
Submitted:
18 October 2023
Posted:
19 October 2023
You are already at the latest version
Abstract
Zinc, being a part of many proteins and enzymes, plays an important role in the vital processes of the body. Racemic α-lipoic acid (LA) has a wide range of biological activities: antioxidant, antidiabetic, anticancer, and it is also used in diseases such as Parkinson's and Alzheimer's. Nowadays, the synthesis and structural studies of new zinc complexes with biologically active compounds are of great importance for drug discovery. This work shows the studies carried out on the synthesis, structure, identification, also biological activity using in silico methods of a complex compound based on LA and zinc. A complex compound of zinc with LA (ZnLA) was synthesized in the presence of LA and zinc acetate in ethanol. The identification of ZnLA by (FT-IR, XRD and TGA-DTA), also DFT calculations at the B3LYP/def2-TZVPP level of theory was performed. The molecular docking investigation involved docking ZnLA with two different protein receptors such us Glycogen phosphorylase (2GPA) and Kv potassium channel beta subunit (1ZSX) using CB-Dock 2 server and Auto-Dock Vina program to determine the mechanism of action and biological activity. This study reveals potential ligand-protein interactions at the atomic level and suggests that the treatment of diabetes (non-insulin dependent) with ZnLA.
Keywords:
Racemic α-lipoic acid
; zinc
; metallcomplex
; in silico
; DFT calculation
; molecular docking simulation
; target proteins
; diabetes
1. Introduction
Zinc is part of various proteins and enzymes and plays an important role in many life processes: metabolism, transmission of genetic information, growth and development of the body. The chemically stable, but stereochemically flexible, non-toxic nature of zinc and its amphoteric properties underlie the production of a number of important biocomplexes [1,2].
In additional, disturbance of zinc homeostasis in the body can cause diabetes [3,4]. Several combinations of zinc with various bioactive ligands with insulinomimetic properties have been obtained [5]. Zinc is effective in brain damage and neurodegenerative diseases at physiological concentrations [6]. Zinc-based complex compounds have anticancer activity [7,8], antiepileptic drug [9], antibacterial activity [10,11], antioxidative property [12,13], gastroprotective activity [14], inorganic photosensitizer [15] and DNA binding activity [16].
Natural enzyme cofactors such as 5-(1,2-dithiolane-3-yl) pentanoic acid or racemic α-lipoic acid (LA), is synthesized from octanoic acid from many prokaryotic, eukaryotic microorganisms, and from mitochondria and plastids of plants and animals [17]. LA is a sulfur-containing heterocyclic compound containing one chiral center and an asymmetric carbon. It has two possible optical isomers, R-LA and S-LA. The R-isomer is synthesized endogenously and binds to proteins. For therapeutic purposes, LA is usually used as a racemic mixture of R-LA and S-LA. Differences in the efficacy of the R-LA and S-LA isomers have not been identified [18]. Lipoic acid exhibits a wide range of therapeutic activities, including hyperlipidemia, antioxidant activity [19,20] anti-inflammatory [21,22], anticancer [23], hypoglycemic [24], protective from diabetic neuropathy [25,26], Parkinson's disease [27], Alzheimer's disease [28,29] and others.
Molecular docking is particularly important for the identification and prediction of new compounds with therapeutic value [30]. Meanwhile, in the 21st century, Kohn-Sham density functional theory emerged as the most popular electronic structure method in computational chemistry [31]. There is an opportunity to explore the limitations and challenges in modeling nuclear electron spectroscopy with DFT [32]. Through these methods, it is possible to determine the multifaceted properties of newly synthesized molecules, especially their biological activity and toxicity.
The purpose of this work is the synthesis, structural, theoretical and molecular docking study of the synthesized complex based on Zn (II) and R-lipoic acid.
2. Results and Discussion
In the present study, a new complex compound of zinc with LA (ZnLA) was synthesized, the structure of diaqua [5-(1,2-dithiolan-3-yl) pentanoato] acetate zinc [Zn(C8H13O2S2)(CH3COO)(H2O)2)] was determined by XRD technique and the spectroscopic properties were investigated by FT-IR. In addition, the thermal behavior of the complex was investigated. Theoretical calculations have been carried out by using Density Functional Theory (DFT) method in the ground state. The frontier orbitals (HOMO-LUMO) and Mulliken population method were obtained computationally. The def2-TZVPP was chosen as basis set for the theoretical calculations. Also docking investigation calculation of ZnLA calculated with using CB-Dock 2 server and Auto Dock Vina program. Comparison of the experimental and theoretical results shows that there is a good agreement between them.
2.1. IR-spectroscopic Analysis Results
Solid-state IR-Fure spectra of LA and ZnLA powders were obtained by using an IR Affinity-1S (Shimadzu, Japan) spectrometer (Figure 1). The specific vibrational frequencies of the complex compound were determined by comparison with those of the ligand (Table 1).
It is notable that new bands appeared at 3266-3000, 1357 and 464 cm−1 and in the spectrum of ZnLA, which were not observed in LA (Figure 1, Table 1). Absorption band frequency between 3266-3000 cm−1 is due to the presence of O-H stretching mode of the hydroxyl group [33]. This is explained by the occurrence of the hydroxyl group of the water molecule in the complex compound. Concurrently, the absorption band at 464 cm−1 corresponds to the stretching vibration. In general, the Zn-O bond absorption band corresponds to the area of 400-600 cm−1. In some studies, the Zn-O bond absorption band was reported to be at 426 [34] and 470 cm−1 [35], which is similar results with our work. Also, the absorption band in the 1357 cm−1 region observed in the IR spectrum of ZnLA corresponds to the valence vibration of the CH3 group in the acetic acid residue. The bands appearing at 2925 and 2852 cm−1 are characteristic to CH2 stretching mode [34,35]. Also, in the spectrum of ZnLA, absorption bands characteristic of associated acids disappeared, and intense absorption bands characteristic of and appeared in the 1535 and 1448 cm−1 regions, respectively. The expression ∆= - is located in the interval 87 (1535-1448) cm−1 in ZnLA. From this, we can conclude that metal ions in ZnLA are coordinated through oxygen atoms in the ligand (LA).
2.2. XRD Analysis Results
To determine the reliability of the analysis results and the fact that the obtained ZnLA is a truly new substance, further studies were carried out by powder X-ray diffractometric (XRD) analysis of the initial substances and the obtained ZnLA. Initially, the starting materials, namely, LA, ZnA, ZnLA, and the mechanical mixture of LA and ZnA were studied separately for the analysis results to be valid. The results of the analysis are presented in Figure 2 and Table 2.
As can be seen from Table 2, the XRD diffractogram of ZnLA has absorption bands at specific values of 13.4, 20.2 and 21.2 2θ diffraction peaks. In contrast to LA, at the XRD diffraction peaks of ZnA, ZnLA and ZnA + LA showed 12.5–12.8 2θ, which is characteristic of Zn metal. A similar result was also observed in XRD of the zinc complex obtained with other ligands [34,36]. A 1:1 mol ratio mixture of LA and ZnA has 8 12.5; 17.8; 23.3; 25.1; 27.5 2θ diffraction peaks in the XRD diffractogram and do not differ from the peaks of LA and ZnA taken separately in XRD diffractograms. These results, in turn, confirm that ZnLA is an individual substance.
2.3. DTA-TGA analysis results
The lipoic acid crystallized melts at 64°C, according to the first endotherm in differential thermal analysis (DTA) analysis (Figure 3A). The thermal gravimetric analysis (TGA) curve shows that the material is stable up to 150°C, experiencing weight loss of 64% at ∼160–320°C. This loss is associated with the second endotherm in the DTA curve. These correspond to the total decomposition of LA [36]. The differential thermal analysis of ZnLA revealed the decomposition of water molecules bound by hydrogen bonds at 115.84°C, and the endothermic effect observed at 158.37°C revealed the decomposition of water coordinated with the central atom (Figure 3B). Decomposition of the LA residue from the compound was observed at 442.96°C. This temperature is close to the melting point of zinc and is used in its identification. The endothermic effect observed at this moment was characterized by anion decomposition (mass loss of 45.124%) and exothermic peaks that arose due to the formation of CO2 and SO2 as a result of combustion and oxidation at a temperature of 458°C. At this moment in 160-300°C decomposition of acetate and from 300 to 840°C it was decomposition of lipoic acid residue. On the derivatogram, the decrease in mass continued up to a temperature of 839.75°C. Starting from a temperature of 840°C, no change in mass was observed. As a final product, ZnO and part of the carbon that remained unoxidized turned into coal (27.815%) and remained a solid residue [33].
Based on the results of the physicochemical analysis, an approximate structural formula of ZnLA was proposed (Figure 4A,B). In the given picture one can see that the coordination number of zinc is 6, and this complex compound consists of molecule LA, molecule of acetic acid residues, also 2 molecules of water, which are associated with zinc by a coordination bond.
