Submitted:
21 October 2023
Posted:
23 October 2023
You are already at the latest version
Abstract
Hearing loss stands as the most prevalent sensory deficit among humans, posing a significant global health challenge. Projections indicate that by 2050, approximately 10% of the world's population will grapple with disabling hearing impairment. While approximately half of these cases have a genetic etiology, traditional interventions such as hearing aids and cochlear implants do not completely restore normal hearing. The absence of biological treatment has prompted significant efforts in recent years, with a strong focus on gene therapy to address hereditary hearing loss. Although several studies have exhibited promising recovery of common forms of genetic deafness in mouse models, existing challenges must be overcome to make gene therapy applicable in the near future. Herein, we summarize the primary gene therapy strategies employed over past years, provide an overview of the recent achievements in the preclinical studies for genetic hearing loss, and outline the current key obstacles to cochlear gene therapy.
Keywords:
genetics
; genomics
; deafness
; inner ear
; cochlea
; delivery
; editing
1. Introduction
Hearing loss affects hundreds of millions of people around the world as the most common human sensory disorder. While deafness already presents a significant global public health concern, with approximately 1 in 500 newborns, its prevalence is projected to escalate markedly [1]. By 2050, the number of individuals who experience disabling hearing impairment is expected to double and surpass 900 million people [2]. About 50% of congenital hearing loss cases can be attributed to genetic causes. These genetic factors can lead to the condition manifesting either at birth or later in life, exhibiting a range of severity levels [3,4]. Over the past decades, significant progress in understanding the genetic basis of deafness has been made, leading to discoveries of more than 150 genes [5]. Despite these significant advancements, the field still lacks clinical interventions that can fully restore natural hearing. Current approaches, like hearing aids and cochlear implants, successfully enhance auditory abilities within specific patient populations, but they fall short of providing therapy to reinstate the inner ear's original functionality completely [5-7].
The imperative need to find biological treatments for genetic deafness has spurred a global undertaking over the past two decades. This effort has led to the development of preclinical therapies for genetic hearing loss, with 46 reported studies including more than 20 deafness genes [8]. These studies have demonstrated success in restoring hearing loss attributed to various mutations and genes across diverse stages of development (Figure 1). While the potential application of these therapeutic strategies to humans is now within closer reach than ever, substantial challenges remain on the horizon. These include optimizing the treatment's transduction efficiency and specificity through more targeted capsids and promoters, identifying superior delivery methods, and a deeper comprehension of potential treatment side effects [9,10] .
Moreover, a notable obstacle lies in the genetic diversity inherent in deafness. Each gene linked to deafness, along with its unique pathogenic variations, represents only a segment within the intricate tapestry of genetic hearing impairments [11]. This diversity necessitates a diverse array of gene-specific therapies and complicates the implementation of treatments tailored to specific variants.
In this review, we provide an overview of the significant advances in gene therapy for hearing loss, shedding light on the prevailing obstacles that requires resolution from the scientific community.
2. Inner Ear Gene Therapy Strategies
Genetic therapy is a research field that aims to modify, replace, or repair the genetic factors responsible for genetic abnormalities. Advancements in gene delivery technologies and therapeutic tools, as well as the increasing knowledge about the genes involved in hearing loss and their functions in the auditory system, have led to a rapid expansion in the field of gene therapy for heredity hearing loss [12]. The inner ear is particularly well-suited for such interventions due to its unique properties. Firstly, the ear comprises distinct compartments that allow for the targeted delivery of therapeutic agents with minimal diffusion beyond the surrounding tissues. Secondly, the ear contains fluids that facilitate the dissemination of therapeutic agents to many target cells if administered locally. Various types of genetic therapies, including gene replacement, genome editing, and RNA-based therapies, can be used for this purpose [6,13,14].
2.1. Gene Replacement
The gene replacement approach, the most common strategy in gene therapy experiments in the inner ear, involves delivering a functional coding sequence of cDNA for a specific gene into targeted cells to replace a nonfunctional mutant gene and restore its normal function. In cases of recessive inheritance, where two copies of the mutated gene are present, gene replacement can supplement the nonfunctional mutant gene with a functional copy. In dominant inheritance with haploinsufficiency, where one copy of the gene is insufficient for normal function, gene replacement can provide an additional functional copy of the gene [15,16]. Delivery timing plays a critical role in the effectiveness of gene replacement. If the expression of the transgene begins during the prenatal stage, the lack of a functional gene can result in significant developmental consequences that cannot be reversed by introducing the functional sequence at later stages. Although many successful studies have been shown in different animal models of genetic deafness for various stages of inner ear maturation, most of these studies have been conducted during early neonatal stages (Table 1) [17].
2.2. Gene Suppression
Gene suppression therapies involve the use of small RNA molecules, such as small interfering (siRNAs), microRNAs (miRNAs), and antisense oligonucleotides (ASOs), to specifically silence genes without affecting the DNA sequence. While ASOs are designed to target a single mRNA molecule and bind to the RNA directly, siRNAs or miRNAs trigger cellular pathways that lead to the degradation of the RNA target. RNA gene suppression therapies are particularly relevant for dominant mutations in heterozygous models (Table 2). In such cases, silencing the mutated gene while retaining the expression of the wild-type gene may be adequate to counteract the effects of the dominant form of hearing loss [37-39].
Gene suppression involving the use of RNAi (RNA interference) was performed in Beethoven (Bth) mice, a mouse model of TMC1 (transmembrane channel-like 1) autosomal dominant hearing loss [40] (Table 2). An AAV vector containing a synthetic microRNA specifically designed to target the dominant allele was inserted through a single intracochlear injection. Some of the treated mice exhibited slower deterioration of the progression of hearing loss, with ABR thresholds similar to wild-type mice [41]. In addition to conventional RNA interference techniques, a novel and highly specific RNA interference tool, the CRISPR-Cas13 RNA editing system, has been used recently in neonatal Bth mice. This proof-of-concept study revealed a remarkable 70% reduction in Tmc1 Bth mRNA in vivo using the CRISPR-CasRx system, accompanied by minimal off-target effects [42] .
One example of the therapeutic potential of ASOs was shown in a mouse model of Usher Syndrome type 1 [43-45]. Systemic administration of antisense oligonucleotides was investigated in a mouse model carrying a c.216G>A variant of the Ush1c gene, with three localized delivery strategies. The study reported a significant improvement in auditory and vestibular function with ASO through inner or middle ear injection [46].
These observations, along with the approval of other nucleic acid drugs and ongoing clinical trials as potential therapeutics for various diseases, have generated significant interest in developing oligonucleotides and nucleic acids for genetic hearing loss [47].
2.3. Gene Editing
Gene editing techniques enable targeted modifications to the genome, including deletion, addition, and replacement of bases to repair mutations and restore the wild-type function. Previously, gene editing methods relied on technologies such as zinc-finger nucleases and TALENs that utilized specific protein motifs to bind to a particular genomic DNA sequence. However, a gradual advancement in genome editing tools has resulted in higher performance levels. Currently, two primary gene editing technologies, CRISPR-Cas and base editing, are widely used for inner ear applications [50,51] (Table 3). The Clustered regularly interspaced short palindromic repeat (CRISPR) system involves using a small piece of RNA to guide a nuclease enzyme, Cas9, to a specific location in the genome where it cuts the DNA. The cut triggers the cell's natural DNA repair mechanisms, which can be harnessed to either insert, delete, or replace DNA at the cut site.
Gene editing for deletions has been applied in a mouse model of human DFNB2 deafness, serving as an example of its practical application (Table 3). This model has a frameshift mutation in the Pcdh15 transcript due to the insertion of a single base. By injecting a gRNA that mainly causes a 1-bp deletion, the frameshift mutation in the hair cells of the mutant mice was corrected. As a result, the auditory and balance functions of the mice were partially rescued [52] .
