
Submitted:
23 October 2023
Posted:
23 October 2023
You are already at the latest version
Abstract
To further understand the role of phosphorylation in ISGF3- and STAT2/IRF9-mediated constitutive and long-term IFN-I-stimulated transcriptional responses, we performed RNA-Seq and ChIP-Seq, in combination with phosphorylation inhibition and anti-viral experiments. First, we identified a group of ISRE-containing ISGs that were commonly regulated in IFNα treated WT and STAT1-KO cells. Thus, in 2fTGH and Huh7.5 WT cells IFNα-inducible transcription and anti-viral activity relied on the recruitment of the ISGF3 components STAT1, STAT2 and IRF9 in a phosphorylation- and time-dependent manner. Likewise, in ST2-U3C and Huh-STAT1KO cells lacking STAT1, ISG expression correlated with DNA-binding of phosphorylated STAT2/IRF9. This pointed to a dominant role of classical ISGF3 and STAT2/IRF9, and not U-ISGF3 or U-STAT2/IRF9, in the regulation of early and prolonged ISG expression and viral protection, in WT and STAT1-KO cells. In addition, comparative experiments in U3C (STAT1-KO) cells overexpressing all ISGF3 components (ST1-ST2-IRF9-U3C), revealed a threshold-dependent role of U-ISFG3, and potentially U-STAT2/IRF9, in the regulation of constitutive and possibly long-term IFNα-treated ISG expression and anti-viral activity.
Keywords:
Interferon type-I
; JAK/STAT signaling
; ISGF3
; U-ISGF3
; STAT2/IRF9
; U-STAT2/IRF9
; Integrative omics approach
; IFN-dependent and -independent transcription
1. Introduction
As the main mediators of cellular homeostatic responses to viral infection, Type I IFNs (IFN-I) are produced by many different cell types. IFN-I predominantly consist of IFNα and IFNβ subtypes- and target responsive cells by interacting with the heterodimeric transmembrane IFN receptor (IFNAR). This activates members of the Janus kinase (Jak) and signal transducer and activator of transcription (STAT) family and the JAK/STAT signaling cascade. In the canonical IFN-I-mediated signaling pathway, Jak1- and Tyk2 phosphorylate STAT1 on Tyr701 and STAT2 on Tyr690, which after heterodimerization interact with IRF9 and form ISGF3. Then this complex translocates to the nucleus and activates transcription of numerous ISRE-containing antiviral IFN-stimulated genes (ISGs) [1,2,3]. For this reason the ISGF3-dependent canonical response forms the fundamental line of defense against viral infections.
In addition, evidence emerged for the existence of a non-canonical IFN-I signaling pathway, in which the ISGF3-like complex STAT2/IRF9 was shown to activate transcription of ISRE-containing genes in response to IFNα in the absence of STAT1 [4]. Under these conditions, the IFNα-induced expression of typical antiviral ISGs correlated with the kinetics of STAT2 phosphorylation, and the presence of a STAT2/IRF9 complex. More important, the STAT2/IRF9 complex triggered the expression of a similar subset of ISGs as ISGF3, although with a more prolonged expression profile [1,5,6]. As a consequence, STAT2/IRF9 was able to induce an antiviral response upon encephalomyocarditis virus (EMCV) and vesicular stomatitis Indiana virus (VSV). Different in vitro and in vivo studies have subsequently pointed to the existence of a STAT1-independent IFN-I signaling pathway, where STAT2/IRF9 can potentially substitute for the role of ISGF3 [7,8,9,10,11].
In accordance with the general paradigm of IFN-I signaling a robust and transient phosphorylation pattern of STAT1 and STAT2 is followed by a similar ISG expression profile that decreases over time. Conversely, recent studies revealed more complexity, with more prolonged ISG expression patterns that were shown to rely on sustained expression of the components of ISGF3 as part of a positive feedback loop [2,12,13,14]. In this context, the existence of an additional ISGF3-like complex has recently been reported, composed of unphosphorylated STAT1 and 2 with IRF9 (named U-ISGF3), which may switch with ISGF3 to drive prolonged expression of ISGs in response to IFN-I [12,13]. Likewise, long-term IFN responses in the absence of STAT1 have shown to depend on expression of STAT2 and IRF9, with a possible regulatory role of U-STAT2/IRF9 in prolonged ISG transcription [2]. Moreover, evidence exists that U-ISGF3 [15] and U-STAT2/IRF9 [16] can be formed independent of IFN-I treatment and regulate basal ISG expression. Accordingly, Platanitis [16] proposed the presence of a molecular switch from STAT2/IRF9 to ISGF3 that underlies IFN-induced transcription in mouse cells, but not in human.
We furthered assessed the genome-wide comparative role of phosphorylated and unphosphorylated ISGF3 and STAT2/IRF9 complexes in connection to constitutive and long-term IFNα-treated ISG expression and anti-viral activity. For this we performed RNA-Seq and ChIP-Seq, in combination with phosphorylation inhibition and anti-viral experiments in WT, STAT1-KO and STAT1, STAT2 and IRF9-overexpressing cells. Collectively, our data provide evidence to suggest that phosphorylated and unphosphorylated ISGF3 and STAT2/IRF9 complexes, jointly direct IFN-dependent and independent transcription of ISRE containing ISGs and protection against viral infection.
2. Results
2.1. Characterisation of time-dependent IFNα responses in WT and STAT1-KO cells
To study the role of phosphorylation of ISGF3 and STAT2/IRF9 in long-term IFN-I signaling, we performed different types of experiments. First we characterized the IFNα-induced expression and phosphorylation of ISGF3 components i.e. STAT1, STAT2 and IRF9 at the protein level in 2fTGH and Huh7.5 WT cells (Figure 1A–D). Therefore, cells were treated with IFNα for 1, 2, 4, 8, 24, 48 and 72h and compared to untreated cells. In 2fTGH phosphorylation of STAT1 and STAT2 was absent in untreated cells, but highly induced upon IFNα treatment with an early, transient increase between 1 and 4 h, after which it rapidly diminished to still detectable levels at 72h (Figure 1A). A similar early and transient IFNα-dependent STAT1 and STAT2 phosphorylation pattern was observed in Huh7.5 (Figure 1B), with a clear drop between 4 and 8h after treatment. Likewise, in
these cells the phosphorylation of STAT1 and STAT2 was still clearly visible after 72h. In contrast to their phosphorylated counterparts, expression of unphosphorylated STAT1 (U-STAT1) and U-STAT2 was already detectable in untreated 2fTGH and Huh7.5 cells, and further induced between 4 and 72h after IFNα addition. The same pattern was seen for IRF9, although it was nearly undetectable in resting 2fTGH and absent in untreated Huh7.5 cells. The IFNα-induced expression of STAT1, STAT2 and IRF9 in both cell types followed the phosphorylation pattern of STAT1 and STAT2 at later time-points and correlated with the formation and action of classical ISGF3 under these conditions. On the other hand, the accumulation of U-STAT1, U-STAT2 and IRF9 in time marks the positive feedback regulation of the ISGF3 components observed in response to IFNα, and may predict an additional role of U-ISGF3 as well in mediating the prolonged IFN Type-I signalling.
Likewise, we treated the STAT1-deficient human fibrosarcoma cell line U3C overexpressing STAT2 (ST2-U3C) [5] and Huh STAT1 knock-out cells (Huh-STAT1KO) [9,14] with IFNα, to examine the role of STAT2/IRF9 in the absence of STAT1 (Figure 1C,D). In case of ST2-U3C (Figure 1C) a subtle change in the STAT2 phosphorylation profile could be observed as compared to 2fTGH (Figure 1A). The profile remained transient, with an early increase between 1 and 2h, but, in contrast to wild-type cells, the level of pSTAT2 only decreased slowly and remained still high until 72h. This also corresponded with a higher IFN-induced expression of IRF9 in ST2-U3C at later time-points, being nearly undetectable in resting cells. And it pointed to a delayed formation of STAT2/IRF9 in these cells as compared to ISGF3 in 2fTGH. As STAT2 is overexpressed in these cells, STAT2 expression was not affected by IFN treatment. Different from ST2-U3C, Huh7.5 STAT1KO (Figure 1D) displayed a rather shifted STAT2 phosphorylation pattern, being delayed and prolonged as compared to Huh7.5 (Figure 1B). Accordingly, in Huh7.5 STAT1KO, pSTAT2 levels reached a maximum after 72h of INFα treatment. Like in ST2-U3C, in Huh STAT1KO IFN-dependent accumulation of IRF9 (absent in untreated cells) and also U-STAT2, was observed, however it predominantly occurred at later time points. This correlated with the time-dependent formation and action of STAT2/IRF9 and marks the reconstitution of the IFN-dependent positive feedback regulation loop of STAT2/IRF9 components. It also confirms the important role of STAT2/IRF9 in the regulation of prolonged ISG expression in the absence of STAT1 in both cell types. How this depends on phosphorylation of STAT2 and if a contribution of U-STAT2/IRF9 under these conditions exists has not been studied.
2.2. Genome-wide characterization of IFNα-induced transcription in WT vs. STAT1KO cells
Next, we performed RNA-Seq on RNA from 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO treated with IFNα for different timepoints (see Material and Methods). Differential gene expression analysis (DEG) was performed and genes upregulated in at least one of the time points were selected based on the cut-off of log2FC > 1 and padj < 0.05. As such, IFNα-induced expression of 1459 genes in 2fTGH and 484 in Huh7.5 cells, of which 158 genes were in common (Figure 1E). In ST2-U3C and Huh STAT1KO, IFNα induced expression of 115 and 943 genes respectively, of which 88 genes were in common (Figure 1E). When compared to the 158 commonly IFNα-induced genes from 2fTGH vs. Huh7.5, a group of 63 IFNα-inducible genes was recognized that showed overlap between WT and STAT1 deficient cell lines (Figure 1E).
GO term enrichment analysis of these genes revealed significant enrichment in biological terms connected to Type-I signalling as well as broadly defined antiviral responses, and recognizes these genes among the core subset of antiviral ISGs (Figure 1F). The expression profiles of the 63 common genes in IFNα treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO cells are presented in the form of a heatmap in Figure 1G. Obviously, a transient expression pattern was observed in both WT cell lines (2fTGH vs. Huh7.5) upon IFNα treatment, with a maximum at 8h followed by a significant drop to still detectable levels over time. On the other hand, STAT1-deficiency, changed their expression profile dramatically, resulting in lower and prolonged expression in case of ST2-U3C, and delayed and prolonged expression in case of Huh STAT1KO (Figure 1G). More important, the gene expression patterns of the 63 commonly IFNα-induced genes in these four different cell lines reflect the phosphorylation profiles and/or production of ISGF3 and STAT2/IRF9 components, as described above (Figure 1A–D).
Closer examination of this group of 63 genes recognized the presence of many known ISRE-containing antiviral ISGs (Figure 1G), including IFIT1, IFIT2, IFIT3, ISG15, IFI6, IFI27, OAS1, OAS2, as well as the components of ISGF3 (STAT1, STAT2 and IRF9). Indeed in 2fTGH, a pre-selection of these 11 genes exhibited a similar IFN-induced expression profile, with a maximum expression level after 8h of treatment, followed by a significant drop to still detectable levels at 72h (Figure 1H). In contrast, in Huh7.5 the expression profiles of these genes were more prolonged, reaching maximum expression between 8 and 72h (Figure 1H). Also, the potency of transcriptional responses in Huh7.5 was higher in comparison to 2fTGH. In ST2-U3C, the expression level of these pre-selected ISGs was much lower and their expression profile more prolonged as compared to 2fTGH (Figure 1H). Likewise, in Huh STAT1KO expression of these ISGs was prolonged, but in addition even more delayed, shifting the expression profiles of all selected ISGs towards later time points while reaching comparable levels as in Huh7.5 (Figure 1H). It also further proves, that in Huh STAT1KO IFNα-mediated ISG expression clearly depends on both STAT2 and IRF9, as early presence of only STAT2 is not sufficient for triggering significant gene expression and absence of IRF9 strongly delays the start of transcription, when compared to Huh7.5. To validate the quality of our RNA-Seq dataset in general and the expression profiles observed for the pre-selected ISRE-containing antiviral ISGs, the expression of a number of these genes, including OAS2, IFIT1, IFI27 and IFI6, was additionally confirmed by qPCR and compared in the four different cell lines (Figure S1).