2.4. Mulliken Population Analysis (MPA)
Mulliken atomic charge plays important role in the quantum chemical calculations of the molecular system. The progression of equalization of electronegativity and transfer of charge in chemical reactions are described by Mulliken atomic charge [37,38]. The Mulliken charge distributions analysis of ZnLA has been achieved by DFT-B3LYP [def2-TZVPP] method and the net atomic charges are furnished in Table 3. According to Mulliken population analysis results, all hydrogen atoms in the ZnLA have a positive charge. The highest negative charge value was predicted by the oxygen atom of the carboxyl group O2=-0.4738, O4=-0.5080, O31=-0.4609, O32=-0.4942, and water O34=-0.3374 and O36=-0.3372, respectively. The carbon atoms which are directly or indirectly integrated to the electronegative atoms such as O2, O4, O31, O32 and S9 accommodate higher positive charges C1=0.4760, C27=0.4705, C8=0.0643, and become more acidic. The carbon present in the negative region depends upon neighboring atoms carbon-negative range of C3=-0.1527, C5=-0.0195, C6=-0.1411, C7=-0.1504, C10=-0.1620, C11=-0.1363, C26=-0.22693. In addition, Mulliken atomic charge values of sulfur atoms in the molecule also have negative values S9=-0.1253 and S12=-0.1221.
2.5. Frontier Molecular Orbital (FMO) Analysis
It is known, both molecular orbitals of highest occupied (HOMO) and lowest unoccupied (LUMO), which are called frontier MO’s are significant quantum chemical factors to take part in the chemical reactivity and kinetic stability of a molecule. Usually, HOMO is known as donor, while LUMO is known acceptor [39,40]. On the basis of this MO’s can be determined significantly helpful quantum chemical parameters, such as chemical hardness (η) and softness (σ), energy bandgap (ΔE), electronegativity (𝜒) and electrophilicity index (ω). According to Koopmans' theorem, while HOMO energy is related to ionization potential, LUMO energy is related to electron affinity [41]. Another significant parameter is energy band gap value (difference between HOMO and LUMO), which is also crucial for the stability of the molecules [42]. The patterns of the HOMO and LUMO of the LA and ZnLA has been computed by a method of B3LYP [def2-TZVPP], which are displayed in Figure 5A–C, and their related energy values are given in Table 4. The positive and negative phases are symbolized with red and blue, respectively.
According to diagrams of FMO are presented in Figure 5, which shows the electronic distribution in HOMO-2, HOMO-1 and HOMO with LUMO, LUMO+1 and LUMO+2 for ZnLA. The FMO phenomenon suggests that the intramolecular charge transfer (ICT) character of the HOMO to LUMO transition exist. The delocalization of HOMO on all the atoms of a molecule except carbonyl and OMe groups while concentration of LUMO at all the atoms except OMe and carbonyl group for ZnLA can be easily observed from Figure 5. It is well known that HOMO orbitals represent bonding character, while LUMO reflects anti-bonding character. In addition to that, HOMO-LUMO gap (∆E) along with calculated energy levels associated to ZnLA are summarized in Table 4.
As Table 4 indicates that the ∆E value in respect of ZnLA is 4.637 eV. In general, compounds hold higher energy gap reveal lower chemical reactivity while compounds possess lower energy gap reveal higher chemical reactivity. EHOMO, ELUMO and energy gaps (∆E) associated to them are effectively employed to predict global reactivity descriptors [43,44,45] which assist in a clear description of the chemical reactivity, internal charge transfer, and stability of ZnLA. The equations employed for calculation of global reactivity descriptors are given in Table 5.
An inverse relation is found between energy gaps, softness as well as reactivity of species. Moreover, it has direct relation to the hardness and stability of the molecule. Consequently, the molecules with larger energy gaps are considered harder molecules which resist any change in electronic configuration and clearly links low reactivity with more kinetic stability. On the other side, the molecules with low energy gaps are soft molecules having tunable character, high reactivity and lower kinetic stability [46]. The ZnLA showed higher ionization potential values as compared to that of electron affinity values as presented in Table 5. The observed values of chemical hardness for ZnLA are higher than their softness. The greater chemical hardness, thus, indicates less reactivity or more stability of ZnLA. The stability and reactivity of compounds also connected with chemical potential of the compound. There is a direct relation between the chemical potential of the compound and stability whereas it is inversely related to reactivity. The result with respect to stability and reactivity of ZnLA on the basis of chemical potential is almost similar to that of the result obtained from chemical hardness data. However, ZnLA quite stable overall as reflected by its chemical hardness.
2.6. Molecular Docking Analysis (Studies)
Molecular docking has been proved as an efficient tool to study and predict the binding mode, the binding affinity of a ligand with the proteins, which is an essential part of drug discovery. This method for drug design is a highly reliable program, cost-effective and time-saving [47,48]. In this work, to explore the biological activity of ZnLA and LA, molecular docking simulations have been performed using CB-Dock 2 server [49] and AutoDock Vina software [50], and the docked structures were visualized by using Biovia Discover Studio Visualizer software [51]. The 3D geometries of ZnLA and LA have been built using Avogadro program package [52]. The target protein structures of 2GPA and 1ZSX were taken from the PDB database [53] as a pdb file.
Docking analysis have been investigated by of CB-Dock 2 server and AutoDock Vina results.
In the first case, AutoDock Vina was used for docking investigation. At the beginning protein structures were cleaned from additional molecules and polar hydrogen atoms were added. Proteins were minimized applying Kollman’s all partial atomic charges. Once minimized, the protein is loaded in MGLTools [54] creating a PDBQT file that contains a protein structure with hydrogens in all polar residues, and it is then used by AutoDock Vina software to obtain the affinity binding values for a ZnLA and LA. The docking site for ZnLA and LA structures on protein targets was defined by establishing a cube with the dimensions 40 × 40 × 40 Å, covering the binding site predicted for CB-Dock 2 with a grid spacing of 0.375 Å centered on the center of mass of the ZnLA and LA. Ten runs with AutoDock Vina were performed in all cases per each ZnLA and LA structures, and for each run the best pose was saved. The average affinity for best poses was taken as the final affinity value for ZnLA and LA.
In the second case, the structures of the cleaned proteins from additional molecules using Biovia DS visualizer program have been used for molecular docking studies by help of the CB-Dock2 server. CB-Dock automatically identifies the binding sites. By default, the number of docking cavities is set to 5. During analysis of docking results, the main attention was paid to the active center, in which tested compounds are localized. The molecular docking results have been visualized by Biovia DS visualizer software.
Molecular docking studies conducted by the authors revealed that α-lipoic acid binds well to 2GPA and 1ZSX and acts as a weak inhibitor [55]. Proteins 2GPA and 1ZSX are proteins responsible for blood glucose levels. By inhibiting them, it is possible to reduce the amount of glucose in the blood. In order to predict the mechanism of action and initial biological activity of the substance synthesized by us, studies were conducted with 2GPA and 1ZSX proteins.
Glycogen phosphorylase - 2GPA, which is the main enzyme regulating glycogen metabolism, catalyzes the process of phosphorolysis, in which glycogen is broken down into glucose-1-phosphate. In muscle, glycogen is converted to glucose-1-phosphate, undergoes glycolysis in the liver to produce metabolic energy and is converted to glucose. By inhibiting the 2GPA enzyme, it is possible to prevent the phosphorylase process, which, in turn, leads to blocking the process of glucose formation from glycogen [56].
The most suitable posture of ZnLa-2GPA, obtained through various interactions at the binding site, was found to have the highest binding energy of -7.950.35 kcal/mol (Table 6) which is 1.35 times greater than the binding energy of LA to 2GPA. This indicates a favorable binding affinity. It should be noted that Arg49, Thr94, Gly135, Leu136, Asn187, Lys574, Asn284, Arg292, Phe285, His341, His377, Tyr573, Lys574, Glu672, Ser674, Gly675 and Gly677 were involved in the interaction with the ZnLA molecule via hydrogen bonds. Arg569 and Ala673 are formed hydrophobic bonds with ZnLa, Tyr185 formed pi-sulfur bonds with ZnLA, Tyr648 pi-sulfur, Glu126, Lys574, Ala383, and Glu672 were involved in the interaction through electrostatic attraction (Figure 6A,B).
The amino acids Ser, Tyr and Phe in 2GPA play an important role in phosphorylation processes. In our study, 14 amino acids in 2GPA were connected with ZnLA by different bonds (Table 6). These include Ser and Tyr. Thus, ZnLA may induce antidiabetic activity by inhibiting its allosteric moiety and binding more tightly to 2GPA. In silico studies have been carried out comparing ZnLA with LA as a ligand, which also inhibits 2GPA, but its binding energy is 5.9 kcal/mol. It can be seen that the complex of zinc ZnLA is connected relatively stronger (BE=--8.3 kcal/mol) with 2GPA than LA. It should be noted that this compound may also be a potential zinc transporter. Of course, this must be confirmed in in vitro and in vivo experiments.
The most suitable posture of ZnLa-1ZSX, obtained through various interactions at the binding site, was found to have the highest binding energy of -7.70.4 kcal/mol (Table 6) which is 1.16 times greater than the binding energy of LA with 1ZSX. It is noteworthy that Trp43, Tyr76, Lys104, Arg175, Ser230 and Arg250 are involved in the interaction with the ZnLA molecule via hydrogen bonds. An electrostatic interaction of Asp71, Lys104, Arg175, Trp43, and Trp229 with the ZnLa molecule was observed. In particular, Trp43 interacts with zinc in the pi-cation and acetate residues in the pi-anion interaction. It can be seen that Trp43 and Trp43, Trp229, Trp258 with sulfur, carbon, and alkyl groups in the molecule create pi-sulfur, pi-sigma, and pi-alkyl hydrophobic effects (Figure 7A,B).
