Submitted:
13 October 2023
Posted:
18 October 2023
You are already at the latest version
Abstract
Systemic sclerosis (also known as scleroderma) is a chronic fibrosing autoimmune disease with both skin and multisystem organ involvement. Scleroderma has the highest mortality among all rheumatic diseases. The pathophysiology mechanism of systemic sclerosis is a progressive self-amplifying process, which implicates the widespread microvascular damage, followed by a dysregulation of innate and adaptive immunity and inflammation, and diffuse fibrosis of the skin and visceral organs. Fibrosis of internal organs is a hint for systemic sclerosis, moreover associated with interstitial lung disease (SSc-ILD) is a complex process. We report a case of a 56 years old female, diagnosed with Systemic Sclerosis 16 years ago. The systemic and clinical manifestations, respiratory functional tests, radiological aspects and specific therapy were discussed.
Keywords:
systemic sclerosis
; interstitial lung disease
; lung diseases
; Raynaud’s syndrome
1. Introduction
Systemic sclerosis (also known as scleroderma) is a rarechronic fibrosing autoimmune disease with both skin and multisystem organ involvement [1,2].Scleroderma has the highest mortality among all rheumatic diseases [3]. Its pathophysiology is complex, an altered balance of the acquired and innate immune system leads to the release of several cytokines and chemokines, as well as autoantibodies, which induce the activation of fibroblasts with the formation of myofibroblasts and the formation of a rigid connective tissue [1].
Fibrosis of internal organs is a hint of systemic sclerosis, moreover associated with interstitial lung disease (SSc-ILD) is a complex process involving inflammation, alveolar epithelial damage, and activation of resident fibroblasts resulting in thickening of the lung interstitium [2,4,5].
In this paper, we have done a case-based literature review of the pulmonary fibrosis related to systemic scleroderma, because the increased risk of developing pulmonary fibrosis and pulmonary artery hypertension is among the leading causes of SSc-related death.
1.1. Epidemiology and Pathophysiology
Systemic sclerosis is a rare rheumatological condition, with a prevalence of 7.2–33.9, 13.5–44.3 and 8.2 per 100,000 individuals in Europe, North America and The United Kingdom, respectively with an annual incidence of 0.6–5.6 per 100,000 individuals. Like many other rheumatological conditions, women are more commonly affected than men, with a reported ratio of between 3:1 to 8:1 [2,6].
Genetic factors likely contribute towards disease susceptibility and could explain some of the clinical heterogeneity of the disease. Also environmental and occupational exposures, specifically silica, solvents, pesticides and epoxy resins, have been implicated as potential causative factors [6,7,8].
The pathophysiology mechanism of systemic sclerosis is a progressive self-amplifying process, which implicates the widespread microvascular damage, which is believed to play a central role, followed by a dysregulation of innate and adaptive immunity and inflammation, and diffuse fibrosis of the skin and visceral organs [6,9,10,11]. Most likely, vascular injury (possibly initiated by viruses, autoantibodies, chemicals, or oxidative products) and dysfunction of the endothelium cause local tissue ischemia which promotes tissue fibrosis [11].
Raynaud’s phenomenon is the clinical consequence of repeated vascular damage and vasospasm of the small arteries and arterioles of the fingers and toes, and it can be triggered by cold or even emotional stress. This is accompanied by an altered expression of adhesion proteins and cytokines [12].
The presence of serum autoantibodies that target multiple intracellular antigens distinguishes SSc. These autoantibodies, which are seen in more than 95% of patients, can be used to screen for SSc. ANA has been shown to occur in 75-95% of SSc patients, with an immunofluorescence sensitivity of 85% and a specificity of 54% [13].
Furthermore, anti-topoisomerase I (ATA) antibodies were previously known as anti-Scl-70 antibodies. ATA was found in 15-42% of SSc patients, with a sensitivity of 90-100%. The sensitivity of the ATA is 34%. ATA is related to diffuse cutaneous SSc (dcSSc) and has a dismal prognosis. In SSc patients with ATA, the risk of severe lung fibrosis and cardiac involvement is enhanced. Additionally, ATA has been linked to the development of digital ulcers and joint involvement [13,14].
Fibroblast to myofibroblast transition is believed to be a key event and is driven by a number of profibrotic factors, in particular transforming growth factor-beta [6,8].
The dysregulation of the innate and adaptive immune system response plays an important role and includes the increased presence and altered functions of inflammatory cells and products in target tissues, such as the skin and lungs, together with a polymorphism in IFN-regulatory factors that confers an increased risk of SSc [1,9,15,16].
The inflammatory profibrogenic cytokines and growth factors lead to the activation of fibroblasts [1,17]. The origin of these fibroblasts has been discussed to derive from the circulation and also from the subcutaneous layer, from transdifferentiation or resident cells in the tissue [1]. Activated fibroblasts produce ET-1, a potent vasoconstrictor that is able to increase fibronectin synthesis in normal and SSc human skin [9,18]. These have the characteristics of myofibroblasts, which have long been regarded as the key culprit in SSc fibrosis [17,19].
Another interesting aspect of SSc is the loss of subcutaneous adipose tissue [1,20]. Subcutaneous adipose mesenchymal stem cells and mature adipocytes are both involved in the transdifferentiation into fibroblast-like cells. The adipocytes from fibrotic locations in SSc are phenotypically different from normal adipocytes [21,22].
1.2. Diagnosis and Classification
Because there is no a single diagnostic test, the diagnosis of systemic sclerosis is usually based on clinical features, but is supported through results from targeted investigations, in consequence of which several sets of classification criteria have been developed such as 2013 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) Criteria [6,26,27].
The most widely used technique divides systemic sclerosis into subsets: limited (LcSSC) and diffuse cutaneous systemic sclerosis (DcSSc), based upon skin involvement [28,29,30]. The term ‘CREST’ (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly and telangiectasis) is a useful hint for some of the dominant features of systemic sclerosis because patients with diffuse disease can also develop all of these manifestations [6].
1.3. Clinical Manifestations
1.3.1. Skin
Skin fibrosis is one of the dominant clinical features of SSc. The extent of skin fibrosis in SSc is most commonly assessed using the modified Rodnan skin score (mRSS), which measures skin thickness on a scale of 0 to 3 at 17 anatomical sites (score range 0–51). [33,34].
1.3.2.Digital Vascular Disease
The fingers are commonly affected, but other sites can be involved too, including the toes and other vascular areas (e.g., lips and ears) [6]. Digital ulcers are a combination of the progressive microangiopathy and digital artery disease, which are commonly meet in systemic sclerosis [35,36]. Often, they occur on the fingertips and over the dorsal (extensor) aspects of the hands, overlying the small joints [35].
1.3.3. Cardiovascular System
In systemic sclerosis, cardiovascular involvement is common and can be life- threatening, because primary cardiac manifestation is often subclinical [37]. Arrhythmias are one of the most severe and potentially fatal complications in SSc, but an increased risk of atherosclerotic disease has also been reported [31,38].
1.3.4. Respiratory Tract
Today, respiratory involvement (pulmonary fibrosis and pulmonary artery hypertension) is among the leading causes of SSc-related death [31,39]. Systemic sclerosis-associated interstitial lung disease is the end result of the interplay between fibrosis, autoimmunity, inflammation, and vascular injury [40]. Clinical symptoms occur late and are nonspecific, but when reported, dyspnea, non-productive cough, and overwhelming fatigue are the most common symptoms [40,41]. Physical examination reveals Velcro like crackles on auscultation in addition to the cutaneous findings, and pulmonary function evaluation often reveals restriction [40,42,43].
HRCT is the gold standard for the early diagnosis of SScILD [4,42]. The most common imaging pattern on HRCT is nonspecific interstitial pneumonia, which is seen in more than 70-80% of patients with SSc-ILD [40,42,44]. It is characterized by peripheral ground-glass opacities with an apical to basal gradient, frequently accompanied by subpleural sparing. Parenchymal changes are defined by the presence of reticulation, traction bronchiectasis, and bronchiolectasis in a similar distribution [42,45]. Also, functional pulmonary testing (spirometry and Dlco), is mandatory to identify patients with developing progressive interstitial lung disease.
