
Submitted:
15 October 2023
Posted:
18 October 2023
You are already at the latest version
Abstract
Age-related macular degeneration, a leading cause of visual loss and dysfunction in the devel-oped world, is a disease initiated by genetic polymorphisms that impairs negative regulation of complement. Proteomic investigation points to altered glycosylation and loss of SIGLEC medi-ated glyco-immune checkpoint parainflammatory homeostasis as a main determinant for the vi-sion impairing complications of macular degeneration. The effect of altered glycosylation on microglial maintained retinal para-inflammatory homeostasis and eventual recruitment and polarization of peripheral blood monocyte derived macrophages (PBMDM) into the retina can explain the phenotypic variability seen in this clinically heterogenous disease. Restoring gly-co-immune checkpoint control with a sialic acid mimetic nanoparticle targeting microgli-al/macrophage SIGLECs to regain retinal para inflammatory homeostasis is a promising thera-peutic that could halt the progression of and improve visual function in all stages of macular de-generation.
Keywords:
Microglia
; Macrophages
; Macular Degeneration
; Sialic Acid
; SIGLECs
; Nanoparticles
; Glycosylation
; Geographic Atrophy
; PolySialic Acid.
1. Age Related Macular Degeneration
1.1. Background
The prevalence of age-related macular degeneration in the US is approximately 18 million. [1] This prevalence represents three times the number of patients with Alzheimer’s disease and equal to all patients with a cancer diagnosis excluding melanoma. [2,3] By 2040, this disease that progressively causes central visual loss, is estimated to affect 288 million people worldwide.[4] Currently macular degeneration represents the third worldwide cause of vision loss secondary to ocular pathology. [5]
1.2. Clinical Stages
The disease of macular degeneration is a disease of stages that can be divided into early, intermediate, and late stage.[6] The hallmark of the early stage is the initial development of macular drusen and pigmentary change. When these changes worsen and reach a certain density, the stage is classified as intermediate. The late stage is reached when geographic atrophy or neovascularization develop. [6] While drusen and RPE changes constitute a common initiating point of clinically detectable disease, the late-stage complications bifurcate into either the development of geographic atrophy, exudative macular degeneration, or both.
1.3. Clinical evidence of inflammation
Clinically these stages affect visual function variably. Early and intermediate stages of macular degeneration are characterized by measurable photoreceptor dysfunction as reflected in age related abnormalities in dark adaptation, visual field, photo stress and electro-retinographic changes.[7,8,9,10] These disease correlated changes in visual function point to inflammation as the underlying pathology behind even the early stages of macular degeneration. Dark adaptation which is a measure of the efficiency of the visual cycle is impaired and indicates development of early stage of macular degeneration.[11,12] Patients with early AMD had qualitative visual changes and symptoms of distortion that could be detected and quantified by visual field analysis. These deficits of form recognition and sensitivity were found in areas of RPE atrophy not defined as geographic atrophy within this patient population.[8] Photo stress recovery time which is a measure of visual pigment recycling time, was inversely correlated with visual acuity and prolongation of recovery time was directly correlated with presence or absence of geographic atrophy and advancing age.[10] In late AMD slowing of implicit time and amplitude reduction on electroretinograms of patients with geographic atrophy were seen in areas bordering fundus autofluorescence defined geographic atrophy.[13] Foveal ERG performed in fellow eyes of patients with wet AMD who did not have severe visual acuity changes demonstrated implicit time prolongation. [9]
1.4. A disease of inflammaging and dysfunctional parainflammation
These electrophysiological and psychophysical studies implicate retinal inflammation as an underlying factor affecting visual function in patients at all stages of AMD. [14] These studies also support the concept that macular degeneration is a disease of inflamaging. During the period these studies were performed the understanding of microglial parainflammatory homeostatic control was still yet to be postulated or described. Medzhitov in 2008 postulated a parainflammatory state that lay between basal homeostatic conditions and true inflammation. [15] This para inflammatory state is considered an adaptive immune response to low level tissue stress such as the age-related accumulation of oxidative byproducts. This acquired dysfunctional para inflammation that occurs with aging, has been termed inflamaging. [16]
1.5. Early Stage AMD dysfunctional para inflammation
The abnormalities in these electroretinographic and psychophysical tests demonstrate that there are differing parainflammatory dysfunctions that occur during the different stages of the disease. The early/intermediate stage is the accumulation of oxidized, metabolic, inflammatory debris that appear as yellow sub retinal deposits called drusen. The pigmentary clumping seen in AMD, represent pigment ladened microglial or macrophage migration towards the retina.[17] The inability to clear pigment or debris in the retinal and subretinal space represents the main pathology of this early/intermediate stage of AMD. (Figure 1) This impaired clearance can only be explained as a dysfunction of the phagocytic housekeeping function of microglia.
1.6. Late stage AMD Overt inflammation
Unlike the early/ intermediate stage which represents an inhibition of phagocytosis with resultant accumulation of debris, geographic atrophy represents an enhancement of phagocytic activity resulting in elimination (atrophy) of retinal cells initially of the outer retina then progressing to the inner retina. (Figure 2) Exudative AMD represents the overproduction of angiogenic and neuroprotective vascular endothelial growth factor which when overexpressed results in the development of neovascularization. The only process that can explain these constellations of pathologic features are microglial cells or macrophages.
2. Pathogenesis of Age-Related Macular Degeneration
2.1. Genomic-Complement Pathway Polymorphisms
Macular degeneration is considered a genetic disease and the development of disease is strongly correlated with multiple polymorphisms that impair the negative regulation of the complement pathway.[18] While complement associated polymorphisms are associated with development of AMD, these polymorphisms are not as strongly correlated with development of late stage vision threatening geographic atrophy or CNV.[19]
2.2. Proteomic- Abberant glycosylation
A large proteomic analysis of serum proteins in patients with macular degeneration was correlated to the early or late stage of disease and identified several proteins that were elevated differentially in early and late AMD. In this analysis elevated serum protein levels of Alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 1 (ST6GALNAC1/ST6) and Alpha-(1,3)-fucosyltransferase(FUT5) were highly correlated to patients with early stage AMD.[20] In late-stage AMD patients only FUT5 was correlated. The expression of FUT5 in both late and early AMD and the expression of ST6 in only early AMD point to alterations of sialic acid glycan expression as a major determinant in the progression to late-stage AMD. It also implicates microglial/macrophage checkpoint regulation via their Siglec immune resolving checkpoint receptors as a critical determinant of late-stage AMD progression.
2.2.1. ST6GALNAC1/ST6- Early/Intermediate AMD
The glycan produced by ST6 is only expressed in early dry AMD patients, and is a tumor associated sTn glycan which is involved in the development of tumor associated macrophages (a non-activated macrophage that cloak and allow tumors to evade immune detection). The expression of sTN diminish phagocytic activity by microglial cells resulting in an accumulation of inflammatory debris. Combined with the impaired negative regulation of complement, which will produce more complement mediated cell damage, and complement pathway derived proteins this inability to clear debris from the retina will produce drusen and pigmentary clumping.
2.2.2. FUT5 – Late AMD
During the lates stage of AMD ST6 is no longer upregulated instead FUT5 becomes the predominant sialyltransferase. FUT5 is a critical glycotransferase that produces Lewis x (3Gal1,4dFuc1,3]GlcNAc-), sialyl Lewis x (sLex,1 NeuAc2, 3Gal1,4[Fuc1,3]GlcNAc-), Lewis a (Lea, 3Gal1,3[Fuc1,4]GlcNAc), sialyl Lewis a (sLea, NeuAc2, 3Gal1,3[Fuc1,4]GlcNAc-), and Lewis b (Leb, Fuc 1,2Gal1,3[Fuc1,4]GlcNAc-). These glycans are the main binding determinants for Selectins in particularly e-selectin which is responsible for localizing monocytes to areas of active inflammation. (Figure 1) Without sTN checkpoint restraint of microglia and recruited macrophages, inflammatory activation will convert to overproduction of cytokines such as VEGF and unbridled phagocytic activity resulting in accelerated atrophy. (Figure 2)
2.2.3. Loss of Glycoimmune Checkpoint control
Clinical and proteomic findings in all stages of macular degeneration point to microglia and macrophages glyco-immune checkpoint dysfunction as the main post translation determinant of development of early/intermediate stage AMD and progression to late-stage geographic atrophy or CNV. Genomics point to oxidation and complement overactivation, as a lifelong stimulator of disease but not the direct determinant of disease development or progression. The major determinant to the development of early intermediate stage AMD is the aberrant glycosylation with sTn which inactivates microglial homeostatic phagocytosis resulting in accumulation of drusen and pigment. The major determinant of conversion to late-stage AMD is the loss of sialic acid microglial/macrophage checkpoint control and the initiation of true inflammation and recruitment of peripheral blood monocytes and macrophages. If checkpoint control could be regained and homeostatic microglial function restored, then the potential of halting late-stage disease and even regaining visual function could be obtained.
2.3. Microglia- Central Role in AMD pathogenesis
2.3.1. Background
Microglia are considered the main immune cell and resident macrophage of the CNS and retina. Their existence was first described and delineated over 100 years ago by Pio del Rio Ortega who described their macrophage like, phagocytic, plastic and heterogenous nature. [21] Microglia constitute between 5-12 % of the CNS and are cells that are mesenchymal in origin. These cells while not bone marrow derived are analogous to macrophages, in that they carry out three critical roles, surveillance, phagocytosis and production-release of soluble factors. [22] Microglia contribute to the key biologic functions of myelination, inflammation, vasculogenesis, neurogenesis, synapse remodeling, neuronal function, and blood brain barrier permeability.
