
Submitted:
18 October 2023
Posted:
19 October 2023
You are already at the latest version
Abstract
Background: Several hereditary-familial syndromes associated with various types of tumors have been identified to date, evidencing that hereditary cancers caused by germline mutations account for 5-10% of all tumors. Advances in genetic technology and the implementation of Next Generation Sequencing (NGS) have accelerated the discovery of several susceptibility cancer genes, allowing the detection of cancer-predisposing mutations in a larger number of cases. The aim of this study is to highlight how the application of an NGS-multi gene panel to a group of oncological patients subsequently leads to the improvement of healthy pathogenic variants/likely pathogenic variants (PVs/LPVs) carriers' identification and prevention of the disease in these cases. Methods: Starting from a total of 110 cancer patients carrying PVs/LPVs in genes involved in cancer susceptibility detected by a customized NGS panel of 27 cancer-associated genes, we enrolled 250 healthy collateral family members from January 2020 to July 2022. The specific PVs/LPVs identified in each proband were tested in healthy collateral family members by Sanger sequencing. Results: 131 out of the 250 cases (52%) weren’t carriers of the mutation detected in the affected relative, while 119 were carriers. Of these, 81/250 patients carried PVs/LPVs on BRCA1/2 (33%), 35/250 harbored PVs/LPVs on other genes beyond BRCA1 and BRCA2 (14%), and 3/250 (1%) were PVs/LPVs carriers both on BRCA1/2 and on another susceptibility gene. Conclusion: Our results show that the analysis of BRCA1/2 genes only would have resulted in a missed diagnosis in a number of cases and in the lack of prevention of the disease in a considerable percentage of healthy carriers with a genetic mutation (14%).
Keywords:
NGS Multigene panel
; hereditary cancer
; healthy collateral family members
1. Introduction
Several hereditary-familial syndromes associated with various types of tumors have been identified to date. The most common are Lynch syndrome (hereditary non-polyposis colorectal cancer, HNPCC) and breast and ovarian cancer syndrome (HBOC). However, there are many other syndromes, such as familial adenomatous polyposis (FAP), Cowden syndrome, and Li Fraumeni syndrome [1,2], related to germline mutations in genes less frequently involved in hereditary cancer but that can be transmitted by inheritance, increasing the risk of cancer within family members. Consequently, the prevalence of hereditary tumors, considered to account for 5-10 percent of all cancers [3], could likely be underestimated.
During the last decades, the basis for such genetic predisposition has been clarified for several hereditary cancer syndromes, and high penetrant/high-risk genes mutated in familial cases are currently subjected to genetic diagnostic screening programs. Mutation testing in these genes has a major impact on genetic counseling, defines the prognosis of carriers, identifies the most appropriate and personalized prophylactic measures, and increases the chance of survival. In HBOC, the high-penetrant BRCA1 and BRCA2 susceptibility genes were discovered between 1994 and 1995 [4]. Subsequent genetic studies based on linkage and positional cloning helped identify additional moderate-risk genes, and genome-wide association studies identified common low-penetrance alleles associated with breast cancer heritability [4]. In Lynch syndrome, germline pathogenic variants in the mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2 play an essential role in carcinogenesis. Importantly, these genes have variable penetrance and different risk rates of endometrial and colon cancer; in particular, MSH6 and PMS2 are estimated to have lower penetrance for colorectal cancer [5]. In this context, multigene panel testing is considered a powerful tool for increasing the detection rate of pathogenic variants in a number of non-BRCA genes and should be routinely supplied to high-risk patients. As a result, the use of NGS technology in clinical practice is expanding.
The majority of hereditary-familial syndromes are inherited in an autosomal dominant manner. Once a causative mutation has been identified in a patient, there's an indication to extend the analysis to first-degree relatives. In fact, each family member of an individual carrying the mutation has a 50% chance of being a carrier of the same mutation. In a few cases, however, mutations in both alleles are required to produce a high oncological risk, autosomal recessive inheritance. In these latter cases, genetic testing is first indicated for the siblings of the index case because each of them has a 25% chance of having inherited both mutations [6].
It is critical to emphasize that if a family member does not inherit the pathogenetic mutation, his/her risk is similar to that of the general population. In contrast, in the presence of the causative mutation, the risk of developing the disease during a lifetime is higher [7].
According to national and international guidelines, surveillance protocols for healthy mutation carriers include imaging and laboratory tests, depending on the genetic mutation detected. For female BRCA mutation carriers, instrumental surveillance for breast and ovarian cancer is suggested, while for male BRCA carriers, surveillance for breast and prostate cancer is planned. Screening protocols allow early diagnosis and prompt treatment in order to have a better prognosis [8].
