
Submitted:
21 October 2023
Posted:
23 October 2023
You are already at the latest version
Abstract
The formation and functioning of membrane-less organelles (MLOs) is one of the main driving forces in the molecular life of the cell. These processes are based on the separation of biopolymers into phases regulated by multiple specific and nonspecific inter- and intramolecular interactions. Among the realm of MLOs, a special place is taken by the promyelocytic leukemia nuclear bodies (PML-NBs or PML bodies), which are the intranuclear compartments involved in the regulation of cellular metabolism, transcription, maintenance of genome stability, response to viral infection, apoptosis, and tumor suppression. According to the accepted models, specific interactions, such as SUMO/SIM, formation of disulfide bonds, etc., play a decisive role in the biogenesis of PML bodies. In this work, a number of bioinformatics approaches were used to study proteins found in the proteome of PML-bodies for their tendency to spontaneous liquid-liquid phase separation (LLPS), which is usually caused by weak nonspecific interactions. 205 proteins found in PML bodies have been identified. It has been suggested that UBC9, P53, HIPK2, and SUMO1 can be considered as the scaffold proteins of PML bodies. It was shown that more than half of the proteins in the analyzed proteome are capable of spontaneous LLPS, with 85% of the analyzed proteins being intrinsically disordered proteins (IDPs), and remaining 15% being proteins with intrinsically disordered protein regions (IDPRs). About 44% of all proteins analyzed in this study contain SUMO binding sites and can potentially be SOMYylated. These data suggest that weak nonspecific interactions play a significantly larger role in the formation and biogenesis of PML bodies than previously expected.
Keywords:
PMLbodies
; intrinsically disordered proteins
; intrinsically disordered regions
; liquidliquid phase separation
; membrane-less organelles
; protein-protein interactions
; posttranslational modifications
; SUMOyltion
1. Introduction
The so-called LLPS (liquid-liquid phase separation) revolution that occurred in the 2010s caused fundamental changes in ideas about the spatiotemporal organization of intracellular space and the organization and control of biochemical processes in the cell [1]. It has become obvious that the formation and functioning of biomolecular condensates – membrane-less organelles (MLOs) – is one of the main driving forces in the molecular life of a cell. These processes are based on the separation of biopolymers, primarily intrinsically disordered proteins (IDPs) and RNA, into phases regulated by multiple specific/nonspecific inter- and intramolecular interactions. IDPs and proteins containing intrinsically disordered protein regions (IDPRs) are known to play a decisive role in LLPS leading to the formation of MLOs [2,3,4]. This is because, as a rule, IDPs/IDPRs contain blocks of the same type of amino acid residues, multiple weak nonspecific interactions between which (electrostatic, π-π, cation-π) under conditions of macromolecular crowding can help overcoming the potential energy barrier required for the transition of IDPs to the condensed liquid-droplet phase [5].
MLOs include nuclear PML-bodies (also known as ND10) – compartments involved in the regulation of cellular metabolism, transcription, maintenance of genome stability, response to viral infection, apoptosis and tumor suppression [6]. PML-bodies are subnuclear spherical and toroidal structures with a diameter of 0.1–2 μm [7]. They are present in most types of mammalian cells and tissues. Depending on the cell type, cell cycle phase, and differentiation stage, there may be about 5-30 PML-bodies per cell [8]. Based on the immunofluorescence analysis, PML-bodies exist as nuclear dot-shaped spherical structures residing in the interchromatin nuclear space [9]. As a rule, large PML-bodies have a toroidal structure formed by a ≈100 nm thick shell at the periphery and more mobile internal core [10]. According to the accepted models, canonical PML bodies are protein assemblages that do not contain nucleic acids [9]. However, specific types of PML bodies are known that are capable of incorporating DNA/RNA into their composition [6]:
- -
- viral DNA-containing PML bodies;
- -
- giant PML bodies containing satellite DNA;
- -
- ALT-associated PML bodies.
The major scaffolding protein of PML bodies is promyelocytic leukemia protein (PML). Due to alternative splicing, the PML protein is represented by seven main isoforms transcribed from one gene: six with nuclear localization (PML-I to PML-VI) due to the presence of the nuclear localization signal (NLS) in exon 6 and one with the cytoplasmic localization (PML-VII). The sequences encoded by the first four exons of the PML gene (418 amino acids with the conserved RBCC motif consisting of a RING finger domain (R) followed by two cysteine-histidine-rich B-box domains (B) and an α-helical coiled-coil domain (CC)) are common to all isoforms and contain structurally conserved domains: Proline-rich (1-45 a.a.), RING (45-105 a.a.), B1-box (124- 166 a.a.), B2-box (184-229 a.a.), and Coiled-Coil (229-323 a.a.) [11].
According to modern understanding, specific interactions play a decisive role in the formation and biogenesis of PML bodies [12]. It is assumed that oxidative formation of disulfide bonds between PML molecules and non-covalent interactions in the region of the ordered RBCC motif of PML trigger the oligomerization of non-sumoylated PML proteins, which subsequently undergo UBC9-mediated (poly-)SUMOylation and recruit PML body client proteins via SUMO-SIM interactions [12].
We have previously shown that a certain role in the formation of PML bodies is played by nonspecific intra- and intermolecular interactions that can lead to spontaneous phase separation of at least the disordered C-terminal domains of the PML-II and PML-V isoforms [13]. In this regard, we decided to analyze the proteins included in the proteome of PML bodies for their tendency to spontaneously separate into phases, and, consequently, for the ability to be nonspecifically included in PML bodies.
2. Results and Discussion
2.1. Finding Proteins Included in the PML Body Proteome
PML bodies take part in a large number of intracellular processes [6]. In this regard, the composition of client proteins of PML bodies varies significantly. In addition, proteins that interact with PML are often included in the PML body proteome [14,15]. However, PML is localized in the cell not only in PML bodies. It is known that several isoforms of the PML protein can be localized in the cytoplasm [11,16], and the PML bodies themselves pass into the cytoplasm during mitosis [17]. Taken together, this complicates the identification of proteins uniquely present in PML bodies.
In 2010, mainly based on an analysis of literature data, the so-called PML-body interactome was proposed, combining 166 proteins, most of which are part of the SUMO conjugation pathway [14]. A 2015 paper [15] discussed a network of 120 proteins that are involved in the direct physical interaction with PML, most of which are part of PML bodies. This set included proteins whose interaction with PML was confirmed by affinity capture followed by Western blotting (BIOGRID data). Proteins associated with PML, identified by high-throughput methods, were not discussed in this work [15].
To date, according to the BIOGRID database (v.4.4) [18], the PML protein molecular interaction network consists of 707 proteins. To identify the proteins that make up PML bodies, we analyzed the PML interactome from the BIOGRID database, selecting proteins involved in the biogenesis of these non-membrane organelles. From 707 proteins included in the network of molecular interactions of the PML protein, we selected 267 proteins for which interaction with PML was shown at least twice. Using information obtained from the resources https://amigo.geneontology.org/amigo/term/GO:0016605 GO:0016605 (PML NB/ND10) and SL-0465 (https://www.uniprot.org/locations/SL -0465), for further analysis, we selected 105 proteins potentially included in human PML bodies. Analysis of literature data allowed us to identify 205 proteins that can be localized in PML bodies according to the fluorescent microscopy or electron microscopy data. The list of these proteins with corresponding evidence is provided in Table S1.