2.3.2. Function
Transcriptomic analysis reveal 3 major function of microglia: 1) sensing, 2) housekeeping and 3) protection against injurious self and non-self-stimuli. [23] Microglia produce nearly 100 gene products that can sense microenvironment change combined with thin processes that scan the area around their cell body every few hours. These gene products such as P2yr12, AXL and MER and tentacles represent the sensome which can rapidly migrate towards focal injury.
The sensome allows microglia to perform its second function of housekeeping such as synaptic remodeling,[25,26,27] migration towards and phagocytoses of dying cells or the debris of dead cells,[28,29] myelin homeostasis,[30] astrocyte interaction,[31] and production of multiple gene products to carry out these functions.[32] Genes expressed by microglia that are involved in housekeeping include genes that encode chemokines, chemoattractant receptors, phagocytic receptors (Scavenger receptors, TREM2) and synaptic pruning and remodeling proteins ( C1q, CxC3R1).[24]
The third microglial function of protection against injurious self and non-self-stimuli, give microglia properties like macrophages and other inflammatory cells. Like macrophages, microglia express Fc receptors, Toll-like receptor (TLRs), viral receptors, and antimicrobial peptides[24] which allow it to provide immune defense against infectious agents, toxic substances like amyloid β, or oxidized superoxide dismutase. These stimuli can elicit a secretory response similar to activated peripheral blood derived macrophages such as secretion of TNF, IL-1 cytokine production[33,34] and CcL2 chemokine production which allows for recruitment of other microglial cells without necessarily recruiting peripheral leukocytes if proper checkpoint modulation is in place.[35]
2.3.3. Role in Retina
Because of the central role of microglia in neuronal homeostasis, microglial overactivation represents a common patho-mechanism in a variety of retinal degenerative diseases and is often overactivated prior to the onset of overt retinal cell death. [36] In the retina, microglia’s dynamic motility allow comprehensive surveillance coverage of the entire retina in a short time period.[37] This motility allows microglia to interact with retinal neurons, macroglia and play a central role in retinal homeostatic maintenance and clearance of cellular and metabolic debris.[38]
Activated microglia in the retina have been implicated in many retinal pathologies. In SD-OCTs hyperreflective spots are visualized in AMD and diabetic retinopathy which represent large amoeboid microglial cells, the activated microglial appearance, as opposed to the resting ramified microglial configuration.[39,40]
Phagocytic cells presumably microglia/macrophages are bloated with engulfed membranous material near areas of drusen suggesting that drusen and its components attract and stimulate microglia to migrate but not phagocytose drusen and the surrounding retina.[41,42] The presence of activated microglia near drusen indicate the inability of microglia alone to remove the drusen deposits. (Figure 1a) While microglia can maintain retinal homeostasis during the first 5 to 6 decades of life, the onset of drusen and pigmentary changes signal the migration of microglia and the recruitment of peripheral blood monocytes to the Bruch’s membrane. [43] This sequence of events indicates the requirement of peripheral blood monocyte recruitment in the progression of the drusen stage to the late stages of AMD.
2.4. Peripheral Blood Derived Macrophages (PBMC)- Central Role in AMD pathogenesis
The major role of peripheral blood monocyte derived macrophages in the progression of early drusen stage to late-stage geographic atrophy and exudative AMD is evidenced by the increase in number of activated macrophages in the choroid and Bruch’s membrane as AMD progresses. The observation that the highest number of activated macrophages are found in eyes with choroidal neovascularization further supports this central role. [43] .
2.4.1. PBMC retinal recruitment
Patho-mechanistically, recruitment of peripheral blood monocytes in clinically evident macular degeneration is mediated my microglial chemokine signaling. Microglial chemokine signaling is responsible for recruitment of monocytes to the choroid and the retina. The chemokine receptors CCR2 and CX3CR1 and their respective ligands CCL2 (monocyte chemotactic protein-1, MCP-1) and CX3CL1 (fractalkine) mediate this recruitment.[22,35,44] This monocyte/macrophage recruitment to the Bruch’s membrane and the increase in peripheral blood derived macrophages in the retina correlate and determines severity of geographic atrophy [45] and exudative AMD.[46] (Figure 3)
2.4.2. Macrophages central role in Geographic Atrophy
The central role of macrophages in the pathogenesis of neovascular wet AMD is widely accepted,[46,47,48,49] but the activated macrophage’s critical role in the pathogenesis of geographic atrophy has been widely overlooked due to the focus on the complement pathway.[50] Prior to the genomic association between complement factors and AMD, histopathologic evidence pointed to macrophage/ mononuclear phagocyte/multinucleated giant cells as the central causative factor in the pathogenesis of dry AMD and the main phagocytic cause of retinal cellular clearance that manifests as RPE and photoreceptor loss in geographic atrophy.[42,45]
One of the earliest demonstrations of this central role was a on electron microscopic/ histopathologic studies of postmortem retinal specimens of 7 patients with geographic atrophy. In this study the association of mononuclear phagocyte series (MPS) cells situated between the basement membrane of the RPE and inner collagenous layer of Bruch’s membrane was found in all patient macular specimens.[51] (Figure 1) Another finding in these specimens, was the presence of multinucleated giant cells within the area of geographic atrophy. These giant cells arise from macrophage fusion caused by the presence of poorly degradable substances such as constituents of Bruch’s membrane or undegraded RPE pigment and show no RPE cell involvement. (Figure 2) This hallmark study implicated microglia, macrophages as the main phagocytic cell in the pathology of geographic atrophy as opposed to RPE cells the only other phagocytic cell in the retina. Clinico-pathologic correlation demonstrated that both MPS and giant cells containing pigment are found typically in the outer edge of the geographic atrophy lesion or in areas of coarse pigmentary mottling which is clinically predictive of the development of geographic atrophy. [52,53] (Figure2) The close apposition of these macrophage derived cells to the leading edge and to areas that will develop GA, indicate that it is monocyte derived macrophages that phagocytose retinal cells that cause the development and growth of geographic atrophy in AMD.[42]
2.5. Sialic acid loss initiates PBMC recruitment
The recruitment of peripheral blood derived macrophages is a major function of retinal microglial cells when injury or toxic material overwhelm the microglia’s ability to phagocytose toxic oxidative byproducts, injured cells, or apoptotic cells.[54] To maintain macular health the microglia must phagocytose oxidative byproducts (oxidized phospholipids, malondialdehyde, carboxyethyl pyrrole), apoptotic cells, complement proteins, abnormal proteins (Amyloid B) and toxic metabolites (A2E). (Figure 2) If microglia do not encounter appropriate checkpoint ligands in the form of sialic acid, then chemokine signaling will recruit peripheral blood monocyte macrophages.[55]
Photooxidative stress which is focused and concentrated onto the macula by the ocular lens system[56] produce oxidized phospholipids which stimulate the RPE to upregulate, MCP-1 which serves as a major chemoattractant to macrophages.[57] After photooxidative stress, fractalkine upregulation from photoreceptor segment and outer nuclear layer mediates the cross talk between photoreceptors and microglia which exclusively express the CX3CR1 receptor for fractalkines. This release of fractalkine serves to attract and activate microglia towards the injured photoreceptors but also appears to exert a neuroprotective role.[44] This dual role of Fractalkine/CX3CR1 interaction, on microglial proinflammatory activation and neuroprotective properties demonstrate the necessity of tight regulation of microglial cells. (Figure 1)
Without appropriate checkpoint regulation of microglial cells, the inflammatory activation state of microglia will promote more photoreceptor degeneration rather than rescue photoreceptors. While fractalkine -CX3CR1 signaling is definitively proinflammatory, it is also critical for progesterone mediated neuroprotection of the retina.[58] If the proinflammatory properties of fractalkine-CX3CR1 activated microglia and macrophages could be checked, but maintain its neuroprotective properties, then polarizing macrophages to the resolution state would eliminate the inflammation while providing neuroprotection.
2.6. Macrophages Polarization Directs AMD pathology.
2.6.1. Macrophage polarization states
The prototypical description of macrophage polarization states as M1 and M2 has been based on biomarker expression and cytokine production but does not reflect the true character of these subtypes. Describing these macrophages based on function better characterizes their role in pathology.[59,60] The M1 polarization state is characterized as the pro-inflammatory phagocytic state. The M2 state can be subdivided into 4 M2 subtypes. The M2 a, b subtype are the anti-inflammatory pro-fibrotic type, the M2d is the pro angiogenic phenotype, the M2c is the anti-inflammatory and neuroprotective type.[60,61] The M2c is also considered the resolution state and can reconvert myofibroblasts into fibroblasts so is considered anti-fibrotic. [62]
2.6.2. Macrophage polarization in CNS
In Vitro, M1 macrophages have been demonstrated to be neurotoxic and only modest axon growth promoting effect in contrast to the m2 macrophages which are not neurotoxic and promote long distance axon growth. [63] While few studies on macrophage polarization have been done in the retina, many studies using traumatic brain injury or spinal cord injury have characterized the polarization properties and the time course of macrophage polarization after injury. From these studies M1-like macrophage versus the M2-like macrophage can be qualitatively described.
