
Submitted:
18 October 2023
Posted:
20 October 2023
You are already at the latest version
Abstract
Abstract: The transforming growth factor beta (TGFβ) and Hippo signaling pathways are evolutionarily conserved pathways that play a critical role in cardiac fibroblasts during embryonic development, tissue repair, and fibrosis. TGFβ and Hippo signaling is also important for cardiac cushion remodeling and septation during embryonic development. Loss of TGFβ2 in mice causes cardiac cushion remodeling defects resulting in congenital heart disease. In this study, we used in vitro molecular and pharmacologic approaches in the cushion mesenchymal cell line (tsA58-AVM) and investigated if Hippo pathway acts as a mediator of TGFβ2 signaling. Immunofluorescence staining showed that TGFβ2 induced nuclear translocation of activated SMAD3 in the cushion mesenchymal cells. In addition, the results indicated increased nuclear localization of Yes associated protein 1 (YAP1) following a similar treatment of TGFβ2. In collagen lattice formation assays, TGFβ2 treatment of cushion cells resulted in an enhanced collagen contraction compared to the untreated cushion cells. Interestingly, verteporfin, a YAP1 inhibitor, significantly blocked the ability of cushion cells to contract collagen gel in absence or presence of by exogenously added TGFβ2. To confirm the molecular mechanisms of verteporfin induced inhibition of TGFβ2-dependent extra-cellular matrix (ECM) reorganization we performed gene expression analysis of key mesenchymal genes involved in ECM remodeling in heart development and disease. Our results confirmed that verteporfin significantly decreased the expression of α-smooth muscle actin (Acta2), collagen 1a1 (Col1a1), Ccn1 (i.e., Cyr61), and Ccn2 (i.e., Ctgf). Western blot analysis indicated that verteporfin treatment significantly blocked TGFβ2-induced activation of SMAD2/3 in cushion mesenchymal cells. Collectively, these results indicate that TGFβ2 regulation of cushion mesenchymal cell be-havior and ECM remodeling is mediated by YAP1. Thus, TGFβ2 and Hippo pathway integration represents an important step in understanding the etiology of congenital heart disease.
Keywords:
Transforming growth factor beta
; YAP
; SMAD3
; Heart Valve
; Extracellular matrix
1. Introduction
Defective heart valve formation is a major cause of congenital heart defects (CHD) (Gittenberger-de Groot et al., 2005). Bicuspid aortic valve (BAV) is found in up to 2% of the general population (Bosse et al., 2013; Grewal et al., 2014; Poelmann et al., 2021). Atrioventricular (AV) septal defects (AVSD) comprise 5-10% of all CHD and is defined as a variable degree of the atrial and ventricular septal defect along with a common or partially separate atrioventricular (AV) orifice (Burns et al., 2016; Calkoen et al., 2016). Cardiac outflow tract (OFT) defects and abnormalities are estimated to cause approximately 30% of all CHD (Neeb et al., 2013). Normal heart development requires that cells integrate signals from various signaling pathways, such as the Hippo and transforming growth factor-beta (TGFβ), which are major regulators of tissue growth, apoptosis, organ size, and differentiation (Attisano and Wrana, 2013; Piersma et al., 2015; Tsai and Martin, 2022). Cell-cell contact inhibition and growth can be regulated by TGFβ but in a Hippo independent manner (Nallet-Staub et al., 2015). These pathways are intricately involved in cardiac fibroblasts during heart development and postnatal cardiac tissue repair and cardiac fibrosis (Tsai and Martin, 2022). There are three TGFβ ligands in mammals (Frangogiannis, 2017; Hanna et al., 2021). Genetic mutations in TGFB2 are found in many syndromic patients of connective tissue disorder with CHD caused by abnormal AV and OFT cushion remodeling resulting in aortic and/or mitral valve malformations (Boileau et al., 2012; Lindsay et al., 2012). All TGFβ ligands bind to heteromeric TGFβ receptor complex leading to phosphorylation of canonical SMAD2 or SMAD3. Phosphorylated SMAD2/3 function as activated SMADs and in conjunction with SMAD4 they translocate to the nucleus. The three multifunctional TGFβ cytokines, which upon interaction with its receptors induces both “canonical,” i.e. SMAD2/3-dependent, and noncanonical, i.e. non-SMAD (p38 and ERK1/2 MAPK-mediated) signaling, is the main inducer of EMT and fibrogenesis (Heldin and Moustakas, 2016). Activated SMAD2/3/4 complex binds to TGFβ response elements in the promoter of target genes and regulated transcription. TGFβ ligands are profibrotic molecules which regulate key genes involved in extracellular matrix (ECM) remodeling and (myo)fibroblast activation, including α-SM actin (Acta2), Collagen 1a1 (Col1a1), and Cellular Communication Network Factor 2 (Ccn2) (also known as CTGF) (Hanna et al., 2021; Sun et al., 2021). The transcriptional regulatory mechanisms by which specific TGFβ ligands regulate ECM remodeling in cardiac cushion remodeling has not been fully resolved.
The Hippo signaling pathway modulates cardiac fibroblast activation and regenerative response by regulating cardiomyocyte and fibroblast proliferation and ECM remodeling in cardiac development and cardiomyopathy (Mia and Singh, 2019, 2022; Tsai and Martin, 2022). The Hippo signaling mechanisms were first investigated in Drosophila. The Hippo signaling pathway is crucial for the development of several organs including heart by controlling cell proliferation, apoptosis, cell fate decisions, and stem cell self-renewal (Ma et al., 2019). The mammalian core components of the Hippo signaling pathway are composed of a kinase cascade, several transcription factors, and coactivators, like Mst1/2, Sav1, Lats1/2, Mob1 and Yap/Taz (Ma et al., 2019) (Misra and Irvine, 2018). Activation of the protein cascade leads to phosphorylation, cytoplasmic retention, and degradation of the transcriptional coactivators, Yes-associated protein 1 (YAP1) and transcriptional coactivator with PDZ-binding motif (TAZ) (Ma et al., 2019). In contrast, inactivation of upstream kinases resulted nuclear translocation of both YAP1 and TAZ, where they further interact with various transcription factors including SMADs family member (Attisano and Wrana, 2013). This results in regulation of transcriptional activity for downstream target genes involved in cell proliferation, survival, differentiation, and ECM remodeling (Ma et al., 2019; Mia and Singh, 2022; Misra and Irvine, 2018; Tsai and Martin, 2022). Cellular Communication Network Factor 1 (CCN1/CYR61) and CCN2 (also known as connective tissue growth factor, CTGF) are among major targets of Hippo pathway involved ECM remodeling, cell proliferation, and cell survival (Ma et al., 2019). There are many studies indicating that TGFβ induced SMAD2/3 localization is mediated by YAP and TAZ (Attisano and Wrana, 2013). The activity of YAP and TAZ is determined by their localization within the cell, with nuclear protein being ‘active’ and cytosolic protein being ‘inactive’. Furthermore, treating cells and mice with verteporfin, a well-characterized YAP1 inhibitor (Liu-Chittenden et al., 2012), blocks TGFβ induced myofibroblast activation.
ECM remodeling represents a key step in cardiac cushion remodeling and maturation and cardiac septation (Lockhart et al., 2011). Given the role of TGFβ and Hippo pathways in fibrogenesis and ECM remodeling, it is plausible that these pathways integrate and control ECM remodeling in heart development. It is thought that there are retention factors in the nucleus that have a higher affinity for phosphorylated SMADs (Labibi et al., 2020). However, the regulation of SMAD nuclear accumulation which governs transcription of target genes is still not very well understood (Gori et al., 2021). Thus, to understand signaling cross talk between TGFβ SMAD and YAP1 signaling pathways in cardiac cushion remodeling in heart development, we have investigated the role of YAP1 in SMAD nuclear accumulation in immortalized embryonic cushion mesenchymal cells. We used cushion mesenchymal cells culture and molecular and biochemical approaches to determine if Hippo signaling via YAP1 is essential for TGFβ2-dependent cushion mesenchymal cell-matrix remodeling. Collectively, our results confirmed that YAP1 dependent pathway regulates TGFβ–induced transcriptional regulatory mechanisms involved in ECM remodeling in cardiac cushion mesenchyme.
2. Material and Methods
2.1. Cushion Mesenchymal Cell Culture and Drug Treatment
The tsA58-AVM cell line was derived from AV cushions of H-2Kb-tsA58 embryos at E9.5 (Peng et al., 2016). In H-2Kb-tsA58 animals, expression of the temperature sensitive SV40 large T antigen mutant gene (tsA58) is driven by the γ-interferon inducible promoter of the H-2Kb gene. Under the permissive condition (33°C, with γ-interferon), cushion mesenchymal cells proliferate continuously like immortalized cells. However, under the restrictive condition (37°C, no γ-interferon), the large T antigen is degraded, and these cells cease to proliferate and resemble primary cell cultures. Thus, cushion mesenchymal cells were grown at 33°C in DMEM media containing 10% FBS, 1% penicillin-streptomycin, Glutamine (1 mM) and γ-interferon (10 units/ml; Peprotechn INC.; catalog: 315-05). Once cells were ~80-90% confluent, they were seeded in 100 mm dishes or well plates for drug treatments at 37°C without γ-interferon. At the end of the treatments, cells were washed thrice in 1xPBS and collected for different experiments.