Since in the last year, an increasing number of studies suggested the use of multigene panel analysis including low, moderate, and high penetrance genes, a crucial point of the present study is to evaluate how this evolution in the detection of cancer-predisposing mutations can affect our ability to identify healthy carriers and prevent the disease in these subjects.
The purpose of this manuscript is to underline the effectiveness of the multigene panel in increasing the detection rate of germline mutations in cancer patients and, as a result, improving the identification of healthy collateral family members. We first collected 110 cancer patients who were carriers of PVs/LPVs detected using a customized NGS panel of 27 cancer-associated genes, and subsequently, we proceeded to the detection of known mutations in healthy collateral family members.
2. Materials and Methods
2.1. Study population
A retrospective study was carried out on 250 subjects (155 women and 95 men) who were relatives of 110 cancer patients carrying a PV/LPV in the BRCA1/2 genes or other cancer susceptibility genes and referred to the Medical Genetic Service of the University "G.d’Annunzio" of Chieti-Pescara - Center of Advanced Studies and Technologies (CAST) from January 2020 to July 2022. Among them, 44 cancer patients entered the study belonging to families in which the mutation was detected in another affected relative. All cases' medical personal and family histories were acquired during genetic counseling in the presence of a clinical multidisciplinary team based on geneticists and psychologists. All patients were informed about the significance of the genetic test and the possible implications of detecting the gene variant related to an increased cancer risk and possible prevention strategies. All subjects signed an informed consent. The results obtained from the analysis and their implications were explained during the post-test counseling.
2.2. Genomic DNA extraction
Buccal swabs or blood samples were collected from all patients. Genomic DNA was extracted by the MagPurix instrument and the Forensic DNA Extraction Kit (Zinexts Life Science Corp.- CatZP01001)/Blood DNA Extraction Kit 200 (Zinexts Life Science Corp.-CatZP02001), according to the manufacturer's protocol.
2.3. Next-Generation Sequencing (NGS)
NGS analysis was carried out with a Thermo-fisher Oncomine custom panel developed in our laboratory, including 27 genes [Table 1]. NGS was performed by the Ion Torrent S5 system (Thermo Fisher Scientific, Waltham, MA, USA) after automatic library preparation using Ion Chef (Thermo Fisher Scientific, Waltham, MA, USA). Ion Chef consists of fragmentation and adapter ligation onto the PCR products, clonal amplification. After quantification of DNA libraries with the Real-Time Step One PCR System (Thermo Fisher Scientific, Waltham, MA, USA), the prepared samples of ion sphere particles (ISP) were loaded onto an Ion 530™ chip with the Ion Chef (Thermo Fisher Scientific, Waltham, MA, USA). Sequencing was performed using the Ion S5™ sequencing reagents (Thermo Fisher Scientific, Waltham, MA, USA). The Torrent Suite 5.14.0 platform and specific plugins were used for NGS data analysis. The uniformity of base coverage was over 98% in all batches, and base coverage was over 20× in all target regions.
2.4. Sanger Sequencing
The specific PVs/LPVs identified in each proband by NGS were tested in healthy collateral family members enrolled in the study, by Sanger sequencing. All DNA samples were amplified by polymerase chain reaction (PCR) performed in 30-μl reaction volume, containing 22,25 μl of H2O, 3 μl of 10X PCR buffer, 2,1 μl of MgCl2 solution 25 mM, 0,5 μl of dNTPs 10 mM, 0,15 μl of AmpliTaq Gold polymerase, 1 μl of DNA and 0,5 μl of Forward and 0,5 μl of Reverse primers. All primers were designed using NCBI designing tools (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
Amplification was performed By SimpliAmpTM thermal cycler (ThermoFisher, Applied Biosystem, CA, USA). FastGene Gel/PCR Extraction (Nippon Genetics Europe) was utilized for purification of PCR products, according to the manufacturer's protocol. The amplification products were submitted to direct sequencing procedure using BigDye Term v3.1 CycleSeq Kit (Life Technologies, Monza, Italy) followed by automatic sequencing analysis. All sequences were purified by "NucleoSEQColumns" purification kit (Macherey-Nagel Colonia, Germany) and analyzed in forward and reverse directions on a SeqstudioGenetic Analyzer (ThermoFisher, Applied Biosystem, CA, USA).