M1-like macrophages release oxidative metabolites and proteases that can kill neurons and glial cells.[63]In contrast M2-like cells facilitate tissue repair.[64] In spinal cord injury models, increased m2c (acquired deactivation) microglia expressed in the first week after injury correlated to better neurological outcome indicating a healing neuroprotective function of M2c microglia /macrophages. [65] The time course study comparing M1 versus M2 levels in this model show that M1 expression is upregulated for at least a month while M2 levels diminish drastically after 1 week post injury. The level of M2a and M2c upregulation within the first week correlated well with neurological recovery. The neurological recovery resulted from the neuroprotective properties of microglia polarized to the M2c or M2a state. These properties were the secretion of trophic factors such as TGF-b, BDNF, and GDNF and the ability to perform controlled phagocytosis that prevents necrosis of surrounding tissue. [66] (Figure 3)
2.6.3. Macrophage polarization in AMD
While macular degeneration may not appear to be a disease of acute CNS injury, the macrophage microglial behavior in the retina is consistent with what is seen in injury models of the CNS. Eyes with advanced AMD, had a higher ratio of M1 to M2 macrophages than age matched normal autopsied eyes. [67] This enhanced chronic expression of neurotoxic phagocytic m1 macrophages with the reduction of M2c neuroprotective macrophages will result in the unchecked photoreceptor, RPE cell degeneration and phagocytosis of these degenerating cells (geographic atrophy) [67] The abundance of m1 polarized macrophages combined with the abundance of adenosine, will produce m2D VEGF producing macrophages resulting in the development of subretinal neovascularization.[68] Like CNS injury, the failure to polarize M1 macrophages towards the M2 c state will result in pathology.
2.6.4. Sialic Acid Determines Macrophage Homeostasis
How is this imbalance between the healing macrophage polarization state of M2c and the neurotoxic, hyper phagocytic M1 and the M1 derived angiogenic VEGF producing M2d develop. The answer lies in the alteration of the glycocalyx “the sugar coat” of retinal cells which in healthy young retinas are decorated with sialic acid caps that serve as self-associated molecular pattern receptor ligands to the global immunosuppressive checkpoint receptors called Siglecs. These sialic acid caps also serve as an activator of CFH whose role is to degrade the complement amplification complex C3 convertase. This overlooked sugar coating that determines immune self-versus non-self, are the master regulators of immune cells and immune function both harmful and protective.[69]
The resident macrophage microglia have multiple functions. In a non-inflammatory environment, microglia are like physicians their job is to detect illness and maintain health of the cells that surround them. This sensome function permits detection of stressed, damaged, sick or apoptosing cells which activate the microglia to M1 like state to eliminate the toxins, and sick cells, but is followed temporally by repolarizing these M1 microglia to the M2a,c like phenotype which produces neuroprotective cytokines such as BDNF or CTGF.[60] In the setting of the cellular environment, normalcy is defined by sensing through their checkpoint receptors the presence of normal sialic acid patterns on the cells that they are tasked to maintain health.
2.6.5. Retinal Stress Chronically Activates Macrophages
Photoreceptors are cells that are constantly being bombarded with oxidative, metabolic, and toxic metabolite induced stress. The retina is constantly exposed to blue/violet light which is photooxidative and with overexposure can cause S-cone retinal degeneration and apoptosis.[56]. The visual cycle is very metabolic and the regeneration of visual pigment following light exposure requires many biochemical reactions that consume energy and create toxic biochemical waste. In the retina a fluorophore called lipofuscin which has been identified as Bis-retinoid N-retinyl-N-retinylidene ethanolamine (A2E) represents an auto fluorescent toxic visual cycle metabolite that has been implicated in the progression of geographic atrophy.[70] A2E cannot be enzymatically degraded and so accumulates in the RPE cells and in healthy youthful eyes are cleared by microglia or macrophages cells.[71] (Figure 2)
The retinal environment is unique amongst all other CNS compartments in that it undergoes constant damaging stress, and the microglia are constantly activated in a parainflammatory state.[72] To maintain this homeostatic retinal parainflammation, the regulators of microglial function have to be tightly controlled to determine the species of microglia that predominate. The different microglial states can be M0 sentinel and resting, M1 pro-inflammatory and phagocytic, M2d anti-inflammatory and proangiogenic, M2a, b anti-inflammatory and profibrotic and M2c anti-inflammatory, neuroprotective and healing. If the balance between these polarization states tilts towards any of these states besides M2c, pathology will ensue. If M0 predominates then housekeeping function of microglia will be impaired and the accumulation of cellular debris will develop producing pigment clumping and drusen. M1 predominates than neurotoxic cytokines will be produced inciting rpe/photoreceptor apoptosis and phagocytic clearance of these injured cells. If m2d predominates then VEGF will be produced resulting in choroidal neovascularization or exudative AMD. If M2a, b predominates then fibrotic scarring will be the hallmark of the pathology which is synonymous with disciform scarring of the macula. (Figure 4)
2.6.6. Age related loss of sialic acid caused unchecked retinal inflammation.
The clinical manifestation of macular degeneration is determined by macrophage polarization, but the loss of cell surface sialic acid and its glycoimmune checkpoint regulation that allows the disease to progress. Overtime, overactivated complement and cumulative oxidative damage, erodes the complex sialylated glycan structures of the cellular glycome of the retina, particularly in the fovea and macula, the glyco-immune checkpoint restraint on microglial cells is released and the microglia transform into their more pro-inflammatory state. This subsequently recruits peripheral blood macrophages by the upregulation of FUT5 that produces lewis x glycan moieties that attract peripheral blood monocytes by binding e-selectin on vascular endothelium of inflamed areas. The recruitment of predominantly M1 polarized macrophages to the retina in the absence of appropriate sialic acid results in unchecked inflammation and clinical progression of macular degeneration.
3. Targeting Microglial/Macrophage SIGLECs to Treat AMD
3.1. M2c polarization a promising therapeutic strategy for AMD
As in spinal cord injury, elevating M2c results in better neuronal recovery. A potential therapeutic strategy that could not only halt all forms of macular degeneration but restore visual function and retinal homeostatic health would be to reintroduce sialic acid control of microglia and macrophages. This strategy would increase M2c and decrease M1, M2d, M2a microglia /macrophages, by repolarizing them to M2c. If this repolarization strategy were to be possible, then not only would stopping progression of the disease be possible, but potentially restoration of visual function and retinal health would also be obtainable.
3.2. Sialic acid can repolarize macrophages to M2c
Tumor’s ability to evade immune attack provides the blue print on how to repolarize microglia and macrophages to the healing M2c state. Many tumors hyper express sialic acid by upregulating Golgi resident sialyltransferases.[73] These sialyltransferases produce sialylation patterns on tumors that allow them to evade immune surveillance.[74] One well characterized tumor derived sialic acid pattern that dictates monocyte/macrophage differentiation is the sialic acid ligands that are produced by pancreatic ductal carcinoma that bind Siglec 7 and 9 and reduce inflammatory signaling, increase PD-L1 a T-cell checkpoint ligand and increases IL-10. This pancreatic cancer produced sialic acid pattern agonizes Siglec-7 and 9 which deactivates monocyte derived macrophages to their quiescent state M2c state which cloaks tumors from both the innate and adaptive immune system and permits tumors to grow unchecked.[75]
Another demonstration of the ability for sialic acid to determine inflammatory state was demonstrated in a glycoprotein called serum amyloid protein a well characterized anti-fibrotic and anti-inflammatory that targets fibrotic and inflammatory macrophages.[76] The innate immune antifibrotic properties of SAP are mediated by the α(2,6)- linked terminally sialylated glycan found on the N32 position of SAP. If the sialic acid is removed from this glycan, it no longer has the anti-fibrotic properties. [77] C-reactive protein which has similar sequence homology to SAP but is a pro-inflammatory molecule can be made to behave like sialylated SAP by mutating CRP at position 32 from an alanine to an asparagine. This mutation resulted in an N-linked glycosylation added to the surface of CRP which functionally made Sialylated CRP A32N behave like sialylated SAP. This enabled CRP A32N to inhibit fibrocyte differentiation in human peripheral blood monocytes. [77]
3.3. Protein sialic acid mimetics not feasible pharmaceutical
This promise of developing immune modulating therapeutic proteins with altered glycosylation is reduced by the difficulty and unpredictability of altering glycosylation by mutating amino acid sequences. When CRP A32N was mutated, only 40% of the recombinant CRP A32N was sialylated. [77] This absence of sialylation prevented the anti-fibrotic gain of function that sialylated CRP A32N produced. This failure can be explained by the post translational nature of cellular glycosylation.
Protein glycosylation is based on O or N-linked glycosylation. O-glycosylation is defined by a sugar attachment to the oxygen atom on amino acids serine or threonine. N-linked glycosylation is defined by a sugar attachment to the nitrogen atom of an asparagine. To create a recombinant glycoprotein with the correct glycan expression pattern the correct sialyltransferases must be upregulated to produce a particular glycan pattern. Since most recombinant proteins are produced in non-human cell lines, the cells do not express the correct glycosyltransferases. Because of these technological hurdles these protein glyco-mimetic immune modulating therapeutics are not druggable.
3.4. Naked PolySialic Acid not feasible pharmaceutical
3.4.1. PSA in Laser CNV model
The concept of just using sialic acid glycans by themselves has been investigated. In a laser induced CNV model of exudative macular degeneration, α(2,8) linked polysialic acid was able to reduce microglial/macrophage activation and recruitment indicating its ability in the eye to agonize appropriate Siglecs which in the wild type mouse is Siglec-E the mouse ortholog of Siglec 7 and 9. The intravitreal injection of this polysialic acid was also able to inhibit terminal complement complex formation and reduce the size and leakage of the neovascular lesions significantly.[78]
3.4.2. PSA in Parkinson’s model
Polysia was also shown to ameliorate inflammatory dopaminergic neurodegeneration in a lipopolysaccharide induced Siglec 11 transgenic mouse model of Parkinson’s disease. Intraperitoneal injection of LPS was followed by either injection of polysia or control and brains were then examined for complement 4(C4) integrin alpha M (Itgam) a subunit of complement receptor 3 and C3. The brains were also probed for oxidative burst pathway enzymes such as nitric oxide synthase 2 (NOS2) and cytochrome b245 alpha and beta chain (Cyba/Cybb) . The polysia only reduced C4 expression but not Itgam or C3 expression in the LPS challenged mice. Polysia could not reduce Nos2 or Cyba. [79] It is unclear why PSA could not suppress these factors in vivo but none the less does not support the use of PSA not presented on a NCAM like protein as a therapeutic.