2.2. Immunofluorescence Staining and Immunoblotting
Cushion mesenchymal cells were cultured on the 2 well BD chamber slides as discussed above. When 80-90% confluent, cells were starved with 0.5% FBS/DMEM media for 24 hr and then treated with TGFβ2 (5 ng/ml, R&D Systems, Inc. Minneapolis, MN) for 12 hr. A set of untreated control was also kept comparing with the TGFβ2 treated cells. Cells were then washed thrice with PBS (1X) and stained with antibodies directed against SMAD3 or YAP1 both at a dilution of 1:500. Primary IgG antibodies of SMAD3 and YAP1 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) and abcam (Cambridge, MA, USA), respectively. The Cy3 goat anti-rabbit secondary IgG antibody (ThermoFisher Scientific, Grand Island, NY, USA) was used at 1µg/ml dilutions to detect the primary IgG antibody. After the addition of secondary antibodies, nuclear counterstaining with DAPI was performed. Immunofluorescence microscopic images were obtained using Leica Thunder imager (Leica Microsystems Inc., Buffalo Grove, IL, United States), and the relative intensities of SMAD3 or YAP1 immunofluorescence was calculated using ImageJ software (National Institutes of Health, Bethesda, MD). We measured 7-8 fields of different images using ImageJ software. The data were analyzed and graphically presented using GraphPad Prism 9.0 software (San Diego, CA). All experiments were done at least three times. The results are presented as bar graph and dots representing individual data points.

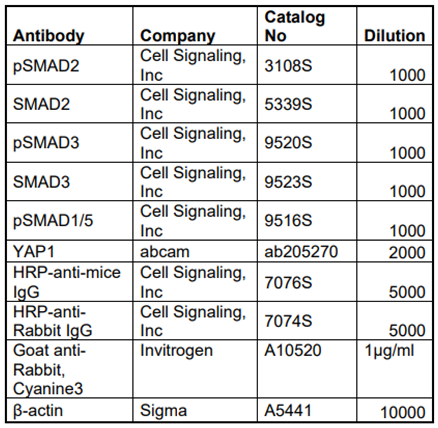
Table 1.
List of antibodies used to determine expression levels of different proteins.
2.3. Collagen Gel Contraction Assays
Cushion mesenchymal cells were maintained in DMEM (Invitrogen) supplemented with 10% bovine serum, 5% fetal calf serum and 1% penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA) at 33 °C/5% CO2 as discussed above. In addition to reorganization, collagen contraction assays are indicative of the capacity for embedded cells to generate mechanical loads. The capability of cushion cells to form lattices in collagen gels was assessed by plating 105 cells in 2 mg/mL collagen type-I (in 18mM acetic acid) prepared in complete media and supplemented with 0.1 M NaOH, as detailed (Carver et al., 2021; Haskett et al., 2012). Free-floating collagen gels were incubated at 37 °C for 5 days, with or without recombinant TGFβ2 (5 ng/ml, R&D Systems, Inc., Minneapolis, MN) and Verteporfin (5 µg/ml, Tocris/Bio-Techne, Minneapolis, MN). Images were acquired and best-fit shapes were used to calculate the diameter of the gels using ImageJ software. The relative change in gel diameter after 5 days denotes the degree of collagen contraction with lower values indicate greater contraction (Carver et al., 2021; Fix et al., 2019). The results are presented as bar graph and dots representing individual data points.
2.4. RNA isolation, cDNA synthesis, and quantitative PCR
mRNA was isolated using Trizol (Invitrogen, Waltham MA) and miRNeasy micro kit (Qiagen, Germantown, MD) according to manufacturer’s protocols and cDNA was generated from 1 µg mRNA using Invitrogen kit according to manufacturer's instructions. 20 ng cDNA was subjected to quantitative PCR amplification (Biorad-CFX) using pre-validated gene specific primers procured from the vendor (Biorad Inc, Hercules, CA). Following PCR analyses, the cycle count threshold (Ct) was normalized to species specific housekeeping genes, β2-microglobulin (B2M; purchased from Biorad Inc.) and the ∆Ct and fold changes in experimental samples over controls were determined. Statistically significant differences in gene expression levels were determined using unpaired one-tailed Student's t-test and indicated in the figures, on at least 3 independent experiments with p < 0.05 considered significant.
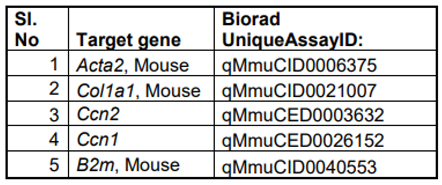
Table 2.
List of qPCR primers used to determine expression levels of different genes.
2.5. Western Blot Analysis
Western blotting was performed with the protein extracted from the cell lysate of cushion mesenchymal cells following different treatments (Carver et al., 2021; Fix et al., 2019). The cells homogenized using Wheaton tapered tissue grinders (Thermo Scientific, Rockford, IL) in M-PER mammalian protein extraction reagent (Thermo Scientific, Rockford, IL) with complete mini protease inhibitor cocktail (Sigma-Aldrich, states. Louis, MO, USA) and Halt protease and phosphatase inhibitor single-use cocktail (ThermoScientific, Rockford, IL, USA) as per the manufacturer’s protocol. Homogenized tissue lysates were subjected to brief sonication for 20 sec on ice and kept at room temperature for 20 min. Then, centrifugation was performed at 15,000 rpm for 20 min at 4 °C and the supernatants were collected. Total protein concentration in the supernatant was determined using Pierce BCA protein assay kit (ThermoScientific, Rockford, IL, USA) and the samples were stored at −80 °C until further use. Western blotting was performed with equal amounts of protein samples and the primary IgG antibodies against phospho-SMAD2 (Cell Signaling Technology, Danvers, MA, USA; cat #3108), SMAD2 (Cell Signaling, Inc; cat #5339), phospho-SMAD3 (Cell Signaling Inc, cat #9520), SMAD3 (Cell Signaling Inc, cat #9523), YAP1 (abcam, Cambridge, MA, USA; cat #ab205270) at a dilution of 1:1000. Primary IgG antibodies against all these proteins were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) or Abcam (Cambridge, MA, USA). The horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary IgG antibody (Cell Signaling, cat#7074) was used at 1:5000 dilutions to detect a primary IgG antibody. In a separate Western blot, the levels of β-actin in all cell lysate samples were determined. Western blots were incubated with Clarity Western ECL detection reagents (Bio-Rad Laboratories, USA) and exposed to X-OMAT AR films (Eastman Kodak, Rochester, NY, USA) for autoradiography. The autoradiograms were scanned on an EPSON scanner using Photoshop software (Adobe Systems, Seattle, WA, USA). β-actin, clone AC-15 monoclonal primary antibody (Sigma-Aldrich, St. Louis, MO, USA) was used as an internal housekeeping control to compare equal loading in the SDS-PAGE. The ratio of both phosphorylated protein/non-phosphorylated protein or β-actin were plotted as scatter dot-plots with the box denoting the mean ± SEM and dots representing individual data points.
2.6. Statistical Analysis
Microsoft Excel was used for recording and managing the raw data. Data were presented as mean ± SEM. Continuous data were presented as bar/dot plots, showing the individual data points together with the average/error bars. Statistical significance was calculated using Student’s t test (one-tailed, independent two-sample t-test) using the GraphPad Prism 8 statistical program (GraphPad, San Diego, CA, USA). Exact p-values were calculated, and the probability values <0.05 were considered as significant. The p-values were indicated on all figures.
3. Results
3.1. Exogenous TGFβ2 induced SMAD3 activation in cushion mesenchymal cells
In the absence of TGFβ2, SMAD2/3 which is not phosphorylated remains mostly in the cytoplasm. TGFβ stimulation triggers TGFβ receptor dependent SMAD2/3 phosphorylation at the C-terminal end which activates SMAD3. Activated forms of both SMAD2/3 then translocate to the nucleus and induce context dependent expression of TGFβ target genes to regulate cellular behavior and ECM remodeling. To determine the effect of TGFβ2 on SMAD3 activation the cushion mesenchymal cells were treated with 1x PBS (control) or TGFβ2 for 12 hr. Equal number of cushion mesenchymal cells were used in all independent experiments. Cushion cells were collected for and immunofluorescence staining using anti-SMAD3 antibody were performed. Nuclei were stained. The levels of SMAD3 in the nucleus for the control and TGFβ2-treated cells were quantified. In control samples, SMAD3 predominantly remained in the cytoplasm (Figure 1A-B, arrowheads in B). The data indicated that there was an increased accumulation of SMAD3 in the nucleus (i.e., activated, or phosphorylated form of SMAD3 or pSMAD3) of cushion mesenchymal cells following TGFβ2 treatment (Figure 1C-D, arrows). Quantitative analysis confirmed that TGFβ2 treatment significantly upregulated the nuclear SMAD3 (i.e., pSMAD2) in cushion mesenchymal cells compared to control cells (Control, n = 8; TGFβ2-treated, n = 5; p=0.0006) (Figure 1E).