A comparison of the data obtained suggested that 52 of 205 proteins capable of being included in PML bodies do not interact directly with PML (Figure 1).
2.2. PML Body Scaffold Proteins
It is known that the scaffold proteins of PML bodies include PML, death domain-associated protein (DAXX), cyclic AMP-responsive element-binding protein (CREB)-binding protein (CREBBP), and speckled 100 kDa (SP100, also known as nuclear dot-associated Sp100 protein or nuclear autoantigen Sp-100 [19]. We analyzed the interactomes of these four proteins and found that, according to the BIOGRID database, the proteins ubiquitin carrier protein 9 (UBC9, also known as SUMO-conjugating enzyme UBC9), small ubiquitin-related modifier 1 (SUMO1), p53, and homeodomain-interacting protein kinase 2 (HIPK2) are included in all four interactomes analyzed (Figure 2).
Analysis of the literature data indicates that these proteins are one way or another included in the biogenesis of PML bodies (UBC9 [21], SUMO1 [22], HIPK2 [23] and p53 [24,25,26]). As is known, PML bodies are a platform for SUMOylation and ubiquitination of various proteins [27]. SUMO-conjugating enzyme UBC9 accepts ubiquitin-like proteins SUMO1, SUMO2, SUMO3, SUMO4, and SUMO1 pseudogene 1 (SUMO1P1/SUMO5) from the ubiquitin-like 1-activating enzyme E1A – ubiquitin-like 1-activating enzyme E1B (UBLE1A-UBLE1B) complex and catalyzes their covalent attachment to other proteins using E3 ligases, such as Ran-binding protein 2 (RANBP2, also known as E3 SUMO-protein ligase RanBP2 or nucleoporin Nup358), chromobox protein homolog 4 (CBX4, also known as E3 SUMO-protein ligase CBX4 or Polycomb 2 homolog, Pc2), and zinc finger protein 451 (ZNF451, also known as E3 SUMO-protein ligase ZNF451) [27,28]. UBC9 can catalyze the formation of poly-SUMO chains. It is known that SUMOylation of proteins included in PML bodies occurs in a UBC9-dependent manner [29]. Conjugation of SUMO substrates often requires an E3 ligase, which provides substrate specificity by simultaneously binding UBC9 and the substrate. E3 SUMO ligases typically use the RING domain to interact with UBC9. The PML protein is considered a possible SUMO ligase [19]. The antiparallel PML dimer model has been proposed to potentially support the accessibility of the PML B-box1 domain for UBC9 binding [29].
PML bodies are also a platform that provides post-translational modifications (PTMs) of the tumor suppressor p53, allowing this protein to take part in the regulation of a number of intracellular processes, in particular, senescence [30]. It is worth noting that one of the p53 regulatory proteins that serves as a part of PML bodies is the ubiquitin carboxyl-terminal hydrolase 7 (USP7) protein [31]. Homeodomain-interacting protein kinase 2 (HIPK2) promotes p300-mediated acetylation of p53 at K382 and influences the selective transactivation of proapoptotic p53 target genes. To cause HIPK2-mediated induction of pro-apoptotic p53 genes, p53 must be modified by both S46 phosphorylation and K382 acetylation [32]. It is suggested that SP100 acts as a coactivator of HIPK2-mediated activation of p53 and thus has an essential role in p53-dependent gene expression [33]. p300 (CREBBP homolog) and CBP (cyclic AMP response element-binding protein-binding protein) directly bind to p53 and acetylate several lysine residues located in the C-terminal domain of this protein: K370, K372, K373, K381, K382, and K386. Furthermore, four additional lysine residues, K101, K139, K164, and K305, can be acetylated by p300/CBP. It has been suggested that acetylation of these sites enhances the DNA-binding ability of p53 and promotes its transcription, leading to growth arrest or apoptosis [32].
Taken together, these data suggest that UBC9, SUMO1, p53, and HIPK2 can be included to the set of the PML scaffolding proteins.
2.3. Proteome Analysis of PML Bodies
We analyzed 205 proteins that were identified as proteins of the PML body proteome for their predisposition for intrinsic disorder and tendency. Results of this analysis are summarized in Figure 3.
This global disorder analysis revealed that proteins in the PML body proteome are characterized by the exceptionally high levels of intrinsic disorder. This is illustrated by Figure 3A representing the results of the classification of the disorder status of these proteins based on the outputs of the per-residue disorder predictor PONDR® VSL2 in a form of the mean disorder score (MDS) vs. percent of predicted disordered residues (PPIDR) dependence. This plot shows distribution of the proteins within MDS-PPIDR blocks containing mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red) proteins. Based on these classifications, none of the moonlighting proteins was predicted as ordered by MDS and PPIDR (dark blue and both light blue segments are empty). Therefore, all these proteins are expected to be either moderately or highly disordered. In fact, according to the Figure 3A 14.6% of these proteins were predicted as moderately disordered and containing noticeable levels of flexible or disordered residues/regions based on both their MDS and PPIDR values (dark pink segment). Additional 17.1% proteins were expected to be moderately disordered based on their MDS values (light pink segment), whereas 68.3% of the proteins from the PML body proteome are expected to be highly disordered (red segment). Importantly, most of these highly disordered proteins (131 of 140) have PPIDR ≥ 50% and MDS ≥ 0.5. This global disorder content in PML body proteome dramatically exceeds that of the human proteome, where the moderately and highly disordered proteins account for 51.7% and 39.8%, respectively [34].
Further support for exceptionally high disorder status of human proteins in PML body proteome was provided by analyzing the combined outputs of two binary disorder predictors charge-hydropathy (CH) plot and cumulative distribution function (CDF) analysis that classify proteins as mostly ordered or mostly disordered (see Figure 3B). The result of the ΔCH-ΔCDF plot provides useful means for more detailed characterization of the global disorder status of proteins, classifying them as mostly ordered, molten globule-like or hybrid, or highly disordered (see Materials and Methods section). Figure 3B shows that only 34.3% (cf. 59.1% in entire human proteome [34]) of these proteins are located within the quadrant Q1 (bottom right corner) containing proteins predicted to be ordered by both predictors, whereas 65.7% of these proteins are located outside the quadrant Q1 and can be considered as proteins with high disorder levels. Figure 3B shows that the quadrant Q2 (bottom left corner) corresponding to molten globular or hybrid proteins predicted to be ordered/compact by the CH-plot and disordered by the CDF analysis includes 42.9% (21.5% in human proteome [34]) of proteins from the PML proteome, 21.5% (12.3% in human proteome [34]) of these proteins are located in the quadrant Q3 (top left corner) and therefore are predicted as highly disordered by both predictors, whereas the quadrant Q4 (top right corner) contains 1.5% proteins (3.1% in human proteome [34]) predicted as disordered by CH-plot and ordered by CDF analysis. Therefore, based on the results of these analyses, one can conclude that the levels of intrinsic disorder in human proteins found in the PML body proteome are considerably higher than those of entire human proteome, suggesting that intrinsic disorder plays important roles in functions of these proteins and is likely to be related to the biogenesis of this MLO.
This hypothesis was supported by the analysis of the predisposition of human proteins from the PML body proteome to undergo spontaneous LLPS. For this purpose, a combined approach was used, based on the multiparametric bioinformatics analysis of the sequences of the proteins under study (Figure 4).