3.4.3. PSA in Multiple Sclerosis model
In a model of multiple sclerosis, polysia dp-24-30 reduced nitric oxide, and recruited arginase-1 positive microglia to enhance remyelination in organotypic cerebellar slice culture of demyelination. Interestingly polysia of dp-8-14 reduced in vitro differentiation and did not help with remyelination. This observation demonstrates the specificity of Siglec receptors to the form of sialic acid and the presentation of the Sialic acid.[80]
3.4.4. PSA in Alzheimer’s model
Polysialic acid fragments have also been delivered through the intranasal route to the brain of mice who are deficient in polysia and two mouse models of Alzheimer’s disease. The diseases of Alzheimer’s and schizophrenia have been associated with a deficit of neural cell adhesion molecule (NCAM) which is the brains main repository of polysialic acid. While mice receiving this treatment were shown to have rescued medial prefrontal cortex tasks. Studies of intracranial pharmacokinetics demonstrated that these particles were only sparsely distributed in the mouse brain after 3 hours and declined rapidly by 24 hours.[81]
3.4.5. PSA by itself not druggable
While exogenous polysialic acid has demonstrated beneficial effect in animal models of wet AMD, Alzheimer’s and Parkinson’s disease, the ability to optimize sialic acid- Siglec immune cell synapse, requires presentation, density, and persistence to mediate true immune cell deactivation. [82] If a therapy were to be developed for geographic atrophy the drug would have to be administered at least monthly so a drug would need to persist. This drug would also need to present sialic acids in a multivalent fashion.
As has been demonstrated by these early proof of concept experiments, PSA alone would not be feasible therapeutic from a biologic, pharmacokinetic and a biodistribution perspective.
3.5. PSA-Nanoparticles promising therapeutic strategy for AMD
Nanoparticles decorated with sialic acid have been used to increase blood circulation time[83] and target tumor lectins for delivery of anti-tumor prodrugs. [84] The concept of decorating a nanoparticle with sialic acid to down modulate inflammation by agonizing SIGLEC receptors was first conceptualized and demonstrated in the laboratory of Professor Chris Scott. His lab decorated a nanoparticle with disialic acid that functionally mimicked a protein covered with sialylated glycans. This disialic acid α(2,8) linked decorated nanoparticle was able to completely abrogate a model of sepsis and a model of acute respiratory distress. [85] This immune modulating demonstration paved the way for a nanoparticle to be decorated with polysialic acid targeting microglial cells and complement to abrogate microglial and macrophage M1 polarization by repolarizing macrophages to the M2c healing macrophage.
3.5.1. PSA- nanoparticle mimics Microglial release of PSA-Proteins.
This PSA-nanoparticle was designed to mimic PSA bearing proteins such as Neuropilin-2 or E-selectin ligand -1. These PSA bearing proteins are secreted by microglia as part of the negative feedback regulation of microglial inflammatory response to injury or toxic stress. [86] After LPS administration in cell culture, PSA-neuropilin-2 and PSA-E-selectin ligand 1 are released in a metalloproteinase -dependent manner into the cell culture media for at least 24 hours after LPS challenge. The release of microglial derived polysia ligand that binds Siglec-E via a trans interaction, in the setting of acute brain injury, consolidates the role of polysialic acid ligands expressed on neuropilin-2 and e selectin ligand 1 as the main agonistic activator of Siglec -E which is negative feedback regulator of microglial activation preventing unbridled inflammatory responses. [87] (Figure 5)
3.5.2. PSA- nanoparticle promising AMD therapeutic
If this nanoparticle can mimics PSA -neuropilin 2 or PSA-e-selectin ligand 1 and can repolarize activated M1 microglia/macrophages to the M2c healing state this will decrease M1 and increase M2c. This repolarization has the potential to restore homeostatic healing microglial/macrophage function. In the setting of macular degeneration, this homeostatic restoration could theoretically cease cellular loss, normalize the parainflammatory environment, clear toxic substances such as auto fluorescent lipofuscin, rescue pre-apoptotic photoreceptors/RPE cells and restore efficient visual pigment regeneration. Potentially a treatment such as this could halt geographic atrophy growth, reduce lipofuscin mediated autofluorescence at the edge of the GA lesion, and recover visual function.(Figure 5)
4. Conclusion
Currently a poly sialic acid nanoparticle has entered human clinical trials for the treatment of geographic atrophy secondary to age related macular degeneration.[88] The PLGA nanoparticle core will be resistant to degradation and should demonstrate prolonged half-life in the vitreous.[89] The individual constituents of the nanoparticle are all biodegradable and have been proven safe for intravitreal injections.
This nanoparticle therapeutic is a novel technology that targets microglia and macrophage cells to repolarize their active state to the resolution healing state in a physiologic manner. This is a paradigm shift in terms of targeting macrophages and microglia by agonizing their main sialic acid binding checkpoint receptors (SIGLECS). If this therapeutic is proven to be effective in age related macular degeneration it will open the door for the development of other sialic acid presenting nanoparticle to treat diseases of acute, chronic inflammation such as arthritis, colitis, Lupus, Alzheimer’s, multiple sclerosis, acute respiratory distress syndrome, liver fibrosis and other inflammatory pathologies.
Supplementary Materials
None
Author Contributions
Conceptualization MJT,AJT,AK,MAG; methodology, X.X.; software, X.X.; validation, X.X., Y.Y. and Z.Z.; formal analysis, X.X.; investigation, X.X.; resources, X.X.; data curation, X.X.; writing—original draft preparation, MJT; writing—review and editing MJT MJT,AJT,AK,MAG; visualization, MJT,AJT; supervision, MJT,AJT,AK,MAG; project administration, MJT,AJT,AK; funding acquisition, MAG. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Aviceda Therapeutics and Tolentino Eye Research Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable
Acknowledgments
Figures created with BioRender.com
Conflicts of Interest
The authors are full time (AK, MAG) or part time (MJT, AJT) employees of Aviceda Therapeutics the funder of this paper.
References
- Rein, D.B.; Wittenborn, J.S.; Burke-Conte, Z.; Gulia, R.; Robalik, T.; Ehrlich, J.R.; Lundeen, E.A.; Flaxman, A.D. Prevalence of Age-Related Macular Degeneration in the US in 2019. JAMA Ophthalmol 2022, 140, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- 2021 Alzheimer's disease facts and figures. Alzheimers Dement 2021, 17, 327–406. [CrossRef]
- Global Burden of Disease Cancer, C.; Kocarnik, J.M.; Compton, K.; Dean, F.E.; Fu, W.; Gaw, B.L.; Harvey, J.D.; Henrikson, H.J.; Lu, D.; Pennini, A.; et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol 2022, 8, 420–444. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Blindness, G.B.D.; Vision Impairment, C.; Vision Loss Expert Group of the Global Burden of Disease, S. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. Lancet Glob Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Keenan, T.D.L.; Cukras, C.A.; Chew, E.Y. Age-Related Macular Degeneration: Epidemiology and Clinical Aspects. Adv Exp Med Biol 2021, 1256, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.J.; Rodrigo-Diaz, E.; Kelly, J.M.F.; Aslam, T.M.; Tahir, H.J.; Carden, D.; Patryas, L.; Parry, N.R.A. The role of dark adaptation in understanding early AMD. Prog Retin Eye Res 2022, 88, 101015. [Google Scholar] [CrossRef]
- Tolentino, M.J.; Miller, S.; Gaudio, A.R.; Sandberg, M.A. Visual field deficits in early age-related macular degeneration. Vision Res 1994, 34, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Remulla, J.F.; Gaudio, A.R.; Miller, S.; Sandberg, M.A. Foveal electroretinograms and choroidal perfusion characteristics in fellow eyes of patients with unilateral neovascular age-related macular degeneration. Br J Ophthalmol 1995, 79, 558–561. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Gaudio, A.R. Slow photostress recovery and disease severity in age-related macular degeneration. Retina 1995, 15, 407–412. [Google Scholar] [CrossRef]
- Owsley, C.; Huisingh, C.; Clark, M.E.; Jackson, G.R.; McGwin, G., Jr. Comparison of Visual Function in Older Eyes in the Earliest Stages of Age-related Macular Degeneration to Those in Normal Macular Health. Curr Eye Res 2016, 41, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Owsley, C.; McGwin, G., Jr.; Clark, M.E.; Jackson, G.R.; Callahan, M.A.; Kline, L.B.; Witherspoon, C.D.; Curcio, C.A. Delayed Rod-Mediated Dark Adaptation Is a Functional Biomarker for Incident Early Age-Related Macular Degeneration. Ophthalmology 2016, 123, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Panorgias, A.; Zawadzki, R.J.; Capps, A.G.; Hunter, A.A.; Morse, L.S.; Werner, J.S. Multimodal assessment of microscopic morphology and retinal function in patients with geographic atrophy. Invest Ophthalmol Vis Sci 2013, 54, 4372–4384. [Google Scholar] [CrossRef] [PubMed]
- Galuszka, M.; Pojda-Wilczek, D.; Karska-Basta, I. Age-Related Macular or Retinal Degeneration? Medicina (Kaunas) 2023, 59. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Goto, M. Inflammaging (inflammation + aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci Trends 2008, 2, 218–230. [Google Scholar]
- Ho, J.; Witkin, A.J.; Liu, J.; Chen, Y.; Fujimoto, J.G.; Schuman, J.S.; Duker, J.S. Documentation of intraretinal retinal pigment epithelium migration via high-speed ultrahigh-resolution optical coherence tomography. Ophthalmology 2011, 118, 687–693. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet 2014, 15, 151–171. [Google Scholar] [CrossRef]
- Seddon, J.M.; Rosner, B. Validated Prediction Models for Macular Degeneration Progression and Predictors of Visual Acuity Loss Identify High-Risk Individuals. Am J Ophthalmol 2019, 198, 223–261. [Google Scholar] [CrossRef]
- Emilsson, V.; Gudmundsson, E.F.; Jonmundsson, T.; Jonsson, B.G.; Twarog, M.; Gudmundsdottir, V.; Li, Z.; Finkel, N.; Poor, S.; Liu, X.; et al. A proteogenomic signature of age-related macular degeneration in blood. Nat Commun 2022, 13, 3401. [Google Scholar] [CrossRef]
- Río Hortega, P. Noticia de un nuevo y fácil método para la coloración.