1.3.5. Gastrointestinal and Renal System
Increased deposition of collagen and other components of extracellular matrix leads to fibrotic changes in the upper and lower GI tract, resulting in dysmotility, malabsorption, malnutrition and dilation of the intestine [46,47]. The commonly reported symptoms of SSc include meteorism (87%), fecal incontinence (23%) and features related to reduced esophageal motility or gastroparesis like dysphagia, heartburn and gastrointestinal reflux symptoms [6,46]. In addition to pulmonary clinical manifestations, coughing and a sore voice can occur [47,48].
Moving forward, among all possible systemic sclerosis internal organ complications, kidney involvement is frequently underestimated, because it is usually attributed to other health problems [49,50]. The primary event of kidney damage is an injury to the endothelial cells, causing intimal thickening and proliferation of intralobular and arcuate arteries [50]. Typical clinical features appear with the onset of accelerated hypertension: severe headache, blurred vision and other encephalopathic symptoms. Otherwise, most patients with scleroderma renal crisis (SRC) complain of non-specific symptoms, including: increased fatigue, dyspnea or just dizziness [51].
1.3.6. Musculoskeletal System
The musculoskeletal system is commonly affected in patients with systemic sclerosis [6]. For example, joint involvement can range from non-specific arthralgia and myalgia to rheumatoid arthritis (RA) [52]. Hand (finger) flexion contractures are an important cause of disability [53]. Bilateral carpal tunnel syndrome (CTS) or median neuropathy at the wrist (MNW) can be seen in patients with early disease and is sometimes the first non-Raynaud’s presenting feature of systemic sclerosis [52,54].
2. Case Scenario
The presented case is from a patient admitted to the Pneumology Department of the Mures Clinical County Hospital. Informed consent was obtained from the patient. This study was conducted in accordance with the Declaration of Helsinki.
We present the case of a 56-year-old female, with prolonged professional exposure (worked in vulcanization), diagnosed with Systemic Sclerosis 16 years ago (anti-Scl-70 antibodies = 7.2, antinuclear antibodies = 21.3 UI/ml), with the onset of symptoms in 2003, with secondary Raynaud's syndrome, digital ulcers, recurrent pneumonia and pericarditis in 2007, respiratory failure and interstitial lung disease progressive pattern, initially on treatment with immunosuppressive agents (Cyclophosphamide 100 mg/day) (stopped in December on her own initiative), later on treatment with Methotrexate 20 mg/week (stopped in June 2012 for administrative reasons). Since December 2015, treatment with endothelin receptor antagonists (Bosentan 2 x 125 mg/day) has been initiated, to which a PDE-5 inhibitor (Sildenafil 3 x 20 mg/day) has been associated since 2019, with the patient being included in the National Treatment Program for patients with Pulmonary Arterial Hypertension. On May 7th 2022, antifibrotic treatment with tyrosine-kinase inhibitors was initiated (Nintedanib 150 mg 2 x 1/day, later 100 mg 2 x 1/day from December 12th 2022). The patient is also known to have viral hepatitis B (under antiviral treatment with Entecavir), L4-L5 spondylolisthesis, cervical-dorso-lumbar spondylodiscarthrosis, hepatic steatosis, reflux esophagitis, pangastric erosive disease and hemorrhoidal disease (upper digestive endoscopy and colonoscopy 2018). Moreover, recently she was diagnosed with osteoporosis (T-score -3.1) and low serum levels of vitamin D, and was initiated on treatment with bisphosphonate medication (Ibandronic Acid 3 mg/day) and high doses of D3 vitamin.
Objective examination and examination of the locomotor system:
Normosthenic patient, BMI 23.78 kg/m2, face with the appearance of a "Byzantine icon", widened palpebral fissures, with deletion of nasolabial folds, decreasing mouth opening and multiplication of peribuccal folds. Indurated, hyper pigmented skin, sclerodactyly, fingers fixed in flexion, bilateral upper limb digital ulcerations, II, III, V toes, multiple stellate scars at the level of the phalanges of the upper limbs, cold, pale feet with an active ulcer in the second toe of the left foot. Active mobilization is accompanied by diffuse joint pain and cramping when mobilizing the knees, accentuated dorsal kyphosis, ante projected shoulders, limitation of lateral flexion, cervical and lumbar spine pain, flattening of the lumbar lordosis, muscle contracture of the upper border trapezium and bilateral dorso-lumbar paravertebral, percussive sensitivity of the spinous apophyses dorso-lumbar spine, painful limited ante flexion, crural plexus elongation positive bilaterally, Schober 10/13 cm, Lasegue positive bilaterally. Kyphotic thorax, vesicular murmur present bilaterally, bilateral basal Velcro rales, oxygen saturation 87% in ambient air, 97% with oxygen mask 6-8 L/min. Apexian shock in the left V intercostal space on the medio-clavicular line, rhythmic heart sounds, sound II accentuated and doubled at the pulmonary area, ventricular allure 68/min, blood pressure 110/80 mmHg, palpable peripheral pulse bilaterally at the pedis artery. Microstomia, abdomen located in the xipho-pubic plane, sensitive to deep palpation in the right hypochondrium, the spleen is not palpable, the Giordano maneuver is negative bilaterally. Temporospatial oriented, symmetrical triggerable osteo tendinous reflexes, without signs of meningeal irritation, overall low muscle strength, low tactile and superficial sensitivity at the level of the affected skin.
2.1. Investigations
2.1.1. Laboratory Investigations (Table 1)
2.1.2. Electrocardiography (Figure 1)
2.1.3. Minutes Walking Test
At start, ventricular rate = 59/min and oxygen saturation is 85% in ambient air, dyspnea (Borg scale) = 2.
At stop, ventricular rate 71/min, saturation is 82%, dyspnea (Borg scale) = 7. Total distance = 50 meters (9 % of the predicted distance = 551m). The test was stopped after one minute and 13 seconds, due to marked dyspnea and the feeling of vertigo manifested by the patient.
2.1.4. Echocardiography (Table 2)
Mitral valve: mobile, moderate atherosclerotic changes, hemodynamically insignificant. Aortic valve: tricuspid, mobile, moderate ATS changes, hemodynamically insignificant. Tricuspid valve: normally inserted, flexible, mobile. Pulmonary valve: supple, mobile. Interatrial septum/interventricular septum: intact. Left ventricle: efficient, without segmental and global kinetic disorders, global EF 55%, diastolic dysfunction grade I. Right cavities: RV 30/34/56 mm, hypokinetic TAPSE = 17 mm, MAPSE = 13 mm.
Conclusions: Efficient, non-dilated LV, global EF 55%, with diastolic dysfunction grade 1, minor mitral regurgitation, moderate pulmonary regurgitation. Hypokinetic RV. Severe systolic pulmonary hypertension, echocardiographic criteria showing very high probability of pulmonary hypertension. GLPS LAX: 11.5; GLPS A4C:10.3; GLPS A2C: 13.1; AVERAGE GLPS: 11.7; Biplane FE: 55%, EDV: 72 ml, ESV 41 ml, SV 31 ml, LVCO: 2.3L/min, GLPS VD: 7.8%.
2.1.5. Bone Densitometry (Table 3)
The BMD measured at the Femur Neck Right is 0.601 g/cm2, with a T score of - 3.1 as it can be seen in Table 3.
2.1.6. High-Resolution Computed Tomography (October 12th 2022) (Figure 2)
Thyroid gland of normal appearance. Advanced fibrotic changes in both lung fields with septal thickening, architectural disorganization and traction bronchiectasis, the changes being more important at the basal level of the bilateral lower lobes. Small diffuse calcified granulomas bilaterally, without areas of pulmonary condensation. Absence of suspicious pulmonary nodules. Trachea and bronchi with free lumens. Absence of mediastinal masses. Absence of pleural fluid accumulations. Mediastinal adenopathies up to 17 mm perivascular, 16 mm pretracheal at the right, 18 mm left hilar, 15 mm right hilar, and multiple subcentimeter, some with punctate calcifications. Esophagus minimally dilated, with liquid content. Cardiomegaly, pericardial blade up to 18 mm in the right ventricle. Accentuation of dorsal kyphosis. Early degenerative changes in the dorsal spine, without suspicious lesions on the scanned bone segment. Conclusions: Pulmonary fibrosis changes with medium-advanced damage. Pericardial minimum. Esophageal stasis, more likely in the context of achalasia. Bilateral mediastinal and hilar adenopathies, some with calcifications.