Another approach to performing gene editing is base editing, a technique for creating targeted single-nucleotide alterations without cutting the DNA. Base editing uses a modified version of Cas9, which cannot cut DNA, and a second enzyme, such as a cytidine or adenine deaminase, to directly convert one DNA base to another [53,54]. One example of base editing with direct repair was conducted on Tmc1 mice, aimed to correct the Baringo mutation to the wild-type sequence using a cytosine base editor. A dual-AAV system was used to deliver the editor to prevent hearing loss. Successfully, the intervention managed to reverse 51% of the point mutation and led to a notable improvement in hearing function[55]. RNA base editors can offer a correction on the RNA level and, therefore, present an elevated safety profile for the remediation of genetic disorders.
A study using RNA-based editors to treat hereditary hearing loss was performed in Myo6 mice carrying a dominant mutation (Table 3). To correct the mutated allele, a mini dCas13X.1 base editor was used, effectively restoring the mice' auditory function for up to three months [56]. Gene correction indeed holds substantial promise in the realm of inner ear therapy. However, several challenges persist that require resolution, notably on the optimization of editing efficiency, as well as reducing off-target effects.
3. Delivery Vectors
3.1. Adeno-Associated Virus
After more than 50 years of research, the adeno-associated virus (AAV) has gained recognition as a highly effective and versatile human gene transfer tool. With more than 170 clinical trials, AAV has demonstrated its tremendous potential and has been utilized as a gene therapeutic, as seen in Luxturna, Glybera, and Zolgensma [60-62]. AAV, a member of the Parvoviridae family, is a small, non-pathogenic virus with a single-stranded DNA genome. AAV can be engineered to target specific cell types and has high transduction efficiency and long-term gene expression in both animal models and humans [63,64]. The AAV capsid exhibits an icosahedral symmetry with a diameter ranging from 18 to 28 nanometers. It is responsible for enclosing a single-stranded (ss) DNA genome that is approximately 4.7 kilobases in size. The DNA is flanked by two inverted terminal repeats (ITRs), crucial for viral genome replication, encapsidation, and integration into the host cell genome [65] . During the production of recombinant AAVs (rAAVs), the only remaining viral sequences are the ITRs, as the therapeutic gene expression cassettes replace all AAV protein-coding sequences within the encapsulated genome. This results in a capsid structure that contains only the ITRs and therapeutic genes, which can be used to minimize the immunogenicity and cytotoxicity of the vector when introduced in vivo [66]. AAV's different serotypes are another critical feature that contributes to their versatility. Researchers have identified thirteen naturally occurring AAV serotypes and over 100 additional variants from various animal species. Each serotype has distinct capsid structures that affect its ability to transduce specific cell types and tissues [67]. AAV vectors exhibit considerable potential as a gene therapy tool for different diseases, and currently, they are the safest and most promising viral vectors for inner ear gene therapy.
To date, the effectiveness of gene delivery to cochlear hair cells has been notably observed with AAV vectors, specifically AAV2/2 and AAV2/9, as well as the synthetic constructs AAV2/Anc80L65 and AAV2/9PHP.eB. These vectors have demonstrated exceptional transduction efficiency and have consequently garnered favor among numerous research groups.
A significant limitation of AAV lies in its cargo capacity (4.7 kb), limiting the size of the therapeutic sequence [68]. Given that several deafness-related genes extend considerably in length, addressing this has led to the concept of co-transduction with two AAVs[30,32,69-71] or even a triple-AAV strategy [72]. A recent study in a mouse model of PCDH15-related hearing loss highlights an alternative strategy aimed at overcoming the restricted cargo capacity of AAV. Given that the full-length Pcdh15 transcript spans 7.9 kb, surpassing the maximum delivery capability of AAV, single-AAV gene replacement becomes unfeasible. To address this, the concept of "mini-PCDH15" proteins was devised, involving the removal of extracellular domains under the assumption that specific domains might be redundant or dispensable for achieving partial hearing restoration. The results included preventing the degeneration of hair cell bundles and partial restoration of auditory brainstem response (ABR), emphasizing that complete protein expression might not be essential [25]. Expanding our knowledge of hair cells' molecular mechanism and dynamics could pave the way for refining mini-gene replacement strategies for treating hereditary hearing loss.
3.2. Lentivirus
Lentiviruses (LVs), belonging to the Retroviridae family, are RNA viruses that use reverse transcriptase to convert their RNA genome into DNA. It comprises envelop virions that contain a single-stranded DNA genome of approximately 9 kb. In addition to the large capacity size, LVs are considered a viable option for gene delivery applications as they can naturally infect various types of cells, including both dividing and non-dividing cells such as neurons and certain types of immune cells [73,74]. However, despite the benefits of using LVs, the risk of insertional mutagenesis, which can potentially disrupt gene function in transduced cells, is one of the main barriers to using lentiviral vectors for in vivo gene therapy [64,75]. Furthermore, LVs have not been extensively used in inner ear applications because they cannot specifically target hair cells [76,77]. However, generating viable lentiviral vector candidates for effective gene delivery in the inner ear may be possible with further advancements and modifications.
3.2. Adenovirus
Adenoviruses (AdVs) are a family of non-enveloped, double-stranded DNA viruses that can infect a wide range of animals, including humans. A major advantage of AdV is its large packaging capacity, with a range of 26 kb to 45 kb, depending on the serotype [78,79]. However, the use of adenovirus vectors derived from the most prevalent serotypes is limited due to its prevalence among healthy individuals. As a result, researchers have instead turned to using rare serotypes such as 2 and 5 to develop AdV vectors for gene therapy. Still, the highly immunogenic nature of all AdV proteins raises additional concerns [80,81]. A further limitation of AdVs is the lack of integration into the host genome, resulting in a short duration of transgene expression that typically lasts only weeks to months [82]. This evidence makes it less suitable for rescuing genetic hearing loss, which typically requires long-term transgene expression, but it is more convenient for regeneration and otoprotection applications.
4. Inner Ear Delivery Approaches
Inner ear gene therapy routes are integral to the development of innovative treatments for hearing and balance-related disorders. Each route has its own advantages and considerations, while choosing the most suitable path depends on factors such as the nature of the disorder and the desired treatment outcome.
4.1. Round-Window Injection
The round-window membrane (RWM) is a three-layered flexible membrane that opens to the perilymph space of the scala tympani. This intracochlear method has become the most commonly used method for delivering genetic agents into the inner ear (Figure 2). Compared to other methods, the RWM approach is considered relatively safe and minimally invasive, with a low risk of hearing damage, as only the membrane needs to be pierced, and the perilymph volume is larger than the endolymph [83]. Additionally, studies have demonstrated the successful transduction of both inner and outer hair cells, supporting cells, and spiral ganglion cells in non-human primates using AAV delivered via the round window [84,85]. One notable drawback of utilizing the RWM approach for gene therapy is the difficulty in achieving even distribution of the viral vector throughout the entire cochlear duct. Consequently, transduction often occurs in a gradient from the cochlear base to the apex, especially in adult mice. To address this challenge, a study utilized a combined approach of RWM injection along with canal fenestration to create an exit pathway [86]. This innovative strategy facilitated viral vectors to achieve comprehensive transduction of both cochlear inner hair cells (IHCs) and vestibular hair cells (VHCs), all the while avoiding any auditory dysfunction.