- de la neuroglia y el tejido conjuntivo. Trab. Lab. Invest. Biol. 1918, 15, 367–378.
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb Perspect Biol 2015, 8, a020560. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat Neurosci 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014, 17, 400–406. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci 2010, 13, 411–413. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O'Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J Immunol 2016, 196, 3375–3384. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Fourgeaud, L.; Traves, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagorska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J Exp Med 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T. Microglia activation in retinal degeneration. J Leukoc Biol 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Coscas, G.; De Benedetto, U.; Coscas, F.; Li Calzi, C.I.; Vismara, S.; Roudot-Thoraval, F.; Bandello, F.; Souied, E. Hyperreflective dots: a new spectral-domain optical coherence tomography entity for follow-up and prognosis in exudative age-related macular degeneration. Ophthalmologica 2013, 229, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Bini, S.; Midena, G.; Berton, M.; Pilotto, E.; Midena, E. Hyperreflective intraretinal spots in diabetics without and with nonproliferative diabetic retinopathy: an in vivo study using spectral domain OCT. J Diabetes Res 2013, 2013, 491835. [Google Scholar] [CrossRef] [PubMed]
- Killingsworth, M.C.; Sarks, J.P.; Sarks, S.H. Macrophages related to Bruch's membrane in age-related macular degeneration. Eye (Lond) 1990, 4 ( Pt 4) Pt 4, 613–621. [Google Scholar] [CrossRef]
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: the involvement of immunocompetent cells. Graefes Arch Clin Exp Ophthalmol 1985, 223, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch's membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol 2010, 94, 918–925. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, G.; Liu, W.; Ni, Y.; Zhou, W. Role of fractalkine/CX3CR1 interaction in light-induced photoreceptor degeneration through regulating retinal microglial activation and migration. PLoS One 2012, 7, e35446. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L.; Bell, B.A.; Hu, J.; Milliner, C.; Pauer, G.J.; Hagstrom, S.A.; Radu, R.A.; Hollyfield, J.G. Geographic Atrophy: Confocal Scanning Laser Ophthalmoscopy, Histology, and Inflammation in the Region of Expanding Lesions. Invest Ophthalmol Vis Sci 2020, 61, 15. [Google Scholar] [CrossRef]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog Retin Eye Res 2021, 82, 100906. [Google Scholar] [CrossRef]
- Marneros, A.G. Role of inflammasome activation in neovascular age-related macular degeneration. FEBS J 2023, 290, 28–36. [Google Scholar] [CrossRef]
- Dieckmann, B.W.; Paguaga, M.E.; McCollum, G.W.; Penn, J.S.; Uddin, I. Role of NLRP3 inflammasomes in monocyte and microglial recruitments in choroidal neovascularization. Res Sq 2023. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, F.; Tang, M.; Yuan, M.; Hu, A.; Zhan, Z.; Li, Z.; Li, J.; Ding, X.; Lu, L. Macrophage polarization in experimental and clinical choroidal neovascularization. Sci Rep 2016, 6, 30933. [Google Scholar] [CrossRef]
- Cruz-Pimentel, M.; Wu, L. Complement Inhibitors for Advanced Dry Age-Related Macular Degeneration (Geographic Atrophy): Some Light at the End of the Tunnel? J Clin Med 2023, 12. [Google Scholar] [CrossRef]
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration. The involvement of giant cells in atrophy of the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1986, 27, 364–371. [Google Scholar]
- Sarks, S.H. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol 1976, 60, 324–341. [Google Scholar] [CrossRef]
- Sarks, S.H. Drusen patterns predisposing to geographic atrophy of the retinal pigment epithelium. Aust J Ophthalmol 1982, 10, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Karlen, S.J.; Miller, E.B.; Wang, X.; Levine, E.S.; Zawadzki, R.J.; Burns, M.E. Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J Neuroinflammation 2018, 15, 344. [Google Scholar] [CrossRef] [PubMed]
- Klaus, C.; Liao, H.; Allendorf, D.H.; Brown, G.C.; Neumann, H. Sialylation acts as a checkpoint for innate immune responses in the central nervous system. Glia 2021, 69, 1619–1636. [Google Scholar] [CrossRef]
- Miralles de Imperial-Ollero, J.A.; Gallego-Ortega, A.; Ortin-Martinez, A.; Villegas-Perez, M.P.; Valiente-Soriano, F.J.; Vidal-Sanz, M. Animal Models of LED-Induced Phototoxicity. Short- and Long-Term In Vivo and Ex Vivo Retinal Alterations. Life (Basel) 2021, 11. [Google Scholar] [CrossRef]
- Suzuki, M.; Tsujikawa, M.; Itabe, H.; Du, Z.J.; Xie, P.; Matsumura, N.; Fu, X.; Zhang, R.; Sonoda, K.H.; Egashira, K.; et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J Cell Sci 2012, 125, 2407–2415. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.L.; Wyse-Jackson, A.C.; Ruiz-Lopez, A.M.; Byrne, A.M.; Cotter, T.G. Fractalkine-CX3CR1 signaling is critical for progesterone-mediated neuroprotection in the retina. Sci Rep 2017, 7, 43067. [Google Scholar] [CrossRef]
- Tolentino, M.J.; Tolentino, A.J. Investigational drugs in clinical trials for macular degeneration. Expert Opin Investig Drugs 2022, 31, 1067–1085. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, X.; Wang, K.; Bao, Y.; Zhang, T.; Ainiwaer, D.; Wang, G.; Li, H.; Sun, Z. Peripheral Klotho protects the kidney and brain by regulating M2a/M2c macrophage polarization in d-gal-treated aged mice. Tissue Cell 2023, 82, 102049. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Karaman, S.; Mohamed, W.K.E.; Garcia-Sabate, A.; Quartey, B.C.; Teo, J.C.M. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. NPJ Regen Med 2021, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Goerdt, S.; Politz, O.; Schledzewski, K.; Birk, R.; Gratchev, A.; Guillot, P.; Hakiy, N.; Klemke, C.D.; Dippel, E.; Kodelja, V.; et al. Alternative versus classical activation of macrophages. Pathobiology 1999, 67, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Kisucka, A.; Bimbova, K.; Bacova, M.; Galik, J.; Lukacova, N. Activation of Neuroprotective Microglia and Astrocytes at the Lesion Site and in the Adjacent Segments Is Crucial for Spontaneous Locomotor Recovery after Spinal Cord Injury. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Shen, D.; Patel, M.M.; Tuo, J.; Johnson, T.M.; Olsen, T.W.; Chan, C.C. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int 2011, 61, 528–535. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Alves, I.; Fernandes, A.; Santos-Pereira, B.; Azevedo, C.M.; Pinho, S.S. Glycans as a key factor in self and nonself discrimination: impact on the breach of immune tolerance. FEBS Lett 2022, 596, 1485–1502. [Google Scholar] [CrossRef]
- Bearelly, S.; Khanifar, A.A.; Lederer, D.E.; Lee, J.J.; Ghodasra, J.H.; Stinnett, S.S.; Cousins, S.W. Use of fundus autofluorescence images to predict geographic atrophy progression. Retina 2011, 31, 81–86. [Google Scholar] [CrossRef]
- Su, N.; Hansen, U.; Plagemann, T.; Gaher, K.; Leclaire, M.D.; Konig, J.; Hohn, A.; Grune, T.; Uhlig, C.E.; Eter, N.; et al. Sub-Retinal Injection of Human Lipofuscin in the Mouse - A Model of "Dry" Age-Related Macular Degeneration? Aging Dis 2023, 14, 184–203. [Google Scholar] [CrossRef]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol 2015, 98, 713–725. [Google Scholar] [CrossRef]
- Bull, C.; Stoel, M.A.; den Brok, M.H.; Adema, G.J. Sialic acids sweeten a tumor's life. Cancer Res 2014, 74, 3199–3204. [Google Scholar] [CrossRef]
- Pietrobono, S.; Stecca, B. Aberrant Sialylation in Cancer: Biomarker and Potential Target for Therapeutic Intervention? Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Eveline Li, R.J.; Kruijssen, L.; Bruijns, S.C.M.; van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; van der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat Commun 2021, 12, 1270. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Gomer, R.H. The Development of Serum Amyloid P as a Possible Therapeutic. Front Immunol 2018, 9, 2328. [Google Scholar] [CrossRef]
- Cox, N.; Pilling, D.; Gomer, R.H. DC-SIGN activation mediates the differential effects of SAP and CRP on the innate immune system and inhibits fibrosis in mice. Proc Natl Acad Sci U S A 2015, 112, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Karlstetter, M.; Kopatz, J.; Aslanidis, A.; Shahraz, A.; Caramoy, A.; Linnartz-Gerlach, B.; Lin, Y.; Luckoff, A.; Fauser, S.; Duker, K.; et al. Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol Med 2017, 9, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Winkler, J.; Wissfeld, J.; Shahraz, A.; Klaus, C.; Neumann, H. Low molecular weight polysialic acid prevents lipopolysaccharide-induced inflammatory dopaminergic neurodegeneration in humanized SIGLEC11 transgenic mice. Glia 2021, 69, 2845–2862. [Google Scholar] [CrossRef]
- Schroder, L.J.; Thiesler, H.; Gretenkort, L.; Mollenkamp, T.M.; Stangel, M.; Gudi, V.; Hildebrandt, H. Polysialic acid promotes remyelination in cerebellar slice cultures by Siglec-E-dependent modulation of microglia polarization. Front Cell Neurosci 2023, 17, 1207540. [Google Scholar] [CrossRef] [PubMed]
- Varbanov, H.; Jia, S.; Kochlamazashvili, G.; Bhattacharya, S.; Buabeid, M.A.; El Tabbal, M.; Hayani, H.; Stoyanov, S.; Sun, W.; Thiesler, H.; et al. Rescue of synaptic and cognitive functions in polysialic acid-deficient mice and dementia models by short polysialic acid fragments. Neurobiol Dis 2023, 180, 106079. [Google Scholar] [CrossRef]
- Gonzalez-Gil, A.; Schnaar, R.L. Siglec Ligands. Cells 2021, 10. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine 2006, 1, 297–315. [Google Scholar]
- Jayant, S.; Khandare, J.J.; Wang, Y.; Singh, A.P.; Vorsa, N.; Minko, T. Targeted sialic acid-doxorubicin prodrugs for intracellular delivery and cancer treatment. Pharm Res 2007, 24, 2120–2130. [Google Scholar] [CrossRef]
- Spence, S.; Greene, M.K.; Fay, F.; Hams, E.; Saunders, S.P.; Hamid, U.; Fitzgerald, M.; Beck, J.; Bains, B.K.; Smyth, P.; et al. Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation. Sci Transl Med 2015, 7, 303ra140. [Google Scholar] [CrossRef]
- Werneburg, S.; Buettner, F.F.; Erben, L.; Mathews, M.; Neumann, H.; Muhlenhoff, M.; Hildebrandt, H. Polysialylation and lipopolysaccharide-induced shedding of E-selectin ligand-1 and neuropilin-2 by microglia and THP-1 macrophages. Glia 2016, 64, 1314–1330. [Google Scholar] [CrossRef] [PubMed]
- Thiesler, H.; Beimdiek, J.; Hildebrandt, H. Polysialic acid and Siglec-E orchestrate negative feedback regulation of microglia activation. Cell Mol Life Sci 2021, 78, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Aviceda Therapeutics, I. A Multiple Dose Study of AVD-104 for Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD) (SIGLEC). Available online: https://clinicaltrials.gov/study/NCT05839041.