2.1.7. Spirometry:
Mixed ventilatory dysfunction predominantly restrictive, decreased vital capacity (VC) by 45%, decreased forced expiratory volume in one second (FEV1) by 45.4%. Normal Tiffneau index.
2.1.8. Bodypletismography (Table 4):
Airway resistance (RAW) and total airway resistance (Rtot) normal values, resistance-volume ratio (R-V) 83% (normal value), low total lung capacity - 67%(TLC) and Functional residual capacity (FRC) – 80%.
2.1.9. Lung Diffusion Capacity (Table 5)
Very low values of diffusing capacity of the lung for carbon monoxide (Dlco) – 25% and carbon monoxide transfer coefficient (Kco) – 41%.
Functional respiratory studies reveal a restrictive type pattern due to pulmonary fibrotic lesions. Pulmonary interstitial damage is caused by the rheumatological pathology, progressive systemic scleroderma coexisting with the etiological context of pulmonary arterial hypertension.
2.1.10. High-Resolution Computed Tomography (March first 2023) (Figure 3)
Predominantly subpleural reticular lesions, with a four-cornered appearance, associated with minimal right anterobasal peribronchovascular extension. Traction bronchiectasis is associated with the reticular beaches above. Subpleural areas of honeycombing more are accentuated in the lower half of the lung. Discrete peripheral organizing masses, especially the left posterior. Fibrous bands with small associated calcifications. The pleural contour is irregularly marked, with numerous spicules on the contour. The esophagus is markedly dilated along its entire length, with a caliber of up to 26 mm, regular walls, liquid stasis in the lower half. Circumferential pericarditis in a small amount. Global cardiomegaly. The pulmonary artery cone has a caliber of 36 mm, in the context of known pulmonary hypertension. Numerous supracarinal bilateral mediastino-hilar adenopathies, some of them with small calcifications, with an inflammatory appearance. Thyroid with normal position and dimensions, inhomogeneous, micropolynodular structure. Conclusions: The CT appearance is an appearance of interstitial lung pneumopathy progressive fibrosing phenotype, examination quasi-identical to the previous CT examination. Dilated pulmonary arteries with the appearance of PAH. Minimal pericarditis. Cardiomegaly. Polynodular goiter. Dorsal spondylarthrosis.
Considering the underlying pathology, progressive systemic sclerosis and interstitial lung disease, the next step in the diagnostic algorithm was progressive evaluation. The restrictive pattern expressed on body plethysmography associated with a significant decrease in DLCO, the clinical deterioration of the patient, and the progression of imaging lesions on HRCT led to the classification of the patient as having an SSC-ILD progressive phenotype.
2.2. Management of SSc
Treatment of SSc can be tough due to its rarity and heterogeneous disease manifestations [55]. Best practice often involves shared medical care and therapy should be implemented to point directly active organ-specific complications of disease.
2.2.1. Cutaneous and Vascular Involvement
In addition to skin thickening, cutaneous disease involves the presence of calcinosis and pruritus, which is common, results as a consequence of small fiber neuropathy [56]. Immunosuppressive therapies include methotrexate, mycophenolate mofetil, with the modified Rodnan skin score (mRSS) routinely used to quantify the extent of cutaneous sclerosis [55,57]. For the peripheral vascular system (Raynaud’s phenomenon, digital ulcers and critical ischemia) the following therapies help reduce the frequency and severity of vascular manifestations: calcium-channel blockers, phosphodiesterase type 5 inhibitors, angiotensin II receptor blockers, endothelin receptor antagonists, prostacyclin analogue, wound care for digital ulcers and antibiotic therapy for infected ulcers [6,55].
Heart involvement:
Heart involvement is a strong prognostic factor in systemic sclerosis and may presents more frequently with diastolic (rather than systolic) dysfunction (heart failure with preserved ejection fraction) [56,58]. Current pharmacological therapies for heart failure include usual drug therapies such as calcium channel blockers for prevention and treatment of left ventricular dysfunction, ACE inhibitors, diuretics or calcium channel blockers for improvement in myocardial perfusion and anti-arrhythmic agents [6,55]. Regarding the inflammatory cardiac profile, immunosuppressive therapy (for example corticosteroid or cyclophosphamide drugs) should be taken into consideration.
2.2.2. Scleroderma Renal Crisis
The use of ACEI to treat SRC, has been associated with a good outcome, and is mandatory for improvement in morbidity and mortality due to scleroderma renal crisis [59]. Additionally, education for those at high risk regarding a proper routine of monitoring blood pressure and close communication of new symptom development (headache, dyspnea, dizziness, syncope) is strongly recommended [56].
2.2.3. Gastrointestinal Disease
2.2.4. Interstitial Lung Disease and Pulmonary Hypertension
Excepting methotrexate, immunosuppressive treatments for cutaneous fibrosis are frequently successful for treating SSc-ILD, highlighting the similar etiology of both symptoms [60]. The recommendations are for cyclophosphamide, mycophenolate, rituximab, or tocilizumab, with priority given to mycophenolate due to its documented effectiveness for interstitial lung disease, skin, and good side effect profile [61,62]. Also, the tyrosine kinase inhibitor Nintedanib is authorized for use in treating progressive pulmonary fibrosis [61].
The progression of interstitial pathology in cases with scleroderma is a serious factor that impacts the prognosis of these patients and it is similar with amiodarone-induced lung injury [63]. Progressive Pulmonary Fibrosis (PPF) is defined as the presence of pulmonary fibrosis, to which two of the following criteria must be added: aggravation of respiratory symptoms; progression of the disease from a functional point of view (decrease in FVC >5% predicted since the previous visit or in the last year or decrease in DLCO (corrected for Hb) >10% since the previous assessment); imaging evidence of disease progression evidenced on HRCT [64].
Cases in which, from a pneumological point of view, the presence of progression has been established, require special monitoring that requires the following investigations: repeating the functional tests and the walking test every 4-6 months or sooner if the symptomatology requires it; repeat HRCT at 1 year or less if there is another suspected diagnosis; performing Angio CT if there are signs of pulmonary embolism.
All patients with SSc are at risk for developing pulmonary arterial hypertension. Phosphodiesterase 5 inhibitors (such as Sildenafil and Tadalafil), endothelin receptor antagonists (such as Bosentan or Ambrisentan), and prostacyclins are all used to attain functional New York Heart Association Class II or higher (light breathlessness) and minimal restriction when performing routine tasks [55,56].
2.2.5. Musculoskeletal Involvement
3. Discussions
Improving the management of this potentially fatal SSc consequence requires early detection to risk stratify, monitor progression, and act when appropriate [61].
Considering HRCT the gold standard for detection of ILD, in our case the patient was examined every 6-12 months, but only 50%-66% of medical experts frequently conduct HRCT in newly diagnosed SSc patients, this demonstrates the wide diversity in global practice [43,66].
The peak age of onset is 55–69 years and women are more commonly affected than men, [6] and while the full spectrum of SSc is seen among those with late-age onset SSc as it can be seen in Manno et al. study [67], our patient, unfortunately, had an early onset of her clinical manifestations (at 36 years old), with cutaneous, pulmonary, cardiac and gastrointestinal involvement.
Moinzadeh et al., in their cohort study from Germany (2020) imply that pulmonary hypertension and cardiac involvement occurred substantially more frequently within the late-onset sample in terms of organ manifestation, which is consistent with prior publications [68]. Also, Veronika K. Jaeger et al., in the largest direct comparison of different ethnicities from EUSTAR 2004-2018 database strengthens the knowledge about the clinical and serological differences between black, asian and white people, in which asian people had higher prevalence of pulmonary hypertension and severe lung involvement [69].
Regarding our case, it is well known that since December 2015, the patient (caucasian female) has been included in the Romanian National Treatment Program for patients with Arterial Hypertension Pulmonary at the age of 48-year-old, with recurrent pneumonia and pericarditis since 2007.