1ZSX - Kv potassium channel beta subunit (KCNAB2) regulates cellular processes such as hormone secretion and repolarization of excitable cells. Prolongation of action potentials by blocking delayed rectifying potassium channels in pancreatic beta cells is known to increase intracellular free calcium and stimulate glucose-dependent insulin release [57]. ZnLA may be a potential antidiabetic drug by blocking 1ZSX.
3. Materials and Methods
3.1. Chemicals
Chemicals and solvents used were of reagent grade and were used without any further purification. Zinc (II) acetate dihydrate, racemic α-lipoic acid, ethanol, acetonitrile, acetone, petroleum ether, heptane, chloroform (Merck, Frankfurter, Germany) and deionized water were used as research materials.
3.2. Synthesis
Synthesis was carried out at room temperature by mixing 1:1 mol (Mn+:LA) solutions of metal salt ZnAc and LA substance in 0.1 mol/L ethyl alcohol. Mixing was carried out on MS7-H550-S equipment at a speed of 250 rpm at a temperature of 60-80°C for 15-20 min. In the end, a heterogeneous solution was obtained. The resulting solution was filtered and the precipitate was dried for 24 h in a SH-DO-54FG (China) drying cabinet at a temperature of 25±2°C. The resulting substance (a complex combination of zinc (II) ions with LA is conditionally named “ZnLA”) was pale yellow in color. Reaction yield was 76.07±3,2%. calculated C 32.81, H 5.47, O 26.37, S 17.48, Zn 17.87; found C 32.67, H 5.49, O 26.61, S 17.31, Zn 17.92, melting point–92.4. Further research focused on determining the solubility of ZnLA in solvents. This process was carried out in comparison with the starting materials obtained for the synthesis. According to it, ZnLA is insoluble in water, acetonitrile, acetone, petroleum ether and heptane, but well soluble in chloroform. Subsequently, the obtained results showed that chloroform can be used in the recrystallization of ZnLA and the purification from starting materials.
3.3. IR-spectroscopy of ZnLA
For IR-spectroscopic analysis, the sample was placed in an FT-IR IRAffinity-1S (Shimadzu Scientific Instruments, Inc., Japan) and measured in the range of 4000 cm−1 to 400 cm−1 at 4 cm−1. To reduce the noise-to-signal ratio, each sample was scanned 10 times and finally integrated to obtain the mean value of the spectra. Before the main absorption band line (correction), the spectra were transferred to the absorption condition, after which the spectra were processed [58].
3.4. Powder X-ray Diffractometry of ZnLA
X-ray powder diffractometry (XRD) analysis results were obtained on an XRD-6100 powder X-ray diffractometer (Shimadzu Scientific Instruments, Inc., Japan) at 30 kV, 30 mA (monochromatic radiation CuKa, λ(CuKa) = 1.54178 Å), spacing size 0.02 2θ, calculation time is 6 s per interval, angle range 4–80◦ 2θ [59].
3.5. TGA-DTA Analysis
Thermogravimetry was performed using a DTG-60 Thermogravimetric Analyzer (Shimadzu Scientific Instruments, Inc., Japan) with samples in the 1.5–3.5 mg mass range placed in alumina crucibles and heated at a rate of 10 K min−1 over the temperature ranges of 25–900°C. The argon gas purge was used at a flow rate of 60 mL min−1 [60]. The ranges for the complexes were selected to avoid possible damage to the DTA thermocouple that can occur with sample spillage due to foaming at higher temperatures. The data were processed with TA Instruments Universal V4.7A. TGA and DTA measurements for the complexes were generally recorded in triplicate, enabling accurate the stoichiometry in complex to be determined.
3.6. Computational Methods
Since no crystals suitable for a full crystallographic study were obtained, molecular modeling studies were applied to optimize the structure of the complex. All theoretical calculations were carried out using density functional theory (DFT) with Becke’s 3-parameters hybrid method [61] and the Lee-Yang-Parr correlation functional (B3LYP) [62] combined with def2-TZVPP method developed by Ahlirichs group [63], which also has been tested in a previous study [64] for description of geometrical structures of the complexes. Input files for the DFT calculations using the ORCA 4.2.0 program package [65] Mulliken population analysis [66], global hardness, chemical potential and electrophilicity [67] of systems were also calculated. The docking studies [68] were performed by CB-Dock 2 server and AutoDock Vina. The molecular docking results have been visualized by Biovia DS visualizer program [69].
4. Conclusions
Our results showed that the complex compound ZnLA was synthesized with high efficiency based on the interaction of LA and ZnAc in an ethanol medium. Its structure and properties have been studied by XRD, IR, and TGA-DTA methods. X-ray phase and IR analyses comparing ZnLA with LA showed that it is an individual substance.
In silico studies were performed with the 2GPA and 1ZSX proteins to determine the mechanism of action and biological activity of ZnLA. This study reveals potential ligand-protein interactions at the atomic level and suggests that the treatment of diabetes (non-insulin dependent) with ZnLA.
Author Contributions
Conceptualization, A.S. and F.J.; methodology, F.J., D.K., A.A. and R.Z.; software, F.J.; validation, D.A., F.J. and A.S.; formal analysis, A.S., F.J., R.Z., A.A. and D.K.; investigation, F.J.; data curation, F.J.; writing—original draft preparation, F.J., R.Z. and A.S.; writing—review and editing, A.S. and F.J.; supervision, A.S. All authors read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Higher Education, Science and Innovations of the Republic of Uzbekistan, Research Grant No. IL-21091385.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The Authors thank Mr. Sunnatullo Fazliev for their critical proofreading of the manuscript and fruitful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Walkup, G.K.; Burdette, S.C.; Lippard, S.J.; Tsien, R.Y. A New Cell-Permeable Fluorescent Probe for Zn 2+. J Am Chem Soc 2000, 122, 5644–5645. [Google Scholar] [CrossRef]
- Psomas, G. Copper(II) and Zinc(II) Coordination Compounds of Non-Steroidal Anti-Inflammatory Drugs: Structural Features and Antioxidant Activity. Coord Chem Rev 2020, 412. [Google Scholar] [CrossRef]
- Dubey, P.; Thakur, V.; Chattopadhyay, M. Role of Minerals and Trace Elements in Diabetes and Insulin Resistance. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Chabosseau, P.; Rutter, G.A. Zinc and Diabetes. Arch Biochem Biophys 2016, 611. [Google Scholar] [CrossRef]