4.2. Canalostomy
An alternative method for delivering genetic agents into the inner ear is the canalostomy, mainly through the posterior semicircular canal (Figure 2). This technique involves creating an opening in the bony canal and introducing the therapeutic agent into the perilymphatic space. While this approach has proven successful in various preclinical studies [20,28,31,36], the challenge associated with this method is in achieving precise injections because of the small size of the semicircular canal, making it difficult to definitively ascertain whether the therapeutic product was delivered to the perilymphatic or endolymphatic compartments [87]. Adapting this method for use in humans could also prove challenging due to the anatomical positioning of the posterior semicircular canal. However, an alternative option could involve injection through the lateral semicircular canal in the human inner ear, provided that this approach has been previously validated as safe through experimentation in larger animal models [88,89].
4.3. Cochleostomy
The cochleostomy route delivers vectors directly into the scala media, a fluid-filled compartment within the cochlea (Figure 2). This is achieved by creating an opening through the basal part of the cochlea, allowing access to the cochlear endolymphatic space in close proximity to the round window. The safety and effectiveness of this approach have been demonstrated in neonatal mice and successfully applied in various preclinical studies [53,56,58,90-92]. Nevertheless, this surgical procedure is more intricate in adult mice because the cochlear bone is less pliable than in neonatal mice, resulting in a lasting elevation of hearing thresholds, particularly at high frequencies [91,93]. Consequently, this technique becomes invasive when applied to adult mice, potentially disrupting the cochlear structure or upsetting inner ear equilibrium by intermixing endolymph and perilymph fluids. Nonetheless, the larger size of the human cochlea could offer possibilities for future interventions in human patients, though additional validation is imperative.
4.4. Utricle
The utricle injection route was developed through the effort to target the endolymphatic fluids specifically (Figure 2). This approach efficiently delivered AAV vectors to all six sensory organs within the inner ear of neonatal mice. Administering injections through this route, readily accessible in neonatal mice, resulted in a remarkable rate of transduction IHCs and various other cell types within the inner ear, approaching nearly 100%. These injections did not cause any harm to auditory or vestibular functions [94]. Notably, when treatment was delivered via utricle injection to a mouse model with a Strc mutation, it led to a significant outcome; namely, partial prevention of hearing loss and restoration of stereocilia morphology in 50% of the treated animals [30]. Nonetheless, a limitation of this method lies in the challenge of definitively confirming whether the therapeutic products remain confined solely to the endolymphatic spaces within the inner ear or whether some reach the perilymphatic fluids. Moreover, it is important to highlight that the endolymphatic utricular area presents more complexities in humans, as it is primarily covered by the facial nerve, making access less straightforward [17].
5. Challenges and limitations
5.1. The Mouse as a Model for Human Deafness
In inner ear research, mice models stand out as the prevailing choice for experimental animals due to their anatomical similarities to the human inner ears and extensively studied genomes [95]. Mice have played a pivotal role in unraveling numerous intricate processes and accumulating a vast wealth of knowledge about this organ, including examining different aspects of gene therapy cure, such as delivery methods, selectivity of vectors, and efficiency. Despite many preclinical trials that have successfully demonstrated the feasibility and potential of gene therapy as a treatment for hearing loss, one critical factor that needs to be considered is the dissimilar developmental timelines observed in mice and humans [96]. While mice are born with underdeveloped cochlea that necessitates an additional two to three weeks to achieve full maturity, in humans, the auditory system is fully operational at birth, with auditory startle reflexes emerging at approximately 24 weeks of gestation. Consequently, most of the cochlear maturation in humans occurs in the prenatal stage [97,98]. Given that a significant portion of preclinical studies have been carried out in mice before the onset of hearing, the translation of these findings to a relevant therapeutic window in humans aligns with the gestational stage of around 20 weeks. This situation introduces a range of possible risk factors, adding complexity to obtaining approval from regulatory bodies such as the FDA and EMA. Despite the promising results obtained from mouse studies for potentially treating genetic causes of deafness, the lack of clinical trials with positive outcomes presents a significant gap. Consequently, it remains a challenge to predict the potential consequences for humans at this juncture confidently.
5.2. Genetic Heterogenicity
One of the significant challenges in addressing genetic hearing loss arises from its inherent heterogeneity. More than 150 genes, including thousands of distinct variants, are known to be associated with hearing loss. Each of these genes presents unique complexities regarding protein function, the optimal timing for therapeutic intervention, and the specific target cell populations for treatment [3]. Genetic factors' diversity necessitates a tailored and precise approach for each case. This complexity makes it challenging to devise a single, standardized gene therapy strategy that effectively addresses the broad spectrum of genetic causes of hearing loss. As medicine trends towards greater personalization, the effectiveness of precision medicine strategies in precisely targeting individual genetic variants remains uncertain.
5.3. Applications in Mature Mice
Most proof-of-concept studies aimed at restoring hearing loss have been conducted on neonatal mice during their early developmental stages. Performing interventions in adult mice is complicated due to two main factors. Firstly, the cochlea, which is the auditory portion of the inner ear, is embedded within the temporal bone, making it structurally difficult to access. Secondly, the effectiveness of gene delivery using viral vectors in the inner ear cells of mature mice is notably low [99]. Nonetheless, a recent study may have provided a potential solution to tackle these obstacles. A recently explored novel approach enables drug delivery to the adult inner ear using cerebrospinal fluid (CSF) [27]. The brain and inner ear fluid are connected via the cochlear aqueduct, which allows the delivery of AAV-Slc17a8 to adult Slc17a8−/− mice. Hearing recovery was observed two weeks post-treatment, with sensory cells in the cochlea effectively transduced. This transduction was more pronounced at the cochlear base and gradually decreased towards the apex. While CSF delivery shows promise in facilitating treatment during later developmental stages, additional actions, such as using nonhuman primate models, are required.
Another critical aspect to consider when addressing hearing loss in adult mice is the variability of hearing upon aging [100]. This includes the presence of a variant allele in the Cdh23 gene, which is associated with age-related hearing loss (ARHL) [101]. The effect of this variant on hearing loss in several mice strains has been evaluated [102]. This allele was corrected by CRISPR in a C57BL strain, to alleviate this confounding problem of ARHL in preclinical studies [103]. The presence of this or other alleles leading to ARHL in mice should be considered when using a particular strain as a model for evaluation of hearing at post-natal stages.
5.3. Implications of the Immune Response
AAV-based approaches to restore auditory and vestibular function are the most promising and safest. Despite this advantage, our current understanding of the host immune responses to AAV within the mammalian inner ear remains limited [104,105]. Several critical questions must be addressed to bridge the gap between preclinical studies and clinical applications: are factors such as viral dose, serotype, and delivery route determining the degree of inflammation? Does pre-existing immunity influence the efficacy and safety of gene therapy within the inner ear? Answers to these questions will maximize the safety and effectiveness of human inner ear therapy. Insights from ongoing clinical trials centered on eye treatments have indicated that viral gene therapy might induce an adaptive immune response. This observation raises optimism for similar prospects in the inner ear domain [9,106,107].
5. Conclusions
Gene therapy holds great promise for restoring auditory function and treating hereditary hearing loss, a feat beyond the reach of conventional medical approaches. The collaborative efforts of numerous teams worldwide have resulted in several successful pre-clinical studies and, more recently, the initiation of phase 1/2 clinical trials for gene therapies targeting otoferlin-related hearing loss. These trials are being conducted by two companies, Akouos and Decibel Therapeutics [108,109]. Undoubtedly, the beginning of clinical trials is a considerable landmark in auditory research. However, it is essential to remain aware that numerous challenges must be addressed before genetic therapeutics can be applied to patients across the board with hereditary hearing loss. These challenges encompass addressing limitations such as transduction efficacy, assessing potential toxicity, and advancing the development of delivery methods that are both feasible and safe for human applications. In addition, a more comprehensive understanding of treatment side effects, including assessing the longevity of the treatment and potential immune responses, is imperative for the progress of gene therapy in this context. Together, the current advancements and forthcoming insights from clinical trials offer fresh optimism for addressing hereditary hearing loss.