The conducted analysis revealed (Figure 5) that there are practically no globular proteins in the analyzed data set, which differs significantly from the human proteome (the proportion of IDPs in which is about 40% [34]). This is also consistent with the data that 52% of the analyzed proteins in the PML body proteome are highly likely to be capable of spontaneous phase separation. Furthermore, according to the FuzDrop analysis, the number of proteins – “LLPS drivers”; i.e., proteins capable of spontaneous phase separation in the proteome of PML bodies is almost twice as large as the number of client proteins; i.e., proteins capable of LLPS upon interaction with some partners (Table S2). However, only 44% of all proteins studied contain SUMO binding sites and can potentially be SUMOylated.
Among the proteins that do not directly interact with PML, there are even fewer potentially SUMOylated proteins—only 34%. The data obtained indicate that the proportion of proteins in the proteome of PML bodies capable of nonspecific inclusion in these structures is at least no less than the proportion of proteins included in these MLOs due to SUMO/SIM interactions.
Proteins prone to spontaneous LLPS are mainly represented by transcription factors, regulatory elements, DNA-Damage Response (DDR) proteins, and proteins involved in alternative telomere elongation (Figure 6). About 10% of LLPS-prone proteins are DNA-binding proteins.
This abundance of DNA-binding proteins in the proteome of PML bodies suggests that DNA is somehow involved in the biogenesis of these MLOs. This assumption is also supported by the fact that more than half of the proteins studied are positively charged, which may indicate their nonspecific interaction with negatively charged nucleic acids. Furthermore, the proportion of such proteins among proteins that do not directly interact with PML is 8% higher than in the full data set.
There are a number of works that discussed the influence of DNA on the biogenesis of PML bodies. For example, in 2004, it was shown [36] that the structural integrity of nuclear PML bodies depends not only on PTMs of PML, but also on the integrity and state of chromatin condensation. In cells not treated with transcription inhibitors, PML bodies are surrounded by chromatin, which explains the stability of their position; direct physical contacts between the protein core and chromatin fibers predominate. Inhibition of transcription causes a reduction in these contacts due to the chromatin condensation.
PML bodies have been shown to degrade in response to changes in chromatin integrity (demonstrated by introducing exogenous nuclease DNase I into cells) [36]. A 2009 study showed that PML bodies are surrounded by decondensed and condensed chromatin [37]. It has been suggested that PML regulates the incorporation of H3.3 between loose and compact chromatin compartments [38]. In doing so, PML regulates the routing of H3.3 to chromatin by diverting the H3.3 pool and limiting its deposition in chromatin. In addition, there is evidence that PML bodies are involved not only in gene silencing, but also as a potential regulator of epigenetic conditions [38].
3. Materials and Methods
3.1. Study Design
Identification of the proteins included in the PML body proteome was carried out using two approaches:
- i)
- analysis of the PML body proteome and the PML protein interactome in the BIOGRID (v.4.4) databases https://thebiogrid.org/, and resources https://amigo.geneontology.org/amigo/term/GO:0016605 GO:0016605 (PML NB/ND10) and SL-0465 (https://www.uniprot.org/locations/SL-0465), and analysis of literature data.
For further analysis, 205 proteins were selected that could be localized in PML bodies according to fluorescent microscopy or electron microscopy data. Figure 7 schematically represents the design of our study.
3.2. Intrinsic Disorder Prediction
Analysis of the intrinsic disorder predispositions of the studied proteins was carried out using the RIDAO online platform [39]. This platform predicts per-residue disorder potential in proteins based on their amino acid sequences. This package combines outputs of several commonly used predictors that utilize different algorithms, such as PONDR® VLS2, PONDR® VL3, PONDR® VLXT, PONDR® FIT, IUPred-Long, IUPred-Short [40,41,42,43,44,45].
In the case of a significant discrepancy between the results obtained by different predictors, the correctness of the prediction of protein structure disorder was determined by predicting the protein structure using the AlphaFold2 algorithm [46]. The PONDR® VSL2 algorithm showed the most correct prediction of disorder for a large data set. In this regard, the PER(VSL2B) indicator was used as a measure of protein structural disorder, which reflects the percentage of residues in the amino acid sequence of a protein for which the prediction of disorder (Disorder Score) is higher than 0.5. Such regions are determined by the algorithm as “probably disordered.” The retrieved PER(VSL2B) values then were used for estimation of the disorder status of query proteins using very conservative criteria, according to which proteins with the PER(VSL2B) values < 50% were classified as ordered; proteins with the values of 50 ≤ PER(VSL2B) < 85% were classified as partially disordered proteins containing IDRPs; whereas proteins with the PER(VSL2B) values of ≥ 85% were classified as intrinsically disordered proteins (IDPs).
In a less conservative analysis, the percent of predicted intrinsically disorder residues (PPIDR) was used to classify each protein based on their level of disorder. Generally, a PPIDR value of less than 10% is taken to correspond to a highly ordered protein, PPIDR between 10% and 30% is ascribed to moderately disordered protein, and PPIDR greater than 30% corresponds to a highly disordered protein [47,48]. In addition to PPIDR, mean disorder score (MDS) was calculated for each query protein as a protein length-normalized sum of all the per-residue disorder scores. Here again, proteins are grouped based on their corresponding MDS values, being classified as highly ordered (MDS < 0.15), moderately disordered of flexible (MDS between 0.15 and 0.5) and highly disordered (MDS ≥ 0.5).
Global intrinsic disorder predisposition of the analyzed dataset was further evaluated by the CH-CDF analysis, data for which were generated using RIDAO [39]. CH-CDF analysis combines results from two binary disorder predictors, charge-hydropathy (CH) plot and cumulative distribution function (CDF) analysis [49,50]. Combination of data generated by these approaches results in the ΔCH-ΔCDF plot allowing discrimination between flavors of disorder based on the positions of proteins within the quadrants of the ΔCH-ΔCDF phase space [51,52]. Here, proteins in the top-left quadrant are predicted to be disordered by both CH and CDF, the ones in the bottom-left are predicted to be compact/ordered by CH and disordered by CDF, the ones in the top-right are predicted to be disordered by CH and ordered by CDF, and in the bottom-right quadrant predicted to be ordered by both predictors [51,52].
3.3. LLPS Predisposion Analysis
The studied data sets were analyzed for the tendency of their proteins to undergo liquid-liquid phase separation using the FuzDrop [53] and PSPredictor [54] predictors. The propensity of the analyzed protein to LLPS was determined based on PSPredcitor score > 0.5 and FuzDrop score > 0.6. In the case of the discrepancy between the results of predicting the propensity for phase separation obtained using both predictors, the analyzed proteins were assigned to the “controversial LLPS” group.
The FuzDrop predictor also allows one to predict the ability of the analyzed protein to undergo spontaneous and interaction-induced LLPS. Proteins, which spontaneously undergo liquid-liquid phase separation are termed as droplet-driving proteins. Proteins, which require additional interactions to form droplets, are termed as clients. Proteins with pLPS ≥ 0.60 likely drive liquid-liquid phase separation. Proteins with droplet-promoting regions, defined as consecutive residues with pDP ≥ 0.60 will likely serve as droplet-clients.
3.4. Evaluation of Protein Charge and Hydrophobicity
The charge of the studied proteins was determined at pH 7.0 as the average charge of the protein over the sequence, taking into account the charge of amino acids: R = 1, K = 1, H = 0.5, D = −1, E = −1, according to [49]. The hydrophobicity of the studied proteins was calculated using the normalized Kite and Doolittle scale [55].