Together, these results provide evidence to suggest, that ISGF3 and STAT2/IRF9 regulate transcription of a common group of ISRE-containing genes in a phosphorylation and time-dependent manner. It also offers further prove for the previous observation that STAT2/IRF9 can take over the role of ISGF3 and generate an antiviral response in the absence of STAT1 [5].
2.3. Genome-wide binding of phosphorylated and unphosphorylated ISGF3 components to ISRE sites of IFNα upregulated genes in WT vs. STAT1KO cells
We subsequently characterized the genome-wide binding of phosphorylated and unphosphorylated ISGF3 components to the regulatory regions of the 63 commonly IFNα-induced genes in WT and STAT1KO cells (Figure 2). As such, ChIP-Seq was performed on chromatin isolated from cells treated with IFNα for 0, 2, 24 and 72h, using antibodies against STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 (2fTGH and Huh7.5) and STAT2, pSTAT2 and IRF9 (ST2-U3C and Huh STAT1KO). After statistical analysis, under these conditions, the peak number distribution in 2fTGH followed a transient pattern for all antibodies, with maximum at 2h of IFNα stimulation, and clear dependence on treatment (no basal binding) (Figure 2A). Moreover, STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 binding peaks could still be detected after 72 hours. The peak number distribution in Huh7.5 exhibited a higher potency and more prolonged pattern for all antibodies, being absent in untreated cells and showing high binding scores at 2, 24 and 72h of IFNα stimulation (Figure 2B). Also, no shift could be detected from binding of phosphorylated STATs to U-STATs at later time-points. Collectively, the IFNα- and time-dependent distribution of STAT1, STAT2, pSTAT1, pSTAT2, IRF9 binding in 2fTGH and Huh7.5 correlated with the expression profile observed for the 63 commonly IFNα-induced genes in the individual cell lines (see Figure 1G,H). The peak number distribution in ST2-U3C and Huh STAT1KO cells followed a prolonged pattern for all antibodies, without binding in untreated cells. The maximal peak number in ST2-U3C was observed at 2h (Figure 2C), while in Huh STAT1KO it was shifted towards 24h (Figure 2D). Moreover, in both cell lines STAT2, pSTAT2 and IRF9 binding peaks could still be detected after 72 hours, without a detectable shift from phosphorylated STAT2 to U-STAT2 after long-term treatment. Additionally, the peak scores in ST2-U3C were generally much lower than in Huh STAT1KO, which correlated with the gene expression pattern observed in these two cell lines (see Figure 1G,H).
Successively, using HOMER software, ISRE and GAS consensus motifs (Figure S2) were mapped to IFNα-induced STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 binding regions of the 63 commonly IFNα-induced genes. Not surprisingly, all of these genes contained an ISRE binding site (not shown). Closer inspection of the 11 pre-selected genes (Figure 1G,H) confirmed a correlation between gene expression and recruitment of STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 in response to IFNα in 2fTGH (Figure 2E) and Huh7.5 (Figure 2F) and STAT2, pSTAT2 and IRF9 in ST2-U3C (Figure 2G) and Huh STAT1KO (Figure 2H). Accordingly, for the majority of the pre-selected genes the binding profiles of the different antibodies resembled the peak distribution as shown in Figure 2A–D. Also, no basal binding of U-ISGF3 components could be observed and no shift could be detected from binding of phosphorylated STATs to U-STATs after long-term treatment.
2.4. U-ISG expression correlates with pSTAT1 and pSTAT2 expression and long-term binding in wild type and STAT1KO cells
According to Cheon et al. [12] a subset of 29 ISRE containing U-ISGs exists that are exclusively regulated by ISGF3 at early time points and further induced by U-ISGF3 at late time points, to maintain the long-term IFN-I response. When compared to the 158 commonly IFNα-induced genes from 2fTGH vs. Huh7.5, 25 of these U-ISGs were shown to be expressed in both cell lines (Figure 3A). Among these 25 genes the presence of 7 previously characterized ISGs could be recognized, including IFIT1, IFIT3, ISG15, IFI27, OAS1, OAS2, and STAT1 (Figure 1G,H). The remaining 18 genes included DDX58, DDX60, BST2, IFI44, IFI44L, XAF1, IFITM1, RTP4, OAS3, IFIH1, HERC5, HERC6, MX1, PLSCR1, OASL, BATF2, TMEM140 and IFI35.
As shown in Figure 3B, these 25 U-ISGs exhibited a similar IFN-induced expression profile in 2fTGH vs. Huh7.5, and in ST2-U3C vs. Huh STAT1KO, which was comparable to the 63 commonly IFNα-induced genes (see Figure 1H). Indeed, a transient expression pattern was observed in both WT cell lines (2fTGH vs. Huh7.5) upon IFNα treatment, with a maximum at 8h followed by a significant drop to still detectable levels over time. On the other hand, STAT1-deficiency, changed their expression profile, resulting in lower and prolonged expression in case of ST2-U3C, and delayed and prolonged expression in case of Huh STAT1KO (Figure 3B). More important, the gene expression patterns of the 25 U-ISGs in these four different cell types reflect the phosphorylation profiles and/or production of ISGF3 and STAT2/IRF9 components, as described above (Figure 1).
Close comparison of the 7 previously characterized (Figure 1G-H) and a pre-selection of the remaining 18 U-ISGs (Figure 3C–F), confirmed a similar correlation between gene expression and recruitment of STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 in response to IFNα in 2fTGH (Figure 3C) and Huh7.5 (Figure 3D), and STAT2, pSTAT2 and IRF9 in ST2-U3C (Figure 3E) vs. Huh STAT1KO (Figure 3F). Accordingly, for all 25 U-ISGs the binding profiles of the different antibodies resembled the peak distribution as shown in Figure 2, with STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 binding peaks still detectable after 72 hours. Also, no basal binding of U-ISGF3 or U-STAT2/IRF9 components could be observed and no shift could be detected from binding of phosphorylated STATs to U-STATs after long-term treatment.
2.5. The role of phosphorylation of ISGF3 and STAT2/IRF9 in the regulation of prolonged ISG expression in wild type and STAT1KO cells
To further address the role of phosphorylation of STAT1 and STAT2, especially in long-term IFN-I activated transcriptional responses of WT and STAT1KO cells, experiments with JAK Inhibitor I (JII) were performed. JII is a potent compound that inhibits activity of all 4 Janus kinases, and because of this, it blocks IFNα-dependent phosphorylation of STAT1 and STAT2.
Accordingly, 2fTGH and Huh7.5 cells treated with 1000U/mL of IFNα alone for 0, 1, 2, 4, 8, 24, 48 and 72h were compared to cells treated with IFNα alone for 0, 1, 2, 4h, and for 8, 24, 48 and 72h together with JII (added after 6h of IFN treatment, at each time-point) (Figure 4A,B). As becomes clear, addition of JII resulted in a complete block of STAT1 and STAT2 phosphorylation in 2fTGH at 8, 24, 48 and 72h. Likewise, in Huh7.5 phosphorylation was inhibited at each of these time-points, except at 8h, where weak phosphorylation of STAT1 and STAT2 was still visible. In both cell types, increased production of total STAT1, STAT2 and IRF9, observed after long-term IFNα-treatment, was also impaired by the addition of JAK Inhibitor I. However, expression was still detectable. This predicts a role of STAT1 and STAT2 phosphorylation in the long-term expression of ISGF3 components and in the action of ISGF3 at later time-points in 2fTGH and Huh7.5 cells.
In a similar fashion, phosphorylation of STAT2 was significantly diminished after treatment with JII and no longer detectable in both ST2-U3C and Huh STAT1KO after 48 and 72h of IFNα treatment (Figure 4C,D, respectively). Like in WT cells, JII addition also significantly hampered the IFN-dependent increased production of STAT2 and IRF9, although more dramatically in Huh STAT1KO cells than in ST2-U3C. In Huh STAT1KO expression of IRF9 was even no longer detectable upon JII treatment.
These observations suggest, that in the STAT1-deficient cell lines, long-term expression of STAT2 and IRF9 is highly dependent on phosphorylation of STAT2 itself. It also points to the crucial role of STAT2/IRF9, in prolonged IFNα signalling in the absence of STAT1 in ST2-U3C and Huh STAT1KO cells.
Under the same conditions, we studied the effect of JII treatment in both WT and STAT1KO cell lines on the long-term IFNα-induced expression of a selection of ISGs, including OAS2, IFIT1, IFI27 and IFI6, by qPCR (Figure 4E–H). In 2fTGH and Huh7.5, addition of JII dramatically decreased expression of these genes after 8h of IFNα treatment, which correlated with a block in STAT1 and STAT2 phosphorylation (Figure 3A,B). In ST2-U3C and Huh STAT1KO, addition of JII dramatically decreased expression of OAS2, IFIT1, IFI27 and IFI6 to nearly undetectable levels at 48 and/or 72h of IFNα treatment (Figure 3C,D). This correlated with a block in STAT2 phosphorylation.
Together, these findings show that phosphorylation is a key factor in the ISGF3-or STAT2/IRF9-mediated regulation of both early and prolonged ISG expression in WT and STAT1-KO cells. At the same time, the impaired expression of unphosphorylated ISGF3 and STAT2/IRF9 components in JII treated cells can’t rule out the involvement of U-ISGF3 and U-STAT2/IRF9 under these conditions.
2.6. The role of unphosphorylated ISGF3 components in the regulation of basal ISG expression in cells overexpressing STAT1, STAT2 and IRF9
A role of unphosphorylated STATs has been proposed in the transcriptional regulation of ISGs in an IFN-independent manner [12,15]. In this respect, we observed increased basal expression of a selection of ISGs in ST2-U3C cells, as compared to U3C in the absence of IFNα treatment [5]. Recently, we generated the ST2-IRF9-U3C variant, overexpressing both STAT2 and IRF9, and studied the IFNα-dependent and -independent ISG expression in comparison to ST2-U3C and U3C (Figure S3). As expected, IFN-induced expression of OAS2, IFI27 and IFI6 was the highest in ST2-IRF9-U3C, correlating with the increased levels of phosphorylated ISGF3 components (not shown). Likewise, IFN-independent expression of these genes was also significantly higher in ST2-IRF9-U3C, which could point to a regulatory role of U-ISGF3 or U-STAT2/IRF9 in basal ISG expression (Figure S3). Subsequently, we generated the U3C-based cell line ST1-ST2-IRF9-U3C, to examine the effect of overexpressing the ISGF3 components STAT1, STAT2 and IRF9 and the possible role of U-ISGF3 in mediating basal ISG expression.
