
Submitted:
25 October 2023
Posted:
26 October 2023
You are already at the latest version
Abstract
Drug discovery utilizes high-throughput screening (HTS) methods applying target and cell-based assays. This review discusses the challenges and benefits associated with these assays in HTS. Discussed the strategies that can be applied for the development of screening assays through primary and secondary screens for target identification. Further discussion on identify-ing the most efficacious drugs following these approaches in cancer. Even though various drugs have been identified to treat cancer, there is high demand of more relevant phenotypic assays to produce desired diseased phenotype to only highlighting the specific targets instead of off-targets. Hopefully, the new developing strategies could provide innovative drugs to treat cancer patients to reduce mortality rate.
Keywords:
target based screening
; phenotypic based screening
; targets
; transcriptional reprogramming
; highthroughput screening
; cancer therapeutics
Introduction
The current drug discovery paradigm is highly focused on high-throughput screening (HTS), where large libraries of compounds are screened against the target of interest to identify suitable and most probable approaches for additional biological or pharmacological targets to get beneficial therapeutic outcomes [1,2]. The lead identified targets are being used as probes to address biological questions in basic research. These days, combinatorial chemistry and genomics data is being highly applied to HTS in the drug discovery process [3]. However, the process of testing the large compound libraries for their binding affinity or biological activity is very expensive to generate a sufficient number of hits for drug[4, 5]. Recent studies are being more likely to believe in information-driven strategy rather than screening huge number of compounds to achieve higher probability of success. It gets more complex and expensive when we reach to mimic more relevant disease phenotype. This emphasizes the demand of methods to develop integrated approach to enhance quantity, quality, and cost efficiency. The idea of applying combinatorial chemistry is to screen huge array of structurally diverse compounds in one screen to allow rapid concurrent screening against specific drug targets. These smart libraries would only be helpful for drug discovery when we know exactly about the druggable target. In this regard particularly genomics comes into the picture, which gives the information on the type of mutation and its burden depending upon the disease and age. A case study on a 10-year-old boy with a multiple recurrent glioblastoma facing challenges in treatment due to inter- and intra-tumoral heterogeneity was solved with the integrated application of genomic, in vitro, and in vivo testing [6]. Many novel potential drug targets have been identified by using the information of genomics. Pharmaceutical industry utilizes various technologies in terms of assay miniaturization, lab automation and methodologies to make the whole process cost and time effective and enhance the efficacy and selectivity [7]. To improve its efficiency, HTS has been miniaturized to ultra-high-throughput screening (uHTS) to enable vigorous testing [8]. HTS efforts resulted Viramune (brand name, nevirapine), a non-nucleoside reverse transcriptase inhibitor against HIV [9] and since 1999, FDA has approved around 61 first-in-class small molecules for the treatment of cancer, cardiovascular and metabolic disorders, gastrointestinal and infectious diseases [10]. Among them, total 46 were through phenotypic screening (e.g., Azacitidine, Fulvestran, Nelarabine, and Vorinostat) and 17 through target-based screening (e.g., Bortezomib, Gefitinib, Imatinib, Sorafenib and Sunitinib).
After all these new implements, though the rate of successful drugs screening is very low due to molecular heterogeneity underlying disease mechanisms within and between patients for many disease types is the limitations of contemporary drug discovery strategies. Therefore, “one drug, one target” paradigm is usually followed to reduce unwanted off-target side effects. However, this approach oversimplifies the mechanisms, which are actually very complicated and controlled by various factors in the real scenario inside the cells. Highly potent and single-target treatments could exhibit weak clinical efficacy as compared to multi-target drugs [11, 12]. Although, most of the effective drugs are multi-target ligands and affect diversity of molecular mechanisms[13-16]. Examples of multi-targeting anti-cancer drugs include Lapatinib and Duvelisib, which is a reversible, ATP-competitive inhibitor of the human epidermal growth factor receptor 2 (HER2) and epidermal growth factor receptor (EGFR) tyrosine kinases [17] and a dual inhibitor of PI3K-δ and PI3K-γ,showed promising clinical efficacy in advanced hematologic malignancies [18]. The dilemma of choosing single or multiple target drug can be solved by prior screening of compounds with biochemical and cell-based assays one by one.
HTS demands the development of robust assays that provide high signal-to-noise ratios, which can be applied to small volumes as well. Therefore, cell based in vitro assay are more relevant to biological phenotypes to predict the therapeutic response in a quick and effective manner. Cell-based assays include in vitro toxicity assay, RNAi, second messenger, cell proliferation, and reporter assays [19]. Cell-based assays have several advantages over cell-free biochemical assays in many aspects such as resemblance to the clinical physiological state, less expensive, and real selectivity of compounds that can cross cell membrane to reach target sites. Both screening methods have immensely contributed to drug discovery, by producing high-quality data (Figure 1) [20]. Depending upon the information available on the disease target and the availability of specific library against that target would decide which stream of assays need to be performed first or both phases can go parallelly. Eventually, all the primary hit compounds passed through the phase I (primary screening) will go to phase II for cell-based assay to see its pharmacokinetics and pharmacodynamics studies.
Here, we review in brief the screening methods and their challenges and benefits in HTS. In addition, several recent methodologies will be discussed to develop successful experimental assay and their implementation via primary and secondary screens, and target identification. Further discussion on identifying the most efficacious drugs following these approaches in cancer.
Target-Based Screening
Biochemical assays are based on target where defined/known molecular targets are used to find a lead compound from the library that can efficiently induces/inhibits the target’s activity [21]. Target-based screening utilizes the information from genomics to identify the targets causing disease, which is already available through prior phenotypic studies. Genomic studies also provide functional targets to understand MOA [22]. Therefore, the target-based approach is quite simpler, direct, and specific because of prior knowledge of drug’s MOA availability, which can be easily utilized to understand the interaction of drug with the target in a relatively easy manner as compared to phenotype-based screening approaches. Several ways have been followed to identify MOA such as protein-protein interactions, structural and receptor mediated protein targets, and regulatory factors. These hallmark biochemical assays involve the assessment of enzymatic activity for cell growth, proliferation, differentiation, and metabolism. Drug discovery also utilizes two most popular target classes, one is enzymes and the other one is G-protein coupled receptors (GPCRs). GPCRs are considered as the largest family of targets for approved drugs due to its direct interaction with numerous chemical entities and instigates molecular interactions in the extracellular milieu [23]. Another druggable target is ion channels because of its coupling with the plethora of physiological consequences [24-31]. Biochemical assays include enzymatic kinase assays, voltage-gated ion channels, and serine/cysteine proteases fluorescence resonance energy transfer (FRET), fluorescence correlation spectroscopy (FCS), fluorescence intensity distribution analysis (FIDA), and in vitro transcription assays [32]. Main purpose of these assays to screen out the compounds within compound libraries having the potential to induce enzyme activity. The critical parameter for kinase assay development is the ability to choose an appropriate “readout” technology and the amount of enzymes, kind of cell lines, type of antibodies and the reference compounds. Moreover, these assays need to be neccessarly optimized for reagents, readout time-points amount of buffer conditions, reagent concentrations, timing, stopping, order of addition, plate type and assay volume [33]. Fragment-based screening method is one of the most popular ways to identify drug’s interaction with the specified target protein of interest, that is ligand binding assay. Here, the target of interest has to either isolate and purified for cell free system or recombinantly expressed in a cellular system. Then the library of compounds is screened through various in vitro assays to find selective chemicals which can modulate the activity of the target proteins. Another approach is measurement of binding activity through Nuclear magnetic resonance (NMR) spectroscopy to screen library of compounds. It is a powerful tool for fragment-based drug discovery to discover high-affinity ligands for target proteins [34]. In 2011, FDA approved the first small molecule inhibitor, Vemurafenib originating from a fragment-based screen [35]. Following this robust screening, final lead compounds are rested for their potency in terms of pharmacokinetic properties, enzyme, and cellular activity of the protein/enzyme [36]. Usually, target-based screening assays are simple and produce less variation due to homogeneous nature of reactions with purified proteins. Importantly, these selected compounds not always translate into the same activity in a cellular context or in vivo. Since, the intracellular environment is very complex and crosstalk between various signaling pathways lead to undesired target or misleading targets, cellular impenetrability of the compound, or compound metabolism causing undesirable toxic effects. In spite of these challenges, 70% of successful drugs results from target-based screening that offer new therapeutic options that are precise and personalize to specific mutations or to counter resistance [37]. Imatinib, a tyrosine kinase inhibitor was the first cancer therapies, which showed targeted specificity against BCR-ABL, c-KIT, and PDGFRA proteins [38]. Not only being successful in chronic myeloid leukemia, but it has also shown benefits to steroid-refractory chronic graft-versus-host disease because of its activity against PDGFR action. However, due to the problem of its resistance through Bcr-Abl-dependent and -independent mechanisms, new drugs, such as dasatinib, nilotinib, bosutinib and ponatinib have been developed[39]. A natural anticancer compound, cryptotanshinone, is an abietane diterpenoid functions as an anticancer agent by inhibiting cell proliferation via dual inhibition of pSTAT5 and pSTAT3 that effectively blocks IL-6-mediated STAT3 activation and reverse Bcr-Abl kinase-independent drug resistance in chronic myelogenous leukemia (CML) [40]. There are therapies to treat TNBC-breast cancer irrespective of BRCA status by targeting the neddylation pathway using a selective inhibitor, MLN4924, a selective inhibitor of neddylation enzyme NEDD8 Activation Enzyme (NAE1). It promotes G2-M arrest and apoptosis in CML cells [41, 42].