2.5. Genetic Variant Classification
According to the guidelines of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) (https://enigmaconsortium.org/) the genetic variants were classified into five classes: benign (C1), likely benign (C2), variant of uncertain significance (VUS, C3), likely pathogenic (C4), and pathogenic (C5). In our study, we focused on the LPVs and PVs that can be used for clinical purposes and cancer prevention. Variants were referred to according to the nomenclature recommendations of the Human Genome Variation Society (https://www.hgvs.org). The clinical significance of the genetic variants found in this study was evaluated according to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Varsome (https://varsome.com), Franklin Genoox (https://franklin.genoox.com) and, for some other susceptibility genes according to LOVD-InSIGHT (https://www.insight-group.org/variants/databases/).
3. Results
A total of 250 cases were analyzed by Sanger sequencing, from January 2020 to July 2022, in order to verify the presence of a PV/LPV already detected in an affected relative.
One-hundred and forty-three subjects aged < 45 years old and 107 aged > 45 years old. One-hundred and nineteen cases were detected to be carriers of the mutation previously evidenced in an affected relative. Of these, 81 cases had PVs/LPVs on BRCA1/2 (33%), 35 in other genes related to cancer susceptibility (14%), and only three patients had PVs/LPVs on both BRCA1/2 and other genes (1%). One-hundred and thirty-one patients did not inherit the pathogenic mutation previously detected in the family (52%). Among the younger group, 53 had BRCA1/2 germline PVs/LPVs (38%), 15 were carriers of other cancer susceptibility genes (10%), primarily APC, NBN, ATM, MUTYH, MLH1, and only 2 patients were carriers of PVs/LPVs in both BRCA1/2 and other susceptibility genes (1%). Seventy-three patients showed no PVs/LPVs (51%). Twenty-nine out of the 53 BRCA1/2 PVs/LPVs carriers were women and 24 were males, meanwhile, among the 15 carriers of other susceptibility genes, 6 were women and 9 males.
In the older group, 28 were carriers of BRCA1/2 germline PVs/LPVs (26%), while 20 had mutations on other genes (18%), such as CHEK2, MUTYH, PALB2 and BRIP1. Only one patient carried a PV/LPV in both BRCA2 and ATM. Fifty-eight cases had no PVs/LPVs (55%). Among the 28 BRCA1/2 carriers, 19 were women and 9 males, meanwhile in the other susceptibility genes carriers’ group (20 patients), 9 were women and 11 were males.
Overall, the most prevalent PV/LPV on BRCA1 was c.5266dupC, while on BRCA2 was c.7007G>A, found respectively in 7 and 4 patients from different families.
Specifically, the BRCA1 variant causes an insertion of one cytosine, resulting in a frameshift mutation with the creation of a novel translational termination codon after 74 amino acid residues [p.(Gln1756Profs*74)]. The protein product thus produced is truncated and non-functional [9].
The BRCA2 pathogenic variant, instead, replaces arginine with histidine at codon 2336 of the protein [p.(Arg2336His)]. RNA analysis indicates that this missense mutation induces altered splicing and may result in an absent or disrupted protein product [10]. Another interesting finding was the presence of germline PVs/LPVs on BRCA2 in 22 out of 45 male carriers (49%).
Our analysis revealed that CHEK2 was the gene with the most recurrent mutations in 11 patients, while the second most mutated gene was MUTYH, found in 5 patients. The most frequent CHEK2 PV/LPV was c.499G>A, observed in 9 individuals (9/11, 82%); this missense variant located in coding exon 3 of the gene, results from a Guanine to Adenine substitution at nucleotide position 499 and has a deleterious impact on protein structure and function [p.(Gly167Arg)] [11]. Two out of 5 patients (40%) with MUTYH mutation showed the c.884C>T p.(Pro267Leu) variant.