It has been shown that late-age onset SSc was surprisingly protective against digital ischemia, early age being previously described as a risk factor for digital ulcers in systemic scleroderma [67,68]. Our case fits this description, digital ulcers involvement being present since the early onset in 2007.
The EULAR Scleroderma Trial and Research cohort revealed 6.6% of deaths from SSc that resulted from Gastrointestinal complications among elderly patients and patients with diffuse skin involvement [46,68]. The fibrosis of oral and perioral tissues, chronic inflammation, deformity of the oral cavity, and misalignment of osseous structures that result in microstomia and malocclusion of the teeth are the causes of oropharyngeal problems [46]. As a result, our patient suffered the gastrointestinal symptomatology detailed in scientific literature, including impaired mastication and deglutition, food leakage, regurgitation, and hoarseness of voice. Furthermore, approximately 50% to 90% of patients with scleroderma experience esophageal manifestations such as acid reflux that further triggers erosive esophagitis [71]. Our patient underwent an upper digestive endoscopy in response to the symptoms mentioned above, and erosive esophagitis and chronic gastritis were ultimately diagnosed. Because of this gastroenterological involvement, digestive intolerance to the initial dose of antifibrotic medication could be justified. The low dose of 200 mg per day divided into 2 doses could be tolerated without adverse effects.
Both the treatment of PAH and the treatment of esophagitis were managed in this case according to the latest European recommendations. The French practical guidelines brought updates to the ATS/ERS recommendations for the management of these comorbidities [72].
The connection between SSc and the risk of osteoporotic fracture did not reach statistical significance so far, according to Chen et al. in their meta-analysis study (2019). However, patients with gastrointestinal involvement have impaired vitamin D absorption, which leads to malnutrition and thickening of the skin or mucosa in SSc patients reduces UV penetration and lowers pre-vitamin D3 production, leading to overall lower bone density [73]. Outlining the points made above, our case demonstrated how malabsorption and skin thickening resulted in secondary osteoporosis, with a T score of -3.1, thus, this patient is considered osteoporotic according to World Health Organization (WHO) criteria [74]. Moreover, vitamin D may interfere with each of the pathobiological processes triggered in SSc, including autoimmunity, peripheral vasculopathy, and fibrosis, due to its immunomodulatory, cardioprotective, and antifibrotic biological actions. Vitamin D levels appear to be considerably reduced as compared to healthy controls, and vitamin D supplementation is mandatory [75].
Interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH), the most common pulmonary symptoms of SSc, have been highlighted by clinical practice and the scientific community as the leading causes of death [76]. When compared to SSc patients without ILD, patients with SSc and ILD (SSc-ILD) had a mortality risk that was almost three times higher. Additionally, patients with Scl-70 (anti-topoisomerase I) antibodies, male sex, and African-American race have a propensity for more severe SSc-ILD and a higher chance of worsening over time [76,77]. Despite the fact that our case does not fit the gender and ethnicity requirements described above, the presence of pulmonary fibrosis and pulmonary hypertension, as well as the presence of anti-Scl-70 antibodies that are positive, place the patient at high risk for morbidity and mortality.
Recent European guidelines for the treatment of SSC-ILD support as a consensus the effectiveness of medication with Mycophenolate Mofetil, Cyclophosphamide and Nintedanib. After identifying the progression, it is recommended to escalate the drug treatment, along with the non-pharmacological adjuvant. According to this algorithm based on modified Delphi process, the patient in the presented case received pharmacological treatment with Nintedanib for ILD, as well as oxygen therapy at home. Immunomodulatory and antifibrotic therapies act mainly on the pathways related to autoimmune or inflammatory processes, respectively, and the pathways related to the production of fibrosis. Even if recent evidence suggests that immunomodulatory medication can also influence the appearance of fibrosis, self-sustaining pulmonary fibrosis requires an effective antifibrotic agent, and in the case of scleroderma, Nintedanib is the only antifibrotic licensed [78]. According to these recent recommendations, the patient in the presented case was initiated on antifibrotic therapy with Nintedanib.
Goh et al. demonstrated that patients with SSc-ILD had a greater risk of eventual death for FVC declines of 10% or for FVC declines of 5-9% combined with a fall of 15% in Dlco [79], as indicated by our patient's repeated respiratory functional tests. Nevertheless, while DLco has been found to be the strongest predictor of HRCT-measured ILD [77], Ryerson et al., additionally, identified that the 6 min walk test distance (6MWT) is an independent predictor of mortality [79]. Taking into consideration that 6MWT is frequently used as a measure of exercise tolerance in patients with SSc-ILD [80,81], we tried to perform the test on our patient, but unfortunately we have been forced to stop the examination after only one minute and 13 seconds, with no more than 9% of the predicted distance completed, because of the exacerbation dyspnea and vertigo.
The particularity of this case consists in the association of multiple comorbidities burdened by a significant gravity. The extremely important systemic damage, the cardiovascular impact of the disease with the presence of severe PAH significantly affects the patient's quality of life. The association with Hepatitis B limits the possibility of using immunosuppressive medications and, at the same time excludes the last step of treatment escalation: lung transplantation.
4. Conclusion
In conclusion, because of its severe circulatory and pulmonary dysfunction, unexpected onset and course, and wide range of clinical manifestations, SSc is an overall challenging condition. Patient demographics, SSc-specific traits such as skin distribution and illness duration, serological markers, pulmonary function tests, and the degree of lung damage on HRCT are all important factors in risk factors for morbidity and mortality. Thus, to emphasize the impact of interstitial lung involvement in systemic scleroderma, we have presented a narrative review and a case from a patient admitted to our Pneumology Department from Mures Clinical County Hospital who was diagnosed with pulmonary fibrosis related to systemic scleroderma. By understanding the mechanism of developing progressive pulmonary fibrosis related to systemic scleroderma, a personalized treatment can pe established to increase the patient’s outcome and by this to increase the quality of life.