- Ranasinghe, P.; Pigera, S.; Galappatthy, P.; Katulanda, P.; Constantine, G.R. Zinc and Diabetes Mellitus: Understanding Molecular Mechanisms and Clinical Implications. DARU, Journal of Pharmaceutical Sciences, 2015; 23. [Google Scholar]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The Neurobiology of Zinc in Health and Disease. Nat Rev Neurosci 2005, 6. [Google Scholar] [CrossRef]
- Pellei, M.; Del Bello, F.; Porchia, M.; Santini, C. Zinc Coordination Complexes as Anticancer Agents. Coord Chem Rev 2021, 445. [Google Scholar] [CrossRef]
- Tu, L.Y.; Pi, J.; Jin, H.; Cai, J.Y.; Deng, S.P. Synthesis, Characterization and Anticancer Activity of Kaempferol-Zinc(II) Complex. Bioorg Med Chem Lett 2016, 26. [Google Scholar] [CrossRef] [PubMed]
- Yuen, W.C.; Whiteoak, R.; Thompson, R.P.H. Zinc Concentrations in Leucocytes of Patients Receiving Antiepileptic Drugs. J Clin Pathol 1988, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Li, J.; Khan, M.Z.H.; Ma, F.; Liu, X. A Novel Zinc Complex with Antibacterial and Antioxidant Activity. BMC Chem 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Abendrot, M.; Chȩcińska, L.; Kusz, J.; Lisowska, K.; Zawadzka, K.; Felczak, A.; Kalinowska-Lis, U. Zinc(II) Complexes with Amino Acids for Potential Use in Dermatology: Synthesis, Crystal Structures, and Antibacterial Activity. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, S.; Yuan, L.; Deng, H.; Zhang, Z. Synthesis, Structure Characterization, and Antioxidant and Antibacterial Activity Study of Iso-Orientin-Zinc Complex. J Agric Food Chem 2021, 69. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Z.Y.; Li, T.R. Synthesis, Characterization, Antioxidative Activity and DNA Binding Properties of the Copper(II), Zinc(II), Nickel(II) Complexes with 1,2-Di(4′-Iminonaringenin)Ethane. Chem Pharm Bull (Tokyo) 2008, 56. [Google Scholar] [CrossRef] [PubMed]
- Salga, M.S.; Ali, H.M.; Abdulla, M.A.; Abdelwahab, S.I.; ElhassanTaha, M.M.; Yagoub, U. Synthesis and Gastroprotective Activities of Some Zinc (II) Complexes Derived from (E)-2-(1-(2-(Piperazin-1-Yl)Ethylimino)Ethyl)Phenol and (E)-4-(1-(2-(Piperazin-1-Yl)Ethylimino)Ethyl)Benzene-1,3-Diol Schiff Bases against Aspirin Induced Ulceration. Arabian Journal of Chemistry 2017, 10. [Google Scholar] [CrossRef]
- Wen, X.; Nowak-Król, A.; Nagler, O.; Kraus, F.; Zhu, N.; Zheng, N.; Müller, M.; Schmidt, D.; Xie, Z.; Würthner, F. Tetrahydroxy-Perylene Bisimide Embedded in a Zinc Oxide Thin Film as an Electron-Transporting Layer for High-Performance Non-Fullerene Organic Solar Cells. Angewandte Chemie - International Edition 2019, 58. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wang, B.; Zhu, L. DNA Binding, Cytotoxicity, Apoptotic Inducing Activity, and Molecular Modeling Study of Quercetin Zinc(II) Complex. Bioorg Med Chem 2009, 17. [Google Scholar] [CrossRef]
- Reed, L.J.; Gunsalus, I.C.; Schnakenberg, G.H.F.; Soper, Q.F.; Boaz, H.E.; Kern, S.F.; Parke, T. V. Isolation, Characterization and Structure of α-Lipoic Acid. J Am Chem Soc 1953, 75. [Google Scholar] [CrossRef]
- Hermann, R.; Niebch, G.; Borbe, H.O.; Fieger-Büschges, H.; Ruus, P.; Nowak, H.; Riethmüller-Winzen, H.; Peukert, M.; Blume, H. Enantioselective Pharmacokinetics and Bioavailability of Different Racemic α-Lipoic Acid Formulations in Healthy Volunteers. European Journal of Pharmaceutical Sciences 1996, 4. [Google Scholar] [CrossRef]
- Scott, B.C.; Aruoma, O.I.; Evans, P.J.; O’neill, C.; Van Der Vliet, A.; Cross, C.E.; Tritschler, H.; Halliwell, B. Lipoic and Dihydrolipoic Acids as Antioxidants. A Critical Evaluation. Free Radic Res 1994, 20. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-Lipoic Acid as a Biological Antioxidant. Free Radic Biol Med 1995, 19. [Google Scholar] [CrossRef]
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and Clinical Relevance of Alpha Lipoic Acid: Antioxidant and Anti-Inflammatory Activity, Molecular Pathways and Therapeutic Potential. Inflammation Research 2017, 66. [Google Scholar] [CrossRef]
- Cores, Á.; Michalska, P.; Pérez, J.M.; Crisman, E.; Gómez, C.; Villacampa, M.; Menéndez, J.C.; León, R. Enantioselective Synthesis and Pharmacological Evaluation of Aza-CGP37157–Lipoic Acid Hybrids for the Treatment of Alzheimer’s Disease. Antioxidants 2022, 11. [Google Scholar] [CrossRef]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Wozniak, M. Lipoic Acid Decreases the Viability of Breast Cancer Cells and Activity of PTP1B and SHP2. Anticancer Res 2017, 37. [Google Scholar] [CrossRef]
- Gora̧ca, A.; Huk-Kolega, H.; Piechota, A.; Kleniewska, P.; Ciejka, E.; Skibska, B. Lipoic Acid - Biological Activity and Therapeutic Potential. Pharmacological Reports 2011, 63. [Google Scholar] [CrossRef]
- Ziegler, D.; Reljanovic, M.; Mehnert, H.; Gries, F.A. α-Lipoic Acid in the Treatment of Diabetic Polyneuropathy in Germany: Current Evidence from Clinical Trials. Experimental and Clinical Endocrinology and Diabetes 1999, 107. [Google Scholar] [CrossRef]
- Ziegler, D.; Hanefeld, M.; Ruhnau, K.J.; Mei\ner, H.P.; Lobisch, M.; Schütte, K.; Gries, F.A. Treatment of Symptomatic Diabetic Peripheral Neuropathy with the Anti-Oxidant α-Lipoic Acid - A 3-Week Multicentre Randomized Controlled Trial (ALADIN Study). Diabetologia 1995, 38. [Google Scholar] [CrossRef]
- De Araújo, D.P.; Lobato, R.D.F.G.; Cavalcanti, J.R.L.D.P.; Sampaio, L.R.L.; Araújo, P.V.P.; Silva, M.C.C.; Neves, K.R.T.; Fonteles, M.M.D.F.; Sousa, F.C.F. De; Vasconcelos, S.M.M. The Contributions of Antioxidant Activity of Lipoic Acid in Reducing Neurogenerative Progression of Parkinson’s Disease: A Review. International Journal of Neuroscience 2011, 121. [Google Scholar] [CrossRef]
- Holmquist, L.; Stuchbury, G.; Berbaum, K.; Muscat, S.; Young, S.; Hager, K.; Engel, J.; Münch, G. Lipoic Acid as a Novel Treatment for Alzheimer’s Disease and Related Dementias. Pharmacol Ther 2007, 113. [Google Scholar] [CrossRef]
- Maczurek, A.; Hager, K.; Kenklies, M.; Sharman, M.; Martins, R.; Engel, J.; Carlson, D.A.; Münch, G. Lipoic Acid as an Anti-Inflammatory and Neuroprotective Treatment for Alzheimer’s Disease. Adv Drug Deliv Rev 2008, 60. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol Phys 2017, 115. [Google Scholar] [CrossRef]
- Besley, N.A. Modeling of the Spectroscopy of Core Electrons with Density Functional Theory. Wiley Interdiscip Rev Comput Mol Sci 2021, 11. [Google Scholar] [CrossRef]
- Salehi, E.; Naderi, R.; Ramezanzadeh, B. Synthesis and Characterization of an Effective Organic/Inorganic Hybrid Green Corrosion Inhibitive Complex Based on Zinc Acetate/Urtica Dioica. Appl Surf Sci 2017, 396. [Google Scholar] [CrossRef]