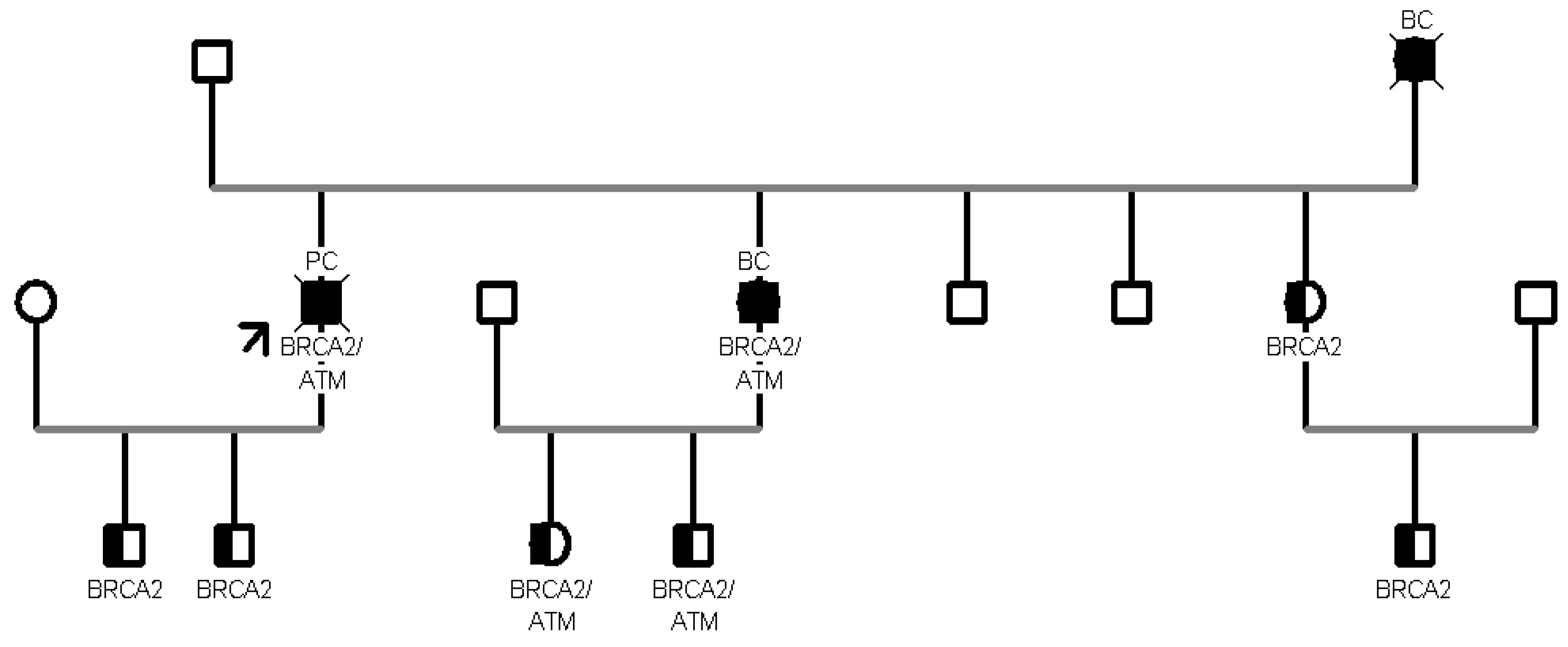
Analyzing the healthy collateral family members, one family showed strong inheritance with a PV on BRCA2, the c.6450dupA one, found in all the 5 family members tested. Specifically, 3 were early onset and 2 were late onset.
In addition, in another family segregation study, 8 patients aged >45 years turned out to carry the same proband PV c.499G>A in CHEK2 gene. Furthermore, in another family with 10 healthy collateral relatives, we tested two variants: BRCA2 c.8487+1G>A and ATM c.6095G>A. Seven patients were carriers of the c.8487+1G>A pathogenic variant in BRCA2, and 3 were carriers of both variants. (Figure 2).
In particular, the BRCA2 intronic variant occurs in the invariant region of the splice consensus sequence and is predicted to cause altered splicing leading to an abnormal or absent protein [12]; the missense variant in ATM causes a G to A nucleotide substitution at the last nucleotide of exon 41 of the ATM gene and replaces arginine with lysine at codon 2032 of the ATM protein [p.(Arg2032Lys)]. The aberrant transcript is expected to result in an absent or non-functional protein product [13].
Figure 1.
Family A.
