
Submitted:
24 October 2023
Posted:
26 October 2023
You are already at the latest version
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly termed nonalcoholic fatty liver disease (NAFLD), is a widespread global health concern that affects around 25% of the global population. Its influence is expanding, and it is anticipated to overtake alcohol as the leading cause of liver failure and liver-related death worldwide. Unfortunately, there are no approved therapies for MASLD; as such, national and international regulatory health agencies undertook strategies and action plans designed to expedite the development of drugs for treatment of MASLD. A sedentary lifestyle and an unhealthy diet intake are important risk factors. Western countries have a greater estimated prevalence of MASLD partly due to lifestyle habits. Mitochondrial dysfunction is strongly linked to the development of MASLD. Further, it has been speculated that mitophagy, a type of mitochondrial quality control, may be impaired in MASLD. Thyroid hormone (TH) coordinates signals from the nuclear and mitochondrial genomes to control mitochondrial biogenesis and function in hepatocytes. Mitochondria are known TH targets and preclinical and clinical studies suggest that TH, thyroid receptor β (TR-β) analogs, and synthetic analogs specific to liver could be of therapeutic benefit in treating MASLD. In this review, we highlight how mitochondrial dysfunction contributes to development of MASLD, and how understanding the role of TH in improving mitochondrial function paved the way for innovative drug development programs of TH-based therapies targeting MASLD.
Keywords:
Liver
; Mitochondrial dysfunction
; FAO
; MASLD
; Thyroid
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) is the new term replacing nonalcoholic fatty liver disease (NAFLD). It is the most prevalent chronic liver disease in the Western world, paralleling the steady rise in the prevalence of obesity and its associated metabolic disorders such as hyperlipidemia and type 2 diabetes [1]. Epidemiological studies reported that MASLD affects ~ 25% of the world's population [2,3]. In many patients with MASLD, the accumulation of toxic amounts of lipids is accompanied by liver injury caused by oxidative stress characterized histologically by hepatocyte injury (ballooning) and foci of inflammation in the surrounding liver tissue. Histologically, the combination of steatosis, in addition to hepatocyte ballooning and/or inflammation, is called metabolic dysfunction-associated steatohepatitis (MASH), previously termed nonalcoholic steatohepatitis (NASH) [4]. Fibrosis is an important endpoint in MASLD/MASH. Progression to cirrhosis and development of hepatocellular cancer (HCC) are the most feared liver-related complications of MASH [5]. In fact, MASH is projected to be the leading indication for liver transplantation, surpassing other competing etiologies [6]. Further, MASH is the fastest growing cause of HCC in the United States [7]. There are several pathophysiological mechanisms that have been described and implicated in the pathogenesis and progression of MASH. Insulin resistance (IR) is an established risk factor for the development and progression of MASLD. Studies have shown that IR due to chronic nutritional overload renders hepatocytes more susceptible to oxidative stress and mitochondrial dysfunction, which can enhance inflammation and promote severe liver damage [8,9].
Published reports from our group and others document that hepatic mitochondrial dysfunction plays a key role in the progression of MASLD [10,11,12,13,14]. Mitochondrial dysfunction is characterized by various levels of structural damage within the mitochondria, ATP depletion, increased permeability in both the outer and inner mitochondrial membranes, reduced respiratory chain activity, excess production of reactive oxygen species (ROS), and the consequent deleterious deletions in mitochondrial DNA (mtDNA) due to oxidative stress [15]. The metabolic changes seen in MASLD are commonly linked to mitochondrial dysfunction in the hepatocytes. The mitochondrial quality control (MQC) system intricately controls mitophagy, proteostasis, biogenesis, dynamics, and other processes that are essential for maintaining cellular homeostasis. Mitochondrial dysfunction due to failure of MQC is one of the known factors known to perpetuate MASLD [16,17].
Thyroid hormone (TH) has an important role in many physiological processes, including homeostasis, mineral, lipid, carbohydrate, and protein metabolism. TH has an effect on nearly all organs in the body, with the liver representing one of the most important sites of TH action [18]. Low TH function can result in hypercholesterolemia, which is believed to be an important step in the pathogenesis of hypothyroidism induced MASLD [19]. TH regulates mitochondrial biogenesis and function in hepatocytes by synchronized nuclear and mitochondrial genome signals [20]. As previously stated, TH performs critical functions in energy and metabolic balance; therefore, it plays a role in the pathophysiology of MASLD. MASLD is closely related to hypothyroidism [21]. TH may play a role in the etiology of MASLD based on studies that suggest disturbances in cellular TH signaling cause MASLD [18,22,23]. This review focuses on the role of TH in mitochondrial dysfunction and potential therapeutic implications of TH in MASLD.
2. Mitochondrial Dysfunction and MASLD
Mitochondria account for up to 18% of a hepatocyte's total volume and carry out vital functions in the hepatic metabolic processes to generate energy [24]. In addition to producing ATP and β-oxidation, mitochondria also generate reactive oxygen species (ROS) and control calcium signaling. Studies have repeatedly shown that impaired mitochondria contribute to the development and progression of MASLD. These derangements include decreased β-oxidation, electron transport chain (ETC) defects, decreased ATP levels, increased ROS generation, cellular damage caused by oxidative stress, and structural alterations in mitochondria [25,26,27]. The alternations in mitochondrial structure and function accentuate the accumulation of lipids in the liver, triggering inflammation and fibrogenesis, and thus contribute to the progression of MASLD [9,25]. During the early stages of MASLD, mitochondria appear to adjust to increased substrate consumption in both humans and mice by boosting β-oxidation, mitochondrial respiration, and ketogenesis [28,29]. However, as MASH progresses, this adaptive response fades, resulting in mitochondrial dysfunction characterized by inefficient β-oxidation, impaired ketogenesis, decreased ATP generation, and electron transport chain leakage [30]. Recent research has established a link between mitochondrial dysfunction and both cell apoptosis and the activation of the inflammasome [31].
2.1. ROS and MASLD
Mitochondrial dysfunction results in decreased oxidative phosphorylation and excess ROS production. Increased ROS can cause oxidative stress by oxidizing proteins and peroxidizing mitochondrial membranes, resulting in mitochondrial dysfunction via reduced respiratory chain activity leading to mitochondrial DNA (mtDNA) damage [32]. Furthermore, excessive ROS production has been shown to increase the influx of cytochrome C and other proapoptotic substances through the mitochondrial permeability transition (MPT) channels, resulting in hepatocyte death, a hallmark feature of MASH progression [33,34]. Moreover, previous studies suggest that ROS and lipid peroxidation may enhance TGF-β synthesis in Kupffer cells, activating hepatic stellate cells and increasing the development of collagen-producing myofibroblasts. Collectively, such pathological changes have been shown to promote hepatic fibrosis and, in extreme situations, liver cirrhosis [35]. Further, it has been postulated that ultrastructural abnormalities in mitochondria and an imbalance in mitochondrial dynamics are associated with more severe MASLD/MASH. Failure to eliminate damaged mitochondria can result in an accumulation of defective mitochondria; as such, the liver's ability to restore normal mitochondrial function deteriorates over time, leading to hepatocyte demise and further progression of MASH [36].