To compare the genome-wide basal gene expression in ST1-ST2-IRF9-U3C and U3C, we performed RNA-Seq on RNA from three independent repeats. Differential gene expression analysis (DEG) was performed and upregulated genes were selected based on the cut-off of log2FC > 1 and padj < 0.05. As such, 413 genes were identified in ST1-ST2-IRF9-U3C with increased basal expression (Figure 5A). After comparing these genes with the 158 commonly IFNα-induced genes in WT cells (Table 1), 43 genes were in common (Figure 5A). GO term enrichment analysis of these genes revealed significant enrichment in biological terms connected to Type-I signalling as well as broadly defined antiviral responses, and recognizes these genes among the core subset of antiviral ISGs (Figure 5B). Among these 43 genes the previously characterized pre-selected 11 ISRE-containing antiviral ISGs could be recognized (Figure 1G,H), as well as many additional known ISRE-containing ISGs, including DDX60, IFITM1 and IFITM 3 as well as BST2 (Table 1). Indeed, as compared to U3C, ST1-ST2-IRF9-U3C cells exhibited higher basal expression of OAS2, IFIT1, IFI27 and IFI6 (Figure 5C). Pre-treatment with JII for 1h did not significantly lower the basal expression of a number of these genes (including OAS2, IFI27 and IFI6) in untreated ST1-ST2-IRF9-U3C (Figure 5D). Treatment with IFNα for 2h increased the expression levels of these genes, returning to basal levels upon JII pre-treatment. This is in agreement with the role of phosphorylation in IFN-I activated transcriptional responses and provides further evidence for a phosphorylation-independent mechanism involved in the regulation of basal ISG expression in ST1-ST2-IRF9-U3C.
Therefore, we characterized the chromatin interactions of the ISGF3 components to the regulatory regions of IFIT1, ISG15, OAS2, IFI27, STAT1, STAT2 and IRF9, by ChIP-PCR (Figure 5E). As such, chromatin was isolated from untreated and IFNα-treated ST1-ST2-IRF9-U3C cells, as well as untreated U3C, using antibodies against STAT1, STAT2, and IRF9. Increased binding of all three ISGF3 components could be detected to the ISRE sites present in the promoter of these genes in untreated ST1-ST2-IRF9-U3C cells as compared to U3C. As expected, binding was even further increased in IFN-treated ST1-ST2-IRF9-U3C cells (Figure 5E).
Next, we studied the ability of ST1-ST2-IRF9-U3C cells to combat viral infection in the absence of IFN treatment. In comparison U3C and 2fTGH, cells were infected with serial 10-fold dilutions of Vesicular stomatitis Indiana virus starting from MOI=10 (Figure 5F). U3C, as well as 2fTGH, were not able to fight with the virus at MOI more than 0.001. Oppositely, ST1-ST2-IRF9-U3C cells were protected 10-100x more against VSV infection (up to MOI=0.1). Pre-treatment with JAK Inhibitor I for 2 or 24h, did not affect this anti-viral potential of ST1-ST2-IRF9-U3C cells (Compare Figure 5F: no JII vs. JII 2h vs. JII 24h).
2.7. The role of unphosphorylated ISGF3 components in prolonged IFNα signalling in cells overexpressing STAT1, STAT2 and IRF9
Finally, we examined the effect of overexpressing the ISGF3 components STAT1, STAT2 and IRF9 and the possible role of U-ISGF3 in mediating IFNα-dependent long-term ISG expression in ST1-ST2-IRF9-U3C cells. Thus, growing these cells in the absence or presence of IFNα revealed that phosphorylation of STAT1 and STAT2 was absent in untreated cells, but highly induced upon IFNα treatment with an early, transient increase between 1 and 4 h, after which it diminished to still detectable levels at 72h (Figure 6A). As STAT1, STAT2 and IRF9 are overexpressed in these cells, their expression was not affected by IFN treatment.
Addition of JII resulted in a dramatic drop in STAT1 and STAT2 phosphorylation at 8, 24, 48 and 72h. On the other hand, no effect was seen on the native levels of STAT1, STAT2 as well as IRF9. Comparison of IFN-stimulated 2fTGH and ST1-ST2-IRF9-U3C cells, treated with JII under the same conditions, revealed a dramatic difference in gene expression levels of IFIT1, OAS2, IFI27 and IFI6 after long-term IFNα and JII treatment (Figure 6B–E). In contrast to 2fTGH, expression of all of these ISGs was still detectable in the presence of JII, even after 72h. Moreover, higher basal expression of these genes could be detected in ST1-ST2-IRF9-U3C cells as compared to 2fTGH, and in the presence of IFNα and JII their expression never dropped below the basal level observed in untreated ST1-ST2-IRF9-U3C cells.
Next we compared the ability of 2fTGH and ST1-ST2-IRF9-U3C cells to generate an IFN-activated anti-viral response, in the presence or absence of JII (Figure 6F). Thus, cells were treated with a 2-fold serial dilution of IFNα for 24 and then infected with Vesicular stomatitis Indiana virus (VSV) for another 24h. During this infection time, cells were treated with JII for 4h or 25h. As a control the STAT1-deficient U3C cell line was used and treated similarly. The results clearly showed that in 2fTGH, anti-viral activity is strongly dependent on phosphorylation. Consequently, addition of JAK Inhibitor I rendered these cells more sensitive to VSV infection. Pre-treatment of cells with IFNα could not effectively protect from virus-dependent lysis. Importantly, 4h of stimulation with JII only partially impaired IFNα-mediated 2fTGH viral protection. However, addition of JII for 25h completely blocked viral protection . On the other hand, in ST1-ST2-IRF9-U3C cells blocking phosphorylation did not have a major effect on IFN-induced anti-viral activity. qPCR results under these conditions indeed confirmed that OAS2, IFI27 and IFI6 were still significantly expressed at later time-points and never dropped below basal levels being sufficient to protect cells from lysis by VSV (Figure 6G).
3. Discussion
In accordance with the general paradigm of IFN-I signaling a robust and transient phosphorylation pattern of STAT1 and STAT2 is followed by a similar ISG expression profile that decreases over time. Conversely, recent studies revealed increasing complexity, with more prolonged ISG expression patterns that were shown to rely on sustained expression of the components of ISGF3 as part of a positive feedback loop [2,12,13,14]. In this context, Cheon et al. [12] postulated a novel model of how anti-viral effects are prolonged after IFNβ exposure. In the early response phase, phosphorylation of STAT1 and STAT2 correlated with formation of ISGF3 and transcriptional regulation of many ISGs, including STAT1, STAT2, and IRF9. A drop in STAT phosphorylation during the course of a few hours, corresponded with a parallel decrease in the expression of a subset of early ISGF3 target genes (e.g., IRF1, ADAR, and MYD88). In contrast, at later times after IFN stimulation, high levels of IRF9 together with U-STAT1 and U-STAT2 proteins increased formation of U-ISGF3 and prolonged expression of a subset of U-ISGs (IFI27, OAS2, MX1, BST2, IFIT1, and IFIT3), which were previously found to be induced by U-STAT1 [17]. Likewise, Sung et al. [13] observed that the level of U-ISGF3, but not tyrosine phosphorylated STAT1, was significantly elevated in response to IFN-λ and IFN-β during chronic HCV infection. Subsequently, U-ISGF3 prolonged the expression of a subset of U-ISGs and restricted HCV chronic replication.
Using RNA-Seq and ChIP-Seq, we further assessed the genome-wide comparative role of these ISGF3 and ISGF3-like complexes in connection to constitutive and long-term IFNα-treated ISG expression and anti-viral activity. First, we identified a group of ISRE-containing ISGs that were commonly regulated in IFNα treated WT and STAT1-KO cells. Thus, in 2fTGH and Huh7.5 WT cells IFNα-inducible transcription and anti-viral activity relied on the recruitment of the ISGF3 components STAT1, STAT2 and IRF9 in a phosphorylation- and time-dependent manner. Indeed, in these cells the phosphorylation and chromatin binding of STAT1 and STAT2 was still clearly visible after 72h. Moreover, no shift could be detected from binding of phosphorylated STATs to U-STATs at later time-points, which is in contrast to the phosphorylation-independent model proposed by Cheon et al. [12]. Along the same lines, our data also disagree with the existence of ISGs and U-ISGs, as suggested by Cheon et al. [12] and Sung et al. [13]. Instead, U-ISG expression profiles followed a similar pattern as the commonly IFNα-inducible ISGs. More important, binding profiles of the different antibodies to these genes resembled the commonly IFNα-inducible ISG peak distribution, with STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 binding peaks still detectable after 72 hours.
Together with our recently published data [2,14], these results are in line with a dominant role of classical ISGF3, and not U-ISGF3, in the regulation of early as well as prolonged ISG expression and viral protection, in 2fTGH and Huh7.5 cells.
The importance of the STAT2/IRF9 complex in long-term IFN responses under conditions of STAT1 deficiency, has been addressed in different studies. For example, Lou et al. [6] reported that STAT2 together with IRF9 can effectively drive the transcription of the RIG-G gene by their functional interaction, even without the tyrosine phosphorylation of STAT2. We showed previously that in the absence of STAT1, STAT2 is capable of forming homodimers when phosphorylated in response to IFN-I [4]. Together with IRF9, these STAT2 homodimers formed STAT2/IRF9 that activated transcription of ISRE-containing genes in response to IFNα [4]. In a more genome-wide setting, this STAT2/IRF9 complex triggered the expression of a similar subset of ISGs as ISGF3, although with a more prolonged expression profile [1,5,6]. As a consequence, STAT2/IRF9 was able to initiate an antiviral response upon EMCV and VSV offering additional proof for the functional overlap between STAT2/IRF9 and ISGF3.
Our data here add to this that in ST2-U3C and Huh-STAT1KO cells lacking STAT1, IFN-I induced expression of a common set of ISRE ISGs was delayed (as compared to WT cells) and associated with DNA-binding of phosphorylated STAT2/IRF9. This delayed kinetics corresponded with the lower DNA-binding affinity of this complex and lower transcriptional potency as compared to ISGF3 [4]. Also, no shift could be detected from binding of phosphorylated STAT2 to U-STAT2 at later time-points. Agreeing with the observation that U-ISG expression and binding profiles in ST2-U3C and Huh-STAT1KO cells displayed a similar pattern as identified for the commonly IFNα-inducible ISGs. In combination with phosphorylation inhibition experiments using the JAK inhibitor JI1, these findings highly suggest that in analogy to ISGF3, phosphorylation is also a key factor in the STAT2/IRF9-mediated regulation of prolonged ISG expression in STAT1-KO cells. It also offers further prove for the previous observation that STAT2/IRF9 can take over the role of ISGF3 and generate an antiviral response in the absence of STAT1 [4,5,9]. This is in agreement with Yamauchi 2016 who observed in HCV-infected Huh-7.5 human hepatoma cells that IFN-α activated transcription of ISRE genes and inhibited HCV replication through a STAT2-dependent but STAT1-independent pathway. In contrast, IFN-λ induced ISG expression and inhibited HCV replication exclusively through a STAT1- and STAT2-dependent pathway. Additional in vivo evidence was provided by Abdul-Sater [7] and Perry [8] for the existence of a STAT2/IRF9-dependent, STAT1-independent host defense mechanism against Dengue virus and Legionella pneumophila, respectively. These studies confirm that IFN-I is able to drive the formation of STAT2/IRF9 that regulates the expression of a subset of ISRE-containing ISGs and offers a back-up response against viral infection.