Author Contributions
Conceptualization, D.E.P. and C.E.B; methodology, E.L. and S.H.K.; software, I.A.R; validation, D.E.P., C.E.B. and A.N.N.; formal analysis, I.E.V; investigation, A.N.N.; resources, I.E.V; data curation, I.A.R. and S.H.K.; writing—original draft preparation, D.E.P.; writing—review and editing, C.E.B.; visualization, G.M.R.; supervision, C.E.B.; All authors have read and agreed to the published version of the manuscript.”.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACR/EULAR | American College of Rheumatology/European League Against Rheumatism |
| ACEI | Angiotensin converting enzyme inhibitors |
| ANA positivity | Positive antinuclear antibody |
| Ao | Aortic |
| ATA | Anti-topoisomerase I |
| BMI | Body mass index |
| BMD | Body mass density |
| CREST | Calcinosis, Raynaud’s phenomenon, oesophageal dysmotility, sclerodactyly and telangiectases |
| CTS | Carpal tunnel syndrome |
| DcSSC | Diffuse cutaneous systemic scleroderma |
| Dlco | Diffusion capacity of the lung for carbon monoxide |
| EDV | End-dyastolic volume |
| EF | Ejection fraction |
| ESV | End-systolic volume |
| FEV1 | Forced expiratory volume in one second |
| FRC | Functional residual capacity |
| GI | Gastrointestinal |
| GLPS | Global longitudinal peak strain |
| HRCT | High-resolution computed tomography |
| IFN | Interferon |
| IL | Interleukin |
| ILD | Interstitial lung disease |
| IVS | Interventricular septum |
| LA | Left atrium |
| LcSSc | Limited cutaneous systemic sclerosis |
| LV | Left ventricle |
| LVCO | Left ventricle cavity obliteration |
| MAPSE | Mitral annular plane systolic excursion; |
| MNW | Median neuropathy of the wrist |
| mRSS | Modified Rodnan skin score |
| MWT | Minutes walk test |
| PA | Pulmonary artery |
| PAH | Pulmonary arterial hypertension |
| PF | Puffy fingers |
| PDE 5 inhibitors | Phosphodiesterase 5 inhibitors |
| PPF | Progressive Pulmonary Fibrosis |
| RA | Rheumatoid arthritis |
| RA | Right atrium |
| RAW | Airway resistance |
| RBBB | Right bundle branch block |
| RP | Raynaud’s phenomenon |
| Rtot | Total airway resistance |
| RV | Right ventricle |
| R-V | Resistance-volume graph |
| SSc | Systemic sclerosis |
| SSc ILD | Systemic sclerosis associated interstitial lungdisease |
| SRC | Scleroderma renal crisis |
| TAPSE | Tricuspid annular plane systolic, excursion |
References
- Rosendahl, Ann-Helen et al. Pathophysiology of systemic sclerosis (scleroderma). The Kaohsiung journal of medical sciences 2022, 38, 187–195. [Google Scholar] [CrossRef]
- Gumkowska-Sroka, Olga et al. Novel Therapeutic Strategies in the Treatment of Systemic Sclerosis. Pharmaceuticals 2023, 16, 1066. [Google Scholar] [CrossRef]
- Volkmann, Elizabeth R et al. Systemic sclerosis. Lancet 2023, 401, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Roofeh, David et al. Management of systemic sclerosis-associated interstitial lung disease. Current opinion in rheumatology 2019, 31, 241–249. [Google Scholar] [CrossRef]
- Wells, AU. Interstitial lung disease in systemic sclerosis. Press Medicale. 2014, 43, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Hughes, Michael, and Ariane L Herrick. Systemic sclerosis. British journal of hospital medicine 2019, 80, 530–536. [Google Scholar] [CrossRef]
- Jerjen, Rebekka et al. Systemic sclerosis in adults. Part I: Clinical features and pathogenesis. Journal of the American Academy of Dermatology 2022, 87, 937–954. [Google Scholar] [CrossRef]
- Denton CP, Khanna D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, Maurizio et al. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert review of clinical immunology 2019, 15, 753–764. [Google Scholar] [CrossRef]
- Cutolo M, Sulli A, Smith V. Assessing microvascular changes in systemic sclerosis diagnosis and management. Nat Rev Rheumatol. 2010, 6, 578–587. [Google Scholar] [CrossRef]
- Fuschiotti, P. Current perspectives on the immunopathogenesis of systemic sclerosis. ImmunotargetsTher. 2016, 5, 21–35. [Google Scholar] [CrossRef]
- Ruaro, Barbara et al. Innovations in the Assessment of Primary and Secondary Raynaud's Phenomenon. Frontiers in pharmacology 2019, 10, 360. [Google Scholar] [CrossRef]
- Almaabdi, Kholoud et al. Advanced Autoantibody Testing in Systemic Sclerosis. Diagnostics 2023, 13, 851. [Google Scholar] [CrossRef]
- Sulli, Alberto et al. Progression of nailfold microvascular damage and antinuclear antibody pattern in systemic sclerosis. The Journal of rheumatology 2013, 40, 634–639. [Google Scholar] [CrossRef]
- Skaug B, Assassi S. Type I interferon dysregulation in systemic sclerosis. Cytokine. 2019. [Google Scholar]
- Lepri, Gemma et al. Systemic sclerosis: one year in review 2022. Clinical and experimental rheumatology 2022, 40, 1911–1920. [Google Scholar] [CrossRef]
- Gabbiani, G. 50 years of myofibroblasts: how the myofibroblast concept evolved. Methods Mol Biol. 2021, 2299, 1–5. [Google Scholar] [PubMed]
- Shi-Wen X, Kennedy L, Renzoni EA, et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor β in human lung fibroblasts. Arthritis Rheum. 2007, 56, 4189–4194. [Google Scholar] [CrossRef] [PubMed]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Taki Z, Gostjeva E, Thilly W, Yaseen B, Lopez H, Mirza M, et al. Pathogenic activation of Mesenchymal stem cells is induced by the disease microenvironment in systemic sclerosis. Arthritis Rheumatol. 2020, 72, 1361–1374. [Google Scholar] [CrossRef]
- Kruglikov, IL. Interfacial adipose tissue in systemic sclerosis. Curr Rheumatol Rep. 2017, 19, 4. [Google Scholar] [CrossRef]
- Avouac J, Lepri G, Smith V, et al. Sequential nailfold videocapillaroscopy examinations have responsiveness to detect organ progression in systemic sclerosis. Semin Arthritis Rheum. 2017, 47, 86–94. [Google Scholar] [CrossRef]
- Smith V, Scirè CA, Talarico R, et al. Systemic sclerosis: state of the art on clinical practice guidelines. RMD Open. 2018, 4 (Suppl. 1). [Google Scholar]
- Burmester GR, Bijlsma JWJ, Cutolo M, et al. Managing rheumatic and musculoskeletal diseases - past, present and future. Nat Rev Rheumatol. 2017, 13, 443–448. [Google Scholar] [CrossRef]
- van den Hoogen, Frank et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis and rheumatism 2013, 65, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Nadashkevich O, Davis P, Fritzler MJ. A proposal of criteria for the classification of systemic sclerosis. Med Sci Monit. 2004, 10, CR615–21. [Google Scholar]
- Parisi, Simone, and Maria Chiara Ditto. Videocapillaroscopy in Connective Tissue Diseases. Systemic Sclerosis, InTech, 4 Oct. 2017. [CrossRef]
- LeRoy EC, Black C, Fleischmajer R et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988, 15, 202–205. [Google Scholar]
- Varga, John, and Monique Hinchcliff. Connective tissue diseases: systemic sclerosis: beyond limited and diffuse subsets? Nature reviews. Rheumatology 2014, 10, 200–202. [Google Scholar] [CrossRef]
- Bellando-Randone S, Matucci-Cerinic M, Very early systemic sclerosis, Best Practice & Research Clinical Rheumatology. [CrossRef]
- Avouac J, Fransen J, Walker U, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Annals of the Rheumatic Diseases 2011, 70, 476–481. [Google Scholar] [CrossRef]
- Pope, Janet E et al. State-of-the-art evidence in the treatment of systemic sclerosis. Nature reviews. Rheumatology 2023, 19, 212–226. [Google Scholar] [CrossRef]
- Khanna D, et al. Standardization of the modified Rodnan skin score for use in clinical trials of systemic sclerosis. J. Scleroderma Relat. Disord. 2017, 2, 11–18. [Google Scholar] [CrossRef]
- Hughes, Michael, and Ariane L Herrick. Digital ulcers in systemic sclerosis. Rheumatology 2017, 56, 14–25. [Google Scholar] [CrossRef]
- Sharp, Charlotte A et al. Differential diagnosis of critical digital ischemia in systemic sclerosis: Report of five cases and review of the literature. Seminars in arthritis and rheumatism 2016, 46, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kahan, A.; Coghlan, G.; McLaughlin, V. Cardiac complications of systemic sclerosis. Rheumatology 2009, 48, iii45–iii48. [Google Scholar] [CrossRef]
- Man, Ada et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Annals of the rheumatic diseases 2013, 72, 1188–1193. [Google Scholar] [CrossRef]
- Nihtyanova, S.I.; Schreiber, B.E.; Ong, V.H.; Rosenberg, D.; Moinzadeh, P.; Coghlan, J.G.; Wells, A.U.; Denton, C.P. Prediction of Pulmonary Complications and Long-Term Survival in Systemic Sclerosis. Arthritis & Rheumatology 2014, 66, 1625–1635. [Google Scholar] [CrossRef]
- Perelas, A.; Silver, R.M.; Arrossi, A.V.; Highland, K.B. Systemic sclerosis-associated interstitial lung disease. The Lancet Respiratory Medicine. 2020. [Google Scholar] [CrossRef]