3.5. Determination of Proteins Targeted for SUMOylation
This was done by visiting the UniProt website (https://www.uniprot.org/, accessed on September 10, 2023) [56] and then searching for mentions of SUMO in the PTM/Processing section of the corresponding UniProt entry.
3.6. Determination of the Biological Processes and Molecular Functions of Proteins
Gene Onthology (GO) was performed using the Enrichr resource (https://maayanlab.cloud/Enrichr/, accessed on September 10, 2023), and 10 terms with the lowest p-values were selected [57]. The p-values were computed from the Fisher exact test, which is used to determine whether or not there are significant nonrandom associations between two categorical variables.
4. Conclusions
In some of our previous works, we have already raised the question of the need to change existing ideas about the formation and biogenesis of PML bodies [11,13]. In particular, our analysis of the amino acid sequences of PML isoforms showed that the variable C-terminal domains of almost all isoforms of this protein have properties characteristic of protein sequences potentially capable of phase separation due to electrostatic interactions. It was shown that the C-terminal domains of the PML-II and PML-V isoforms are capable of not only being incorporated into endogenous PML bodies, but also forming dynamic liquid-droplet compartments under conditions of endogenous PML knockout. Furthermore, the exclusion of SUMO/SIM interactions from the forces driving LLPS of the C-terminal domains of PML-II and PML-V by disrupting the SUMOylation sites of these proteins using the K490R substitution led to an increase in the liquid-like properties of the condensates formed by these proteins.
The results obtained indicate a high content of disordered proteins in the composition of PML bodies, potentially capable of spontaneous separation into phases, and, therefore, allow us to raise the question of changing ideas about the role of nonspecific protein-protein interactions and, possibly, protein-DNA interactions in the formation and biogenesis of PML bodies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: The list of 205 human proteins that can be localized in PML bodies according to the fluorescent microscopy or electron microscopy data; Table S2: Results of the multifactorial bioinformatics analysis of 205 human proteins localized in nuclear PML bodies.
Author Contributions
Conceptualization, A.V.F. and V.N.U.; methodology, A.V.F. and V.N.U.; validation, I.M.K., K.K.T., V.N.U., and A.V.F.; formal analysis, S.A.S., Y.I.M., E.M.N., E.Y.S., V.N.U., and A.V.F.; investigation, S.A.S., Y.I.M., E.M.N., E.Y.S., V.N.U., and A.V.F.; writing—original draft preparation, S.A.S., Y.I.M., E.M.N., E.Y.S., I.M.K., K.K.T., V.N.U., and A.V.F.; writing—review and editing, S.A.S., Y.I.M., E.M.N., E.Y.S., I.M.K., K.K.T., V.N.U., and A.V.F.; visualization, S.A.S., Y.I.M., E.M.N., E.Y.S., V.N.U., and A.V.F.; supervision, A.V.F. and V.N.U.; funding acquisition, A.V.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 22-15-00429 (K.K.T.).
Data Availability Statement
Data are contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid-liquid phase separation in human health and diseases. Signal Transduct Target Ther 2021, 6, 290. [Google Scholar] [CrossRef]
- Brangwynne, Clifford P. ; Tompa, P.; Pappu, Rohit V. Polymer physics of intracellular phase transitions. Nat Phys 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem Sci 2019, 44, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Current opinion in structural biology 2016, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Dignon, G.L.; Best, R.B.; Mittal, J. Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu Rev Phys Chem 2020, 71, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: catch me if you can! Nucleic Acids Res 2020, 48, 11890–11912. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb Perspect Biol 2010, 2, a000661. [Google Scholar] [CrossRef]
- Prikrylova, T.; Pachernik, J.; Kozubek, S.; Bartova, E. Epigenetics and chromatin plasticity in embryonic stem cells. World J Stem Cells 2013, 5, 73–85. [Google Scholar] [CrossRef]
- Boisvert, F.M.; Hendzel, M.J.; Bazett-Jones, D.P. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol 2000, 148, 283–292. [Google Scholar] [CrossRef]
- Hands, K.J.; Cuchet-Lourenco, D.; Everett, R.D.; Hay, R.T. PML isoforms in response to arsenic: high-resolution analysis of PML body structure and degradation. J Cell Sci 2014, 127, 365–375. [Google Scholar] [CrossRef]
- Fonin, A.V.; Silonov, S.A.; Shpironok, O.G.; Antifeeva, I.A.; Petukhov, A.V.; Romanovich, A.E.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. The Role of Non-Specific Interactions in Canonical and ALT-Associated PML-Bodies Formation and Dynamics. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Wu, W.; Chen, Z.; Meng, G. PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends Cancer 2020, 6, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Fonin, A.V.; Silonov, S.A.; Fefilova, A.S.; Stepanenko, O.V.; Gavrilova, A.A.; Petukhov, A.V.; Romanovich, A.E.; Modina, A.L.; Zueva, T.S.; Nedelyaev, E.M. , et al. New Evidence of the Importance of Weak Interactions in the Formation of PML-Bodies. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 2010, 6, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Kao, H.Y. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci 2015, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Gao, Y.; Lin, H.K. Cytoplasmic PML: from molecular regulation to biological functions. J Cell Biochem 2014, 115, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Lang, E.; Boe, S.O. PML Bodies in Mitosis. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F. , et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Xu, P.; Roizman, B. The SP100 component of ND10 enhances accumulation of PML and suppresses replication and the assembly of HSV replication compartments. Proc Natl Acad Sci U S A 2017, 114, E3823–E3829. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wang, P.; Benhenda, S.; Wu, H.; Lallemand-Breitenbach, V.; Zhen, T.; Jollivet, F.; Peres, L.; Li, Y.; Chen, S.J.; Chen, Z. , et al. RING tetramerization is required for nuclear body biogenesis and PML sumoylation. Nat Commun 2018, 9, 1277. [Google Scholar] [CrossRef]