Phenotype Based Screening
Despite the continued discovery of therapeutic compounds based on target-based drug discovery technologies, recent drug approval list follows cell-based assays, which represent approximately half of all high-throughput screens currently performed. Phenotype base screening offers a way advantages over target-based methods due to discovery of multiple protein-level targets or hits and leads in complex biological systems without prior knowledge of a direct molecular target [20].
In recent years, cell-based assays focused on the modulation of a cellular phenotype in a way that has biological relevance to the disease. Instead, these assays identify modulators of a pathway of interest in more physiological environment that mimics some aspect of disease. This approach facilitates to understand the MOA for the disease with additional layer of information instead of single-target based biochemical approach [43]. Multiple endpoints at various levels are determined by primary endpoints and secondary endpoints. Primary endpoints e.g., cell viability and proliferation are followed by secondary endpoints. Once primary endpoints are successfully determined, then functional assays such as second messenger mobilization (intracellular calcium fluxes or cAMP) after GPCR activation [44, 45], reporter gene assays, cell migration, cytokinesis are performed [46-48].
A big question arise is that how do we know the phenotypic drug screen is a good one and what makes a good phenotypic drug screen (Figure 2)? To answer this, in 2015, Vincent et al. coined the phenotypic screening “rule of 3” that emphasized on three specific criteria to design the most predictive phenotypic assay [49]. First is the relevance of the assay system to the disease that highlights the importance of physiological relevance in the assay systems. Advance in vitro preclinical studies results translate to clinical outcomes by widely using the primary cells and induced pluripotent stem cell (iPSC)–derived cells that are considered to be more representative of human physiology [50]. First HTS study by the group McNeish et al, utilizing murine embryonic stem cell-derived neurons to screen around 2 million compound-library for α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) subtype glutamate receptors showed human translatability by showing a similar rank order in hit potency [51]. Hence, the phenotypic assays predict more translatability to clinical outcome and helps to predict the clinical therapeutic response to a drug. Second criteria is the relevance of the stimulus to achieve the production of the desired phenotype. The purpose of stimuli is to engage specific signaling pathways related to the disease caused by the imbalance of the crosstalk between signaling. This can be achieved by focusing on the highly disease-relevant biological systems possessing the same genetic background as depicted by iPSCs [52-54]. Lee et al., 2012, have tested 6,912 small-molecule compounds screening in neural crest precursors derived-iPSCs from individuals with familial dysautonomia (FD). FD is caused by a single point mutation in the I-κ-B kinase complex-associated protein (IKBKAP) gene. They found a potential hit SKF-86466, was able to rescue IKBKAP transcription through intracellular cAMP levels modulation [54]. Another study on Cystic fibrosis that is the result of mutations in the CF transmembrane conductance regulator (CFTR) gene required for an epithelial chloride channel has aimed to correct the processing of F508del-CFTR with small molecules e.g., VX-809, a CFTR corrector. It improves F508del-CFTR processing and folding in the endoplasmic reticulum and trafficking to the cell surface [53].
If the mechanism and the root cause of the disease is not well understood, then the efforts should be made to induce such effect by using inflammatory or cytotoxic agents to recreate a cellular injury of interest. The last criterion is to determine the assay readout endpoint relevant to the clinical end point. To address this concern, more complex cell models and a range of microphysiological systems have emerged that better recapitulate in vivo physiological characteristics. In this regard, patient-derived organoids give hope to personalized medicine because of their ability to mimic at least some function of an organ and can be derived from iPSCs or donor tissue. In extension to this model, whole organism-based screens provide an additional level of physiological relevance beyond cell-based assays. For example, utilizing those organisms that can readily adapted to growth e.g., yeast and bacteria have been conducted extensively in the pharmaceutical and biotechnology industries [55-59]. Secondary to this step is utilizing multicellular organisms such as zebrafish, worms, and plants, where cell-to-cell communication and multi-dimensional tissue organization could implicate more relevance to human [59-63]. For example, a large-scale screening of 16,000 compound library has identified a compound, persynthamide (psy, 1), that delayed S-phase of the cell-cycle and suppressed bmyb-dependent mitotic defects [64]. These models not only help to understand the developmental process but also pull outs the mechanisms of the disease e.g., chemical genetic analysis using zebrafish embryos to identify the role of PI3K and ERK in artery/vein specification to decide the arteries or veins cell-fate from angioblasts by using GS4898, a known PI3K inhibitor [65]. This study utilized zebrafish model very beautifully to screen small molecules against PI3K inhibitors from the DiverSet E library (Chembridge). Screening was performed in 96-well plates containing 1, 2-celled stage and 6-somite stage embryo derived from zebrafish with a mutation (grlm145) in hey2/gridlock that lack trunk and tail circulation because of an aortic dysmorphogenesis that resembles congenital aortic coarctation in humans.
Translational biomarkers can be considered as an clinically end point assay readouts to predict efficacy in patients [66] but the limitation is biomarkers are not a remedy because of difficulty to identify and validate for a range of target hits [67, 68]. Most recent advancements in new disciplines such as genomics, proteomics, transcriptomics, metabonomics, bioinformatics and system biology could impact the understanding of disease due to either loss or gain of function or activation of genes, that can be measured by CRISPR technologies [69, 70]. Another approach that supports drug discovery is RNAi that helps in hit selection, lead optimization and development of animal models. For gene editing, CRISPR-Cas9 technology is being used without altering the DNA. Integrated genomic screening approach using CRISPR–Cas9 and RNAi to induce loss of function has been used to identify new potential therapeutic targets in a rare sarcoma [71]. In this study, Hong et. al., has developed a patient-derived cancer cell line, CLF-PED-015-T from a patient with a rare undifferentiated sarcoma that holds most of the somatic genetic alterations found in a metastatic lesion isolated. They used other complimentary methods such as CRISPR-Cas9 and RNAi mediated loss-of-function screens to identify CDK4 and XPO1 as druggable cancer targets. They performed small-molecule screen in CLF-PED-015-T using a library of small molecules from Broad Institute Informer Set containing 72 drugs approved by the US Food and Drug Administration (FDA), 100 clinical candidates and 268 biologically active chemical probes to screen for antiproliferative effects in a cell line derived from the sarcoma.
Challenges to Phenotypic Screening
Phenotypic screening often suffers from a very potential disadvantage that is target deconvolution, which is an important step for understanding compound MOA. Phenotypic screening provides identification of multiple off-targeted proteins or pathways, which might not link to a given biological output. These multiple target identification can generate a spectrum of possible other associated targets. This process is termed as ‘target deconvolution’ that arises due to possible interaction of small molecules with multiple cellular and/or extracellular targets or non-protein targets [72, 73]. However, target deconvolution is not a primary condition or demand for passing any candidate drug from phenotypic screens into late-stage preclinical or even clinical development [74-76]. Advances in the field of chemical proteomics, coupled with in silico approaches, genome sequencing, have greatly improved our ability to determine protein targets and underlying hits from phenotypic screens [77-79]. On one hand, single target based approach is more useful to understand MOA of the drug and bridge the link between modulation of target through a MOA while on the other hand, it could lead to off target drug activity and toxicity and suggest the need of multi-targeted therapies to address complex multifactorial disease mechanisms [80, 81].
