suture site (Figure 11D,E; [25]) confirmed the explanation: the numbers of the motoneurons regenerating their axons just distal to the surgical repair site was just a few 4 days after the repair, the remaining motoneurons regenerating their axons over the protracted period of 4 weeks (Figure 11F). Our silver-staining of staggered regeneration of axons in the distal stump and the tortuous paths taken by the axons within the proximal stump (Figure 11G,H) are in accord with Cajal’s findings [1] and with later observations of the outgrowth of fluorescent axons from the proximal stump of thy-1-YFP transgenic mice into a denervated distal nerve stump of a non-transgenic wild-type litter mate [34,66].
5c. Low Frequency Electrical Stimulation Accelerates Axon Outgrowth
Continuous 20 Hz ES accelerated rat soleus muscle reinnervation [23]) suggesting but not proving, that the ES accelerated nerve regeneration. Alternatively, the finding could be explained by the ES accelerating the formation of functional connections between regenerated soleus nerve fibres and the denervated soleus muscle fibres rather than by accelerating nerve regeneration. A similar explanation might suffice for the accelerated appearance of the plantar extensor reflex after sciatic nerve crush injury and applied brief {(15–60 min) 20-Hz ES [29]}. To address whether ES accelerates nerve regeneration and not simply the formation of target contacts, we implanted a stimulator to deliver continuous 20 Hz ES to axotomized femoral motoneurons in adults and retrogradely labeled them to count how many had regenerated their axons (Figure 12A-C; [24]). The 20 Hz-frequency of ES was chosen because it is the mean frequency of action potential generation in motoneurons [395].
Continuous 20 Hz ES delivered for two weeks promoted a striking acceleration of femoral nerve regeneration such that all the axotomized motoneurons regenerated their axons 25 mm from the site of transection and repair at three weeks as compared to 10 weeks after sham ES (the stimulator implanted but not turned on; Figure 12A-G; [24]). Initially, the femoral motoneurons regenerated their axons randomly into their motor and sensory branches. Thereafter, the regeneration was specific: the numbers of femoral motoneurons regenerating their axons into the inappropriate sensory nerve branch remained constant whilst the numbers of the motoneurons regenerating their axons specifically into the appropriate motor nerve branch increased steadily to their maximum, accounting for the steady increase in the total numbers over 10 and 3 weeks for the femoral nerves after sham and ES, respectively (Figure 12D-F). The data for the 2-week femoral nerve ES was not significantly different from the data obtained when the duration of the ES was progressively shortened from two weeks to one hour [24]. Hence, the data shown in Figure 12D-G,J and K are those obtained for all the stimulated femoral nerves irrespective of the ES durations. These data were used for direct comparison with the corresponding data for sham ES in Figure 12D,E.
The efficacy of ES in promoting nerve regeneration was attributed to acceleration of axonal outgrowth and not to an increase in the regeneration rate because (1) ES did not change the rate of slow axonal transport that concurs with regeneration rate, and (2) ES accelerated the regeneration of axons across the suture site, elucidated by injecting fluororuby (another name for FR) into the femoral nerve 1.5 mm distal to the suture line (Figure 12I; [25]). This effect of ES accelerating axon outgrowth only contrasts with that of a conditioning lesion that accelerates both axonal outgrowth and regeneration rate as discussed below in 5f. Conditioning lesion and conditioning ES. The accelerated axonal outgrowth, illustrated in the cartoon in Figure 12L, is mediated by action potentials that are conducted to the neuronal somas [24,27]. This was demonstrated in experiments in which local application of tetrodotoxin proximal to the site of ES that blocked action potential propagation to the soma of the axotomized motoneurons, eliminated the ES effect of accelerating motor and sensory nerve growth into their appropriate nerve branches (Figure 12N,O). Although the sensory neurons did not show a preference for the cutaneous sensory branch of the femoral nerve in the experiments of Geremia et al. [27], significant preference was demonstrated by those of Brushart et al. [26].
We demonstrated that the BNDF that was upregulated with its receptor trkB by ES (Figure 9D,E; [86]) is essential for the efficacy of the ES in accelerating nerve regeneration. A 3-day infusion of a BDNF antibody into the spinal fluid prior to ES and surgical repair of a transected femoral nerve (Figure 13A), followed by retrograde labelling of the branches of the nerve with FR and FG (Figure 13B), completely blocked the ES effect of accelerating specific regeneration of motor axons into the appropriate quadriceps motor branch: the number of motoneurons that regenerated their axons into the motor branch was not elevated three weeks after surgical repair, ES, and administration of the BDNF antibody (Figure 13C). The same efficacy of ES on nerve regeneration was associated with BDNF or NT-4/5 upregulation in thy-1- YFP-H transgenic mice where the regenerating CP axons, a subset of highly visible YFP-positive axons, were visualized in a wild-type mouse nerve graft [31,34,66] that provided little or no upregulated sources of the neurotrophic factors in denervated SCs [244,396] or in denervated muscle [397].
The positive effects of ES on peripheral nerve regeneration in animals reported by Brushart, Gordon, and colleagues in the 2000’s [24,25,26,27,28] have now been replicated many times [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56] and reviewed by Gordon et al. [5,6,17,62,63,64,65,66,67,69,70,248,301,319,320,322] and more recently by Zuo et al [70] and Juckett et al. [398]. The two recent reviews outline the origins of this century’s studies of ES with the 1900 studies of enhanced neurite and nerve outgrowth in response to electrical fields. Now, the same 1-hour ES demonstrated in animal studies to promote nerve regeneration after nerve transection and surgical repair, have been shown to be effective in promoting nerve regeneration and target reinnervation in human subjects [399,400,401].
The 1-hour ES was also shown to accelerate nerve regeneration through a nerve isograft of 10 mm in Lewis rats [52], and through nerve autografts of 10 and 20 mm in Sprague Dawley rats [53]. Macrophages and neutrophils normally accumulate in denervated distal nerve stumps after ES, the ES of the proximal nerve stump increasing the numbers of M2 macrophages within the autografts [52]. Similarly, ES shifted the macrophage phenotype in a locally demyelinated peripheral nerve, from the proinflammatory M1 toward the predominantly pro-repair M2 type [402]. ES also accelerates Wallerian degeneration, promoting nerve degeneration and clearance of the axons and their myelin, and upregulating BDNF and NGF in the denervated SCs [403].
The efficacy in promoting muscle reinnervation and functional recovery, of a 1½ hour period of ES, repeated daily for one to 6-8 weeks after nerve transection and surgical repair [28,54,404,405] or after sciatic nerve isograft repair [55], was in accord with the efficacy of the 2-week period of continuous ES and the 1 hour ES in accelerating motor nerve regeneration, muscle reinnervation and functional recovery [24,28,33]. Transcutaneous ES, however, was less effective in promoting nerve regeneration than direct ES of a transected nerve [406]. A 20-minute period of continuous ES increased numbers of sciatic neurons regenerating axons through a 5-mm long hollow conduit after delayed repair [50] but, the increase was only 20% and 10% of the numbers of stimulated motoneurons and sensory neurons, respectively, that regenerated their axons after delayed cross-suture of CP and TIB nerve stumps [28]. Also, the reported effective 10-minute protocol of 20 Hz ES [406,407,408] was not supported by a study that used several measures of functional recovery to show that this 10-minute protocol did not promote significant recovery after a sciatic nerve isograft repair [409]. Bioluminescent optogenetics were used recently to excite mouse sciatic neurons and to demonstrate acceleration of sciatic nerve regeneration and muscle reinnervation after transection and resuture [410]. The neurons were excited with an intraperitoneal injection of a luciferase substrate, coelenterase (CTZ), two weeks after injection of a viral vector encoding an excitatory luminopsin, lLMO3 into the sciatic nerve [410]. The ILMOS injection to excite CP neurons was also effective in accelerating gastrocnemius muscle reinnervation a 1-month after chronic axotomy of CP motoneurons and cross-suture of the CP proximal nerve stump to the freshly denervated TIB distal nerve stump [410].
It is important to note that daily administration of the androgen, testosterone, alone or together with daily ES after crush injury in castrated male rats, promotes facial nerve regeneration and functional recovery of both whisking and blink reflexes [39,42]. The beneficial effect of the testosterone-ES combination elicited a more rapid and sustained upregulation of BDNF than either alone [42]. The effects of both ES and exercise (see 5f. Exercise and axonal regeneration) increasing the length of regenerating axons, were blocked by treating both male and female mice with the androgen receptor blocker flutamide [411]. While androgen receptors are found in all motoneurons [412] and on astrocytes and microglia [413], it is not clear whether the release of BDNF from reactive glial cells after nerve injury, including spinal cord injury, assists or hinders nerve regeneration after injury [414].
5d. Preferential Reinnervation and Growth Factors
The preferential upregulation of the growth factors, PTN (pleiotrophin) and GDNF in denervated SCs (Figure 15C; [245,247]) accounts for the preferential growth of regenerating femoral motor nerve fibres into their appropriate motor nerve branch (Figure 14B,D; [415]). The expression of both growth factors is on the rise by 4 days after denervation at the time when few motoneurons that regenerated their axons across the surgical site after repair, and had randomly reinnervated both appropriate and inappropriate branches (Figure 11F and Figure 14A-C). The upregulation of both growth factors is maximal in the motor SCs at 14 days (Figure 14C) when ~70% of the femoral motoneurons had regenerated their axons across the suture line (Figure 14A,D) and, by 4 weeks, the axons grow preferentially into the endoneurial tubes that previously surrounded only motor nerve fibres (Figure 14B,D). When the remaining motoneurons regenerate their axons across the repair site by 28 days, the axons continue to enter the endoneurial ‘motor’ tubes preferentially (see the purple interrupted line in Figure 14D) because their specific growth factors likely remain in the SCs lining these tubes even though their expression is fading (Figure 14C). A similar explanation of the preferential reinnervation of sensory pathways by sensory neurons has been elaborated (data not shown but see [415]). Studies of the growth of motor axons into femoral nerve branches in NCAM (neural cell adhesion molecule) knockout mice (NCAM-/-) demonstrated that N-CAM was required for preferential reinnervation of the motor branch [416]. Moreover, the preferential reinnervation was not observed when the polysialic acid (PSA) moiety was removed enzymatically. This indicated that regenerating motor axons must express polysialylated NCAM that reduces axon-axon adhesion and that enables the axons to grow selectively into the appropriate motor pathway [416].
5e. ES Promotes Axon Outgrowth after Delayed Surgery
The regression of the regenerative capacity of chronically axotomized neurons and the regenerative support of chronically denervated SCs with the concomitant regression of expression of GAGs in both the neurons and SCs with time, described above in 3b. Transient expression of regeneration associated genes, begged the question of whether ES of the proximal stump of a chronically axotomized transected nerve could counteract these negative effects of delayed surgical repair of injured nerves. We addressed this question in experiments in which we systematically evaluated the effects of chronic motor and sensory neuronal axotomy and/or the chronic denervation of the distal nerve stump on the ability of ES to accelerate nerve regeneration after nerve transection and surgical repair (Figure 15, [28]). The experimental paradigm of chronic axotomy and/or chronic denervation of hindlimb nerve in rats (Figure 15A-D) was designed to address this question and the data collected demonstrated that, indeed, ES was able to counteract the effects of delayed surgery [28]. After ligating either the CP proximal nerve stump and/or the distal TIB stump for two months prior to cross-suture repair, the numbers of motor and sensory neurons that regenerated their axons was elevated to the same levels as after immediate cross-suture (Figure 15B,C; [28]). The ES increased both the number of regenerated nerves and those that reinnervated muscle (Figure 15B,C, the lowest two histograms), demonstrating that ES of the chronically injured nerves accelerated both nerve regeneration and target reinnervation. It is important to note that the surgical release of the CP proximal stump for the cross-suture repair constituted a second injury. The resulting RAG upregulation [337,338] likely promotes nerve regeneration after chronic injuries despite the more rapid decline in expression after the second injury as compared to the first. This is illustrated in Figure 5D where the decay in the expression of α-tubulin mRNA in axotomized sciatic motoneurons from 7d to 6 months was shown to be reversed by a second injury. The effectiveness of ES in promoting nerve regeneration after chronic denervation of the distal nerve stump (Figure 15B,C) is likely consequent to the release of neuregulin from the stimulated proximal nerve stump.
5f. Exercise and Axonal Regeneration
The accelerated rise in BDNF and trkB expression in axotomized motoneurons in response to brief ES (Figure 9D,E; [86]) or exercise [417,418,419] as well as the elevated NT/5 expression by ES [34], was the rationale for investigating whether exercise might also promote nerve regeneration as did ES [420,421,422,423]. In 2009, it was reported that daily exercise, like ES, accelerates muscle reinnervation with the combination of exercise and ES being the most effective during the early phase of nerve regeneration [421]. Extending these studies, English and colleagues reported that a 2-week period of moderate daily exercise 3 days after surgical repair promoted axon outgrowth that was significantly greater than that after brief, low frequency ES [420,423,424]. This efficacy of exercise confirmed previous findings of enhanced axon regeneration of both motor and sensory neurons using several different exercise protocols [30,425,426,427,428,429,430,431,432].
The exercise effect and that of ES also involves androgens in addition to BDNF. That androgen receptor signalling regulates expression of BDNF mRNAs [433] spurred the laboratories of Jones and of English to investigate the effects of daily administration of testosterone on nerve regeneration and the combination of testosterone and daily ES and to compare the effects of either testosterone or ES and both testosterone and ES together [39,42,411]. Exogenous testosterone or ES was effective in promoting the regeneration of facial nerve and the functional recovery of the whiskers [39,42]. It was the combination of both testosterone and ES that had the most prolonged and the maximum effect [42]. English and colleagues demonstrated that the androgen blocker, flutamide, eliminated both the ES and the exercise effects on regeneration after nerve transection and surgical repair [411]. The findings are consistent with the regulation of the expression of BDNF mRNAs by androgens [423].
The effect of exercise on the regeneration of the femoral nerve after surgical reunion of the transected nerve stumps has not evaluated to this author’s knowledge. As described above, preferential reinnervation of the quadriceps motor branch by the regenerating motor nerves does occur with time after the surgery and this reinnervation is accelerated by low frequency ES [24,379,434]. However, after transection and microsurgical repair of the sciatic nerve, the reinnervation of the distal stump is random [435] and ES exacerbated misdirection of regenerating nerve fibres with reduced TIB nerve contribution to the innervation of the triceps surae muscles [435]. Also, the more rostral and normal position of CP motoneurons was shifted to a more caudal position after sciatic nerve transection and repair, a shift that was exacerbated when the transected sciatic nerve was subjected to ES [436], but not when the rat was subjected to daily exercise on a treadmill [437]. However, irrespective of whether the sciatic nerve was subjected to ES, or the rats were exercised daily, or whether the misdirection of the regenerating axons was reduced by transecting and surgically repairing the CP and TIB nerve branches of the sciatic nerve, the normal reciprocal activation of flexor and extensor muscles was irretrievably altered by coactivation of the antagonistic muscles [438,439,440]. This misdirection and abnormal activation of antagonistic muscles remains a problem for functional recovery after nerve repairs that are performed on large nerves, despite the use of physiotherapy to improve movement. One study of the misdirection of regenerating axons after sciatic nerve injury in rats, reported that 71% of regenerating CP motoneurons were directed correctly into two muscles after crush injury, 42% after autograft repair, and 25% after autograft repair but recovery of ankle motion and balance was incomplete in all cases [441].
5g. Conditioning Lesion and Conditioning ES
There is now the relatively new approach of a CES (conditioning ES) paradigm to promote nerve regeneration and elevated expression of RAGs, even after chronic nerve injuries [442,443,444,445,446,447,448]. The CES is the 1-hour 20 Hz ES of an intact peripheral nerve prior to nerve transection and surgical repair, an approach that is promising and is being advocated for clinical use. The conditioning lesion (CL), a crush of an intact nerve 1-7 days prior to a more proximal nerve crush or transection injury, increases both the outgrowth of regenerating axons and their regeneration rate [442,449,450,451,452,453,454,455,456] in association with elevated levels of cAMP [457]. The rate of the slow component of axonal transport that largely governs regeneration rate, is accelerated by the CL with increased transport and amount of SCb proteins that include tubulin and actin [453,454,455,456]. Based on our finding that ES of the intact sciatic nerve performed 7 days prior to excision of DRGs, enhanced neurite outgrowth from the sensory neurons, akin to that seen after a CL (Figure 16; [458]), the effects of a CL and CES on how many and how far CP nerves regenerated their axons into a denervated nerve stump were compared in vivo [442,443,444,446]. These comparisons revealed the superiority the CES (referred to in their publications as functional ES) in (1) increasing the numbers of neurofilament-positive regenerating axons and how far these axons grew into the distal stump in the studies of crushed nerves [442,444] and transected and coapted nerves [444,446] after immediate [443], and delayed nerve repairs [448], (2) superior return of muscle and intraepidermal skin reinnervation [444,445,446], and (3) superior recovery of motor and sensory function [444,445,446].
Senger et al. pointed out that CES could be applied in chronic nerve injuries which are common after major polytrauma that necessitate emergency management of life or limb threatening injuries. Indeed, with the exception of immediate primary repair of sharp injuries from knives or razors, most surgeries are delayed for three to six months [287,289]. Presumably, the application of CES relies on placing the stimulating electrodes on the nerve proximal to the injury and days prior to the surgical repair, a requirement of two consecutive surgeries. In addition, CES could be applied, if open surgery is not required to place stimulating electrodes before distal nerve transfer surgery, the transection of a ‘donor’ nerve branch of a redundant muscle and its suture to the distal end of a non-functional ‘recipient’ nerve to restore function [398,447]. The Oberlin’s transfer, as first described in 2004 [459], is the classical example. This transfer to restore elbow flexion is one of several nerve transfers [459,460,461,462,463,464,465]. It is the suture a transected ulnar nerve fascicle that had supplied the flexor carpi ulnaris muscle, to the distal stump of the musculocutaneous nerve branch to biceps brachii muscle.
Whether CES could be applied successfully in end-to-side neurorrhaphies remains to be determined. The end-to-side neurorrhaphy is where a denervated nerve stump is inserted into an intact nerve to ‘borrow’ innervation for a denervated muscle [326,462,463,464,465,466,467,468,469,470,471,472]. It is performed to, ‘protect’ denervated distal stumps and in turn, increase the numbers of motoneurons that regenerate their axons through them [471,472]. Serial in vivo imaging of the regeneration of facial nerve axons identified with GFP (green fluorescent protein) in Thy1-GFP rats [473], demonstrated enhanced nerve regeneration through a denervated cross-face nerve autograft that was ‘protected’ by adding end-to-side sensory nerves [469]. Would CES of the intact nerve accelerate the reinnervation of chronically denervated distal nerve stumps by the end-to-side neurorrhaphy and improve muscle reinnervation? The surgical approach of side-to side neurorrhaphy [474,475,476,477,478] with one or 3 nerve autografts placed as cross-bridges between an intact nerve and a chronically denervated distal nerve stump, significantly increased the numbers of axotomized motor and sensory neurons that regenerated their axons after delayed nerve repair as compared with the neurons regenerated through chronically denervated nerve stumps that were not ‘protected’ by cross-bridges from an adjacent intact nerve [477]. The question remains as to whether ES might accelerate axon outgrowth after the side-to side neurorrhaphy and in turn, promote target reinnervation, especially for the nerve injuries that are distant from their denervated target organs.
5g. Drug-Induced cAMP Elevation Mimics the Effect of Electrical Stimulation (ES) on Nerve Regeneration
ES at 20 Hz for 1-hour raises intracellular cAMP in DRG neurons to the same levels as a CL does (Figure 18B; [458]). If cAMP plays a role in mediating the effect of ES of accelerating axon outgrowth across a peripheral nerve transection and repair site, we reasoned that elevating cAMP by inhibiting PDE4 (phosphodiesterase-4), the main enzyme responsible for cAMP hydrolysis, would mimic ES in promoting axon outgrowth across the injury site (Figure 17B;[28]). Additional evidence that PDE4 in combination with dbcAMP administration increased regeneration of spinal cord axons through a SC graft placed within the cord [479], prompted our study. We administered rolipram, a specific PDE4 inhibitor [480], via a mini-osmotic pump to the site of CP transection and microsurgical repair in rats (Figure 17B). The CP motoneurons that regenerated their axons across and 10 mm distal to the surgical repair site, were counted after retrograde dye labeling (Figure 17C-E; [28]). In addition, the number of motoneurons that reinnervated the target TA muscle was calculated by counting the number of reinnervated MUs (MUNE, Figure 17E;[28]). The significant increase in all three of the outcome measures demonstrated that, indeed, the cAMP elevation mediated by PDE4 inhibition, was sufficient to mimic the positive effects of ES of accelerating nerve regeneration across the surgical site, through the denervated distal nerve stump, and reinnervating denervated muscle fibres. Simply elevating cAMP with forskolin that stimulates adenylate cyclase, was found previously to increase the rate of regeneration of transected sciatic nerve fibres [481].
5h. ES Promotes Sensory Nerve Outgrowth in the Central Nervous System
Filbin and colleagues had noted high levels of cAMP in neonatal CNS neurons and that these high levels were associated with the capacity of these neonatal CNS neurons in contrast to adult CNS neurons, to regenerate their axons after injury ([482; reviewed by [483]). Qui et al. [484] and Neumann et al [457] discovered that it was the elevated cAMP that accounted for the effectiveness of the CL of the sciatic nerve to promote regeneration of transected sensory nerve fibres in the CNS. Indeed, elevated cAMP mimicked the effect of a CL both in vivo and in vitro. Later it was shown that the transcription factor, CREB acts as the primary mediator of cAMP-induced transcription [120] via PKA and MEK/Erk pathways [122]. Neurotrophins also play a role, BDNF-mediated activation of Erk producing a transient inhibition of PDE4 (phosphodiesterase-4), the main enzyme responsible for cAMP hydrolysis. Thereby, the neurotrophins promote the elevated intracellular cAMP [485]. That a CL of peripheral sensory nerve fibres promotes regeneration of their axons in the CNS [457,486] prompted us to ask whether ES of the intact sciatic nerve containing these nerves might also be effective in promoting their regeneration in the CNS.
Because sensory neurons may fire at high frequencies, albeit transiently [395], we asked whether either or both 20 and 200 Hz ES promote outgrowth of the central axons of DRG neurons [458]. The central axons were transected at thoracic level 8 (T8) in the spinal cord (Figure 18A,B; [457]) and the sciatic nerve was subjected to a CL, ES at 20 Hz, or ES at 200 Hz, the 200 Hz frequency studied as a control because only 20 Hz ES raised cAMP levels to those after a CL (Figure 18B; [457]). The DRG sensory nerve fibres were labelled with cholera toxin B (CTB) 14 weeks after the spinal cord lesion to visualize the outgrowth of axons across the lesion site in vivo (Figure 18B; [457]). As shown previously [457,486], the CL was very effective in promoting the outgrowth of sensory axons across the lesion site (Figure 18D). The sprouted axons regenerated further into the spinal cord and more rapidly than the control axons at the T8 lesion site that received sham ES (cf Figure 18D and C). This finding is consistent with the efficacy of the CL accelerating axon outgrowth and elongation in the PNS [442,449,450,451,452,453,454,455,456]. The 20 Hz ES but not the 200 Hz ES of the sciatic nerve, promoted significant axon outgrowth across the T8 lesion site, the axons not elongating as much as the outgrowth after a CL (cf Figure 18E,D). This was consistent again with ES promoting outgrowth without increasing the overall rate of regeneration after peripheral nerve injury [24,25]. In the comparisons of mean values of the percent of the regenerated DRG central axons as a function of the distance from the T8 lesion site, the 20 Hz ES promoted axon outgrowth but not axon lengthening in the CNS in contrast to the CL that promotes both outgrowth and lengthening of the transected axons (Figure 18G-I).
The effect of the CL promoting central sensory axonal regeneration was related directly to the levels of intracellular levels of cAMP in the DRG neurons [457,486]. However, cAMP injection into the DRG in vivo or in vitro did not match the CL effect, the effect being smaller [457]. That the elevated cAMP, either by injection [487] or in response to ES [458], was insufficient to promote the lengthening of the axon sprouts across the lesion site, is consistent with findings that elevated cAMP does not promote elongation of transected DRG central axons into a peripheral nerve transplant [488]. Moreover, a CL and ES elevates cAMP to the same level (Figure 18B) but the CL promotes axonal outgrowth and lengthening [143,452,453,454]. This discrepancy made it likely that the CL effect may be mediated at least in part, by other mechanisms that include the contribution of non-neural cells. A possible molecular candidate is oncomodulin, the small calcium binding protein whose mRNA is highly expressed in neutrophils, the first responders of the innate immune system [489,490]. The neutrophils are recruited into the injury site of the eye within a day [491]. The oncomodulin binds to retinal ganglion cells with high affinity in a cAMP-dependent manner, increases the level of phosphorylated CREB (P-CREB), the activated form of the transcriptional activator, and stimulates the cells to regenerate long axons beyond the site of optic nerve injury [492]. The outgrowth was more extensive than other known trophic agents. Moreover, oncomodulin also acts upon peripheral sensory neurons to promote neurite outgrowth although neutrophils do not participate in the response of the neurons to injury [492].
5i. ES Signalling Pathways
Our findings to date of the effectiveness of ES in accelerating axon outgrowth and target reinnervation are summarized in a schematic representation of the intracellular signaling pathways activated by ES in Figure ES elevates intracellular cAMP that, via PKA, initiates transcription by CREB that, in turn, elevates expression of growth associated proteins, including neurotrophins and their trk receptors, and the cytoskeletal proteins, tubulin and actin. Thereby, ES promotes axon outgrowth after nerve injury to accelerate the regenerating nerves towards their denervated targets. The neurotrophins also feed back to amplify the cAMP pathway, in part by a transient inhibition of PDE4 activity that elevates cAMP to threshold levels and in turn, promotes axon outgrowth after immediate or delayed nerve repair of injured peripheral nerves. The positive effect of ES in promoting axonal regeneration after delayed nerve surgery, involving both chronic axotomy of motor and sensory neurons and the chronic denervation of the distal nerve stump is likely due to the release of mitogens such as neuregulin from the stimulated proximal nerve stump. These mitogens promote SC proliferation and migration, shifting the SCs from their atrophic state to a growth permissive state. The latter state includes the expression of neurotrophic factors and p75NTR that may present the factors to the regenerating axons to further enhance their growth. Further details of the pathways involved in the efficacy of ES in promoting nerve regeneration and target reinnervation await further experimentation.
5j. ES Accelerates Human Nerve Regeneration and Muscle Reinnervation
Having demonstrated the effect of ES accelerating axon outgrowth, and reinnervation of targets in rats, we asked whether ES promotes functional recovery after nerve injuries in patients. Could we accelerate target reinnervation? In patients with carpal tunnel syndrome (CTS), the number of functionally intact MUs, MUNE (MU number estimation), was determined after ascertaining that the syndrome was not the result of a conduction block at the wrist but rather the result of the loss of functional MUs [399]. We established that the CTS was due to MU loss by stimulating the medial nerve above and below the carpal tunnel (Figure 20A-C) and ascertaining that the size of the CMAP (compound motor action potential) in response to maximal stimulation of the median nerve above the tunnel equaled the CMAP recorded in response to stimulation of the nerve below the carpal tunnel (Figure 20B,C). We recruited only those patients with severe chronic CTS for the study of the effects of ES on the reinnervation of the partially denervated median eminence musculature after the CTRS (carpal tunnel release surgery) of cutting the ligament overlying the carpal tunnel (Figure 20D). Prior to the surgery, the number of intact MUs was estimated by electrically stimulating the median nerve maximally to evoke the CMAP in the muscle and then evoking at least 20 all-or-none MUAPs (MU action potentials) in response to electrical stimuli to the median nerve as the stimulator was moved progressively along the course of the nerve from the wrist up to the upper arm (Figure 20E). Thereafter, the patients were divided into two groups, either receiving or not receiving 20 Hz ES for one hour immediately after CTRS surgery (Figure 20F). MUNE was determined in a similar fashion to that in the rats (Figure 1C) namely from the ratio of CMAP and mean MUAP amplitudes (Figure 20G).
The number of intact MUs averaged ~150 which was 50% of the ~300 intact MUs in individuals who did not suffer CTS (Figure 20H). The effect of the ES was dramatic especially when contrasted with the very slow progression of reinnervation of the partially denervated median eminence musculature by the median nerve in the control patients who did not receive ES. Although there was a trend for the number of reinnervated MUs to increase over a period of 12 months in the unstimulated control group of patients, the increase was not significant ((p>0.05). In contrast there was already a significant increase at 3 months (p<0.05) with the number reaching their normal values by 6-8 months (Figure 20H). These findings were particularly striking when the distance of regeneration from the site of crush injury to the musculature of 100 mm (see Figure 1A) is considered. With the reported regeneration rate of 1 mm/day [290,291], the expected time of reinnervation of the median eminence musculature would be ~ 3-4 months and yet there was no significant increase in MUNE in the patients whose median nerve was not subjected to ES. Thus, the accelerated reinnervation of the musculature was particularly striking with all the crushed medial motor nerves reinnervating the muscles within 6-8 months. The effect of the ES on axon outgrowth after CTRS is shown figuratively as the regenerating axons staggering across the crush site 3 months after CTRS and, at 6 months, failing to reinnervate the muscles of the median eminence without ES application (Figure 20I,K), and the staggering axons progressing further after ES and reinnervating all the denervated muscle fibres (Figure 20L,M).
Chan and his colleagues went on to demonstrate the significant acceleration of sensory nerve regeneration in human subjects after surgical repair of transected digital nerves in the hand [400] as well as after ulnar nerve compression at the elbow [401]. In summary, the demonstrated efficacy of low frequency ES to accelerate nerve regeneration in human subjects as well as in animals, indicates the potential for its utility in promoting successful outcomes after injuries in patients.
6. Conclusions
Neural activity in motor and sensory neurons declines rapidly during the first month of nerve injury. The decline in motoneurons is associated with and likely caused by the loss of both excitatory and inhibitory synapses on the motoneurons in the spinal cord. The decline in activity of sensory neurons to unmeasurable levels is due to the disconnection of the neurons from their sensory input from the skin and muscles. The progressive decline in regenerative capacity of axotomized neurons and the regenerative support of the denervated distal nerve stumps accounts for progressive decline in functional recovery with time and distance. The loss occurs concurrent with the progressive fall in the growth associated genes (GAGs) that are upregulated in both the axotomized neurons and the SCs in the denervated distal nerve stumps within the first week after nerve injury. Brief (1 hour) low frequency (20 Hz) electrical stimulation (ES), by promoting axon outgrowth, accelerates nerve regeneration and target reinnervation after both acute and chronic nerve injuries in animals and in humans. Recent studies demonstrate that the same brief 20 Hz ES applied prior to a nerve injury acts as a conditioning ES to promote both axon outgrowth as well as the rate of nerve regeneration. The effectiveness of the ES after acute injury is accounted for by (1) preventing the loss of synaptic contacts that occurs on axotomized motoneurons, (2) the upregulation of GAGs in response to increased intracellular cAMP, and (3) the feedback of expressed neurotrophins including BDNF, that amplify the expression of cAMP. After chronic nerve injuries where synaptic contacts on the motoneurons have already been withdrawn, the increase in cAMP and the feedback loops of the neurotrophic factors transiently inhibit cAMP breakdown. The elevated cAMP, via CREB and expression of GAGs, allows increased axonal outgrowth with resultant promotion of axonal regeneration and target reinnervation. The contribution of the release of mitogens from regenerating motor and sensory axons, including neuregulin, increases the proliferation of SCs and promotes their growth permissive state. This state includes the synthesis and release of neurotrophic factors that likely amplify the effects of the neuronal expression of the factors to, in turn, promote the synthesis of growth associated proteins, including the cytoskeletal proteins that are essential for axon outgrowth and nerve regeneration.
That brief low frequency ES is effective in accelerating axon outgrowth from the proximal stump of an injured nerve in animals and human patients indicates its potential for clinical application. The conditioning ES may be applicable to enhance the regenerative capacity in nerve transfer surgeries and end-to-side neurorrhaphies, should open surgery be unnecessary in applying the electrodes for the ES. Finally, our findings that delayed nerve repair is deleterious with respect to the prospects of functional recovery, emphasize the importance of ensuring minimal delay between the diagnosis of nerve injury and nerve repair and the application of brief ES. Thereby, we might anticipate improved outcomes of nerve surgeries in the future.
Acknowledgments
My sincere thanks to my many students and postdoctoral fellows, my collaborators, and my technologist, who worked with me in our research toward the ultimate goal of promoting nerve regeneration and functional recovery after peripheral nerve injuries in patients. Thanks too to the Canadian granting agents, the Canadian Institutes for Heath Research, the Muscular Dystrophy Association of Canada, and the Amyotrophic Lateral Sclerosis Society of Canada for their continued support of my research.
References
- Cajal, S.R. Degeneration and Regeneration of the Nervous System (translated by R.M. May). 1928, Oxford University Press, New York, Oxford.
- Sunderland, S. Nerve and Nerve Injuries. 1978, Livingstone, Edinburgh.
- Kline, D.G.; Hudson, A.R. Nerve Injuries: Operative Results for Major Nerve Injuries, Entrapments and Tumors. 1995, W.B. Saunders, Philadelphia, PA.
- Fenrich, K.; Gordon, T. Canadian Association of Neuroscience review: axonal regeneration in the peripheral and central nervous systems – current issues and advances. Can. J. Neurol. Sci. 2004, 31, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Nerve Repair. 2011, Oxford University Press, Oxford.
- Sulaiman, O.A.R.; Midha, R.; Gordon, T. Pathophysiology of surgical nerve disorders. In Youmans’ Neurological Surgery 6th ed., Part 2, 2011, 2368-Ed Winn HR. Philadelphia, PA.
- Ruijs, A.C.J.; Jaquet, J.-B.; Kalmign, S.; Giele, H.; Hovius, S.E.R. Median and ulnar nerve injuries: a meta-analysis of predictors of motor and sensory recovery after modern microsurgical nerve repair. Plast. Reconstr. Surg. 2005, 116, 484–94. [Google Scholar] [CrossRef] [PubMed]
- Scholz, T.; Krichevsky, A.; Sumarito, A.; Jaffurs, D.; Wirth, G.A.; Paydar, K.; Evans, G.R.D. Peripheral nerve injuries: An international survey of current treatments and future perspectives. J. Reconstr. Microsurg 2009, 25, 339–344. [Google Scholar] [CrossRef]
- Park, H.R.; Lee, G.S.; Kim, I.S.; Chang, J.-C. Brachial plexus injury in adults. The Nerve 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Smania, N.; Berto, G.; La Marchina, E.; Melotti, C.; Midiri, A.; Roncori, L.; Zenorini, A.; Ianes, P.; Picelli, A.; Waldner, A.; Faccioli, S.; Gandolfi, M. Rehabilitation of brachial plexus injuries in adults and children. Eur. J. Phys. Rehabil. Med. 2012, 48, 483–506. [Google Scholar]
- Tian, A.; Sachar, R.; Ray, W.Z.; Brogan, D.M.; Dy, C.J. Indirect cost of traumatic brachial plexus injuries in the United States. J. Bone Joint Surg. Am. 2019, 101, e80. [Google Scholar]
- Rosberg, H.E.; Carlsson, K.S.; Hojgard, S.; Lindgren, B.; Lundborg, G.; Dahlin, L.B. Injury to the human median and ulnar nerves in the forearm- analysis of costs for treatment and rehabilitation of 69 patients in southern Sweden. J Hand Surg (Edinburgh, Scotland) 2005, 30, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Bergmeister, K.D.; Groβe-Hartlage, L.; Daeschler, S.C.; et al. Acute and long-term costs of 268 peripheral nerve injuries in the upper extremity. Plos One 2020, 15, e0229530. [Google Scholar] [CrossRef]
- Jaquet, J.B.; Luijsterburg, A.J.; Kalmijn, S.; Kuypers, P.D.; Hofman, A.; Hovius, S.E. Median, ulnar, and combined medium-ulnar nerve injuries: Functional outcome and return to productivity. J. Trauma. 2001, 51, 687–692. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. The cellular and molecular basis of peripheral nerve regeneration. Mol. Neurobiol. 1997, 14, 67–116. [Google Scholar] [CrossRef]
- Boyd, J.G.; Gordon, T. Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury Mol. Neurobiol. 2003, 27, 277–324. [Google Scholar]
- Chan, K.M.; Gordon, T.; Zochodne, D.W.; Power, H.A. Improving peripheral nerve regeneration: From molecular mechanisms to potential therapeutic targets. Exp. Neurol. 2014, 261, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.J.; Coleman, M.P. Lessons from injury: How nerve injury studies reveal basic biological mechanisms and therapeutic opportunities for peripheral nerve diseases. Neurotherapeutics 2021, 2200–2221. [Google Scholar] [CrossRef] [PubMed]
- Daeschler, S.C.; Feinberg, K.; Harhaus, L.; Kneser, U.; Gordon, T.; Borschel, G.H. Advancing nerve regeneration: Translational perspectives of tacrolimus (FK506). Int. J. Mol. Sci. 2023, 24, 12771. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, E.L; Houghton, P.; Anthony, J.; Rennie, S.; Shay, B.L; Hoens, A.M. Neuromuscular electrical stimulation of muscle impairment: Critical review and recommendations for clinical practice. Physiother. Can. 2017, 69, 1–78. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Hsiao, H.-F.; Li, L.-F.; Chen, N.-H.; Huang, C.-C. Effects of electrical muscle stimulation in subjects undergoing prolonged mechanical ventilation. Respir. CRE 2019, 64, 262–271. [Google Scholar] [CrossRef]
- Huang, Y.; Gong, Y.; Liu, Y.; Lu, J. Global trends and hot topics in electrical stimulation of skeletal muscle research over the past decade: A bibliometric analysis. Frontiers Neurol. 2022, 13, 991099. [Google Scholar] [CrossRef]
- Nix, W.A.; Hopf, H.C. Electrical stimulation of regenerating nerve and its effect on motor recovery. Brain Res. 1983, 272, 21–25. [Google Scholar] [CrossRef]
- Al-Majed, A.A.; Neumann, C.M.; Brushart, T.M.; Gordon, T. Brief electrical stimulation promotes the speed and accuracy of motor axonal regeneration. J. Neurosci. 2000, 20, 2602–2608. [Google Scholar] [CrossRef]
- Brushart, T.M.; Hoffman, P.N.; Royall, R.M.; Murinson, B.B.; Witzel, C.; Gordon, T. Electrical stimulation promotes motoneuron regeneration without increasing its speed or conditioning the neuron. J. Neurosci. 2002, 22, 6631–6638. [Google Scholar] [CrossRef]
- Brushart, T.M.; Jari, R.; Verge, V.; Rohde, C.; Gordon, T. Electrical stimulation restores the specificity of sensory axon regeneration. Exp. Neurol. 2005, 194, 221–229. [Google Scholar] [CrossRef]
- Geremia, N.M.; Gordon, T.; Brushart, T.M.; Al-Majed, A.A.; Verge, V.M. Electrical stimulation promotes sensory neuron regeneration and growth-associated gene expression. Exp. Neurol. 2007, 205, 347–359. [Google Scholar] [CrossRef]
- Elzinga, K.; Tyreman, N.; Ladak, A.; Savaryn, B.; Olson, J.; Gordon, T. Brief electrical stimulation improves nerve regeneration after delayed repair in Sprague Dawley rats. Exp. Neurol. 2015, 269, 142–153. [Google Scholar] [CrossRef]
- Pockett, S.; Gavin, R.M. Acceleration of peripheral nerve regeneration after crush injury in rat. Neurosci. Lett. 1985, 59, 221–224. [Google Scholar] [CrossRef]
- Marqueste, T.; Alliez, J.R.; Alluin, O.; Jammes, Y.; Decherchi, P. Neuromuscular rehabilitation by treadmill running or electrical stimulation after peripheral nerve injury and repair. J. Appl. Physiol. 2004, 96, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- English, A.W. Enhancing axon regeneration in peripheral nerves also increases functionally inappropriate reinnervation of targets. J. Comp. Neurol. 2005, 490, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rouabhia, M.; Wang, Z.; Roberge, C.; Shi, G.; Roche, P.; Li, J.; Dao, L.H. Electrically conductive biodegradable polymer composite for nerve regeneration: electricity-stimulated neurite outgrowth and axon regeneration. Artificial Organs 2006, 31, 13–22. [Google Scholar] [CrossRef]
- Ahlborn, P.; Schachner, M.; Irintchev, A. One hour electrical stimulation accelerates functional recovery after femoral nerve repair. Exp. Neurol. 2007, 208, 137–144. [Google Scholar] [CrossRef] [PubMed]
- English, A.W.; Schwartz, G.; Meador, W.; Sabatier, M.J.; Mulligan, A. Electrical stimulation promotes peripheral axon regeneration by enhanced neuronal neurotrophin signaling. Dev. Neurobiol. 2007, 67, 18–172. [Google Scholar] [CrossRef] [PubMed]
- Hetzler, L.E.; Sharma, N.; Tanzer, L.; Wurter, R.D.; Leonetti, J.; Marco, S.J.; Jones, K.J.; Foecking, E.M. Accelerating functional recovery after rat facial nerve injury: effects of gonadal steroids and electrical stimulation. Otolaryng Head. Neck 2008, 139, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Hetzler, L.T.; Sharma, N.; Wurster, R.D.; Marzo, S.J. Jones, K.J.; Foecking, E.M. Electrical stimulation facilitates rat facial nerve recovery from a crush injury. Otolaryng Head. Neck 2008, 139, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vivó, M.; Puigdemasa, A.; Casals, L.; Asensio, E.; Udina, E.; Navarro, X. Immediate electrical stimulation enhances regeneration and reinnervation and modulates spinal plastic changes after sciatic nerve injury and repair. Exp. Neurol. 2008, 211, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Pinilla, E.; Udina, E.; Jaramillo, J.; Navarro, X. Electrical stimulation combined with exercise increase axonal regeneration after peripheral nerve injury. Exp. Neurol. 2009, 219, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Coughlin, L.; Porter, R.G.; Tanzer, L.; Wurster, R.D.; Marzo, S.J.; Jones, K.J.; Foecking, E.M. Effects of electrical stimulation and gonadal steroids on rat facial nerve regenerative properties. Restor. Neurol. Neurosci. 2009, 27, 633–644. [Google Scholar] [CrossRef]
- Alrashdan, M.S.; Park, J.C.; Sung, M.A.; Yoo, S.B.; Jahng, J.W.; Lee, T.H.; Kim, S.J; Lee, J.H. Thirty minutes of low intensity electrical stimulation promotes nerve regeneration after sciatic nerve crush injury in a rat model. Acta Neurol. Belg. 2010, 110, 168–179. [Google Scholar]
- Huang, J.; Lu, L.; Hu, X.; Ye, Z.; Peng, Y.; Yan, X.; Geng, D.; Luo, Z. Electrical stimulation accelerates motor functional recovery in the rat model of 15-mm sciatic nerve gap bridged by scaffolds with longitudinally oriented microchannels. Neurorehabil Neural Repair. 2010, 24, 736–745. [Google Scholar] [CrossRef]
- Sharma, N.; Marzo, S.J.; Jones, K.J.; Foecking, E.M. Electrical stimulation and testosterone differentially enhance expression of regeneration-associated genes. Exp. Neurol. 2010, 223, 183–191. [Google Scholar] [CrossRef]
- Wan, L.; Xia, R.; Ding, W. Short-term low-frequency electrical stimulation enhanced remyelination of injured peripheral nerves by inducing the promyelination effect of brain-derived neuro- trophic factor on Schwann cell polarization. J. Neurosci. Res. 2010, 88, 2578–2587. [Google Scholar] [CrossRef]
- Yeh, C.C.; Lin, Y.C.; Tsai, F.J.; Huang, C.Y.; Yao, C.H.; Chen, Y.S. Timing of applying electrical stimulation is an important factor deciding the success rate and maturity of regenerating rat sciatic nerves. Neurorehabil Neural Repair. 2010, 24, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Alrashdan, M.S.; Sung, M.A.; Kwon, Y.K.; Chung, H.J.; Kim, S.J.; Lee, J.H. Effects of combining electrical stimulation with BDNF gene transfer on the regeneration of crushed rat sciatic nerve. Acta Neurol (Wien) 2011, 153, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Han, S.J.; Shin, D.H.; Lee, W.S.; Choi, J.Y. Subthreshold continuous electrical stimulation facilitates functional recovery of facial nerve after crush injury in rabbit. Muscle Nerve 2011, 43, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Foecking, E.M.; Fargo, K.N.; Coughlin, L.M.; Kim, J.T.; Marzo, S.J.; Jones, K.J. Single session of brief electrical stimulation immediately following crush injury enhances functional recovery of rat facial nerve. J. Rehabil. Res. Dev. 2012, 49, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Xu, Q. G.; Franz, C. K.; Zhang, R.; Dalton, C.; Gordon, T.; Verge, V.M.K.; Midha, R.; Zochodne, D.W. Accelerated axon outgrowth, guidance, and target reinnervation across nerve transection gaps following a brief electrical stimulation paradigm. J. Neurosurg. 2012, 116, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Chang, R.L.; Chang, S.L.; Tsai, C.C.; Tsai, F.J.; Chen, Y.S. Electrical stimulation improves peripheral nerve regeneration in streptozotocin-induced diabetic rats. J. Trauma. Acute Care Surg. 2012, 72, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, Y.; Lu, L.; Hu, X.; Luo, Z. Electrical stimulation accelerates nerve regeneration and functional recovery in delayed peripheral nerve injury in rats Europ J. Neurosci. 2013, 38, 3691–3701. [Google Scholar]
- Koppes, A.N.; Nordberg, A.L.; Paolillo, G.M.; Goodsell, N.M.; Darwish, H.A.; Zhang, L.; Thompson, D.M. Electrical stimulation of Schwann cells promotes sustained increases in neurite outgrowth. Tiss. Eng. Pt. A 2014, 20, 494–506. [Google Scholar] [CrossRef]
- Keane, G.C.; Pan, D.; Roh, J.; Larson, E.L.; Schellhardt, L.; Hunter, D.A.; Snyder-Warwick, A.K.; Moore, A.M.; Mackinnon, S.E.; Wood, M.D. The effects of intraoperative electrical stimulation on regeneration and recovery after nerve isograft repair in a rat model. Hand 2020, 17, 540–548. [Google Scholar] [CrossRef]
- Zuo, K.J.; Shafa, G.; Antonyshyn, K.; Chan, K.; Gordon, T.; Borschel, G.H. A single session of brief electrical stimulation enhances axon regeneration through nerve autografts. Exp. Neurol. 2020, 323, 113074. [Google Scholar] [CrossRef]
- Javeed, S.; Faraji, A.J.; Dy, C.; Ray, W.Z.; MacEwan, M.R. Application of electrical stimulation for peripheral nerve regeneration: Stimulation parameters and future horizons. Interdiscipl Neurosurg. 2021, 24, 101117. [Google Scholar] [CrossRef]
- Birenbaum, N.K.; Yan, Y.; Odabas, A.; Chandra, N.S.; Ray, W.Z.; MacEwan, M.R. Multiple sessions of therapeutic electrical stimulation using implantable thin-film wireless nerve stimulators improve functional recovery after sciatic nerve isograft repair. Muscle & Nerve 2023, 67, 244–251. [Google Scholar]
- Keane, G.C.; Marsh, E.B.; Hunter, D.A.; Schelhardt, L.; Walker, E.R.; Wood, M.D. Lidocaine nerve block diminishes the effects of therapeutic electrical stimulation to enhance nerve regeneration in rats. Hand 2023, 18 (1_suppl), 119S–125S. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Voda, J.; Gold, B.G.; Gordon, T. FK506 increases peripheral nerve regeneration after chronic axotomy but not after chronic Schwann cell denervation. Exp. Neurol. 2002, 175, 127–137. [Google Scholar] [CrossRef]
- Tajdaran, K.; Shoichet, M.S.; Gordon, T.; Borschel, G.H. A novel polymeric drug delivery system for localized and sustained release of tacrolimus (FK506). Biotech. and Bioeng. 2015, 112, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Tajdaran, K.; Chan, K.; Zhang, J.; Gordon, T.; Borschel, G.H. Local FK506 dose-dependent study using a novel three-dimensional organotypic assay. Biotech. Bioengin 2019, 116, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Tajdaran, K.; Chan, K.; Shoichet, M.; Gordon, T.; Borschel, G.H. Local delivery of FK506 to injured peripheral nerve enhances axon regeneration after surgical nerve repair in rats. Acta Biomater 2019, 96, 211–221. [Google Scholar] [CrossRef]
- Zuo, K.J.; Shafa, G.; Chan, K.; Zhang, J.; Hawkins, C.; Tajdaran, K.; Gordon, T.; Borschel, G.H. Local FK506 drug delivery enhances nerve regeneration through fresh, unprocessed peripheral nerve allografts. Exp. Neurol. 2021, 341, 113680. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Sulaiman, O.A.R.; Boyd, J.G. Experimental strategies to promote functional recovery after peripheral nerve injuries. J. Periph Nerv. System 2003, 236–250. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Cellular and molecular interactions after peripheral and central nerve injury. Biomed. Rev. 2003, 14, 51–62. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Neurobiology of peripheral nerve regeneration and functional recovery: from bench top research to bedside application. Ochsner J. 2013, 13, 100–180. [Google Scholar]
- Gordon, T. Electrical stimulation to enhance axon regeneration after peripheral nerve injuries in animal models and humans. Neurotherapeutics 2016, 13, 295–310. [Google Scholar] [CrossRef]
- Gordon, T.; English, A.W. Strategies to promote functional recovery after nerve injury: Electrical stimulation and/or exercise. Eur. J. Neurosci. 2016, 43, 336–350. [Google Scholar] [CrossRef]
- Gordon, T. The neurotrophic factor rationale for using brief electrical stimulation to promote peripheral nerve regeneration in animal models and human patients. In “Conductive Polymers: Electrical interactions in Cell Biology and Medicine”: Eds Zhang, Z.; Rouabhia, M.; Moulton, S.E. CRC Press, New York, 2017, Ch 11, pp 231-249.
- Tajdaran, K.; Chan, K.; Gordon, T.; Borschel, G.H. Matrices, scaffolds, and carriers for protein and molecule delivery peripheral nerve regeneration. Exp. Neurol. 2019, 319, 112817. [Google Scholar] [CrossRef]
- Gordon, T. Peripheral nerve regeneration and muscle reinnervation Int. J. Mol. Sci. 2020, 21, 8652. [Google Scholar] [CrossRef]
- Zuo, K.J.; Gordon, T.; Chan, K.M.; Borschel, G.H. Electrical stimulation to enhance peripheral nerve regeneration: Update in molecular investigations and clinical translation. Exp. Neurol. 2020, 332, 113397. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J. Neurosci. 1995, 15, 3886–3895. [Google Scholar] [CrossRef]
- Lieberman, A.R. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 1971, 14, 49–124. [Google Scholar] [PubMed]
- Grafstein, B. The nerve cell body response to axotomy. Exp. Neurol. 1975, 48, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.E. Cellular responses to axotomy and to related procedures. Br. Med. Bull. 1974, 30, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. Dependence of peripheral nerves on their target organs, in Somatic and Autonomic Nerve-Muscle Interactions (Bumstock G., Vrbova G., and O'Brien R. A., eds.), Elsevier, New York, 1993, pp. 289–323.
