
Submitted:
17 October 2023
Posted:
18 October 2023
You are already at the latest version
Abstract
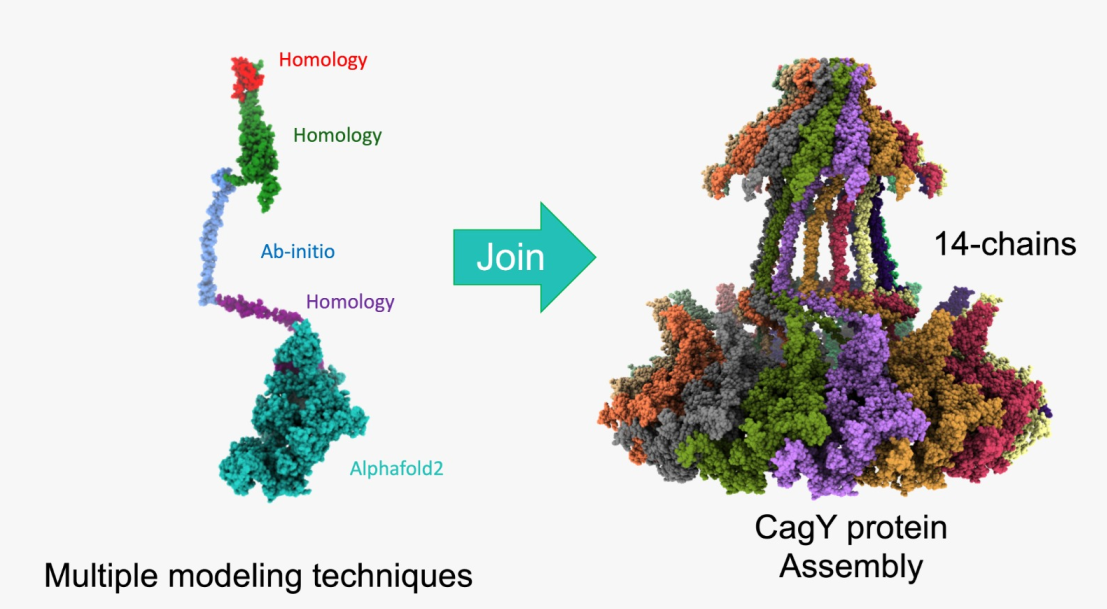
CagY is the largest and most complex protein from Helicobacter pylori's type IV secretion system (T4SS) and may participate in the modulation of gastric tissue inflammation. A three-dimensional structure has been reported for only two segments of the protein. To build a more complete model, particularly the region that spans between the outer membrane (OM) and the inner membrane (IM), we employed different approaches, including homology modeling, ab initio, and deep learning techniques. For the long-middle repeat region (MRR), modeling was performed using deep learning techniques and Molecular Dynamics. The modeled segments were assembled into a chain of 1595 aa, and a 14-chain CagY multimer structure was composed by structural alignment. The final multimer structure correlated with previously published struc-tures and allows to show how the multimer may form the T4SS channel through which CagA and other molecules are translocated to gastric epithelial cells. The model further confirmed that MRR, the most polymorphic and complex region of CagY, presents numerous cysteine residues forming disulfide bonds that stabilize the protein and suggest this domain probably functions as a contractile region that may play an essential role in the modulatory activity of CagY on tissue inflammation.
Keywords:
bioinformatics
; structural biology
; CagY protein
; T4SS
; deep learning
1. Introduction
Helicobacter pylori (Hp) is a bacterium that colonizes the stomach of more than 50% of the human population and has evolved to specifically grow in the harsh environment of the human gastric mucosa [1]. Gastric colonization is possible due to its spiral-shaped, multiple unipolar flagella and to the production of urease that counteracts the extreme acidity of the stomach [2,3].
Hp is usually acquired during early childhood, probably by intimate oral-oral contact with the mother [4] to co-exist for life with human gastric cells in most cases (over 80%), representing a clear example of microbiota vertically transmitted from mother to child. However, in a few cases, Hp might cause peptic ulcers and may become a risk factor for gastric cancer (GC) [5]. Hp strains with increased capacity to cause GC to encode virulence genes like those of the Type IV Secretion System (T4SS), adhesins, and a cytotoxin [6,7]. The Hp T4SS translocate the CagA protein, DNA, and heptose into the cytoplasm of gastric cells. CagA has multiple effects in the host cells, activating several pathways, and is the first bacterial protein recognized as an oncoprotein [6,7,8,9,10]. The Hp T4SS is a complex secretion system with partial homology to proteins from other bacterial T4SS [11,12]. The largest protein in Hp T4SS is CagY, a protein of about 2000 amino acids (aa) and a carboxylic terminal region homologous to VirB10, but the rest of the protein has no homology to other known proteins [9,10].
The CagY protein is widely variable in length and sequence, and its structure is highly complex, presenting two repeated regions (RR), one known as the 5′ region (FRR) (at the amino terminus), which is an intrinsically disordered region (IDR), and the other in the middle of the protein, the unusually long Middle Repeat Region (MRR). The MRR repeats are classified into A and B modules, which are composed of three distinct motifs: delta, mu, and alpha for module A, and epsilon, lambda, and beta for module B [13,14] (Figure 1a,b). It has been suggested that the variation in the number and location of the repeats in the MRR may be involved in the regulation of the translocation of Cag A throughout the T4SS and may also help to modulate the host immune response. CagY may also regulate gastric tissue inflammation by interacting with the Toll-like receptor 5 (TLR5) [15,16,17,18,19,20,21]. The middle repeat region is susceptible to rearrangements (by modification, deletion, and insertion of modules) that may affect the structure of CagY and hence its function, including modulation of the inflammatory response in the gastric mucosa [22] and the phosphorylation of CagA [23].
However, the complex and large size of CagY makes its study challenging, particularly in a high number of strains. Up to date, a total of 3446 cagY/CagY coding genes/proteins exists in the NCBI database, but most of them are truncated or incomplete (NCBI, 2023), mainly due to problems in sequencing and assembling the repetitive regions by short-read sequencing techniques. Only the recent long-read sequencing technologies (SMRT/PacBio or Oxford-Nanopore) have allowed us to get more accurate and reliable complete CagY sequences to study their complexity and possible function better.
Due to the complex characteristics of the CagY protein, its tridimensional structure is not completely elucidated yet, with only parts of the VirB10 homologous region recently reported [24,25], although this region represents only a fraction of the protein (approximately 20% of the sequence). Elucidating the structure of CagY and of the whole T4SS is a challenge, but it is essential to better understand the interaction and mechanisms of function of the SS proteins. The most recently reported structure elucidated by Cryo-EM shows the core of T4SS, composed of an Outer Membrane Complex (OMC), a Periplasmic Ring (PR), and a Stalk, where fragments of the proteins CagY (Cag 7), CagX (Cag 8), CagT (Cag 12), CagM (Cag 16) and CagD (Cag 3) have been identified. The pilus of the T4SS was found to be composed mainly of CagY and CagL [26]. However, the previous works have helped to partially decipher the assembly of T4SS. Still, most of the tridimensional structure of CagY is unknown. CagY seems to play a major role in the structure and function of T4SS, probably spanning the inner Hp membrane, the periplasmic space, and the outer membrane facing the host cell [11,12]. It is then of the utmost importance to clarify the structure of the whole protein and the function of this major protein in the T4SS.
As a complementary tool to the experimental techniques to elucidate the protein structure, different bioinformatic methods can be used to model and predict the structure of proteins [27]. For years, the most accurate method for protein structure prediction was homology modeling, where a tridimensional known structure is used as a template for modeling the structure of a close sequence homolog [28]. Until recently, these computer methods had limitations for predicting the structure of proteins with low identity to homologous with known tridimensional structure. However, with the advent of Deep learning approaches such as AlphaFold2, bioinformatic methods for structure prediction reached unprecedented accuracy [29,30]. Threading methods, also known as fold recognition methods, are currently used to search potential low-similarity templates for remote homologous [31]. In ab initio modeling, methods are used to build the structure of fragments from the amino acid sequence with the help of short tridimensional templates from known structures or molecular dynamics modeling and posterior simulation and optimization for their assembly [32]. On the other hand, deep learning-based modeling uses neural networks to calculate the tridimensional structure from all available information in structural databases [33]. These methods can be combined to achieve the highest quality and efficiency in protein modeling [34]. Nevertheless, even these methods still have limitations for complex targets such as CagY. In this work, we combine different approaches to elucidate a theoretical structure for the CagY protein, which may allow us to understand how it interacts with the remaining proteins of the T4SS.
We propose a tridimensional model for most of the CagY protein based on all available data on the protein and theoretical data presented in multiple works on CagY. We used tridimensional models available for fragments of CagY in the PDB database for homology modeling and ab initio methods to model segments of CagY with missing tridimensional structures. Moreover, deep learning methods (DL) were used to predict the structure of the MRR of CagY.
2. Results
2.1. Building the CagY model.
For modeling purposes, the CagY protein was divided into different motifs/segments, as described in Figure 1a and Table 1. We divided the protein into three functional domains: the Five repeat region (FRR), the Middle Repeat Region (MRR), and the homologous VirB10 Region, and highlighted the motifs corresponding to the two predicted transmembranal regions and the antenna projections (AP) [35]. Figure 1b summarizes the predictions for the secondary structure of the protein using the PsiPred server. Figure 1c shows the logos for modules A and B from the MRR, calculated from the consensus sequences of the repeats present in the CagY sequence (WP_103414807).
A model based on the symmetric 14-chain CagY model was constructed, including most of the protein. For the purposes of this work, the suggested asymmetric complex was not considered for the construction of the model.
2.2. Modeling of the homologous VirB10 Region
The first step for building the tridimensional model was the homology modeling of the CagY protein for the Hp 26695 (sequence accession WP_103414807) using the CagY chains from 6ODI and 6X6J structures as templates. The model was based on the symmetric model 6ODI containing only 14 subunits of CagY. The 6ODI template only contains the residues of segments of CagY 1677 to 1816 and 1850 to 1907, and the 6X6J template includes the residues from 1469 to 1603. These templates were helpful in the alignment of residues 1469 to 1603 and 1677 to 1907 from 6ODI and 6X6J, respectively (Figure 2a).