Second major challenge is metagenes influencing multiple signaling. Genome-wide gene expression profiling measures gene activities (expressions) in the given cell for a certain time frame that helps to understand human diseases at the molecular level [82, 83]. Molecular targets can be recognized by the consistent pattern of gene expression across large cohorts of human samples, which provide insights to evaluate treatment-related phenotypic changes on the molecular basis. Indeed, distinct tumor subtypes in the same histological environment may present differential response that can cause remarkable hurdle for drug development and treatment implications [84]. The reason behind the differential response to therapies could be the heterogeneous patterns of gene expression called metagenes, which influence multiple physiological properties tumor via various signaling e.g., RAS, Myc pathways regulates the degree of epithelial-to-mesenchymal transition [85-87]. Rising incidences of drug resistance, and limited efficacy against complex diseases exacerbate the difficulties of drug discovery. To ease these bottlenecks, drug discovery started thinking of using polypharmacology as an alternative new therapeutic strategy, where therapeutic molecules are being used that can be recognized by multiple biomolecular targets [88]. However, the limitation of using this strategy involve toxicological concerns or side-effects. These complexities in molecular patterns reveal the need of more efficient understanding of drug MOA at phenotypic and pathway levels for drawing correct diagnostic and prognostic decisions.
Screening for Genotoxic Compounds
Genotoxicity in the cancer cells can be induced by the treatment of chemotherapeutic agents, e.g., chlorambucil, cyclophosphamide, cisplatin, oxalaplatin, 5-fluorouracil, anthracyclines, methotrexate, and topoisomerase inhibitors, to induce cell death [89, 90]. In 2009, Ji et al., has developed a DNA-repair pathways-deficient chicken DT40 cell lines for high-throughput genotoxicity screening that have effective gene targeting, stable phenotype and karyotype and are easy to maintain in suspension culture [91-94]. However, there several other phenotypic assays for screening out genotoxic compounds include GreenScreen HC GADD45a-green fluorescent protein (GFP) (BlueScreen HC , CellCiphr p53, and CellSensor p53-bla [95-98]. A study tested 320 predominantly pesticide active compounds in Phase I of US. Environmental Protection Agency’s ToxCast research project. These systems utilize p53 or DNA-damage associated genes due to their role in a genotoxic stress causing the arrest of cell cycle at the G1/S phase until repair is effective, otherwise cells will go under apoptosis. Gentronix ‘GreenScreen HC’ assay utilizes the fact of genotoxin-induced transcription of GADD45a (growth arrest and DNA damage) gene in human lymphoblastoid TK6, a karyotypically stable cell line[99]. This assay measures cell’s response to genotoxic stress, which is linked to the GFP reporter having p53 regulatory elements that ensure specific and dose-dependent response from the gene reporter [97]. The cellular systems biology (CSB™) approach (Cellumen ‘CellCiphr’ profile) is used to measure DNA damage-induced cytotoxicity in human cellular hepatic cell line HepG2 by activating p53 via a fluorescent anti-p53 antibody [100]. Another p53 dependent beta-lactamase reporter assay is Invitrogen ‘CellSensor’ assay to measure cytotoxicity in HCT-116 cells [101, 102]. Lack of metabolic activation and the removal of genotoxic lesions by the DNA repair system could lead to high false-negative [96]. Therefore, ATAD5-luciferase assay developed by Fox et al. 2012, which is based on the ATAD5 protein expression levels upon DNA damage [103, 104]. ATAD5 is a suppressor of direct repeat recombination [105]. Though this assay, 22 antioxidant-compounds have been identified including potential chemotherapeutic agents (resveratrol, genistein, and baicalein), that offer improvements over conventional cancer drugs [103]. In 2018, Sylora et al., has developed an advanced the single cell gel electrophoresis (SCGE) assay coupled with data processing software (CometChip Platform) to identify and characterize genotoxic agents in large compound libraries. This tool can be applied to determine sensitivity to genotoxic exposures in human populations for epidemiological studies [106].
Application in Tumor Chemoresistance and Cancer Treatment
Cancer treatment suffers an obstacle due to resistance to chemotherapeutic agents because tumor cells develop a multidrug resistant (MDR) phenotype through changing the expression of transporter proteins e.g., ATP-binding cassette (ABC) superfamily that regulate intracellular drug concentrations or increasing repair of drug induced damage [107-110]. It has been observed in a study on mdr1a(-/-) mice that P-glycoprotein (P-gp) hampers the oral uptake of paclitaxel [111]. There were several efforts made to generate inhibitors to reverse chemoresistance caused by overexpression of high molecular weight surface P-gp [112, 113]. In clinical trials with third generation modulators such as LY335979, R101933 and XR9576, showed better accumulation of Pgp substrate Tc-99m Sestamibi, an imaging agent in a subset of patients [114-116]. However, these inhibitors didn’t perform well in the clinical trials due to low bioavailability, unexpected secondary physiological effects, and unanticipated drug-drug interactions [108, 117]. Therefore, several assays have been designed using human MDR cell lines to aid in the discovery of novel inhibitors against efflux pump P-gp coded by the MDR1/ABCB1 gene and other ABC transporters. Drug sensitive and resistant human myeloma cell lines, 8226/S and 8226/Dox6 were used to demonstrate association of P-gp overexpression with resistance to glucocorticoid, etoposide, doxorubicin, and vincristine [118]. Studies have identified several compounds such as mometasone furoate, NSC23925, NSC77037, pimozide, acacetin and loxapine as tolerable and effective therapy to reverse MDR [119-122].
Cancer stem cell (CSC) can differentiate into other cell types of cells in the body due to phenotypic plasticity following transcriptional reprogramming driven by an evolutionarily conserved starvation response causing cancer progression and recurrence [123]. This leads to high heterogeneity among tumor tissue environments causing drug resistance [124]. Transcriptional reprogramming helps cancer cells not only to escape from host immune defense system but also stimulate invasion, proliferation and metastasis [125, 126]. This heterogeneity in the tumor microenvironment creates complexity in the signaling pathways due to unstable and altered genetic and epigenetic profiles that cause drug discovery process more difficult [127, 128]. Genetical modifications and several key pathways such as Wnt, phosphoinositide 3’-kinase signaling pathway (PI3K) signaling, interferons (IFNs), and Erk/MAPK signaling are involved in this reprogramming [129-133]. A detailed study by Van Keymeulen et al., 2015, has demonstrated how tumor heterogeneity is determined by the cancer cell of origin using Cre-inducible Pik3caH1047R knock-in mice, specifically in basal cells (BCs) using K5-CreERT2 mice [129]. They showed deletion of p53 and expression of Pik3ca(H1047R) accelerated more aggressive tumor development. Also, they determined its role in cancer cell fate switches depending upon its expression in basal and luminal cells. Stemness of cancer cells is also controlled by Wnt signaling that promotes migration and invasion of nasopharyngeal carcinoma (NPC) by upregulating expression of WNT5A in biopsies samples of metastatic NPC tissues [134]. To study Wnt signaling role in cancer treatment, cell lines were prepared to constitutively show the activated Wnt signaling [135, 136]. To make this cell line, inducible HEK cell line was used, which express Dvl2 fusion with estrogen receptor (ER) and luciferase with GFP genes under the control of a minimal c-Fos promoter with TCF-binding sites and Xnr3 enhancer. This cell line helps to screen β-catenin inhibitor such as CCT031374, CCT036477, and CCT7070535 that block β-catenin induction upon estradiol addition. These compounds were not only able to block the induction of β-catenin activity but also reduced the proliferation of colorectal cancer cell lines (HCT116, HT29 and SW480), and induced caspase 3 activation in HCT116 cells.
EGFR signaling controls cellular and molecular pathways and its alteration leads to carcinogenesis. This receptor tyrosine kinase belongs to ERBB family of receptors, and its activation regulates KRAS and PI3K/PTEN pathways.
HTS study to screen Ras-MAPK inhibitors against mitogen-activated protein/extracellular signal-regulated kinase 1 (MEK1) identified two series of substituted 4-anilino-3-quinolinecarbonitriles as potent MEK1 inhibitors [137]. Torrance et al., identified Triphenyl tetrazolium (TPT) and a sulfinyl cytidine derivative (SC-D) as novel KRAS pathways inhibitors in DLD-1 colorectal cancer cells [138].
A most recent study by Liu et al., has shown how to overcome the resistance to a MEK inhibitor trametinib through epigenetic reprogramming using histone deacetylase (HDAC) inhibitors e.g., trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), LBH589, and PXD101 in advanced ovarian cancer. They performed combinatorial drug screen with a customized epigenetics compound library from Selleck Chemicals in A2780-R and SKOV3 cells. LBH589 (Panobinostat, HDAC inhibitor)) in combination with trametinib retarded tumor growth in SKOV3 xenograft model [139]. Their findings demonstrated that HDAC inhibitors overcome resistance through the suppression of ERK restoration.