Author Contributions
Writing—original draft preparation, R.H., K.B.A.; writing—review and editing, R.H., K.B.A.; visualization, R.H.; funding acquisition, K.B.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the United States-Israel Binational Science Foundation (BSF), grant number 01027150 and the Israel Science Foundation within the Israel Precision Medicine Partnership Program, grant number 3499/19.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sheffield, A.M.; Smith, R.J.H. The epidemiology of deafness. Cold Spring Harb Perspect Med 2019, 9.
- Olusanya, B.O.; Davis, A.C.; Hoffman, H.J. Hearing loss: Rising prevalence and impact. Bull World Health Organ 2019, 97, 646-646A. [CrossRef]
- Carpena, N.T.; Lee, M.Y. Genetic hearing loss and gene therapy. Genomics Inform 2018, 16, e20.. [CrossRef]
- Alford, R.L.; Arnos, K.S.; Fox, M.; Lin, J.W.; Palmer, C.G.; Pandya, A.; Rehm, H.L.; Robin, N.H.; Scott, D.A.; Yoshinaga-Itano, C.; et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 2014, 16, 347–355. [CrossRef]
- Jiang, L.; Wang, D.; He, Y.; Shu, Y. Advances in gene therapy hold promise for treating hereditary hearing loss. Mol. Ther. 2023, 31, 934–950. [CrossRef]
- Geleoc, G.S.; Holt, J.R. Sound strategies for hearing restoration. Science 2014, 344. 1241062. [CrossRef]
- Delmaghani, S.; El-Amraoui, A. Inner ear gene therapies take off: Current promises and future challenges. J. Clin. Med. 2020, 9, 2309. [CrossRef]
- Petit, C.; Bonnet, C.; Safieddine, S. Deafness: from genetic architecture to gene therapy. Nat. Rev. Genet. 2023, 24, 665–686. [CrossRef]
- Amariutei, A.E.; Jeng, J.-Y.; Safieddine, S.; Marcotti, W. Recent advances and future challenges in gene therapy for hearing loss. R. Soc. Open Sci. 2023, 10, 230644. [CrossRef]
- Klimara, M.J.; Smith, R.J.H. Advances in cochlear gene therapies. Curr Opin Pediatr 2023, 35, 631–640. [CrossRef]
- Dror, A.A.; Avraham, K.B. Hearing Impairment: A Panoply of Genes and Functions. Neuron 2010, 68, 293–308. [CrossRef]
- Taiber, S.; Gwilliam, K.; Hertzano, R.; Avraham, K.B. The Genomics of Auditory Function and Disease. Annu. Rev. Genom. Hum. Genet. 2022, 23, 275–299. [CrossRef]
- Müller, U.; Barr-Gillespie, P.G. New treatment options for hearing loss. Nat. Rev. Drug Discov. 2015, 14, 346–365. [CrossRef]
- Ahmed, H.; Shubina-Oleinik, O.; Holt, J.R. Emerging Gene Therapies for Genetic Hearing Loss. J. Assoc. Res. Otolaryngol. 2017, 18, 649–670. [CrossRef]
- Taiber, S.; Avraham, K.B. Genetic Therapies for Hearing Loss: Accomplishments and Remaining Challenges. Neurosci. Lett. 2019, 713, 134527. [CrossRef]
- Askew, C.; Chien, W.W. Adeno-associated virus gene replacement for recessive inner ear dysfunction: Progress and challenges. Hear. Res. 2020, 394, 107947. [CrossRef]
- Lahlou, G.; Calvet, C.; Giorgi, M.; Lecomte, M.-J.; Safieddine, S. Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. J. Clin. Med. 2023, 12, 1046. [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Hathaway, D.M.; Klein, A.J.; Peters, C.W.; Li, Y.; Tamvakologos, P.I.; Nammour, J.; Maguire, C.A.; Corey, D.P. AAV-S: A versatile capsid variant for transduction of mouse and primate inner ear. Mol. Ther. - Methods Clin. Dev. 2021, 21, 382–398. [CrossRef]
- Guo, J.; Ma, X.; Skidmore, J.M.; Cimerman, J.; Prieskorn, D.M.; Beyer, L.A.; Swiderski, D.L.; Dolan, D.F.; Martin, D.M.; Raphael, Y. GJB2 gene therapy and conditional deletion reveal developmental stage-dependent effects on inner ear structure and function. Mol. Ther. - Methods Clin. Dev. 2021, 23, 319–333. [CrossRef]
- Wu, X.; Zhang, L.; Li, Y.; Zhang, W.; Wang, J.; Cai, C.; Lin, X. Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen syndrome type 2. Nat. Commun. 2021, 12, 1–12. [CrossRef]
- Chang, Q.; Wang, J.; Li, Q.; Kim, Y.; Zhou, B.; Wang, Y.; Li, H.; Lin, X. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 2015, 7, 1077–1086. [CrossRef]
- György, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of Hearing by Gene Delivery to Inner-Ear Hair Cells Using Exosome-Associated AAV. Mol. Ther. 2016, 25, 379–391. [CrossRef]
- Kim, M.-A.; Cho, H.-J.; Bae, S.-H.; Lee, B.; Oh, S.-K.; Kwon, T.-J.; Ryoo, Z.-Y.; Kim, H.-Y.; Cho, J.-H.; Kim, U.-K.; et al. Methionine Sulfoxide Reductase B3-Targeted In Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxidants Redox Signal. 2016, 24, 590–602. [CrossRef]
- Tang, H.; Wang, H.; Wang, S.; Hu, S.W.; Lv, J.; Xun, M.; Gao, K.; Wang, F.; Chen, Y.; Wang, D.; et al. Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum. Genet. 2022, 142, 289–304. [CrossRef]
- Ivanchenko, M.V.; Hathaway, D.M.; Klein, A.J.; Pan, B.; Strelkova, O.; De-La-Torre, P.; Wu, X.; Peters, C.W.; Mulhall, E.M.; Booth, K.T.; et al. Mini-PCDH15 gene therapy rescues hearing in a mouse model of Usher syndrome type 1F. Nat. Commun. 2023, 14, 1–21. [CrossRef]
- Lu, Y.-C.; Tsai, Y.-H.; Chan, Y.-H.; Hu, C.-J.; Huang, C.-Y.; Xiao, R.; Hsu, C.-J.; Vandenberghe, L.H.; Wu, C.-C.; Cheng, Y.-F. Gene therapy with a synthetic adeno-associated viral vector improves audiovestibular phenotypes in Pjvk-mutant mice. J. Clin. Investig. 2022, 7. [CrossRef]
- Mathiesen, B.K.; Miyakoshi, L.M.; Cederroth, C.R.; Tserga, E.; Versteegh, C.; Bork, P.A.R.; Hauglund, N.L.; Gomolka, R.S.; Mori, Y.; Edvall, N.K.; et al. Delivery of gene therapy through a cerebrospinal fluid conduit to rescue hearing in adult mice. Sci. Transl. Med. 2023, 15, eabq3916. [CrossRef]
- Zhao, X.; Liu, H.; Liu, H.; Cai, R.; Wu, H. Gene Therapy Restores Auditory Functions in an Adult Vglut3 Knockout Mouse Model. Hum. Gene Ther. 2022, 33, 729–739. [CrossRef]
- Kim, M.-A.; Kim, S.H.; Ryu, N.; Ma, J.-H.; Kim, Y.-R.; Jung, J.; Hsu, C.-J.; Choi, J.Y.; Lee, K.-Y.; Wangemann, P.; et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics 2019, 9, 7184–7199. [CrossRef]
- Shubina-Oleinik, O.; Nist-Lund, C.; French, C.; Rockowitz, S.; Shearer, A.E.; Holt, J.R. Dual-vector gene therapy restores cochlear amplification and auditory sensitivity in a mouse model of DFNB16 hearing loss. Sci. Adv. 2021, 7, eabi7629. [CrossRef]
- Taiber, S.; Cohen, R.; Yizhar-Barnea, O.; Sprinzak, D.; Holt, J.R.; Avraham, K.B. Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol. Med. 2020, 13, e13259. [CrossRef]
- Wu, J.; Solanes, P.; Nist-Lund, C.; Spataro, S.; Shubina-Oleinik, O.; Marcovich, I.; Goldberg, H.; Schneider, B.L.; Holt, J.R. Single and Dual Vector Gene Therapy with AAV9-PHP.B Rescues Hearing in Tmc1 Mutant Mice. Mol. Ther. 2021, 29, 973–988. [CrossRef]
- Marcovich, I.; Baer, N.K.; Shubina-Oleinik, O.; Eclov, R.; Beard, C.W.; Holt, J.R. Optimized AAV vectors for Tmc1 gene therapy in a humanized mouse model of DFNB7/11. Biomolecules 2022, 12.