3.2. TGFβ2 treatment leads to nuclear translocation of YAP1 in cushion mesenchymal cells
Cushion mesenchymal cells were cultured and treated with TGFβ2 for 12 hr and immunofluorescence analysis was done using YAP1 antibody to determine the levels of cytoplasmic and nuclear YAP1. Cytoplasmic YAP1 is predominantly phosphorylated form of YAP1, whereas nuclear YAP1 is de-phosphorylated. The fluorescence signal was quantified and the ratio of cytoplasmic to nuclear YAP1 levels were compared in both control and TGFβ2-treated cushion cells. Equal numbers of cushion cells were used in the independent experiments. The data showed higher expression of YAP1 in the cytoplasm of control cushion cells compared to TGFβ2 treated cushion cells (Figure 2A-B, arrowheads in B). Following TGFβ2 treatment, the levels of cytoplasmic YAP1 were decreased, but most of the YAP1 was translocated into the nucleus (Figure 2C-D, arrows). Quantitative analysis comparing the ratio of cytoplasmic to nuclear YAP1 in control and TGFβ2-treated cushion mesenchymal cells indicated that the overall ratio of cytoplasmic to nuclear YAP1 was significantly decreased in TGFβ2-treated cultures compared to control cushion cells (Control, n = 7; TGFβ2-treated, n = 7; p = 0.0004) (Figure 2E).
3.3. Suppression of YAP1 inhibits TGFβ2-induced cell-matrix remodeling in cushion mesenchymal cells
The cushion mesenchymal cells were cultured in a collagen gel with or without exogenous TGFβ2-stimulation in the presence or absence of verteporfin, a well-characterized inhibitor of YAP1. Cushion cells were grown for 5 days and TGFβ2 and/or verteporfin were replenished every day. Equal number of cushion mesenchymal cells were used in all independent experiments (n = 3). At the end of the experiment, the % area of the collagen gels was measured for all four treatment groups (control, TGFβ2 treatment, verteporfin treatment, TGFβ2 and verteporfin treatment). The data indicated that TGFβ2 treatment significantly reduced collagen gel % area compared to control (n = 3, p=0.0001) (Figure 3A-B, E). Verteporfin significantly suppressed contraction of the collagen gel in cushion mesenchymal cells both in the absence (n = 3, p=0.0001) or presence of TGFβ2 (n = 3, p=0.0001) (Figure 3A-E).
3.4. YAP1 inhibitor attenuated TGFβ2-induced expression of genes involved in cushion mesenchymal cell-matrix remodeling.
Given the effect of verteporfin in mediating the TGFβ2-dependent collagen gel organization and contraction, qPCR was used to quantify the alteration in expression of TGFβ2-induced mesenchymal genes following YAP1 inhibitor (verteporfin) treatment. The cushion mesenchymal cells were treated with TGFβ2 and/or verteporfin for 12 hr, and total cellular mRNA were extracted. Quantitative RT-PCR was performed to determine the expression of α-smooth muscle actin (Acta2), collagen I (Col1a1), connective tissue growth factor/cell communication network factor 2 (Ctgf or Ccn2), and cysteine-rich angiogenic inducer 61/cell communication network factor 1(Cyr61 or Ccn1). Equal numbers of cells were used for various treatment and for RNA extraction in all independent experiments (n = 3). The data indicated that TGFβ2 significantly induced expression of Acta2 (n = 3, p = 0.02) (Figure 4A), Col1a1 (n = 3, p = 0.04) (Figure 4B), Ccn2 (n = 3, p = 0.018) (Figure 4C), and Ccn1 (n = 3, p = 0.003) (Figure 4D). Verteporfin treatment significantly inhibited expression of Acta2, Col1a1, and Ccn1. However, effect of verteporfin on Ccn2 was not significant (Figure 4A-D). Importantly, verteporfin significantly reduced the TGFβ2-induced upregulation of Acta2 (n = 3, p = 0.003) (Figure 4A), Col1a1 (n = 3, p = 0.003) (Figure 4B), Ccn2 (n = 3, p = 0.002) (Figure 4C), and Ccn1 (n = 3, p = 0.002) (Figure 4D). Collectively, the results showed that verteporfin effectively decreased the expression of key TGFβ2-induced genes involved in cushion mesenchymal cell-matrix remodeling.
Gene expression was normalized by B2M and the numerical data were presented as a scatter plot with bar showing individual values. The error bars indicate mean±SD. Statistically significant differences of gene expression between two individual groups (one-tailed Student’s t test with unpaired; n = 3 each group) were determined and p-values were indicated on the scatter plot.
3.5. YAP1 inhibition decreases activation of SMAD2 and SMAD3
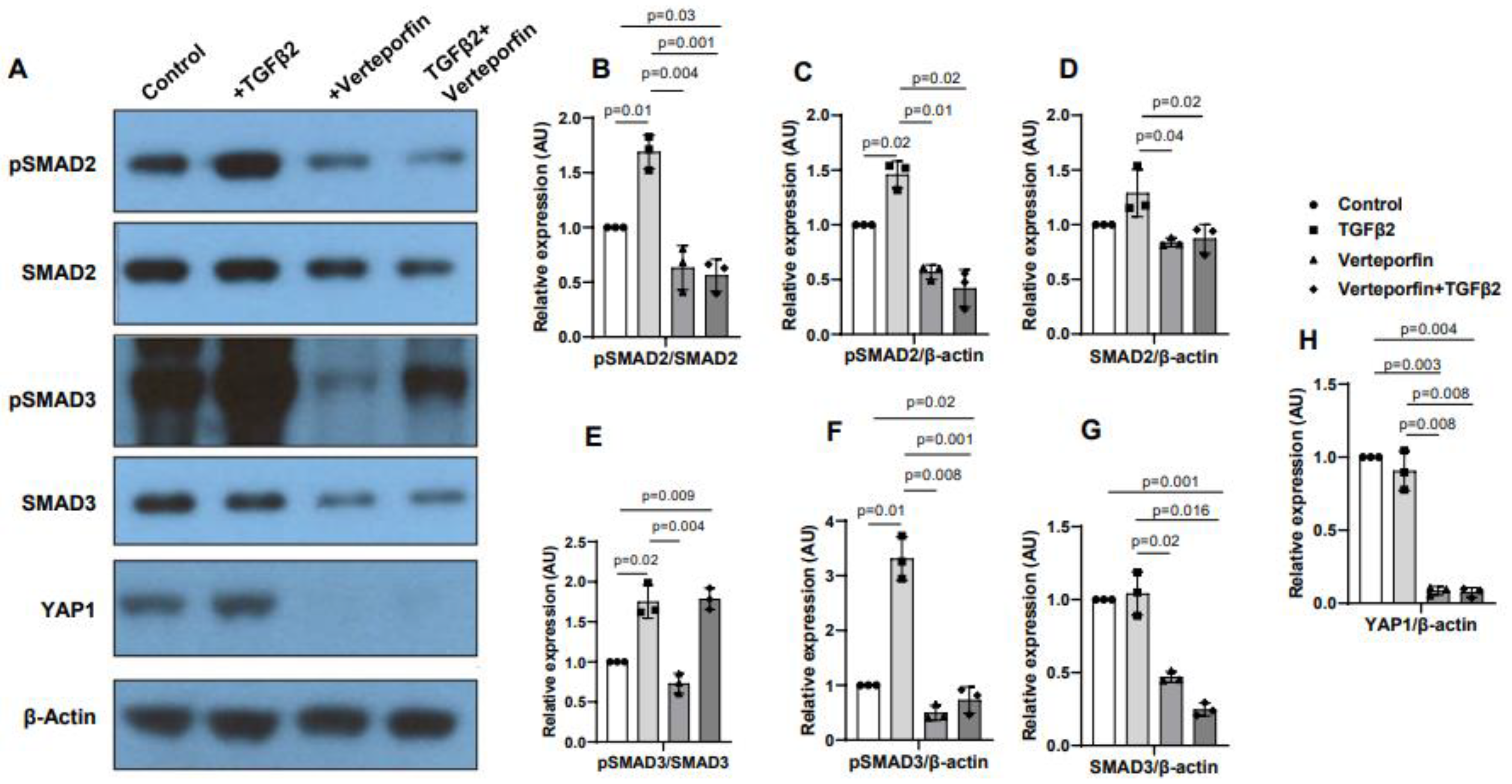
Cushion mesenchymal cells were treated with TGFβ2 in the absence and/or presence of verteporfin for 12 hr. Western blot analyses were performed to quantify the changes in both phosphorylated (pSMAD2, pSMAD3) and total SMAD2 and SMAD3 proteins. Total SMAD2 or SMAD3 proteins were used to normalize the expression of pSMAD2 or pSMAD3. YAP1 antibodies were used to measure the effect of verteporfin on Hippo pathway inhibition. Housekeeping β-actin protein was used to normalize the protein levels of total SMAD2, SMAD3, and YAP1. Equal number of cushion mesenchymal cells were used in all experiments (n = 3). Densitometric quantification of proteins from western blots was done. The data indicated higher levels of pSMAD2 (n = 3, p = 0.01) (Figure 5A-C) and pSMAD3 (n = 3, p = 0.02) (Figure 5A, E-F) following TGFβ2 treatment in cushion mesenchymal cells. The total SMAD2 or SMAD3 levels were not affected by TGFβ2 (Figure 5 D, G). Verteporfin decreased the levels of YAP1 in the absence of TGFβ2 (n=3, p=0.003) or presence of TGFβ2 (n=3, p=0.004) compared to control (Figure 5A, H). Densitometric data confirmed that verteporfin significantly blocked the phosphorylation of pSMAD2 (Figure 5. A-C) and pSMAD3 (Figure 5A, E-F). Finally, there was no significant effect of TGFβ2 and/or verteporfin on β-actin (Figure 5A). However, verteporfin treatment resulted in significant downregulation of SMAD2 or SMAD3 in absence or presence of TGFβ2 compared to the control or TGFβ2-treated cushion mesenchymal cells (Figure 5A, D, G). Collectively, the results indicate that verteporfin blocked YAP1 and both phosphorylated and unphosphorylated forms of SMAD2 and SMAD3.