The accumulation of U-STAT1, U-STAT2 and IRF9 in time, marking the positive feedback regulation of the ISGF3 components observed in 2fTGH and Huh7.5 cells in response to IFNα, raised the possibility of an additional role of U-ISGF3 in mediating the prolonged IFN Type-I signalling. Similarly, in STAT1KO cells accumulation of IRF9 and U-STAT2 was observed (predominantly at later time points) and pointed to a potential role of U-STAT2/IRF9 in long-term IFN Type-I signalling in ST2-U3C and Huh-STAT1KO cells. Moreover, the impaired expression of unphosphorylated ISGF3 and STAT2/IRF9 components in JII treated cells, proofs the importance of phosphorylated ISGF3 and STAT2/IRF9 complexes, but can’t rule out the involvement of U-ISGF3 and U-STAT2/IRF9 under these conditions. To address this issue we generated the U3C-based cell line ST1-ST2-IRF9-U3C, to examine the effect of overexpressing the ISGF3 components STAT1, STAT2 and IRF9 and the possible role of U-ISGF3 in mediating basal ISG expression. Indeed, comparative experiments in U3C (STAT1-KO) cells overexpressing all ISGF3 components (ST1-ST2-IRF9-U3C), revealed increased expression of ISRE genes and anti-viral activity independent of phosphorylation and IFN treatment. Moreover, binding of all three ISGF3 components could be detected to the ISRE sites present in the promoter of pre-selected ISGs, in untreated ST1-ST2-IRF9-U3C cells as compared to U3C.
Our data agree with a model proposed by Wang et al. [15], in which constitutive expression of ISGs in immortalized cell lines, primary intestinal and liver organoids, and liver tissues was shown to depend on U-ISGF3. Moreover, their analysis of a pre-existing ChIP-Seq data set (GSE31477)[15], claimed that STAT1 specifically bound, although with very low affinity, to the promoters of ISGs even in the absence of IFNs. In contrast, analysis of our ChIP-Seq data sets here on untreated 2fTGH, Huh7.5, ST2U3C and Huh7.5 STAT1KO, provide no proof of basal binding of U-STAT1, U-STAT2 and IRF9. Likewise, no basal DNA binding of unphosphorylated ISGF3 components could be detected in human THP1 cells as opposed to mouse macrophages under physiological conditions [16]. However, ISRE binding of STAT1, STAT2 and IRF9 in untreated ST1-ST2-IRF9-U3C cells, overexpressing all ISGF3 components, provided clear proof for a role of U-ISGF3 in basal ISG transcription. Likewise, in Wang’s study, simultaneous overexpression of all ISGF3 components, but not any single factor, induced the expression of ISGs and inhibited viral replication; however, no phosphorylated STAT1 and STAT2 were detected. Moreover, a phosphorylation-deficient STAT1 mutant was comparable to the wild-type protein in mediating the IFN-independent expression of ISGs and antiviral activity. Collectively, this offers evidence for a scenario, in which a certain threshold of STAT1, STAT2 and especially IRF9 expression and levels of U-ISGF3 has to be reached to be able to trigger basal ISG transcription. A similar mechanism would account for a role of U-STAT2/IRF9 in U3C cells overexpressing STAT2 or STAT2+IRF9, displaying increased basal ISG expression as compared to U3C cells. In this respect, Platanitis [16] showed that preformed STAT2/IRF9 complexes control basal ISG expression in murine macrophages. So far this has not been shown in human cells. It would be interesting to see if differences in basal expression levels of ISGF3 components in mouse and human cells could account for these contrasting observations. At the same time, overexpressing the ISGF3 components STAT1, STAT2 and IRF9 provided conditions for a possible role of U-ISGF3 in mediating IFNα-dependent long-term ISG expression in ST1-ST2-IRF9-U3C cells. Indeed, in ST1-ST2-IRF9-U3C cells blocking phosphorylation did not have a major effect on IFN-induced anti-viral activity, whereas ISG transcription never dropped below basal levels being sufficient to protect cells from lysis by VSV. Based on this it is tempting to suggest that in addition to the dominant role of classical ISGF3 in the regulation of early as well as prolonged ISG expression and viral protection, U-ISGF3 (and possibly U-STAT2/IRF9) could have an additional involvement. This is consistent with studies published by Majoros et al. [18], which show a potential contribution of Y701-unphosphorylated STAT1 to innate antibacterial immunity.
Collectively, our data are in line with a dominant role of classical ISGF3 and STAT2/IRF9, in the IFN-dependent regulation of early as well as prolonged ISG expression and viral protection. However, we and others also provide evidence for an additional role of U-ISFG3, and possibly U-STAT2/IRF9, in the regulation of constitutive and possibly IFN-dependent ISG expression. In this respect, a certain threshold of STAT1, STAT2 and IRF9 expression and levels of U-ISGF3 has to be reached to be able to drive ISG transcription and viral protection. As a consequence, together with phosphorylated ISGF3 and STAT2/IRF9 complexes, U-ISGF3 and possibly U-STAT2/IRF9 could be instrumental in IFN-dependent and -independent ISG transcription and anti-viral activity.
4. Materials and Methods
4.1. Cell lines
The human fibrosarcoma 2fTGH cell line and the STAT1-deficient U3C cell line were kind gifts from Dr Sandra Pellegrini (Institute Pasteur, Paris, France) [5]. The U3C cell lines stably overexpressing combinations of ISGF3-components (STAT2-U3C, STAT2-IRF9-U3C, STAT1-STAT2-IRF9-U3C) were generated in our laboratory, by using MigR1 vectors containing genes of interests and pcDNA6/TR (blasticidin-resistance; ThermoFisher Scientific, V102520), that were transfected with Xtreme HP reagent (Roche, 6366236001).
The human hepatocellular carcinoma cell line Huh7.5 WT and Huh7.5 STAT1 K.O., that was generated using the CRISPR/Cas9 system [9,14], were kind gifts from Prof. Kiyonao Sada (Department of Genome Science and Microbiology, University of Fukui, Fukui, Japan).
Cell culture and treatment
All cell lines were cultured in DMEM (IITD PAN Wrocław, 11) supplemented with 10% fetal bovine serum (FBS) (ThermoFisher Scientific, 10500-064), 2mM L-glutamine (BioWest, X0550) and 100 IU/ml penicillin/100 µg/ml streptomycin/250 ng/ml amphotericin B (Sigma-Aldrich, A5955) and, in case of Huh7.5 WT and Huh7.5 STAT1 K.O., also with 1% MEM NEAA (ThermoFisher Scientific, 11140-035). Both media are referred later in the manuscript as full culture media. Cells were starved with medium containing lowered to 1% FBS concentration and treated with IFNα (1000 U/ml, Merck-Milipore, IF007) up to 72h and with JAK Inhibitor I (JII, Pyronide 6, 5uM, Bio-techne, 6577/10) up to 25h depending on the experiment. For clarifying the presented RNA-Seq and ChIP-Seq data, on heatmaps and boxplots on Figure 1 and 3 we showed unified timepoints. For STAT1-KO cell lines we used later timepoints as in these cell types the response is prolonged.
4.2. Western blotting
Protein isolation, quantification and immunoblotting were performed as described before by Piaszyk-Borychowska [19] with the use of primary antibodies anti: -pSTAT1 (CST, 7649, D4A7, 1:200), -pSTAT2 (CST, 88410, D3P2P, 1:500), -tSTAT1 (CST, 14994, D1K9Y, 1:500), -STAT2 (CST, 72604, D9J7L, 1:500), -IRF9 (CST, 76684, D2T8M, 1:400 in case of 2fTGH and U3C-based cells and 1:300 in case of Huh cells) as well as -α-Tubulin (Abcam, AB52866, 1:2000) and secondary HRP-conjugated goat anti-rabbit antibodies (Sigma-Aldrich, A9169, 1:20000).
4.3. RNA isolation and qPCR
Total RNA was extracted using the GeneMATRIX Universal RNA Purification Kit (EurX, E3598-02), according to the protocol provided by the manufacturer with minor changes. 500 ng of purified total RNA was then reverse-transcribed using ThermoFisher Scientific reagents (K1622). Quantification of transcripts was performed by qPCR with Maxima SYBR Green/ROX qPCR Master Mix (K0223, TFS) using the CFX Connect Thermal Cycler System (Bio-Rad). Target gene levels were normalized to - glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and quantified as described by Willems et al. [20]. Sequences of used primers are shown in Table 2. Relative gene expression results, if not indicated differently, are presented as mean +/- SEM for two independent biological repeats. Graphs were prepared using GraphPad Prism 7.01 [21].
4.4. RNA-Seq library preparation and sequencing
RNA was isolated from cells treated with IFNα up to 72h and JII depending on the experiment type and quantified using Qubit RNA BR assay kit (Q10210 ThermoFisher Scientific) and quality was assessed by the Agilent 2100 Bioanalyzer using the RNA 6000 Nano kit (5067-1511, Agilent Technologies), according to the protocols provided by manufacturers. RNA degradation was assessed by RIN (RNA integrity number) and samples with RIN higher than 9 were then used for further analysis. RNA libraries were prepared in three biological repeats from 1μg of total RNA using NEBNext® Ultra™ or Ultra™ II RNA Library Prep Kit for Illumina® (New England Biolabs, NEB) together with NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) and NEBNext® Multiplex Oligos for Illumina® (NEB), according to the manufacturer’s protocol. Quality and fragment distribution of prepared libraries were estimated using the Agilent High Sensitivity DNA kit (5067-4626, Agilent Technologies) and quantity was assessed by the Qubit dsDNA HS assay kit (Q32851, ThermoFisher Scientific). Sequencing (HighOutput SR75, v2 chemistry) was performed on the NextSeq500 (Illumina) in Lexogen, BioCenter in Vienna, Austria.
4.5. RNA-Seq data analysis
For ST1-ST2-IRF9-U3C Salmon v1.10.2 [22] have been used to count transcript-level abundance estimates with gencode.v43.basic.annotation GTF file. For the rest of cell lines Fastq files were aligned using STAR v2.7.3a [23] against the Homo_sapiens.GRCh38.dna.primary_assembly genome build (release-100). Gene counts (reads aligned to each gene of each sample) were generated using FeatureCounts v1.6.2 with default parameters [24]. Quality control assessments for raw and mapped read counts were made using FastQC [25] and reports combined with MultiQC [26]. Genes with low counts (below 10 in any time point) were considered as “non-expressed” and filtered out for the downstream testing.
4.5.1. Differential gene expression analysis (DEG)
Counts were normalized and DEG analysis were performed using the DESeq2 v1.30.1 package [27] in R v4.0.3 software [28]. The Wald Test (for ST1-ST2-IRF9-U3C and U3C) or likelihood ratio test (LRT; for the rest of the cell lines) was used to identify genes that respond to IFNα treatment over time. Relationships between replicates and time of treatment were assessed through a principal component analysis (PCA) plot generated using DESeq2. False discovery rate (FDR)‒adjusted q-values (5% threshold) were calculated by Benjamini-Hochberg procedure. The log2FC (fold change) was also calculated for each gene. Genes with adjusted p-values (padj) less than 0.05 and log2FC > 0.5 (for ST2-U3C and ST1-ST2-IRF9-U3C) or log2FC > 1 (for the rest of the cell lines) were considered as DEGs.
4.5.2. Heatmap generation
Heatmap visualizing transcriptional response to IFNα stimulation was prepared using pheatmap v1.0.12 [29] and ComplexHeatmap v2.10.0 [30] packages (R). For selected genes, normalized counts obtained from DESeq2 were extracted and subjected to hierarchical clustering (by row) with default clustering method: complete, euclidean distance. For plotting, row scaling with Z-scores was performed. Colour scale (from blue to red, that represents low and high normalized intensity, respectively) indicates the expression change over time for each sample compared to the expression of the non-stimulated control.
4.5.3. Gene ontology term enrichment analysis
The GO term enrichment analysis was performed using R package clusterProfiler 4.6.0 [31,32] with following settings: 1) all differentially expressed genes from 2fTGH (14 920) as background 2) p-value cutoff 0.01 and q-value cutoff 0.05 on enrichment tests 3) p-values adjustment method Benjamini & Hochberg 4) sub-ontology Biological Process (BP). To reduce redundancy of enriched GO terms “simplify” function was applied, with default parameters and cutoff 0.6. 15 terms with highest statistical significance were used for visualization as barplot (ggplot2 3.4.2) [33]. Enrichment was defined as -log10(adjusted p-value).