The amino acids 1817 to 1849, missing in the 6ODI structure, are part of a region called Antenna Projections (AP), a channel-like domain formed by multiple helix-loops [35]. The AP region of CagY is located as a “crown” structure at the upper portion, above the OM. [11,12] Its structure was modeled for the 26695 WT by homology modeling with Swiss-Model/Users-template, using the 6ODI structure as a 14-chain multimeric template.
There is also a missing region, corresponding to residues 1604 to 1677, which joins CagY segments from the structures 6ODI and 6X6J. Secondary structure predictions of this connecting segment, belonging to the PR of the T4SS, were compatible with an alpha-helix motif (See Figure 1b).Independent ab initio modeling of this region with I-Tasser and Robetta yielded similar alpha-helix models with the proper dimensions for fitting the space between the 6ODI and the 6X6J regions, varying only slightly in the overall conformation of the strand. We selected the model provided by the I-TASSER server.
Therefore, a monomer model of residues 1469 to 1833 was calculated using a single chain of the 6ODI, one from the 6X6J, and the I-Tasser model as templates, and the assembled model of the three regions is shown in Figure 2b. The resulting model was used to align each of the 14 chains of the 6ODI structure, including the AP structure models predicted by Swiss-Model, to produce the symmetric multimeric model shown in Figure 2c. Interestingly, in this model, the region corresponding to the 6X6J structure fits a space similar to that filled by this protein in the asymmetric 6X6J structure, but with a slightly higher interspace between the subunits, despite this structure has 17 alternated units of CagY and CagX, respectively.
2.3. Modeling of the Middle Region of CagY
The most challenging region of CagY, which includes part of the transmembrane motif and the MRR (from residues 366 through 1469), was initially modeled with programs Robetta, I-Tasser, and ColabFold2/AlphaFold2. However, the final structural conformation of the single chain resultant models differed considerably between programs, except by a strong coincidence in the secondary structure assignation (Figure 1Sa). This assignation was like the secondary structure prediction with Psipred (Figure 1b y Figure S1b.- models of I-Tasser and Robetta for MRR). The structure predicted with ColabFold2/AlphaFold2 had considerable differences with the anterior models (Figure S2a) but got reasonable support, according to its predicted IDDT (Figure S2b, supplementary material).
To improve the modeling of the MRR, we calculated a dimer model of this region with ColabFold2/AlphaFold2-Multimer. Memory limitations in our computer equipment prevented the calculation of a high order multimer. It has been reported that the multimer option in ColabFold2/AlphaFold2 may result in a more efficient prediction of the structure of multimeric proteins [33]. Confidence values (predicted IDDT values) for the dimer structure were lower than for the monomer, and it was only possible to calculate two dimer models. However, both models showed remarkable similarities with the monomer, with a more compact packing (Figure S3a), and an IDDT value above 60, which implies reasonable confidence for the dimer modeling (Figure S3b). In addition, both chains in the dimer had similar conformations and were oriented in parallel from the C to the N terminus (Figure 3a). By aligning the chains, A and B of the two tridimensional structures for the best dimer model, we built a trimer ABB’ (Figure 3b), which was subject to an EMD of 10 ns in explicit solvent (Figure. 3c). The global RMSD values for the trimer showed that its conformation did not stabilize during this period. However, a closer examination of the RMSD values of the individual chains shows that chains at the extremes of the model (A and B’) have higher structural variation.
In contrast, the central structure (B) presented lower variation and showed a tendency to stabilize. These results suggest that the interactions of the central chain with the neighboring chains are essential for maintaining the conformation in the multimer. Therefore, we considered the trimer’s central chain (B) as the representative conformation of CagY (Figure S4).
The representative structure of the trimer, optimized by EMD, was used to calculate their stereochemical properties with the PDBsum-EBI server. The Ramachandran plot showed that over 95% of the residues had psi and phi angle values in the most favored regions (Figure S5a). Moreover, the analysis also suggested that the 58 Cys residues in this region participate in disulfide bond formation (Figure S5b). The disulfide bonds were also found in the non-optimized structures for the monomer and the dimer. The PDBSum information for the trimer structure indicated only potential disulfide intrachain bonds, but there was no support for interchain bonds.
A detailed examination of the reported structure analysis showed that the 29 disulfide bonds are homogeneously distributed in the repeated modules, with two cysteines per module (Figure S5b). Each disulfide bond was consistently placed between a short alpha-helix (10 aa) and a consecutive long alpha-helix (17 aa), with a spacing of 3 aa. It was outstanding to find that cysteine was found only in this region of the protein.
2.4. Final assembly of CagY
In the structure of the dimer and the optimized structure of the trimers, the MRR fragments of CagY were oriented parallelly with the N and C extremes placed in opposite positions (Figure. 3a and 3b). Therefore, joining the optimized central structure of the trimer with the previous model of the OM and PR regions was simple, as shown in Figure 4a. Similarly, this structure was aligned with each of the 14 chains of the 6ODI structure, yielding the final model shown in Figure. 4b.
We previously mentioned that CagY has several disulfide bonds. It is interesting that only in the MRR of CagY protein are present cysteines. The cysteines are distributed in the 14-chain complex and appear to outline two rings of these residues in the CagY model (Figure 4c).
A close view of these cysteine residues in the MRR of the protein, as well as a bottom view of the entire CagY complex, are shown in Figure. 4c. We can see that the series of disulfide bonds seem to be homogeneously disposed of, describing apparent concentric “rings” to the central channel of the multimeric complex.
This bell-shaped structure of the ordered component of CagY fits well with previously reported microphotographs of the structures of OMC in the membrane and with the electron microscopy maps of the OMCC as described below (Figure 5).
The electron density map for the T4SS OMCC of Hp, obtained by Single-Particle Electron Microscopy, with a resolution of 3.8 Å (EMDB, access EMD-20020) is shown In Figure. 5a, which shows a good fit with the structures PDB 6odi, 6oeg, 6oee, 6oef and 6oeh of the PDB as shown by Chung et al., 2019. The section with the largest volume, the upper crown, corresponds to the structures previously reported for the OMCC. Furthermore, two diffuse clouds were visible beneath this structure, as we show in the Figure 5b. The best-outlined cloud corresponded to the part of the complex that is located below the internal membrane IM, which includes the potentially disordered region of CagY, the proteins of the internal membrane and the energy complexes. Above this region, another, less-defined cloud was identified in the area where the previously missing part of the CagY that includes the MRR would be expected to be found. Although the resolution of this technique is not sufficient to calculate the structure that occupies this region, the predicted model fits correctly with this region (see Figure. 5b upper figure). It is important to highlight that the Cryo-EM map shows a tunnel/pore in the middle of the image that fits adequately with our model (Figure 5b).
On the other hand, the assembled multimer of CagY was aligned with the 6X6S and 6ODI structures (see Figure 6a), which corresponds to the OMC, PR, and stalk of the Hp wild-type strain [12] .The structures have a good fit, and it is interesting to see that some portions of the MRR in the modeled CagY were at a close distance (about 5-6 Å) from each of the 14 regions of the OMC that authors reported as unknown protein fragments, i.e., fragments observed in the experimental maps of 6X6S but not identified at the sequence level (Figure 6b). It is possible that some of these fragments may cor-respond to portions of CagY in the experimental maps. However, they may also belong to a different protein that mediates the contact between these regions [12].
2.5. Changes in the AP region of CagY alters the conformation of the OMC
We also built variants of the CagY corresponding to the AP described in a recently published work [47] to see if the present model can provide insights regarding the trans-location properties of the CagA protein described in this work. The AP region of CagY corresponds to a loop in the 1820 to 1851 aa, which is not visible in the 6ODI model and is reported as a missing structure. This region is flanked by two α-helix motifs that include the residues 1763 to 1863. We modeled this region by homology with Swiss-Model (Figure 7a) and deep learning with ColabFold/AlphaFold2 (Figure 7b). Whereas the structure models for the WT sequence look similar for both methods, we see topologic differences in the remaining models. In the models based on homology (Swiss-Model), a crown-like shape is observed, which increases in diameter as the length of the deleted AP region increases. By comparison, the deep learning multimer models show considerable changes in the conformation of the pore with respect to the homology models. The pore diameter for WT (Figure 2b) and Glicine-Serine mutant (GS20) (Figure 7d) of the deep learning models is similar and corresponds to the largest diameter for AP regions. The pore diameter for the Xc CagY is considerably shorter. The DL model, which corresponds to the mutation of 20 residues to glycine and serine (Fig7d) in this section, shows an entirely closed pore with a considerable change in conformation. By contrast, Swiss-Model models for the CagY Xc (Figure 7e) and GS20 (Figure 7c) are shorter and have more extended pore overture. The structure in which the AP region was completely deleted shows a pore with an even larger diameter than those of GS20 (Figure 7g) and Xc model, but a significant structural change is also observed with respect to the 6odi structure used as a template in this region. In summary, the DL models predict pores with smaller diameters when the AP length decreases and changes the amino acid sequence, while the Swiss-Model models predict shorter AP models and larger pore diameters when the AP length decreases.
3. Discussion
The CagY protein is an essential structural component of the T4SS of Hp with a key participation in its regulation [13,14,20,22,36,37]. The length of the protein from the Hp 26695 reference strain is 1927 amino acids and has unusually long repetitive elements (close to 900 amino acids) that might be related to their immunogenicity and pathogenicity properties [15,20,38].
It has been reported that the CagY protein completely spans the cell envelope of Hp from the Outer Membrane (OM) to the Internal Membrane (IM). This arrangement agrees with bioinformatic predictions of the secondary structure of CagY, which show two transmembrane regions (Figure 1a). Considering its length and extension through the membrane, it is suggested that CagY plays an essential role in the translocation of the Cag A oncoprotein [11,12,39].
CagY is an unusually long protein that seems to have evolved to perform multiple activities associated with a specific region of the protein. Thus, in order to better understand its functions, we need to thoroughly study its sequence and structure, which, because of its large size and complexity, has not been an easy task. Preliminary analysis of CagY secondary structure showed that residues 5 to 343 correspond to a repeated region domain in the amino terminal (Figure 1a), also known as FRR. The next region (344 to 365) corresponds to a transmembrane region, which may allow anchoring the protein to the IM, although it is unknown if this domain associates with other proteins [13]. Positions 366 to 1458 correspond to a region that includes the Middle Repetitive Region (MRR), almost 1,000 aa long, sometimes described as a conserved region [13,36]. In fact, it is the most polymorphic region of CagY among different Hp strains [13,36].
