
Submitted:
19 October 2023
Posted:
19 October 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in Wuhan, China in December 2019, it has spread rapidly, and many coronavirus disease (COVID-19) cases have occurred in Gwangju, South Korea. Viral mutations following the COVID-19 epidemic have increased interest in the characteristics of epidemics in this region, and pathogen genetic analysis is required for infection control and prevention. In this study, SARS-CoV-2 whole-genome analysis was performed on samples from patients with COVID-19 in Gwangju from 2020 to 2022 to identify the trends in COVID-19 prevalence and to analyze the phylogenetic tree of dominant variants. B.41 and B.1.497 prevailed in 2020, the early stage of the COVID-19 outbreak; then, B.1.619.1 mainly occurred until June 2021. B.1.617.2, classified as sublineage AY.69 and AY.122, occurred continuously from July to December 2021. Since strict measures to strengthen national quarantine management had been implemented in South Korea until this time, mutations phylogenetic analysis was also able to infer the epidemiological relationship between infection transmission routes. Since the first identification of the Omicron variant in late December 2021, the spread of infection has been very rapid, and weekly whole-genome analysis of specimens has enabled us to monitor new Omicron sublineage occurring in Gwangju. Our study suggests that conducting reginal surveillance in addition to nation-level genomic surveillance will enable more rapid and detailed variant surveillance, which will be helpful in the overall prevention and management of infectious diseases.
Keywords:
COVID-19
; SARS-CoV-2
; whole-genome sequencing
; variants
; lineages
1. Introduction
Since coronavirus disease (COVID-19) cases were first reported in Wuhan, Hubei Province, China, in December 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) rapidly spread worldwide, and the World Health Organization (WHO) officially declared COVID-19 a pandemic with the highest alert level on March 11, 2020 [1]. The first confirmed COVID-19 case in South Korea was identified as a Chinese entry from Wuhan on January 20, 2020. Starting with the first reported case of a Korean patient with COVID-19 patient who returned from Thailand and was diagnosed on February 3, 2020, there have been many cases of COVID-19 infection through imported cases and local outbreaks in Gwangju, South Korea [2,3].
+All viruses, including SARS-CoV-2 (the causative agent of COVID-19), have constantly evolved. Mutations resulting from viral genetic changes have little effect on viral characteristics. However, some mutations can affect transmission, pathogenicity, and vaccine and therapeutic efficacy [4]. The WHO monitors the occurrence of significant changes in COVID-19 variants through amino acid substitution and classifies them into variants of concern (VOCs) and variants of interest, updates them regularly, and recommends public health measures [5].
Viral mutations can be identified through genetic analysis. Whole-genome sequencing has been widely used recently, and genetic information associated with COVID-19 variants has been shared worldwide through the Global Initiative on Sharing All Influenza Data (GISAID) and Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) [6]. Accordingly, we established a whole-genome analysis in May 2021 to trace the source of infection and confirm genetic mutations in COVID-19 cases.
As the lineage transitions of SARS-CoV-2 increase, several institutions have constructed naming systems to establish standardized nomenclature. GISAID, NextStrain, and PANGOLIN are the most common naming conventions used by scientific communities around the world [7,8,9].
The GISAID system is named based on a large-scale clade defined by the marker variant of the reference genome WIV04 (Genbank: MN996528.1), which is a simple system, but it is inconsistent and classified only by a few mutations. The SARS-CoV-2 sequence in GISAID can be easily analyzed using NextStrain, a system that uses phylogenetic analysis to identify evolutionarily stable lineages and sublineages [10]. Once identified, they are named based on their year of appearance and consecutive characters. Specific sublineages are identified as additional information; however, there are no systematic rules. To overcome this problem, the dynamic PANGOLIN system based on evolutionary relations also considers the mechanical relevance of lineages. According to this system, each lineage name consists of an alphabetical prefix and a numeric suffix separated by a period or point. The PANGOLIN system provides detailed and informative outbreak cluster information [6].
In this study, we identified COVID-19 variants, including VOCs, and performed mutational and phylogenetic analyses of the dominant strain using whole-genome sequencing of COVID-19 confirmed cases in Gwangju, Korea, from February 2020 to December 2022. SARS-CoV-2 can be continuously monitored to prevent the spread of infectious diseases by tracking new variants that affect pathogenicity, transmissibility, and vaccine efficacy. In addition, a survey of the evolutionary history of mutant viruses will help prepare countermeasures against newly emerging variants of concern [11].
2. Materials and methods
2.1. Sample collection
Samples were collected using nasopharyngeal and oropharyngeal swabs in universal transport medium between February 2020 and December 2022 from people visiting public centers in five districts of Gwangju Metropolitan City, including suspected patients with COVID-19, close contacts of confirmed cases, and those seeking testing. Real-time PCR was performed to detect SARS-CoV-2 viral RNA using a PowerChek™ SARS-CoV-2 Real-time PCR Kit (Kogenebiotech, Seoul, South Korea), and samples with high virus copy numbers were selected for sequencing. Most samples were randomly selected; however, some positive samples were classified based on epidemiological information. We selected samples representing each case to confirm the variants using whole-genome sequencing.
2.2. Library preparation and sequencing
Total RNA was extracted from 140 µL of nasopharyngeal and oropharyngeal swab fluid using a QIAamp viral RNA mini kit (QIAGEN, Hilden, Germany) according to the manufacturer’s manual. Libraries were prepared using an Illumina COVIDSeq assay kit (Illumina, San Diego, CA, USA) according to manufacturer’s instructions. The RNA was reverse-transcribed to synthesize cDNA using a random hexamer primer. The cDNA was amplified using two primers based on ARTIC, which cover the entire SARS-CoV-2 genome. The amplified PCR product was fragmented, and the PCR amplicons were tagged using the IDT-ILMN Nextera DNA UD Index Set A. The libraries were pooled, cleaned, and quantified using Qubit™ 1X dsDNA HS (High Sensitivity) Assay kits on a Qubit 3 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The final loading concentration was 9 pM. Sequencing was performed on a Miseq instrument (Illumina, San Diego, CA, USA) using an Illumina Miseq reagent kit v2 (300-cycles) with dual-indexed paired-end 2*151 bp reads.
2.3. Sequence data and analysis
FASTQ sequencing files were generated from MiSeq and imported to the CLC genomics workbench ver.21.0.3 (CLC bio, QIAGEN, Aarhus, Denmark) to run the workflow. The workflow involved the mapping, alignment, and generation of consensus sequences. The sequence reads were mapped to the SARS-CoV-2 reference genome (NCBI: NC045512). Only high-coverage whole-genome sequences were used for analysis. The PANGOLIN (https://pangolin.cog-uk.io/) web application and the Nextclade tool (https://clades.nextstrain.org/) were used to identify the SARS-CoV-2 lineage.