4. Discussion
Although TGFβ and Hippo pathways are involved in heart development and heart disease, the transcriptional regulatory mechanisms and how these pathways interact and regulate ECM remodeling remains to be fully understood. Several earlier studies reported that YAP1 and its transcriptional coactivator TAZ promote TGFβ signaling via retaining activated SMAD2/3 in the nucleus (Attisano and Wrana, 2013; Labibi et al., 2020). YAP1 remains inactive in cytoplasm by activated Hippo pathway. In the absence of Hippo activation, the YAP1 stabilizes and subsequently translocate to the nucleus, where YAP1 acts as a transcriptional cofactor and induces expression of several genes involved differentiation and ECM remodeling during organ development (Ma et al., 2019). This study is the first to provide evidence that YAP1 acts as a mediator of TGFβ2 function in cushion mesenchymal cells. In this study, cushion mesenchymal cells (tsA58-AVM cell line) are treated with TGFβ2 and/or verteporfin to determine if YAP1 inhibition interferes with SMAD3 activation and nuclear translocation. The cushion mesenchymal cell line used in this study is a very well-characterized and unique cell line to perform in vitro investigation to study cushion mesenchymal cell proliferation, apoptosis, ECM remodeling; and cardiac cushion remodeling, differentiation, and maturation (Peng et al., 2016). We have chosen TGFβ2 for in vitro investigation in this study because loss of TGFβ2 leads to major cardiovascular defects, including cardiac cushion remodeling defects (Bartram et al., 2001; Bhattacharya et al., 2021; Ishtiaq Ahmed et al., 2014). Zhang et al (Zhang et al., 2014) studied the role of YAP1 in ‘endothelial cells’ in TGFβ1-induced endothelial to mesenchymal transition (EMT). Several studies have suggested that Tgfb2 knockout but not Tgfb1 knockout mice develop congenital heart defects (Azhar et al., 2009a; Azhar et al., 2009b). Our study focuses on the role of TGFβ2 in embryonic cushion mesenchymal cells and not cushion endothelial cells and investigates the function of YAP1 in TGFβ2-induced cushion mesenchymal cell remodeling and ECM expression. Thus, the findings presented in this study are novel and not confirmatory of previously reported developmental analysis in mouse embryos (Zhang et al., 2014).
Firstly, the immunofluorescence analysis indicates that TGFβ2 induces a robust nuclear accumulation of SMAD3 as well as YAP1 in cushion mesenchymal cells. Biochemical analysis confirmed that YAP1 inhibition effectively blocked the activation of SMAD2 and SMAD3 in cushion mesenchymal cells. The data also indicate that YAP1 inhibition results in decreased levels of both total SMAD2 and SMAD3 proteins suggesting that Hippo signaling acts as a negative mediator of canonical TGFβ-SMAD signaling in cushion mesenchymal cells. Together, the data suggest a role of Hippo pathway in controlling TGFβ-SMAD signaling by regulating the transcription of Smad2 or Smad3 and/or post-translational (phosphorylation) changes and nuclear translocation of pSMAD2 and pSMAD3. Verteporfin has been shown to induce a dramatic reduction in YAP and TAZ that is associated not only with reduced SMAD2/3 nuclear accumulation, but also diminished SMAD2/3 levels after TGFβ stimulation in Immortalized normal rat kidney interstitial fibroblasts (NRK49F)(Szeto et al., 2016). It is also known that YAP is a Smad7 partner that facilitates the recruitment of cytoplasmic YAP to activated TGFβR1 and enhances the inhibitory activity of Smad7 against TGFβ signaling in HaCaT keratinocytes and COS-7 cells (Ferrigno et al., 2002). Collectively, our results suggest that YAP1 can induce the expression and physically interact with activated SMAD2/3 within the cytoplasm and together they migrate to the nucleus in cushion mesenchymal cells. While in the nucleus, YAP1 independently or in conjunction with SMAD2/3 and in the presence of other transcriptional coactivators could regulate transcription of key target genes in cushion cells. One of the limitations of the current study is that this study has not addressed if there is any interaction between noncanonical TGFβ signaling (e.g., p38 MAPK, ERK1/2 MAPK) and Hippo pathway. Since verteporfin significantly inhibits total SMAD3 levels, we were not able to use the pSMAD3/SMAD3 to monitor changes in pSMAD3. Instead, we used pSMAD3/β-actin and found that TGFβ2-induced pSMAD3 is significantly decreased in the presence of verteporfin. It is possible that verteporfin through transcriptional and/or post-transcriptional epigenetic mechanism can also have an impact on overall pSMAD3. This has not been investigated. Further investigation utilizing co-immunoprecipitation studies is required to determine if SMAD2/3 and YAP1 physically interacts in cushion cells. It also remains unclear if TAZ and YAP1 responds redundantly to TGFβ2 in cushion mesenchymal cells. Finally, this is an in vitro investigation, and therefore future in vivo studies based on combinatorial genetics approaches are required to establish the functional interaction between TGFβ2 and Hippo signaling in ECM remodeling in cardiac development. given the focus Taken together, these results suggest that Hippo signaling could negatively modulate cell cushion mesenchymal cell behavior by interacting with TGFβ pathway in heart valve development.
Secondly, the direct role of YAP1 in the ECM organization mediated by cushion mesenchymal cells in response to TGFβ2 and/or verteporfin is also investigated in the present study. TGFβ2 stimulated cushion mesenchymal cells in reorganization of ECM in three-dimensional collagen gel contraction assays and YAP1 inhibition has blocked the TGFβ2-induced collagen contraction response. Since active Hippo signaling (Hippo-ON) normally de-activates YAP1 (YAP1-OFF), our data identify a negative role of Hippo pathway in mediating the ability of cushion mesenchymal cells in reorganizing collagens in response to the exogenously added TGFβ2. The effect of TGFβ2 on collagen contraction seen in this study is comparable to the collagen contraction ability of mouse embryonic fibroblasts or cardiac fibroblasts by exogenously added TGFβ1, as reported by us (Carver et al., 2021; Chakrabarti et al., 2020; Fix et al., 2019; Haskett et al., 2012). Further investigation to elucidate the role of TGFβ2 and Hippo pathway in vivo in cushion mesenchymal differentiation and maturation and ECM remodeling and reorganization process will lead to a better understanding heart development and heart disease.
Finally, given the importance of both TGFβ and Hippo pathway in regulating genes involved cardiac fibroblasts activation and ECM remodeling during development and cardiac disease, the expression of mesenchymal genes such as Acta2, Col1a1, Ccn2, and Ccn1 was regulated by both TGFβ2 and/or Hippo signaling. TGFβ2 induces the expression of these ECM genes, whereas YAP1 inhibition blocks TGFβ2 regulation of these genes. These observations are consistent with the negative role of Hippo signaling in directly reorganizing collagen by exogenously added TGFβ2 in a three-dimensional gel contraction assay. Increased Acta2, Ctgf (Ccn2), and Col1a1 expression is involved in tissue fibrosis (Mia and Singh, 2022; Tsai and Martin, 2022) and tissue regeneration (Wang et al., 2018). The loss of Ccn1 or Tgfb2 causes cardiac septal defects in humans and mice (Bhattacharya et al., 2021; Ishtiaq Ahmed et al., 2014; Mo and Lau, 2006; Perrot et al., 2015), suggesting a potential genetic and molecular interaction between TGFβ2 and Hippo pathway. Mechanosensitive and profibrotic signaling pathways such as YAP/TGFβ are also implicated in multiple aspects of valvular heart disease, and therefore considered potential targets for therapeutic interventions and prognostic biomarkers with the implications to improve the management of valvular heart disease(Dayawansa et al., 2022). Moreover, through the repurposing of a clinically used small molecule inhibitor verteporfin, it is possible to alter TGFβ/SMAD signaling in vivo to investigate the role TGFβ/Hippo pathway integration in heart development and heart disease.