PI3K signaling is involved in multiple cellular processes including cell survival and cell death mechanisms. In a study by Li et al., identified N’-[(1-benzyl-1H-indol-3-yl)methylene]benzenesulfonohydrazide (CID1340132) as a novel compound to induce apoptosis preferentially in PTEN and PIK3CA mutant human cancer cells through cell based drug screening [140].
IFNs (IFN-α and IFN-β) plays key role in cancer and several chronic diseases after binding to its receptors IFNAR1 and IFNAR2. This receptor complex activates Janus activated kinases (JAKs) and signal transducers and activators of transcription (STAT) pathways. HTS against type-I IFN with the secreted embryonic alkaline phosphatase (SEAP) reporter gene assay involving 32,000 compounds resulted 25 confirmed hits. Further screening by cytotoxicity assay in neuroblastoma cell line SH-SY5Y showed two hits (CD2093-G007 and RUS0903-C006) that decreased viability to less than 50%. Finally, RUS0910-G009 exhibited inhibition of STAT1 and STAT3 phosphorylation and suppression of IRF7 mRNA transcription [141].
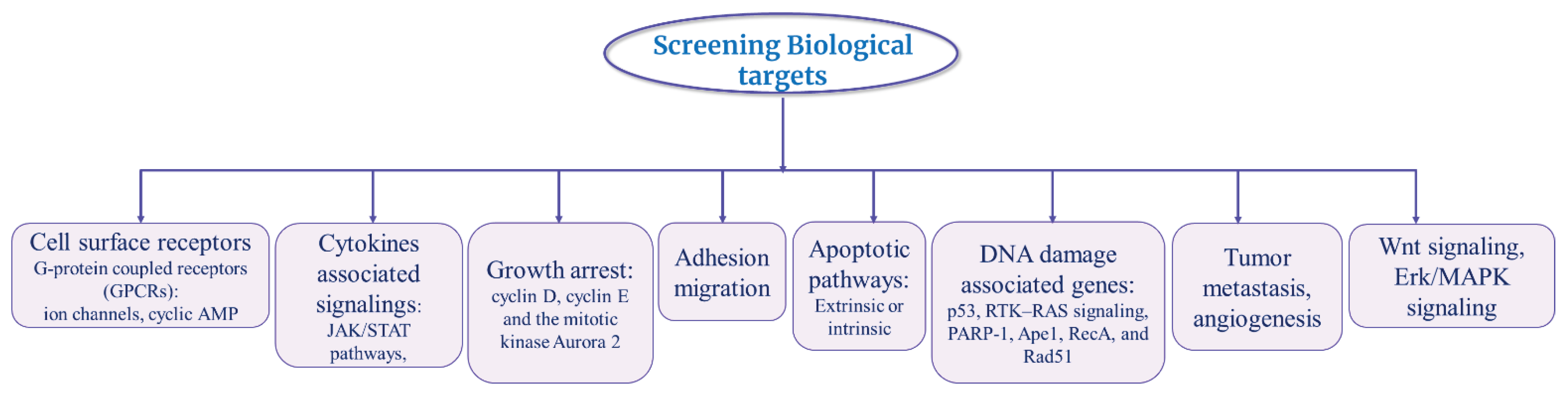
Several investigations have been going on finding DNA repair factors through HTS assays to screen inhibitors targeting PARP-1, Ape1, RecA, and Rad51 [142-146]. Dillon et al., has developed a novel FlashPlate scintillation proximity assay HTS of large compound libraries to identify inhibitors of PARP-1 [142]. Further on, Peterson et al., has developed a robust method to screen large scale of small molecules in HTS against RecA using commercial reagents (Transcreener(®) adenosine 5’-O-diphosphate [ADP](2) fluorescence polarization assay) [144]. Therefore, identification of biomarkers of these reprogramming at the level of gene and protein could help to explain MOA of different tumor variant types in different individual depending on the case (Figure 3) [147].
Additionally, there are other relevant targets or the pathways that can be assessed. For instance, cancer signaling pathways involving the p53, RTK–RAS signaling [148-150] or various cell-cycle regulation targets such as cyclin D, cyclin E and the mitotic kinase Aurora 2 [151, 152]. Mortality rate is higher in breast cancer patients with overexpressing cyclin-dependent kinase 2/cyclin E2 (CDK2/cyclin E2) via r via estrogen receptor pathway [153, 154] Another serine/threonine kinase oncogene, STK15/BTAK (approved gene symbols are Aurora 2, ARK1, AIK1) required for the formation of the mitotic bipolar spindle has been reported to be overexpressed in breast cancers [152]. These targets could be used in future to find more potential and selective cancer drugs. However, a study has shown that targeting Aurora proteins subfamily members such as Aurora A and Aurora B in combination would be ineffective rather each should be treated as autonomously because they have different biological response [155].
Cell cycle is tightly controlled by the regulation of p53 gene on cell cycle kinases that is cyclin-dependent kinase (CDK4/6 and CDK2). Its inactivation or mutation causes dysregulation in proliferation and apoptosis leading to cancer. Therefore, an attempt has been made in drug development to restore its activity by using small molecules modulators (CP-31398, PRIMA1 and Nutlins) in p53 deficient human colon tumor xenografts (HCT116/p53(-/-) or DLD1) [156]. These compounds were able to reactivate p53 reporter activity via increasing its targeted genes such as p21 (WAF1) and death receptor 5 (DR5).
Similarly, heat-shock protein 90 (HSP90) is an important molecular chaperone found to be overexpressed in patients with cancer [157, 158]. HIF-1 is considered to be an important signaling pathway playing a crucial role in tumor progression and angiogenesis via upregulating kinases and hypoxia-inducible factor-1 (HIF-1) [159-161]. Other enzymes playing role in DNA modification such as histone acetyltransferases and histone deacetylases enzymes play critical role in chromatin modification required for post-translational modifications to catalyze acetylation and deacetylation of histones on specific lysine residues. It has possible role in the pathology of cancer, asthma, and viral infection [162-165].
Moreover, genome-based strategy has already been successfully highlighted the use of Herceptin, a humanized antibody to the ErbB2 receptor to treat breast cancer because breast cancer patients overexpress [166]. Other example is farnesyltransferase inhibitor R115777 that showed efficacy in the treatment of human breast cancer xenografts [167] and in patients with advanced breast cancer [168]. Hopefully, further understanding of genetic versatility of breast cancer would highlight more druggable targets for HTS. On the same note, compounds either in combination or alone has demonstrated superior efficacy and tolerability than standard endocrine therapy, e.g., tamoxifen, in clinical trials for the treatment of breast cancer e.g., aromatase inhibitor Arimidex (anastrazole) in hormone receptor-positive breast cancer in post-menopausal women [169, 170]. All these studies suggest HTS should be designed very carefully to determine effective compounds for clinical usage.
Conclusion
Different kind of drug screening methods in HTS have provided several potent compounds that are being used in treatment procedures. Even though these drug selection assays are not fully well developed to produce desired endpoint relevant to the disease phenotype. Therefore, this review highlighted the importance of choosing appropriate endpoints to develop and mimic more to the desired phenotype to select efficacious drug. However, new technologies such as genomics and proteomics provides abundant knowledge of the targets and their signaling but we still need to find out clear specific signaling axis that led to correct clinical outcome and provide innovative new agents to the cancer clinic for patient benefit.

Table.
List of targets and compounds/ inhibitors screened through high-throughput screen against oncogenic pathways.
Table.
List of targets and compounds/ inhibitors screened through high-throughput screen against oncogenic pathways.
| Druggable target | Compounds | Ref. |
|---|---|---|
| P-glycoprotein | LY335979, R101933 and XR9576 | [114-116] |
| Mometasone furoate, NSC23925, NSC77037, pimozide, acacetin and loxapine | [119-122] | |
| β-catenin (Wnt signaling) | CCT031374, CCT036477, and CCT7070535 | [135, 136] |
| MEK1 | Class 1 and class 2 substituted 4-anilino-3-quinolinecarbonitriles | [137] |
| HDAC | Trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), LBH589, and PXD101 | [139] |
| KRAS | Demethoxyviridin, mithramycin, triphenyl tetrazolium (TPT), sulfinyl cytidine derivative (SC-D) | [138] |
| PI3K/PTEN | N’-[(1-benzyl-1H-indol-3-yl)methylene]benzenesulfonohydrazide (CID1340132) | [140] |
| JAK/STAT | RUS0910-G009 | [141] |
| p53 | CP-31398, PRIMA1 and Nutlins | [156] |
Author Contributions
Conceptualization, RR and SKS; writing-original draft preparation, RR and SKS; writing—review and editing, RR, SKS and SM. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
No possible conflict of interest.