2.4. Phylogenetic analysis
Phylogenetic analysis was conducted using Molecular Evolutionary Genetics (MEGA-11 ver.11.0.13). A phylogenetic tree was created using the maximum-likelihood method and Tamura and Nei 1993(TN93), and the general time reversible model with gamma distribution and invariant sites (G+ I) parameters as the best-fit model of nucleotide substitution with 1000 bootstrap replications. FASTQ sequencing files were generated from MiSeq and imported to the CLC genomics workbench ver.21.0.3 (CLC bio, QIAGEN, Aarhus, Denmark) to run the workflow. The workflow involved the mapping, alignment, and generation of consensus sequences. The sequence reads were filtered at or above 30, trimmed for quality, and mapped to the SARS-CoV-2 reference genome (NCBI: NC045512). Sequenced samples with ≥95% at 20X depth genome coverage were used for analysis. PANGOLIN and NextStrain were used to identify the SARS-CoV-2 lineage.
3. Results
3.1. SARS-CoV-2 cases during pandemic waves in Gwangju, South Korea
By the end of December 2022, a total of 850,707 cases have been reported in Gwangju since the first COVID-19 infection in February 2020. When classified by epidemic period, there were 36 cases in the first wave, including the first imported case of COVID-19 and occurrence in metropolitan and Seoul, Daegu, and Gyeongbuk (February 2020 to May 2020). In the second wave, 725 cases were confirmed in a rapid spread throughout the Seoul Metropolitan Area, including outbreaks in assemblies and religious organizations (June 2020 to November 2020). During the third wave, when a new COVID-19 variant appeared and spread nationwide (December 2020 to June 2021), 2285 patients were diagnosed with COVID-19. In addition, 17,123 cases occurred in the fourth wave, spreading the Delta variant (July 2021 to January 2022). In the fifth wave, the number of confirmed cases increased explosively nationwide to 518,055 patients due to the Omicron variant, and 312,483 cases were confirmed during the period of continued emergence of new Omicron variants (July 2022 to December 2022) [12]. We randomly selected 2399 COVID-19 cases by classifying patients with or without a confirmed infection route. Then, whole-genome sequencing was performed to assign lineages. The sequences identified 147 lineages based on the PANGOLIN criteria (v.4.3) (Table 1 and Table 2). According to the NextClade classification [13], 16 different clades were detected in Gwangju: 19A, 20A, 20H, 20I, 21A, 21I, 21J, 21K, 21L, 22A, 22B, 22C, 22D, 22E, 22F, and the recombinant strain. Figure 1 shows the phylogenetic tree constructed using the NextClade online tool (accessed on 30th June 2023).
3.2. Dominant SARS-CoV-2 lineages during waves
Sequences from Gwangju were included along with the global sequences to determine where they lie in the tree. Figure 2 shows a phylogenetic tree based on the PANGOLIN classification (Figure 2a) and indicates the epidemic period classified by sample collection date and variants relevant to that period (Figure 2b).
During the first wave, two cases were identified as B.41, and 35 cases were identified as B.1.497 during the second wave. In the third epidemic period, we revealed 50.9% B.1.619.1 (n = 84), 37.6% B.1.497 (n = 62), 9.1% B.1.1.7 (n = 15), 1.8% B.1.617.2 (n = 3), and 0.6% B.1.351 (n = 1). In the fourth wave, the prevalence rate was as follows: 53.8% 1.617.2 (n = 168) > 30.8% BA.1 (n = 96) > 11.2% B.1.619.1 (n = 35) > 3.2% BA.2 (n = 10), with Delta becoming the dominant SARS-CoV-2 variant. Omicron variants were as follows: 66.6% BA.2 (n = 504), 23.0% BA.1 (n = 174), 8.6% BA.5 (n = 65), and 1.5% BA.4 (n = 11) during the fifth wave. In the sixth epidemic, when Omicron sublineages continued to appear, 82.6% BA.5 (n = 932) > 16.2% BA.2 (n = 183) > 0.7% recombinant variant (n = 8) > 0.5% BA.4 (n = 6) were identified.
3.3. B.1.619.1 genome sequences
In the early days of the COVID-19 outbreak, B.1.497 with a D614G mutation in the Spike protein and B.1.619.1 with additional mutations, such as N440K and E484K, were prevalent. We focused on the B.1.619.1 variant, which was dominant in Gwangju until the fourth wave when the B.1.617.2 (Delta variant) spread. Among the sequences corresponding to B.1.619.1, the FASTA files of 86 sequences, excluding those with low-quality data, were used for analysis. We observed 125 nucleotide mutations and 61 amino acid mutations after analyzing the mutation sites and numbers of 86, B.1.619 mutant strains in this study and comparing them with the phylogenetic analysis reference strain (Wuhan-Hu 1, NC045512) provided by NextClade. Regarding the distribution of amino acid mutations in individual genes, ORF1ab had the highest number of mutations (30), followed by 12 mutations in S, eight in N, five in ORF7a, three in ORF3a, two in ORF8, and one in M.
Among a total of 61 amino acid mutations, 20 mutations were identified in all 86 sequences: eight mutations in ORF1ab (A2123V, E2607L, S3675del, G3676del, F3677del, M3752I, K3929R, and P4715L), seven mutations in S (I210T, N440K, E484K, D614G, D936N, S939F, and T1027I), three mutations in N (P13L, S201I, T205I), I82T in M (I82T), and E22D in ORF7a (Supplementary Table S2).
To analyze the genetic relationship among the 86, B.1.619.1 cases identified in this study, we performed phylogenetic analysis using MEGA 11software (Figure 3a). The sequences were grouped into three clusters. Group A comprised a cluster of 24 cases in May and June 2021, including 18 where the route of infection was confirmed. Group B consisted of nine patients who had been in contact with confirmed cases in different regions of Korea. Group C comprised 33 cases detected in July and August 2021, 31 of which had an additional F548S amino acid change in ORF1ab.
3.4. B.1.617.2 genome sequences
B.1.619.1 gradually disappeared as B.1.617.2 was introduced in the second half of 2021, and became the dominant variant from July to December 2021 after its confirmation in June 2021. In particular, the sublineages of B.1.617.2, namely, AY.69 and AY.122, accounted for most of the cases (Figure 4a). To analyze the genetic relationships of B.1.617.2 identified in this study, we created a phylogenetic tree with 108 sequences of B.1.617.2 using MEGA-11(ver.11.0.13) software (Figure 3b). Samples collected from COVID-19 patients in Gwangju were classified into three clades: 21A, 21I, and 21J.
Clade 21A included five samples related to the inflow of the metropolitan area, clade 21J consisted of 27 samples related to imported cases and enumeration of foreigners, and clade 21I included 76 cases of various infection groups that occurred sporadically in Gwangju. Thus, they were grouped based on the common route of infection and classified into sublineages.
3.5. Omicron variant spread in Gwangju in 2022
By the end of December 2021, Omicron had spread rapidly against Delta, becoming the dominant variant within several weeks. This result appeared two weeks after the first report of Omicron in Gwangju, and its spread was faster than that of the previous variants. As the prevalence of Omicron prolonged, its sublineages continued to appear. Figure 4b shows the distribution of the COVID-19 Omicron variants in 2022. BA.1 and BA.2 were identified as the dominant variants from January to May 2022. BA.1 was the dominant variant for 10 weeks starting from the 1st week of 2022, and the Omicron sublineages BA.1.1 and BA.1.1.5 accounted for a large proportion. BA.2 was the dominant variant in the 12th and 13th weeks of 2022, and the proportion of sublineage BA.2.3 was also high. Subsequently, both BA.4 and BA.5 were detected, and BA.5 became the dominant variant in June 2022, indicating a tendency for various sublineages to occur simultaneously. BF.7, BQ.1, BQ.1.1, XBB, and XBB.1 were detected since November 2022, and sublineages of BA.2.75, BN.1, and CH.1.1 were confirmed.