2.2. Impaired Mitochondrial Quality Control (MQC) and MASLD
MQC involves several processes such as fission, fusion, biogenesis and mitophagy. When exposed to oxidative stress, mitochondria use mechanisms such as antioxidants, DNA repair, protein folding and degradation to maintain their function. Mitochondrial biogenesis, fusion, and fission all serve to compensate for mitochondrial dysfunction in normal physiological and pathological states [37,38]. In the event of cellular injury, mitochondria can be repaired by fusing with healthy counterparts, whereas severely damaged mitochondria undergo fission and eventually are degraded by mitophagy [39,40]. The onset of MASLD is greatly impacted by mitochondrial dysfunction caused by MQC failure [11]. Mitophagy, or mitochondrial autophagy, is an important preventative mechanism against the development and progression of MASLD, as it is responsible for eliminating damaged mitochondria that are highly concentrated inside the cytosol. Mitophagy disorders, on the other hand, have been reported in both MASLD mice models and patients with MASLD. Studies have shown that defects in PINK1 or Parkin cause defective mitochondrial engulfment, thereby worsening MASLD [41]. The inactivation of mitofusin 2 (Mfn2) activity caused by inflammation can have additional deleterious effects on mitophagy, thereby reducing the production of autophagosomes, increasing hepatic steatosis, and accelerating the progression of MASH [42]. Collectively, these data underscore that mitochondrial activity and antioxidant levels in the liver are critical in the pathogenesis of MASLD. A recent study has reported that defective mitochondria is associated with greater risk of MASLD development in those with obesity. In this study, increased formation of ROS in the liver and decreased MQC were shown to be associated with significant decrease in β-oxidation in ~ 50% of patients with MASH [14]. Mitophagy is influenced by hormonal factors. TH has been shown to reduce the severity of MASLD by increasing FAO and stimulating mitophagy and mitochondrial biogenesis [43,44]. TH has also been shown to increase the expression of BNIP3, NIX, ULK1, p62, and LC3 mRNA expression [45]. Mitophagy and mitochondrial biogenesis work synergistically to fine-tune the mitochondrial homeostasis, enabling cells to modify their mitochondrial composition in accordance with cellular metabolic status, stress, and various signals originating from the intracellular environment and hormones.
3. Hypothyroidism and MASLD
Hypothyroidism affects 0.2% to 5.3% of the US and European population. It is characterized by an increase in thyroid-stimulating hormone (TSH) levels and a reduction in thyroxine and triiodothyronine hormones. Clinical or overt hypothyroidism occurs when these alterations emerge as hypothyroidism-related symptoms. Subclinical hypothyroidism occurs when TSH levels are increased but TH levels remain normal [46]. It is well-established that hypothyroidism is associated with hypometabolism, commonly manifested as an increase in body weight, decrease in basal metabolic rate, gluconeogenesis, and lipolysis. TH impairment can result in metabolic disorders such as obesity, low lipid metabolism, and insulin resistance, which are commonly associated with MASLD [47]. Both clinical and subclinical hypothyroidism have been linked to MASLD [48]. Several factors contribute to the progression of MASLD in subclinical hypothyroidism, including direct TSH-hepatocyte interaction via the TSH receptor on the cell membrane, which influences hepatic triglyceride metabolism, resulting in increased hepatic lipogenesis, which is accomplished by increasing SREBP-1c activity in response to TSH receptor stimulation [49]. In addition, reduced TH levels cause decreased glucose-sensing receptors in pancreatic β cells, which results in decreased insulin secretion. This results in increased lipolysis in adipose tissue with increased hepatic FFA influx [50,51,52].
Hyperlipidemia in hypothyroidism has been thought to be due to inadequate lipid metabolism due to decreased number of low-density lipoprotein (LDL) receptors on hepatic cell and an increase in intestinal cholesterol absorption [53]. Increased total and LDL cholesterol levels are thus seen in hypothyroid individuals. MASLD caused by hypothyroidism may occur as a result of increased triglyceride accumulation in the hepatic tissue [51,54]. Lipid accumulation causes oxidative stress and inflammatory reactions within the liver [48]. Leptin has also been implicated in the thyroid-liver complex interactions. Leptin levels are elevated in hypothyroid individuals as well as in MASLD patients [23]. Leptin increases insulin resistance in liver and can contribute to fibrogenesis [23]. Due to mitochondrial dysfunction, individuals with hypothyroidism are also more vulnerable to increased oxidative stress [55,56]. Hypothyroidism is a significant risk factor for MASLD and it has been shown to be associated with impaired glucose and insulin metabolism [57]. Hypothyroid patients have elevated oxidative stress markers; thus, oxidative stress could be the source of hepatocellular damage by decreasing FAO and increasing lipid peroxidation [50,58]. Collectively, these data suggest that TH therapy could be of benefit in hypothyroidism induced MASLD, and therapeutic application of TH (or its analogs) in the treatment of MASLD/MASH offers tremendous promise.
4. Thyroid Hormone and MASLD: From Underlying Mechanisms to Therapeutic Implications
TH regulates multiple metabolic activities within cells linked to metabolism and breakdown of macromolecules, such as carbohydrates, proteins, lipids, and damaged cellular organelles, to maintain homeostasis in various conditions [59,60]. TH plays an important role in hepatic lipid metabolism [61]. According to published reports, individuals with obesity are more likely to have hypothyroidism than those with a normal BMI [62,63,64]. These findings strengthen the hypothesis that TH therapy might be of therapeutic benefit in patients with MASLD with or without hypothyroidism [51].
4.1. Mechanisms of Action of TH
The thyroid hormone receptor (TR), a nuclear receptor, controls T3 activity by acting as a T3-inducible transcription factor. TR has two main isoforms: TRα and TRβ, and its expression varies by tissue. TRα receptor is commonly found in heart, brain, and bone, whereas TRβ is predominantly found in the liver and kidney. TR interacts with thyroid hormone response elements (TREs) in target gene regulatory domains as a heterodimer with another nuclear receptor, the retinoid X receptor (RXR). TR's recruitment of coregulator proteins regulates target gene expression. The TR/RXR heterodimer binds nuclear receptor corepressor and silencing mediator of retinoid and TR to inhibit gene transcription via histone deacetylation in the absence of T3. Coactivators are enrolled when T3 is present, whereas corepressors are dismissed, and TH-responsive gene expression occurs [65].
4.2. TH and Its Isoform
THs are synthesized and secreted by the thyroid gland and are required for the control of numerous metabolic processes. The thyroid gland utilizes thyroid follicles as fundamental structures to concentrate iodide and produce the primary THs, namely 3,3',5,5'-tetraiodo-L-thyronine (T4) and 3,5,3'-triiodo-L-thyronine (T3) [66]. The anterior pituitary's thyrotrophs, which release thyroid-stimulating hormone (TSH), regulate TH (mainly T4) secretion from the thyroid gland. By synthesizing and releasing iodinated THs into the bloodstream, the thyroid controls numerous physiological processes in the liver, adipose tissue, central nervous system, cardiovascular system, and musculoskeletal system [67]. Iodothyronine deiodinases (DIO1, DIO2, and DIO3) in extrathyroidal tissue regulate T4 to T3 conversion. Both DIO1 and DIO2 convert circulating T4 to the bioactive TH form, i.e., T3. DIO3 reduces intracellular thyroid by converting T4 and T3 to reverse T3 (rT3) and T2 [68]. Recent studies suggest that T2 has tissue specific TH activity [18,69].
T3, the most active form of TH, binds to two nuclear hormone receptor isoforms (TRα and TRβ). These receptors function as ligand-inducible transcription factors that interact with TH response elements (TREs) found in target gene promoters, enhancers, and intronic regions [59,70]. TRα is the predominant isoform found in the heart, brain, and bone, whereas TRβ is present dominantly in the liver, accounting for more than 90% of TRs in this tissue [60]. TR subtypes are encoded by two genes. The first gene, TRα, is linked to the c-erbA gene on chromosome 17 [71]. The transcription of C-erbA produces three mRNAs: one full-length TRα1, and the other two variants that code for proteins which do not bind with TH [72,73]. The second gene THRβ, is located on chromosome 3 and shares DNA with c-erbA. TRα1 mRNA contains numerous alternative start sites, resulting in the translation of three shorter isoforms (p43, p30, and p28) based on their kilo dalton sizes (Figure 1) [71,74,75]. The mitochondrion is a primary site of TH accumulation within the cell [76,77,78]. TR1α p43 is directed to the mitochondrial matrix, whereas TR1α p28 is particularly directed to the mitochondrial inner membrane [76,79,80].