- Suliman, Y.A.; Dobrota, R.; Huscher, D.; Nguyen-Kim TD, L.; Maurer, B.; Jordan, S.; Distler, O. Brief Report: Pulmonary Function Tests: High Rate of False-Negative Results in the Early Detection and Screening of Scleroderma-Related Interstitial Lung Disease. Arthritis & Rheumatology 2020, 67, 3256–3261. [Google Scholar] [CrossRef]
- Khanna, Dinesh et al. Systemic Sclerosis-Associated Interstitial Lung Disease: How to Incorporate Two Food and Drug Administration-Approved Therapies in Clinical Practice. Arthritis & rheumatology (Hoboken, N.J.) 2022, 74, 13–27. [Google Scholar] [CrossRef]
- Rahaghi, Franck F et al. Expert consensus on the management of systemic sclerosis-associated interstitial lung disease. Respiratory research 2023, 24. [CrossRef]
- Okamoto, Masaki et al. A retrospective cohort study of outcome in systemic sclerosis-associated interstitial lung disease. Respiratory investigation 2016, 54, 445–453. [Google Scholar] [CrossRef]
- Hoffmann-Vold, Anna-Maria et al. Survival and causes of death in an unselected and complete cohort of Norwegian patients with systemic sclerosis. The Journal of rheumatology 2013, 40, 1127–1133. [Google Scholar] [CrossRef]
- Nassar, Mahmoud MD, PhDa; Ghernautan, Victoria MDa; Nso, Nso MD, MPHa; Nyabera, Akwe MDa; Castillo, Francisco Cuevas MDa; Tu, Wan MDa; Medina, Luis MDa; Ciobanu, Camelia MDb; Alfishawy, Mostafa MDc; Rizzo, Vincent MDa; Eskaros, Saphwat MDd; Mahdi, Mamdouh PhDe; Khalifa, Mohamed PhDf; El-Kassas, Mohamed MD, PhDg,*. Gastrointestinal involvement in systemic sclerosis: An updated review. Medicine 2022, 101, e31780. [Google Scholar] [CrossRef]
- Schmeiser T, Saar P, Jin D, et al. Profile of gastrointestinal involvement in patients with systemic sclerosis. Rheumatol Int. 2012, 32, 2471–2478. [Google Scholar] [CrossRef]
- Szamosi, Szilvia, Zoltán Szekanecz, and Gabriella Szűcs. Gastrointestinal manifestations in Hungarian scleroderma patients. Rheumatology international 2006, 26, 1120–1124. [Google Scholar] [CrossRef]
- Bruni, C.; Cuomo, G.; Rossi, F.W.; Praino, E.; Bellando-Randone, S. Kidney involvement in systemic sclerosis: From pathogenesis to treatment. Journal of Scleroderma and Related Disorders 2018, 3, 43–52. [Google Scholar] [CrossRef]
- Steen, Virginia D. Kidney involvement in systemic sclerosis. Presse medicale 2014, 43 Pt 2, e305–14. [Google Scholar] [CrossRef]
- Steen VD and Medsger Jr, TA. Long-term outcomes of scleroderma renal crisis. Ann Intern Med. 2000, 133, 600–603. [Google Scholar] [CrossRef]
- Robert David Sandler, Marco Matucci-Cerinic, Michael Hughes, Musculoskeletal hand involvement in systemic sclerosis. Seminars in Arthritis and Rheumatism 2020, 50, 329–334. [CrossRef]
- Avouac, J.; Walker, U.; Tyndall, A.; Kahan, A.; Matucci-Cerinic, M.; Allanore, Y. , et al. Characteristics of Joint Involvement and Relationships with Systemic Inflammation in Systemic Sclerosis: Results from the EULAR Scleroderma Trial and Research Group (EUSTAR) Database. J Rheumatol 2010, 37, 1488–1501. [Google Scholar] [CrossRef]
- Barr, Walter G. , and Sidney J. Blair. Carpal tunnel syndrome as the initial manifestation of scleroderma. The Journal of hand surgery 1988, 13, 366–368. [Google Scholar] [CrossRef]
- Heather Bukiri, Elizabeth R. Volkmann, Current advances in the treatment of systemic sclerosis. Current Opinion in Pharmacology 2022, 64, 102211. [Google Scholar] [CrossRef] [PubMed]
- Roofeh, D.; Khanna, D. Management of systemic sclerosis. Current Opinion in Rheumatology 2020, 32, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Khanna, Dinesh et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis & rheumatology 2020, 72, 125–136. [Google Scholar] [CrossRef]
- Meune, Christophe et al. A right ventricular diastolic impairment is common in systemic sclerosis and is associated with other target-organ damage. Seminars in arthritis and rheumatism 2016, 45, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, Vivek. Management of scleroderma renal crisis. Current opinion in rheumatology 2019, 31, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Jerjen, Rebekka et al. Systemic sclerosis in adults. Part II: management and therapeutics. Journal of the American Academy of Dermatology 2022, 87, 957–978. [Google Scholar] [CrossRef] [PubMed]
- Roofeh, David et al. Treatment for systemic sclerosis-associated interstitial lung disease. Current opinion in rheumatology 2021, 33, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, Donald P et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. The Lancet. Respiratory medicine 2016, 4, 708–719. [Google Scholar] [CrossRef]
- Budin, Corina Eugenia et al. Pulmonary Fibrosis Related to Amiodarone-Is It a Standard Pathophysiological Pattern? A Case-Based Literature Review. Diagnostics 2022, 12, 3217. [Google Scholar] [CrossRef]
- Raghu, Ganesh et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Avouac, Jerome et al. Characteristics of joint involvement and relationships with systemic inflammation in systemic sclerosis: results from the EULAR Scleroderma Trial and Research Group (EUSTAR) database. The Journal of rheumatology 2010, 37, 1488–1501. [Google Scholar] [CrossRef]
- Bernstein EJ, Khanna D, Lederer DJ. Screening High-Resolution Computed Tomography of the Chest to Detect Interstitial Lung Disease in Systemic Sclerosis: A Global Survey of Rheumatologists. Arthritis Rheumatol. 2018, 70, 971–972. [Google Scholar] [CrossRef]
- Manno, R.L.; Wigley, F.M.; Gelber, A.C.; Hummers, L.K. Late-age Onset Systemic Sclerosis. The Journal of Rheumatology 2011, 38, 1317–1325. [Google Scholar] [CrossRef]
- Moinzadeh, P.; Kuhr, K.; Siegert, E.; Mueller-Ladner, U.; Riemekasten, G.; Günther, C.; Hunzelmann, N. Older age onset of systemic sclerosis – accelerated disease progression in all disease subsets. Rheumatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, V.K.; Tikly, M.; Xu, D.; Siegert, E.; Hachulla, E.; Airò, P.; Cozzi, F. Racial differences in systemic sclerosis disease presentation: a European Scleroderma Trials and Research group study. Rheumatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Lin YT, Chuang Y-S, Wang J-W, et al. High risk of gastrointestinal hemorrhage in patients with systemic sclerosis. Arthritis Res Ther. 2019, 21, 301. [Google Scholar] [CrossRef] [PubMed]
- Frech TM, Mar D. Gastrointestinal and hepatic disease in systemic sclerosis. Rheum Dis Clin North Am. 2018, 44, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Cottin, Vincent. Treatment of progressive fibrosing interstitial lung diseases: a milestone in the management of interstitial lung diseases. European respiratory review: an official journal of the European Respiratory Society 2019, 28, 190109. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lei, L.; Pan, J.; Zhao, C. A meta-analysis of fracture risk and bone mineral density in patients with systemic sclerosis. Clinical Rheumatology 2019, 39, 1181–1189. [Google Scholar] [CrossRef]
- World Health Organization. WHO scientific group on the assessment of osteoporosis at primary health care level: summary meeting report. http://www.who.int/chp/topics/Osteoporosis.pdf.
- Diaconu, Alexandra-Diana et al. Role of Vitamin D in Systemic Sclerosis: A Systematic Literature Review. Journal of immunology research 2021, 2021, 9782994. [Google Scholar] [CrossRef]
- Highland, K.B.; Distler, O.; Kuwana, M.; Allanore, Y.; Assassi, S.; Azuma, A.; Maher, T.M. Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: a subgroup analysis of the SENSCIS trial. The Lancet Respiratory Medicine 2021, 9, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Patel, N.M.; Volkmann, E.R. Interstitial Lung Disease in Systemic Sclerosis: Focus on Early Detection and Intervention. Open Access Rheumatol. 2019, 11, 283–307. [Google Scholar] [CrossRef] [PubMed]
- et al. The indentification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol 2020, 2, e71–83. [Google Scholar] [CrossRef]
- Goh, N.S.; Hoyles, R.K.; Denton, C.P.; et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017, 69, 1670–1678. [Google Scholar] [CrossRef]
- Ryerson, Christopher J et al. Predicting Mortality in Systemic Sclerosis-Associated Interstitial Lung Disease Using Risk Prediction Models Derived From Idiopathic Pulmonary Fibrosis. Chest 2015, 148, 1268–1275. [CrossRef] [PubMed]
- Rizzi M, Sarzi-Puttini P, Airoldi A, Antivalle M, Battellino M, Atzeni F. Performance capacity evaluated using the 6-minute walk test: 5-year results in patients with diffuse systemic sclerosis and initial interstitial lung disease. Clin Exp Rheumatol. 2015, 33 (Suppl. 91), S142–147. [Google Scholar]
Figure 1.