- Patodia, S.; Raivich, G. Role of transcription factors in peripheral nerve regeneration. Frontiers Mol. Neurosci. 2012, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, W.; Bisby, M.A.; Kreutzberg, G.W. Changes in cytoskeletal proteins in the rat facial nucleus following axotomy. J. Neurosci. 1988, 8, 3181–3189. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.; Tetzlaff, W.; Bisby, M.; Fawcett, J.W.; Milner, R. Rapid induction of the major embryonic alpha-tubulin mRNA, T alpha 1, during nerve regeneration in adult rats. J. Neurosci. 1989, 9, 1452–1463. [Google Scholar] [CrossRef]
- Kato, S.; Ide, C. Axonal sprouting at the node of Ranvier of the peripheral nerve disconnected with the cell body. Restor. Neurol. Neurosci. 1994, 6, 181–187. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Thompson, G.W.; Griffen, J.W.; Price, D.L. Changes in neurofilament transport coincide temporally with alterations in the caliber of axons in regenerating motor fibers. J. Cell Biol. 1985, 101, 1332–1340. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, R.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: a major determinant of axon caliber. Proc. Natl. Acad. Sci. USA 1997, 84, 3472–3476. [Google Scholar] [CrossRef]
- Gordon, T.; Stein, R.B. Time course and extent of recovery of reinnervated motor units of cat triceps surae muscles. J. Physiol. 1982, 323, 307–323. [Google Scholar] [CrossRef]
- Gordon, T.; Gillespie, J.; Orozco, R.; Davis, L. Axotomy-induced changes in rabbit hindlimb nerves and the effects of chronic electrical stimulation. J. Neurosci. 1991, 11, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.M.; Danielsen, N.; Holmquist, B.; Kanje, M.; Lundborg, G. The influence of predegeneration on regeneration through peripheral nerve grafts in the rat. Exp. Neurol. 1993, 122, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.R.; Bedard, A.M.; Hincke, M.T.; Tetzlaff, W. Increased expression of BDNF and trkB mRNA in rat facial motoneurons after axotomy. Eur. J. Neurosci. 1996, 8, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Al-Majed, A.A.; Brushart, T.M.; Gordon, T. Electrical stimulation accelerates and increases expression of BDNF and TrkB mRNA in regenerating rat femoral motoneurons. Eur. J. Neurosci. 2000, 12, 4381–4390. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.C.; Li, E.; Kawaguchi, R.; Gerschwind, D.H.; Woolf, C.J. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 2020, 108, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.-Q.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.L.; Fainzilber, M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Rishal, I.; Fainziber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Saito, A.; Cavelli, V. Signaling over distances. Mol. Cell Proteomics 2016, 15, 382–393. [Google Scholar] [CrossRef]
- Spira, M. E.; Benbassat, D.; Dormann, A. Resealing of the proximal and distal cut ends of transected axons: electrophysiological and ultrastructural analysis. J Neurobiol. 1993, 24, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Madeddu, F.; Bozzi, Y.; Maffei, L.; Ratto, G.M. Acute physiological response of mammalian central neurons to axotomy: ionic regulation and electrical activity. FASEB J. 2004, 18, 1934–1936. [Google Scholar] [CrossRef]
- Kershensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, c572–c557. [Google Scholar] [CrossRef]
- Kamber, D.; Erez, H.; Spira, M.E. Local calcium-dependent mechanisms determine whether a cut axonal end assembles a retarded endbulb or competent growth cone. Exp. Neurol. 2009, 219, 112–125. [Google Scholar] [CrossRef]
- Bradke, F.; Fawcett, J.W.; Spira, M.E. Assembly of a new growth cone after axotomy: The precursor to axon regeneration. Nat. Rev. Neurosci. 2012, 13, 183–193. [Google Scholar] [CrossRef]
- Ambron, R. T.; Walters, E. T. Priming events and retrograde injury signals. A new perspective on the cellular and molecular biology of nerve regeneration. Mol. Neurobiol. 1996, 13, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Chierzi, S.; Codd, A.M.; Campbell, D.S.; Meyer, R.L; Holt, C.E.; Fawcett, J.W. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 2005, 25, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Vogelaar, C. F.; Gervasi, N.M.; Gumy, L.F.; Story, D.J.; Raja-Chowdhury, R.; Leung, K.-M.; Hold, C.E.; Fawcett, J.W. Axonal mRNAs: characterisation and role in the growth and regeneration of dorsal root ganglion axons and growth cones. Mol. Cell Neurosci. 2009, 42, 102–115. [Google Scholar] [CrossRef]
- Yan, D.; Wu, Z.; Chisholm, A. D.; Jin, Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 2009, 138, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Omotade, O.F.; Pollitte, S.F.; Zheng, J.Q. Actin-based growth cone motility and guidance. Mol. Cell Neurosci. 2017, 84, 4–10. [Google Scholar] [CrossRef]
- Liu, M.; Li, P.; Jia, Y.; Cui, Q.; Zhang, K.; Jiang, J. Role of non-coding RNAs in axon regeneration after peripheral nerve injury. In J. Biol. Sci. 2022, 18, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Willis, D.E. What makes a RAG regeneration associated? Front. Mol. Neurosci. 2015, 8, 43. [Google Scholar] [CrossRef]
- Hao, Y.; Frey, E.; Yoon, C.; Wong, H.; Nestorovski, D.; Holtzman, L.B.; Giger, R.J.; DiAntonio, A.; Collins, C. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 2016, 5, e14048. [Google Scholar] [CrossRef]
- Shin, J.E.; Cho, Y.; Beirowski, B.; Milbrandt, J.; Cavalli, V.; DiAntonio, A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 2012, 74, 1015–1022. [Google Scholar] [CrossRef]
- Kiryu-Seo, S.; Kato, R.; Ogawa, T.; Nakagomi, S.; Nagata, K.; Kiyama, H. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J. Biol. Chem. 2008, 283, 6988–6996. [Google Scholar] [CrossRef]
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 2015, 77, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Ha, H.; Kim, Y.K.; Cho, Y.; DiAntonio, A. DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury. Neurobiol. Dis. 2019, 127, 178. [Google Scholar] [CrossRef] [PubMed]
- George, E.B.; Glass, J.D.; Griffen, J.W. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J. Neurosci. 1995, 15, 6445–6452. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflamm 2001, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Sievers, C.; Platt, N.; Perry, V.H.; Coleman, M.P.; Conforti, L. Neurites undergoing Wallerian degeneration show an apoptotic-like process with annexin V positive staining and loss of mitochondrial membrane potential. Neurosci. Res. 2003, 46, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kershensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, c572–557. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R. Janeckova, L; Vargas, M.E.; Worrell, L..A.; Loreto, A.; Tickle, J.; Patrick, J.; Webster, J.R.M.; Marangoni, M.; Carpi,, F.M.; Pucciarelli, S.; Rossi, ;Meng, W.; Sagasti, A.; Ribchester, R.R.; Magni, G.; Coleman, M.P.; Conforti, L. A rise in NAD precursor nicotinamide mono-nucleoride (NMN) after injury promotes axon degeneration. Cell Death Differen 2015, 22, 731–742. [Google Scholar]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martines, A.; Whitworth, A.J.; Conforti, L.; Dajas-Bailador, F.; Coleman, M.P. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 2020, 134, 104678. [Google Scholar] [CrossRef]
- Waller, A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Phil Trans. Royal Soc. Lond. 1850, 140, 423–429. [Google Scholar]
- Küry, P.; Stoll, G.; Müller, H.W. Molecular mechanisms of cellular interactions in peripheral nerve regeneration. Curr. Opin. Neurol. 2001, 14, 635–639. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Myers, R.R. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller’s observations to neuroinflammation. J. Peripher. Nerv. Syst. 2002, 7, 13–27. [Google Scholar] [CrossRef]
- Ziv, N.E.; Spira, M.E. Axotomy induces a transient and localized elevation of the free intracellular calcium concentration to the millimolar range. J. Neurophysiol. 1995, 74, 2625–2537. [Google Scholar] [CrossRef] [PubMed]
- Ziv, N.E.; Spira, M.E. Localized and Transient Elevations of Intracellular Ca21 Induce the Dedifferentiation of Axonal Segments into Growth Cones. J. Neurosci. 1997, 3568–3579. [Google Scholar] [CrossRef] [PubMed]
- Spira, M.E.; Oren, R.; Dormann, A.; Gitler, D. Critical calpain-dependent ultrastructural alterations underlie the transformation of an axonal segment into a growth cone after axotomy of cultured Aplysia neurons. J. Comp. Neurol. 2003, 457, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Ginty, D.D. Function and regulation: Review of CREB family transcription factors in the nervous system. Neuron 2002, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Roy, A.; Wu, Z.; Concharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 2010, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Shin, J.E.; Ewan, E.E.; Oh, Y.M.; Pita-Thomas, W.; Cavalli, V. Activating injury-responsive genes with hypoxia enhances axon regeneration through neuronal HIF-1alpa. Neuron 2015, 88, 720–734. [Google Scholar] [CrossRef]
- Song, W.; Cho, Y.; Watt, D.; Cavalli, V. Tubulin-tyrosine ligase (TTL) mediated increase in tyrosinated α-tubulin in injured axons is required for retrograde injury signaling and axon regeneration. J. Biol. Chem. 2015, 290, 14765–14775. [Google Scholar] [CrossRef]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.F.; Fainzilber, M. Axoplasmic importins enable retrograde injury blot analysis. Signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Perlson, E.; Hanz, S.; Ben-Yaakov, K.; Segal-Ruder, Y.; Seger, R.; Fainzilber, M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 2005, 45, 715–726. [Google Scholar] [CrossRef]
- Lezana, J.P.; Dagan, S.Y.; Robinson, A.; Goldstein, R.S.; Fainzilber, M.; Bronfman, F.C.; B1onfman, M. Axonal PPARγ promotes neuronal regeneration after injury. Dev. Neurobiol. 2016, 76, 688–701. [Google Scholar] [CrossRef]
- Terenzio, M.; Koley, S.; Samra, N.; Rishal, I.; Zhao, Q.; Sahoo, P.K.; Uirsman, A.; Marvaldi, L.; Oses-Priesto, J.A.; Forester, C.; Goes, C.; Kliniski, A.L.; Di Pizio, A.; Doron-Mandel, E.D.; Perry, R.B-T.; Koppel, I.; Twiss, J.L.; Burlingame, A.L.; Fainzilber, M. Locally translated mTOR controls axonal local translation in nerve injury. Science 2018, 359, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavelli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Misra, V.; Verge, V.M.K. Sensing nerve injury at the axonal ER: activated Luman/CREB3 serves as a novel axonally synthesized retrograde regeneration signal. PNAS 2014, 16142–16147. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Zhai, R.; McLean, N.A.; Johnson, J.M.; Misra, V.; Verge, V.M.K. The unfolded protein response and cholesterol biosynthesis link Lumen/CREB3 to regenerative axon growth in sensory neurons. J. Neurosci. 2015, 35, 4557–14570. [Google Scholar] [CrossRef] [PubMed]
- Hasmatali, J.C.D.; De Guzman, J.; Zhai, R.; Yang, L.; McLean, N.A.; Hutchinson, C. ; Johnston.; Misra, V.; Verge, V.M.K. Axotomy induces phasic alterations in Luman/CREB3 expression and nuclear localization in injured and contralateral uninjured sensory neurons: correlation with intrinsic axon growth capacity. Neuropathol. Exp. Neurol. 2019, 78, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Schoeps, T.; Haubrock, M.; Dönitz, J. TFClass: a classification of human transcription factors and their rodent orthologs. Nucleic Acids Res 2015, 43, D97–D102. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.; Hunt, S.P. (1991) Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci. Lett. 1991, 129, 107–110. [Google Scholar] [CrossRef]
- Van Kesteren, R.E.; Mason, M.R.; Macgillavry, H.D.; Smit, A.B.; Verhaagen, J. A gene network perspective on axonal regeneration. Front. Mol. Neurosci. 2011, 4, 46. [Google Scholar] [CrossRef]
- Song, J.; Singh, M. From hub proteins to hub modules: the relationship between essentiality and centrality in the yeast interactome at different scales of organization. PLoS Comput. Biol. 2013, 9, 1002910. [Google Scholar] [CrossRef]
- Fontana, X.; Hristova, M.; Costa, C.D.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.R.; Klein, R.; Raivich, G.; Behrens, A. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef]
- Jessen, K. R.; Mirsky, R.; Arthur-Farraj, P. The role of cell plasticity in tissue repair: adaptive cellular reprogramming. Devel Cell 2005, 34, 613–620. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia 2008, 56, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Herdegen, T.; Skene, P.; Bähr, M. The c-Jun transcription factor–bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Michaeleviski, I.; Segal-Ruder, Y.; Rozenbaum, M.; Medzihradszky, K.F.; Shalem, O.; Coppola, G.; Horn-Saban, S.; Ben-Yaakov, K.; Dagan, S.Y.; Rishal, I.; Geschwind, D.H.; Pilpel, Y.l.; Burlingame, A.L.; Fainzilber, M. Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 2010, 3, ra53. [Google Scholar] [CrossRef] [PubMed]
- Blesch, A.; Lu, P.; Tsukada, S.; Alto, L.T.; Roet, K.; Coppola, G.; Geschwind, D.; Tuszynski, M.H. Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to cAMP-mediated effects. Exp. Neurol. 2012, 235, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, F. W.; Hager, G.; Schmitt, A. B.; Horvat, A.; Streif, R.; Spitzer, C.; Gamal, S.; Breuer, S.; Brook, G.A.; Nacimiento, W.; Kreutzberg, G.W. Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 2000, 12, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, H.; Kondo, E.; Fukuoka, T.; Dai, Y. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol. Cell Neurosci. 2000, 15, 170–182. [Google Scholar] [CrossRef]
- Seijffers, R.; Allchorne, A.J.; Woolf, C.J. The transcription factor ATF3 promotes neurite outgrowth. Mol. Cell Neurosci. 2006, 143–154. [Google Scholar] [CrossRef]
- Seijffers, R.; Mills, C.D.; Woolf, C.J. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 2007, 27, 7911–7920. [Google Scholar] [CrossRef]
- Lindwall, C.; Kanje, M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell Neurosci. 2005, 29, 269–282. [Google Scholar] [CrossRef]
- Schweizer, U.; Gunnersen, J.; Karch, C.; Wiese, S.; Holtmann, B.; Takeda, K.; Akira, S.; Sendtner, M. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J. Cell Biol. 2002, 156, 287–297. [Google Scholar] [CrossRef]
- Bareyre, F.M.; Garzorz, N.; Lang, C.; Misgeld, T.; Buning, H.; Kerschensteiner, M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U S A 2011, 108, 6282–6287. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.P.; Cornuet, P.K.; McIlwrath, S.; Koerber, H.R.; Albers, K.M. SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neurosci. 2006, 143, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.P.; McIlwrath, S.L.; Jing, X.; Cornuet, P.K.; Salerno, K.M.; Koerber, H.R.; Albers, K.M. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 2009, 1256, 43–54. [Google Scholar] [CrossRef]
- Jankowski, M.P.; Miller, L.; Koerber, H.R. Increased expression of transcription factor SRY-box-containing gene 11 (Sox11) enhances neurite growth by regulating neurotrophic factor responsiveness. Neurosci. 2018, 382, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Wang, T.; Huang, S.; Glorioso, J.C.; Albers, K.M. The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp. Neurol. 2012, 233, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Ho, C.; Wong, K.; Tessier-Lavigne, M. Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J. Neurosci. 2009, 29, 7116–7123. [Google Scholar] [CrossRef] [PubMed]
- Finelli, M.G.; Wong, J.K.; Zou, H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J. Neurosci. 2013, 33, 19664–19676. [Google Scholar] [CrossRef] [PubMed]
- Saijilafu, E.-M.H.; Hur, C.-M.; Liu, C.-M.; Jiao, Z.; Xu, W.-L; Zhou, F.-Q. P13K-GSK3 signalling regulates mammalian axon regeneration by inducing expression of Smad. Nat Commun. 2013, 4, 2690. [Google Scholar] [CrossRef]
- Saijilafu, Z.B.Y.; Zhou, F.Q. Signaling pathways that regulate axon regeneration. Neurosci. Bull. 2013, 29, 411–420. [Google Scholar] [CrossRef]
- Okuyama, N.; Kiryu-Seo, S.; Kiyama, H. Altered expression of Smad family members in injured motor neurons of rat. Brain Res. 2007, 1132, 36–41. [Google Scholar] [CrossRef]
- Jiang, J.J.; Liu, C.M.; Zhang, B.Y.; Wang, W.X.; Zhang, M.; Saijilafu, E.-M.H. MicroRNA-26a supports mammalian axon regeneration in vivo by suppressing GSK3beta expression. Cell Death Dis. 2015, 6, e1865. [Google Scholar] [CrossRef]
- Nadeau, S.; Hein, P.; Fernandes, K.J.; Peterson, A.C.; Miller, F.D. A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol. Cell Neurosci. 2005, 29, 525–535. [Google Scholar] [CrossRef]
- Ma, J.J.; Ju, X.; Xu, R.J.; Wang, W.H.; Luo, Z.-P.; Liu, C.-M.; Yang, L.; Li, B.; Chen, J.-Q.; Meng, B.; Yang, H.-L.; Zhou, F.-Q. ; Saijilafu. Telomerase reverse transcriptase and p53 regulate mammalian peripheral nervous system and CNS axon regeneration downstream of c-Myc. J Neurosci 2019, 39, 9107–9118. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Knights, C.D.; Rao, M.; Yakovlev, A.; Beers, J.; Catania, J.; Avantaggiati, M. L.; Faden, A. I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006, 25, 4084–4096. [Google Scholar] [CrossRef]
- Moore, D. L.; Blackmore, M. G.; Hu, Y.; Kaestner, K. H.; et al. KLF family members regulate intrinsic axon regeneration ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef]
- Xu, J.H.; Qin, X.Z.; Zhang, H.N.; Ma, Y.X.; Qi, S.-B.; Zhang, H.-C.; Ma, J.-J.; Fu, X.-Y.; Xie, J.-L. Saijilafu. Deletion of Kruppel-like factor-4 promotes axonal regeneration in mammals. Neural Regen. Res. 2021, 16, 166–171. [Google Scholar]
- Palmisano, I.; Di Giovanni, S. Advances and limitations of current epigenetic studies investigating mammalian axonal regeneration. Neurotherapeutics 2018, 15, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series. Pulm. Circ. 2014, 169–174. [Google Scholar] [CrossRef]
- Zhou, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef]
- Puttagunta, R.; Tedeschi, A.; Soria, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; Laskowski, C.J.; Boutillier, A.-L.; Bradke, F.; Giovanni, S.D. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nature Comm. 2014, 5, 3527. [Google Scholar] [CrossRef]
- Karpranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Liu, X.; Yu, X.; He, Y.; Wang, L. Long noncoding RNA nuclear enriched abundant transcript 1 promotes the proliferation and migration of Schwann cells by regulating the miR-34a/Satb1 axis. Glia 2021, 234, 15357–16366. [Google Scholar] [CrossRef]
- Strickland, I.T; Richards, L.; Holes, F.E.; Wynick, D.; Uney, J.B.; Wong, L.F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One 2011, 6, e23423. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, F.; Gu, X. Non-coding RNAs as emerging regulators of neural injury response and regeneration. Neurosci. Bull. 2016, 32, 253–264. [Google Scholar] [CrossRef]
- Dorrier, C.E.; Jones, H.E.; Pintarić, L.; Siegenthaler, J.A.; Daneman, R. Emerging roles for CNS fibroblasts in health, injury and disease. Nature Rev. Neurosci. 2022, 23, 23–34. [Google Scholar] [CrossRef]
- Knöferle, J.; Koch, J.C.; Ostendorf, T.; Michel, V.; Planchamp, V.; Vutova, P.; Tönges, L.; Stadelmann, C.; Brück, W.; Bähr, M.; Lingor, P. Mechanisms of acute axonal degeneration in the optic nerve in vivo. PNAS 2010, 107, 6064–6069. [Google Scholar] [CrossRef]
- Mårtensson, L.B.; Blom, C.L.; Dahlin, L.B. Ca2+ involvement in activation of extracellular-signal-regulated-kinase 1/2 and m-calpain after axotomy of the sciatic nerve. Neural Reg Res B 2017, 12, 623–628. [Google Scholar]
- Miledi, R.; Slater, C.R. On the degeneration of rat neuromuscular junctions after nerve section. J. Physiol. 1970, 207, 507–528. [Google Scholar] [CrossRef]
- Gilliatt, R. W.; Horth, R.J. Nerve conduction during Wallerian degeneration in the baboon. J. Neurol. Neurosurg. Psychiatry 1972, 35, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lubinska, L. Early course of Wallerian degeneration in myelinated fibres of the rat phrenic nerve. Brain Res. 1977, 130, 47–63. [Google Scholar] [CrossRef]
- Mackenzie, S.J.; Smirnov, I.; Calancie, B. Cauda equina repair in the rat: part Time course of ventral root conduction failure. J. Neurotrauma 2012, 29, 1683–1690. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Perez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef]
- Napoli, I.; Noon, L.A.; Ribeiro, S.; Kerai, A.P.; Parrinello, S.; Rosenberg, L.H.; Collins, M.J.; Harrisingh, M.C.; White, I.J.; Woodhoo, A.; Lloyd, A.C. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration. Neuron 2012, 73, 729–742. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J. A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M..; Griffith, M.; Hantke, J.; Maciias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; De Juan, V.G.; Jefferies, H.B.; Aspichueta, P.; Elortza, F.; Aransay, A.M.; Martínez-Chantar, M.L.; Baas, F.; Mato, J.M.; Mirsky, R.; Woodhoo, A.; Jessen, K.R. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef]
- Jang, S. Y.; Shin, Y. K.; Park, S. Y.; Park, J. Y.; Lee, H. J.; Yoo, Y. H.; Kim, J.K.; Park, H.W. Autophagic myelin destruction by Schwann cells during Wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Brosius Lutz, A.; Chung, W. S.; Sloan, S. A.; Carson, G. A.; Zhou, L.; Lovelett, E.; Posada, S.; Zuchero, J.B.; Barres, B.A. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. USA 2017, 114, E8072–E8080. [Google Scholar] [CrossRef] [PubMed]
- Perry, V. H.; Tsao, J. W.; Fearn, S.; Brown, M. C. Radiation-induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur. J. Neurosci. 1995, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Gregson, N.A.; Hall, S.M. A quantitative analysis of the effects of the intraneural injection of lysophosphatidyl choline. J. Cell Sci. 1973, 13, 257–277. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Tigueros, M.A.; Kalvas, A.; David, S. Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol. Cell Neurosci. 2003, 24, 753–765. [Google Scholar] [CrossRef]
- Banner, L.R.; Patterson, P.H. Major changes in the expression of the mRNAs for cholinergic differentiation factor/leukemia inhibitory factor and its receptor after injury to adult peripheral nerves and ganglia. Proc Natl Acad Sci U S A 1994, 91, 7109–7113. [Google Scholar] [CrossRef]
- Bolin, M.; Verity, A.N.; Silver, J.E.; Shooter, E.M.; Abrams, J.S. Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem 1995, 64, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bourde, O.; Kiefer, R.; Toyka, K.V.; Hartung, H.P. Quantification of interleukin-6 mRNA in Wallerian degeneration by competitive reverse transcription polymerase chain reaction. J. Neuroimmunol. 1996, 69, 135–140. [Google Scholar] [PubMed]
- Kurek, J.B.; Austin, L.; Cheema, S.S.; Bartlett, P.F.; Murphy, M. Up-regulation of leukaemia inhibitory factor and interleukin-6 in transected sciatic nerve and muscle following denervation. Neuromuscul. Disorders 1996, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, H.S.; Roytta, M. Increased expression of chemokines (MCP-1, MIP-1alpha, RANTES) after peripheral nerve transection. J Periph Nerv Syst 2000, 5, 75–81. [Google Scholar] [CrossRef]
- Subang, M.C.; Richardson, P.M. Influence of injury and cytokines on synthesis of monocyte chemoattractant protein-1 mRNA in peripheral nervous tissue. Eur. J. Neurosci. 2001, 13, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Perrin, F.E.; Lacroix, S.; Aviles-Trigueros, M.; David, S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 2005, 128, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Boivin, A.; Pineau, I.; Barrette, B.; Filali, M.; Vallieres, N.; Rivest, S.; Lacroix, S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 2007, 27, 12565–12576. [Google Scholar] [CrossRef]
- Shubayev, V.I.; Angert., M.; Dolkas, J.; Campana, W.M.; Palenscar, K.; Myers, R.R. TNFalpha-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol Cell Neurosci 2006, 31, 407–415. [Google Scholar] [CrossRef]
- Cattopadhyay, S.; Meyers, R.R.; Janes, J. Shubayev, V. Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerve. Brain Behav Immun 2007, 21, 561–568. [Google Scholar] [CrossRef]
- Beuche, W.; Friede, R.L. The role of non-resident cells in Wallerian degeneration. J. Neurocytol. 1984, 13, 767–796. [Google Scholar] [CrossRef]
- Avellino, A.M.; Hart, D.; Dailey, A.T.; MacKinnon, M.; Ellegala, D.; Kliot, M. Differential macrophage responses in the peripheral and central nervous system during allerian degeneration of axons. Exp. Neurol. 1995, 136, 183–198. [Google Scholar] [CrossRef]
- Brück, W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997, 7, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Griffin, J.W.; Li, C.Y.; Trapp, B.D. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J. Neurocytol. 1989, 18, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kubes, P.; Zochodne, D.W. Delayed peripheral nerve degeneration, regeneration, and pain in mice lacking inducible nitric oxide synthase. J. Neuropathol. Exp. Neurol. 2001, 60, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Friede, R.L. The role of complement in myelin phagocytosis during PNS wallerian degeneration. J. Neurol. Sci. 1991, 103, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E. Phagocytosis of myelin in demyelinative disease: a review. Neurochem. Res. 1999, 24, 261–268. [Google Scholar] [CrossRef] [PubMed]
- da Costa, C.C.; van der Laan, L.J.; Dijkstra, C.D.; Bruck, W. The role of the mouse macrophage scavenger receptor in myelin phagocytosis. Eur. J. Neurosci. 1997, 9, 2650–2657. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, R.E.; Echevarria, F.D. Macrophage biology in the peripheral nervous system after injury Prog. Neurobiol. 2019, 102–121. [Google Scholar]
- Nadeau, S.; Filali, M.; Zhang, J.; Kerr, B.J.; Rivest, S.; Soulet, D.; Iwakura, Y.; de Rivero Vaccari, J.; Keane, R.W.; Lacroix, S. Functional recovery after peripheral nerve injury in dependent n the proinflammatory cytokines IL-1beta and TNF: implications for neuropathic pain. J. Neurosci. 2011, 31, 12533–12542. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; et al. Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef]
- Liu, P.; Peng, J.; Han, G.H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar]
- Hall, S.M. The response of the (myelinating) Schwann cell population to multiple episodes of demyelination. J. Neurocytol. 1983.12, 1–12. [CrossRef]
- Doree, M.; Hunt, T. From Cdc3 to Cdk1: when did the cell cycle kinase join its cyclin partner? Cell Sci. 2002, 2451–2464. [Google Scholar]
- Han, S.; Seo, T.B.; Kim, K.-H.; Yoon, J.-H.; Yoon, S.-J.; Namgung, U.K. Cdc2-mediated Schwann cell migration during peripheral nerve regeneration. J. Cell Sci. 2007, 120, 246–255. [Google Scholar] [CrossRef]
- Namgung, U.K. The role of Schwann cell-axon interaction in peripheral nerve regeneration. Cell Tiss. Organs 2014, 4, 6–12. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; Wicher, G.K.; Mitter, R.; Greensmith, L.; Behrens, A.; Raivich, G.; Mirsky, R.; Jessen, K.R. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Arthur-Farraj, M.P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.; Welcher, A.A.; Suter, U.; Shooter, E.M. Identification of transcriptionally regulated genes after sciatic nerve injury. J. Neurosci. Res. 1991, 29, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wu, J.; Zhou, S.; Wang, Y.; Qin, J.; Yu, B.; Gu, X.; Yao, C. Global analysis of transcriptome in dorsal root ganglia following peripheral nerve injury in rats. Biochem. Biophys. Res. Commun. 2016, 478, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: tumor necrosis factor-α, Interleukin-1α, and Interleukin-1β. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Patterson, P.H.; Jessen, K.R.; Mirsky, R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci 2002, 2002 22, 6696–6703. [Google Scholar] [CrossRef]
- Marchionni, M.A.; Goodearl, A.D.; Chen, M.S.; Bermingham-McDonogh, O.; Kirk, C.; Hendricks, M.; Danehy, F.; Misumi, D.; Sudhalta, J.; Kobayashi, K.; Wroblewski, D.; Lynch, C.; Baldassare, M.; Hiles, I.; Davis, J.B.; Hsuan, J.J.; Totty, N.F.; Otsu, M.l.; McBurney, R.N.; Waterfield, M.D.; Strooband, P.; Gwynne, D. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 1993, 362, 312–318. [Google Scholar] [CrossRef]
- Falls, D.L. Neuregulins and the neuromuscular system: 10 years of answers and questions. J. Neurocytol. 2003, 32, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.M. The biology of chronically denervated Schwann cells. Ann. NY Acad. Sci. 1999, 883, 215–233. [Google Scholar] [CrossRef]
- Carroll, S.L.; Miller, M.L.; Frohnert, P.W.; Kim, S.S.; Corbett, J.A. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J. Neurosci. 1997, 17, 1642–1659. [Google Scholar] [CrossRef]
- Stassart, R. M.; Fledrich, R.; Velanac, V.; Brinkmann, B. G.; Schwab, M. H.; Meijer, D.; Swerenda, M.W.; Nave, K.-A. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat. Neurosci. 2013, 16, 48–54. [Google Scholar] [CrossRef]
- Ronchi, G.; Haastert-Talini, K.; Fornasari, B. E.; Perroteau, I.; Geuna, S.; Gambarotta, G. The Neuregulin1/ErbB system is selectively regulated during peripheral nerve degeneration and regeneration. Eur. J. Neurosci. 2016, 43, 351–364. [Google Scholar] [CrossRef]
- El Soury, M.; Fornasari, B.; Morano, M.; Grazio, E.; Ronchi, G.; Incarnato, D.; Giacobini, M.; Geuna, S. ; Provero. Gambarotta, G. Soluble Neuregulin1 down-regulates myelination genes. Frontiers Cell Neurosci. 2018, 11, 157. [Google Scholar]
- Salzer, J.L; Bunge, R.P.; Glaser, L. Studies of Schwann cell proliferation. III. Evidence for the surface localization of the neurite mitogen. J. Cell Biol. 1980, 84, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.; Spencer, P.S. Schwann cell mitosis in response to regenerating peripheral axons in vivo. Brain Res. 1985, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Clemence, A.; Mirsky, R.; Jessen, K.R. Non-myelin-forming Schwann cells proliferate rapidly during Wallerian degeneration in the rat sciatic nerve. J. Neurocytol. 1989, 18, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Rahmatullah, M.; Schroering, A.; Rothblum, K.; Stahl, R.C.; Urban, B.; Carey, D.J. Synergistic regulation of Schwann cell proliferation by heregulin and forskolin. Mol. Cell Biol. 1998, 18, 6245–6252. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.J.; Eccleston, P.A.; Jessen, K.R.; Mirsky, R. Interaction between cAMP elevation, identified growth factors, and serum components in regulating Schwann cell growth. J. Neurosci. Res. 1991, 30, 346–352. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Lin, P.X.; Chang, Y.; Webster, H.D. Effects of nerve segment supernatants on cultured Schwann cell proliferation and laminin production. J. Neurosci. Res. 1994, 37, 612–622. [Google Scholar] [CrossRef]
- Morrissey, T.K.; Levi, A.D.; Nuijens, A.; Sliwkowski, M.X.; Bunge, R.P. Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbBProc Natl. Acad. Sci. USA 1995, 92, 1431–1435. [Google Scholar] [CrossRef]
- Shin, Y.K.; Jang, S.Y.; Yun, S.H.; Choi, Y.Y.; Yoon, B.-A.; Jo, Y.R.; Park, S.Y.; Pak, M.G.; Park, J.I.; Park, H.T. Cooperative interaction of hepatocyte growth factor and neuregulin regulates Schwann cell migration and proliferation through Grb2-associated binder-2 in peripheral nerve repair. Glia 2017, 65, 794–1808. [Google Scholar] [CrossRef]
- Adams, S. J.; Aydin, I. T.; Celebi, J. T. GAB2-A scaffolding protein in cancer. Mol. Cancer Res. 2012, 10, 1265–1270. [Google Scholar] [CrossRef]
- Wu, G.; Li, X.; Li, M.; Zhang, Z. Long non-coding RNA MALAT1 promotes the proliferation and migration of Schwann cells by elevating BDNF through sponging miR-129-5p. Exp. Cell Res. 2020, 390, 111937. [Google Scholar] [CrossRef]
- Yao, C.; Wang, Q.; Wang, Y.; Wu, J.; Cao, X.; Lu, Y.; Chen, Y.; Feng, W.; Gu, X.; Dun, X.-P.; Yu, B. Loc6802454 regulates Schwann cell proliferation through Psrc1 and Ska1-a as a microRNA sponge following sciatic nerve injury. Glia 2021, 69, 2391–2403. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Zhang, X.; Ding, L.; Zeng, Q. Microanalysis of the expression profile of IncRNA BC088327 as an agonist to heregulin-1β-induced cell proliferation in the peripheral nervous system. Int. J. Mol. Med. 2018, 41, 3477–3484. [Google Scholar]
- Yao, C.; Chen, Y.; Wang, J.; Wu, J.; Cao, X.; Lu, Y.; Chen, Y.; Feng, W.; Gu, X.; Dun, X.-P.; Yu, B. LncRNA BC088259 promotes Schwann cell migration through vimentin following peripheral nerve injury. Glia 2020, 68, 670–679. [Google Scholar] [CrossRef]
- Ma, Y.; Zhai, D.; Zhang, W.; Zhang, H.; Dong, I.; Zhou, Y.; Feng, D.; Zheng, W.; Wang, T.; Mao, C.; Wang, W. Down-regulation of long non-coding RNA MEG3 promotes Schwann cell proliferation and migration and repairs sciatic nerve injury in rats. J. Cell Mol. Med. 2020, 24, 7460–7469. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Huang, T.; Chen, Y.; Shen, L.; Zhou, S.; Zhang, S.; Yu, B. Circ-Spidr enhances axon regeneration after peripheral nerve injury. Cell Death Dis. 2019, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, H.; Frisen, J.; Barbany, G.; Timmusk, T.; Zachrisson, O.; Verge, V.M.; Persson, H. Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J. Cell Biol. 1993, 123, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hoke, A.; Redett, R.; Hameed, H.; Jari, R.; Zhou, C.; Li, Z.B.; Griffin, J.W.; Brushart, T.M. Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J. Neurosci. 2006, 26, 9646–9655. [Google Scholar] [CrossRef] [PubMed]
- Mi, R.; Chen, W.; Hoke, A. Pleiotrophin is a neurotrophic factor for spinal motor neurons. PNAS 2007, 104, 4664–4669. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M.; Aspalter, M.; Griffin, J.W.; Redett, R.; Hameed, H.; Zhou, C.; Wright, M.; Vyas, A.; Hoke, A. Schwann cell phenotype is regulated by axon modality and central–peripheral location, and persists in vitro. Exp. Neurol. 2013, 247, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Sulaiman, A.R. Nerve regeneration in the peripheral nervous system. Neuroglia (3 edn), Ch 55, 2012, 701-714.
- Wang, X. Pleiotrophin: activity and mechanism. Adv. Clin. Chem. 2020, 98, 1–89. [Google Scholar]
- Heumann, R.; Lindholm, D.; Bandtlow, C.; Meyer, M.; Radeke, M.J.; Misko, T.P.; Shooter, E.; Thoenen, H. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerves during development, degeneration and regeneration: role of macrophages. Proc. Natl. Acad. Sci. USA 1987, 84, 8735–8739. [Google Scholar] [CrossRef]
- Meyer, M.; Matsuoka, I.; Wetmore, C.; Olson, L.; Thoenen, H. Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J. Cell Biol. 1992, 119, 45–54. [Google Scholar] [CrossRef]
- Naveilhan, P.; ElShamy, W.E.; Ernsfors, P. Differential regulation of mRNAs for GDNF and its receptors Ret and GDFRα after sciatic nerve lesion in the mouse. Eur. J. Neurosci. 1997, 9, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Petrov, T.; Chung, P.H.; Gordon, T. The expression of the low affinity nerve growth factor receptor in long-term denervated Schwann cells. Glia 1997, 20, 87–100. [Google Scholar] [CrossRef]
- Seo, T.B.; Oh, M.J.; You, B.G.; Kwon, K.B.; Chang, I.A.; Yoon, J.H.; Lee, C.-Y; Namgung, U.K. Erk1/2-mediated Schwann cell proliferation in the regenerating sciatic nerve by treadmill training. J. Neurotrauma 2009, 26, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Jang, S.Y.; Park, J.Y.; Park, S.Y.; Lee, H.J.; Suh, D.J.; Park, H.T. The Neuregullin-RacMKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia 2013, 61, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Blom, C.L.; Martensson, L.B.; Dahlin, L.B. Nerve injury-induced c-activation in Schwann cells is JNK independent. Biomed. Res. Int. 2014, 2014, 392971. [Google Scholar]
- Friede, R.L.; Bischhausen, R. The fine structure of stumps of transected nerve fibers in subserial sections. J NeuroI Sci. 1980, 44, 181–203. [Google Scholar] [CrossRef]
- McQuarrie, I.G. Effect of a conditioning lesion on axonal sprout formation at nodes of Ranvier I. Comp. Neurol. 1985, 231, 239–249. [Google Scholar] [CrossRef]
- McQuarrie, I.G.; Lasek, R.J. Transport of cytoskeletal elements from parent axons into regenerating daughter axons. J. Neurosci. 1989, 9, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ranson, S.W. Degeneration and regeneration of nerve fibres J. Comp. Neurol. 1912, 22, 487–546. [Google Scholar]
- Aitken J., T.; Sherman, M.; Young J., Z. Maturation of peripheral nerve fibres with various peripheral connections. J. Anat. 1947, 81, 1–22. [Google Scholar]
- Mackinnon, S.E.; Dellon, A.L.; O-Brien, J.P. Changes in nerve fiber numbers distal to a nerve repair in the rat sciatic nerve model. Muscle Nerve 2001, 14, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Toft, P.B.; Fugleholm, K.; Schmalbruch, H. Axonal branching following crush lesions of peripheral nerves of rats. Muscle Nerve 1988, 11, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S.; Napoli, I.; Ribeiro, S.; Digby, P.W.; Fedorova, M.; Parkinson, D.B.; Doddrell, R.D.; Nakayama, M.; Adams, R.H.; Lloyd, A.C. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 2010, 143, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bixby, J. l.; Lilien, J.; Reichardt, l. F. Identification of the major proteins that promote neuronal process outgrowth on Schwann cells in vitro. J. Cell Biol. 1988, 107, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, their firbillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef]
- Gonzalez Perez, F.; Udina, E.; Navarro, X. Chapter 10 Extracellular matrix components in peripheral nerve regeneration. Int. Rev. Neurobiol. 2013, 108, 257–275. [Google Scholar]
- Lefcort, F.; Venstrom, K.; McDonald, J.A.; Reichardt, L.F. Regulation of expression of fibronectin and its receptor, α5β1, during development and regeneration of peripheral nerve. Development 1992, 116, 767–782. [Google Scholar] [CrossRef]
- Mathews, G.A.; Ffrench-Constant, C. Embryonic fibronectins are up-regulated following peripheral nerve injury in rats J. Neurobiol. 1995, 26, 171–188. [Google Scholar] [CrossRef]
- Gardiner, N. Integrins and the extracellular matrix: key mediators of development and regeneration of the sensory nervous system. Dev Neurobiol 2011, 71, 1054–1072. [Google Scholar] [CrossRef]
- Gonzalez Perez, F.; Alé, A.; Santos, D.; Barwig, C.; Freier, T.; Navarro, X.; Udina, E. Substratum preferences of motor and sensory neurons in postnatal and adult rats. Europ J. Neurosci. 2016, 43, 431–442. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Ann Rev Cell Dev Biol 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Kloss, C. U. A.; Werner, A.; Klein, M.A.; Shen, J.; Menuz, K.; Probst, J.C.; Kreutzberg, G.W.; Raivich, G. Integrin family of cell adhesion molecules in the injured brain: regulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J Comp Neurol 1999, 411, 162–178. [Google Scholar] [CrossRef]
- Yanagida, H.; Tanaka, J.; Maruo, S. Immunocytochemical localization of a cell adhesion molecule, integrin α5β1, in nerve growth cones. J. Orthop. Sci. 1999, 4, 353–360. [Google Scholar] [CrossRef]
- Hammarberg, H.; Wallquist, W.; Piehl, F.; Risling, M.; Cullheim, S. Regulation of laminin-associated integrin subunit mRNAs in rat spinal motoneurons during postnatal development and after axonal injury. J. Comp. Neurol. 2000, 428, 294–304. [Google Scholar] [CrossRef]
- Vogelezang, M.G.; Liu, Z.; Relvas, J.B.; Raivich, G.; Scherer, S.S.; Charles ffrench-Constant, C. α4 Integrin Is expressed during peripheral nerve regeneration and enhances neurite outgrowth. J. Neurosci. 2001, 21, 6732–6744. [Google Scholar] [CrossRef] [PubMed]
- Wallquist, W.; Zelano, J.; Plantman, S.; Kaufman, S.J.; Cullheim, S.; Hammarberg, H. Dorsal root ganglion neurons up-regulate the expression of laminin-associated integrins after peripheral but not central axotomy. J. Comp. Neurol. 2004, 480, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, N.J.; Fernyhough, P.; Tomlinson, D.R.; Mayer, U.; von der, M.H.; Streuli, C.H. Alpha7 integrin mediates neurite outgrowth of distinct populations of adult sensory neurons. Mol. Cell Neurosci. 2005, 28, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Strittmatter, S.M. The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J. Neurosci. 2008, 28, 1262–1269. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Haenzi, B.; Andrews, M.R.; Verhaagen, J.; Fawcett, J.W. Integrins promote axonal regeneration after injury of the nervous system. Biol. Rev. Camb. Philos. Soc. 2018, 1339–1362. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, C.; Bow, C.M.; Remahl, I.N. Myelination and myelin sheath remodeling in normal and pathological PNS nerve fibres. Prog. Neurobiol. 1994, 43, 85–141. [Google Scholar] [CrossRef] [PubMed]
- Fricker, F.R.; Bennett, D.A. The role of neuregulin-1 in the response to nerve injury. Future Neurol. 2011, 6, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Fricker, F.R.; Antunes-Martins, A.; Jorge Galino, J.; Paramsothy, R.; La Russa, F.; Perkins, J.; Goldberg, R.; Brelstaff, J.; Zhu, N.; McMahan, S.B.; Orengo, C.; Garratt, A.N.; Birchmeier, C.; Bennett, D.L.H. Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 2013, 136, 2279–2297. [Google Scholar] [CrossRef]
- Haftek, J.; Thomas, P. K. Electron-microscope observation on the effects of localized crush injuries on the connective tissues of the peripheral nerve. J. Anat. 1968, 103, 233–243. [Google Scholar]
- Devor, M.; Govrin-Lippman, R. Maturation of axonal sprouts after nerve crush. Exp. Neurol. 1979, 64, 260–270. [Google Scholar] [CrossRef]
- Gordon, T.; Stein, R.B. Time course and extent of recovery in reinnervated motor units of cat triceps surae muscles. J. Physiol. 1982, 323, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Houdek, M.T.; Shin, A.Y. Management and complications of traumatic peripheral nerve injuries. Hand Clin. 2015, 31, 151–163. [Google Scholar] [CrossRef]
- Ransom, S.C.; Shahrestani, S.; Lien, B.V.; Tafreshi, A.R.; Brown, N.J.; Hanst, B.; Lehrich, B.M.; Ransom, R.C.; Sahyouni, R. Translational approaches to electrical stimulation for peripheral nerve regeneration. Neurorehabil Neural Repair. 2020, 34, 979–985. [Google Scholar] [CrossRef]
- Rasulić, L.; Simić, V.; Savić, A.; Lepić, M.; Kovačević, V.; Puzović, V’.; Vitošević, F.; Novaković, N.; Samardžić, M.; Rotim, K. Management of brachial plexus missile injuries. Acta Clin. Croat. 2018, 57, 487–496. [Google Scholar] [CrossRef]
- Sunderland, S. Rate of regeneration of motor fibers in the ulnar and sciatic nerves. Arch. Neurol. Psych. 1947, 58, 7–13. [Google Scholar] [CrossRef]
- Sunderland, S. Rate of regeneration of sensory nerve fibers. Arch. Neurol. Psychiat 1947, 58, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, H.J.; Spencer, P.S. The fate of Schwann cells isolated from axonal contact J. Neurocytol. 1978, 7, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Röyttä, M.; Salonen, V. Long-term endoneurial changes after nerve transection. Acta Neuropathol. 1988, 76, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Salonen, V.; Peltonen, J.; Röyttä, M.; Virtanen, I. Laminin in traumatized peripheral nerve: basement membrane changes during degeneration and regeneration. J. Neurocytol. 1987, 16, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Salonen, V.; Röyttä, M.; Peltonen, J. The effects of nerve transection on the endoneurial collagen fibril sheaths. Acta Neuropathol. 1987, 74, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Abernethy, D.A.; King, R.H.M.; Muddle, J.R.; Thomas, P.K. Neural architecture I transected rabbit sciatic nerve after prolonged nonreinnervation. J. Anat. 1998, 192, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Dedkov, E.I.; Kostrominova, T. Y.; Borisov, A. B.; Carlson, B. M. Survival of Schwann cells in chronically denervated skeletal muscles. Acta Neuropath 2002, 103, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Wood, P.; Sulaiman, O.A.R. Long-term denervated rat Schwann cells retain their capacity to proliferate and to myelinate axons in vitro. Frontiers Cell Neurosci. 2019, 12, 511. [Google Scholar] [CrossRef]
- Qian, T.; Wang, P.; Chen, Q.; Yi, S.; Liu, Q.; Wang, H.; Wang, s.; Geng, W.; Liu, Z.; Li, S. The dynamic changes of main cell types in the microenvironment of sciatic nerves following sciatic nerve injury and the influence of let-7 on their distribution. RSC Adv. 2018, 8, 41181–41191. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Effects of short- and long-term Schwann cell denervation on peripheral nerve regeneration, myelination, and size. Glia 2000, 32, 234–246. [Google Scholar] [CrossRef]
- Gordon, T. The biology, limits, and promotion of peripheral nerve regeneration in rats and human. Nerves and Nerve Injuries. Elsevier, New York, 2015, pp. 903–1019.
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmain, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 controls the long-term survival and phenotype of repair Schwann cells during nerve regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Dong, Z.; Bunting, H.; Whitfield, J.; Meier, C.; Marie, H.; Mirsky, R.; Jessen, K.R. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci. 2001, 21, 8572–8585. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Bhaskaran, A.; Droggiti, A.; Dickinson, S.; D-Antonio, M.; Mirsky, R.; Jessen, K.R. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol. 2004, 164, 385–394. [Google Scholar] [CrossRef]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef]
- Lundborg, G. Nerve Injury and Repair. 2004, Churchill Livingstone, New York.
- Adhihetty, PJ.; O’Leary, M.F.N.; Chabi, B.; Wicks, K.L.; Hood, D.A. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 2007, 102, 1143–1151. [Google Scholar] [CrossRef]
- O′Leary, M.F.N.; Vainshtein, A.; Carter, H.N.; Zhang, Y.; Hood, D.A. Denervation-induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis-deficient animals. Am. J. Physiol. Physiol. 2012, 303, 447–454. [Google Scholar] [CrossRef]
- Yang, X.; Xue, P.; Chen, H.; Yuan, M.; Kang, Y.; Duscher, D.; Machens, H.-G.; Chen, Z. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics 2020, 10, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Warwick, D.; Srinivasan, H.; Solomon, L. Peripheral Nerve Disorder. In: Arnold, H., editor. Apley’s system of Orthopaedics and Fracture. 9th ed. 2010. pp. 269–United Kingdom.
- Holmes, W.; Young J., Z. Nerve regeneration after immediate and delayed suture. J. Anat. 1942, 77, 63–I06. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged denervation. J. Neurosci. 1995, 15, 3876–3885. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J. Neurobiol. 2001, 49, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain derived neurotrophic factor. Eur. J. Neurosci. 2002, 15, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 2003, 183, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.A.R.; Gordon, T. Transforming growth factor-β and forskolin attenuate the adverse effects of long-term Schwann cell denervation on peripheral nerve regeneration in vivo. Glia 2002, 37, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.A.R.; Midha, R.; Munro, C.A.; Matusyama, T.; Al-Majed, A.A.; Gordon, T. Chronic Schwann cell denervation and the presence of a sensory nerve reduce motor axonal denervation. Exp. Neurol. 2002, 176, 342–354. [Google Scholar] [CrossRef]
- Gordon, T.; Tyreman, N.; Raji, M.A. The basis for diminished functional recovery after delayed peripheral nerve repair. J. Neurosci. 2011, 31, 5325–5334. [Google Scholar] [CrossRef]
- Gordon, T.; Brushart, T.M.; Chan, M. Augmenting nerve regeneration with electrical stimulation. Neurol. Res. 2008, 30, 1012–1022. [Google Scholar] [CrossRef]
- Gordon, T. The role of neurotrophic factors in nerve regeneration. Invited review. Neurosurg. Focus. 2009, 26, E3. [Google Scholar] [CrossRef]
- Pfister, B.J.; Gordon, T.; Loverde, J.R.; Mackinnon, S.E.; Cullen, D.K. Biomedical engineering strategies for peripheral nerve repair: Surgical applications, state of the art, and future challenges. Crit. Rev. Biomed. Eng. 2011, 39, 5–48. [Google Scholar] [CrossRef]
- Gordon, T. The Biology, Limits, and Promotion of Peripheral Nerve Regeneration in Rats and Humans. In Nerves and Nerve Injuries. Eds: Tubbs RS, Rizk E, Shoja M, Loukas M, and Spinner RJ. Elsevier, Vol. 2 Ch 61, 2015, pp. 993–1019.
- Rafuse, V.R.; Gordon, T. Self-reinnervated cat medial gastrocnemius muscles. I. Comparisons of the capacity for regenerating nerves to form enlarged motor units after extensive peripheral nerve Injuries. J. Neurophysiol. 1996, 75, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.R.; Gordon, T. Self-reinnervated cat medial gastrocnemius muscles. II. Analysis of the mechanisms and significance of fiber type grouping in reinnervated muscles. J. Neurophysiol. 1996, 75, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.F.; Gordon, T.; Orozco, R. Proportional enlargement of motor units after partial denervation of cat triceps surae muscles. J. Neurophysiol. 1992, 68, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Borschel, G.H. The use of the rat as a model for studying peripheral nerve regeneration and sprouting after complete and partial nerve injuries. Invited review for a special issue on in vivo models of neural regeneration. Exp. Neurol. 2017, 287, 331–347. [Google Scholar] [CrossRef]
- Dyck, P.J.; Boes, C.J.; Mulder, D.; Millikan, C.; Windebank, A.J.; Dyck, J.B.; Espinosa, R. History of standard scoring, notation, and summation of neuromuscular signs. A current survey and recommendation. J. Peripher. Nerv. Syst. 2005, 10, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, E.K.; Faber, C.G.; van Nes, S.I.; Jacobs, B.C.; van Doorn, P.A.; van Doorn, P.A; van Koningsveld, R.; Cornblath, D.R.; van der Kooi, A.J.; Cats, E.A.; van den Berg, L.H.; Notermans, N.C.; van der Pol, W.L.; Hermans, M.C.E.; van der Beek, N.A.M.E.; Gorson, K.C.; Eurelings, M.; Jeroen Engelsman, J.; Hendrik Boot, H.; Meijer, R.J.; Lauria, G.; Tennant, A.; Merkies, I.A.J.; PeriNomS Study Group. Modifying the Medical Research Council grading system through Rasch analyses. Brain 2012, 135, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.R.; Gordon, T. Size of myelinated nerve fibres is not increased by expansion of the peripheral field in cats. J. Physiol. 1998, 532, 835–849. [Google Scholar]
- Dedkov, E.L.; Kostrominova, T.Y.; Borisov, A.B.; Carlson, B.M. Reparative myogenesis in long-term denervated skeletal muscles of adult rats results in a reduction of the satellite cell population. Anat. Rec. 2001, 263, 139–154. [Google Scholar] [CrossRef]
- Schmalbruch, H.; Lewis, D.M. A comparison of the morphology of denervated with aneurally regenerated soleus muscle of rat. Muscle Res. Cell Motil. 1994, 15, 256–266. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Marcelo, C.L.; Kuzon, W.M., Jr. Skeletal muscle denervation increases satellite cell susceptibility to apoptosis. Plast. Reconstr. Surg. 2002, 110, 160–168. [Google Scholar] [CrossRef]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Le Grand, F.; Rudnicki, M.A. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 2007, 19, 628–633. [Google Scholar] [CrossRef]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS 2013, 280, 3991–4003. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Suroto, H.; Wardana, G.R.; Sugianto, J.A.; Aprilya, D.; Samijo, S. Time to surgery and myo-D expression in biceps muscle of adult brachial plexus injury: a preliminary study. BMC Res. Notes 2023, 16, 51. [Google Scholar] [CrossRef]
- Anzil, A.P.; Wernig, A. Muscle fibre loss and reinnervation after long-term denervation. J. Neurocytol. 1989, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Schmalbruch, H.; Lewis, D.M. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and degenerated rat muscles. Muscle Nerve 2000, 23, 617–626. [Google Scholar] [CrossRef]
- Gutmann, E.; Zelena, J. Morphological changes in the denervated muscle. In The Denervated Muscle (edited by Gutmann, E.), 1962, pp. 57–Prague: Publishing House of the Czechoslovak Academy of Sciences.
- Gordon, T.; Tetzlaff, W. Regeneration associated genes decline in injured rat axotomized sciatic motoneurons. Eur. J. Neurosci. 2015, 42, 2783–2791. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; You, S.; Cassar, S.L.; Tetzlaff, W. Reduced expression of regeneration associated genes in chronically axotomized facial motoneurons. Exp. Neurol. 2015, 264, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hoke, A.; Gordon, T.; Zochodne, D.W.; Sulaiman, O.A.R. A decline in glial cell-line-derived neurotrophic factor expression is associated with impaired regeneration after long-term Schwann cell denervation. Exp. Neurol. 2002, 173, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, L.J.; Gomez-Sanchez, J.A.; Fazal, S.V.; Otto, G.W.; et al. Failures of nerve regeneration caused by aging or chronic denervation are rescued by restoring Schwann cell c-Jun. eLife 2021, 10e. [Google Scholar] [CrossRef]
- Hashimoto, M.; Ishii, K.; Nakamura, Y.; Watabe, K.; Kohsaka, S.; Akazawa, C. Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J. Neurochem. 2008, 24, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.R.; Yuk, D.; Alberta, J.A.; Zhu, Z.; Pawlitzky, I.; Chan, J.; McMahon, C.D.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding BHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Trakanant, S.; Nihara, J.; Kudo, T.; Seo, K.l.; Saeki, M.; Kurose, M.; Matsumaru, D.; Maeda, T.; Ohazama, A. Gli3 is a key factor in the Schwann cells from both intact and injured peripheral nerves. Neurosci. 2020, 432, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Shapiro, R.I.; Wang, S.; Chakraborty, A.; Gill, A.; Lepage, D.J.; Wen, D.; Rayhorn, R.; Horan, G.S.B.; Taylor, F.R.; Garber, E.A.; Galdes, A.; Engber, T.M. Long-acting forms of sonic hedgehog with improved pharmacokinetic and pharmacodynamic properties are efficacious in a nerve injury model. J Pharm Sci 2002, 91, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.W.; Angeloni, N.; Harrington, D.; Stupp, S.; Podlasek, C.A. Sonic hedgehog regulates brain-derived neurotrophic factor in normal and regenerating cavernous nerves. J. Sex. Med. 2013, 10, 730–737. [Google Scholar] [CrossRef]
- Martinez, J.A.; Kobayashi, M.; Krishnan, A.; Webber, C.; Christie, K.; Guo, GF.; Singh, V.; Zouchodne, D.W. Intrinsic facilitation of adult peripheral nerve regeneration by the sonic hedgehog morphogen. Exp. Neurol. 2015, 271, 493–505. [Google Scholar] [CrossRef]
- Ma, K.H.; Hung, H.A.; Srinivasan, R.; Xie, H.; Orkin, S.H.; Svaren, J. Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex J. Neurosci. 2015, 35, 8640–8652. [Google Scholar] [CrossRef]
- Boerboom, A.; Dion, V.; Chariot, A.; Franzen, R. Molecular mechanisms involved in Schwann cell plasticity. Frontiers Mol. Neurosci. 2017, 10, 38. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Schaefer, S.A.; Hall, R.A. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. PNAS 2013, 110, 9529–9534. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Campana, W.M.; Mizisin, A.P.; Mohiuddin, L.; O’Brien, J.S. Prosaposin: A myelinotrophic protein that promotes expression of myelin constituents and is secreted after nerve injury. Glia 1999, 26, 353–360. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, R.; Wang, Z.; Lin, G.; Lue, T.F.; Lin, C. Adipose tissue-derived stem cells secrete CXCL5 cytokine with neurotrophic effects on cavernous nerve regeneration. J. Sex. Med. 2011, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Eskin, A.; Chareyre, F.; Jessen, W.J.; Manent, J.; Niwa-Kawakita, M.; Chen, R.; White, C.M.; Jaffer, Z.M.; Nelson, S.F.; Rubenstein, A.E.; Giovannini, M. Therapeutic potential of HSP90 inhibition for neurofibromatosis type Clin. Cancer Res. 2013, 19, 3856–3870. [Google Scholar]
- Weiss, T.; Taschner-Mandl, S.; Bileck, A.; Slany, A.; Kromp, F.; Rifatbegovic, D.; Frech, C.; Windhager, R.; Kitzinger, H.; Tzou, C.-H.; Ambros, P.F.; Gerner, C.; Ambros, I.M. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 2016, 64, 2133–2153. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.H.; Hung, H.A.; Svaren, J. Epigenomic regulation of Schwann cell reprogramming in peripheral nerve injury. J. Neurosci. 2016, 36, 9135–9147. [Google Scholar] [CrossRef] [PubMed]
- Brosius Lutz, A.; Chung, W.S.; Sloan, S.A.; Carson, G.A.; et al. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. PNAS 2017, 114, E8072–E8080. [Google Scholar] [CrossRef]
- Wolter, P.; Schmitt, K.; Fackler, M.; Kremling, H.; Probst, L.; Hauser, S.; Gruss, O.J.; Gaubatz, S. GAS2L3, a target gene of the DREAM complex, is required for proper cytokinesis and genomic stability. J. Cell Sci. 2012, 125, 2393–2406. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; Smith, S.J.; Barres, B.A. A. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Calissano, P. The nerve-growth factor. Sci. Am. 1979, 240, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P. B.; Young, J. Z. Fibrin suture of peripheral nerves. Lancet 1940, 239, 126–128. [Google Scholar]
- Carlsen, R.C. Delayed induction of the cell body response and enhancement of regeneration following a condition/test lesion of frog peripheral nerve at 15 °C. Brain Res. 1983, 279, 9–18. [Google Scholar] [CrossRef]
- Kilmer, S.L.; Carlsen, R.C. Forskolin activation of adenylate cyclase in vivo stimulates nerve regeneration. Nature 1984, 307, 455–457. [Google Scholar] [CrossRef]
- Sjoberg, J.; Kanje, M.; Edstrom, A. Influence of non-neuronal cells on regeneration of the rat sciatic nerve. Brain Res. 1988, 453, 221–226. [Google Scholar] [CrossRef]
- Kanje, M.; Skottner, A.; Sjöberg, J.; Lundborg, G. Insulin-like growth factor I (IGF-I) stimulates regeneration of the rat sciatic nerve. Brain Res. 1989, 486, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, J.; Kanje, M. The initial period of peripheral nerve regeneration and the importance of the local environment for the conditioning lesion effect. Brain Res. 1990, 529, 79–84. [Google Scholar] [CrossRef]
- Holmquist, B.; Kanje, M.; Kerns, J.M.; Danielsen, N. A mathematical model for regeneration rate and initial delay following surgical repair of peripheral nerves. J. Neurosci. Meth 1993, 48, 27–33. [Google Scholar] [CrossRef]
- Wood, M.D.; Kemp, S.W.P; Weber, C.; Borschel, G.H.; Gordon, T. Outcome measures of peripheral nerve regeneration. Ann. Anat. 2011, 193, 321–333. [Google Scholar] [PubMed]
- Fouad, K.; Pearson, K. Restoring walking after spinal cord injury. Prog. Neurobiol. 2004, 73, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Vrbova, G.; Gordon, T.; Jones, R. Nerve-Muscle Interaction 1978, Chapman and Hall, Ltd, Ch 6, p 113.
- Tajdaran, K.; Gordon, T.; Wood, M.D.; Shoichet, M.S.; Borschel, G.H. A glial cell line-derived neurotrophic factor delivery system enhances nerve regeneration across acellular nerve allografts. Acta Biomat 2016, 29, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Hoffer, J.A.; Jhamandas, J.; Stein, R.B. Long-term effects of axotomy on neural activity during cat locomotion. J. Physiol. 1980, 303, 243–263. [Google Scholar] [CrossRef]
- Mendell, L.M.; Munson, J.B.; Scott, J.G. Alterations of synapses on axotomized motoneurons. J. Physiol. 1976, 255, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Titmus, M.; Faber, D. Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog. Neurobiol. 1990, 35, 1–51. [Google Scholar] [CrossRef]
- Moran, L.B.; Graeber, M.B. The facial nerve axotomy model. Brain Res. Rev. 2004, 44, 154. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J.; Bullinger, K.L.; Titus, H.E.; Nardelli, P.; Cope, T.C. Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Ann. NY Acad. Sci. 2010, 1198, 231–241. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Titus-Mitchel, H.E.; Bullinger, K.L.; Kraszpulski, M.; Nardelli, P.; Cope, T.C. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J. Neurophysiol. 2011, 106, 2450–2470. [Google Scholar] [CrossRef]
- Sumner, B.E. A quantitative analysis of boutons with different types of synapse in normal and injured hypoglossal nuclei. Exp. Neurol. 1975, 49, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, D. H. Qualitative and quantitative study of synaptic displacement in chromatolyzed spinal motoneurons of the cat. J. Comp. Neurol. 1978, 177, 635–664. [Google Scholar] [CrossRef]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef]
- Brannstrom, T.; Kellerth, J. O. Changes in synaptology of adult cat spinal alpha-motoneurons after axotomy. Exp. Brain Res. 1998, 118, 1–13. [Google Scholar]
- Lindå, H.; Shupliakov, O.; Ornung, G.; Ottersen, O. P.; Storm-Mathisen, J.; Risling, M.; Cullheim, S. Ultrastructural evidence for a preferential elimination of glutamate-immunoreactive synaptic terminals from spinal motoneurons after intramedullary axotomy. J. Comp. Neurol. 2000, 425, 10–23. [Google Scholar] [CrossRef]
- Oliveira, A. L. R.; Thams, S.; Lidman, O.; Piehl, F.; Hökfelt, T.; Kärre, K.; Lindå, H.; Cullheim, S.A. role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. USA 2004, 101, 17843–17848. [Google Scholar] [CrossRef]
- Rotterman, T.M.; Nardelli, P.; Cope, T.C.; Alvarez, F.J. Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. J. Neurosci. 2014, 34, 3475–3492. [Google Scholar] [CrossRef]
- Liu, C.; Ward, P.J.; English, A.W. The effects of exercise on synaptic stripping require androgen receptor signaling. PLoS One 2014, 9, e98633. [Google Scholar] [CrossRef]
- Brandt, J.; Evans, J.T.; Mildenhall, T.; Mulligan, A.; Konieczny, A.; Rose, S.J; English, A.W. Delaying the onset of treadmill exercise following peripheral nerve injury has different effects on axon regeneration and motoneuron synaptic plasticity. J. Neurophysiol. 2015, 113, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Aldskogius, H.; Liu, L.; Svensson, M. Glial responses to synaptic damage and plasticity. J. Neurosci. Res. 1999, 58, 33–41. [Google Scholar] [CrossRef]
- Krakowiak, J.; Liu, C.; Papudesu, C.; Ward, P.J.; Wilhelm, J.C.; English, A.W. Neuronal BDNF signalling is necessary for the effects of treadmill exercise on synaptic stripping of axotomized motoneurons. Neural Plast. 2015, 11, 392591. [Google Scholar]
- Brushart, T.M.; Seiler, W.A. Selective reinnervation of distal motor stumps by peripheral motor axons. Exp. Neurol. 1987, 97, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Motor axons preferentially reinnervate motor pathways. J. Neurosci. 1993, 13, 2730–2738. [Google Scholar] [CrossRef]
- Gutmann, E.; Gutmann, L.; Medawar, P.K.; Young, J. Z. The rate of regeneration. J. Exp. Biol. 1942, 19, 14–44. [Google Scholar] [CrossRef]
- Witzel, C.; Rohde, C.; Brushart, T.M. Pathway sampling by regenerating peripheral axons. J. Comp. Neurol. 2005, 485, 183–190. [Google Scholar] [CrossRef]
- Burke, R.E. Motor units: anatomy, physiology and functional organization. In Handbook of physiology. Sect. I. Vol. II. The Nervous System: Motor Control. Part I. Edited by V.B. Brooks. American Physiological Society, Washington, DC. 1981, pp. 345–422.
