
Submitted:
23 October 2023
Posted:
23 October 2023
Read the latest preprint version here
Abstract
Tumor heterogeneity is a major obstacle to achieving consistent outcomes in cancer therapy. Here, we present a new perspective on tumor heterogeneity. The foundation for understanding tumor heterogeneity lies in recognizing that tumor evolution is a process driven by cells responding to survival challenges, and this cellular response precipitates the development of hallmark cancer capabilities. We posit that tumor heterogeneity arises from cells’ continued development of hallmark cancer capabilities, which ultimately leads to increased molecular chaos. Therapeutic inhibition of the essential genes driving the disease can disrupt tumor evolution, leading to cell death, a phenomenon termed oncogene addiction. Drawing from the same idea that cells respond to survival challenges, we introduce the notion that the increasing primitiveness of cancer during its progress stems from the cellular response to the continuous need for regeneration due to sustained cellular damage. To expand our concept of tumor heterogeneity, we further explore redefining key genes based on heterogeneity levels, and we propose a potential treatment strategy for advanced-stage cancers based on inducing aging-related deterioration. Taken together, the notions explored herein pave the way for novel cancer therapies and the betterment of human health.
Keywords:
tumor heterogeneity
; cancer progression
; oncogene addiction
; cancer stem cell
Main Concept
Tumor heterogeneity refers to the diversity of tumor cells in terms of various cellular and molecular characteristics [1,2]. This variation is observed among different tumors, called inter-tumor heterogeneity, and within a single tumor, termed intra-tumor heterogeneity. Such heterogeneity poses significant challenges in developing effective treatment strategies [3,4,5]. Meticulously examining and characterizing the driving forces behind this heterogeneity can bolster our comprehension of the disease’s origins and progression and guide the development of more effective treatment methods.
Tumor progression necessitates a continuous process whereby most, if not all, cancer hallmarks are activated once a triggering event occurs [6,7]. Conversely, if only a limited number of cancer hallmark phenotypes could emerge, tumor progression would be non-existent, and the condition wouldn’t be classified as cancer. Once triggered, the uninterrupted process hinges on the interconnectedness of these phenotypes, with each one arising from the previous. This cascade prompts the notion that activation of more phenotypes can exacerbate the situation, ushering in additional phenotypes or an increased heterogeneity of capabilities at both single-cell and population levels. This continuous interplay of phenotypes must result from the advantage of each behavior that, once initiated, leads to another advantageous phenotype. The presence of common cancer hallmarks in most, if not all, various human cancers suggests that these traits are unlikely to be mere random occurrences caused by random mutations. Instead, they may represent necessary, non-random acquisitions of specific characteristics that enhance disease progression in a process of continuous adaptation for survival and promoting regeneration. Thus, tumor progression can be seen as a continuous evolutionary process at each step invoked, then acquired, and perpetuated, irrespective of any specific mutations or cellular alternations that may arise as a result and potentially intensify each step. The chaotic situation disrupts the molecular networks that underpin these capacities. The molecular-level heterogeneity seen in cancer, which often becomes more evident as the disease progresses, stems from this continuous escalation as cells persistently grapple to respond with more advantageous capabilities (Figure 1).
Inherent/inherited cellular diversity undoubtedly leads to varying levels of susceptibility to survival threats. However, it is the heterogeneous molecular responses to the survival threat - response based on the inherent/inherited cellular diversity and triggered by the activation of the cell’s own genomic machinery, that ultimately determine survivability. The previously presumed cause of tumor heterogeneity, genomic instability [1,8,9,10,11,12], is less likely the root cause but perhaps a parallel process or even an outcome of the continuous evolution of advantageous capacities. As cells strive to gain a competitive edge, beneficial mutations and other cellular alterations that support essential tumor capacities keep arising despite genomic instability. The diversity generated at this juncture will be selectively favored by environmental pressure, and cells with more advantageous characteristics will proceed to the next beneficial capability. Nonetheless, simplistically speaking, the essence of the disease lies not just in the initial trigger but more critically in the fact that cellular evolution necessitates the amplification of cancer hallmark capacities, which in turn causes molecular chaos within and among different tumors.
Metastasis represents the most perilous phase of cancer progression, where cancer cells migrate from their original location and establish themselves in different parts of the body [13,14]. Chemotaxis may guide cells to new locations that offer resources that have become scarce at their original site, or attract the cells to environments beneficial for their survival. Initially, new implantation sites may offer beneficial conditions. Once situated in this new environment, the cells often revert to a non-aggressive state as they obtain what is lacking. This halts their evolutionary trajectory and suppresses the emergence of further essential mutations. This phenomenon aligns with findings from numerous studies that have observed that metastatic tumors often harbor key mutations that reflect those found in the primary site [18,19,20,21].
Moreover, despite these cells carrying mutations that induce aggression at the original site, they often remain dormant in their new environment [22,23,24,25,26], reinforcing the concept that cellular aggression arises from a necessity for survival. When the cells’ needs are adequately met in the new location, it diminishes the imperative for aggression, even in the presence of critical gene mutations. However, these cells may eventually face new survival challenges due to the unfamiliar conditions of the new environment. This leads to a temporary setback; however, these cells frequently pursue survival by reverting to a more aggressive state, as noted in numerous studies [23,27,28,29]. Consequently, the same evolutionary process of the disease and the molecular chaos recurs at the distant site, potentially in unlimited cycles.
There is an urgent need for advancements in cancer treatments. The innate immune system functions as an individual’s primary line of defense against foreign invaders and abnormal cells. While a robust immune system presumably mounts a defensive response against both primary and metastatic tumors [30,31], the concept we’re exploring underscores that human cells, upon transforming into cancerous states, adeptly elude the body’s regular immune defenses—a phenomenon corroborated as observed [30,31,32]. This evasion stems from the fact that tumors, despite their mutations, aren’t seen as external threats. Rather, their emergence is grounded in the cells’ intrinsic adaptive response. This understanding may explain a critical observation that boosting the efficacy of cancer immunotherapy frequently results in autoimmune-like side effects and damage to normal tissues [33,34,35,36,37]. Clearly, tumor disease needs to be treated by addressing the intricacies of tumor evolution. As our understanding of disease progression navigates the evolving clinical landscape, several critical considerations emerge for future applications:
- Rethinking Oncogene Addiction: As our grasp of cellular evolution and tumor heterogeneity deepens, the necessity to re-evaluate oncogene addiction becomes evident. The phenomena of oncogene addiction might be better understood as a cell fate outcome, occurring when cells are detached from the evolutionary process when the target oncogene is inhibited. This fresh perspective could herald innovative treatment paradigms.
- Deepening Insights into Oncogenes and Tumor Suppressors: Reclassifying genes according to their heterogeneity levels has promise. Treatment strategies resulting from this approach could focus on amplifying the homogeneity of critical genes and specifying precise levels of homogeneity, which in turn is likely to enhance the predictability and efficacy of therapeutic outcomes.
- Exploring Aging’s Role in Cancer Development: A better understanding of aging’s role in cancer will allow us to probe the links between aging and cancer onset. A focus here should be distinguishing between normal aging decline and tumor formation. Such insights will contribute to innovative strategies to enhance broader human health.