Electrocardiogram of the patient. Sinus rhythm, right axis deviation, right bundle branch block (RBBB), negative T-wave in DIII, V1-V4.
Figure 1.
Electrocardiogram of the patient. Sinus rhythm, right axis deviation, right bundle branch block (RBBB), negative T-wave in DIII, V1-V4.
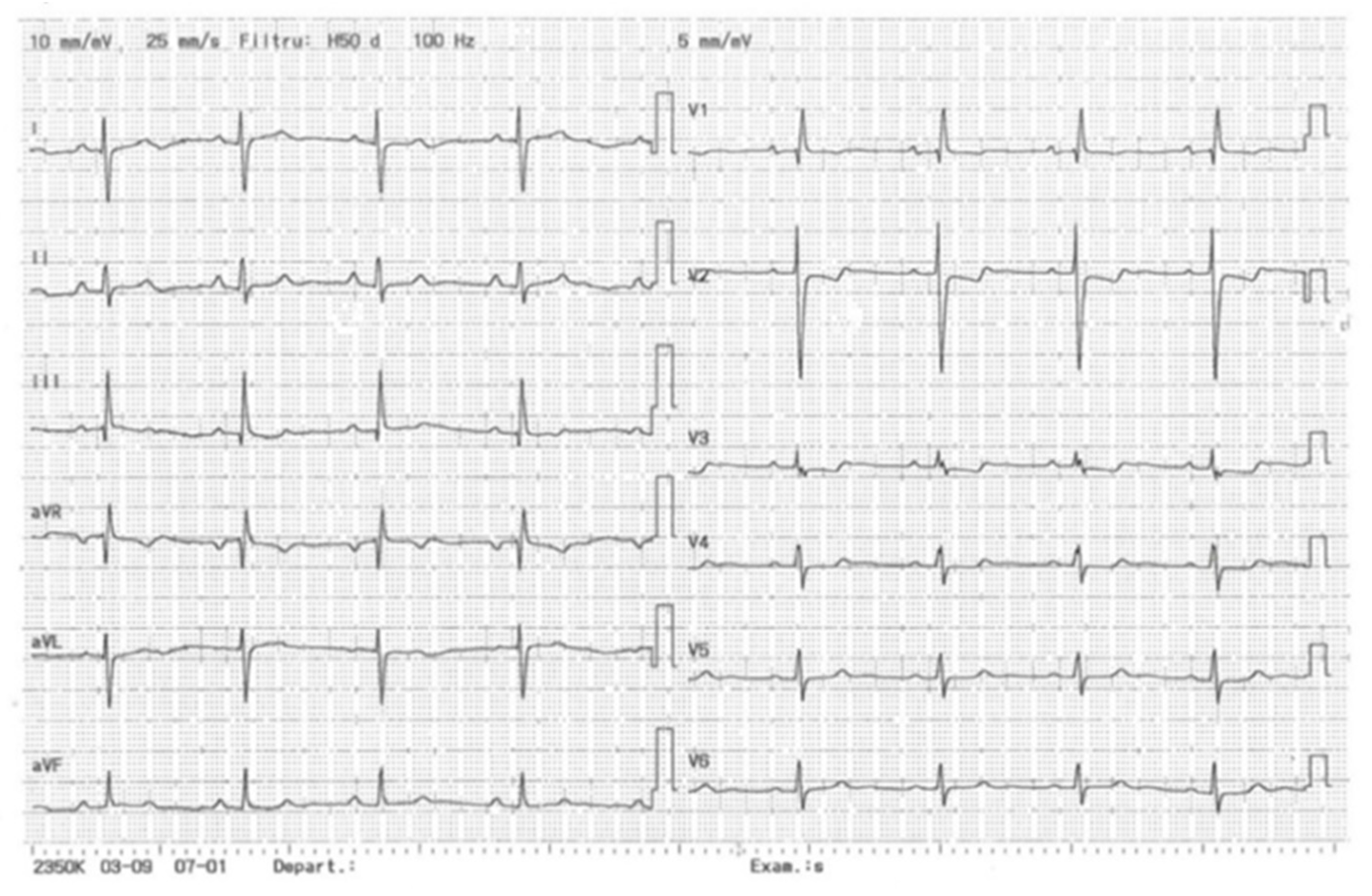
Figure 2.
a), b): Computed Tomography of the Thorax.
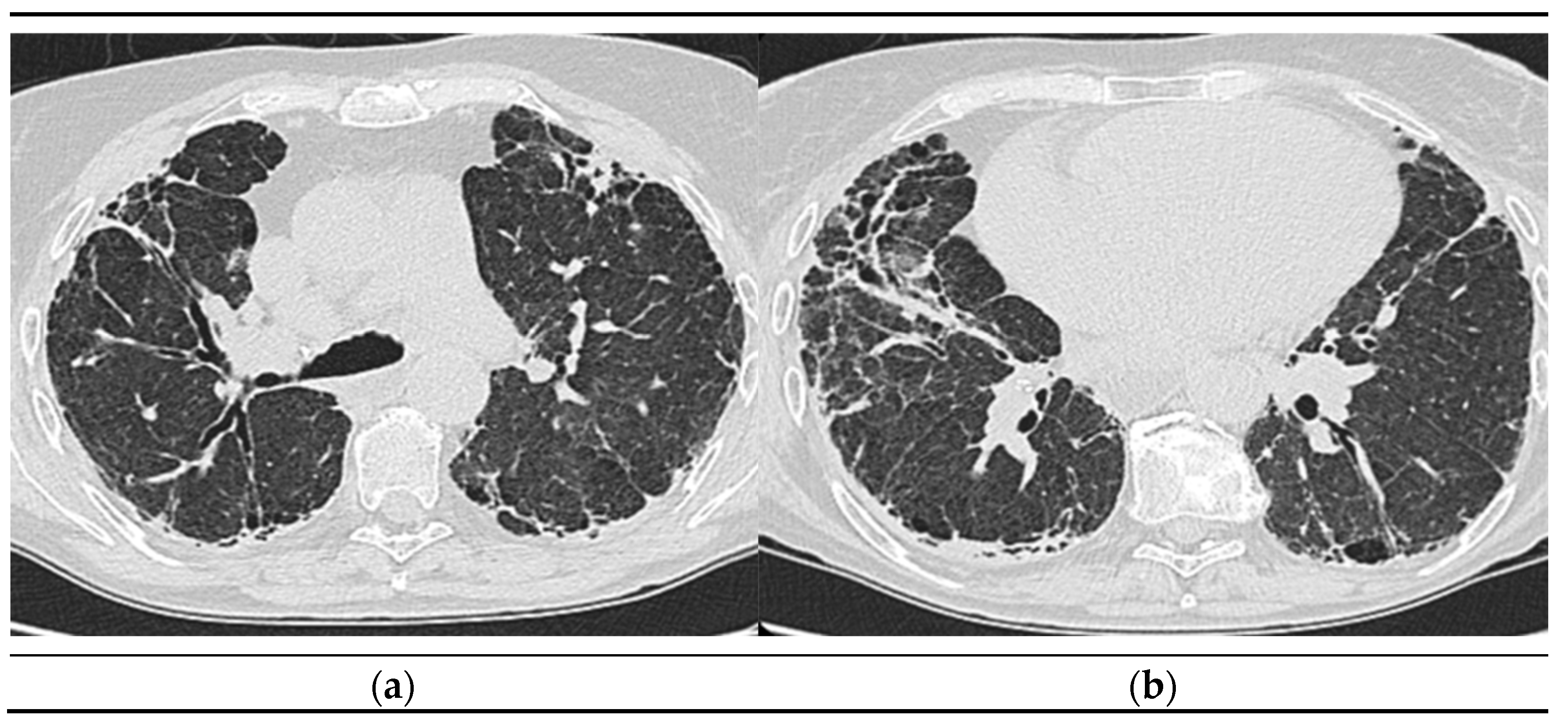
Figure 3.
a), b): Reevaluation of Lung Tomography.