4.3. TH and MASLD
TH has been linked to the pathogenesis of MASLD; reduced TH levels have been frequently reported in MASLD patients [90]. According to animal studies, moderate hypothyroidism has been shown to be associated with a higher risk of MASLD [81]. Translational studies of liver transcriptomes from individuals with MASLD undergoing bariatric surgery revealed a decrease in the expression of genes involved in RNA metabolism, protein catabolism, and energy metabolism. These genes, which are controlled by THs under normal physiological conditions, have been shown to have lower expression levels in individuals with MASLD [82]. In rats and humans, lower intrahepatic TH levels in MASLD has been reported [83,84]. TH not only increases de novo lipogenesis (DNL) and improves hepatic insulin sensitivity, but also decreases hepatic gluconeogenesis in hepatocytes. Furthermore, TH promotes lipid export and oxidation [85,86]. The regulation of lipid and glucose metabolism is carried out by the TH receptors, which have direct and indirect effects by interacting with other nuclear receptors such as the peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR), and bile acid signaling pathways [85]. Several observational studies reported a link between increased serum TSH levels and the presence and severity of MASLD [22,87]. According to recently published study, the regulation of hepatic autophagy and mitochondrial metabolism by TH have been described as crucial steps in hepatic triglyceride metabolism [88,89].
4.3.1. TH and FAO
TH has been shown to increase fatty acid import, FAO and oxygen uptake in isolated mitochondria [90]. Previous work suggests that T2 treatment increased FAO when palmitoyl-CoA was utilized as a substrate rather than palmitoyl-carnitine, suggesting that carnitine-palmitoyltransferase 1 (CPT1) could be a potential T2 target, which was further verified by assessing CPT activity [91]. In isolated mitochondria, TH treatment increased the activity of mitochondrial thioesterase, an enzyme responsible for converting acyl-CoA to fatty acid and CoA [92]. We have recently shown that low dose T3 treatment in mice increased FAO in both Chow- and western diet fed-animals [93]. The ETC and the tricarboxylic acid (TCA) cycle also play key roles in FAO [89,94], and CPT1 levels are increased indirectly by TH via surtuin 1 (SIRT1) and PPARα [85,95]. TH further increases the amounts of other mitochondrial enzymes required for FAO such as medium-chain acyl-CoA dehydrogenase (MCAD), pyruvate dehydrogenase kinase, and mitochondrial uncoupling protein 2 (UCP2) [96,97,98]. T2 treatment has been shown to promote hepatic FAO in liver mitochondria, increase downstream respiratory activity, increase proton leak, and reduce oxidative stress in the liver mitochondria without causing thyrotoxicity [99,100].
4.3.2. TH and Mitochondrial Biogenesis
TH exerts multiple actions at a molecular level aimed at increasing the number of mitochondria. T3 promotes mitophagy and mitochondrial biogenesis via peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) [45,101]. TH stimulates mitochondrial biogenesis by inducing PGC1α gene expression which stimulates the transcription of nuclear respiratory factor 1 (NRF1) and mitochondrial transcription factor A (mtTFA) [20]. SIRT1 is activated by TH, which deacetylates PGC1α and increases its ability to bind the regulatory areas of mitochondrial synthesis and function genes [95]. Further, T3 also increases the expression and activation of Unc-51-like autophagy activating kinase 1 (ULK1), which improves dynamin-related protein 1 (DRP1)-mediated mitochondrial fission, activation and association of FUN14 domain-containing 1 (FUNDC1) with LC3B, and p62 translocation to mitochondria in hepatic cells [101,102]. T3-mediated mitophagy activation is required for mitochondrial oxidative phosphorylation system (OXPHOS) stimulation [101]. Additionally, T3 promotes mitochondrial biogenesis [102] and increases the rates of mitophagy and mitochondrial synthesis, both of which contribute to enhanced mitochondrial activity and fatty acid β-oxidation.
4.3.3. TH and Mitophagy
To minimize cellular harm caused by reactive oxygen species (ROS), TH induces protective autophagy of mitochondria, a process called mitophagy [101]. Mitophagy is initiated by excessive ROS production from mitochondria, leading to the release of intracellular Ca2+, activation of calcium/calmodulin-dependent protein kinase 2 (CAMMK2), phosphorylation of AMP-activated protein kinase (AMPK), and subsequent activation and translocation of ULK1 to the mitochondria after AMPK phosphorylation [101]. Elevated ROS levels are perpetuators of mitophagy, which ensures the preservation of mitochondrial quality required for β-oxidation of fatty acids and oxidative phosphorylation. Mitophagy induced by ROS generation selectively sequesters damaged mitochondria to be removed from the cell, preventing additional oxidative damage and cell death [101].
4.4. Potential Therapeutic Use of TH and Its Analogs in MASLD
In recent years, THs as well as the TR-β agonists and additional liver specific analogs have been studied as a potential MASLD treatment [22]. Research from previous studies reported the use of T3 to promote weight loss in patients with obesity and to treat hypercholesterolemia [103]. T3 injections given daily intraperitoneally (ip) to ob/ob mice have been found to decrease both body weight and fat while increasing oxygen intake and oxidative metabolism [104]. GC-1, a novel TR-β agonist, has been reported to reduce the development of hepatic steatosis and lipid peroxidation in mice [43] and decrease hepatic TG levels with no major side effects [105,106]. MB07811, another orally administered TR-β agonist, has been shown to prevent hepatic steatosis in rats and mice via boosting β-oxidation and mitochondrial respiration rates, lowering hepatic TG levels and stimulating CPT1α expression [44]. Resmetirom (MGL-3196) has been shown to be effective in lowering hepatic TG, lipid peroxidation, ALT, steatosis, inflammation, and fibrosis in animal models [107,108]. Increased mitochondrial β-oxidation has been suggested to be one of the mechanisms by which Resmetirom decreases liver fat [109]. VK2809 therapy has been shown to reduce hepatic lipid accumulation in a glycogen storage disease Ia (GSD1a) mouse model by restoring autophagy, mitochondrial biogenesis, and β-oxidation of fatty acids [110]. KB2115, a TR-β agonist, has been reported to decrease total and low-density lipoprotein (LDL) cholesterol levels in the blood and prevent the development of hepatic steatosis [111]. Ongoing research in our laboratory has demonstrated that low dose T3 is effective in increasing hepatic mitochondrial FAO and reversing MASLD in mice [112]. Based on these results, our group recently initiated a randomized double-blinded placebo-controlled clinical trial to test whether low dose thyroxine (T4) is effective in improving the histological features in Veterans with biopsy proven MASH (National Library of Medicine NCT05526144) [113]. Taken all together, the results of the above studies demonstrate that TH therapy might be an effective strategy in treating MASLD/MASH. The use of TH and its analogs in preclinical and clinical research is summarized in Table 1.
5. Conclusions
MASLD poses a considerable public health problem with major socioeconomic impact. The onset and progression of MASLD is a multifactorial process influenced by genetic, epigenetic, and environmental factors. The development of MASLD is directly linked to mitochondrial dysfunction, as mitochondria play an important role in the β-oxidation of FFAs and are the principle intracellular generators of ROS. Recent studies focusing on understanding the role of TH in hepatic lipid metabolism and defective autophagy, mitophagy, and mitochondrial function in health and disease have shed light on the role of TH and mitochondrial dysfunction in the development and progression of MASLD. TH therapy, TRβ1 analogs, and liver-specific synthetic analogs in preclinical models and preliminary studies in patients with MASLD have shown promise as safe and potentially useful strategies in the treatment of MASLD.
Author Contributions
Review of literature: RR, SAP, and AHA; Conception: JAI; Drafting the article: RR, JAI; Revision: RR, SAP, AHA, JAI; Critical revision and final editing: JAI; Final approval of the version to be Published: RR, SAP, AHA and JAI.
Funding
Supported by Veterans Administration Merit Review awards BX004710 and CX002436 to JAI.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
All authors declare no conflict of interest.