- Zhang, J.Y.; Luo, X.G.; Xian, C.J.; Liu, Z.H.; Zhou, X.F. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents, Eur. J. Neurosci. 2000, 12, 4171–4180. [Google Scholar]
- Griesbeck, O.; Parsadanian, A.S.; Sendtner, M.; Thoenen, H. Expression of neurotrophins in skeletal muscle: Quantitative comparison and significance for motoneuron survival and maintenance of function. J. Neurosci. Res. 1995, 42, 21–33. [Google Scholar] [CrossRef]
- Juckett, L.; Saffari, T.M.; Ormseth, B.; Senger, J.-L.; Moore, A.M. The effect of electrical stimulation on nerve regeneration following peripheral nerve injury. Biomolec 2022, 12, 1856. [Google Scholar] [CrossRef]
- Gordon, T.; Amirjani, N.; Edwards, D.C.; Chan, K. M. Brief post-surgical electrical stimulation accelerates axon regeneration and muscle reinnervation without affecting the functional measures in carpal tunnel syndrome patients. Exp. Neurol. 2010, 223, 192–202. [Google Scholar] [CrossRef]
- Wong, J.N.; Olson, J.L.; Morhart, M.J.; Chan, K.M. Electrical stimulation enhances sensory recovery: a randomized controlled trial. Ann. Neurol. 2015, 77, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Power, H.A.; Morhart, M.J.; Olson, J.L.; Chan, K.M. Postsurgical electrical stimulation enhances recovery following surgery for severe Cubital Tunnel Syndrome: A Double-blind randomized controlled trial. Neurosurg. 2020, 86, 769–777. [Google Scholar] [CrossRef] [PubMed]
- McLean, N.A.; Verge, V. Dynamic impact of brief electrical stimulation on the neural immune axis-polarization of macrophages toward a pro-repair phenotype in demyelinated peripheral nerve. Glia 2016, 64, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, T.; Li, C.; Xu, W.; Guan, Y.; Li, X.; Cheng, H.; Chen, S.; Yang, B.; Liu, Y.; Ren, Z.; Song, X.; Jia, Z.; Wang, Y.; Tang, J. Electrical stimulation accelerates Wallerian degeneration and promotes nerve regeneration after sciatic nerve injury. GLIA 2023, 71, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; McEwan, S.-K.; Kang, S.M.; Won, S.M.; Stephen, M.; Gamble, P.; Xie, Z.; Yan, Y.; Chen, Y.-Y.; Shin, J.; Birenbaum, N.; Chung, S.; Kim, S.B.; Khalifeh, J.; Harburg, D.V.; Bean, K.; Paskett, M.; Kim, J.; Zohny, Z.S.; Lee, S.M.; Zhang, R.; Luo, K.; Ji, B.; Banks, A.; Lee, H.M.; Huang, Y.; Ray, W.Z.; Rogers, J.A. Wireless bioresorbable electronic system enables sustained non-pharmacological neuroregenerative therapy. Nature Med. 2018, 24, 1830–1836. [Google Scholar] [CrossRef]
- Ju, C.; Park, E.; Kim, T.; Kim, T.; Kang, M.; Lee, K.; Park, S.-M. Effectiveness of electrical stimulation on nerve regeneration after crush injury: comparison between invasive and non-invasive stimulation. Plos One 2020, 15, e0233531. [Google Scholar] [CrossRef]
- Yan, X.; Ye, Z.; Huang, J.; He, F.; Xiao, W.; Hu, X.; Luo, Z. CaMKI-mediated CREB phosphorylation is involved in Ca2+-induced BDNF mRNA transcription and neurite outgrowth mediated by electrical stimulation. Plos One 2016, 11, e0162784. [Google Scholar] [CrossRef] [PubMed]
- Sayanagi, J.; Jesus, A.; Acevedo-Cintrog, B.S; Pan, D.; Schellhardt, L.; Snyder-Warwick, A.K.; Mackinnon, S.E.; Wood, M.D. Brief electrical stimulation accelerates axon regeneration and promotes recovery following nerve transection and repair in mice. J. Bone Joint Surg. Am. 2021, 103, e80. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Schellhardt, L.; Keane, G.C.; Hunter, D.A.; Moore, A.M.; Snyder-Warwick, A.K.; Mackinnon, S.E.; Wood, M.D. Short-duration, pulsatile, electrical stimulation therapy accelerates axon regeneration and recovery following tibial nerve injury and repair in rats. Plast. Reconstr. Surg. 2022, 149, 681e–690e. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.P.C.; Lanzinger, W.; Willits, R.K. Effect of intraoperative electrical stimulation on recovery after rat sciatic nerve isograft repair. Neurotrauma Rep 2020, 1, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ecanow, A.; Berglund, K.; Carrasco, D.; Isaacson, R.; English, A.W. Enhancing motor and sensory axon regeneration after peripheral nerve injury using bioluminescent optogenetics. Int. J. Mol. Sci. 2022, 23, 16084. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.J.; Sengelaub, D.R.; English, A.W. Enhancement of peripheral nerve regeneration due to treadmill training and electrical stimulation is dependent on androgen receptor signaling. Dev. Neurobiol. 2014, 74, 531–540. [Google Scholar] [CrossRef]
- Hosli, E.; Jurasin, K.; Ruhl, W.; Luthy, R.; Hosli, L. Colocalization of androgen, estrogen and cholinergic receptors on cultured astrocytes of rat central nervous system. Int. J. Dev. Neurosci. 2001, 19, 11–19. [Google Scholar] [CrossRef]
- Garcia-Ovejero, D.; Veiga, S.; Garcia-Segura, L.; Doncarlo, S.L. Glial expression of estrogen and androgen receptors after rat brain injury. J. Comp. Neurol. 2002, 450, 256–271. [Google Scholar] [CrossRef]
- English, A.W.; Wilhelm, J.C.; Ward, P.J. Exercise, neurotrophins, and axon regeneration in the PNS. Physiol. 2014, 29, 437–445. [Google Scholar] [CrossRef]
- 415 Gordon, T. Neurotrophic factor expression in denervated motor and sensory Schwann cells: Relevance to specificity of peripheral nerve regeneration. Exp. Neurol. 2014, 25, 499–108. [Google Scholar] [CrossRef]
- Franz, C.K.; Rutishauser, U.; Rafuse, V.F. Polysialylated neural cell adhesion molecule is necessary for selective targeting of regenerating motor neurons. J. Neurosci. 2005, 25, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Ying, Z.; Roy, R.R.; Molteni, R.; Edgerton, V.R. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J. Neurophysiol. 2002, 88, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Exercise induces BDNF and synapsin I to specific hippocampal subfields. J. Neurosci. Res. 2004, 76, 356–362. [Google Scholar] [CrossRef]
- Vaynman, S.; Gomez-Pinilla, F. License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehab Neural Res. 2005, 19, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, M.J.; Redmon, N.; Schwartz, G.; English, A.W. Treadmill training promotes axon regeneration in injured peripheral nerves. Exp. Neurol. 2008, 211, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Pinilla, E.; Udina, E.; Jaramillo, J.; Navarro, X. Electrical stimulation combined with exercise increase axonal regeneration after peripheral nerve injury. Exp. Neurol. 2009, 219, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, J.C.; Xu, M.; Cucoranu, D.; Chmielewski, S.; Holmes, T.; Lau, S.L.; Bassell, G.J.; English, A.W. Cooperative roles of BDNF expression in neurons and Schwann cells are modulated by exercise to facilitate nerve regeneration. J. Neurosci. 2012, 32, 5002–5009. [Google Scholar] [CrossRef]
- Wood, K.; Wilhelm, J.C.; Sabatier, M.J.; Liu, K.; Gu, J.; English, A.W. Sex differences in the effectiveness of treadmill training in enhancing axon regeneration in injured peripheral nerves. Dev. Neurobiol. 2012, 72, 688–698. [Google Scholar] [CrossRef]
- English, A.W.; Mulligan, A.; Cucoranu, D.; Rodriguez, J.A.; Sabatier, M.J. Neurotrophin 4/5 is implicated in the enhancement of axon regeneration produced by treadmill training following peripheral nerve injury. Erup J. Neurosci. 2011 33, 2265–2271. [CrossRef]
- Gutmann, E.; Jakoubek, B. Effect of increased motor activity on regeneration of the peripheral nerve in young rats. Physiol. Bohemoslov. 1963, 12, 463–468. [Google Scholar]
- Herbison, G.J.; Jaweed, M.M.; Ditunno, J.F.; Scott, C.M. Effect of overwork during reinnervation of rat muscle. Exp. Neurol. 1973, 41, 1–14. [Google Scholar] [CrossRef]
- Van Meeteren, N.L.; Brakkee, J.H.; Hamers, F.P.; Helders, P.J.; Gispen, W.H. Exercise training improves functional recovery and motor nerve conduction velocity after sciatic nerve crush lesion in the rat. Arch. Phys. Med. Rehab 1997, 78, 70–77. [Google Scholar] [CrossRef]
- Van Meeteren, N.L.; Brakkee, J.H.; Helders, P.J.; Gispen, W.H. The effect of exercise training on functional recovery after sciatic nerve crush in the rat. J. Peripher. Nerv. Syst. 1998, 3, 277–282. [Google Scholar]
- Hutchinson, K.J.; Gomez-Pinilla, F.; Crowe, M.J.; Ying, Z.; Basso, D.M. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain 2004, 127, 1403–1414. [Google Scholar] [CrossRef]
- Molteni, R.; Zheng, J.Q.; Ying, Z.; Gomez-Pinilla, F.; Twiss, J.L. Voluntary exercise increases axonal regeneration from sensory neurons. Proc. Natl. Acad. Sci. USA 2004, 101, 8473–8478. [Google Scholar] [CrossRef]
- Teodori, R.M.; Betini, J.; de Oliveira, L.S.; Sobral, L.L.; Takeda, S.Y.; de Lima Montebelo, M.I. Swimming exercise in the acute or late phase after sciatic nerve crush accelerates nerve regeneration. Neural Plast. 2011, 783901. [Google Scholar] [CrossRef]
- Udina, E.; Puigdemasa, A.; Navarro, X. Passive and active exercise improve regeneration and muscle reinnervation after peripheral nerve injury in the rat. Muscle Nerve 2011, 43, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Ottem, E.N.; Poort, J.E.; Wang, H.; Jordan, C.L; Breedlove, S.M. Differential expression and regulation of brain-derived neurotrophic factor (BDNF) mRNA isoforms in androgen-sensitive motoneurons of the rat lumbar spinal cord. Mol. Cell Endocrinol. 2010, 328, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Preferential reinnervation of motor nerves by regenerating motor axons. J. Neurosci. 1988, 8, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M.; Mesulam, M.M. Alteration in connections between muscle and anterior horn motoneurons after peripheral nerve repair. Science 1980, 208, 603–605. [Google Scholar] [CrossRef] [PubMed]
- English, A.W. Enhancing axon regeneration in peripheral nerves also increases functionally inappropriate reinnervation of targets. Exp. Neurol. 2005, 490, 421–441. [Google Scholar] [CrossRef]
- English, A.W.; Cucoranu, D.; Mulligan, M.; Sabatier, M. Treadmill training enhances axon regeneration in injured mouse peripheral nerves without increased loss of topographic specificity. J. Comp. Neurol. 2009, 517, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.K.; Hinkle, M.L.; Nicolini, J.; Rambo, L.N.; Rexwinkle, A.M.; Rose, S.J.; Sabatier, M.J.; Backus, D.; English, A.W. Misdirection of regenerating axons and functional recovery following sciatic nerve injury in rats. J. Comp. Neurol. 2011, 519, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, M.J.; To, B.N.; Nicolini, J.; English, A.W. Effect of axon misdirection on recovery of electromyographic activity and kinematics after peripheral nerve injury. Cells Tiss. Organs 2011, 193, 298–309. [Google Scholar] [CrossRef]
- Sabatier, M.J.; To, B.N.; Nicolini, J.; English, A.W. Effect of slope and sciatic nerve injury on ankle muscle recruitment and hindlimb kinematics during walking in the rat. J. Exp. Biol. 2011, 214, 1007–1016. [Google Scholar] [CrossRef]
- de Ruiter, G.C.W.; Malessy, M.J.A.; Alaid, A.O.; Spinner, R.J.; Englestad, J.K.; Sorenson, E.J.; Kaufman, K.R.; Dyck, P.J.; Windebank, A.J. Misdirection of regenerating motor axons after nerve injury and repair in the rat sciatic nerve model. Exp. Neurol. 2008, 211, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Senger, J.L.B.; Verge, W.M.K.; Olson, J.L.; Chan, K.M.; Webber, C.A. Electrical stimulation as a conditioning strategy for promoting and accelerating peripheral nerve regeneration. Exp. Neurol. 2017, 302, 75–84. [Google Scholar] [CrossRef]
- Senger, J.L.B.; Verge, W.M.K.; Chan, K.M.; Webber, C.A. The nerve conditioning lesion – a strategy to enhance nerve regeneration. Annals Neurol. 2018, 83, 691–702. [Google Scholar] [CrossRef]
- Senger, J.-L.; Chan, K.M.; Macandili, H.; Chan, A.W.M.; Verge, V.M.K.; Jones, K.E.; Webber, C.A. Conditioning electrical stimulation promotes functional nerve regeneration. Exp. Neurol. 2019, 315, 60–71. [Google Scholar] [CrossRef]
- Senger, J.B.; Chan, A.W.M.; Chan, K.M.; Kwon-Wong, T.; Acton, L.; Olson, J.; Webber, C.A. Conditioning electrical stimulation is superior to postoperative electrical stimulation in enhanced nerve regeneration and functional recovery following nerve graft repair. Neurorehabil Neural Repair. 2020, 34, 299–308. [Google Scholar] [CrossRef]
- Senger, J.B.; Chan, K.M.; Webber, C.A. Conditioning electrical stimulation is superior to postoperative electrical stimulation, resulting in enhanced nerve regeneration and functional recovery. Exp. Neurol. 2020, 325, 113147. [Google Scholar] [CrossRef]
- Senger, J.B.; Rabey, K.N.; Morhart, M.J.; Chan, K.M.; Webber, C.A. Conditioning electrical stimulation accelerates regeneration in nerve transfers. Ann. Neurol. 2020, 88, 363–374. [Google Scholar] [CrossRef]
- Senger, J.-L.B.; Rabey, K.N.; Acton, L.; Lin, Y.-H.; Lingrell, S.; Chan, K.M.; Webber, C.A. Recovering the regenerative potential in chronically injured nerves by using conditioning electrical stimulation. J. Neurosurg. 2021, 136, 1442–1454. [Google Scholar] [CrossRef]
- Richardson, P.M.; Verge, V.M. Axonal regeneration in dorsal spinal roots is accelerated by peripheral axonal transection. Brain Res. 1987, 411, 406–408. [Google Scholar] [CrossRef]
- Arntz, C.; Kanje, M.; Lundborg, G. Regeneration of the rat sciatic nerve after different conditioning lesions: effects of the conditioning interval. Microsurg. 1989, 10, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.M.; McQuarrie, I.G. Acceleration of axonal outgrowth in rat sciatic nerve at one week after axotomy. J. Neurobiol. 1993, 24, 356–367. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I.G.; Grafstein, B. Axon outgrowth enhanced by a previous nerve injury. Arch. Neurol. 1973, 29, 53–55. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I.G. Structural protein transport in elongation of motor axons after sciatic nerve crush: Effect of a conditioning lesion. Neurochem. Pathol. 1986, 5, 5,153–164. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.E.; McQuarrie, I.G. Increased slow transport in axons of regenerating Newt limbs after a nerve conditioning lesion. Dev. Biol. 1990, 140, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, K.; Hashimoto, K.; Lundborg, G. A role of migratory Schwann cells in a conditioning effect of peripheral nerve regeneration Exp. Neurol. 1999, 160, 99–108. [Google Scholar]
- Hoffman, P.N. A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp. Neurol. 2010, 223, 11–18. [Google Scholar] [CrossRef]
- Neumann, S.; Bradke, F.; Tessier-Lavigne, M.; Basbaum, A.I. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 2002, 34, 885–893. [Google Scholar] [CrossRef]
- Udina, E.; Furey, M.; Busch, S.; Silver, J.; Gordon, T.; Fouad, K. Electrical stimulation of intact peripheral sensory axons in rats promotes outgrowth of their central projections. Exp. Neurol. 2008, 210, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, C.; Beal, D.; Leechavengvongs, S.; Salon, A.; Dauge, M.C.; Sarcy, J.J. Nerve transfer to biceps muscle using a part of ulnar nerve for C5-C6 avulsion of the brachial plexus: anatomical study and report of four cases. J. Hand Surg. Am. 1994, 19, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Teboul, F.; Kakkar, R.; Ameur, N.; Beaulieu, J.-Y.; Oberlin, C. Transfer of fascicles from the ulnar nerve to the nerve to the biceps in the treatment of upper brachial plexus palsy. J. Bone J. Surg. Am. 2004, 86, 1485–1590. [Google Scholar] [CrossRef] [PubMed]
- Midha, R. Nerve transfers for severe brachial plexus injuries: a review. Neurosurg. Focus. 2004, 16, 1–10. [Google Scholar] [CrossRef]
- Midha, R. Techniques and options in nerve reconstruction and repair. in Youmans Neurological Surgery, 6th Ed, 2011, Ch 239, 2456-3464.
- Mackinnon, S.E. Donor distal, recipient proximal and other personal perspectives on nerve transfers. Hand Clin. 2016, 32, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.M.; Wagner, I.J.; Fox, I.K. Principles of nerve repair in complex wounds of the upper extremity. Semin. Plastic Surg. 2015, 29, 40–47. [Google Scholar] [CrossRef]
- Peters, B.R.; Van Handel, A.C.; Russo, S.A.; Moore, A.M. five reliable nerve transfers for the treatment of isolated upper extremity nerve injuries. Plast. Reconstr. Surg. 2021, 147, 830e. [Google Scholar] [CrossRef]
- Viterbo, F.; Trindade, J.C.; Hoshino, K.; Mazzoni, A. Two end-to-side neurorrhaphies and nerve graft with removal of the epineural sheath: experimental study in rats. Br. J. Plast. Surg. 1994, 47, 75–80. [Google Scholar] [CrossRef]
- Viterbo, F.; Amr, A.H.; Stipp, E.J.; Reis, F.J. End-to-side neurorrhaphy: past, present, and future. Plast. Reconstr. Surg. 2009, 124, e351–e358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fischer, K.A. End-to-side neurorrhaphy. Microsurgery 2002, 22, 122–127, 109. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, Y.; Abe, H. Hypoglossal-facial nerve side-to-end anastomosis for preservation of hypoglossal function: results of delayed treatment with a new technique. J. Neurosurg. 1997, 86, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Soucacos, P.N.; Bo, J.; Beris, A.E. Evaluation of collateral sprouting after end-to-side nerve coaptation using a fluorescent double-labeling technique. Microsurg. 1999, 19, 281–286. [Google Scholar] [CrossRef]
- Placheta, E.; Wood, M.D.; Lafontaine, C.; Liu, E.H.; Hendry, J.M.; Angelov, D.N.; Frey, M.; Gordon, T.; Borschel, G.H. Enhancement of facial nerve motoneuron regeneration through cross-face nerve grafts by adding end-to-side sensory axons. Plast. Reconstr. Surg. 2015, 135, 460–71. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.; Gordon, T. A rat study of the use of an end-to-side repair as a ‘baby-sitting’ technique to reduce the deleterious effect of chronic denervation. J. Neurosurg. 2018, 131, 6221–632. [Google Scholar]
- Placheta, E.; Wood, M.D.; Lafontaine, C.; Frey, M.; Gordon, T.; Borschel, G.H. Macroscopic in vivo imaging of facial nerve regeneration in Thy1-GFP rats. JAMA Facial Plast. Surg. 2015, 17, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Placheta, E.; Borschel, G.H. Delayed peripheral nerve repair: methods, including surgical ‘cross-bridging’, to promote nerve regeneration. Neural Regen. Res. 2015, 10, 1540–1544. [Google Scholar] [CrossRef]
- Yuksel, F.; Karacaoglu, E.; Guler, M.M. Nerve regeneration through side-to-side neurorrhaphy sites in a rat model: a new concept in peripheral nerve surgery. Plast. Reconstr. Surg. 1999, 104, 2092–2099. [Google Scholar] [CrossRef]
- Ladak, A.l.; Schembri, P.; Olson, J.; Udina, E.; Tyreman, N.; Gordon, T. Side-to-side nerve grafts sustain chronically denervated peripheral nerve pathways during axon regeneration and result in improved functional reinnervation. Neurosurg. 2011, 68, 1654–65. [Google Scholar] [CrossRef]
- Gordon, T.; Hendry, M.; Lafontaine, C.A.; Cartar, H.; Zhang, J.J.; Borschel, G.H. Nerve cross-bridging to enhance nerve regeneration in a rat model of delayed nerve repair. PLoS One 2015, 10, e0127397. [Google Scholar] [CrossRef]
- Hendry, J.M.; Alvarez-Veronesi, C.; Snyder-Warwick, A.; Gordon, T.; Borschel, G.H. Side-to-side nerve bridges support donor axon regeneration Into chronically denervated nerves and are associated with characteristic changes in Schwann cell phenotype. Neurosurg. 2015, 77, 803–813. [Google Scholar] [CrossRef]
- Pearse, D.D.; Pereira, F.C.; Marcillo, A.E.; Bates, M.L.; Berrocal, Y.A.; Filbin, M.T; Bunge, M.B. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004, 10, 610–616. [Google Scholar] [CrossRef]
- Krause, W.; Kulne, G. Pharmacokinetics of rolipram in rhesus and cynomolgus monkeys, the rat and the rabbit. Studies on species differences. Xenobiotica 1988, 18, 561–571. [Google Scholar] [CrossRef]
- Kilmer, S.L.; Carlsen, R.C. Forskolin activation of adenylate cyclase in vivo stimulates nerve regeneration. Nature 1984, 307, 455–457. [Google Scholar] [CrossRef]
- Cai, D.; Cao, Z.; McAtee, M.; Bregman, B.S.; Filbin, M.T. Neuronal cyclic AMP controls the developmental loss of ability of axons to regenerate. J. Neurosci. 2001, 21, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Hannila, S.S.; Filbin, M.T. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp. Neurol. 2008, 209, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Cai, D.; Dai, H.; McAtee, M.; Hoffman, P.N.; Bregman, B.S.; Filbin, M.T. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 2002, 34, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nikulina, E.; Mellado, W.; Filbin, M.T. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J. Neurosci. 2003, 23, 11770–11777. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Woolf, C.J. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 1999, 23, 83–91. [Google Scholar] [CrossRef]
- Oudega, M.; Varon, S.; Hagg, T. Distribution of corticospinal motor neurons in the postnatal rat: quantitative evidence for massive collateral elimination and modest cell death. J. Comp. Neurol. 1994, 347, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, Y.; Bo, X.; Huang, W.; Fang, X.; Zhang, X.; Miao, T.; Subang, M.C.; Richardson, P.M. Actions of neuropoietic cytokines and cyclic AMP in regenerative conditioning of rat primary sensory neurons. Exp. Neurol. 2007, 204, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Han, P.J.; Shukla, S.; Subramanian, P.S.; Hoffman, P.N. Cyclic AMP elevates tubulin expression without increasing intrinsic axon growth capacity. Exp. Neurol. 2004, 189, 293–302. [Google Scholar] [CrossRef]
- Dusart, I.; Schwab, M.E. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur. J. Neurosci. 1994, 6, 712–724. [Google Scholar] [CrossRef]
- Popovich, P.G.; Longbrake, E.E. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 2008, 9, 481–493. [Google Scholar] [CrossRef]
- Kurimoto, T.; Yin, Y.; Habboub, G.; Gilbert, H.Y.; Li, Y.; Nakao, S.; Hafezi-Moghadam, A.; Benowitz, L.I. Neutrophils express oncomodulin and promote optic nerve regeneration. J. Neurosci. 2013, 33, 14816–14824. [Google Scholar] [CrossRef]
- Yin, Y.; Henzl, M.T.; Lorber, B.; Nakazawa, T.; Thomas, T.T.; Jiang, F.; Langer, R.; Benowitz, L.I. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat. Neurosci. 2006, 9, 843–852. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs. Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs. Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
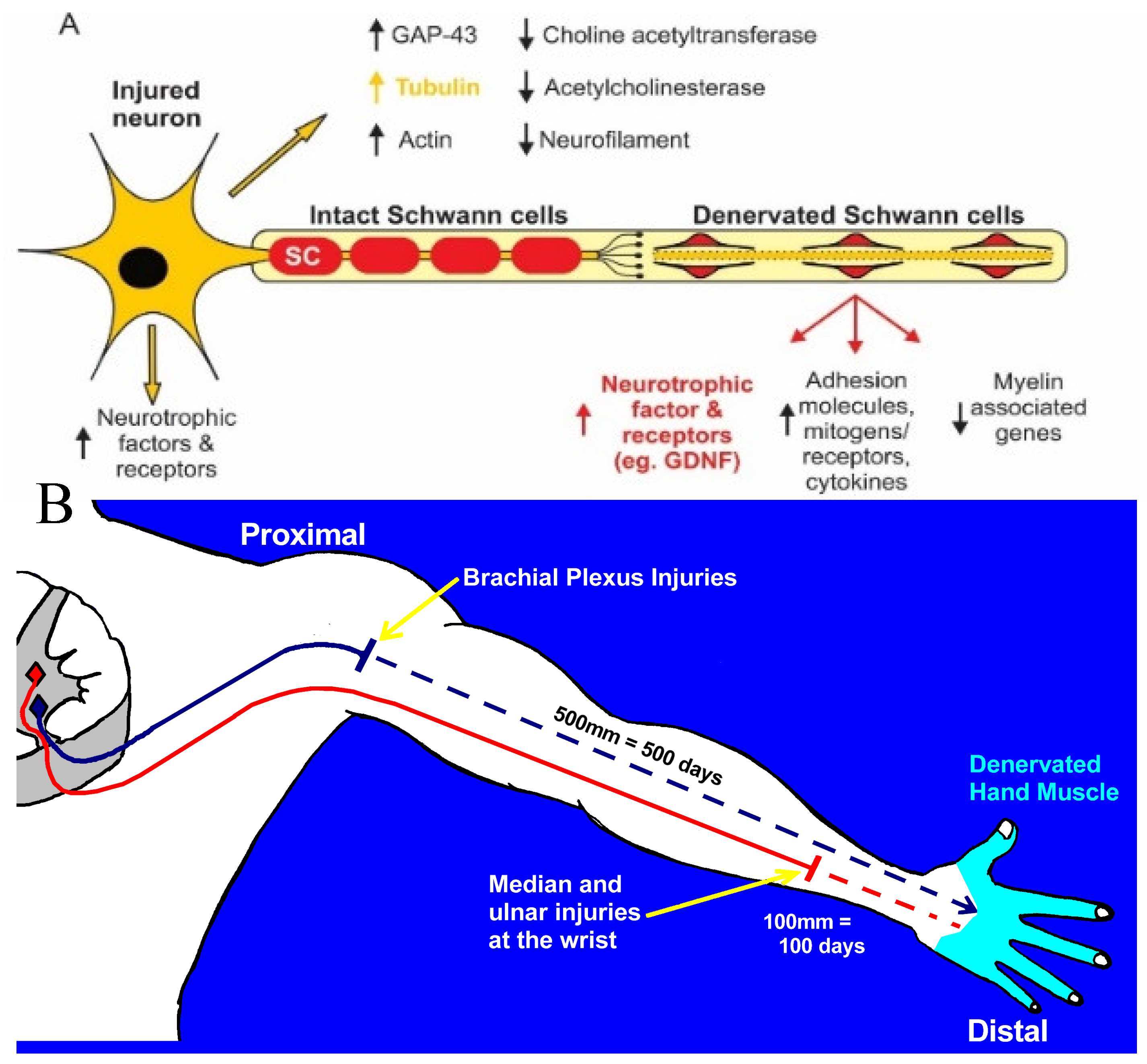
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
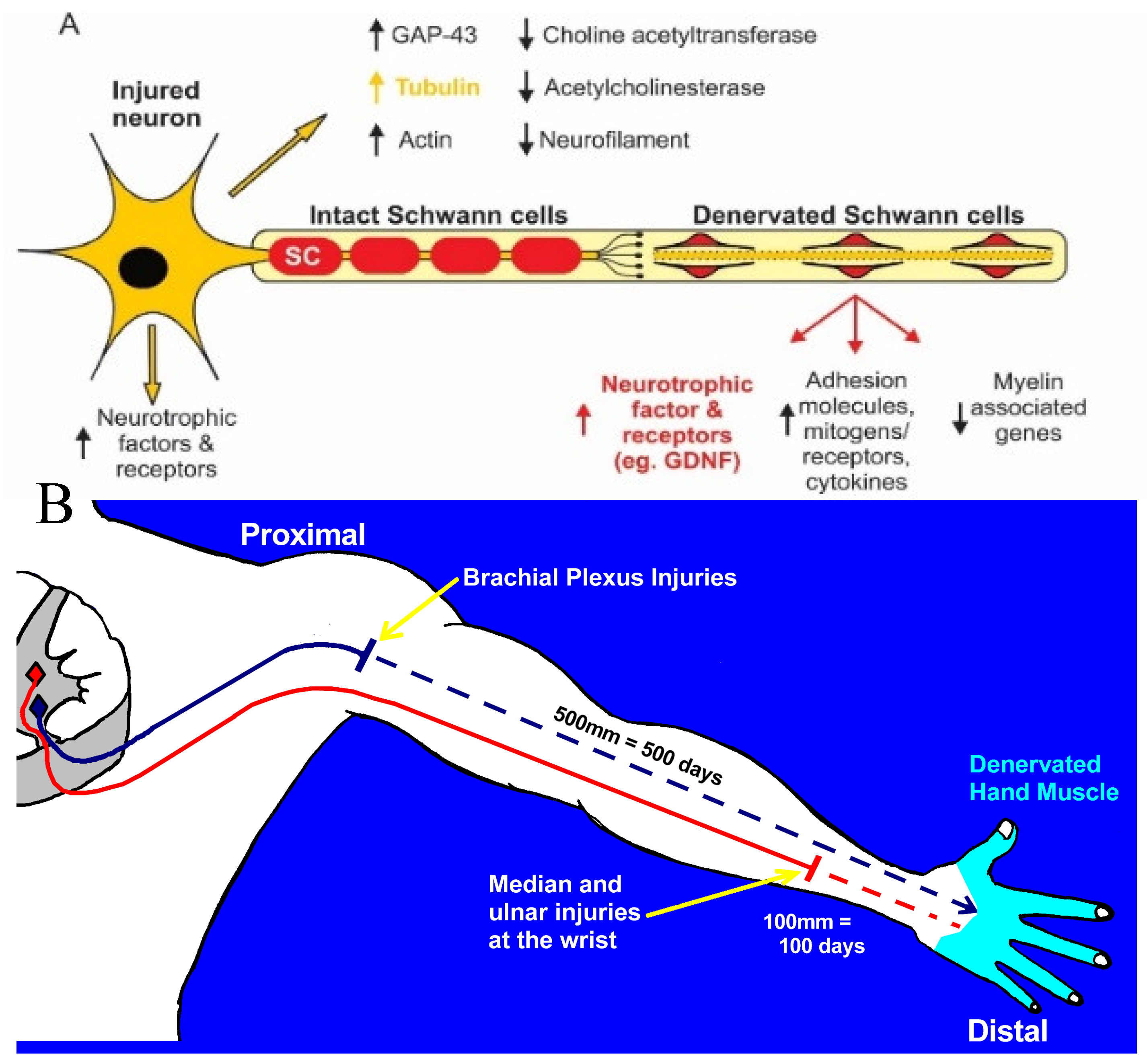
Figure 2.
Survival of chronically denervated Schwann cells (SCs) and their capacity to proliferate, myelinate neurites and nerve fibres in vitro and in vivo, respectively, and to support recovery of reinnervated nerve fibre size. (A) Sterile surgery to prolong SC denervation in the rat sciatic distal nerve stump by cutting the nerve and ligating the proximal and distal nerve stumps to innervated muscles, (B) The left sciatic distal nerve stump remained denervated for either 7 days, 7 weeks, or 17 months and the right sciatic nerve was denervated for 7 days for direct comparison of the data of acutely denervated nerves with the data from chronically denervated nerves. Histograms of (C) the numbers of non-neural cells in the sciatic distal nerve stump, their numbers after exposure to the mitogen vitamin C, and the percentage of the dorsal root ganglion neurons myelinated by SCs in cocultures. (D) Myelination of tibial nerves that regenerated into 1-3 month chronically denervated common peroneal distal nerve stumps in vivo.
Figure 2.
Survival of chronically denervated Schwann cells (SCs) and their capacity to proliferate, myelinate neurites and nerve fibres in vitro and in vivo, respectively, and to support recovery of reinnervated nerve fibre size. (A) Sterile surgery to prolong SC denervation in the rat sciatic distal nerve stump by cutting the nerve and ligating the proximal and distal nerve stumps to innervated muscles, (B) The left sciatic distal nerve stump remained denervated for either 7 days, 7 weeks, or 17 months and the right sciatic nerve was denervated for 7 days for direct comparison of the data of acutely denervated nerves with the data from chronically denervated nerves. Histograms of (C) the numbers of non-neural cells in the sciatic distal nerve stump, their numbers after exposure to the mitogen vitamin C, and the percentage of the dorsal root ganglion neurons myelinated by SCs in cocultures. (D) Myelination of tibial nerves that regenerated into 1-3 month chronically denervated common peroneal distal nerve stumps in vivo.

Figure 3.
Physiological and morphological measures of regenerative success after nerve injuries. Diagrammatic illustration of surgeries in Sprague Dawley rats for (A) prolonging TIB (tibial) motoneuron axotomy and (B) CP (common peroneal) distal nerve stump and TA (tibialis anterior) muscle denervation. Each of the nerve stumps were sutured to innervated muscle to prevent nerve regeneration for up to 12 months prior to refreshing the proximal TIB nerve stump to cross-suture it to the distal nerve stump of a freshly cut CP nerve. (C) Recordings made at least 5 months later of muscle and single MUs (motor units) in response to stimulation of isolated ventral root filaments and (D) retrograde labeling of TIB motoneurons that regenerated their nerve fibres into the CP distal nerve stump. (E) Examples of sagittal sections of the lumbosacral spinal cord in which TIB motoneurons were retrogradely labelled with fluorescent dyes, FG (fluorogold) and FR (fluororuby), and (F) the number of the motoneurons retrogradely labelled with FG or FR were equal such that either dye could be used for counting motoneurons as well as sensory neurons that regenerate their axons after experimental nerve injuries.
Figure 3.
Physiological and morphological measures of regenerative success after nerve injuries. Diagrammatic illustration of surgeries in Sprague Dawley rats for (A) prolonging TIB (tibial) motoneuron axotomy and (B) CP (common peroneal) distal nerve stump and TA (tibialis anterior) muscle denervation. Each of the nerve stumps were sutured to innervated muscle to prevent nerve regeneration for up to 12 months prior to refreshing the proximal TIB nerve stump to cross-suture it to the distal nerve stump of a freshly cut CP nerve. (C) Recordings made at least 5 months later of muscle and single MUs (motor units) in response to stimulation of isolated ventral root filaments and (D) retrograde labeling of TIB motoneurons that regenerated their nerve fibres into the CP distal nerve stump. (E) Examples of sagittal sections of the lumbosacral spinal cord in which TIB motoneurons were retrogradely labelled with fluorescent dyes, FG (fluorogold) and FR (fluororuby), and (F) the number of the motoneurons retrogradely labelled with FG or FR were equal such that either dye could be used for counting motoneurons as well as sensory neurons that regenerate their axons after experimental nerve injuries.

Figure 4.
Decline in regenerative capacity of rat lumbosacral motoneurons (MNs) after chronic axotomy and prolonged Schwann cell denervation. (A,B) Plots of the number of reinnervated MUs (motor units) and TIB (tibial) motoneurons that regenerated their axons into the CP (common peroneal) distal nerve stump as a function of days of (A) chronic axotomy and (B) chronic denervation after delayed cross-suture of (A) axotomized proximal TIB nerve to freshly denervated CP distal nerve stump or (B) freshly axotomized TIB nerve to chronically denervated CP stump. Both MU and MN numbers declined exponentially as a function of days after chronic axotomy or chronic denervation. The effects are shown figuratively in (C,D) respectively for regeneration 4 months after TIB-CP cross suture. The methods used to determine regenerative success are shown in Figure 1.
Figure 4.
Decline in regenerative capacity of rat lumbosacral motoneurons (MNs) after chronic axotomy and prolonged Schwann cell denervation. (A,B) Plots of the number of reinnervated MUs (motor units) and TIB (tibial) motoneurons that regenerated their axons into the CP (common peroneal) distal nerve stump as a function of days of (A) chronic axotomy and (B) chronic denervation after delayed cross-suture of (A) axotomized proximal TIB nerve to freshly denervated CP distal nerve stump or (B) freshly axotomized TIB nerve to chronically denervated CP stump. Both MU and MN numbers declined exponentially as a function of days after chronic axotomy or chronic denervation. The effects are shown figuratively in (C,D) respectively for regeneration 4 months after TIB-CP cross suture. The methods used to determine regenerative success are shown in Figure 1.
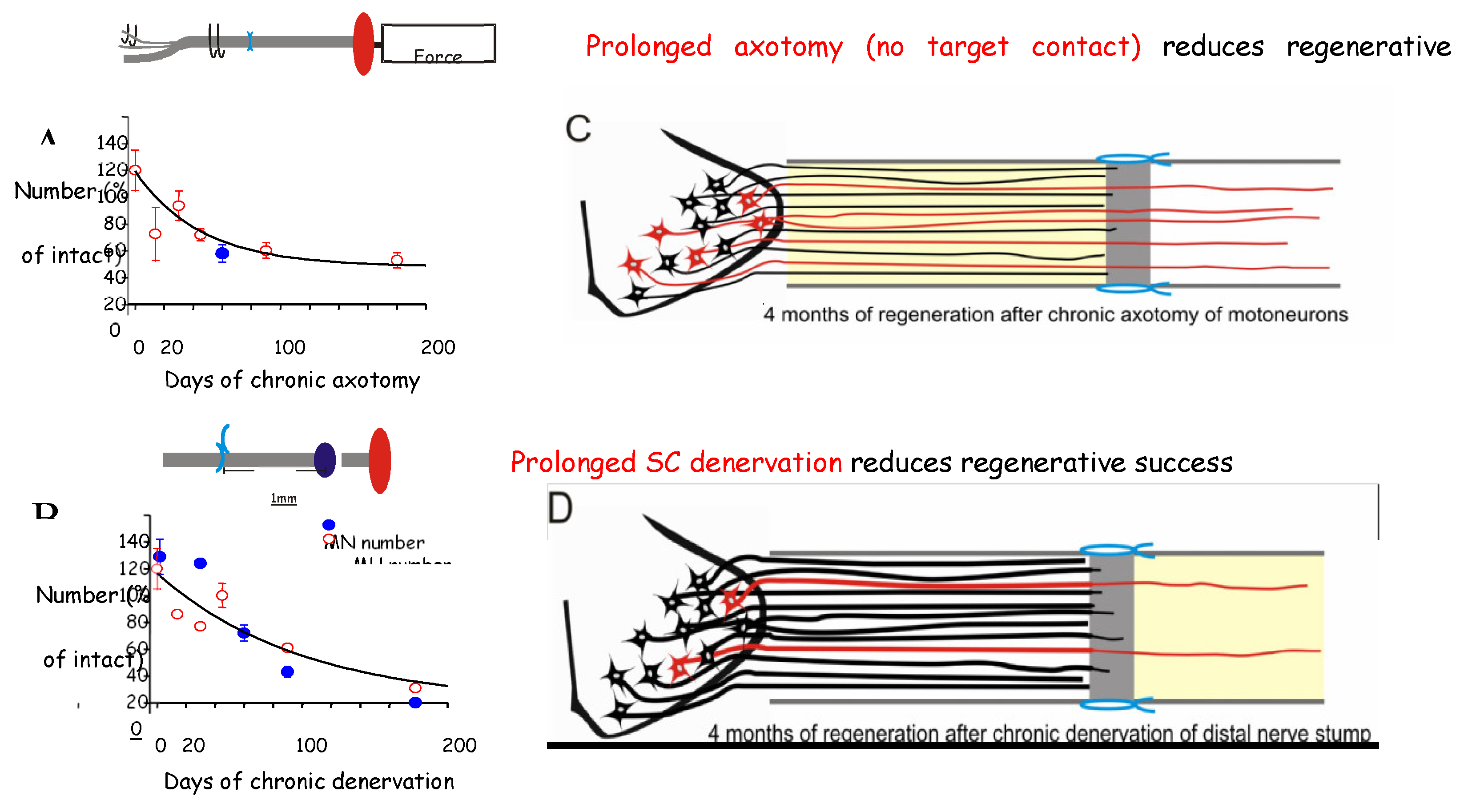
Figure 5.
Expression of mRNA of cytoskeletal proteins in motoneurons decays with time after axotomy. (A) Sciatic nerve section and ligation of the proximal nerve stump to nearby innervated muscle to prevent nerve regeneration. (B,C) Time-dependent decay in the in-situ hybridization signals of tubulin mRNA in the axotomized motoneurons. (D) Decay in expression of tubulin with time and the accelerated decay after elevation of expression after a 1 to 6-month refreshment injury. Histograms of expression of (E) tubulin, (F) actin, (G) GAP-43 with time after axotomy and (H-J) months after axotomy and a refreshment injury. Mean values + standard error bars are shown with significance at p<0.05 and p<0.01 denoted by 1 and 2 *s.
Figure 5.
Expression of mRNA of cytoskeletal proteins in motoneurons decays with time after axotomy. (A) Sciatic nerve section and ligation of the proximal nerve stump to nearby innervated muscle to prevent nerve regeneration. (B,C) Time-dependent decay in the in-situ hybridization signals of tubulin mRNA in the axotomized motoneurons. (D) Decay in expression of tubulin with time and the accelerated decay after elevation of expression after a 1 to 6-month refreshment injury. Histograms of expression of (E) tubulin, (F) actin, (G) GAP-43 with time after axotomy and (H-J) months after axotomy and a refreshment injury. Mean values + standard error bars are shown with significance at p<0.05 and p<0.01 denoted by 1 and 2 *s.
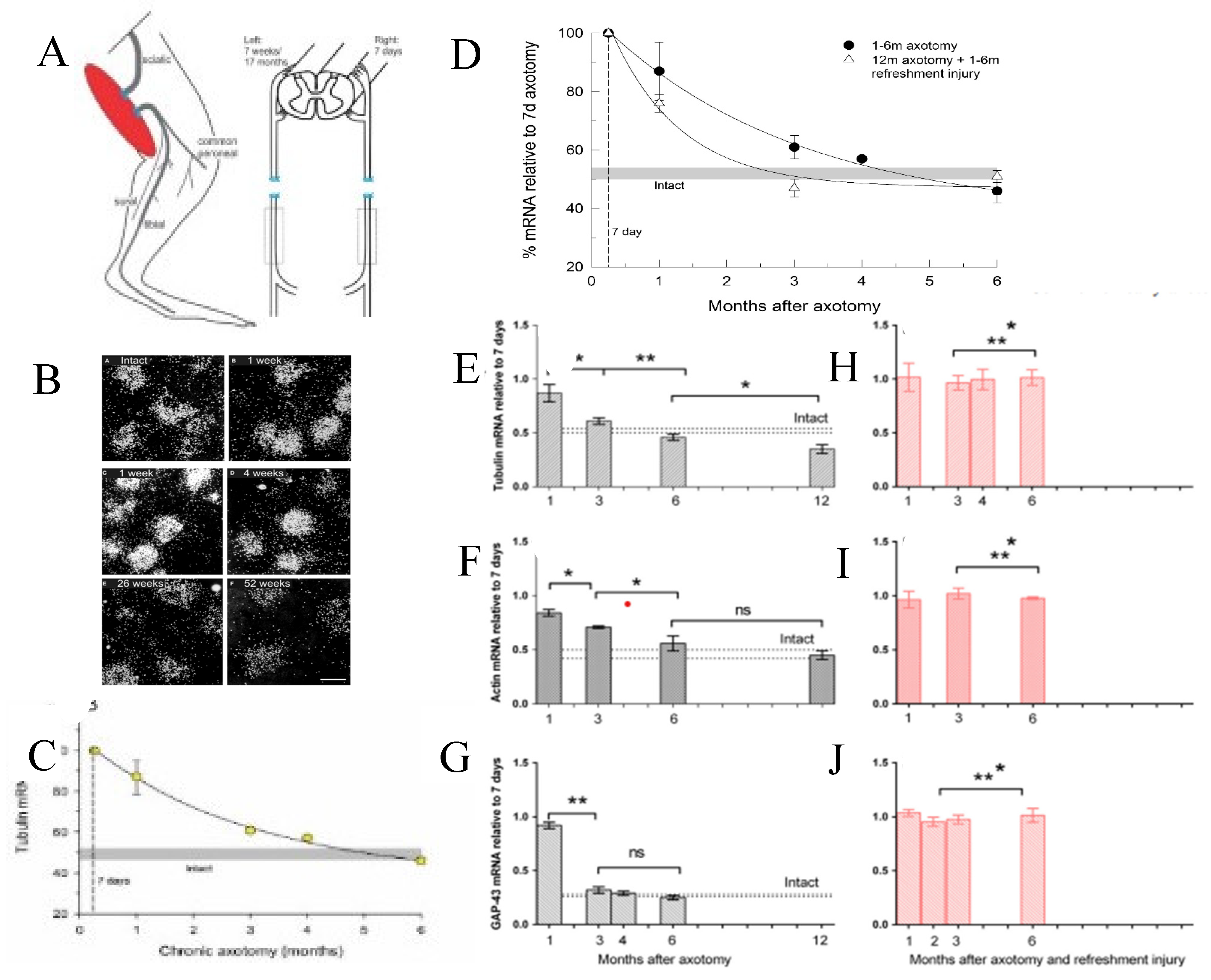
Figure 6.
Expression of mouse Schwann cell (SC) transcription factor c-Jun and the neurotrophic factor receptor (p75NTR) with time after chronic denervation. (A) Representative Western blots of c-Jun and the standard, Calnexin. (B) The quantification of the c-Jun protein data with densitometry normalized to the data at 1-week post-injury in uninjured (UI) nerve and distal nerve stumps after 1-,3-,6-,8-, and 10-weeks of chronic denervation. C. Western blots of c-Jun expression 1 week after injury in wild-type (WT) and c-Jun knockout (cKO) mice, and (D) the quantification of the data with densitometry. (E) Densitometry data of p75NTR with time after injury. (F) Representative Western blots of c-Jun and p75NTR in SC cultures after two and nine passages and. the quantification of the data with densitometry of (G) c-Jun and (H) p75NTR. Mean values + one standard error bar are shown with significance at p<0.05, <0.01 and <0.001 denoted by 1,2, and 3 *s Adapted from [340].
Figure 6.
Expression of mouse Schwann cell (SC) transcription factor c-Jun and the neurotrophic factor receptor (p75NTR) with time after chronic denervation. (A) Representative Western blots of c-Jun and the standard, Calnexin. (B) The quantification of the c-Jun protein data with densitometry normalized to the data at 1-week post-injury in uninjured (UI) nerve and distal nerve stumps after 1-,3-,6-,8-, and 10-weeks of chronic denervation. C. Western blots of c-Jun expression 1 week after injury in wild-type (WT) and c-Jun knockout (cKO) mice, and (D) the quantification of the data with densitometry. (E) Densitometry data of p75NTR with time after injury. (F) Representative Western blots of c-Jun and p75NTR in SC cultures after two and nine passages and. the quantification of the data with densitometry of (G) c-Jun and (H) p75NTR. Mean values + one standard error bar are shown with significance at p<0.05, <0.01 and <0.001 denoted by 1,2, and 3 *s Adapted from [340].
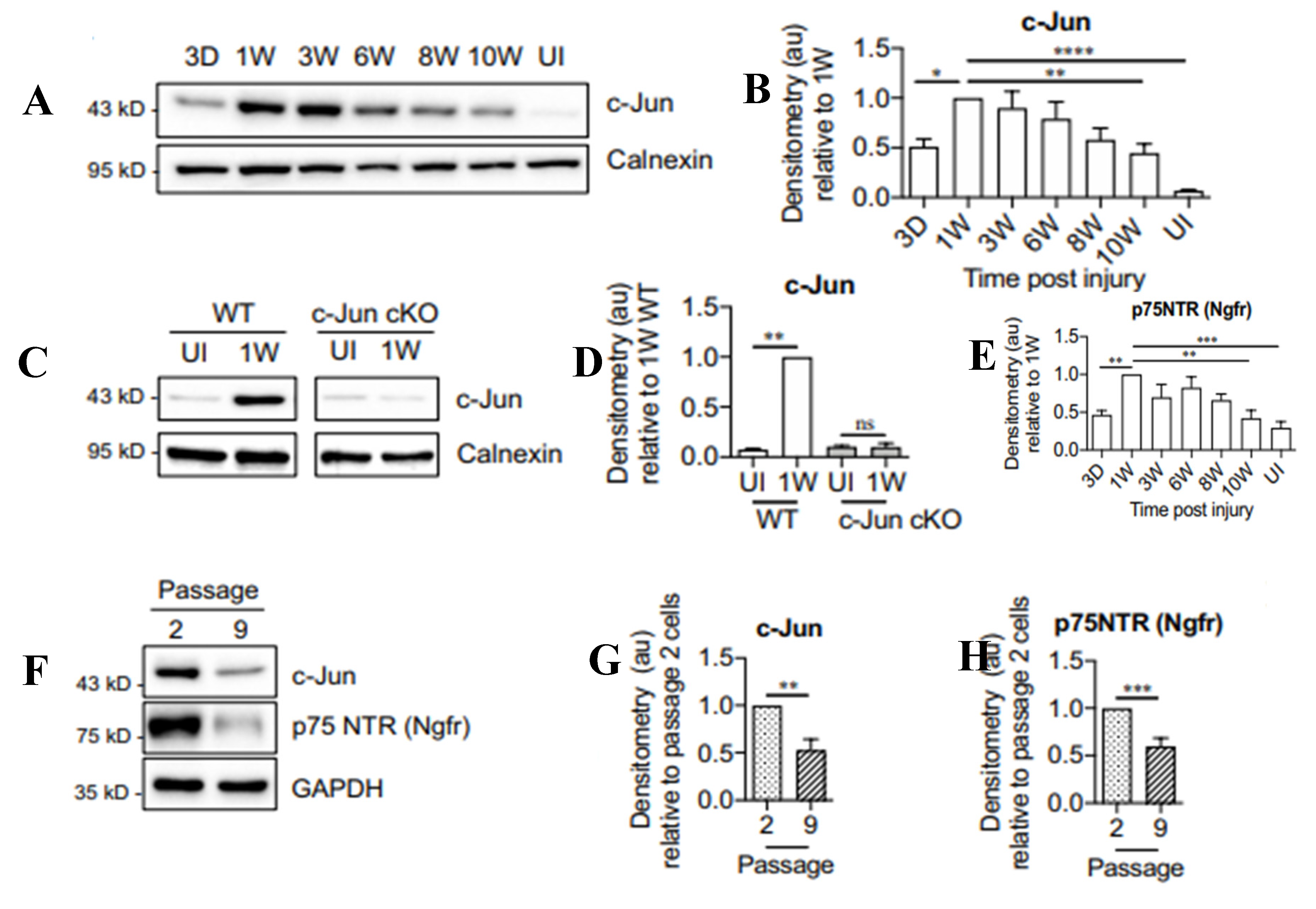
Figure 7.
The neurotrophic factor receptor (p75NTR) mRNA and protein in rat Schwann cells (SCs) and the expression of glial cell neurotrophic factor (GDNF) in the denervated distal nerve stump as a function of time after chronic denervation (A) Rat sciatic nerve section and ligation of the proximal and distal nerve stumps to nearby innervated muscle to prevent nerve regeneration. (B) p75NTR mRNA, depicted as dark grains following in situ hybridization histochemistry in intact sciatic nerve and transected distal nerve stumps 7 days and 4 months after chronic denervation, with a plot of the p75NTR mRNA as a function of the duration of chronic denervation. (C) The relative density of p75NTR immunohistochemistry [shown in with (D) the DAB reaction in short-term (1 week) and long-term (>1 month) denervated distal nerve stumps] plotted as a function of time. (E) Changes in mRNA levels of GDNF, Neurturin (NTN), Persiphin (PSP), and Artemin (ART) mRNAs, as measured by semiquantitative PCR with GAPDH as an internal control and expressed as the change in the transected distal nerve stumps vs the sham-operated control sciatic nerves, as a function of chronic denervation. The data are plotted in (F) as histograms and in (G) as a plot of the mean values of the GDNF mRNA as a function of time of chronic distal nerve denervation. Mean values + one standard error bar are shown with significance at p<0.005 denoted by a *.
Figure 7.
The neurotrophic factor receptor (p75NTR) mRNA and protein in rat Schwann cells (SCs) and the expression of glial cell neurotrophic factor (GDNF) in the denervated distal nerve stump as a function of time after chronic denervation (A) Rat sciatic nerve section and ligation of the proximal and distal nerve stumps to nearby innervated muscle to prevent nerve regeneration. (B) p75NTR mRNA, depicted as dark grains following in situ hybridization histochemistry in intact sciatic nerve and transected distal nerve stumps 7 days and 4 months after chronic denervation, with a plot of the p75NTR mRNA as a function of the duration of chronic denervation. (C) The relative density of p75NTR immunohistochemistry [shown in with (D) the DAB reaction in short-term (1 week) and long-term (>1 month) denervated distal nerve stumps] plotted as a function of time. (E) Changes in mRNA levels of GDNF, Neurturin (NTN), Persiphin (PSP), and Artemin (ART) mRNAs, as measured by semiquantitative PCR with GAPDH as an internal control and expressed as the change in the transected distal nerve stumps vs the sham-operated control sciatic nerves, as a function of chronic denervation. The data are plotted in (F) as histograms and in (G) as a plot of the mean values of the GDNF mRNA as a function of time of chronic distal nerve denervation. Mean values + one standard error bar are shown with significance at p<0.005 denoted by a *.
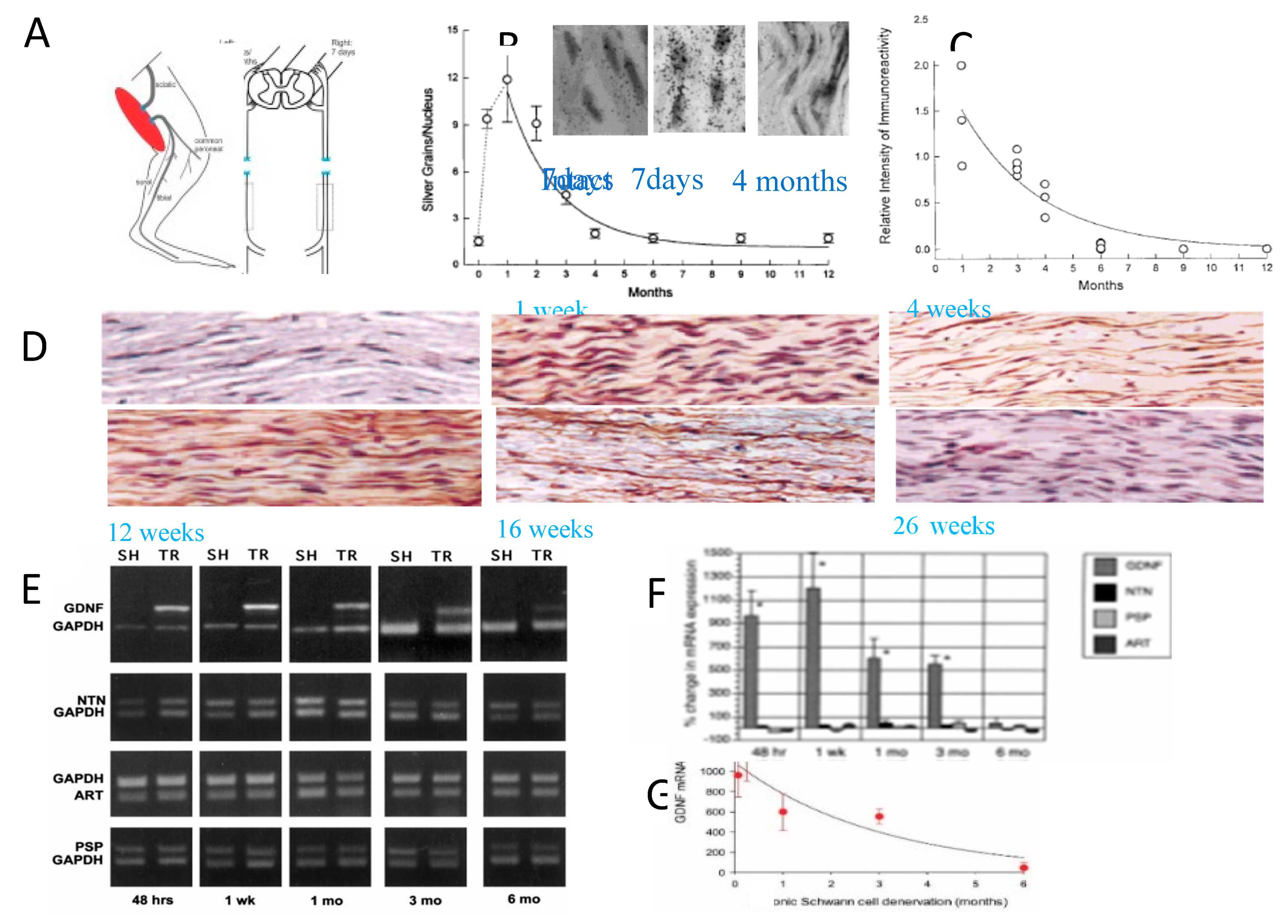
Figure 8.
Exogenous delivery of brain- and glial cell-derived neurotrophic factors, GDNF and BDNF respectively, was effective in improving nerve regeneration after chronic axotomy but, increased axon sprouting in the distal nerve stump. (A) The TIB (tibial nerve) in the rat was cut and ligated. (B) After 2 months, the proximal stump of the TIB nerve was cross-sutured to the freshly denervated CP (common peroneal) nerve stump and (C) a mini-osmotic pump was placed on the back of the rat for constant infusion of low dose BDNF (2ug/day for 28 days). (D) Following a 2-month period of chronic axotomy, fluororuby was administered to the CP distal nerve stump 20 mm from the cross-suture site for retrograde labeling of the TIB motoneurons that had regenerated their axons into the denervated CP distal nerve stump. (E) The numbers of motoneurons that regenerated their axons and were counted in 50 µm longitudinal sections of the ventral horn of the spinal cord [all sections were counted and a correction factor of 0.6 applied [313,314,315]. The number of motoneurons that regenerated their axons after 2 months of chronic axotomy was significantly reduced. GDNF and BDNF were both effective in increasing the motoneuron regeneration to normal levels and the combination elevated the levels even more. (F) Local delivery of GDNF to the suture sites of an acellular nerve allograft (ANA) interposed between the CP proximal and distal nerve stumps. GDNF was encapsulated with an efficiency of 78 + 3% and GDNF loading of 0.72 + 0.08 µg/mg of microspheres (MS; diameters of 45 + 5 µm), placed onto the suture sites and held in place by surrounding them with 2 µL gels placed below and above the suture sites that adhered to one another. (G) Retrograde labeling of the CP nerve 10 mm from the ASA 8 weeks later. (H) Numbers of motoneurons that regenerated their axons through the ANA plotted for control conditions of no delivery system (DDS) and empty MS and experimental conditions of GDNF delivery in combinations of 2- and 4-week formulations. (I) Numbers of sensory neurons counted in every fifth dorsal root ganglia 20 µm section. Irrespective of the nature of GDNF delivery, all motor and sensory neurons regenerated their axons through the ANA in 8 weeks as compared to ~50% of them that regenerated through the ANAs that did not deliver GDNF.(J) Electron micrographs of Schwann cells (SCs) and their axons in intact nerves, regenerating nerves in the distal nerve stump 2 months after TIB-CP cross-suture with saline administration via a cuff around the suture site linked to a mini-osmotic pump, GDNF delivery to the cross-suture site via the pump, and delivery of both GDNF and BDNF via to pump. (K) The mean + standard error values of the number of axons within each endoneurial sheath associated with a single SC in the control (A and B), C. GDNF and D. GDNF + BDNF.
Figure 8.
Exogenous delivery of brain- and glial cell-derived neurotrophic factors, GDNF and BDNF respectively, was effective in improving nerve regeneration after chronic axotomy but, increased axon sprouting in the distal nerve stump. (A) The TIB (tibial nerve) in the rat was cut and ligated. (B) After 2 months, the proximal stump of the TIB nerve was cross-sutured to the freshly denervated CP (common peroneal) nerve stump and (C) a mini-osmotic pump was placed on the back of the rat for constant infusion of low dose BDNF (2ug/day for 28 days). (D) Following a 2-month period of chronic axotomy, fluororuby was administered to the CP distal nerve stump 20 mm from the cross-suture site for retrograde labeling of the TIB motoneurons that had regenerated their axons into the denervated CP distal nerve stump. (E) The numbers of motoneurons that regenerated their axons and were counted in 50 µm longitudinal sections of the ventral horn of the spinal cord [all sections were counted and a correction factor of 0.6 applied [313,314,315]. The number of motoneurons that regenerated their axons after 2 months of chronic axotomy was significantly reduced. GDNF and BDNF were both effective in increasing the motoneuron regeneration to normal levels and the combination elevated the levels even more. (F) Local delivery of GDNF to the suture sites of an acellular nerve allograft (ANA) interposed between the CP proximal and distal nerve stumps. GDNF was encapsulated with an efficiency of 78 + 3% and GDNF loading of 0.72 + 0.08 µg/mg of microspheres (MS; diameters of 45 + 5 µm), placed onto the suture sites and held in place by surrounding them with 2 µL gels placed below and above the suture sites that adhered to one another. (G) Retrograde labeling of the CP nerve 10 mm from the ASA 8 weeks later. (H) Numbers of motoneurons that regenerated their axons through the ANA plotted for control conditions of no delivery system (DDS) and empty MS and experimental conditions of GDNF delivery in combinations of 2- and 4-week formulations. (I) Numbers of sensory neurons counted in every fifth dorsal root ganglia 20 µm section. Irrespective of the nature of GDNF delivery, all motor and sensory neurons regenerated their axons through the ANA in 8 weeks as compared to ~50% of them that regenerated through the ANAs that did not deliver GDNF.(J) Electron micrographs of Schwann cells (SCs) and their axons in intact nerves, regenerating nerves in the distal nerve stump 2 months after TIB-CP cross-suture with saline administration via a cuff around the suture site linked to a mini-osmotic pump, GDNF delivery to the cross-suture site via the pump, and delivery of both GDNF and BDNF via to pump. (K) The mean + standard error values of the number of axons within each endoneurial sheath associated with a single SC in the control (A and B), C. GDNF and D. GDNF + BDNF.
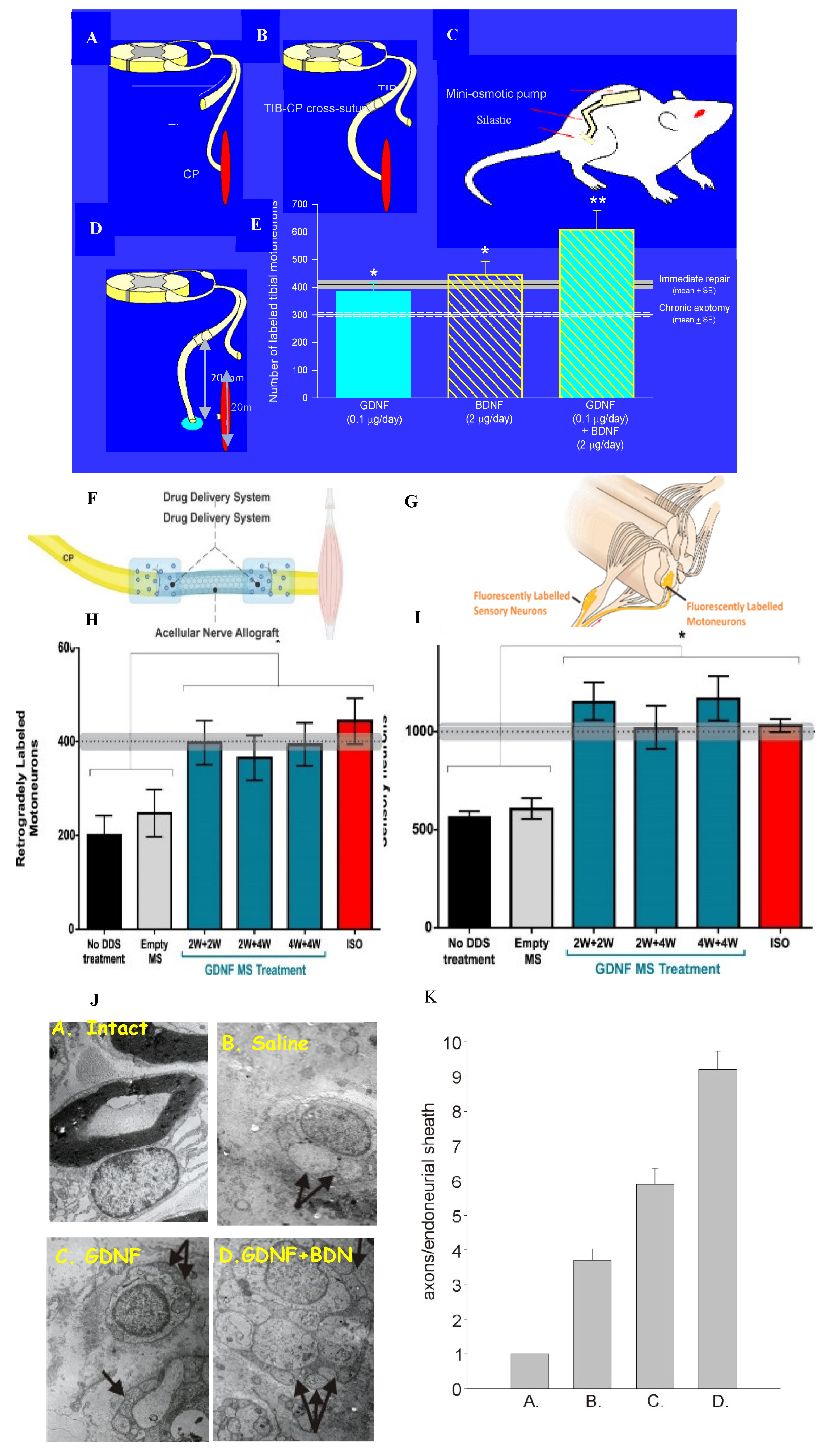
Figure 9.
Gene expression of BDNF (brain derived neurotrophic factor) and its trkB receptor, and of the regeneration associated genes, tubulin and GAP-43, after axotomy. Transection and surgical repair of the rat femoral nerve and (A) sham or (B) 1 hour 20 Hz electrical stimulation (stim) of the proximal stump. (C) Dark-field micrographs of in situ hybridization (ISH) with 35S-labeled oligonucleotides complementary to tubulin after sham and Stim. Means + standard deviation of ISH signals/motoneuron of (D) BDNF, (E) trkB, (F) tubulin, (G) GAP-43, and (H) neurofilament in individual rats are plotted as a function of time after femoral nerve axotomy after Sham and Stim.
Figure 9.
Gene expression of BDNF (brain derived neurotrophic factor) and its trkB receptor, and of the regeneration associated genes, tubulin and GAP-43, after axotomy. Transection and surgical repair of the rat femoral nerve and (A) sham or (B) 1 hour 20 Hz electrical stimulation (stim) of the proximal stump. (C) Dark-field micrographs of in situ hybridization (ISH) with 35S-labeled oligonucleotides complementary to tubulin after sham and Stim. Means + standard deviation of ISH signals/motoneuron of (D) BDNF, (E) trkB, (F) tubulin, (G) GAP-43, and (H) neurofilament in individual rats are plotted as a function of time after femoral nerve axotomy after Sham and Stim.
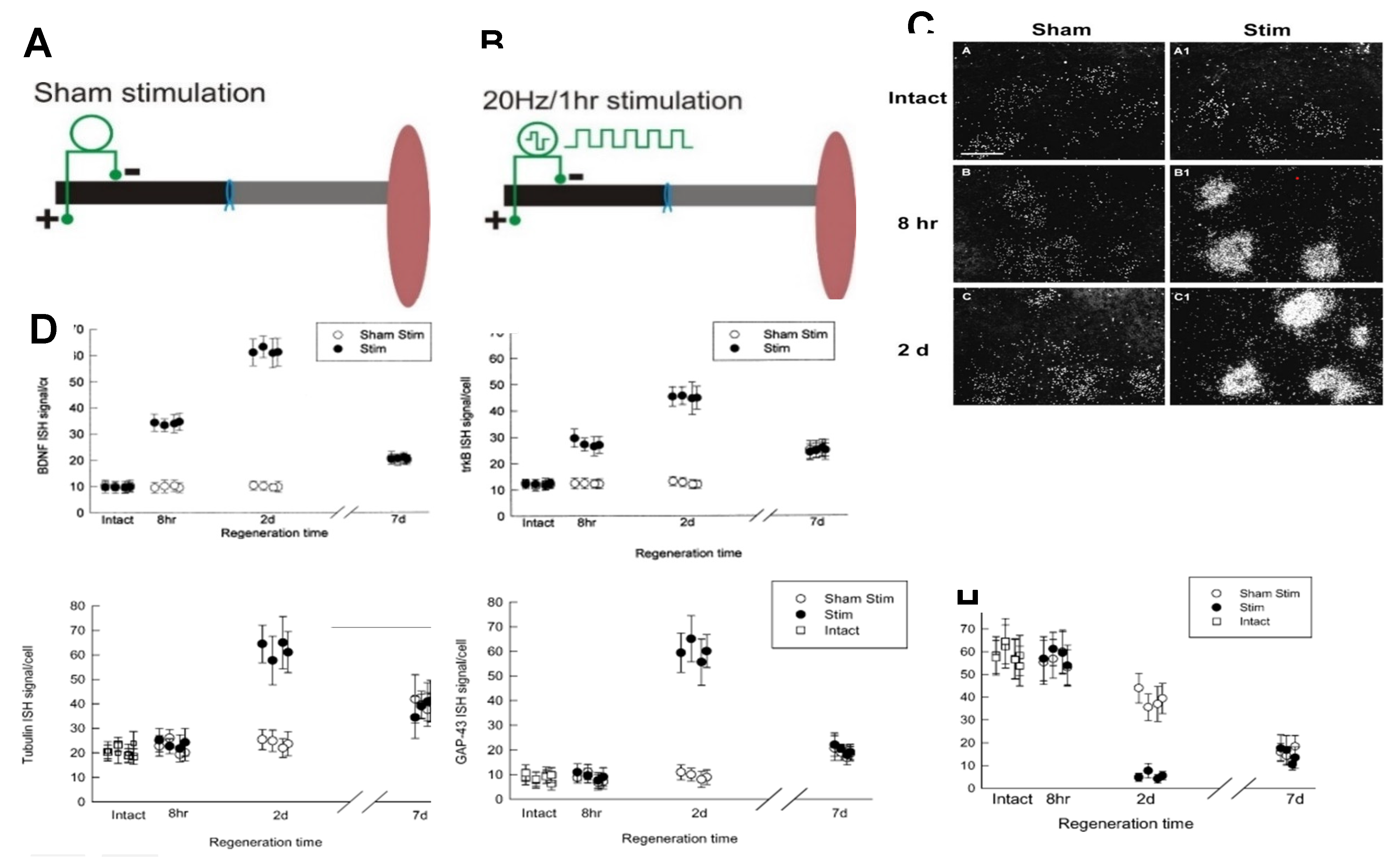
Figure 10.