4. Discussion
Viruses change constantly, and new variants continuously emerge during SARS-CoV-2 proliferation and propagation [14]. New virus variants may affect the infectivity, virulence, and immune evasion [15,16]. SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus, which is more pathogenic than SARS-CoV (2002) and Middle East respiratory syndrome coronavirus (2013) [17]. The SARS-CoV-2 genome is approximately 30 kb long (GenBank number MN908947), and it encodes 9860 amino acids [18,19]. Gene fragments encode structural and nonstructural proteins involved in viral replication. The nucleocapsid (N), spike (S), membrane (M), and envelope (E) genes encode structural proteins, whereas non-structural proteins, such as 3-chymotrypsin-like protease, papain-like protease, and RNA-dependent RNA polymerase are encoded by ORF regions [20,21]. Among them, the most notable mutations are those in the S gene, which is involved in viral entry into cells [22]. It has been widely reported that SARS-CoV-2 has undergone several genetic variants, leading to various mutations such as substitutions, reversions, and deletions throughout the genome [23]. Therefore, analyzing its impact on public health by monitoring variants is of utmost importance, which is performed through genetic composition change analysis, also known as genome sequencing.
We used whole-genome sequencing to monitor variants that could be new inflows into Gwangju, South Korea, or propagate within the region. Genome analysis of SARS-CoV-2 in Gwangju showed that B.1.497 was prevalent in the early days of the COVID-19 outbreak. Since then, B.1.1.7 (Alpha variant), first reported in the U.K in September 2020, was partially confirmed through infection in the region, but did not spread significantly. In addition, all cases of B.1.351 (Beta variant), first reported in South Africa in May 2020, were imported, and there were few cases of local transmission. P.1 (Gamma variant), which was first identified in Brazil in November 2020, was not identified, and mainly B.1.619.1 occurred during this time [24]. B.1.617.2 (Delta variant) was identified as the dominant strain from the 2nd week of July 2021, and the sublineages AY.69 and AY.122 were predominantly observed. Since the Delta variant was first identified in India in October 2020, it rapidly increased since April 2021 and became prevalent worldwide since July 2021. It accounts for more than 90% of the data registered in GISAID (analysis update, 10/01/2021) [25]. After the end of December 2021, the Delta variant decreased owing to the appearance of the omicron variant, and as the prevalence of the omicron variant continued for a prolonged period, various Omicron sublineages, such as BA.1, BA.2, BA.4, and BA.5 were detected. Omicron is now the dominant strain worldwide, accounting for more than 98% of viruses in GISAID beyond 2022, and its transmission continues [26]. Therefore, in order to track the public health risks that may arise from variants in the various Omicron sublineages, WHO has defined “Omicron subvariants under monitoring” as a new category to monitor variants [5].
In this study, we noted the existence of the B.1.619.1 variant, which is not classified as a VOC by the WHO, but was identified as the dominant strain in Gwangju, South Korea until the introduction of the Delta variant. Our results match those of previous studies on the distribution of variant strains in South Korea; B.1.497 predominated from March 2020 to January 2021, and the prevalence of B.1.619 and B.1.620 increased rapidly from March 2021 [27]. According to WHO data, the B.1.619 variant was first reported in May 2020, managed as a variant under monitoring on July 14, 2021, and reclassified as a formerly monitored variant on November 9, 2021. In addition, it is difficult to find a reference for the B.1.619 variant, but we confirmed that the mutant virus, which was widespread in Central Africa, spread to Europe using GISAID data [28].
The B.1.619 variant in South Korea was first confirmed in immigrants from Cameroon in February 2021, but was later confirmed to be distinct from those of European countries based on sequence phylogenetic analysis of the B.1.619 variant isolated in Korea. It has been reported that ORF1ab is reclassified as sub-lineage B.1.619.1 due to the additional presence of the K3929R mutation [27]. Most variants are subdivided according to the processes of propagation and spread to new countries. In this study, as previously reported in South Korea, the B.1.619.1 variant was reclassified as a sub-lineage of the B.1.619 variant.
We focused on the B.1.619.1 variant because of its mutations, specifically E484K and N440K, which affect the antigenicity of the spike protein. The E484K mutation was identified in both the B.1.351 (Beta variant) and P.1 (Gamma variant) strains, making it an escape mutation that weakens the binding affinity between the neutralizing antibody and the RBD. This reduces the antibody effectiveness and is a significant factor in the ability to evade vaccine-induced antibodies [29,30]. The N440K mutation increases the affinity for angiotensin-converting enzyme 2 and confers resistance to monoclonal antibodies [31]. The emergence of variants associated with contagiousness and immune evasion can lead to more cases of reinfection and negatively affect vaccines and therapeutic effects [32,33]. Notably, common mutations and deletions were observed in the B.1.619.1 and B.1.351 variants in this study, namely S3675-3677del and P4715L in ORF1ab, T205I in N, E484K and D614G in S, and S84L in ORF8.
Like the B.1.619.1 variant, AY.69 and AY.122 were identified as the B.1.617.2 (Delta) sublineage that occurred in large numbers in South Korea, including Gwangju. According to previously reported, AY.69 was specifically distributed in South Korea, and AY.122 was reported to have spread widely in Russia as well [34,35]. As new mutations occur through intra-regional transmission or new variants are introduced from other regions, conducting surveillance in each region along with national genome surveillance will increase the efficiency of infectious disease prevention and management.
During the early stages of the COVID-19 pandemic, large outbreaks were mostly associated with specific groups. However, the spread of the virus was also closely related to everyday activities, small gatherings, and contact with infected individuals. Accordingly, measures to strengthen quarantine management, such as an order to ban gatherings, were implemented, and an epidemiological investigation was systematically carried out, which we used to identify the route of infection [36]. For patients whose route of infection was unclear, an epidemiological link was inferred through whole-genome sequencing. Phylogenetic tree analysis of the sequences corresponding to B.1.619.1 and B.1.617.2 (Delta) showed that cases with unidentified infection routes formed a cluster with other cases, and those with confirmed contact with other regions formed a single cluster. COVID-19 cases in May–June 2021 and July–August 2021 formed different clusters in terms of time. In addition, groups sharing a common route of infection could be classified into the same lineage. These results will provide useful data for tracking the source of infection and blocking the spread of infectious diseases; it is considered necessary to examine the relevance of various mutations to changes in countermeasures against infectious diseases, including vaccination [37,38].