References
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The Natural History of Nonalcoholic Fatty Liver Disease: A Population-Based Cohort Study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat Rev Gastroenterol Hepatol 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat Med 2018, 24, 908–922. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between Nonalcoholic Fatty Liver Disease and Risk for Hepatocellular Cancer, Based on Systematic Review. Clin Gastroenterol Hepatol 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int J Mol Sci 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial Dysfunction-Related Lipid Changes Occur in Nonalcoholic Fatty Liver Disease Progression. J Lipid Res 2018, 59, 1977–1986. [Google Scholar] [CrossRef]
- Li, R.; Toan, S.; Zhou, H. Role of Mitochondrial Quality Control in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Aging (Albany NY) 2020, 12, 6467–6485. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice Heterozygous for a Defect in Mitochondrial Trifunctional Protein Develop Hepatic Steatosis and Insulin Resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef]
- Rector, R.S.; Morris, E.M.; Ridenhour, S.; Meers, G.M.; Hsu, F.F.; Turk, J.; Ibdah, J.A. Selective Hepatic Insulin Resistance in a Murine Model Heterozygous for a Mitochondrial Trifunctional Protein Defect. Hepatology 2013, 57, 2213–2223. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised Hepatic Mitochondrial Fatty Acid Oxidation and Reduced Markers of Mitochondrial Turnover in Human NAFLD. Hepatology 2022. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Zhang, H.X.; Guo, J.R.; Lam, C.W.K.; Wang, C.Y.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol Nutr Food Res 2019, 63, e1900043. [Google Scholar] [CrossRef]
- Vazquez-Calvo, C.; Suhm, T.; Büttner, S.; Ott, M. The Basic Machineries for Mitochondrial Protein Quality Control. Mitochondrion 2020, 50, 121–131. [Google Scholar] [CrossRef]
- Sheldon, R.D.; Meers, G.M.; Morris, E.M.; Linden, M.A.; Cunningham, R.P.; Ibdah, J.A.; Thyfault, J.P.; Laughlin, M.H.; Rector, R.S. eNOS Deletion Impairs Mitochondrial Quality Control and Exacerbates Western Diet-Induced NASH. Am J Physiol Endocrinol Metab 2019, 317, E605–E616. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Columbano, A.; Perra, A. Thyroid Hormones, Thyromimetics and Their Metabolites in the Treatment of Liver Disease. Front Endocrinol (Lausanne) 2018, 9, 382. [Google Scholar] [CrossRef]
- Chung, G.E.; Kim, D.; Kim, W.; Yim, J.Y.; Park, M.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S. Non-Alcoholic Fatty Liver Disease across the Spectrum of Hypothyroidism. J Hepatol 2012, 57, 150–156. [Google Scholar] [CrossRef]
- Weitzel, J.M.; Iwen, K.A. Coordination of Mitochondrial Biogenesis by Thyroid Hormone. Mol Cell Endocrinol 2011, 342, 1–7. [Google Scholar] [CrossRef]
- Lonardo, A.; Mantovani, A.; Lugari, S.; Targher, G. NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, M.; Han, B.; Qi, X. Association of Non-Alcoholic Fatty Liver Disease with Thyroid Function: A Systematic Review and Meta-Analysis. Dig Liver Dis 2018, 50, 1153–1162. [Google Scholar] [CrossRef]
- Mandato, C.; D’Acunzo, I.; Vajro, P. Thyroid Dysfunction and Its Role as a Risk Factor for Non-Alcoholic Fatty Liver Disease: What’s New. Dig Liver Dis 2018, 50, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Shum, M.; Ngo, J.; Shirihai, O.S.; Liesa, M. Mitochondrial Oxidative Function in NAFLD: Friend or Foe? Mol Metab 2021, 50, 101134. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Prasun, P.; Ginevic, I.; Oishi, K. Mitochondrial Dysfunction in Nonalcoholic Fatty Liver Disease and Alcohol Related Liver Disease. Transl Gastroenterol Hepatol 2021, 6, 4. [Google Scholar] [CrossRef]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol Metab 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA Cycle Function in the Pathology of Diet-Induced Hepatic Insulin Resistance and Fatty Liver. J Lipid Res 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Mansouri, A.; Fromenty, B. Nonalcoholic Steatosis and Steatohepatitis. V. Mitochondrial Dysfunction in Steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2002, 282, G193-9. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the Regulation of the NLRP3 Inflammasome. Trends Mol Med 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Pessayre, D. Role of Mitochondria in Non-Alcoholic Fatty Liver Disease. J Gastroenterol Hepatol 2007, 22 Suppl 1, S20-7. [Google Scholar] [CrossRef]
- Haouzi, D.; Lekéhal, M.; Moreau, A.; Moulis, C.; Feldmann, G.; Robin, M.A.; Lettéron, P.; Fau, D.; Pessayre, D. Cytochrome P450-Generated Reactive Metabolites Cause Mitochondrial Permeability Transition, Caspase Activation, and Apoptosis in Rat Hepatocytes. Hepatology 2000, 32, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Ricchelli, F.; Sileikytė, J.; Bernardi, P. Shedding Light on the Mitochondrial Permeability Transition. Biochim Biophys Acta 2011, 1807, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Robin, M.A.; Igoudjil, A.; Mansouri, A.; Pessayre, D. The Ins and Outs of Mitochondrial Dysfunction in NASH. Diabetes Metab 2004, 30, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 Controls Mitochondrial Etiology of Fatty Liver Diseases, Linking Defective Mitophagy to Steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial Quality Control: From Molecule to Organelle. Cell Mol Life Sci 2021, 78, 3853–3866. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, Y.; Gooz, M.; Li, L.; Lemasters, J.J.; Zhong, Z. Role of Mitochondrial Depolarization and Disrupted Mitochondrial Homeostasis in Non-Alcoholic Steatohepatitis and Fibrosis in Mice. Int J Physiol Pathophysiol Pharmacol 2019, 11, 190–204. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Edmunds, L.R.; Xie, B.; Mills, A.M.; Huckestein, B.R.; Undamatla, R.; Murali, A.; Pangburn, M.M.; Martin, J.; Sipula, I.; Kaufman, B.A.; et al. Liver-Specific Prkn Knockout Mice Are More Susceptible to Diet-Induced Hepatic Steatosis and Insulin Resistance. Mol Metab 2020, 41, 101051. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef]
- Perra, A.; Simbula, G.; Simbula, M.; Pibiri, M.; Kowalik, M.A.; Sulas, P.; Cocco, M.T.; Ledda-Columbano, G.M.; Columbano, A. Thyroid Hormone (T3) and TRbeta Agonist GC-1 Inhibit/Reverse Nonalcoholic Fatty Liver in Rats. FASEB J 2008, 22, 2981–2989. [Google Scholar] [CrossRef]
- Cable, E.E.; Finn, P.D.; Stebbins, J.W.; Hou, J.; Ito, B.R.; van Poelje, P.D.; Linemeyer, D.L.; Erion, M.D. Reduction of Hepatic Steatosis in Rats and Mice after Treatment with a Liver-Targeted Thyroid Hormone Receptor Agonist. Hepatology 2009, 49, 407–417. [Google Scholar] [CrossRef]
- Sinha, R.A.; Yen, P.M. Thyroid Hormone-Mediated Autophagy and Mitochondrial Turnover in NAFLD. Cell Biosci 2016, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Chaker, L.; Bianco, A.C.; Jonklaas, J.; Peeters, R.P. Hypothyroidism. Lancet 2017, 390, 1550–1562. [Google Scholar] [CrossRef]
- Notariza, K.R.; Wisnu, W. The Risk of Developing Non-Alcoholic Fatty Liver Disease in Adult Patients with Subclinical Hypothyroidism Compared to Euthyroid: An Evidence-Based Case Report. Acta Med Indones 2019, 51, 179–188. [Google Scholar] [PubMed]