). Secondary Structure predictions of this region show essentially short alpha-helix motifs, mainly associated with the repeated modules (Figs. 1a and b). CagY proteins from different Hp strains show changes in the arrangement and number of these modules, affecting the length of CagY (although the FRR also contributes to some extent) [13,36].
The repeated modules have been classified as A or B (Figure 1a,c). It is worth noting that this is the only region in the protein where the amino acid cysteine is present. In an unusual number and distribution, e.g., in Hp 26695 strain, it has a total of 42 residues, two for each repeated module. It has been suggested these residues may be involved in the formation of disulfide bonds, playing an essential role in the folding stability of CagY, probably also needed for maintaining the multimer structure, and function as a modulator of the T4SS activity [13,14].
The region from residues 1469 to 1927 has been majorly described in different structural studies with cryo-EM and cryo-ET techniques [11,12,24,39]. This is also the only region of CagY that has homology with the VirB10 proteins from other bacterial T4SS, such as Escherichia coli and Agrobacterium tumefaciens [12,13,24]. Experimental structures for this region are reported in the RCSB-PDB database (Table 1). However, recent studies have reported an asymmetric complex with 17 chains of CagY/Cag X (6X6J) and a symmetric complex with only 14 CagY chains (6ODI), which is known as the symmetry mismatch [11,12]. Previous studies with cryo-ET have only detected a 14-chain structure complex thought [12,24].
Due to the complexity of this protein, its complete structure has not been experimentally elucidated yet. Recent structural works with cryo-ET and cryo-EM have shown an impressive complex assembly for the T4SS complex, with several proteins coded in the Cag PAI, like Cag X, CagY, CagT, CagM, and Cag3. However, most of the CagY protein structure remains unsolved. Bioinformatic sequence analysis predicts that the C-terminal of this protein corresponds to an Intrinsically Disordered Region (IDR), consistent with sequence composition with amino acids that promote sequence disorder. This region is on the interior side of the Hp IM, and its structure is difficult to predict because of its disordered character and possible interaction with other proteins, like the energetic protein complexes of T4SS (Figure S6) [26]. Proteins to which CagY interacts, according to STRING, are CagE virB homologs involved in DNA transfer and required for induction of IL-8 in gastric epithelial cells, CagT or TrwB, an inner-membrane nucleoside-triphosphate-binding protein. The periplasmic region of CagY interacts with VirB9 and VirB10. This family also includes the conjugal transfer protein family TrbF, a family of proteins known to be involved in conjugal transfer. The TrbF protein is thought to compose part of the pilus required for transfer. This domain has a similar fold to the NTF2 protein and CagX, a conjugation protein.
Starting from the structures obtained from cryo-ET and cryo-EM studies, we built an initial model by homology modeling. Despite the structural discrepancies between the models, we used the 6ODI and 6X6J structures, focusing on building a model with only 14 chains of CagY, consistent with cryo-EM studies. A segment of 32 amino acids between 6ODI and 6X6J corresponds to an “empty zone” not solved in the cryo-EM studies. We filled this space with an ab initio model for the corresponding residues at this zone. This step was relatively simple because most bioinformatic methods for secondary prediction and ab initio and threading modeling methods showed agreement with an alpha-helix conformation.
The symmetry mismatch, previously described in cryo-EM studies, arises from detecting only 14 CagY chains in the structures closer to the OM and 17 units in structures from the PR region, without an evident connection between the 3 CagY chains in excess. Authors of the cryo-EM works admitted that it was not possible to explain the causes of such structural discrepancies. Possible explanations include the presence of truncated versions of CagY (and Cag X) in the T4SS, conformational isomers of these proteins, or early or incomplete assemblies of the T4SS. [11,12,39]. However, previous cryo-ET studies have reported only 14 chains for the same complex. Therefore, we decided to deduce the CagY structure model using the symmetric 14 chains of CagY. As was demonstrated in the built model (Figure 2c), the space occupied by the “asymmetric complex zone” was similar, with a slightly wider space between the chains.
More challenging for modeling is the zone that includes the MRR, as it corresponds to the most polymorphic and complex zone of CagY. This zone is of particular interest since previous studies associate sequence variations with the modulation of the T4SS activity for CagA translocation, the immunogenicity response, and the phosphorylation of the CagA oncoprotein [5,15,36].
Sequence variations include losses and gains of the repeated modules A and B and single residue substitutions. Some authors have also highlighted the high degree of sequence conservation of the modules, even at the nucleotide coding level.
Previous studies suggest that the initial T4SS assembly should start by placing CagY and CagX in the bacterial envelope [40]. Because the multimer structure of CagY and Cag X can be remarkably stable and independent of other proteins, the absence of Cag3 in mutant strains interferes with the incorporation of other proteins of the Cag PAI. Still, it does not avoid the assembly of CagY and CagX. [12,41]. CagX is shorter than CagY and seems to interact only with the OM section of CagY but is absent in the IM region. Therefore, we consider a reasonable assumption to build multimer models of this protein domain in the presence of the CagX protein.
Our first attempts to build structure models for this domain produced results with considerable differences in conformation. Nevertheless, usually, there was good agreement on the secondary structure. The ColabFold2/AlphaFold2 DL has been recognized as the most confident approach for predicting protein structure, reaching “experimental confidence” in some cases [33,42,43]. However, the method still has limitations for challenging targets, which include complex and large proteins and those for which there are not enough templates for modeling, like CagY [44]. When trying to model the structure for the whole sequence of CagY, ColabFold2 yielded a structure that was mainly disordered and difficult to align with the known partial structures from cryo-EM studies. Therefore, we divide the problem by identifying the previously determined domain structures and sequence motifs to build hybrid structure models. On the other hand, recent works have remarked that DL approaches are bioinformatic methods that produce highly confident structure models for proteins with repetitive modules [45,46] and disulfide bonds [47].
We focus on the DL approaches for modeling the MRR with ColabFold2/AlphaFold2. The modeling as a monomer of this domain had a more compact structure than those obtained with ab initio methods such as I-Tasser and Robetta (Figure S1). However, it was not possible to establish its correct orientation to the remaining part of the CagY structure. The modeling as a multimer was challenging because of memory limitations due to this domain’s large size and complexity. Still, it was possible to estimate models for the dimer. Despite the low confidence values for these predictions, the dimer model showed a more compact CagY structure placed parallelly, allowing us to deduce their correct orientation regarding the remaining part of the model. Possible causes of the low confidence values are probably an insufficient refinement of the conformation of individual chains in the dimer, which showed considerable differences (RMSD > 2Å). On the other hand, stereochemical and structure analysis of the monomer and dimer models showed that all the cysteine residues were grouped in pairs at the correct distance to participate in disulfide bonds (typically less than 2.5 Å), further supporting the accuracy of the model [48].
We refine the conformation by EMD by building a trimer from two dimer models to optimize the conformation of the central chain and including restrictions for the predicted disulfide bonds. The RMSD behavior of the trimer dynamics was consistent with the hypothesis that the neighboring chains help maintain the multimer’s conformation. Notably, the chains with only a single neighbor did not stabilize in conformation. Therefore, we consider the conformation of this central chain as the most appropriate for assembling the remaining part of the model.
A key observation was the presence of unusually large numbers of Cys (58 in Hp 26695 strain) separated at very regular intervals in the MRR region, a number rarely observed in any protein. It has been suggested that Cys content is correlated with evolution, and prokaryotes have the lower content and mammals the highest [49]. Small human proteins represent some of the richer Cys proteins, like in metallothionein domain proteins (MTs) or in granulins (GRNs) with over 20% of Cys [50]; however, we are unaware of other large proteins with such a high number of Cys concentrated in a region of the protein like in MRR of CagY. Besides, Cys usually presents as C-(X)2-C motifs, with disorders domains interspersed with Cys. The above highly suggests a particular evolution history of MRR, different from the rest of CagY protein and probably different from most Cys-rich proteins.
Previous studies already reported the presence of Cys in the MRR of CagY, and authors suggested they may participate in the formation of disulfide bonds and the stability of the region [13,14]. The process involved in their formation and the functions of the disulfide bonds in prokaryotes have been less studied than in eukaryotes [47]. Of note, these motifs are commonly found in transmembrane and extracellular proteins. In our study, the structural analysis of the trimer showed evidence of intrachain disulfide bonds but not interchain bonds. Therefore, these bonds do not seem to play an essential role in the quaternary structure but seem to be most relevant for the stability of the complex structure of CagY, particularly in the large MRR region.
It has been previously described that Hp has numerous Cysteine-rich proteins and that many of them possibly form disulfide bonds. [51]. For many prokaryotic organisms, the enzymes DsbA and DsbB are the most frequently involved in disulfide bond formation [47]. However, these enzymes are absent in Hp, and instead, the enzyme DsbK (HP0231 gene) is present and is most probably involved in this process [52,53].
The finding that CagY has cysteines forming disulfide bonds is essential because several studies have remarked that this covalent interaction can play an important role in the dynamics and modulation of proteins [54,55]. We propose a scenario where CagY modifies its structure to modulate the translocation of the CagA protein. These disulfide bonds can act as a Redox switch or mediate the contraction of this protein region to modulate the T4SS function and Cag A translocation. Cys-rich proteins are reported to be important to respond to oxidative stress. They may then be essential in the response and regulation of inflammation [56]. It is intriguing to suggest that the redox-sensitive potential of Cys residues in the MRR region of CagY may play a role in its well-described regulation of inflammation in animal models [53,55,57].
The model of the CagY structure proposed in this work can be used to understand how this protein interacts with CagA, DNA and heptose during their transit through the T4SS and the importance of the MRR repeat modules in the reported CagY functions. The tridimensional model will also help better understand its interaction with other proteins of T4SS, such as CagX, CagM, CagT, and Cag3, which are part of the core complex of the secretion system (see Figure 6b). Our model suggests that CagY constitutes the tunnel that spans from the inner membrane of Hp to the membrane of the gastric cell in its host, and the assembly of the T4SS components illustrates the role of CagY as the “skeleton” of the system around which all proteins assemble.
Some studies have suggested that changes in the AP regions and in the MRR are associated with the translocation capacity of the CagA protein, the immunogenic response, and the phosphorylation capacity [35].
The models obtained by DL and homology for the AP in this work allow us to propose, in relation to the recent work of Tran et al. 2023, that the shortening of the AP region may also affect the translocation capacity. by altering its interaction with the OM of Hp. In contrast, the decrease in the pore channel’s diameter may obstruct CagA’s passage. It is important to emphasize that the size of the CagA protein is considerably larger than the AP pore, so the translocation process is more complex and may involve major structural changes.