The decline in electrical activity in motor nerves after axotomy coincides with that of the expression of regeneration associated genes (RAGs) and loss of presynaptic contacts. Regeneration after nerve repair and/or 20 Hz electrical stimulation (ES) restores synaptic contacts. (A) Motor and (B) sensory cross-correlation peaks normalized to pre-operative values decline with an exponential time course over the first month of axotomy and begin to recover with nerve regeneration. (C)The decline in the relative amplitude of the motor potentials occurs with (D) the decline in expression of GDNF (glial cell-derived neurotrophic factor). (E) A l µm-thick confocal image of a retrogradely CTB-AF545-labeled motoneuron with immunohistochemical visualization of VGLUT1 (vesicular glutamate transporter 1)-containing excitatory synapses, arising mainly from primary afferent neurons, and GAD67 (glutamic acid decarboxylase 67)-containing inhibitory synapses, were used to measure the percentages of the neuronal perimeter in contact with the synapses. The silencing of motoneurons, namely the reduced motor activity, is due to the withdrawal of (F) glutamatergic and (G) GABAergic synaptic contacts from the neurons. The withdrawal is prevented by treadmill exercise either (H) continuous exercise in males or (I) ‘interval exercise’ in female mice.
Figure 10.
The decline in electrical activity in motor nerves after axotomy coincides with that of the expression of regeneration associated genes (RAGs) and loss of presynaptic contacts. Regeneration after nerve repair and/or 20 Hz electrical stimulation (ES) restores synaptic contacts. (A) Motor and (B) sensory cross-correlation peaks normalized to pre-operative values decline with an exponential time course over the first month of axotomy and begin to recover with nerve regeneration. (C)The decline in the relative amplitude of the motor potentials occurs with (D) the decline in expression of GDNF (glial cell-derived neurotrophic factor). (E) A l µm-thick confocal image of a retrogradely CTB-AF545-labeled motoneuron with immunohistochemical visualization of VGLUT1 (vesicular glutamate transporter 1)-containing excitatory synapses, arising mainly from primary afferent neurons, and GAD67 (glutamic acid decarboxylase 67)-containing inhibitory synapses, were used to measure the percentages of the neuronal perimeter in contact with the synapses. The silencing of motoneurons, namely the reduced motor activity, is due to the withdrawal of (F) glutamatergic and (G) GABAergic synaptic contacts from the neurons. The withdrawal is prevented by treadmill exercise either (H) continuous exercise in males or (I) ‘interval exercise’ in female mice.
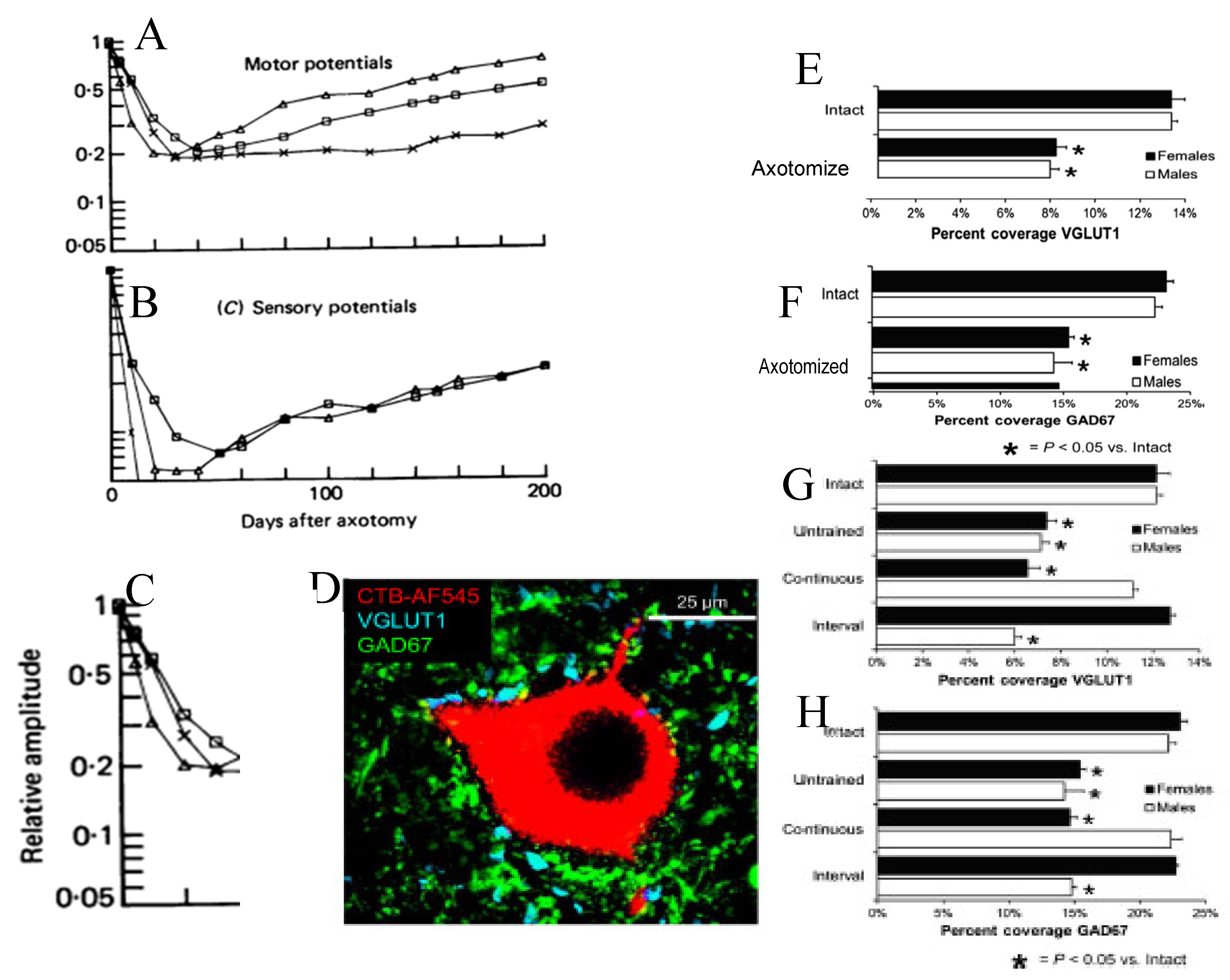
Figure 11.
Motor nerve regeneration is staggered. (A) The femoral nerve in the rat was cut 20 mm from its branches to the quadriceps muscle and to the skin. It was repaired immediately with microsurgical technique. The regeneration of axons into the two branches was evaluated 2 to 10 weeks later by application of fluorescent dyes, RR (rubyred) for 1-2 hours to one branch and FG (fluorogold) to the other branch, 5 mm from the point of their division from the femoral nerve. (B) Motoneurons that were retrogradely labelled with either or both dyes are the sum of those that regenerated their axons into either branch as well as both the branches. The number of these motoneurons that regenerated their axons over the distance of 25 mm was ~55% of all the motoneurons in the femoral motoneuron pool, the numbers increasing with an exponential time course to the reach the 350 motoneurons of the control intact nerve (shown by the shaded horizontal lines that represent + standard errors of the mean values). The 10 weeks for all the motoneurons to regenerate the 25 mm distance to the site of dye application was surprisingly long when a latency of at least 2 days and the regeneration rate of 3 mm/day is considered. On the basis of the latency and regeneration rate, a 2-to-3-week period of regeneration would be expected for all the motoneurons to regenerate their axons. (C) The possible explanation that axon outgrowth across the surgical site is staggered with some axons even growing backwards into the proximal nerve stump, proved to be correct. The Schwann cells that multiply after nerve injury and line the distal nerve stump are known to migrate into the injury site and are shown in red. (D) To determine whether indeed, this explanation accounts for the observed slow regeneration of nerve fibres, those axons that had regenerated just across the suture line of the femoral nerve repair site, were retrogradely labelled by crushing the distal nerve stump 1.5 mm from the surgical site. (E) The microinjection of FR and (F) the counts of the retrograde labelled motoneurons that regenerated the axons, plotted as a function of days after femoral nerve repair surgery revealed the ~one month period of outgrowth of the femoral motor axons across the injury site with (G) staggering of axons regenerating within the distal nerve stump and across the site as (H) some regenerating axons grow into the proximal nerve stump in straight lines or or take a spiralling course. The axons in (G,H) were visualized with silver staining.
Figure 11.
Motor nerve regeneration is staggered. (A) The femoral nerve in the rat was cut 20 mm from its branches to the quadriceps muscle and to the skin. It was repaired immediately with microsurgical technique. The regeneration of axons into the two branches was evaluated 2 to 10 weeks later by application of fluorescent dyes, RR (rubyred) for 1-2 hours to one branch and FG (fluorogold) to the other branch, 5 mm from the point of their division from the femoral nerve. (B) Motoneurons that were retrogradely labelled with either or both dyes are the sum of those that regenerated their axons into either branch as well as both the branches. The number of these motoneurons that regenerated their axons over the distance of 25 mm was ~55% of all the motoneurons in the femoral motoneuron pool, the numbers increasing with an exponential time course to the reach the 350 motoneurons of the control intact nerve (shown by the shaded horizontal lines that represent + standard errors of the mean values). The 10 weeks for all the motoneurons to regenerate the 25 mm distance to the site of dye application was surprisingly long when a latency of at least 2 days and the regeneration rate of 3 mm/day is considered. On the basis of the latency and regeneration rate, a 2-to-3-week period of regeneration would be expected for all the motoneurons to regenerate their axons. (C) The possible explanation that axon outgrowth across the surgical site is staggered with some axons even growing backwards into the proximal nerve stump, proved to be correct. The Schwann cells that multiply after nerve injury and line the distal nerve stump are known to migrate into the injury site and are shown in red. (D) To determine whether indeed, this explanation accounts for the observed slow regeneration of nerve fibres, those axons that had regenerated just across the suture line of the femoral nerve repair site, were retrogradely labelled by crushing the distal nerve stump 1.5 mm from the surgical site. (E) The microinjection of FR and (F) the counts of the retrograde labelled motoneurons that regenerated the axons, plotted as a function of days after femoral nerve repair surgery revealed the ~one month period of outgrowth of the femoral motor axons across the injury site with (G) staggering of axons regenerating within the distal nerve stump and across the site as (H) some regenerating axons grow into the proximal nerve stump in straight lines or or take a spiralling course. The axons in (G,H) were visualized with silver staining.
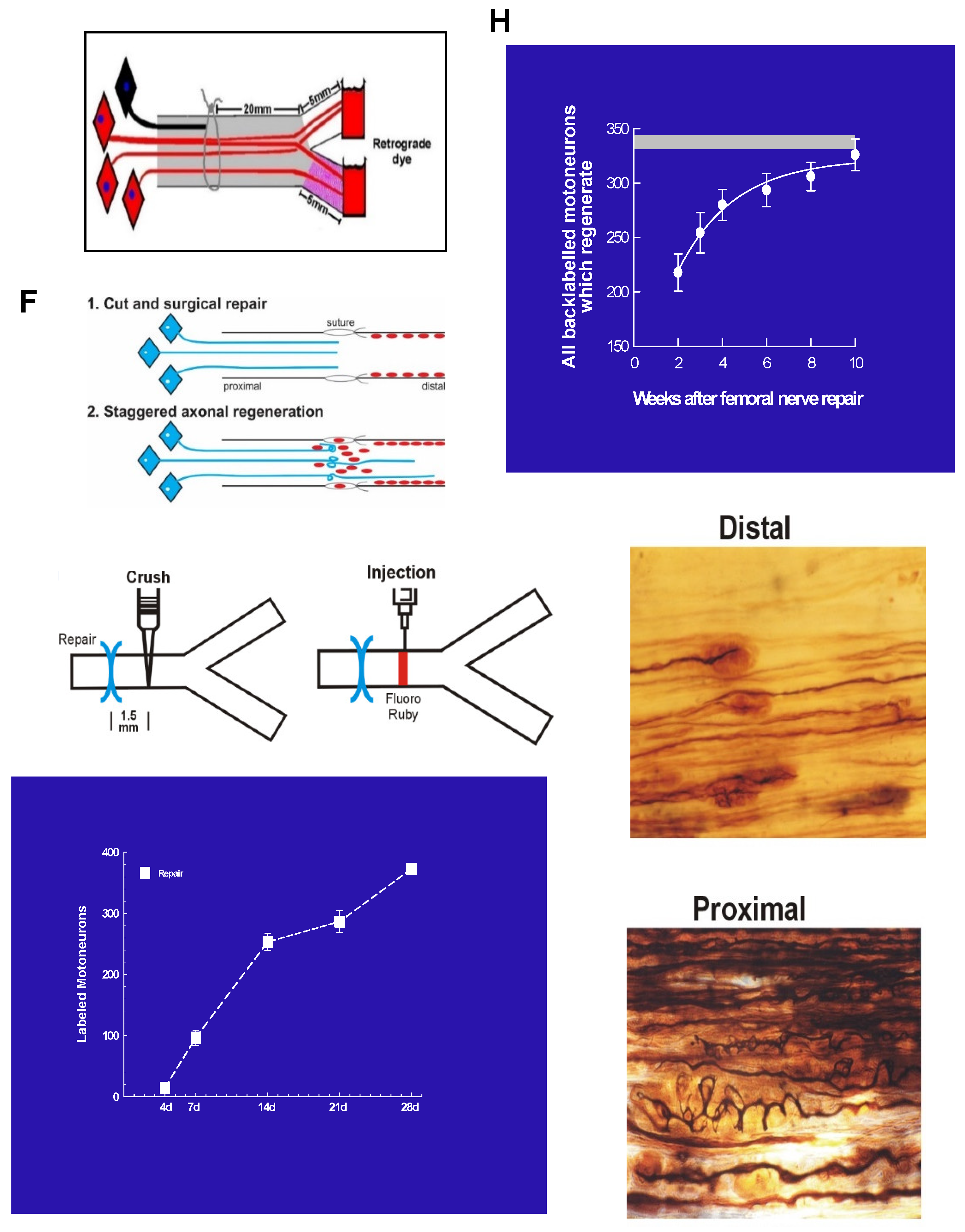
Figure 12.
Low frequency electrical stimulation (ES) accelerates nerve outgrowth across the site of transection and surgical repair. Figurative illustrations of femoral motoneurons and their (A) projection to the motor branch to the quadriceps muscle and not into the sensory saphenous branch to the skin, (B) transection, surgical repair, and application of dyes fluororuby (FR: analogous to rubyred) and fluorogold to each branch to retrogradely label the motoneurons that regenerated their axons 25 mm into each femoral nerve branch (see also (H)), and (C) ES with a 20 Hz frequency, proximal to the site of repair. Histograms and plots of the mean + standard error (SE) of (D,E) the number of femoral motoneurons that regenerated their axons into motor and sensory branches and those that regenerated into both branches after sham ES and (F,G) ES. (I) FR was injected 1.5 mm distal to the suture site to count the motoneurons that regenerated across the site. (J,K) Comparison of the effects of sham and ES on the mean + SE of all the femoral motoneurons that regenerated their axons into the motor and sensory branches and those that regenerated across the suture site. Diagrammatic illustration of (L) the effect of ES in ‘straightening’ out the contorted progression of regenerating axons across the suture site and. (M) application of tetrodotoxin (TTX, 60 mg/ml) to the proximal stump of the transected and surgically repaired femoral nerve to block the action potentials elicited by ES, from progressing to the cell bodies of the motor and sensory neurons. After a three-week period of nerve regeneration, FG and FR were applied to count those motor and sensory neurons that had regenerated their axons 25 mm from the surgical and repair site. The striking increased regeneration after 3 weeks of (N) motor and (O) sensory nerves into the motor and sensory branches of the cut and repaired femoral motoneurons by the 1 hour 20 Hz ES was blocked by TTX.
Figure 12.
Low frequency electrical stimulation (ES) accelerates nerve outgrowth across the site of transection and surgical repair. Figurative illustrations of femoral motoneurons and their (A) projection to the motor branch to the quadriceps muscle and not into the sensory saphenous branch to the skin, (B) transection, surgical repair, and application of dyes fluororuby (FR: analogous to rubyred) and fluorogold to each branch to retrogradely label the motoneurons that regenerated their axons 25 mm into each femoral nerve branch (see also (H)), and (C) ES with a 20 Hz frequency, proximal to the site of repair. Histograms and plots of the mean + standard error (SE) of (D,E) the number of femoral motoneurons that regenerated their axons into motor and sensory branches and those that regenerated into both branches after sham ES and (F,G) ES. (I) FR was injected 1.5 mm distal to the suture site to count the motoneurons that regenerated across the site. (J,K) Comparison of the effects of sham and ES on the mean + SE of all the femoral motoneurons that regenerated their axons into the motor and sensory branches and those that regenerated across the suture site. Diagrammatic illustration of (L) the effect of ES in ‘straightening’ out the contorted progression of regenerating axons across the suture site and. (M) application of tetrodotoxin (TTX, 60 mg/ml) to the proximal stump of the transected and surgically repaired femoral nerve to block the action potentials elicited by ES, from progressing to the cell bodies of the motor and sensory neurons. After a three-week period of nerve regeneration, FG and FR were applied to count those motor and sensory neurons that had regenerated their axons 25 mm from the surgical and repair site. The striking increased regeneration after 3 weeks of (N) motor and (O) sensory nerves into the motor and sensory branches of the cut and repaired femoral motoneurons by the 1 hour 20 Hz ES was blocked by TTX.
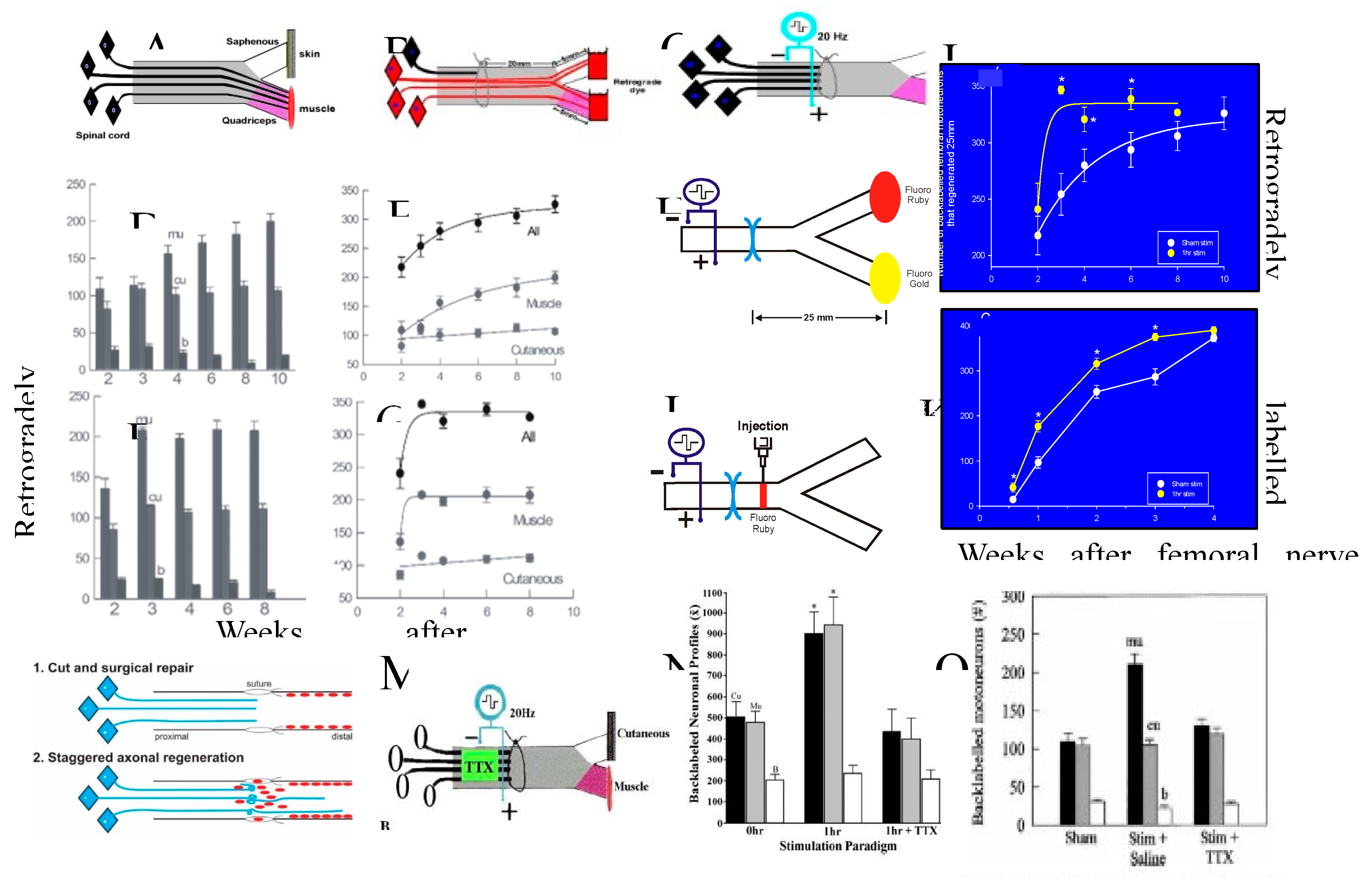
Figure 13.
BDNF (brain derived neurotrophic factor is essential for the efficacy of ES (20 Hz electrical stimulation for one hour) in accelerating nerve regeneration. (A) The infusion of the BDNF antibody, α-BDNF, into the intrathecal space of the lumbar spinal cord 3-days prior to (B) ES via electrodes placed proximal to the surgical site of femoral nerve transection and microsurgical repair site and the application of the fluorescent dyes, fluororuby and fluorogold to each of the femoral motor and cutaneous sensory branches for retrograde labeling of those motoneurons that regenerated their axons into either or both the branches at 3 weeks after surgical repair. (C) The histogram of mean motoneuron numbers (+ standard errors) that were corrected as described previously [24], that regenerated their axons 25 mm into the appropriate and inappropriate motor and sensory nerve branches, respectively were plotted for the motoneurons subjected to ES (stim) and sham ES. ES accelerated the regeneration of motor nerves into the appropriate motor branch without affecting either the regeneration into the inappropriate motor branch or into both branches (see also Figure 12D,E).This accelerated motor nerve regeneration was blocked by infusion of the α-BDNF. The numbers of motoneurons that regenerated their axons inappropriately into the sensory branch of the femoral nerve or those that regenerated their axons into both branches were not changed by the infusion of α-BDNF.
Figure 13.
BDNF (brain derived neurotrophic factor is essential for the efficacy of ES (20 Hz electrical stimulation for one hour) in accelerating nerve regeneration. (A) The infusion of the BDNF antibody, α-BDNF, into the intrathecal space of the lumbar spinal cord 3-days prior to (B) ES via electrodes placed proximal to the surgical site of femoral nerve transection and microsurgical repair site and the application of the fluorescent dyes, fluororuby and fluorogold to each of the femoral motor and cutaneous sensory branches for retrograde labeling of those motoneurons that regenerated their axons into either or both the branches at 3 weeks after surgical repair. (C) The histogram of mean motoneuron numbers (+ standard errors) that were corrected as described previously [24], that regenerated their axons 25 mm into the appropriate and inappropriate motor and sensory nerve branches, respectively were plotted for the motoneurons subjected to ES (stim) and sham ES. ES accelerated the regeneration of motor nerves into the appropriate motor branch without affecting either the regeneration into the inappropriate motor branch or into both branches (see also Figure 12D,E).This accelerated motor nerve regeneration was blocked by infusion of the α-BDNF. The numbers of motoneurons that regenerated their axons inappropriately into the sensory branch of the femoral nerve or those that regenerated their axons into both branches were not changed by the infusion of α-BDNF.
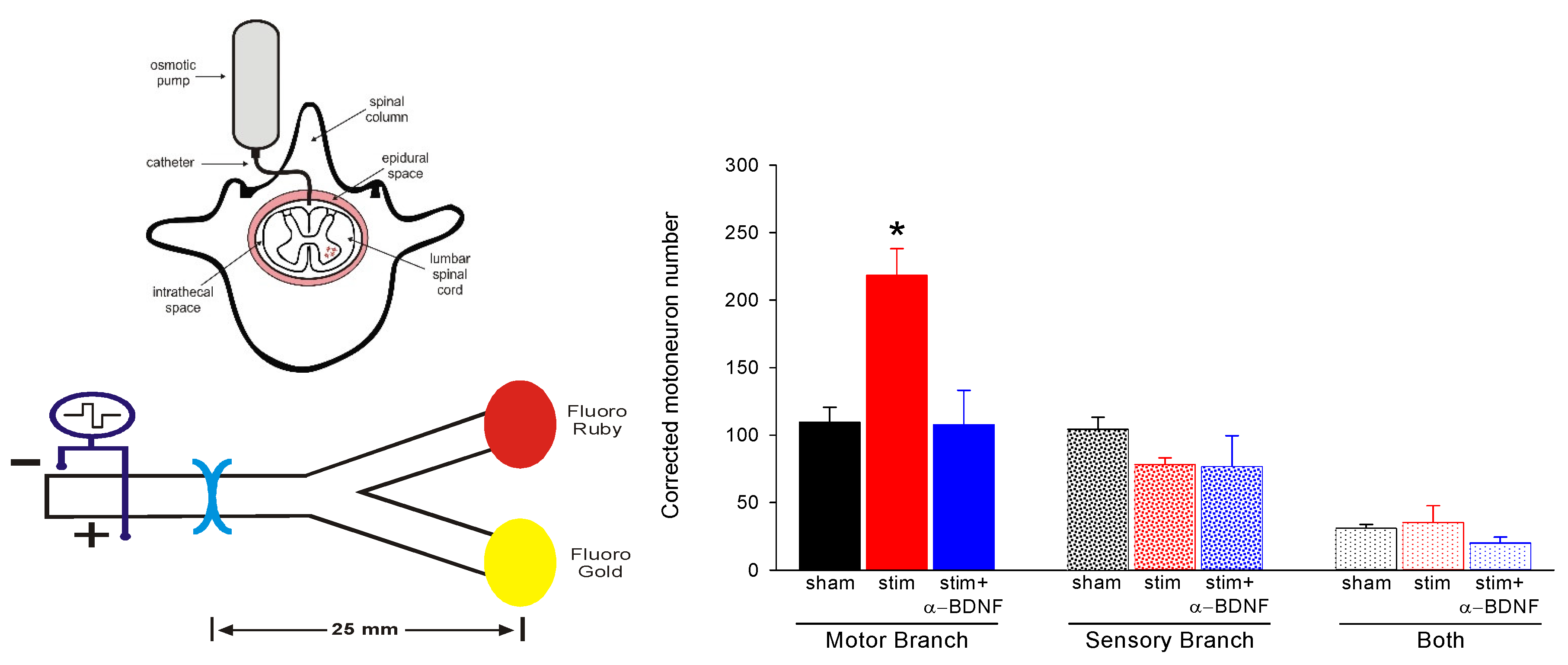
Figure 14.
Preferential motor reinnervation of the motor branch of the femoral nerve is accounted for by differential expression of neurotrophic factors in the denervated distal nerve stump. Numbers of rat femoral motoneurons that regenerate their axons (A) across the site of transection (injury) and surgical repair and (B) 25 mm from the injury site into appropriate muscle (black bars), inappropriate sensory cutaneous branches (white bars), and both branches (grey bars). (C) The fold-increase in the mRNA of PTN (pleiotrophin) and GDNF (glial cell-derived neurotrophic factor) in the distal nerve stumps of transected and repaired femoral nerve relative to that of intact nerves as a function of days of denervation. (D) Diagrammatic representation of motoneurons regenerating their nerve fibres into appropriate and inappropriate muscle and cutaneous pathways, respectively as well as into both. Further detail is provided in the text.
Figure 14.
Preferential motor reinnervation of the motor branch of the femoral nerve is accounted for by differential expression of neurotrophic factors in the denervated distal nerve stump. Numbers of rat femoral motoneurons that regenerate their axons (A) across the site of transection (injury) and surgical repair and (B) 25 mm from the injury site into appropriate muscle (black bars), inappropriate sensory cutaneous branches (white bars), and both branches (grey bars). (C) The fold-increase in the mRNA of PTN (pleiotrophin) and GDNF (glial cell-derived neurotrophic factor) in the distal nerve stumps of transected and repaired femoral nerve relative to that of intact nerves as a function of days of denervation. (D) Diagrammatic representation of motoneurons regenerating their nerve fibres into appropriate and inappropriate muscle and cutaneous pathways, respectively as well as into both. Further detail is provided in the text.
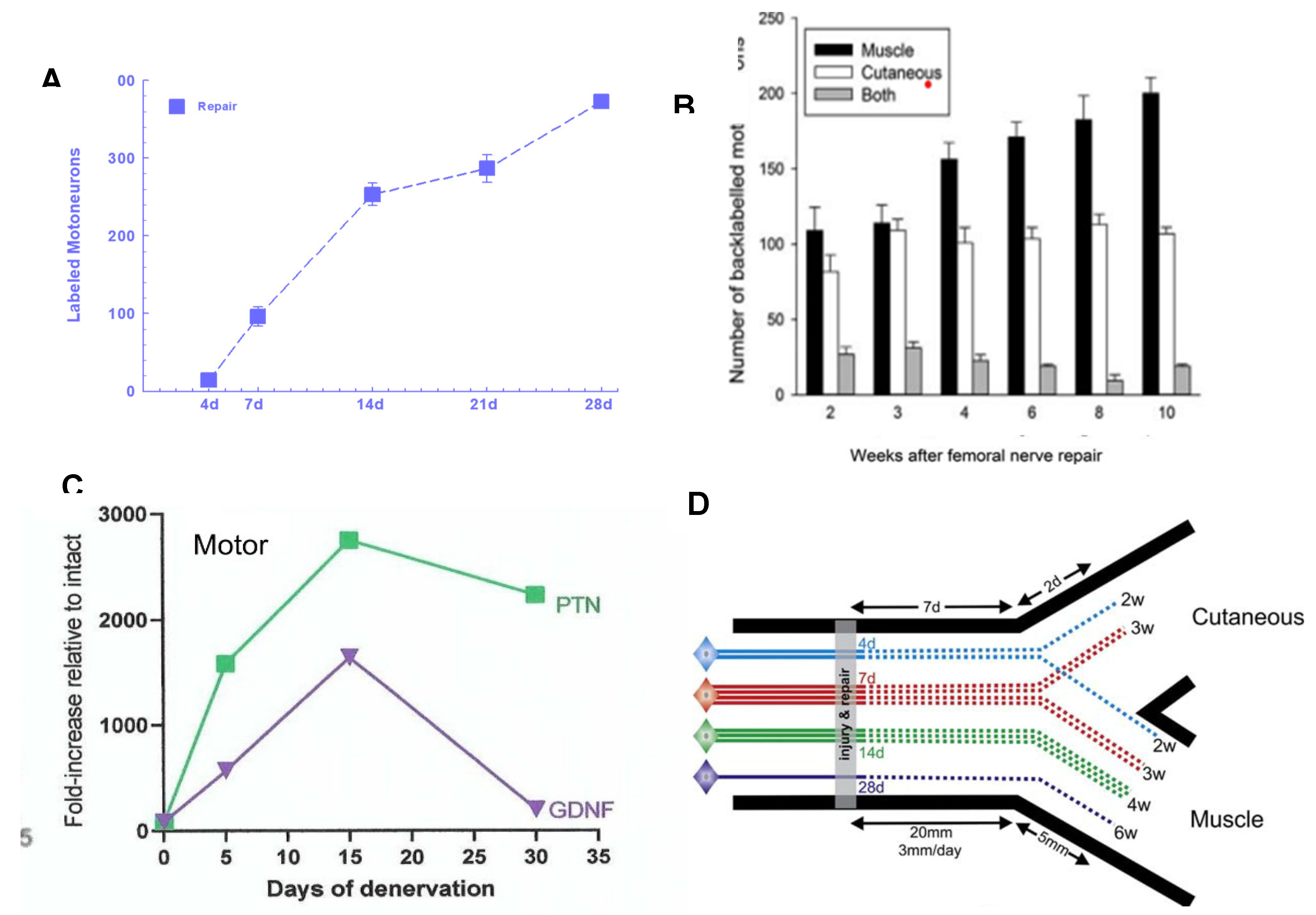
Figure 15.
Brief 1 hour 20 Hz electrical stimulation (ES) promotes nerve regeneration and target reinnervation after delayed nerve repair. (A) Surgeries: The common peroneal nerve (CP), and/ or tibial (TIB) nerve were cut and cross-sutured either a. immediately or 2 months after b. prolonged CP axotomy and/or distal TIB nerve denervation (c, d) and the proximal nerve stump subjected to either sham or ES. The mean numbers (+ standard error SE) and the numbers from different experiments (shown as symbols) are plotted in histograms 2 and 4 weeks after cross-suture for (B) motoneurons and (C) sensory neurons that regenerated their axons 20 mm into the distal nerve stump. (D) Examples of the in-situ hybridization of α-tubulin mRNA in sciatic motoneurons whose axons were either intact (0 days), transected previously for 7 days (7d acute axotomy) or transected previously for 6 months (6m chronic axotomy), and transected previously for 6 months and then subjected to a refreshment injury thereafter for 7 days (6m+1w chronic axotomy and refreshment injury). Details are provided in the text.
Figure 15.
Brief 1 hour 20 Hz electrical stimulation (ES) promotes nerve regeneration and target reinnervation after delayed nerve repair. (A) Surgeries: The common peroneal nerve (CP), and/ or tibial (TIB) nerve were cut and cross-sutured either a. immediately or 2 months after b. prolonged CP axotomy and/or distal TIB nerve denervation (c, d) and the proximal nerve stump subjected to either sham or ES. The mean numbers (+ standard error SE) and the numbers from different experiments (shown as symbols) are plotted in histograms 2 and 4 weeks after cross-suture for (B) motoneurons and (C) sensory neurons that regenerated their axons 20 mm into the distal nerve stump. (D) Examples of the in-situ hybridization of α-tubulin mRNA in sciatic motoneurons whose axons were either intact (0 days), transected previously for 7 days (7d acute axotomy) or transected previously for 6 months (6m chronic axotomy), and transected previously for 6 months and then subjected to a refreshment injury thereafter for 7 days (6m+1w chronic axotomy and refreshment injury). Details are provided in the text.
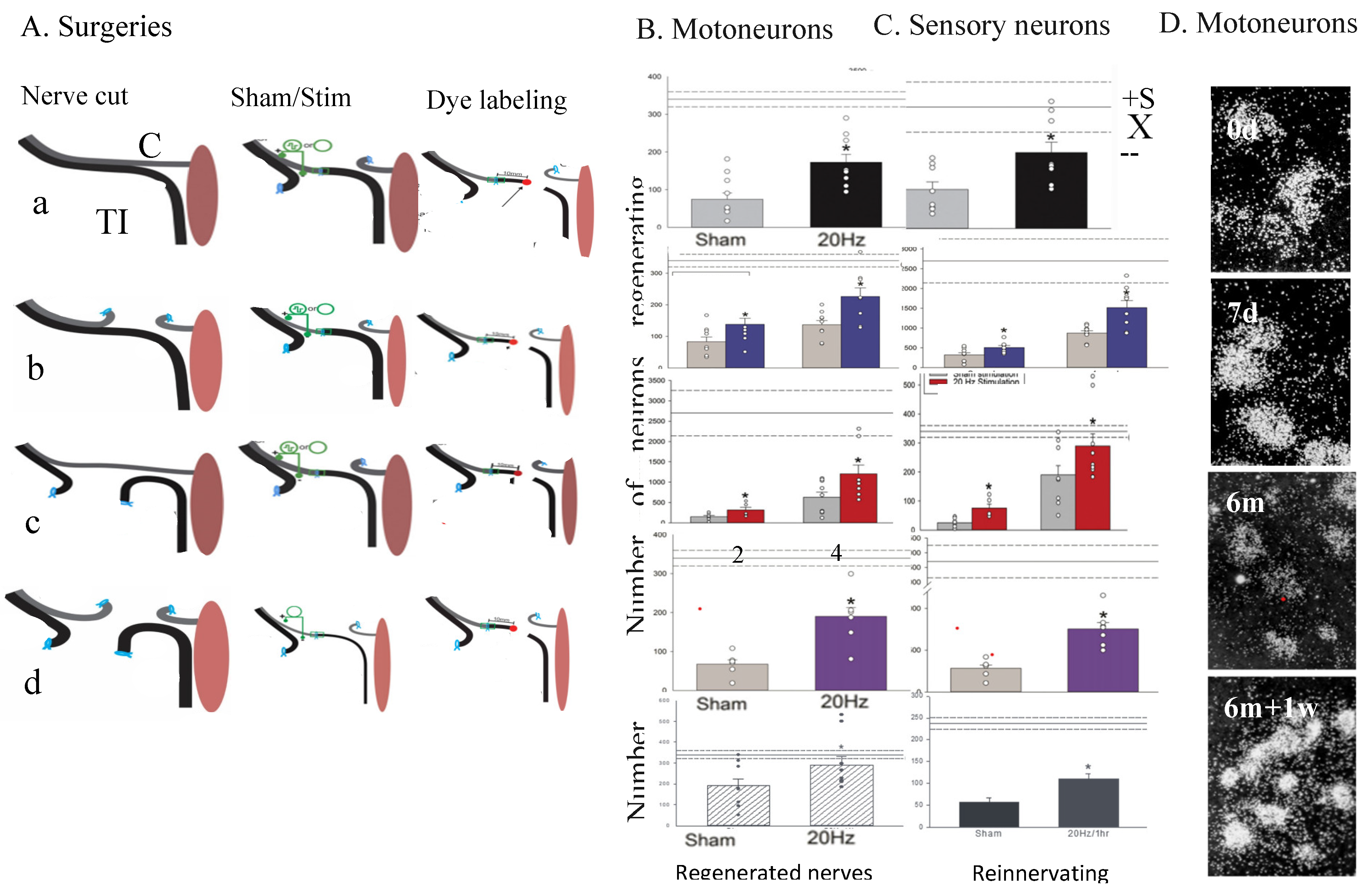
Figure 16.
Electrical stimulation of the intact sciatic nerve of the rat at 20 Hz for 1 hour (ES) in vivo before excising dorsal root ganglion (DRG) sensory neurons and plating them on a laminin substrate in vitro, promotes their neurite outgrowth. An intact sciatic nerve 1 day after (A) no ES and (B) ES. An intact sciatic nerve 7 days after (C) no ES and (D) ES. (E) The mean + standard error of the lengths of the longest neurites (in µm) for the control and experimental DRG neurons that were not and were stimulated, respectively. The ES of intact sciatic nerves promoted neurite extension from plated DRG neurons. This promoted neurite extension is akin to the effect of a nerve crush conditioning lesion (CL) that is made 7 days prior to the transection and suture of the nerve proximal to the CL, that accelerates nerve regeneration. The neurons were immunostained with β-tubulin III. The calibration bar = 50 µm. *Statistical significance p<0.05.
Figure 16.
Electrical stimulation of the intact sciatic nerve of the rat at 20 Hz for 1 hour (ES) in vivo before excising dorsal root ganglion (DRG) sensory neurons and plating them on a laminin substrate in vitro, promotes their neurite outgrowth. An intact sciatic nerve 1 day after (A) no ES and (B) ES. An intact sciatic nerve 7 days after (C) no ES and (D) ES. (E) The mean + standard error of the lengths of the longest neurites (in µm) for the control and experimental DRG neurons that were not and were stimulated, respectively. The ES of intact sciatic nerves promoted neurite extension from plated DRG neurons. This promoted neurite extension is akin to the effect of a nerve crush conditioning lesion (CL) that is made 7 days prior to the transection and suture of the nerve proximal to the CL, that accelerates nerve regeneration. The neurons were immunostained with β-tubulin III. The calibration bar = 50 µm. *Statistical significance p<0.05.
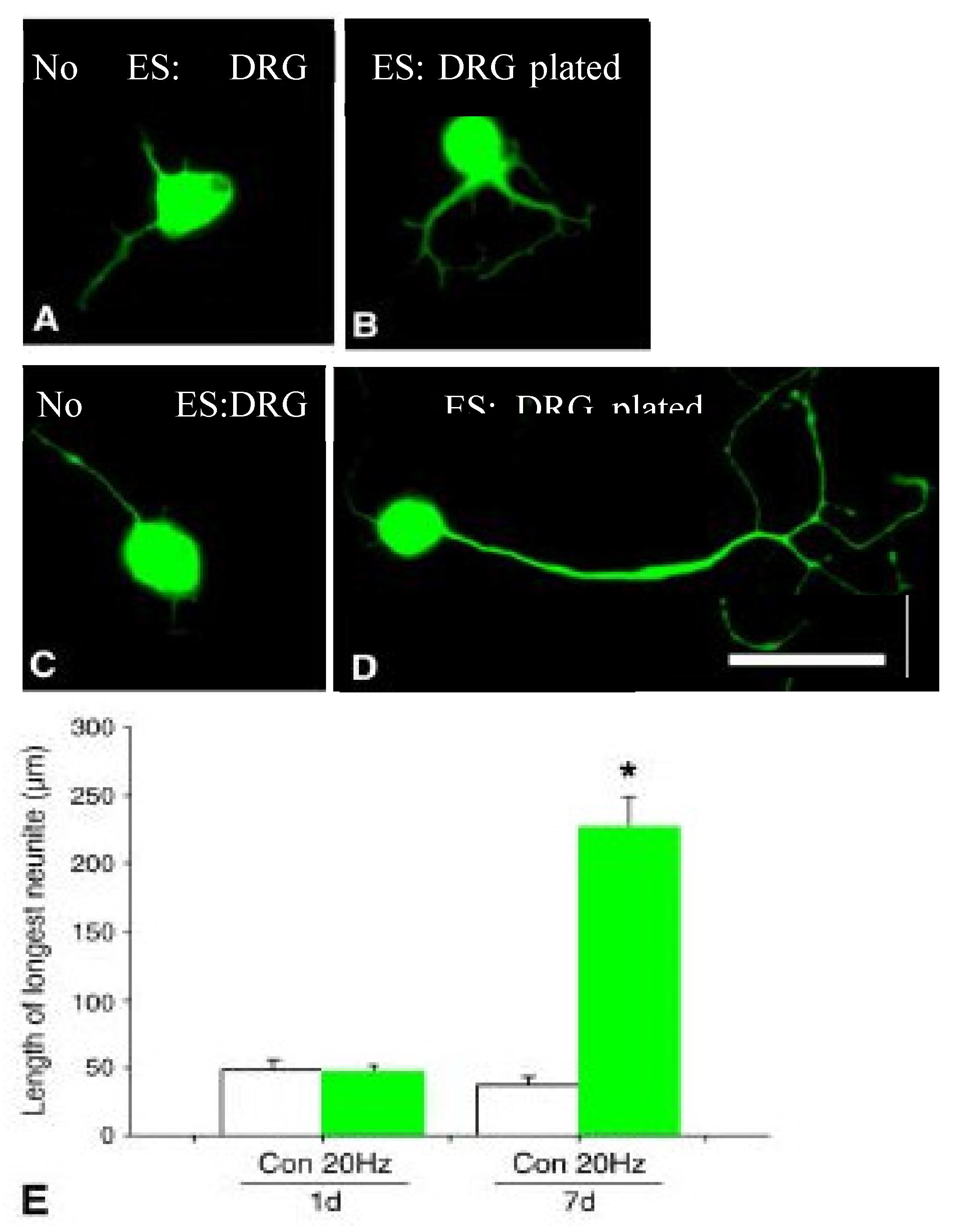
Figure 17.
(A) We asked the question as to whether cAMP mediates the effect of ES on axon regeneration by administering rolipram to the site of injury and surgical repair to block phosphodiesterase 4 that hydrolyses cAMP. (B) Rolipram (1:1 saline/dimethyl sulfoxide [DSMO]) or vehicle (1:1 saline/DMSO) was delivered via an Alzet pump at a rate of 0.4 µmol/g/h to the site of the cut and sutured the CP (common peroneal) nerve that was surrounded by a 3 mm long silicone silastic cuff of 0.7 mm interior diameter. The mean (+ standard error) number of CP motoneurons that regenerated across the suture site (expressed as a percentage of the numbers of intact CP motoneurons on the contralateral side) were retrogradely labelled with rubyred (RR) (C) 3 mm, and (D) 10 mm from the suture site. (E) MUNE [the estimated number of MUs (motor units) that equals the ratio of the muscle and mean MU twitch forces of the reinnervated TA (tibialis anterior) muscle after the CP nerve suture repair] expressed as a percentage of MUNE in muscles with intact innervation. The significant increase in the number of CP motoneurons that (D,E) regenerated their axons (C) 3 mm past the CP nerve suture site after 7 (*** p<0.01) and after 14 days (*** p<0.01), (D) 10 mm past the suture site after 14 days and E. reinnervated TA muscle after 5 months.
Figure 17.
(A) We asked the question as to whether cAMP mediates the effect of ES on axon regeneration by administering rolipram to the site of injury and surgical repair to block phosphodiesterase 4 that hydrolyses cAMP. (B) Rolipram (1:1 saline/dimethyl sulfoxide [DSMO]) or vehicle (1:1 saline/DMSO) was delivered via an Alzet pump at a rate of 0.4 µmol/g/h to the site of the cut and sutured the CP (common peroneal) nerve that was surrounded by a 3 mm long silicone silastic cuff of 0.7 mm interior diameter. The mean (+ standard error) number of CP motoneurons that regenerated across the suture site (expressed as a percentage of the numbers of intact CP motoneurons on the contralateral side) were retrogradely labelled with rubyred (RR) (C) 3 mm, and (D) 10 mm from the suture site. (E) MUNE [the estimated number of MUs (motor units) that equals the ratio of the muscle and mean MU twitch forces of the reinnervated TA (tibialis anterior) muscle after the CP nerve suture repair] expressed as a percentage of MUNE in muscles with intact innervation. The significant increase in the number of CP motoneurons that (D,E) regenerated their axons (C) 3 mm past the CP nerve suture site after 7 (*** p<0.01) and after 14 days (*** p<0.01), (D) 10 mm past the suture site after 14 days and E. reinnervated TA muscle after 5 months.
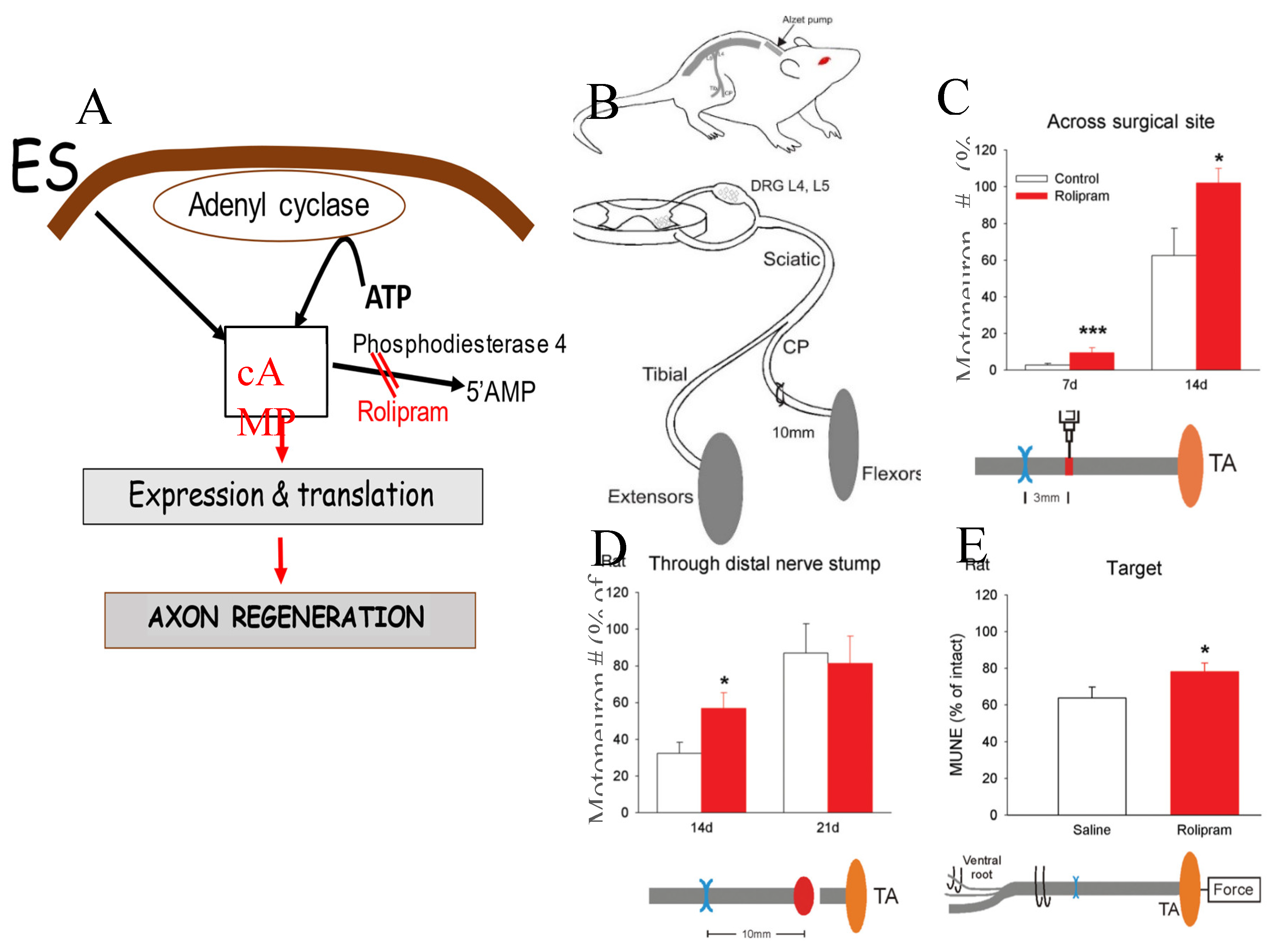
Figure 18.
Electrical stimulation (ES) promotes regeneration of transected sensory nerves in the central nervous system (CNS). (A) Figurative drawing of surgical T8 transection of the sciatic sensory nerves in the CNS, the three experimental groups of 1) conditioning lesion (CL, 1), 20 Hz ES (2), and 200 Hz (3), and the injection a 1% solution of cholera Toxin B (CTB) [487] into the sciatic nerve 14 weeks later to label CNS sensory nerves in vivo. (B) Mean ± standard error (SE) of cAMP levels in L4 to 6 dorsal root ganglion neurons, 24 hours after CLs and ES. Camara lucida drawings of spinal cord 25 µm sagittal sections of CTB-immunocytochemical identification with examples of DAB staining of DRG sensory axons of (C) sham ES, (D) CL, (E) 20 Hz ES, and (F) 200 Hz ES The lesion site in the (C-F) drawings is denoted by the transition from a white to a grey background and the arrows correspond with the visualization of the CTB stained axons. The cumulative sum of the mean (± SE) numbers of regenerated axons that advanced into the lesion site as a percent of all the labeled axons proximal to the lesion in the (G) sham control group compared to the CL group, (H) the 20 Hz ES group compared to Sham group, (I) 200 Hz ES group compared to the sham group. Significant values (p<0/05) are denoted by s. See the text for details.
Figure 18.
Electrical stimulation (ES) promotes regeneration of transected sensory nerves in the central nervous system (CNS). (A) Figurative drawing of surgical T8 transection of the sciatic sensory nerves in the CNS, the three experimental groups of 1) conditioning lesion (CL, 1), 20 Hz ES (2), and 200 Hz (3), and the injection a 1% solution of cholera Toxin B (CTB) [487] into the sciatic nerve 14 weeks later to label CNS sensory nerves in vivo. (B) Mean ± standard error (SE) of cAMP levels in L4 to 6 dorsal root ganglion neurons, 24 hours after CLs and ES. Camara lucida drawings of spinal cord 25 µm sagittal sections of CTB-immunocytochemical identification with examples of DAB staining of DRG sensory axons of (C) sham ES, (D) CL, (E) 20 Hz ES, and (F) 200 Hz ES The lesion site in the (C-F) drawings is denoted by the transition from a white to a grey background and the arrows correspond with the visualization of the CTB stained axons. The cumulative sum of the mean (± SE) numbers of regenerated axons that advanced into the lesion site as a percent of all the labeled axons proximal to the lesion in the (G) sham control group compared to the CL group, (H) the 20 Hz ES group compared to Sham group, (I) 200 Hz ES group compared to the sham group. Significant values (p<0/05) are denoted by s. See the text for details.
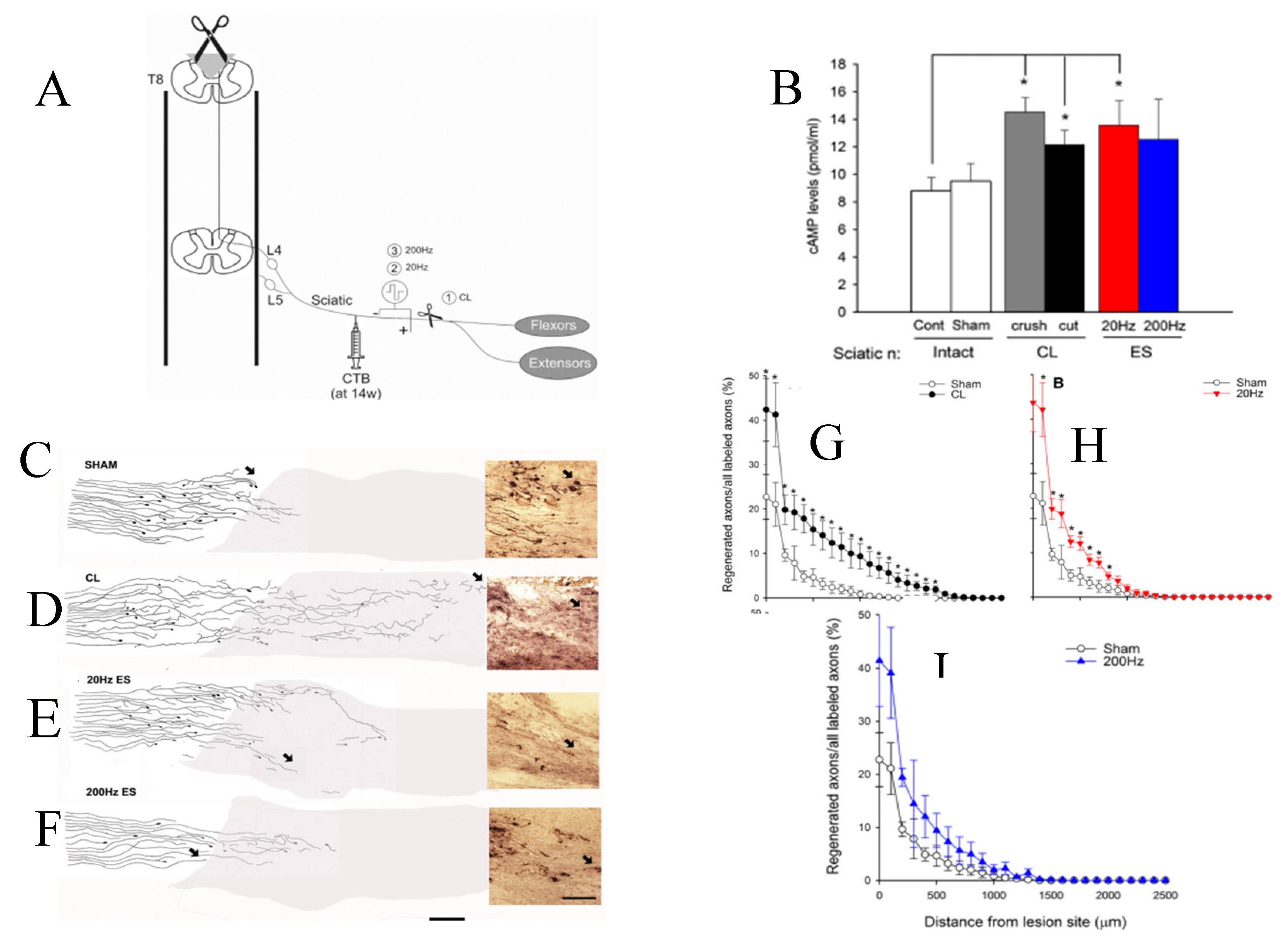
Figure 19.
Schematic representation of the intracellular signalling pathways activated by brief (1-hour 20 Hz electrical stimulation (ES). ES increases calcium influx (not shown) and in turn, activates adenyl cyclase to elevate cAMP levels. Cyclic AMP, in turn, acts via PKA to phosphorylate CREB in the nucleus leading to transcription of proteins, including neurotrophins and the cytoskeletal proteins tubulin and actin that are essential for axonal elongation during regeneration. The neurotrophins also feedback to further increase cAMP (via ERK with transient inhibition of phosphodiesterase 4 to prevent hydrolysis of cAMP to 5’AMP–not shown). Thereby the transcription of regeneration associated genes is amplified to promote axonal outgrowth. The ES also promotes axonal regeneration after chronic denervation of distal nerve stumps. This positive effect is likely mediated by the release of mitogens including neuregulin that promote Schwann cell proliferation and expression of their growth permissive state. This state includes the transcription of neurotrophic factors that likely potentiate axon regeneration via their amplification of neurotrophic factor expression by the neurons. These feedback loops likely aid in promoting the accelerated regeneration and target reinnervation. The ES of the injured nerve proximal to the site of nerve repair likely leads to the release of mitogens such as neuregulin that promotes Schwann cell proliferation and transition from the myelinating to a growth permissive state. These in turn, account for the positive effect of ES in promoting nerve regeneration after chronic denervation of the distal nerve stumps (Figure 15).
Figure 19.
Schematic representation of the intracellular signalling pathways activated by brief (1-hour 20 Hz electrical stimulation (ES). ES increases calcium influx (not shown) and in turn, activates adenyl cyclase to elevate cAMP levels. Cyclic AMP, in turn, acts via PKA to phosphorylate CREB in the nucleus leading to transcription of proteins, including neurotrophins and the cytoskeletal proteins tubulin and actin that are essential for axonal elongation during regeneration. The neurotrophins also feedback to further increase cAMP (via ERK with transient inhibition of phosphodiesterase 4 to prevent hydrolysis of cAMP to 5’AMP–not shown). Thereby the transcription of regeneration associated genes is amplified to promote axonal outgrowth. The ES also promotes axonal regeneration after chronic denervation of distal nerve stumps. This positive effect is likely mediated by the release of mitogens including neuregulin that promote Schwann cell proliferation and expression of their growth permissive state. This state includes the transcription of neurotrophic factors that likely potentiate axon regeneration via their amplification of neurotrophic factor expression by the neurons. These feedback loops likely aid in promoting the accelerated regeneration and target reinnervation. The ES of the injured nerve proximal to the site of nerve repair likely leads to the release of mitogens such as neuregulin that promotes Schwann cell proliferation and transition from the myelinating to a growth permissive state. These in turn, account for the positive effect of ES in promoting nerve regeneration after chronic denervation of the distal nerve stumps (Figure 15).
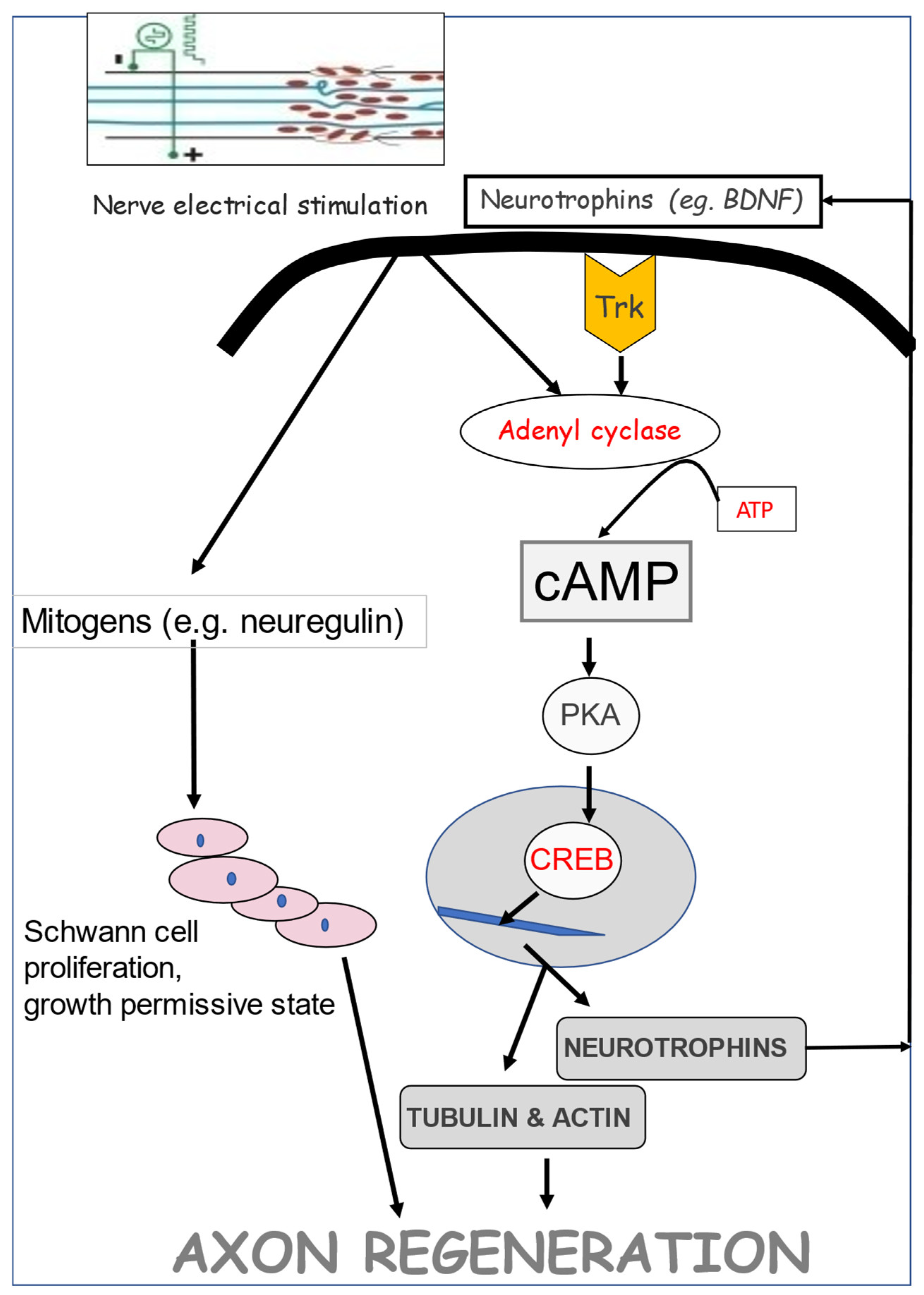
Figure 20.