Incorporating these perspectives into clinical protocols will result in a more refined approach to cancer treatment. Beyond oncology, these insights will also help direct efforts to bolster overall health and longevity.
Future Perspectives
- 1)
- rethinking oncogene addiction.
Oncogene addiction refers to the reliance of cancer cells on one or more genes for their survival [38,39]. As a result, inhibiting the proteins corresponding to these oncogenes can lead to a complete halt or regression in tumor growth [38,39]. This concept of “addiction” explains the selectivity and effectiveness seen in certain molecularly targeted therapies currently used in clinical practice despite the common occurrence of resistance at later stages of treatment [38,39,40,41,42].
The concept outlined here suggests that cells continuously develop advantageous capacities in response to environmental pressures. Consequently, beneficial gene mutations keep arising, including but not limited to oncogenes. This process contributes to the evolution of adaptive capabilities, with survival being a primary objective of this evolution. When treatment agents inhibit these advantageous genes, the evolutionary process is disrupted, leading to cell death, a phenomenon observed as oncogene addiction [38,39]. Moreover, the occurrence of oncogene addiction further reinforces the notion that survival is one of the key objectives driving the evolution. It is crucial to understand that any genes that contribute to cellular aggressiveness are essential for the cells to respond to and overcome environmental pressures, thereby facilitating survival. As such, when oncogenes are inhibited, or tumor suppressors are activated, the initial observation post-treatment is a decrease in the cells’ aggressiveness, and this is almost immediately followed by cell death [43,44,45] when the cells are removed from their evolutionary trajectory. Intriguingly, if the evolutionary process becomes unnecessary, the cells may revert to a normal state by ceasing the generation of aggressive traits, irrespective of the presence or absence of the oncogene. Though this is rare due to constant environmental pressures, such occurrences have been documented in some studies [46,47,48,120].
Each tumor capability represents a step in the evolutionary process; when the oncogene fosters cell aggressiveness, the more reliant cells are on this particular gene for aggressiveness and continued evolution, the more significant the evolutionary loss will be when the gene is inhibited. The cessation of the evolutionary process, in turn, leads to cell death (Figure 2, right side). Activating more tumor capabilities and displaying increased aggressiveness are the primary factors determining the degree of cell addiction to the genes that drive this aggressiveness, in the sense that any cells in the tumor are addicted when there is a necessity to be aggressive. Suppose the gene is not the primary or sole contributor to the cells’ aggressiveness, and several other contributing factors could emerge. In that case, inhibiting this oncogene is unlikely to result in oncogene addiction unless multiple targets are inhibited, as observed [58,59,60]. This may explain why, in the advanced stage or any phase of the disease, when it is more likely that cells will construct intricate networks to support their continued evolution, many cells develop resistance to the inhibitor treatments administered at the beginning of the disease [81,82,83].
Drug-tolerant persister cells constitute a rare, slowly proliferating population that can withstand drug treatments [49,50,51]. It’s widely recognized that drug therapies impose significant pressure on cells, often prompting some cells to develop new survival mechanisms, a phenomenon known as adaptive drug resistance [52,53]. It is reasonable to presume that when cells lose advantageous genes and grapple with adapting to the environment during targeted gene inhibition, they’re likely to compensate for this loss by seeking alternative pathways or proceeding with new advantageous genetic changes. After treatment, any residual cells not only adapt to the loss of advantageous genes and are able to survive but also develop alternative mechanisms that enable them to continue their evolutionary process, fostering aggressive adaptive capabilities. This response is likely amplified by the added survival pressure from the treatment, which tends to make these remaining cells both aggressive and diverse, as observed [54,55,56,57].
It is important to note that oncogene mutations typically become the catalyst for tumorigenesis when cells begin to evolve, meaning without a necessity for cellular evolution at any phase of a disease, cells won’t transition into or continue as tumor cells despite the presence of an oncogene. Thus, another way to improve current therapy based on this concept is to relieve the cells from the survival pressures that need to be identified and investigated. This will be particularly essential for inherited genetic defects in crucial genes that are prevalent in families [61,62,63]. These genetic defects may cause cell damage already or may enhance the cell’s potential to develop tumorigenic capabilities [61,62,63] when necessary. Vigilantly monitoring and alleviating cellular stress is crucial in preventing disease development in such individuals.
During the development of tumor capabilities, advantageous genes are selected and remain activated to cope with environmental pressures. Cancer stem cells are known to be the slow-cycling cells that are often found to be resistant to target therapies by numerous studies [64,65,66]. In the inherently chaotic environment within cells, it’s reasonable to presume that not all cells will proceed with the same advantageous genes. It’s plausible that certain cells, not limited to cancer stem cells, might not express a specific advantageous oncogene. In these instances, the oncogene may not be the singular advantageous factor, and these cells could depend on or be addicted to other networks to sustain their tumorigenic capacities and drive disease progression, and this is observed as resistance in target inhibition therapies [64,65,66].
Cancer stem cells share a similarity with embryonic and tissue stem cells known as replicative immortality [67,68]. There are two sources for cancer stem cells. First, when stem cells with ‘stemness’ attributes originate from tissue stem cells [69,70,71], they probably don’t face the same survival pressures as more differentiated cells due to their inherent relative immortality. As such, they can remain dormant yet viable and be resistant under treatment. Secondly, cancer stem cells can derive from more differentiated tumor cells through a process referred to as dedifferentiation or, as observed, dynamic stemness [72,73,74,75].
Pathologists commonly grade cancers based on their differentiation status, suggesting that many human cancers are in a more primitive state at the time of diagnosis. In this paper, cancer is perceived as an inherent cellular response to survival threats. It is plausible to view cancer dedifferentiation as a mechanism triggered when cellular damage necessitates regeneration as part of this adaptive response. From this viewpoint, any human cells that possess oncogenic potential inherently can revert to a more stem-like state, just as a pathologist grades the tumors. Supporting this idea, existing research suggests normal fibroblasts can be induced to adopt a more primitive state [76]. Interestingly, a striking resemblance exists between fibrosarcoma cells, which activate specific key genes to sustain their stem cell component [77,78,79], and the stem-like characteristics observed in normal fibroblasts when they are experimentally exposed to these same genes [76]. Yamanaka factors typically should not be expressed in tissues that can give rise to fibrosarcoma, indicating the cells in fibrosarcoma that do express Yamanaka factors likely originate from a dedifferentiation process. We suggest that the shift toward a more stem-like state is intrinsically initiated by the cell in response to cellular damage aiming for regeneration (Figure 2, left side). Cancer cells are observed to increasingly revert to a more primitive state as cancer advances [79,80], likely because the accumulation of cellular damage triggers a more intensified regeneration process. Once they develop stemness, they can become resistant to the necessity of evolution for innate survivability, they can temporarily shift to a dormancy state even under treatment. Conversely, during treatment, cells that can still activate their regeneration process might not succumb to the drug. Hence, it’s not that ‘stemness’ is inherently resistant to treatment; rather, stemness emerges as a response to the treatment because of the increased pressure and the intensified demand for regeneration.