On the other hand, the models calculated by homology tend to fit the dimensions of the template (in this case, the 6odi structure), which may explain the opposite prediction of the DL methods with respect to the pore diameter. DL methods consider a multitude of factors, and it is generally accepted that they model more efficiently the interactions between the chains [33], suggesting they can offer a more accurate description of the conformational changes of the pore. However, to obtain more precise data, it will be necessary to use other techniques, such as molecular dynamics of these complexes, preferentially including the membrane, and our model may be used as a starting point for such simulations.
The structural prediction of the MRR region may provide essential clues regarding its role in modulating the translocation and phosphorylation of CagA and the immunogenicity associated with the polymorphisms reported in this region. The presence of disulfide bonds in this region, as well as the similarity with some contractile proteins as detected by I-Tasser (Table 2), suggest that MRR has contractile properties, and given the interaction of the disordered region of CagY with the energy complex beneath the IM, its function might be similar to that of flagellar or pilins proteins [58,59].
Contractile properties may play a critical role in modulating the translocation capacity of CagA and other molecules. We plan to conduct MD studies to predict better conformational changes in the MRR and AP regions and their possible associations with the phenotypes produced by CagY variants present in Hp strains of patients with different gastric conditions.
We recognize that a limitation of our proposed model is that we did not include the 17/14 CagY asymmetric model previously suggested [12] and considered only the symmetric 14 CagY subunits to build the model. However, with the available data, it is not possible to determine whether the asymmetry corresponds to incomplete versions of CagY or to important conformational topology and arrangements of the CagY multimer.
In conclusion, using a combination of informatics and analytic tools, we provide a structural model for almost the complete CagY protein, particularly the complex and highly diverse MRR region, and present evidence of the role of the unusually abundant Cys residues in stabilizing the protein. We also modeled the AP region and provided proof of the role of sequence variants in CagA translocation. Finally, we offer a more detailed model for the assembly of the T4SS focused on the central role of CagY and show the agreement of our model with cryo-ET and cryo-EM studies.
4. Materials and Methods
4.1. CagY model sequence and structure models.
The amino acid sequence used to model the structure of the CagY protein corresponded to the wild-type (WT) strain Hp 26695 and was obtained from the NCBI database (WP_103414807), where the cagY gene Structure models 6X6J and 6ODI, obtained by Cryo-EM, are available in the Protein Data Bank (PDB) and were used as homology modeling templates.
4.2. Determination of Secondary Structure and Transmembrane Regions
PSIPRED workbench v4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) was used to predict structural domains and secondary structure of the CagY protein. The modules for predicting secondary structure (PSIPRED 4.0), sequence disorder (DISOPRED3), and transmembrane regions (MEMSAT-SVM) were employed.
4.3. Modeling of the Protein Monomer and Construction of the Multimer
The Modeller v10.3 program [60] was used for homology modeling of the CagY individual chains covered in 3D models 6X6J and 6ODI, which correspond to part of the OM and PR regions of the T4SS. These structures were obtained by cryo-EM at atomic resolutions of 3.50 and 3.80 Å, respectively. The 60DI corresponds to a 14-chain multimer structure composed exclusively of CagY structures (residues X to Y). The 6X6J structure belongs to a 34-chain structure; half correspond to CagY (residues X to Y), and the remaining 17 chains correspond to CagX. Due to the odd number of chains of both CagY and CagX, this multimer is known as the asymmetric complex.
The positions of the atoms in the structures were preserved in the models relative to the T4SS structure. Modeling of the missing segments of CagY was performed with the Swiss-Model [61], I-Tasser [62], and Robetta servers [63]. Also, Modeller’s multi-template scripts for homology modeling were used for the assembly of models derived from homology and segments from ab initio, as is described later. The 3D alignment and assembly of the CagY multimers for the OM and PR regions were made with UCSF ChimeraX 1v.6.1 [64] using the coordinates of CagY present in the 6ODI and 6X6J structures as templates.
4.4. Modeling of the Middle Repeat Region (MRR) of CagY
The ColabFold2/AlphaFold2 platform was used for the calculation of the conformation of the segment, including the xx aa long (aa positions a to b) MRR of the CagY protein. The AlphaFold2/MMseqs2 method [42] was selected using the pdb70 template mode, with the mmseqs2_uniref_env and unpaired_paired options, which suggested five models of the structure, which were evaluated using the IDDT confidence coefficient [65]. The MRR segment was also tested for dimer modeling (AB) using the ColabFold2/AlphaFold2-Multimer option. To improve the multimer building, a trimer model was generated from the alignment with ChimeraX of two identical dimers (labeled AB and A’B’) through the aligning of the chains B and A’ of the dimers, and posterior remotion of one of the superimposed chains (A’), to produce an ABB’ trimer. The 3D conformation of the trimer structure was optimized using a 10 ns Equilibrium Molecular Dynamics (EMD) in explicit solvent, and the system was configured using the CHARMM-GUI platform [[66] The trimer model from the protein surface to the edges was then immersed in a solvation box, with edges at a minimum distance of 10 Å from the protein, in an aqueous solution with KCl, and Charmm36m was selected as the force field potential [67]. The dynamic optimization was done using theNAMD3 program [68] with a canonical ensemble (isothermal-isochoric ensemble) in combination with an NPT ensemble (isothermal-isobaric ensemble) and a temperature of 303.15 K. The RMSD of the final EMD trajectories was computed with VMD 1.9.4 [69]. A PDB representative for the cluster of structures in the stable trajectory of the trimer central chain (frame 95) was selected with ChimeraX. The stereochemical and optimized structural properties of the monomer, dimer, and trimer were analyzed with the PDBsum-EBI server [70].
4.5. Final Assembly of CagY multimer
A CagY multimer structure with 14 chains was assembled with models from ab initio, homology modeling, and the central chain (B) of the trimer model obtained by the EMD. CagY coordinates in the 6ODI and 6X6J structures were used as a reference, as described below. The assembly was completed with ChimeraX and a manual edition of PDB files. The final CagY multimer model was assembled comprising amino acids 363 to 1927, and the IDR of the protein (residues 1 to 362) was not included.
4.6. Modeling of the Antenna Projections (AP) of CagY
Additionally, we modeled the structure corresponding to the AP sequences described in the work of Tran et al., 2023, and built the structures described as CagY ΔAP without the AP (residues 1763 to 1863 aa), CagY GS20 by replacing several residues in the AP region with glycine and serine (residues 1820 to 1851 aa), and CagY Xc, by replacing the AP region with the AP region of Xanthomonas citri (1820 to 1851 aa). These regions were modeled with Swiss-Model/DeepView by using the previously built multi-template model and Colabfold/AlphaFold2 multimer.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.M-T., J.T., E.E.S-P., and M.A.L-L; methodology, A.M-T., J.C.P.-F., E. E. S.-P. and M. A. L.-L.; writing—original draft preparation, A.M.-T., M.A.L.-L., and J.C.P.-F.; writing—review and editing, J.C.P.-F., M.A.L-L., A.M-T., E.E.S-P., J.T., and R.M.-R.; supervision, A. M.-T. and J. T.; project administration, A. M.-T.
Funding
This research received no external funding, postdoctoral scholarship 387325 to J.C.P-F., doctoral scholarship 988721 to M.A.L.-L., and doctoral scholarship 739485 to E. E. S.-P.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data found in this work is currently being uploaded in the Model Archive Database.
Acknowledgments
We thankful to Dr. Jay and Dr. Nina for the suggestions to the realization of this work.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of human migrations in Helicobacter pylori populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 2006, 19, 449–490. [Google Scholar] [CrossRef]
- Reshetnyak, V.I.; Reshetnyak, T.M. Significance of dormant forms of Helicobacter pylori in ulcerogenesis. World J Gastroenterol 2017, 23, 4867–4878. [Google Scholar] [CrossRef]
- Kayali, S.; Manfredi, M.; Gaiani, F.; Bianchi, L.; Bizzarri, B.; Leandro, G.; Di Mario, F.; De’ Angelis, G.L. Helicobacter pylori, transmission routes and recurrence of infection: state of the art. Acta Biomed 2018, 89, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M. Molecular Mechanism of Helicobacter pylori-Induced Gastric Cancer. J Gastrointest Cancer 2021, 52, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun Signal 2011, 9, 28. [Google Scholar] [CrossRef]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Skoog, E.C.; Morikis, V.A.; Martin, M.E.; Foster, G.A.; Cai, L.P.; Hansen, L.M.; Li, B.; Gaddy, J.A.; Simon, S.I.; Solnick, J.V. CagY-Dependent Regulation of Type IV Secretion in Helicobacter pylori Is Associated with Alterations in Integrin Binding. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Akopyants, N.S.; Clifton, S.W.; Kersulyte, D.; Crabtree, J.E.; Youree, B.E.; Reece, C.A.; Bukanov, N.O.; Drazek, E.S.; Roe, B.A.; Berg, D.E. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol 1998, 28, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Banta, L.M.; Bohne, J.; Lovejoy, S.D.; Dostal, K. Stability of the Agrobacterium tumefaciens VirB10 protein is modulated by growth temperature and periplasmic osmoadaption. J Bacteriol 1998, 180, 6597–6606. [Google Scholar] [CrossRef]
- Chung, J.M.; Sheedlo, M.J.; Campbell, A.M.; Sawhney, N.; Frick-Cheng, A.E.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structure of the Helicobacter pylori Cag type IV secretion system. Elife 2019, 8. [Google Scholar] [CrossRef]
- Sheedlo, M.J.; Chung, J.M.; Sawhney, N.; Durie, C.L.; Cover, T.L.; Ohi, M.D.; Lacy, D.B. Cryo-EM reveals species-specific components within the Helicobacter pylori Cag type IV secretion system core complex. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Delahay, R.M.; Balkwill, G.D.; Bunting, K.A.; Edwards, W.; Atherton, J.C.; Searle, M.S. The highly repetitive region of the Helicobacter pylori CagY protein comprises tandem arrays of an alpha-helical repeat module. J Mol Biol 2008, 377, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; McDaniel, T.K.; Falkow, S.; Karlin, S. Sequence anomalies in the Cag7 gene of the Helicobacter pylori pathogenicity island. Proc Natl Acad Sci U S A 1999, 96, 7011–7016. [Google Scholar] [CrossRef]
- Barrozo, R.M.; Hansen, L.M.; Lam, A.M.; Skoog, E.C.; Martin, M.E.; Cai, L.P.; Lin, Y.; Latoscha, A.; Suerbaum, S.; Canfield, D.R.; et al. CagY Is an Immune-Sensitive Regulator of the Helicobacter pylori Type IV Secretion System. Gastroenterology 2016, 151, 1164–1175 e1163. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 1996, 93, 14648–14653. [Google Scholar] [CrossRef]
- Grohmann, E.; Christie, P.J.; Waksman, G.; Backert, S. Type IV secretion in Gram-negative and Gram-positive bacteria. Mol Microbiol 2018, 107, 455–471. [Google Scholar] [CrossRef]
- Hayashi, T.; Senda, M.; Morohashi, H.; Higashi, H.; Horio, M.; Kashiba, Y.; Nagase, L.; Sasaya, D.; Shimizu, T.; Venugopalan, N.; et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 2012, 12, 20–33. [Google Scholar] [CrossRef]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef]
- Sierra, J.C.; Suarez, G.; Piazuelo, M.B.; Luis, P.B.; Baker, D.R.; Romero-Gallo, J.; Barry, D.P.; Schneider, C.; Morgan, D.R.; Peek, R.M., Jr.; et al. alpha-Difluoromethylornithine reduces gastric carcinogenesis by causing mutations in Helicobacter pylori cagY. Proc Natl Acad Sci U S A 2019, 116, 5077–5085. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Rappuoli, R.; Covacci, A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A 2000, 97, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Neddermann, M.; Lind, J.; Pachathundikandi, S.K.; Sharafutdinov, I.; Gutierrez-Escobar, A.J.; Bronstrup, M.; Tegge, W.; Hong, M.; Rohde, M.; et al. Toll-like Receptor 5 Activation by the CagY Repeat Domains of Helicobacter pylori. Cell Rep 2020, 32, 108159. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L.; Lacy, D.B.; Ohi, M.D. The Helicobacter pylori Cag Type IV Secretion System. Trends Microbiol 2020, 28, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Sheedlo, M.J.; Ohi, M.D.; Lacy, D.B.; Cover, T.L. Molecular architecture of bacterial type IV secretion systems. PLoS Pathog 2022, 18, e1010720. [Google Scholar] [CrossRef]
- Fischer, W.; Tegtmeyer, N.; Stingl, K.; Backert, S. Four Chromosomal Type IV Secretion Systems in Helicobacter pylori: Composition, Structure and Function. Front Microbiol 2020, 11, 1592. [Google Scholar] [CrossRef]
- Kondabala, R.; Kumar, V. Computational Intelligence Tools for Protein Modeling. Singapore, 2019; pp. 949-956.