Brief (1-hour) 20 Hz electrical stimulation (ES) of injured human nerves accelerates muscle reinnvation. (A) Diagrammatic representation of the recording of CMAPs (compound muscle action potentials) in the muscles of the median eminence above and below the ligament of the carpal tunnel. Only patients with severe CTS (carpal tunnel syndrome) with (B) reduced numbers of innervated MUs (motor units) exemplified by reduced CMAP amplitudes recorded both above and below the nerve were included in the study of the regenerative effect of ES (C) Patients with a reduced CMAP amplitude recorded below in the wrist but not above the released nerve, evident of conduction block and not MU loss, were excluded. (D) The ligament over the median nerve was released (CTRS: carpal tunnel release surgery). (F) Immediately after surgery, the median nerve was subjected to ES. (E. A maximum (2x threshold) electrical stimulus above the carpal tunnel evoked (G. an CMAP and the minimal stimuli evoked all-or-none single MU action potentials (S-MUAP) at points along the median nerve to estimate MU number (MUNE) from the ratio of the CMAP and mean of at least 20 recorded S-MUAPs. (H) Histograms of the mean (+ standard error) MUNE recorded before and after CTRS with and without ES (Stimulation and Control, respectively). Diagrammatic illustration of (I) the sluggish growth of axons across the nerve crush site that likely accounts for the (J) the regenerating nerve fibres not reaching the muscle to increase the numbers of reinnervated MUs and (L) axonal growth accelerated across the carpal tunnel crush site by ES at 3 months and (M) regenerating nerves progressing through the distal stumps to reinnervated denervated muscle fibres in the thenar eminence by 6 months, accounting for the restoration of the full complement of MUs by 12 months after CTRS.
Figure 20.
Brief (1-hour) 20 Hz electrical stimulation (ES) of injured human nerves accelerates muscle reinnvation. (A) Diagrammatic representation of the recording of CMAPs (compound muscle action potentials) in the muscles of the median eminence above and below the ligament of the carpal tunnel. Only patients with severe CTS (carpal tunnel syndrome) with (B) reduced numbers of innervated MUs (motor units) exemplified by reduced CMAP amplitudes recorded both above and below the nerve were included in the study of the regenerative effect of ES (C) Patients with a reduced CMAP amplitude recorded below in the wrist but not above the released nerve, evident of conduction block and not MU loss, were excluded. (D) The ligament over the median nerve was released (CTRS: carpal tunnel release surgery). (F) Immediately after surgery, the median nerve was subjected to ES. (E. A maximum (2x threshold) electrical stimulus above the carpal tunnel evoked (G. an CMAP and the minimal stimuli evoked all-or-none single MU action potentials (S-MUAP) at points along the median nerve to estimate MU number (MUNE) from the ratio of the CMAP and mean of at least 20 recorded S-MUAPs. (H) Histograms of the mean (+ standard error) MUNE recorded before and after CTRS with and without ES (Stimulation and Control, respectively). Diagrammatic illustration of (I) the sluggish growth of axons across the nerve crush site that likely accounts for the (J) the regenerating nerve fibres not reaching the muscle to increase the numbers of reinnervated MUs and (L) axonal growth accelerated across the carpal tunnel crush site by ES at 3 months and (M) regenerating nerves progressing through the distal stumps to reinnervated denervated muscle fibres in the thenar eminence by 6 months, accounting for the restoration of the full complement of MUs by 12 months after CTRS.
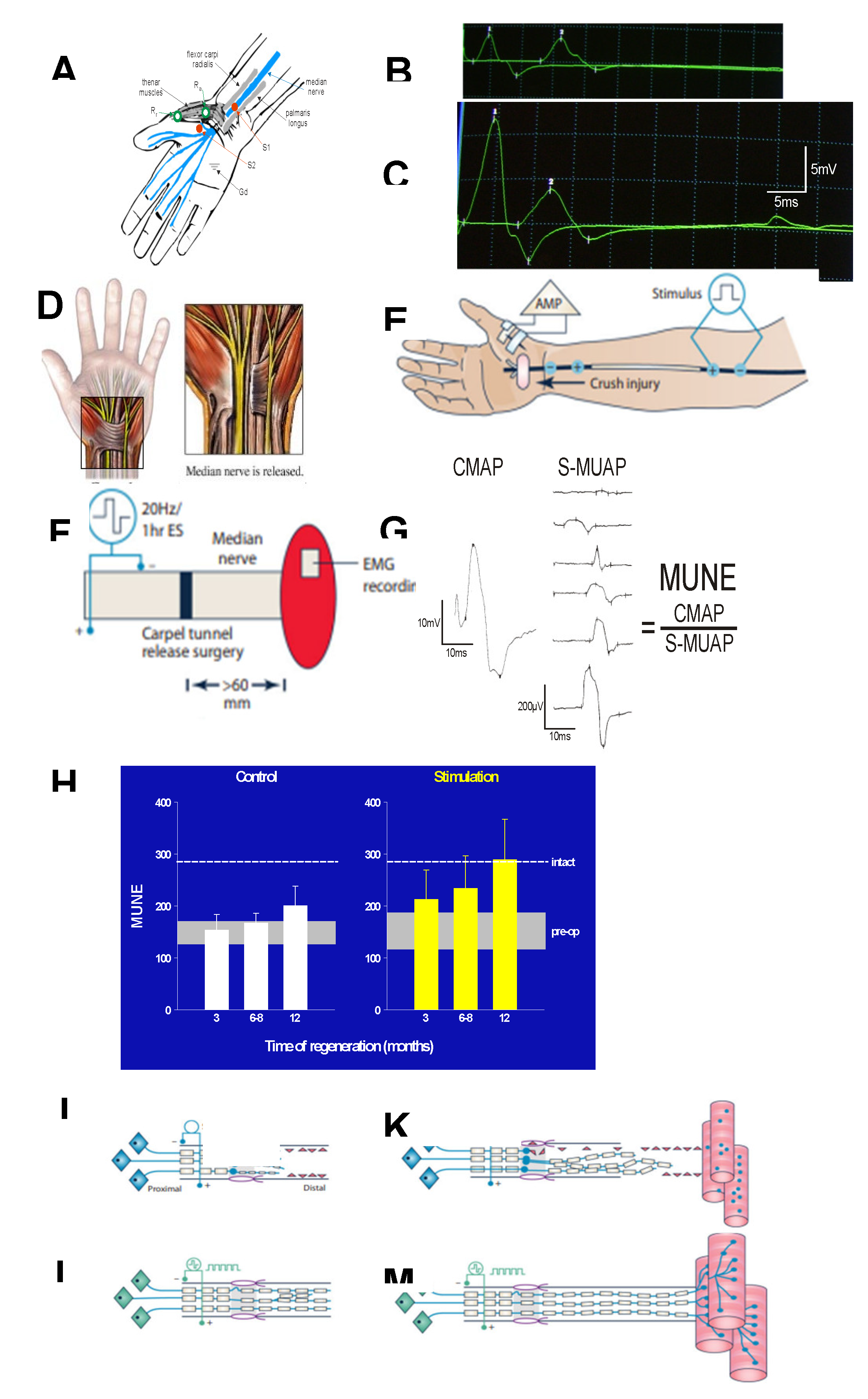
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Submitted:
14 October 2023
Posted:
17 October 2023
You are already at the latest version
Alerts
A peer-reviewed article of this preprint also exists.
This version is not peer-reviewed
Submitted:
14 October 2023
Posted:
17 October 2023
You are already at the latest version
Alerts
Abstract
Injured peripheral nerves regenerate their axons in contrast to those in the central nervous system. However, functional recovery after surgical repair is often disappointing. The basis for the poor recovery is the progressive deterioration with time and distance, of the growth capacity of the neurons that lose their contact with targets (chronic axotomy) and the growth support of the chronically denervated Schwann cells (SC) in the distal nerve stumps. This is despite the retained capacity of chronically denervated and atrophic muscle to accept reinnervation. Progressive decline in regeneration associated genes in both axotomized neurons and denervated SCs accounts for the decline in regenerative success in association with silencing of neural activity in sensory neurons due to their disconnection from their sense organs and, in motoneurons due to loss of their synaptic contacts in the spinal cord. Whilst exogenous neurotrophic factors promote nerve regeneration, the profuse axonal outgrowth and difficulties in delivery are avoided by promoting their endogenous expression with brief (1 hour) low frequency (20Hz) electrical stimulation (ES) proximal to the injury site. ES accelerates axon outgrowth and in turn, target reinnervation in both animals and human subjects. Applying ES to intact nerve days prior to nerve injury, conditional ES (CES) increases axonal outgrowth and regeneration rate with the potential for application in nerve transfer surgeries and end-to-side neurorrhaphies. However, the additional surgery for applying CES electrodes may be a hurdle. ES is applicable in all surgeries with excellent outcomes.
Keywords:
Subject: Medicine and Pharmacology - Neuroscience and Neurology
1. Introduction
Romon Y Cajal [1] recognized the contrasting capacity of injured nerves in the peripheral nervous system (PNS) and central nervous system (CNS) to regenerate. Yet, recovery of function is generally disappointing despite the contrasting support of the regeneration, the regrowth of the nerve fibres, by the Schwann cells (SCs) of the PNS and lack of support by the oligodendrocytes of the CNS [2,3,4,5,6]. As an example, fewer than 50% of patients regain adequate motor or sensory function after surgical repair of injured median or ulnar nerves [7]. Indeed, only ~10% of the two million Americans who suffer some form of peripheral nerve injury, demonstrate limited functional recovery, many with impaired motor and sensory function, and frequently suffering pain [8]. Functional recovery varies with location and severity. The recovery is the most severe for the more proximal nerve injuries [8], brachial plexus injury being the most disabling with severe functional impairment and poor quality of life [9]. Hand function is rarely restored after these injuries, occurring in only 1.2% of patients with multiple-traumatic injuries with an incidence of 1.64 cases out of 100,000 patients [10]. Patients are typically healthy, young adults who are economically productive [11]. They cannot work for ~21-31 weeks after upper extremity injuries, requiring long periods of rehabilitation [11,12,13]. Many of these patients, being unable to fulfill their former functions, must make changes in their careers [11,14].
The poor functional recovery has been relatively neglected in the scientific literature. Scientists, primarily clinical scientists, have addressed the issue in studies of whether administration of drugs and or various growth factors have a positive effect on nerve regeneration [15,16,17,18,19]. Electrical stimulation (ES) of muscles, used for many years by physiotherapists, is now a standard manipulation with the goals of preventing denervation atrophy and joint fixation [20,21,22]. In contrast, the use of ES for injured nerves has been a relatively recent approach introduced in the 1980s by Nix and Hopf [23] and furthered to promote muscle and sensory reinnervation by Gordon, Brushart, and colleagues in the 2000’s [24,25,26,27,28], their findings, largely confirmed by many investigators [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56]. The efficacy of localized tacrolimus (FK506) treatment demonstrated in in vivo and in vitro animal studies [57,58,59,60,61], and its potential use in human subjects, is considered in detail in a recent review [19] and hence, will not be considered further in this review. This review considers how and why functional recovery is poor after peripheral nerve injury and the efficacy of neurotrophic factors and/or brief low frequency ES for counteracting the negative effects of delayed surgical repair as a prelude for advocating ES to promote nerve regeneration and functional recovery after nerve injuries in patients. Previous reviews have concerned one or more of these issues [6,15,62,63,64,65,66,67,68,69,70].
2. Peripheral Nerve Injury
After crush (axonotmesis) or transection (neurotmesis) nerve injuries, the neurons, and their nerve fibres proximal to the injury are separated from their peripheral targets, a state of axotomy [15,71]. The axotomized neurons, the SCs in the denervated nerve stump distal to the injury, and the denervated muscle fibres and sense organs, undergo growth-associated changes as precursors to nerve regeneration and target reinnervation. These changes are reviewed prior to consideration of their declining capabilities with time after and distance from the nerve injury.
2a. Chromatolysis and Gene Expression in Axotomized Neurons
The classical morphological changes of the movement of the nucleus of axotomized neurons to an eccentric position and the dispersion of the Nissl bodies in the cytoplasm of the cell body, are referred to as chromatolysis [72,73]. The changes reflect the increased neuronal metabolism and protein synthesis [74] as the neurons transition from their normal transmitting state to a growth/regenerative mode [75]. This transition is usually driven by the transcriptome in which transcription factors, discussed in Section 2a. Neuronal molecular signaling of nerve injury, are responsible for coordinating the expression of multiple regeneration associated genes (RAGs) [76]. Genes that translate proteins for neurotransmitter synthesis including choline acetyl transferase in motoneurons, are downregulated whilst the RAGs, including those that transcribe the cytoskeletal proteins, tubulin, and actin, are upregulated (Figure 1A [77,78] reviewed by [75]). The cytoskeletal proteins are essential for the transport of materials from the cell body to the growth cones for elongation of the growing axons [79]. The corresponding downregulation of neurofilament protein that controls the calibre of the axons in motor and sensory nerves [15,77,80,81], accounts for the reduced size of the nerve fibres in the proximal nerve stump of injured nerves, as measured electrophysiologically [15,75,82] and morphologically [15,80,81,83,84]. The expression of neurotrophic factors including BDNF (brain derived neurotrophic factor) and its receptors trkB and p75, in motoneurons, is upregulated (Figure 1A: [15,16,85,86]. The several phenotypes of the heterogeneous DRG sensory neurons and their Ia, Ib, and cutaneous afferent nerve fibres that supply muscle spindles, tendons, and skin, respectively, transition to a more homogenous phenotype after axotomy [87].
2b. Neuronal Molecular Signaling of Nerve Injury
There are two distinct phases of the neuronal signalling of the nerve injury: (i) a rapid phase dictated by a retrograde calcium wave to the neuronal soma and (ii) a later slow signaling phase characterized by the retrograde transport of signaling molecules by motor proteins [88,89,90].
In the first rapid phase, the disruption of the axonal membrane at the site of the nerve crush or transection, exposes the axonal cytoplasm to external ionic concentrations. Within seconds of the injury, calcium ions enter the proximal nerve stump from the external fluid and, as membrane depolarization activates voltage-gated calcium channels, the intracellular calcium concentrations rise and trigger rapid sealing of the axon membrane at the injury site [91,92,93,94,95]. Local protein translation proceeds, long-range retrograde signaling is activated, and the local cytoskeleton contributes to the formation of the growth cone [96,97,98,99]. The growth cone is composed primarily of a microtubule cytoskeleton and F-actin (filamentous actin) with the central domain containing microtubules, organelles, and vesicles, and the P domain, composed of dynamic microtubules and F-actin [100,101]. The F-actin bundles comprise the finger-like projections of the filopodia.
The axonal calcium activates the nucleotide cAMP (cyclic adenosine monophosphate), via the calcium-dependent adenylyl cyclase enzyme which, in turn, activates the pro-regenerative kinase DLK (dual leucine zipper-bearing kinase) via PKA (protein kinase A), and the transcription factor, CREB1 (cAMP-responsive element-binding protein 1), amongst others [102,103]. DLK is a key sensor of local injury that informs the soma about the injury [102,103]. JNK (JUN N-terminal kinase) downstream of DLK signaling, is also transported to the soma where it activates STAT3 (signal transducer and activator of transcription factor 3) and c-Jun {an immediate early gene of the AP-1 (activator protein-1 transcription factor) transcription complex of Jun, c-Fos, and the ATF/CREB families [105]}, to promote nerve regeneration [106,107]. A wavefront of the calcium ions propagates anterograde to the cell body to reach millimolar concentrations in the soma. Calcium is also raised throughout the stump by additional calcium entry that follows the reversal of the sodium-calcium exchange pump by the calcium load [92]. The accumulating calcium ions activate local intracellular calpain (a serine-threonine protein) and oxidative species, resulting in axon swelling, rapid granular disintegration of the axonal cytoskeleton [108,109,110,111,112,113], and dieback to the first node of Ranvier [114,115,116]. The local calpain at the sealed end of the stump cleaves the submembranous spectrin complex, restructures the cytoskeleton by microtubule and actin depolymerization, and, in turn, allows the elaboration of the growth cone [95,117,118,119].
The local calcium activates many other signaling pathways. These include CaMK (Ca2+/calmodulin-dependent protein kinase) that phosphorylates CREB in the nucleus. The phosphorylated CREB later influences gene expression directly. It does so by mediating cAMP-induced transcription [120,121] via PKA and MEK/Erk (extracellular signal-regulated kinase) pathways [122], and the translation of the transcription factor, HIF-1a (hypoxia-inducible factor), that, in turn, activates HIF-1a responsive genes for axonal regeneration [123].
There is an initial and local translational burst of mTOR (mammalian target of rapamycin) that controls ~250 localized mRNAs. These in turn, transcribe several proteins that are transported to the cell body via retrograde axonal transport. The transport is facilitated by the local increase in tyrosinated α-tubulin in the microtubular cytoskeleton [124]. The transported proteins include importin-β1, the adaptor protein that transports cytoplasmic proteins via dynein, the retrograde motor on the microtubules [125], vimentin (the intermediate filament protein), STAT3, ZBP-1 (Z-DNA-binding protein 1), RanBP1 (Ran binding protein 1), Ran being the Ras-related nuclear protein ligand-activated nuclear receptor, and PPARƔ, the peroxisome proliferator-activated receptor ([126,127,128], reviewed by [129]). Luman/ATF3, an endoplasmic reticulum (ER) transmembrane basic leucine zipper transcription factor, is also transported in an importin-dependent manner to the cell body where it is critical regulator of sensory axon regeneration, linking the unfolded protein response and the ensuing endoplasmic stress response to axon repair [130,131]. There is an interesting coordination of the temporal phases of the luman protein levels. They are co-ordinated with the three phases of the growth responsive response of sensory neurons, (1) the stress response phase immediately after injury, (2) the pre-regenerative phase, 9 to 24 hours thereafter when transcription factor activity regulates DNA replication and transcription, and (3) the regenerative phase at 4 days [132].
The transported proteins activate pro-regenerative pathways in the second slow signaling phase of nerve injury. These pathways include the translation and activation of transcription factors, specific epigenetic modifiers, and additional signaling molecules [129].
Transcription factors There are more than 1500 transcription factors in the genome [133] of which c-Jun is markedly increased in axotomized motor and sensory neurons [134]. The genes that coordinate the transcription of many other genes, often the transcription factors, are referred to as Hub genes [102,135,136]. The transcription factors bind to selective DNA promotor regions [137,138,139,140] to increase or repress the transcription of specific target genes, thereby coordinating the expression of multiple RAGs [76,129]. The transcription factor c-JUN was the first to be identified in a RAG network [134,141). CREB serves to coordinate the transcription of many RAGs because the CREB protein not only regulates transcription of BDNF and arginase 1, amongst other genes, but also drives the transcription of both the AP1 and the ATF3 hub genes, being a highly connected node [101]. One study distilled the RAG network to ~40 transcription factors downstream of multiple parallel signaling pathways [142], of which the calcium-dependent cAMP activates only a fraction of injury-induced genes, at least in sensory neurons [143].
Transcription factors that were identified independently, include ATF3 (cAMP-dependent transcription factor 3) [144,145,146,147,148], STAT3 [149,150], SOX11 {(SRY-Box Transcription Factor 11) [151,152,153,154]}, SMAD1 (Suppressor of Mothers against Decapentaplegic family member 1) [155,156,157,158,159], C/EBPβ (CCAAT/enhancer binding protein β) [160,161], p53 (tumor protein p53) [162,162], GSK3β (glycogen synthase kinase 3β [163]), c-Myc [151,152,153,154], and KLF4 (Krüppel-like factor 4) [163,164]. ATF-3, a cAMP-dependent transcription factor, is upregulated rapidly in injured motor and sensory neurons following JNK signalling [87,146,147,148]. The factor is used frequently and reliably as a biomarker of axotomy [27;87,145]. Both Jun and ATF3 mediate peripheral nerve regeneration in vitro [151] and in vivo [152]. Phosphorylated STAT3 stimulates growth initiation but does not perpetuate axonal growth [150]. SOX11 is also elevated in injured nerves and promotes their regeneration [151,153]. It does so via the activation of the transcription factors, ATF3 and c-Jun, and the RAGs, Arpc3 and Sprr1a [151,153], and by increasing the responsiveness of neurotrophic factors [153]. Elevated SMADs are phosphorylated and accumulate in the nuclei of injured DRGs and motoneurons, SMAD1 in the sensory neurons [155,156,157,158] and SMADs 1,2, and 4 in the motoneurons [159]. They act as modulators of activated GSK3β (glycogen synthase kinase 3) downstream of P13K (phophoinositide-3-kinase)/Akt pathway, by interacting with the transcriptional coregulator p300 HAT (histone acetyltransferase p300), to promote expression of several pro-regenerative target genes and in turn, nerve regeneration [155,156,158]. The transcription factor C/EBP (CCAAT/enhancer binding protein) that is induced in injured neurons, is also an essential transcription factor for nerve regeneration. It binds to the promoters of Tα1-tubulin of the microtubules and the growth cone protein, GAP-43 [161]. It was reported that p53 stimulates regeneration of transected sciatic nerve fibres with reduced reinnervation of target muscle fibres after facial nerve injury in p53 knockout mice [162,163]. The c-Myc transcription factor stimulates peripheral nerve regeneration by elevating expression of the RAGs arpc3 and Sprr1a and the transcription factors c-Jun and Atf3, in the nucleus of axotomized DRG neurons, and by increasing the responsiveness of the neurons to neurotrophic factors [151,152,153,154]. In contrast to the other factors, KLF4 is downregulated after injury, thereby negating its inhibitory effect on nerve regeneration [164,165].
Epigenetic modifications, or “tags,” regulate patterns of gene expression by altering DNA accessibility and chromatin structure without altering the DNA sequence [166]. They include the miRNA (microRNA), and the non-coding RNAs, namely the long chain and circular RNAs. They can affect nerve regeneration by altering the access of transcription factors to DNA by DNA methylation, post-translational modification of the histone protein that wraps around nuclear DNA, or by controlling ncRNAs (noncoding RNA) that silence genes [101,130,167].
DNA methylation generally, is associated with repression of transcription [129]. There are >200 identified histone modifications [168]. An important example is the p300/CBP-associated factor (PCAF)-dependent acetylation of H3K9ac (histone 3 lysine 9) that is paralleled by the reduction in methylation of H3K9 (H3K9me2). This reduced methylation relaxes the chromatin environment surrounding the promoters of several pro-regenerative genes. In DRG sensory neurons, this modification results in the expression of the growth-associated genes GAP-43, galanin, and BDNF [169]. The acetylation requires retrograde ERK signaling. Also, PCAF overexpression promotes axonal regeneration of the central axons of the DRG neurons across injured spinal cord.
MicroRNAs (miRNA), of which lncRNA (long chain non-coding RNAs) account for 60 to 80% of the mammalian genome transcriptome [170], are differentially expressed after peripheral nerve injury [101,171]. They target specific mRNAs with resulting repression or degradation of their translation. For example, miR-21 is upregulated after sciatic nerve injury and by targeting Sprouty2 (a specific inhibitor of the Ras/Raf/Erk pathway), promotes axonal growth from adult DRG neurons [172,173]. A second example is the nerve regeneration that results from miR-26a that specifically targets GSK3β to rescue axon regeneration, the miR26a-GSK3β pathway regulating axon regeneration at the neuronal soma by controlling the expression of the regeneration-associated transcription factor, SMAD1 [160].
2c. Wallerian Degeneration
The peripheral nerve fibres distal to the site of crush or transection, are isolated from their neuronal cell bodies and, in turn are deprived, for all intents and purposes, of their source of synthesis of proteins, lipids, glycoproteins, and carbohydrates [15,72,109]. They undergo the self-destructive process of Wallerian degeneration of nerve fibres and their myelin, including the axons that die back to the first node of Ranvier in the proximal nerve stump that prevents the scarring that occurs in CNS injury [114,115,116,174]. Ramon Y Cajal [1], using his silver staining technique, elucidated the degeneration of the axons distal to the nerve injury, myelin breakdown, and the proliferation of the remaining SCs. The calcium ions that enter the nerve via calcium channels, activate calpain proximal and distal to the injury site, that mediates proteolysis with degeneration of axon segments several hundred micrometers from the injury site [175,176]. The expression of SARM1 (sterile alpha TIR motif-containing protein 1), the essential protein for axon degeneration in the distal nerve stump, is elevated due to the loss of the anterograde transport by the calcium-dependent proteolysis of the cytoskeletal structures in the stump, followed by a rapid depletion of NMNAT2 (NAD-synthesizing enzyme, nicotinamide mononucleotide acetyltransferase 2; reviewed by [18]). The downstream steps to axon degeneration remain to be determined. Meanwhile, the remaining fast axonal transport allows for continued propagation of action potentials in the distal stump for hours and even days [108,177,178,179,180].
Ras/raf/ERK signaling in SCs is evident immediately after injury with ERK levels returning to lower levels just prior to SC proliferation [181]. Activation of Raf-kinase drives SC dedifferentiation as well as inducing much of the inflammatory response important for nerve repair, including breakdown of the blood nerve barrier and the delayed recruitment of macrophages into the denervated nerve stump [182]. SCs break their myelin sheaths down through autophagy [183,184,185] with ~50% of the total myelin thought to be broken down by the SCs [186]. The SC expression of proinflammatory cytokines and chemokines within 3 to 5 hours of nerve injury, contribute to myelin and axon breakdown and the phagocytosis of their debris within the first 3 days of injury [187]. The cytokines, including IL-1α (interleukin 1α), and LIF (leukemia inhibitory factor), their receptors IL-6R and gp130 respectively, and tumor necrosis factor α (TNF-α), stimulate the expression of the two cystolic forms of PLA2 (phospholipase enzyme-A2) [188]. These remain high for two weeks [187]. TNF-α hydrolyses the phosphatidylcholines in the myelin membranes, thereby releasing the potent myelinolytic agent, lysophosphotidylcholine. Lysophosphotidylcholine also feeds back to sustain cytokine expression and the expression of the chemokines, including MIP-1 (macrophages inflammatory protein-1), MMP-1α or CCL2 (monocyte chemoattractant protein-1α) and IL-lβ [17,189,190,191,192,193,194,195,196]. Within a day of the nerve injury, IL-6 and TNF-α induce the expression of MMP-9 (matrix metallopeptidase 9) that contributes to myelinolysis [197,198].
The recruited macrophages also express these cytokines and chemokines and are responsible for the bulk of the myelin and axonal phagocytosis over the following ~3 weeks [116,199,200,201,202]. They play the major role in removing inhibitory molecules, including MAGs (myelin associated glycoproteins), from the degenerating axons [202]. Their release of nitric oxide has been implicated in the myelin breakdown [203] and their phagocytosis of the myelin debris includes the myelin that is opsonized by complement components binding to the complement receptor type 3 on the macrophages [204]. Antibodies to non-opsonized myelin are phagocytosed via the macrophage Fc receptor [205]. The third macrophage receptor used is the scavenger receptor-AI,II [206]. The spectrum of macrophages has been separated into two “polarizing” phenotypes, M1 and M2 with the M1 macrophages associated with pro-inflammatory and neurodegenerative functions and the M2 macrophages broadly viewed as anti-inflammatory and promoting cellular repair [207]. Although an early increase in M1 macrophages 1-2 days after injury with M2 macrophages replacing these from days 3 to 7, the majority of the accumulating M2 macrophages may be of a mixed phenotype because these mixed type macrophages were not included in their analysis [207,208]. In addition to their essential role in phagocytosis of degenerating axons and their myelin, the macrophages sense hypoxic conditions and stimulate angiogenesis for a polarized vasculature that guides SCs and elongating axons [209,210].
2d. Schwann Cell Response to Nerve Injury
Once the SCs lose their myelin, they re-enter the cell cycle, undergo mitosis, and differentiate toward a state supportive of nerve regeneration [5,15,75,108,139,211]. The transient expression of Cdc2 (cyclin-dependent kinase, cell cycle division 2) by the denervated SCs, possibly induced by c-Jun [212], is involved in their proliferation and migration [213]. The SC genes that transcribe myelin proteins, including MAP (myelin associated protein), MBP (myelin basic protein), P0 (myelin protein zero), and PLP (proteolipid protein), are downregulated in the denervated distal stump with the RAGs associated with nerve regeneration, upregulated as the SCs dedifferentiate and acquire the ability to survive without axonal interactions (Figure 1A; [214]). This change in SC transcription occurs with their transition from their myelinating state to the growth supportive state [75]. The transcription factor, c-Jun, programs the SCs to generate the repair cells that are essential for nerve regeneration, c-Jun accelerating the downregulation of myelin genes, promoting myelin breakdown, and amplifying the upregulation of a broad spectrum of repair-supportive features, including the expression of trophic factors ([137,215]; reviewed by Jessen and Arthur-Farraj [216]). As early as 1991, the upregulation of hundreds of growth-supportive RAGs was reported in denervated SCs [217] with more genes differentially expressed in the SCs than in sensory neurons [218].
IL-6, synthesized in the SCs within 24 hours [190,192,219,220], signals the expression of RAGs via its receptor [191]. The SCs express NRG-1 (neuregulin-1), a member of the family of glial growth factors, and its ErbB2/3 receptor [221,222,223,224]. The NRG-1 levels remain elevated for at least 30 days [225,226,227]. NRG-1 strongly inhibits the expression of genes involved in myelination and in glial cell differentiation, suggesting that it might be involved in the dedifferentiation process of SCs from the myelinating to the repair phenotype [228]. In addition, NRG-1 likely mediates, at least in part, the second phase of SC proliferation that is stimulated when regenerating axons contact the SCs in the Bands of Büngner [229,230,231,232,233,234,235]. The scaffolding oncoprotein Gab2 (Grb2-associated binder-2) is required for SC proliferation after nerve injury, its activation leading to the migration of the SCs [236], possibly through actin modulation [237]. In the model based on their findings, autocrine/paracrine activation by NRG of its erbB2 receptor on the SCs, promotes the SC migration by leading to transcriptional Gab2 expression via the Rac-JNK-cJun pathway, and the phosphorylation of Gab2 via the paracrine HGF (hepatocyte growth factor) from fibroblasts [236].
The non-coding RNAs, namely the long chain and circular RNAs, play important roles in SC proliferation and migration [101]. Examples of the long chain RNAs include NEAT1 (nuclear enriched abundant transcript 1) that promotes SC proliferation and migration [101], MALAT1 (metastasis associated lung adenocarcinoma transcript 1) that elevates BDNF [238], and Loc680254 that promotes nerve regeneration by inducing SC proliferation [239]. BC088259 which showed the most significant upregulation after sciatic nerve injury, interacts with vimentin to regulate SC migration [240,241]. Downregulation of some long chain RNAs also enhances SC proliferation and migration. An example is MEG-3 (maternally expressed gene 3) that increases SC proliferation and migration and facilitates nerve regeneration through the PTEN/P13K/ADT pathway [242]. Some circular RNAs are also upregulated in the denervated nerve stumps and are associated with SC proliferation. For example, cirRNA-Spidr targets P13K-Akt to promote nerve regeneration after rat sciatic nerve crush injury [243].
Within the first week of nerve injury, the denervated SC’s express several neurotrophic factors and their receptors, their levels peaking within a month. The factors include the neurotrophins, NGF (nerve growth factor), BDNF, and their p75 receptor, NT-3 and NT-4/5 (neurotrophin-3 and 4/5) and the TrkC receptor, the GDNF (glial derived neurotrophic factor) family and their receptors, GDFRα1 and ret, and other factors including IGF-1 and IGF-II (insulin-like neurotrophic factor I and II), VEGF (vascular endothelial growth factor), HGF (hepatocyte growth factor), platelet-rederived growth factor-BB, FGF (fibroblast growth factor), TFG-β (transforming growth factor β), and their receptors, and pleiotrophin [16,244,245,246,247,248,249] and Xu et al 2023 unpublished data. The expression of GDNF and pleiotrophin is specific for the denervated SCs in the motor pathways of the quadriceps nerve branch of the femoral nerve, whereas the remaining neurotrophic factors, HGF, BDNF, NGF, and IGF I and II, are more specific for the denervated SCs located in the sensory pathways of the saphenous nerve branch [245,246,247]. The time course of expression varies for different trophic factors. NGF rises rapidly, then declines prior to a 5-fold upregulation possibly in response to macrophage release of IL-1β and persisting for at least 3 weeks [250,251]. The upregulation of BDNF is much slower, detectable at 7 days and increasing up to 28 days after nerve injury to levels much higher than NGF [16,251]. On the other hand, GDNF and its GDFRα1, but not its coreceptor Ret, are upregulated and reach a peak within 7 days [16,252]. The expression is not sustained, declining with time when the denervation period of the distal nerve stump is prolonged for more than a month [16,244,245,247,252]. NT-3 upregulates c-Jun [215] and regulates the levels of the upregulated p75 receptor in the SCs [182,254,255,256]. The transcription factor, Notch, is also upregulated in the SCs [137,138,139,140].
2e. Axonal Regeneration
Regenerating axonal sprouts emanate from the first node of Ranvier proximal to the injury site [1,257,258]. The formation of the growth cones occurs without direct support from the cell body and depending on material that is locally available in the axons in the proximal nerve stump that includes the preexisting cytoskeletal elements of actin and tubulin [258,259]. The major source of materials for subsequent axonal elongation comes from the cell body via anterograde axonal transport [79]. As described above in Section 2d. Schwann cell response to nerve injury, the growth cones link to ECM glycoproteins that become organized at the injury site, navigate across the site, and regenerate axons along the bands of Bungner in the denervated distal stumps. A single regenerating axon can give rise to as many of 50-100 branches [260] but, it is an average of 5 daughter axons that regenerate with more regenerating into the distal stump after crush than after transection injuries [261,262,263].
Following the nerve injury, the SCs migrate to form the repair SC layer of the Bands of Bungner on the endothelial laminal sheath [214]. They do so by their sorting through intercellular N-cadherin linkage mediated by EphB-Sox signaling [264]. The growing axons contact the SC basement membrane glycoproteins that are secreted by both SCs and fibroblasts. These glycoproteins include collagen, fibronectin, tenascin C, and laminin ([265,266,268,269]; reviewed by [5,214,267,270,272]). Via adaptor molecules on the SC membranes, including N-cadherin and integrins, the interaction of the axons with the glycoproteins mediates the progression of the growth cones as the axons regenerate [271,273,274,275,276,277,278,279,280].
Contact between the axolemma and SCs initiates remyelination of regenerating axons with the SCs forming myelin layers (lamellae) in proportion to the size of the regenerating axons [281]. Both NRG-III from axons and NRG1-I from SCs play a pivotal role in remyelination [226,282,283] with tyrosine phosphorylation of GAB1 in the denervated SCs, principally regulated by NRG-1, being essential for remyelination [236]. The internodal distances between the formed myelin sheaths are shorter than normal as a result of the 3-fold increase in SC numbers after denervation [284] but they lengthen with time [281]. The regenerating axons increase their diameters in proportion to the size of their parent nerve [285], recovering their normal size if and when they make functional connections [286].
3. Poor Recovery of Function after Peripheral Nerve Injury and Repair
The clinical examinations and overall patient health being considered, decisions to operate after nerve injuries are not as straight forward as the surgeons would like. The simplest classification of nerve injuries is whether the injury is closed or open. Traumatic injuries that are classified as open injuries result in neurotmesis (nerve transection), but the timing of surgical intervention depends on whether the nerve transection is ragged or sharp [287,288]. The primary repair of sharp injuries from knives or razors should be repaired within 3 days of trauma but the repair of ragged nerve transections from blast, gunshot, fracture, or crush injuries is usually delayed for at least 3 weeks to allow the nerve ends to be demarcated [288]. Frequently, an early surgical evaluation is made of whether any of the nerve remains in continuity. When the nerve is transected, the nerve stumps may be sutured to a nearby local structure to prevent their retraction and thereby, to facilitate later surgical repair. Especially when a nerve injury is associated with comorbidities that include vascular problems, the later repair is undertaken once the local inflammation has subsided [287,289]. Irrespective of whether or not surgical repairs are delayed, proximal injuries in particular, result in long delays before regenerating nerve fibres contact distal denervated targets (Figure 1B). Even after early surgical repair after proximal injuries such as the brachial plexus nerve injuries, many axotomized neurons remain chronically axotomized without target contacts for periods of 2 or more years as they regenerate their axons slowly over long distances at the regeneration rate of the estimated 1 mm/day using the Tinel sign that identifies the site at which a tap on the regenerating nerve elicits a tingling sensation in the conscious patient [290,291]. The SCs in the nerve stump distal to the injury are subjected progressively to chronic denervation especially those far distal to the microsurgical suture of the proximal and distal nerve stumps.
Based on morphological evidence, the predominant view of the denervated distal nerve stump is that the SCs progressively die as the endoneurial tubes break down and are replaced by connective tissue that likely is impenetrable to regenerating axons. It was reported in rat and rabbit hindlimb nerves that, over periods of 1 to 26 months, denervated SCs undergo progressive atrophy and loss with disruption of their basement membranes, shrinkage of their columns, and filling of the columns with densely packed longitudinally orientated collagen fibrils [292,293,294,295,296]. Yet, SCs do remain 6 -25 months after denervation of rat and rabbit sciatic distal nerve stumps and intramuscular nerve trunks, although they are reduced in number [224,296,297]. The numbers decline to less than 10% but, the surviving SCs proliferate in response to mitogens, and they myelinate neurites normally (Figure 2A-C; [298,299]). Indeed, these SCs support the full recovery of the size of regenerated nerve fibres in vivo [300,301] The transcription factor STAT3, has been implicated in the long-term maintenance of SCs [302] and c-Jun influences both apoptosis and proliferation of denervated SCs [303,304,305]. The most accepted interpretation of poor functional recovery remains the irreversible atrophy of muscle and sensory targets and their replacement by fat [3,306]. Muscle mass is known to decline rapidly within two weeks after denervation, to decline more gradually thereafter [307,308,309], and to undergo permanent damage after two years [310].
3a. Time- and Distance-Related Decline in Nerve Regenerative Capacity
A cross-suture technique was pioneered by Holmes and Young in 1942 to examine their hypothesis that chronic motoneuron axotomy accounts for the poor functional recovery after surgical repair [311]. Their finding that reinnervated muscle contractile force was restored to normal levels negated their hypothesis. leading them to conclude that indeed, denervated targets are replaced by fat. The conclusion of fat replacement of denervated muscle accounting for poor functional recovery after nerve injury remained the accepted view for the next 50 years. More recently, we systematically examined each of the contributions of chronic axotomy, chronic denervation of the distal nerve stump, and chronic denervation of target musculature, to poor recovery after nerve transection (Figure 2; [28,71,300,312,313,314,315,316,317,318]; reviewed by [6,15,16,62,64,69,248,319,320,321,322]. Our surgical technique, illustrated in Figure 3A,B, was to prolong the axotomy of TIB (tibial) motoneurons or to prolong denervation of the CP (common peroneal) distal nerve stump prior to the cross-suture of TIB and CP proximal and distal nerve stumps, respectively. After at least 5 months, we determined how many motoneurons (1) reinnervated tibialis anterior (TA) muscle fibres by estimating the number of reinnervated motor units (MUs) by the ratio of the whole muscle to mean MU isometric contractile forces; Figure 3C) and (2) regenerated their nerve fibers into the CP nerve stump by applying retrograde dyes of either FG (fluorogold) or FR (fluororuby) to the regenerated nerves in the CP nerve stump. Either dye was used in the experiments that followed because the motoneuron counts using either dye, were not significantly different (Figure 3D,E).
Contrary to the conclusions made in 1942 [311], chronic axotomy of motoneurons had a pronounced negative effect on the regeneration of their nerve fibres: both the numbers of (MN) motoneurons that regenerated their axons into the freshly denervated nerve stumps and the numbers of MUs, the motoneurons whose nerves reinnervated muscle fibres, declined exponentially with a time constant (Ƭ) of 40 days to reach a plateau of 33% of the numbers after immediate nerve repair (Figure 4A,C; [71,312,313]). It was the capacity of regenerating motor nerve fibres to reinnervate 3-5 times as many denervated fibres as they normally do [323,324], the same capacity as that of intact nerve fibres sprouting in partially denervated muscles [325,326]. This 3- 5- fold increase in MU size resulted in the reinnervation of all the denervated muscle fibres and, with their full recovery from muscle atrophy, the full recovery of the reinnervated muscle contractile force [71]. Complete recovery of muscle force by reinnervation by chronically axotomized motoneurons, was the basis for the faulty conclusion made previously in 1942 [311] and is the reason why the clinical assessments of reinnervated skeletal muscle strength by the Medical Research Council (MRC) 5-point scale [327] or by revised scales [328]. are frequently misleading. These clinical assessments continue to underestimate the deleterious effects of chronic axotomy as well as chronic denervation of distal nerve stumps and muscles on nerve regeneration and muscle reinnervation. It is interesting, as an aside, that the size of the nerve supplying the enlarged MUs does not increase [329], the regenerated nerve fibres recovering their normal size once they made functional connection with denervated muscle fibres [300].
Chronic denervation of the distal nerve stump and the muscles that the intact nerve normally supplies, is even more deleterious to nerve regeneration than chronic motoneuron axotomy (Figure 4B; D;300,312.318]. The exponential decline in numbers of MUs and freshly axotomized motoneurons that regenerate their axons into the chronically denervated nerve stumps plateaued at ~5% at 180 days of chronic denervation (Figure 4B). To determine whether the poor regeneration was due to chronic SC denervation within the distal nerve stump and/or prolonged muscle denervation, a chronically denervated autograft was inserted between a freshly cut TIB proximal nerve stump and a chronically denervated CP distal nerve stump [318]. The experiments demonstrated that it is the chronic denervation of the SCs and not the chronic denervation of the TA muscle that was the causative factor of the poor nerve regeneration through chronically denervated distal nerve stumps [318]. It is noteworthy that the chronically denervated SCs that do survive support myelination in vitro [298] and in vivo [318].
The reduced numbers of nerve fibres that regenerated through the chronically denervated nerve stumps reinnervated as many as 3-times as many TA muscle fibres as normal [312]. However, this capacity of the nerves to expand the size of the reinnervated MUs was insufficient to reinnervate all the remaining denervated muscle fibres. In addition, the reinnervate muscle fibres did not recover their normal size [312]. As a result, the partially reinnervated muscles failed to recover their normal contractile force after their chronic denervation, [312].
Chronically denervated muscle fibres undergo extreme atrophy within a year of denervation, declining rapidly within the first two weeks and falling more gradually thereafter [306,307,308,309]. Mitochondrial dysfunction with upregulation of the miRNA, miR142a-5p and subsequent lysosomal degradation, accompany this muscle denervation atrophy [309]. Nonetheless, the fibres survive with 98% reduction in their size after two years of chronic denervation [330]. They also conserve their fascicular arrangement and their functionally important proteins, including acetylcholine receptors and neural-cell adhesion molecules [330]. Although the few freshly axotomized motoneurons (~5% of the normal number) that regenerated their axons through chronically denervated distal nerve stumps, did reinnervate chronically denervated muscle fibres, the reinnervated muscle fibres failed to recover their former size [312]. We suggested that this incomplete recovery was the result of declining numbers of their resident satellite cells able to provide nuclei to the fibres [312]. This was demonstrated in morphological studies [331,332] and later evidence of apoptosis of the cells [332]. The satellite cells are a population of heterogenous stem cells that lie between the sarcolemma of the myofibres and the ensheathing basal lamina [333]. Normally, they are mitotically quiescent and express the paired box protein, Pax7 [334,335,336]. They are activated upon nerve and/or muscle injury [333]. They divide by asymmetrical division to produce myogenic progenitor cells, and by symmetrical division, they produce multiple satellite cells that contribute nuclei to the atrophic muscle fibres driving reinnervated muscle fibres to increase their size [334,335,336]. As the activated satellite cells differentiate into myocytes, they co-express Pax7 and the muscle-specific basic helix-loop-helix protein, MyoD (myoblast determination protein1) of the MRF (myogenic regulatory factor) family of transcription factors [334,335,336]. A linear regression line was fitted to the decline in the numbers of MyD positive cells in chronically denervated biceps muscle of patients as a function of the time to the surgical repair of their brachial plexus injury [337]. Examination of their data, however, indicates that the data are more readily fitted by an exponential rather than a linear decline with the MyD expressing cells declining to a low plateau rather than to zero. Earlier morphological studies of chronically denervated frog muscles, suggested that the proliferative capacity of the satellite cell pool is exhaustible [338] and evidence of repeated cycles of muscle fibre degeneration and regeneration [339] indicated but did not prove that the ultimate fate of chronically denervated muscle fibres is their replacement by fatty and connective tissues [340]. The question remains as to whether the explanation for incomplete recovery of reinnervated muscle fibre diameters after chronic denervation, is that the satellite population of the denervated muscles is depleted with time and/or that the cells fail to divide and express MyD.
3b. Transient Expression of Regeneration Associated Genes
We addressed the question of why the regenerative capacity of the motoneurons and the growth support of the SCs decline with time and distance. We reasoned that elevation of RAG expression by axotomized motoneurons and/or denervated SCs declines with time after chronic nerve injury, a hypothesis that proved to be correct. The parallel decline in expression of RAGs that likely accounts at least in part, for poor functional recovery, is illustrated by the exponential decline of the expression of tubulin in chronically axotomized motoneurons (Figure 5), and of c-jun (Figure 6), p75 (Figure 6,Figure 7), and GNDF (Figure 7), in chronically denervated SCs.
Chronic axotomy After section and ligation of the sciatic nerve in rats (Figure 2A,B and A), the mRNA of cytoskeletal proteins, tubulin and actin, and of GAP-43, was detected and quantified in the axotomized sciatic motoneurons by in situ hybridization [341]. The mRNA levels in the chronically axotomized motoneurons were expressed relative to the high levels of the proteins that were detected at a week after axotomy of the contralateral sciatic nerve (Figure 5B-J) They declined exponentially to the low levels of intact motoneurons by 6 months (Figure 5B-G). Yet, a refreshment injury of a second sciatic nerve section after 1,3,4, and 6 months of chronic axotomy resulted in upregulation of the genes to the high levels detected after the first axotomy (Figure 5D,H-J). This elevation however, decayed more rapidly than after the first axotomy of the motoneurons (Figure 5D). Similar findings were reported for facial motoneurons [342].
Chronic denervation Gene expression and protein translation in denervated SCs also declines progressively after chronic denervation of the distal nerve stump [16,253,342,343,344]. The transcription factor c-Jun which is central to reprograming SCs to the repair phenotype and is the major determinant of effective repair [215], is upregulated in mouse denervated SCs within a week of injury but, the protein levels decline progressively after chronic denervation (Figure 6A,B; [344]). The location of c-Jun in the denervated SCs was verified using mice in which c-Jun was inactivated selectively in SCs in c-Jun knockout mice (Figure 6C,D; [215]). The p75 receptor (p75NTR) protein declined with time in chronically denervated nerves (Figure 6E in mice and Figure 7C,D. in rats; [253]) in parallel with the decline in its mRNA (Figure 7B; [253]). These in vivo findings were replicated in SC cultures where both c-Jun and p75NTR proteins were reduced in the 9th passage of the SCs (Figure 6F-H; [344]). Downstream of c-Jun, the expression of GDNF in the chronically denervated nerve stumps declines exponentially (Figure 7E-G; [339]). The decline is specific for the neurotrophic factors and not other proteins in the distal stump, including NTN (Neurturin), PSP (Persiphin), and ART (Artemin) (Figure 7E-F). The transient nature of gene expression of neurotrophins and their receptors in motoneurons is summarized in Figure 2 of the extensive review of Boyd and Gordon [16].
Shh, a gene that is not expressed in either developing SCs or in intact nerves, is upregulated strongly in SCs after injury [214,345,346,347] and improves nerve regeneration in several settings [347,348,349,350]. Applied Shh elevated c-Jun in cultured SCs and the expression of both Shh and c-Jun declined during chronic denervation [344]. Furthermore, the activation of c-Jun is reduced in the SCs of injured nerves together with reduced expression of its target p75NTR when the Shh gene was knocked out conditionally in the SCs [344]. Inhibiting Shh signaling also reduces SC expression of BDNF, motoneuron survival after injury, and axon regeneration [345,347].
The identified gene set of Aqp5, Gpr37L1, Igfbp2, and Olig1, regulated by c-Jun in injured nerves [214], is highly enriched amongst the genes affected by chronic denervation as well as in aging and it is positively correlated with both c-Jun levels and regeneration [344]. Igfbp2 for example, promotes phosphorylation of Akt, a pathway that is linked to SC proliferation and differentiation (reviewed in several publications [215,351,353]). Gpr37L1 is a receptor for prosaposin and prosapeptide [353] that are secreted after nerve injury and facilitate regeneration [354]. In experiments on chronic denervation, this gene group encompasses Cxcl5, Egfl8, Gas2I3, Megf10, and Pcdh20, all of which are upregulated in SCs after injury ([355,356,357] reviewed by [358,359]). Cxcl5 activates STAT3 [355], the transcription factor shown to be important for maintaining repair cells during chronic denervation [302]. Gas2I3 has a role in the cell cycle, and Megf10 in phagocytosis [360,361].
4. Neurotrophic Factors to Sustain Their Levels
4a. Exogenous Application of Neurotrophic Factors
The demonstration of dramatic neurite outgrowth from sympathetic and DRG (dorsal root ganglion) neurons in response to NGF [362]) resulted in many in vitro and in vivo studies which advocated that growth factors, including BDNF and GDNF, promote nerve regeneration [62,67,248,301,320]. Prior to 2000, the evaluations of the effects of exogenous neurotrophic factors on peripheral nerve regeneration relied on several outcome measures. These included axon counts distal to the site of injury, the ‘pinch test’ that evaluates sensory nerve regeneration in animals by identifying the most distal point of the regenerating nerves at which a nerve pinch with fine forceps elicits an intake of breath in the lightly anesthetized animal [84,363,364,365,366,367,368,369], and walking track analysis to evaluate motor nerve regeneration [15,370]. While these evaluations may have clinical relevance, their relevance for nerve regeneration is confounded by their failure to consider the outgrowth of several axonal sprouts from the proximal stump of injured nerves in both the PNS [15,67,261,262,372] and the CNS [371]. As described in 2e. Axonal regeneration, each nerve fibre in the proximal stump regenerates an average of 5 or more axons into the denervated distal nerve stump [261,262,372]. As a result, the effects of neurotrophic factors on the numbers of axotomized neurons that (1) regenerate their axons, (2) reinnervate denervated targets, and (3) result in functional recovery, have been overestimated. This led us to adopt our retrograde labelling technique to examine how many neurons regenerate their axons into the distal nerve stump in response to exogenous application of neurotrophic factors in addition to counting the number of regenerated axons in the distal stump.
BDNF and/or GDNF When an Alzet mini-osmotic pump was implanted to deliver BDNF and/or GDNF to the site of TIB-CP nerve cross-suture in rats, either immediately or after a 2 month period of chronic axotomy of TIB neurons, retrograde labeling revealed that the reduced numbers of chronically axotomized TIB motoneurons regenerating their axons into the freshly denervated CP distal nerve stump were elevated significantly by the exogenous growth factors (Figure 8A-E; [314]). BDNF was effective only in low doses and not at high doses due to the binding of this neurotrophic factor to both the trkB and p75NTR receptors on the motor nerve membranes, the former receptor mediating a positive effect on nerve regeneration and the latter, an inhibitory effect. Further details of the receptors and the dose-dependent effects of BDNF can be found elsewhere [313]. GDNF on the other hand, was effective at all doses, acting via two receptors ret and GFR-α1, with only positive effects [314]. GDNF administration to the cross-sutured TIB-CP nerves after immediate repair did not promote nerve regeneration as it did after prolonged axotomy [314]. However, a 2- and/or 4- week delivery of GDNF encased in polymeric microspheres and placed on the suture sites of a 10 mm long ANA (acellular nerve graft) between CP nerve stumps (Figure 8F), was effective in elevating the numbers of CP motor and sensory neurons that regenerated their axons to normal levels by 8 weeks (Figure 8H,I; [373]).
Unfortunately, the regeneration enhancing effects of GDNF and BDNF were confounded by increased axonal sprouting from the site of the TIB-CP coaptation [314]. Transmitting electron micrographs of SCs and their regenerated axons in the distal nerve stump revealed a regenerated SC unit of a SC with four daughter axons when saline was administered to the suture site (Figure 8I-B) rather than the one-to-one relationship of SCs and myelinated axons in the intact nerve (Figure 8I-A; [314]). BDNF alone increased the number of daughter axons (Figure 8I-C) and the combination of BDNF and GDNF increased the number even more (Figure 8I-D). The numbers increased to as high as nine axons in each endothelial sheath (Figure 8K-A,B,C,D), This increased axonal outgrowth led us to consider increasing endogenous rather than exogenous neurotrophic factors to promote nerve regeneration.
4b. Endogenous Neurotrophic Factors
The slow upregulation of neurotrophins and cytoskeletal proteins after axotomy, described above in Section 2a. Chromatolysis and gene expression in axotomized neurons, was dramatically accelerated and amplified by a 1-hour period of low frequency (20Hz) electrical stimulation (ES; Figure 9A-G [86]). The accelerated RAG upregulation of tubulin and GAP-43 (Figure 9F,G) and the decline in expression of neurofilament (Figure 9H) are preceded by the striking accelerated expression of BDNF and its trkB receptor (Figure 9D,E). That ES increases and accelerates RAG expression after elevating BDNF and trkB expression, suggests causation between neurotrophic factor upregulation, RAG expression, and accelerated nerve regeneration as discussed below in Section 5.
5. Neuronal Activity and Nerve Regeneration
5a. Reduced Activity and Synapse Withdrawal
We addressed the question of whether the decline in regenerative capacity of chronically axotomized neurons over time/distance (described above in Section POOR RECOVERY OF FUNCTION AFTER PERIPHERAL NERVE INJURY AND REPAIR) is due to reduced neuronal activity and, in turn, to their reduced interaction with denervated SCs in the distal nerve stump.
When hindlimb motor and sensory neural activity after ligation, nerve-nerve or nerve-muscle anastomoses of transected sciatic nerve branches, was recorded on implanted electrodes during cat treadmill locomotion and separated by cross-correlation (Figure 10A,B; [374]), the motor potentials were found to decline within the first month and the sensory potentials to decline to levels below resolution (Figure 10B; [374]). The latter decline was due to the disconnection of the sensory neurons from their sense organs. The reported synaptic depression in axotomized motoneurons [375,376,377,378,379] and the decline in their motor activity levels were accounted for by the withdrawal of synapses from the neurons, the excitatory VGLUT1 glutamatergic synapses declining by ~50% and the coverage by inhibitory GAD67 GABAergic synapses declining by ~35% (Figure 10C-F; [380,381,382,383,384,385,386,387,388]). The activation of astrocytes after motoneuron axotomy likely is responsible, at least in part, for the displacement of the synaptic boutons [381,389]. The loss of synapses was reversed by daily treadmill exercise implemented 3 days after nerve transection and surgical repair, the reversal depending on slow, continuous exercise in males and interval exercise (short high-speed sprints followed by rest periods) in female mice (Figure 10G, H; [387,388]). The effectiveness of the exercise was reduced when the exercise program was delayed [388]. These findings elucidated (1) the profound effect of synapse withdrawal on neural activity during the period when the expression of RAGs, including the cytoskeletal proteins and neurotrophic factors, decline after axotomy and (2) how appropriate patterned treadmill exercise prevents this withdrawal. The exercise effect likely depends on BDNF because the synaptic contacts on intact motoneurons were reduced in conditional BDNF knock-out transgenic mice [390].
5b. Staggered Nerve Regeneration
In 1987, Brushart and Seiler [391] introduced the rat femoral nerve as a model to study peripheral nerve regeneration. It was used later in studies of Brushart and Gordon that used retrograde labeling of the motoneurons that regenerated their axons to demonstrate (1) ‘staggering’ of regenerating axons across the site of femoral nerve transection and microsurgical repair [24], and (2) preferential reinnervation of the quadiceps motor branch by motor nerves and of the cutaneous sensory branch by sensory nerves [24,26,392]. The staggering became evident when only ~220 of a total of 350 femoral motoneurons were found to regenerate axons the distance of 25 mm into the distal nerve stump (Figure 11A,B; [24]). At the accepted regeneration rate of 3 mm/day in animals [393], measured primarily with the pinch test (as described in 4a. Exogenous application of neurotrophic factors; [86,363,364,365,366,367,368,369], and including estimates of latencies of ~1-2 days prior to axonal outgrowth [81,381,382,383,384,385,386,387,388,389], all 350 motoneurons would have been expected to regenerate their axons over the distance of 25 mm. Yet, the time for all the axotomized femoral motoneurons to regenerate their axons to the site of application of the fluorescent dye(s) was 8-10 weeks (Figure 11B; [24]) The regeneration rates have been measured with the widely used pinch test in animals [86,363,364,365,366,367,368,369].
Our explanation for the protracted period of regeneration after nerve transection and repair was that growth cones ‘stagger’ across the suture site prior to entering into the denervated nerve stump [24]. That longitudinal alignment of laminin and the SCs across the suture site was observed ~10 days after nerve transection and surgical repair (Figure 11C; [394]) supported this explanation. More directly, the results of experiments in which the retrograde dye FR was applied just distal to the suture to determine when regenerating axons cross the suture site (Figure 11D,E; [25]) confirmed the explanation: the numbers of the motoneurons regenerating their axons just distal to the surgical repair site was just a few 4 days after the repair, the remaining motoneurons regenerating their axons over the protracted period of 4 weeks (Figure 11F). Our silver-staining of staggered regeneration of axons in the distal stump and the tortuous paths taken by the axons within the proximal stump (Figure 11G,H) are in accord with Cajal’s findings [1] and with later observations of the outgrowth of fluorescent axons from the proximal stump of thy-1-YFP transgenic mice into a denervated distal nerve stump of a non-transgenic wild-type litter mate [34,66].
5c. Low Frequency Electrical Stimulation Accelerates Axon Outgrowth
Continuous 20 Hz ES accelerated rat soleus muscle reinnervation [23]) suggesting but not proving, that the ES accelerated nerve regeneration. Alternatively, the finding could be explained by the ES accelerating the formation of functional connections between regenerated soleus nerve fibres and the denervated soleus muscle fibres rather than by accelerating nerve regeneration. A similar explanation might suffice for the accelerated appearance of the plantar extensor reflex after sciatic nerve crush injury and applied brief {(15–60 min) 20-Hz ES [29]}. To address whether ES accelerates nerve regeneration and not simply the formation of target contacts, we implanted a stimulator to deliver continuous 20 Hz ES to axotomized femoral motoneurons in adults and retrogradely labeled them to count how many had regenerated their axons (Figure 12A-C; [24]). The 20 Hz-frequency of ES was chosen because it is the mean frequency of action potential generation in motoneurons [395].
Continuous 20 Hz ES delivered for two weeks promoted a striking acceleration of femoral nerve regeneration such that all the axotomized motoneurons regenerated their axons 25 mm from the site of transection and repair at three weeks as compared to 10 weeks after sham ES (the stimulator implanted but not turned on; Figure 12A-G; [24]). Initially, the femoral motoneurons regenerated their axons randomly into their motor and sensory branches. Thereafter, the regeneration was specific: the numbers of femoral motoneurons regenerating their axons into the inappropriate sensory nerve branch remained constant whilst the numbers of the motoneurons regenerating their axons specifically into the appropriate motor nerve branch increased steadily to their maximum, accounting for the steady increase in the total numbers over 10 and 3 weeks for the femoral nerves after sham and ES, respectively (Figure 12D-F). The data for the 2-week femoral nerve ES was not significantly different from the data obtained when the duration of the ES was progressively shortened from two weeks to one hour [24]. Hence, the data shown in Figure 12D-G,J and K are those obtained for all the stimulated femoral nerves irrespective of the ES durations. These data were used for direct comparison with the corresponding data for sham ES in Figure 12D,E.
The efficacy of ES in promoting nerve regeneration was attributed to acceleration of axonal outgrowth and not to an increase in the regeneration rate because (1) ES did not change the rate of slow axonal transport that concurs with regeneration rate, and (2) ES accelerated the regeneration of axons across the suture site, elucidated by injecting fluororuby (another name for FR) into the femoral nerve 1.5 mm distal to the suture line (Figure 12I; [25]). This effect of ES accelerating axon outgrowth only contrasts with that of a conditioning lesion that accelerates both axonal outgrowth and regeneration rate as discussed below in 5f. Conditioning lesion and conditioning ES. The accelerated axonal outgrowth, illustrated in the cartoon in Figure 12L, is mediated by action potentials that are conducted to the neuronal somas [24,27]. This was demonstrated in experiments in which local application of tetrodotoxin proximal to the site of ES that blocked action potential propagation to the soma of the axotomized motoneurons, eliminated the ES effect of accelerating motor and sensory nerve growth into their appropriate nerve branches (Figure 12N,O). Although the sensory neurons did not show a preference for the cutaneous sensory branch of the femoral nerve in the experiments of Geremia et al. [27], significant preference was demonstrated by those of Brushart et al. [26].