- Azam, M.; Wabaidur, S.M.; Alam, M.J.; Trzesowska-Kruszynska, A.; Kruszynski, R.; Alam, M.; Al-Resayes, S.I.; Dwivedi, S.; Khan, M.R.; Islam, M.S.; et al. Synthesis, Structural Investigations and Pharmacological Properties of a New Zinc Complex with a N4-Donor Schiff Base Incorporating 2-Pyridyl Ring. Inorganica Chim Acta 2019, 487. [Google Scholar] [CrossRef]
- Halevas, E.; Mavroidi, B.; Pelecanou, M.; Hatzidimitriou, A.G. Structurally Characterized Zinc Complexes of Flavonoids Chrysin and Quercetin with Antioxidant Potential. Inorganica Chim Acta 2021, 523. [Google Scholar] [CrossRef]
- Da Silva Portela, A.; Almeida, M.D.G.; Gomes, A.P.B.; Correia, L.P.; Da Silva, P.C.D.; Montenegro Neto, A.N.; De Medeiros, A.C.D.; Simões, M.O.S. Vapor Pressure Curve Determination of α-Lipoic Acid Raw Material and Capsules by Dynamic Thermogravimetric Method. Thermochim Acta 2012, 544. [Google Scholar] [CrossRef]
- Dobson, J.C.; Hinchliffe, A. Mulliken Population Analysis and Quantum Mechanical Probability. J Mol Struct 1975, 27. [Google Scholar] [CrossRef]
- Demircioʇlu, Z.; Kaştaş, Ç.A.; Büyükgüngör, O. Theoretical Analysis (NBO, NPA, Mulliken Population Method) and Molecular Orbital Studies (Hardness, Chemical Potential, Electrophilicity and Fukui Function Analysis) of (E)-2-((4-Hydroxy-2-Methylphenylimino)Methyl)-3-Methoxyphenol. J Mol Struct 2015, 1091. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science (1979) 1982, 218. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105. [Google Scholar] [CrossRef]
- Rauk, A. Orbital Interaction Theory of Organic Chemistry. 2000. [Google Scholar]
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions, Reference Edition. 2010. [Google Scholar]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The Density Functional Viewpoint. J Chem Phys 1977, 68. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity Index. Chem Rev 2006, 106. [Google Scholar]
- Lesar, A.; Milošev, I. Density Functional Study of the Corrosion Inhibition Properties of 1,2,4-Triazole and Its Amino Derivatives. Chem Phys Lett 2009, 483. [Google Scholar] [CrossRef]
- Haroon, M.; Khalid, M.; Akhtar, T.; Tahir, M.N.; Khan, M.U.; Saleem, M.; Jawaria, R. Synthesis, Spectroscopic, SC-XRD Characterizations and DFT Based Studies of Ethyl2-(Substituted-(2-Benzylidenehydrazinyl))Thiazole-4-Carboxylate Derivatives. J Mol Struct 2019, 1187. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem Biol 2003, 10. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D. Bioinformatics Software Resources. Brief Bioinform 2004, 5. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved Protein-Ligand Blind Docking by Integrating Cavity Detection, Docking and Homologous Template Fitting. Nucleic Acids Res 2022, 50. [Google Scholar] [CrossRef]
- Sandeep, G.; Nagasree, K.P.; Hanisha, M.; Kumar, M.M.K. AUDocker LE: A GUI for Virtual Screening with AUTODOCK Vina. BMC Res Notes 2011, 4. [Google Scholar] [CrossRef]
- Miyata, T. Discovery Studio Modeling Environment. Ensemble 2015, 17. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J Cheminform 2012, 4. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J Mol Graph Model 1999, 17. [Google Scholar]
- Maldonado-Rojas, W.; Olivero-Verbel, J.; Ortega-Zuñiga, C. Searching of Protein Targets for Alpha Lipoic Acid. In Proceedings of the Journal of the Brazilian Chemical Society; 2011; Vol. 22. [Google Scholar]
- Oikonomakos, N.G.; Tsitsanou, K.E.; Zographos, S.E.; Skamnaki, V.T.; Goldmann, S.; Bischoff, H. Allosteric Inhibition of Glycogen Phosphorylase a by the Potential Antidiabetic Drug 3-Isopropyl 4-(2-Chlorophenyl)-1,4-Dihydro-1-Ethyl-2-Methyl-Pyridine-3,5,6-Tricarboxylate. Protein Science 1999, 8. [Google Scholar] [CrossRef]
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M.G. Expression of Voltage-Gated Potassium Channels in Human and Rhesus Pancreatic Islets. Diabetes 2004, 53. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, N.; Endo, T.; Hosomi, S.; Setou, K.; Tanaka, S.; Ogawa, N.; Yamamoto, H.; Mizukami, T.; Arai, S.; Okuno, M.; et al. Structural Analysis of Crystalline R(+)-α-Lipoic Acid-α-Cyclodextrin Complex Based on Microscopic and Spectroscopic Studies. Int J Mol Sci 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Sharipov, A.; Boboev, Z.; Fazliev, S.; Gulyamov, S.; Yunuskhodjayev, A.; Razzokov, J. Development of an Improved Method for the Determination of Iodine/β-Cyclodextrin by Means of Hplc-Uv: Validation and the Thyroid-Stimulating Activity Revealed by in Vivo Studies. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dang, L.; Wei, H. Thermodynamic Analysis of Lipoic Acid Crystallized with Additives. J Therm Anal Calorim 2013, 111. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys 1993, 98. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys Rev B 1988, 37. [Google Scholar] [CrossRef]
- Weigend, F. Extending DFT-Based Genetic Algorithms by Atom-to-Place Re-Assignment via Perturbation Theory: A Systematic and Unbiased Approach to Structures of Mixed-Metallic Clusters. Journal of Chemical Physics 2014, 141. [Google Scholar] [CrossRef]
- Vovna, V.I.; Korochentsev, V. V.; Dotsenko, A.A. Electronic Structures and Photoelectron Spectra of Zinc(II) Bis-β-Diketonates. Russian Journal of Coordination Chemistry/Koordinatsionnaya Khimiya 2012, 38. [Google Scholar] [CrossRef]
- Xu, P.; Du, H.; Peng, X.; Tang, Y.; Zhou, Y.; Chen, X.; Fei, J.; Meng, Y.; Yuan, L. Degradation of Several Polycyclic Aromatic Hydrocarbons by Laccase in Reverse Micelle System. Science of the Total Environment 2020, 708. [Google Scholar] [CrossRef]
- Huizar, L.H.M.; Rios-Reyes, C.H.; Olvera-Maturano, N.J.; Robles, J.; Rodriguez, J.A. Chemical Reactivity of Quinclorac Employing the HSAB Local Principle - Fukui Function. Open Chem 2015, 13. [Google Scholar] [CrossRef]
- Ramirez-Balderrama, K.; Orrantia-Borunda, E.; Flores-Holguin, N. Calculation of Global and Local Reactivity Descriptors of Carbodiimides, a DFT Study. J Theor Comput Chem 2017, 16. [Google Scholar] [CrossRef]
- Azam, S.S.; Abbasi, S.W. Molecular Docking Studies for the Identification of Novel Melatoninergic Inhibitors for Acetylserotonin-O-Methyltransferase Using Different Docking Routines. Theor Biol Med Model 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, D.S. Discovery Studio Visualizer V21.1.0.20298. BIOVIA, Dassault Systèmes, 2005. [Google Scholar]
Figure 1.
IR spectra of LA (in blue) and ZnLA (in red).
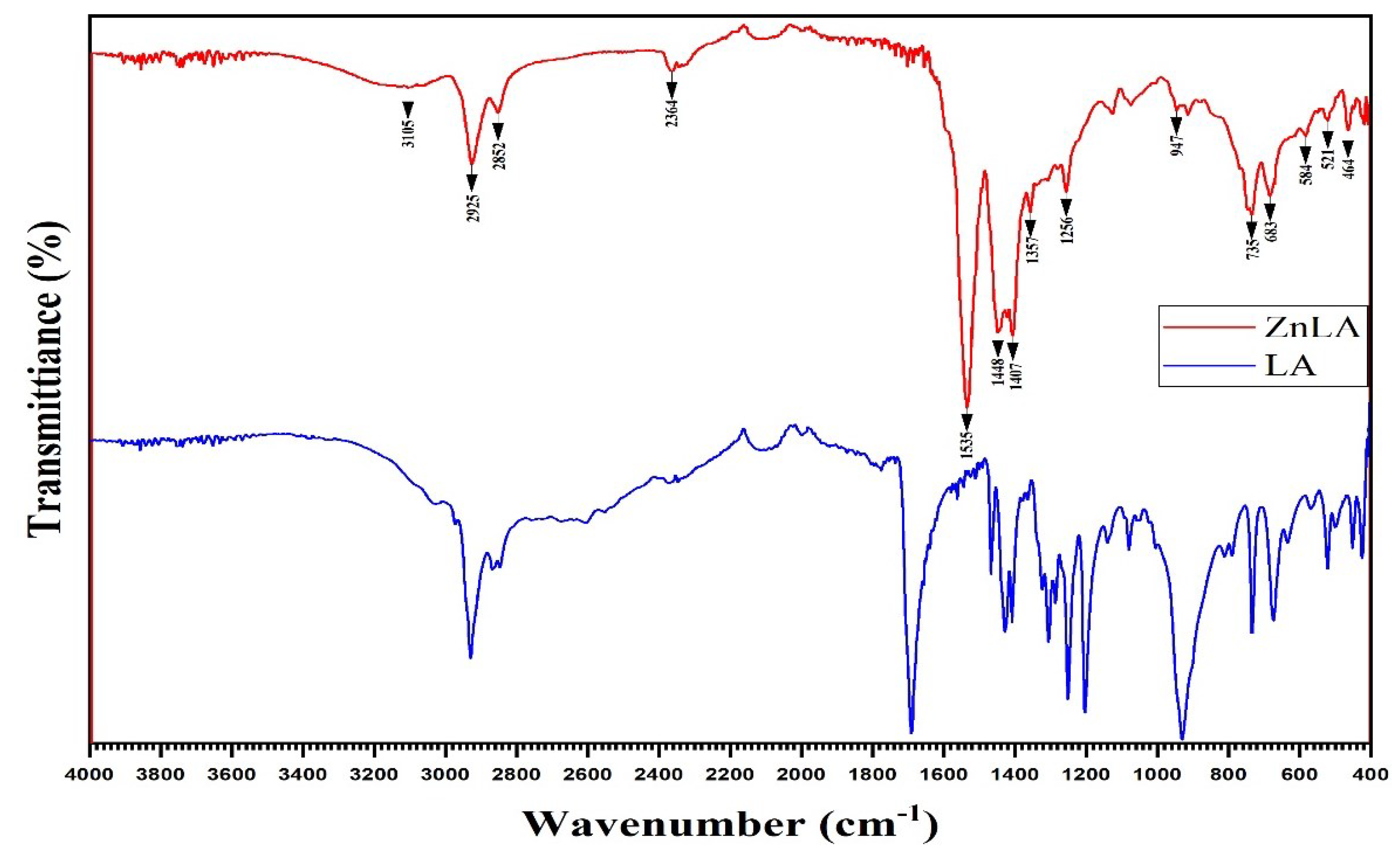
Figure 2.
XRD diffractograms: LA (in blue), ZnAc (in green), mechanical mixture of ZnAc and LA (1:1) (in pink) and ZnLA (in violet).
Figure 2.
XRD diffractograms: LA (in blue), ZnAc (in green), mechanical mixture of ZnAc and LA (1:1) (in pink) and ZnLA (in violet).