In this respect, cancer is not merely viewed as a process where cells acquire stemness solely for enhanced survivability, leaning towards a dormant state. Instead, because survivability remains a prerequisite for regeneration, cancer is perceived as a continuous progression in a chaotic environment where most cells keep responding to pressures and the imperative for regeneration. Cells persistently revert to a more primitive state, a shift driven by continuous survival challenges that push them to continue into earlier stages. While offspring cells integrate into the population, the prevailing trend sees this population increasingly gravitating towards the primitive state rather than undergoing differentiation and regeneration. Within this context, cancer manifests as a progressively deeper regression into primitiveness under ongoing challenges, and cancer stem cells become a direct result - and hence, a sign - of the increased risk for survival.
As survival threats escalate, there’s a heightened compulsion in cells to develop stemness, and for the same reason, these cells exhibit a stronger predisposition towards tumorigenesis, a trend substantiated by studies [84,85,86]. Clearly, in these scenarios, the possible enhanced survivability associated with stemness is offset by persistent survival threats. Any cells that can respond will become tumorigenic. Once cancer stem cells are activated, their tumorigenic potential, coupled with the molecular alterations derived and accumulated in the later phase of the disease when they are derived, is further amplified by their capacity to display one of the primary characteristics of tumors – uncontrolled proliferation. This proliferation then sets off a cascade of subsequent steps more attainable and driven by heightened survival threats; thus, the cells become aggressive, and once they become aggressive, they will be addicted to the genes that drive the aggressiveness.
- 2)
- Deepening Insights into Oncogenes and Tumor Suppressors.
During the early stages of the disease, it is widely observed that invasive cells constitute only a minority of the cell population within the primary tumor [87,88,89]. The limited growth potential observed in these invasive cells suggests that they acquire their proliferative capacity at a distant metastatic site. This underscores that a disease cannot be deemed aggressive if a cell can only proliferate without the ability to invade, or if a cell can only invade without the ability to proliferate. It is reasonable to assume that different cellular behaviors, such as proliferation and invasion, necessitate distinct oncogenic networks, signaling pathways, and oncogenic mutations for optimal activation, as observed [92,93,94,95]. The apparent inhibited growth ability in invasive cells may exemplify how one oncogenic network supporting invasion reduces the intensity of another oncogenic network that supports growth. This concept can be extended to other cancer hallmark behaviors, as each of them is regulated by various signaling pathways, oncogenic networks, and mutations.
Consequently, cells must adjust these driving factors for each behavior to achieve the ideal level needed for disease progression. At the population level, this modulation manifests as heterogeneity. At the single-cell level, it is seen as cell plasticity. Conversely, an increase in population homogeneity restricts the number of oncogenic behaviors activated to achieve the optimal level, resulting in a reduction of disease aggressiveness by lessening the intensity of at least some oncogenic behaviors.
The traditional understanding of an oncogene or tumor suppressor suggests that when a gene is activated, it can either safeguard against or facilitate cellular aggression during tumor progression. These tumor suppressors and oncogenes are likely the primary drivers of identified oncogenic behaviors, and these genes probably play a role in influencing downstream network regulations [90,91]. Based on the concept in this paper, significant alterations in oncogene expression might reflect how cells progress through the disease by modifying their own genome machinery to cope with survival pressures. Optimal levels of each behavior likely involve a distinct gene network or require a specific intensity of the oncogene [92,93,94,95]. Fine-tuning the heterogeneity of oncogene levels allows cells to transition towards states characterized by a full range of tumorigenic behaviors by increasing diversity, reducing the intensity of specific behaviors through more homogeneity, or even diminishing overall tumorigenicity (Figure 3). It’s crucial to note that not all cell states lead to metastasis, which typically occurs at a later stage when a broader range of aggressive behaviors is enhanced [96,97]. The concepts discussed here propose a shift in defining oncogenes and tumor suppressors, focusing on the heterogeneity of the leading genes. The heterogeneity can be categorized as high (oncogene) or low (tumor suppressor). This concept may shed light on the dual roles of oncogenes and tumor suppressors. Notably, some genes with dual functions have already been observed [98,99,100,101]. A deeper exploration of the relationship between the heterogeneity of these crucial genes and their dual nature warrants further investigation.
We propose a focus on regulating target oncogene fluctuation as an alternative method to implementing a strict inhibition on the advantageous genes. The treatment-induced pressure often leads the cells to activate a wider spectrum of tumorigenic capacities [102,103] to cope with the enhanced stress caused by inhibiting advantageous genes. Thus, when the complete eradication of all residual cancer cells proves challenging, causing a shift to an aggressive state, it becomes necessary to develop treatment strategies that extend beyond merely aiming at the strenuous inhibition of the oncogenes. The aim should be to achieve homogeneity of the oncogene across cells. This strategy can be coupled with other debulking methods, such as surgery or radiation, for effective disease control. On the other side, in tumors with greater homogeneity, we propose to emphasize the identification of the precise total oncogene levels within the tumor. The goal is to adjust these levels such that individual tumorigenic behaviors experience a significant decrease from their peak, suggesting that the cells have shifted away from their utmost aggressiveness.
In contrast to existing treatment paradigms, these approaches give precedence to halting disease progression rather than attempting to eliminate cells outright. This reduces the risk of provoking an aggressive cell state in the residual cells post-treatment. This perspective suggests that human cancer treatment strategies could go beyond methods that intensify or modulate oncogene addiction. They could also encompass approaches to stabilize the expression state and to achieve precise levels of the addicted oncogene, leading to new drug designs.
- 3)
- Exploring Aging’s Role in Cancer Development.
Aging is a process marked by a progressive deterioration in normal cellular function, an increased susceptibility to diseases, and elevated mortality rates [104]. As we age, our cells accrue DNA damage due to a mixture of environmental exposure to mutagens and natural errors in DNA replication. Although cells possess repair mechanisms to correct these errors, these systems can also become less efficient as we age, leading to an accumulation of mutations [104,105]. This paper suggests that accumulating irreparable cellular damage can have dire implications for cells. When such damage surpasses a critical point that threatens cell survival, cells may respond adaptively. This adaptation often results in tumor formation or prolonged tumor progression if the threats continue. Thus, the emergence of cancer may be an almost inevitable outcome for any cells during their lifetime, especially within the context of aging. The rate at which this happens is largely influenced by the extent of damage these cells endure. Any form of cellular damage, aging included, elevates the likelihood of tumor emergence [106,107]. The initiation of tumor transformation signifies the onset of a cellular response and regeneration process stemming from internal or external inherent defects.
If without the ability to form tumors, cells will just continue a path of decline. In essence, the usual decline observed in aging could be perceived as the converse of tumor development, and both these processes, aging and tumor formation, originate from the same premise of cellular damage [104,105,106,107]. This suggests that certain mechanisms that contribute to the decline in healthy aging might also reduce the potential for tumor development. When tumor cells tend to evolve beneficial genetic mutations and other cellular modifications in response to damage, the loss of control over normal aging processes likely becomes a key element in this evolution. Notably, several prominent tumor suppressors have key roles in both the aging process and tumor suppression [108,109]. Despite the predisposition to tumorigenesis due to cellular damage and the presence of advantageous oncogenes, it is frequently observed that an additional ‘hit’ on these tumor suppressors is necessary for tumor initiation [110,111]. In situations where there’s an existing risk of tumor formation, cells might counteract this by amplifying their natural aging processes despite the activation of oncogenes, and this can be observed as senescence, a typical hallmark in aging [104]. Interestingly, proliferation stands in contrast to senescence and is one of the earliest hallmarks of most cancer to manifest, likely resulting from a cell fate decision.