In conclusion, Hippo signaling via YAP1 acts as a mediator of TGFβ2 function in cardiac cushion mesenchymal cells to regulate extracellular matrix remodeling. ECM organization is the hallmark of cushion mesenchymal differentiation and maturation process during heart development. Thus, understanding the molecular regulation of TGFβ2-dependent ECM remodeling by YAP1 is an important step in elucidating underlying mechanisms involved in etiology of heart disease.
Author Contributions
Conceptualization, M.A.; methodology, M.C., A.C., A.N., K.J., J. D. P., M.A.; formal analysis, M.C., L.W., M.A.; writing—original draft preparation, M.C., M.A.; writing—review and editing, M.C., K. J., L. W., J. D. P., S. B., S.H., S.K., M.A.; supervision, M.A.; funding acquisition, M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by, in parts, funds from the University of South Carolina, School of Medicine (Bob Price-Instrumentation Resource Facility Endowment Fund), and ASPIRE-II (Advanced Support Program for Integration of Research Excellence-II- Office of the Vice President for Research) from the University of South Carolina, KOR Life Sciences/ Vikor Scientific, and the National Institutes of Health grants - R01HL126705, R01HL157017-01A1, 5P20GM109091, and 1P20GM103641.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Animal Care and Use Committee of UNIVERSITY OF SOUTH CAROLINA (protocol code: 2605-101718-060222 and date of approval: 02 June 2022).
Acknowledgments
We thank Ms. Charity Fix in the Cell and Tissue Culture Facility for assistance in providing media and other supplies for the experiments. We also thank the Instrumentation Resource Facility at the University of South Carolina, School of Medicine, and the Dorn VA Medical Center for the instrumentation support.
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- Attisano, L., and Wrana, J.L. (2013). Signal integration in TGF-beta, WNT, and Hippo pathways. F1000Prime Rep 5, 17.
- Azhar, M.; Runyan, R.B.; Gard, C.; Sanford, L.P.; Miller, M.L.; Andringa, A.; Pawlowski, S.; Rajan, S.; Doetschman, T. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev. Dyn. 2009, 238, 431–442. [CrossRef]
- Azhar, M.; Yin, M.; Bommireddy, R.; Duffy, J.J.; Yang, J.; Pawlowski, S.A.; Boivin, G.P.; Engle, S.J.; Sanford, L.; Grisham, C.; et al. Generation of mice with a conditional allele for transforming growth factor beta 1 gene. genesis 2009, 47, 423–431. [CrossRef]
- Bartram, U.; Molin, D.G.M.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Groot, A.C.G.-D. Double-Outlet Right Ventricle and Overriding Tricuspid Valve Reflect Disturbances of Looping, Myocardialization, Endocardial Cushion Differentiation, and Apoptosis in TGF-β 2 –Knockout Mice. Circ. 2001, 103, 2745–2752. [CrossRef]
- Bhattacharya, A., Al-Sammarraie, N., Gebere, M.G., Johnson, J., Eberth, J.F., and Azhar, M. (2021). Myocardial TGFbeta2 Is Required for Atrioventricular Cushion Remodeling and Myocardial Development. J Cardiovasc Dev Dis 8.
- Boileau, C.; Guo, D.-C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; D'Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [CrossRef]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 2013, 60, 27–35. [CrossRef]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [CrossRef]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Groot, A.C.G.-D.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2015, 202, 784–795. [CrossRef]
- Carver, W., Fix, E., Fix, C., Fan, D., Chakrabarti, M., and Azhar, M. (2021). Effects of emodin, a plant-derived anthraquinone, on TGF-beta1-induced cardiac fibroblast activation and function. J Cell Physiol 236, 7440-7449.
- Chakrabarti, M.; Al-Sammarraie, N.; Gebere, M.G.; Bhattacharya, A.; Chopra, S.; Johnson, J.; Peña, E.A.; Eberth, J.F.; Poelmann, R.E.; Groot, A.C.G.-D.; et al. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 19. [CrossRef]
- Dayawansa, N.H.; Baratchi, S.; Peter, K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2022, 9, 783543. [CrossRef]
- Ferrigno, O., Lallemand, F., Verrecchia, F., L'Hoste, S., Camonis, J., Atfi, A., and Mauviel, A. (2002). Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-beta/Smad signaling. Oncogene 21, 4879-4884.
- Fix, C., Carver-Molina, A., Chakrabarti, M., Azhar, M., and Carver, W. (2019). Effects of the isothiocyanate sulforaphane on TGF-beta1-induced rat cardiac fibroblast activation and extracellular matrix interactions. J Cell Physiol 234, 13931-13941.
- Frangogiannis, N.G. (2017). The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J Thorac Dis 9, S52-S63.
- Groot, A.C.G.-D.; Bartelings, M.M.; Deruiter, M.C.; E Poelmann, R. Basics of Cardiac Development for the Understanding of Congenital Heart Malformations. Pediatr. Res. 2005, 57, 169–176. [CrossRef]
- Gori, I., George, R., Purkiss, A.G., Strohbuecker, S., Randall, R.A., Ogrodowicz, R., Carmignac, V., Faivre, L., Joshi, D., Kjaer, S., et al. (2021). Mutations in SKI in Shprintzen-Goldberg syndrome lead to attenuated TGF-beta responses through SKI stabilization. Elife 10.
- Grewal, N.; DeRuiter, M.C.; Jongbloed, M.R.M.; Goumans, M.J.; Klautz, R.J.M.; Poelmann, R.E.; Groot, A.C.G.-D. Normal and abnormal development of the aortic wall and valve: correlation with clinical entities. Neth. Hear. J. 2014, 22, 363–369. [CrossRef]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell. Signal. 2020, 77, 109826–109826. [CrossRef]
- Haskett, D.; Doyle, J.J.; Gard, C.; Chen, H.; Ball, C.; Estabrook, M.A.; Encinas, A.C.; Dietz, H.C.; Utzinger, U.; Geest, J.P.V.; et al. Altered tissue behavior of a non-aneurysmal descending thoracic aorta in the mouse model of Marfan syndrome. Cell Tissue Res. 2011, 347, 267–277. [CrossRef]
- Heldin, C.H., and Moustakas, A. (2016). Signaling Receptors for TGF-beta Family Members. Cold Spring Harb Perspect Biol 8.
- Ahmed, A.S.I.; Bose, G.C.; Huang, L.; Azhar, M. Generation of mice carrying a knockout-first and conditional-ready allele of transforming growth factor beta2 gene. genesis 2014, 52, 817–826. [CrossRef]
- Labibi, B., Bashkurov, M., Wrana, J.L., and Attisano, L. (2020). Modeling the Control of TGF-beta/Smad Nuclear Accumulation by the Hippo Pathway Effectors, Taz/Yap. iScience 23, 101416.
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [CrossRef]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. Part A: Clin. Mol. Teratol. 2011, 91, 535–550. [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [CrossRef]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 211. [CrossRef]
- Mia, M.M.; Singh, M.K. New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells 2022, 11, 2065. [CrossRef]
- Misra, J.R.; Irvine, K.D. The Hippo Signaling Network and Its Biological Functions. Annu. Rev. Genet. 2018, 52, 65–87. [CrossRef]
- Mo, F.-E.; Lau, L.F The matricellular protein CCN1 is essential for cardiac development. Circ. Res. 2006, 99, 961–969. [CrossRef]
- Nallet-Staub, F., Yin, X., Gilbert, C., Marsaud, V., Ben Mimoun, S., Javelaud, D., Leof, E.B., and Mauviel, A. (2015). Cell density sensing alters TGF-beta signaling in a cell-type-specific manner, independent from Hippo pathway activation. Dev Cell 32, 640-651.
- Neeb, Z., Lajiness, J.D., Bolanis, E., and Conway, S.J. (2013). Cardiac outflow tract anomalies. Wiley Interdiscip Rev Dev Biol 2, 499-530.
- Peng, Y., Song, L., Li, D., Kesterson, R., Wang, J., Wang, L., Rokosh, G., Wu, B., Wang, Q., and Jiao, K. (2016). Sema6D acts downstream of bone morphogenetic protein signalling to promote atrioventricular cushion development in mice. Cardiovasc Res 112, 532-542.
- Perrot, A.; Schmitt, K.R.; Roth, E.-M.G.; Stiller, B.; Posch, M.G.; Browne, E.N.L.; Timmann, C.; Horstmann, R.D.; Berger, F.; Özcelik, C. CCN1 mutation is associated with atrial septal defect. Pediatr. Cardiol. 2014, 36, 295–299. [CrossRef]
- Piersma, B., Bank, R.A., and Boersema, M. (2015). Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med (Lausanne) 2, 59.