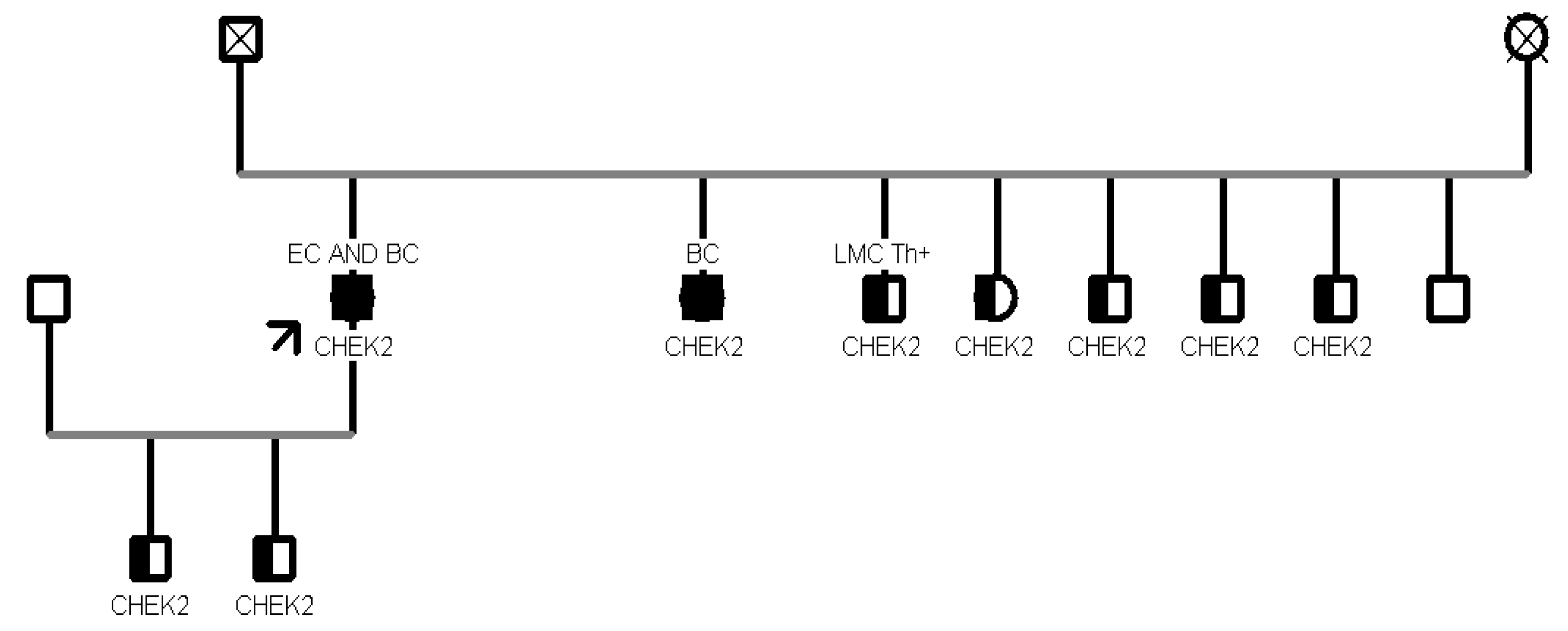
Figure 2.
Family B.
4. Discussion
Hereditary tumors caused by germline mutations account for 5-10% of all cancers, with increased prevalence in some specific cancers such as breast, ovary, colon, and others. Advances in genetic technology and the implementation of Next Generation Sequencing (NGS) have accelerated the simultaneous analysis of several susceptibility cancer genes. In fact, even though the BRCA1/2 genes are known to explain up to 25% of all the suspected hereditary forms [14,15] , several other non BRCA genes are known to be involved in cancer predisposition, as evidenced by the continuous updating of the National Comprehensive Cancer Network’s (NCCN) guidelines for hereditary cancers [16]. As a consequence of this improvement in the diagnosis of hereditary cancers, a larger number of cancer patients are at present identified as carriers of genetic mutations, increasing their risk of developing cancer during their lifetime [17,18]. In turn, this leads to an increased number of healthy relatives in which the presence of the mutation must be assessed to prevent the disease's development. The aim of the present study is to highlight how the application of the multigene panel on cancer probands can subsequently improve the healthy non-BRCA PVs/LPVs carriers' identification.
Starting from 110 affected probands, tested with a NGS multigene panel based on 27 cancer susceptibility genes and carriers of mutations in cancer susceptibility genes, we analyzed 250 healthy collaterals and detected 119 cases harboring at least one PVs/LPVs. Among them, 35 patients carried germline PVs/LPVs on non-BRCA1/2 genes (29%) involving CHEK2, MUTYH, ATM, APC, MSH2, PALB2, MLH1, TP53, RAD51C, NBN, BRIP1 and CDH1.
A crucial point in applying information about the gene variant in cancer prevention is related to the different risks associated with each single gene. In other words, the prevention strategies to use in patients with non BRCA1/2 PVs/LPVs are different from those typically adopted in BRCA1/2 carriers.
CHEK2 was the most frequently mutated gene in our population since two different pathogenic variants (c.349A>G and c.499G>A) in 10 patients from 3 families were detected. CHEK2 is a tumor suppressor gene conferring a predisposition to sarcoma, breast cancer, and brain tumors. CHEK2, a protein kinase activated in response to DNA damage, is involved in cell cycle arrest, and heterozygous germline mutations in this gene have been reported in patients with Li-Fraumeni syndrome-2 [19].
The second most frequently mutated gene in our population is MUTYH, found in 5 patients from 2 families. Mammalian MutY homologue (MUTYH) encodes a DNA glycosylase involved in base excision repair during DNA replication and damage repair. PVs/LPVs in MUTYH are associated with autosomal recessive colorectal adenomatous polyposis, but interestingly, monoallelic variants on this gene have been reported by our and other groups as associated with cancer predisposition in several patients [20,21].