We demonstrated that the BNDF that was upregulated with its receptor trkB by ES (Figure 9D,E; [86]) is essential for the efficacy of the ES in accelerating nerve regeneration. A 3-day infusion of a BDNF antibody into the spinal fluid prior to ES and surgical repair of a transected femoral nerve (Figure 13A), followed by retrograde labelling of the branches of the nerve with FR and FG (Figure 13B), completely blocked the ES effect of accelerating specific regeneration of motor axons into the appropriate quadriceps motor branch: the number of motoneurons that regenerated their axons into the motor branch was not elevated three weeks after surgical repair, ES, and administration of the BDNF antibody (Figure 13C). The same efficacy of ES on nerve regeneration was associated with BDNF or NT-4/5 upregulation in thy-1- YFP-H transgenic mice where the regenerating CP axons, a subset of highly visible YFP-positive axons, were visualized in a wild-type mouse nerve graft [31,34,66] that provided little or no upregulated sources of the neurotrophic factors in denervated SCs [244,396] or in denervated muscle [397].
The positive effects of ES on peripheral nerve regeneration in animals reported by Brushart, Gordon, and colleagues in the 2000’s [24,25,26,27,28] have now been replicated many times [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56] and reviewed by Gordon et al. [5,6,17,62,63,64,65,66,67,69,70,248,301,319,320,322] and more recently by Zuo et al [70] and Juckett et al. [398]. The two recent reviews outline the origins of this century’s studies of ES with the 1900 studies of enhanced neurite and nerve outgrowth in response to electrical fields. Now, the same 1-hour ES demonstrated in animal studies to promote nerve regeneration after nerve transection and surgical repair, have been shown to be effective in promoting nerve regeneration and target reinnervation in human subjects [399,400,401].
The 1-hour ES was also shown to accelerate nerve regeneration through a nerve isograft of 10 mm in Lewis rats [52], and through nerve autografts of 10 and 20 mm in Sprague Dawley rats [53]. Macrophages and neutrophils normally accumulate in denervated distal nerve stumps after ES, the ES of the proximal nerve stump increasing the numbers of M2 macrophages within the autografts [52]. Similarly, ES shifted the macrophage phenotype in a locally demyelinated peripheral nerve, from the proinflammatory M1 toward the predominantly pro-repair M2 type [402]. ES also accelerates Wallerian degeneration, promoting nerve degeneration and clearance of the axons and their myelin, and upregulating BDNF and NGF in the denervated SCs [403].
The efficacy in promoting muscle reinnervation and functional recovery, of a 1½ hour period of ES, repeated daily for one to 6-8 weeks after nerve transection and surgical repair [28,54,404,405] or after sciatic nerve isograft repair [55], was in accord with the efficacy of the 2-week period of continuous ES and the 1 hour ES in accelerating motor nerve regeneration, muscle reinnervation and functional recovery [24,28,33]. Transcutaneous ES, however, was less effective in promoting nerve regeneration than direct ES of a transected nerve [406]. A 20-minute period of continuous ES increased numbers of sciatic neurons regenerating axons through a 5-mm long hollow conduit after delayed repair [50] but, the increase was only 20% and 10% of the numbers of stimulated motoneurons and sensory neurons, respectively, that regenerated their axons after delayed cross-suture of CP and TIB nerve stumps [28]. Also, the reported effective 10-minute protocol of 20 Hz ES [406,407,408] was not supported by a study that used several measures of functional recovery to show that this 10-minute protocol did not promote significant recovery after a sciatic nerve isograft repair [409]. Bioluminescent optogenetics were used recently to excite mouse sciatic neurons and to demonstrate acceleration of sciatic nerve regeneration and muscle reinnervation after transection and resuture [410]. The neurons were excited with an intraperitoneal injection of a luciferase substrate, coelenterase (CTZ), two weeks after injection of a viral vector encoding an excitatory luminopsin, lLMO3 into the sciatic nerve [410]. The ILMOS injection to excite CP neurons was also effective in accelerating gastrocnemius muscle reinnervation a 1-month after chronic axotomy of CP motoneurons and cross-suture of the CP proximal nerve stump to the freshly denervated TIB distal nerve stump [410].
It is important to note that daily administration of the androgen, testosterone, alone or together with daily ES after crush injury in castrated male rats, promotes facial nerve regeneration and functional recovery of both whisking and blink reflexes [39,42]. The beneficial effect of the testosterone-ES combination elicited a more rapid and sustained upregulation of BDNF than either alone [42]. The effects of both ES and exercise (see 5f. Exercise and axonal regeneration) increasing the length of regenerating axons, were blocked by treating both male and female mice with the androgen receptor blocker flutamide [411]. While androgen receptors are found in all motoneurons [412] and on astrocytes and microglia [413], it is not clear whether the release of BDNF from reactive glial cells after nerve injury, including spinal cord injury, assists or hinders nerve regeneration after injury [414].
5d. Preferential Reinnervation and Growth Factors
The preferential upregulation of the growth factors, PTN (pleiotrophin) and GDNF in denervated SCs (Figure 15C; [245,247]) accounts for the preferential growth of regenerating femoral motor nerve fibres into their appropriate motor nerve branch (Figure 14B,D; [415]). The expression of both growth factors is on the rise by 4 days after denervation at the time when few motoneurons that regenerated their axons across the surgical site after repair, and had randomly reinnervated both appropriate and inappropriate branches (Figure 11F and Figure 14A-C). The upregulation of both growth factors is maximal in the motor SCs at 14 days (Figure 14C) when ~70% of the femoral motoneurons had regenerated their axons across the suture line (Figure 14A,D) and, by 4 weeks, the axons grow preferentially into the endoneurial tubes that previously surrounded only motor nerve fibres (Figure 14B,D). When the remaining motoneurons regenerate their axons across the repair site by 28 days, the axons continue to enter the endoneurial ‘motor’ tubes preferentially (see the purple interrupted line in Figure 14D) because their specific growth factors likely remain in the SCs lining these tubes even though their expression is fading (Figure 14C). A similar explanation of the preferential reinnervation of sensory pathways by sensory neurons has been elaborated (data not shown but see [415]). Studies of the growth of motor axons into femoral nerve branches in NCAM (neural cell adhesion molecule) knockout mice (NCAM-/-) demonstrated that N-CAM was required for preferential reinnervation of the motor branch [416]. Moreover, the preferential reinnervation was not observed when the polysialic acid (PSA) moiety was removed enzymatically. This indicated that regenerating motor axons must express polysialylated NCAM that reduces axon-axon adhesion and that enables the axons to grow selectively into the appropriate motor pathway [416].
5e. ES Promotes Axon Outgrowth after Delayed Surgery
The regression of the regenerative capacity of chronically axotomized neurons and the regenerative support of chronically denervated SCs with the concomitant regression of expression of GAGs in both the neurons and SCs with time, described above in 3b. Transient expression of regeneration associated genes, begged the question of whether ES of the proximal stump of a chronically axotomized transected nerve could counteract these negative effects of delayed surgical repair of injured nerves. We addressed this question in experiments in which we systematically evaluated the effects of chronic motor and sensory neuronal axotomy and/or the chronic denervation of the distal nerve stump on the ability of ES to accelerate nerve regeneration after nerve transection and surgical repair (Figure 15, [28]). The experimental paradigm of chronic axotomy and/or chronic denervation of hindlimb nerve in rats (Figure 15A-D) was designed to address this question and the data collected demonstrated that, indeed, ES was able to counteract the effects of delayed surgery [28]. After ligating either the CP proximal nerve stump and/or the distal TIB stump for two months prior to cross-suture repair, the numbers of motor and sensory neurons that regenerated their axons was elevated to the same levels as after immediate cross-suture (Figure 15B,C; [28]). The ES increased both the number of regenerated nerves and those that reinnervated muscle (Figure 15B,C, the lowest two histograms), demonstrating that ES of the chronically injured nerves accelerated both nerve regeneration and target reinnervation. It is important to note that the surgical release of the CP proximal stump for the cross-suture repair constituted a second injury. The resulting RAG upregulation [337,338] likely promotes nerve regeneration after chronic injuries despite the more rapid decline in expression after the second injury as compared to the first. This is illustrated in Figure 5D where the decay in the expression of α-tubulin mRNA in axotomized sciatic motoneurons from 7d to 6 months was shown to be reversed by a second injury. The effectiveness of ES in promoting nerve regeneration after chronic denervation of the distal nerve stump (Figure 15B,C) is likely consequent to the release of neuregulin from the stimulated proximal nerve stump.
5f. Exercise and Axonal Regeneration
The accelerated rise in BDNF and trkB expression in axotomized motoneurons in response to brief ES (Figure 9D,E; [86]) or exercise [417,418,419] as well as the elevated NT/5 expression by ES [34], was the rationale for investigating whether exercise might also promote nerve regeneration as did ES [420,421,422,423]. In 2009, it was reported that daily exercise, like ES, accelerates muscle reinnervation with the combination of exercise and ES being the most effective during the early phase of nerve regeneration [421]. Extending these studies, English and colleagues reported that a 2-week period of moderate daily exercise 3 days after surgical repair promoted axon outgrowth that was significantly greater than that after brief, low frequency ES [420,423,424]. This efficacy of exercise confirmed previous findings of enhanced axon regeneration of both motor and sensory neurons using several different exercise protocols [30,425,426,427,428,429,430,431,432].
The exercise effect and that of ES also involves androgens in addition to BDNF. That androgen receptor signalling regulates expression of BDNF mRNAs [433] spurred the laboratories of Jones and of English to investigate the effects of daily administration of testosterone on nerve regeneration and the combination of testosterone and daily ES and to compare the effects of either testosterone or ES and both testosterone and ES together [39,42,411]. Exogenous testosterone or ES was effective in promoting the regeneration of facial nerve and the functional recovery of the whiskers [39,42]. It was the combination of both testosterone and ES that had the most prolonged and the maximum effect [42]. English and colleagues demonstrated that the androgen blocker, flutamide, eliminated both the ES and the exercise effects on regeneration after nerve transection and surgical repair [411]. The findings are consistent with the regulation of the expression of BDNF mRNAs by androgens [423].
The effect of exercise on the regeneration of the femoral nerve after surgical reunion of the transected nerve stumps has not evaluated to this author’s knowledge. As described above, preferential reinnervation of the quadriceps motor branch by the regenerating motor nerves does occur with time after the surgery and this reinnervation is accelerated by low frequency ES [24,379,434]. However, after transection and microsurgical repair of the sciatic nerve, the reinnervation of the distal stump is random [435] and ES exacerbated misdirection of regenerating nerve fibres with reduced TIB nerve contribution to the innervation of the triceps surae muscles [435]. Also, the more rostral and normal position of CP motoneurons was shifted to a more caudal position after sciatic nerve transection and repair, a shift that was exacerbated when the transected sciatic nerve was subjected to ES [436], but not when the rat was subjected to daily exercise on a treadmill [437]. However, irrespective of whether the sciatic nerve was subjected to ES, or the rats were exercised daily, or whether the misdirection of the regenerating axons was reduced by transecting and surgically repairing the CP and TIB nerve branches of the sciatic nerve, the normal reciprocal activation of flexor and extensor muscles was irretrievably altered by coactivation of the antagonistic muscles [438,439,440]. This misdirection and abnormal activation of antagonistic muscles remains a problem for functional recovery after nerve repairs that are performed on large nerves, despite the use of physiotherapy to improve movement. One study of the misdirection of regenerating axons after sciatic nerve injury in rats, reported that 71% of regenerating CP motoneurons were directed correctly into two muscles after crush injury, 42% after autograft repair, and 25% after autograft repair but recovery of ankle motion and balance was incomplete in all cases [441].
5g. Conditioning Lesion and Conditioning ES
There is now the relatively new approach of a CES (conditioning ES) paradigm to promote nerve regeneration and elevated expression of RAGs, even after chronic nerve injuries [442,443,444,445,446,447,448]. The CES is the 1-hour 20 Hz ES of an intact peripheral nerve prior to nerve transection and surgical repair, an approach that is promising and is being advocated for clinical use. The conditioning lesion (CL), a crush of an intact nerve 1-7 days prior to a more proximal nerve crush or transection injury, increases both the outgrowth of regenerating axons and their regeneration rate [442,449,450,451,452,453,454,455,456] in association with elevated levels of cAMP [457]. The rate of the slow component of axonal transport that largely governs regeneration rate, is accelerated by the CL with increased transport and amount of SCb proteins that include tubulin and actin [453,454,455,456]. Based on our finding that ES of the intact sciatic nerve performed 7 days prior to excision of DRGs, enhanced neurite outgrowth from the sensory neurons, akin to that seen after a CL (Figure 16; [458]), the effects of a CL and CES on how many and how far CP nerves regenerated their axons into a denervated nerve stump were compared in vivo [442,443,444,446]. These comparisons revealed the superiority the CES (referred to in their publications as functional ES) in (1) increasing the numbers of neurofilament-positive regenerating axons and how far these axons grew into the distal stump in the studies of crushed nerves [442,444] and transected and coapted nerves [444,446] after immediate [443], and delayed nerve repairs [448], (2) superior return of muscle and intraepidermal skin reinnervation [444,445,446], and (3) superior recovery of motor and sensory function [444,445,446].
Senger et al. pointed out that CES could be applied in chronic nerve injuries which are common after major polytrauma that necessitate emergency management of life or limb threatening injuries. Indeed, with the exception of immediate primary repair of sharp injuries from knives or razors, most surgeries are delayed for three to six months [287,289]. Presumably, the application of CES relies on placing the stimulating electrodes on the nerve proximal to the injury and days prior to the surgical repair, a requirement of two consecutive surgeries. In addition, CES could be applied, if open surgery is not required to place stimulating electrodes before distal nerve transfer surgery, the transection of a ‘donor’ nerve branch of a redundant muscle and its suture to the distal end of a non-functional ‘recipient’ nerve to restore function [398,447]. The Oberlin’s transfer, as first described in 2004 [459], is the classical example. This transfer to restore elbow flexion is one of several nerve transfers [459,460,461,462,463,464,465]. It is the suture a transected ulnar nerve fascicle that had supplied the flexor carpi ulnaris muscle, to the distal stump of the musculocutaneous nerve branch to biceps brachii muscle.
Whether CES could be applied successfully in end-to-side neurorrhaphies remains to be determined. The end-to-side neurorrhaphy is where a denervated nerve stump is inserted into an intact nerve to ‘borrow’ innervation for a denervated muscle [326,462,463,464,465,466,467,468,469,470,471,472]. It is performed to, ‘protect’ denervated distal stumps and in turn, increase the numbers of motoneurons that regenerate their axons through them [471,472]. Serial in vivo imaging of the regeneration of facial nerve axons identified with GFP (green fluorescent protein) in Thy1-GFP rats [473], demonstrated enhanced nerve regeneration through a denervated cross-face nerve autograft that was ‘protected’ by adding end-to-side sensory nerves [469]. Would CES of the intact nerve accelerate the reinnervation of chronically denervated distal nerve stumps by the end-to-side neurorrhaphy and improve muscle reinnervation? The surgical approach of side-to side neurorrhaphy [474,475,476,477,478] with one or 3 nerve autografts placed as cross-bridges between an intact nerve and a chronically denervated distal nerve stump, significantly increased the numbers of axotomized motor and sensory neurons that regenerated their axons after delayed nerve repair as compared with the neurons regenerated through chronically denervated nerve stumps that were not ‘protected’ by cross-bridges from an adjacent intact nerve [477]. The question remains as to whether ES might accelerate axon outgrowth after the side-to side neurorrhaphy and in turn, promote target reinnervation, especially for the nerve injuries that are distant from their denervated target organs.
5g. Drug-Induced cAMP Elevation Mimics the Effect of Electrical Stimulation (ES) on Nerve Regeneration
ES at 20 Hz for 1-hour raises intracellular cAMP in DRG neurons to the same levels as a CL does (Figure 18B; [458]). If cAMP plays a role in mediating the effect of ES of accelerating axon outgrowth across a peripheral nerve transection and repair site, we reasoned that elevating cAMP by inhibiting PDE4 (phosphodiesterase-4), the main enzyme responsible for cAMP hydrolysis, would mimic ES in promoting axon outgrowth across the injury site (Figure 17B;[28]). Additional evidence that PDE4 in combination with dbcAMP administration increased regeneration of spinal cord axons through a SC graft placed within the cord [479], prompted our study. We administered rolipram, a specific PDE4 inhibitor [480], via a mini-osmotic pump to the site of CP transection and microsurgical repair in rats (Figure 17B). The CP motoneurons that regenerated their axons across and 10 mm distal to the surgical repair site, were counted after retrograde dye labeling (Figure 17C-E; [28]). In addition, the number of motoneurons that reinnervated the target TA muscle was calculated by counting the number of reinnervated MUs (MUNE, Figure 17E;[28]). The significant increase in all three of the outcome measures demonstrated that, indeed, the cAMP elevation mediated by PDE4 inhibition, was sufficient to mimic the positive effects of ES of accelerating nerve regeneration across the surgical site, through the denervated distal nerve stump, and reinnervating denervated muscle fibres. Simply elevating cAMP with forskolin that stimulates adenylate cyclase, was found previously to increase the rate of regeneration of transected sciatic nerve fibres [481].
5h. ES Promotes Sensory Nerve Outgrowth in the Central Nervous System
Filbin and colleagues had noted high levels of cAMP in neonatal CNS neurons and that these high levels were associated with the capacity of these neonatal CNS neurons in contrast to adult CNS neurons, to regenerate their axons after injury ([482; reviewed by [483]). Qui et al. [484] and Neumann et al [457] discovered that it was the elevated cAMP that accounted for the effectiveness of the CL of the sciatic nerve to promote regeneration of transected sensory nerve fibres in the CNS. Indeed, elevated cAMP mimicked the effect of a CL both in vivo and in vitro. Later it was shown that the transcription factor, CREB acts as the primary mediator of cAMP-induced transcription [120] via PKA and MEK/Erk pathways [122]. Neurotrophins also play a role, BDNF-mediated activation of Erk producing a transient inhibition of PDE4 (phosphodiesterase-4), the main enzyme responsible for cAMP hydrolysis. Thereby, the neurotrophins promote the elevated intracellular cAMP [485]. That a CL of peripheral sensory nerve fibres promotes regeneration of their axons in the CNS [457,486] prompted us to ask whether ES of the intact sciatic nerve containing these nerves might also be effective in promoting their regeneration in the CNS.
Because sensory neurons may fire at high frequencies, albeit transiently [395], we asked whether either or both 20 and 200 Hz ES promote outgrowth of the central axons of DRG neurons [458]. The central axons were transected at thoracic level 8 (T8) in the spinal cord (Figure 18A,B; [457]) and the sciatic nerve was subjected to a CL, ES at 20 Hz, or ES at 200 Hz, the 200 Hz frequency studied as a control because only 20 Hz ES raised cAMP levels to those after a CL (Figure 18B; [457]). The DRG sensory nerve fibres were labelled with cholera toxin B (CTB) 14 weeks after the spinal cord lesion to visualize the outgrowth of axons across the lesion site in vivo (Figure 18B; [457]). As shown previously [457,486], the CL was very effective in promoting the outgrowth of sensory axons across the lesion site (Figure 18D). The sprouted axons regenerated further into the spinal cord and more rapidly than the control axons at the T8 lesion site that received sham ES (cf Figure 18D and C). This finding is consistent with the efficacy of the CL accelerating axon outgrowth and elongation in the PNS [442,449,450,451,452,453,454,455,456]. The 20 Hz ES but not the 200 Hz ES of the sciatic nerve, promoted significant axon outgrowth across the T8 lesion site, the axons not elongating as much as the outgrowth after a CL (cf Figure 18E,D). This was consistent again with ES promoting outgrowth without increasing the overall rate of regeneration after peripheral nerve injury [24,25]. In the comparisons of mean values of the percent of the regenerated DRG central axons as a function of the distance from the T8 lesion site, the 20 Hz ES promoted axon outgrowth but not axon lengthening in the CNS in contrast to the CL that promotes both outgrowth and lengthening of the transected axons (Figure 18G-I).
The effect of the CL promoting central sensory axonal regeneration was related directly to the levels of intracellular levels of cAMP in the DRG neurons [457,486]. However, cAMP injection into the DRG in vivo or in vitro did not match the CL effect, the effect being smaller [457]. That the elevated cAMP, either by injection [487] or in response to ES [458], was insufficient to promote the lengthening of the axon sprouts across the lesion site, is consistent with findings that elevated cAMP does not promote elongation of transected DRG central axons into a peripheral nerve transplant [488]. Moreover, a CL and ES elevates cAMP to the same level (Figure 18B) but the CL promotes axonal outgrowth and lengthening [143,452,453,454]. This discrepancy made it likely that the CL effect may be mediated at least in part, by other mechanisms that include the contribution of non-neural cells. A possible molecular candidate is oncomodulin, the small calcium binding protein whose mRNA is highly expressed in neutrophils, the first responders of the innate immune system [489,490]. The neutrophils are recruited into the injury site of the eye within a day [491]. The oncomodulin binds to retinal ganglion cells with high affinity in a cAMP-dependent manner, increases the level of phosphorylated CREB (P-CREB), the activated form of the transcriptional activator, and stimulates the cells to regenerate long axons beyond the site of optic nerve injury [492]. The outgrowth was more extensive than other known trophic agents. Moreover, oncomodulin also acts upon peripheral sensory neurons to promote neurite outgrowth although neutrophils do not participate in the response of the neurons to injury [492].
5i. ES Signalling Pathways
Our findings to date of the effectiveness of ES in accelerating axon outgrowth and target reinnervation are summarized in a schematic representation of the intracellular signaling pathways activated by ES in Figure ES elevates intracellular cAMP that, via PKA, initiates transcription by CREB that, in turn, elevates expression of growth associated proteins, including neurotrophins and their trk receptors, and the cytoskeletal proteins, tubulin and actin. Thereby, ES promotes axon outgrowth after nerve injury to accelerate the regenerating nerves towards their denervated targets. The neurotrophins also feed back to amplify the cAMP pathway, in part by a transient inhibition of PDE4 activity that elevates cAMP to threshold levels and in turn, promotes axon outgrowth after immediate or delayed nerve repair of injured peripheral nerves. The positive effect of ES in promoting axonal regeneration after delayed nerve surgery, involving both chronic axotomy of motor and sensory neurons and the chronic denervation of the distal nerve stump is likely due to the release of mitogens such as neuregulin from the stimulated proximal nerve stump. These mitogens promote SC proliferation and migration, shifting the SCs from their atrophic state to a growth permissive state. The latter state includes the expression of neurotrophic factors and p75NTR that may present the factors to the regenerating axons to further enhance their growth. Further details of the pathways involved in the efficacy of ES in promoting nerve regeneration and target reinnervation await further experimentation.
5j. ES Accelerates Human Nerve Regeneration and Muscle Reinnervation
Having demonstrated the effect of ES accelerating axon outgrowth, and reinnervation of targets in rats, we asked whether ES promotes functional recovery after nerve injuries in patients. Could we accelerate target reinnervation? In patients with carpal tunnel syndrome (CTS), the number of functionally intact MUs, MUNE (MU number estimation), was determined after ascertaining that the syndrome was not the result of a conduction block at the wrist but rather the result of the loss of functional MUs [399]. We established that the CTS was due to MU loss by stimulating the medial nerve above and below the carpal tunnel (Figure 20A-C) and ascertaining that the size of the CMAP (compound motor action potential) in response to maximal stimulation of the median nerve above the tunnel equaled the CMAP recorded in response to stimulation of the nerve below the carpal tunnel (Figure 20B,C). We recruited only those patients with severe chronic CTS for the study of the effects of ES on the reinnervation of the partially denervated median eminence musculature after the CTRS (carpal tunnel release surgery) of cutting the ligament overlying the carpal tunnel (Figure 20D). Prior to the surgery, the number of intact MUs was estimated by electrically stimulating the median nerve maximally to evoke the CMAP in the muscle and then evoking at least 20 all-or-none MUAPs (MU action potentials) in response to electrical stimuli to the median nerve as the stimulator was moved progressively along the course of the nerve from the wrist up to the upper arm (Figure 20E). Thereafter, the patients were divided into two groups, either receiving or not receiving 20 Hz ES for one hour immediately after CTRS surgery (Figure 20F). MUNE was determined in a similar fashion to that in the rats (Figure 1C) namely from the ratio of CMAP and mean MUAP amplitudes (Figure 20G).
The number of intact MUs averaged ~150 which was 50% of the ~300 intact MUs in individuals who did not suffer CTS (Figure 20H). The effect of the ES was dramatic especially when contrasted with the very slow progression of reinnervation of the partially denervated median eminence musculature by the median nerve in the control patients who did not receive ES. Although there was a trend for the number of reinnervated MUs to increase over a period of 12 months in the unstimulated control group of patients, the increase was not significant ((p>0.05). In contrast there was already a significant increase at 3 months (p<0.05) with the number reaching their normal values by 6-8 months (Figure 20H). These findings were particularly striking when the distance of regeneration from the site of crush injury to the musculature of 100 mm (see Figure 1A) is considered. With the reported regeneration rate of 1 mm/day [290,291], the expected time of reinnervation of the median eminence musculature would be ~ 3-4 months and yet there was no significant increase in MUNE in the patients whose median nerve was not subjected to ES. Thus, the accelerated reinnervation of the musculature was particularly striking with all the crushed medial motor nerves reinnervating the muscles within 6-8 months. The effect of the ES on axon outgrowth after CTRS is shown figuratively as the regenerating axons staggering across the crush site 3 months after CTRS and, at 6 months, failing to reinnervate the muscles of the median eminence without ES application (Figure 20I,K), and the staggering axons progressing further after ES and reinnervating all the denervated muscle fibres (Figure 20L,M).
Chan and his colleagues went on to demonstrate the significant acceleration of sensory nerve regeneration in human subjects after surgical repair of transected digital nerves in the hand [400] as well as after ulnar nerve compression at the elbow [401]. In summary, the demonstrated efficacy of low frequency ES to accelerate nerve regeneration in human subjects as well as in animals, indicates the potential for its utility in promoting successful outcomes after injuries in patients.
6. Conclusions
Neural activity in motor and sensory neurons declines rapidly during the first month of nerve injury. The decline in motoneurons is associated with and likely caused by the loss of both excitatory and inhibitory synapses on the motoneurons in the spinal cord. The decline in activity of sensory neurons to unmeasurable levels is due to the disconnection of the neurons from their sensory input from the skin and muscles. The progressive decline in regenerative capacity of axotomized neurons and the regenerative support of the denervated distal nerve stumps accounts for progressive decline in functional recovery with time and distance. The loss occurs concurrent with the progressive fall in the growth associated genes (GAGs) that are upregulated in both the axotomized neurons and the SCs in the denervated distal nerve stumps within the first week after nerve injury. Brief (1 hour) low frequency (20 Hz) electrical stimulation (ES), by promoting axon outgrowth, accelerates nerve regeneration and target reinnervation after both acute and chronic nerve injuries in animals and in humans. Recent studies demonstrate that the same brief 20 Hz ES applied prior to a nerve injury acts as a conditioning ES to promote both axon outgrowth as well as the rate of nerve regeneration. The effectiveness of the ES after acute injury is accounted for by (1) preventing the loss of synaptic contacts that occurs on axotomized motoneurons, (2) the upregulation of GAGs in response to increased intracellular cAMP, and (3) the feedback of expressed neurotrophins including BDNF, that amplify the expression of cAMP. After chronic nerve injuries where synaptic contacts on the motoneurons have already been withdrawn, the increase in cAMP and the feedback loops of the neurotrophic factors transiently inhibit cAMP breakdown. The elevated cAMP, via CREB and expression of GAGs, allows increased axonal outgrowth with resultant promotion of axonal regeneration and target reinnervation. The contribution of the release of mitogens from regenerating motor and sensory axons, including neuregulin, increases the proliferation of SCs and promotes their growth permissive state. This state includes the synthesis and release of neurotrophic factors that likely amplify the effects of the neuronal expression of the factors to, in turn, promote the synthesis of growth associated proteins, including the cytoskeletal proteins that are essential for axon outgrowth and nerve regeneration.
That brief low frequency ES is effective in accelerating axon outgrowth from the proximal stump of an injured nerve in animals and human patients indicates its potential for clinical application. The conditioning ES may be applicable to enhance the regenerative capacity in nerve transfer surgeries and end-to-side neurorrhaphies, should open surgery be unnecessary in applying the electrodes for the ES. Finally, our findings that delayed nerve repair is deleterious with respect to the prospects of functional recovery, emphasize the importance of ensuring minimal delay between the diagnosis of nerve injury and nerve repair and the application of brief ES. Thereby, we might anticipate improved outcomes of nerve surgeries in the future.
Acknowledgments
My sincere thanks to my many students and postdoctoral fellows, my collaborators, and my technologist, who worked with me in our research toward the ultimate goal of promoting nerve regeneration and functional recovery after peripheral nerve injuries in patients. Thanks too to the Canadian granting agents, the Canadian Institutes for Heath Research, the Muscular Dystrophy Association of Canada, and the Amyotrophic Lateral Sclerosis Society of Canada for their continued support of my research.
References
- Cajal, S.R. Degeneration and Regeneration of the Nervous System (translated by R.M. May). 1928, Oxford University Press, New York, Oxford.
- Sunderland, S. Nerve and Nerve Injuries. 1978, Livingstone, Edinburgh.
- Kline, D.G.; Hudson, A.R. Nerve Injuries: Operative Results for Major Nerve Injuries, Entrapments and Tumors. 1995, W.B. Saunders, Philadelphia, PA.
- Fenrich, K.; Gordon, T. Canadian Association of Neuroscience review: axonal regeneration in the peripheral and central nervous systems – current issues and advances. Can. J. Neurol. Sci. 2004, 31, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Nerve Repair. 2011, Oxford University Press, Oxford.
- Sulaiman, O.A.R.; Midha, R.; Gordon, T. Pathophysiology of surgical nerve disorders. In Youmans’ Neurological Surgery 6th ed., Part 2, 2011, 2368-Ed Winn HR. Philadelphia, PA.
- Ruijs, A.C.J.; Jaquet, J.-B.; Kalmign, S.; Giele, H.; Hovius, S.E.R. Median and ulnar nerve injuries: a meta-analysis of predictors of motor and sensory recovery after modern microsurgical nerve repair. Plast. Reconstr. Surg. 2005, 116, 484–94. [Google Scholar] [CrossRef] [PubMed]
- Scholz, T.; Krichevsky, A.; Sumarito, A.; Jaffurs, D.; Wirth, G.A.; Paydar, K.; Evans, G.R.D. Peripheral nerve injuries: An international survey of current treatments and future perspectives. J. Reconstr. Microsurg 2009, 25, 339–344. [Google Scholar] [CrossRef]
- Park, H.R.; Lee, G.S.; Kim, I.S.; Chang, J.-C. Brachial plexus injury in adults. The Nerve 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Smania, N.; Berto, G.; La Marchina, E.; Melotti, C.; Midiri, A.; Roncori, L.; Zenorini, A.; Ianes, P.; Picelli, A.; Waldner, A.; Faccioli, S.; Gandolfi, M. Rehabilitation of brachial plexus injuries in adults and children. Eur. J. Phys. Rehabil. Med. 2012, 48, 483–506. [Google Scholar]
- Tian, A.; Sachar, R.; Ray, W.Z.; Brogan, D.M.; Dy, C.J. Indirect cost of traumatic brachial plexus injuries in the United States. J. Bone Joint Surg. Am. 2019, 101, e80. [Google Scholar]
- Rosberg, H.E.; Carlsson, K.S.; Hojgard, S.; Lindgren, B.; Lundborg, G.; Dahlin, L.B. Injury to the human median and ulnar nerves in the forearm- analysis of costs for treatment and rehabilitation of 69 patients in southern Sweden. J Hand Surg (Edinburgh, Scotland) 2005, 30, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Bergmeister, K.D.; Groβe-Hartlage, L.; Daeschler, S.C.; et al. Acute and long-term costs of 268 peripheral nerve injuries in the upper extremity. Plos One 2020, 15, e0229530. [Google Scholar] [CrossRef]
- Jaquet, J.B.; Luijsterburg, A.J.; Kalmijn, S.; Kuypers, P.D.; Hofman, A.; Hovius, S.E. Median, ulnar, and combined medium-ulnar nerve injuries: Functional outcome and return to productivity. J. Trauma. 2001, 51, 687–692. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. The cellular and molecular basis of peripheral nerve regeneration. Mol. Neurobiol. 1997, 14, 67–116. [Google Scholar] [CrossRef]
- Boyd, J.G.; Gordon, T. Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury Mol. Neurobiol. 2003, 27, 277–324. [Google Scholar]
- Chan, K.M.; Gordon, T.; Zochodne, D.W.; Power, H.A. Improving peripheral nerve regeneration: From molecular mechanisms to potential therapeutic targets. Exp. Neurol. 2014, 261, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.J.; Coleman, M.P. Lessons from injury: How nerve injury studies reveal basic biological mechanisms and therapeutic opportunities for peripheral nerve diseases. Neurotherapeutics 2021, 2200–2221. [Google Scholar] [CrossRef] [PubMed]
- Daeschler, S.C.; Feinberg, K.; Harhaus, L.; Kneser, U.; Gordon, T.; Borschel, G.H. Advancing nerve regeneration: Translational perspectives of tacrolimus (FK506). Int. J. Mol. Sci. 2023, 24, 12771. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, E.L; Houghton, P.; Anthony, J.; Rennie, S.; Shay, B.L; Hoens, A.M. Neuromuscular electrical stimulation of muscle impairment: Critical review and recommendations for clinical practice. Physiother. Can. 2017, 69, 1–78. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Hsiao, H.-F.; Li, L.-F.; Chen, N.-H.; Huang, C.-C. Effects of electrical muscle stimulation in subjects undergoing prolonged mechanical ventilation. Respir. CRE 2019, 64, 262–271. [Google Scholar] [CrossRef]
- Huang, Y.; Gong, Y.; Liu, Y.; Lu, J. Global trends and hot topics in electrical stimulation of skeletal muscle research over the past decade: A bibliometric analysis. Frontiers Neurol. 2022, 13, 991099. [Google Scholar] [CrossRef]
- Nix, W.A.; Hopf, H.C. Electrical stimulation of regenerating nerve and its effect on motor recovery. Brain Res. 1983, 272, 21–25. [Google Scholar] [CrossRef]
- Al-Majed, A.A.; Neumann, C.M.; Brushart, T.M.; Gordon, T. Brief electrical stimulation promotes the speed and accuracy of motor axonal regeneration. J. Neurosci. 2000, 20, 2602–2608. [Google Scholar] [CrossRef]
- Brushart, T.M.; Hoffman, P.N.; Royall, R.M.; Murinson, B.B.; Witzel, C.; Gordon, T. Electrical stimulation promotes motoneuron regeneration without increasing its speed or conditioning the neuron. J. Neurosci. 2002, 22, 6631–6638. [Google Scholar] [CrossRef]
- Brushart, T.M.; Jari, R.; Verge, V.; Rohde, C.; Gordon, T. Electrical stimulation restores the specificity of sensory axon regeneration. Exp. Neurol. 2005, 194, 221–229. [Google Scholar] [CrossRef]
- Geremia, N.M.; Gordon, T.; Brushart, T.M.; Al-Majed, A.A.; Verge, V.M. Electrical stimulation promotes sensory neuron regeneration and growth-associated gene expression. Exp. Neurol. 2007, 205, 347–359. [Google Scholar] [CrossRef]
- Elzinga, K.; Tyreman, N.; Ladak, A.; Savaryn, B.; Olson, J.; Gordon, T. Brief electrical stimulation improves nerve regeneration after delayed repair in Sprague Dawley rats. Exp. Neurol. 2015, 269, 142–153. [Google Scholar] [CrossRef]
- Pockett, S.; Gavin, R.M. Acceleration of peripheral nerve regeneration after crush injury in rat. Neurosci. Lett. 1985, 59, 221–224. [Google Scholar] [CrossRef]
- Marqueste, T.; Alliez, J.R.; Alluin, O.; Jammes, Y.; Decherchi, P. Neuromuscular rehabilitation by treadmill running or electrical stimulation after peripheral nerve injury and repair. J. Appl. Physiol. 2004, 96, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- English, A.W. Enhancing axon regeneration in peripheral nerves also increases functionally inappropriate reinnervation of targets. J. Comp. Neurol. 2005, 490, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rouabhia, M.; Wang, Z.; Roberge, C.; Shi, G.; Roche, P.; Li, J.; Dao, L.H. Electrically conductive biodegradable polymer composite for nerve regeneration: electricity-stimulated neurite outgrowth and axon regeneration. Artificial Organs 2006, 31, 13–22. [Google Scholar] [CrossRef]
- Ahlborn, P.; Schachner, M.; Irintchev, A. One hour electrical stimulation accelerates functional recovery after femoral nerve repair. Exp. Neurol. 2007, 208, 137–144. [Google Scholar] [CrossRef] [PubMed]
- English, A.W.; Schwartz, G.; Meador, W.; Sabatier, M.J.; Mulligan, A. Electrical stimulation promotes peripheral axon regeneration by enhanced neuronal neurotrophin signaling. Dev. Neurobiol. 2007, 67, 18–172. [Google Scholar] [CrossRef] [PubMed]
- Hetzler, L.E.; Sharma, N.; Tanzer, L.; Wurter, R.D.; Leonetti, J.; Marco, S.J.; Jones, K.J.; Foecking, E.M. Accelerating functional recovery after rat facial nerve injury: effects of gonadal steroids and electrical stimulation. Otolaryng Head. Neck 2008, 139, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Hetzler, L.T.; Sharma, N.; Wurster, R.D.; Marzo, S.J. Jones, K.J.; Foecking, E.M. Electrical stimulation facilitates rat facial nerve recovery from a crush injury. Otolaryng Head. Neck 2008, 139, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Vivó, M.; Puigdemasa, A.; Casals, L.; Asensio, E.; Udina, E.; Navarro, X. Immediate electrical stimulation enhances regeneration and reinnervation and modulates spinal plastic changes after sciatic nerve injury and repair. Exp. Neurol. 2008, 211, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Pinilla, E.; Udina, E.; Jaramillo, J.; Navarro, X. Electrical stimulation combined with exercise increase axonal regeneration after peripheral nerve injury. Exp. Neurol. 2009, 219, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Coughlin, L.; Porter, R.G.; Tanzer, L.; Wurster, R.D.; Marzo, S.J.; Jones, K.J.; Foecking, E.M. Effects of electrical stimulation and gonadal steroids on rat facial nerve regenerative properties. Restor. Neurol. Neurosci. 2009, 27, 633–644. [Google Scholar] [CrossRef]
- Alrashdan, M.S.; Park, J.C.; Sung, M.A.; Yoo, S.B.; Jahng, J.W.; Lee, T.H.; Kim, S.J; Lee, J.H. Thirty minutes of low intensity electrical stimulation promotes nerve regeneration after sciatic nerve crush injury in a rat model. Acta Neurol. Belg. 2010, 110, 168–179. [Google Scholar]
- Huang, J.; Lu, L.; Hu, X.; Ye, Z.; Peng, Y.; Yan, X.; Geng, D.; Luo, Z. Electrical stimulation accelerates motor functional recovery in the rat model of 15-mm sciatic nerve gap bridged by scaffolds with longitudinally oriented microchannels. Neurorehabil Neural Repair. 2010, 24, 736–745. [Google Scholar] [CrossRef]
- Sharma, N.; Marzo, S.J.; Jones, K.J.; Foecking, E.M. Electrical stimulation and testosterone differentially enhance expression of regeneration-associated genes. Exp. Neurol. 2010, 223, 183–191. [Google Scholar] [CrossRef]
- Wan, L.; Xia, R.; Ding, W. Short-term low-frequency electrical stimulation enhanced remyelination of injured peripheral nerves by inducing the promyelination effect of brain-derived neuro- trophic factor on Schwann cell polarization. J. Neurosci. Res. 2010, 88, 2578–2587. [Google Scholar] [CrossRef]
- Yeh, C.C.; Lin, Y.C.; Tsai, F.J.; Huang, C.Y.; Yao, C.H.; Chen, Y.S. Timing of applying electrical stimulation is an important factor deciding the success rate and maturity of regenerating rat sciatic nerves. Neurorehabil Neural Repair. 2010, 24, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Alrashdan, M.S.; Sung, M.A.; Kwon, Y.K.; Chung, H.J.; Kim, S.J.; Lee, J.H. Effects of combining electrical stimulation with BDNF gene transfer on the regeneration of crushed rat sciatic nerve. Acta Neurol (Wien) 2011, 153, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Han, S.J.; Shin, D.H.; Lee, W.S.; Choi, J.Y. Subthreshold continuous electrical stimulation facilitates functional recovery of facial nerve after crush injury in rabbit. Muscle Nerve 2011, 43, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Foecking, E.M.; Fargo, K.N.; Coughlin, L.M.; Kim, J.T.; Marzo, S.J.; Jones, K.J. Single session of brief electrical stimulation immediately following crush injury enhances functional recovery of rat facial nerve. J. Rehabil. Res. Dev. 2012, 49, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Xu, Q. G.; Franz, C. K.; Zhang, R.; Dalton, C.; Gordon, T.; Verge, V.M.K.; Midha, R.; Zochodne, D.W. Accelerated axon outgrowth, guidance, and target reinnervation across nerve transection gaps following a brief electrical stimulation paradigm. J. Neurosurg. 2012, 116, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Chang, R.L.; Chang, S.L.; Tsai, C.C.; Tsai, F.J.; Chen, Y.S. Electrical stimulation improves peripheral nerve regeneration in streptozotocin-induced diabetic rats. J. Trauma. Acute Care Surg. 2012, 72, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, Y.; Lu, L.; Hu, X.; Luo, Z. Electrical stimulation accelerates nerve regeneration and functional recovery in delayed peripheral nerve injury in rats Europ J. Neurosci. 2013, 38, 3691–3701. [Google Scholar]
- Koppes, A.N.; Nordberg, A.L.; Paolillo, G.M.; Goodsell, N.M.; Darwish, H.A.; Zhang, L.; Thompson, D.M. Electrical stimulation of Schwann cells promotes sustained increases in neurite outgrowth. Tiss. Eng. Pt. A 2014, 20, 494–506. [Google Scholar] [CrossRef]
- Keane, G.C.; Pan, D.; Roh, J.; Larson, E.L.; Schellhardt, L.; Hunter, D.A.; Snyder-Warwick, A.K.; Moore, A.M.; Mackinnon, S.E.; Wood, M.D. The effects of intraoperative electrical stimulation on regeneration and recovery after nerve isograft repair in a rat model. Hand 2020, 17, 540–548. [Google Scholar] [CrossRef]
- Zuo, K.J.; Shafa, G.; Antonyshyn, K.; Chan, K.; Gordon, T.; Borschel, G.H. A single session of brief electrical stimulation enhances axon regeneration through nerve autografts. Exp. Neurol. 2020, 323, 113074. [Google Scholar] [CrossRef]
- Javeed, S.; Faraji, A.J.; Dy, C.; Ray, W.Z.; MacEwan, M.R. Application of electrical stimulation for peripheral nerve regeneration: Stimulation parameters and future horizons. Interdiscipl Neurosurg. 2021, 24, 101117. [Google Scholar] [CrossRef]
- Birenbaum, N.K.; Yan, Y.; Odabas, A.; Chandra, N.S.; Ray, W.Z.; MacEwan, M.R. Multiple sessions of therapeutic electrical stimulation using implantable thin-film wireless nerve stimulators improve functional recovery after sciatic nerve isograft repair. Muscle & Nerve 2023, 67, 244–251. [Google Scholar]
- Keane, G.C.; Marsh, E.B.; Hunter, D.A.; Schelhardt, L.; Walker, E.R.; Wood, M.D. Lidocaine nerve block diminishes the effects of therapeutic electrical stimulation to enhance nerve regeneration in rats. Hand 2023, 18 (1_suppl), 119S–125S. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Voda, J.; Gold, B.G.; Gordon, T. FK506 increases peripheral nerve regeneration after chronic axotomy but not after chronic Schwann cell denervation. Exp. Neurol. 2002, 175, 127–137. [Google Scholar] [CrossRef]
- Tajdaran, K.; Shoichet, M.S.; Gordon, T.; Borschel, G.H. A novel polymeric drug delivery system for localized and sustained release of tacrolimus (FK506). Biotech. and Bioeng. 2015, 112, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Tajdaran, K.; Chan, K.; Zhang, J.; Gordon, T.; Borschel, G.H. Local FK506 dose-dependent study using a novel three-dimensional organotypic assay. Biotech. Bioengin 2019, 116, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Tajdaran, K.; Chan, K.; Shoichet, M.; Gordon, T.; Borschel, G.H. Local delivery of FK506 to injured peripheral nerve enhances axon regeneration after surgical nerve repair in rats. Acta Biomater 2019, 96, 211–221. [Google Scholar] [CrossRef]
- Zuo, K.J.; Shafa, G.; Chan, K.; Zhang, J.; Hawkins, C.; Tajdaran, K.; Gordon, T.; Borschel, G.H. Local FK506 drug delivery enhances nerve regeneration through fresh, unprocessed peripheral nerve allografts. Exp. Neurol. 2021, 341, 113680. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Sulaiman, O.A.R.; Boyd, J.G. Experimental strategies to promote functional recovery after peripheral nerve injuries. J. Periph Nerv. System 2003, 236–250. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Cellular and molecular interactions after peripheral and central nerve injury. Biomed. Rev. 2003, 14, 51–62. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Neurobiology of peripheral nerve regeneration and functional recovery: from bench top research to bedside application. Ochsner J. 2013, 13, 100–180. [Google Scholar]
- Gordon, T. Electrical stimulation to enhance axon regeneration after peripheral nerve injuries in animal models and humans. Neurotherapeutics 2016, 13, 295–310. [Google Scholar] [CrossRef]
- Gordon, T.; English, A.W. Strategies to promote functional recovery after nerve injury: Electrical stimulation and/or exercise. Eur. J. Neurosci. 2016, 43, 336–350. [Google Scholar] [CrossRef]
- Gordon, T. The neurotrophic factor rationale for using brief electrical stimulation to promote peripheral nerve regeneration in animal models and human patients. In “Conductive Polymers: Electrical interactions in Cell Biology and Medicine”: Eds Zhang, Z.; Rouabhia, M.; Moulton, S.E. CRC Press, New York, 2017, Ch 11, pp 231-249.
- Tajdaran, K.; Chan, K.; Gordon, T.; Borschel, G.H. Matrices, scaffolds, and carriers for protein and molecule delivery peripheral nerve regeneration. Exp. Neurol. 2019, 319, 112817. [Google Scholar] [CrossRef]
- Gordon, T. Peripheral nerve regeneration and muscle reinnervation Int. J. Mol. Sci. 2020, 21, 8652. [Google Scholar] [CrossRef]
- Zuo, K.J.; Gordon, T.; Chan, K.M.; Borschel, G.H. Electrical stimulation to enhance peripheral nerve regeneration: Update in molecular investigations and clinical translation. Exp. Neurol. 2020, 332, 113397. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J. Neurosci. 1995, 15, 3886–3895. [Google Scholar] [CrossRef]
- Lieberman, A.R. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 1971, 14, 49–124. [Google Scholar] [PubMed]
- Grafstein, B. The nerve cell body response to axotomy. Exp. Neurol. 1975, 48, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.E. Cellular responses to axotomy and to related procedures. Br. Med. Bull. 1974, 30, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. Dependence of peripheral nerves on their target organs, in Somatic and Autonomic Nerve-Muscle Interactions (Bumstock G., Vrbova G., and O'Brien R. A., eds.), Elsevier, New York, 1993, pp. 289–323.