- Lang, M.; Jegou, T.; Chung, I.; Richter, K.; Munch, S.; Udvarhelyi, A.; Cremer, C.; Hemmerich, P.; Engelhardt, J.; Hell, S.W. , et al. Three-dimensional organization of promyelocytic leukemia nuclear bodies. J Cell Sci 2010, 123, 392–400. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Moller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 2002, 4, 1–10. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. Regulating the p53 Tumor Suppressor Network at PML Biomolecular Condensates. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Willms, A.; Schupp, H.; Poelker, M.; Adawy, A.; Debus, J.F.; Hartwig, T.; Krichel, T.; Fritsch, J.; Singh, S.; Walczak, H. , et al. TRAIL-receptor 2-a novel negative regulator of p53. Cell Death Dis 2021, 12, 757. [Google Scholar] [CrossRef]
- Kwek, S.S.; Derry, J.; Tyner, A.L.; Shen, Z.; Gudkov, A.V. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene 2001, 20, 2587–2599. [Google Scholar] [CrossRef]
- Sha, Z.; Blyszcz, T.; Gonzalez-Prieto, R.; Vertegaal, A.C.O.; Goldberg, A.L. Inhibiting ubiquitination causes an accumulation of SUMOylated newly synthesized nuclear proteins at PML bodies. J Biol Chem 2019, 294, 15218–15234. [Google Scholar] [CrossRef]
- Erker, Y.; Neyret-Kahn, H.; Seeler, J.S.; Dejean, A.; Atfi, A.; Levy, L. Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Mol Cell Biol 2013, 33, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Bregnard, T.; Ahmed, A.; Semenova, I.V.; Weller, S.K.; Bezsonova, I. The B-box1 domain of PML mediates SUMO E2-E3 complex formation through an atypical interaction with UBC9. Biophys Chem 2022, 287, 106827. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. Crosstalk between p53 modifiers at PML bodies. Mol Cell Oncol 2018, 5, e1074335. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Tang, Y.; Tang, S.; Liu, J.; Wu, K.; Yao, S.; Sun, Y.; Zhou, L.; Deng, T.; Chen, Y. , et al. STIP is a critical nuclear scaffolding protein linking USP7 to p53-Mdm2 pathway regulation. Oncotarget 2015, 6, 34718–34731. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Miyajima, C.; Aoki, H.; Aoyama, M.; Morishita, D.; Inoue, Y.; Hayashi, H. Insights into Regulators of p53 Acetylation. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Sirma, H.; Hofmann, T.G.; Staege, H.; Gresko, E.; Ludi, K.S.; Klimczak, E.; Droge, W.; Will, H.; Schmitz, M.L. Sp100 is important for the stimulatory effect of homeodomain-interacting protein kinase-2 on p53-dependent gene expression. Oncogene 2003, 22, 8731–8737. [Google Scholar] [CrossRef]
- Mohammed, A.S.; Uversky, V.N. Intrinsic Disorder as a Natural Preservative: High Levels of Intrinsic Disorder in Proteins Found in the 2600-Year-Old Human Brain. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of Proceedings of the 9th Python in Science Conference; pp. 51–56. [CrossRef]
- Eskiw, C.H.; Dellaire, G.; Bazett-Jones, D.P. Chromatin contributes to structural integrity of promyelocytic leukemia bodies through a SUMO-1-independent mechanism. J Biol Chem 2004, 279, 9577–9585. [Google Scholar] [CrossRef] [PubMed]
- Torok, D.; Ching, R.W.; Bazett-Jones, D.P. PML nuclear bodies as sites of epigenetic regulation. Front Biosci (Landmark Ed) 2009, 14, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Ivanauskiene, K.; Spirkoski, J.; Shah, A.; Vekterud, K.; Moskaug, J.O.; Boe, S.O.; Wong, L.H.; Kuntziger, T.; Collas, P. PML protein organizes heterochromatin domains where it regulates histone H3.3 deposition by ATRX/DAXX. Genome Res 2017, 27, 913–921. [Google Scholar] [CrossRef]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci 2022, 31, e4496. [Google Scholar] [CrossRef]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins: Structure, Function, and Bioinformatics 2005, 61, 176–182. [Google Scholar] [CrossRef]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC bioinformatics 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. Journal of bioinformatics and computational biology 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A. , et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Uversky, V.N. Analyzing IDPs in interactomes. In Intrinsically Disordered Proteins, Kragelund, B.B., Skriver, K., Eds. Humana New York, NY, 2020; Vol. Methods in Molecular Biology, pp. 895–945.
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar] [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 2012, 128–139. [Google Scholar]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid-liquid phase separating proteins using machine learning. BMC bioinformatics 2022, 23, 72. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consotrium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A. , et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Venn diagram of three sets of proteins potentially included in PML bodies. Blue circle represents the set based on the analysis of the PML interactome in the BIOGRID database. Green circle represents the set according to the analysis of the GO:0016605 and SL-0465 databases. Yellow circle represents the set collected during the analysis of literary sources.
Figure 1.
Venn diagram of three sets of proteins potentially included in PML bodies. Blue circle represents the set based on the analysis of the PML interactome in the BIOGRID database. Green circle represents the set according to the analysis of the GO:0016605 and SL-0465 databases. Yellow circle represents the set collected during the analysis of literary sources.
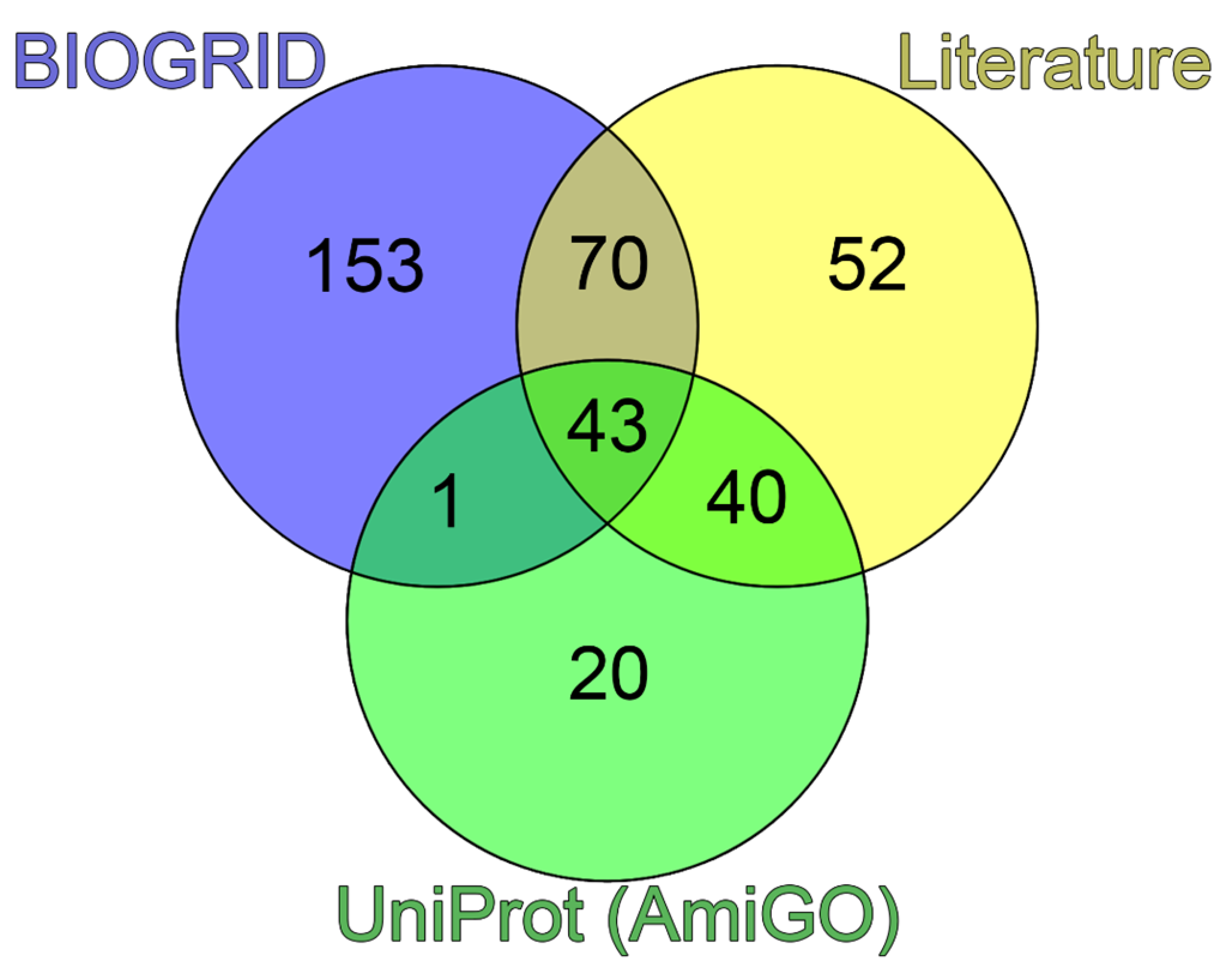
Figure 2.