Another vital response of organisms to environmental threats to their survival is the state of dormancy [112]. This phase in an organism’s life cycle encompasses a temporary pause in growth, development, and physical activity [112]. Used as a survival strategy during unfavorable environmental conditions, dormancy is observed in various organisms and on a cellular level, including human cells [113]. When a cell enters a dormant state, it likely hasn’t succumbed to cell death, even in the face of damage. It’s reasonable to infer that dormant cells are resilient enough to endure existing harm. But if adverse conditions or damage escalate, the cell might be unable to sustain its dormant state and counteract the damage.
Similarly, if the initial damage is sufficiently severe, cells may bypass dormancy as their initial response. It’s essential to differentiate between dormancy and senescence, a characteristic of aging [104]. In the face of survival threats, cells might initially opt for dormancy. If the threat persists, cells are then at a crossroads: either continue their decline or give rise to tumors, paths that are opposed (Figure 4). Dormant cells are often observed at the onset of tumor formation, after specific targeted therapies, and when they first colonize a distant location [114,115,116]. These observations suggest that cells, at first, could handle the stresses stemming from the endurance to existing damage. Most often, the eventual reactivation signifies that, even in a dormant state, cells continually face survival threats unless they reach permanent senescence and then decline as in the aging process.
Knowledge of the mechanisms underlying aging could profoundly influence the treatment strategies for metastatic tumors. Typically, metastatic tumors manifest during the disease’s advanced stages. By this phase, cancer cells have already undergone complex transformations, enhancing their tumorigenic capabilities [23,27,28,29]. As a result, they are less likely to depend on single or limited genes for survival compared to their predecessors, giving these cancers resistance to targeted treatments [82,83]. Treating at this stage requires multifaceted strategies. First, there is a need to mitigate environmental stresses that contribute to tumor progression. Second, we can consider inducing a state of cellular dormancy by deciphering what was provided at the distant site that mitigates the cells. However, these approaches may offer only temporary solutions. As a potential last resort, encouraging a cell’s natural aging deterioration might be more advantageous, especially compared to continuing to inhibit advantageous genes, as that risks further cell evolution. This strategy depends on understanding and targeting the key drivers of natural aging to facilitate cellular decline, particularly for those cancers typically untreatable by other means. Future research should address the links between aging and cancer biology, thereby contributing to therapeutic interventions for advanced-stage cancer patients.
Conversely, if we consider tumorigenesis as a cellular response to threats to survival and possibly lead to regeneration, mutations in oncogenes and tumor suppressors that enhance tumor capabilities might slow down natural aging, which leads to more cellular damage and increases the risk of tumorigenesis. In the beginning, after transformation, most tumor cells do not exhibit tumor-promoting capabilities [6,7,117,118,119]. It’s typically after prolonged exposure to stressful environments that they begin to manifest aggressive behaviors [6,7,117,118,119]. Thus, there must be a pivotal moment when this transition occurs. While the same mechanisms can result in uncontrolled disease progression, there might be an opportunity to intervene and control it. If experimentally manipulating this tumorigenic process may be feasible, it can lead to enhanced resilience to cellular damage and improved cell survivability. Essentially, if there is a method to induce cellular transformation without eliciting aggressive behavior, it could offer significant advantages to cell health. This could complement existing strategies related to cellular repair and even traditional aging processes in the management of the disease. This area undoubtedly calls for further investigation and extensive research.
Final remark
Cancer heterogeneity poses a significant challenge for many clinical therapies, leading to varied responses from cancer cells and allowing aggressive strains to endure as residual tumors. This review seeks to shed light on the origins of cancer heterogeneity and offers suggestions to bolster existing therapeutic strategies. We suggest that tumor heterogeneity originates from the chaotic molecular responses when cells evolute beneficial tumor capacities under survival pressure. We touch upon the term oncogene addiction as a cellular phenomenon that arises when cellular evolution is halted. Drawing from the same idea that cells respond to survival challenges, we introduce the notion that cancer stem cells arise from the necessity to regenerate under continuous cellular damage.
Furthermore, we advocate for a strategy aimed at enhancing oncogene homogeneity across cells. We also present the intriguing juxtaposition of the standard aging decline with tumorigenesis and propose a method for patients with advanced-stage cancer that seeks to induce normal cellular deterioration. By introducing these innovative perspectives, we hope to pave the way for new treatment strategies, furthering the overarching mission of healing patients and ultimately enhancing human health.
Acknowledgment
This paper was generously funded by the Elsa U. Pardee Foundation (CA-0122861 to YZ). I extend my gratitude to Robert Judson-Torres for his invaluable insights and editing that significantly enhanced the manuscript. I also acknowledge William Weiss for his comments and Elliott Peterson for his editing, which further improved the paper. Assistance with English editing was provided by American Manuscript Editors.
Author information
This paper was generated and written by YZ.
Contributions
YZ generated the concept and did the writing.
Footnote
English editing was assisted by ChatGPT.