4.5.4. Selection of commonly upregulated genes
4.6. Chromatin immunoprecipitation (ChIP) and sequencing (ChIP-Seq)
ChIP experiments were prepared with chromatin isolated from cells treated with IFNα for 2, 24 and 72h or left untreated. ChIP was performed as previously described [5] using antibodies listed in Western blotting paragraph. Immunoprecipitated chromatin concentration was measure using a Qubit dsDNA HS assay kit (Q32851, ThermoFisher Scientific). Chromatin was diluted 13x and quantified using qPCR method and primers listed in Suppl. Table 3. Relative gene expression was calculated over GAPDH and using U3C Ct values as a control.
4.7. ChIP-Seq data analysis
For Huh7.5 and Huh ST1 K.O. ChIP-Seq data was analyzed according to ENCODE Transcription Factor processing pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2) with default parameters [36] as described previously [14]. The analysis for the rest of the cell lines has been carried out as follows. After quality assessment of raw reads with FastQC the alignment to the GRCh38.106 human genome assembly was done with Bowtie2 v2.4.1 [37] and BAM files were created and sorted by SAMTools v1.6 [38]. PCR duplicates were removed using BamUtil v1.0.15 [39]. The read enrichments (peaks) of transcription factors were predicted by MACS v2.2.6 [40]. During the secondary analysis artefacts were determined and removed based on the blacklisted genes list from the ENCODE project [36] using BEDTools v2.30.0 [41].
To annotate the location of a given peak in terms of important genomic features and to associate peaks with nearby genes annotatePeaks.pl program from HOMER v4.11 was used [42].
4.7.1. Visualization in the Integrative Genomics Viewer
4.7.2. Binding profiles
In order to generate STAT1, STAT2, IRF9, pSTAT1 and pSTAT2 binding profiles upon IFNα treatment by time-point, the matrices that represent binding signal over selected genomic intervals have been quantified prepared as follows. For each treatment, p-value signal tracks from pooled replicates per each time-points and bed file with peaks annotated to promoters of selected 63 (commonly upregulated genes in wild-type vs. STAT1 knock-out cell lines) genes were used. Signals were computed across 4 kb region centered on ChIP-Seq peak summits with computeMatrix function from deepTools v3.5.0 (reference-point TSS, upstream region 3000 bp, downstream region 1000 bp) [43]. Further steps were performed with profileplyr_1.10.0 package [45]. First, k-means clustering of the signal across the genomic intervals of gene promoters was performed with pheatmap package [29]. Mean range signal for each cluster was subsequently visualized as boxplots with ggplot2 [33].
4.7.3. Binding site motifs identification
Peaks annotated for 63 commonly upregulated genes in wild-type vs. STAT1 knock-out cell lines were used to identify enriched transcription-factor-binding motifs by Hypergeometric Optimization of Motif EnRichment (HOMER) v. 4.9.1 [42]. Matrices selected in Sekrecka et al. [14] for binding elements annotation (4 for GAS and 3 for ISRE), shown in Figure S1, were applied for binding site annotation using annotatePeaks.pl program (HOMER). Motif logos were generated using universalmotif package v1.12.1 [46].
4.8. Deposited sequencing data
RNA sequencing and/or ChIP sequencing data for 2fTGH, ST2-U3C, Huh STAT1KO, ST1-ST2-IRF9-U3C and U3C are under submission at NCBI GEO DataSets. For Huh7.5 RNA-Seq and ChIP-Seq data have been submitted to GEO under the accession number SuperSeries GSE222668.
4.9. Antiviral assay
Two different schemes were used for the antiviral assay. One, classical, was done as described before [47,48] on cells pre-treated with or without 2-fold serial dilutions of IFNα, starting from 50 U/ml for 24, 48 and 72h. The second was done on untreated cells. Both procedures, besides the treatments, were performed in the same way: vesicular stomatitis Indiana virus (VSV) at a multiplicity of infection (MOI) of 1.0 was added to the cells using serum-free DMEM. Twenty or twenty four hours post-infection, the medium was removed and cells were fixed with the 10% formaldehyde solution for 20 minutes at room temperature. After fixation, cells were visualized by the crystal violet staining. Excess dye was removed by immersing the plate in water.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Confirmation of RNA-Seq results obtained from 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO performed by qPCR for selected ISGs, n =2; mean ± SEM. Figure S2. Logos representing matrices used for promoter analysis of the 63 genes commonly upregulated in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO generated using HOMER software. Figure S3. Overexpression of different combinations of ISGF3 components increases ISG expression in untreated cells. qPCR results demonstrating increased basal expression of selected ISGs, n =2; mean ± SEM.
Author Contributions
Conceptualization, HN and HB; Methodology, HN, KB, KK; Data analysis, KK, KB, HB; Investigation, HN, AS, KB and C-YC, C-KL; Writing—original draft preparation, HN and HB; Writing—review and editing HN, JW and KK.; Visualization, HN and KK; Supervision, HB; Funding acquisition, HN and HB; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The National Research Center Poland, grant number UMO-2016/21/N/NZ2/01720 (HN), UMO-2016/23/B/NZ2/00623 (HB) and The National Centre for Research and Development grant: “Passport to the future - Interdisciplinary doctoral studies at the Faculty of Biology UAM" POWR.03.02.00-00-I006/17 (HN); Project-based Personnel Exchange Program (PPP) between the MOST (Ministry of Science and Technology), Taiwan and the PAS (Polish Academy of Science), Poland (Project #104-2911-I-002-522) (HN).
Acknowledgments
We would like to thank Prof. Sandra Pellegrini, Institute Pasteur, Paris, France – who kindly provided 2fTGH and U3C cell lines; Prof. Kiyonao Sada, Department of Genome Science and Microbiology, University of Fukui, Fukui, Japan – who kindly shared Huh7.5 and Huh STAT1KO cell lines.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blaszczyk, K.; Nowicka, H.; Kostyrko, K.; Antonczyk, A.; Wesoly, J.; Bluyssen, H.A.R. The Unique Role of STAT2 in Constitutive and IFN-Induced Transcription and Antiviral Responses. Cytokine & growth factor reviews 2016, 29, 71–81. [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Frontiers in Immunology 2018, 9, 1–17. [CrossRef]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional Control and Biological Impact. Nature Reviews Molecular Cell Biology 2002, 3, 651–662. [CrossRef]
- Bluyssen, H.A.R.; Levy, D.E. Stat2 Is a Transcriptional Activator That Requires Sequence-Specific Contacts Provided by Stat1 and P48 for Stable Interaction with DNA. Journal of Biological Chemistry 1997, 272, 4600–4605. [CrossRef]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 Directs a Prolonged ISGF3-like Transcriptional Response and Antiviral Activity in the Absence of STAT1. Biochemical Journal 2015, 466, 511–524. [CrossRef]
- Lou, Y.J.; Pan, X.R.; Jia, P.M.; Li, D.; Xiao, S.; Zhang, Z.L.; Chen, S.J.; Chen, Z.; Tong, J.H. Ifr-9/Stat2 Functional Interaction Drives Retinoic Acid-Induced Gene g Expression Independently of Stat1. Cancer Research 2009, 69, 3673–3680. [CrossRef]
- Abdul-Sater, A.A.; Majoros, A.; Plumlee, C.R.; Perry, S.; Gu, A.-D.; Lee, C.; Shresta, S.; Decker, T.; Schindler, C. Different STAT Transcription Complexes Drive Early and Delayed Responses to Type I IFNs. The Journal of Immunology 2015, 195, 210–216. [CrossRef]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 Mediates Innate Immunity to Dengue Virus in the Absence of STAT1 via the Type I Interferon Receptor. PLoS Pathogens 2011, 7. [CrossRef]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 Is Essential for the Inhibition of Hepatitis C Virus Replication by Interferon-λ but Not by Interferon-α. Scientific Reports 2016, 6, 1–11. [CrossRef]
- Kraus, T.A.; Lau, J.F.; Parisien, J.P.; Horvath, C.M. A Hybrid IRF9-STAT2 Protein Recapitulates Interferon-Stimulated Gene Expression and Antiviral Response. Journal of Biological Chemistry 2003, 278, 13033–13038. [CrossRef]
- Poat, B.; Hazari, S.; Chandra, P.K.; Gunduz, F.; Alvarez, X.; Balart, L.A.; Garry, R.F.; Dash, S. Intracellular Expression of IRF9 Stat Fusion Protein Overcomes the Defective Jak-Stat Signaling and Inhibits HCV RNA Replication. Virology Journal 2010, 7, 265. [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-Dependent Increases in STAT1, STAT2, and IRF9 Mediate Resistance to Viruses and DNA Damage. EMBO Journal 2013, 32, 2751–2763. [CrossRef]
- Sung, P.S.; Cheon, H.J.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of Unphosphorylated ISGF3 in HCV Infection and Interferon Responsiveness. Proceedings of the National Academy of Sciences of the United States of America 2015, 112, 10443–10448. [CrossRef]
- Sekrecka, A.; Kluzek, K.; Sekrecki, M.; Boroujeni, M.E.; Hassani, S.; Yamauchi, S.; Sada, K.; Wesoly, J.; Bluyssen, H.A.R. Time-Dependent Recruitment of GAF, ISGF3 and IRF1 Complexes Shapes IFNα and IFNγ-Activated Transcriptional Responses and Explains Mechanistic and Functional Overlap. Cell. Mol. Life Sci. 2023, 80, 187. [CrossRef]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 Drives Constitutive Expression of Interferon-Stimulated Genes to Protect against Viral Infections. Science Signaling 2017, 10, 1–13. [CrossRef]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Müller, M.; Novatchkova, M.; Decker, T. A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription. Nature Communications 2019, 10, 1–17. [CrossRef]
- Cheon, H.J.; Stark, G.R. Unphosphorylated STAT1 Prolongs the Expression of Interferon-Induced Immune Regulatory Genes. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 9373–9378. [CrossRef]
- Majoros, A.; Platanitis, E.; Szappanos, D.; Cheon, H.; Vogl, C.; Shukla, P.; Stark, G.R.; Sexl, V.; Schreiber, R.; Schindler, C.; et al. Response to Interferons and Antibacterial Innate Immunity in the Absence of Tyrosine-Phosphorylated STAT1. EMBO reports 2016, 17, 367–382. [CrossRef]
- Piaszyk-Borychowska, A.; Széles, L.; Csermely, A.; Chiang, H.C.; Wesoły, J.; Lee, C.K.; Nagy, L.; Bluyssen, H.A.R. Signal Integration of IFN-I and IFN-II with TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFκB to Enhance pro-Inflammatory Transcription. Frontiers in Immunology 2019, 10, 1–20. [CrossRef]
- Willems, E.; Leyns, L.; Vandesompele, J. Standardization of Real-Time PCR Gene Expression Data from Independent Biological Replicates. Analytical Biochemistry 2008, 379, 127–129. [CrossRef]
- GraphPad Software, Inc. GraphPad Prism 7.01.
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat Methods 2017, 14, 417–419. [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics (Oxford, England) 2013, 29, 15–21. [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics (Oxford, England) 2014, 30, 923–930. [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online] 2010.
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome biology 2014, 15, 550. [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing 2021.
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0. 12. CRAN. R-project. org/package= pheatmap 2019.
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation (Camb) 2021, 2, 100141. [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag New York, 2016;
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An Interactive Venn Diagram Viewer. BMC Bioinformatics 2014, 15, 293. [CrossRef]
- Inkscape Project. Inkscape 2020.
- ENCODE Project consortium Transcription Factor ChIP-Seq Data Standards and Processing Pipeline – ENCODE Available online: https://www.encodeproject.org/chip-seq/transcription_factor/(accessed on 13 October 2023).
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nature Methods 2012, 9, 357–359. [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, 1–4. [CrossRef]
- Jun, G.; Wing, M.K.; Abecasis, G.R.; Kang, H.M. An Efficient and Scalable Analysis Framework for Variant Extraction and Refinement from Population-Scale DNA Sequence Data. Genome Research 2015, 25, 918–925. [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biology 2008, 9, R137. [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 2010, 38, 576–589. [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Research 2016, 44, W160–W165. [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genome Viewer. Nature Biotechnology 2011, 29, 24–26. [CrossRef]
- Carroll, T.; Barrows, D. Profileplyr: Visualization and Annotation of Read Signal over Genomic Ranges with Profileplyr 2021.
- Tremblay, B.J.-M. Universalmotif: Import, Modify, and Export Motifs with R 2021.
- Wang, W.-B.; Levy, D.E.; Lee, C.-K. STAT3 Negatively Regulates Type I IFN-Mediated Antiviral Response. The Journal of Immunology 2011, 187, 2578–2585. [CrossRef]
- Costa-Pereira, A.P.; Williams, T.M.; Strobl, B.; Watling, D.; Briscoe, J.; Kerr, I.M. The Antiviral Response to Gamma Interferon. 2002. [CrossRef]
Figure 1.
ISGF3 and STAT2/IRF9 regulate transcription of a common group of ISRE-containing genes in a phosphorylation- and time-dependent manner. (A-D) Protein synthesis and phosphorylation patterns in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO cells evaluated by immunoblotting; p – phosphorylated, t - total; (E) The 63 commonly upregulated genes are presented using Venn diagram based on RNA-Seq results of 2fTGH (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC >1 or 0.5; padj < 0.05; (F) Gene Ontology terms enrichment analysis of 63 commonly upregulated genes in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO (significant enrichment considered with FDR < 0.05). (G) Heatmaps generated from the expression values of commonly upregulated genes in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO. Counts were normalized using Z-score. Each row represents one of 63 genes, preselected ISGs are highlighted; (H) Boxplots representing the expression profiles of the pre-selected 11 commonly upregulated genes (see text) in 2fTGH (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC > 1 or 0.5 and padj < 0.05.
Figure 1.
ISGF3 and STAT2/IRF9 regulate transcription of a common group of ISRE-containing genes in a phosphorylation- and time-dependent manner. (A-D) Protein synthesis and phosphorylation patterns in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO cells evaluated by immunoblotting; p – phosphorylated, t - total; (E) The 63 commonly upregulated genes are presented using Venn diagram based on RNA-Seq results of 2fTGH (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC >1 or 0.5; padj < 0.05; (F) Gene Ontology terms enrichment analysis of 63 commonly upregulated genes in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO (significant enrichment considered with FDR < 0.05). (G) Heatmaps generated from the expression values of commonly upregulated genes in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO. Counts were normalized using Z-score. Each row represents one of 63 genes, preselected ISGs are highlighted; (H) Boxplots representing the expression profiles of the pre-selected 11 commonly upregulated genes (see text) in 2fTGH (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC > 1 or 0.5 and padj < 0.05.
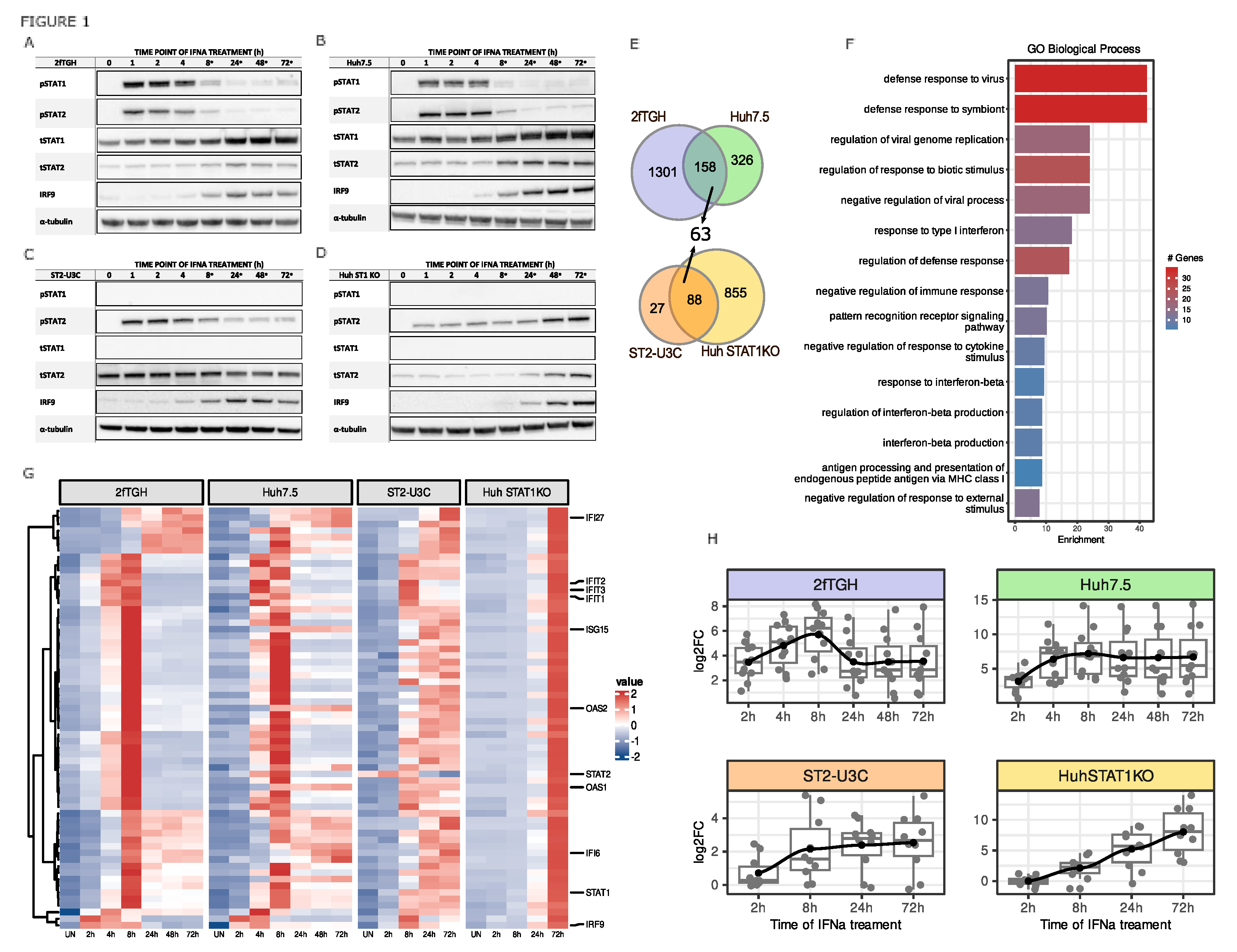
Figure 2.
Genome-wide promoter interactions of phosphorylated and unphosphorylated ISGF3 and STAT2/IRF9 components reflect ISG transcription profiles. (A) ChIP-Seq peak distribution for STAT1, pSTAT1, STAT2, pSTAT2 and IRF9 in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO presented as boxplots showing average peak signal generated using Macs2 (corresponding values—left y axis); (B) Representative ChIP peaks graphically presented in IGV software. Scale was set to 0–200, except for 2fTGH STAT1, ST2-U3C STAT2 and IRF9 were it was set 0-100; p - phosphorylated.
Figure 2.
Genome-wide promoter interactions of phosphorylated and unphosphorylated ISGF3 and STAT2/IRF9 components reflect ISG transcription profiles. (A) ChIP-Seq peak distribution for STAT1, pSTAT1, STAT2, pSTAT2 and IRF9 in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO presented as boxplots showing average peak signal generated using Macs2 (corresponding values—left y axis); (B) Representative ChIP peaks graphically presented in IGV software. Scale was set to 0–200, except for 2fTGH STAT1, ST2-U3C STAT2 and IRF9 were it was set 0-100; p - phosphorylated.
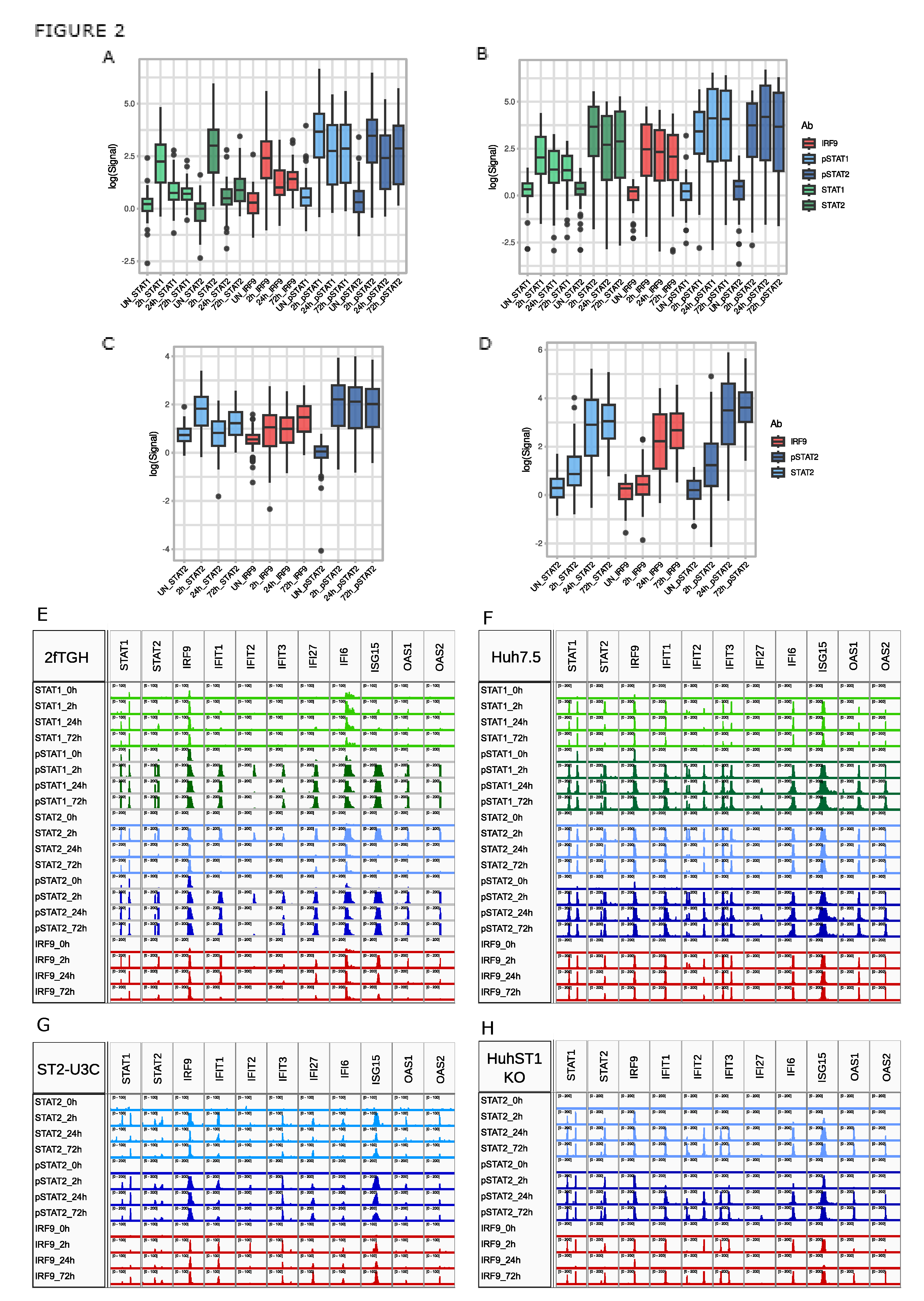
Figure 3.
Long-term U-ISG expression profiles follow the chromatin binding patterns of pSTAT1 and pSTAT2. (A) Venn diagram showing U-ISGF3 target genes described by Cheon et al. [12] (pink) upregulated in 2fTGH and Huh7.5 (WTs, turquoise); (B) RNA-Seq result-based boxplots representing U-ISG expression profiles in 2fTHG (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC >1 or 0.5; padj < 0.05; (C) Binding of STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 to U-ISG promoters demonstrated as ChIP peaks in IGV software. Scale was set to 0–200, except for 2fTGH STAT1 (60), STAT2 (100) and IRF9 (100), Huh7.5 STAT1 (100) and ST2-U3C STAT2 (100), pSTAT2 (100) and IRF9 (50); p - phosphorylated. .
Figure 3.
Long-term U-ISG expression profiles follow the chromatin binding patterns of pSTAT1 and pSTAT2. (A) Venn diagram showing U-ISGF3 target genes described by Cheon et al. [12] (pink) upregulated in 2fTGH and Huh7.5 (WTs, turquoise); (B) RNA-Seq result-based boxplots representing U-ISG expression profiles in 2fTHG (blue), Huh7.5 (green), ST2-U3C (orange) and Huh STAT1KO (yellow), log2FC >1 or 0.5; padj < 0.05; (C) Binding of STAT1, STAT2, pSTAT1, pSTAT2 and IRF9 to U-ISG promoters demonstrated as ChIP peaks in IGV software. Scale was set to 0–200, except for 2fTGH STAT1 (60), STAT2 (100) and IRF9 (100), Huh7.5 STAT1 (100) and ST2-U3C STAT2 (100), pSTAT2 (100) and IRF9 (50); p - phosphorylated. .
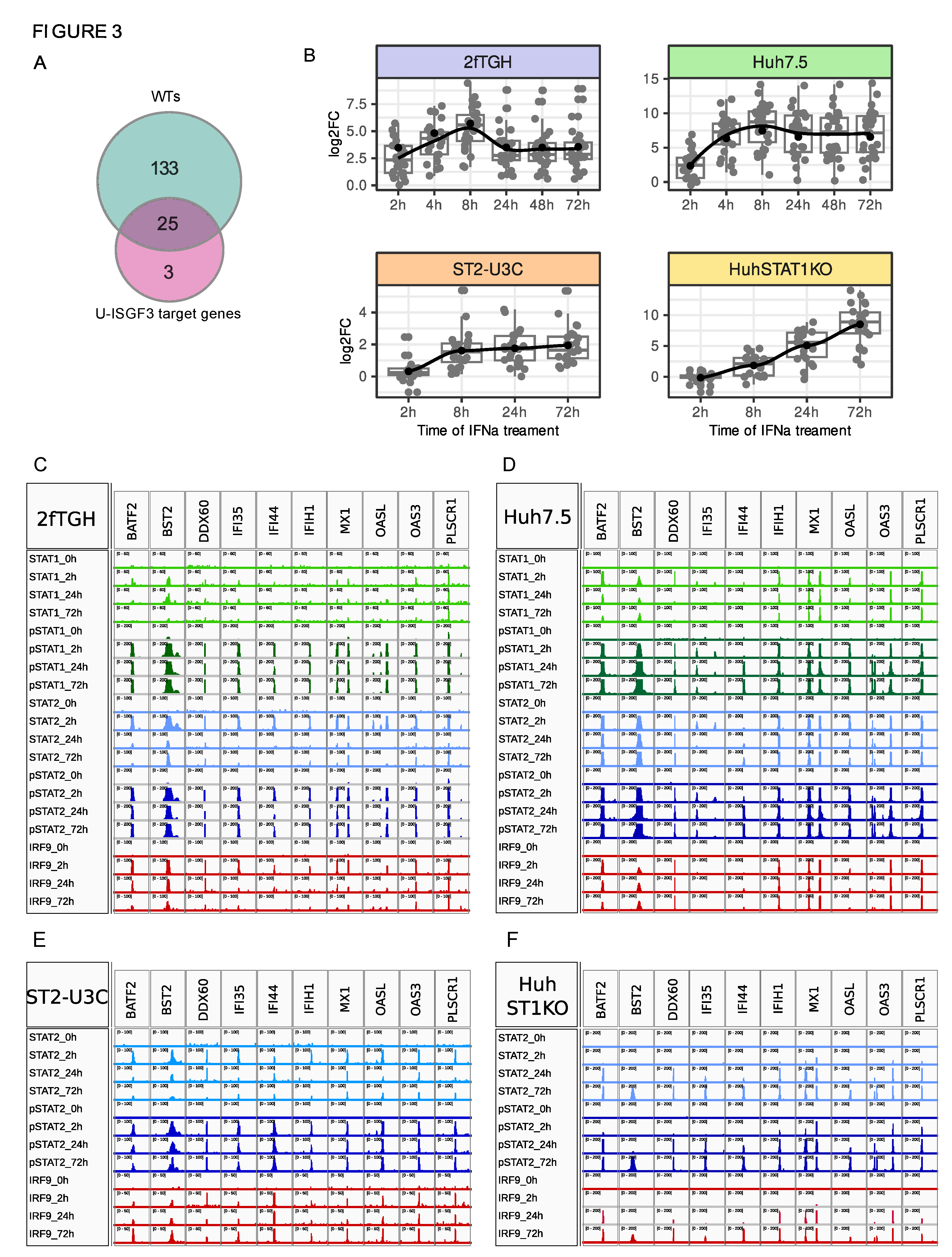
Figure 4.
Long-term ISG expression in the WT and STAT1-KO cell lines depends on the phosphorylation of STAT2 and/or STAT1. (A-D) Immunoblot results representing protein production and phosphorylation profiles in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO cells additionally treated or not with JAK Inhibitor I (JII) for 6h; p – phosphorylated, t - total; (E-H) qPCR results demonstrating expression profiles of selected ISGs in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO treated with IFNα and JII. Relative expression over GAPDH was estimated; n = 2; mean ± SEM.
Figure 4.
Long-term ISG expression in the WT and STAT1-KO cell lines depends on the phosphorylation of STAT2 and/or STAT1. (A-D) Immunoblot results representing protein production and phosphorylation profiles in IFNα-treated 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO cells additionally treated or not with JAK Inhibitor I (JII) for 6h; p – phosphorylated, t - total; (E-H) qPCR results demonstrating expression profiles of selected ISGs in 2fTGH, Huh7.5, ST2-U3C and Huh STAT1KO treated with IFNα and JII. Relative expression over GAPDH was estimated; n = 2; mean ± SEM.
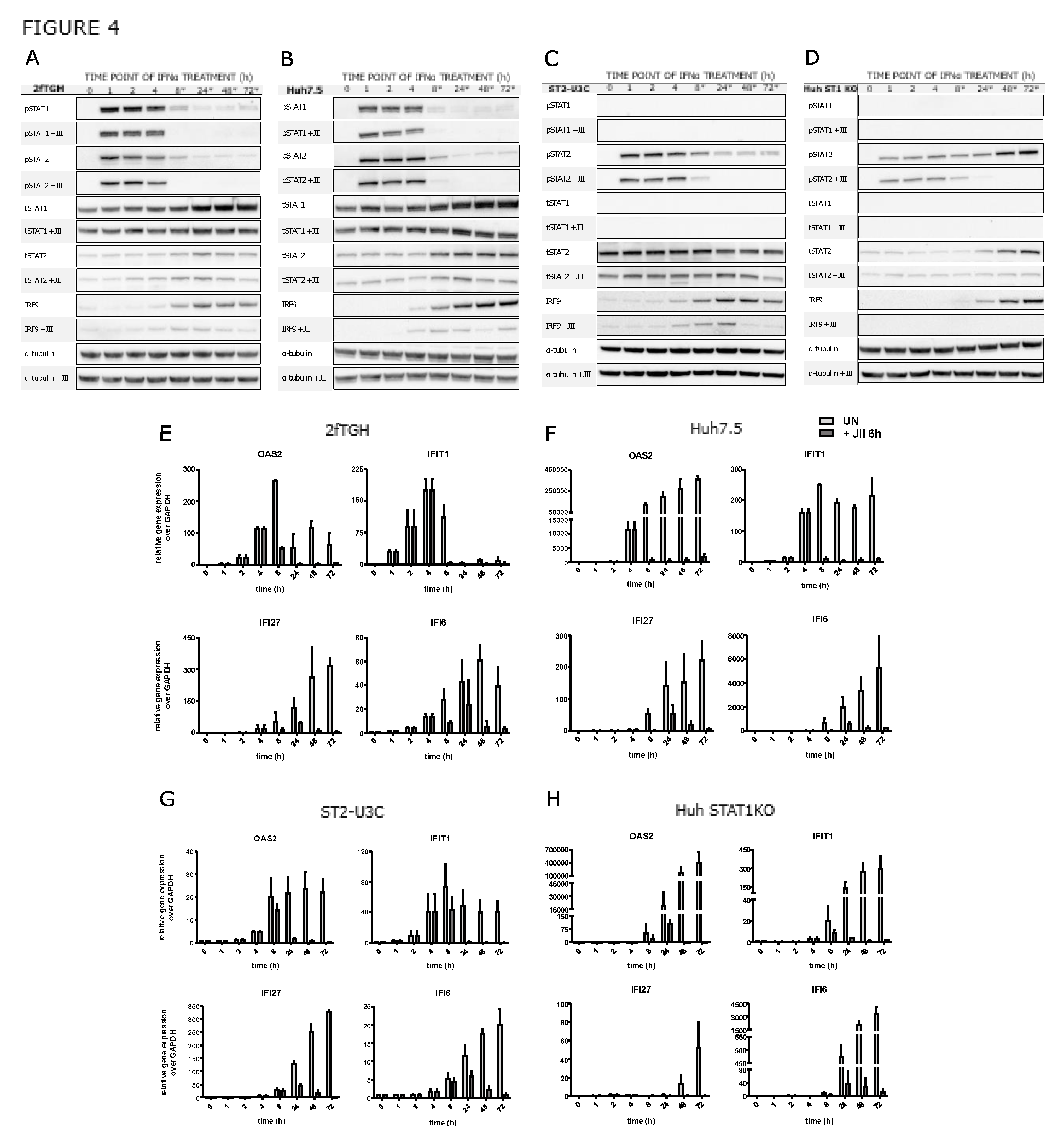
Figure 5.
Overexpression of STAT1, STAT2 and IRF9 is sufficient to drive transcriptional responses and viral protection in the absence of IFNα treatment. (A) Venn diagram based on RNA-Seq results demonstrating commonly upregulated ISGs in ST1-ST2-IRF9-U3C vs. WTs (2fTGH vs. Huh7.5). (B) Gene Ontology terms enrichment analysis of the 43 commonly upregulated genes in ST1-ST2-IRF9-U3C and WT cells, significant enrichment considered with FDR < 0.05; (C) Representative ISG expression levels in untreated ST1-ST2-IRF9-U3C vs. untreated U3C based on RNA-Seq results (upper panel, log2FC > 0.5; padj < 0.05) validated by qPCR (lower panel, n = 2; mean ± SEM). (D) Phosphorylation independence of ISG expression studied by treatment with IFNα and JAK Inhibitor I (JII) demonstrated by using qPCR, n = 2; mean ± SEM; (E) Chromatin interactions of STAT1, STAT2 and IRF9 with regulatory regions of selected ISGs examined by ChIP-PCR, n = 2; mean ± SEM. (F) Ability of combating viral infection of JII-treated ST1-ST2-IRF9-U3C cells (and U3C and 2fTGH as a control) in the absence of IFNα, examined using antiviral assay.