- Coluzza, I. Computational protein design: a review. J Phys Condens Matter 2017, 29, 143001. [Google Scholar] [CrossRef]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein J 2021, 40, 522–544. [Google Scholar] [CrossRef]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Floristean, C.; Kharkar, A.; Roy, K.; Rochereau, C.; Ahdritz, G.; Zhang, J.; Church, G.M.; et al. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol 2022, 40, 1617–1623. [Google Scholar] [CrossRef]
- Soding, J.; Remmert, M. Protein sequence comparison and fold recognition: progress and good-practice benchmarking. Curr Opin Struct Biol 2011, 21, 404–411. [Google Scholar] [CrossRef]
- Xiao, X.; Lin, W.Z.; Chou, K.C. Recent advances in predicting protein classification and their applications to drug development. Curr Top Med Chem 2013, 13, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Mahajan, S.P.; Sulam, J.; Gray, J.J. Deep Learning in Protein Structural Modeling and Design. Patterns (N Y) 2020, 1, 100142. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, V.K. Current directions in combining simulation-based macromolecular modeling approaches with deep learning. Expert Opin Drug Discov 2021, 16, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.C.; McClain, M.S.; Cover, T.L. Role of the CagY antenna projection in Helicobacter pylori Cag type IV secretion system activity. Infect Immun 2023, 91, e0015023. [Google Scholar] [CrossRef]
- Barrozo, R.M.; Cooke, C.L.; Hansen, L.M.; Lam, A.M.; Gaddy, J.A.; Johnson, E.M.; Cariaga, T.A.; Suarez, G.; Peek, R.M., Jr.; Cover, T.L.; et al. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 2013, 9, e1003189. [Google Scholar] [CrossRef]
- Jackson, L.K.; Potter, B.; Schneider, S.; Fitzgibbon, M.; Blair, K.; Farah, H.; Krishna, U.; Bedford, T.; Peek, R.M., Jr.; Salama, N.R. Helicobacter pylori diversification during chronic infection within a single host generates sub-populations with distinct phenotypes. PLoS Pathog 2020, 16, e1008686. [Google Scholar] [CrossRef]
- Della Bella, C.; Soluri, M.F.; Puccio, S.; Benagiano, M.; Grassi, A.; Bitetti, J.; Cianchi, F.; Sblattero, D.; Peano, C.; D’Elios, M.M. The Helicobacter pylori CagY Protein Drives Gastric Th1 and Th17 Inflammation and B Cell Proliferation in Gastric MALT Lymphoma. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Chang, Y.W.; Shaffer, C.L.; Rettberg, L.A.; Ghosal, D.; Jensen, G.J. In Vivo Structures of the Helicobacter pylori cag Type IV Secretion System. Cell Rep 2018, 23, 673–681. [Google Scholar] [CrossRef]
- Hu, B.; Khara, P.; Song, L.; Lin, A.S.; Frick-Cheng, A.E.; Harvey, M.L.; Cover, T.L.; Christie, P.J. In Situ Molecular Architecture of the Helicobacter pylori Cag Type IV Secretion System. mBio 2019, 10. [Google Scholar] [CrossRef]
- Frick-Cheng, A.E.; Pyburn, T.M.; Voss, B.J.; McDonald, W.H.; Ohi, M.D.; Cover, T.L. Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. mBio 2016, 7, e02001–02015. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nat Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M.; Zhou, H.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J Chem Inf Model 2021, 61, 4827–4831. [Google Scholar] [CrossRef] [PubMed]
- Aras, R.A.; Fischer, W.; Perez-Perez, G.I.; Crosatti, M.; Ando, T.; Haas, R.; Blaser, M.J. Plasticity of repetitive DNA sequences within a bacterial (Type IV) secretion system component. J Exp Med 2003, 198, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Bassot, C.; Elofsson, A. Accurate contact-based modelling of repeat proteins predicts the structure of new repeats protein families. PLoS Comput Biol 2021, 17, e1008798. [Google Scholar] [CrossRef]
- Hatahet, F.; Boyd, D.; Beckwith, J. Disulfide bond formation in prokaryotes: history, diversity and design. Biochim Biophys Acta 2014, 1844, 1402–1414. [Google Scholar] [CrossRef]
- Dombkowski, A.A. Disulfide by Design: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics 2003, 19, 1852–1853. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Bhopatkar, A.A.; Uversky, V.N.; Rangachari, V. Disorder and cysteines in proteins: A design for orchestration of conformational see-saw and modulatory functions. Prog Mol Biol Transl Sci 2020, 174, 331–373. [Google Scholar] [CrossRef]
- Dumrese, C.; Slomianka, L.; Ziegler, U.; Choi, S.S.; Kalia, A.; Fulurija, A.; Lu, W.; Berg, D.E.; Benghezal, M.; Marshall, B.; et al. The secreted Helicobacter cysteine-rich protein A causes adherence of human monocytes and differentiation into a macrophage-like phenotype. FEBS Lett 2009, 583, 1637–1643. [Google Scholar] [CrossRef]
- Bocian-Ostrzycka, K.M.; Lasica, A.M.; Dunin-Horkawicz, S.; Grzeszczuk, M.J.; Drabik, K.; Dobosz, A.M.; Godlewska, R.; Nowak, E.; Collet, J.F.; Jagusztyn-Krynicka, E.K. Functional and evolutionary analyses of Helicobacter pylori HP0231 (DsbK) protein with strong oxidative and chaperone activity characterized by a highly diverged dimerization domain. Front Microbiol 2015, 6, 1065. [Google Scholar] [CrossRef] [PubMed]
- Lester, J.; Kichler, S.; Oickle, B.; Fairweather, S.; Oberc, A.; Chahal, J.; Ratnayake, D.; Creuzenet, C. Characterization of Helicobacter pylori HP0231 (DsbK): role in disulfide bond formation, redox homeostasis and production of Helicobacter cystein-rich protein HcpE. Mol Microbiol 2015, 96, 110–133. [Google Scholar] [CrossRef] [PubMed]
- Silvers, R.; Sziegat, F.; Tachibana, H.; Segawa, S.; Whittaker, S.; Gunther, U.L.; Gabel, F.; Huang, J.R.; Blackledge, M.; Wirmer-Bartoschek, J.; et al. Modulation of structure and dynamics by disulfide bond formation in unfolded states. J Am Chem Soc 2012, 134, 6846–6854. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, C.; Kumar, A.; Lang, A.; Ohlenschlager, O. Cysteines and Disulfide Bonds as Structure-Forming Units: Insights From Different Domains of Life and the Potential for Characterization by NMR. Front Chem 2020, 8, 280. [Google Scholar] [CrossRef]
- Erdos, G.; Meszaros, B.; Reichmann, D.; Dosztanyi, Z. Large-Scale Analysis of Redox-Sensitive Conditionally Disordered Protein Regions Reveals Their Widespread Nature and Key Roles in High-Level Eukaryotic Processes. Proteomics 2019, 19, e1800070. [Google Scholar] [CrossRef]
- Ryu, S.E. Structural mechanism of disulphide bond-mediated redox switches. J Biochem 2012, 151, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem Rev 2018, 118, 1169–1198. [Google Scholar] [CrossRef]
- Giganti, D.; Yan, K.; Badilla, C.L.; Fernandez, J.M.; Alegre-Cebollada, J. Disulfide isomerization reactions in titin immunoglobulin domains enable a mode of protein elasticity. Nat Commun 2018, 9, 185. [Google Scholar] [CrossRef]
- Sali, A. Comparative protein modeling by satisfaction of spatial restraints. Mol Med Today 1995, 1, 270–277. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 2015, 43, W174–181. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J Comput Chem 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Henin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 2020, 153, 044130. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J Mol Graph 1996, 14, 33–38, 27. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci 2018, 27, 129–134. [Google Scholar] [CrossRef]
Figure 1.