- He, W.; An, X.; Li, L.; Shao, X.; Li, Q.; Yao, Q.; Zhang, J.A. Relationship between Hypothyroidism and Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front Endocrinol (Lausanne) 2017, 8, 335. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Q.; Lu, M.; Chen, W.; Song, Y.; Jing, F.; Guan, Y.; Wang, L.; Lin, Y.; Bo, T.; et al. Thyrotropin Increases Hepatic Triglyceride Content through Upregulation of SREBP-1c Activity. J Hepatol 2014, 61, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in Endocrine Diseases. Endocr Connect 2021, 10, R52–R65. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Mantovani, A.; Nascimbeni, F.; Lugari, S.; Targher, G. Pathogenesis of Hypothyroidism-Induced NAFLD: Evidence for a Distinct Disease Entity? Dig Liver Dis 2019, 51, 462–470. [Google Scholar] [CrossRef]
- Liebe, R.; Esposito, I.; Bock, H.H.; Vom Dahl, S.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and Management of Secondary Causes of Steatohepatitis. J Hepatol 2021, 74, 1455–1471. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.N. Update in Lipid Alterations in Subclinical Hypothyroidism. J Clin Endocrinol Metab 2012, 97, 326–333. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Gusdon, A.M.; Qu, S. Cross-Talk between the Thyroid and Liver: A New Target for Nonalcoholic Fatty Liver Disease Treatment. World J Gastroenterol 2013, 19, 8238–8246. [Google Scholar] [CrossRef] [PubMed]
- Nanda, N.; Bobby, Z.; Hamide, A. Inflammation and Oxidative Stress in Hypothyroids: Additive Effects on Cardiovascular Risk. Indian J Physiol Pharmacol 2011, 55, 351–356. [Google Scholar]
- Baskol, G.; Atmaca, H.; Tanriverdi, F.; Baskol, M.; Kocer, D.; Bayram, F. Oxidative Stress and Enzymatic Antioxidant Status in Patients with Hypothyroidism before and after Treatment. Exp Clin Endocrinol Diabetes 2007, 115, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Kizivat, T.; Maric, I.; Mudri, D.; Curcic, I.B.; Primorac, D.; Smolic, M. Hypothyroidism and Nonalcoholic Fatty Liver Disease: Pathophysiological Associations and Therapeutic Implications. J Clin Transl Hepatol 2020, 8, 347–353. [Google Scholar] [CrossRef]
- Lugari, S.; Mantovani, A.; Nascimbeni, F.; Lonardo, A. Hypothyroidism and Nonalcoholic Fatty Liver Disease - a Chance Association? Horm Mol Biol Clin Investig 2018, 41. [Google Scholar] [CrossRef]
- Yen, P.M. Physiological and Molecular Basis of Thyroid Hormone Action. Physiol Rev 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct Effects of Thyroid Hormones on Hepatic Lipid Metabolism. Nat Rev Endocrinol 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Cordeiro, A.; Souza, L.L.; Einicker-Lamas, M.; Pazos-Moura, C.C. Non-Classic Thyroid Hormone Signalling Involved in Hepatic Lipid Metabolism. J Endocrinol 2013, 216, R47–57. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, Y.; Gagné, D.J.; Papasavas, P.; Hayetian, F.; Maurer, J.; Bononi, P.; Caushaj, P.F. Improvement of Hypothyroidism after Laparoscopic Roux-En-Y Gastric Bypass for Morbid Obesity. Obes Surg 2004, 14, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Loomis, A.K.; Kabadi, S.; Preiss, D.; Hyde, C.; Bonato, V.; St Louis, M.; Desai, J.; Gill, J.M.; Welsh, P.; Waterworth, D.; et al. Body Mass Index and Risk of Nonalcoholic Fatty Liver Disease: Two Electronic Health Record Prospective Studies. J Clin Endocrinol Metab 2016, 101, 945–952. [Google Scholar] [CrossRef]
- Mahdavi, M.; Amouzegar, A.; Mehran, L.; Madreseh, E.; Tohidi, M.; Azizi, F. Investigating the Prevalence of Primary Thyroid Dysfunction in Obese and Overweight Individuals: Tehran Thyroid Study. BMC Endocr Disord 2021, 21, 89. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Hollenberg, A.N. Thyroid Hormone Signaling and the Liver. Hepatology 2020, 72, 742–752. [Google Scholar] [CrossRef]
- Sinha, R.A.; Bruinstroop, E.; Singh, B.K.; Yen, P.M. Nonalcoholic Fatty Liver Disease and Hypercholesterolemia: Roles of Thyroid Hormones, Metabolites, and Agonists. Thyroid 2019, 29, 1173–1191. [Google Scholar] [CrossRef]
- Gereben, B.; McAninch, E.A.; Ribeiro, M.O.; Bianco, A.C. Scope and Limitations of Iodothyronine Deiodinases in Hypothyroidism. Nat Rev Endocrinol 2015, 11, 642–652. [Google Scholar] [CrossRef]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the Local Control of Thyroid Hormone Action. J Clin Invest 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Cioffi, F.; de Lange, P.; Goglia, F.; Lanni, A. Thyroid: Biological Actions of “nonclassical” Thyroid Hormones. J Endocrinol 2014, 221, R1–12. [Google Scholar] [CrossRef]
- Ortiga-Carvalho, T.M.; Sidhaye, A.R.; Wondisford, F.E. Thyroid Hormone Receptors and Resistance to Thyroid Hormone Disorders. Nat Rev Endocrinol 2014, 10, 582–591. [Google Scholar] [CrossRef]
- Weinberger, C.; Thompson, C.C.; Ong, E.S.; Lebo, R.; Gruol, D.J.; Evans, R.M. The C-Erb-A Gene Encodes a Thyroid Hormone Receptor. Nature 1986, 324, 641–646. [Google Scholar] [CrossRef]
- Mitsuhashi, T.; Tennyson, G.E.; Nikodem, V.M. Alternative Splicing Generates Messages Encoding Rat C-erbA Proteins That Do Not Bind Thyroid Hormone. Proc Natl Acad Sci U S A 1988, 85, 5804–5808. [Google Scholar] [CrossRef]
- Mitsuhashi, T.; Nikodem, V.M. Regulation of Expression of the Alternative mRNAs of the Rat Alpha-Thyroid Hormone Receptor Gene. J Biol Chem 1989, 264, 8900–8904. [Google Scholar] [CrossRef]
- Bigler, J.; Eisenman, R.N. C-erbA Encodes Multiple Proteins in Chicken Erythroid Cells. Mol Cell Biol 1988, 8, 4155–4161. [Google Scholar] [CrossRef]
- Bigler, J.; Hokanson, W.; Eisenman, R.N. Thyroid Hormone Receptor Transcriptional Activity Is Potentially Autoregulated by Truncated Forms of the Receptor. Mol Cell Biol 1992, 12, 2406–2417. [Google Scholar] [CrossRef]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Mitochondrial T3 Receptor and Targets. Mol Cell Endocrinol 2017, 458, 112–120. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic Actions of Thyroid Hormone. Nat Rev Endocrinol 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Bassett, J.H.; Harvey, C.B.; Williams, G.R. Mechanisms of Thyroid Hormone Receptor-Specific Nuclear and Extra Nuclear Actions. Mol Cell Endocrinol 2003, 213, 1–11. [Google Scholar] [CrossRef]
- Kalyanaraman, H.; Schwappacher, R.; Joshua, J.; Zhuang, S.; Scott, B.T.; Klos, M.; Casteel, D.E.; Frangos, J.A.; Dillmann, W.; Boss, G.R.; et al. Nongenomic Thyroid Hormone Signaling Occurs through a Plasma Membrane-Localized Receptor. Sci Signal 2014, 7, ra48. [Google Scholar] [CrossRef]
- Carazo, A.; Levin, J.; Casas, F.; Seyer, P.; Grandemange, S.; Busson, M.; Pessemesse, L.; Wrutniak-Cabello, C.; Cabello, G. Protein Sequences Involved in the Mitochondrial Import of the 3,5,3’-L-Triiodothyronine Receptor P43. J Cell Physiol 2012, 227, 3768–3777. [Google Scholar] [CrossRef] [PubMed]
- Ferrandino, G.; Kaspari, R.R.; Spadaro, O.; Reyna-Neyra, A.; Perry, R.J.; Cardone, R.; Kibbey, R.G.; Shulman, G.I.; Dixit, V.D.; Carrasco, N. Pathogenesis of Hypothyroidism-Induced NAFLD Is Driven by Intra- and Extrahepatic Mechanisms. Proc Natl Acad Sci U S A 2017, 114, E9172–E9180. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamäki, J.; Boes, T.; Kim, E.Y.; Dearie, F.; Kim, B.W.; Schroeder, J.; Mun, E.; Nasser, I.; Park, P.J.; Bianco, A.C.; et al. Thyroid Hormone-Related Regulation of Gene Expression in Human Fatty Liver. J Clin Endocrinol Metab 2009, 94, 3521–3529. [Google Scholar] [CrossRef]