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [CrossRef]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; Boutet de Monvel, J.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc Natl Acad Sci U S A 2017, 114, 9695–9700. [CrossRef]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; Chien, W.W. Gene therapy restores balance and auditory functions in a mouse model of Usher syndrome. Mol Ther 2017, 25, 780-791. [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol 2017, 35, 222-229.
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol 2018, 18, 126-131. [CrossRef]
- Maeda, Y.; Fukushima, K.; Nishizaki, K.; Smith, R.J. In vitro and in vivo suppression of Gjb2 expression by RNA interference. Hum Mol Genet 2005, 14, 1641-1650. [CrossRef]
- Shibata, S.B.; Ranum, P.T.; Moteki, H.; Pan, B.; Goodwin, A.T.; Goodman, S.S.; Abbas, P.J.; Holt, J.R.; Smith, R.J.H. Rna interference prevents autosomal-dominant hearing loss. Am J Hum Genet 2016, 98, 1101-1113. [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Moteki, H.; Smith, R.J.H. Targeted allele suppression prevents progressive hearing loss in the mature murine model of human TMC1 deafness. Mol Ther 2019, 27, 681-690. [CrossRef]
- Zheng, Z.; Li, G.; Cui, C.; Wang, F.; Wang, X.; Xu, Z.; Guo, H.; Chen, Y.; Tang, H.; Wang, D.; et al. Preventing autosomal-dominant hearing loss in Bth mice with CRISPR/CasRx-based RNA editing. Signal Transduct Target Ther 2022, 7, 79.
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med 2013, 19, 345-350. [CrossRef]
- Vijayakumar, S.; Depreux, F.F.; Jodelka, F.M.; Lentz, J.J.; Rigo, F.; Jones, T.A.; Hastings, M.L. Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum Mol Genet 2017, 26, 34823494.
- Robillard, K.N.; de Vrieze, E.; van Wijk, E.; Lentz, J.J. Altering gene expression using antisense oligonucleotide therapy for hearing loss. Hear Res 2022, 426, 108523.
- Lentz, J.J.; Pan, B.; Ponnath, A.; Tran, C.M.; Nist-Lund, C.; Galvin, A.; Goldberg, H.; Robillard, K.N.; Jodelka, F.M.; Farris, H.E.; et al. Direct delivery of antisense oligonucleotides to the middle and inner ear improves hearing and balance in Usher mice. Mol Ther 2020, 28, 2662-2676. [CrossRef]
- Halloy, F.; Biscans, A.; Bujold, K.E.; Debacker, A.; Hill, A.C.; Lacroix, A.; Luige, O.; Stromberg, R.; Sundstrom, L.; Vogel, J.; Ghidini, A. Innovative developments and emerging technologies in RNA therapeutics. RNA Biol 2022, 19, 313-332. [CrossRef]
- Ponnath, A.; Depreux, F.F.; Jodelka, F.M.; Rigo, F.; Farris, H.E.; Hastings, M.L.; Lentz, J.J. Rescue of outer hair cells with antisense oligonucleotides in Usher mice is dependent on age of treatment. J Assoc Res Otolaryngol 2018, 19, 1–16. [CrossRef]
- Wang, L.; Kempton, J.B.; Jiang, H.; Jodelka, F.M.; Brigande, A.M.; Dumont, R.A.; Rigo, F.; Lentz, J.J.; Hastings, M.L.; Brigande, J.V. Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res 2020, 48, 5065-5080. [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013, 31, 397-405. [CrossRef]
- Niggemann, P.; Gyorgy, B.; Chen, Z.Y. Genome and base editing for genetic hearing loss. Hear Res 2020, 394, 107958. [CrossRef]
- Liu, L.; Zou, L.; Li, K.; Hou, H.; Hu, Q.; Liu, S.; Li, J.; Song, C.; Chen, J.; Wang, S.; et al. Template-independent genome editing in the Pcdh15av-3j mouse, a model of human DFNB23 nonsyndromic deafness. Cell Rep 2022, 40, 111061.
- Cui, C.; Wang, D.; Huang, B.; Wang, F.; Chen, Y.; Lv, J.; Zhang, L.; Han, L.; Liu, D.; Chen, Z.Y.; et al. Precise detection of CRISPR-Cas9 editing in hair cells in the treatment of autosomal dominant hearing loss. Mol Ther Nucleic Acids 2022, 29, 400-412.
- Abdelnour, S.A.; Xie, L.; Hassanin, A.A.; Zuo, E.; Lu, Y. The potential of CRISPR/Cas9 gene editing as a treatment strategy for inherited diseases. Front Cell Dev Biol 2021, 9, 699597. [CrossRef]
- Yeh, W.H.; Shubina-Oleinik, O.; Levy, J.M.; Pan, B.; Newby, G.A.; Wornow, M.; Burt, R.; Chen, J.C.; Holt, J.R.; Liu, D.R. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci Transl Med 2020, 12. [CrossRef]
- Xue, Y.; Hu, X.; Wang, D.; Li, D.; Li, Y.; Wang, F.; Huang, M.; Gu, X.; Xu, Z.; Zhou, J.; et al. Gene editing in a Myo6 semi-dominant mouse model rescues auditory function. Mol Ther 2022, 30, 105-118. [CrossRef]
- Noh, B.; Rim, J.H.; Gopalappa, R.; Lin, H.; Kim, K.M.; Kang, M.J.; Gee, H.Y.; Choi, J.Y.; Kim, H.H.; Jung, J. In vivo outer hair cell gene editing ameliorates progressive hearing loss in dominant-negative Kcnq4 murine model. Theranostics 2022, 12, 2465–2482. [CrossRef]
- Xiao, Q.; Xu, Z.; Xue, Y.; Xu, C.; Han, L.; Liu, Y.; Wang, F.; Zhang, R.; Han, S.; Wang, X.; et al. Rescue of autosomal dominant hearing loss by in vivo delivery of mini dCas13X-derived RNA base editor. Sci Transl Med 2022, 14, eabn0449. [CrossRef]
- Gyorgy, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat Med 2019, 25, 1123-1130. [CrossRef]
- Grimm, D.; Zolotukhin, S. E Pluribus Unum: 50 years of research, millions of viruses, and one goal-tailored acceleration of aav evolution. Mol Ther 2015, 23, 1819-1831.
- Keeler, A.M.; Flotte, T.R. Recombinant adeno-associated virus gene therapy in light of luxturna (and zolgensma and glybera): Where are we, and how did we get here? Annu Rev Virol 2019, 6, 601-621. [CrossRef]
- Fakhiri, J.; Landegger, L.D.; Grimm, D. Breaking the sound barrier: Towards next-generation AAV vectors for gene therapy of hearing disorders. Hear Res 2022, 413, 108092. [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017, 31, 317-334.
