Submitted:
24 October 2023
Posted:
26 October 2023
You are already at the latest version
Abstract
The chromatographic conditions were optimized using headspace gas chromatography, and a simple and rapid method was established for the simultaneous determination of five residual solvents in ursodeoxycholic acid raw materials. The corresponding quality standards were revised. The results showed that the concentration of methanol ranged from 0.06mg/ml to 0.3mg/ml, and the concentrations of acetone, tert butanol, ethyl acetate, and triethylamine showed a good linear relationship with the peak area within the range of 0.1mg/ml to 0.5mg/ml (r=0.999); The quantitation limits for methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were 4.2, 0.9, 1.5, 1, and 0.1 μg/ml, respectively, with detection limits of 1.2, 0.25, 0.025, 0.3, and 0.025 μg/ml, respectively. The recovery rates of each solvent ranged from 92.9% to 106.0%, with RSD% (n=9) less than 3.8%; The method exhibited good repeatability, with RSD% (n=6) less than 2.5%. Furthermore, the durability is good. The established method is simple, accurate, specific, and highly sensitive, and can be used for the simultaneous and rapid determination of five residual solvents in ursodeoxycholic acid raw materials.
Keywords:
headspace-gas chromatography
; ursodeoxycholic acid
; residual solvents
; triethylamine
; tert butanol
; ethyl acetate
1. Introduction
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid extracted from the precious traditional Chinese medicine material - bear bile. This compound has been widely used to treat various liver and gallbladder diseases and has shown significant therapeutic effects in clinical practice. The main therapeutic mechanism involves accelerating the generation of lipophilic bile acids such as chenodeoxycholic acid, which are toxic to the liver, reducing their reabsorption and thereby reducing their damage to liver cells [1,2,3,4]. UDCA has the characteristics of non-toxicity and hydrophilicity, so it can competitively prevent the absorption of harmful bile acid in the ileum and replace it to reduce the damage of bile acid to liver cells. Ursodeoxycholic acid was first discovered as a gallstone dissolving drug in 1920, and its molecular structure was determined in 1937. It began to be used in clinical trials in 1957 and has been widely included in pharmacopoeias of various countries [5,6,7].
The methods for preparing ursodeoxycholic acid include traditional bile drainage method [8,9], chemical synthesis method [10,11,12], and biosynthesis method [13,14,15]. The chemical synthesis method is widely used in industrial production because of its simple operation and low reagent cost. In the synthesis process, it is usually necessary to use some organic solvents, such as methanol, acetone, ethyl acetate, etc. [16,17]. However, organic solvents are difficult to completely remove in synthesis reactions. if their residual amount exceeds a certain safety standard, it may be harmful to human health. Although Li Ji [18] determined three residual organic solvents in ursodeoxycholic acid, the number of residual solvents detected was relatively small. A simple methodological description was provided, which was lacking accurate method for durability data.
According to the preparation process of ursodeoxycholic acid raw material and the requirements of ICHQ3C [19], the solvent residue inspection items of ursodeoxycholic acid raw material in the standard was revised. According to the technical guidelines for the study of residual solvents in chemical drugs [20], a headspace gas chromatography method for the simultaneous and rapid analysis of five organic solvent residues was established in China for the first time, and comprehensive methodological research was conducted. This will provide new data support to ensure the quality of raw materials, while also providing reliable raw material guarantee for the subsequent preparation of new formulations. This is crucial for ensuring the quality and safety of drugs.
2. Materials and Methods
2.1. Materials
Analytical-grade reagents such as dimethyl sulfoxide, methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were purchased from Shanghai Maclean Biochemical Technology Co. Deionized water was used throughout. Ursodeoxycholic acid API were purchased from Nykom Pharmaceuticals Co.
2.2. Apparatus and Operations
The quantitative HS-GC analysis was performed on an auto-headspace sampler (Aglient GC 7697A, US) connected to a GC system (Agilent GC 7890B, US) equipped with a hydrogen flame ionization detector (FID) and a HP-5 capillary column (30m×0.32mm×1.0μm, Agilent, US). A linear temperature program was used: initial from 45°C increased to 60°C at a rate of 5°C per minute, then ramped to 100°C at a rate of 10°C per minute, at last, it was raised to 200°C for 10 minutes at a rate of 40°C per minute. At a flow rate of 1.0 mL/min, nitrogen was used as the carrier, the split ratio is 10:1. The GC injector temperature was 200 °C and the flame ionization detector (FID) was operated at 220 °C. Conditions in headspace sampler: vial temperature = 100°C; vial equilibration time = 45 min; loop temperature = 110 °C; transfer line temperature = 115 °C; and pressurization pressure = 0.10 MPa.