- Bohinc, B.N.; Michelotti, G.; Xie, G.; Pang, H.; Suzuki, A.; Guy, C.D.; Piercy, D.; Kruger, L.; Swiderska-Syn, M.; Machado, M.; et al. Repair-Related Activation of Hedgehog Signaling in Stromal Cells Promotes Intrahepatic Hypothyroidism. Endocrinology 2014, 155, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Bruinstroop, E.; Dalan, R.; Cao, Y.; Bee, Y.M.; Chandran, K.; Cho, L.W.; Soh, S.B.; Teo, E.K.; Toh, S.A.; Leow, M.K.S.; et al. Low-Dose Levothyroxine Reduces Intrahepatic Lipid Content in Patients With Type 2 Diabetes Mellitus and NAFLD. J Clin Endocrinol Metab 2018, 103, 2698–2706. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol Rev 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Nascimbeni, F.; Lonardo, A.; Zoppini, G.; Bonora, E.; Mantzoros, C.S.; Targher, G. Association Between Primary Hypothyroidism and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Thyroid 2018, 28, 1270–1284. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Reciprocal Crosstalk Between Autophagic and Endocrine Signaling in Metabolic Homeostasis. Endocr Rev 2017, 38, 69–102. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid Hormone Stimulates Hepatic Lipid Catabolism via Activation of Autophagy. J Clin Invest 2012, 122, 2428–2438. [Google Scholar] [CrossRef]
- Lombardi, A.; De Matteis, R.; Moreno, M.; Napolitano, L.; Busiello, R.A.; Senese, R.; de Lange, P.; Lanni, A.; Goglia, F. Responses of Skeletal Muscle Lipid Metabolism in Rat Gastrocnemius to Hypothyroidism and Iodothyronine Administration: A Putative Role for FAT/CD36. Am J Physiol Endocrinol Metab 2012, 303, E1222–33. [Google Scholar] [CrossRef]
- Lombardi, A.; de Lange, P.; Silvestri, E.; Busiello, R.A.; Lanni, A.; Goglia, F.; Moreno, M. 3,5-Diiodo-L-Thyronine Rapidly Enhances Mitochondrial Fatty Acid Oxidation Rate and Thermogenesis in Rat Skeletal Muscle: AMP-Activated Protein Kinase Involvement. Am J Physiol Endocrinol Metab 2009, 296, E497–502. [Google Scholar] [CrossRef]
- Chocron, E.S.; Sayre, N.L.; Holstein, D.; Saelim, N.; Ibdah, J.A.; Dong, L.Q.; Zhu, X.; Cheng, S.Y.; Lechleiter, J.D. The Trifunctional Protein Mediates Thyroid Hormone Receptor-Dependent Stimulation of Mitochondria Metabolism. Mol Endocrinol 2012, 26, 1117–1128. [Google Scholar] [CrossRef]
- Raghu Ramanathan, S.A.J.; Jamal, A. Ibdah THYROID HORMONE INCREASES HEPATIC MITOCHONDRIAL FATTY ACID OXIDATION AND RESCUES NAFLD IN MICE.; WILEY, 2021; Vol. 74, pp. 1095A-1095A.
- Harper, M.E.; Seifert, E.L. Thyroid Hormone Effects on Mitochondrial Energetics. Thyroid 2008, 18, 145–156. [Google Scholar] [CrossRef]
- Thakran, S.; Sharma, P.; Attia, R.R.; Hori, R.T.; Deng, X.; Elam, M.B.; Park, E.A. Role of Sirtuin 1 in the Regulation of Hepatic Gene Expression by Thyroid Hormone. J Biol Chem 2013, 288, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Lammel Lindemann, J.A.; Angajala, A.; Engler, D.A.; Webb, P.; Ayers, S.D. Thyroid Hormone Induction of Human Cholesterol 7 Alpha-Hydroxylase (Cyp7a1) in Vitro. Mol Cell Endocrinol 2014, 388, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bonde, Y.; Plösch, T.; Kuipers, F.; Angelin, B.; Rudling, M. Stimulation of Murine Biliary Cholesterol Secretion by Thyroid Hormone Is Dependent on a Functional ABCG5/G8 Complex. Hepatology 2012, 56, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cheng, S.Y. New Insights into Regulation of Lipid Metabolism by Thyroid Hormone. Curr Opin Endocrinol Diabetes Obes 2010, 17, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Lombardi, A.; de Lange, P.; Silvestri, E.; Ragni, M.; Farina, P.; Baccari, G.C.; Fallahi, P.; Antonelli, A.; et al. 3,5-Diiodo-L-Thyronine Powerfully Reduces Adiposity in Rats by Increasing the Burning of Fats. FASEB J 2005, 19, 1552–1554. [Google Scholar] [CrossRef]
- Cavallo, A.; Taurino, F.; Damiano, F.; Siculella, L.; Sardanelli, A.M.; Gnoni, A. Acute Administration of 3,5-Diiodo-L-Thyronine to Hypothyroid Rats Stimulates Bioenergetic Parameters in Liver Mitochondria. J Bioenerg Biomembr 2016, 48, 521–529. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid Hormone Induction of Mitochondrial Activity Is Coupled to Mitophagy via ROS-AMPK-ULK1 Signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid Hormone Receptor and ERRα Coordinately Regulate Mitochondrial Fission, Mitophagy, Biogenesis, and Function. Sci Signal 2018, 11. [Google Scholar] [CrossRef]
- Krotkiewski, M. Thyroid Hormones and Treatment of Obesity. Int J Obes Relat Metab Disord 2000, 24 Suppl 2, S116-9. [Google Scholar] [CrossRef]
- Oh, S.S.; Kaplan, M.L. Early Treatment of Obese (Ob/Ob) Mice with Triiodothyronine Increases Oxygen Consumption and Temperature and Decreases Body Fat Content. Proc Soc Exp Biol Med 1994, 207, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J.; Egan, D.M.; Sleph, P.G.; Beehler, B.C.; Chiellini, G.; Nguyen, N.H.; Baxter, J.D.; Scanlan, T.S. Effects of the Thyroid Hormone Receptor Agonist GC-1 on Metabolic Rate and Cholesterol in Rats and Primates: Selective Actions Relative to 3,5,3’-Triiodo-L-Thyronine. Endocrinology 2004, 145, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Trost, S.U.; Swanson, E.; Gloss, B.; Wang-Iverson, D.B.; Zhang, H.; Volodarsky, T.; Grover, G.J.; Baxter, J.D.; Chiellini, G.; Scanlan, T.S.; et al. The Thyroid Hormone Receptor-Beta-Selective Agonist GC-1 Differentially Affects Plasma Lipids and Cardiac Activity. Endocrinology 2000, 141, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Wohlfart, P.; Madsen, A.N.; Veidal, S.S.; Feigh, M.; Schmoll, D. Activation of Thyroid Hormone Receptor-β Improved Disease Activity and Metabolism Independent of Body Weight in a Mouse Model of Non-Alcoholic Steatohepatitis and Fibrosis. Br J Pharmacol 2021, 178, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Deng, Z.; Wang, W.; Liao, G.; Zhao, Y.; Zhong, H.; Zhang, Q.; Liu, J.; Mao, X.; Chen, B.; et al. CS27109, A Selective Thyroid Hormone Receptor-β Agonist Alleviates Metabolic-Associated Fatty Liver Disease in Murine Models. Int J Endocrinol 2023, 2023, 4950597. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Genomic and Non-Genomic Mechanisms of Action of Thyroid Hormones and Their Catabolite 3,5-Diiodo-L-Thyronine in Mammals. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Zhou, J.; Waskowicz, L.R.; Lim, A.; Liao, X.H.; Lian, B.; Masamune, H.; Refetoff, S.; Tran, B.; Koeberl, D.D.; Yen, P.M. A Liver-Specific Thyromimetic, VK2809, Decreases Hepatosteatosis in Glycogen Storage Disease Type Ia. Thyroid 2019, 29, 1158–1167. [Google Scholar] [CrossRef]
- Berkenstam, A.; Kristensen, J.; Mellström, K.; Carlsson, B.; Malm, J.; Rehnmark, S.; Garg, N.; Andersson, C.M.; Rudling, M.; Sjöberg, F.; et al. The Thyroid Hormone Mimetic Compound KB2115 Lowers Plasma LDL Cholesterol and Stimulates Bile Acid Synthesis without Cardiac Effects in Humans. Proc Natl Acad Sci U S A 2008, 105, 663–667. [Google Scholar] [CrossRef]
- Ramanathan, R.; Johnson, S.; Ibdah, J.A. Ramanathan, R.; Johnson, S.; Ibdah, J.A. Low Dose Thyroid Hormone Improves Hepatic Mitochondrial Fatty Acid Oxidation and Rescues Non-Alcoholic Fatty Liver Disease in Mice.; J Hep 2022, Vol. 77, pp. S692–S693 (Elsevier: London).