- Giordano, G.G.; Chevez-Barrios, P.; Refojo, M.F.; Garcia, C.A. Biodegradation and tissue reaction to intravitreous biodegradable poly(D,L-lactic-co-glycolic)acid microspheres. Curr Eye Res 1995, 14, 761–768. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Early/Intermediate Stage AMD. Constant photooxidative blue light release produces oxidative byproducts (MDA, CEP) and reactive oxygen species, which activate microglial cells to secrete cytokines. These activated macrophages are hindered from becoming phagocytic due to the upregulation ST6 that produces sTN on the surface of retinal cells and agonizes Siglecs to control phagocytosis. Reactive oxygen species also causes photoreceptors to secrete CX3CL1 which promotes migration of microglia and macrophages to the retina. Oxidized phospholipids stimulate RPE cells to produce MCP—1 which recruits peripheral blood monocytes along with upregulation of FUT5 on PBMCs that localize it to areas that are secreting chemokines such as CX3CL1.
Figure 1.
Early/Intermediate Stage AMD. Constant photooxidative blue light release produces oxidative byproducts (MDA, CEP) and reactive oxygen species, which activate microglial cells to secrete cytokines. These activated macrophages are hindered from becoming phagocytic due to the upregulation ST6 that produces sTN on the surface of retinal cells and agonizes Siglecs to control phagocytosis. Reactive oxygen species also causes photoreceptors to secrete CX3CL1 which promotes migration of microglia and macrophages to the retina. Oxidized phospholipids stimulate RPE cells to produce MCP—1 which recruits peripheral blood monocytes along with upregulation of FUT5 on PBMCs that localize it to areas that are secreting chemokines such as CX3CL1.
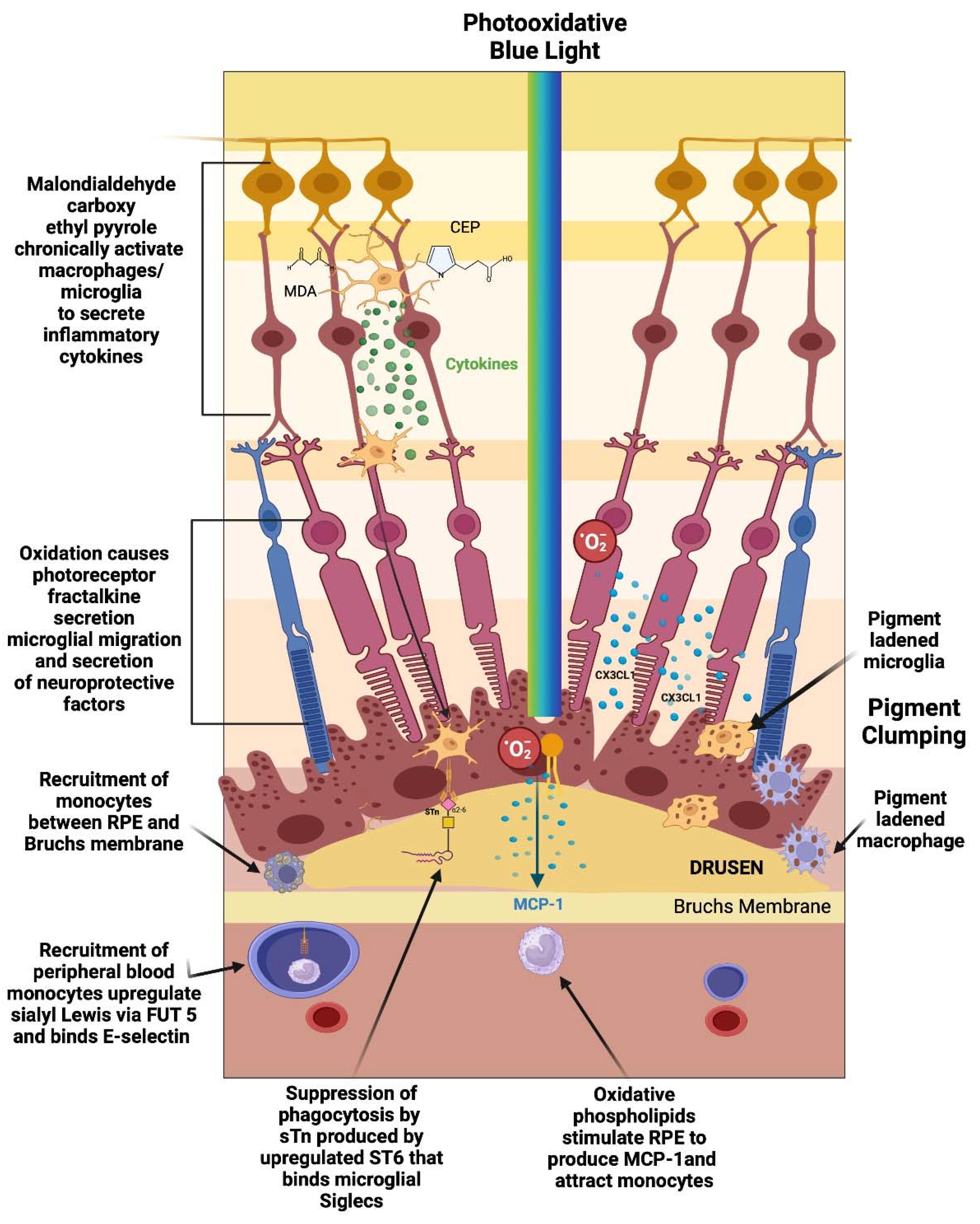
Figure 2.
Geographic Atrophy: Oxidative byproducts (CEP, MDA) and complement chronically activate microglia which secrete neuraminidase and desialylate photoreceptors and RPE cells. This loss of sialylation prevents restoration of homeostasis. Chronically overactivated phagocytosis and inflammation recruits peripheral blood macrophages by upregulation of the fucosyltransferase FUT5 that produces Lewis X glycosylation on monocytes to bind E-selectin and localize monocytes to sites of inflammation. These monocytes polarize to M1 macrophages when they enter the retina and are not able to clear substances like lipofuscin and other undegradeable substances. The macrophages form multinucleated giants cells because they phagocytose structures that are undegradable. Since there is reduced sialic acid and altered sialic acid on the chronically inflamed retina, the macrophages are unchecked and result in elimination first of the RPE cells than the photoreceptors. The unchecked macrophages are the main determinant of growth of geographic atrophy.
Figure 2.
Geographic Atrophy: Oxidative byproducts (CEP, MDA) and complement chronically activate microglia which secrete neuraminidase and desialylate photoreceptors and RPE cells. This loss of sialylation prevents restoration of homeostasis. Chronically overactivated phagocytosis and inflammation recruits peripheral blood macrophages by upregulation of the fucosyltransferase FUT5 that produces Lewis X glycosylation on monocytes to bind E-selectin and localize monocytes to sites of inflammation. These monocytes polarize to M1 macrophages when they enter the retina and are not able to clear substances like lipofuscin and other undegradeable substances. The macrophages form multinucleated giants cells because they phagocytose structures that are undegradable. Since there is reduced sialic acid and altered sialic acid on the chronically inflamed retina, the macrophages are unchecked and result in elimination first of the RPE cells than the photoreceptors. The unchecked macrophages are the main determinant of growth of geographic atrophy.