- Patodia, S.; Raivich, G. Role of transcription factors in peripheral nerve regeneration. Frontiers Mol. Neurosci. 2012, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, W.; Bisby, M.A.; Kreutzberg, G.W. Changes in cytoskeletal proteins in the rat facial nucleus following axotomy. J. Neurosci. 1988, 8, 3181–3189. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.; Tetzlaff, W.; Bisby, M.; Fawcett, J.W.; Milner, R. Rapid induction of the major embryonic alpha-tubulin mRNA, T alpha 1, during nerve regeneration in adult rats. J. Neurosci. 1989, 9, 1452–1463. [Google Scholar] [CrossRef]
- Kato, S.; Ide, C. Axonal sprouting at the node of Ranvier of the peripheral nerve disconnected with the cell body. Restor. Neurol. Neurosci. 1994, 6, 181–187. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Thompson, G.W.; Griffen, J.W.; Price, D.L. Changes in neurofilament transport coincide temporally with alterations in the caliber of axons in regenerating motor fibers. J. Cell Biol. 1985, 101, 1332–1340. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, R.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: a major determinant of axon caliber. Proc. Natl. Acad. Sci. USA 1997, 84, 3472–3476. [Google Scholar] [CrossRef]
- Gordon, T.; Stein, R.B. Time course and extent of recovery of reinnervated motor units of cat triceps surae muscles. J. Physiol. 1982, 323, 307–323. [Google Scholar] [CrossRef]
- Gordon, T.; Gillespie, J.; Orozco, R.; Davis, L. Axotomy-induced changes in rabbit hindlimb nerves and the effects of chronic electrical stimulation. J. Neurosci. 1991, 11, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.M.; Danielsen, N.; Holmquist, B.; Kanje, M.; Lundborg, G. The influence of predegeneration on regeneration through peripheral nerve grafts in the rat. Exp. Neurol. 1993, 122, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.R.; Bedard, A.M.; Hincke, M.T.; Tetzlaff, W. Increased expression of BDNF and trkB mRNA in rat facial motoneurons after axotomy. Eur. J. Neurosci. 1996, 8, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Al-Majed, A.A.; Brushart, T.M.; Gordon, T. Electrical stimulation accelerates and increases expression of BDNF and TrkB mRNA in regenerating rat femoral motoneurons. Eur. J. Neurosci. 2000, 12, 4381–4390. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.C.; Li, E.; Kawaguchi, R.; Gerschwind, D.H.; Woolf, C.J. Transcriptional reprogramming of distinct peripheral sensory neuron subtypes after axonal injury. Neuron 2020, 108, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.-Q.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.L.; Fainzilber, M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Rishal, I.; Fainziber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Saito, A.; Cavelli, V. Signaling over distances. Mol. Cell Proteomics 2016, 15, 382–393. [Google Scholar] [CrossRef]
- Spira, M. E.; Benbassat, D.; Dormann, A. Resealing of the proximal and distal cut ends of transected axons: electrophysiological and ultrastructural analysis. J Neurobiol. 1993, 24, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Madeddu, F.; Bozzi, Y.; Maffei, L.; Ratto, G.M. Acute physiological response of mammalian central neurons to axotomy: ionic regulation and electrical activity. FASEB J. 2004, 18, 1934–1936. [Google Scholar] [CrossRef]
- Kershensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, c572–c557. [Google Scholar] [CrossRef]
- Kamber, D.; Erez, H.; Spira, M.E. Local calcium-dependent mechanisms determine whether a cut axonal end assembles a retarded endbulb or competent growth cone. Exp. Neurol. 2009, 219, 112–125. [Google Scholar] [CrossRef]
- Bradke, F.; Fawcett, J.W.; Spira, M.E. Assembly of a new growth cone after axotomy: The precursor to axon regeneration. Nat. Rev. Neurosci. 2012, 13, 183–193. [Google Scholar] [CrossRef]
- Ambron, R. T.; Walters, E. T. Priming events and retrograde injury signals. A new perspective on the cellular and molecular biology of nerve regeneration. Mol. Neurobiol. 1996, 13, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Chierzi, S.; Codd, A.M.; Campbell, D.S.; Meyer, R.L; Holt, C.E.; Fawcett, J.W. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci. 2005, 25, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Vogelaar, C. F.; Gervasi, N.M.; Gumy, L.F.; Story, D.J.; Raja-Chowdhury, R.; Leung, K.-M.; Hold, C.E.; Fawcett, J.W. Axonal mRNAs: characterisation and role in the growth and regeneration of dorsal root ganglion axons and growth cones. Mol. Cell Neurosci. 2009, 42, 102–115. [Google Scholar] [CrossRef]
- Yan, D.; Wu, Z.; Chisholm, A. D.; Jin, Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 2009, 138, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Omotade, O.F.; Pollitte, S.F.; Zheng, J.Q. Actin-based growth cone motility and guidance. Mol. Cell Neurosci. 2017, 84, 4–10. [Google Scholar] [CrossRef]
- Liu, M.; Li, P.; Jia, Y.; Cui, Q.; Zhang, K.; Jiang, J. Role of non-coding RNAs in axon regeneration after peripheral nerve injury. In J. Biol. Sci. 2022, 18, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Willis, D.E. What makes a RAG regeneration associated? Front. Mol. Neurosci. 2015, 8, 43. [Google Scholar] [CrossRef]
- Hao, Y.; Frey, E.; Yoon, C.; Wong, H.; Nestorovski, D.; Holtzman, L.B.; Giger, R.J.; DiAntonio, A.; Collins, C. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 2016, 5, e14048. [Google Scholar] [CrossRef]
- Shin, J.E.; Cho, Y.; Beirowski, B.; Milbrandt, J.; Cavalli, V.; DiAntonio, A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 2012, 74, 1015–1022. [Google Scholar] [CrossRef]
- Kiryu-Seo, S.; Kato, R.; Ogawa, T.; Nakagomi, S.; Nagata, K.; Kiyama, H. Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J. Biol. Chem. 2008, 283, 6988–6996. [Google Scholar] [CrossRef]
- Valakh, V.; Frey, E.; Babetto, E.; Walker, L.J.; DiAntonio, A. Cytoskeletal disruption activates the DLK/JNK pathway, which promotes axonal regeneration and mimics a preconditioning injury. Neurobiol. Dis. 2015, 77, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Ha, H.; Kim, Y.K.; Cho, Y.; DiAntonio, A. DLK regulates a distinctive transcriptional regeneration program after peripheral nerve injury. Neurobiol. Dis. 2019, 127, 178. [Google Scholar] [CrossRef] [PubMed]
- George, E.B.; Glass, J.D.; Griffen, J.W. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J. Neurosci. 1995, 15, 6445–6452. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Popovich, P.G.; Ramer, M.S. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflamm 2001, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Sievers, C.; Platt, N.; Perry, V.H.; Coleman, M.P.; Conforti, L. Neurites undergoing Wallerian degeneration show an apoptotic-like process with annexin V positive staining and loss of mitochondrial membrane potential. Neurosci. Res. 2003, 46, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kershensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, c572–557. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R. Janeckova, L; Vargas, M.E.; Worrell, L..A.; Loreto, A.; Tickle, J.; Patrick, J.; Webster, J.R.M.; Marangoni, M.; Carpi,, F.M.; Pucciarelli, S.; Rossi, ;Meng, W.; Sagasti, A.; Ribchester, R.R.; Magni, G.; Coleman, M.P.; Conforti, L. A rise in NAD precursor nicotinamide mono-nucleoride (NMN) after injury promotes axon degeneration. Cell Death Differen 2015, 22, 731–742. [Google Scholar]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martines, A.; Whitworth, A.J.; Conforti, L.; Dajas-Bailador, F.; Coleman, M.P. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 2020, 134, 104678. [Google Scholar] [CrossRef]
- Waller, A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Phil Trans. Royal Soc. Lond. 1850, 140, 423–429. [Google Scholar]
- Küry, P.; Stoll, G.; Müller, H.W. Molecular mechanisms of cellular interactions in peripheral nerve regeneration. Curr. Opin. Neurol. 2001, 14, 635–639. [Google Scholar] [CrossRef]
- Stoll, G.; Jander, S.; Myers, R.R. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller’s observations to neuroinflammation. J. Peripher. Nerv. Syst. 2002, 7, 13–27. [Google Scholar] [CrossRef]
- Ziv, N.E.; Spira, M.E. Axotomy induces a transient and localized elevation of the free intracellular calcium concentration to the millimolar range. J. Neurophysiol. 1995, 74, 2625–2537. [Google Scholar] [CrossRef] [PubMed]
- Ziv, N.E.; Spira, M.E. Localized and Transient Elevations of Intracellular Ca21 Induce the Dedifferentiation of Axonal Segments into Growth Cones. J. Neurosci. 1997, 3568–3579. [Google Scholar] [CrossRef] [PubMed]
- Spira, M.E.; Oren, R.; Dormann, A.; Gitler, D. Critical calpain-dependent ultrastructural alterations underlie the transformation of an axonal segment into a growth cone after axotomy of cultured Aplysia neurons. J. Comp. Neurol. 2003, 457, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Ginty, D.D. Function and regulation: Review of CREB family transcription factors in the nervous system. Neuron 2002, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Roy, A.; Wu, Z.; Concharov, A.; Jin, Y.; Chisholm, A.D. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J. Neurosci. 2010, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Shin, J.E.; Ewan, E.E.; Oh, Y.M.; Pita-Thomas, W.; Cavalli, V. Activating injury-responsive genes with hypoxia enhances axon regeneration through neuronal HIF-1alpa. Neuron 2015, 88, 720–734. [Google Scholar] [CrossRef]
- Song, W.; Cho, Y.; Watt, D.; Cavalli, V. Tubulin-tyrosine ligase (TTL) mediated increase in tyrosinated α-tubulin in injured axons is required for retrograde injury signaling and axon regeneration. J. Biol. Chem. 2015, 290, 14765–14775. [Google Scholar] [CrossRef]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.F.; Fainzilber, M. Axoplasmic importins enable retrograde injury blot analysis. Signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Perlson, E.; Hanz, S.; Ben-Yaakov, K.; Segal-Ruder, Y.; Seger, R.; Fainzilber, M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 2005, 45, 715–726. [Google Scholar] [CrossRef]
- Lezana, J.P.; Dagan, S.Y.; Robinson, A.; Goldstein, R.S.; Fainzilber, M.; Bronfman, F.C.; B1onfman, M. Axonal PPARγ promotes neuronal regeneration after injury. Dev. Neurobiol. 2016, 76, 688–701. [Google Scholar] [CrossRef]
- Terenzio, M.; Koley, S.; Samra, N.; Rishal, I.; Zhao, Q.; Sahoo, P.K.; Uirsman, A.; Marvaldi, L.; Oses-Priesto, J.A.; Forester, C.; Goes, C.; Kliniski, A.L.; Di Pizio, A.; Doron-Mandel, E.D.; Perry, R.B-T.; Koppel, I.; Twiss, J.L.; Burlingame, A.L.; Fainzilber, M. Locally translated mTOR controls axonal local translation in nerve injury. Science 2018, 359, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Mahar, M.; Cavelli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Misra, V.; Verge, V.M.K. Sensing nerve injury at the axonal ER: activated Luman/CREB3 serves as a novel axonally synthesized retrograde regeneration signal. PNAS 2014, 16142–16147. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Zhai, R.; McLean, N.A.; Johnson, J.M.; Misra, V.; Verge, V.M.K. The unfolded protein response and cholesterol biosynthesis link Lumen/CREB3 to regenerative axon growth in sensory neurons. J. Neurosci. 2015, 35, 4557–14570. [Google Scholar] [CrossRef] [PubMed]
- Hasmatali, J.C.D.; De Guzman, J.; Zhai, R.; Yang, L.; McLean, N.A.; Hutchinson, C. ; Johnston.; Misra, V.; Verge, V.M.K. Axotomy induces phasic alterations in Luman/CREB3 expression and nuclear localization in injured and contralateral uninjured sensory neurons: correlation with intrinsic axon growth capacity. Neuropathol. Exp. Neurol. 2019, 78, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Schoeps, T.; Haubrock, M.; Dönitz, J. TFClass: a classification of human transcription factors and their rodent orthologs. Nucleic Acids Res 2015, 43, D97–D102. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.; Hunt, S.P. (1991) Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci. Lett. 1991, 129, 107–110. [Google Scholar] [CrossRef]
- Van Kesteren, R.E.; Mason, M.R.; Macgillavry, H.D.; Smit, A.B.; Verhaagen, J. A gene network perspective on axonal regeneration. Front. Mol. Neurosci. 2011, 4, 46. [Google Scholar] [CrossRef]
- Song, J.; Singh, M. From hub proteins to hub modules: the relationship between essentiality and centrality in the yeast interactome at different scales of organization. PLoS Comput. Biol. 2013, 9, 1002910. [Google Scholar] [CrossRef]
- Fontana, X.; Hristova, M.; Costa, C.D.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.R.; Klein, R.; Raivich, G.; Behrens, A. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef]
- Jessen, K. R.; Mirsky, R.; Arthur-Farraj, P. The role of cell plasticity in tissue repair: adaptive cellular reprogramming. Devel Cell 2005, 34, 613–620. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia 2008, 56, 1552–1565. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Herdegen, T.; Skene, P.; Bähr, M. The c-Jun transcription factor–bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Michaeleviski, I.; Segal-Ruder, Y.; Rozenbaum, M.; Medzihradszky, K.F.; Shalem, O.; Coppola, G.; Horn-Saban, S.; Ben-Yaakov, K.; Dagan, S.Y.; Rishal, I.; Geschwind, D.H.; Pilpel, Y.l.; Burlingame, A.L.; Fainzilber, M. Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 2010, 3, ra53. [Google Scholar] [CrossRef] [PubMed]
- Blesch, A.; Lu, P.; Tsukada, S.; Alto, L.T.; Roet, K.; Coppola, G.; Geschwind, D.; Tuszynski, M.H. Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to cAMP-mediated effects. Exp. Neurol. 2012, 235, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, F. W.; Hager, G.; Schmitt, A. B.; Horvat, A.; Streif, R.; Spitzer, C.; Gamal, S.; Breuer, S.; Brook, G.A.; Nacimiento, W.; Kreutzberg, G.W. Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 2000, 12, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, H.; Kondo, E.; Fukuoka, T.; Dai, Y. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol. Cell Neurosci. 2000, 15, 170–182. [Google Scholar] [CrossRef]
- Seijffers, R.; Allchorne, A.J.; Woolf, C.J. The transcription factor ATF3 promotes neurite outgrowth. Mol. Cell Neurosci. 2006, 143–154. [Google Scholar] [CrossRef]
- Seijffers, R.; Mills, C.D.; Woolf, C.J. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 2007, 27, 7911–7920. [Google Scholar] [CrossRef]
- Lindwall, C.; Kanje, M. Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell Neurosci. 2005, 29, 269–282. [Google Scholar] [CrossRef]
- Schweizer, U.; Gunnersen, J.; Karch, C.; Wiese, S.; Holtmann, B.; Takeda, K.; Akira, S.; Sendtner, M. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J. Cell Biol. 2002, 156, 287–297. [Google Scholar] [CrossRef]
- Bareyre, F.M.; Garzorz, N.; Lang, C.; Misgeld, T.; Buning, H.; Kerschensteiner, M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U S A 2011, 108, 6282–6287. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.P.; Cornuet, P.K.; McIlwrath, S.; Koerber, H.R.; Albers, K.M. SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neurosci. 2006, 143, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, M.P.; McIlwrath, S.L.; Jing, X.; Cornuet, P.K.; Salerno, K.M.; Koerber, H.R.; Albers, K.M. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 2009, 1256, 43–54. [Google Scholar] [CrossRef]
- Jankowski, M.P.; Miller, L.; Koerber, H.R. Increased expression of transcription factor SRY-box-containing gene 11 (Sox11) enhances neurite growth by regulating neurotrophic factor responsiveness. Neurosci. 2018, 382, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Wang, T.; Huang, S.; Glorioso, J.C.; Albers, K.M. The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp. Neurol. 2012, 233, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Ho, C.; Wong, K.; Tessier-Lavigne, M. Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J. Neurosci. 2009, 29, 7116–7123. [Google Scholar] [CrossRef] [PubMed]
- Finelli, M.G.; Wong, J.K.; Zou, H. Epigenetic regulation of sensory axon regeneration after spinal cord injury. J. Neurosci. 2013, 33, 19664–19676. [Google Scholar] [CrossRef] [PubMed]
- Saijilafu, E.-M.H.; Hur, C.-M.; Liu, C.-M.; Jiao, Z.; Xu, W.-L; Zhou, F.-Q. P13K-GSK3 signalling regulates mammalian axon regeneration by inducing expression of Smad. Nat Commun. 2013, 4, 2690. [Google Scholar] [CrossRef]
- Saijilafu, Z.B.Y.; Zhou, F.Q. Signaling pathways that regulate axon regeneration. Neurosci. Bull. 2013, 29, 411–420. [Google Scholar] [CrossRef]
- Okuyama, N.; Kiryu-Seo, S.; Kiyama, H. Altered expression of Smad family members in injured motor neurons of rat. Brain Res. 2007, 1132, 36–41. [Google Scholar] [CrossRef]
- Jiang, J.J.; Liu, C.M.; Zhang, B.Y.; Wang, W.X.; Zhang, M.; Saijilafu, E.-M.H. MicroRNA-26a supports mammalian axon regeneration in vivo by suppressing GSK3beta expression. Cell Death Dis. 2015, 6, e1865. [Google Scholar] [CrossRef]
- Nadeau, S.; Hein, P.; Fernandes, K.J.; Peterson, A.C.; Miller, F.D. A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol. Cell Neurosci. 2005, 29, 525–535. [Google Scholar] [CrossRef]
- Ma, J.J.; Ju, X.; Xu, R.J.; Wang, W.H.; Luo, Z.-P.; Liu, C.-M.; Yang, L.; Li, B.; Chen, J.-Q.; Meng, B.; Yang, H.-L.; Zhou, F.-Q. ; Saijilafu. Telomerase reverse transcriptase and p53 regulate mammalian peripheral nervous system and CNS axon regeneration downstream of c-Myc. J Neurosci 2019, 39, 9107–9118. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Knights, C.D.; Rao, M.; Yakovlev, A.; Beers, J.; Catania, J.; Avantaggiati, M. L.; Faden, A. I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006, 25, 4084–4096. [Google Scholar] [CrossRef]
- Moore, D. L.; Blackmore, M. G.; Hu, Y.; Kaestner, K. H.; et al. KLF family members regulate intrinsic axon regeneration ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef]
- Xu, J.H.; Qin, X.Z.; Zhang, H.N.; Ma, Y.X.; Qi, S.-B.; Zhang, H.-C.; Ma, J.-J.; Fu, X.-Y.; Xie, J.-L. Saijilafu. Deletion of Kruppel-like factor-4 promotes axonal regeneration in mammals. Neural Regen. Res. 2021, 16, 166–171. [Google Scholar]
- Palmisano, I.; Di Giovanni, S. Advances and limitations of current epigenetic studies investigating mammalian axonal regeneration. Neurotherapeutics 2018, 15, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series. Pulm. Circ. 2014, 169–174. [Google Scholar] [CrossRef]
- Zhou, Y.; Garcia, B.A. Comprehensive catalog of currently documented histone modifications. Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef]
- Puttagunta, R.; Tedeschi, A.; Soria, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; Laskowski, C.J.; Boutillier, A.-L.; Bradke, F.; Giovanni, S.D. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nature Comm. 2014, 5, 3527. [Google Scholar] [CrossRef]
- Karpranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Liu, X.; Yu, X.; He, Y.; Wang, L. Long noncoding RNA nuclear enriched abundant transcript 1 promotes the proliferation and migration of Schwann cells by regulating the miR-34a/Satb1 axis. Glia 2021, 234, 15357–16366. [Google Scholar] [CrossRef]
- Strickland, I.T; Richards, L.; Holes, F.E.; Wynick, D.; Uney, J.B.; Wong, L.F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS One 2011, 6, e23423. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, F.; Gu, X. Non-coding RNAs as emerging regulators of neural injury response and regeneration. Neurosci. Bull. 2016, 32, 253–264. [Google Scholar] [CrossRef]
- Dorrier, C.E.; Jones, H.E.; Pintarić, L.; Siegenthaler, J.A.; Daneman, R. Emerging roles for CNS fibroblasts in health, injury and disease. Nature Rev. Neurosci. 2022, 23, 23–34. [Google Scholar] [CrossRef]
- Knöferle, J.; Koch, J.C.; Ostendorf, T.; Michel, V.; Planchamp, V.; Vutova, P.; Tönges, L.; Stadelmann, C.; Brück, W.; Bähr, M.; Lingor, P. Mechanisms of acute axonal degeneration in the optic nerve in vivo. PNAS 2010, 107, 6064–6069. [Google Scholar] [CrossRef]
- Mårtensson, L.B.; Blom, C.L.; Dahlin, L.B. Ca2+ involvement in activation of extracellular-signal-regulated-kinase 1/2 and m-calpain after axotomy of the sciatic nerve. Neural Reg Res B 2017, 12, 623–628. [Google Scholar]
- Miledi, R.; Slater, C.R. On the degeneration of rat neuromuscular junctions after nerve section. J. Physiol. 1970, 207, 507–528. [Google Scholar] [CrossRef]
- Gilliatt, R. W.; Horth, R.J. Nerve conduction during Wallerian degeneration in the baboon. J. Neurol. Neurosurg. Psychiatry 1972, 35, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lubinska, L. Early course of Wallerian degeneration in myelinated fibres of the rat phrenic nerve. Brain Res. 1977, 130, 47–63. [Google Scholar] [CrossRef]
- Mackenzie, S.J.; Smirnov, I.; Calancie, B. Cauda equina repair in the rat: part Time course of ventral root conduction failure. J. Neurotrauma 2012, 29, 1683–1690. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Perez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef]
- Napoli, I.; Noon, L.A.; Ribeiro, S.; Kerai, A.P.; Parrinello, S.; Rosenberg, L.H.; Collins, M.J.; Harrisingh, M.C.; White, I.J.; Woodhoo, A.; Lloyd, A.C. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration. Neuron 2012, 73, 729–742. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J. A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M..; Griffith, M.; Hantke, J.; Maciias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; De Juan, V.G.; Jefferies, H.B.; Aspichueta, P.; Elortza, F.; Aransay, A.M.; Martínez-Chantar, M.L.; Baas, F.; Mato, J.M.; Mirsky, R.; Woodhoo, A.; Jessen, K.R. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef]
- Jang, S. Y.; Shin, Y. K.; Park, S. Y.; Park, J. Y.; Lee, H. J.; Yoo, Y. H.; Kim, J.K.; Park, H.W. Autophagic myelin destruction by Schwann cells during Wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Brosius Lutz, A.; Chung, W. S.; Sloan, S. A.; Carson, G. A.; Zhou, L.; Lovelett, E.; Posada, S.; Zuchero, J.B.; Barres, B.A. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. USA 2017, 114, E8072–E8080. [Google Scholar] [CrossRef] [PubMed]
- Perry, V. H.; Tsao, J. W.; Fearn, S.; Brown, M. C. Radiation-induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur. J. Neurosci. 1995, 7, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Gregson, N.A.; Hall, S.M. A quantitative analysis of the effects of the intraneural injection of lysophosphatidyl choline. J. Cell Sci. 1973, 13, 257–277. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Tigueros, M.A.; Kalvas, A.; David, S. Phospholipase A2 plays an important role in myelin breakdown and phagocytosis during Wallerian degeneration. Mol. Cell Neurosci. 2003, 24, 753–765. [Google Scholar] [CrossRef]
- Banner, L.R.; Patterson, P.H. Major changes in the expression of the mRNAs for cholinergic differentiation factor/leukemia inhibitory factor and its receptor after injury to adult peripheral nerves and ganglia. Proc Natl Acad Sci U S A 1994, 91, 7109–7113. [Google Scholar] [CrossRef]
- Bolin, M.; Verity, A.N.; Silver, J.E.; Shooter, E.M.; Abrams, J.S. Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem 1995, 64, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bourde, O.; Kiefer, R.; Toyka, K.V.; Hartung, H.P. Quantification of interleukin-6 mRNA in Wallerian degeneration by competitive reverse transcription polymerase chain reaction. J. Neuroimmunol. 1996, 69, 135–140. [Google Scholar] [PubMed]
- Kurek, J.B.; Austin, L.; Cheema, S.S.; Bartlett, P.F.; Murphy, M. Up-regulation of leukaemia inhibitory factor and interleukin-6 in transected sciatic nerve and muscle following denervation. Neuromuscul. Disorders 1996, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, H.S.; Roytta, M. Increased expression of chemokines (MCP-1, MIP-1alpha, RANTES) after peripheral nerve transection. J Periph Nerv Syst 2000, 5, 75–81. [Google Scholar] [CrossRef]
- Subang, M.C.; Richardson, P.M. Influence of injury and cytokines on synthesis of monocyte chemoattractant protein-1 mRNA in peripheral nervous tissue. Eur. J. Neurosci. 2001, 13, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Perrin, F.E.; Lacroix, S.; Aviles-Trigueros, M.; David, S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 2005, 128, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Boivin, A.; Pineau, I.; Barrette, B.; Filali, M.; Vallieres, N.; Rivest, S.; Lacroix, S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 2007, 27, 12565–12576. [Google Scholar] [CrossRef]
- Shubayev, V.I.; Angert., M.; Dolkas, J.; Campana, W.M.; Palenscar, K.; Myers, R.R. TNFalpha-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol Cell Neurosci 2006, 31, 407–415. [Google Scholar] [CrossRef]
- Cattopadhyay, S.; Meyers, R.R.; Janes, J. Shubayev, V. Cytokine regulation of MMP-9 in peripheral glia: implications for pathological processes and pain in injured nerve. Brain Behav Immun 2007, 21, 561–568. [Google Scholar] [CrossRef]
- Beuche, W.; Friede, R.L. The role of non-resident cells in Wallerian degeneration. J. Neurocytol. 1984, 13, 767–796. [Google Scholar] [CrossRef]
- Avellino, A.M.; Hart, D.; Dailey, A.T.; MacKinnon, M.; Ellegala, D.; Kliot, M. Differential macrophage responses in the peripheral and central nervous system during allerian degeneration of axons. Exp. Neurol. 1995, 136, 183–198. [Google Scholar] [CrossRef]
- Brück, W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997, 7, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Griffin, J.W.; Li, C.Y.; Trapp, B.D. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J. Neurocytol. 1989, 18, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kubes, P.; Zochodne, D.W. Delayed peripheral nerve degeneration, regeneration, and pain in mice lacking inducible nitric oxide synthase. J. Neuropathol. Exp. Neurol. 2001, 60, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Friede, R.L. The role of complement in myelin phagocytosis during PNS wallerian degeneration. J. Neurol. Sci. 1991, 103, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E. Phagocytosis of myelin in demyelinative disease: a review. Neurochem. Res. 1999, 24, 261–268. [Google Scholar] [CrossRef] [PubMed]
- da Costa, C.C.; van der Laan, L.J.; Dijkstra, C.D.; Bruck, W. The role of the mouse macrophage scavenger receptor in myelin phagocytosis. Eur. J. Neurosci. 1997, 9, 2650–2657. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, R.E.; Echevarria, F.D. Macrophage biology in the peripheral nervous system after injury Prog. Neurobiol. 2019, 102–121. [Google Scholar]
- Nadeau, S.; Filali, M.; Zhang, J.; Kerr, B.J.; Rivest, S.; Soulet, D.; Iwakura, Y.; de Rivero Vaccari, J.; Keane, R.W.; Lacroix, S. Functional recovery after peripheral nerve injury in dependent n the proinflammatory cytokines IL-1beta and TNF: implications for neuropathic pain. J. Neurosci. 2011, 31, 12533–12542. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; et al. Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef]
- Liu, P.; Peng, J.; Han, G.H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar]
- Hall, S.M. The response of the (myelinating) Schwann cell population to multiple episodes of demyelination. J. Neurocytol. 1983.12, 1–12. [CrossRef]
- Doree, M.; Hunt, T. From Cdc3 to Cdk1: when did the cell cycle kinase join its cyclin partner? Cell Sci. 2002, 2451–2464. [Google Scholar]
- Han, S.; Seo, T.B.; Kim, K.-H.; Yoon, J.-H.; Yoon, S.-J.; Namgung, U.K. Cdc2-mediated Schwann cell migration during peripheral nerve regeneration. J. Cell Sci. 2007, 120, 246–255. [Google Scholar] [CrossRef]
- Namgung, U.K. The role of Schwann cell-axon interaction in peripheral nerve regeneration. Cell Tiss. Organs 2014, 4, 6–12. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; Wicher, G.K.; Mitter, R.; Greensmith, L.; Behrens, A.; Raivich, G.; Mirsky, R.; Jessen, K.R. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Arthur-Farraj, M.P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.; Welcher, A.A.; Suter, U.; Shooter, E.M. Identification of transcriptionally regulated genes after sciatic nerve injury. J. Neurosci. Res. 1991, 29, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wu, J.; Zhou, S.; Wang, Y.; Qin, J.; Yu, B.; Gu, X.; Yao, C. Global analysis of transcriptome in dorsal root ganglia following peripheral nerve injury in rats. Biochem. Biophys. Res. Commun. 2016, 478, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: tumor necrosis factor-α, Interleukin-1α, and Interleukin-1β. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Patterson, P.H.; Jessen, K.R.; Mirsky, R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci 2002, 2002 22, 6696–6703. [Google Scholar] [CrossRef]
- Marchionni, M.A.; Goodearl, A.D.; Chen, M.S.; Bermingham-McDonogh, O.; Kirk, C.; Hendricks, M.; Danehy, F.; Misumi, D.; Sudhalta, J.; Kobayashi, K.; Wroblewski, D.; Lynch, C.; Baldassare, M.; Hiles, I.; Davis, J.B.; Hsuan, J.J.; Totty, N.F.; Otsu, M.l.; McBurney, R.N.; Waterfield, M.D.; Strooband, P.; Gwynne, D. Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 1993, 362, 312–318. [Google Scholar] [CrossRef]
- Falls, D.L. Neuregulins and the neuromuscular system: 10 years of answers and questions. J. Neurocytol. 2003, 32, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Falls, D.L. Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.M. The biology of chronically denervated Schwann cells. Ann. NY Acad. Sci. 1999, 883, 215–233. [Google Scholar] [CrossRef]
- Carroll, S.L.; Miller, M.L.; Frohnert, P.W.; Kim, S.S.; Corbett, J.A. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J. Neurosci. 1997, 17, 1642–1659. [Google Scholar] [CrossRef]
- Stassart, R. M.; Fledrich, R.; Velanac, V.; Brinkmann, B. G.; Schwab, M. H.; Meijer, D.; Swerenda, M.W.; Nave, K.-A. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat. Neurosci. 2013, 16, 48–54. [Google Scholar] [CrossRef]
- Ronchi, G.; Haastert-Talini, K.; Fornasari, B. E.; Perroteau, I.; Geuna, S.; Gambarotta, G. The Neuregulin1/ErbB system is selectively regulated during peripheral nerve degeneration and regeneration. Eur. J. Neurosci. 2016, 43, 351–364. [Google Scholar] [CrossRef]
- El Soury, M.; Fornasari, B.; Morano, M.; Grazio, E.; Ronchi, G.; Incarnato, D.; Giacobini, M.; Geuna, S. ; Provero. Gambarotta, G. Soluble Neuregulin1 down-regulates myelination genes. Frontiers Cell Neurosci. 2018, 11, 157. [Google Scholar]
- Salzer, J.L; Bunge, R.P.; Glaser, L. Studies of Schwann cell proliferation. III. Evidence for the surface localization of the neurite mitogen. J. Cell Biol. 1980, 84, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.; Spencer, P.S. Schwann cell mitosis in response to regenerating peripheral axons in vivo. Brain Res. 1985, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Clemence, A.; Mirsky, R.; Jessen, K.R. Non-myelin-forming Schwann cells proliferate rapidly during Wallerian degeneration in the rat sciatic nerve. J. Neurocytol. 1989, 18, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Rahmatullah, M.; Schroering, A.; Rothblum, K.; Stahl, R.C.; Urban, B.; Carey, D.J. Synergistic regulation of Schwann cell proliferation by heregulin and forskolin. Mol. Cell Biol. 1998, 18, 6245–6252. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.J.; Eccleston, P.A.; Jessen, K.R.; Mirsky, R. Interaction between cAMP elevation, identified growth factors, and serum components in regulating Schwann cell growth. J. Neurosci. Res. 1991, 30, 346–352. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Lin, P.X.; Chang, Y.; Webster, H.D. Effects of nerve segment supernatants on cultured Schwann cell proliferation and laminin production. J. Neurosci. Res. 1994, 37, 612–622. [Google Scholar] [CrossRef]
- Morrissey, T.K.; Levi, A.D.; Nuijens, A.; Sliwkowski, M.X.; Bunge, R.P. Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbBProc Natl. Acad. Sci. USA 1995, 92, 1431–1435. [Google Scholar] [CrossRef]
- Shin, Y.K.; Jang, S.Y.; Yun, S.H.; Choi, Y.Y.; Yoon, B.-A.; Jo, Y.R.; Park, S.Y.; Pak, M.G.; Park, J.I.; Park, H.T. Cooperative interaction of hepatocyte growth factor and neuregulin regulates Schwann cell migration and proliferation through Grb2-associated binder-2 in peripheral nerve repair. Glia 2017, 65, 794–1808. [Google Scholar] [CrossRef]
- Adams, S. J.; Aydin, I. T.; Celebi, J. T. GAB2-A scaffolding protein in cancer. Mol. Cancer Res. 2012, 10, 1265–1270. [Google Scholar] [CrossRef]
- Wu, G.; Li, X.; Li, M.; Zhang, Z. Long non-coding RNA MALAT1 promotes the proliferation and migration of Schwann cells by elevating BDNF through sponging miR-129-5p. Exp. Cell Res. 2020, 390, 111937. [Google Scholar] [CrossRef]
- Yao, C.; Wang, Q.; Wang, Y.; Wu, J.; Cao, X.; Lu, Y.; Chen, Y.; Feng, W.; Gu, X.; Dun, X.-P.; Yu, B. Loc6802454 regulates Schwann cell proliferation through Psrc1 and Ska1-a as a microRNA sponge following sciatic nerve injury. Glia 2021, 69, 2391–2403. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Zhang, X.; Ding, L.; Zeng, Q. Microanalysis of the expression profile of IncRNA BC088327 as an agonist to heregulin-1β-induced cell proliferation in the peripheral nervous system. Int. J. Mol. Med. 2018, 41, 3477–3484. [Google Scholar]
- Yao, C.; Chen, Y.; Wang, J.; Wu, J.; Cao, X.; Lu, Y.; Chen, Y.; Feng, W.; Gu, X.; Dun, X.-P.; Yu, B. LncRNA BC088259 promotes Schwann cell migration through vimentin following peripheral nerve injury. Glia 2020, 68, 670–679. [Google Scholar] [CrossRef]
- Ma, Y.; Zhai, D.; Zhang, W.; Zhang, H.; Dong, I.; Zhou, Y.; Feng, D.; Zheng, W.; Wang, T.; Mao, C.; Wang, W. Down-regulation of long non-coding RNA MEG3 promotes Schwann cell proliferation and migration and repairs sciatic nerve injury in rats. J. Cell Mol. Med. 2020, 24, 7460–7469. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Huang, T.; Chen, Y.; Shen, L.; Zhou, S.; Zhang, S.; Yu, B. Circ-Spidr enhances axon regeneration after peripheral nerve injury. Cell Death Dis. 2019, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, H.; Frisen, J.; Barbany, G.; Timmusk, T.; Zachrisson, O.; Verge, V.M.; Persson, H. Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J. Cell Biol. 1993, 123, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hoke, A.; Redett, R.; Hameed, H.; Jari, R.; Zhou, C.; Li, Z.B.; Griffin, J.W.; Brushart, T.M. Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J. Neurosci. 2006, 26, 9646–9655. [Google Scholar] [CrossRef] [PubMed]
- Mi, R.; Chen, W.; Hoke, A. Pleiotrophin is a neurotrophic factor for spinal motor neurons. PNAS 2007, 104, 4664–4669. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M.; Aspalter, M.; Griffin, J.W.; Redett, R.; Hameed, H.; Zhou, C.; Wright, M.; Vyas, A.; Hoke, A. Schwann cell phenotype is regulated by axon modality and central–peripheral location, and persists in vitro. Exp. Neurol. 2013, 247, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Sulaiman, A.R. Nerve regeneration in the peripheral nervous system. Neuroglia (3 edn), Ch 55, 2012, 701-714.
- Wang, X. Pleiotrophin: activity and mechanism. Adv. Clin. Chem. 2020, 98, 1–89. [Google Scholar]
- Heumann, R.; Lindholm, D.; Bandtlow, C.; Meyer, M.; Radeke, M.J.; Misko, T.P.; Shooter, E.; Thoenen, H. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerves during development, degeneration and regeneration: role of macrophages. Proc. Natl. Acad. Sci. USA 1987, 84, 8735–8739. [Google Scholar] [CrossRef]
- Meyer, M.; Matsuoka, I.; Wetmore, C.; Olson, L.; Thoenen, H. Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J. Cell Biol. 1992, 119, 45–54. [Google Scholar] [CrossRef]
- Naveilhan, P.; ElShamy, W.E.; Ernsfors, P. Differential regulation of mRNAs for GDNF and its receptors Ret and GDFRα after sciatic nerve lesion in the mouse. Eur. J. Neurosci. 1997, 9, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Petrov, T.; Chung, P.H.; Gordon, T. The expression of the low affinity nerve growth factor receptor in long-term denervated Schwann cells. Glia 1997, 20, 87–100. [Google Scholar] [CrossRef]
- Seo, T.B.; Oh, M.J.; You, B.G.; Kwon, K.B.; Chang, I.A.; Yoon, J.H.; Lee, C.-Y; Namgung, U.K. Erk1/2-mediated Schwann cell proliferation in the regenerating sciatic nerve by treadmill training. J. Neurotrauma 2009, 26, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Jang, S.Y.; Park, J.Y.; Park, S.Y.; Lee, H.J.; Suh, D.J.; Park, H.T. The Neuregullin-RacMKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia 2013, 61, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Blom, C.L.; Martensson, L.B.; Dahlin, L.B. Nerve injury-induced c-activation in Schwann cells is JNK independent. Biomed. Res. Int. 2014, 2014, 392971. [Google Scholar]
- Friede, R.L.; Bischhausen, R. The fine structure of stumps of transected nerve fibers in subserial sections. J NeuroI Sci. 1980, 44, 181–203. [Google Scholar] [CrossRef]
- McQuarrie, I.G. Effect of a conditioning lesion on axonal sprout formation at nodes of Ranvier I. Comp. Neurol. 1985, 231, 239–249. [Google Scholar] [CrossRef]
- McQuarrie, I.G.; Lasek, R.J. Transport of cytoskeletal elements from parent axons into regenerating daughter axons. J. Neurosci. 1989, 9, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ranson, S.W. Degeneration and regeneration of nerve fibres J. Comp. Neurol. 1912, 22, 487–546. [Google Scholar]
- Aitken J., T.; Sherman, M.; Young J., Z. Maturation of peripheral nerve fibres with various peripheral connections. J. Anat. 1947, 81, 1–22. [Google Scholar]
- Mackinnon, S.E.; Dellon, A.L.; O-Brien, J.P. Changes in nerve fiber numbers distal to a nerve repair in the rat sciatic nerve model. Muscle Nerve 2001, 14, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Toft, P.B.; Fugleholm, K.; Schmalbruch, H. Axonal branching following crush lesions of peripheral nerves of rats. Muscle Nerve 1988, 11, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S.; Napoli, I.; Ribeiro, S.; Digby, P.W.; Fedorova, M.; Parkinson, D.B.; Doddrell, R.D.; Nakayama, M.; Adams, R.H.; Lloyd, A.C. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 2010, 143, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bixby, J. l.; Lilien, J.; Reichardt, l. F. Identification of the major proteins that promote neuronal process outgrowth on Schwann cells in vitro. J. Cell Biol. 1988, 107, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, their firbillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef]
- Gonzalez Perez, F.; Udina, E.; Navarro, X. Chapter 10 Extracellular matrix components in peripheral nerve regeneration. Int. Rev. Neurobiol. 2013, 108, 257–275. [Google Scholar]
- Lefcort, F.; Venstrom, K.; McDonald, J.A.; Reichardt, L.F. Regulation of expression of fibronectin and its receptor, α5β1, during development and regeneration of peripheral nerve. Development 1992, 116, 767–782. [Google Scholar] [CrossRef]
- Mathews, G.A.; Ffrench-Constant, C. Embryonic fibronectins are up-regulated following peripheral nerve injury in rats J. Neurobiol. 1995, 26, 171–188. [Google Scholar] [CrossRef]
- Gardiner, N. Integrins and the extracellular matrix: key mediators of development and regeneration of the sensory nervous system. Dev Neurobiol 2011, 71, 1054–1072. [Google Scholar] [CrossRef]
- Gonzalez Perez, F.; Alé, A.; Santos, D.; Barwig, C.; Freier, T.; Navarro, X.; Udina, E. Substratum preferences of motor and sensory neurons in postnatal and adult rats. Europ J. Neurosci. 2016, 43, 431–442. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Ann Rev Cell Dev Biol 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Kloss, C. U. A.; Werner, A.; Klein, M.A.; Shen, J.; Menuz, K.; Probst, J.C.; Kreutzberg, G.W.; Raivich, G. Integrin family of cell adhesion molecules in the injured brain: regulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J Comp Neurol 1999, 411, 162–178. [Google Scholar] [CrossRef]
- Yanagida, H.; Tanaka, J.; Maruo, S. Immunocytochemical localization of a cell adhesion molecule, integrin α5β1, in nerve growth cones. J. Orthop. Sci. 1999, 4, 353–360. [Google Scholar] [CrossRef]
- Hammarberg, H.; Wallquist, W.; Piehl, F.; Risling, M.; Cullheim, S. Regulation of laminin-associated integrin subunit mRNAs in rat spinal motoneurons during postnatal development and after axonal injury. J. Comp. Neurol. 2000, 428, 294–304. [Google Scholar] [CrossRef]
- Vogelezang, M.G.; Liu, Z.; Relvas, J.B.; Raivich, G.; Scherer, S.S.; Charles ffrench-Constant, C. α4 Integrin Is expressed during peripheral nerve regeneration and enhances neurite outgrowth. J. Neurosci. 2001, 21, 6732–6744. [Google Scholar] [CrossRef] [PubMed]
- Wallquist, W.; Zelano, J.; Plantman, S.; Kaufman, S.J.; Cullheim, S.; Hammarberg, H. Dorsal root ganglion neurons up-regulate the expression of laminin-associated integrins after peripheral but not central axotomy. J. Comp. Neurol. 2004, 480, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, N.J.; Fernyhough, P.; Tomlinson, D.R.; Mayer, U.; von der, M.H.; Streuli, C.H. Alpha7 integrin mediates neurite outgrowth of distinct populations of adult sensory neurons. Mol. Cell Neurosci. 2005, 28, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Strittmatter, S.M. The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J. Neurosci. 2008, 28, 1262–1269. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Haenzi, B.; Andrews, M.R.; Verhaagen, J.; Fawcett, J.W. Integrins promote axonal regeneration after injury of the nervous system. Biol. Rev. Camb. Philos. Soc. 2018, 1339–1362. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, C.; Bow, C.M.; Remahl, I.N. Myelination and myelin sheath remodeling in normal and pathological PNS nerve fibres. Prog. Neurobiol. 1994, 43, 85–141. [Google Scholar] [CrossRef] [PubMed]
- Fricker, F.R.; Bennett, D.A. The role of neuregulin-1 in the response to nerve injury. Future Neurol. 2011, 6, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Fricker, F.R.; Antunes-Martins, A.; Jorge Galino, J.; Paramsothy, R.; La Russa, F.; Perkins, J.; Goldberg, R.; Brelstaff, J.; Zhu, N.; McMahan, S.B.; Orengo, C.; Garratt, A.N.; Birchmeier, C.; Bennett, D.L.H. Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 2013, 136, 2279–2297. [Google Scholar] [CrossRef]
- Haftek, J.; Thomas, P. K. Electron-microscope observation on the effects of localized crush injuries on the connective tissues of the peripheral nerve. J. Anat. 1968, 103, 233–243. [Google Scholar]
- Devor, M.; Govrin-Lippman, R. Maturation of axonal sprouts after nerve crush. Exp. Neurol. 1979, 64, 260–270. [Google Scholar] [CrossRef]
- Gordon, T.; Stein, R.B. Time course and extent of recovery in reinnervated motor units of cat triceps surae muscles. J. Physiol. 1982, 323, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Houdek, M.T.; Shin, A.Y. Management and complications of traumatic peripheral nerve injuries. Hand Clin. 2015, 31, 151–163. [Google Scholar] [CrossRef]
- Ransom, S.C.; Shahrestani, S.; Lien, B.V.; Tafreshi, A.R.; Brown, N.J.; Hanst, B.; Lehrich, B.M.; Ransom, R.C.; Sahyouni, R. Translational approaches to electrical stimulation for peripheral nerve regeneration. Neurorehabil Neural Repair. 2020, 34, 979–985. [Google Scholar] [CrossRef]
- Rasulić, L.; Simić, V.; Savić, A.; Lepić, M.; Kovačević, V.; Puzović, V’.; Vitošević, F.; Novaković, N.; Samardžić, M.; Rotim, K. Management of brachial plexus missile injuries. Acta Clin. Croat. 2018, 57, 487–496. [Google Scholar] [CrossRef]
- Sunderland, S. Rate of regeneration of motor fibers in the ulnar and sciatic nerves. Arch. Neurol. Psych. 1947, 58, 7–13. [Google Scholar] [CrossRef]
- Sunderland, S. Rate of regeneration of sensory nerve fibers. Arch. Neurol. Psychiat 1947, 58, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, H.J.; Spencer, P.S. The fate of Schwann cells isolated from axonal contact J. Neurocytol. 1978, 7, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Röyttä, M.; Salonen, V. Long-term endoneurial changes after nerve transection. Acta Neuropathol. 1988, 76, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Salonen, V.; Peltonen, J.; Röyttä, M.; Virtanen, I. Laminin in traumatized peripheral nerve: basement membrane changes during degeneration and regeneration. J. Neurocytol. 1987, 16, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Salonen, V.; Röyttä, M.; Peltonen, J. The effects of nerve transection on the endoneurial collagen fibril sheaths. Acta Neuropathol. 1987, 74, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Abernethy, D.A.; King, R.H.M.; Muddle, J.R.; Thomas, P.K. Neural architecture I transected rabbit sciatic nerve after prolonged nonreinnervation. J. Anat. 1998, 192, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Dedkov, E.I.; Kostrominova, T. Y.; Borisov, A. B.; Carlson, B. M. Survival of Schwann cells in chronically denervated skeletal muscles. Acta Neuropath 2002, 103, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Wood, P.; Sulaiman, O.A.R. Long-term denervated rat Schwann cells retain their capacity to proliferate and to myelinate axons in vitro. Frontiers Cell Neurosci. 2019, 12, 511. [Google Scholar] [CrossRef]
- Qian, T.; Wang, P.; Chen, Q.; Yi, S.; Liu, Q.; Wang, H.; Wang, s.; Geng, W.; Liu, Z.; Li, S. The dynamic changes of main cell types in the microenvironment of sciatic nerves following sciatic nerve injury and the influence of let-7 on their distribution. RSC Adv. 2018, 8, 41181–41191. [Google Scholar] [CrossRef]
- Sulaiman, O.A.R.; Gordon, T. Effects of short- and long-term Schwann cell denervation on peripheral nerve regeneration, myelination, and size. Glia 2000, 32, 234–246. [Google Scholar] [CrossRef]
- Gordon, T. The biology, limits, and promotion of peripheral nerve regeneration in rats and human. Nerves and Nerve Injuries. Elsevier, New York, 2015, pp. 903–1019.
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmain, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 controls the long-term survival and phenotype of repair Schwann cells during nerve regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Dong, Z.; Bunting, H.; Whitfield, J.; Meier, C.; Marie, H.; Mirsky, R.; Jessen, K.R. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci. 2001, 21, 8572–8585. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Bhaskaran, A.; Droggiti, A.; Dickinson, S.; D-Antonio, M.; Mirsky, R.; Jessen, K.R. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol. 2004, 164, 385–394. [Google Scholar] [CrossRef]
- Parkinson, D.B.; Bhaskaran, A.; Arthur-Farraj, P.; Noon, L.A. c-Jun is a negative regulator of myelination. J. Cell Biol. 2008, 181, 625–637. [Google Scholar] [CrossRef]
- Lundborg, G. Nerve Injury and Repair. 2004, Churchill Livingstone, New York.
- Adhihetty, PJ.; O’Leary, M.F.N.; Chabi, B.; Wicks, K.L.; Hood, D.A. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 2007, 102, 1143–1151. [Google Scholar] [CrossRef]
- O′Leary, M.F.N.; Vainshtein, A.; Carter, H.N.; Zhang, Y.; Hood, D.A. Denervation-induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis-deficient animals. Am. J. Physiol. Physiol. 2012, 303, 447–454. [Google Scholar] [CrossRef]
- Yang, X.; Xue, P.; Chen, H.; Yuan, M.; Kang, Y.; Duscher, D.; Machens, H.-G.; Chen, Z. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics 2020, 10, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Warwick, D.; Srinivasan, H.; Solomon, L. Peripheral Nerve Disorder. In: Arnold, H., editor. Apley’s system of Orthopaedics and Fracture. 9th ed. 2010. pp. 269–United Kingdom.
- Holmes, W.; Young J., Z. Nerve regeneration after immediate and delayed suture. J. Anat. 1942, 77, 63–I06. [Google Scholar] [CrossRef]
- Fu, S.Y.; Gordon, T. Contributing factors to poor functional recovery after delayed nerve repair: prolonged denervation. J. Neurosci. 1995, 15, 3876–3885. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J. Neurobiol. 2001, 49, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain derived neurotrophic factor. Eur. J. Neurosci. 2002, 15, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.G.; Gordon, T. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 2003, 183, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.A.R.; Gordon, T. Transforming growth factor-β and forskolin attenuate the adverse effects of long-term Schwann cell denervation on peripheral nerve regeneration in vivo. Glia 2002, 37, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.A.R.; Midha, R.; Munro, C.A.; Matusyama, T.; Al-Majed, A.A.; Gordon, T. Chronic Schwann cell denervation and the presence of a sensory nerve reduce motor axonal denervation. Exp. Neurol. 2002, 176, 342–354. [Google Scholar] [CrossRef]
- Gordon, T.; Tyreman, N.; Raji, M.A. The basis for diminished functional recovery after delayed peripheral nerve repair. J. Neurosci. 2011, 31, 5325–5334. [Google Scholar] [CrossRef]
- Gordon, T.; Brushart, T.M.; Chan, M. Augmenting nerve regeneration with electrical stimulation. Neurol. Res. 2008, 30, 1012–1022. [Google Scholar] [CrossRef]
- Gordon, T. The role of neurotrophic factors in nerve regeneration. Invited review. Neurosurg. Focus. 2009, 26, E3. [Google Scholar] [CrossRef]
- Pfister, B.J.; Gordon, T.; Loverde, J.R.; Mackinnon, S.E.; Cullen, D.K. Biomedical engineering strategies for peripheral nerve repair: Surgical applications, state of the art, and future challenges. Crit. Rev. Biomed. Eng. 2011, 39, 5–48. [Google Scholar] [CrossRef]
- Gordon, T. The Biology, Limits, and Promotion of Peripheral Nerve Regeneration in Rats and Humans. In Nerves and Nerve Injuries. Eds: Tubbs RS, Rizk E, Shoja M, Loukas M, and Spinner RJ. Elsevier, Vol. 2 Ch 61, 2015, pp. 993–1019.
- Rafuse, V.R.; Gordon, T. Self-reinnervated cat medial gastrocnemius muscles. I. Comparisons of the capacity for regenerating nerves to form enlarged motor units after extensive peripheral nerve Injuries. J. Neurophysiol. 1996, 75, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.R.; Gordon, T. Self-reinnervated cat medial gastrocnemius muscles. II. Analysis of the mechanisms and significance of fiber type grouping in reinnervated muscles. J. Neurophysiol. 1996, 75, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.F.; Gordon, T.; Orozco, R. Proportional enlargement of motor units after partial denervation of cat triceps surae muscles. J. Neurophysiol. 1992, 68, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Borschel, G.H. The use of the rat as a model for studying peripheral nerve regeneration and sprouting after complete and partial nerve injuries. Invited review for a special issue on in vivo models of neural regeneration. Exp. Neurol. 2017, 287, 331–347. [Google Scholar] [CrossRef]
- Dyck, P.J.; Boes, C.J.; Mulder, D.; Millikan, C.; Windebank, A.J.; Dyck, J.B.; Espinosa, R. History of standard scoring, notation, and summation of neuromuscular signs. A current survey and recommendation. J. Peripher. Nerv. Syst. 2005, 10, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, E.K.; Faber, C.G.; van Nes, S.I.; Jacobs, B.C.; van Doorn, P.A.; van Doorn, P.A; van Koningsveld, R.; Cornblath, D.R.; van der Kooi, A.J.; Cats, E.A.; van den Berg, L.H.; Notermans, N.C.; van der Pol, W.L.; Hermans, M.C.E.; van der Beek, N.A.M.E.; Gorson, K.C.; Eurelings, M.; Jeroen Engelsman, J.; Hendrik Boot, H.; Meijer, R.J.; Lauria, G.; Tennant, A.; Merkies, I.A.J.; PeriNomS Study Group. Modifying the Medical Research Council grading system through Rasch analyses. Brain 2012, 135, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Rafuse, V.R.; Gordon, T. Size of myelinated nerve fibres is not increased by expansion of the peripheral field in cats. J. Physiol. 1998, 532, 835–849. [Google Scholar]
- Dedkov, E.L.; Kostrominova, T.Y.; Borisov, A.B.; Carlson, B.M. Reparative myogenesis in long-term denervated skeletal muscles of adult rats results in a reduction of the satellite cell population. Anat. Rec. 2001, 263, 139–154. [Google Scholar] [CrossRef]
- Schmalbruch, H.; Lewis, D.M. A comparison of the morphology of denervated with aneurally regenerated soleus muscle of rat. Muscle Res. Cell Motil. 1994, 15, 256–266. [Google Scholar] [CrossRef]
- Jejurikar, S.S.; Marcelo, C.L.; Kuzon, W.M., Jr. Skeletal muscle denervation increases satellite cell susceptibility to apoptosis. Plast. Reconstr. Surg. 2002, 110, 160–168. [Google Scholar] [CrossRef]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Le Grand, F.; Rudnicki, M.A. Skeletal muscle satellite cells and adult myogenesis. Curr. Opin. Cell Biol. 2007, 19, 628–633. [Google Scholar] [CrossRef]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS 2013, 280, 3991–4003. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Suroto, H.; Wardana, G.R.; Sugianto, J.A.; Aprilya, D.; Samijo, S. Time to surgery and myo-D expression in biceps muscle of adult brachial plexus injury: a preliminary study. BMC Res. Notes 2023, 16, 51. [Google Scholar] [CrossRef]
- Anzil, A.P.; Wernig, A. Muscle fibre loss and reinnervation after long-term denervation. J. Neurocytol. 1989, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Schmalbruch, H.; Lewis, D.M. Dynamics of nuclei of muscle fibers and connective tissue cells in normal and degenerated rat muscles. Muscle Nerve 2000, 23, 617–626. [Google Scholar] [CrossRef]
- Gutmann, E.; Zelena, J. Morphological changes in the denervated muscle. In The Denervated Muscle (edited by Gutmann, E.), 1962, pp. 57–Prague: Publishing House of the Czechoslovak Academy of Sciences.
- Gordon, T.; Tetzlaff, W. Regeneration associated genes decline in injured rat axotomized sciatic motoneurons. Eur. J. Neurosci. 2015, 42, 2783–2791. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; You, S.; Cassar, S.L.; Tetzlaff, W. Reduced expression of regeneration associated genes in chronically axotomized facial motoneurons. Exp. Neurol. 2015, 264, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hoke, A.; Gordon, T.; Zochodne, D.W.; Sulaiman, O.A.R. A decline in glial cell-line-derived neurotrophic factor expression is associated with impaired regeneration after long-term Schwann cell denervation. Exp. Neurol. 2002, 173, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, L.J.; Gomez-Sanchez, J.A.; Fazal, S.V.; Otto, G.W.; et al. Failures of nerve regeneration caused by aging or chronic denervation are rescued by restoring Schwann cell c-Jun. eLife 2021, 10e. [Google Scholar] [CrossRef]
- Hashimoto, M.; Ishii, K.; Nakamura, Y.; Watabe, K.; Kohsaka, S.; Akazawa, C. Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J. Neurochem. 2008, 24, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.R.; Yuk, D.; Alberta, J.A.; Zhu, Z.; Pawlitzky, I.; Chan, J.; McMahon, C.D.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding BHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Trakanant, S.; Nihara, J.; Kudo, T.; Seo, K.l.; Saeki, M.; Kurose, M.; Matsumaru, D.; Maeda, T.; Ohazama, A. Gli3 is a key factor in the Schwann cells from both intact and injured peripheral nerves. Neurosci. 2020, 432, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Shapiro, R.I.; Wang, S.; Chakraborty, A.; Gill, A.; Lepage, D.J.; Wen, D.; Rayhorn, R.; Horan, G.S.B.; Taylor, F.R.; Garber, E.A.; Galdes, A.; Engber, T.M. Long-acting forms of sonic hedgehog with improved pharmacokinetic and pharmacodynamic properties are efficacious in a nerve injury model. J Pharm Sci 2002, 91, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.W.; Angeloni, N.; Harrington, D.; Stupp, S.; Podlasek, C.A. Sonic hedgehog regulates brain-derived neurotrophic factor in normal and regenerating cavernous nerves. J. Sex. Med. 2013, 10, 730–737. [Google Scholar] [CrossRef]
- Martinez, J.A.; Kobayashi, M.; Krishnan, A.; Webber, C.; Christie, K.; Guo, GF.; Singh, V.; Zouchodne, D.W. Intrinsic facilitation of adult peripheral nerve regeneration by the sonic hedgehog morphogen. Exp. Neurol. 2015, 271, 493–505. [Google Scholar] [CrossRef]
- Ma, K.H.; Hung, H.A.; Srinivasan, R.; Xie, H.; Orkin, S.H.; Svaren, J. Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex J. Neurosci. 2015, 35, 8640–8652. [Google Scholar] [CrossRef]
- Boerboom, A.; Dion, V.; Chariot, A.; Franzen, R. Molecular mechanisms involved in Schwann cell plasticity. Frontiers Mol. Neurosci. 2017, 10, 38. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Schaefer, S.A.; Hall, R.A. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. PNAS 2013, 110, 9529–9534. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Campana, W.M.; Mizisin, A.P.; Mohiuddin, L.; O’Brien, J.S. Prosaposin: A myelinotrophic protein that promotes expression of myelin constituents and is secreted after nerve injury. Glia 1999, 26, 353–360. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, R.; Wang, Z.; Lin, G.; Lue, T.F.; Lin, C. Adipose tissue-derived stem cells secrete CXCL5 cytokine with neurotrophic effects on cavernous nerve regeneration. J. Sex. Med. 2011, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Eskin, A.; Chareyre, F.; Jessen, W.J.; Manent, J.; Niwa-Kawakita, M.; Chen, R.; White, C.M.; Jaffer, Z.M.; Nelson, S.F.; Rubenstein, A.E.; Giovannini, M. Therapeutic potential of HSP90 inhibition for neurofibromatosis type Clin. Cancer Res. 2013, 19, 3856–3870. [Google Scholar]
- Weiss, T.; Taschner-Mandl, S.; Bileck, A.; Slany, A.; Kromp, F.; Rifatbegovic, D.; Frech, C.; Windhager, R.; Kitzinger, H.; Tzou, C.-H.; Ambros, P.F.; Gerner, C.; Ambros, I.M. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 2016, 64, 2133–2153. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.H.; Hung, H.A.; Svaren, J. Epigenomic regulation of Schwann cell reprogramming in peripheral nerve injury. J. Neurosci. 2016, 36, 9135–9147. [Google Scholar] [CrossRef] [PubMed]
- Brosius Lutz, A.; Chung, W.S.; Sloan, S.A.; Carson, G.A.; et al. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. PNAS 2017, 114, E8072–E8080. [Google Scholar] [CrossRef]
- Wolter, P.; Schmitt, K.; Fackler, M.; Kremling, H.; Probst, L.; Hauser, S.; Gruss, O.J.; Gaubatz, S. GAS2L3, a target gene of the DREAM complex, is required for proper cytokinesis and genomic stability. J. Cell Sci. 2012, 125, 2393–2406. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; Smith, S.J.; Barres, B.A. A. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Calissano, P. The nerve-growth factor. Sci. Am. 1979, 240, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P. B.; Young, J. Z. Fibrin suture of peripheral nerves. Lancet 1940, 239, 126–128. [Google Scholar]
- Carlsen, R.C. Delayed induction of the cell body response and enhancement of regeneration following a condition/test lesion of frog peripheral nerve at 15 °C. Brain Res. 1983, 279, 9–18. [Google Scholar] [CrossRef]
- Kilmer, S.L.; Carlsen, R.C. Forskolin activation of adenylate cyclase in vivo stimulates nerve regeneration. Nature 1984, 307, 455–457. [Google Scholar] [CrossRef]
- Sjoberg, J.; Kanje, M.; Edstrom, A. Influence of non-neuronal cells on regeneration of the rat sciatic nerve. Brain Res. 1988, 453, 221–226. [Google Scholar] [CrossRef]
- Kanje, M.; Skottner, A.; Sjöberg, J.; Lundborg, G. Insulin-like growth factor I (IGF-I) stimulates regeneration of the rat sciatic nerve. Brain Res. 1989, 486, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Sjoberg, J.; Kanje, M. The initial period of peripheral nerve regeneration and the importance of the local environment for the conditioning lesion effect. Brain Res. 1990, 529, 79–84. [Google Scholar] [CrossRef]
- Holmquist, B.; Kanje, M.; Kerns, J.M.; Danielsen, N. A mathematical model for regeneration rate and initial delay following surgical repair of peripheral nerves. J. Neurosci. Meth 1993, 48, 27–33. [Google Scholar] [CrossRef]
- Wood, M.D.; Kemp, S.W.P; Weber, C.; Borschel, G.H.; Gordon, T. Outcome measures of peripheral nerve regeneration. Ann. Anat. 2011, 193, 321–333. [Google Scholar] [PubMed]
- Fouad, K.; Pearson, K. Restoring walking after spinal cord injury. Prog. Neurobiol. 2004, 73, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Vrbova, G.; Gordon, T.; Jones, R. Nerve-Muscle Interaction 1978, Chapman and Hall, Ltd, Ch 6, p 113.
- Tajdaran, K.; Gordon, T.; Wood, M.D.; Shoichet, M.S.; Borschel, G.H. A glial cell line-derived neurotrophic factor delivery system enhances nerve regeneration across acellular nerve allografts. Acta Biomat 2016, 29, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Hoffer, J.A.; Jhamandas, J.; Stein, R.B. Long-term effects of axotomy on neural activity during cat locomotion. J. Physiol. 1980, 303, 243–263. [Google Scholar] [CrossRef]
- Mendell, L.M.; Munson, J.B.; Scott, J.G. Alterations of synapses on axotomized motoneurons. J. Physiol. 1976, 255, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Titmus, M.; Faber, D. Axotomy-induced alterations in the electrophysiological characteristics of neurons. Prog. Neurobiol. 1990, 35, 1–51. [Google Scholar] [CrossRef]
- Moran, L.B.; Graeber, M.B. The facial nerve axotomy model. Brain Res. Rev. 2004, 44, 154. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J.; Bullinger, K.L.; Titus, H.E.; Nardelli, P.; Cope, T.C. Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Ann. NY Acad. Sci. 2010, 1198, 231–241. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Titus-Mitchel, H.E.; Bullinger, K.L.; Kraszpulski, M.; Nardelli, P.; Cope, T.C. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J. Neurophysiol. 2011, 106, 2450–2470. [Google Scholar] [CrossRef]
- Sumner, B.E. A quantitative analysis of boutons with different types of synapse in normal and injured hypoglossal nuclei. Exp. Neurol. 1975, 49, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, D. H. Qualitative and quantitative study of synaptic displacement in chromatolyzed spinal motoneurons of the cat. J. Comp. Neurol. 1978, 177, 635–664. [Google Scholar] [CrossRef]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef]
- Brannstrom, T.; Kellerth, J. O. Changes in synaptology of adult cat spinal alpha-motoneurons after axotomy. Exp. Brain Res. 1998, 118, 1–13. [Google Scholar]
- Lindå, H.; Shupliakov, O.; Ornung, G.; Ottersen, O. P.; Storm-Mathisen, J.; Risling, M.; Cullheim, S. Ultrastructural evidence for a preferential elimination of glutamate-immunoreactive synaptic terminals from spinal motoneurons after intramedullary axotomy. J. Comp. Neurol. 2000, 425, 10–23. [Google Scholar] [CrossRef]
- Oliveira, A. L. R.; Thams, S.; Lidman, O.; Piehl, F.; Hökfelt, T.; Kärre, K.; Lindå, H.; Cullheim, S.A. role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. USA 2004, 101, 17843–17848. [Google Scholar] [CrossRef]
- Rotterman, T.M.; Nardelli, P.; Cope, T.C.; Alvarez, F.J. Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. J. Neurosci. 2014, 34, 3475–3492. [Google Scholar] [CrossRef]
- Liu, C.; Ward, P.J.; English, A.W. The effects of exercise on synaptic stripping require androgen receptor signaling. PLoS One 2014, 9, e98633. [Google Scholar] [CrossRef]
- Brandt, J.; Evans, J.T.; Mildenhall, T.; Mulligan, A.; Konieczny, A.; Rose, S.J; English, A.W. Delaying the onset of treadmill exercise following peripheral nerve injury has different effects on axon regeneration and motoneuron synaptic plasticity. J. Neurophysiol. 2015, 113, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Aldskogius, H.; Liu, L.; Svensson, M. Glial responses to synaptic damage and plasticity. J. Neurosci. Res. 1999, 58, 33–41. [Google Scholar] [CrossRef]
- Krakowiak, J.; Liu, C.; Papudesu, C.; Ward, P.J.; Wilhelm, J.C.; English, A.W. Neuronal BDNF signalling is necessary for the effects of treadmill exercise on synaptic stripping of axotomized motoneurons. Neural Plast. 2015, 11, 392591. [Google Scholar]
- Brushart, T.M.; Seiler, W.A. Selective reinnervation of distal motor stumps by peripheral motor axons. Exp. Neurol. 1987, 97, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Motor axons preferentially reinnervate motor pathways. J. Neurosci. 1993, 13, 2730–2738. [Google Scholar] [CrossRef]
- Gutmann, E.; Gutmann, L.; Medawar, P.K.; Young, J. Z. The rate of regeneration. J. Exp. Biol. 1942, 19, 14–44. [Google Scholar] [CrossRef]
- Witzel, C.; Rohde, C.; Brushart, T.M. Pathway sampling by regenerating peripheral axons. J. Comp. Neurol. 2005, 485, 183–190. [Google Scholar] [CrossRef]
- Burke, R.E. Motor units: anatomy, physiology and functional organization. In Handbook of physiology. Sect. I. Vol. II. The Nervous System: Motor Control. Part I. Edited by V.B. Brooks. American Physiological Society, Washington, DC. 1981, pp. 345–422.
- Zhang, J.Y.; Luo, X.G.; Xian, C.J.; Liu, Z.H.; Zhou, X.F. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents, Eur. J. Neurosci. 2000, 12, 4171–4180. [Google Scholar]
- Griesbeck, O.; Parsadanian, A.S.; Sendtner, M.; Thoenen, H. Expression of neurotrophins in skeletal muscle: Quantitative comparison and significance for motoneuron survival and maintenance of function. J. Neurosci. Res. 1995, 42, 21–33. [Google Scholar] [CrossRef]
- Juckett, L.; Saffari, T.M.; Ormseth, B.; Senger, J.-L.; Moore, A.M. The effect of electrical stimulation on nerve regeneration following peripheral nerve injury. Biomolec 2022, 12, 1856. [Google Scholar] [CrossRef]
- Gordon, T.; Amirjani, N.; Edwards, D.C.; Chan, K. M. Brief post-surgical electrical stimulation accelerates axon regeneration and muscle reinnervation without affecting the functional measures in carpal tunnel syndrome patients. Exp. Neurol. 2010, 223, 192–202. [Google Scholar] [CrossRef]
- Wong, J.N.; Olson, J.L.; Morhart, M.J.; Chan, K.M. Electrical stimulation enhances sensory recovery: a randomized controlled trial. Ann. Neurol. 2015, 77, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Power, H.A.; Morhart, M.J.; Olson, J.L.; Chan, K.M. Postsurgical electrical stimulation enhances recovery following surgery for severe Cubital Tunnel Syndrome: A Double-blind randomized controlled trial. Neurosurg. 2020, 86, 769–777. [Google Scholar] [CrossRef] [PubMed]
- McLean, N.A.; Verge, V. Dynamic impact of brief electrical stimulation on the neural immune axis-polarization of macrophages toward a pro-repair phenotype in demyelinated peripheral nerve. Glia 2016, 64, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, T.; Li, C.; Xu, W.; Guan, Y.; Li, X.; Cheng, H.; Chen, S.; Yang, B.; Liu, Y.; Ren, Z.; Song, X.; Jia, Z.; Wang, Y.; Tang, J. Electrical stimulation accelerates Wallerian degeneration and promotes nerve regeneration after sciatic nerve injury. GLIA 2023, 71, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; McEwan, S.-K.; Kang, S.M.; Won, S.M.; Stephen, M.; Gamble, P.; Xie, Z.; Yan, Y.; Chen, Y.-Y.; Shin, J.; Birenbaum, N.; Chung, S.; Kim, S.B.; Khalifeh, J.; Harburg, D.V.; Bean, K.; Paskett, M.; Kim, J.; Zohny, Z.S.; Lee, S.M.; Zhang, R.; Luo, K.; Ji, B.; Banks, A.; Lee, H.M.; Huang, Y.; Ray, W.Z.; Rogers, J.A. Wireless bioresorbable electronic system enables sustained non-pharmacological neuroregenerative therapy. Nature Med. 2018, 24, 1830–1836. [Google Scholar] [CrossRef]
- Ju, C.; Park, E.; Kim, T.; Kim, T.; Kang, M.; Lee, K.; Park, S.-M. Effectiveness of electrical stimulation on nerve regeneration after crush injury: comparison between invasive and non-invasive stimulation. Plos One 2020, 15, e0233531. [Google Scholar] [CrossRef]
- Yan, X.; Ye, Z.; Huang, J.; He, F.; Xiao, W.; Hu, X.; Luo, Z. CaMKI-mediated CREB phosphorylation is involved in Ca2+-induced BDNF mRNA transcription and neurite outgrowth mediated by electrical stimulation. Plos One 2016, 11, e0162784. [Google Scholar] [CrossRef] [PubMed]
- Sayanagi, J.; Jesus, A.; Acevedo-Cintrog, B.S; Pan, D.; Schellhardt, L.; Snyder-Warwick, A.K.; Mackinnon, S.E.; Wood, M.D. Brief electrical stimulation accelerates axon regeneration and promotes recovery following nerve transection and repair in mice. J. Bone Joint Surg. Am. 2021, 103, e80. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Schellhardt, L.; Keane, G.C.; Hunter, D.A.; Moore, A.M.; Snyder-Warwick, A.K.; Mackinnon, S.E.; Wood, M.D. Short-duration, pulsatile, electrical stimulation therapy accelerates axon regeneration and recovery following tibial nerve injury and repair in rats. Plast. Reconstr. Surg. 2022, 149, 681e–690e. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.P.C.; Lanzinger, W.; Willits, R.K. Effect of intraoperative electrical stimulation on recovery after rat sciatic nerve isograft repair. Neurotrauma Rep 2020, 1, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ecanow, A.; Berglund, K.; Carrasco, D.; Isaacson, R.; English, A.W. Enhancing motor and sensory axon regeneration after peripheral nerve injury using bioluminescent optogenetics. Int. J. Mol. Sci. 2022, 23, 16084. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.J.; Sengelaub, D.R.; English, A.W. Enhancement of peripheral nerve regeneration due to treadmill training and electrical stimulation is dependent on androgen receptor signaling. Dev. Neurobiol. 2014, 74, 531–540. [Google Scholar] [CrossRef]
- Hosli, E.; Jurasin, K.; Ruhl, W.; Luthy, R.; Hosli, L. Colocalization of androgen, estrogen and cholinergic receptors on cultured astrocytes of rat central nervous system. Int. J. Dev. Neurosci. 2001, 19, 11–19. [Google Scholar] [CrossRef]
- Garcia-Ovejero, D.; Veiga, S.; Garcia-Segura, L.; Doncarlo, S.L. Glial expression of estrogen and androgen receptors after rat brain injury. J. Comp. Neurol. 2002, 450, 256–271. [Google Scholar] [CrossRef]
- English, A.W.; Wilhelm, J.C.; Ward, P.J. Exercise, neurotrophins, and axon regeneration in the PNS. Physiol. 2014, 29, 437–445. [Google Scholar] [CrossRef]
- 415 Gordon, T. Neurotrophic factor expression in denervated motor and sensory Schwann cells: Relevance to specificity of peripheral nerve regeneration. Exp. Neurol. 2014, 25, 499–108. [Google Scholar] [CrossRef]
- Franz, C.K.; Rutishauser, U.; Rafuse, V.F. Polysialylated neural cell adhesion molecule is necessary for selective targeting of regenerating motor neurons. J. Neurosci. 2005, 25, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Ying, Z.; Roy, R.R.; Molteni, R.; Edgerton, V.R. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J. Neurophysiol. 2002, 88, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Exercise induces BDNF and synapsin I to specific hippocampal subfields. J. Neurosci. Res. 2004, 76, 356–362. [Google Scholar] [CrossRef]
- Vaynman, S.; Gomez-Pinilla, F. License to run: exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehab Neural Res. 2005, 19, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, M.J.; Redmon, N.; Schwartz, G.; English, A.W. Treadmill training promotes axon regeneration in injured peripheral nerves. Exp. Neurol. 2008, 211, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Asensio-Pinilla, E.; Udina, E.; Jaramillo, J.; Navarro, X. Electrical stimulation combined with exercise increase axonal regeneration after peripheral nerve injury. Exp. Neurol. 2009, 219, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, J.C.; Xu, M.; Cucoranu, D.; Chmielewski, S.; Holmes, T.; Lau, S.L.; Bassell, G.J.; English, A.W. Cooperative roles of BDNF expression in neurons and Schwann cells are modulated by exercise to facilitate nerve regeneration. J. Neurosci. 2012, 32, 5002–5009. [Google Scholar] [CrossRef]
- Wood, K.; Wilhelm, J.C.; Sabatier, M.J.; Liu, K.; Gu, J.; English, A.W. Sex differences in the effectiveness of treadmill training in enhancing axon regeneration in injured peripheral nerves. Dev. Neurobiol. 2012, 72, 688–698. [Google Scholar] [CrossRef]
- English, A.W.; Mulligan, A.; Cucoranu, D.; Rodriguez, J.A.; Sabatier, M.J. Neurotrophin 4/5 is implicated in the enhancement of axon regeneration produced by treadmill training following peripheral nerve injury. Erup J. Neurosci. 2011 33, 2265–2271. [CrossRef]
- Gutmann, E.; Jakoubek, B. Effect of increased motor activity on regeneration of the peripheral nerve in young rats. Physiol. Bohemoslov. 1963, 12, 463–468. [Google Scholar]
- Herbison, G.J.; Jaweed, M.M.; Ditunno, J.F.; Scott, C.M. Effect of overwork during reinnervation of rat muscle. Exp. Neurol. 1973, 41, 1–14. [Google Scholar] [CrossRef]
- Van Meeteren, N.L.; Brakkee, J.H.; Hamers, F.P.; Helders, P.J.; Gispen, W.H. Exercise training improves functional recovery and motor nerve conduction velocity after sciatic nerve crush lesion in the rat. Arch. Phys. Med. Rehab 1997, 78, 70–77. [Google Scholar] [CrossRef]
- Van Meeteren, N.L.; Brakkee, J.H.; Helders, P.J.; Gispen, W.H. The effect of exercise training on functional recovery after sciatic nerve crush in the rat. J. Peripher. Nerv. Syst. 1998, 3, 277–282. [Google Scholar]
- Hutchinson, K.J.; Gomez-Pinilla, F.; Crowe, M.J.; Ying, Z.; Basso, D.M. Three exercise paradigms differentially improve sensory recovery after spinal cord contusion in rats. Brain 2004, 127, 1403–1414. [Google Scholar] [CrossRef]
- Molteni, R.; Zheng, J.Q.; Ying, Z.; Gomez-Pinilla, F.; Twiss, J.L. Voluntary exercise increases axonal regeneration from sensory neurons. Proc. Natl. Acad. Sci. USA 2004, 101, 8473–8478. [Google Scholar] [CrossRef]
- Teodori, R.M.; Betini, J.; de Oliveira, L.S.; Sobral, L.L.; Takeda, S.Y.; de Lima Montebelo, M.I. Swimming exercise in the acute or late phase after sciatic nerve crush accelerates nerve regeneration. Neural Plast. 2011, 783901. [Google Scholar] [CrossRef]
- Udina, E.; Puigdemasa, A.; Navarro, X. Passive and active exercise improve regeneration and muscle reinnervation after peripheral nerve injury in the rat. Muscle Nerve 2011, 43, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Ottem, E.N.; Poort, J.E.; Wang, H.; Jordan, C.L; Breedlove, S.M. Differential expression and regulation of brain-derived neurotrophic factor (BDNF) mRNA isoforms in androgen-sensitive motoneurons of the rat lumbar spinal cord. Mol. Cell Endocrinol. 2010, 328, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M. Preferential reinnervation of motor nerves by regenerating motor axons. J. Neurosci. 1988, 8, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Brushart, T.M.; Mesulam, M.M. Alteration in connections between muscle and anterior horn motoneurons after peripheral nerve repair. Science 1980, 208, 603–605. [Google Scholar] [CrossRef] [PubMed]
- English, A.W. Enhancing axon regeneration in peripheral nerves also increases functionally inappropriate reinnervation of targets. Exp. Neurol. 2005, 490, 421–441. [Google Scholar] [CrossRef]
- English, A.W.; Cucoranu, D.; Mulligan, M.; Sabatier, M. Treadmill training enhances axon regeneration in injured mouse peripheral nerves without increased loss of topographic specificity. J. Comp. Neurol. 2009, 517, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.K.; Hinkle, M.L.; Nicolini, J.; Rambo, L.N.; Rexwinkle, A.M.; Rose, S.J.; Sabatier, M.J.; Backus, D.; English, A.W. Misdirection of regenerating axons and functional recovery following sciatic nerve injury in rats. J. Comp. Neurol. 2011, 519, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, M.J.; To, B.N.; Nicolini, J.; English, A.W. Effect of axon misdirection on recovery of electromyographic activity and kinematics after peripheral nerve injury. Cells Tiss. Organs 2011, 193, 298–309. [Google Scholar] [CrossRef]
- Sabatier, M.J.; To, B.N.; Nicolini, J.; English, A.W. Effect of slope and sciatic nerve injury on ankle muscle recruitment and hindlimb kinematics during walking in the rat. J. Exp. Biol. 2011, 214, 1007–1016. [Google Scholar] [CrossRef]
- de Ruiter, G.C.W.; Malessy, M.J.A.; Alaid, A.O.; Spinner, R.J.; Englestad, J.K.; Sorenson, E.J.; Kaufman, K.R.; Dyck, P.J.; Windebank, A.J. Misdirection of regenerating motor axons after nerve injury and repair in the rat sciatic nerve model. Exp. Neurol. 2008, 211, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Senger, J.L.B.; Verge, W.M.K.; Olson, J.L.; Chan, K.M.; Webber, C.A. Electrical stimulation as a conditioning strategy for promoting and accelerating peripheral nerve regeneration. Exp. Neurol. 2017, 302, 75–84. [Google Scholar] [CrossRef]
- Senger, J.L.B.; Verge, W.M.K.; Chan, K.M.; Webber, C.A. The nerve conditioning lesion – a strategy to enhance nerve regeneration. Annals Neurol. 2018, 83, 691–702. [Google Scholar] [CrossRef]
- Senger, J.-L.; Chan, K.M.; Macandili, H.; Chan, A.W.M.; Verge, V.M.K.; Jones, K.E.; Webber, C.A. Conditioning electrical stimulation promotes functional nerve regeneration. Exp. Neurol. 2019, 315, 60–71. [Google Scholar] [CrossRef]
- Senger, J.B.; Chan, A.W.M.; Chan, K.M.; Kwon-Wong, T.; Acton, L.; Olson, J.; Webber, C.A. Conditioning electrical stimulation is superior to postoperative electrical stimulation in enhanced nerve regeneration and functional recovery following nerve graft repair. Neurorehabil Neural Repair. 2020, 34, 299–308. [Google Scholar] [CrossRef]
- Senger, J.B.; Chan, K.M.; Webber, C.A. Conditioning electrical stimulation is superior to postoperative electrical stimulation, resulting in enhanced nerve regeneration and functional recovery. Exp. Neurol. 2020, 325, 113147. [Google Scholar] [CrossRef]
- Senger, J.B.; Rabey, K.N.; Morhart, M.J.; Chan, K.M.; Webber, C.A. Conditioning electrical stimulation accelerates regeneration in nerve transfers. Ann. Neurol. 2020, 88, 363–374. [Google Scholar] [CrossRef]
- Senger, J.-L.B.; Rabey, K.N.; Acton, L.; Lin, Y.-H.; Lingrell, S.; Chan, K.M.; Webber, C.A. Recovering the regenerative potential in chronically injured nerves by using conditioning electrical stimulation. J. Neurosurg. 2021, 136, 1442–1454. [Google Scholar] [CrossRef]
- Richardson, P.M.; Verge, V.M. Axonal regeneration in dorsal spinal roots is accelerated by peripheral axonal transection. Brain Res. 1987, 411, 406–408. [Google Scholar] [CrossRef]
- Arntz, C.; Kanje, M.; Lundborg, G. Regeneration of the rat sciatic nerve after different conditioning lesions: effects of the conditioning interval. Microsurg. 1989, 10, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.M.; McQuarrie, I.G. Acceleration of axonal outgrowth in rat sciatic nerve at one week after axotomy. J. Neurobiol. 1993, 24, 356–367. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I.G.; Grafstein, B. Axon outgrowth enhanced by a previous nerve injury. Arch. Neurol. 1973, 29, 53–55. [Google Scholar] [CrossRef] [PubMed]
- McQuarrie, I.G. Structural protein transport in elongation of motor axons after sciatic nerve crush: Effect of a conditioning lesion. Neurochem. Pathol. 1986, 5, 5,153–164. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.E.; McQuarrie, I.G. Increased slow transport in axons of regenerating Newt limbs after a nerve conditioning lesion. Dev. Biol. 1990, 140, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, K.; Hashimoto, K.; Lundborg, G. A role of migratory Schwann cells in a conditioning effect of peripheral nerve regeneration Exp. Neurol. 1999, 160, 99–108. [Google Scholar]
- Hoffman, P.N. A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp. Neurol. 2010, 223, 11–18. [Google Scholar] [CrossRef]
- Neumann, S.; Bradke, F.; Tessier-Lavigne, M.; Basbaum, A.I. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 2002, 34, 885–893. [Google Scholar] [CrossRef]
- Udina, E.; Furey, M.; Busch, S.; Silver, J.; Gordon, T.; Fouad, K. Electrical stimulation of intact peripheral sensory axons in rats promotes outgrowth of their central projections. Exp. Neurol. 2008, 210, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, C.; Beal, D.; Leechavengvongs, S.; Salon, A.; Dauge, M.C.; Sarcy, J.J. Nerve transfer to biceps muscle using a part of ulnar nerve for C5-C6 avulsion of the brachial plexus: anatomical study and report of four cases. J. Hand Surg. Am. 1994, 19, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Teboul, F.; Kakkar, R.; Ameur, N.; Beaulieu, J.-Y.; Oberlin, C. Transfer of fascicles from the ulnar nerve to the nerve to the biceps in the treatment of upper brachial plexus palsy. J. Bone J. Surg. Am. 2004, 86, 1485–1590. [Google Scholar] [CrossRef] [PubMed]
- Midha, R. Nerve transfers for severe brachial plexus injuries: a review. Neurosurg. Focus. 2004, 16, 1–10. [Google Scholar] [CrossRef]
- Midha, R. Techniques and options in nerve reconstruction and repair. in Youmans Neurological Surgery, 6th Ed, 2011, Ch 239, 2456-3464.