- Ibdah, J.A. Thyroid Hormone for Treatment of Nonalcoholic Steatohepatitis in Veterans. Available online: https://clinicaltrials.gov/study/NCT05526144.
- Grasselli, E.; Voci, A.; Canesi, L.; De Matteis, R.; Goglia, F.; Cioffi, F.; Fugassa, E.; Gallo, G.; Vergani, L. Direct Effects of Iodothyronines on Excess Fat Storage in Rat Hepatocytes. J Hepatol 2011, 54, 1230–1236. [Google Scholar] [CrossRef]
- Jonas, W.; Lietzow, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-L-Thyronine (3,5-T2) Exerts Thyromimetic Effects on Hypothalamus-Pituitary-Thyroid Axis, Body Composition, and Energy Metabolism in Male Diet-Induced Obese Mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; De Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-Diiodo-l-Thyronine, by Modulating Mitochondrial Functions, Reverses Hepatic Fat Accumulation in Rats Fed a High-Fat Diet. J Hepatol 2009, 51, 363–370. [Google Scholar] [CrossRef]
- Iannucci, L.F.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Yen, P.M.; Sinha, R.A. Metabolomic Analysis Shows Differential Hepatic Effects of T2 and T3 in Rats after Short-Term Feeding with High Fat Diet. Sci Rep 2017, 7, 2023. [Google Scholar] [CrossRef]
- Bruinstroop, E.; Zhou, J.; Tripathi, M.; Yau, W.W.; Boelen, A.; Singh, B.K.; Yen, P.M. Early Induction of Hepatic Deiodinase Type 1 Inhibits Hepatosteatosis during NAFLD Progression. Mol Metab 2021, 53, 101266. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Wohlfart, P.; Madsen, A.N.; Veidal, S.S.; Feigh, M.; Schmoll, D. Activation of Thyroid Hormone Receptor-β Improved Disease Activity and Metabolism Independent of Body Weight in a Mouse Model of Non-Alcoholic Steatohepatitis and Fibrosis. Br J Pharmacol 2021, 178, 2412–2423. [Google Scholar] [CrossRef]
- Vatner, D.F.; Weismann, D.; Beddow, S.A.; Kumashiro, N.; Erion, D.M.; Liao, X.H.; Grover, G.J.; Webb, P.; Phillips, K.J.; Weiss, R.E.; et al. Thyroid Hormone Receptor-β Agonists Prevent Hepatic Steatosis in Fat-Fed Rats but Impair Insulin Sensitivity via Discrete Pathways. Am J Physiol Endocrinol Metab 2013, 305, E89–100. [Google Scholar] [CrossRef]
- Caddeo, A.; Kowalik, M.A.; Serra, M.; Runfola, M.; Bacci, A.; Rapposelli, S.; Columbano, A.; Perra, A. TG68, a Novel Thyroid Hormone Receptor-β Agonist for the Treatment of NAFLD. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Cioffi, F.; Zambad, S.P.; Chhipa, L.; Senese, R.; Busiello, R.A.; Tuli, D.; Munshi, S.; Moreno, M.; Lombardi, A.; Gupta, R.C.; et al. TRC150094, a Novel Functional Analog of Iodothyronines, Reduces Adiposity by Increasing Energy Expenditure and Fatty Acid Oxidation in Rats Receiving a High-Fat Diet. FASEB J 2010, 24, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the Treatment of Non-Alcoholic Steatohepatitis: A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Taub, R.; Chiang, E.; Chabot-Blanchet, M.; Kelly, M.J.; Reeves, R.A.; Guertin, M.C.; Tardif, J.C. Lipid Lowering in Healthy Volunteers Treated with Multiple Doses of MGL-3196, a Liver-Targeted Thyroid Hormone Receptor-β Agonist. Atherosclerosis 2013, 230, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ladenson, P.W.; McCarren, M.; Morkin, E.; Edson, R.G.; Shih, M.-C.; Warren, S.R.; Barnhill, J.G.; Churby, L.; Thai, H.; O’Brien, T.; et al. Effects of the Thyromimetic Agent Diiodothyropropionic Acid on Body Weight, Body Mass Index, and Serum Lipoproteins: A Pilot Prospective, Randomized, Controlled Study. J Clin Endocrinol Metab 2010, 95, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
TR-α isoforms. Nuclear localization signals (NLS), nuclear export signals (NES), and mitochondrial targeting signals (MT) are shown in TRα1, TRβ1, and TRβ2. Localization signals are positioned in reference to the individual TR domains: N-terminal A/B domain (A/B), DNA-binding domain (DBE), Ligand-binding domain (LBD).
Figure 1.
TR-α isoforms. Nuclear localization signals (NLS), nuclear export signals (NES), and mitochondrial targeting signals (MT) are shown in TRα1, TRβ1, and TRβ2. Localization signals are positioned in reference to the individual TR domains: N-terminal A/B domain (A/B), DNA-binding domain (DBE), Ligand-binding domain (LBD).