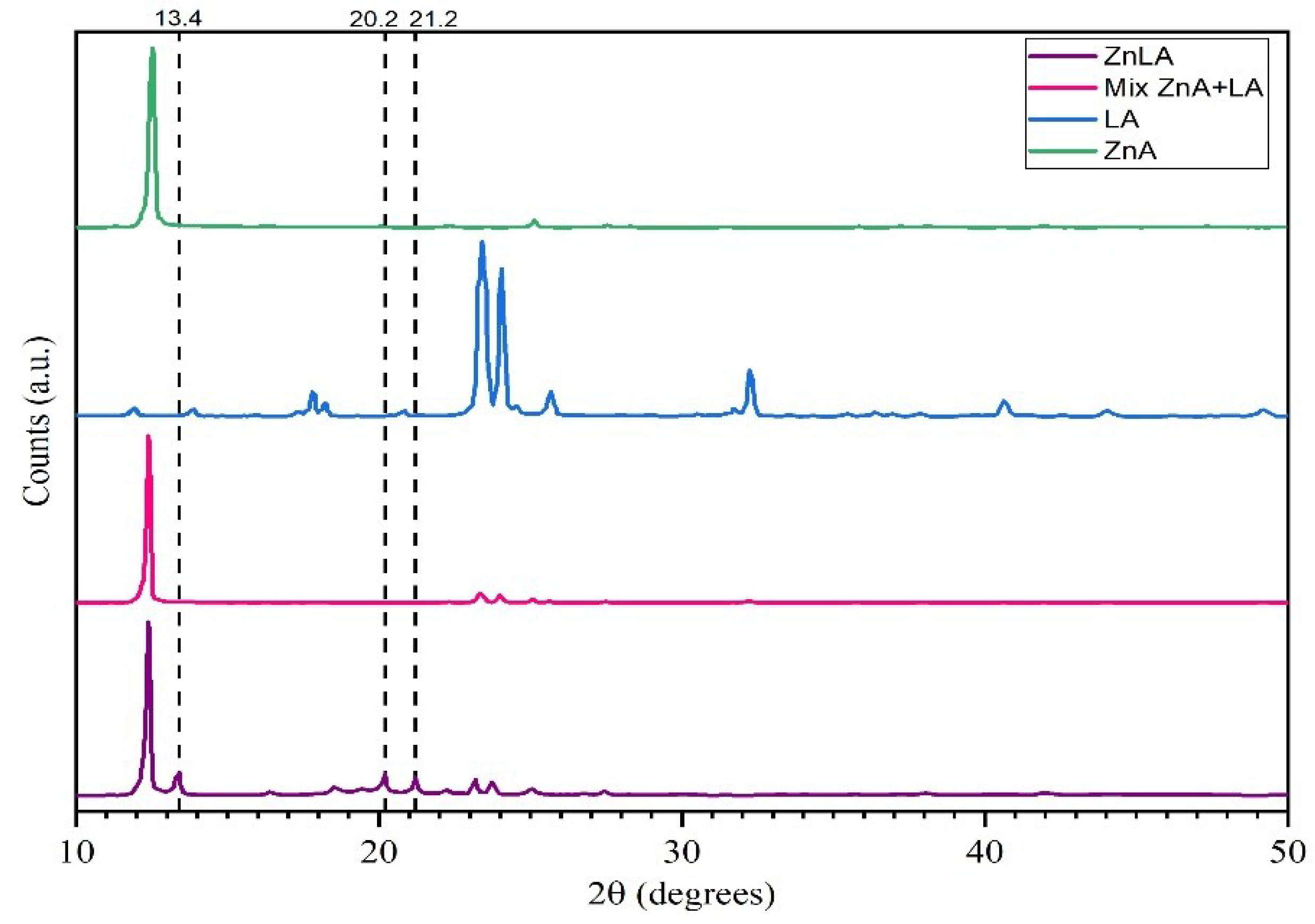
Figure 3.
Thermal analysis curves (in blue) thermogravimetric analysis (TGA) and (in red) differential thermal analysis (DTA) curves of LA (A) and ZnLA (B).
Figure 3.
Thermal analysis curves (in blue) thermogravimetric analysis (TGA) and (in red) differential thermal analysis (DTA) curves of LA (A) and ZnLA (B).
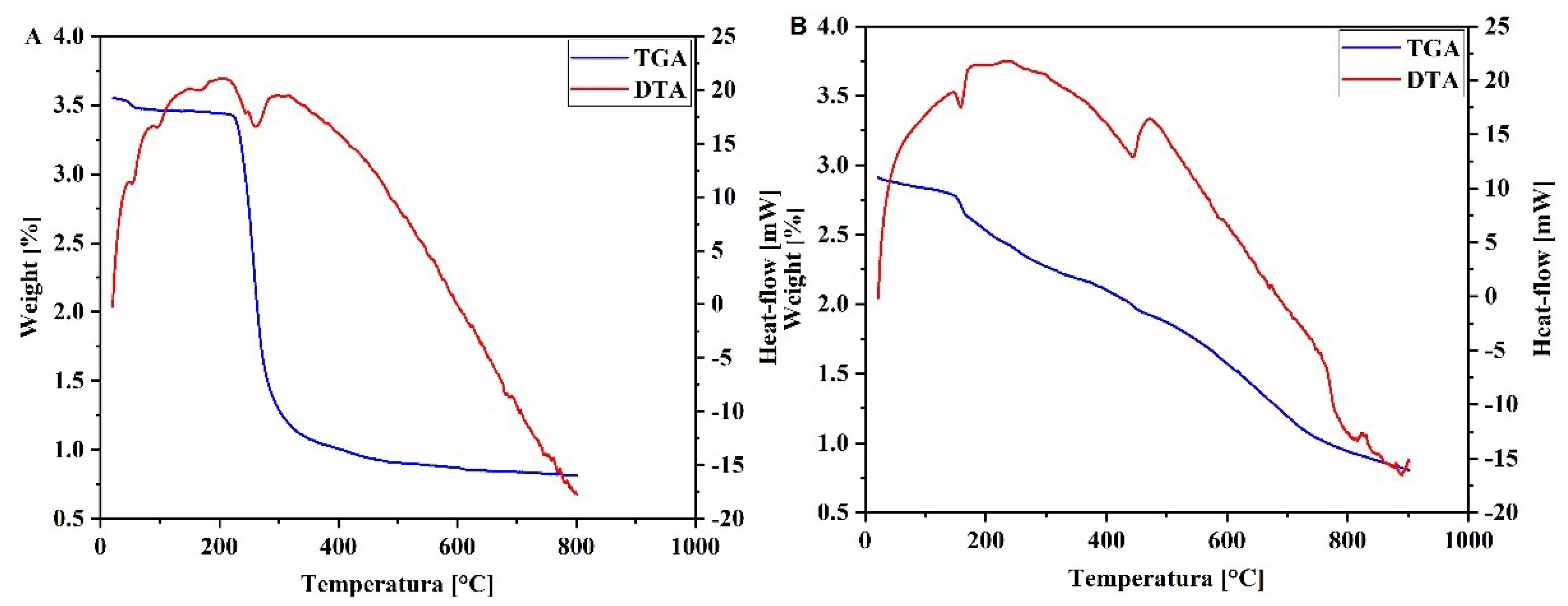
Figure 4.
(A) ORTEP structure of ZnLA. (B) Optimized geometrical diagram of ZnLA obtain by B3LYP/ def2-TZVPP basis set.
Figure 4.
(A) ORTEP structure of ZnLA. (B) Optimized geometrical diagram of ZnLA obtain by B3LYP/ def2-TZVPP basis set.
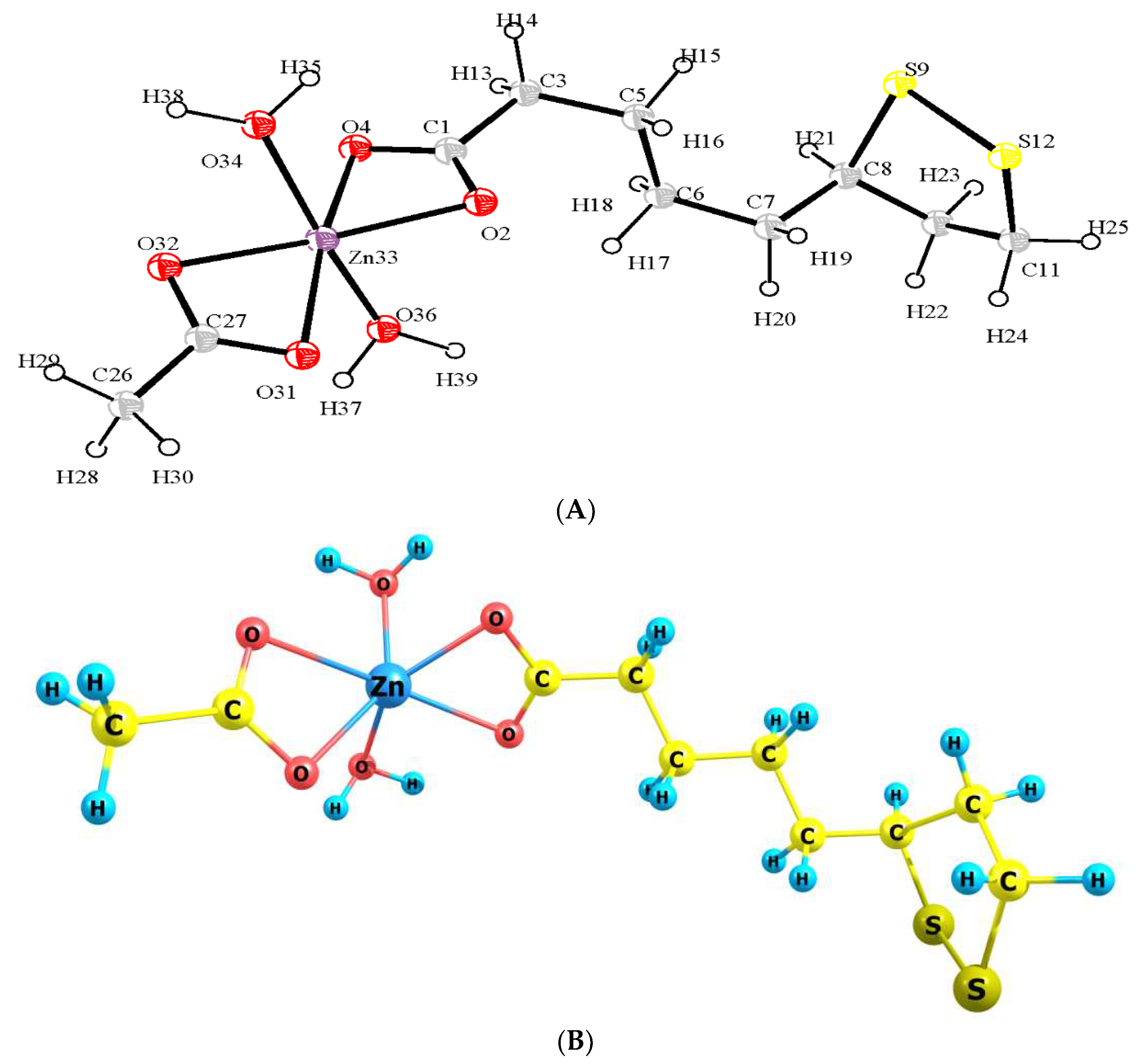
Figure 5.