In the absence of vaccines and treatments in the early stages of the pandemic, strengthened quarantine policies, such as social distancing, were implemented to prevent a surge in the number of COVID-19 patients responding to SARS-CoV-2 [39]. Subsequently, the national vaccination rate reached 70%, and gradual daily recovery increased; however, with mitigated controls and the emergence of the highly contagious Omicron variant, the number of COVID-19 patients has soared, stopping daily recovery [40]. Due to the rapid increase in confirmed Omicron cases, epidemiological investigation to trace the source of infection was halted, making it virtually meaningless. The Omicron variant was first reported in South Africa in November 2021, and it is known for its lower fatality rate than the Delta variant, but also for its very high propagation power [41,42]. As the Omicron variant has spread since the fifth wave, the number of infected people has soared nationwide, regardless of the region or route of infection. Thus, owing to the various Omicron sublineages, real-time reverse transcription PCR targeting some genes could not predict gene mutations and deletion patterns. Therefore, the difficulty in identifying various sublineages was addressed using whole-genome sequencing [43].
After Omicron was first detected in Gwangju in December 2021, it spread rapidly and became the dominant strain in a short period of time, and BA.1, BA.1.1, BA.1.1.5, BA.2, BA.2.3. dominated in the first half of 2022. In the second half of 2022, BA.5 with mutations such as R346X, K444X, and N460X in the S gene was prevalent, and sublineages of BA.5 occurred simultaneously, typically including BF.7, BQ.1, and others [44,45]. Moreover, we identified BA.2.75, CH.1.1, and BN.1, which have additional mutations in BA.2. A recombinant variant of BJ.1 and BM.1.1.1 known as XBB was detected recently [46].
This study has limitations. Owing to resource constraints, only selectively analyzed samples were included, thus limiting the representation of the entire case cohort. However, despite this limitation, the analysis enabled the determination of variant distribution and trends within the analyzed subset, offering valuable insights within the scope of the study’s conditions.
We monitored mutations in Gwangju that may be newly introduced or propagating in the area via whole-genome sequencing analysis. We estimated the epidemiological link between patients with COVID-19 and unknown sources of infection using phylogenetic analysis. Continuous surveillance of SARS-CoV-2 variants tracks the influence of new variants on the pathogenicity, propagation power, and vaccine efficacy; ultimately, it can be used as evidence to prevent the spread of infectious diseases. Additional research on the evolutionary history of variants will help to prepare countermeasures against new variants in the future [47,48].
Supplementary Materials
Not applicable.
Author Contributions
Conceptualization, Y.-U.L., S.-H.K. and Y.-S.C.; Methodology, Y.-U.L. and K.L.; Investigation, Y.-U.L., K.L., H.L., J.P., S.-J.C., J.-S.P., S.P., C.-m.L., J.L., M.S. and E.K.; Writing—original draft preparation, Y.-U.L.; Writing—review and editing, S.-H.K. and Y.-S. C.; Project administration, S.-H.K., J.S. and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all participants involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank the authors of the originating and submitting laboratories that have deposited and shared genome data on GISAID EpiCoV.
Conflicts of Interest
The authors declare that this research was conducted in the absence of commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Mohan, B.S.; Vinod, N. COVID-19: An Insight into SARS-CoV2 Pandemic Originated at Wuhan City in Hubei Province of China. J. Infect. Dis. Epidemiol. 2020, 6, 146. [Google Scholar] [CrossRef]
- Kim, J.M., et al. Identification of Coronavirus Isolated from a Patient in Korea with COVID-19. Osong Public Health Res. Perspect. 2020, 11, 3–7. [CrossRef] [PubMed]
- Kim, M.J., et al. Isolation and Genomic Analyses of an Early SARS-CoV-2 Strains from the 2020 Epidemic in Gwangju, South Korea. J. Bacteriol. Virol. 2021, 51, 138–147. [CrossRef]
- Markov, P.V., et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [CrossRef] [PubMed]
- (WHO). W.H.O. Tracking SARS-CoV-2 variants. Available from: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants.
- O'Toole, A., et al. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2022, 23, 121.
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A., et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef]
- Flores-Vega, V.R., et al. SARS-CoV-2: Evolution and Emergence of New Viral Variants. Viruses 2022, 14, 653. [CrossRef] [PubMed]
- Rando, H.M., et al. Pathogenesis, Symptomatology, and Transmission of SARS-CoV-2 through Analysis of Viral Genomics and Structure. mSystems 2021, 6, e0009521. [CrossRef]
- Ha, J.H., et al. COVID-19 Waves and Their Characteristics in the Seoul Metropolitan Area (Jan 20, 2020-Aug 31, 2022). Public Health Wkly. Rep. 2023, 16, 111–136.
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Mishra, P.M., et al. SARS-CoV-2 Spike mutations modify the interaction between virus Spike and human ACE2 receptors. Biochem. Biophys. Res. Commun. 2022, 620, 8–14. [CrossRef] [PubMed]
- Carabelli, A.M., et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177.
- Cosar, B., et al. SARS-CoV-2 Mutations and their Viral Variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.R., et al. Structure genomics of SARS-CoV-2 and its Omicron variant: Drug design templates for COVID-19. Acta Pharmacol. Sin. 2022, 43, 3021–3033. [CrossRef] [PubMed]
- Huang, Y., et al. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [CrossRef]
- Brant, A.C., et al. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [CrossRef] [PubMed]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Lekana-Douki, S.E., et al. Screening and Whole Genome Sequencing of SARS-CoV-2 Circulating During the First Three Waves of the COVID-19 Pandemic in Libreville and the Haut-Ogooue Province in Gabon. Front. Med. 2022, 9, 877391. [CrossRef] [PubMed]
- Zhou, D., et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348–2361. [CrossRef] [PubMed]
- GISAID. GISAID Analysis Update (10/01/2021). 2021; Available from: http://gisaid.org.
- Yu, C.Y., et al. Whole genome sequencing analysis of SARS-CoV-2 from Malaysia: From alpha to Omicron. Front. Med. 2022, 9, 1001022. [CrossRef] [PubMed]
- Park, A.K., et al. SARS-CoV-2 B.1.619 and B.1.620 Lineages, South Korea, 2021. Emerg. Infect. Dis. 2022, 28, 415–419. [CrossRef] [PubMed]
- Park, A.K., et al. Genomic Surveillance of SARS-CoV-2: Distribution of Clades in the Republic of Korea in 2020. Osong Public Health Res. Perspect. 2021, 12, 37–43. [CrossRef] [PubMed]
- Harvey, W.T., et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [CrossRef]
- Wang, W.B., et al. E484K mutation in SARS-CoV-2 RBD enhances binding affinity with hACE2 but reduces interactions with neutralizing antibodies and nanobodies: Binding free energy calculation studies. J. Mol. Graph Model 2021, 109, 108035. [CrossRef] [PubMed]
- Borkotoky, S.; Dey, D.; Hazarika, Z. Interactions of angiotensin-converting enzyme-2 (ACE2) and SARS-CoV-2 spike receptor-binding domain (RBD): A structural perspective. Mol. Biol. Rep. 2023, 50, 2713–2721. [Google Scholar] [CrossRef]
- Zhang, Y., et al. Immune Evasive Effects of SARS-CoV-2 Variants to COVID-19 Emergency Used Vaccines. Front. Immunol. 2021, 12, 771242. [CrossRef]
- Winger, A. and T. Caspari, The Spike of Concern-The Novel Variants of SARS-CoV-2. Viruses 2021, 13. [CrossRef] [PubMed]
- Lee, S., et al., Phylodynamic analysis revealed that human mobility and vaccination were correlated to the local spread of SARS-CoV-2 in Republic of Korea. Emerg. Microbes Infect. 2023, 12, 2228934. [CrossRef]
- Klink, G.V. , et al., The rise and spread of the SARS-CoV-2 AY.122 lineage in Russia. Virus Evol. 2022, 8, veac017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, K.; Zha, S.; Wang, L.; Chen, R. Epidemiological Characteristics of COVID-19 Resurgence in Areas Initially Under Control. Front. Public Health 2021, 9, 749294. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gong, D.; Xiao, J.; Hu, J.; He, G.; Rong, Z.; Ma, W. Cluster infections play important roles in the rapid evolution of COVID-19 transmission: A systematic review. Int. J. Infect. Dis. 2020, 99, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.E.F.; Anderson, D.E.; E Wei, W.; Pang, J.; Ni Chia, W.; Tan, C.W.; Teoh, Y.L.; Rajendram, P.; Toh, M.P.H.S.; Poh, C.; et al. Connecting clusters of COVID-19: An epidemiological and serological investigation. Lancet Infect. Dis. 2020, 20, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Choi, J.H.; Cho, J.H. Estimation of the Effectiveness of a Tighter, Reinforced Quarantine for the Coronavirus Disease 2019 (COVID-19) Outbreak: Analysis of the Third Wave in South Korea. J. Pers. Med. 2023, 13, 402. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Strategy to Achieve Global Covid-19 Vaccination by mid-2022. Available from: https://cdn.who.int/media/docs/default-source/immunization/covid-19/strategy-to-achieve-global-covid-19-vaccination-by-mid-2022.pdf?sfvrsn=5a68433c_5.