An interesting case in the present study is represented by the detection of a MUTYH c.734G>A variant in one female patient with a personal history of breast cancer diagnosed at the age of 44 years, previously tested for BRCA1/2 variants at another institute and turned out to be negative. In this case, the identification of the pathogenic variant was allowed by using the multigene panel testing in her sister, suggesting the usefulness of multigene panel analysis in affected patients negative for BRCA1/2 testing in the presence of strong familiarity.
In addition, another female patient was detected to harbor only the c.650G>A in the MUTYH gene while being negative for the second variant (c.884C>T) found in the proband (a son affected by colon cancer). The patient had a personal history of cancer, at first she had a diagnosis of breast cancer at the age of 50, then of colon cancer at an older age (82 years). Due to the time of disease onset, she had never received the indication for genetic testing, representing a further case of detection of a germline mutation through the analysis of an affected relative using a multigene panel. To date, the cancer risk associated with germline variants in individuals carrying only one MUTYH defective allele is controversial. Studies have shown that the risks of colorectal cancer for carriers of monoallelic variants in MUTYH with a first-degree relative with colorectal cancer are sufficiently high to warrant more intensive screening than for the general population, as a consequence NCCN guidelines propose colonoscopy every five years beginning at age 40 [22,23]. Nevertheless, there is no strong evidence of an association between increased BC risk and carriers of monoallelic variants in MUTYH [24]. More research is needed to confirm the cancer risks linked with these heterozygous MUTYH mutations. Some considerations should also be made about the ATM variants encountered in our cohort. Focusing on family B (see Figure 2), the proband who initiated the segregation analysis was diagnosed with pancreatic tail and body cancer at the age of 55 and was found to carry PVs/LPVs in two distinct genes, specifically the c.6095G>A in ATM and the c.8487+1G>A in BRCA2. Sanger sequencing in healthy collaterals highlighted the presence of the same compound heterozygosity in the proband’s daughter and in his two male grandchildren, with a negative personal oncological anamnesis.
5. Conclusions
In conclusion, from the analysis of our data it emerges that, without the application of the NGS multigene panel in the probands, a considerable percentage of healthy collaterals, carriers of PVs/LPVs in the other susceptibility genes, would have been lost (14%). This percentage corresponds to 35 healthy carriers that, due to the presence of germline variants, will be included in the clinical and instrumental surveillance protocols.
The identification of hereditary forms, related to germline, inherited DNA variants, is thereby crucial to admit patients and their at-risk family members to the most proper surveillance and therapeutic programs [25].
The NCCN Clinical Practice Guidelines in Oncology have specific recommendations for patients found to have pathogenic variants that confer an increased risk of breast cancer, including imaging modalities, frequency of evaluation and risk-reducing surgery. Genetic testing and NCCN guidelines for patients with pathogenic variants have changed the clinical landscape for breast oncologists, who routinely address the relevance of genetics, the criteria for testing, and recommendations for radiographic and/or operative follow-up during patient consultations [16,26].
To mention some examples, the status of carriers of PVs/LPVs in the CHEK2 gene leads to the setting up of a surveillance plan, even if the subjects have not developed a first tumor yet. Specifically, based on the NCCN and AIOM (Italian Association of Medical Oncology) guidelines, the suggested prevention protocol provides, for female patients, breast clinical and instrumental surveillance, with an annual mammography and magnetic resonance imaging (MRI) starting at the age of 40. Instead, for men, the prevention protocol requires an annual Prostate Specific Antigen (PSA) dosage for prostate cancer surveillance, starting at the age of 40.
For both female and male patients, the protocol provides Colorectal Cancer Surveillance based on a colonoscopy every 5 years starting at 40 years of age. Furthermore, patients with a CHEK2 mutation must be followed by a multidisciplinary team with expertise in the fields of gastroenterology and urology.
MUTYH is defined as a “high penetrance” gene whose pathogenetic variants are transmitted in an autosomal recessive manner. Based on NCCN Guidelines, the prevention protocol for carriers includes a colonoscopy every 5 years starting at 40 years of age, even if they suggest a specific surveillance, tailored to each patient, considering the personal medical and familial history.