- Chien, W.W.; Monzack, E.L.; McDougald, D.S.; Cunningham, L.L. Gene therapy for sensorineural hearing loss. Ear Hear 2015, 36, 1-7. [CrossRef]
- Yang, J.; Zhou, W.; Zhang, Y.; Zidon, T.; Ritchie, T.; Engelhardt, J.F. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J Virol 1999, 73, 9468-9477. [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 2019, 18, 358–378. [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol Ther 2006, 14, 316-327. [CrossRef]
- Marrone, L.; Marchi, P.M.; Azzouz, M. Circumventing the packaging limit of AAV-mediated gene replacement therapy for neurological disorders. Expert Opin Biol Ther 2022, 22, 1163-1176. [CrossRef]
- Omichi, R.; Yoshimura, H.; Shibata, S.B.; Vandenberghe, L.H.; Smith, R.J.H. Hair cell transduction efficiency of single- and dual-AAV serotypes in adult murine cochleae. Mol Ther Methods Clin Dev 2020, 17, 1167-1177. [CrossRef]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; Boutet de Monvel, J.; Hardelin, J.P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci U S A 2019, 116, 4496-4501. [CrossRef]
- Chen, Z.R.; Guo, J.Y.; He, L.; Liu, S.; Xu, J.Y.; Yang, Z.J.; Su, W.; Liu, K.; Gong, S.S.; Wang, G.P. Co-transduction of dual-adeno-associated virus vectors in the neonatal and adult mouse utricles. Front Mol Neurosci 2022, 15, 1020803. [CrossRef]
- Akil, O. Dual and triple AAV delivery of large therapeutic gene sequences into the inner ear. Hear Res 2020, 394, 107912. [CrossRef]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol 2013, 36, 1-22.
- Wanisch, K.; Yanez-Munoz, R.J. Integration-deficient lentiviral vectors: A slow coming of age. Mol Ther 2009, 17, 1316-1332. [CrossRef]
- Milone, M.C.; O'Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529-1541. [CrossRef]
- Wang, Y.; Sun, Y.; Chang, Q.; Ahmad, S.; Zhou, B.; Kim, Y.; Li, H.; Lin, X. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J Gene Med 2013, 15, 123-133. [CrossRef]
- Han, J.J.; Mhatre, A.N.; Wareing, M.; Pettis, R.; Gao, W.Q.; Zufferey, R.N.; Trono, D.; Lalwani, A.K. Transgene expression in the guinea pig cochlea mediated by a lentivirus-derived gene transfer vector. Hum Gene Ther 1999, 10, 1867–1873. [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet 2003, 4, 346-358. [CrossRef]
- Shu, Y.; Tao, Y.; Li, W.; Shen, J.; Wang, Z.; Chen, Z.Y. Adenovirus vectors target several cell subtypes of mammalian inner ear in vivo. Neural Plast 2016, 2016, 1–8. [CrossRef]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part I. Gene delivery technologies. Discov Med 2014, 18, 67-77.
- Lasaro, M.O.; Ertl, H.C. New insights on adenovirus as vaccine vectors. Mol Ther 2009, 17, 1333-1339. [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis 2017, 4, 43-63. [CrossRef]
- Plontke, S.K.; Hartsock, J.J.; Gill, R.M.; Salt, A.N. Intracochlear drug injections through the round window membrane: Measures to improve drug retention. Audiol Neurootol 2016, 21, 72-79. [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of hearing in the vglut3 knockout mouse using virally mediated gene therapy. Neuron 2012, 75, 283-293. [CrossRef]
- Xia, L.; Yin, S.; Wang, J. Inner ear gene transfection in neonatal mice using adeno-associated viral vector: A comparison of two approaches. PLoS One 2012, 7, e43218. [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci Rep 2018, 8, 1–10. [CrossRef]
- Kawamoto, K.; Oh, S.H.; Kanzaki, S.; Brown, N.; Raphael, Y. The functional and structural outcome of inner ear gene transfer via the vestibular and cochlear fluids in mice. Mol Ther 2001, 4, 575-585. [CrossRef]
- Zhu, J.; Choi, J.W.; Ishibashi, Y.; Isgrig, K.; Grati, M.; Bennett, J.; Chien, W. Refining surgical techniques for efficient posterior semicircular canal gene delivery in the adult mammalian inner ear with minimal hearing loss. Sci Rep 2021, 11, 18856.
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci Rep 2017, 7, 45524.
- Chien, W.W.; Isgrig, K.; Roy, S.; Belyantseva, I.A.; Drummond, M.C.; May, L.A.; Fitzgerald, T.S.; Friedman, T.B.; Cunningham, L.L. Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol Ther 2016, 24, 17-25. [CrossRef]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther 2011, 18, 569–578. [CrossRef]
- Shu, Y.; Tao, Y.; Wang, Z.; Tang, Y.; Li, H.; Dai, P.; Gao, G.; Chen, Z.Y. Identification of adeno-associated viral vectors that target neonatal and adult mammalian inner ear cell subtypes. Hum Gene Ther 2016, 27, 687–699. [CrossRef]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope 2015, 125, 2557-2564. [CrossRef]
- Lee, J.; Nist-Lund, C.; Solanes, P.; Goldberg, H.; Wu, J.; Pan, B.; Schneider, B.L.; Holt, J.R. Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear Res 2020, 394, 107882. [CrossRef]
- Dror, A.A.; Avraham, K.B. Hearing loss: Mechanisms revealed by genetics and cell biology. Annu Rev Genet 2009, 43, 411-437. [CrossRef]
- Wang, L.; Kempton, J.B.; Brigande, J.V. Gene therapy in mouse models of deafness and balance dysfunction. Front Mol Neurosci 2018, 11, 300. [CrossRef]
- Litovsky, R. Development of the auditory system. Handb Clin Neurol 2015, 129, 55-72.
- Lim, R.; Brichta, A.M. Anatomical and physiological development of the human inner ear. Hear Res 2016, 338, 9-21. [CrossRef]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; Chen, Z.Y. Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum Gene Ther 2018, 29, 492-506. [CrossRef]
- Zheng, Q.Y.; Johnson, K.R.; Erway, L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res 1999, 130, 94-107. [CrossRef]
- Kane, K.L.; Longo-Guess, C.M.; Gagnon, L.H.; Ding, D.; Salvi, R.J.; Johnson, K.R. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear Res 2012, 283, 80-88. [CrossRef]
- Johnson, K.R.; Tian, C.; Gagnon, L.H.; Jiang, H.; Ding, D.; Salvi, R. Effects of Cdh23 single nucleotide substitutions on age-related hearing loss in C57BL/6 and 129S1/Sv mice and comparisons with congenic strains. Sci Rep 2017, 7, 44450.
- Mianne, J.; Chessum, L.; Kumar, S.; Aguilar, C.; Codner, G.; Hutchison, M.; Parker, A.; Mallon, A.M.; Wells, S.; Simon, M.M.; et al. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med 2016, 8, 16. [CrossRef]
- Arjomandnejad, M.; Dasgupta, I.; Flotte, T.R.; Keeler, A.M. Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs 2023, 37, 311-329. [CrossRef]
- Ishibashi, Y.; Sung, C.Y.W.; Grati, M.; Chien, W. Immune responses in the mammalian inner ear and their implications for AAV-mediated inner ear gene therapy. Hear Res 2023, 432, 108735. [CrossRef]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 can induce innate and adaptive immune response in the primate eye. Mol Ther 2017, 25, 2648-2660. [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med 2015, 372, 1887-1897. [CrossRef]
- "Gene Therapy Trial for Otoferlin Gene-Mediated Hearing Loss." https://classic.clinicaltrials.gov/show/NCT05821959, May 2023.