Figure 5.
Overexpression of STAT1, STAT2 and IRF9 is sufficient to drive transcriptional responses and viral protection in the absence of IFNα treatment. (A) Venn diagram based on RNA-Seq results demonstrating commonly upregulated ISGs in ST1-ST2-IRF9-U3C vs. WTs (2fTGH vs. Huh7.5). (B) Gene Ontology terms enrichment analysis of the 43 commonly upregulated genes in ST1-ST2-IRF9-U3C and WT cells, significant enrichment considered with FDR < 0.05; (C) Representative ISG expression levels in untreated ST1-ST2-IRF9-U3C vs. untreated U3C based on RNA-Seq results (upper panel, log2FC > 0.5; padj < 0.05) validated by qPCR (lower panel, n = 2; mean ± SEM). (D) Phosphorylation independence of ISG expression studied by treatment with IFNα and JAK Inhibitor I (JII) demonstrated by using qPCR, n = 2; mean ± SEM; (E) Chromatin interactions of STAT1, STAT2 and IRF9 with regulatory regions of selected ISGs examined by ChIP-PCR, n = 2; mean ± SEM. (F) Ability of combating viral infection of JII-treated ST1-ST2-IRF9-U3C cells (and U3C and 2fTGH as a control) in the absence of IFNα, examined using antiviral assay.
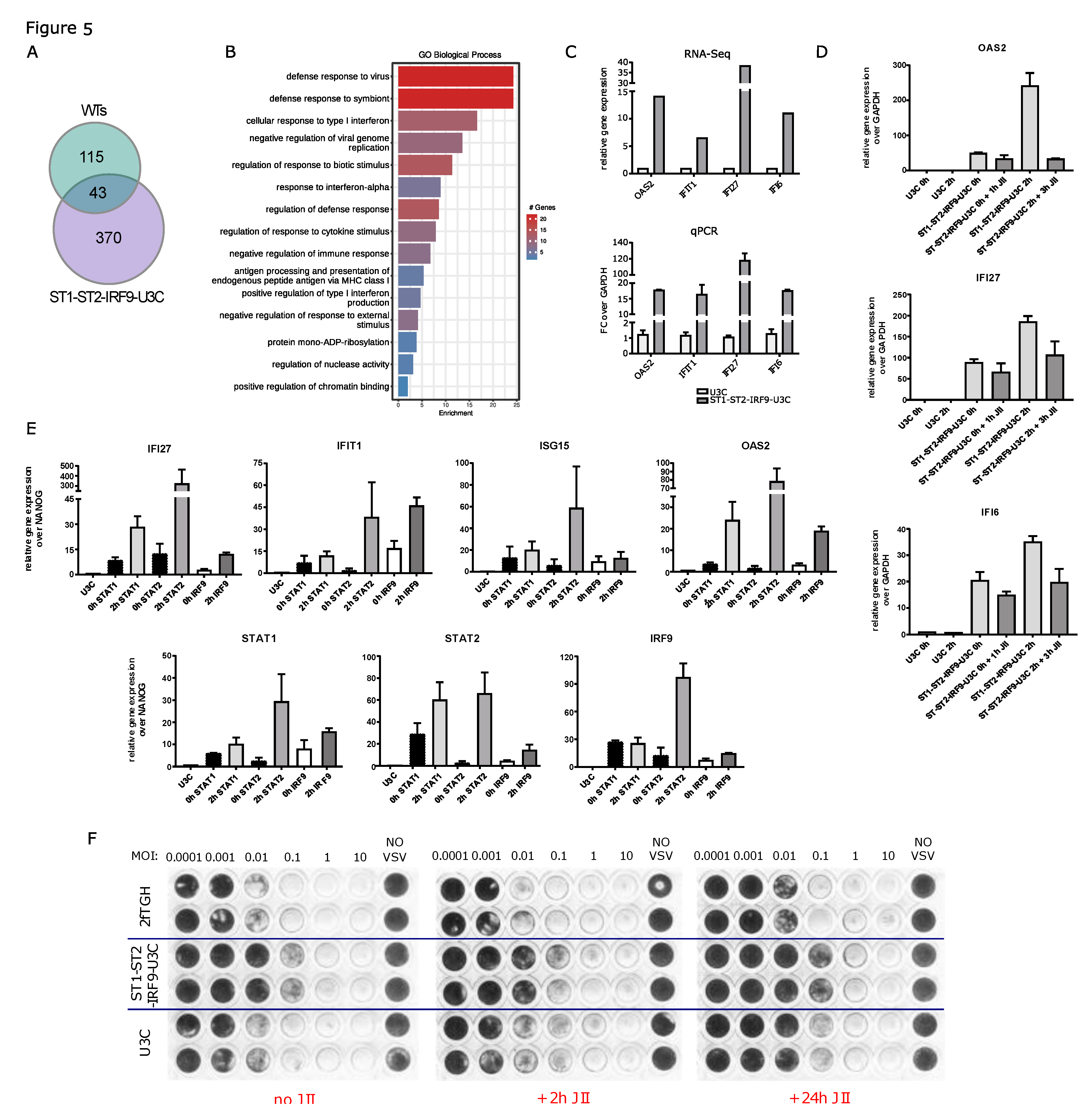
Figure 6.
Long-term ISG expression upon abundance of STAT1, STAT2 and IRF9 depends on phosphorylation process. (A) Immunoblot results representing protein production and phosphorylation profiles in the IFNα-treated ST1-ST2-IRF9-U3C additionally treated or not with JAK Inhibitor I (JII) for 6h. (B-E) qPCR results demonstrating expression profile of selected ISGs in ST1-ST2-IRF9-U3C and 2fTGH treated with IFNα and JII. Relative expression over GAPDH was estimated; n = 2; mean ± SEM. (F) Ability to combating viral infection of the IFNα- and JII-treated ST1-ST2-IRF9-U3C cells (and U3C and 2fTGH as a control) examined using antiviral assay, (G) validated by examination of selected ISG expression profiles using qPCR, n = 2; mean ± SEM.
Figure 6.
Long-term ISG expression upon abundance of STAT1, STAT2 and IRF9 depends on phosphorylation process. (A) Immunoblot results representing protein production and phosphorylation profiles in the IFNα-treated ST1-ST2-IRF9-U3C additionally treated or not with JAK Inhibitor I (JII) for 6h. (B-E) qPCR results demonstrating expression profile of selected ISGs in ST1-ST2-IRF9-U3C and 2fTGH treated with IFNα and JII. Relative expression over GAPDH was estimated; n = 2; mean ± SEM. (F) Ability to combating viral infection of the IFNα- and JII-treated ST1-ST2-IRF9-U3C cells (and U3C and 2fTGH as a control) examined using antiviral assay, (G) validated by examination of selected ISG expression profiles using qPCR, n = 2; mean ± SEM.

Table 1.
Genes commonly upregulated in 2fTGH vs. Huh7.5 (WTs, log2FC>=1) and ST1-ST2-IRF9-U3C (log2FC>=0,5).
Table 1.
Genes commonly upregulated in 2fTGH vs. Huh7.5 (WTs, log2FC>=1) and ST1-ST2-IRF9-U3C (log2FC>=0,5).
| Gene name | Log2FC | padj |
|---|---|---|
| APOL1 | 1,179548695 | 0,004684412 |
| APOL6 | 1,004077196 | 0,038984999 |
| BST2 | 6,135203905 | 1,35927E-50 |
| C1R | 1,408098465 | 0,031410471 |
| CASP1 | 1,910053199 | 0,015147452 |
| DDX60 | 1,821430803 | 0,000316359 |
| DENND2D | 2,789095461 | 0,048039125 |
| DTX3L | 1,352686939 | 0,000116872 |
| EIF2AK2 | 1,52332203 | 3,34411E-07 |
| ERAP1 | 0,965079469 | 0,000499671 |
| ERAP2 | 1,130778979 | 0,003692999 |
| HELZ2 | 0,96070728 | 0,014920517 |
| HERC6 | 1,618186178 | 0,000600321 |
| HLA-B | 1,178511842 | 0,001660318 |
| HLA-C | 1,456434161 | 1,75465E-07 |
| IFI27 | 4,788538368 | 0,000413159 |
| IFI6 | 4,034905117 | 3,7793E-25 |
| IFIT1 | 3,212109227 | 1,06397E-13 |
| IFIT2 | 2,829436266 | 5,97437E-05 |
| IFIT3 | 1,906823009 | 4,21176E-05 |
| IFITM1 | 2,733004839 | 5,38304E-20 |
| IFITM3 | 0,752739796 | 0,007072815 |
| IRF9 | 7,842346026 | 1,87668E-80 |
| ISG15 | 1,562830472 | 2,90124E-05 |
| NRP2 | 0,870136135 | 0,002302707 |
| OAS1 | 1,471623318 | 0,002759109 |
| OAS2 | 4,639318222 | 1,20916E-26 |
| OAS3 | 0,941039617 | 0,028400103 |
| PARP10 | 1,009511582 | 0,001614949 |
| PARP14 | 1,66043833 | 3,31686E-08 |
| PARP9 | 1,619836156 | 4,81902E-05 |
| PDGFRL | 2,314318252 | 0,013940482 |
| RTP4 | 3,339158993 | 0,001009013 |
| SAMHD1 | 1,619836156 | |
| STAT1 | 3,374870457 | 2,50965E-06 |
| STAT2 | 5,052303916 | 1,9208E-92 |
| THEMIS2 | 1,173305564 | 0,035610056 |
| TRANK1 | 2,347593899 | 1,32527E-06 |
| UBA7 | 1,029286496 | 0,013001475 |
| UBE2L6 | 1,165570135 | 0,00053531 |
| USP18 | 3,111223801 | 4,5583E-06 |
| XAF1 | 3,694263817 | 1,24333E-05 |
| ZBTB42 | 1,070772287 | 0,031039847 |
Table 2.
qPCR primers.
| Gene name | Primer sequence | |
| Forward | Reverse | |
| GAPDH | CAATATGATTCCACCCATGGCAA | GATCTCGCTCCTGGAAGATGG |
| IFI27 | GTCACTGGGAGCAACTGGAC | GGGCAGGGAGCTAGTAGAAC |
| IFI6 | ATCCTGAATGGGGGCGG | AGATACTTGTGGGTGGCGTAG |
| OAS2 | CAATCAGCGAGGCCAGTAAT | TCCAGGTTGGGAGAAGTCAA |
| IFIT1 | CTTGCAGGAAACACCCACTT | CCTCTAGGCTGCCCTTTTGT |
Table 3.
ChIP-PCR primers.
| Gene name | Primer sequence | |
| Forward | Reverse | |
| NANOG | TGGTAGACGGGATTAACTGAG | GAAGGCTCTATCACCTTAGA |
| OAS2 | CGCTGCAGTGGGTGGAGAGA | GCCGGCAAGACAGTGAATGG |
| IFI27 | CTTCTGGACTGCGCATGAGG | CCACCCCGACTGAAGCACTG |
| IFIT1 | GCAGGAATTCCGCTAGCTTT | GCTAAACAGCAGCCAATGGT |
| ISG15 | AGGGAAACCGAAACTGAAGC | TGAGGCACACACGTCAGG |
| STAT1 | CGCTCAGCCAATTAGACGC | GTAAACAGAACGCCAGTTCCC |
| STAT2 | TGTCACCAAGCAGGCTGTC | TCTGTTCTGTTAGGCTCAGGC |
| IRF9 | AGATGCTGCTGCCCTCTAGT | CCCCTTTCTACAGTCCCCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