- Chavda, V.P. , et al., The Delta and Omicron Variants of SARS-CoV-2: What We Know So Far. Vaccines (Basel), 2022. 10(11).
- Viana, R. , et al., Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature, 2022. 603, 679-686.
- Boudet, A. , et al., Limitation of Screening of Different Variants of SARS-CoV-2 by RT-PCR. Diagnostics (Basel), 2021. 11(7).
- Focosi, D. , et al., Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int J Mol Sci, 2023. 24(3).
- I.H Kim, A.K.P., J. I.H Kim, A.K.P., J.S. No, H.J. Lee, J.A Kim, C.Y. Lee, Y.J. Ahn, J.E. Rhee, E.J Kim, Omicron Subvariants (BQ.1, BQ.1.1, etc.) Outbreak Status. Public Health Weekly Report, 2022. 15, 2917-2924.
- Dijokaite-Guraliuc, A. , et al., Rapid escape of new SARS-CoV-2 Omicron variants from BA.2-directed antibody responses. Cell Rep, 2023. 42, 112271.
- Zhang, Y.; Zhang, H.; Zhang, W. SARS-CoV-2 variants, immune escape, and countermeasures. Front. Med. 2022, 16, 196–207. [Google Scholar] [CrossRef]
- DeGrace, M.M. , et al. , Defining the risk of SARS-CoV-2 variants on immune protection. Nature 2022, 605, 640–652. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of the MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Figure 1.
Phylogenetic tree was created using NextClade online software and visualized using Auspice online tool from metadata produced by NextClade (http://clades.nextstrain.org, accessed on 30 June 2023). 2,399 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) cases detected in Gwangju between February 2020 and December 2022 were uploaded in order to visualize the phylogenetic placement in comparison with published sequences from all over the world. NextStrain clades are broken down according to the indicated color code.
Figure 1.
Phylogenetic tree was created using NextClade online software and visualized using Auspice online tool from metadata produced by NextClade (http://clades.nextstrain.org, accessed on 30 June 2023). 2,399 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) cases detected in Gwangju between February 2020 and December 2022 were uploaded in order to visualize the phylogenetic placement in comparison with published sequences from all over the world. NextStrain clades are broken down according to the indicated color code.
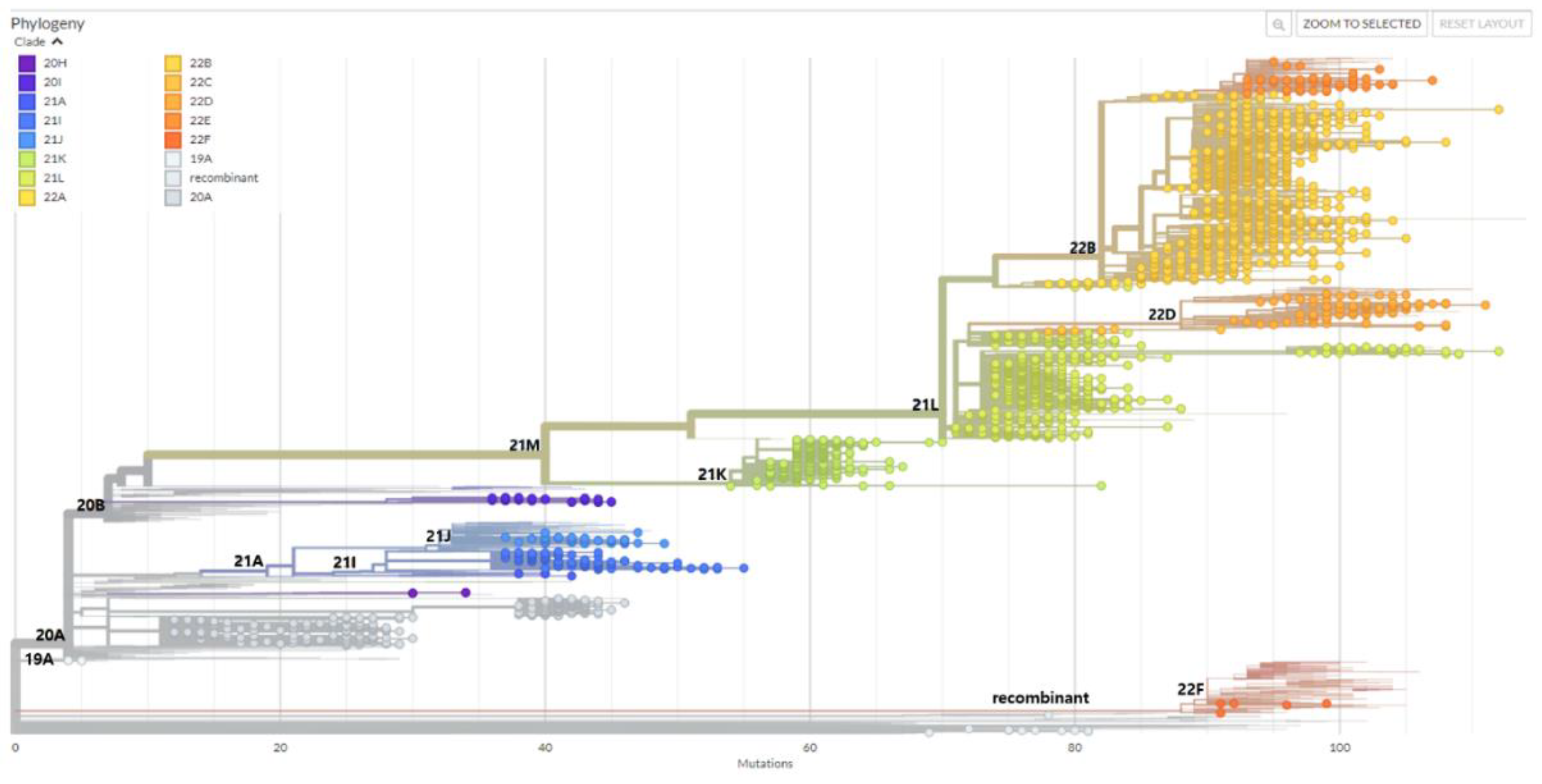
Figure 2.
Investigation of SARS-CoV-2 variants in Gwangju from 2020 to 2022. (a) The number of identified lineages by month of specimen collection date from February, 2020 to December, 2022. A total of 2399 specimen were assigned into 11 Pango lineages and 16 NextStrain clades. (b) Phylogenetic tree showing the relationship between the 596 Gwangju SARS-CoV-2 genome sequences. The list of sequences used to create the tree presented in Supplementary Table S1. The tree was created with the maximum-likelihood method as implemented by MEGA-11(ver.11.0.13) under the general time reversible plus gamma nucleotide substitution model and decorated with iTOL v6. Variants were highlighted in 16 colors. The outer circle colored strip displays the specimen collection date, sorted from first wave to sixth wave. A newick (plain text) version of the phylogenetic tree, with branch lengths and support values at nodes, is reported in Supplementary text files.
Figure 2.
Investigation of SARS-CoV-2 variants in Gwangju from 2020 to 2022. (a) The number of identified lineages by month of specimen collection date from February, 2020 to December, 2022. A total of 2399 specimen were assigned into 11 Pango lineages and 16 NextStrain clades. (b) Phylogenetic tree showing the relationship between the 596 Gwangju SARS-CoV-2 genome sequences. The list of sequences used to create the tree presented in Supplementary Table S1. The tree was created with the maximum-likelihood method as implemented by MEGA-11(ver.11.0.13) under the general time reversible plus gamma nucleotide substitution model and decorated with iTOL v6. Variants were highlighted in 16 colors. The outer circle colored strip displays the specimen collection date, sorted from first wave to sixth wave. A newick (plain text) version of the phylogenetic tree, with branch lengths and support values at nodes, is reported in Supplementary text files.
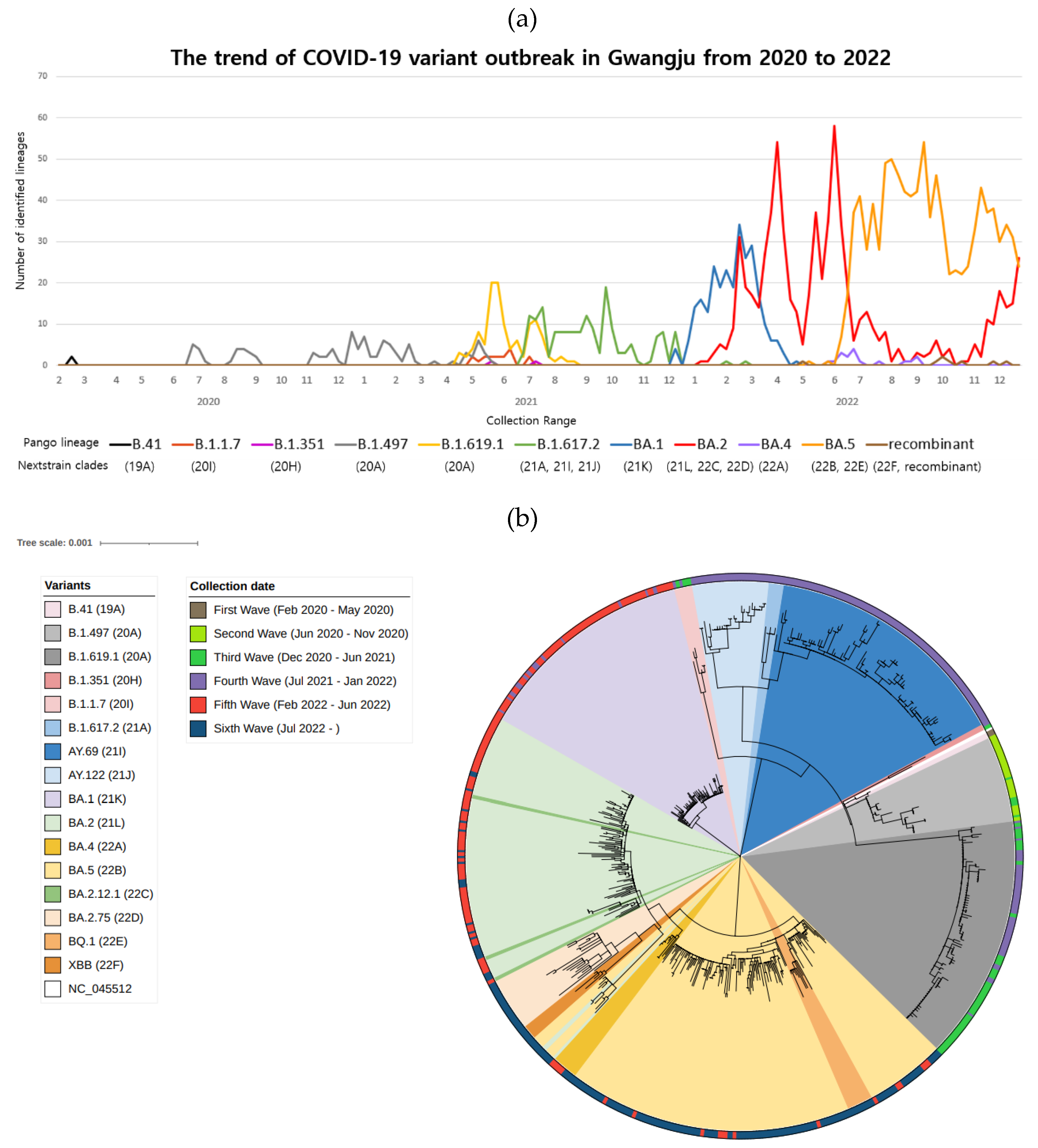
Figure 3.
Analysis of relationship among specific variants identified in this study. (a) Maximum-likelihood phylogenetic tree of B.1.619.1 sequence was generated. The sequences were divided into three groups. Group A was indicated light green triangles, Group B indicated blue squares, and Group C indicated red circles. (b) Phylogenetic tree of B.1.617.2 sequences were created using the maximum-likelihood method and the general time reversible model with gamma distribution and invariant sites parameter as the best-fit model of nucleotide substitution. Each sequence was applied to NextStrain online tool, and divided clades were confirmed. The sequences were divided into three clades; 21A (purple square), 21I (green circle), and 21J (orange triangle).
Figure 3.
Analysis of relationship among specific variants identified in this study. (a) Maximum-likelihood phylogenetic tree of B.1.619.1 sequence was generated. The sequences were divided into three groups. Group A was indicated light green triangles, Group B indicated blue squares, and Group C indicated red circles. (b) Phylogenetic tree of B.1.617.2 sequences were created using the maximum-likelihood method and the general time reversible model with gamma distribution and invariant sites parameter as the best-fit model of nucleotide substitution. Each sequence was applied to NextStrain online tool, and divided clades were confirmed. The sequences were divided into three clades; 21A (purple square), 21I (green circle), and 21J (orange triangle).
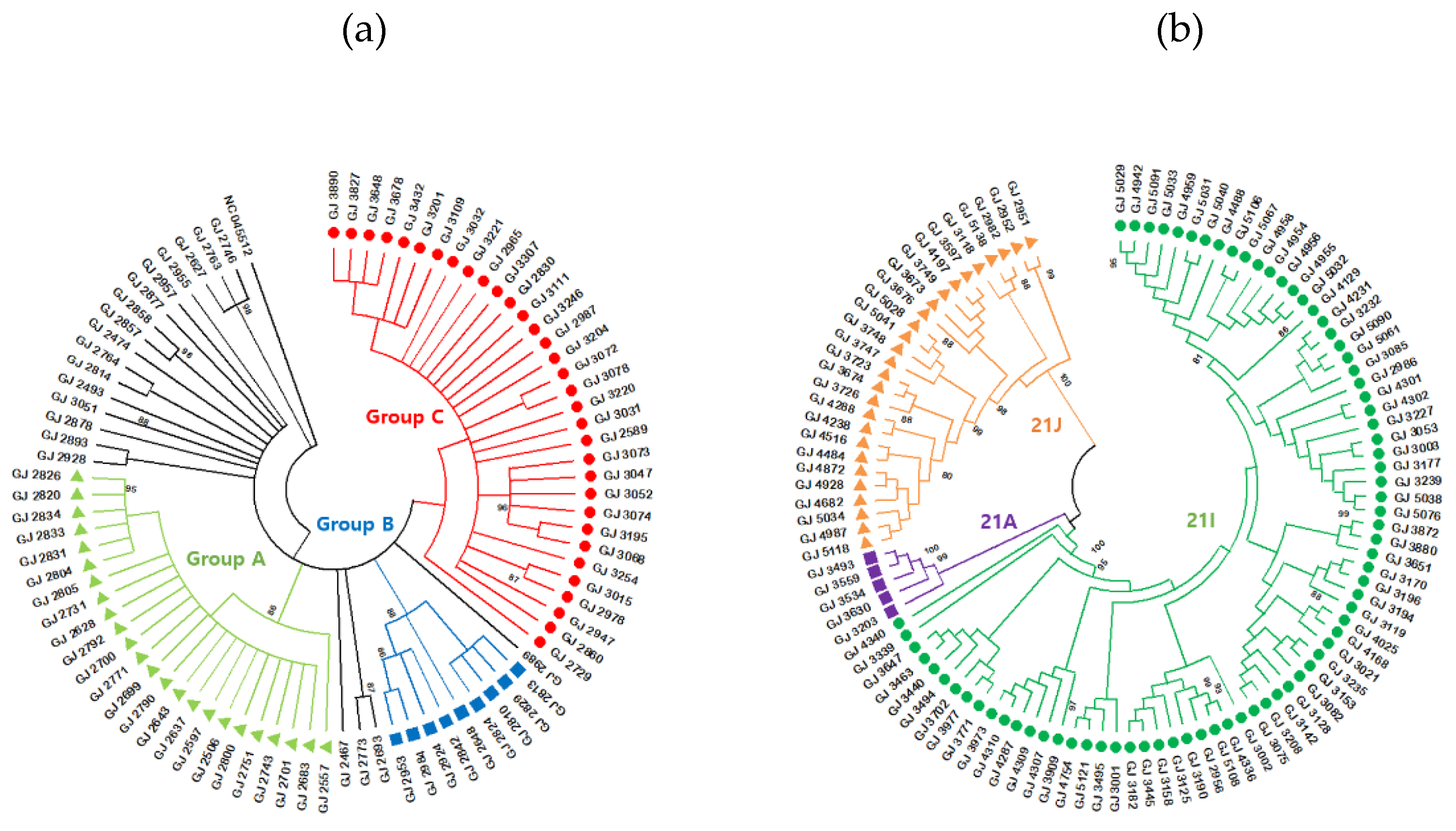
Figure 4.
Distribution of COVID-19 variants by the most prevalent lineages in Gwangju, 2020-2022. (a) Percentage of SARS-CoV-2 lineages during the first to fourth epidemic period. Data are shown for lineages B.1.497, B.1.1.7, B.1.619.1, B.1.620, B.1.617.2, AY.69 and AY.122. (b) Percentage of SARS-CoV-2 omicron sub-lineages during the fifth to sixth epidemic period. Stacked area plot showed the monthly detection rate of various omicron sub-lineages.
Figure 4.
Distribution of COVID-19 variants by the most prevalent lineages in Gwangju, 2020-2022. (a) Percentage of SARS-CoV-2 lineages during the first to fourth epidemic period. Data are shown for lineages B.1.497, B.1.1.7, B.1.619.1, B.1.620, B.1.617.2, AY.69 and AY.122. (b) Percentage of SARS-CoV-2 omicron sub-lineages during the fifth to sixth epidemic period. Stacked area plot showed the monthly detection rate of various omicron sub-lineages.
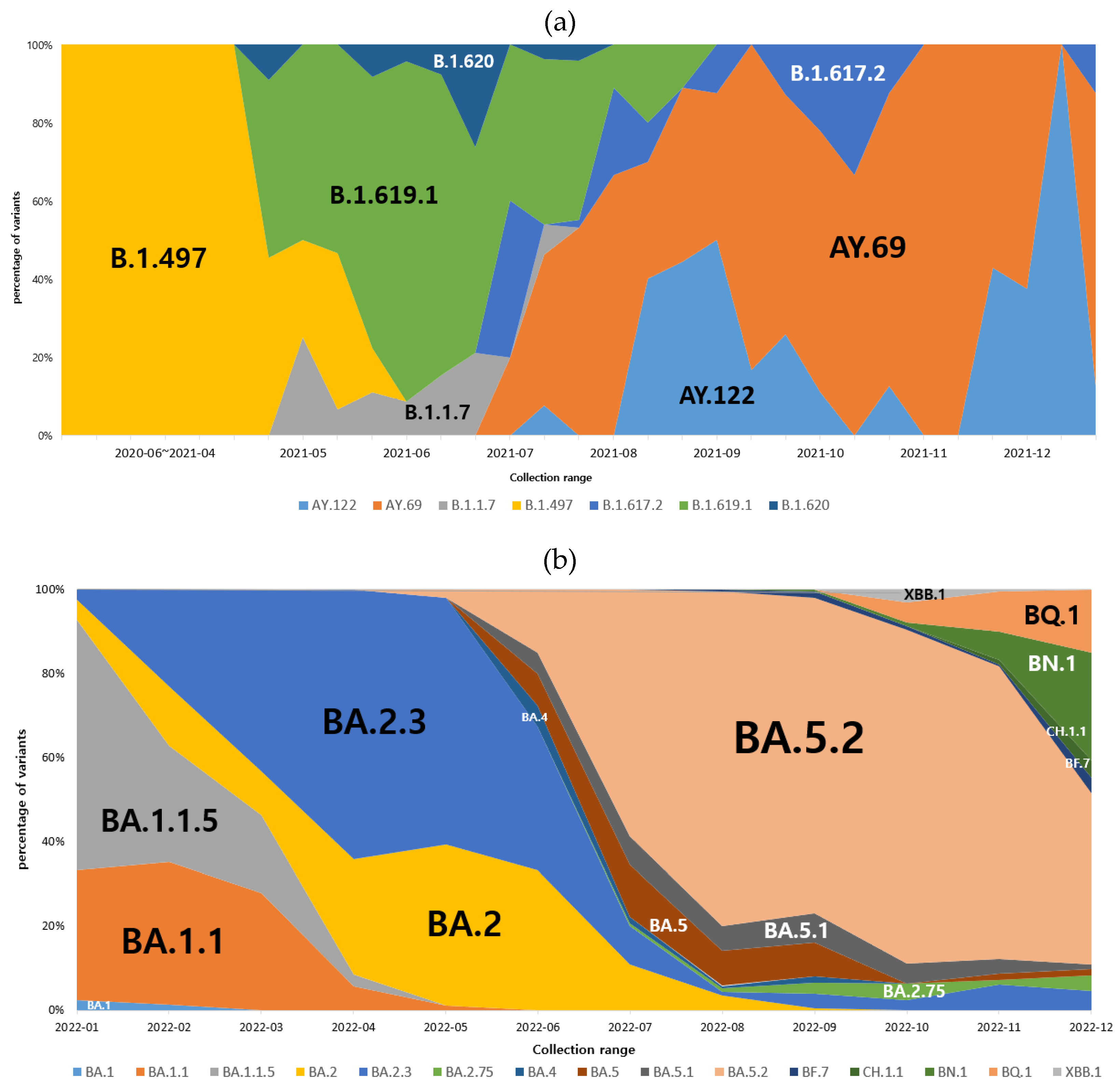

Table 1.
Number of COVID-19 cases and number of sequencing cases for representative surveillance during several waves of COVID-19 in Gwangju from 2020 to 2022.
Table 1.
Number of COVID-19 cases and number of sequencing cases for representative surveillance during several waves of COVID-19 in Gwangju from 2020 to 2022.
| Period | Number of COVID-19 cases |
Number of sequencing cases |
|---|---|---|
| Total | 850,707 | 2399 |
| First Wave (Feb 2020 – May 2020) |
36 | 2 |
| Second Wave (Jun 2020 – Nov 2020) |
725 | 35 |
| Third Wave (Dec 2020 – Jun 2021) |
2285 | 165 |
| Fourth Wave (Jul 2021 – Jan 2022) |
17,123 | 312 |
| Fifth Wave (Feb 2022 – Jun 2022) |
518,055 | 757 |
| Sixth Wave (Jul 2022 - ) |
312,483 | 1128 |
Table 2.
Number of SARS-CoV-2 lineage distribution in Gwangju from 2020 to 2022.
| Pango lineage | Total | Sub-lineages (number of variants) |
|---|---|---|
| B.41 | 2 | B.41 (2) |
| B.1.497 | 97 | B.1.497 (97) |
| B.1.1.7(Alpha) | 17 | B.1.1.7 (17) |
| B.1.351(Beta) | 2 | B.1.351 (2) |
| B.1.619.1 | 119 | B.1.619.1 (119) |
| B.1.617.2(Delta) | 173 | B.1.617.2 (7), AY.20 (1), AY.43 (1), AY.69 (117), AY.75.2 (1), AY.106 (1), AY.122 (42), AY.122.5 (3) |
| BA.1(Omicron) | 270 | BA.1 (2), BA.1.1 (128), BA.1.1.5 (136), BA.1.9 (1), BA.1.15 (1), BA.1.17 (1), BA.1.17.2 (1) |
| BA.2(Omicron) | 697 | BA.2 (114), BA.2.1 (1), BA.2.2 (1), BA.2.3 (302), BA.2.3.1 (1), BA.2.3.2 (5), BA.2.3.7 (1), BA.2.3.8 (4), BA.2.3.10 (1), BA.2.3.12 (19), BA.2.3.13 (4), BA.2.3.14 (12), BA.2.3.20 (10), BA.2.5 (1), BA.2.9 (3), BA.2.10 (13), BA.2.12.1 (12), BA.2.17 (2), BA.2.18 (1), BA.2.38.1 (2), BA.2.40.1 (1), BA.2.51 (1), BA.2.56 (5), BA.2.65 (17), BA.2.68 (39), BA.2.74 (1), BA.2.75 (1), BA.2.75.2 (3), BA.2.75.3 (1), BA.2.75.4 (4), BA.2.75.5 (2), BA.2.76 (1), BG.5 (1), BH.1 (1), BL.1 (7), BM.1.1 (1), BM.1.1.3 (3), BM.4.1.1 (1), BN.1.1 (4), BN.1.2 (15), BN.1.3 (44), BN.1.4 (1), BN.4 (1), BS.1.1 (4), CH.1.1 (10), CM.1 (1), CM.3 (1), CM.4 (13), CM.5 (1), CM.6 (1), CM.8.1 (2) |
| BA.4(Omicron) | 17 | BA.4 (1), BA.4.1 (7), BA.4.1.1 (6), BA.4.1.8 (1), BA.4.4 (1), BA.4.6 (1) |
| BA.5(Omicron) | 997 | BA.5 (2), BA.5.1 (39), BA.5.1.1 (1), BA.5.1.3 (3), BA.5.1.5 (1), BA.5.1.10 (7), BA.5.1.22 (2), BA.5.1.23 (4), BA.5.1.28 (7), BA.5.2 (388), BA.5.2.1 (225), BA.5.2.2 (3), BA.5.2.3 (2), BA.5.2.6 (10), BA.5.2.9 (5), BA.5.2.12 (1), BA.5.2.16 (2), BA.5.2.19 (23), BA.5.2.20 (14), BA.5.2.22 (12), BA.5.2.25 (1), BA.5.2.26 (3), BA.5.2.27 (9), BA.5.2.28 (3), BA.5.2.32 (4), BA.5.2.34 (3), BA.5.2.44 (2), BA.5.5 (34), BA.5.6 (7), BA.5.6.1 (1), BA.5.8 (1), BA.5.9 (3), BA.5.10 (7), BA.5.10.1 (2), BE.1 (6), BE.1.1 (10), BE.1.1.2 (1), BE.1.4 (1), BE.4 (3), BE.4.1.1 (1), BF.3 (1), BF.5 (43), BF.7 (10), BF.7.4.2 (2), BF.7.5 (1), BF.10 (7), BF.11 (2), BF.21 (11), BF.24 (1), BF.26 (1), BF.27 (1), BF.28 (7), BK.1 (1), BQ.1 (1), BQ.1.1 (19), BQ.1.1.1 (1), BQ.1.1.4 (1), BQ.1.1.17 (7), BQ.1.1.18 (2), BQ.1.2 (9), BQ.1.3 (4), BQ.1.5 (5), BQ.1.11 (4), BQ.1.15 (1), BQ.1.23 (1), CK.3 (1) |
| recombinant | 8 | XAH (1), XAZ (1), XBB (1), XBB.1 (4), XBC.1 (1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