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Blanke, C.D.; Demetri, G.D.; Von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; et al. Long-Term Results From a Randomized Phase II Trial of Standard- Versus Higher-Dose Imatinib Mesylate for Patients With Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing KIT. JCO 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef]
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discovery 2015, 5, 1271–1281. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Sun, X.; Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin 2015, 36, 1219–1227. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Landau, D.A. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb Perspect Med 2017, 7, a029611. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. CARCIN 2017, 38, 2–11. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Li, L.; Wang, X. An algorithm to quantify intratumor heterogeneity based on alterations of gene expression profiles. Commun Biol 2020, 3, 1–19. [Google Scholar] [CrossRef]
- Geiger, T.R.; Peeper, D.S. Metastasis mechanisms. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2009, 1796, 293–308. [Google Scholar] [CrossRef]
- Ha, N.-H.; Faraji, F.; Hunter, K.W. Mechanisms of Metastasis. In Cancer Targeted Drug Delivery: An Elusive Dream; Bae, Y.H., Mrsny, R.J., Park, K., Eds.; Springer: New York, NY, USA, 2013; pp. 435–458. [Google Scholar]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- Yokota, J. Tumor progression and metastasis. Carcinogenesis 2000, 21, 497–503. [Google Scholar] [CrossRef]
- Lee, W.-C.; Kopetz, S.; Wistuba, I.I.; Zhang, J. Metastasis of cancer: When and how? Annals of Oncology 2017, 28, 2045–2047. [Google Scholar] [CrossRef]
- Patel, S.A.; Rodrigues, P.; Wesolowski, L.; Vanharanta, S. Genomic control of metastasis. Br J Cancer 2021, 124, 3–12. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef]
- Sherwood, J.; Dearden, S.; Ratcliffe, M.; Walker, J. Mutation status concordance between primary lesions and metastatic sites of advanced non-small-cell lung cancer and the impact of mutation testing methodologies: A literature review. Journal of Experimental & Clinical Cancer Research 2015, 34, 92. [Google Scholar]
- Xie, T.; Cho, Y.B.; Wang, K.; Huang, D.; Hong, H.K.; et al. Patterns of somatic alterations between matched primary and metastatic colorectal tumors characterized by whole-genome sequencing. Genomics 2014, 104, 234–241. [Google Scholar] [CrossRef]
- Townson, J.L.; Chambers, A.F. Dormancy of Solitary Metastatic Cells. Cell Cycle 2006, 5, 1744–1750. [Google Scholar] [CrossRef]
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp Mol Med 2020, 52, 569–581. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Molecular Oncology 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Summers, M.A.; McDonald, M.M.; Croucher, P.I. Cancer Cell Dormancy in Metastasis. Cold Spring Harb Perspect Med 2020, 10, a037556. [Google Scholar] [CrossRef]
- Yadav, A.S.; Pandey, P.R.; Butti, R.; Radharani, N.N.V.; Roy, S.; et al. The Biology and Therapeutic Implications of Tumor Dormancy and Reactivation. Frontiers in Oncology 2018, 8. [Google Scholar] [CrossRef]
- Pradhan, S.; Sperduto, J.L.; Farino, C.J.; Slater, J.H. Engineered In Vitro Models of Tumor Dormancy and Reactivation. J Biol Eng 2018, 12, 37. [Google Scholar] [CrossRef]
- Singh, D.K.; Patel, V.G.; Oh, W.K.; Aguirre-Ghiso, J.A. Prostate Cancer Dormancy and Reactivation in Bone Marrow. Journal of Clinical Medicine 2021, 10, 2648. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J Immunother Cancer 2017, 5, 79. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune responses to malignancies. J Allergy Clin Immunol 2010, 125, S272–S283. [Google Scholar] [CrossRef]
- Caspi, R.R. Immunotherapy of autoimmunity and cancer: The penalty for success. Nat Rev Immunol 2008, 8, 970–976. [Google Scholar] [CrossRef]
- Ofuji, K.; Hiramatsu, K.; Nosaka, T.; Naito, T.; Takahashi, K.; et al. Pembrolizumab-induced autoimmune side effects of colon and pancreas in a patient with lung cancer. Clin J Gastroenterol 2021, 14, 1692–1699. [Google Scholar] [CrossRef]
- Amos, S.M.; Duong, C.P.M.; Westwood, J.A.; Ritchie, D.S.; Junghans, R.P.; et al. Autoimmunity associated with immunotherapy of cancer. Blood 2011, 118, 499–509. [Google Scholar] [CrossRef]
- Yang, S.; Yu, K.; Palmer, N.; Fox, K.; Kou, S.C.; Kohane, I.S. Autoimmune Effects of Lung Cancer Immunotherapy Revealed by Data-Driven Analysis on a Nationwide Cohort. Clin Pharma and Therapeutics 2020, 107, 388–396. [Google Scholar] [CrossRef]
- Cheng, F.; Loscalzo, J. Autoimmune Cardiotoxicity of Cancer Immunotherapy. Trends in Immunology 2017, 38, 77–78. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A. Oncogene Addiction. Cancer Research 2008, 68, 3077–3080. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A.K. Mechanisms of Disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat Rev Clin Oncol 2006, 3, 448–457. [Google Scholar] [CrossRef]
- McCormick, F. Cancer therapy based on oncogene addiction. Journal of Surgical Oncology 2011, 103, 464–467. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.-J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Reports 2015, 16, 280–296. [Google Scholar] [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; et al. Small molecule inhibition of the KRAS–PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Kopp, F.; Wagner, E.; Roidl, A. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 2013, 5, 185–195. [Google Scholar] [CrossRef]
- Kim, Y.; Yoon, J.W.; Xiao, X.; Dean, N.M.; Monia, B.P.; Marcusson, E.G. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res 2007, 67, 3583–3593. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Schmidt, A.V.; Monga, S.P.; Prochownik, E.V.; Goetzman, E.S. A Novel Transgenic Mouse Model Implicates Sirt2 as a Promoter of Hepatocellular Carcinoma. Int J Mol Sci 2023, 24, 12618. [Google Scholar] [CrossRef]
- Pollack, R.; Wolman, S.; Vogel, A. Reversion of Virus-transformed Cell Lines: Hyperploidy accompanies Retention of Viral Genes. Nature 1970, 228, 938–938. [Google Scholar] [CrossRef]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-Tolerant Persister Cells in Cancer Therapy Resistance. Cancer Res 2022, 82, 2503–2514. [Google Scholar] [CrossRef]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Álvarez-Varela, A.; Novellasdemunt, L.; Barriga, F.M.; Hernando-Momblona, X.; Cañellas-Socias, A.; et al. Mex3a marks drug-tolerant persister colorectal cancer cells that mediate relapse after chemotherapy. Nat Cancer 2022, 3, 1052–1070. [Google Scholar] [CrossRef]
- Kumar, U.; Castellanos-Uribe, M.; May, S.T.; Yagüe, E. Adaptive resistance is not responsible for long-term drug resistance in a cellular model of triple negative breast cancer. Gene 2023, 850, 146930. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Casadesús, J. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 355–360. [Google Scholar] [CrossRef]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. Journal of Hematology & Oncology 2019, 12, 134. [Google Scholar]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. Journal of Biomedical Science 2018, 25, 20. [Google Scholar] [CrossRef]
- Klein, C.A.; Blankenstein, T.J.F.; Schmidt-Kittler, O.; Petronio, M.; Polzer, B.; et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 2002, 360, 683–689. [Google Scholar] [CrossRef]
- Berger, N.; Kim-Schulze, S.; Parekh, S. Minimal Residual Disease in Multiple Myeloma: Impact on Response Assessment, Prognosis and Tumor Heterogeneity. In Biological Mechanisms of Minimal Residual Disease and Systemic Cancer; Aguirre-Ghiso, J.A., Ed.; Springer International Publishing: Cham, 2018; pp. 141–159. [Google Scholar]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S.V.U.M. Dual or multi-targeting inhibitors: The next generation anticancer agents. European Journal of Medicinal Chemistry 2018, 143, 1277–1300. [Google Scholar] [CrossRef]
- Eder, J.P.; Shapiro, G.I.; Appleman, L.J.; Zhu, A.X.; Miles, D.; et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res 2010, 16, 3507–3516. [Google Scholar] [CrossRef]
- Petrelli, A.; Giordano, S. From single- to multi-target drugs in cancer therapy: When aspecificity becomes an advantage. Curr Med Chem 2008, 15, 422–432. [Google Scholar]
- Foulkes, W.D. Inherited Susceptibility to Common Cancers. N Engl J Med 2008, 359, 2143–2153. [Google Scholar] [CrossRef]
- Fearon, E.R. Human Cancer Syndromes: Clues to the Origin and Nature of Cancer. Science 1997, 278, 1043–1050. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; et al. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Moore, N.; Lyle, S. Quiescent, Slow-Cycling Stem Cell Populations in Cancer: A Review of the Evidence and Discussion of Significance. Journal of Oncology 2010, 2011, e396076. [Google Scholar] [CrossRef]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Shiokawa, D.; Sakai, H.; Ohata, H.; Miyazaki, T.; Kanda, Y.; et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res 2020, 80, 4451–4464. [Google Scholar] [CrossRef]
- Robinson, N.J.; Taylor, D.J.; Schiemann, W.P. Stem cells, immortality, and the evolution of metastatic properties in breast cancer: Telomere maintenance mechanisms and metastatic evolution. J Cancer Metastasis Treat 2019, 5, 39. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase in normal and cancer stem cells. FEBS Letters 2010, 584, 3819–3825. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Reports 2014, 15, 244–253. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat Rev Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. Journal of Clinical Medicine 2019, 8, 1542. [Google Scholar] [CrossRef]
- Zhou, F.; Aroua, N.; Liu, Y.; Rohde, C.; Cheng, J.; et al. A Dynamic rRNA Ribomethylome Drives Stemness in Acute Myeloid Leukemia. Cancer Discov 2023, 13, 332–347. [Google Scholar] [CrossRef]
- Jain, P.; Duddu, A.S.; Jolly, M.K. Stochastic population dynamics of cancer stemness and adaptive response to therapies. Essays Biochem 2022, 66, 387–398. [Google Scholar]
- Ma, H.; He, M.; Wei, M. [Research progress on targeting effect and regulating mechanisms of the stemness of cancer stem cells]. Yao Xue Xue Bao 2016, 51, 189–196. [Google Scholar]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Chico, M.A.; Mesas, C.; Doello, K.; Quiñonero, F.; Perazzoli, G.; et al. Cancer Stem Cells in Sarcomas: In Vitro Isolation and Role as Prognostic Markers: A Systematic Review. Cancers 2023, 15, 2449. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, T.; Gu, W.; Li, P.; Cheng, X.; et al. The ALDH1+ subpopulation of the human NMFH-1 cell line exhibits cancer stem-like characteristics. Oncol Rep 2015, 33, 2291–2298. [Google Scholar] [CrossRef]
- Feng, B.-H.; Liu, A.-G.; Gu, W.-G.; Deng, L.; Cheng, X.-G.; et al. CD133+ subpopulation of the HT1080 human fibrosarcoma cell line exhibits cancer stem-like characteristics. Oncol Rep 2013, 30, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Cell Dedifferentiation—An overview | ScienceDirect Topics. www.sciencedirect.com.