- Poelmann, R.E., Gittenberger-de Groot, A.C., Goerdajal, C., Grewal, N.; De Bakker, M.A.G.; Richardson, M.K. Ventricular Septation and Outflow Tract Development in Crocodilians Result in Two Aortas with Bicuspid Semilunar Valves. J. Cardiovasc. Dev. Dis. 2021, 8, 132. [CrossRef]
- Sun, C.; Zhang, H.; Liu, X. Emerging role of CCN family proteins in fibrosis. J. Cell. Physiol. 2020, 236, 4195–4206. [CrossRef]
- Szeto, S.G., Narimatsu, M., Lu, M., He, X., Sidiqi, A.M., Tolosa, M.F., Chan, L., De Freitas, K., Bialik, J.F., Majumder, S., et al. (2016). YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J Am Soc Nephrol 27, 3117-3128.
- Tsai, C.R., and Martin, J.F. (2022). Hippo signaling in cardiac fibroblasts during development, tissue repair, and fibrosis. Curr Top Dev Biol 149, 91-121.
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [CrossRef]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.-L.; et al. Yap1 is required for endothelial to mesenchymal transition of the atrioventricular cushion. J. Biol. Chem. 2014, 289, 18681–18692. [CrossRef]
Figure 1.
TGFβ2 induces nuclear translocation of SMAD3 in cushion mesenchymal cells. (A-D) Cushion mesenchymal cells were grown on 2 well chamber slides at 80-90% confluency and were untreated (control) (A-B) or treated with TGFβ2 (5 ng/mL) for 12 hr (C-D). Cells were stained with primary SMAD3 antibodies, followed by secondary Cy3 goat anti rabbit IgG (pink color) and counterstained with DAPI (teal color) to identify nuclei. Representative images confirmed enhanced nuclear accumulation of SMAD3 following treatment with TGFβ2 (D). The nuclear SMAD3 fluorescence intensity was measured for the untreated control and TGFβ2 treated cells and presented graphically by a scatter plot with bar showing individual values. The error bars indicate mean±SD. Arrows (A-D) indicate nuclei and arrowhead (B) denotes cytoplasm. (E). Statistically significant differences between control and TGFβ2 treated cells (p = 0.0006, two-tailed Student’s t test with Welch’s correction; Control, n = 8: TGFβ2-treated, n = 5) were determined and indicated on the scatter plot.
Figure 1.
TGFβ2 induces nuclear translocation of SMAD3 in cushion mesenchymal cells. (A-D) Cushion mesenchymal cells were grown on 2 well chamber slides at 80-90% confluency and were untreated (control) (A-B) or treated with TGFβ2 (5 ng/mL) for 12 hr (C-D). Cells were stained with primary SMAD3 antibodies, followed by secondary Cy3 goat anti rabbit IgG (pink color) and counterstained with DAPI (teal color) to identify nuclei. Representative images confirmed enhanced nuclear accumulation of SMAD3 following treatment with TGFβ2 (D). The nuclear SMAD3 fluorescence intensity was measured for the untreated control and TGFβ2 treated cells and presented graphically by a scatter plot with bar showing individual values. The error bars indicate mean±SD. Arrows (A-D) indicate nuclei and arrowhead (B) denotes cytoplasm. (E). Statistically significant differences between control and TGFβ2 treated cells (p = 0.0006, two-tailed Student’s t test with Welch’s correction; Control, n = 8: TGFβ2-treated, n = 5) were determined and indicated on the scatter plot.
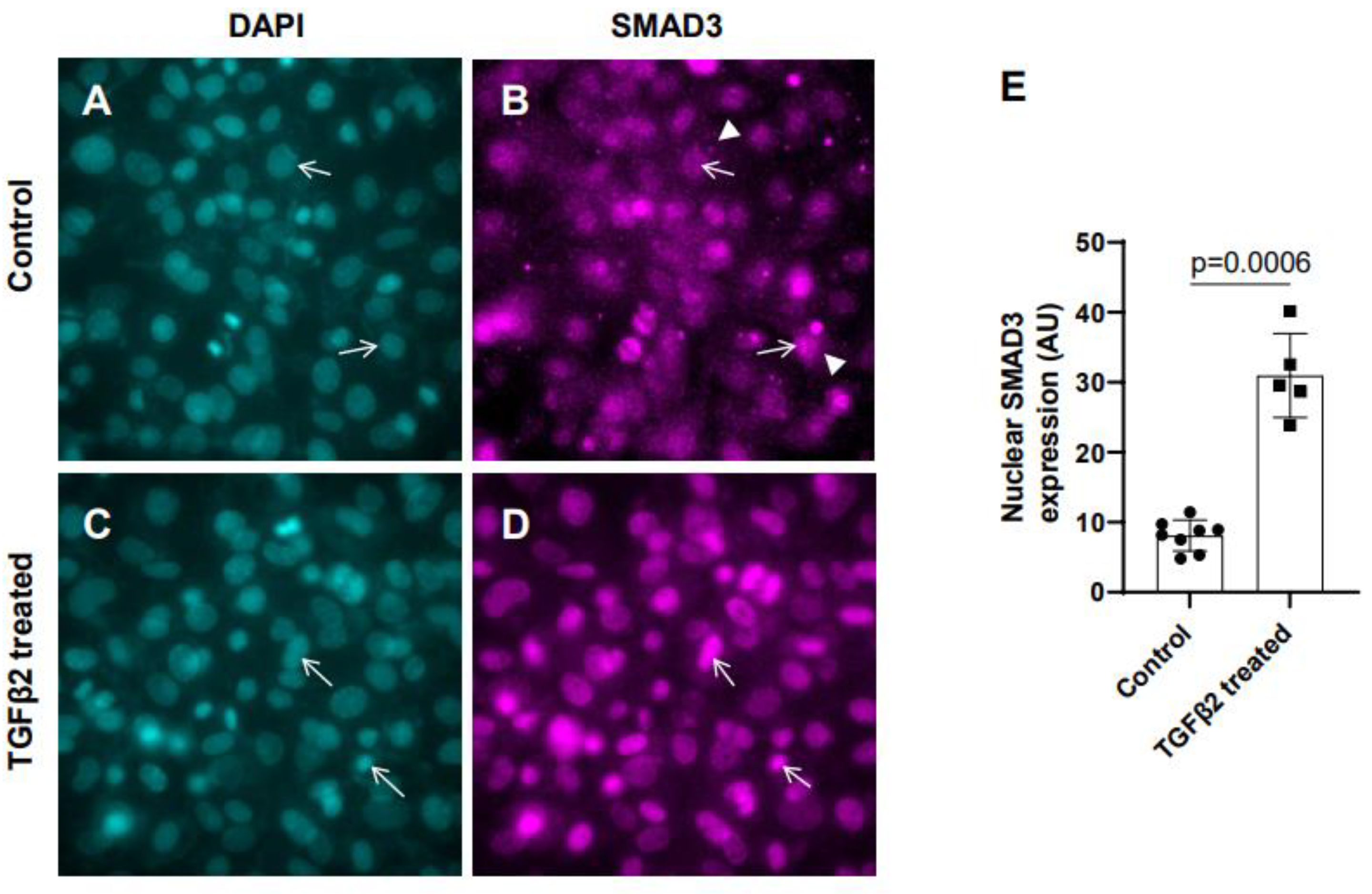
Figure 2.
TGFβ2 treatment alters cytoplasmic and nuclear localization of YAP1 protein in cushion mesenchymal cells. (A-D) Cushion mesenchymal cells were grown on 2 well chamber slides at 80-90% confluency and were untreated (control) (A-B) treated with TGFβ2 (5 ng/mL) for 12 hr. Cells were stained with primary YAP1 antibodies followed by secondary Cy3 goat anti rabbit IgG (pink color) and counterstained with DAPI (teal color) to identify nuclei. Representative images confirmed altered cytoplasmic/nuclear localization of YAP1 following treatment with TGFβ2. Arrows (A-D) indicate nuclei and arrowhead (B) denotes cytoplasm. (E) The YAP1 fluorescence intensity in cytoplasm and nucleus was measured in 7-8 fields of different images from the untreated control and TGFβ2 treated cells by using NIH ImageJ software. The ratio of cytoplasmic/nuclear fluorescence intensity was presented graphically by a scatter plot with bar showing individual values. The error bars indicate mean±SD. Statistically significant differences between control and TGFβ2 treated cells (p = 0.0004, two-tailed Student’s t test with Welch’s correction; Control, n = 7; TGFβ2-treated, n = 7) were determined and indicated on the scatter plot.
Figure 2.
TGFβ2 treatment alters cytoplasmic and nuclear localization of YAP1 protein in cushion mesenchymal cells. (A-D) Cushion mesenchymal cells were grown on 2 well chamber slides at 80-90% confluency and were untreated (control) (A-B) treated with TGFβ2 (5 ng/mL) for 12 hr. Cells were stained with primary YAP1 antibodies followed by secondary Cy3 goat anti rabbit IgG (pink color) and counterstained with DAPI (teal color) to identify nuclei. Representative images confirmed altered cytoplasmic/nuclear localization of YAP1 following treatment with TGFβ2. Arrows (A-D) indicate nuclei and arrowhead (B) denotes cytoplasm. (E) The YAP1 fluorescence intensity in cytoplasm and nucleus was measured in 7-8 fields of different images from the untreated control and TGFβ2 treated cells by using NIH ImageJ software. The ratio of cytoplasmic/nuclear fluorescence intensity was presented graphically by a scatter plot with bar showing individual values. The error bars indicate mean±SD. Statistically significant differences between control and TGFβ2 treated cells (p = 0.0004, two-tailed Student’s t test with Welch’s correction; Control, n = 7; TGFβ2-treated, n = 7) were determined and indicated on the scatter plot.
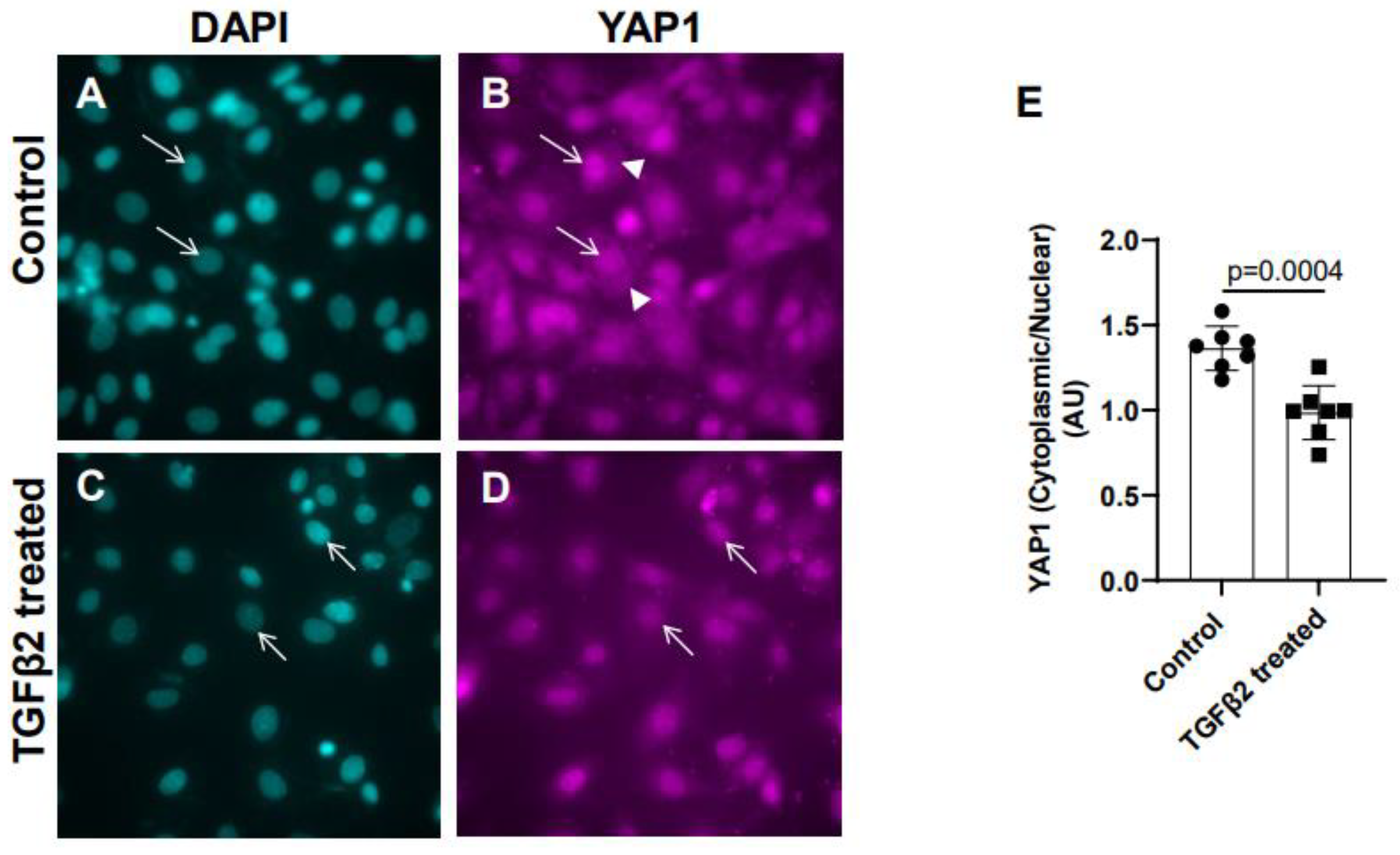
Figure 3.
Verteporfin blocks the ability of cushion mesenchymal cells to reorganize and contract collagen gel by exogenously added TGFβ2. (A-D) Collagen lattice formation assay. Representative images of cushion mesenchymal cells-laden collagen gels after 5-days after treatment with TGFβ2 (5 ng/mL) and/or Verteporfin (5 µg/mL). TGFβ2 treated cushion cells showed enhanced ability to contract collagen gel (B) compared to the untreated cells (A). Verteporfin significantly blocked the collagen contraction in the absence (C) and presence (D) of TGFβ2 (D). (E) Collagen gel area (%) was measured using NIH ImageJ software and the numerical data were presented as a scatter plot with bar showing individual values. White dotted circles outlined the collagen gels to reinforce the changes in collagen gel circumference. The error bars indicate mean±SD. Statistically significant differences of collagen gel diameter between two individual groups (two-tailed Student’s t test with Welch’s correction; n = 3 each group) were determined and p-values were indicated on the scatter plot.
Figure 3.
Verteporfin blocks the ability of cushion mesenchymal cells to reorganize and contract collagen gel by exogenously added TGFβ2. (A-D) Collagen lattice formation assay. Representative images of cushion mesenchymal cells-laden collagen gels after 5-days after treatment with TGFβ2 (5 ng/mL) and/or Verteporfin (5 µg/mL). TGFβ2 treated cushion cells showed enhanced ability to contract collagen gel (B) compared to the untreated cells (A). Verteporfin significantly blocked the collagen contraction in the absence (C) and presence (D) of TGFβ2 (D). (E) Collagen gel area (%) was measured using NIH ImageJ software and the numerical data were presented as a scatter plot with bar showing individual values. White dotted circles outlined the collagen gels to reinforce the changes in collagen gel circumference. The error bars indicate mean±SD. Statistically significant differences of collagen gel diameter between two individual groups (two-tailed Student’s t test with Welch’s correction; n = 3 each group) were determined and p-values were indicated on the scatter plot.
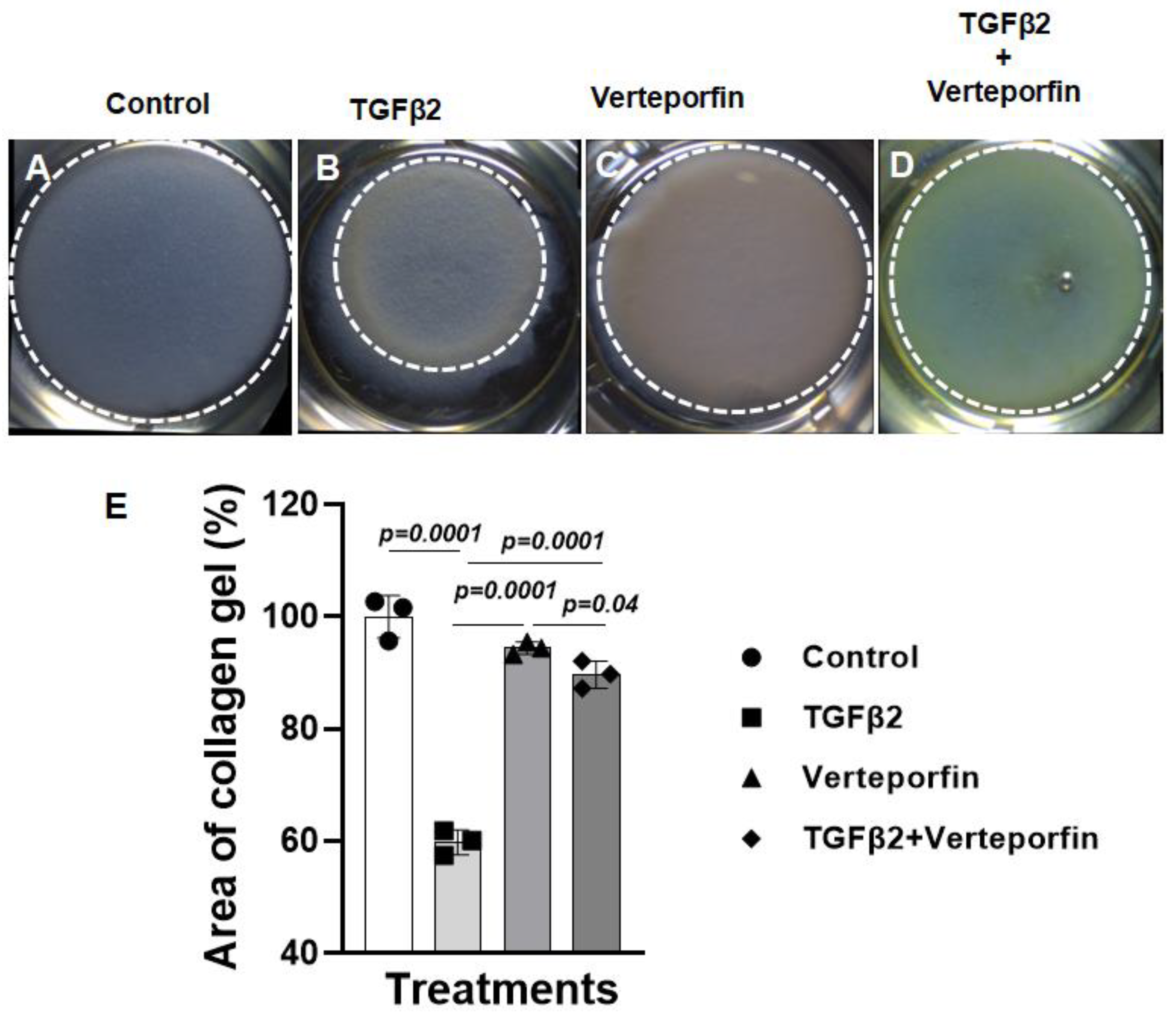
Figure 4.