- Mackinnon, S.E. Donor distal, recipient proximal and other personal perspectives on nerve transfers. Hand Clin. 2016, 32, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.M.; Wagner, I.J.; Fox, I.K. Principles of nerve repair in complex wounds of the upper extremity. Semin. Plastic Surg. 2015, 29, 40–47. [Google Scholar] [CrossRef]
- Peters, B.R.; Van Handel, A.C.; Russo, S.A.; Moore, A.M. five reliable nerve transfers for the treatment of isolated upper extremity nerve injuries. Plast. Reconstr. Surg. 2021, 147, 830e. [Google Scholar] [CrossRef]
- Viterbo, F.; Trindade, J.C.; Hoshino, K.; Mazzoni, A. Two end-to-side neurorrhaphies and nerve graft with removal of the epineural sheath: experimental study in rats. Br. J. Plast. Surg. 1994, 47, 75–80. [Google Scholar] [CrossRef]
- Viterbo, F.; Amr, A.H.; Stipp, E.J.; Reis, F.J. End-to-side neurorrhaphy: past, present, and future. Plast. Reconstr. Surg. 2009, 124, e351–e358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fischer, K.A. End-to-side neurorrhaphy. Microsurgery 2002, 22, 122–127, 109. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, Y.; Abe, H. Hypoglossal-facial nerve side-to-end anastomosis for preservation of hypoglossal function: results of delayed treatment with a new technique. J. Neurosurg. 1997, 86, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Soucacos, P.N.; Bo, J.; Beris, A.E. Evaluation of collateral sprouting after end-to-side nerve coaptation using a fluorescent double-labeling technique. Microsurg. 1999, 19, 281–286. [Google Scholar] [CrossRef]
- Placheta, E.; Wood, M.D.; Lafontaine, C.; Liu, E.H.; Hendry, J.M.; Angelov, D.N.; Frey, M.; Gordon, T.; Borschel, G.H. Enhancement of facial nerve motoneuron regeneration through cross-face nerve grafts by adding end-to-side sensory axons. Plast. Reconstr. Surg. 2015, 135, 460–71. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, O.; Gordon, T. A rat study of the use of an end-to-side repair as a ‘baby-sitting’ technique to reduce the deleterious effect of chronic denervation. J. Neurosurg. 2018, 131, 6221–632. [Google Scholar]
- Placheta, E.; Wood, M.D.; Lafontaine, C.; Frey, M.; Gordon, T.; Borschel, G.H. Macroscopic in vivo imaging of facial nerve regeneration in Thy1-GFP rats. JAMA Facial Plast. Surg. 2015, 17, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Placheta, E.; Borschel, G.H. Delayed peripheral nerve repair: methods, including surgical ‘cross-bridging’, to promote nerve regeneration. Neural Regen. Res. 2015, 10, 1540–1544. [Google Scholar] [CrossRef]
- Yuksel, F.; Karacaoglu, E.; Guler, M.M. Nerve regeneration through side-to-side neurorrhaphy sites in a rat model: a new concept in peripheral nerve surgery. Plast. Reconstr. Surg. 1999, 104, 2092–2099. [Google Scholar] [CrossRef]
- Ladak, A.l.; Schembri, P.; Olson, J.; Udina, E.; Tyreman, N.; Gordon, T. Side-to-side nerve grafts sustain chronically denervated peripheral nerve pathways during axon regeneration and result in improved functional reinnervation. Neurosurg. 2011, 68, 1654–65. [Google Scholar] [CrossRef]
- Gordon, T.; Hendry, M.; Lafontaine, C.A.; Cartar, H.; Zhang, J.J.; Borschel, G.H. Nerve cross-bridging to enhance nerve regeneration in a rat model of delayed nerve repair. PLoS One 2015, 10, e0127397. [Google Scholar] [CrossRef]
- Hendry, J.M.; Alvarez-Veronesi, C.; Snyder-Warwick, A.; Gordon, T.; Borschel, G.H. Side-to-side nerve bridges support donor axon regeneration Into chronically denervated nerves and are associated with characteristic changes in Schwann cell phenotype. Neurosurg. 2015, 77, 803–813. [Google Scholar] [CrossRef]
- Pearse, D.D.; Pereira, F.C.; Marcillo, A.E.; Bates, M.L.; Berrocal, Y.A.; Filbin, M.T; Bunge, M.B. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004, 10, 610–616. [Google Scholar] [CrossRef]
- Krause, W.; Kulne, G. Pharmacokinetics of rolipram in rhesus and cynomolgus monkeys, the rat and the rabbit. Studies on species differences. Xenobiotica 1988, 18, 561–571. [Google Scholar] [CrossRef]
- Kilmer, S.L.; Carlsen, R.C. Forskolin activation of adenylate cyclase in vivo stimulates nerve regeneration. Nature 1984, 307, 455–457. [Google Scholar] [CrossRef]
- Cai, D.; Cao, Z.; McAtee, M.; Bregman, B.S.; Filbin, M.T. Neuronal cyclic AMP controls the developmental loss of ability of axons to regenerate. J. Neurosci. 2001, 21, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Hannila, S.S.; Filbin, M.T. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp. Neurol. 2008, 209, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Cai, D.; Dai, H.; McAtee, M.; Hoffman, P.N.; Bregman, B.S.; Filbin, M.T. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 2002, 34, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nikulina, E.; Mellado, W.; Filbin, M.T. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J. Neurosci. 2003, 23, 11770–11777. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Woolf, C.J. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 1999, 23, 83–91. [Google Scholar] [CrossRef]
- Oudega, M.; Varon, S.; Hagg, T. Distribution of corticospinal motor neurons in the postnatal rat: quantitative evidence for massive collateral elimination and modest cell death. J. Comp. Neurol. 1994, 347, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, Y.; Bo, X.; Huang, W.; Fang, X.; Zhang, X.; Miao, T.; Subang, M.C.; Richardson, P.M. Actions of neuropoietic cytokines and cyclic AMP in regenerative conditioning of rat primary sensory neurons. Exp. Neurol. 2007, 204, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Han, P.J.; Shukla, S.; Subramanian, P.S.; Hoffman, P.N. Cyclic AMP elevates tubulin expression without increasing intrinsic axon growth capacity. Exp. Neurol. 2004, 189, 293–302. [Google Scholar] [CrossRef]
- Dusart, I.; Schwab, M.E. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur. J. Neurosci. 1994, 6, 712–724. [Google Scholar] [CrossRef]
- Popovich, P.G.; Longbrake, E.E. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 2008, 9, 481–493. [Google Scholar] [CrossRef]
- Kurimoto, T.; Yin, Y.; Habboub, G.; Gilbert, H.Y.; Li, Y.; Nakao, S.; Hafezi-Moghadam, A.; Benowitz, L.I. Neutrophils express oncomodulin and promote optic nerve regeneration. J. Neurosci. 2013, 33, 14816–14824. [Google Scholar] [CrossRef]
- Yin, Y.; Henzl, M.T.; Lorber, B.; Nakazawa, T.; Thomas, T.T.; Jiang, F.; Langer, R.; Benowitz, L.I. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat. Neurosci. 2006, 9, 843–852. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs. Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs. Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 1.
Diagrammatic illustrations of (A) the downregulation of transmitter associated enzymes and the upregulation of regeneration associated genes in axotomized motoneurons and the downregulation of myelin associated genes and other molecules of the intact nerve after denervation of the distal stump. The denervated Schwann cells elongate and support the regeneration of axons after they sprout from the proximal stump of the transected nerve. (B) Diagrammatic illustration of axonal regeneration over long distances in human limbs Axons that regenerate at 1 mm/day will not reach denervated target muscles and sense organs for at least 500 days. Even median and ulnar nerve injuries at the level of the wrist will require months to reach the denervated targets.
Figure 2.
Survival of chronically denervated Schwann cells (SCs) and their capacity to proliferate, myelinate neurites and nerve fibres in vitro and in vivo, respectively, and to support recovery of reinnervated nerve fibre size. (A) Sterile surgery to prolong SC denervation in the rat sciatic distal nerve stump by cutting the nerve and ligating the proximal and distal nerve stumps to innervated muscles, (B) The left sciatic distal nerve stump remained denervated for either 7 days, 7 weeks, or 17 months and the right sciatic nerve was denervated for 7 days for direct comparison of the data of acutely denervated nerves with the data from chronically denervated nerves. Histograms of (C) the numbers of non-neural cells in the sciatic distal nerve stump, their numbers after exposure to the mitogen vitamin C, and the percentage of the dorsal root ganglion neurons myelinated by SCs in cocultures. (D) Myelination of tibial nerves that regenerated into 1-3 month chronically denervated common peroneal distal nerve stumps in vivo.
Figure 2.
Survival of chronically denervated Schwann cells (SCs) and their capacity to proliferate, myelinate neurites and nerve fibres in vitro and in vivo, respectively, and to support recovery of reinnervated nerve fibre size. (A) Sterile surgery to prolong SC denervation in the rat sciatic distal nerve stump by cutting the nerve and ligating the proximal and distal nerve stumps to innervated muscles, (B) The left sciatic distal nerve stump remained denervated for either 7 days, 7 weeks, or 17 months and the right sciatic nerve was denervated for 7 days for direct comparison of the data of acutely denervated nerves with the data from chronically denervated nerves. Histograms of (C) the numbers of non-neural cells in the sciatic distal nerve stump, their numbers after exposure to the mitogen vitamin C, and the percentage of the dorsal root ganglion neurons myelinated by SCs in cocultures. (D) Myelination of tibial nerves that regenerated into 1-3 month chronically denervated common peroneal distal nerve stumps in vivo.
Figure 3.
Physiological and morphological measures of regenerative success after nerve injuries. Diagrammatic illustration of surgeries in Sprague Dawley rats for (A) prolonging TIB (tibial) motoneuron axotomy and (B) CP (common peroneal) distal nerve stump and TA (tibialis anterior) muscle denervation. Each of the nerve stumps were sutured to innervated muscle to prevent nerve regeneration for up to 12 months prior to refreshing the proximal TIB nerve stump to cross-suture it to the distal nerve stump of a freshly cut CP nerve. (C) Recordings made at least 5 months later of muscle and single MUs (motor units) in response to stimulation of isolated ventral root filaments and (D) retrograde labeling of TIB motoneurons that regenerated their nerve fibres into the CP distal nerve stump. (E) Examples of sagittal sections of the lumbosacral spinal cord in which TIB motoneurons were retrogradely labelled with fluorescent dyes, FG (fluorogold) and FR (fluororuby), and (F) the number of the motoneurons retrogradely labelled with FG or FR were equal such that either dye could be used for counting motoneurons as well as sensory neurons that regenerate their axons after experimental nerve injuries.
Figure 3.
Physiological and morphological measures of regenerative success after nerve injuries. Diagrammatic illustration of surgeries in Sprague Dawley rats for (A) prolonging TIB (tibial) motoneuron axotomy and (B) CP (common peroneal) distal nerve stump and TA (tibialis anterior) muscle denervation. Each of the nerve stumps were sutured to innervated muscle to prevent nerve regeneration for up to 12 months prior to refreshing the proximal TIB nerve stump to cross-suture it to the distal nerve stump of a freshly cut CP nerve. (C) Recordings made at least 5 months later of muscle and single MUs (motor units) in response to stimulation of isolated ventral root filaments and (D) retrograde labeling of TIB motoneurons that regenerated their nerve fibres into the CP distal nerve stump. (E) Examples of sagittal sections of the lumbosacral spinal cord in which TIB motoneurons were retrogradely labelled with fluorescent dyes, FG (fluorogold) and FR (fluororuby), and (F) the number of the motoneurons retrogradely labelled with FG or FR were equal such that either dye could be used for counting motoneurons as well as sensory neurons that regenerate their axons after experimental nerve injuries.
Figure 4.
Decline in regenerative capacity of rat lumbosacral motoneurons (MNs) after chronic axotomy and prolonged Schwann cell denervation. (A,B) Plots of the number of reinnervated MUs (motor units) and TIB (tibial) motoneurons that regenerated their axons into the CP (common peroneal) distal nerve stump as a function of days of (A) chronic axotomy and (B) chronic denervation after delayed cross-suture of (A) axotomized proximal TIB nerve to freshly denervated CP distal nerve stump or (B) freshly axotomized TIB nerve to chronically denervated CP stump. Both MU and MN numbers declined exponentially as a function of days after chronic axotomy or chronic denervation. The effects are shown figuratively in (C,D) respectively for regeneration 4 months after TIB-CP cross suture. The methods used to determine regenerative success are shown in Figure 1.
Figure 4.
Decline in regenerative capacity of rat lumbosacral motoneurons (MNs) after chronic axotomy and prolonged Schwann cell denervation. (A,B) Plots of the number of reinnervated MUs (motor units) and TIB (tibial) motoneurons that regenerated their axons into the CP (common peroneal) distal nerve stump as a function of days of (A) chronic axotomy and (B) chronic denervation after delayed cross-suture of (A) axotomized proximal TIB nerve to freshly denervated CP distal nerve stump or (B) freshly axotomized TIB nerve to chronically denervated CP stump. Both MU and MN numbers declined exponentially as a function of days after chronic axotomy or chronic denervation. The effects are shown figuratively in (C,D) respectively for regeneration 4 months after TIB-CP cross suture. The methods used to determine regenerative success are shown in Figure 1.
Figure 5.
Expression of mRNA of cytoskeletal proteins in motoneurons decays with time after axotomy. (A) Sciatic nerve section and ligation of the proximal nerve stump to nearby innervated muscle to prevent nerve regeneration. (B,C) Time-dependent decay in the in-situ hybridization signals of tubulin mRNA in the axotomized motoneurons. (D) Decay in expression of tubulin with time and the accelerated decay after elevation of expression after a 1 to 6-month refreshment injury. Histograms of expression of (E) tubulin, (F) actin, (G) GAP-43 with time after axotomy and (H-J) months after axotomy and a refreshment injury. Mean values + standard error bars are shown with significance at p<0.05 and p<0.01 denoted by 1 and 2 *s.
Figure 5.
Expression of mRNA of cytoskeletal proteins in motoneurons decays with time after axotomy. (A) Sciatic nerve section and ligation of the proximal nerve stump to nearby innervated muscle to prevent nerve regeneration. (B,C) Time-dependent decay in the in-situ hybridization signals of tubulin mRNA in the axotomized motoneurons. (D) Decay in expression of tubulin with time and the accelerated decay after elevation of expression after a 1 to 6-month refreshment injury. Histograms of expression of (E) tubulin, (F) actin, (G) GAP-43 with time after axotomy and (H-J) months after axotomy and a refreshment injury. Mean values + standard error bars are shown with significance at p<0.05 and p<0.01 denoted by 1 and 2 *s.
Figure 6.
Expression of mouse Schwann cell (SC) transcription factor c-Jun and the neurotrophic factor receptor (p75NTR) with time after chronic denervation. (A) Representative Western blots of c-Jun and the standard, Calnexin. (B) The quantification of the c-Jun protein data with densitometry normalized to the data at 1-week post-injury in uninjured (UI) nerve and distal nerve stumps after 1-,3-,6-,8-, and 10-weeks of chronic denervation. C. Western blots of c-Jun expression 1 week after injury in wild-type (WT) and c-Jun knockout (cKO) mice, and (D) the quantification of the data with densitometry. (E) Densitometry data of p75NTR with time after injury. (F) Representative Western blots of c-Jun and p75NTR in SC cultures after two and nine passages and. the quantification of the data with densitometry of (G) c-Jun and (H) p75NTR. Mean values + one standard error bar are shown with significance at p<0.05, <0.01 and <0.001 denoted by 1,2, and 3 *s Adapted from [340].
Figure 6.
Expression of mouse Schwann cell (SC) transcription factor c-Jun and the neurotrophic factor receptor (p75NTR) with time after chronic denervation. (A) Representative Western blots of c-Jun and the standard, Calnexin. (B) The quantification of the c-Jun protein data with densitometry normalized to the data at 1-week post-injury in uninjured (UI) nerve and distal nerve stumps after 1-,3-,6-,8-, and 10-weeks of chronic denervation. C. Western blots of c-Jun expression 1 week after injury in wild-type (WT) and c-Jun knockout (cKO) mice, and (D) the quantification of the data with densitometry. (E) Densitometry data of p75NTR with time after injury. (F) Representative Western blots of c-Jun and p75NTR in SC cultures after two and nine passages and. the quantification of the data with densitometry of (G) c-Jun and (H) p75NTR. Mean values + one standard error bar are shown with significance at p<0.05, <0.01 and <0.001 denoted by 1,2, and 3 *s Adapted from [340].
Figure 7.
The neurotrophic factor receptor (p75NTR) mRNA and protein in rat Schwann cells (SCs) and the expression of glial cell neurotrophic factor (GDNF) in the denervated distal nerve stump as a function of time after chronic denervation (A) Rat sciatic nerve section and ligation of the proximal and distal nerve stumps to nearby innervated muscle to prevent nerve regeneration. (B) p75NTR mRNA, depicted as dark grains following in situ hybridization histochemistry in intact sciatic nerve and transected distal nerve stumps 7 days and 4 months after chronic denervation, with a plot of the p75NTR mRNA as a function of the duration of chronic denervation. (C) The relative density of p75NTR immunohistochemistry [shown in with (D) the DAB reaction in short-term (1 week) and long-term (>1 month) denervated distal nerve stumps] plotted as a function of time. (E) Changes in mRNA levels of GDNF, Neurturin (NTN), Persiphin (PSP), and Artemin (ART) mRNAs, as measured by semiquantitative PCR with GAPDH as an internal control and expressed as the change in the transected distal nerve stumps vs the sham-operated control sciatic nerves, as a function of chronic denervation. The data are plotted in (F) as histograms and in (G) as a plot of the mean values of the GDNF mRNA as a function of time of chronic distal nerve denervation. Mean values + one standard error bar are shown with significance at p<0.005 denoted by a *.
Figure 7.
The neurotrophic factor receptor (p75NTR) mRNA and protein in rat Schwann cells (SCs) and the expression of glial cell neurotrophic factor (GDNF) in the denervated distal nerve stump as a function of time after chronic denervation (A) Rat sciatic nerve section and ligation of the proximal and distal nerve stumps to nearby innervated muscle to prevent nerve regeneration. (B) p75NTR mRNA, depicted as dark grains following in situ hybridization histochemistry in intact sciatic nerve and transected distal nerve stumps 7 days and 4 months after chronic denervation, with a plot of the p75NTR mRNA as a function of the duration of chronic denervation. (C) The relative density of p75NTR immunohistochemistry [shown in with (D) the DAB reaction in short-term (1 week) and long-term (>1 month) denervated distal nerve stumps] plotted as a function of time. (E) Changes in mRNA levels of GDNF, Neurturin (NTN), Persiphin (PSP), and Artemin (ART) mRNAs, as measured by semiquantitative PCR with GAPDH as an internal control and expressed as the change in the transected distal nerve stumps vs the sham-operated control sciatic nerves, as a function of chronic denervation. The data are plotted in (F) as histograms and in (G) as a plot of the mean values of the GDNF mRNA as a function of time of chronic distal nerve denervation. Mean values + one standard error bar are shown with significance at p<0.005 denoted by a *.
Figure 8.
Exogenous delivery of brain- and glial cell-derived neurotrophic factors, GDNF and BDNF respectively, was effective in improving nerve regeneration after chronic axotomy but, increased axon sprouting in the distal nerve stump. (A) The TIB (tibial nerve) in the rat was cut and ligated. (B) After 2 months, the proximal stump of the TIB nerve was cross-sutured to the freshly denervated CP (common peroneal) nerve stump and (C) a mini-osmotic pump was placed on the back of the rat for constant infusion of low dose BDNF (2ug/day for 28 days). (D) Following a 2-month period of chronic axotomy, fluororuby was administered to the CP distal nerve stump 20 mm from the cross-suture site for retrograde labeling of the TIB motoneurons that had regenerated their axons into the denervated CP distal nerve stump. (E) The numbers of motoneurons that regenerated their axons and were counted in 50 µm longitudinal sections of the ventral horn of the spinal cord [all sections were counted and a correction factor of 0.6 applied [313,314,315]. The number of motoneurons that regenerated their axons after 2 months of chronic axotomy was significantly reduced. GDNF and BDNF were both effective in increasing the motoneuron regeneration to normal levels and the combination elevated the levels even more. (F) Local delivery of GDNF to the suture sites of an acellular nerve allograft (ANA) interposed between the CP proximal and distal nerve stumps. GDNF was encapsulated with an efficiency of 78 + 3% and GDNF loading of 0.72 + 0.08 µg/mg of microspheres (MS; diameters of 45 + 5 µm), placed onto the suture sites and held in place by surrounding them with 2 µL gels placed below and above the suture sites that adhered to one another. (G) Retrograde labeling of the CP nerve 10 mm from the ASA 8 weeks later. (H) Numbers of motoneurons that regenerated their axons through the ANA plotted for control conditions of no delivery system (DDS) and empty MS and experimental conditions of GDNF delivery in combinations of 2- and 4-week formulations. (I) Numbers of sensory neurons counted in every fifth dorsal root ganglia 20 µm section. Irrespective of the nature of GDNF delivery, all motor and sensory neurons regenerated their axons through the ANA in 8 weeks as compared to ~50% of them that regenerated through the ANAs that did not deliver GDNF.(J) Electron micrographs of Schwann cells (SCs) and their axons in intact nerves, regenerating nerves in the distal nerve stump 2 months after TIB-CP cross-suture with saline administration via a cuff around the suture site linked to a mini-osmotic pump, GDNF delivery to the cross-suture site via the pump, and delivery of both GDNF and BDNF via to pump. (K) The mean + standard error values of the number of axons within each endoneurial sheath associated with a single SC in the control (A and B), C. GDNF and D. GDNF + BDNF.
Figure 8.
Exogenous delivery of brain- and glial cell-derived neurotrophic factors, GDNF and BDNF respectively, was effective in improving nerve regeneration after chronic axotomy but, increased axon sprouting in the distal nerve stump. (A) The TIB (tibial nerve) in the rat was cut and ligated. (B) After 2 months, the proximal stump of the TIB nerve was cross-sutured to the freshly denervated CP (common peroneal) nerve stump and (C) a mini-osmotic pump was placed on the back of the rat for constant infusion of low dose BDNF (2ug/day for 28 days). (D) Following a 2-month period of chronic axotomy, fluororuby was administered to the CP distal nerve stump 20 mm from the cross-suture site for retrograde labeling of the TIB motoneurons that had regenerated their axons into the denervated CP distal nerve stump. (E) The numbers of motoneurons that regenerated their axons and were counted in 50 µm longitudinal sections of the ventral horn of the spinal cord [all sections were counted and a correction factor of 0.6 applied [313,314,315]. The number of motoneurons that regenerated their axons after 2 months of chronic axotomy was significantly reduced. GDNF and BDNF were both effective in increasing the motoneuron regeneration to normal levels and the combination elevated the levels even more. (F) Local delivery of GDNF to the suture sites of an acellular nerve allograft (ANA) interposed between the CP proximal and distal nerve stumps. GDNF was encapsulated with an efficiency of 78 + 3% and GDNF loading of 0.72 + 0.08 µg/mg of microspheres (MS; diameters of 45 + 5 µm), placed onto the suture sites and held in place by surrounding them with 2 µL gels placed below and above the suture sites that adhered to one another. (G) Retrograde labeling of the CP nerve 10 mm from the ASA 8 weeks later. (H) Numbers of motoneurons that regenerated their axons through the ANA plotted for control conditions of no delivery system (DDS) and empty MS and experimental conditions of GDNF delivery in combinations of 2- and 4-week formulations. (I) Numbers of sensory neurons counted in every fifth dorsal root ganglia 20 µm section. Irrespective of the nature of GDNF delivery, all motor and sensory neurons regenerated their axons through the ANA in 8 weeks as compared to ~50% of them that regenerated through the ANAs that did not deliver GDNF.(J) Electron micrographs of Schwann cells (SCs) and their axons in intact nerves, regenerating nerves in the distal nerve stump 2 months after TIB-CP cross-suture with saline administration via a cuff around the suture site linked to a mini-osmotic pump, GDNF delivery to the cross-suture site via the pump, and delivery of both GDNF and BDNF via to pump. (K) The mean + standard error values of the number of axons within each endoneurial sheath associated with a single SC in the control (A and B), C. GDNF and D. GDNF + BDNF.
Figure 9.
Gene expression of BDNF (brain derived neurotrophic factor) and its trkB receptor, and of the regeneration associated genes, tubulin and GAP-43, after axotomy. Transection and surgical repair of the rat femoral nerve and (A) sham or (B) 1 hour 20 Hz electrical stimulation (stim) of the proximal stump. (C) Dark-field micrographs of in situ hybridization (ISH) with 35S-labeled oligonucleotides complementary to tubulin after sham and Stim. Means + standard deviation of ISH signals/motoneuron of (D) BDNF, (E) trkB, (F) tubulin, (G) GAP-43, and (H) neurofilament in individual rats are plotted as a function of time after femoral nerve axotomy after Sham and Stim.
Figure 9.
Gene expression of BDNF (brain derived neurotrophic factor) and its trkB receptor, and of the regeneration associated genes, tubulin and GAP-43, after axotomy. Transection and surgical repair of the rat femoral nerve and (A) sham or (B) 1 hour 20 Hz electrical stimulation (stim) of the proximal stump. (C) Dark-field micrographs of in situ hybridization (ISH) with 35S-labeled oligonucleotides complementary to tubulin after sham and Stim. Means + standard deviation of ISH signals/motoneuron of (D) BDNF, (E) trkB, (F) tubulin, (G) GAP-43, and (H) neurofilament in individual rats are plotted as a function of time after femoral nerve axotomy after Sham and Stim.
Figure 10.
The decline in electrical activity in motor nerves after axotomy coincides with that of the expression of regeneration associated genes (RAGs) and loss of presynaptic contacts. Regeneration after nerve repair and/or 20 Hz electrical stimulation (ES) restores synaptic contacts. (A) Motor and (B) sensory cross-correlation peaks normalized to pre-operative values decline with an exponential time course over the first month of axotomy and begin to recover with nerve regeneration. (C)The decline in the relative amplitude of the motor potentials occurs with (D) the decline in expression of GDNF (glial cell-derived neurotrophic factor). (E) A l µm-thick confocal image of a retrogradely CTB-AF545-labeled motoneuron with immunohistochemical visualization of VGLUT1 (vesicular glutamate transporter 1)-containing excitatory synapses, arising mainly from primary afferent neurons, and GAD67 (glutamic acid decarboxylase 67)-containing inhibitory synapses, were used to measure the percentages of the neuronal perimeter in contact with the synapses. The silencing of motoneurons, namely the reduced motor activity, is due to the withdrawal of (F) glutamatergic and (G) GABAergic synaptic contacts from the neurons. The withdrawal is prevented by treadmill exercise either (H) continuous exercise in males or (I) ‘interval exercise’ in female mice.
Figure 10.
The decline in electrical activity in motor nerves after axotomy coincides with that of the expression of regeneration associated genes (RAGs) and loss of presynaptic contacts. Regeneration after nerve repair and/or 20 Hz electrical stimulation (ES) restores synaptic contacts. (A) Motor and (B) sensory cross-correlation peaks normalized to pre-operative values decline with an exponential time course over the first month of axotomy and begin to recover with nerve regeneration. (C)The decline in the relative amplitude of the motor potentials occurs with (D) the decline in expression of GDNF (glial cell-derived neurotrophic factor). (E) A l µm-thick confocal image of a retrogradely CTB-AF545-labeled motoneuron with immunohistochemical visualization of VGLUT1 (vesicular glutamate transporter 1)-containing excitatory synapses, arising mainly from primary afferent neurons, and GAD67 (glutamic acid decarboxylase 67)-containing inhibitory synapses, were used to measure the percentages of the neuronal perimeter in contact with the synapses. The silencing of motoneurons, namely the reduced motor activity, is due to the withdrawal of (F) glutamatergic and (G) GABAergic synaptic contacts from the neurons. The withdrawal is prevented by treadmill exercise either (H) continuous exercise in males or (I) ‘interval exercise’ in female mice.
Figure 11.
Motor nerve regeneration is staggered. (A) The femoral nerve in the rat was cut 20 mm from its branches to the quadriceps muscle and to the skin. It was repaired immediately with microsurgical technique. The regeneration of axons into the two branches was evaluated 2 to 10 weeks later by application of fluorescent dyes, RR (rubyred) for 1-2 hours to one branch and FG (fluorogold) to the other branch, 5 mm from the point of their division from the femoral nerve. (B) Motoneurons that were retrogradely labelled with either or both dyes are the sum of those that regenerated their axons into either branch as well as both the branches. The number of these motoneurons that regenerated their axons over the distance of 25 mm was ~55% of all the motoneurons in the femoral motoneuron pool, the numbers increasing with an exponential time course to the reach the 350 motoneurons of the control intact nerve (shown by the shaded horizontal lines that represent + standard errors of the mean values). The 10 weeks for all the motoneurons to regenerate the 25 mm distance to the site of dye application was surprisingly long when a latency of at least 2 days and the regeneration rate of 3 mm/day is considered. On the basis of the latency and regeneration rate, a 2-to-3-week period of regeneration would be expected for all the motoneurons to regenerate their axons. (C) The possible explanation that axon outgrowth across the surgical site is staggered with some axons even growing backwards into the proximal nerve stump, proved to be correct. The Schwann cells that multiply after nerve injury and line the distal nerve stump are known to migrate into the injury site and are shown in red. (D) To determine whether indeed, this explanation accounts for the observed slow regeneration of nerve fibres, those axons that had regenerated just across the suture line of the femoral nerve repair site, were retrogradely labelled by crushing the distal nerve stump 1.5 mm from the surgical site. (E) The microinjection of FR and (F) the counts of the retrograde labelled motoneurons that regenerated the axons, plotted as a function of days after femoral nerve repair surgery revealed the ~one month period of outgrowth of the femoral motor axons across the injury site with (G) staggering of axons regenerating within the distal nerve stump and across the site as (H) some regenerating axons grow into the proximal nerve stump in straight lines or or take a spiralling course. The axons in (G,H) were visualized with silver staining.
Figure 11.
Motor nerve regeneration is staggered. (A) The femoral nerve in the rat was cut 20 mm from its branches to the quadriceps muscle and to the skin. It was repaired immediately with microsurgical technique. The regeneration of axons into the two branches was evaluated 2 to 10 weeks later by application of fluorescent dyes, RR (rubyred) for 1-2 hours to one branch and FG (fluorogold) to the other branch, 5 mm from the point of their division from the femoral nerve. (B) Motoneurons that were retrogradely labelled with either or both dyes are the sum of those that regenerated their axons into either branch as well as both the branches. The number of these motoneurons that regenerated their axons over the distance of 25 mm was ~55% of all the motoneurons in the femoral motoneuron pool, the numbers increasing with an exponential time course to the reach the 350 motoneurons of the control intact nerve (shown by the shaded horizontal lines that represent + standard errors of the mean values). The 10 weeks for all the motoneurons to regenerate the 25 mm distance to the site of dye application was surprisingly long when a latency of at least 2 days and the regeneration rate of 3 mm/day is considered. On the basis of the latency and regeneration rate, a 2-to-3-week period of regeneration would be expected for all the motoneurons to regenerate their axons. (C) The possible explanation that axon outgrowth across the surgical site is staggered with some axons even growing backwards into the proximal nerve stump, proved to be correct. The Schwann cells that multiply after nerve injury and line the distal nerve stump are known to migrate into the injury site and are shown in red. (D) To determine whether indeed, this explanation accounts for the observed slow regeneration of nerve fibres, those axons that had regenerated just across the suture line of the femoral nerve repair site, were retrogradely labelled by crushing the distal nerve stump 1.5 mm from the surgical site. (E) The microinjection of FR and (F) the counts of the retrograde labelled motoneurons that regenerated the axons, plotted as a function of days after femoral nerve repair surgery revealed the ~one month period of outgrowth of the femoral motor axons across the injury site with (G) staggering of axons regenerating within the distal nerve stump and across the site as (H) some regenerating axons grow into the proximal nerve stump in straight lines or or take a spiralling course. The axons in (G,H) were visualized with silver staining.
Figure 12.
Low frequency electrical stimulation (ES) accelerates nerve outgrowth across the site of transection and surgical repair. Figurative illustrations of femoral motoneurons and their (A) projection to the motor branch to the quadriceps muscle and not into the sensory saphenous branch to the skin, (B) transection, surgical repair, and application of dyes fluororuby (FR: analogous to rubyred) and fluorogold to each branch to retrogradely label the motoneurons that regenerated their axons 25 mm into each femoral nerve branch (see also (H)), and (C) ES with a 20 Hz frequency, proximal to the site of repair. Histograms and plots of the mean + standard error (SE) of (D,E) the number of femoral motoneurons that regenerated their axons into motor and sensory branches and those that regenerated into both branches after sham ES and (F,G) ES. (I) FR was injected 1.5 mm distal to the suture site to count the motoneurons that regenerated across the site. (J,K) Comparison of the effects of sham and ES on the mean + SE of all the femoral motoneurons that regenerated their axons into the motor and sensory branches and those that regenerated across the suture site. Diagrammatic illustration of (L) the effect of ES in ‘straightening’ out the contorted progression of regenerating axons across the suture site and. (M) application of tetrodotoxin (TTX, 60 mg/ml) to the proximal stump of the transected and surgically repaired femoral nerve to block the action potentials elicited by ES, from progressing to the cell bodies of the motor and sensory neurons. After a three-week period of nerve regeneration, FG and FR were applied to count those motor and sensory neurons that had regenerated their axons 25 mm from the surgical and repair site. The striking increased regeneration after 3 weeks of (N) motor and (O) sensory nerves into the motor and sensory branches of the cut and repaired femoral motoneurons by the 1 hour 20 Hz ES was blocked by TTX.
Figure 12.
Low frequency electrical stimulation (ES) accelerates nerve outgrowth across the site of transection and surgical repair. Figurative illustrations of femoral motoneurons and their (A) projection to the motor branch to the quadriceps muscle and not into the sensory saphenous branch to the skin, (B) transection, surgical repair, and application of dyes fluororuby (FR: analogous to rubyred) and fluorogold to each branch to retrogradely label the motoneurons that regenerated their axons 25 mm into each femoral nerve branch (see also (H)), and (C) ES with a 20 Hz frequency, proximal to the site of repair. Histograms and plots of the mean + standard error (SE) of (D,E) the number of femoral motoneurons that regenerated their axons into motor and sensory branches and those that regenerated into both branches after sham ES and (F,G) ES. (I) FR was injected 1.5 mm distal to the suture site to count the motoneurons that regenerated across the site. (J,K) Comparison of the effects of sham and ES on the mean + SE of all the femoral motoneurons that regenerated their axons into the motor and sensory branches and those that regenerated across the suture site. Diagrammatic illustration of (L) the effect of ES in ‘straightening’ out the contorted progression of regenerating axons across the suture site and. (M) application of tetrodotoxin (TTX, 60 mg/ml) to the proximal stump of the transected and surgically repaired femoral nerve to block the action potentials elicited by ES, from progressing to the cell bodies of the motor and sensory neurons. After a three-week period of nerve regeneration, FG and FR were applied to count those motor and sensory neurons that had regenerated their axons 25 mm from the surgical and repair site. The striking increased regeneration after 3 weeks of (N) motor and (O) sensory nerves into the motor and sensory branches of the cut and repaired femoral motoneurons by the 1 hour 20 Hz ES was blocked by TTX.
Figure 13.
BDNF (brain derived neurotrophic factor is essential for the efficacy of ES (20 Hz electrical stimulation for one hour) in accelerating nerve regeneration. (A) The infusion of the BDNF antibody, α-BDNF, into the intrathecal space of the lumbar spinal cord 3-days prior to (B) ES via electrodes placed proximal to the surgical site of femoral nerve transection and microsurgical repair site and the application of the fluorescent dyes, fluororuby and fluorogold to each of the femoral motor and cutaneous sensory branches for retrograde labeling of those motoneurons that regenerated their axons into either or both the branches at 3 weeks after surgical repair. (C) The histogram of mean motoneuron numbers (+ standard errors) that were corrected as described previously [24], that regenerated their axons 25 mm into the appropriate and inappropriate motor and sensory nerve branches, respectively were plotted for the motoneurons subjected to ES (stim) and sham ES. ES accelerated the regeneration of motor nerves into the appropriate motor branch without affecting either the regeneration into the inappropriate motor branch or into both branches (see also Figure 12D,E).This accelerated motor nerve regeneration was blocked by infusion of the α-BDNF. The numbers of motoneurons that regenerated their axons inappropriately into the sensory branch of the femoral nerve or those that regenerated their axons into both branches were not changed by the infusion of α-BDNF.
Figure 13.
BDNF (brain derived neurotrophic factor is essential for the efficacy of ES (20 Hz electrical stimulation for one hour) in accelerating nerve regeneration. (A) The infusion of the BDNF antibody, α-BDNF, into the intrathecal space of the lumbar spinal cord 3-days prior to (B) ES via electrodes placed proximal to the surgical site of femoral nerve transection and microsurgical repair site and the application of the fluorescent dyes, fluororuby and fluorogold to each of the femoral motor and cutaneous sensory branches for retrograde labeling of those motoneurons that regenerated their axons into either or both the branches at 3 weeks after surgical repair. (C) The histogram of mean motoneuron numbers (+ standard errors) that were corrected as described previously [24], that regenerated their axons 25 mm into the appropriate and inappropriate motor and sensory nerve branches, respectively were plotted for the motoneurons subjected to ES (stim) and sham ES. ES accelerated the regeneration of motor nerves into the appropriate motor branch without affecting either the regeneration into the inappropriate motor branch or into both branches (see also Figure 12D,E).This accelerated motor nerve regeneration was blocked by infusion of the α-BDNF. The numbers of motoneurons that regenerated their axons inappropriately into the sensory branch of the femoral nerve or those that regenerated their axons into both branches were not changed by the infusion of α-BDNF.
Figure 14.
Preferential motor reinnervation of the motor branch of the femoral nerve is accounted for by differential expression of neurotrophic factors in the denervated distal nerve stump. Numbers of rat femoral motoneurons that regenerate their axons (A) across the site of transection (injury) and surgical repair and (B) 25 mm from the injury site into appropriate muscle (black bars), inappropriate sensory cutaneous branches (white bars), and both branches (grey bars). (C) The fold-increase in the mRNA of PTN (pleiotrophin) and GDNF (glial cell-derived neurotrophic factor) in the distal nerve stumps of transected and repaired femoral nerve relative to that of intact nerves as a function of days of denervation. (D) Diagrammatic representation of motoneurons regenerating their nerve fibres into appropriate and inappropriate muscle and cutaneous pathways, respectively as well as into both. Further detail is provided in the text.
Figure 14.
Preferential motor reinnervation of the motor branch of the femoral nerve is accounted for by differential expression of neurotrophic factors in the denervated distal nerve stump. Numbers of rat femoral motoneurons that regenerate their axons (A) across the site of transection (injury) and surgical repair and (B) 25 mm from the injury site into appropriate muscle (black bars), inappropriate sensory cutaneous branches (white bars), and both branches (grey bars). (C) The fold-increase in the mRNA of PTN (pleiotrophin) and GDNF (glial cell-derived neurotrophic factor) in the distal nerve stumps of transected and repaired femoral nerve relative to that of intact nerves as a function of days of denervation. (D) Diagrammatic representation of motoneurons regenerating their nerve fibres into appropriate and inappropriate muscle and cutaneous pathways, respectively as well as into both. Further detail is provided in the text.
Figure 15.
Brief 1 hour 20 Hz electrical stimulation (ES) promotes nerve regeneration and target reinnervation after delayed nerve repair. (A) Surgeries: The common peroneal nerve (CP), and/ or tibial (TIB) nerve were cut and cross-sutured either a. immediately or 2 months after b. prolonged CP axotomy and/or distal TIB nerve denervation (c, d) and the proximal nerve stump subjected to either sham or ES. The mean numbers (+ standard error SE) and the numbers from different experiments (shown as symbols) are plotted in histograms 2 and 4 weeks after cross-suture for (B) motoneurons and (C) sensory neurons that regenerated their axons 20 mm into the distal nerve stump. (D) Examples of the in-situ hybridization of α-tubulin mRNA in sciatic motoneurons whose axons were either intact (0 days), transected previously for 7 days (7d acute axotomy) or transected previously for 6 months (6m chronic axotomy), and transected previously for 6 months and then subjected to a refreshment injury thereafter for 7 days (6m+1w chronic axotomy and refreshment injury). Details are provided in the text.
Figure 15.
Brief 1 hour 20 Hz electrical stimulation (ES) promotes nerve regeneration and target reinnervation after delayed nerve repair. (A) Surgeries: The common peroneal nerve (CP), and/ or tibial (TIB) nerve were cut and cross-sutured either a. immediately or 2 months after b. prolonged CP axotomy and/or distal TIB nerve denervation (c, d) and the proximal nerve stump subjected to either sham or ES. The mean numbers (+ standard error SE) and the numbers from different experiments (shown as symbols) are plotted in histograms 2 and 4 weeks after cross-suture for (B) motoneurons and (C) sensory neurons that regenerated their axons 20 mm into the distal nerve stump. (D) Examples of the in-situ hybridization of α-tubulin mRNA in sciatic motoneurons whose axons were either intact (0 days), transected previously for 7 days (7d acute axotomy) or transected previously for 6 months (6m chronic axotomy), and transected previously for 6 months and then subjected to a refreshment injury thereafter for 7 days (6m+1w chronic axotomy and refreshment injury). Details are provided in the text.
Figure 16.
Electrical stimulation of the intact sciatic nerve of the rat at 20 Hz for 1 hour (ES) in vivo before excising dorsal root ganglion (DRG) sensory neurons and plating them on a laminin substrate in vitro, promotes their neurite outgrowth. An intact sciatic nerve 1 day after (A) no ES and (B) ES. An intact sciatic nerve 7 days after (C) no ES and (D) ES. (E) The mean + standard error of the lengths of the longest neurites (in µm) for the control and experimental DRG neurons that were not and were stimulated, respectively. The ES of intact sciatic nerves promoted neurite extension from plated DRG neurons. This promoted neurite extension is akin to the effect of a nerve crush conditioning lesion (CL) that is made 7 days prior to the transection and suture of the nerve proximal to the CL, that accelerates nerve regeneration. The neurons were immunostained with β-tubulin III. The calibration bar = 50 µm. *Statistical significance p<0.05.
Figure 16.
Electrical stimulation of the intact sciatic nerve of the rat at 20 Hz for 1 hour (ES) in vivo before excising dorsal root ganglion (DRG) sensory neurons and plating them on a laminin substrate in vitro, promotes their neurite outgrowth. An intact sciatic nerve 1 day after (A) no ES and (B) ES. An intact sciatic nerve 7 days after (C) no ES and (D) ES. (E) The mean + standard error of the lengths of the longest neurites (in µm) for the control and experimental DRG neurons that were not and were stimulated, respectively. The ES of intact sciatic nerves promoted neurite extension from plated DRG neurons. This promoted neurite extension is akin to the effect of a nerve crush conditioning lesion (CL) that is made 7 days prior to the transection and suture of the nerve proximal to the CL, that accelerates nerve regeneration. The neurons were immunostained with β-tubulin III. The calibration bar = 50 µm. *Statistical significance p<0.05.
Figure 17.
(A) We asked the question as to whether cAMP mediates the effect of ES on axon regeneration by administering rolipram to the site of injury and surgical repair to block phosphodiesterase 4 that hydrolyses cAMP. (B) Rolipram (1:1 saline/dimethyl sulfoxide [DSMO]) or vehicle (1:1 saline/DMSO) was delivered via an Alzet pump at a rate of 0.4 µmol/g/h to the site of the cut and sutured the CP (common peroneal) nerve that was surrounded by a 3 mm long silicone silastic cuff of 0.7 mm interior diameter. The mean (+ standard error) number of CP motoneurons that regenerated across the suture site (expressed as a percentage of the numbers of intact CP motoneurons on the contralateral side) were retrogradely labelled with rubyred (RR) (C) 3 mm, and (D) 10 mm from the suture site. (E) MUNE [the estimated number of MUs (motor units) that equals the ratio of the muscle and mean MU twitch forces of the reinnervated TA (tibialis anterior) muscle after the CP nerve suture repair] expressed as a percentage of MUNE in muscles with intact innervation. The significant increase in the number of CP motoneurons that (D,E) regenerated their axons (C) 3 mm past the CP nerve suture site after 7 (*** p<0.01) and after 14 days (*** p<0.01), (D) 10 mm past the suture site after 14 days and E. reinnervated TA muscle after 5 months.
Figure 17.
(A) We asked the question as to whether cAMP mediates the effect of ES on axon regeneration by administering rolipram to the site of injury and surgical repair to block phosphodiesterase 4 that hydrolyses cAMP. (B) Rolipram (1:1 saline/dimethyl sulfoxide [DSMO]) or vehicle (1:1 saline/DMSO) was delivered via an Alzet pump at a rate of 0.4 µmol/g/h to the site of the cut and sutured the CP (common peroneal) nerve that was surrounded by a 3 mm long silicone silastic cuff of 0.7 mm interior diameter. The mean (+ standard error) number of CP motoneurons that regenerated across the suture site (expressed as a percentage of the numbers of intact CP motoneurons on the contralateral side) were retrogradely labelled with rubyred (RR) (C) 3 mm, and (D) 10 mm from the suture site. (E) MUNE [the estimated number of MUs (motor units) that equals the ratio of the muscle and mean MU twitch forces of the reinnervated TA (tibialis anterior) muscle after the CP nerve suture repair] expressed as a percentage of MUNE in muscles with intact innervation. The significant increase in the number of CP motoneurons that (D,E) regenerated their axons (C) 3 mm past the CP nerve suture site after 7 (*** p<0.01) and after 14 days (*** p<0.01), (D) 10 mm past the suture site after 14 days and E. reinnervated TA muscle after 5 months.
Figure 18.
Electrical stimulation (ES) promotes regeneration of transected sensory nerves in the central nervous system (CNS). (A) Figurative drawing of surgical T8 transection of the sciatic sensory nerves in the CNS, the three experimental groups of 1) conditioning lesion (CL, 1), 20 Hz ES (2), and 200 Hz (3), and the injection a 1% solution of cholera Toxin B (CTB) [487] into the sciatic nerve 14 weeks later to label CNS sensory nerves in vivo. (B) Mean ± standard error (SE) of cAMP levels in L4 to 6 dorsal root ganglion neurons, 24 hours after CLs and ES. Camara lucida drawings of spinal cord 25 µm sagittal sections of CTB-immunocytochemical identification with examples of DAB staining of DRG sensory axons of (C) sham ES, (D) CL, (E) 20 Hz ES, and (F) 200 Hz ES The lesion site in the (C-F) drawings is denoted by the transition from a white to a grey background and the arrows correspond with the visualization of the CTB stained axons. The cumulative sum of the mean (± SE) numbers of regenerated axons that advanced into the lesion site as a percent of all the labeled axons proximal to the lesion in the (G) sham control group compared to the CL group, (H) the 20 Hz ES group compared to Sham group, (I) 200 Hz ES group compared to the sham group. Significant values (p<0/05) are denoted by s. See the text for details.
Figure 18.
Electrical stimulation (ES) promotes regeneration of transected sensory nerves in the central nervous system (CNS). (A) Figurative drawing of surgical T8 transection of the sciatic sensory nerves in the CNS, the three experimental groups of 1) conditioning lesion (CL, 1), 20 Hz ES (2), and 200 Hz (3), and the injection a 1% solution of cholera Toxin B (CTB) [487] into the sciatic nerve 14 weeks later to label CNS sensory nerves in vivo. (B) Mean ± standard error (SE) of cAMP levels in L4 to 6 dorsal root ganglion neurons, 24 hours after CLs and ES. Camara lucida drawings of spinal cord 25 µm sagittal sections of CTB-immunocytochemical identification with examples of DAB staining of DRG sensory axons of (C) sham ES, (D) CL, (E) 20 Hz ES, and (F) 200 Hz ES The lesion site in the (C-F) drawings is denoted by the transition from a white to a grey background and the arrows correspond with the visualization of the CTB stained axons. The cumulative sum of the mean (± SE) numbers of regenerated axons that advanced into the lesion site as a percent of all the labeled axons proximal to the lesion in the (G) sham control group compared to the CL group, (H) the 20 Hz ES group compared to Sham group, (I) 200 Hz ES group compared to the sham group. Significant values (p<0/05) are denoted by s. See the text for details.
Figure 19.
Schematic representation of the intracellular signalling pathways activated by brief (1-hour 20 Hz electrical stimulation (ES). ES increases calcium influx (not shown) and in turn, activates adenyl cyclase to elevate cAMP levels. Cyclic AMP, in turn, acts via PKA to phosphorylate CREB in the nucleus leading to transcription of proteins, including neurotrophins and the cytoskeletal proteins tubulin and actin that are essential for axonal elongation during regeneration. The neurotrophins also feedback to further increase cAMP (via ERK with transient inhibition of phosphodiesterase 4 to prevent hydrolysis of cAMP to 5’AMP–not shown). Thereby the transcription of regeneration associated genes is amplified to promote axonal outgrowth. The ES also promotes axonal regeneration after chronic denervation of distal nerve stumps. This positive effect is likely mediated by the release of mitogens including neuregulin that promote Schwann cell proliferation and expression of their growth permissive state. This state includes the transcription of neurotrophic factors that likely potentiate axon regeneration via their amplification of neurotrophic factor expression by the neurons. These feedback loops likely aid in promoting the accelerated regeneration and target reinnervation. The ES of the injured nerve proximal to the site of nerve repair likely leads to the release of mitogens such as neuregulin that promotes Schwann cell proliferation and transition from the myelinating to a growth permissive state. These in turn, account for the positive effect of ES in promoting nerve regeneration after chronic denervation of the distal nerve stumps (Figure 15).
Figure 19.
Schematic representation of the intracellular signalling pathways activated by brief (1-hour 20 Hz electrical stimulation (ES). ES increases calcium influx (not shown) and in turn, activates adenyl cyclase to elevate cAMP levels. Cyclic AMP, in turn, acts via PKA to phosphorylate CREB in the nucleus leading to transcription of proteins, including neurotrophins and the cytoskeletal proteins tubulin and actin that are essential for axonal elongation during regeneration. The neurotrophins also feedback to further increase cAMP (via ERK with transient inhibition of phosphodiesterase 4 to prevent hydrolysis of cAMP to 5’AMP–not shown). Thereby the transcription of regeneration associated genes is amplified to promote axonal outgrowth. The ES also promotes axonal regeneration after chronic denervation of distal nerve stumps. This positive effect is likely mediated by the release of mitogens including neuregulin that promote Schwann cell proliferation and expression of their growth permissive state. This state includes the transcription of neurotrophic factors that likely potentiate axon regeneration via their amplification of neurotrophic factor expression by the neurons. These feedback loops likely aid in promoting the accelerated regeneration and target reinnervation. The ES of the injured nerve proximal to the site of nerve repair likely leads to the release of mitogens such as neuregulin that promotes Schwann cell proliferation and transition from the myelinating to a growth permissive state. These in turn, account for the positive effect of ES in promoting nerve regeneration after chronic denervation of the distal nerve stumps (Figure 15).
Figure 20.
Brief (1-hour) 20 Hz electrical stimulation (ES) of injured human nerves accelerates muscle reinnvation. (A) Diagrammatic representation of the recording of CMAPs (compound muscle action potentials) in the muscles of the median eminence above and below the ligament of the carpal tunnel. Only patients with severe CTS (carpal tunnel syndrome) with (B) reduced numbers of innervated MUs (motor units) exemplified by reduced CMAP amplitudes recorded both above and below the nerve were included in the study of the regenerative effect of ES (C) Patients with a reduced CMAP amplitude recorded below in the wrist but not above the released nerve, evident of conduction block and not MU loss, were excluded. (D) The ligament over the median nerve was released (CTRS: carpal tunnel release surgery). (F) Immediately after surgery, the median nerve was subjected to ES. (E. A maximum (2x threshold) electrical stimulus above the carpal tunnel evoked (G. an CMAP and the minimal stimuli evoked all-or-none single MU action potentials (S-MUAP) at points along the median nerve to estimate MU number (MUNE) from the ratio of the CMAP and mean of at least 20 recorded S-MUAPs. (H) Histograms of the mean (+ standard error) MUNE recorded before and after CTRS with and without ES (Stimulation and Control, respectively). Diagrammatic illustration of (I) the sluggish growth of axons across the nerve crush site that likely accounts for the (J) the regenerating nerve fibres not reaching the muscle to increase the numbers of reinnervated MUs and (L) axonal growth accelerated across the carpal tunnel crush site by ES at 3 months and (M) regenerating nerves progressing through the distal stumps to reinnervated denervated muscle fibres in the thenar eminence by 6 months, accounting for the restoration of the full complement of MUs by 12 months after CTRS.
Figure 20.
Brief (1-hour) 20 Hz electrical stimulation (ES) of injured human nerves accelerates muscle reinnvation. (A) Diagrammatic representation of the recording of CMAPs (compound muscle action potentials) in the muscles of the median eminence above and below the ligament of the carpal tunnel. Only patients with severe CTS (carpal tunnel syndrome) with (B) reduced numbers of innervated MUs (motor units) exemplified by reduced CMAP amplitudes recorded both above and below the nerve were included in the study of the regenerative effect of ES (C) Patients with a reduced CMAP amplitude recorded below in the wrist but not above the released nerve, evident of conduction block and not MU loss, were excluded. (D) The ligament over the median nerve was released (CTRS: carpal tunnel release surgery). (F) Immediately after surgery, the median nerve was subjected to ES. (E. A maximum (2x threshold) electrical stimulus above the carpal tunnel evoked (G. an CMAP and the minimal stimuli evoked all-or-none single MU action potentials (S-MUAP) at points along the median nerve to estimate MU number (MUNE) from the ratio of the CMAP and mean of at least 20 recorded S-MUAPs. (H) Histograms of the mean (+ standard error) MUNE recorded before and after CTRS with and without ES (Stimulation and Control, respectively). Diagrammatic illustration of (I) the sluggish growth of axons across the nerve crush site that likely accounts for the (J) the regenerating nerve fibres not reaching the muscle to increase the numbers of reinnervated MUs and (L) axonal growth accelerated across the carpal tunnel crush site by ES at 3 months and (M) regenerating nerves progressing through the distal stumps to reinnervated denervated muscle fibres in the thenar eminence by 6 months, accounting for the restoration of the full complement of MUs by 12 months after CTRS.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Enhancing Motor and Sensory Axon Regeneration after Peripheral Nerve Injury Using Bioluminescent Optogenetics
Anna Ecanow
et al.
IJMS,
2022
A Therapeutic Strategy for Lower Motor Neuron Disease and Injury Integrating Neural Stem Cell Transplantation and Functional Electrical Stimulation in a Rat Model
Katsuhiro Tokutake
et al.
IJMS,
2022
Bioluminescent Optogenetics: A Novel Experimental Therapy to Promote Axon Regeneration after Peripheral Nerve Injury
Arthur English
et al.
IJMS,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated