
Submitted:
12 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
Age-related diseases place an intense and growing stress on healthcare systems. Numerous theories of ageing exist. Yet prediction, prevention and treatment limitations of age-related disease persist because we lack a complete aetiological understanding of their origins. Here we propose a new theory that aims to provide a blueprint-type understanding of ageing and disease aetiology, working across multiple scientific disciplines. Supported by specific examples, we demonstrate that the primary driver of much of age-related diseases is the futile triggering of specific biological pathways in a pathological manner, which we term pathophysiological pathways or patho-pathways. Patho-pathways induce pathological cell, tissue and system changes in the form of dysactivity (out of context activity that is at the wrong time and/or place), which is characteristic of age related disease. As part of patho-pathways, all wild-type (normal) genes have the potential to contribute to age-related diseases (though specific risk varies and can be formularised). In a domino-style effect, one patho-pathway typically triggers others, resulting in complex, but traceable, cascades. Utility, in terms of changing clinical and scientific practice, is derived from mapping these cascades to create Blueprint Maps that allow identification of biomarkers and therapeutic targets far upstream in the causal chain of events. Finally, our theory provides testable hypotheses concerning predictions of links between otherwise seemingly disparate diseases as well as new interventions to reduce ageing.
Keywords:
ageing
; age-related disease
; antagonistic pleiotropy
; biological mechanisms
; Blueprint Maps
; evolutionary biology
; dysactivity
; new ageing theory
; patho-pathway
; patho-pathway cascades
; prediction
; prevention
; network constraint
; treatment
; triggers
Introduction
Regardless of whether one subscribes to the view that ageing is a collection of diseases or whether one sees the ageing process as distinct from the diseases arising in later life, there can be little argument that it is the diseases associated with growing old that present the greatest challenge for 21st-century medicine and are a growing burden on the economy.
To date, multiple theories of ageing have enhanced our understanding; however, none provide complete explanations for the cause of age-related diseases that are backed up by specific disease examples. Consequently, distinctive features of ageing (as typified by the Strategies for Engineered Negligible Senescence (SENS) or “Hallmarks of Ageing” typology) are relied upon as linchpins to guide study of the ageing process. And these are viewed as producing chaotic and intractable changes which are often, but not always, classified as “age-related diseases”. This disjunction is often captured by labelling age-related disease aetiologies as “unknown” or illustrating the relationship between ageing and age-related disease with the familiar “black box”. This is a vital gap, which this manuscript aims to fill.
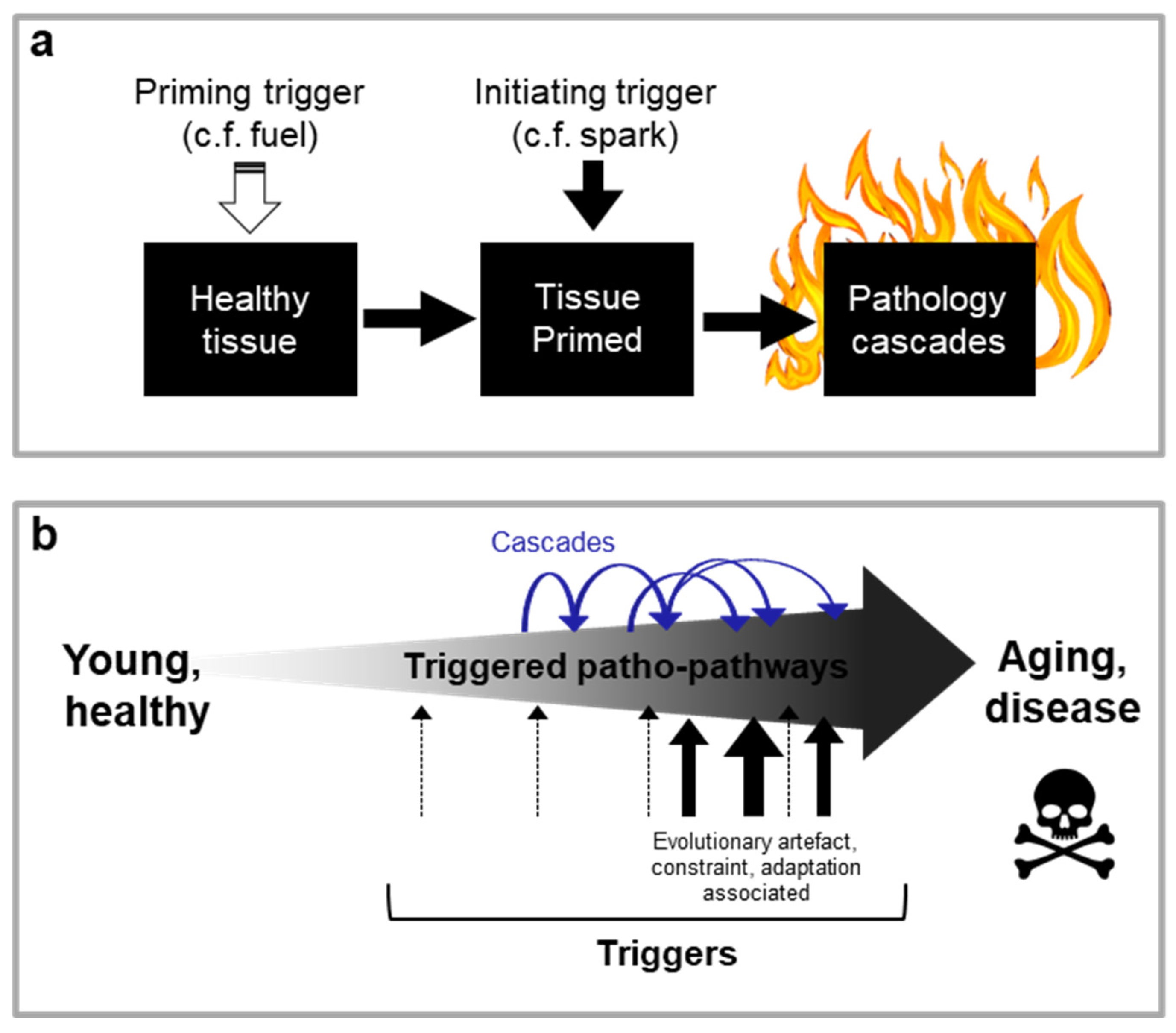
New theory. In this paper, we propose a radically new explanation of ageing and related disease, bridging multiple scientific fields, including evolutionary biology and medicine. We also take account of specific biological mechanisms (genetics, molecular, cellular, physiological) involved. Supported by multiple specific disease examples, we demonstrate that the primary driver of ageing is the futile triggering of certain molecular pathways later in life that we term patho-pathways (pathological pathways; for a glossary see Table 1). Patho-pathways cause pathology via inducing cell, tissue and system dysactivity (any abnormal or faulty activity, e.g. too much, too little, or wrong type) (Figure 1a). Patho-pathways arise from constraints associated with the interconnected nature of multiple interacting biological pathways within organisms, what we term network constraint. More specifically, network constraint arises where optimisation of function (at the molecular, cell, tissue and system level) in all contexts is not possible, risking off-target side effects in the form of pathology when a “trigger” is present. Triggers can include extrinsic (e.g. infection, mechanical injury, molecular damage, behavioural influencers/stress including dietary insults, smoking, and toxins like air pollutants) and intrinsic (somatic mutations) factors, which converge to give rise to multifactorial aetiology. Some triggers act directly, while others act as primers to increase susceptibility to other triggers. Notably, triggers may have an evolutionary origin, and as such induce pathology at only a time point later in the life history. Notably, in a domino style effect, patho-pathways can trigger the futile activation of other patho-pathways, resulting in complex cascades of patho-pathways (Figure 1b). Significantly, these cascades are tractable. In this paper we classify and map these patho-pathway cascades to create Blueprint Maps that explain the aetiology of multiple diseases, which in turn has utility in improving disease prediction, and prevention and treatment therapeutics. One example of the latter goes so far as identifying a new class of drugs that is a first-in-class means to block cellular necrosis (unprogrammed, accidental cell death), see (Kern et al., 2024).
New Theory: Triggered Patho-Pathways
For ease in introducing our new theory, we have tied it to a specific example. In the prostate gland, long-term exposure to normal levels of dihydrotestosterone (DHT) promotes benign prostatic hyperplasia (Nacusi and Tindall, 2011; Waters et al., 2000) and increases the risk of prostate cancer (Untergasser et al., 2005). But what is the exact aetiology of the disease? Clearly here there is some link between pathology (hyperplasia) and long-term stimulus of DHT but what is the nexus of cause and effect? Is it merely the very slow cumulative action of DHT throughout life that drives pathology either directly or indirectly via random damage accumulation? If our hypothesis is correct, and the key driver of pathology is the induction of patho-pathways, i.e. molecular pathways initiated in the wrong place and/or at the wrong time, we need to be able to identify the “trigger”. In other words, something has to change in the ageing prostate, leading to a novel pathogenic response to DHT. Indeed a trigger does exist: increased local, late-life inflammation disrupts the balance between cell proliferation and cell death, such that patho-pathways of proliferation predominate to induce hyperplasia (Tong and Zhou, 2020). Notably, here the DHT-driven patho-pathways are triggered during adulthood rather than simply being something present all along the life cycle.
In principle, multiple causes could trigger patho-pathways, both intrinsic and extrinsic factors (e.g. infection, somatic mutation) (Figure 1a). Patho-pathway triggers can also originate within the affected tissue, as in the case of prostrate hyperplasia, or beyond. An example of the latter is endometrial hyperplasia. This condition is a precursor to endometrial carcinoma, one of the most common gynaecological malignancies (Hannemann et al., 2007). The hyperplasia is caused by chronic oestrogen stimulation of endometrial cell growth, unopposed by progesterone that would otherwise cause cell shedding. Hyperplasia is triggered by factors that increase circulating oestrogen relative to progesterone, including high BMI, late age at menopause and nulliparity (Epplein et al., 2008; Hannemann et al., 2007) (Figure 1c).
Patho-Pathways Induce Pathology Through Cell, Tissue and System Dysactivity
In the two examples of patho-pathways above, pathology has resulted from normal pathways having been initiated to a higher than normal level in a futile manner. In other words, biological function has been increased above the level of what is optimal to maintain healthy physiology. [Note here the definition of the word “function”, which is an activity that is natural to or the purpose of a thing]. But this is not the only way in which age-related disease presents, nor is it the only means through which patho-pathways can induce pathology. Patho-pathways frequently lead to an alteration in biological activity, which includes, not just increased function, but also cells taking on new unwanted activities. This can be as extreme as a change in cellular identity. As such, triggered patho-pathways can explain diseases involving dysactivity commonly seen during ageing, including pathological cellular trans-differentiation. One example of this is vascular calcification, where active extraskeletal ossification (bone formation) takes place with age as vascular smooth muscle cells transdifferentiate into pseudo-osteoblasts, and this is triggered by high blood calcium, phosphate and glucose levels and proinflammatory factors that initiate osteogenic patho-pathways (Abedin et al., 2004; Chen and Moe, 2012; Demer, 2002; Virchow, 1863). As another example, mature medial smooth muscle cells undergoing clonal expansion can pathologically transdifferentiate into macrophage-like cells causing inflammation and atherosclerotic lesions, and this is triggered by patho-pathways induced by changes regulated at the mRNA level (Feil et al., 2014; Rong et al., 2003).
Disease Onset May Be Dependent on the Convergence of Multiple Patho-Pathway Triggers, Some of Which Have Evolutionary Origins
In the examples so far, pathology has been initiated by a single trigger. But this is not necessarily always the case. The multifactorial complexity of many age-related diseases likely arises from the interaction of multiple triggers. Some of these triggers may not directly induce pathology, and rather only prime cells, tissue and systems - while other triggers then act to kick-start destruction once the tissue is primed. As an analogy, for a fire you need both the spark, i.e. kick-starting trigger, and fuel, i.e. priming (Figure 2a). An example of this is seen in chronic obstructive pulmonary disease (COPD). During infections of the lung, innate immune cells like neutrophils secrete elastases to enable them to burrow through tissue to sites of infection. In youth, pathways associated with neutrophil migration are overall protective as neutrophil migration is accurate, with a high degree of effectiveness at clearing infection and minimising collateral damage to lung tissue from elastase secretion. Inflammageing (increased sterile inflammation with age) in later life, however, disrupts neutrophil migration capability, likely via triggering hyper-activation of phosphoinositide 3-kinase (PI3K) in neutrophils (i.e. priming has taken place). When the trigger of an infection is then present later in life (spark), this translates to both poor immune clearance and severe tissue injury, to the point of contributing to COPD due to aberrant neutrophil migration (Sapey et al., 2014; Voynow and Shinbashi, 2021). Timing of pathology onset in this case is determined by the timing of the spark and timing of priming.
Dependence on multiple triggers goes towards explaining why cells, tissues and organ systems can age at different rates in the same individual. Additionally, this dependence on multiple triggers for pathology onset could explain why many age-related diseases take a given number of years to appear (e.g. vascular calcification and cardiovascular disease especially prevalent >50 yrs). This is especially the case if a trigger has an evolutionary origin, where appearance of the trigger is associated with a particular time point in the life history (Figure 2b). In the case of COPD one example of such a trigger is thymic involution which occurs later in life and contributes to inflammaging.
Regarding the exact evolutionary origins behind a particular trigger, these are typically hard to pinpoint owing to difficulty in finding definitive proof of the presence of one evolutionary mechanism over another. That said, possibilities boil down to one of three mechanisms. And we can illustrate this using the menopause as an example. Menopause arises due to oocyte depletion at a particular time point in the life history in women, and this in turn causes a hormonal milieu that mimics lactation, only in a more severe form and particularly through a large dip in oestrogen levels (Kovacs, 2016; Pollycove et al., 2011). This results in triggering of lactational patho-pathways that cause osteoclast dysactivity and consequently severe trabecular bone resorption to the point of inducing osteoporosis. Evolutionary origins of menopause can include any one of the following. (i) Adaptive benefit, where menopause evolved because it confers a selective benefit. For example, the grandmother hypothesis for menopause postulates that the menopause evolved because ancestral middle-aged women gained greater reproductive success from investing in their grandchildren compared to their own fertility. (ii) Biological constraint, where it may simply be impossible to produce a particular trait, or there are inextricable linkages between different traits and/or the environment resulting in trade-offs to fitness (Acerenza, 2016). For example, menopause may have arisen due to the need to select only the fittest oocytes for reproduction, with consequently high levels of follicular atresia resulting in an unavoidable loss of all oocytes by the age of around 50 yrs. (iii) Evolutionary artefact where, either one trait changes (e.g. to cause human lifespan to increase), and there just has not been enough time for selection pressure to have a reciprocal effect on other traits (e.g. increasing female reproductive span), or the trait expresses at a time when selection is attenuated (i.e. menopause occurs at a time when there is a selection shadow) (Hamilton, 1966; Medawar, 1952).
As a third example, to highlight the often complex nature of interactions between different triggers, we demonstrated some years ago that vascular calcification is in part the result of replicative senescence in vascular smooth muscle cells where there is some type of biological constraint or evolutionary artefact at play. This is based on the finding that individuals with cardiovascular disease appear to start off with a lower cellular replicative capacity in early life, than non-diseased individuals (Karavassilis and Faragher, 2013); the SASP contributes to a pro-inflammatory and pro-adhesive milieu which primes tissue for transdiferentiation (Fang et al., 2024) when other (spark) triggers like a high calcium, and glucose blood levels are present.
Figure 2.
Disease onset may be dependent on the convergence of multiple patho-pathway triggers. a, Some triggers may not directly induce pathology, and rather only prime cells, tissue and systems. Other triggers then act to kick-start destruction once the tissue is primed. As an analogy, for a fire you need both the spark, i.e. kick-starting trigger, and fuel, i.e. priming. b, Cascades of primary and secondary patho-pathways promote organismal ageing (hypothetical scheme). More severe patho-pathways are seen in later life due to the occurrence of triggers with an evolutionary origin at that time.
Figure 2.
Disease onset may be dependent on the convergence of multiple patho-pathway triggers. a, Some triggers may not directly induce pathology, and rather only prime cells, tissue and systems. Other triggers then act to kick-start destruction once the tissue is primed. As an analogy, for a fire you need both the spark, i.e. kick-starting trigger, and fuel, i.e. priming. b, Cascades of primary and secondary patho-pathways promote organismal ageing (hypothetical scheme). More severe patho-pathways are seen in later life due to the occurrence of triggers with an evolutionary origin at that time.
Links between Disparate Age-Related Diseases Can Be Explained Through Patho-Pathway Cascades
Triggered patho-pathways can act as, not only as primary, but also as secondary and tertiary drivers of pathogenesis. This is via patho-pathways acting as triggers for other patho-pathways, which in turn may result in a domino-effect of chains or cascades of patho-pathways driving pathogenesis (Figure 1b, 2b).
Broadly, two forms of triggered patho-pathway cascades may be distinguished. First, in a multi-stage patho-pathway cascade a chain reaction based on a single underlying trigger drives pathology onset and progression, but multiple stages can be distinguished. Second, a discrete stage patho-pathway cascade, where individual patho-pathways are temporally or spatially separated from one another by different triggers, and do not necessarily follow one to the next in all patients. To be precise, multi-stage and discrete stage patho-pathway cascades represent opposite ends of a continuum more so than discrete categories, with many cascades corresponding most closely to points in between.
As an example of a multi-stage cascade is the progression of rheumatoid arthritis, where the single underlying trigger driving the patho-pathways that cause the disease is inflammation, and multiple distinguishable stages are as follows (Figure 3a). STAGE 1: A trigger such as inflammaging causes patho-pathways in immune cells to be hyperactivated. This causes immune cells to attack a healthy joint in a futile manner. STAGE 2: Immune cell attack of the joint triggers patho-pathways in the synovial tissue surrounding the joint. This causes the synovial tissue to undergoe vigorous proliferation and vascularisation, and to transform into a hypertrophied rheumatoid pannus (from the Latin pannus for table-cloth, reflecting the presence of a thickened layer of tissue). STAGE 3: The pannus secretes proinflammatory cytokines that triggers patho-pathways in osteoclasts, inducing osteoclast dysactivity that cause bone resorption and loss (Distler et al., 2004; Karmakar et al., 2010). Thus, in this complex disease a localised succession of futile inflammation driven patho-pathways leads to painful and disabling pathology in a multi-stage cascade.
This is the core aetiology in a wider, multifactorial process. Variations in the presentation and progression of the disease may be attributed to: (i) differences in the wild-type genome (that influence the inflammatory predisposition and profile); (ii) occurrence of other extrinsic or intrinsic factors (e.g. injury, infection) that, like tributaries which contribute to the volume of a river, contribute to the severity of the disease; and (iii) distributary like branches of the core pathology that incur their own independent cost, e.g. the hyperplastic synovium independently causes damage by pointlessly invading cartilage and bone, and increased synovial fluid causes damage by increasing intra-articular pressure.
Moving to discrete stage cascades, the relationship between primary and secondary patho-pathway initiation is looser and less inevitable. As one example, any disorder that weakens bone and increases susceptibility to mechanical injury, such as rheumatoid arthritis, greatly increases risk of osteoarthritis. This is because in osteoarthritis, chondrocyte dysactivity is triggered by mechanical stress alongside cellular senescence and associated inflammation. Here patho-pathways of new bone formation (osteogenesis) result in the thickening of subchondral bone, including the development of bone marrow pockets and a blood supply. The resulting new and futile bone growth in the form of bone spurs (or osteophytes) restrict joint movement and cause pain (Glyn-Jones et al., 2015). For other examples of multi-stage and discrete stage patho-pathway cascades, such as insulin-resistance patho-pathways in fatty liver disease, see Table 2 and Figure 3b.
Patho-Pathway Cascades as a Tool in Disease Prediction
One utility of a more complete understanding of disease aetiology is the ability to predict the likelihood of disease occurrence and time of onset via identifying good biomarkers. This can be via identifying disease predisposing factors (e.g. high blood calcium, phosphate, glucose and a pro-inflammatory profile for vascular calcification) or going a step further to identify what the cause of those predisposing factors are in the first place, and so pre-empt disease occurrence far in advance. For this purpose, knowledge of patho-pathway cascades are a useful tool. As an example, we predicted that in general - due to a discrete stage cascade - bone loss disorders should increase susceptibility to arterial pathology, especially where those bone loss disorders are driven by a high inflammatory profile. As previously described, 2 key triggers of pathological arterial transdifferentiation and ossification are (i) high blood calcium and phosphate levels, and (ii) inflammation. Bone loss disorders will increase blood levels of calcium and phosphate, at least transiently. To test this, we looked through the literature, and indeed there are strong correlations between bone resorption diseases, like rheumatoid arthritis, and arterial calcification, particularly when inflammation is also present (Giles et al., 2009) (Figure 3c). Thus tracing out cascades of patho-pathways allows one to make testable hypotheses in the form of predicted links between otherwise seemingly distinct diseases. Such links are of immense use when pre-empting disease occurrence.
Existing Theories of Ageing in the Context of Our New Theory: The Role of Nutrient Signalling in Ageing
Existing theories. Many existing theories of ageing emphasise the nutrient signalling network, including the mTOR, IIS, PI3K-AKT and the Ras-MEK-ERK pathways, as key proximate drivers of the ageing process. One view is that ageing is driven by the futile continuation (or “run-on”) of growth and developmental programmes associated with nutrient signalling into late adulthood (like a tap that has been opened and not switched off), i.e. the Developmental/Hyperfunction Theory (Blagosklonny, 2006; de Magalhães, 2005; Gems, 2022; Maklakov and Chapman, 2019). When seeking specific disease examples, proponents of this theory tend to lean heavily on presbyopia (long-sightedness with age). Presbyopia results in part from the very gradual, continued growth of the eye lens during adulthood, which increases lens thickness thereby impairing ocular function (Blagosklonny, 2012; de Magalhães and Church, 2005; Gems, 2021; Strenk et al., 2005). Presbyopia is presumably alluded to so frequently because it is easy-to-grasp. But to date we have found no other clear examples cited concerning run-on in humans. An alternative view is that nutrient signalling is an “antagonistic hallmark”, whereby damage accumulation occurs throughout life, and increase in nutrient signalling is a response to this damage that initially mitigates the damage, but eventually, if chronic or exacerbated, becomes deleterious and contributes to ageing (López-Otín et al., 2023). Both these views stem from the observation that across laboratory models, inhibition of nutrient signalling via genetic mutations or interventions such as caloric/dietary restriction results in robust lifespan extension. We recently provided evidence, however, that the mechanisms of ageing limiting lifespan need not be the same across species. And mechanisms associated with very large magnitude increases in lifespan following suppression of nutrient signalling, such as the 10-fold lifespan extension seen in the common ageing model Caenorhabditis elegans, are unlikely to be influencing human ageing (Kern and Gems, 2022; Kern et al., 2023; Kern et al., 2021).
Our new theory. Our new theory predicts that nutrient signalling is not an immediate driver of ageing, and instead is only a secondary mediator of triggered patho-pathways. Let us look at mTOR function in specific disease examples to illustrate this. In all the examples of age-related disease described above and in Table 2, mTOR is not the key driver of the disease. As an example, in endometrial hyperplasia, the immediate cause is an increase in the ratio of oestrogen to progesterone. Yes, mTOR excess function is a critical factor for such hyperplasia, but one that is downstream in the chain of events leading to pathology. To emphasise just how “downstream” the role of mTOR is, one can look at what happens when conversely there is too much progesterone vs oestrogen, as with the progesterone only birth control pill: atrophy of the endometrium. As other examples, in prostrate hyperplasia immediate drivers of pathology are DHT and inflammation. In the progression of fatty liver (see Table 2), immediate drivers are insulin resistance and lipotoxicity. In osteoporosis, immediate drivers are disrupted signalling cues to osteoclasts, like disrupted oestrogen levels. In diabetic cataracts (see Table 2) the immediate driver is too much glucose along with constraints on enzymatic conversion rates of its by-product sorbitol. In these examples, mTOR is a player in the development of patho-pathways. Yet mTOR has little to do with the initial cause of the patho-pathways – mTOR is merely one mediator of upstream triggers. Thus, nutrient signalling activity in late life, such as mTOR activity, is a secondary/downstream cause of age-related disease. In terms of testable predictions associated with this, in contrast to previous theories, our new framework predicts that nutrient signalling dysactivity in ageing will not occur gradually throughout life but instead will occur in mid to late life. And nutrient signalling dysactivity will typically originate from triggers outside the cells being affected, rather than from triggers originating within the affected cells (i.e. dysactivity will typically originate cell non-autonomously, rather than cell autonomously).
Evolutionary Theory in the Context of Our New Theory
A biomedical lens is not the only one through which age-related disease aetiology can be perceived. Theories that explain why ageing exists at the evolutionary level can also provide insights. The theory of Antagonistic Pleiotropy (AP) argues ageing is the consequence of a subset of wild-type genes that are pleiotropic, i.e. they influence two or more traits. These traits may have opposing (i.e. antagonistic) effects, such that their presence is associated with a trade-off. The combined costs of these trade-offs is “ageing” (Williams, 1957). AP genes associated with trade-offs where the cost appears later in life are more likely to be selected for during the course of evolution. This is because evolution works on the basis of survival of the fittest, and an earlier acting trait is more likely to have a larger impact on fitness (i.e. the ability of one organism to outcompete the other via reproductive success). Hence ageing (from trade-off costs) appears later in life. Data consistent with AP has been produced through a variety of studies such as laboratory artificial selection experiments, where animals (e.g. fruit fly Drosophila) when selected for enhanced lifespan, presented with reduced early life reproduction and vice versa (Stearns et al., 2001).
The concept of triggered patho-pathways is consistent with the theory of AP. Normal functioning of molecular pathways early in life is the benefit – the trade-off cost is the patho-pathway variants.
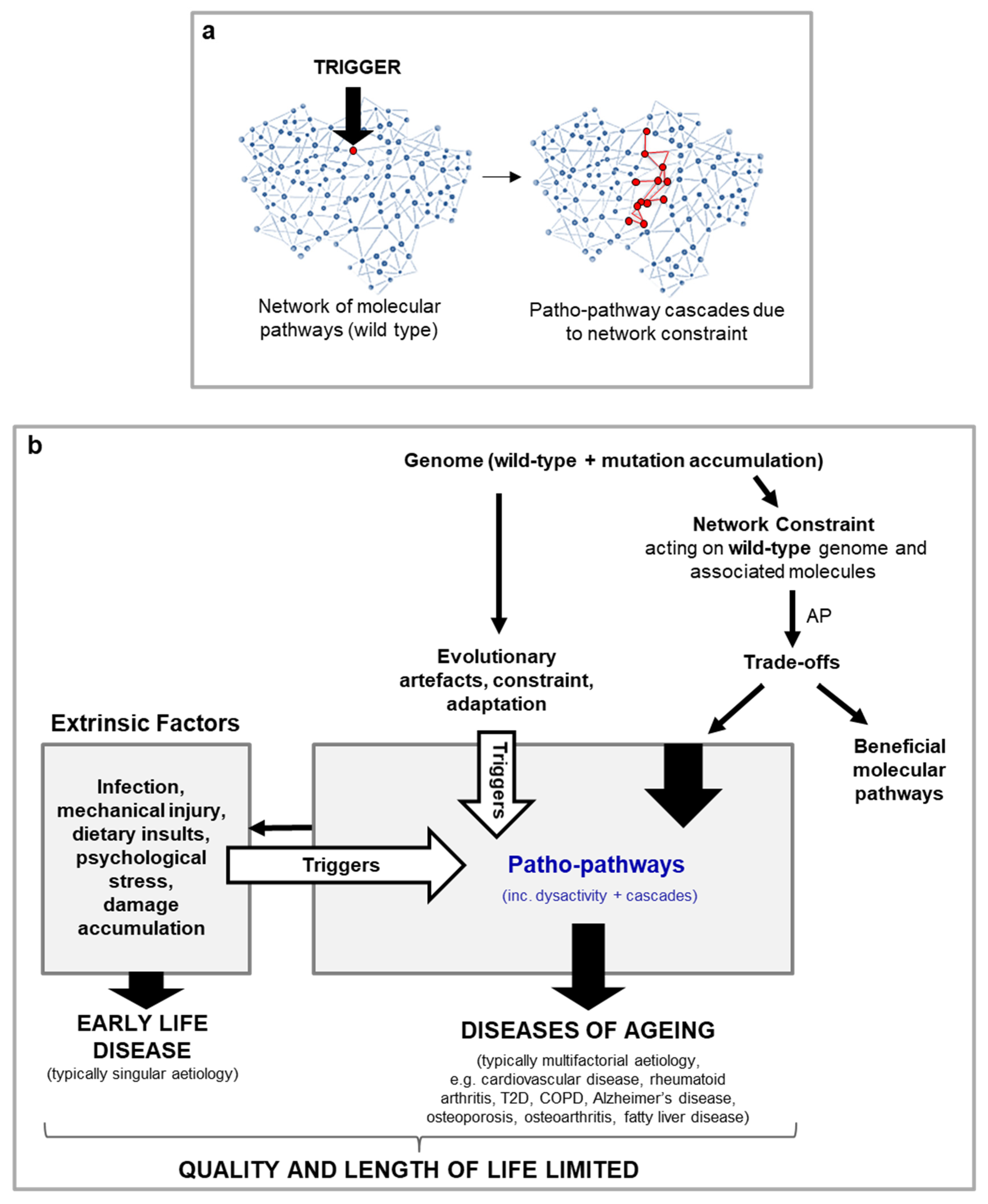
Why are patho-pathways so common such that they give rise to much of deleterious age-related disease, as in the examples above? What is the nature of the trade-off between beneficial molecular pathways vs their patho-pathway variants, such that benefit could not be uncoupled from cost over the course of evolution? The likely answer is that patho-pathways arise from an inability to optimise the function of all molecular pathway networks (at the molecular, cell, tissue and system level) across an organism in all contexts (cell and tissue types, genotypes, phenotypes and environmental conditions) – we term this network constraint. Network constraint translates to a risk of off-target side effects in the form of pathology when an additional “trigger” is present (Figure 4).
While in line with AP, the new concept of triggered patho-pathways requires an adjustment to the evolutionary theory. It is not simply a subset of genes that are AP genes, rather any and all genes in the human body (and their corresponding proteins and associated molecules) have the potential to be triggered (/activated) in a futile manner, and so incur cost in the form of patho-pathways. In other words, AP is feature of all wild-type genes and corresponding molecules. As an example, consider transformed breast epithelial tissues in metastatic breast cancer: here all the genes and molecules supporting cellular function have become pathogenic - all exhibit AP (Figure 4).
Whilst AP is a feature of all wild-type genes and molecules, all do not pose equal risk of generating age-related pathology. There are two key determinants of risk. First, presence of an appropriate trigger, such that dysactivity arises. Second, the cost to normal physiology after the gene/molecule is activated resulting in triggering of a patho-pathway. As such, we suggest a simple risk equation as follows
Equation 1. Calculating risk of pathology from futile triggering of wild-type genes and molecules as part of patho-pathways. Risk of pathology (Rp) is a function of the cost incurred when the gene/molecule is triggered in error (Ct), and the likelihood of the gene/molecule being triggered in error, i.e. the probability of the cost (pCt). Genes/molecules associated with regulatory and signalling proteins like mTOR and IIS are at high risk of contributing to pathology. If triggered in a futile manner, such genes/molecules will have multiple knock on effects, making Ct high. And, as such genes/molecules typically have multiple upstream mediators, pCt is also high.
- where Rp is risk of pathology, Ct is cost incurred when the gene is triggered in error and pCt the probability of the cost, which in turn is dependent on the probability of the trigger. Ct is predicted to be higher when a given gene/molecule plays a role in many different contexts (responding to environment, regulating reproductive status etc). An example of this is regulatory and signalling proteins, like the oestrogen receptor, that may serve multiple functions which, similar to building the circuitry of a machine or software coding, are at higher risk of signal cross-talk. Given triggered patho-pathways arise from trade-offs, trade-off functions are another useful means of mathematical representation. And to put the above another way, if one were to plot level of cost on the y axis and benefit on the x axis, network constraint would determine the shape of the trade-off function. Ecology, by determining fitness strategy, will determine trade-off intercept (and the presence of the trigger(s) determines when cost is incurred).
Figure 4.
A blueprint model for the causes of ageing and late-life disease (hypothetical scheme). a, Illustration of network constraint demonstrating how the presence of a trigger induces patho-pathways due to unavoidable interlinkages within and between molecular pathways. Blue: normal function. Red: pathological function. b, Overview of blueprint model working across scientific disciplines, from evolutionary biology through to medicine and including biological mechanisms (genetics, molecular, cellular, physiological). Patho-pathways are the futile activity (dysactivity) of molecular pathways in the wrong time and/or place. The beneficial action of the same molecular pathways in their adapted and intended form (i.e. function in the right time and at the right place) cannot be uncoupled from their patho-pathway variants due to network constraint, underlying the basis of an AP trade-off. Patho-pathway dysactivity causes dysactivity at higher orders of biological organisations, e.g. cell, tissue, organ and system. At these higher levels of biological organisations, dysactivity translates to pathology phenotypes in the form of hypertrophy, hypersecretion, dystrophy, dysplasia, transdifferentiation etc. And in turn this disrupts organ and system structure and function to cause disease.
Figure 4.
A blueprint model for the causes of ageing and late-life disease (hypothetical scheme). a, Illustration of network constraint demonstrating how the presence of a trigger induces patho-pathways due to unavoidable interlinkages within and between molecular pathways. Blue: normal function. Red: pathological function. b, Overview of blueprint model working across scientific disciplines, from evolutionary biology through to medicine and including biological mechanisms (genetics, molecular, cellular, physiological). Patho-pathways are the futile activity (dysactivity) of molecular pathways in the wrong time and/or place. The beneficial action of the same molecular pathways in their adapted and intended form (i.e. function in the right time and at the right place) cannot be uncoupled from their patho-pathway variants due to network constraint, underlying the basis of an AP trade-off. Patho-pathway dysactivity causes dysactivity at higher orders of biological organisations, e.g. cell, tissue, organ and system. At these higher levels of biological organisations, dysactivity translates to pathology phenotypes in the form of hypertrophy, hypersecretion, dystrophy, dysplasia, transdifferentiation etc. And in turn this disrupts organ and system structure and function to cause disease.
Blueprint Maps of Triggered Patho-Pathways in Wider Aetiological Webs
Complex though they are, triggered patho-pathway cascades represent small parts of much wider webs of disease aetiology. The highly complex and interconnected nature of late-life disease can present ageing as a hopelessly intractable condition. Yet, in principle, it ought to be possible to map out the interconnected webs of causation, thereby creating a Blueprint Map. For an example of a Blueprint Map see case study 1 and Figure 1b.
Such Blueprint Maps are a means to go beyond studying distinctive features, i.e. Hallmarks, of ageing, as they allow for hallmarks to be placed into the context of age-related disease. Hallmarks may act as causal drivers of a disease (acting as triggers of patho-pathways, being part of patho-pathways or being part of dysactivity associated with patho-pathways (see steps 1-4 in Figure 5)). Hallmarks may also play more of a contributor role or play no causal role and merely be symptomatic outcomes that may or may not worsen disease outcome. Notably, different Hallmarks likely play different roles in different age-related diseases as well as at different stages of age-related disease. As specific examples, genomic instability (from replication errors or environmental insults) is a trigger in tumour and cancer formation, inducing patho-pathways and dysactivity associated with unregulated cell division and metastasis. Genomic instability can also lead to senescence which can contribute to chronic fibrotic diseases(Yang et al., 2010). Dysbiosis, via contributing to systemic inflammation, plays more of a trigger role in neurodegerative disease associated with neuroinflammation that contributes to Alzhmier’s and Parkinson’s disease. For instance, neuroinflamation via patho-pathways inducing microglia hyperactivation contribute to Alzhmier’s (Table 2). Deregulated nutrient-signalling is often a mediator of patho-pathways as detailed earlier. Loss of proteostasis is an outcome of lipotoxicity, following patho-pathways of insulin resistance and associated dysactivity in NAFLD (Baiceanu et al., 2016) (Table 2).
Changing Clinical Practice and Scientific Approach: Triggered Patho-Pathway Cascades Are Amenable to Medical Intervention
The newly identified pathological principle of “triggered patho-pathways” serves as a tool to create Blueprint Maps in intricate detail. By understanding the aetiological webs that drive ageing, opportunities for interventions to improve health come into view. For example, using case study 1, we predict that hormone replacement therapy in post-menopausal women will be an effective treatment against periodontitis. When searching the literature, this is supported by correlation data although causality has not been established. As such, Blueprint Maps of triggered patho-pathways are a potential means for rational design of long-distance intervention trials that may potentially lead to therapeutic approaches.
More broadly, there are three types of approaches one can take to utilise Blueprint Maps. (i) Identify and target the initiating triggers of patho-pathways, i.e. prevent a cause far upstream in the cascade of the immediate cause of a disease. This may be particularly effective where trigger amplification has occurred, as in the triggering of metabolic syndrome by inflammageing (Table 2). (ii) Eliminate as many secondary systemic triggered patho-pathways as possible - given that our new theory reveals that much of age-related disease is a secondary consequence of initial triggers due to patho-pathway cascades that arise in a cell non-autonomous manner. One way to do this is via readjusting cell signalling cues in blood; this is likely why parabiosis (linking of the blood supplies) of young and old mice works to alleviate some age-related pathology - at least as far as clonal animals, like clonal laboratory mice, are concerned. Another, likely more practical, approach here is to use drugs like rapamycin (sirolimus) that target key mediators of signal transduction (i.e. target genes/molecules where the likelihood of pathology is high, i.e. Ct is high in Equation 1). Such an approach can be likened to chemotherapy. Chemotherapy can target genes associated with cellular proliferation. The consequence is that highly proliferative cells not involved in the cancer, e.g. skin and hair, are most affected, while there is a smaller effect on more vital and slow dividing cells. General suppression of IIS/mTOR with a drug like rapamycin to treat ageing is likely to be beneficial in the same way - a balance has to be struck between preventing age-related pathological change and dampening all cellular function. But this will also come with side effects like reduced immunosuppression and wound healing (from mTOR hypofunction). As a specific example, rapamycin dampens bone remodelling and so delays fracture healing (Holstein et al., 2008), but multiple papers also suggest it is beneficial against osteoporosis. In other words, beneficial effects will be highly disease-specific, and there is risk of off-target side effects. (iii) Indentify and traget critical nodes, i.e. key patho-pathways, that lie at the heart of patho-pathway cascades to devise interventions that are effective across multiple multifactorial diseases. By strategically targeting these "key patho-pathways" within the system, the risk of systemic collapse can be mitigated (i.e. one can therapeutically decouple the trade-offs underlying network constraint). Notably, for all of these, Blueprint Maps enable bigger picture understanding of disease links and off-target side effects of therapeutics. Blueprint Maps can lead to pre-emptive steps to avoid unwanted off-target effects.
Predictions of the New Theory
An operative theory of ageing must be able to explain age-relate disease aetiology, as the new blueprint-theory does for many diseases. A further means to validate our new theory in this article is to see if it makes testable predictions, some of which may involve explaining past observations. Table 3 contains a number of such predictions, including the utility of BluePrint Maps in uncovering a new class of drugs to block cellular necrosis via inhibiting two patho-pathways triggered upon stress upstream of patho-pathway cascade changes detaild 50 years ago by (Fleckenstein et al., 1974) which predominantly drive cellular, and so tissue and organ, destruction(Kern et al., 2024) (Figure 6).
Conclusions
In this paper we describe a new blueprint-style theory of ageing, spanning multiple scientific disciplines. Our theory is supported by specific disease examples and testable predictions. The theory predicts that the primary driver of much of age-related diseases is the futile triggering of specific molecular pathways in a pathological manner, which we term patho-pathways. Patho-pathways cause pathology via inducing cell, tissue and system dysactivity (i.e. increase or change in normal biological activity). All wild-type (normal) genes and corresponding proteins (and associated molecules) have the potential to contribute to age-related diseases via patho-pathways. Specific risk of a wild-type gene or product giving rise to age-related disease can be formularised. In a domino-style effect, one patho-pathway typically triggers others, resulting in complex, but tractable, cascades. Significantly, mapping of these cascades is achievable, and the resulting Blueprint Maps enable identification of biomarkers and therapeutic targets far upstream in the causal chain of events. The approach may enable better predictions of disease occurrence far in advance, and provide explanations for unexplained correlations between seemingly disparate diseases. The approach can enable development of more effective prevention and treatment measures, where off-target effects side effects are minimised.
Appendix A

Table 1.
Glossary of key terms.
| Antagonistic pleiotropy (AP) | Where action of a given gene is both beneficial and detrimental to fitness. If the latter occurs later in life and is therefore subject to weaker selection, such a gene may be favoured by natural selection, and promote ageing (Williams, 1957). |
| Biological constraint | A property of organisms and/or their ecology that prevents the evolution of traits that would increase fitness. |
| Discrete stage patho-pathway cascade (New term) | A series of patho-pathways triggered in a causal chain by different, but linked, triggers (c.f. multi-stage patho-pathway cascade). |
|
Dysactivity (New term) |
Out of context abnormal or faulty cell activity, i.e. at the wrong time and/or place, where too much, too little, or the wrong type of activity occurs. Notably, this can include not only an increase in normal function such that it is above optimal for health, but also cells and tissues taking on new, unwanted activities, e.g. pathological cellular transdifferentiation. |
| Multi-stage patho-pathway cascade (New term) | Where a single underlying trigger drives multiple patho-pathways in succession, such that multiple stages can be distinguished (c.f. discrete stage patho-pathway cascade). |
| Network constraint (new term) | Where a given molecule acts in diverse contexts (cell and tissue types, genotypes, phenotypes, and environmental conditions) across signalling networks within an organism, such that optimisation of function in all contexts is not possible. (This is a type of biological constraint, c.f. biological constraint). |
| Patho-pathway cascade (New term) | A causal chain of patho-pathways, in which each triggers the next in the chain (c.f. multi-stage patho-pathway cascade and discrete patho-pathway cascade). |
| Wild-type | Genes maintained in the population, i.e. non atypical mutant. |
| Case Study 1: A Blueprint Map of T2D and periodontitis |
| As an illustration of a triggered patho-pathway aetiological web, consider the relationship between type II diabetes (T2D) and periodontitis, where working backwards we find both may be related to menopause-related endocrine changes in women >50 yrs. T2D can suppress the immune system via several different discrete stage patho-pathway cascades. This suppression of the immune system by T2D goes on to lead to infection such as periodontitis (severe gum infection that can destroy teeth and the bone that supports them). As an example, hyperglycaemia due to T2D leads to formation of advanced glycation end-products (AGEs). And this in turn induces patho-pathways in macrophages, causing a shift of class frequency. M1 macrophages that encourage inflammation are reduced, and M2 macrophages that favour tissue repair are increased (He et al., 2020). This leads to a reduction in microbicidal capacity which, along with reduced blood flow, increases risk of infection and impairs wound healing. Reduction in blood flow results from several causes, including increased blood viscosity from the hyperglycaemia and change in arterial wall tension from AGEs and triggered patho-pathway inflammation that causes vascular endothelial cell dysfunction (Cinar et al., 2001; Hadi and Suwaidi, 2007). To illustrate how complex the picture is, infection risk from T2D-associated immune suppression in the mouth is further compounded by hyposalivation (possibly due to T2D associated impaired blood flow to the salivary glands) and an increase in salivary glucose levels (resulting from elevated plasma glucose) which promotes bacterial growth (Al-Maskari et al., 2011; Pérez-Ros et al., 2021). Hyposalivation contributes to infection risk as saliva contains antimicrobial proteins and peptides, and acts as a mechanical barrier preventing adhesion of microbes to the surface of the oral mucosa. Moreover, a lack of saliva production causes stagnation in flow; any stagnation in flow increases risk of infection via bacterial build-up from a lack of mechanical washing (Iwabuchi et al., 2012). Bacterial build-up both directly destroys tissue, and has indirect effects such as lodging in between the gum and enamel thereby reducing adherence of gum to the enamel (leading to gum recession). When tracing out the cascades, what becomes apparent is that one might expect that periodontitis must be especially high in post-menopausal women. This is because the menopause increases risk of T2D as well as can directly contribute to periodontitis. Regarding menopause and its relation to T2D, endocrine changes during menopause initiate patho-pathways of insulin resistance that closely mimic the cascades of insulin resistance seen in more general inflammaging (Table 2, Figure 3b). Oestrogen is known to play a role in regulating insulin sensitivity and glucose metabolism. A decline in oestrogen causes elevated blood sugar levels that give rise to T2D via patho-pathways (Ou et al., 2023) with a decline in pancreatic insulin production (Godsland, 2005), preventing glucose from entering stores like muscle and the adipose tissue. Simultaneously, low oestrogen triggers patho-pathways of liver gluconeogenesis while decreasing glucose catabolism (Shen and Shi, 2015). Regarding menopause’s direct contribution to periodontitis, oestrogen deficiency induces triggered patho-pathways of bone resorption contributing to loss of bone that supports teeth, and oestrogen decline affects gingiva (gum) integrity (Bhardwaj and Bhardwaj, 2012; Kovacs, 2016). The latter arises from yet another triggered patho-pathway change: oestrogen controls proliferation and differentiation of keratinocytes and fibroblasts in the gingiva, with low oestrogen favouring cell shedding (Bhardwaj and Bhardwaj, 2012). This complex example illustrates how diverging pathological outcomes from one condition in an aetiological web can reconverge to generate another disease (here periodontitis). In other words, many diseases can converge to form one disease and one disease can proliferate to generate many more in a domino-style effect. |
Table 2.
Multi-stage vs discrete stage patho-pathway cascade examples.
| Multi-stage patho-pathway cascade example | Discrete stage patho-pathway cascade example |
|---|---|
|
Non-alcoholic fatty liver disease (NAFLD) Key trigger: insulin resistance The progression of NAFLD entails the sequential action of insulin resistance-triggered patho-pathways (Figure 3b). A major initiator of these is inflammation in adipose tissue, typically a consequence of obesity and/or inflammaging. [Obesity, with fat accumulation causes adipocytes to reach their limit of expansion. This induces hypoxia which triggers proinflammatory patho-pathways (Sears and Perry, 2015) that mimic effects of inflammaging]. Steps are as follows: (i) Inflammation triggers patho-pathways of adipocyte insulin resistance. This causes increased export of free fatty acids (FFA) from the adipocytes and into the circulation (stage 1). (ii) FFA are then taken up by other organs that are not adapted to store large amounts of fat, such as the liver. This in turn leads to lipotoxicity (lipid-induced dysfunction) in those organs, which triggers localised patho-pathways of insulin resistance at those sites (stage 2) (Adams et al., 2005; Sears and Perry, 2015; Unger, 2003). It is here that the systemic consequences of insulin resistance really start to take effect, as hepatic insulin resistance and lipid accumulation trigger a patho-pathway positive feedback loop, driving the development of the metabolic syndrome (Figure 3b). To compound these changes, insulin resistance impairs control of blood glucose levels, further triggering type II diabetes (T2D) (Adams et al., 2005; Chen et al., 2017; Unger, 2003). Specifically, in the liver insulin resistance-associated patho-pathways cause excess glucose secretion into the blood, and insulin resistance patho-pathways in muscle reduce glucose uptake, which together pathologically increase blood glucose levels, to induce T2D. In conclusion, Although NAFLD involves different organs (particularly adipose tissue and liver), the tight causal relationship between patho-pathway changes define a multi-stage patho-pathway cascade. |
Non-alcoholic steatohepatitis (NASH) Key trigger: Lipotoxicity associated injury Returning to NAFLD (Figure 3b), lipotoxicity increases the susceptibility of the liver to injury (i.e. primes the liver c.f. Figure 2a). As a discrete stage cascade, upon injury, severe inflammation and fibrosis patho-pathways are triggered as an exaggerated wound healing response. (Specifically, proinflammatory cytokines induce patho-pathways of dysactivity in innate immune cells which directly damage tissue. Concomitantly these cytokines trigger patho-pathways of dysactivity in fibroblasts which causes the fibroblasts to secrete too much extracellular matrix including collagen - the resulting fibrosis contributes to liver failure via disruption of the liver architecture and function) (Figure 3d). Overall, this can result in disease progression to NASH, an aggressive form of fatty liver disease marked by liver inflammation, and advanced scarring and fibrosis (i.e. cirrhosis) (Adams et al., 2005). |
|
Neurodegeneration Key trigger: inflammation General systemic inflammation, from causes such as those that give rise to NAFLD (Figure 3b), can then go on to further trigger neurodegeneration via patho-pathways associated with microglia dysactivity. As a specific example, increasing evidence indicates that systemic inflammation might drive the initiation and progression of Alzheimer’s disease as immune cues can pass into the brain through various routes (e.g. via the circumventricular organs, across the brain barriers, through activating vascular cells at the brain barriers and even neural routes like cytokines from the thoracic-abdominal cavity (e.g. Kupffer cells in the liver) directly activating the vagal nerve which signals to the brain (Xie et al., 2021)). Neuroinflammation in turn contributes to Alzheimer’s disease (Heppner et al., 2015; Sala Frigerio et al., 2019) via microglia hyperactivation as it exacerbates amyloid beta and tau pathologies that in turn cause cognitive decline (Ising et al., 2019). Likewise Parkinson’s disease is associated with neuroinflammation that systemic inflammation contributes to (Wang et al., 2015). | |
|
Diabetic cataracts Key trigger: excessive glucose Insulin resistance leading to T2D (Unger, 2003), can in turn cause diabetic cataracts as a part of a discrete stage patho-pathway cascade. Diabetic cataracts develop due to patho-pathways associated with excessive conversion of glucose into sorbitol, i.e. at a rate higher than sorbitol can be converted into fructose by the enzyme sorbitol dehydrogenase. The resulting build-up of sorbitol then generates increased osmotic stress in the lens fibres, causing them to swell and rupture (Pollreisz and Schmidt-Erfurth, 2010). | |
|
Cellular Senescence accumulation Key trigger: inflammation Another example of a discrete-stage patho-pathway cascade involves immunosenescence and accumulation of cells that have undergone cellular senescence (cell cycle arrest, and entry into a hypertrophic, hypersecretory state) (Prata et al., 2018) due to insulin resistance. Throughout life, fibroblasts undergo these type of differentiative changes to support wound healing (Demaria et al., 2014). Such “senescent” fibroblasts are subsequently cleared by the immune system, triggered by signals within the fibroblast senescence-associated secretory phenotype (SASP). Any factor that compromises efficiency of immune clearance of senescent fibroblasts, such as the insulin resistance that contributes to and aggravates systemic inflammation (see Figure 3b), results in path-pathway variants of this - and the accumulation of senescent fibroblasts that go on to disrupt cellular microenvironments which, in mice at least, promotes diverse diseases of ageing (van Deursen, 2014). Secondary cellular senescence Key trigger: autocrine and paracrine signals In an additional discrete stage cascade, accumulating senescent cells can trigger patho-pathways in neighbouring cells that cause them to also undergo senescence (secondary senescence via autocrine and paracrine signals) (Admasu et al., 2021). |
Table 3.
Predictions of the new theory. Emphasis on Popperian ‘risky predictions’ (one that is improbable and would not be made in the absence of the theory).
Table 3.
Predictions of the new theory. Emphasis on Popperian ‘risky predictions’ (one that is improbable and would not be made in the absence of the theory).
| Predictions of the new theory | |
|---|---|
| Prediction | Supporting or Falsifying Evidence |
| It should be possible to create and utilise BluePrint Maps to uncover new therapeutic targets across age-related disease | As detailed above, creation of Blueprint Maps will allow identification of key endpoints where multiple triggers converge prior to initiating follow-on patho-pathway cascades. And identification of therapeutic targets is likely to be most effective where the same endpoint is prevalent across multiple age-related diseases and conditions. We examined multiple age-related diseases and conditions, and cellular necrosis came up as one such common endpoint. To focus on one organ here for simplicity and to illustrate the point, we can look at the kidney. Multiple stressors in acute and chronic kidney disease converge on the same endpoint which is cellular necrosis. This includes stressors like inflammation, oxidative stress, ischemia and nutrient deprivation (from reduced or lost blood flow), and nephrotoxic drugs. Once necrosis is initiated direct tissue and organ failure ensue, as well as secondary damage from cascades that trigger patho-pathways associated with cellular senescence, vascular rarefaction, presence of a chronic inflammatory infiltrate, glomerulosclerosis and fibrosis that in turn cause loss of kidney structure, function and accelerated kidney ageing (Ferenbach and Bonventre, 2015). More specifically, we uncovered that there are two common convergent endpoints of stressors during necrosis¸ with two associated patho-pathways triggered upstream to that detailed 50 years ago by(Fleckenstein et al., 1974). Here patho-pathway cascades drive cellular destruction, with our Blueprint Mapping revealing that to effectively block necrosis, the two patho-pathways must be simultaneously inhibited – which can be done using repurposed drug combinations. When used alongside different stressors, up to 90% necrosis inhibition was observed in vitro and ex-vivo, with the drug combinations representing the discovery of a new drug class: anti-necrosis agents(Kern et al., 2024). Notably, these poatho-pathways do not have a genetic source (i.e. they are not genetic programmes triggered) but result from molecular level changes with downstream loss of ion gradients triggering further patho-pathways in cascades that culminate in destruction. This includes what we detailed back in 1996, including phospholipase, calpain and cathepsin futile dysactivity (Bonventre, 1996; Yamashima et al., 1996)). Anti-necrosis agents represent the first of likely many drug discoveries using the Blueprint theory. Notably, similar mapping can predict GLP-1 agonsists, such as semaglutide’s (Ozempic, Rybelsus, Wegovy) role in alleviating accelerated aging observed with obesity or potentially inflammaging (c.f. Figure 3e; Table 2). |
|
Nutrient signalling pathway dysactivity in-vivo will occur in mid to late life, rather than increasing gradually throughout life |
To be tested |
|
Nutrient signalling pathway dysactivity will typically originate cell non-autonomously, rather than cell autonomously |
To be tested |
|
Ageing and age-related disease is not restricted to old ages – ageing can be “triggered” in the young If age-related disease can indeed be “triggered”, age-related disease alongside their associated patho-pathway cascades, should be observable in the young |
Supporting evidence:
|
|
Resolving previous paradoxes: calcium paradox and reperfusion paradox at menopause The calcium paradox in disease and menopause dates back to the 1990s(Persy et al., 2006) and details the relationship between bones resorption and bone calcium loss alongside simultaneous ectopic calcification, particularly in the arteries. Why would evolution allow for this? Adding to the paradox is the fact that frequently blood calcium is not necessarily that elevated at the time of ectopic calcification, e.g. in menopause a lack of increased PTH/PTHrP results in calcium wasting, where calcium is excisively lost in urine (Kovacs, 2016). The reperfusion paradox in surgery and organ preservation details high levels of apoptosis following the return of temperature and/or oxygen to tissue following stress. What is paradoxical is the lack of apoptosis during stress, but the apoptosis induced injury post reperfusion. In contrast necrosis typically arises during stress. |
Network constraint explains the calcium paradox through the triggering of futile patho-pathways that trigger osteoclast bone resoprption and simultaneous calcification in areas like arteries. Notably, it is not passive calcium deposition in arteries, but arterial cells pathologically transdifferentiate and become “bone like”. The link between the two disorders stems only in part from bone loss pushing calcium into blood, which aggravates ectopic calcification (it is one of the patho-pathway triggers). Why does blood calcium and arterial calcification not always correlate? Because, as previously detailed, other triggers that “prime tissue” and “spark” pathology (Figure 2a) are what ultimately drive the condition (even when blood calcium is not that elevated), and many of these are the same triggers that cause bone loss: inflammation, high blood glucose and high blood lipid (Abedin et al., 2004; Chen and Moe, 2012; Demer, 2002; Virchow, 1863). Conditions like menopause and obesity result in systemic inflammation, and osteoclasts and are monocyte-macrophage lineage progenitor cells designed to respond to inflammatory cues, but this risks off target side effects; the phenotypic plasticity of smooth muscle cells opens them to the risk of pathological transdifferentiation. High blood glucose and lipid are typically part and parcel of systemic infmation (Figure 3b). Reperfusion paradox: By mapping out the patho-pathways associated with necrosis and identifying the key patho-pathway underlying cell death (Kern et al., 2024), this paradox can be resolved. In short, the triggering of this necrosis patho-pathway during stress may cause necrosis or milder damage that goes on to be picked up by apoptotic pathways. Specifically, when the necrosis patho-pathway is triggered in a severe enough manner during stress, cells secumb to necrosis. When the necrosis patho-pathway is triggered to a milder extent, the damage is not severe to cause immediate cell death necrosis. This milder damage lies latent, until reperfusion (readminstration of temeperature and blood supply) causes an awakening of cell activity and the damage is picked up by apoptotoic pathways (geared to prevent damaged cells form replicating and increasing risk of tumours) and so apoptosis ensues. By blocking the necrosis patho-pathway both necrosis and the “necrosis patho-pathway damage-induced apoptosis”, i.e. much of the reperfusion injury, can be mitigated(Kern et al., 2024). |
|
Observation of age-related increases in genetic variance for fitness components and in inbreeding load Genetic variance. Genetic variance is the difference in DNA sequences between individuals within a population. The more genes involved in generating ageing and related disease, the greater the likelihood of genetic variance being observed for fitness components. Previous proximate mechanism models of Antagonistic Pleiotropy (AP) assume ageing stems from a subset of AP genes. In contrast, triggered patho-pathways and associated cascades do not assume that a small number of AP genes control fitness changes at both early and late stages of life. Cascades arising in later life entail the futile, unregulated triggering of whole hosts of genes and activation of molecules (and many genes have the potential to be a pathology-inducing AP gene). Inbreeding load. If only a subset of AP genes are driving ageing, age-related increases in inbreeding load (the negative consequences of recessive allele build-up) is not expected. In our new theory, however, recessive alleles can act as any other extrinsic or extrinsic factor that triggers patho-pathways due to network constraint. This means that an age-related increase in inbreeding load is expected. Additionally, we should be able to find specific medical examples of mutated alleles inducing disease via triggering patho-pathways. |
Supporting evidence: Observations of age-related change in genetic variance for fitness components and in inbreeding load has mostly been done in invertebrates such as Drosophila. The results show that genetic variation and inbreeding effects increase dramatically with age (Borash et al., 2007; Charlesworth, 2001; Escobar et al., 2008; Everman and Morgan, 2018; Gong et al., 2006; Hughes et al., 2002; Keller et al., 2008; Lesser et al., 2006; Reynolds et al., 2007; Swindell and Bouzat, 2006), in support of our new theory. And there are specific medical examples of mutated alleles inducing disease via triggering patho-pathways. As one such example, the frequency of the mutated allele in haemophilia and so the incidence of the disorder, was greater among the royal families of Europe due to the high levels of royal inbreeding. Haemophilia causes recurring bleeding including into joints that triggers haemophilic arthropathy due to consequential joint inflammation. This joint inflammation initiates patho-pathways of synovial hypertrophy and other associated cascades as in Figure 3a, with haemophilic arthropathy closely resembling the pathological mechanisms seen in rheumatoid arthritis (Knobe and Berntorp, 2011). |
|
Common versus rare alleles as drivers of ageing Previous proximate mechanism models of AP assume ageing stems from a subset of AP genes. If true, genetic variation in ageing should be mainly due to alleles that segregate at intermediate frequencies. This contrasts our new theory. Not just common alleles, but also rare alleles could be triggered with increase over time in an individual as part of patho-pathway cascades and so contribute to ageing. Expression of rare alleles may also act as patho-pathway triggers directly or indirectly via their knock-on effects. If true, there should be medical examples of this. |
Testing the prediction that genetic variation in ageing should be mainly due to alleles that segregate at intermediate frequencies has proved to be challenging, so there is no consensus (Carbone et al., 2006; De Luca et al., 2003; Kelly, 1999; Kelly, 2003; Kelly, 2008; Macdonald and Long, 2007). But in terms of medical examples of rare alleles accelerating age-related disease, there are examples as in the case of Haemophilia above. |
|
Do similar or different genomic regions control variance at early and late ages? Previous proximate mechanism models of AP assume that similar genomic regions will control variance in the phenotype at early and late ages, due to a small number of AP genes. In contrast, late life action of triggered patho-pathways and cascades would mean that different genomic regions would, in fact, be predicted to contribute to variation at early and late ages. Presence of triggered patho-pathways means genes exerting effects at only very specific times during youth for fitness benefits may be more frequently activated in a futile manner late in life. For example, genes and molecules associated with an immune response will only be triggered when an infection is sensed in youth, but may be constantly expressed later in life as part of an autoimmune disease such as rheumatoid arthritis. |
Supporting evidence: Results of quantitative trait locus (QTL) mapping studies in Drosophila and mice were consistent with our new theory (Curtsinger and Khazaeli, 2002; Leips et al., 2006; Miller et al., 2005; Nuzhdin et al., 1997). There is also more recent work that is looking at using human genome wide association studies (GWAS) to test these predictions, with particular focus on long-term longitudinal studies, but sufficiently large data sets are required to attain reliable results here (Long and Zhang, 2019), which we currently lack. |
|
If network constraint is the reason behind why evolution is unable to uncouple trade-offs (i.e. separate cost from benefit) associated with patho-pathways, we should see evolutionary conservation of network constraint (i.e. all similar species should be subject to network constraint). While this is difficult to test for directly, we should see indirect evidence in the form of conservation of triggered patho-pathways and cascades across species |
Supporting evidence: This prediction is consistent with the presence of similar diseases of ageing in many mammalian species. As a specific example, canine rheumatoid arthritis shows patho-pathway cascades and stages that are very similar to those seen in human rheumatoid arthritis (Figure 3a) (Bennett, 1987; Innes & Clegg, 2010). |
|
If patho-pathways stem from network constraint, a prediction that follows is more broadly we should see a pattern where elements of biology associated with greater plasticity of function are expected to be at greater risk of inducing pathology (because they are subject to greater levels of network constraint). This prediction should hold true at all levels, from the molecular level up to cells and whole tissues and organs An easy means to test this is at the tissue and organ level – are tissue types and organs that show greater plasticity of strategy (such as those associated with active tissue remodelling during adulthood, e.g. to respond to infection, or for reproduction) more likely to be associated with disease? |
Supporting evidence: (i) In line with this prediction, structurally more static tissues e.g. myocardial and neurological, are less prone to pathology (Cooper et al., 2009; NCI, 2023). (ii) Sexual dimorphism in pathology presentation is also in line with this prediction. Typically the more “plastic” sex, i.e. females that undergo greater change in tissue status for reproduction, show greater susceptibility to some diseases of ageing both in laboratory model animals and humans. One example of this is intestinal pathology, where the female intestine undergoes remodelling for reproduction and lactation and as a consequence tends to be more prone to patho-pathways and pathology. For example, in the fruit fly Drosophila melanogaster intestinal degeneration and tumours are seen females but not males due to stem cell hyperproliferation in the female intestine (Regan et al., 2016). Female mice are also susceptible to more severe intestinal inflammation and autoimmune disorders compared to males (Livingston et al., 2004; McGee and Huttenhower, 2021). Similar trends are seen in humans, where women are more susceptible to certain gastrointestinal autoimmune diseases (e.g. irritable bowel syndrome) and gastrointestinal cancer (Chang and Heitkemper, 2002; Jemal et al., 2011; Kim et al., 2015; McGee and Huttenhower, 2021). |
References
- Abedin, M., Tintut, Y. and Demer, L.L., 2004. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 24, 1161-70.
- Acerenza, L., 2016. Constraints, Trade-offs and the Currency of Fitness. J Mol Evol. 82, 117-27. [CrossRef]
- Adams, L.A., Angulo, P. and Lindor, K.D., 2005. Nonalcoholic fatty liver disease. Canad. Med. Assoc. J. 172, 899-905.
- Adinolfi, L.E., Rinaldi, L., Guerrera, B., Restivo, L., Marrone, A., Giordano, M. and Zampino, R., 2016. NAFLD and NASH in HCV Infection: Prevalence and Significance in Hepatic and Extrahepatic Manifestations. Int J Mol Sci. 17. [CrossRef]
- Admasu, T.D., Rae, M. and Stolzing, A., 2021. Dissecting primary and secondary senescence to enable new senotherapeutic strategies. Ageing Res Rev. 70, 101412. [CrossRef]
- Al-Maskari, A.Y., Al-Maskari, M.Y. and Al-Sudairy, S., 2011. Oral Manifestations and Complications of Diabetes Mellitus: A review. Sult. Qab. Uni. med. J. 11, 179-186.
- Baiceanu, A., Mesdom, P., Lagouge, M. and Foufelle, F., 2016. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol. 12, 710-722. [CrossRef]
- Bhardwaj, A. and Bhardwaj, S.V., 2012. Effect of menopause on women’s periodontium. J. Midliife Health. 3, 5-9. [CrossRef]
- Blagosklonny, M.V., 2006. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 5, 2087-102. [CrossRef]
- Blagosklonny, M.V., 2012. Answering the ultimate question "what is the proximal cause of aging?". Aging (Albany NY). 4, 861-877.
- Bonventre, J.V., 1996. Roles of phospholipases A2 in brain cell and tissue injury associated with ischemia and excitotoxicity. Journal of Lipid Mediators and Cell Signalling. 14, 15-23. [CrossRef]
- Borash, D.J., Rose, M.R. and Mueller, L.D., 2007. Mutation accumulation affects male virility in Drosophila selected for later reproduction. Physiological and Biochemical Zoology. 80, 461-472. [CrossRef]
- Brown, R.J., Araujo-Vilar, D., Cheung, P.T., Dunger, D., Garg, A., Jack, M., Mungai, L., Oral, E.A., Patni, N., Rother, K.I., von Schnurbein, J., Sorkina, E., Stanley, T., Vigouroux, C., Wabitsch, M., Williams, R. and Yorifuji, T., 2016. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 101, 4500-4511. [CrossRef]
- Carbone, M.A., Jordan, K.W., Lyman, R.F., Harbison, S.T., Leips, J., Morgan, T.J., DeLuca, M., Awadalla, P. and Mackay, T.F., 2006. Phenotypic variation and natural selection at Catsup, a pleiotropic quantitative trait gene in Drosophila. Current Biology. 16, 912-919. [CrossRef]
- Chang, L. and Heitkemper, M.M., 2002. Gender differences in irritable bowel syndrome. Gastroenterology. 123, 1686-1701. [CrossRef]
- Charlesworth, B., 2001. Patterns of age-specific means and genetic variances of mortality rates predicted by the mutation-accumulation theory of ageing. Journal of Theoretical Biology. 210, 47-65. [CrossRef]
- Chen, N.X. and Moe, S.M., 2012. Vascular calcification: pathophysiology and risk factors. Curr. Hypertens. Rep. 14, 228-237. [CrossRef]
- Chen, Z., Yu, R., Xiong, Y., Du, F. and Zhu, S., 2017. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids in Health and Disease. 16, 203. [CrossRef]
- Cinar, Y., Senyol, A.M. and Duman, K., 2001. Blood viscosity and blood pressure: role of temperature and hyperglycemia. Am J Hypertens. 14, 433-8. [CrossRef]
- Cooper, G.S., Bynum, M.L. and Somers, E.C., 2009. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 33, 197-207. [CrossRef]
- Curtsinger, J.W. and Khazaeli, A.A., 2002. Lifespan, QTLs, age-specificity, and pleiotropy in Drosophila. Mechanisms of ageing and development. 123, 81-93.
- De Luca, M., Roshina, N.V., Geiger-Thornsberry, G.L., Lyman, R.F., Pasyukova, E.G. and Mackay, T.F., 2003. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nature genetics. 34, 429-433. [CrossRef]
- de Magalhães, J.P., 2005. Open-minded scepticism: inferring the causal mechanisms of human ageing from genetic perturbations. Ageing Res. Rev. 4, 1–22.
- de Magalhães, J.P. and Church, G.M., 2005. Genomes optimize reproduction: aging as a consequence of the developmental program. Physiology. 20, 252–259.
- Demaria, M., Ohtani, N., Youssef, S., Rodier, F., Toussaint, W., Mitchell, J., Laberge, R., Vijg, J., Van Steeg, H., Dollé, M., Hoeijmakers, J., de Bruin, A., Hara, E. and Campisi, J., 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 31, 722-733. [CrossRef]
- Demer, L.L., 2002. Vascular calcification and osteoporosis: inflammatory responses to oxidized lipids. International Journal of Epidemiology. 31, 737-741. [CrossRef]
- Distler, J.H.W., Wenger, R.H., Gassmann, M., Kurowska, M., Hirth, A., Gay, S. and Distler, O., 2004. Physiologic responses to hypoxia and implications for hypoxia-inducible factors in the pathogenesis of rheumatoid arthritis. Arthritis & Rheumatism. 50, 10-23. [CrossRef]
- Dørum, A., Tonstad, S., Liavaag, A.H., Michelsen, T.M., Hildrum, B. and Dahl, A.A., 2008. Bilateral oophorectomy before 50 years of age is significantly associated with the metabolic syndrome and Framingham risk score: a controlled, population-based study (HUNT-2). Gynecol Oncol. 109, 377-83. [CrossRef]
- Epplein, M., Reed, S.D., Voigt, L.F., Newton, K.M., Holt, V.L. and Weiss, N.S., 2008. Risk of Complex and Atypical Endometrial Hyperplasia in Relation to Anthropometric Measures and Reproductive History. Am. J Epidem. 168, 563-570. [CrossRef]
- Escobar, J.S., Jarne, P., Charmantier, A. and David, P., 2008. Outbreeding alleviates senescence in hermaphroditic snails as expected from the mutation-accumulation theory. Current Biology. 18, 906-910. [CrossRef]
- Everman, E.R. and Morgan, T.J., 2018. Antagonistic pleiotropy and mutation accumulation contribute to age-related decline in stress response. Evolution. 72, 303-317. [CrossRef]
- Fang, Y.P., Zhao, Y., Huang, J.Y., Yang, X., Liu, Y. and Zhang, X.L., 2024. The functional role of cellular senescence during vascular calcification in chronic kidney disease. Front Endocrinol (Lausanne). 15, 1330942. [CrossRef]
- Feil, S., Fehrenbacher, B., Lukowski, R., Essmann, F., Schulze-Osthoff, K., Schaller, M. and Feil, R., 2014. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circulation Research. 115, 662-667. [CrossRef]
- Ferenbach, D.A. and Bonventre, J.V., 2015. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nature Reviews Nephrology. 11, 264-276. [CrossRef]
- Fleckenstein, A., Janke, J., Döring, H.J. and Leder, O., 1974. Myocardial fiber necrosis due to intracellular Ca overload-a new principle in cardiac pathophysiology. Recent Adv Stud Cardiac Struct Metab. 4, 563-80.
- Gems, D., 2021. Understanding hyperfunction: an emerging paradigm for the biology of aging. Preprints. [CrossRef]
- Gems, D., 2022. The hyperfunction theory: an emerging paradigm for the biology of aging. Ageing. Res. Rev. 74, 101557. [CrossRef]
- Giles, J.T., Szklo, M., Post, W., Petri, M., Blumenthal, R.S., Lam, G., Gelber, A.C., Detrano, R., Scott, W.W., Kronmal, R.A. and Bathon, J.M., 2009. Coronary arterial calcification in rheumatoid arthritis: comparison with the Multi-Ethnic Study of Atherosclerosis. Arthritis Research & Therapy. 11, R36. [CrossRef]
- Glyn-Jones, S., Palmer, A.J., Agricola, R., Price, A.J., Vincent, T.L., Weinans, H. and Carr, A.J., 2015. Osteoarthritis. Lancet. 386, 376-387.
- Godsland, I.F., 2005. Oestrogens and insulin secretion. Diabetologia. 48, 2213-2220. [CrossRef]
- Gong, Y., Thompson Jr, J.N. and Woodruff, R., 2006. Effect of deleterious mutations on life span in Drosophila melanogaster. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 61, 1246-1252.
- Hadi, H.A.R. and Suwaidi, J.A., 2007. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 3, 853-876.
- Hamilton, W.D., 1966. The moulding of senescence by natural selection. J. Theor. Biol. 12, 12-45. [CrossRef]
- Hannemann, M.M., Alexander, H.M., Cope, N.J. and Acheson, N., 2007. Endometrial hyperplasia: a clinician’s review. Obst. Gyn. & Reproduct. Med. 17, 169-172.
- He, S., Hu, Q., Xu, X., Niu, Y., Chen, Y., Lu, Y., Su, Q. and Qin, L., 2020. Advanced glycation end products enhance M1 macrophage polarization by activating the MAPK pathway. Biochem Biophys Res Commun. 525, 334-340. [CrossRef]
- Heppner, F.L., Ransohoff, R.M. and Becher, B., 2015. Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience. 16, 358-372. [CrossRef]
- Hibler, E.A., Kauderer, J., Greene, M.H., Rodriguez, G.C. and Alberts, D.S., 2016. Bone loss after oophorectomy among high-risk women: an NRG oncology/gynecologic oncology group study. Menopause (New York, N.Y.). 23, 1228-1232. [CrossRef]
- Holstein, J.H., Klein, M., Garcia, P., Histing, T., Culemann, U., Pizanis, A., Laschke, M.W., Scheuer, C., Meier, C., Schorr, H., Pohlemann, T. and Menger, M.D., 2008. Rapamycin affects early fracture healing in mice. British journal of pharmacology. 154, 1055-1062. [CrossRef]
- Hughes, K.A., Alipaz, J.A., Drnevich, J.M. and Reynolds, R.M., 2002. A test of evolutionary theories of aging. Proceedings of the National Academy of Sciences. 99, 14286-14291.
- Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S.V., Vieira-Saecker, A., Schwartz, S., Albasset, S., McManus, R.M. and Tejera, D., 2019. NLRP3 inflammasome activation drives tau pathology. Nature. 575, 669-673. [CrossRef]
- Iwabuchi, H., Fujibayashi, T., Yamane, G., Imai, H. and Nakao, H., 2012. Relationship between Hyposalivation and Acute Respiratory Infection in Dental Outpatients. Gerontology. 58, 205-211. [CrossRef]
- Jemal, A., Bray, F., Center, M.M., Ferlay, J., Ward, E. and Forman, D., 2011. Global cancer statistics. CA: a cancer journal for clinicians. 61, 69-90.
- Karavassilis, M.E. and Faragher, R., 2013. A relationship exists between replicative senescence and cardiovascular health. Longev Healthspan. 2, 3. [CrossRef]
- Karmakar, S., Kay, J. and Gravallese, E.M., 2010. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum. Dis. Clin. North Am. 36, 385-404. [CrossRef]
- Keller, L., Reid, J. and Arcese, P., 2008. Testing evolutionary models of senescence in a natural population: age and inbreeding effects on fitness components in song sparrows. Proceedings of the Royal Society B: Biological Sciences. 275, 597-604. [CrossRef]
- Kelly, J.K., 1999. An experimental method for evaluating the contribution of deleterious mutations to quantitative trait variation. Genetics Research. 73, 263-273. [CrossRef]
- Kelly, J.K., 2003. Deleterious mutations and the genetic variance of male fitness components in Mimulus guttatus. Genetics. 164, 1071-1085. [CrossRef]
- Kelly, J.K., 2008. Testing the rare-alleles model of quantitative variation by artificial selection. Genetica. 132, 187-198. [CrossRef]
- Kern, C.C. and Gems, D., 2022. Semelparous Death as one Element of Iteroparous Aging Gone Large. Front Genet. 13, 880343. [CrossRef]
- Kern, C.C., Justin, A.W., Davis, B., Faragher, R., Peterson, J., Roch, A., Anstett, V., Denney, S., Grzesiak, N., Winfield, I., Cronk, D., Winfield, N., Brandenberg, N., Stebbing, J. and Bonventre, J., 2024. Anti-necrosis agents as a new class of drugs. In preperation.
- Kern, C.C., Srivastava, S., Ezcurra, M., Hsiung, K.C., Hui, N., Townsend, S., Maczik, D., Zhang, B., Tse, V., Konstantellos, V., Bähler, J. and Gems, D., 2023. C. elegans ageing is accelerated by a self-destructive reproductive programme. Nature Communications. 14, 4381. [CrossRef]
- Kern, C.C., Townsend, S., Salzmann, A., Rendell, N., Taylor, G., Comisel, R.M., Foukas, L., Bähler, J. and Gems, D., 2021. C. elegans feed yolk to their young in a form of primitive lactation. Nature Communications. 12, 5801. [CrossRef]
- Kim, S.-E., Paik, H.Y., Yoon, H., Lee, J.E., Kim, N. and Sung, M.-K., 2015. Sex-and gender-specific disparities in colorectal cancer risk. WJG. 21, 5167.
- Knobe, K. and Berntorp, E., 2011. Haemophilia and joint disease: pathophysiology, evaluation, and management. J Comorb. 1, 51-59. [CrossRef]
- Kovacs, C.S., 2016. Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiological reviews. [CrossRef]
- Leips, J., Gilligan, P. and Mackay, T.F., 2006. Quantitative trait loci with age-specific effects on fecundity in Drosophila melanogaster. Genetics. 172, 1595-1605. [CrossRef]
- Lesser, K.J., Paiusi, I.C. and Leips, J., 2006. Naturally occurring genetic variation in the age-specific immune response of Drosophila melanogaster. Aging cell. 5, 293-295. [CrossRef]
- Livingston, R.S., Myles, M.H., Livingston, B.A., Criley, J.M. and Franklin, C.L., 2004. Sex influence on chronic intestinal inflammation in Helicobacter hepaticus-infected A/JCr mice. Comp Med. 54, 301-8.
- Long, E. and Zhang, J., 2019. Retesting the influences of mutation accumulation and antagonistic pleiotropy on human senescence and disease. Nat Ecol Evol. 3, 992-993. [CrossRef]
- López-Otín, C., Blasco, M.A., Partridge, L., Serrano, M. and Kroemer, G., 2023. Hallmarks of aging: An expanding universe. Cell. [CrossRef]
- Macdonald, S.J. and Long, A.D., 2007. Joint estimates of quantitative trait locus effect and frequency using synthetic recombinant populations of Drosophila melanogaster. Genetics. 176, 1261-1281. [CrossRef]
- Maklakov, A.A. and Chapman, T., 2019. Evolution of ageing as a tangle of trade-offs: energy versus function. Proc Biol Sci. 286, 20191604. [CrossRef]
- Martin, G., 1978. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. 14, 5–39.
- McGee, J.S. and Huttenhower, C., 2021. Of Mice and Men and Women: Sexual Dimorphism of the Gut Microbiome. IJW Derma. [CrossRef]
- Medawar, P.B., 1952. An Unsolved Problem Of Biology, H.K. Lewis, London.
- Miller, R.A., Berger, S.B., Burke, D.T., Galecki, A., Garcia, G.G., Harper, J.M. and Sadighi Akha, A.A., 2005. T cells in aging mice: genetic, developmental, and biochemical analyses. Immunological reviews. 205, 94-103. [CrossRef]
- Nacusi, L.P. and Tindall, D.J., 2011. Targeting 5alpha-reductase for prostate cancer prevention and treatment. Nat Rev Urol. 8, 378-84. [CrossRef]
- NCI, 2023. Surveillance, Epidemiology, and End Results Program http://seer.cancer.gov/.
- Nuzhdin, S.V., Pasyukova, E.G., Dilda, C.L., Zeng, Z.-B. and Mackay, T.F., 1997. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proceedings of the national academy of Sciences. 94, 9734-9739. [CrossRef]
- Ou, Y.J., Lee, J.I., Huang, S.P., Chen, S.C., Geng, J.H. and Su, C.H., 2023. Association between Menopause, Postmenopausal Hormone Therapy and Metabolic Syndrome. J Clin Med. 12. [CrossRef]
- Pérez-Ros, P., Navarro-Flores, E., Julián-Rochina, I., Martínez-Arnau, F.M. and Cauli, O., 2021. Changes in Salivary Amylase and Glucose in Diabetes: A Scoping Review. Diagnostics (Basel). 11. [CrossRef]
- Persy, V., De Broe, M. and Ketteler, M., 2006. Bisphosphonates prevent experimental vascular calcification: Treat the bone to cure the vessels? Kidney Int. 70, 1537-8.
- Pollreisz, A. and Schmidt-Erfurth, U., 2010. Diabetic cataract-pathogenesis, epidemiology and treatment. J Ophthalmol. 2010, 608751.
- Pollycove, R., Naftolin, F. and Simon, J.A., 2011. The evolutionary origin and significance of menopause. Menopause. 18. [CrossRef]
- Prata, L.G.P.L., Ovsyannikova, I.G., Tchkonia, T. and Kirkland, J.L., 2018. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Seminars in immunology. 40, 101275-101275. [CrossRef]
- Regan, J., Khericha, M., Dobson, A., Bolukbasi, E., Rattanavirotkul, N. and Partridge, L., 2016. Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. Elife. 5, e10956. [CrossRef]
- Reynolds, R.M., Temiyasathit, S., Reedy, M.M., Ruedi, E.A., Drnevich, J.M., Leips, J. and Hughes, K.A., 2007. Age specificity of inbreeding load in Drosophila melanogaster and implications for the evolution of late-life mortality plateaus. Genetics. 177, 587-595.
- Rong, J.X., Shapiro, M., Trogan, E. and Fisher, E.A., 2003. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 100, 13531-6. [CrossRef]
- Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., Mancuso, R., Chen, W., Woodbury, M. and Srivastava, G., 2019. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep 27: 1293–1306. e6.
- Sapey, E., Greenwood, H., Walton, G., Mann, E., Love, A., Aaronson, N., Insall, R.H., Stockley, R.A. and Lord, J.M., 2014. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 123, 239-48. [CrossRef]
- Sears, B. and Perry, M., 2015. The role of fatty acids in insulin resistance. Lipids Health Dis. 14, 121-121. [CrossRef]
- Shen, M. and Shi, H., 2015. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. International Journal of Endocrinology. 2015, 294278. [CrossRef]
- Stearns, S., Partridge, L., Masoro, E. and Austad, S., 2001. Handbook of the Biology of Aging.
- Strenk, S.A., Strenk, L.M. and Koretz, J.F., 2005. The mechanism of presbyopia. Prog. Retin. Eye Res. 24, 379-393. [CrossRef]
- Swindell, W.R. and Bouzat, J.L., 2006. Inbreeding depression and male survivorship in Drosophila: implications for senescence theory. Genetics. 172, 317-327. [CrossRef]
- Tong, Y. and Zhou, R.Y., 2020. Review of the Roles and Interaction of Androgen and Inflammation in Benign Prostatic Hyperplasia. Mediators Inflamm. 2020, 7958316. [CrossRef]
- Unger, R.H., 2003. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 144, 5159-65. [CrossRef]
- Untergasser, G., Madersbacher, S. and Berger, P., 2005. Benign prostatic hyperplasia: age-related tissue-remodeling. Exp Gerontol. 40, 121-128. [CrossRef]
- van Deursen, J.M., 2014. The role of senescent cells in ageing. Nature. 509, 439-446.
- Virchow, R., 1863. Cellular Pathology, as Based upon Physiological and Pathological Histology, Dover Publications, Inc, New York, NY. [CrossRef]
- Voynow, J.A. and Shinbashi, M., 2021. Neutrophil Elastase and Chronic Lung Disease. Biomolecules. 11. [CrossRef]
- Wang, Q., Liu, Y. and Zhou, J., 2015. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 4, 19. [CrossRef]
- Waters, D.J., Shen, S. and Glickman, L.T., 2000. Life expectancy, antagonistic pleiotropy, and the testis of dogs and men. The Prostate. 43, 272-277.
- Whiteside, S.K., Snook, J.P., Williams, M.A. and Weis, J.J., 2018. Bystander T Cells: A Balancing Act of Friends and Foes. Trends Immunol. 39, 1021-1035. [CrossRef]
- Williams, G.C., 1957. Pleiotropy, natural selection and the evolution of senescence. Evolution. 11, 398-411.
- Xie, J., Van Hoecke, L. and Vandenbroucke, R.E., 2021. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front Immunol. 12, 796867. [CrossRef]
- Yamashima, T., Saido, T.C., Takita, M., Miyazawa, A., Yamano, J., Miyakawa, A., Nishijyo, H., Yamashita, J., Kawashima, S. and Ono, T., 1996. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. European journal of neuroscience. 8, 1932-1944.
- Yang, L., Besschetnova, T.Y., Brooks, C.R., Shah, J.V. and Bonventre, J.V., 2010. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 16, 535-43, 1p following 143.
Figure 1.
Patho-pathways are triggered. a, Patho-pathways induce pathology via dysactivity . Triggers of patho-pathways include environmental factors as well as genetic, accumulative and evolutionary. Patho-pathways are the futile activation of molecular pathways at the wrong time and/or wrong place. Dysactivity is unwanted activity by cells, tissues and/or whole systems. b, Once triggered, one patho-pathway can trigger others, resulting in cascades of patho-pathways. Patho-pathways may directly induce pathology or converge to induce pathology. c, An example of a triggered patho-pathway. Listed triggers: elevated oestrogen to progesterone ratio causing endometrial hyperplasia, increasing the risk of endometrial carcinoma.
Figure 1.
Patho-pathways are triggered. a, Patho-pathways induce pathology via dysactivity . Triggers of patho-pathways include environmental factors as well as genetic, accumulative and evolutionary. Patho-pathways are the futile activation of molecular pathways at the wrong time and/or wrong place. Dysactivity is unwanted activity by cells, tissues and/or whole systems. b, Once triggered, one patho-pathway can trigger others, resulting in cascades of patho-pathways. Patho-pathways may directly induce pathology or converge to induce pathology. c, An example of a triggered patho-pathway. Listed triggers: elevated oestrogen to progesterone ratio causing endometrial hyperplasia, increasing the risk of endometrial carcinoma.
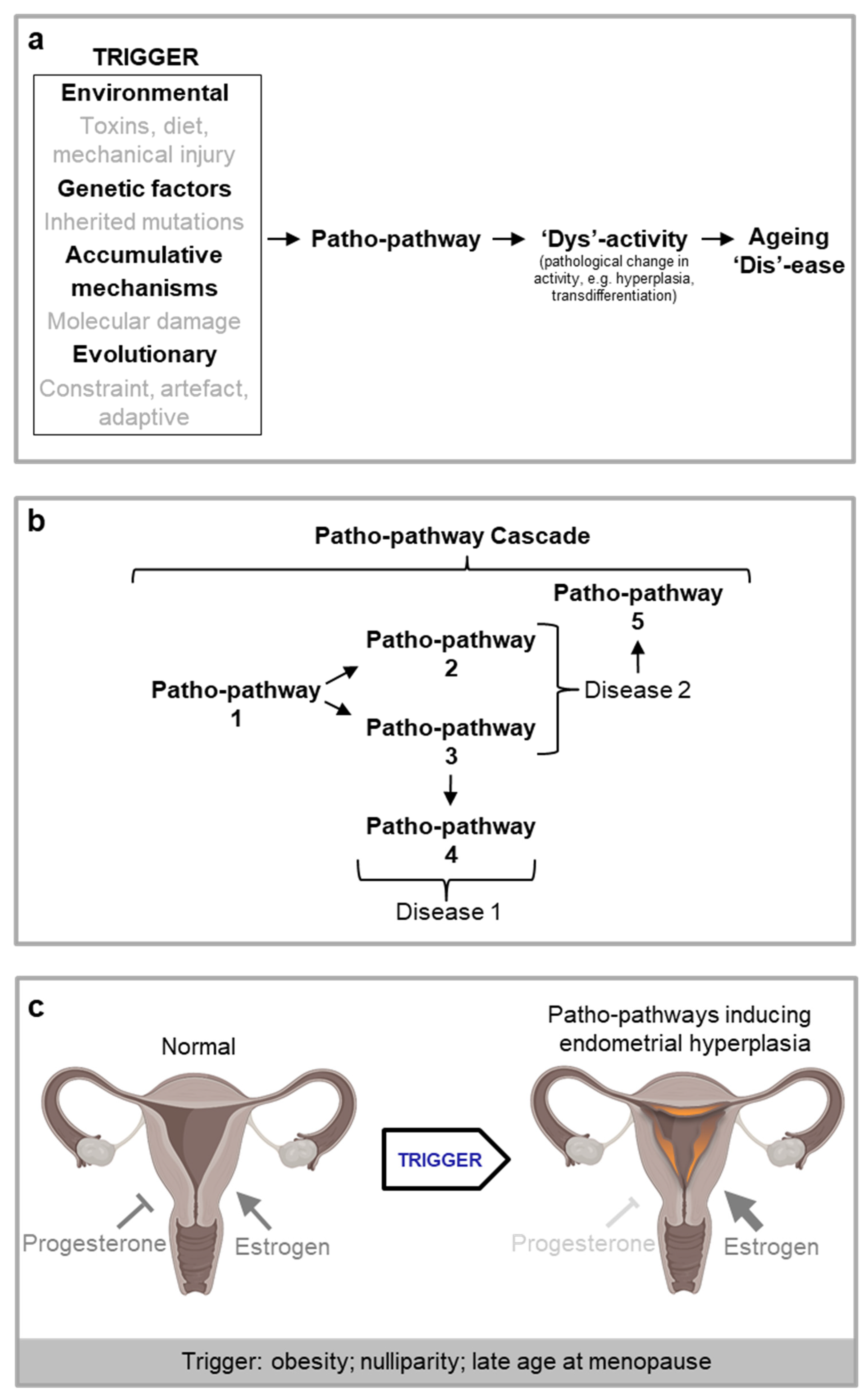
Figure 3.
Multi-stage and discrete-stage patho-pathway cascades. a, b, Multi-stage patho-pathway cascades. a, Rheumatoid arthritis driven by patho-pathways of inflammation. b, Non-alcoholic fatty liver disease (NAFLD) driven by patho-pathways of insulin resistance. c, d, Discrete stage patho-pathway cascades. c, Calcium and phosphate movement into blood triggers patho-pathways of vascular transdifferentiation and ossification, which cause vascular calcification. d, Lipotoxicity in NAFLD increases susceptibility of the liver to injury and causes inflammation, which in turn triggers fibrosis patho-pathways and an exaggerated wound healing response. Specifically, proinflammatory cytokines induce patho-pathways of dysactivity in innate immune cells which directly damage tissue. Concomitantly these cytokines trigger patho-pathways of dysactivity in fibroblasts which causes the fibroblasts to secrete too much extracellular matrix including collagen. The resulting fibrosis contributes to liver failure via disruption of the liver architecture and function.e, Example of a cascade (see Taable 2 for details on patho-pathways and Figure 3d).
Figure 3.
Multi-stage and discrete-stage patho-pathway cascades. a, b, Multi-stage patho-pathway cascades. a, Rheumatoid arthritis driven by patho-pathways of inflammation. b, Non-alcoholic fatty liver disease (NAFLD) driven by patho-pathways of insulin resistance. c, d, Discrete stage patho-pathway cascades. c, Calcium and phosphate movement into blood triggers patho-pathways of vascular transdifferentiation and ossification, which cause vascular calcification. d, Lipotoxicity in NAFLD increases susceptibility of the liver to injury and causes inflammation, which in turn triggers fibrosis patho-pathways and an exaggerated wound healing response. Specifically, proinflammatory cytokines induce patho-pathways of dysactivity in innate immune cells which directly damage tissue. Concomitantly these cytokines trigger patho-pathways of dysactivity in fibroblasts which causes the fibroblasts to secrete too much extracellular matrix including collagen. The resulting fibrosis contributes to liver failure via disruption of the liver architecture and function.e, Example of a cascade (see Taable 2 for details on patho-pathways and Figure 3d).