2.3. Measurement Procedures
In this work, the HS-GC measurement is conducted with automated instrumental systems in which reaction (thermostatting) time and temperature can be accurately controlled. 1.0 ml of sample sheet was immediately placed into a headspace vial and sealed. The headspace vial was placed in the auto-sampler oven at 100 °C for 45 min for the phase equilibration, followed measuring the signals (peak areas) of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine released to vapor phase by HS-GC technique.
2.4. Preparation of Reference Solution and Sample Solution
2.4.1. Preparation of Residual Solvent Localization Solution
Accurately weigh 15mg of methanol, 12.5mg of acetone, tert butanol, ethyl acetate, and triethylamine respectively, and place them in a 50ml volumetric flask. Dilute with dimethyl sulfoxide to the mark, shake well, and serve as the localization solution for each residual solvent.
2.4.2. Preparation of Reference Solution
Accurately weigh 30 mg of methanol, 50 mg of acetone, 50 mg of tert-butanol, 50 mg of ethyl acetate, and 50 mg of triethylamine into a 10 ml volumetric flask. Dilute with dimethyl sulfoxide to the mark, shake well, and use as a reference stock solution. Accurately measure 1ml of the reference stock solution into a 10 ml volumetric flask, dilute with dimethyl sulfoxide to the mark, shake well, and use it as the reference solution.
2.4.3. Preparation of Sample Solution
Take 1g of this product, accurately weigh it, and add 10 ml of dimethyl sulfoxide to dissolve as the sample solution.
2.5. Method Validation
Following the ICH-Q2-R1 recommendations, the proposed HS-GC approach for the simultaneous assessment of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine was validated for repeatability, linearity range, detection (LOD) and quantification (LOQ) limits, accuracy and Durability.
The repeatability of the present method was investigated through triplicate tests on six different samples. The linearity range for methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were assessed by plotting the concentrations of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine against measured HS-GC response. The linearity for methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were assessed using six different concentrations for both of the compounds in triplicates (n=3). Using a multiple dilution method, the sensitivity of the proposed HS-GC approach for the simultaneous assessment of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine was evaluated in terms of “detection (LOD) and quantification (LOQ) limits”.
The accuracy for the proposed HS-GC approach for the simultaneous determination of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were determined as the percent recovery. For every residue solvent, the accuracy was tested using low-quantity control (LQC), middle-quantity control (MQC), and high-quantity control (HQC) samples. The accuracy was determined in standard compounds. The percent recovery was calculated for each residue solvent quality level (n=3). Durability for the proposed HS-GC approach for the simultaneous determination of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine were determined by the slight changes of the flow rates of carrier gas and column temperature.
3. Results and Discussion
3.1. Optimization of Chromatographic Conditions
3.1.1. The Effect of Column Temperature
Different column temperatures have a significant impact on the detection of impurities in the test sample. Therefore, it is necessary to determine the optimal column temperature for the detection of this product. The test sample solution is measured at three column temperatures of 60 ℃, 80 ℃, and 100 ℃, and the chromatogram is recorded. The separation and detection of each impurity are shown in Table 1.
The results showed that the test results of different column temperatures met the system suitability requirements, and there was no significant difference in retention time. However, under the condition of column temperature of 100℃, the peak symmetry, separation degree, and column efficiency are better, which is more conducive to the separation of products.
3.1.2. The Effect of Equilibrium Time
Different equilibrium times have a significant impact on the detection of impurities in the test sample. Therefore, it is necessary to determine the optimal equilibrium time conditions for the detection of this product. The test sample solution is taken and measured under five equilibrium time conditions of 5 minutes, 10 minutes, 20 minutes, 30 minutes, and 45 minutes, and the chromatogram is recorded. The separation and detection of each impurity are shown in Table 2.
The results indicate that the test results of different equilibrium times meet the system suitability requirements, and there is no significant difference in retention time. However, under the condition of equilibrium time of 45 minutes, the peak symmetry, separation degree, and column efficiency are better, which is more conducive to the separation of the product.
3.2. Method Validation
To confirm the suitability of the method for its intended purpose, the method was validated in accordance with the ICH guidelines, for system suitability and specificity.
3.2.1. System Suitability
System-suitability test was an integral part of method development and has been used to ensure adequate performance of the chromatographic system. Take 1ml of the localization solution for each residual solvent, the reference solution and sample solution, respectively. Then inject it into the gas chromatograph through headspace injection. Record the chromatogram. resolution (R), number of theoretical plates (N) were evaluated for six replicate injections of the drug.