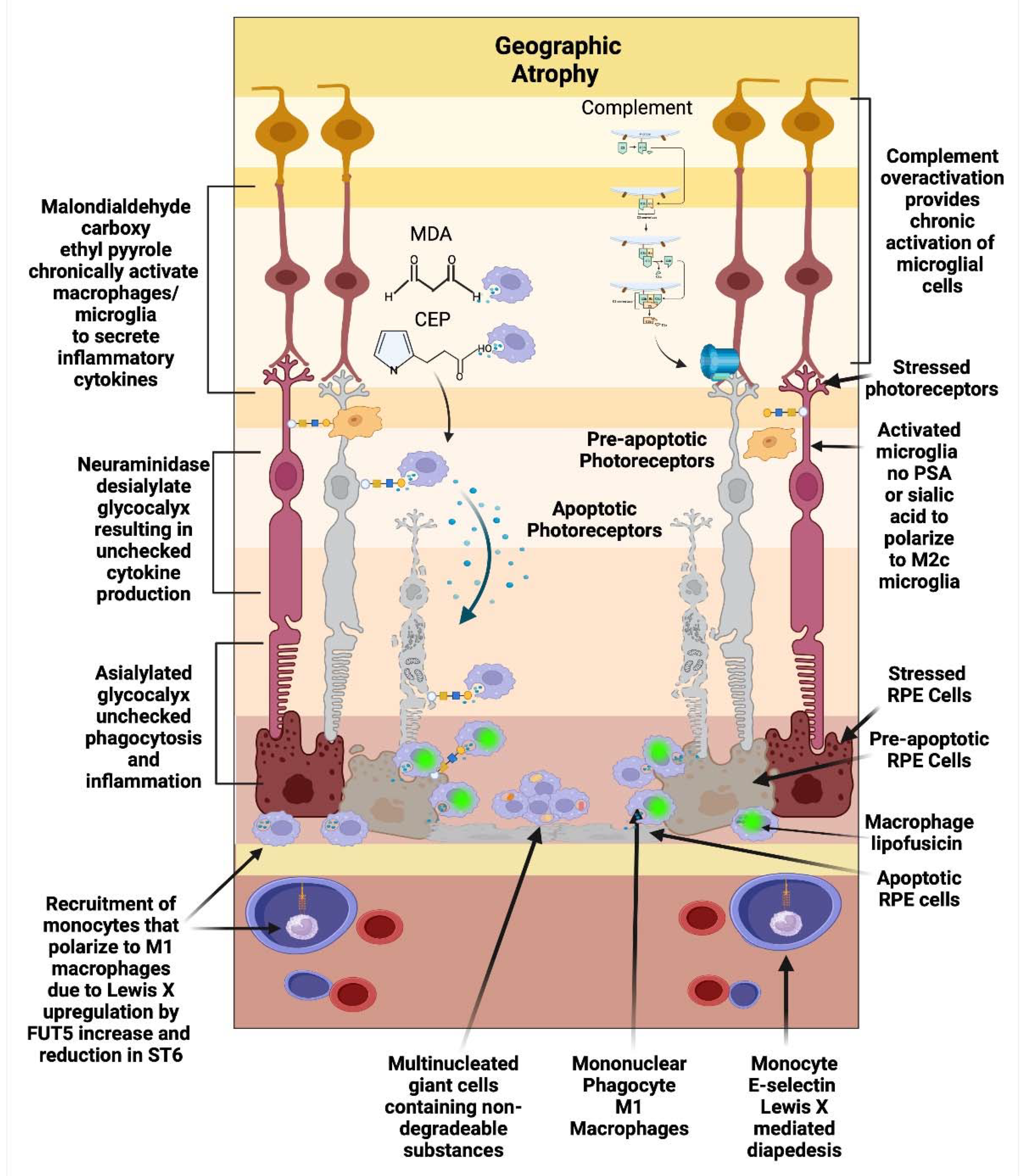
Figure 3.
Microglial Regulation: Microglia remove toxic metabolites, apoptotic cells and oxidative debris while maintaining the health of photoreceptors and retinal cells in a process called para inflammation. Parainflammation is initated through activation receptors such as TLR, TREM2, CX3CR1, NLRP3 and CD36 which sense and bind these substances. Once activated they secrete proinflammatory cytokines and migrate to the areas of stressed RPE cells and photoreceptors. Upon activation, neuropilin-2-PSA and E-selectin ligand 1-PSA are secreted by these activated microglia. The PSA on these glycoproteins binds Siglec’s on activated microglial cells to polarize them to the M2C healing state which releases, growth factors to protect and regenerate the stressed photoreceptors and RPE cells. This homeostatic maintenance function of microglial para inflammation if not adequately modulated with sialic acid checkpoint regulation will result in recruitment of peripheral blood monocytes and AMD disease progression.
Figure 3.
Microglial Regulation: Microglia remove toxic metabolites, apoptotic cells and oxidative debris while maintaining the health of photoreceptors and retinal cells in a process called para inflammation. Parainflammation is initated through activation receptors such as TLR, TREM2, CX3CR1, NLRP3 and CD36 which sense and bind these substances. Once activated they secrete proinflammatory cytokines and migrate to the areas of stressed RPE cells and photoreceptors. Upon activation, neuropilin-2-PSA and E-selectin ligand 1-PSA are secreted by these activated microglia. The PSA on these glycoproteins binds Siglec’s on activated microglial cells to polarize them to the M2C healing state which releases, growth factors to protect and regenerate the stressed photoreceptors and RPE cells. This homeostatic maintenance function of microglial para inflammation if not adequately modulated with sialic acid checkpoint regulation will result in recruitment of peripheral blood monocytes and AMD disease progression.
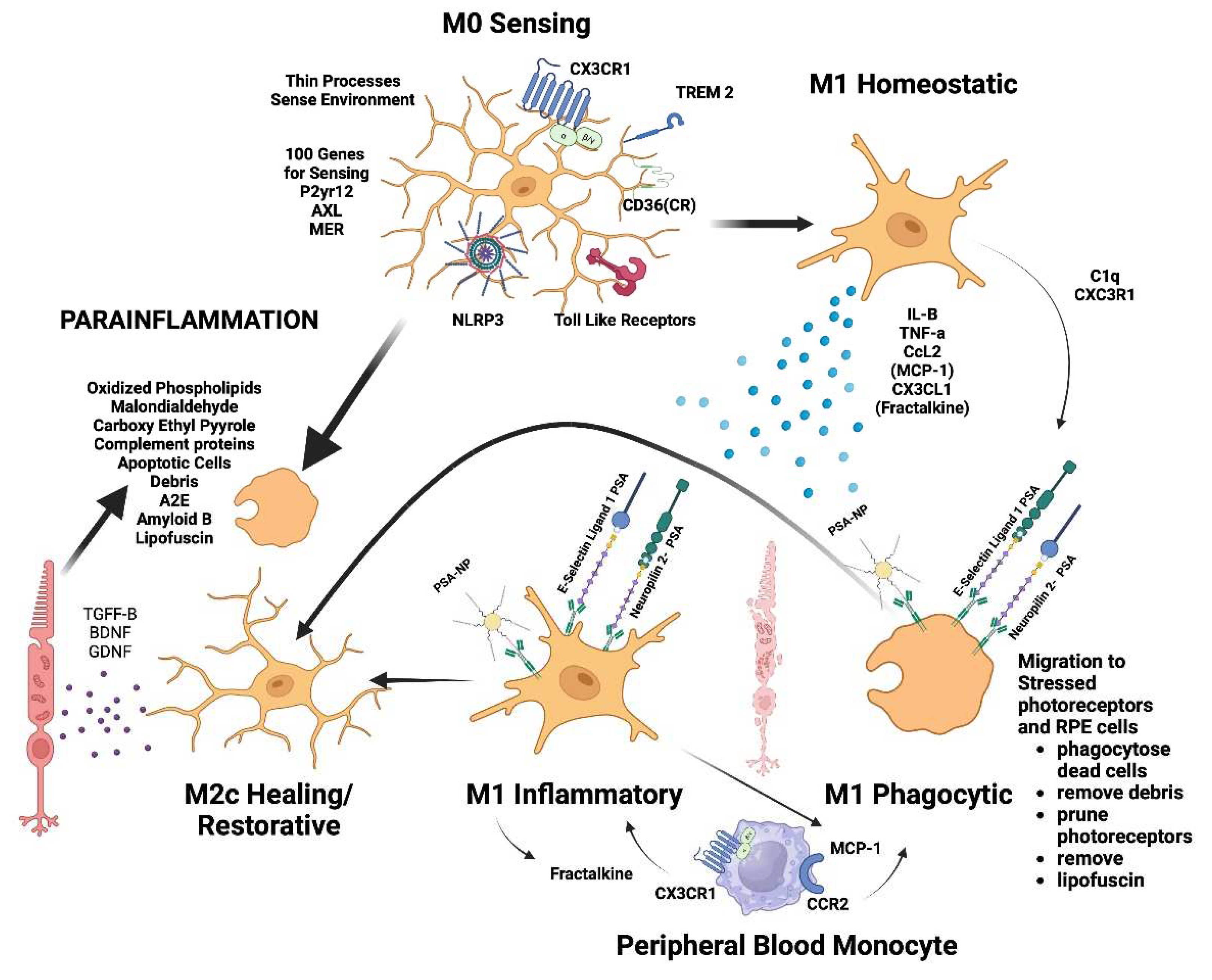
Figure 4.