References
- Macarron, R. , et al., Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 2011, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Wigglesworth, M.J. , et al., Increasing the delivery of next generation therapeutics from high throughput screening libraries. Curr Opin Chem Biol 2015, 26, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A. and J.A. Williams, Origin and evolution of high throughput screening. Br J Pharmacol 2007, 152, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Shun, T.Y. , et al., Identifying actives from HTS data sets: practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J Biomol Screen 2011, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dreiman, G.H.S. , et al., Changing the HTS Paradigm: AI-Driven Iterative Screening for Hit Finding. SLAS Discov 2021, 26, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M. , et al., Integration of genomics, high throughput drug screening, and personalized xenograft models as a novel precision medicine paradigm for high risk pediatric cancer. Cancer Biol Ther 2018, 19, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M. and D. Bojanic, Novel trends in high-throughput screening. Curr Opin Pharmacol 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Szymański, P., M. Markowicz, and E. Mikiciuk-Olasik, Adaptation of high-throughput screening in drug discovery-toxicological screening tests. Int J Mol Sci 2012, 13, 427–452. [Google Scholar] [PubMed]
- Milinkovic, A. and E. Martínez, Nevirapine in the treatment of HIV. Expert Rev Anti Infect Ther 2004, 2, 367–373. [Google Scholar] [CrossRef]
- FDA approved drugs generated from high throughput screening efforts I - 20190719. 2019.
- Margineanu, D.G. , Systems biology, complexity, and the impact on antiepileptic drug discovery. Epilepsy Behav 2014, 38, 131–42. [Google Scholar] [CrossRef]
- Kell, D.B. , Finding novel pharmaceuticals in the systems biology era using multiple effective drug targets, phenotypic screening and knowledge of transporters: where drug discovery went wrong and how to fix it. Febs j 2013, 280, 5957–5980. [Google Scholar] [CrossRef] [PubMed]
- Ericson, E. , et al., Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet 2008, 4, e1000151. [Google Scholar] [CrossRef] [PubMed]
- Lim, H. , et al., Large-Scale Off-Target Identification Using Fast and Accurate Dual Regularized One-Class Collaborative Filtering and Its Application to Drug Repurposing. PLoS Comput Biol 2016, 12, e1005135. [Google Scholar] [CrossRef] [PubMed]
- Lin, A. , et al., Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 2019. 11(509).
- Koeberle, A. and O. Werz, Multi-target approach for natural products in inflammation. Drug Discov Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E. , et al., Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W. , et al., Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Murai, R. , et al., A novel screen using the Reck tumor suppressor gene promoter detects both conventional and metastasis-suppressing anticancer drugs. Oncotarget 2010, 1, 252. [Google Scholar] [CrossRef]
- An, W.F. and N. Tolliday, Cell-based assays for high-throughput screening. Mol Biotechnol 2010, 45, 180–186. [Google Scholar] [CrossRef]
- Takenaka, T. , Classical vs reverse pharmacology in drug discovery. BJU Int 2001. 88 Suppl 2: p. 7-10; discussion 49-50.
- Lage, O.M. , et al., Current Screening Methodologies in Drug Discovery for Selected Human Diseases. Mar Drugs 2018, 16. [Google Scholar]
- Sriram, K. and P.A. Insel, G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Kenny, C.H. , et al., Development of a fluorescence polarization assay to screen for inhibitors of the FtsZ/ZipA interaction. Analytical biochemistry 2003, 323, 224–233. [Google Scholar] [CrossRef]
- Burns, S. , et al., Identification of Small-Molecule Inhibitors of Protein Kinase B (PKB/AKT) in an AlphaScreen™ High-Throughput Screen. SLAS Discovery 2006, 11, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K. , et al., High-throughput screening of low molecular weight NS3-NS4A protease inhibitors using a fluorescence resonance energy transfer substrate. Antiviral Chemistry and Chemotherapy 2005, 16, 385–392. [Google Scholar] [CrossRef]
- Swaney, S. , et al., Characterization of a high-throughput screening assay for inhibitors of elongation factor P and ribosomal peptidyl transferase activity. Journal of biomolecular screening 2006, 11, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Allen, M., J. Reeves, and G. Mellor, High throughput fluorescence polarization: a homogeneous alternative to radioligand binding for cell surface receptors. Journal of biomolecular screening 2000, 5, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. , et al., Ion-channel assay technologies: quo vadis? Drug Discovery Today 2001, 6, 1278–1287. [Google Scholar] [CrossRef]
- Parker, G.J. , et al., Development of high throughput screening assays using fluorescence polarization: nuclear receptor-ligand–binding and kinase/phosphatase assays. SLAS Discovery 2000, 5, 77–88. [Google Scholar] [CrossRef]
- Bagal, S.K. , et al. , Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem 2013, 56, 593–624. [Google Scholar]
- Morachis, J.M., R. Huang, and B.M. Emerson, Identification of kinase inhibitors that target transcription initiation by RNA polymerase II. Oncotarget 2011, 2, 18–28. [Google Scholar] [CrossRef]
- Glickman, J.F. , Assay Development for Protein Kinase Enzymes, in Assay Guidance Manual, S. Markossian, et al., Editors. 2004, Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda (MD).
- Harner, M.J., A. O. Frank, and S.W. Fesik, Fragment-based drug discovery using NMR spectroscopy. J Biomol NMR 2013, 56, 65–75. [Google Scholar] [CrossRef]
- Bollag, G. , et al., Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. , Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J Med Chem 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Chen, G.Q. , et al., Phenotype and target-based chemical biology investigations in cancers. Natl Sci Rev 2019, 6, 1111–1127. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N. and N. Iqbal, Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract 2014, 2014, 357027. [Google Scholar] [PubMed]
- Rossari, F., F. Minutolo, and E. Orciuolo, Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol 2018, 11, 84. [Google Scholar] [PubMed]
- Dong, B. , et al., Cryptotanshinone suppresses key onco-proliferative and drug-resistant pathways of chronic myeloid leukemia by targeting STAT5 and STAT3 phosphorylation. Sci China Life Sci 2018, 61, 999–1009. [Google Scholar] [CrossRef]
- Liu, C. , et al., Antitumor Effects of Blocking Protein Neddylation in T315I-BCR-ABL Leukemia Cells and Leukemia Stem Cells. Cancer Res 2018, 78, 1522–1536. [Google Scholar] [CrossRef]
- Misra, S. , et al., Both BRCA1-wild type and -mutant triple-negative breast cancers show sensitivity to the NAE inhibitor MLN4924 which is enhanced upon MLN4924 and cisplatin combination treatment. Oncotarget 2020, 11, 784–800. [Google Scholar] [CrossRef]
- Krejci, P., K. Pejchalova, and W.R. Wilcox, Simple, mammalian cell-based assay for identification of inhibitors of the Erk MAP kinase pathway. Investigational New Drugs 2007, 25, 391–395. [Google Scholar] [CrossRef]
- Chambers, C. , et al., Measuring intracellular calcium fluxes in high throughput mode. Combinatorial chemistry & high throughput screening 2003, 6, 355–362. [Google Scholar]
- Kariv, I. , et al., High throughput quantitation of cAMP production mediated by activation of seven transmembrane domain receptors. Journal of biomolecular screening 1999, 4, 27–32. [Google Scholar] [CrossRef]
- Eggert, U.S. , et al. , Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS biology 2004, 2, e379. [Google Scholar]
- Beck, V., A. Pfitscher, and A. Jungbauer, GFP-reporter for a high throughput assay to monitor estrogenic compounds. Journal of biochemical and biophysical methods 2005, 64, 19–37. [Google Scholar] [CrossRef]
- Yarrow, J.C. , et al., Screening for cell migration inhibitors via automated microscopy reveals a Rho-kinase inhibitor. Chemistry & biology 2005, 12, 385–395. [Google Scholar]
- Vincent, F. , et al., Developing predictive assays: the phenotypic screening "rule of 3". Sci Transl Med 2015, 7, 293ps15. [Google Scholar] [CrossRef]
- Engle, S.J. and F. Vincent, Small molecule screening in human induced pluripotent stem cell-derived terminal cell types. J Biol Chem 2014, 289, 4562–4570. [Google Scholar] [CrossRef]
- McNeish, J. , et al., High-throughput Screening in Embryonic Stem Cell-derived Neurons Identifies Potentiators of α-Amino-3-hydroxyl-5-methyl-4-isoxazolepropionate-type Glutamate Receptors. Journal of Biological Chemistry 2010, 285, 17209–17217. [Google Scholar] [CrossRef]
- Ashlock, M.A. and E.R. Olson, Therapeutics development for cystic fibrosis: a successful model for a multisystem genetic disease. Annu Rev Med 2011, 62, 107–25. [Google Scholar] [CrossRef]
- Van Goor, F. , et al., Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Lee, G. , et al., Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol 2012, 30, 1244–1248. [Google Scholar] [CrossRef]
- Barberis, A. , et al., Yeast as a screening tool. Drug Discovery Today: Technologies 2005, 2, 187–192. [Google Scholar] [CrossRef]
- Balgi, A.D. and M. Roberge, Screening for chemical inhibitors of heterologous proteins expressed in yeast using a simple growth-restoration assay, in Cell-Based Assays for High-Throughput Screening. 2009, Springer. p. 125-137.