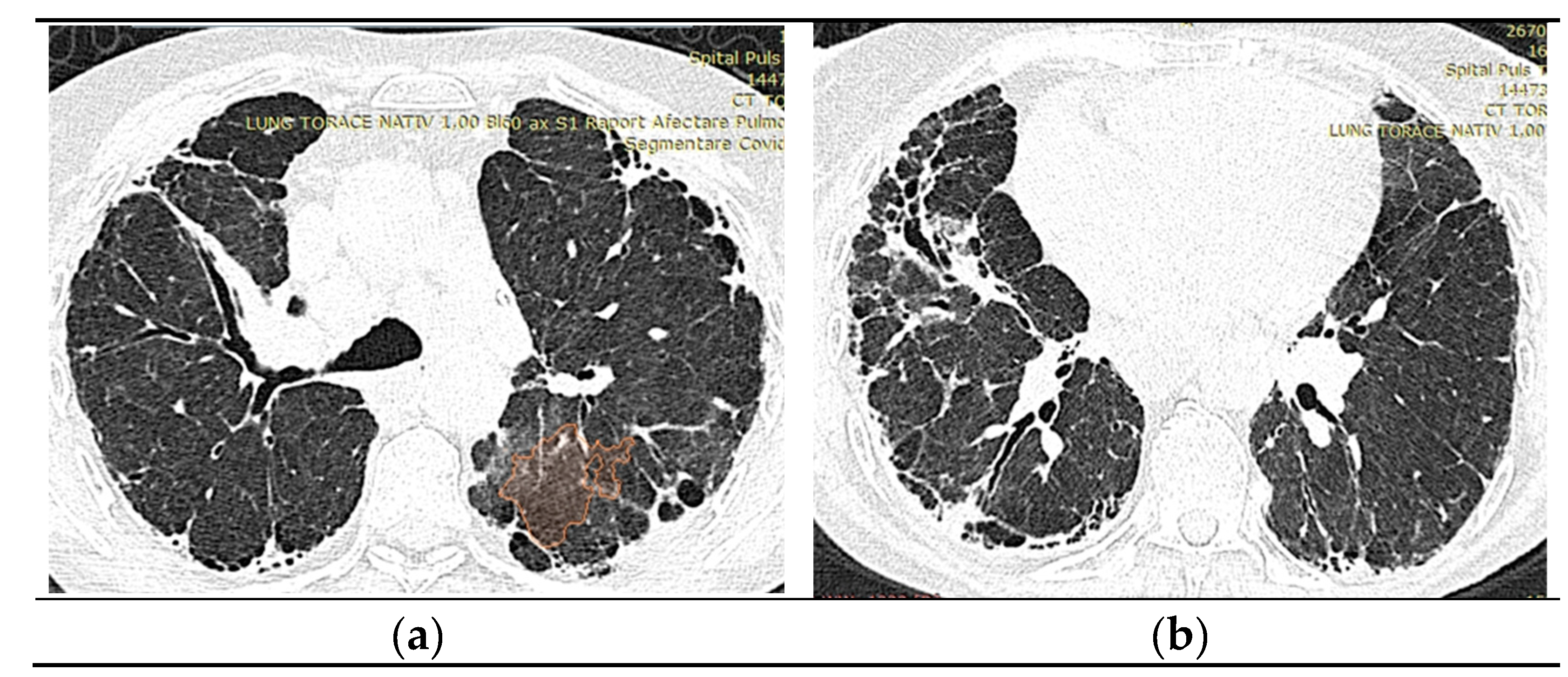

Table 1.
Laboratory Analysis.
| Parameter | Values |
|---|---|
| White blood cells | 4630/µl |
| Hemoglobin | 12.5 g/dl |
| Hematocrit | 38.8% |
| Platelets | 254000/µl |
| Uric acid | 303 µmol/L |
| Alanine transaminase | 12 U/L |
| Aspartate aminotransferase | 20 U/L |
| Total cholesterol | 4.37 mmol/l |
| Creatinine | 0.82 mg/dl |
| Alkaline phosphatase | 210 U/L |
| Glucose | 124 mg/dl |
| Lactate dehydrogenase | 233 U/L |
| C-reactive protein | 5.13 MG/L |
| Fibrinogen | 345 mg/dl |
| Total proteins | 7.2 g/dl |
| Anti-Scl-70 antibodies | 7.2 |
| Antinuclear antibodies | 25.3 UI/ml |
Table 2.
Echocardiography of the patient.
| Dimensions | Values in mm | Normal values |
|---|---|---|
| RV | 32 | 22-44 |
| LV | 36 | 35-60/21-40 |
| IVS | 11 | 6-11 |
| Post. wall | 10 | 6-11 |
| LA | 32 | 23-45 |
| LA dimension in cm2 | 15 | |
| LA volume | - | |
| RA | - | |
| RA dimension in cm2 | 16 | |
| Ao ring | 19 | 14-26 |
| Ascendent Ao | 23 | 21-34 |
| PAring | 21 | 10-22 |
| PAtrunk | 22 | 9-29 |
| EF | 55% | 60% |
RV: right ventricle; LV: left ventricle; IVS: interventricular septum; LA: left atrium; RA: right atrium; Ao: aortic; PA: pulmonary artery; EF: ejection fraction.
Table 3.
Densitometry evaluation.
| Densitometry Trend: Total Mean | ||||
|---|---|---|---|---|
|
Measured Date |
Age (years) |
BMD (g/cm2) |
Change vs. Previous (g/cm2) | Change vs Previous (%) |
| 28-Oct-2022 | 55.4 | 0.693 | -0.138* | -16.6* |
| 19-June-2018 | 51.0 | 0.831 | -0.031* | -3.6* |
| 12-July-2016 | 49.1 | 0.862 | - | - |
| Osteoporosis: YA t-score: -3.1 | ||||
Table 4.
Bodypletismography.
| Bodyplethysmography/ Flow-Volume | |||
|---|---|---|---|
| Pred | Pre | % (Pre/Pred) | |
| R tot | 0.30 | 0.35 | 116 |
| sG tot | 1.04 | 1.10 | 106 |
| R eff | 0.30 | 0.28 | 93 |
| FRCpl | 2.75 | 2.19 | 80 |
| RV | 1.87 | 1.55 | 83 |
| TLC | 5.10 | 3.4, | 67 |
| VC IN | 3.09 | 1.44 | 47 |
| FVC | 2.99 | 1.87 | 63 |
| FEV 1 | 2.54 | 1.46 | 57 |
| FEV1%M | 78.65 | 77.87 | 99 |
| FEV1%F | 78.65 | 77.87 | 99 |
| PEF | 6.32 | 3.54 | 58 |
| FEV6 | 4 | ||
| MEF 75 | 5.54 | 2.57 | 46 |
| MEF 50 | 3.63 | 1.74 | 45 |
| MEF 25 | 1.47 | 0.55 | 37 |
Table 5.
DLCO.
| Diffusion SB | |||
|---|---|---|---|
| Pred | Best | %(Best/Pred) | |
| DLCO_SB mmol/(min*kPa) | 8.06 | 2.04 | 25 |
| KCO_SB mmol/(min*kPa*L) | 1.58 | 0.65 | 41 |
| VA_SB (L) | 4.95 | 3.12 | 63 |
| Hb g(Hb)/dL | 13.50 | 13.40 | 99 |
| DLCO mmol/(min*kPa) | 8.06 | 2.04 | 25 |
| KCOc_SB mmol/(min*kPa*L) | 1.58 | 0.65 | 41 |
| VIN (L) | 0.00 | ||
| TLC_SB (L) | 5.10 | 3.26 | 34 |
| FRC_SB (L) | 2.75 | 2.20 | 90 |
| ERV_SB (L) | 0.88 | 0.72 | 91 |
| RV_SB (L) | 1.87 | 1.48 | 90 |
| RV%TLC_SB (%) | 38 | 46 | 121 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