- Li, M.-M.; Tang, Y.-Q.; Gong, Y.-F.; Cheng, W.; Li, H.-L.; et al. Development of an oncogenic dedifferentiation SOX signature with prognostic significance in hepatocellular carcinoma. BMC Cancer 2019, 19, 851. [Google Scholar] [CrossRef]
- Komarova, N.L.; Wodarz, D. Drug resistance in cancer: Principles of emergence and prevention. Proc. Natl. Acad. Sci. USA 2005, 102, 9714–9719. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.-E.; Fabbro, M.; Theillet, C.; Bibeau, F.; Rouanet, P.; Ray-Coquard, I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Critical Reviews in Oncology/Hematology 2014, 89, 207–216. [Google Scholar] [CrossRef]
- Zhu, P.; Fan, Z. Cancer stem cells and tumorigenesis. Biophys Rep 2018, 4, 178–188. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Ma, S.; Chan, K.; Hu, L.; Lee, T.K.; Wo, J.Y.; et al. Identification and Characterization of Tumorigenic Liver Cancer Stem/Progenitor Cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Wakimoto, H.; Mohapatra, G.; Kanai, R.; Curry, W.T.; Yip, S.; et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol 2012, 14, 132–144. [Google Scholar] [CrossRef]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Oncogene—An overview | ScienceDirect Topics. www.sciencedirect.com.
- Endo, Y.; Lyon, S.; Shen, Y.; Mohan, N.; Wu, W.J. Cell proliferation and invasion are regulated differently by EGFR and MRP1 in T-DM1-resistant breast cancer cells. Sci Rep 2019, 9, 16383. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 2010, 29, 405–434. [Google Scholar] [CrossRef]
- De Donatis, A.; Ranaldi, F.; Cirri, P. Reciprocal control of cell proliferation and migration. Cell Communication and Signaling 2010, 8, 20. [Google Scholar] [CrossRef]
- Swami, P.; Thiyagarajan, S.; Vidger, A.; Indurthi, V.S.K.; Vetter, S.W.; Leclerc, E. RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. International Journal of Molecular Sciences 2020, 21, 7723. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the Origin of Cancer Metastasis. Crit Rev Oncog 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef]
- Robbs, B.K.; Cruz, A.L.S.; Werneck, M.B.F.; Mognol, G.P.; Viola, J.P.B. Dual Roles for NFAT Transcription Factor Genes as Oncogenes and Tumor Suppressors. Molecular and Cellular Biology 2008, 28, 7168–7181. [Google Scholar] [CrossRef] [PubMed]
- Uribesalgo, I.; Benitah, S.A.; Di Croce, L. From oncogene to tumor suppressor: The dual role of Myc in leukemia. Cell Cycle 2012, 11, 1757–1764. [Google Scholar] [CrossRef]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. Journal of Biomedical Science 2018, 25, 20. [Google Scholar] [CrossRef]
- Damen, M.P.F.; Van Rheenen, J.; Scheele, C.L.G.J. Targeting dormant tumor cells to prevent cancer recurrence. The FEBS Journal 2021, 288, 6286–6303. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Guimera, A.M.; Hodgson, D.; Mcdonald, N.; Mooney, K.M.; et al. Modelling the molecular mechanisms of aging. Biosci Rep 2017, 37, BSR20160177. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu Rev Physiol 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Laconi, E.; Marongiu, F.; DeGregori, J. Cancer as a disease of old age: Changing mutational and microenvironmental landscapes. Br J Cancer 2020, 122, 943–952. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Mori, H.; Funahashi, Y.; Yoshino, Y.; Kumon, H.; Ozaki, Y.; et al. Blood CDKN2A Gene Expression in Aging and Neurodegenerative Diseases. J Alzheimers Dis 2021, 82, 1737–1744. [Google Scholar] [CrossRef]
- Zeng, H.; Jorapur, A.; Shain, A.H.; Lang, U.E.; Torres, R.; et al. Bi-allelic loss of CDKN2A initiates melanoma invasion via BRN2 activation. Cancer Cell 2018, 34, 56–68.e9. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci 2019, 110, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Dormancy—An overview | ScienceDirect Topics. www.sciencedirect.com.
- Neophytou, C.M.; Kyriakou, T.-C.; Papageorgis, P. Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. Int J Mol Sci 2019, 20, 6158. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp Mol Med 2020, 52, 569–581. [Google Scholar] [CrossRef]
- Blasco, M.T.; Espuny, I.; Gomis, R.R. Ecology and evolution of dormant metastasis. Trends Cancer 2022, 8, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Afifi, N.; Barrero, C.A. Understanding Breast Cancer Aggressiveness and Its Implications in Diagnosis and Treatment. J Clin Med 2023, 12, 1375. [Google Scholar] [CrossRef]
- Beckwith, J.B.; Zuppan, C.E.; Browning, N.G.; Moksness, J.; Breslow, N.E. Histological analysis of aggressiveness and responsiveness in Wilms’ tumor. Med. Pediatr. Oncol. 1996, 27, 422–428. [Google Scholar] [CrossRef]
- El Zein, D.; Hughes, M.; Kumar, S.; Peng, X.; Oyasiji, T.; et al. Metaplastic Carcinoma of the Breast Is More Aggressive Than Triple-negative Breast Cancer: A Study From a Single Institution and Review of Literature. Clinical Breast Cancer 2017, 17, 382–391. [Google Scholar] [CrossRef]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF Mutations Are Common Somatic Events in Melanocytic Nevi. Journal of Investigative Dermatology 2004, 122, 342–348. [Google Scholar] [CrossRef]
Figure 1.