Analysis of the interactome of PML body scaffold proteins. (A) Venn diagram represents the intersection of the interactomes of four PML body scaffold proteins (PML, DAXX, SP100, and CREBB(CBP)). Data were obtained using BioGRID v4.4. (B) Interaction map for eight potential PML body envelope proteins based on the statistical representation in the BIOGRID (Evidence score) database. Yellow shows results with weak statistics (data from one experiment), blue shows data with strong statistics (> 10 experiments). The heat-map was constructed using CytoScape [20]. (C) Table of results for predicting the propensity of detected proteins to undergo spontaneous phase separation using FuzDrop and PSPrediction scores.
Figure 2.
Analysis of the interactome of PML body scaffold proteins. (A) Venn diagram represents the intersection of the interactomes of four PML body scaffold proteins (PML, DAXX, SP100, and CREBB(CBP)). Data were obtained using BioGRID v4.4. (B) Interaction map for eight potential PML body envelope proteins based on the statistical representation in the BIOGRID (Evidence score) database. Yellow shows results with weak statistics (data from one experiment), blue shows data with strong statistics (> 10 experiments). The heat-map was constructed using CytoScape [20]. (C) Table of results for predicting the propensity of detected proteins to undergo spontaneous phase separation using FuzDrop and PSPrediction scores.
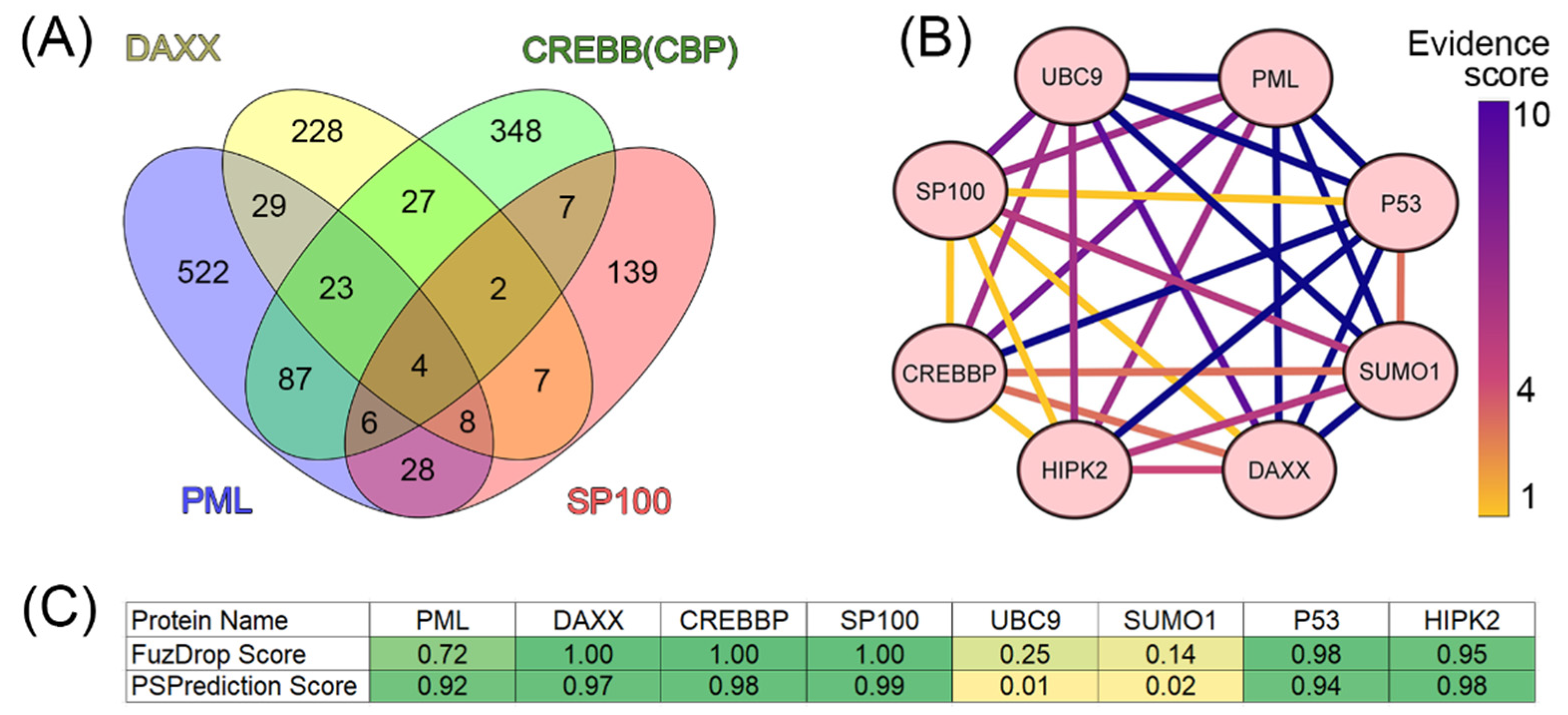
Figure 3.
Global disorder analysis of proteins in the PML body proteome. (A) PONDR® VSL2 output. PONDR® VSL2 score is the mean disorder score (MDS) for a protein, which is calculated as a sequence-length normalized sum of its per-residue disorder scores. PONDR VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR); i.e., the percent of residues with disorder scores above 0.5. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which only one of these criteria applies. The boundaries of the colored regions represent arbitrary and accepted cutoffs for MDS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). (B) Charge-hydropathy and cumulative distribution function (ΔCH-ΔCDF) plot for 205 proteins from the nuclear PML body proteome. The Y-coordinate is calculated as the distance of the corresponding protein from the boundary in the CH plot. The X-coordinate is calculated as the average distance of the corresponding protein’s CDF curve from the CDF boundary. The quadrant where the proteins are located determines their classification. Q1, 111 proteins (67.3%) predicted to be ordered by CDF and compact/ordered by CH-plot. Q2, 30 proteins (18.2%) predicted to be ordered/compact by CH-plot and disordered by CDF. Q3, 16 proteins (9.7%) predicted to be disordered by CH-plot and CDF. Q4, 8 proteins (4.8%) predicted to be disordered by CH-plot and ordered by CDF.
Figure 3.
Global disorder analysis of proteins in the PML body proteome. (A) PONDR® VSL2 output. PONDR® VSL2 score is the mean disorder score (MDS) for a protein, which is calculated as a sequence-length normalized sum of its per-residue disorder scores. PONDR VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR); i.e., the percent of residues with disorder scores above 0.5. Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which only one of these criteria applies. The boundaries of the colored regions represent arbitrary and accepted cutoffs for MDS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). (B) Charge-hydropathy and cumulative distribution function (ΔCH-ΔCDF) plot for 205 proteins from the nuclear PML body proteome. The Y-coordinate is calculated as the distance of the corresponding protein from the boundary in the CH plot. The X-coordinate is calculated as the average distance of the corresponding protein’s CDF curve from the CDF boundary. The quadrant where the proteins are located determines their classification. Q1, 111 proteins (67.3%) predicted to be ordered by CDF and compact/ordered by CH-plot. Q2, 30 proteins (18.2%) predicted to be ordered/compact by CH-plot and disordered by CDF. Q3, 16 proteins (9.7%) predicted to be disordered by CH-plot and CDF. Q4, 8 proteins (4.8%) predicted to be disordered by CH-plot and ordered by CDF.