Another example of surveillance protocols is the ATM gene. ATM is defined as a "moderate-penetrance" gene whose pathogenic mutations confer an increased risk of developing breast, ovarian, and pancreatic cancers over the course of a lifetime. Germline alterations in the ATM gene are transmitted in an autosomal dominant manner, and all offspring, regardless of sex, have a 50 percent chance of inheriting the genetic abnormality from a parent who carries it. The presence of a pathogenic mutation in the ATM gene requires carriers to establish a prevention pathway, even if such individuals have already developed a malignancy. Specifically, according to the NCCN guidelines, the suggested prevention protocol includes: an annual mammography and magnetic resonance imaging (MRI) starting from the age of 40 for breast cancer; a clinical instrumental surveillance regarding annual gynecologic examination with transvaginal ultrasound from age 40 years and annual CA-125 assay from the age of 40 years for the ovarian cancer; gastroscopy and endoscopic ultrasound from age 40 years for gastric cancer; colonoscopy every 5 years starting at age 40 years for colorectal cancer and for pancreatic cancer ultrasound and possibly MRI if there have been relatives with pancreatic cancer in the family, starting at age 45 years or 10 years earlier if there have been cases of juvenile pancreatic cancer.
Importantly, fifteen out of 250 collaterals tested had cancer but no pathogenic mutation; for these kinds of clinical patients, the first hypothesis is that it is a sporadic tumor since these account for 90% of all cancers. Finally, in the future, impacted collaterals not harboring the found family pathogenic variant could be perfect candidates for an extended molecular analysis to detect additional susceptibility genes and potential target therapeutics for better clinical illness management.
Author Contributions
Writing original draft preparation, SD and RG; writing review and editing, SD, RG, LP, FA, MC, MR and AD; performed the genetic analysis, SD; data curation, AD; conceptualization and supervision, IA, PB and LS; clinical investigation, AG, SG, NC, RG, MC, MR and IA.
Funding
This research received no external funding.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- M. Magaña, A. P. Landeta-Sa, e Y. López-Flores, «Cowden Disease: A Review», Am. J. Dermatopathol., vol. 44, n. 10, pagg. 705–717, ott. 2022. [CrossRef]
- S. K. Aedma e A. Kasi, «Li-Fraumeni Syndrome», in StatPearls, Treasure Island (FL): StatPearls Publishing, 2023. Consultato: 10 ottobre 2023. [Online]. Disponibile su: http://www.ncbi.nlm.nih.gov/books/NBK532286/.
- «Lo sai che circa il 10% dei tumori è ereditario?» Consultato: 10 ottobre 2023. [Online]. Disponibile su: https://www.airc.it/cancro/informazioni-tumori/lo-sai-che/il-10-per-cento-circa-dei-tumori-ha-origine-da-fattori-di-rischio-ereditari.
- R. Kamps et al., «Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification», Int. J. Mol. Sci., vol. 18, n. 2, pag. 308, gen. 2017. [CrossRef]
- G. Cerretelli, A. Ager, M. J. Arends, e I. M. Frayling, «Molecular pathology of Lynch syndrome», J. Pathol., vol. 250, n. 5, pagg. 518–531, apr. 2020. [CrossRef]
- «Consulenza genetica e test genetici in oncologia», AIOM. Consultato: 10 ottobre 2023. [Online]. Disponibile su: https://www.aiom.it/consulenza-genetica-e-test-genetici-in-oncologia-aspetti-critici-e-proposte-di-aiom-sigu-2021/.
- F. Baudi, «Hereditary Tumours», BioMed Res. Int., vol. 2013, pag. 490357, 2013. [CrossRef]
- P. Teller e R. K. Kramer, «Management of the asymptomatic BRCA mutation carrier», Appl. Clin. Genet., vol. 3, pagg. 121–131, 2010. [CrossRef]
- M. Koczkowska et al., «Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients», Cancers, vol. 10, n. 11, pag. 442, nov. 2018. [CrossRef]
- A. Coppa et al., «Novel and recurrent BRCA2 mutations in Italian breast/ovarian cancer families widen the ovarian cancer cluster region boundaries to exons 13 and 14», Breast Cancer Res. Treat., vol. 148, n. 3, pagg. 629–635, dic. 2014. [CrossRef]
- N. Tung et al., «Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer», J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol., vol. 34, n. 13, pagg. 1460–1468, mag. 2016. [CrossRef]
- R. L. S. Mesman et al., «Alternative mRNA splicing can attenuate the pathogenicity of presumed loss-of-function variants in BRCA2», Genet. Med. Off. J. Am. Coll. Med. Genet., vol. 22, n. 8, pagg. 1355–1365, ago. 2020. [CrossRef]
- D.-S. Huang et al., «Prevalence of deleterious ATM germline mutations in gastric cancer patients», Oncotarget, vol. 6, n. 38, pagg. 40953–40958, dic. 2015. [CrossRef]
- Antonucci, M. Provenzano, L. Sorino, M. Rodrigues, G. Palka, e L. Stuppia, «A new case of “de novo” BRCA1 mutation in a patient with early-onset breast cancer», Clin. Case Rep., vol. 5, n. 3, pagg. 238–240, mar. 2017. [CrossRef]
- Antonucci et al., «Comparison between CaGene 5.1 and 6.0 for BRCA1/2 mutation prediction: a retrospective study of 150 BRCA1/2 genetic tests in 517 families with breast/ovarian cancer», J. Hum. Genet., vol. 62, n. 3, pagg. 379–387, mar. 2017. [CrossRef]
- «Detection, Prevention, and Risk Reduction», NCCN. Consultato: 10 ottobre 2023. [Online]. Disponibile su: https://www.nccn.org/guidelines/category_2.