Frontier molecular orbitals of the ZnLA obtained at DFT/B3LYP/def2-TZVPP level of theory.
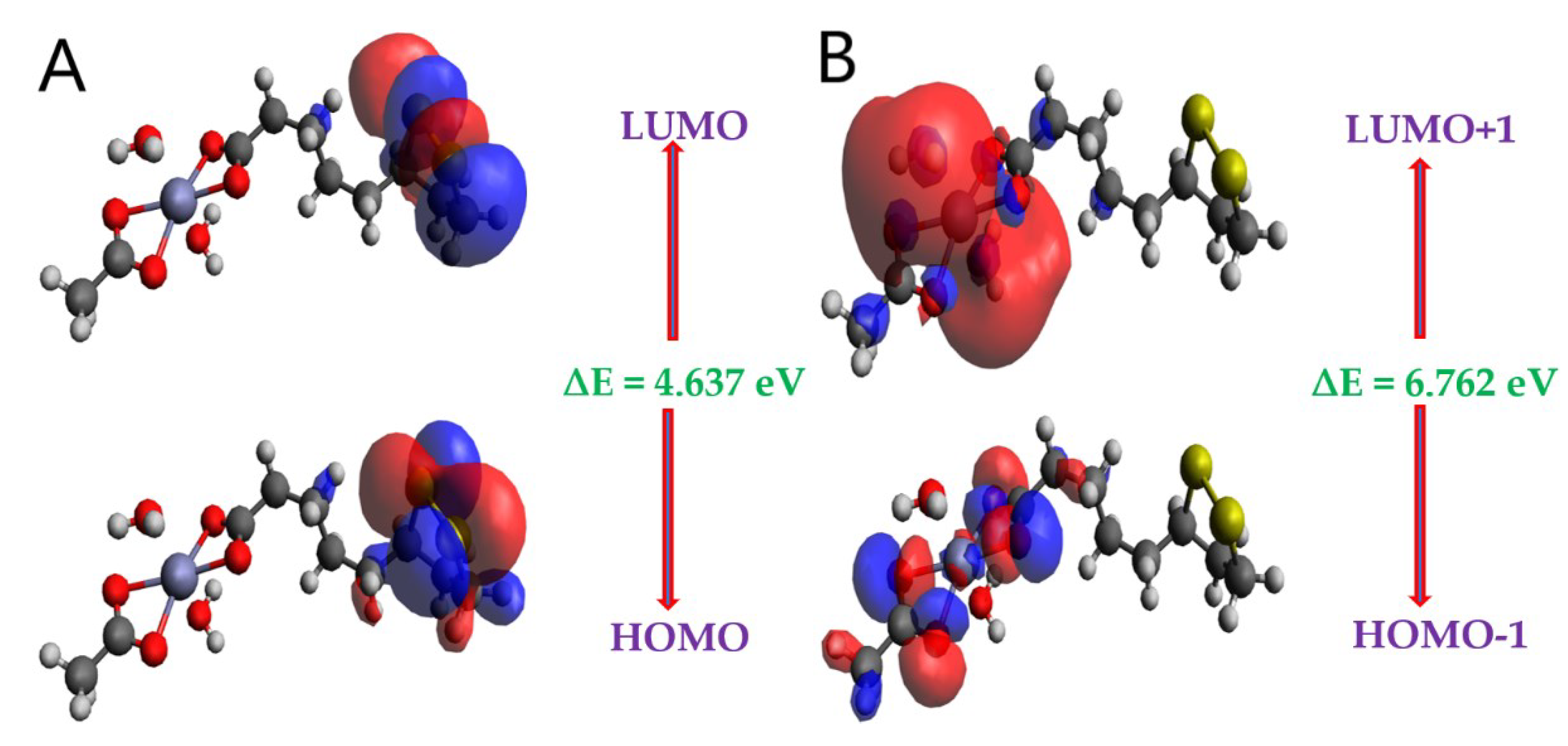
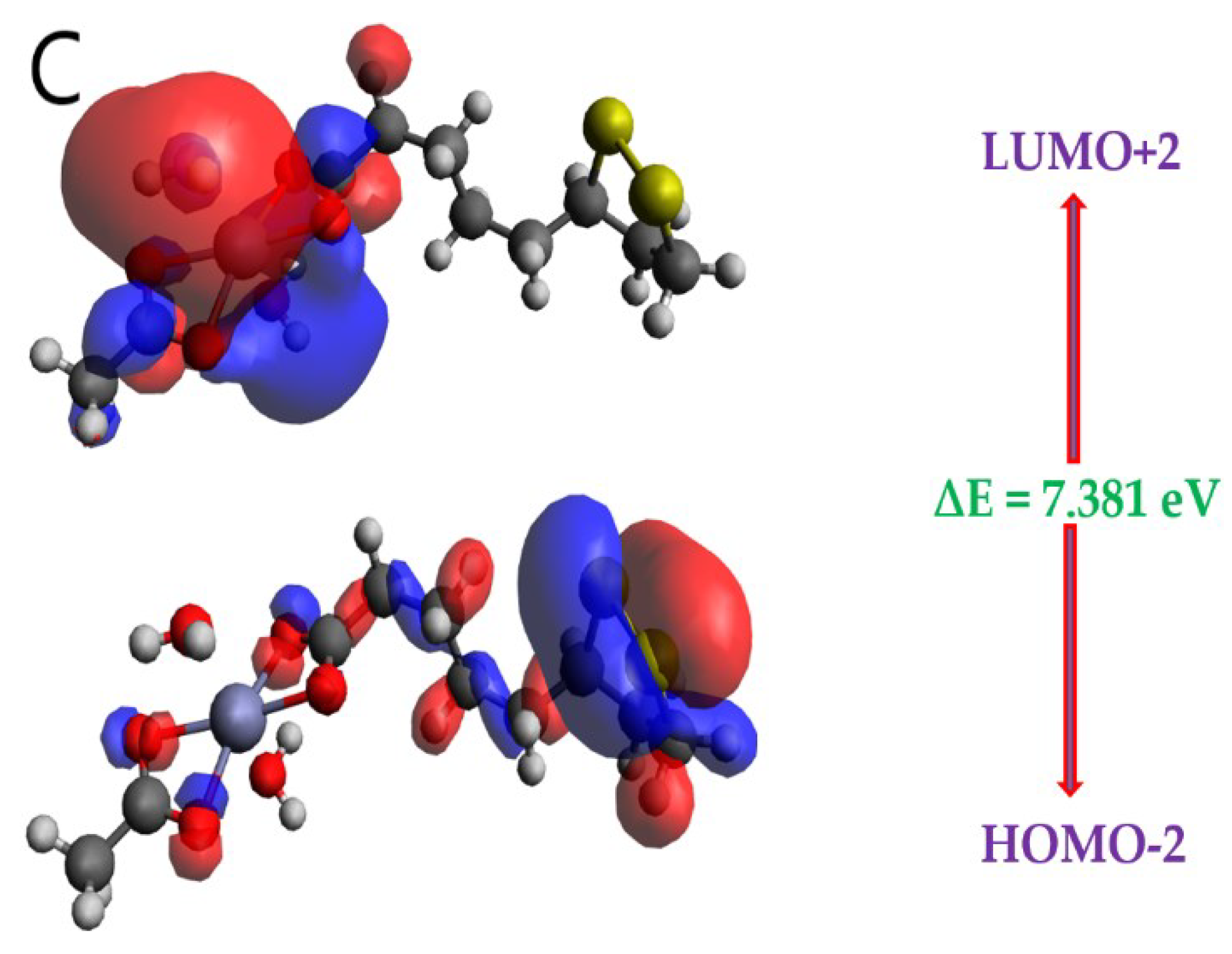
Figure 6.