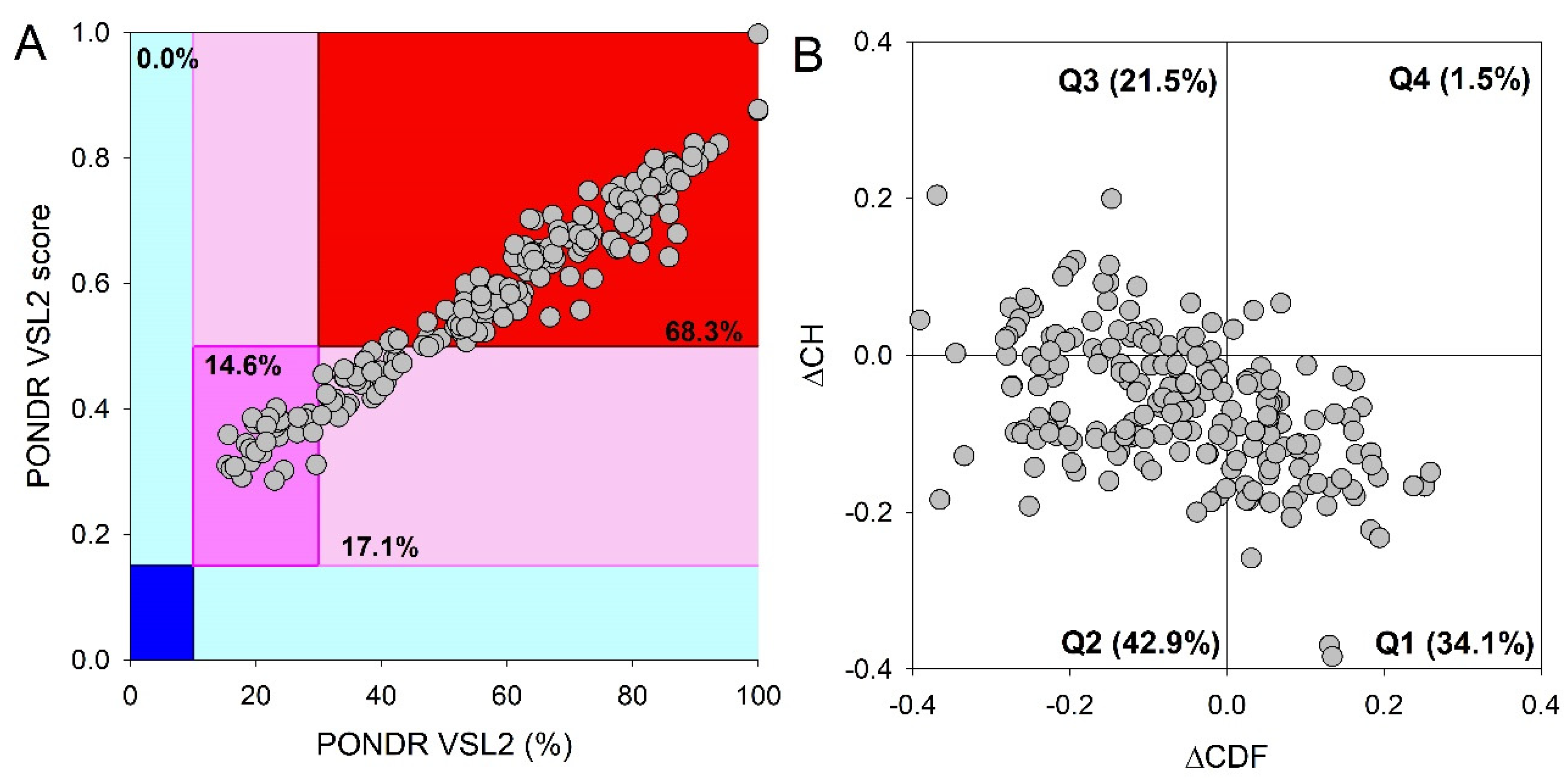
Figure 4.
Correlation of the analyzed parameters using the Pandas package [35]. List of parameters: amino acid sequence length (Length, a.a.); PONDR® VSL2-based content of disordered residues PER(VSL2B); FuzDrop and PSPredictor scores; droplet-promoting regions (DPRs) number and length; aggregation “hot spot” (AHS) number and length; DPRs length (LDPRs), and AHS length (LAHS) per amino acid number; Hydrophobicity and Charge of proteins.
Figure 4.
Correlation of the analyzed parameters using the Pandas package [35]. List of parameters: amino acid sequence length (Length, a.a.); PONDR® VSL2-based content of disordered residues PER(VSL2B); FuzDrop and PSPredictor scores; droplet-promoting regions (DPRs) number and length; aggregation “hot spot” (AHS) number and length; DPRs length (LDPRs), and AHS length (LAHS) per amino acid number; Hydrophobicity and Charge of proteins.
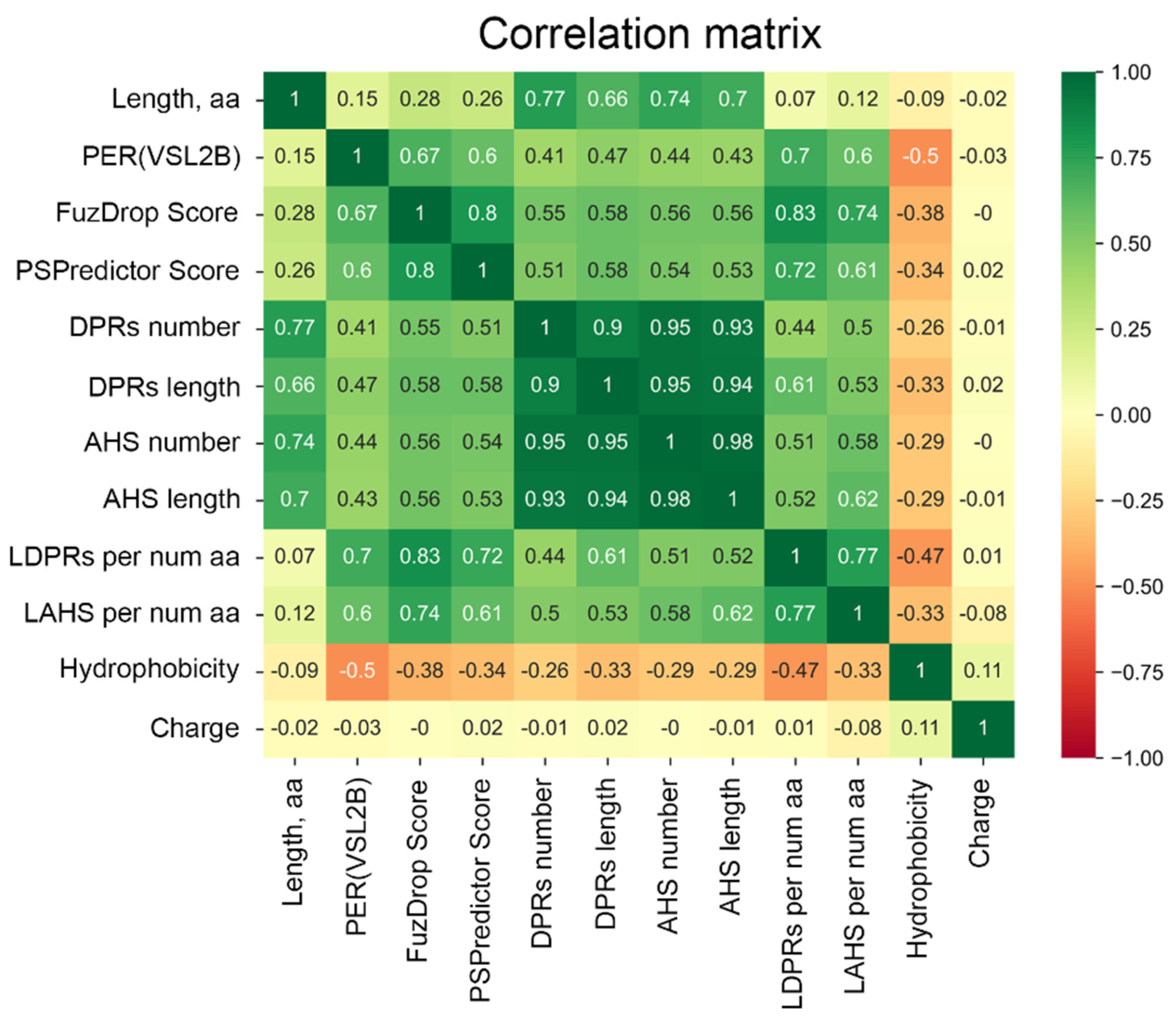
Figure 5.