- Puri, A.W. and M. Bogyo, Using small molecules to dissect mechanisms of microbial pathogenesis. ACS chemical biology 2009, 4, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Zlitni, S., J. E. Blanchard, and E.D. Brown, High-throughput screening of model bacteria, in Cell-Based Assays for High-Throughput Screening. 2009, Springer. p. 13-27.
- Hong, C.C. , Large-scale small-molecule screen using zebrafish embryos, in Cell-Based Assays for High-Throughput Screening. 2009, Springer. p. 43-55.
- Zon, L.I. and R.T. Peterson, In vivo drug discovery in the zebrafish. Nature reviews Drug discovery 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J., A. L. Conery, and T.I. Moy, Whole-animal high-throughput screens: the C. elegans model, in Cell-Based Assays for High-Throughput Screening. 2009, Springer. p. 57-75.
- Agee, A. and D. Carter, Whole-organism screening: plants, in Cell-Based Assays for High-Throughput Screening. 2009, Springer. p. 77-95.
- Norambuena, L., N. V. Raikhel, and G.R. Hicks, Chemical genomics approaches in plant biology. Plant Systems Biology, 345–354.
- Stern, H.M. , et al., Small molecules that delay S phase suppress a zebrafish bmyb mutant. Nat Chem Biol 2005, 1, 366–370. [Google Scholar] [CrossRef]
- Hong, C.C. , et al., Artery/Vein Specification Is Governed by Opposing Phosphatidylinositol-3 Kinase and MAP Kinase/ERK Signaling. Current Biology 2006, 16, 1366–1372. [Google Scholar] [CrossRef]
- Swinney, D.C. , The value of translational biomarkers to phenotypic assays. Front Pharmacol 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P. , Biomarker failures. Clin Chem 2013, 59, 202–204. [Google Scholar] [CrossRef]
- Chau, C.H. , et al., Validation of analytic methods for biomarkers used in drug development. Clin Cancer Res 2008, 14, 5967–5976. [Google Scholar] [CrossRef]
- Spreafico, R. , et al., Advances in Genomics for Drug Development. Genes (Basel) 2020, 11. [Google Scholar] [CrossRef]
- Guo, J.B. and X.J. Li, [Impacts of modern biology on drug discovery]. Sheng Li Ke Xue Jin Zhan 2007, 38, 25–31. [Google Scholar]
- Hong, A.L. , et al., Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun 2016, 7: p. 11987.
- Lin, X. , et al., Life beyond kinases: structure-based discovery of sorafenib as nanomolar antagonist of 5-HT receptors. Journal of medicinal chemistry 2012, 55, 5749–5759. [Google Scholar] [CrossRef] [PubMed]
- Schneidewind, T. , et al., Morphological profiling identifies a common mode of action for small molecules with different targets. ChemBioChem 2020, 21, 3197–3207. [Google Scholar] [CrossRef] [PubMed]
- Han, X. , et al., Discovery of RG7834: the first-in-class selective and orally available small molecule hepatitis B virus expression inhibitor with novel mechanism of action. Journal of Medicinal Chemistry 2018, 61, 10619–10634. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G. , et al., Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nature reviews Drug discovery 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F. , et al., Hit triage and validation in phenotypic screening: considerations and strategies. Cell Chemical Biology 2020, 27, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Warchal, S.J., A. Unciti-Broceta, and N.O. Carragher, Next-generation phenotypic screening. Future medicinal chemistry 2016, 8, 1331–1347. [Google Scholar] [CrossRef] [PubMed]
- Comess, K.M. , et al., Emerging approaches for the identification of protein targets of small molecules-a practitioners’ perspective. Journal of medicinal chemistry 2018, 61, 8504–8535. [Google Scholar] [CrossRef] [PubMed]
- Lee, J. and M. Bogyo, Target deconvolution techniques in modern phenotypic profiling. Curr Opin Chem Biol 2013, 17, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-J. , et al., Multi-target drugs: the trend of drug research and development. PloS one 2012, 7, e40262. [Google Scholar]
- Rena, G., D. G. Hardie, and E.R. Pearson, The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Keller, M.P. and A.D. Attie, Physiological insights gained from gene expression analysis in obesity and diabetes. Annual review of nutrition 2010, 30, 341–364. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J. and R. Bernards, Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature 2008, 452, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R. and M. Park, Breast cancer—one term, many entities? The Journal of clinical investigation 2011, 121, 3789–3796. [Google Scholar] [CrossRef] [PubMed]
- Huang, E. , et al., Gene expression phenotypic models that predict the activity of oncogenic pathways. Nature genetics 2003, 34, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A. , et al., A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC medical genomics 2010, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A. , et al., EMT is the dominant program in human colon cancer. BMC medical genomics 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Méndez-Lucio O, N.J. , Vite-Caritino H, Prieto-Martínez FD, Medina-Franco JL., One Drug for Multiple Targets: A Computational Perspective. J Mex Chem Soc. 2016, 60, 168–181. [Google Scholar]
- Hsieh, J.H. , et al., Identifying Compounds with Genotoxicity Potential Using Tox21 High-Throughput Screening Assays. Chem Res Toxicol 2019, 32, 1384–1401. [Google Scholar] [CrossRef]
- Michod, D. and C. Widmann, DNA-damage sensitizers: potential new therapeutical tools to improve chemotherapy. Critical reviews in oncology/hematology 2007, 63, 160–171. [Google Scholar] [CrossRef]
- Evans, T.J. , et al., Mutant cells defective in DNA repair pathways provide a sensitive high-throughput assay for genotoxicity. DNA repair 2010, 9, 1292–1298. [Google Scholar] [CrossRef]
- Ji, K. , et al., A Novel Approach Using DNA-Repair–Deficient Chicken DT40 Cell Lines for Screening and Characterizing the Genotoxicity of Environmental Contaminants. Environmental health perspectives 2009, 117, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Buerstedde, J.-M. and S. Takeda, Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 1991, 67, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, M. , et al., Reverse genetic studies of the DNA damage response in the chicken B lymphocyte line DT40. DNA repair 2004, 3, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, R.M. and M. Tate, The GADD45a-GFP GreenScreen HC assay. Genetic Toxicology: Principles and Methods, 231–250.