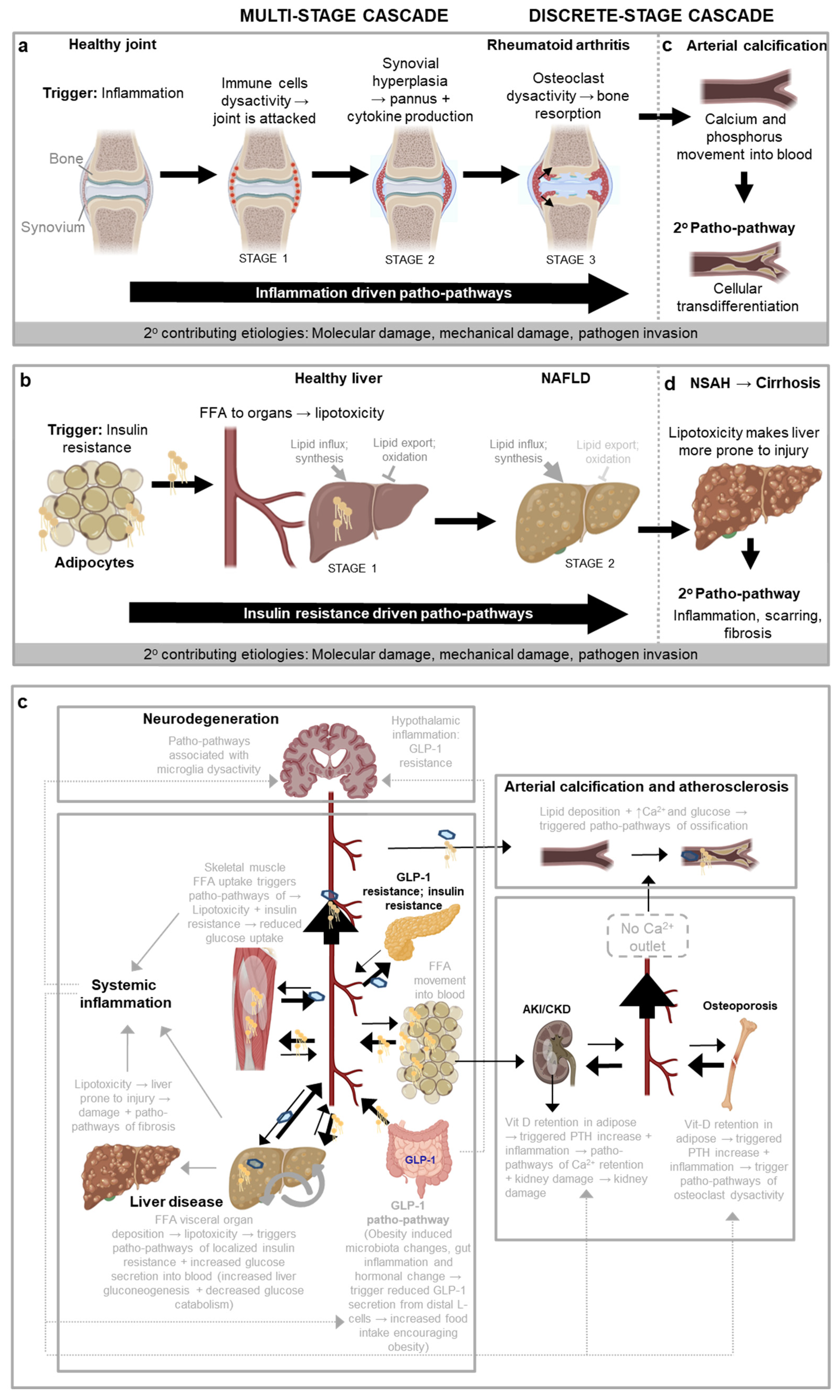
Figure 5.
Blueprint Maps allow for Hallmarks of ageing to be placed into the context of disease aetiology. Hallmarks may fall into any 1-4 steps dhown in blue, or overlap across steps. 12 recent Hallmarks of ageing shown from (López-Otín et al., 2023). Hallmarks may act as causal drivers of a disease, acting as triggers of patho-pathways, being part of patho-pathways or being part of dysactivity associated with patho-pathways. Hallmarks may also play a causal/contributor role or play no causal role and merely be symptomatic outcomes that may or may not worsen disease outcome. Hallmarks may also play no role altogether. Different Hallmarks likely play different roles across different age-related diseases.
Figure 5.
Blueprint Maps allow for Hallmarks of ageing to be placed into the context of disease aetiology. Hallmarks may fall into any 1-4 steps dhown in blue, or overlap across steps. 12 recent Hallmarks of ageing shown from (López-Otín et al., 2023). Hallmarks may act as causal drivers of a disease, acting as triggers of patho-pathways, being part of patho-pathways or being part of dysactivity associated with patho-pathways. Hallmarks may also play a causal/contributor role or play no causal role and merely be symptomatic outcomes that may or may not worsen disease outcome. Hallmarks may also play no role altogether. Different Hallmarks likely play different roles across different age-related diseases.
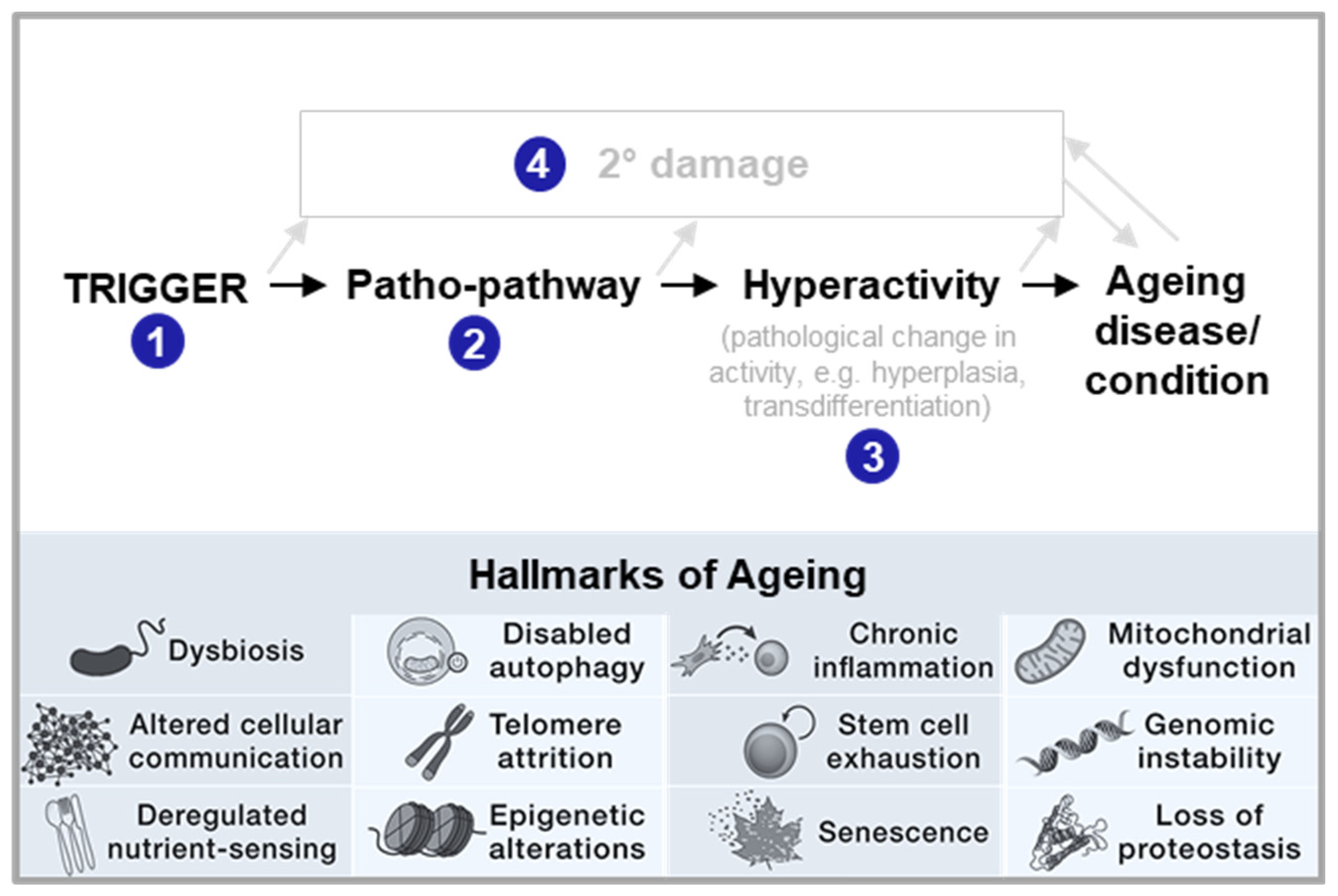
Figure 6.
Intervention into key patho-pathways can have far reaching effects: opening the avenue for new classes of drugs. Cascades of patho-pathways are interconnected within constrained biological networks, meaning that maintaining optimal function across all systems simultaneously is unattainable. Pharmaceuticals can intervene in critical nodes at the centre of these cascades, i.e. key patho-pathways, with the potential to alleviate widespread disease. By strategically targeting these "key patho-pathways" within the system, the risk of systemic collapse can be mitigated (by decoupling the trade-offs that normally constrain biological function).b, One example of key patho-pathways is that governing necrosis where intervention into the correct patho-pathways can have far reaching potential beneficial effects(Kern et al., 2024).
Figure 6.
Intervention into key patho-pathways can have far reaching effects: opening the avenue for new classes of drugs. Cascades of patho-pathways are interconnected within constrained biological networks, meaning that maintaining optimal function across all systems simultaneously is unattainable. Pharmaceuticals can intervene in critical nodes at the centre of these cascades, i.e. key patho-pathways, with the potential to alleviate widespread disease. By strategically targeting these "key patho-pathways" within the system, the risk of systemic collapse can be mitigated (by decoupling the trade-offs that normally constrain biological function).b, One example of key patho-pathways is that governing necrosis where intervention into the correct patho-pathways can have far reaching potential beneficial effects(Kern et al., 2024).
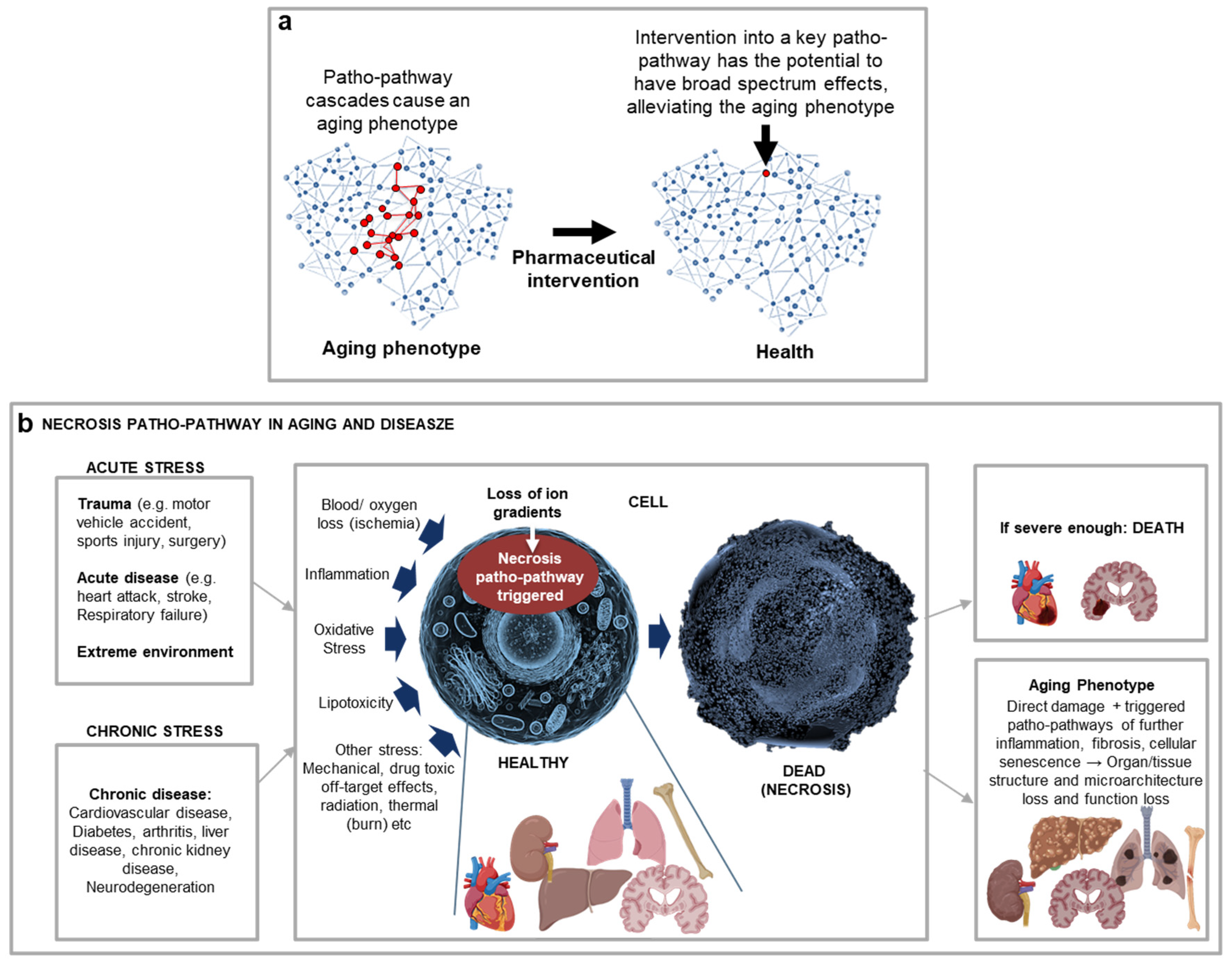
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