The results in Figure 1 showed that the resolution between impurity peaks of methanol, acetone, tert butanol, ethyl acetate, and triethylamine, as well as between impurities and dimethyl sulfoxide solvent, is greater than 2.0. The column efficiency of methanol is greater than 5000, and the column efficiency of other components is greater than 10000, respectively, indicating good column efficiency and optimum mobile phase composition.
3.2.2. Repeatability Test
Repeatability was calculated from six replicate injections of freshly prepared solution in the same equipment on the same day.
The result from Table 3 showed that the RSD of peak area were 2.38%, 1.39%, 2.29%, 1.49% and 0.03%, respectively. The results were in accordance with acceptance criteria for precision (<10%), indicating that the method was precise.
3.2.3. Linearity
The linearity range for five residue solvents was assessed by plotting the concentrations of them against measured HS-GC response. Methanol, acetone, tert-butanol, ethyl acetate, and triethylamine stock solutions were prepared separately by dissolving the prescribed amounts of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine in the requisite volume of dimethyl sulfoxide to provide a final solution of 3 mg/ml for methanol and 5 mg/ml for other compounds. After that, serial dilutions of this solution were created by diluting with dimethyl sulfoxide with different volumes of methanol, acetone, tert-butanol, ethyl acetate, or triethylamine solution to obtain different concentrations range for both substances.
The linearity for methanol was assessed using six different concentrations, including 0.06, 0.09, 0.12, 0.18, 0.24, and 0.3 mg/g (range 0.06–0.3 mg/ml). The linearity for other four residue solvents were assessed using six different concentrations, including 0.1, 0.15, 0.2, 0.3, 0.4, and 0.5 mg/g (range 0.1–0.5 mg/ml) for both of the compounds in triplicates (n = 3).
Approximately 1mL of each concentration of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine was injected into HS-GC system, and HS-GC response for each concentration of methanol, acetone, tert-butanol, ethyl acetate, and triethylamine was recorded. The calibration curve for methanol, acetone, tert-butanol, ethyl acetate, and triethylamine was constructed by plotting the concentrations of each compound against measured HS-GC response.
The results of the linear regression analysis in Table 4 revealed a strong linear association between residue solvents concentrations and the measured HS-GC responses. For five residue solvents, the regression equations obtained were Y = 981.35X + 10.04, Y = 4517.4X + 78.569, Y = 2756.5X + 45.711, Y = 3507.5X + 56.446 and Y = 14349X + 252.25, respectively, where Y is peak area and X is concentration of residue solvent (mg・mL-1). In addition, the values of regression coefficient (R) were calculated to be 0.9993, 0.9991, 0.9990, 0.9990 and 0.9990, respectively. Good linearity was evident by the high value of the correlation coefficient and the low intercept value. These findings showed that the suggested HS-GC approach was suitable for the simultaneous detection of residue solvents in ursodeoxycholic acid raw materials.
3.2.4. Detection (LOD) and Quantitation (LOQ) Limits
The detection limit (LOD) was defined as signal-to-noise ratio of 3:1. The quantitation limit (LOD) was defined as signal-to-noise ratio of 10:1. Therefore, peaks heights were evaluated for this validation parameter. The LOD values of five residue solvents were 1.2 μg/ml, 0.25 μg/ml, 0.025 μg/ml, 0.3μg/ml, and 0.025 μg/ml, respectively. On the other hand, The LOQ values of five residue solvents were calculated as 4.2 μg/ml, 0.9 μg/ml, 1.5 μg/ml, 1.0 μg/ml, and 0.1 μg/ml, respectively. These findings demonstrated the sensitivity of the proposed HS-GC approach for the simultaneous measurement of five residue solvents in ursodeoxycholic acid raw materials.
3.2.5. Accuracy
The accuracy for the proposed HS-GC approach for the simultaneous determination of five residue solvents was determined as the percent recovery. For methanol, the accuracy was tested using low-quantity control (LQC; 0.18 mg/ml), middle-quantity control (MQC; 0.24 mg/ml), and high-quantity control (HQC; 0.3 mg/ml) samples. for other four residue solvents, the accuracy was tested using low-quantity control (LQC; 0.3 µg/g), middle-quantity control (MQC; 0.4 µg/g), and high-quantity control (HQC; 0.5 µg/g) samples. The accuracy was determined in standard compounds. The percent recovery was calculated for each solvent quality level (n = 3) shown in Table 5.
The results showed that the average recovery rate of methanol was 100.6%, the RSD of nine test results was 3.45%, the average recovery rate of acetone was 98.2%, the RSD was 2.60%, the average recovery rate of tert butanol was 106.0%, the RSD was 3.28%, the average recovery rate of ethyl acetate was 98.8%, and the RSD was 2.72%, The average recovery rate of triethylamine is 92.9%, and the RSD was 3.80%. The recovery rate measurement results indicate that the method has good accuracy and the relative deviation meets the requirements.
3.2.6. Durability
When the flow rates of carrier gas was 0.9ml/min, 1.0ml/min and 1.1 ml/min, respectively, the impact of the slight changes of flow rate of carrier gas was investigated on the test result. When the column temperature changes to 90℃, the impact was investigated.
The results showed in Table 6 that the acetone, tert butanol and triethylamine were detected in the test sample. the concentration of acetone changes from 0.2068 mg/ml to 0.1999 mg/ml, the concentration of acetone changes from 0.2068 mg/ml to 0.1999 mg/ml, the concentration of acetone changes from 0.2068 mg/ml to 0.1999 mg/ml, at the column temperature of 100℃.The slightly changing of flow rate and column temperature had no effect on the determination of the test sample. The method had good durability.
4. Conclusions
After the development and verification of the established method, we successfully established a simple, accurate and highly sensitive method for the determination of solvent residues. The method provides a reliable quantitative analysis for five target solvent residues in ursodeoxycholic acid raw material. Through this method, we can effectively control and monitor the quality of ursodeoxycholic acid and provide a reliable means for establishing their quality standards. The research method has obvious specificity and application value. Its simplicity and high accuracy make it suitable for quality control in industrial production and analytical work in drug testing laboratories. The results provide reliable data for the determination of solvent residues in ursodeoxycholic acid and ensure the quality and safety of the products.
Author Contributions
Conceptualization, supervision—R.Z. and F.W.; methodology—R.Z., F.W. and S.J.L.; validation—R.Z., Y.S. and X.Y.G.; data curation—F.W.; funding acquisition—J.Y.H. and S.J.L.; project administration—J.Y.H.; writing, original draft—R.Z.; writing, review and editing—R.Z., F.W. and S.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Research Project on Human Resources and Social Security in Hebei Province (JRSHZ-2023-02316) and Shijiazhuang University Teaching Reform Research and Practice Project (International Construction Project, GJZX-2023009).
Data Availability Statement
Not applicable.
Acknowledgments
Authors are thankful to the Research Project on Human Resources and Social Security in Hebei Province (JRSHZ-2023-02316) and Shijiazhuang University Teaching Reform Research and Practice Project (International Construction Project, GJZX-2023009), which made this study possible.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gu, H.Z. The clinical application of ursodeoxycholic acid. Inn. Mong. Tradit. Chin. Med. 2012, 2, 52–53. [Google Scholar]
- Qi, Z.; Xu, M. L.; Zheng, Y.Y. Clinical use and research progress of ursodeoxycholic acid. J. Clin. Ration. Drug Use 2016, 9, 84–85. [Google Scholar]
- Wan, J.F.; Chu, S.F.; Zhou, X.; Li, Y.T.; He, W.B.; Tan, F.; Luo, P.; Ai, Q.D.; Wang, Q.; Chen, N.N. Ursodeoxycholic acid protects interstitial Cajal-like cells in the gallbladder from undergoing apoptosis by inhibiting TNF-α expression. Acta Pharmacol Sin 2018, 39, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Gazda, J.; Drazilova, S.; Gazda, M.; Martin Janicko, M.; Koky, T.; Macej, M.; Carbone, M.; Jarcuska, P. Treatment response to ursodeoxycholic acid in primary biliary cholangitis: A systematic review and meta-analysis. Dig. Liver Dis. 2023, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- National Pharmacopoeia Commission. Pharmacopoeia of the People's Republic of China: Part II. China Medical Science and Technology Press, China, 2020.
- United States Pharmacopoeia Commission. Ursodeoxycholic acid, US Pharmacopoeia: USP43, 2020.
- European Pharmacopoeia Commission, Ursodeoxycholic acid, European Pharmacopoeia: EP10.0, 2020.
- Dawkins, M. S. From an animal's point of view: motivation, fitness, and animal welfare. Behav. Brain Sci. 1990, 13, 1–9. [Google Scholar] [CrossRef]
- Kim, O.Y.; Lee, S.Y.; Lee, D.Y.; Hur, S.J. Developing a procedure to extract chenodeoxycholic acid and synthesize ursodeoxycholic acid from pig by-products. Heliyon 2023, 9, e18313. [Google Scholar] [CrossRef] [PubMed]
- Lu, M. F.; Yin, W. C.; Wang, F. D.; Peng, D.M. Improvement of the synthesis process of ursodeoxycholic acid. Chin. J. Pharm. Ind. 2015, 46, 1058–1059. [Google Scholar]
- Chen, W.; Hu, D.; Feng, Z.; Liu, Z. An effective synthesis of ursodeoxycholic acid from dehydroepiandrosterone. Steroids 2021, 172, 108870. [Google Scholar] [CrossRef] [PubMed]
- Shen, J. X.; Dong, D. D.; Wang, Z. F.; Wan, J. F.; Cao, X. J. Synthesis of ursodeoxycholic acid by electrochemical stereoselective reduction of 7-ketolithocholic acid in aprotic solvents. Sci. Rep. 2021, 1, 16273–16273. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.W.; Fu, J.J.; Li, J.J.; Tang, Y.; Shao, Z.W.; Zhou, D.Y.; Song, L. Efficient encapsulation of curcumin into spent brewer’s yeast using a pH-driven method. Food Chem. 2022, 394, 133537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H. Construction and application of a recombinant yeast strain for efficient synthesis of ursodeoxycholic acid. Tianjin: Tianjin University of Science and Technology, 2022.