- L. Lombardi et al., «Psychological aspects, risk and protective factors related to BRCA genetic testing: a review of the literature», Support. Care Cancer Off. J. Multinatl. Assoc. Support. Care Cancer, vol. 27, n. 10, pagg. 3647–3656, ott. 2019. [CrossRef]
- S. M. Bramanti et al., «Uncertainty following an inconclusive result from the BRCA1/2 genetic test: A review about psychological outcomes», World J. Psychiatry, vol. 11, n. 5, pagg. 189–200, mag. 2021. [CrossRef]
- D. W. Bell et al., «Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome», Science, vol. 286, n. 5449, pagg. 2528–2531, dic. 1999. [CrossRef]
- F. Anaclerio et al., «Clinical usefulness of NGS multigene panel testing in hereditary cancer analysis», Front. Genet., vol. 14, pag. 1060504, 2023. [CrossRef]
- A. Dell’Elice et al., «Filling the gap: A thorough investigation for the genetic diagnosis of unsolved polyposis patients with monoallelic MUTYH pathogenic variants», Mol. Genet. Genomic Med., vol. 9, n. 12, pag. e1831, dic. 2021. [CrossRef]
- R. S. C. Guindalini et al., «Detection of germline variants in Brazilian breast cancer patients using multigene panel testing», Sci. Rep., vol. 12, n. 1, pag. 4190, mar. 2022. [CrossRef]
- K. Win et al., «Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer», Gastroenterology, vol. 146, n. 5, pagg. 1208-1211.e1–5, mag. 2014. [CrossRef]
- Breast Cancer Association Consortium et al., «Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women», N. Engl. J. Med., vol. 384, n. 5, pagg. 428–439, feb. 2021. [CrossRef]
- M. Nunziato et al., «Multigene panel testing increases germline predisposing mutations' detection in a cohort of breast/ovarian cancer patients from Southern Italy», Front. Med., vol. 9, pag. 894358, 2022. [CrossRef]
- «Recently Updated Guidelines», NCCN. Consultato: 10 ottobre 2023. [Online]. Disponibile su: https://www.nccn.org/guidelines/recently-published-guidelines.

Table 1.
Oncomine NGS panel containing 27 cancer susceptibility genes.
| Gene | Omim | Refseq | Gene | Omim | Refseq |
|---|---|---|---|---|---|
| ATM | 607585 | NM_000051.3 | PALB2 | 610355 | NM_024675.3 |
| EPCAM | 185535 | NM_002354.2 | MLH1 | 120436 | NM_000249.3 |
| MSH2 | 609309 | NM_000251.2 | MSH6 | 600678 | NM_000179.2 |
| PMS2 | 600259 | NM_000535.6 | RAD51C | 602774 | NM_058216.2 |
| BRIP1 | 605882 | NM_03204.2 | RAD51D | 602954 | NM_002878.3 |
| TP53 | 191170 | NM_000546.5 | CHEK2 | 604373 | NM_007194.3 |
| CDH1 | 192090 | NM_004360.4 | PTEN | 601728 | NM_000314.6 |
| MUTYH | 608456 | NM_001128425.2 | APC | 611731 | NM_000038.6 |
| SMAD4 | 600993 | NM_005359.6 | POLE | 174762 | NM_006231.3 |
| POLD1 | 174761 | NM_001256849.1 | CDK4 | 123829 | NM_000075.3 |
| BARD1 | 601593 | NM_000465.3 | CDKN2A | 600160 | NM_000077.5 |
| CDK12 | 615514 | NM_016507.3 | NBN | 6026667 | NM_002485.4 |
| BRCA1 | 113705 | NM_007294.4 | BRCA2 | 164757 | NM_000059.3 |
| NF1 | 162200 | NM_001042492.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