Evolution of Cancer in Response to Survival Threats Resulting in Molecular Heterogeneity. When cells sustain damage that jeopardizes their viability, they respond by activating essential hallmark capabilities to ensure survival. This triggers a cascade of molecular activation. In advanced stages, under continuous stress, this results in molecular disarray as interconnected cancer hallmark capabilities become increasingly activated.
Figure 1.
Evolution of Cancer in Response to Survival Threats Resulting in Molecular Heterogeneity. When cells sustain damage that jeopardizes their viability, they respond by activating essential hallmark capabilities to ensure survival. This triggers a cascade of molecular activation. In advanced stages, under continuous stress, this results in molecular disarray as interconnected cancer hallmark capabilities become increasingly activated.

Figure 2.
Oncogene Addiction and Cancer Stem Cells. Right side: when advantageous oncogenes are inhibited, cells lose their survival response mechanism. This evolutionary disruption results in cell death, a phenomenon termed “oncogene addiction”. Left side: Cancer stemness arises from the cell’s imperative to repair the incurred damage. The ability to initiate the regeneration process is contingent upon cell survival, and this regeneration, in turn, enhances the prospects of cell survival.
Figure 2.
Oncogene Addiction and Cancer Stem Cells. Right side: when advantageous oncogenes are inhibited, cells lose their survival response mechanism. This evolutionary disruption results in cell death, a phenomenon termed “oncogene addiction”. Left side: Cancer stemness arises from the cell’s imperative to repair the incurred damage. The ability to initiate the regeneration process is contingent upon cell survival, and this regeneration, in turn, enhances the prospects of cell survival.
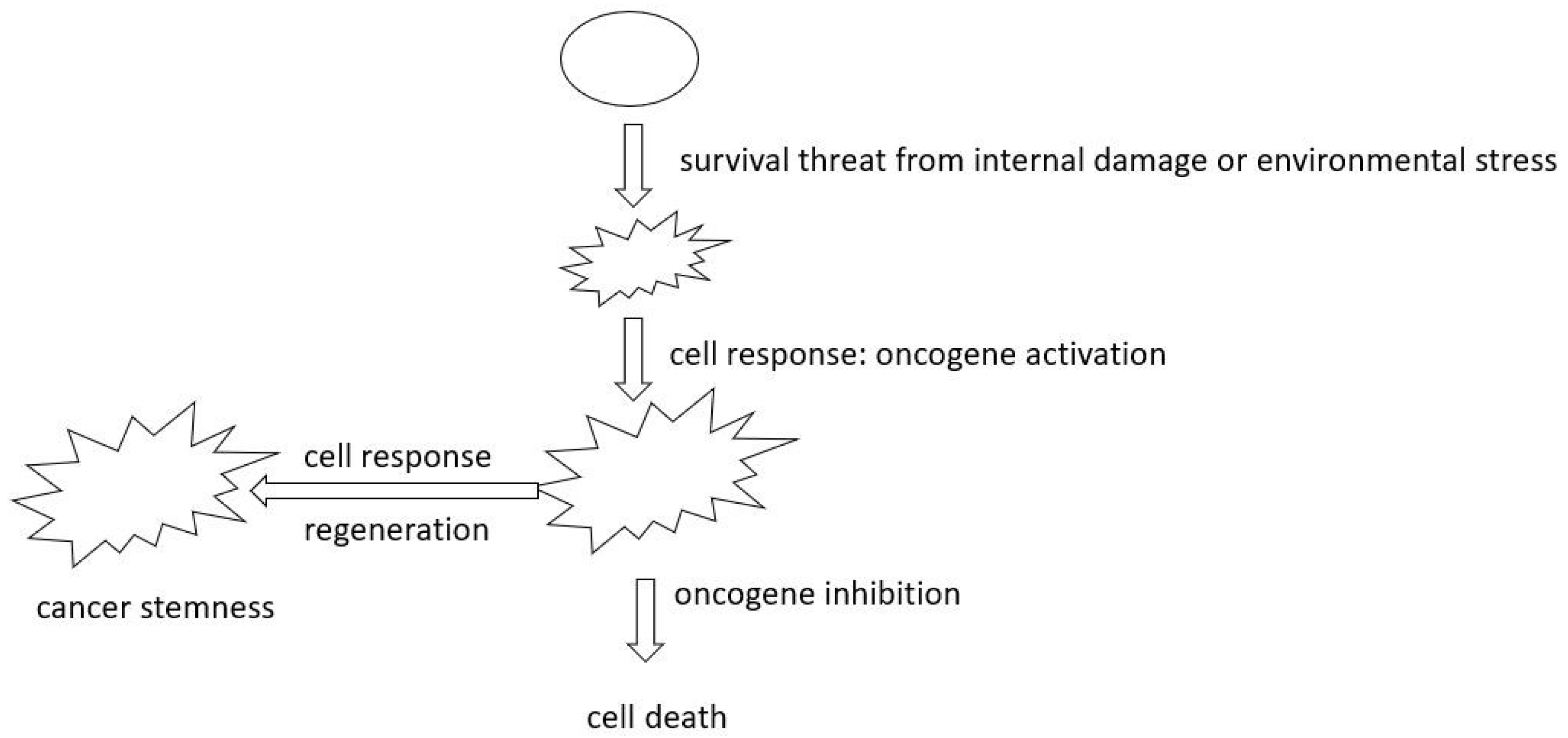
Figure 3.
Heterogeneity in Oncogene Expression. Each hallmark capability of cancer requires varying levels of oncogene activation, leading to heterogeneity in oncogene expression. Consequently, when there’s greater homogeneity in oncogene expression, cells may experience reduced intensity in certain hallmark capabilities, transitioning to a less aggressive phenotype.
Figure 3.
Heterogeneity in Oncogene Expression. Each hallmark capability of cancer requires varying levels of oncogene activation, leading to heterogeneity in oncogene expression. Consequently, when there’s greater homogeneity in oncogene expression, cells may experience reduced intensity in certain hallmark capabilities, transitioning to a less aggressive phenotype.