- Yang, B.Y.; You, Z.N.; Xue, J.T.; Pan, J.; Li, C.X.; Xu, J.H. Clean enzymatic production of ursodeoxycholic acid enabled by a newly identified NADH- dependent 7β-hydroxysteroid dehydrogenase. Mol. Catal. 2023, 537, 112946. [Google Scholar] [CrossRef]
- Zhang, C.L; Li, C.S.; Yu, X.L. Progress in Chinese patents for the effective ingredient ursodeoxycholic acid in bear bile. Chin. J. Tradit. Chin. Med. 2012, 37, 2851–2854. [Google Scholar]
- Luo, L.L.; Yu, J.; Wei, H.Q.; Hou, W.B.; Li, W.L. Research progress in the synthesis of ursodeoxycholic acid. Synth. Chem. 2021, 29, 986–996. [Google Scholar]
- Li, J.; Li, X.; Wang, F.; et al. Determination of three residual organic solvents in ursodeoxycholic acid by headspace gas chromatography. Prog. Mod. Biomed. Sci. 2015, 31, 6156–6159. [Google Scholar]
- Zhou, H. J. International Technical Requirements for Drug Registration Q3C. Beijing: People's Health Publishing House, 2000, 1-8.
- State Food and Drug Administration. Technical guidelines for the study of residual solvents in chemical drugs. 2005, 6-7.
Figure 1.
The chromatogram of system suitability. (a) blank solvent; (b)-(f) localization solutions of five residual solvent (methanol, acetone, tertbutanol, ethyl acetate, triethylamine); (g) reference solution; (h) sample solution.
Figure 1.
The chromatogram of system suitability. (a) blank solvent; (b)-(f) localization solutions of five residual solvent (methanol, acetone, tertbutanol, ethyl acetate, triethylamine); (g) reference solution; (h) sample solution.
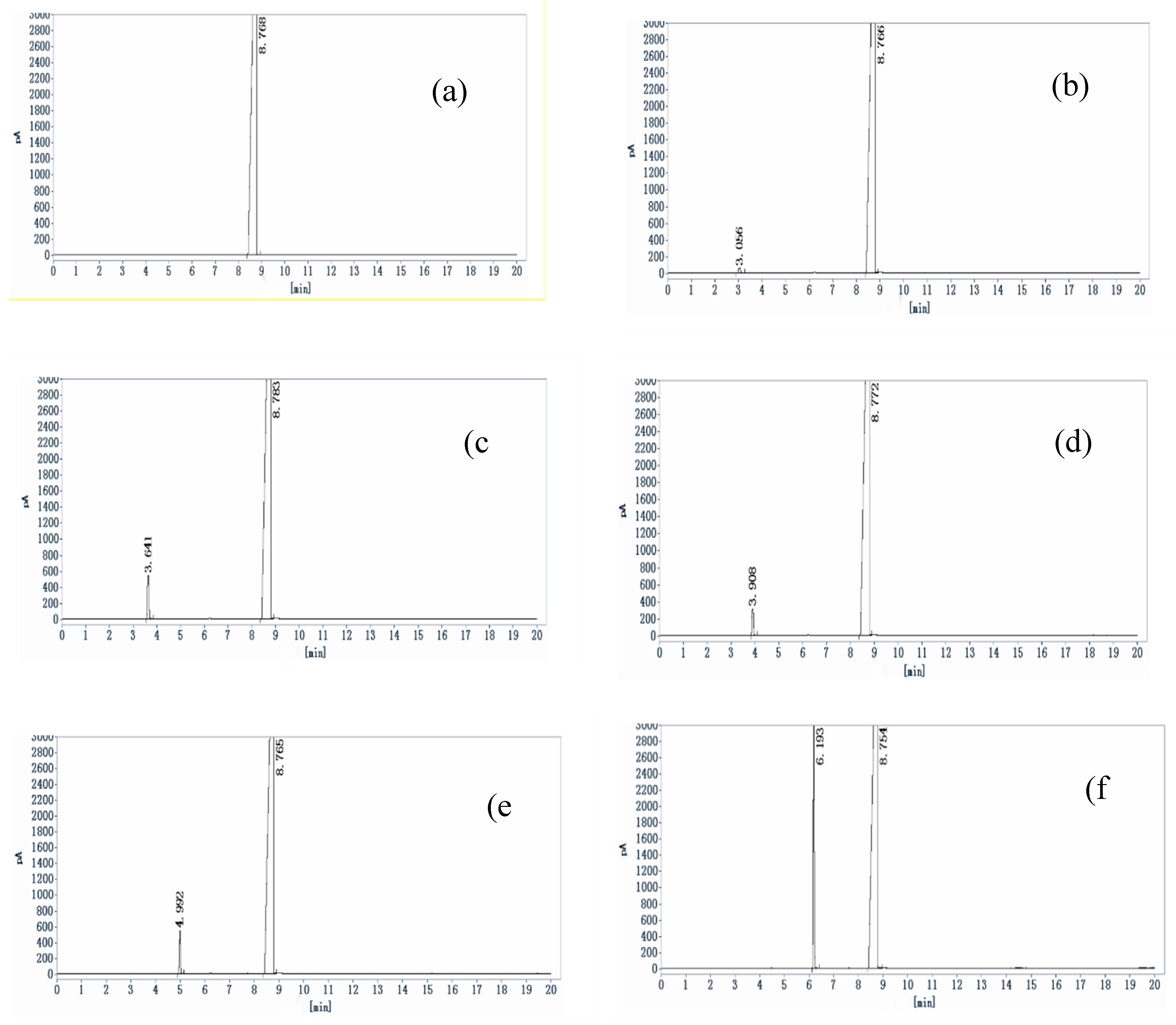
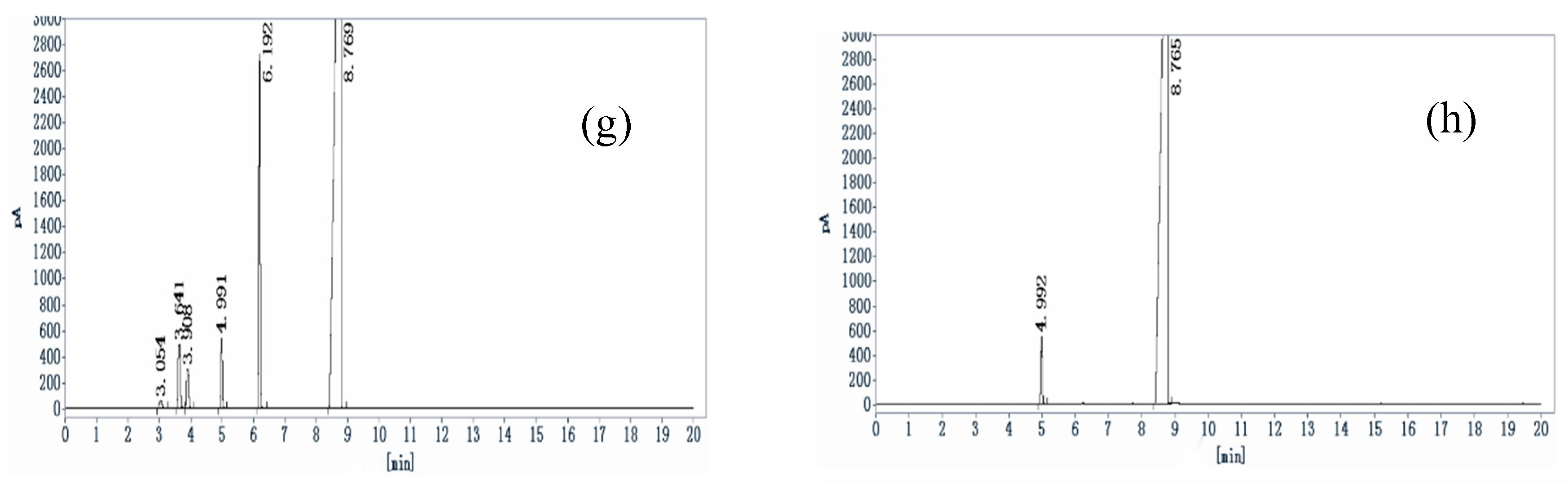

Table 1.