Proteome analysis of PML bodies. (A) Correlation of the results of two predictors FuzDrop and PSPredictor and the result in the form of a Pie diagram. (B) Proportion of proteins potentially capable (SUMO) and incapable (NoSUMO) of sumoylation in the analyzed set of proteins. (C) Share of LLPS drivers/clients. (D) Proportion of disordered proteins (IDP) and proteins with intrinsically disordered ptotein regions (IDPR). (E) Proportion of proteins potentially carrying a negative charge, a weak positive charge, and a positive charge. Proportion of proteins potentially capable (SUMO) and incapable (NoSUMO) of sumoylation for LLPS (F), IDP (G) and weakly positive (H) drivers.
Figure 5.
Proteome analysis of PML bodies. (A) Correlation of the results of two predictors FuzDrop and PSPredictor and the result in the form of a Pie diagram. (B) Proportion of proteins potentially capable (SUMO) and incapable (NoSUMO) of sumoylation in the analyzed set of proteins. (C) Share of LLPS drivers/clients. (D) Proportion of disordered proteins (IDP) and proteins with intrinsically disordered ptotein regions (IDPR). (E) Proportion of proteins potentially carrying a negative charge, a weak positive charge, and a positive charge. Proportion of proteins potentially capable (SUMO) and incapable (NoSUMO) of sumoylation for LLPS (F), IDP (G) and weakly positive (H) drivers.
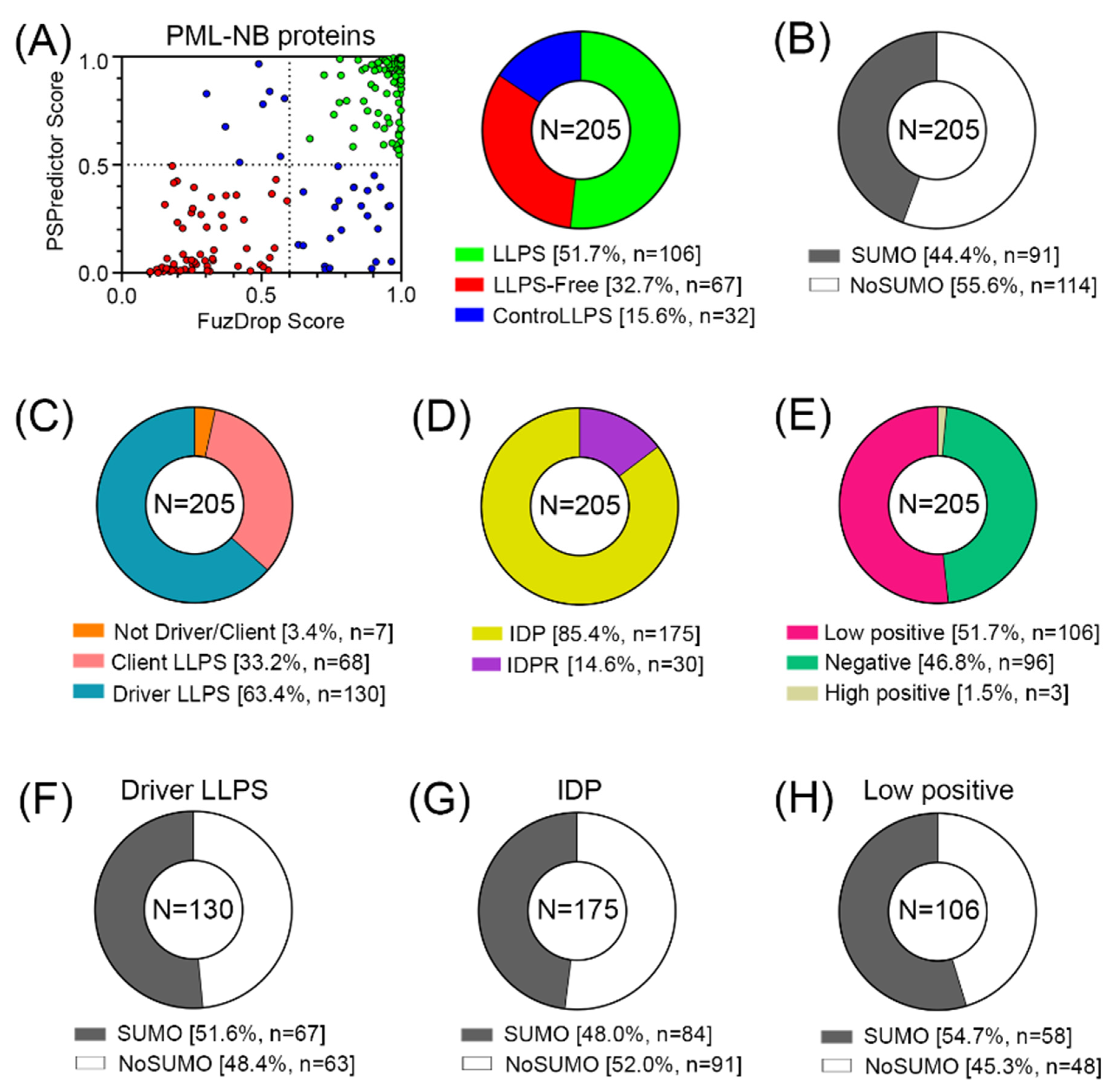
Figure 6.
Result of GO analysis for proteins potentially prone to LLPS. Biological process and molecular functions are shown.
Figure 6.
Result of GO analysis for proteins potentially prone to LLPS. Biological process and molecular functions are shown.
Figure 7.
Scheme illustrating the design of our study.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