- Knight, A.W. , et al., Evaluation of high-throughput genotoxicity assays used in profiling the US EPA ToxCast™ chemicals. Regulatory Toxicology and Pharmacology 2009, 55, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Hastwell, P.W. , et al., High-specificity and high-sensitivity genotoxicity assessment in a human cell line: Validation of the GreenScreen HC GADD45a-GFP genotoxicity assay. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 2006, 607, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.W. , et al., Evaluation of high-throughput genotoxicity assays used in profiling the US EPA ToxCast chemicals. Regul Toxicol Pharmacol 2009, 55, 188–199. [Google Scholar] [CrossRef]
- Walmsley, R.M. and M. Tate, The GADD45a-GFP GreenScreen HC assay. Methods Mol Biol 2012, 817, 231–250. [Google Scholar] [PubMed]
- Giuliano, K.A. , et al., Early safety assessment using cellular systems biology yields insights into mechanisms of action. J Biomol Screen 2010, 15, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Brattain, M.G. , et al., Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res 1981, 41, 1751–1756. [Google Scholar]
- Xing, J.Z. , et al., Microelectronic cell sensor assay for detection of cytotoxicity and prediction of acute toxicity. Toxicology in Vitro 2006, 20, 995–1004. [Google Scholar] [CrossRef]
- Fox, J.T. , et al., High-throughput genotoxicity assay identifies antioxidants as inducers of DNA damage response and cell death. Proceedings of the National Academy of Sciences 2012, 109, 5423–5428. [Google Scholar] [CrossRef] [PubMed]
- Sikdar, N. , et al., DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle 2009, 8, 3199–3207. [Google Scholar] [CrossRef] [PubMed]
- Scholes, D.T. , et al., Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics 2001, 159, 1449–1465. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P. , et al., Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep 2018, 8, 2771. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L. and F. Zunino, Overview of tumor cell chemoresistance mechanisms. Methods Mol Med 2005, 111, 127–48. [Google Scholar]
- Gottesman, M.M., T. Fojo, and S.E. Bates, Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. and V. Ling, The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 2006, 580, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. , ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A. , et al., Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A 1997, 94, 2031–2035. [Google Scholar] [CrossRef]
- Kartner, N., J. R. Riordan, and V. Ling, Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. [Google Scholar] [CrossRef]
- Shen, D.W. , et al., Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem 1986, 261, 7762–7770. [Google Scholar] [CrossRef] [PubMed]
- Roe, M. , et al., Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg Med Chem Lett 1999, 9, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Dantzig, A.H. , et al., Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P-450 activities. J Pharmacol Exp Ther 1999, 290, 854–862. [Google Scholar] [PubMed]
- Mistry, P. , et al., In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res 2001, 61, 749–758. [Google Scholar] [PubMed]
- Bates, S.F. , et al., Reversal of multidrug resistance: lessons from clinical oncology. Novartis Found Symp 2002, 243, 83–96. [Google Scholar] [PubMed]
- Dalton, W. , Detection of multidrug resistance gene expression in multiple myeloma. Leukemia 1997, 11, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Susa, M. , et al., Multidrug resistance reversal agent, NSC77037, identified with a cell-based screening assay. J Biomol Screen 2010, 15, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.S. , et al., High-throughput screening for daunorubicin-mediated drug resistance identifies mometasone furoate as a novel ABCB1-reversal agent. J Biomol Screen 2008, 13, 185–193. [Google Scholar] [CrossRef]
- Duan, Z., E. Choy, and F.J. Hornicek, NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One 2009, 4, e7415. [Google Scholar] [CrossRef]
- Ivnitski-Steele, I. , et al., High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters. Assay Drug Dev Technol 2008, 6, 263–276. [Google Scholar] [CrossRef]
- Gurdon, J.B., T. R. Elsdale, and M. Fischberg, Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature 1958, 182, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Hass, R., J. von der Ohe, and H. Ungefroren, The Intimate Relationship among EMT, MET and TME: AT (ransdifferentiation) E (nhancing) M (ix) to Be Exploited for Therapeutic Purposes. Cancers, 3674. [Google Scholar]
- Saito, S. , et al., Potential application of cell reprogramming techniques for cancer research. Cellular and Molecular Life Sciences.
- Welch, D.R. , Tumor heterogeneity—a ‘contemporary concept’founded on historical insights and predictions. Cancer research 2016, 76, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Denny, S.K. , et al., Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Teng, S. , et al., Tissue-specific transcription reprogramming promotes liver metastasis of colorectal cancer. Cell Res 2020, 30, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A. , et al., Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. , Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.W., Y. Z. Nie, and H. Taniguchi, Cellular reprogramming and hepatocellular carcinoma development. World J Gastroenterol 2013, 19, 8850–8860. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. and P.W. Laird, Cancer epigenetics comes of age. Nat Genet 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, D. , et al., Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef]
- Qin, L. , et al., WNT5A promotes stemness characteristics in nasopharyngeal carcinoma cells leading to metastasis and tumorigenesis. Oncotarget 2015, 6, 10239–10252. [Google Scholar] [CrossRef]
- Ewan, K. , et al., A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res 2010, 70, 5963–5973. [Google Scholar] [CrossRef]
- Bialkowska, A.B. and V.W. Yang, High-throughput screening strategies for targeted identification of therapeutic compounds in colorectal cancer. Future Oncol 2012, 8, 259–272. [Google Scholar] [CrossRef]
- Mallon, R. , et al., Identification of 4-anilino-3-quinolinecarbonitrile inhibitors of mitogen-activated protein/extracellular signal-regulated kinase 1 kinase. Mol Cancer Ther 2004, 3, 755–762. [Google Scholar] [CrossRef]
- Torrance, C.J. , et al. , Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol 2001, 19, 940–945. [Google Scholar]
- Liu, S. , et al., Targeting enhancer reprogramming to mitigate MEK inhibitor resistance in preclinical models of advanced ovarian cancer. J Clin Invest 2021, 131. [Google Scholar]
- Li, H.F. , et al., A high-throughput screen with isogenic PTEN+/+ and PTEN-/- cells identifies CID1340132 as a novel compound that induces apoptosis in PTEN and PIK3CA mutant human cancer cells. J Biomol Screen 2011, 16, 383–393. [Google Scholar] [CrossRef]
- Yuliantie, E. , et al., High-throughput screening for small molecule inhibitors of the type-I interferon signaling pathway. Acta Pharm Sin B 2018, 8, 889–899. [Google Scholar] [CrossRef]
- Dillon, K.J., G. C. Smith, and N.M. Martin, A FlashPlate assay for the identification of PARP-1 inhibitors. J Biomol Screen 2003, 8, 347–352. [Google Scholar] [CrossRef]
- Bapat, A. , et al., Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J Pharmacol Exp Ther 2010, 334, 988–998. [Google Scholar] [CrossRef]
- Peterson, E.J. , et al., High-throughput screening for RecA inhibitors using a transcreener adenosine 5’-O-diphosphate assay. Assay Drug Dev Technol 2012, 10, 260–268. [Google Scholar] [CrossRef]
- Sexton, J.Z. , et al. , Novel Inhibitors of E. coli RecA ATPase Activity. Curr Chem Genomics 2010, 4, 34–42. [Google Scholar]
- Huang, F. , et al., Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 2011, 6, 628–635. [Google Scholar] [CrossRef]
- Huang, L. , et al., High-Throughput Strategies for the Discovery of Anticancer Drugs by Targeting Transcriptional Reprogramming. Front Oncol 2021, 11, 762023. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Ding, L. , et al., Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Jones, S. , et al., Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Steeg, P.S. and Q. Zhou, Cyclins and breast cancer. Breast Cancer Res Treat 1998, 52, 17–28. [Google Scholar] [CrossRef]
- Miyoshi, Y. , et al. , Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer 2001, 92, 370–373. [Google Scholar]
- Dhillon, N.K. and M. Mudryj, Cyclin E overexpression enhances cytokine-mediated apoptosis in MCF7 breast cancer cells. Genes Immun 2003, 4, 336–342. [Google Scholar] [CrossRef]
- Niu, D., G. Wang, and X. Wang, Up-regulation of cyclin E in breast cancer via estrogen receptor pathway. Int J Clin Exp Med 2015, 8, 910–915. [Google Scholar]
- Warner, S.L. , et al., Comparing Aurora A and Aurora B as molecular targets for growth inhibition of pancreatic cancer cells. Mol Cancer Ther 2006, 5, 2450–2458. [Google Scholar] [CrossRef]
- Wang, W., S. H. Kim, and W.S. El-Deiry, Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc Natl Acad Sci U S A 2006, 103, 11003–11008. [Google Scholar] [CrossRef]
- Maloney, A. and P. Workman, HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2002, 2, 3–24. [Google Scholar] [CrossRef]
- Magwenyane, A.M. , et al., Heat Shock Protein 90 (HSP90) Inhibitors as Anticancer Medicines: A Review on the Computer-Aided Drug Discovery Approaches over the Past Five Years. Comput Math Methods Med 2022, 2022, 2147763. [Google Scholar] [CrossRef]
- Bos, R. , et al., Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J Natl Cancer Inst 2001, 93, 309–314. [Google Scholar] [CrossRef]
- Semenza, G.L. , HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002, 8 (Suppl. S4), S62–S67. [Google Scholar] [CrossRef]
- Masoud, G.N. and W. Li, HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Workman, P. , Scoring a bull’s-eye against cancer genome targets. Curr Opin Pharmacol 2001, 1, 342–352. [Google Scholar] [CrossRef]
- Mahlknecht, U. and D. Hoelzer, Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med 2000, 6, 623–644. [Google Scholar] [CrossRef]
- Turlais, F. , et al., High-throughput screening for identification of small molecule inhibitors of histone acetyltransferases using scintillating microplates (FlashPlate). Anal Biochem 2001, 298, 62–68. [Google Scholar] [CrossRef]
- Dekker, F.J. and H.J. Haisma, Histone acetyl transferases as emerging drug targets. Drug Discov Today 2009, 14, 942–948. [Google Scholar] [CrossRef]
- Miles, D.W. , Update on HER-2 as a target for cancer therapy: herceptin in the clinical setting. Breast Cancer Res 2001, 3, 380–384. [Google Scholar] [CrossRef]
- Kelland, L.R. , et al., Preclinical antitumor activity and pharmacodynamic studies with the farnesyl protein transferase inhibitor R115777 in human breast cancer. Clin Cancer Res 2001, 7, 3544–3550. [Google Scholar]
- Johnston, S.R. , Farnesyl transferase inhibitors: a novel targeted tnerapy for cancer. Lancet Oncol 2001, 2, 18–26. [Google Scholar] [CrossRef]
- Nicholls, H. , Aromatase inhibitors continue their ATAC on tamoxifen. Trends Mol Med 2002, 8 (Suppl. S4), S12–3. [Google Scholar] [CrossRef]
- Buzdar, A.U. , Anastrozole (Arimidex)--an aromatase inhibitor for the adjuvant setting? Br J Cancer 2001, 85 (Suppl. S2), 6–10. [Google Scholar]
Figure 1.