The effect of different column temperature on test samples.
| Column temperature | Test samples | ||||
|---|---|---|---|---|---|
| Unknown peak (2.758min) | Acetone | Tert butanol | Triethylamine | ||
| 60 °C | Rt (min) | 2.758 | 3.646 | 3.948 | 6.218 |
| Resolution | —— | 5.709 | 2.191 | 22.044 | |
| Peak area | 45.542 | 411.721 | 2.783 | 12.881 | |
| Tailing factor | 1.533 | 0.934 | 1.056 | 1.718 | |
| Number of theoretical plates | 4268.713 | 10628.133 | 13918.130 | 119444.528 | |
| 80 °C | Rt (min) | 2.758 | 3.645 | 3.949 | 6.212 |
| Resolution | —— | 5.702 | 2.215 | 22.277 | |
| Peak area | 39.932 | 641.844 | 3.963 | 26.420 | |
| Tailing factor | 1.479 | 1.092 | 0.923 | 1.685 | |
| Number of theoretical plates | 4334.966 | 10402.865 | 14348.943 | 123072.631 | |
| 100 °C | Rt (min) | 2.740 | 3.633 | 3.935 | 6.193 |
| Resolution | —— | 6.345 | 2.283 | 22.551 | |
| Peak area | 3.255 | 712.048 | 9.883 | 36.692 | |
| Tailing factor | 1.349 | 1.145 | 0.985 | 1.500 | |
| Number of theoretical plates | 5707.791 | 11286.903 | 15125.859 | 118511.515 | |
Table 2.
The effect of different equilibrium time on test samples.
| Equilibrium time | Test samples | ||||
|---|---|---|---|---|---|
| Unknown peak (2.758min) | Acetone | Tert butanol | Triethylamine | ||
| 5min | Rt (min) | 2.742 | 3.633 | 3.934 | 6.191 |
| Resolution | —— | 6.172 | 2.272 | 22.403 | |
| Peak area | 3.988 | 676.439 | 4.905 | 52.018 | |
| Tailing factor | 1.718 | 1.026 | 1.145 | 1.528 | |
| Number of theoretical plates | 5218.119 | 11320.143 | 14921.887 | 116516.392 | |
| 10min | Rt (min) | 2.740 | 3.633 | 3.935 | 6.193 |
| Resolution | —— | 6.345 | 2.283 | 22.551 | |
| Peak area | 3.255 | 712.048 | 9.883 | 36.692 | |
| Tailing factor | 1.349 | 1.145 | 0.985 | 1.500 | |
| Number of theoretical plates | 5707.791 | 11286.903 | 15125.859 | 118511.515 | |
| 20min | Rt (min) | 2.756 | 3.643 | 3.946 | 6.203 |
| Resolution | —— | 6.032 | 2.332 | 22.725 | |
| Peak area | 51.373 | 965.891 | 14.979 | 56.512 | |
| Tailing factor | 1.545 | 0.962 | 0.947 | 1.585 | |
| Number of theoretical plates | 4788.073 | 11795.839 | 15771.493 | 116968.328 | |
| 30min | Rt (min) | 2.756 | 3.643 | 3.946 | 6.208 |
| Resolution | —— | 6.025 | 2.314 | 23.045 | |
| Peak area | 56.805 | 936.573 | 19.543 | 34.205 | |
| Tailing factor | 1.544 | 0.929 | 1.021 | 1.594 | |
| Number of theoretical plates | 4789.232 | 11711.901 | 15593.435 | 126957.442 | |
| 45min | Rt (min) | 2.757 | 3.644 | 3.947 | 6.212 |
| Resolution | —— | 6.036 | 2.326 | 23.145 | |
| Peak area | 62.264 | 929.488 | 26.532 | 23.802 | |
| Tailing factor | 1.569 | 0.989 | 1.027 | 1.701 | |
| Number of theoretical plates | 4790.395 | 11793.688 | 15744.882 | 127107.633 | |
Table 3.
Test results for injection repeatability.
| Injection times | Peak area | ||||
|---|---|---|---|---|---|
| Methanol | Acetone | Tert butanol | Ethyl acetate | Triethylamine | |
| 1 | 308.7 | 2277.3 | 1422.6 | 1826.5 | 7043.8 |
| 2 | 313.5 | 2330.6 | 1439.6 | 1865.4 | 7199.6 |
| 3 | 311.6 | 2312.1 | 1439.6 | 1851.9 | 7079.7 |
| 4 | 317.5 | 2352.2 | 1463.2 | 1885 | 7354.7 |
| 5 | 319.7 | 2370.5 | 1487.3 | 1907.6 | 7395 |
| 6 | 329.9 | 2330.5 | 1511.1 | 1875.3 | 7026.9 |
| Average | 316.8 | 2328.9 | 1460.6 | 1868.6 | 7183.2 |
| RSD% | 2.38 | 1.39 | 2.29 | 1.49 | 2.24 |
Table 4.