- "A Study of DB-OTO, an AAV Based Gene Therapy, in Children/Infants with Hearing Loss Due to Otoferlin Mutations." https://classic.clinicaltrials.gov/show/NCT05788536, Updated May 12, 2023.
Figure 1.
Schematic illustration of the organ of Corti, including the expression sites of causal genes of hearing loss. All genes are part of proof-of-concept studies for inner ear gene therapy.
Figure 1.
Schematic illustration of the organ of Corti, including the expression sites of causal genes of hearing loss. All genes are part of proof-of-concept studies for inner ear gene therapy.
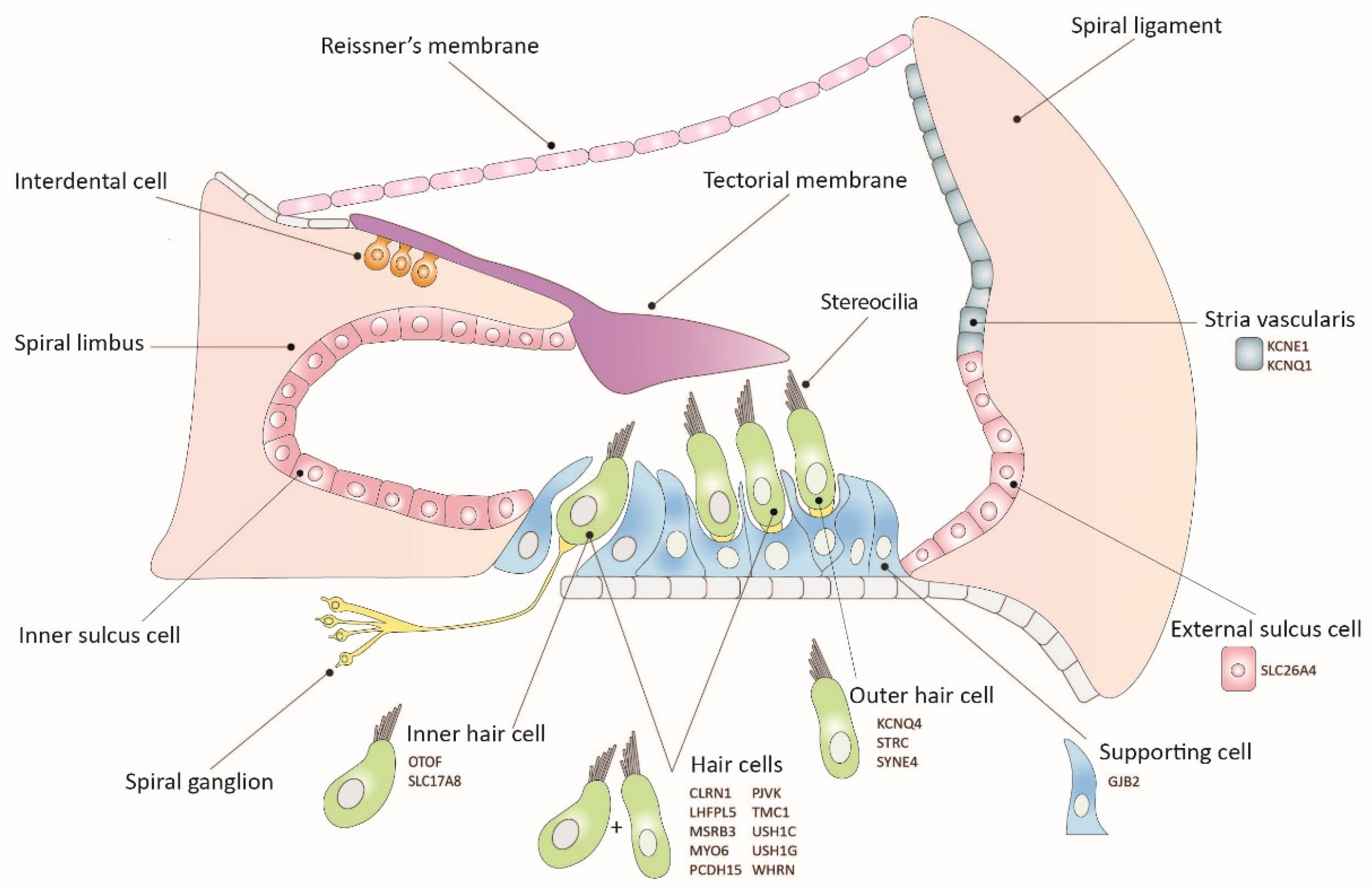
Figure 2.
Schematic illustration of inner-ear gene therapy routes.
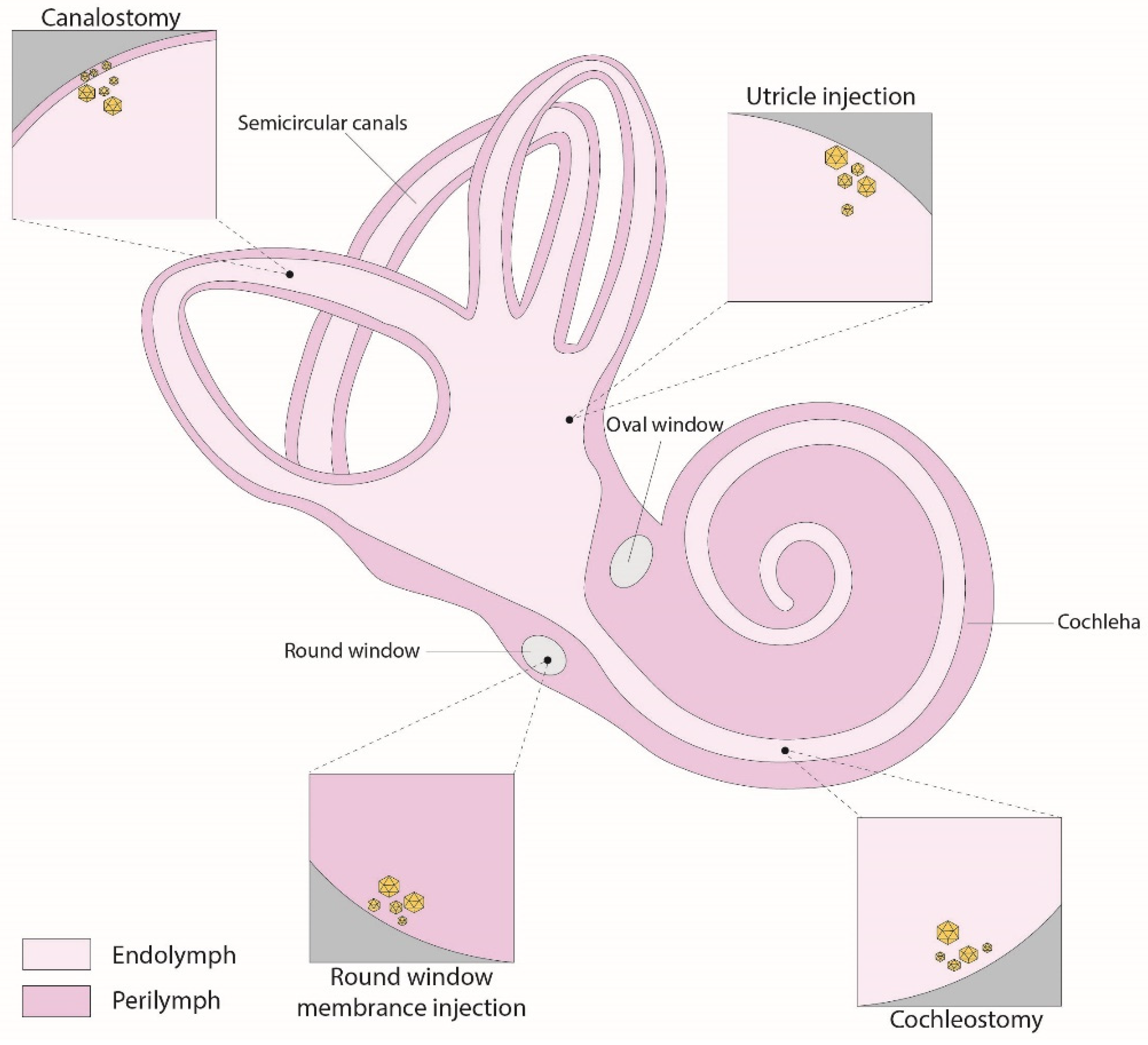

Table 1.