Motifs, secondary structure, and repeated modules of Cag Y. a) Cag Y sequence of 266695 is 1927 residues length, structural domains frequently cited are the 5′ Repeat Region, FRR (cian), the Transmembrane regions (TM) (green), the Middle Repeat Region (MRR) (blue) and the VirB10 homologous region (purple). Also, the figure identifies motifs in which structure was divided for modeling: Intrinsically disordered region (IDR), Middle Repeats Region (MRR), 6odi and 6x6j modeled regions, and ab-initio modeled. b) Secondary structure, disordered regions, and transmembrane regions of Cag Y predicted by the PSIPRED server. c) Sequence of repeated modules A and B from the MRR.
Figure 1.
Motifs, secondary structure, and repeated modules of Cag Y. a) Cag Y sequence of 266695 is 1927 residues length, structural domains frequently cited are the 5′ Repeat Region, FRR (cian), the Transmembrane regions (TM) (green), the Middle Repeat Region (MRR) (blue) and the VirB10 homologous region (purple). Also, the figure identifies motifs in which structure was divided for modeling: Intrinsically disordered region (IDR), Middle Repeats Region (MRR), 6odi and 6x6j modeled regions, and ab-initio modeled. b) Secondary structure, disordered regions, and transmembrane regions of Cag Y predicted by the PSIPRED server. c) Sequence of repeated modules A and B from the MRR.
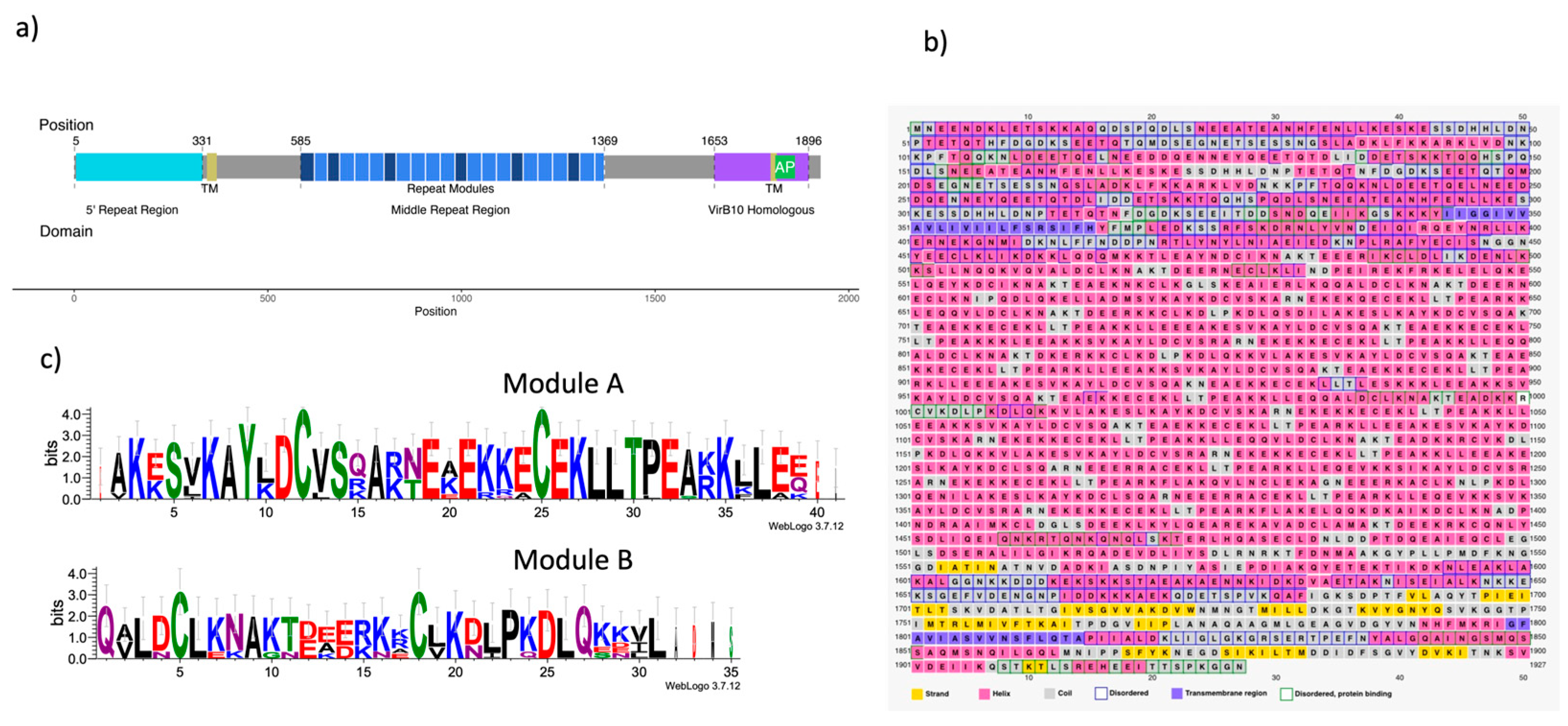
Figure 2.
Homology modeling of the VirB10 homologous region. a) PDB structures 6ODI and 6X6J were used for templates of the OM, Periplasmic regions. b) Monomer model (virB10 homology region) and alignment with template structures, 6odi, 6x6j, and I-tasser model of residues 1604 to 1677 (model calculated with Modeller10v8). c) Multimer 14-chain model of virB10 homology region. Chains aligned with respect 6odi as a template.
Figure 2.
Homology modeling of the VirB10 homologous region. a) PDB structures 6ODI and 6X6J were used for templates of the OM, Periplasmic regions. b) Monomer model (virB10 homology region) and alignment with template structures, 6odi, 6x6j, and I-tasser model of residues 1604 to 1677 (model calculated with Modeller10v8). c) Multimer 14-chain model of virB10 homology region. Chains aligned with respect 6odi as a template.
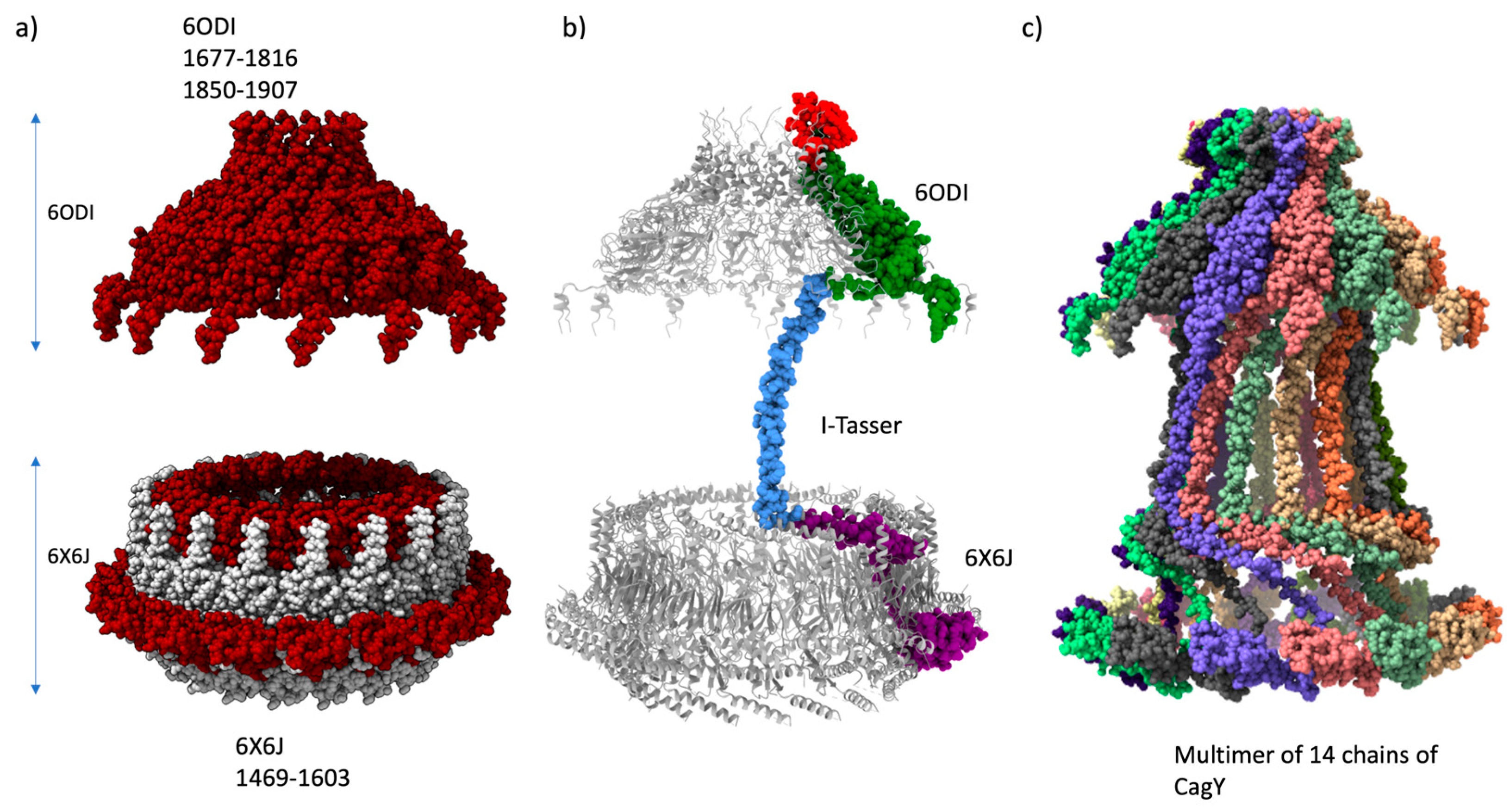
Figure 3.
AlphaFold2 dimer modeling of MRR, trimer building, and Equilibrium Molecular Dynamics (EMD) for the trimer. a) Best Colabfold/AlphaFold2 model dimer structure for the MRR and IDDT values. b) Alignment of two identical dimers, chains labeled as AB (blue and red) and A’B’ (gray and green) respectively, for producing an ABB’ trimer (blue, red, and gray) structure (A’ chain was removed from superposed B/A’ structures). c) RMSD plot of 10 ns Molecular Dynamics Plot for the trimer. Individual plots for chains A (red), B (blue), and B’ (green) are displayed, as well as the plot for the whole ABB’ trimer. Only chain B stabilized its rmsd values (gray).