Linear results for each residual solvent(n=3).
| Residue solvent | Regression equations | R | Linearity range (mg/ml) |
|---|---|---|---|
| Methanol | Y = 981.35X + 10.04 | 0.9993 | 0.06-0.3 |
| Acetone | Y = 4517.4X + 78.569 | 0.9991 | 0.1-0.5 |
| Tert butanol | Y = 2756.5X + 45.711 | 0.9990 | 0.1-0.5 |
| Ethyl acetate | Y = 3507.5X + 56.446 | 0.9990 | 0.1-0.5 |
| Triethylamine | Y = 14349X + 252.25 | 0.9990 | 0.1-0.5 |
Table 5.
Results of recovery tests for each residual solvent(n=9).
| Residue solvents | Theoretical concentration (mg/ml) |
Actual concentration (mg/ml) | Recovery (%) | Average recovery (%) | RSD (%) |
|---|---|---|---|---|---|
| Methanol | 0.18 | 0.1808 | 100.4 | 100.6 | 3.45 |
| 0.18 | 0.1803 | 100.2 | |||
| 0.18 | 0.1773 | 98.5 | |||
| 0.24 | 0.2336 | 97.3 | |||
| 0.24 | 0.2410 | 100.4 | |||
| 0.24 | 0.2409 | 100.4 | |||
| 0.3 | 0.2974 | 99.1 | |||
| 0.3 | 0.2983 | 99.4 | |||
| 0.3 | 0.3282 | 109.4 | |||
| Acetone | 0.3 | 0.3004 | 100.1 | 98.2 | 2.60 |
| 0.3 | 0.2990 | 99.7 | |||
| 0.3 | 0.2894 | 96.5 | |||
| 0.4 | 0.3779 | 94.5 | |||
| 0.4 | 0.3962 | 99.1 | |||
| 0.4 | 0.3890 | 97.2 | |||
| 0.5 | 0.4746 | 94.9 | |||
| 0.5 | 0.4966 | 99.3 | |||
| 0.5 | 0.5107 | 101.8 | |||
| Tert butanol | 0.3 | 0.3205 | 106.8 | 106.0 | 3.28 |
| 0.3 | 0.3185 | 106.2 | |||
| 0.3 | 0.3096 | 103.2 | |||
| 0.4 | 0.4074 | 101.9 | |||
| 0.4 | 0.4251 | 106.3 | |||
| 0.4 | 0.4270 | 106.7 | |||
| 0.5 | 0.5163 | 103.3 | |||
| 0.5 | 0.5284 | 105.7 | |||
| 0.5 | 0.5695 | 113.9 | |||
| Ethyl acetate | 0.3 | 0.3027 | 100.9 | 98.8 | 2.72 |
| 0.3 | 0.3005 | 100.2 | |||
| 0.3 | 0.2897 | 96.6 | |||
| 0.4 | 0.3804 | 95.1 | |||
| 0.4 | 0.3991 | 99.8 | |||
| 0.4 | 0.3946 | 98.6 | |||
| 0.5 | 0.4762 | 95.2 | |||
| 0.5 | 0.5002 | 100.0 | |||
| 0.5 | 0.5152 | 103.0 | |||
| Triethylamine | 0.3 | 0.2939 | 98.0 | 92.9 | 3.80 |
| 0.3 | 0.2922 | 97.4 | |||
| 0.3 | 0.2744 | 91.5 | |||
| 0.4 | 0.3527 | 88.2 | |||
| 0.4 | 0.3733 | 93.3 | |||
| 0.4 | 0.3572 | 89.3 | |||
| 0.5 | 0.4485 | 89.7 | |||
| 0.5 | 0.4755 | 95.1 | |||
| 0.5 | 0.4694 | 93.9 |
Table 6.
Test results of durability.
| Residue solvents | Conditions | |||
|---|---|---|---|---|
| 0.9ml/min | 1.0 ml/min | 1.1ml/min | 90℃ | |
| Acetone | 0.2068 | 0.2036 | 0.1999 | 0.1999 |
| Tert butanol | 0.0026 | 0.0024 | 0.0024 | 0.0024 |
| Triethylamine | 0.0056 | 0.0057 | 0.0043 | 0.0043 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