Summary of gene replacement preclinical therapy studies, using mouse models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
Table 1.
Summary of gene replacement preclinical therapy studies, using mouse models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
| Gene (form of hearing loss) | Animal model | Hearing impairment |
Strategy | Injection age | Injection route | Vector | Reference |
|---|---|---|---|---|---|---|---|
| CLRN1 (USH3A) | TgAC1/Clrn1 KO* | Delayed onset progressive | Replacement | P1 | RWM | AAV-S | [18] |
| GJB2 (DFNA3A/DFNB1A) | Gjb2 iCKO | Severe to profound | Replacement | P28 | RWM | AAV2/Anc80L65 | [19] |
| KCNE1 (JLNS2) | Kcne1 KO | Severe (balance defect) | Replacement | P0–P2 | PSCC | AAV1 | [20] |
| KCNQ1 (JLNS1) | Kcnq1 KO | Severe (balance defect) | Replacement | P0–P2 | RWM, Scala media | AAV1 | [21] |
| LHFPL5 (DFNB66/67) | Lhfpl5 KO | Profound (balance defect) | Replacement | P1-P2 | RWM, Scala media | exo-AAV1 | [22] |
| MSRB3 (DFNB74) | Msrb3 KO | Profound | Replacement | E12.5 | EUGO | AAV2/1 | [23] |
| OTOF (DFNB9) | Otof KO | Profound | Replacement | P0-P2 | RWM | Dual vector: AAV9/PHP.eB | [24] |
| PCDH15 (DFNB23/USH1F) | Pcdh15 KO | Profound (balance defect) | Mini-gene replacement | P1 | RWM | AAV2/9-PHP.B | [25] |
| PJVK (DFNB59) | Pjvk KO | Progressive (balance defect) | Replacement | P0-P1 | RWM | AAV2/Anc80L65 | [26] |
| SLC17A8 (DFNA25) | VGlut3 KO | Profound | Replacement | 6-12 weeks | Cisterna magna | AAV2/9-PHP.B | [27] |
| VGlut3 KO | Profound | Replacement | 5, 8, and 20 weeks | PSCC | AAV8 | [28] |
|
| SLC26A4 (DFNB4) | Slc26a4 KO | Profound (balance defect) | Replacement | E12.5 | EUGO | AAV2/1 | [29] |
| STRC (DFNB16) | Strc KO | Severe | Replacement | P0–P1 | Utricle | Dual vector: AAV9/PHP.B | [30] |
| SYNE4 (DFNB76) | Syne4 KO | Severe to profound progressive | Replacement | P0-P1.5 | PSCC | AAV2/9.PHP.B | [31] |
| TMC1 (DFNB7/11) | Tmc1 KO,Tmc1-Baringo | Profound | Replacement | P1, P7 | Utricle | AAV2/9-PHP.B | [32] |
| Tmc1 KO, Tmc1N1931/N1931 | Profound | Replacement | P1 | Utricle | AAV2/9-PHP.B | [33] | |
| USH1C/ (DFNB18/USH1C) | Ush1c c.216G>A | Severe (balance defect) | Replacement | P0-P1; P10-P12 | RWM | AAV2/Anc80L65 | [34] |
| USH1G/ (USH1G) | Ush1g KO | Profound (balance defect) | Replacement | P2.5 | RWM | AAV2/8 | [35] |
| WHRN (DFNB31) | Whrn wi/wi | Profound (balance defect) | Replacement | P1-P5 | PSCC | AAV2/8 | [36] |
* EUGO- electroporation-mediated transuterine gene transfer into otocysts KO, Knock-out (gene-targeted mutagenesis), iCKO- inducible conditional Knock-out, PSCC- posterior semicircular canal, RWM- round window membrane.
Table 2.
Summary of gene suppression preclinical therapy studies, using mice models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
Table 2.
Summary of gene suppression preclinical therapy studies, using mice models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
| Gene (Deafness form) | Animal model | Hearing impairment | Strategy | Injection age | Injection Route | Vector | Reference |
|---|---|---|---|---|---|---|---|
| GJB2 (DFNA3A) | Gjb2 p.R75W | Severe to profound | RNAi | P42-45 | RWM* | siRNAs | [39] |
| TMC1 (DFNA36) | Tmc1-Bth | Progressive | miRNA | P0-P2 | RWM | rAAV2/9 | [40] |
| Tmc1-Bth | Progressive | miRNA | P15-P16; P56-P60; P84-P90 | RWM, PSCC* | rAAV2/9 | [41] | |
| Tmc1-Bth | Progressive | RNA editing (CasRx) | P1-P2 | RWM | AAV2/9-PHP.eB | [42] | |
| USH1C (USH1C) | Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | P1; P3; P5; P7 | Intraperitoneal | ASO-29 | [48] |
| Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | P1; P5; P10; P20 | RWM | ASO-29 | [46] | |
| Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | E12.5 | EUGO | ASO-29 | [49] |
* EUGO- electroporation-mediated transuterine gene transfer into otocysts, RWM- round window membrane, PSCC - posterior semicircular canal.
Table 3.
Summary of gene editing preclinical therapy studies, using mouse models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
Table 3.
Summary of gene editing preclinical therapy studies, using mouse models. For genes that have been the subject of multiple previous studies, only studies published after 2020 have been incorporated.
| Gene (Deafness form) | Animal model | Hearing impairment | Strategy | Injection age | Injection Route | Vector | Reference |
|---|---|---|---|---|---|---|---|
| KCNQ4 (DFNA2A) | Kcnq4 c.827G>C | Progressive | Disruption | P0-P1 | PSCC*, RWM*, utricle, scala media | Dual vector: AAV2/Anc80L65 | [57] |
| Kcnq4 c.683G>A | Progressive | Disruption | P1–P2 | Scala media | AAV-PHP.eB | [53] | |
| MYO6 (DFNA22) | Myo6 p.C442Y | Progressive | Disruption | P0-P2 | Scala media | AAV-PHP.eB- | [56] |
| Myo6 p.C442Y | Progressive | RNA base editing | P0-P2 | Scala media | AAV-PHP.eB (RNA ABE) | [58] | |
| PCDH15 (DFNB23/USH1F) | Pcdh15av−3J | Profound (balance defect) | Frame restoration | P0-P2 | Scala media | AAV2/9 | [52] |
| TMC1 (DFNA36) | Tmc1-Bth | Progressive | Disruption | P1 | Utricle | Dual vector: AAV9-PHP.B | [32] |
| Tmc1-Bth | Progressive | Disruption | P1–P2 | Inner ear | Dual vector: AAV2/Anc80 L65 | [59] | |
| TMC1 (DFNB7/11) | Tmc1-Baringo | Profound | Base editing | P1 | Inner ear | Dual vector: AAV2/Anc80 L65 (CBE) | [55] |
*PSCC - posterior semicircular canal, RWM- round window membrane.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