The molecular docking results, surface (A) around and docking interactions (B) of the ZnLA with 2GPA protein.
Figure 6.
The molecular docking results, surface (A) around and docking interactions (B) of the ZnLA with 2GPA protein.

Figure 7.
The molecular docking results, surface around (A) and docking interactions (B) of the ZnLA with 1ZSX protein.
Figure 7.
The molecular docking results, surface around (A) and docking interactions (B) of the ZnLA with 1ZSX protein.
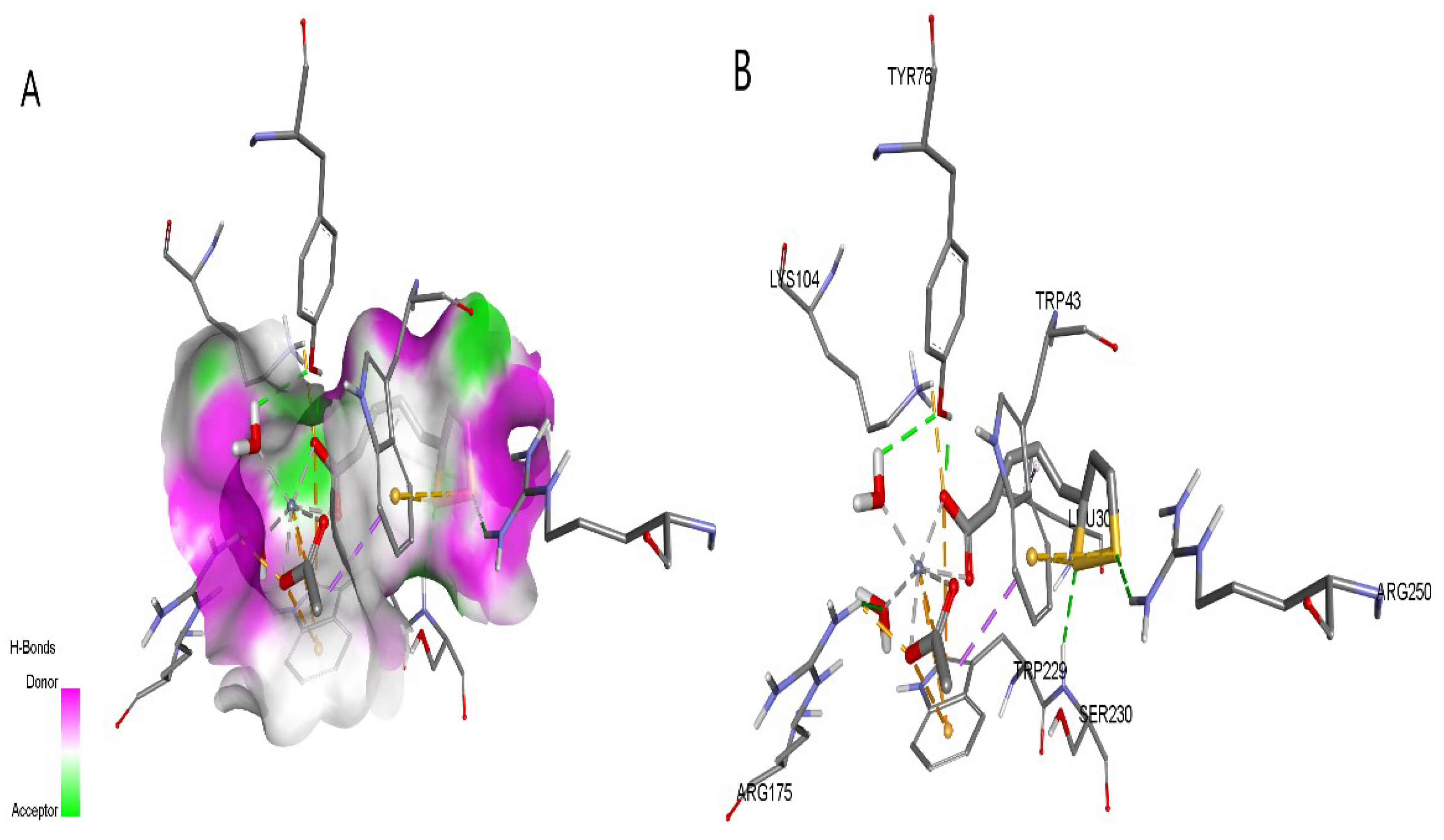

Table 1.
Functional groups in LA and ZnLA and their corresponding vibrational frequencies.
| LA, cm–1 | ZnLA, cm –1 | Functional Groups |
|---|---|---|
| - | 3266-3000 | |
| 2928, 2868 | 2925, 2852 | |
| 2369, 1690, 1426, 1407 | 2364, 1535, 1448, 1407 |
|
| - | 1357 | |
| - | 464 |
Table 2.
Comparison of the results of the XRD analysis of ZnLA and reaction starting substances.
| Compound | 2θ |
|---|---|
| LA | 8; 17.8; 23.3; 23.9; 32.3 |
| ZnA | 12.8; 16.3; 22.3; 25.1; 27.7 |
| Mix. ZnA + LA(1:1 mole ratio) | 8; 12.5; 17.8; 23.3; 25.1; 27.5 |
| ZnLA | 8; 12.5; 13.4; 18.6; 19.5; 20.2; 21.2; 22.3; 25.1; 27.5 |
Table 3.
Calculated Mulliken atomic charges of the atoms for ZnLA by B3LYP/ def2-TZVPP method.
| Atom | Charge | Atom | Charge | Atom | Charge | Atom | Charge |
|---|---|---|---|---|---|---|---|
| C1 | 0.4760 | C11 | -0.1363 | H21 | 0.0838 | O31 | -0.4609 |
| O2 | -0.4738 | S12 | -0.1221 | H22 | 0.0722 | O32 | -0.4942 |
| C3 | -0.1527 | H13 | 0.0772 | H23 | 0.0983 | Zn33 | 0.6539 |
| O4 | -0.5080 | H14 | 0.0904 | H24 | 0.1112 | O34 | -0.3374 |
| C5 | -0.0195 | H15 | 0.0601 | H25 | 0.1096 | H35 | 0.2217 |
| C6 | -0.1411 | H16 | 0.0623 | C26 | -0.2693 | O36 | -0.3372 |
| C7 | -0.1504 | H17 | 0.0684 | C27 | 0.4705 | H37 | 0.2221 |
| C8 | 0.0643 | H18 | 0.0418 | H28 | 0.1006 | H38 | 0.2242 |
| S9 | -0.1253 | H19 | 0.0845 | H29 | 0.1028 | H39 | 0.2242 |
| C10 | -0.1620 | H20 | 0.0632 | H30 | 0.1068 |
Table 4.
Molecular frontier orbital energies for ZnLA.
| MO(s) | E (eV) | ∆E (eV) |
|---|---|---|
| HOMO | -5.426 | 4.637 |
| LUMO | -0.789 | |
| HOMO-1 | -7.033 | 6.762 |
| LUMO+1 | -0.271 | |
| HOMO-2 | -7.444 | 7.381 |
| LUMO+2 | -0.063 |
E= energy, ∆E (eV) = ELUMO-EHOMO; HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital, MO, molecular orbital.
Table 5.
HOMO and LUMO Energy Values and other related parameters of ZnLA using DFT/B3LYP/def2-TZVPP level.
Table 5.
HOMO and LUMO Energy Values and other related parameters of ZnLA using DFT/B3LYP/def2-TZVPP level.
| Parametres | ZnLA | |
|---|---|---|
| HOMO energy | EHOMO (eV) | -5.426 |
| LUMO energy | ELUMO (eV) | -0.789 |
| Energy bandgap (ΔE) | ΔE = EHOMO - ELUMO (eV) | 4.637 |
| Ionization potential (I) | I = - EHOMO (eV | 5.426 |
| Electron affinity (A) | A = - ELUMO (eV) | 0.789 |
| Electronegativity (χ) | χ = (I+A)/2 (eV) | 3.107 |
| Global hardness (η) | η = (I-A)/2 (eV) | 2.318 |
| Global softness (σ or S) | S = 1/2 η (eV) | 0.216 |
| Global electrophilicity (ω) | ω = (µ2/2η) (eV) | 2.0823 |
| Chemical potential (µ) | µ = - (I+A)/2 (eV) | -3.107 |
Table 6.
The molecular docking values of LA and ZnLA with protein targets.
| Protein (PDB ID) | Autodock Vina results | CB-Dock2 results | Contact residues with H-bonds | ||
|---|---|---|---|---|---|
| Binding energy, kcal/mol | Binding energy, kcal/mol | ||||
| LA | ZnLA | LA | ZnLA | ||
| 2GPA | -5.9 | -7.6 | -5.9 | -8.3 | ARG49, THR94, GLY135, LEU136, ASN187, ASN284, ARG292, HIS341, HIS377, TYR573, LYS574, SER674, GLY675, GLY677 |
| 1ZSX | -6.6 | -7.3 | -6.6 | -8.1 | TRP43, TYR76, LYS104, ARG175, SER230, ARG250 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