Macrophage polarization determines AMD phenotype. The plasticity and different polarization states correlate with the clinical picture seen in late-stage macular degeneration. IN geographic atrophy RPE cells and photoreceptors are phagocytosed a function of the M1 polarized phagocytic macrophage. Exudative AMD is neovascularization produced by overexpression of VEGF the main cytokine of the M2D polarization state. The sequelae of exudative AMD untreated with anti-VEGF therapies is a disciform fibrotic scar in the control of pro-fibrotic M2 A, B state. With appropriate sialic acid signaling such as PSA, all polarization states can transform into the healing M2C state.
Figure 4.
Macrophage polarization determines AMD phenotype. The plasticity and different polarization states correlate with the clinical picture seen in late-stage macular degeneration. IN geographic atrophy RPE cells and photoreceptors are phagocytosed a function of the M1 polarized phagocytic macrophage. Exudative AMD is neovascularization produced by overexpression of VEGF the main cytokine of the M2D polarization state. The sequelae of exudative AMD untreated with anti-VEGF therapies is a disciform fibrotic scar in the control of pro-fibrotic M2 A, B state. With appropriate sialic acid signaling such as PSA, all polarization states can transform into the healing M2C state.
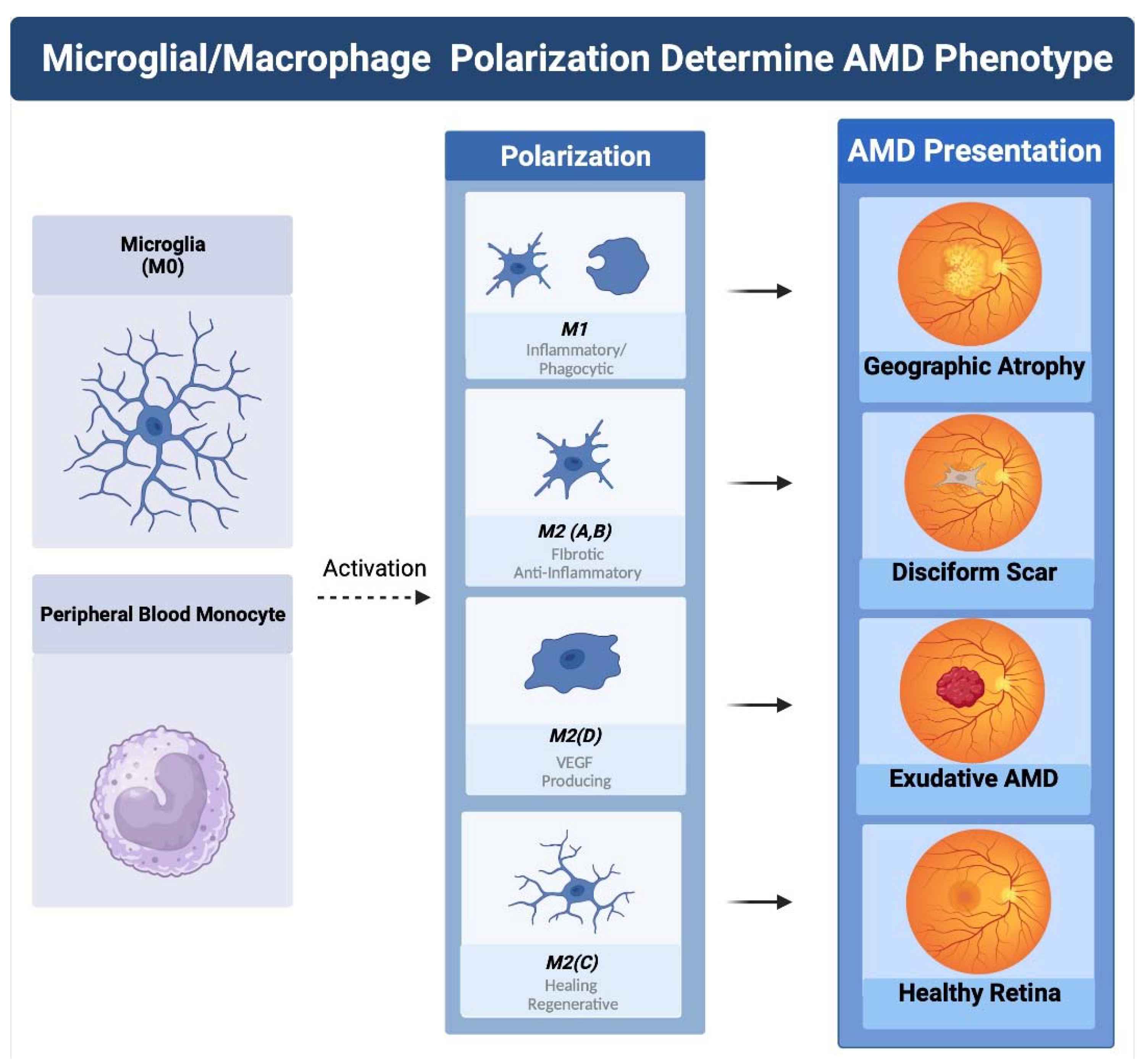
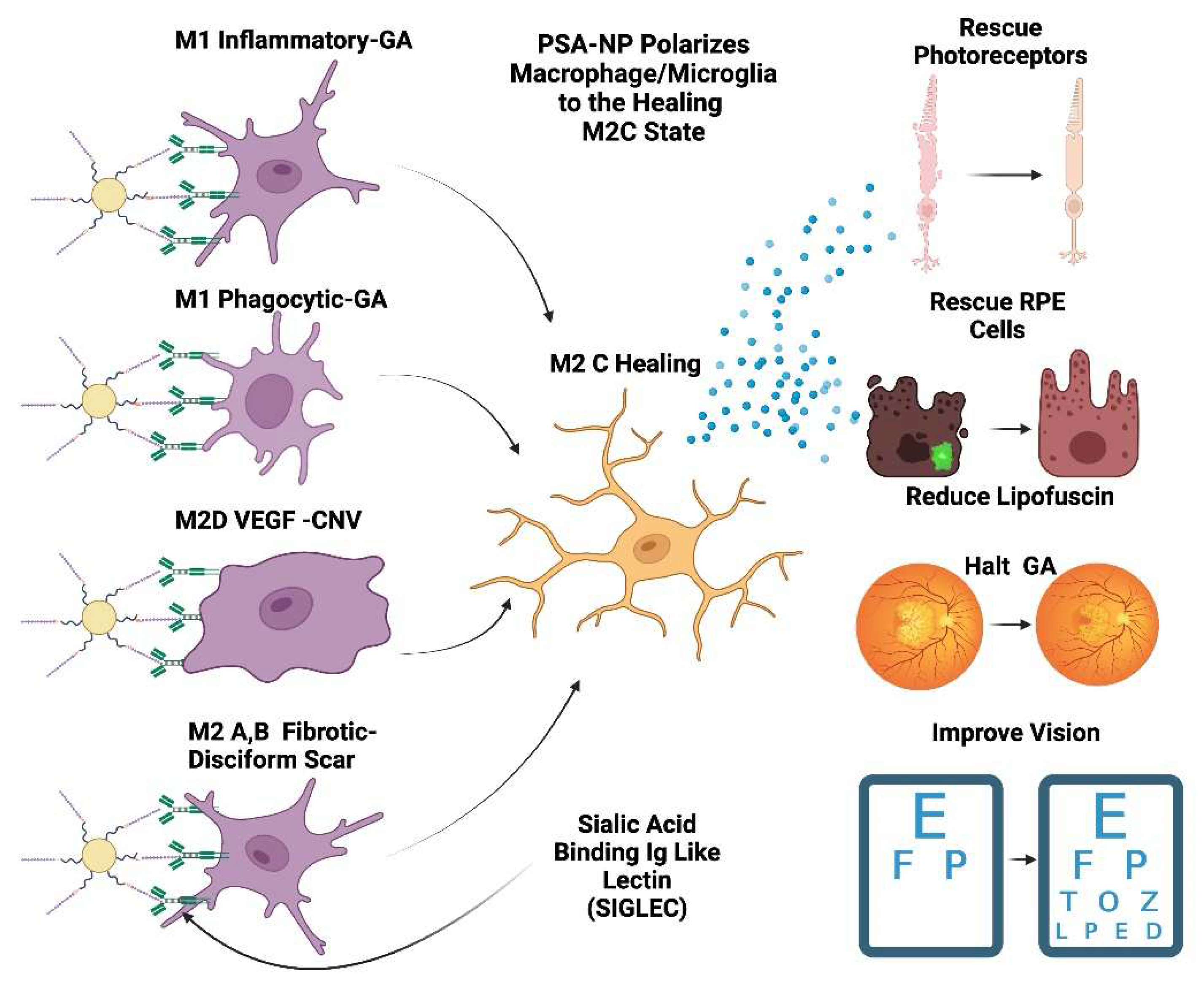
Figure 5.
PSA-NP can restore retinal health PSA-NP can multivalently bind Siglecs on polarized macrophages and microglia which will transform them into the M2 C healing state. This M2 C healing macrophage/microglia will reduce activated macrophages, secrete neuroprotective and healing factors such as BDNF and GDNF. This in turn will rescue photoreceptors, RPE cells and restore the homeostatic parainflammatory function of microglia and potentially can halt GA progression and improve visual function and vision.
Figure 5.
PSA-NP can restore retinal health PSA-NP can multivalently bind Siglecs on polarized macrophages and microglia which will transform them into the M2 C healing state. This M2 C healing macrophage/microglia will reduce activated macrophages, secrete neuroprotective and healing factors such as BDNF and GDNF. This in turn will rescue photoreceptors, RPE cells and restore the homeostatic parainflammatory function of microglia and potentially can halt GA progression and improve visual function and vision.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