Verteporfin reduces the transcript levels of TGFβ2-induced cushion mesenchymal genes involved in extracellular matrix remodeling. (A-D) Quantitative real time PCR analysis showing gene transcript levels of α-smooth muscle actin (Acta2) (A), collagen Ia1 (Col1a1) (B), connective tissue growth factor/cell communication network 2 (Ctgf/Ccn2) (C), and cysteine-rich angiogenic inducer 61/cell communication network 1 (Cyr61/Ccn1) (D) in cushion mesenchymal cells in the absence or presence of TGFβ2 (5 ng/mL) and/or verteporfin (5 µg/mL) for 12 hr.
Figure 4.
Verteporfin reduces the transcript levels of TGFβ2-induced cushion mesenchymal genes involved in extracellular matrix remodeling. (A-D) Quantitative real time PCR analysis showing gene transcript levels of α-smooth muscle actin (Acta2) (A), collagen Ia1 (Col1a1) (B), connective tissue growth factor/cell communication network 2 (Ctgf/Ccn2) (C), and cysteine-rich angiogenic inducer 61/cell communication network 1 (Cyr61/Ccn1) (D) in cushion mesenchymal cells in the absence or presence of TGFβ2 (5 ng/mL) and/or verteporfin (5 µg/mL) for 12 hr.
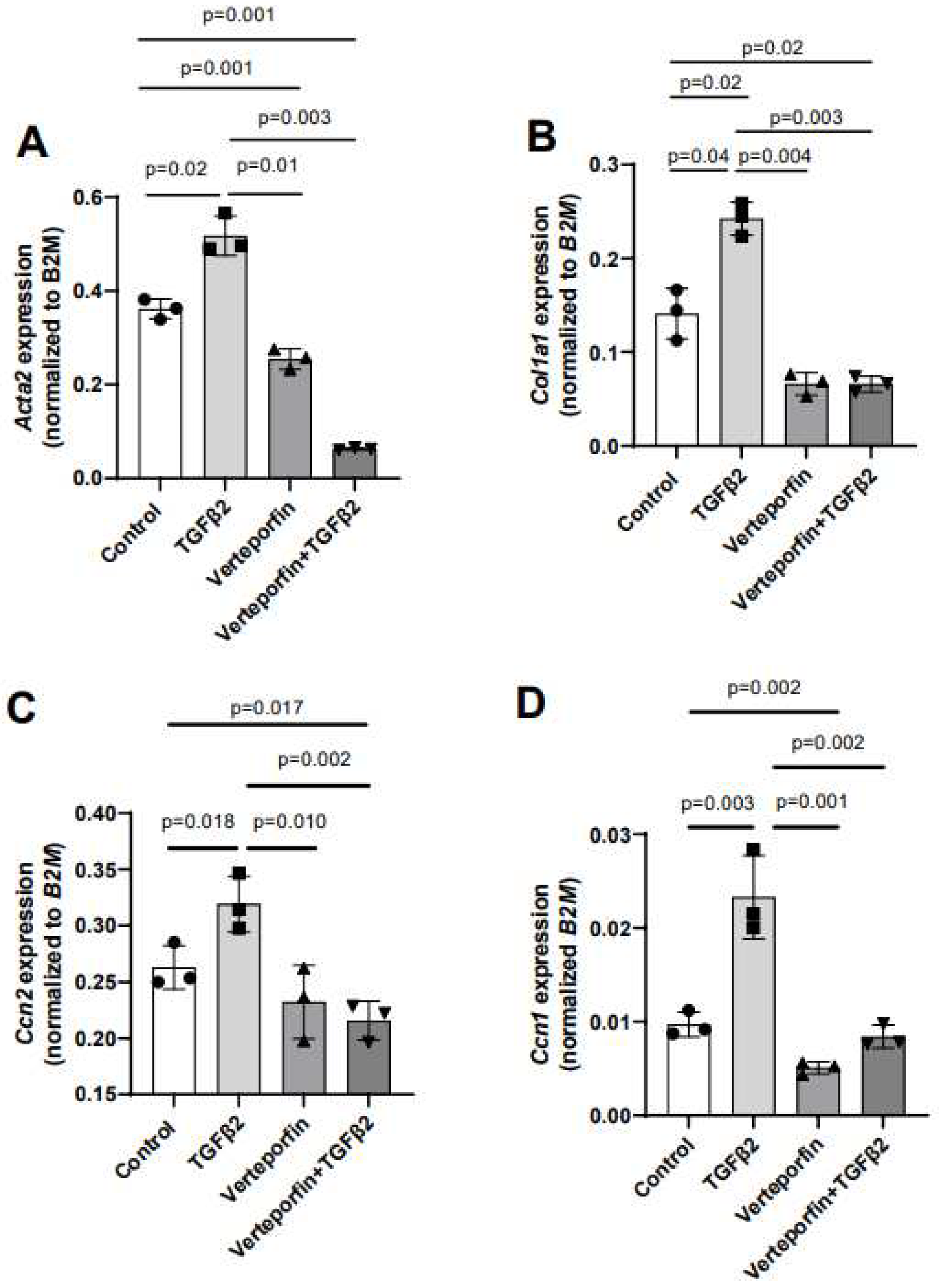
Figure 5.
Verteporfin blocks YAP1 and TGFβ2-dependent activation of SMAD2 and SMAD3 in cushion mesenchymal cells. (A) Western blot analysis of control and TGFβ2 and/or verteporfin treated cushion mesenchymal cells showing protein levels of the phosphorylated SMADs (pSMAD2, pSMAD3), total SMADs (SMAD2, SMAD3), YAP1, and β-actin. (B-H) Densitometric quantification of pSMAD2 (B-C), SMAD2 (D), pSMAD3 (E-F), SMAD3 (G), and YAP1 (H). The phosphorylated SMAD2 and SMAD3 proteins were normalized by both total SMAD2 (B) and SMAD3 (E), respectively, and β-actin (C, F). The levels of ‘total’ phosphorylated and unphosphorylated SMAD2 (D) and SMAD3 (G) were normalized by β-actin. YAP1 protein was normalized with β-actin (H). Three independent western blots were used for protein quantification and statistical analysis (one-tailed Student’s t test with unpaired; n = 3 each group). p-values are shown in the figure. Numerical data from multiple individual samples are presented as scatter dot-plots with error bars denoting the mean±SD.
Figure 5.
Verteporfin blocks YAP1 and TGFβ2-dependent activation of SMAD2 and SMAD3 in cushion mesenchymal cells. (A) Western blot analysis of control and TGFβ2 and/or verteporfin treated cushion mesenchymal cells showing protein levels of the phosphorylated SMADs (pSMAD2, pSMAD3), total SMADs (SMAD2, SMAD3), YAP1, and β-actin. (B-H) Densitometric quantification of pSMAD2 (B-C), SMAD2 (D), pSMAD3 (E-F), SMAD3 (G), and YAP1 (H). The phosphorylated SMAD2 and SMAD3 proteins were normalized by both total SMAD2 (B) and SMAD3 (E), respectively, and β-actin (C, F). The levels of ‘total’ phosphorylated and unphosphorylated SMAD2 (D) and SMAD3 (G) were normalized by β-actin. YAP1 protein was normalized with β-actin (H). Three independent western blots were used for protein quantification and statistical analysis (one-tailed Student’s t test with unpaired; n = 3 each group). p-values are shown in the figure. Numerical data from multiple individual samples are presented as scatter dot-plots with error bars denoting the mean±SD.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