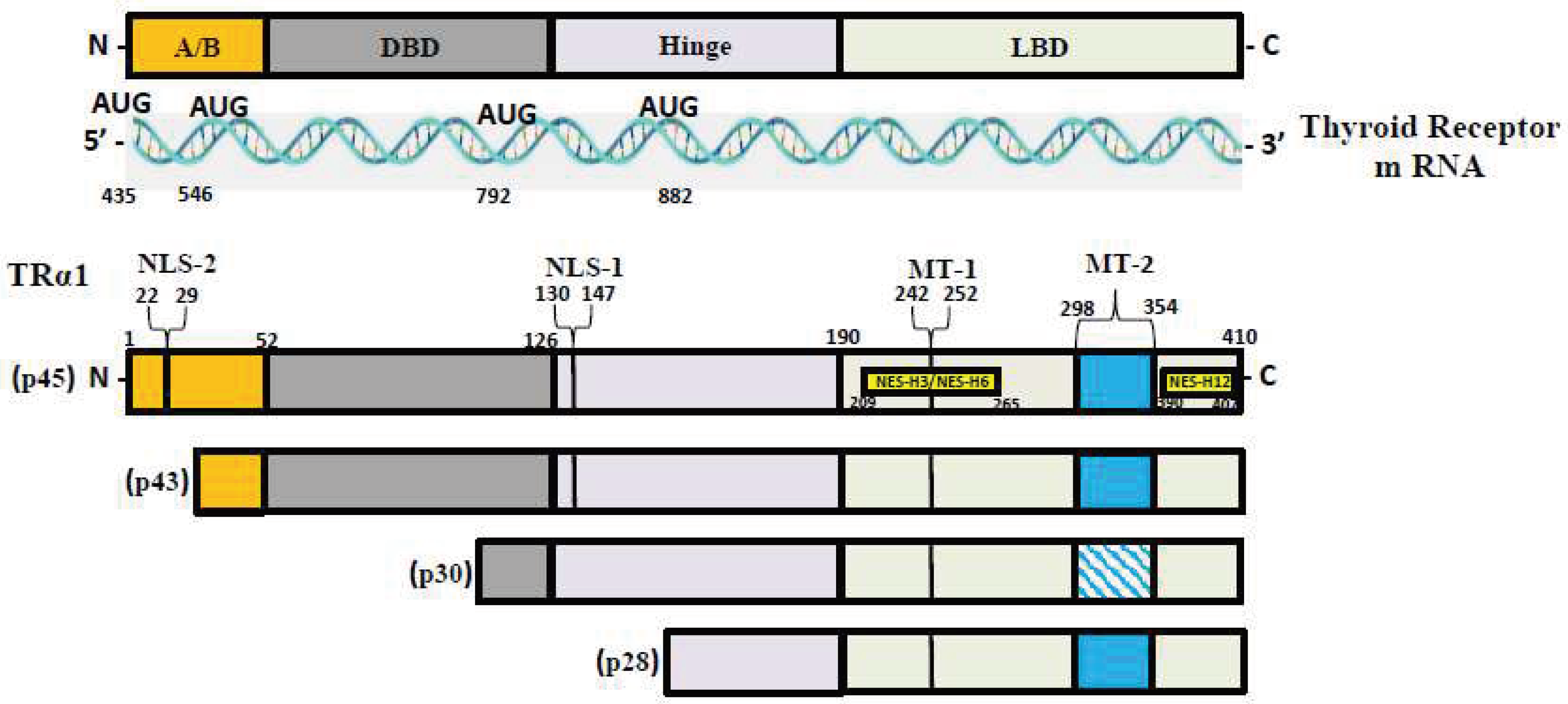

Table 1.
Effects of TH and its analogues in MASLD.
| Compound | Model and dose | Study findings | MASLD impact | References |
|---|---|---|---|---|
| Animal studies | ||||
| TH | ||||
| T2 | Hepatocyte isolated from Wistar rats; 10-7 to 10-5 M | Reduction of acyl-CoA oxidase and peroxisomal β-oxidation | Reduction of hepatic lipid accumulation | [114] |
| T2 | C57BL/6J mice; 2.5 µg/100g; ip | Increased fatty acid oxidation and decreased lipogenesis | Inhibition of fat accumulation in liver | [115] |
| T2 | Male wistar rats; 25 µg/100g; ip |
Reduced hepatic fatty accumulation, enhanced fatty acid oxidation rate and carnitine palmitoyl transferase activity | Activates mitochondrial processes, reverses hepatic steatosis | [116] |
| T2 | Rats injected with 25 µg/100g; ip |
Reduction in Serum TG and cholesterol | Prevents fatty liver by increasing fatty oxidation | [99] |
| T3 | ob/ob mice; 25µg/100g; ip | Lowered body weight and fat, increased oxidative metabolism | Increased oxidative metabolism in brown adipose tissue and liver. | [104] |
| T3 | Male wistar rats; 25µg/100g; ip | Promotes fatty acid peroxisomal and mitochondrial β-oxidation | Prevents hepatic fat accumulation by increasing β-oxidation | [43] |
| T2 and T3 | Wistar rats; 25 and 2.5 µg/100g; ip | Increased CPT-1 levels | Lowering hepatic lipid content, induced autophagy and intra-hepatic acylcarnitine flux. | [117] |
| T4 | Male C57BI/6J mice; | Decreased hepatic triglyceride and cholesterol | Reduce hepatosteatosis and prevent MASH progression. | [118] |
| Thyroid hormome analogues | ||||
| T3 and TRβ agonist GC-1 | Male fischer rats; 4 and 5 mg/kg; ip |
Marked fatty liver with mild hepatitis | Prevents fat accumulation by increasing mitochondrial and peroxisomal oxidation, complete regression of liver steatosis | [43] |
| TRβ agonist GC-1 | Male sprague Dawley rats; 1µg/kg; oral gavage | Reduction in hepatic TG levels | Treatment of obesity and hypercholesterolemia | [105] |
| MB07811 | Male sprague Dawley rats, ob/ob mice; 1 to 50 mg/kg; oral gavage |
Prevents hepatic steatosis, reduced plasma FFA and triglycerides | Increased hepatic fatty acid β-oxidation and mitochondrial respiration rates, as well as lower hepatic triglyceride levels and stimulation of CPT1α expression | [44] |
| Resmetirom (MGL-3196) | C57BI/6J mice; 3mg/kg for 8 weeks by oral gavage |
Lower hepatic triglycerides, lipid peroxidation, steatosis, inflammation and fibrosis |
Improvement in systemic and hepatic metabolism | [119] |
| VK2809 | GSDIa mouse model; 10mg/kg; Subcutaneously |
Restoring autophagy, mitochondrial biogenesis, and β-oxidation of fatty acids | Reduced hepatic lipid accumulation | [110] |
| GC-1 and KB-2115 | Male Sprague-Dawley rats; 164 and 100 µg/kg; ip |
Increased white adipose tissue lipolysis | Reduced hepatic steatosis | [120] |
| TG68 | C57BL mice; 2.8mg/kg in drinking water |
Reduction in liver weight, hepatic steatosis and triglycerides. | Can be used in MASLD | [121] |
| TRC150094 | Male wistar rats for 8 weeks; ip injection (0.750mg/100g b wgt | Reduction of Fat accumulation | Can be used in MASLD | [122] |
| Clinical Trials | ||||
| TH | ||||
| MGL-3196 (Resmetirom) | 36 weeks randomized trial in patients with biopsy proven MASH with fibrosis given 80mg orally daily | Significant reduction of hepatic fat, liver enzymes, lipoprotein, inflammation and fibrosis. | Patients showed reduction of hepatic fat compared to placebo, adverse events were mild and moderate. | [123] |
| 2 weeks randomized trial with 0.25 to 200mg/day | Significant reduction of total cholesterol and triglycerides |
Safe and showed beneficial effect on lipid parameters. | [124] | |
| KB2115 (eprotirome) |
5-day randomized trial in patients given 50 to 2000 µg orally daily |
Reduction in serum TC and LDL in overweight patients |
Reduced body weight | [111] |
| VK2809 | 12-week study of low dose of 5 mg in patients |
Reduction in LDL levels | Improvements in liver fat content in patients with MASLD | [18] |
| Levothyroxine (T4) | Patients with type 2 diabetes and steatosis given 18.75µg/day | Low dose T4 decreased lipid content in euthyroid male patients with type 2 diabetes mellitus. |
Safety and efficacy of TH therapy for MASLD in men | [84] |
| DITPA | 8-week randomized trial in patients with dose from 90 till 360mg/d | Lowered serum cholesterol and decrease in triglycerides |
Reduced body weight | [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