Figure 3.
AlphaFold2 dimer modeling of MRR, trimer building, and Equilibrium Molecular Dynamics (EMD) for the trimer. a) Best Colabfold/AlphaFold2 model dimer structure for the MRR and IDDT values. b) Alignment of two identical dimers, chains labeled as AB (blue and red) and A’B’ (gray and green) respectively, for producing an ABB’ trimer (blue, red, and gray) structure (A’ chain was removed from superposed B/A’ structures). c) RMSD plot of 10 ns Molecular Dynamics Plot for the trimer. Individual plots for chains A (red), B (blue), and B’ (green) are displayed, as well as the plot for the whole ABB’ trimer. Only chain B stabilized its rmsd values (gray).
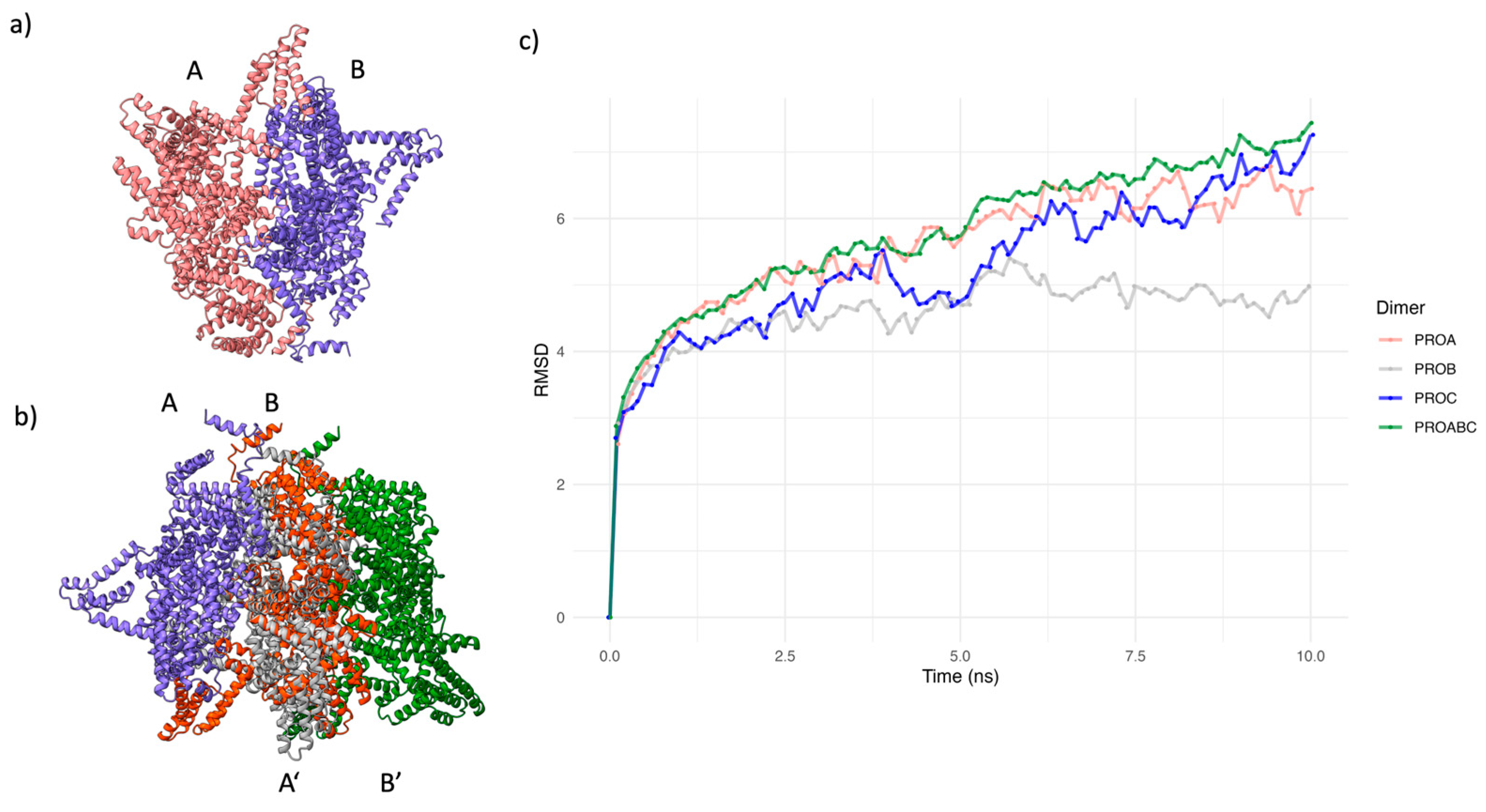
Figure 4.
Cag Y monomer and multimer assembly. a) Summary of the Cag Y whole monomer assembly, structure templates, and methods employed are displayed (6odi, 6x6j, I-tasser, ColabFold2/AlphaFold2-multimer, this monomer was constructed with ChimeraX and a alignment of the missing structure b) Final assembly of the 14-chain multimer. 6odi structure was used as a template. It is showing the side view, the bottom view, and the upper view of the assembled multimer of CagY protein. The missing region corresponding to AP of CagY was completed by homology modeling with Swiss-Model and 6ODI template was used. c) All the 58 Cys residues present in the MRR participate in the formation of 29 disulfide bonds, here highlighted in yellow. A bottom view of the multimer complex seemed to describe about 4 concentric rings of these bonds.
Figure 4.
Cag Y monomer and multimer assembly. a) Summary of the Cag Y whole monomer assembly, structure templates, and methods employed are displayed (6odi, 6x6j, I-tasser, ColabFold2/AlphaFold2-multimer, this monomer was constructed with ChimeraX and a alignment of the missing structure b) Final assembly of the 14-chain multimer. 6odi structure was used as a template. It is showing the side view, the bottom view, and the upper view of the assembled multimer of CagY protein. The missing region corresponding to AP of CagY was completed by homology modeling with Swiss-Model and 6ODI template was used. c) All the 58 Cys residues present in the MRR participate in the formation of 29 disulfide bonds, here highlighted in yellow. A bottom view of the multimer complex seemed to describe about 4 concentric rings of these bonds.
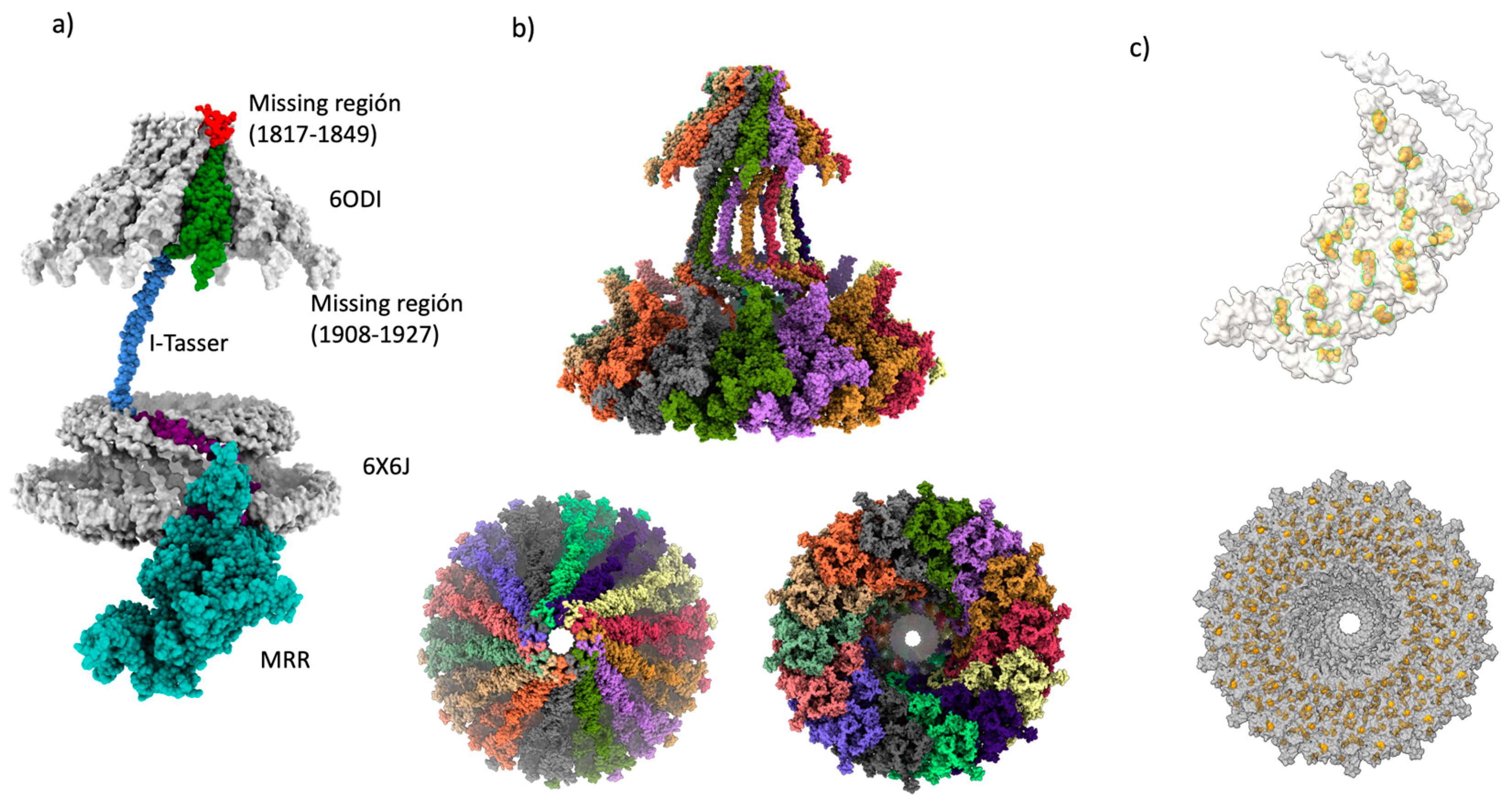
Figure 5.
Comparison of CagY with the Cryo-EM electrography of T4SS. a) Up figure represents the surface of the Cryo-EM dispersion maps, the down figure represents the overlaying T4SS cryo-EM dispersion maps with our model of CagY protein, showing a good fitting with the surface of the Cryo-EM b) Up figure represents the backbone of the Cryo-EM dispersion maps of the T4SS, down figure represents the overlaying of our model of CagY.