Overview of the drug selection process in HTS and workflow to select effective drug.
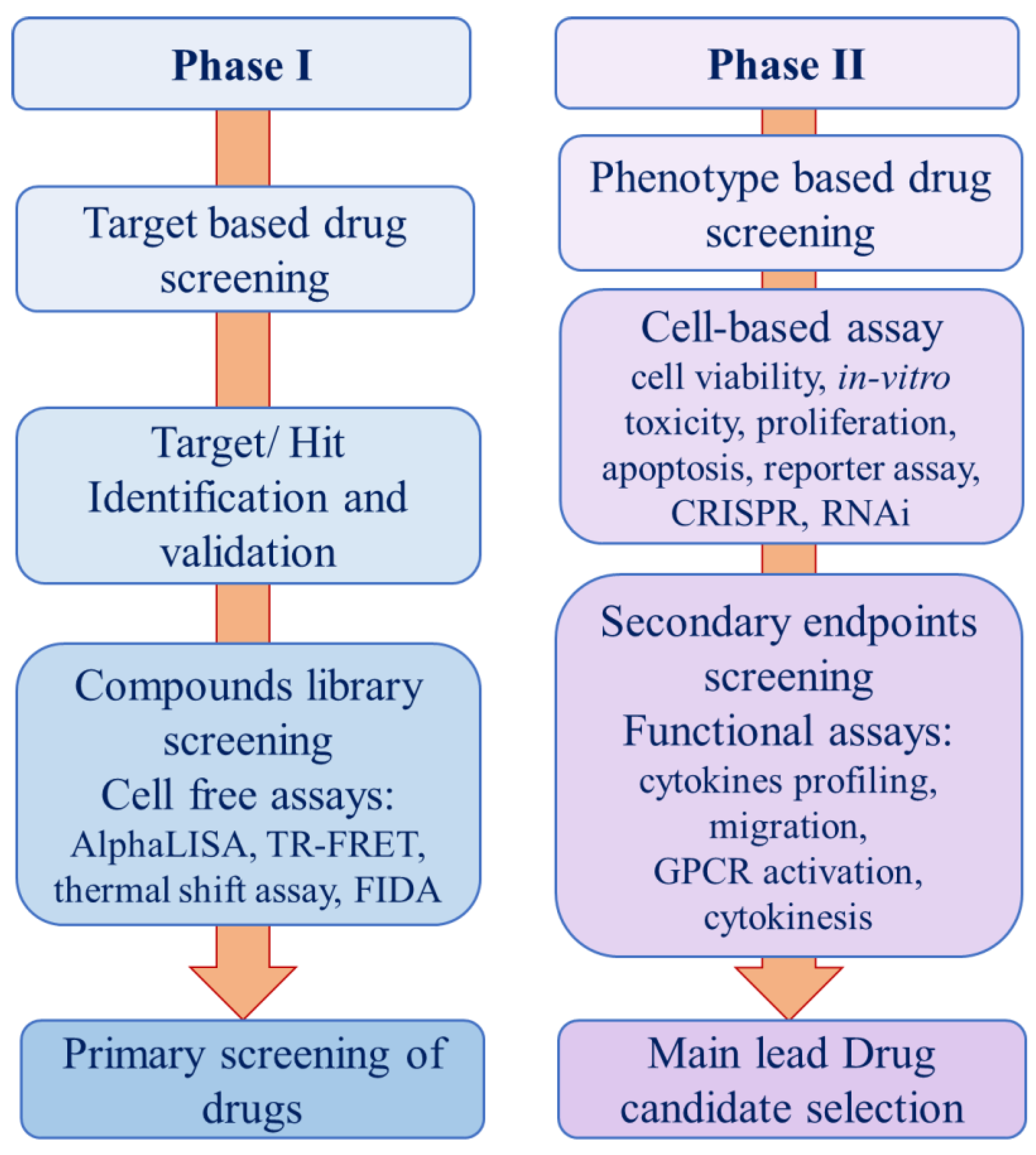
Figure 2.
Drug development challenges and models can be used to identify and validate targets before clinical trials.
Figure 2.
Drug development challenges and models can be used to identify and validate targets before clinical trials.
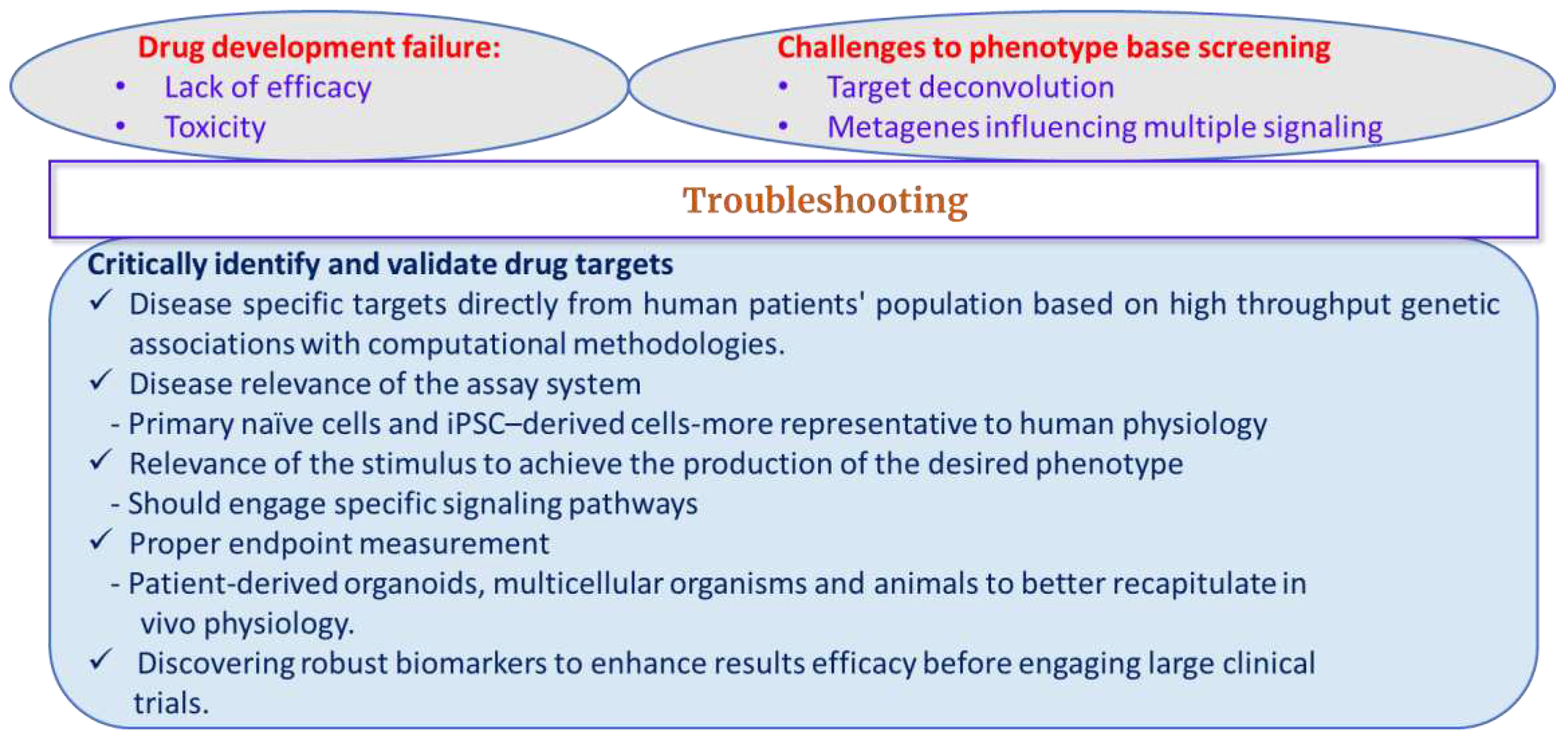
Figure 3.
Promising biological targets to be screened for drug screening for cancer treatment.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