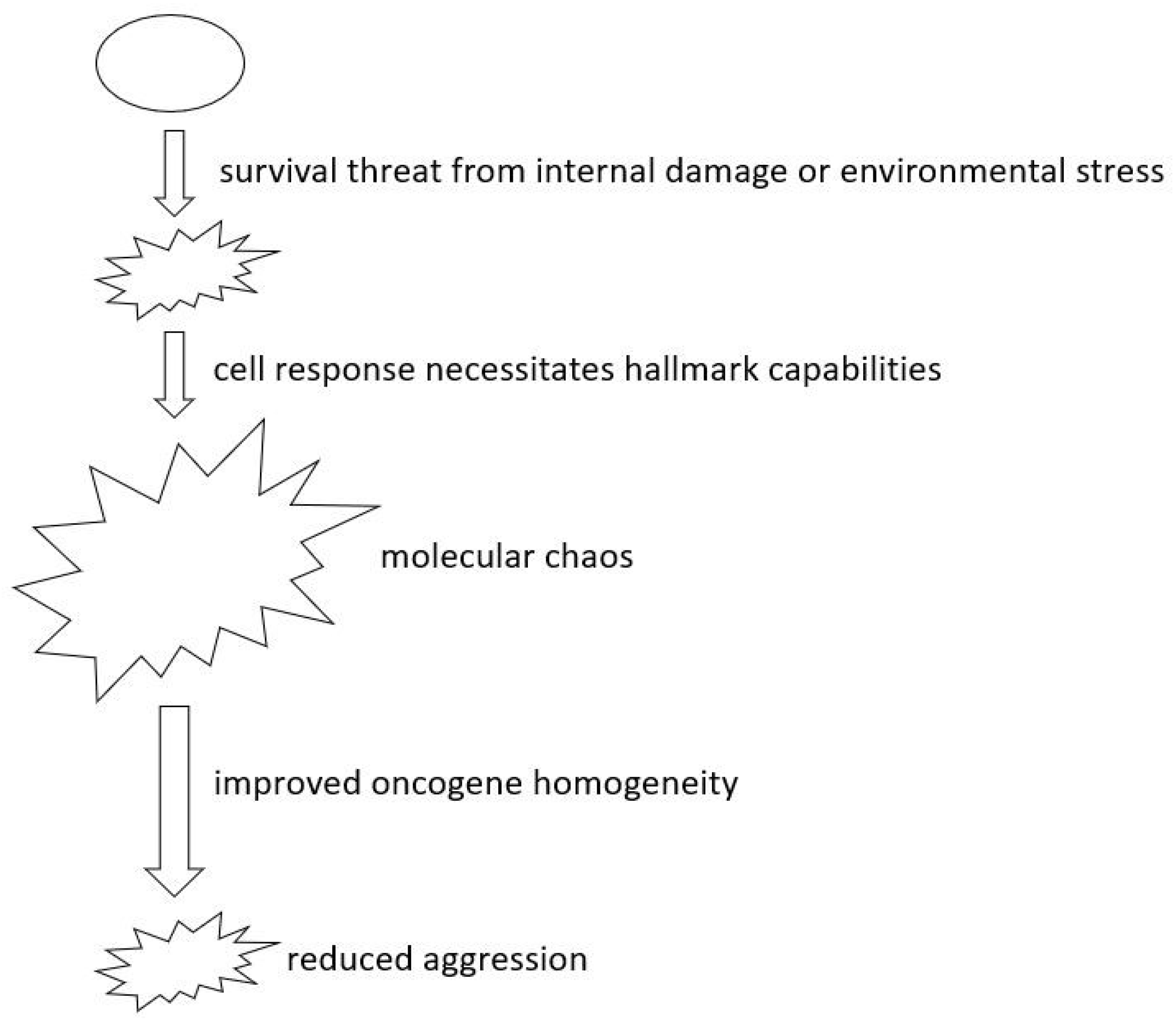
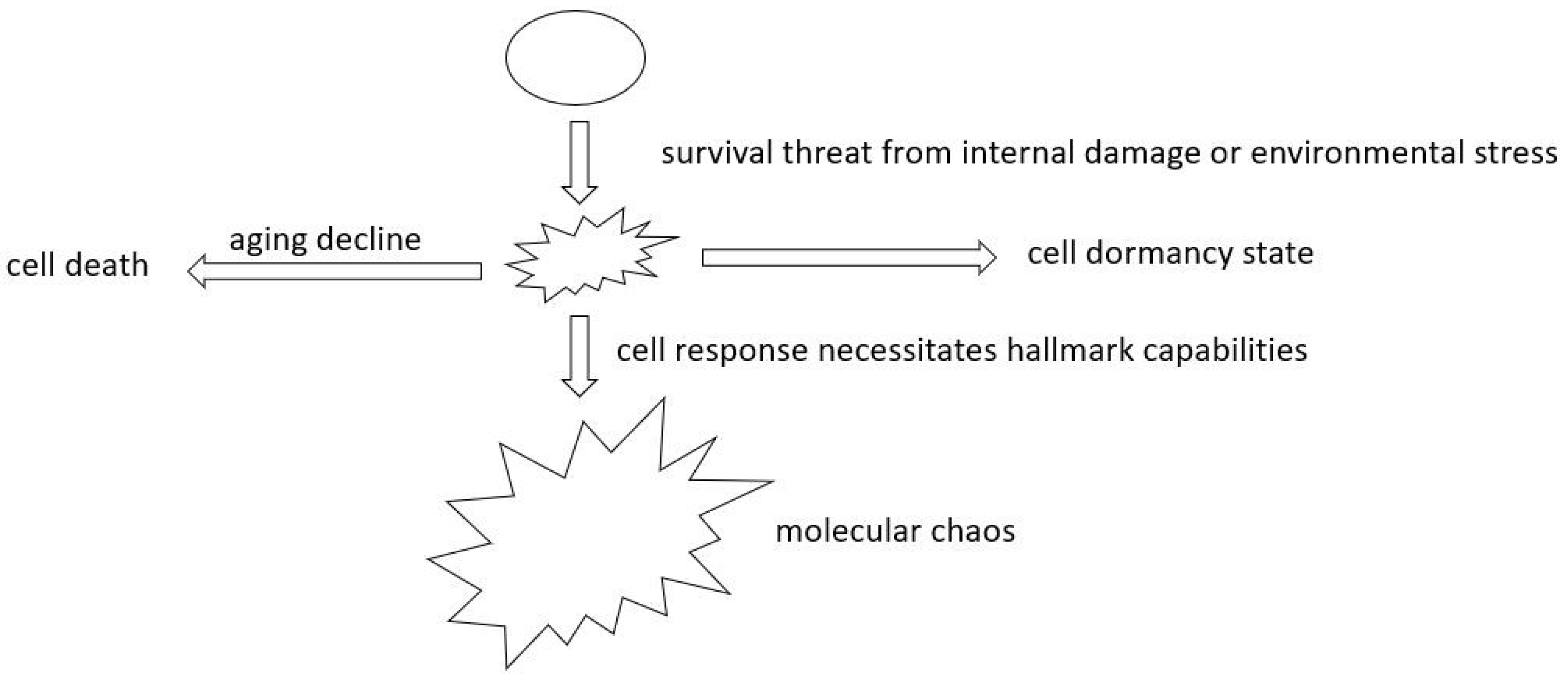
Figure 4.
Cell Fate Choices. Upon facing survival vulnerabilities, cells have several options: 1), Enter a dormancy state, 2), Undergo deterioration through the aging decline process, or 3), respond by activating essential cancer hallmark capabilities, progressing to tumorigenesis.
Figure 4.
Cell Fate Choices. Upon facing survival vulnerabilities, cells have several options: 1), Enter a dormancy state, 2), Undergo deterioration through the aging decline process, or 3), respond by activating essential cancer hallmark capabilities, progressing to tumorigenesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