Figure 5.
Comparison of CagY with the Cryo-EM electrography of T4SS. a) Up figure represents the surface of the Cryo-EM dispersion maps, the down figure represents the overlaying T4SS cryo-EM dispersion maps with our model of CagY protein, showing a good fitting with the surface of the Cryo-EM b) Up figure represents the backbone of the Cryo-EM dispersion maps of the T4SS, down figure represents the overlaying of our model of CagY.
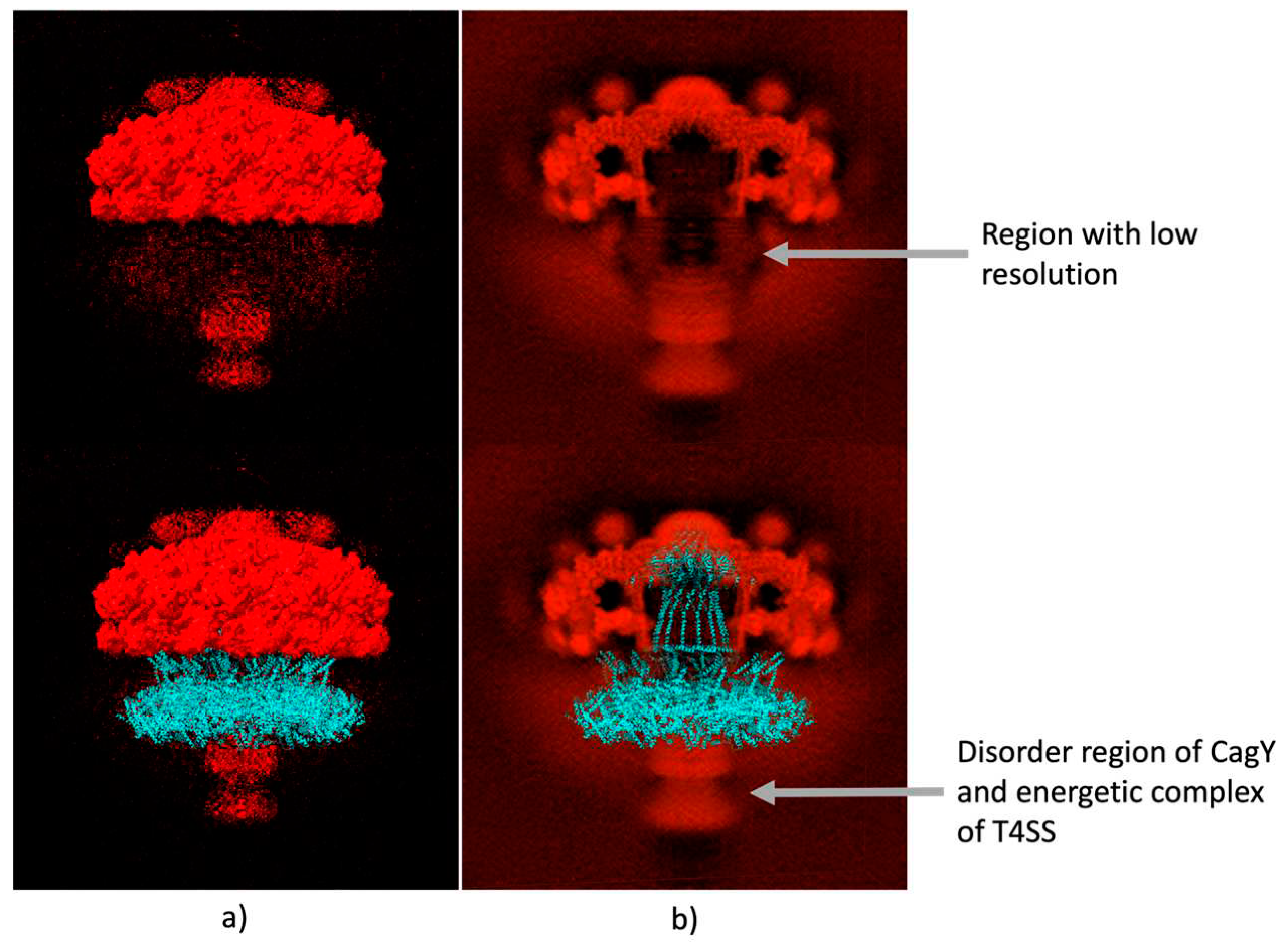
Figure 6.
Overlapping of the CagY protein with other Cag proteins. a) Build of the CagYm multimer with the 6X6S model b) Transversal view of the components of 6X6S model and CagY multimer, it shows how the core complex of T4SS of Hp is assembled.
Figure 6.
Overlapping of the CagY protein with other Cag proteins. a) Build of the CagYm multimer with the 6X6S model b) Transversal view of the components of 6X6S model and CagY multimer, it shows how the core complex of T4SS of Hp is assembled.
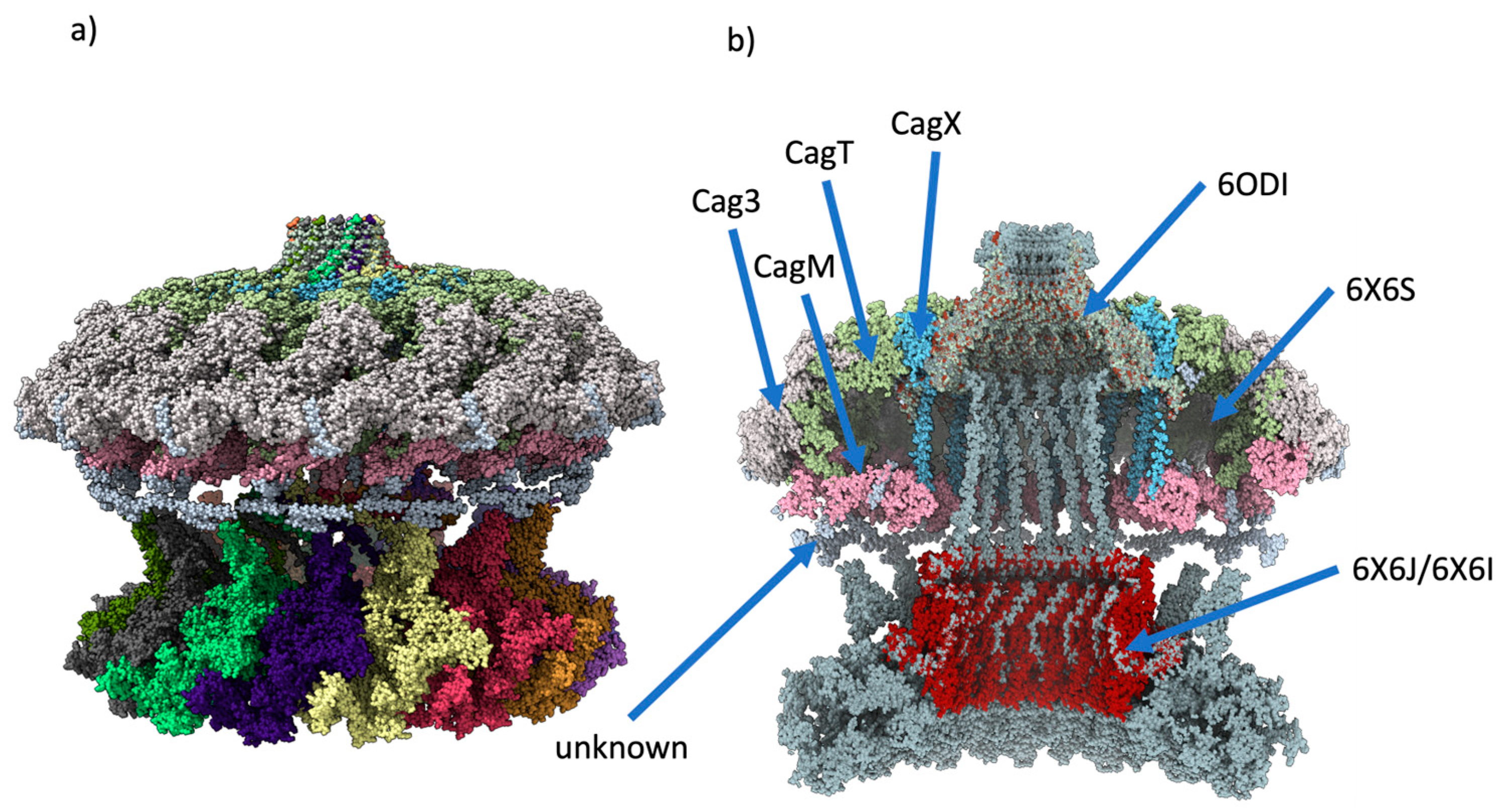
Figure 7.
Antenna projections (AP) of multiples modifications of CagY. a) Wildtype AP of CagY modelled by homology, shows a pore open with a crest-shape. b) Wildtype AP of CagY modelled by alphafold2, this shows a dent in the pore c) CagY GS20, with a modification of the AP region with glycine and serine of 20 amino acids of length, modelled by homology show a pore with up crest. d) CagY GS20 modeled by alphafold2, this structure displays a deformed pore. e) CagY Xc wich has the AP region replaced with AP region of X. citri, modeled by homology, shows a greater overture in comparison with CagY WT. f) CagY Xc, modeled by alphafold2, shows a closer pore g) CagY model with deltaAP lacking both flanking alpha-helix modeled by homology.
Figure 7.
Antenna projections (AP) of multiples modifications of CagY. a) Wildtype AP of CagY modelled by homology, shows a pore open with a crest-shape. b) Wildtype AP of CagY modelled by alphafold2, this shows a dent in the pore c) CagY GS20, with a modification of the AP region with glycine and serine of 20 amino acids of length, modelled by homology show a pore with up crest. d) CagY GS20 modeled by alphafold2, this structure displays a deformed pore. e) CagY Xc wich has the AP region replaced with AP region of X. citri, modeled by homology, shows a greater overture in comparison with CagY WT. f) CagY Xc, modeled by alphafold2, shows a closer pore g) CagY model with deltaAP lacking both flanking alpha-helix modeled by homology.


Table 1.
Functional/Structural domains of CagY protein.
| Region | inicio | fin |
|---|---|---|
| FRR | 1 | 331 |
| TM | 343 | 368 |
| MRR | 585 | 1369 |
| TM | 1798 | 1814 |
| AP | 1820 | 1851 |
| VirB10 | 1653 | 1896 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



